

Abstract
Bone remodeling consists of resorption by osteoclasts (OCs) and formation by osteoblasts (OBs). Precise coordination of these activities is required for the resorbed bone to be replaced with an equal amount of new bone in order to maintain skeletal mass throughout the lifespan. This coordination of remodeling processes is referred to as the “coupling” of resorption to bone formation. In this review, we discuss the essential role for OCs in coupling resorption to bone formation, mechanisms for this coupling, and how coupling becomes less efficient or disrupted in conditions of bone loss. Lastly, we provide perspectives on targeting coupling to treat human bone disease.
Keywords: Osteoclast, coupling factor, resorption, bone remodeling, bone formation
1. Introduction
The adult skeleton undergoes bone remodeling to maintain mineral homeostasis and bone quality [1,2]. Bone remodeling consists of resorption by multi-nucleated, myeloid-derived osteoclasts (OCs) followed by formation by mesenchymal-derived osteoblasts (OBs)[3]. OCs and OBs are organized together with canopy, bone lining, and reversal cells within the basic multicellular unit (BMU)[4–6]. The term BMU helps to describe the sequential steps of bone remodeling that occur over several weeks as one individual, coordinated process [4,7]. Precise coordination of these activities is necessary to ensure replacement of bone at sites of resorption and the overall maintenance of skeletal mass. This coordination of bone remodeling activities is commonly referred to as the coupling of bone resorption to bone formation[8]. With aging and other conditions, such as gonadal hormone loss, the rate of resorption exceeds formation resulting in reduced bone mass [9]. In other pathological conditions, resorption occurs independently from subsequent bone formation leading to destructive bone loss[7]. A better understanding of the mechanisms coupling resorption to bone formation may lead to novel therapeutic strategies to maintain skeletal mass throughout the lifespan and potentially drive anabolic bone accrual in conditions of severe bone loss.
In this review, we focus specifically on the mechanisms by which OCs and resorption are coupled to subsequent bone formation. In addition, we highlight emerging areas of interest, including how the coupling of resorption to formation is impacted by systemic energy metabolism and inflammation. Lastly, we provide perspective on the therapeutic targeting of coupling mechanisms to treat human bone diseases.
2. OCs couple bone resorption to bone formation
The essential role for OCs in coupling resorption and bone formation was largely elucidated through the study of osteopetrosis. Osteopetrosis, which translates to “stone bone,” is a rare disease characterized by increased bone mass resulting from a lack of OC resorption[10,11]. Osteopetrosis patients exhibit an accumulation of bone density, which leads to altered bone structure and bone fragility. While there are several different forms of osteopetrosis, with variable degrees of clinical manifestations and severity, it can be broadly classified into one of two categories: OC-poor and OC-rich. OC-poor osteopetrosis patients exhibits few OCs due to mutations in genes required for normal OC differentiation, such as TNFSF11 and TNFRSF11A, which encode for Receptor Activator of NF-kappa-B Ligand (RANKL) and its receptor RANK, respectively[11]. These patients also have reduced OB numbers and bone formation[12]. In contrast, OC-rich osteopetrosis patients exhibit normal or increased numbers of OC that have impaired resorption capacity due to mutations in genes required for the generation of the characteristic OC ruffled border and acidification of the resorption lacunae (e.g. TCIRG1, CLCN7, OSTM1, and PLEKHM1)[11]. In contrast to OC-poor, OC-rich osteopetrosis patients exhibit normal or increased OBs and bone formation markers[13,14], showing that the presence of OCs even without functional resorption contributes to OB differentiation and bone formation.
Similarly to in humans, mutations in mice that inhibit OC differentiation or resorptive function leads to OC-poor and OC-rich osteopetrosis phenotypes, respectively. The op/op mouse model of osteopetrosis results from a spontaneous loss of function mutation to Csf1, the gene encoding macrophage-colony stimulating factor (M-CSF)[15]. M-CSF is required for macrophage and OC differentiation, and op/op mice lack OCs and bone resorption. Similar to OC-poor osteopetrosis patients, op/op mice exhibit reduced OBs and mineralization [16]. On the other hand, the oc/oc osteopetrosis mouse model results from spontaneous mutation to Tcirg1, which encodes a subunit of the vacuolar proton pump required for acidification of the resorption lacunae. Similar to TCIRG1 mutation in humans, the oc/oc mice exhibit normal or increased numbers of OCs; however these OCs have impaired bone resorption activity[17]. While oc/oc mice die 3–4 weeks after birth, transplant of fetal-liver derived hematopoietic stem cells from oc/oc mice into wild type adult mice leads to a significant reduction in bone resorption indices, but increased bone formation rate [18]. In addition to these spontaneous models of osteopetrosis, OC-poor and -rich phenotypes can be modeled through targeted deletion of specific genes required for OC differentiation or resorption, respectively [19,20].
The coupling-related effects of modulating OC numbers and/or function is also evident following treatment with pharmacologic anti-resorptive therapies. The biotherapeutic Denosumab (DMAb) is a neutralizing antibody to human RANKL that blocks OC differentiation and function[21], and drives fission of multinucleated OCs[22], overall leading to decreased OCs. Bisphosphonates (BPs) are small molecule anti-resorptive drugs that reduce the number of functional OCs on the bone surface through a variety of mechanisms [23], including effects on OC differentiation and maturation [24] and apoptosis. In addition, certain BPs also impair OC G-protein signaling and cytoskeletal functions, leading to the presence of detached OCs within the bone marrow compartment [25]. While these anti-resorptive therapies effectively reduce bone loss, they are linked to a coupling-related reduction in OBs and bone formation [26–29]- although there are also possible direct effects of certain BPs to inhibit new bone formation [29]. The coupling-related reduction in OBs following anti-resorptive therapy is also associated with blunted induction of bone formation by bone anabolic parathyroid hormone (PTH) [30–33].
On the other hand, odanacatib is an anti-resorptive drug that inhibits the OC-specific protease cathepsin K (CTSK), thus inhibiting resorption-mediated protein degradation without impairing other OC processes [21]. Loss of function mutations to CTSK in humans results in pycnodystosis, which has many similarities to OC-rich osteopetrosis[34]. Inhibition of CTSK activity via odanacatib does not reduce OCs, and unlike BPs and DMAb, OB numbers and bone formation are sustained [35,36]. Despite positive effects on bone mass, odanacatib was pulled from development during phase 3 trials due to an unexpected increase in cardiovascular events[37]. Regardless, odanacatib provides proof-of-concept pharmacological data that inhibition of resorption while maintaining functional OCs protects bone formation. Further understanding of the mechanisms linking OCs and resorption to subsequent bone formation may lead to the development of new anti-resorptives that protect and/or stimulate bone formation.
3. Mechanisms coupling bone resorption to bone formation
Several mechanisms have been proposed for the coupling of OCs and resorption to OB-mediated bone formation, and it is highly likely that multiple mechanisms act together to coordinate optimal coupling (Figure 1A). These mechanisms involve local, paracrine signals derived from OCs or bone matrix acting on cells within the bone remodeling compartment (BRC). The majority of identified coupling mediators are soluble factors; however, membrane coupling factors have also been identified. In addition to proposed coupling factors, optimal coupling may also depend on extracellular matrix (ECM) signals and surface topologies exposed or created during bone resorption.
Figure 1. Mechanisms by which OCs couple bone resorption to bone formation.
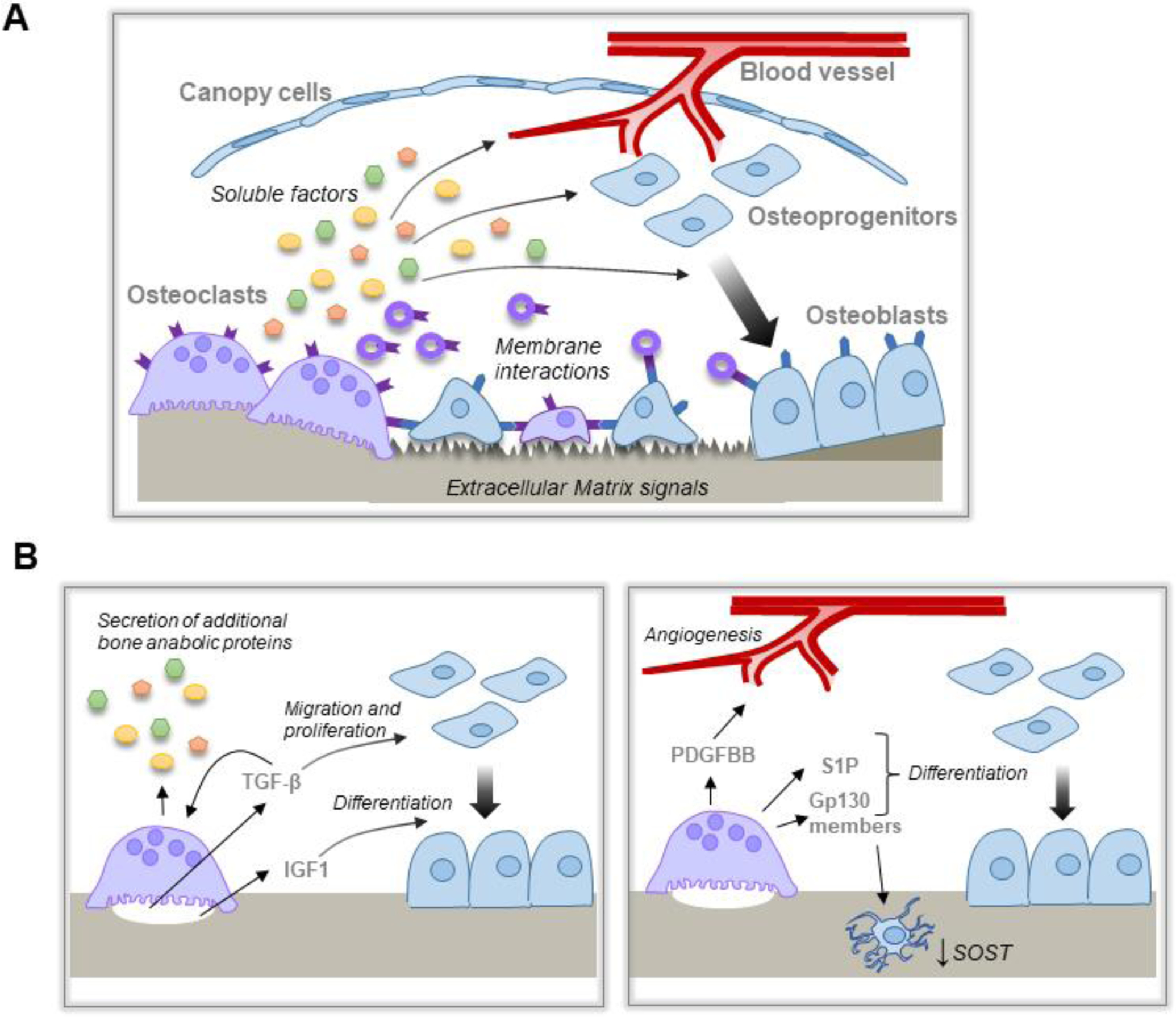
A, Schematic overview of the proposed mechanisms by which OC and resorption are coupled to bone formation. OC resorption releases matrix derived and OC-derived soluble factors and/or EVs that act on multiple cell lineages within the BRC to promote bone formation. OC membrane factors, including on EVs, interact with cognate receptors on cells within the BRC. In addition, altered bone matrix promotes optimal bone formation at sites of bone resorption. B, Matrix derived factors (left panel) released during bone resorption stimulate differential effects on osteoblast migration and proliferation vs differentiation. Matrix derived TGF-β also induces expression of other bone anabolic proteins. OC expression of additional factors (right panel) induce angiogenesis, osteoblast differentiation, and indirect effects on bone formation through the osteocytes.
3.1. Soluble factors
Soluble factors, either released from the bone matrix during resorption or secreted by OCs (Figure 1B), were the first mechanisms proposed for the coupling of resorption and formation, and these coupling factors have been reviewed extensively[7,38–40]. In one of the earliest studies investigating coupling, Howard, et al., demonstrated that PTH-induced acute resorption was followed by an extended period of bone formation. Following dialysis of conditioned media (CM), the authors revealed the presence of soluble factors larger than 12,000 Daltons derived from bone and/or OCs. CM from the resorbing cultures also stimulated bone formation in separate organ cultures[41]. While this work did not result in the identification of specific coupling factors, it provided evidence for OC resorption derived soluble factors that mediate the coupling of resorption to bone formation.
Transforming Growth Factor (TGF)-β was one of the first specific soluble factors identified coupling resorption to bone formation. Bone matrix is rich in TGF-β protein[42], in particular TGF-β1[43], which is bound with Latency Associated Peptide (LAP) and Latent TGF-β Binding Protein (LTBP)[44]. OCs release and activate TGF-β during bone resorption[45]. Matrix-derived TGF-β was first shown to act as a coupling factor by Tang, et al., who documented a low bone formation phenotype in Tgfb1 null mice that was driven by the loss of TGF-β-induced OB progenitor migration[46]. However, TGF-β also has known inhibitory effects on bone formation, suggesting additional coupling factors must promote OB differentiation and bone formation[46,47]. A second matrix derived protein released during resorption is Insulin Growth Factor 1 (IGF1)[47]. Deletion of Igf1r in OB lineage cells reduced bone formation, consistent with a role for matrix IGF1 in coupling resorption to formation. Xian, et al., further demonstrated that IGF1 signaling in OB progenitors activated mammalian target of rapamycin (mTOR) signaling and OB differentiation to drive bone formation at the site of resorption[47].
Subsequently it was proposed that matrix-derived TGF-β may also act directly on the OC lineage to induce the expression of coupling factors[48]. Treatment of mature OCs with TGF-β1 induces gene expression of several secreted factors, including Cxcl16[49], Lif[49,50], Sphk1[48], Wnt1[51], and Wnt10b[48] that have been linked to bone formation and/or coupling[52–63]. Of these genes, we confirmed induction of Wnt1 in OCs cultured on murine bone chips. Induction of Wnt1 gene expression was blocked by treatment with a TGF-β receptor inhibitor, confirming that the effects are mediated through TGF-β signaling and not an indirect effect of bone matrix[51]. TGF-β signaling activates SMAD2/3 phosphorylation; phosphorylated SMAD2 or SMAD3 bind to SMAD4, leading to nuclear translocation and regulation of gene expression. In contrast, bone morphogenetic proteins (BMPs), which belong to the TGF-β superfamily, activate SMAD1/5/8 phosphorylation. Phosphorylated SMADs bind to SMAD4 to translocate to the nucleus and regulate gene expression[64]. In the presence of TGF-β and BMP, SMAD2/3 and SMAD1/5/8 compete for available SMAD4. In this capacity, SMAD1/5 activation has been shown to antagonize OC coupling factor expression[65]. Thus, it is possible that resorption-derived TGF-β drives coupling factor expression to promote bone formation; as OBs differentiate, the concentration of BMPs increases and antagonizes TGF-β-induced coupling factor expression to limit bone formation.
While matrix derived TGF-β and IGF1 provide mechanistic links between resorption and bone formation, it is well established that OC-rich osteopetrosis patients with impaired resorption exhibit normal or increased bone formation[11,13,14]. This has been recapitulated in several mouse models of OC-rich osteopetrosis[20,66], as well as transplant models in which hematopoietic cells from OC-rich mice[67], but not OC-poor, increases bone formation[68]. Further, CM generated from OCs on plastic promotes OB differentiation and bone nodule formation in vitro, although less potently than OCs on bone [69,70]. Thus, OCs must secrete signals to promote OB differentiation independently of resorption.
Several gp130 signaling members have been identified as potential OC-derived coupling factors. The first of these was cardiotrophin (CT)-1. CT-1 is expressed by OCs, and deletion of CT-1 results in large OCs with reduced resorptive activity; in contrast to OC-rich osteopetrosis models, CT-1 deficient mice exhibit reduced bone formation. Treatment with CT-1 promotes OB differentiation and bone formation, consistent with a potential role as an OC-derived coupling factor[71]. In further support for gp130 members in coupling OCs to OBs, Fernandes, et al., found that cord blood-derived OCs promote bone formation, and this effect was ablated by neutralization of gp130 or OncostatinM (OSM)[72]. Similarly, Leukemia Inhibitory Factor (LIF), an Interleukin-6 family cytokine also secreted by OCs, drives increased bone formation[52,53]. Adult LIF knockout mice have decreased bone formation with no significant changes to OC numbers[73]. Our genetic analysis of human bone needle biopsies from placebo and DMAb treated participants revealed that LIF expression correlates with OC and OB coupling at the gene expression level, and LIF was significantly reduced in bone biopsies isolated from DMAb treated participants, consistent with a potential role in coupling[74]. In addition to directly stimulating OB progenitor migration and mitogenic activity[49,53], LIF, CT-1, and OSM may activate bone formation indirectly through modulation of osteocyte (OT) gene expression. Walker, et al., showed that treatment of the UMR 106–01 OT-like cell line with LIF, CT-1, and OSM downregulate expression of sclerostin, a Wnt signaling inhibitor[61]. Consistent with this, Koide, et al., revealed that OC-CM downregulates OT sclerostin, and this effect is mediated via LIF[75]. Mutations to the sclerostin gene, SOST, are associated with the high bone mass diseases sclerosteosis and van Buchem disease[76]; downregulation of this soluble inhibitor of Wnt signaling may thus increase Wnt-induced bone formation. Thus, OC-derived factors may also couple bone resorption to bone formation indirectly through OTs.
Another OC-derived factor proposed to couple resorption and bone formation is sphingosine-1-phosphate (S1P). S1P is a sphingolipid that is involved in several cell functions, including migration, differentiation, and apoptosis[77]. S1P is produced by sphingosine kinase (Sphk)-1 and −2. OCs have been shown to express SPHK enzymes and produce S1P to promote OB migration and survival[56,62]. A role for S1P in coupling was supported by Lotinun, et al., who showed that deletion of Ctsk in the OC lineage increases bone formation through elevated OC lineage S1P production[57]. However, the role for S1P in bone remodeling is multifaceted, including an essential role in OC precursor migration from blood vessels to bone[78,79]. Treatment of mice with S1P receptor agonist[79] or an inhibitor of S1P lyase, which increases S1P by inhibiting its degradation[80], both improve bone mass; however, these positive effects appear to be through an anti-resorptive mechanism, since mice exhibit significant reductions in OCs[79,80]. It is important to note that the mice maintain bone formation despite the reduction in OCs. Thus, while the primary function for S1P in remodeling may be as a regulator of OCs, increasing S1P may support OB and prevent a coupling-related decrease in bone formation.
In addition to OC-derived factors acting on the OB lineage, OCs also promote bone formation by driving angiogenesis to the bone remodeling compartment. Blood vessels have long been recognized as a component of the BMU[4]. These vessels deliver essential metabolic nutrients for remodeling and provide a source of stem cell progenitors for OB differentiation. Several reports demonstrated a specific role for type H vessels (CD31hiEmcnhi) in supporting osteogenesis[81,82]. Subsequently, Xie, et al., demonstrated that OC-lineage secreted Platelet-Derived Growth Factor (PDGF)-BB induces endothelial progenitor cell migration to drive angiogenesis of CD31hiEmcnhi vessels to support bone formation[83]. Conditional knockout of Pdgfb in the OC-lineage resulted in lower bone mass, along with reduced CD31hiEmcnhi vessels compared to wildtype. CD31hiEmcnhi vessels were also reduced in an ovariectomy (OVX) mouse model of post-menopausal bone loss; CD31hiEmcnhi vessels and bone formation were rescued by exogenous PDGF-BB or CTSK inhibition, which increased pre-OC derived PDGF-BB. In addition to effects on angiogenesis, Xie, et al.[83], and others have demonstrated PDGF-BB also drives chemotaxis of mesenchymal OB progenitors[84,85]. OC lineage expression of PDGFB has now been reported in human bone sections, along with expression of PDGF receptors PDGFRA and PDGFRB by mesenchymal lineage canopy and reversal cells adjacent to resorbing OCs[86]. OC-lineage expression of PDGFB appears to be downstream of G protein-coupled receptor kinase 2 interacting protein (GIT)-1, as knockout of GIT1 leads to reduced bone formation and CD31hiEmcnhi vessels through reduced pre-OC PDGF-BB[87]. PDGFB expression is also induced by TGF-β[88], suggesting a role for matrix-derived TGF-β in activating this coupling activity.
Other proposed OC-derived soluble coupling factors include BMPs[89], collagen triple helix repeat containing 1 (CTHRC1)[90], Complement 3a (C3)[91], and SLIT3[92]. Of interest, many of these are induced by treatment with TGF-β, as discussed above. This suggests that matrix-derived TGF-β may polarize OC lineage cells towards a “coupling” phenotype, leading OC to express several bone anabolic proteins concentrated within the BRC; this phenotype may then be antagonized by increasing OB-derived BMPs to prevent excess bone formation[65]. However, several of these soluble factors may be present at a level sufficient for driving coupling activity, even in the absence of bone resorption activity. In that case, increasing the presence of non-resorbing OC-lineage cells promotes coupling activity.
3.2. Membrane factors
In addition to soluble factors, cell-cell interactions between OCs and mesenchymal lineage cells via membrane factors have been proposed as mechanisms for coupling resorption to bone formation. Cell-cell interactions between OC and OB lineage cells are an established mechanism for the OB lineage to stimulate of OC differentiation. OB lineage cells express membrane bound RANKL, which activates RANK on the OC lineage cell membrane. However, resorption and formation are spatially and temporally restricted during the bone remodeling cycle[6], suggesting that OC membrane factors more likely contribute coupling-effects on reversal and/or canopy cells[7].
The first cell-cell interaction proposed for coupling of resorption to formation was the EphrinB2-EphB4 membrane interaction[93]. Zhao, et al., demonstrated OC and OB expression of Efnb2 and OB expression of Ephb4. The authors used antibodies to activate bidirectional signaling in OCs and OBs. However, myeloid lineage specific knockdown of EphrinB2 did not impact the bone phenotype, suggesting that EphrinB2 from other cells, or other Ephrin family members, compensates in the absence of OC EphrinB2[93]. Further study revealed that OB EphrinB2 activates anti-apoptotic signaling in neighboring OBs to drive bone formation[94]. Given evidence that OCs do express EphrinB2 in vivo[95], it remains possible that this cell-cell interaction contributes to anti-apoptotic effects in the OB lineage during the reversal phase.
The second membrane factor proposed for coupling resorption to formation via cell-cell interactions is semaphorin4D (Sema4D). OC lineage Sema4D activates PlexinB1 on OBs. Rather than promoting coupling, Sema4D acts as a negative regulator of OB differentiation, and knockout of either Sema4D or PlexinB1 increases bone formation and improves skeletal mass. Neutralizing antibody to Sema4D delivered either prophylactically or therapeutically similarly improved bone mass in OVX mice by increasing bone formation[96]. In humans, serum Sema4D positively correlates with markers of resorption and negatively correlates with bone mineral density in postmenopausal osteoporosis[97].
The most recent observation of membrane interactions coupling OCs and OBs is reverse signaling through the established signaling partners RANK and RANKL. Reverse signaling via RANKL is the signaling pathway activating within RANKL expressing cells following interaction with OC lineage RANK. Furuya, et al., utilized a peptide (W9) to block RANKL-induced OC differentiation and found that W9 induced bone anabolic effects in vivo. In vitro studies confirmed that this was dependent on OB RANKL expression, suggesting reverse signaling via RANKL in OBs[98]. Reverse RANKL signaling could also be activated via treatment with soluble RANK[99] and this effect was dependent on a proline-rich motif in the RANKL cytoplasmic tail. Mutation of a proline residue within this motif to alanine (Pro29Ala) prevented remodeling-induced bone formation in two models of elevated bone turnover without affecting resorption[100].
Importantly, activation of RANKL reverse signaling does not require cell-cell interactions, as RANK expressed on OC-derived extracellular vesicles (EVs) can also activate reverse signaling in RANKL expressing OBs[100–102]. EVs are membrane-derived vesicular bodies that are released into extracellular space[103]. EVs provide a mechanism for cells to communicate with both neighboring and distant cells, allowing communication of spatially restricted OCs and OBs during bone remodeling. Ikebuchi, et al., documented RANK protein on EVs derived from RAW 264.7 OC-like cells. These RANK positive EVs stimulated OB gene expression in OB cell lines and primary cultures. In vivo administration of an inhibitor of EV secretion or an antibody against a murine OC-derived EV marker (immunoglobulin superfamily member 8, IGSF8) both prevented remodeling induced bone formation, demonstrating that OC derived EVs may promote bone formation[100].
There are several subtypes of EVs classified by origin, function, and/or biogenesis. Three main classes of EVs are exosomes, microvesicles, and apoptotic bodies. Exosomes and microvesicles exhibit lipid bilayers and range in diameter from 40–120 and 50–1,000nm, respectively[103]. Exosomes more closely reflect the membrane composition of their cell of origin and exhibit a biogenesis mechanism distinct from microvesicles. Ikebuchi, et al., showed that RANK positive EVs co-precipitated with exosome markers CD9, CD63, and CD81, suggesting that these could in fact be OC-derived exosomes[100]. Apoptotic bodies (ABs) are a larger subtype of EV, ranging in diameter from 500–2,000nm, generated via blebbing of an apoptotic membrane[103]. Ma, et al., used flow cytometry to separate EVs based on size, with exosomes and microvesicles distinct from ABs[104]. Through separation of these populations, Ma, et al., demonstrated that OC-derived ABs, but not BMM or pre-OC ABs, had increased RANK content and increased ability to drive OB differentiation in vitro compared to BMM, pre-OC, and OC-derived exosomes and microvesicles. In both studies[100,104], the effects on OB differentiation were blocked through the addition of soluble RANKL which competes with OBs for binding the RANK positive EVs.
In addition to activating reverse signaling through RANKL, Liang, et al., revealed that OC-derived small EVs (~90nm size) were enriched with four miRNAs: miR128–3p, miR-324, miR-130b-ep, and miR-1187. Through in vitro overexpression in OB cell line, they found that miR-324 activated, while miR-130b-3p inhibited OB differentiation. Thus, in addition to activation of reverse RANKL signaling, RANK-RANKL membrane interaction can target microRNAs and other EV content specifically to the RANKL positive OB lineage cells to regulate OB differentiation and bone formation[105].
In contrast to the report by Liang, et al.[105], two additional studies showed that OC-derived EVs contain miR-214 and inhibits of osteogenesis[106,107]. Mice transgenic for OC-derived miR-214 exhibited reduced bone formation and bone mass; further, osteoporosis patients exhibited increased serum exosome miR-214[106,107]. Treatment with a miR-214 antagomir promoted bone formation in aging mice[106]. While these results are report a negative effect of OC-derived EVs on OB differentiation, they similarly identified OC-OB membrane interactions that may mediate the targeting of OC-derived EVs to OBs. Li, et al., show that OC-derived EVs are positive for Sema4D, and OB uptake of these EVs could be blocked via anti-Sema4D antibody[106]. Sun, et al., demonstrated an Eprhin-mediated mechanism for targeting OC-derived EVs to OBs[107]. Thus, while the overall effect of OC-derived EVs on bone formation remains uncertain and may be context dependent, the membrane factors mediating OC-OB interactions likely play a crucial role in specifying signaling cargo for target cells. The potential for OC-derived EV also extend the role for membrane factors in coupling beyond the spatial and temporal restrictions of these cellular interactions within the BRC.
3.3. Resorption effects on bone matrix
Bone is composed of both organic and inorganic extracellular matrix (ECM) that act as a physical scaffold and provide mechanical and biochemical signals to regulate cell proliferation, adhesion, and differentiation[108–110]. Changes to bone ECM through either deletion of organic protein components in vivo or in vitro modifications to scaffold content and/or surface topography has a major impact on bone formation[108]. OC resorption alters the bone ECM and surface topography[111–113]. Because of the role for ECM and surface topography in regulating OB differentiation, it is likely that OC resorption-induced changes to the bone matrix contributes to the propensity for OB to differentiate and form bone at resorption sites.
The role of surface topography in regulating OB differentiation and bone formation has been thoroughly evaluated in the context of osteointegration which depends on optimal interactions between biomedical devices and host tissue. Several studies have shown that surface roughness regulates OB proliferation, adhesion, and differentiation, with OB-like cells exhibiting decreased proliferation and increased differentiation on rough surfaces[114–117]. Animal and clinical studies support these findings, showing that rough surfaces improve long-term integration of implants as compared to smooth surfaces[118–120]. Of interest, a combination of micron and sub-micron surface roughness results in three-dimensional topographies similar to the surfaces created by OC resorption during normal remodeling[111,112,121]. Culturing of OBs on dentine or bone chips shows that osteoprogenitors preferentially differentiate and form bone at sites of mechanical defects or prior resorption[112,113], suggesting that surface properties generated may contribute to coupling of resorption to formation.
While performing gene expression analyses of human bone biopsies to identify potential OC-derived coupling factors[74], we stumbled upon a link between the bone matrix and coupling. We performed RNA-sequencing of centrifuged human bone biopsies from participants treated with a single dose of placebo or DMAb, and identified OC and OB signatures that were significantly reduced in DMAb-treated biopsies. Rank means of the OC and OB signatures showed a high degree of correlation, demonstrating OC-OB coupling at the gene expression level[74]. Although not part of our study, we postulated that correlation of OC and OB signatures across the entire expression dataset may reveal novel pathways associated with resorption and formation, respectively. As a proof-of-concept, we ran a correlation analysis of the OC and OB gene sets across a comprehensive list of 555 bone genes, compiled from bone-related gene ontology (GO) gene lists. Genes that significantly correlated with OC or OB signatures were then evaluated by iPathway Analysis, using the positive or negative slope of the correlation in place of fold change. We reasoned that pathways overlapping between OC and OB signatures are pathways involved in coupling. Further, pathways also downregulated in DMAb biopsies, in which patients exhibit a coupling-related decrease in bone formation[21,26,27], may represent specific pathways mediating the coupling of resorption and formation (Figure 2).
Figure 2. Pathways mediating the coupling of bone resorption and bone formation.
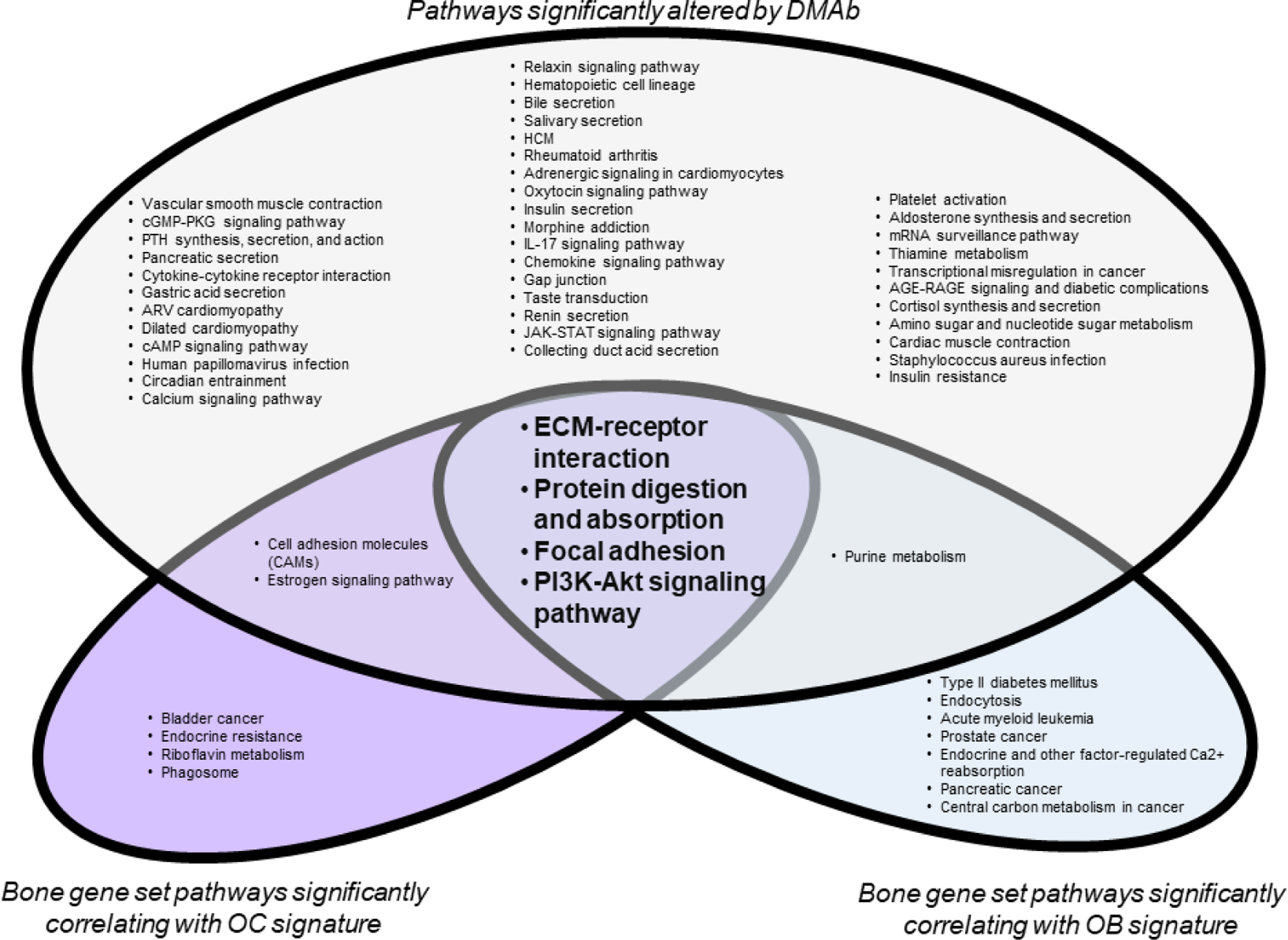
Centrifuged human bone biopsies were subjected to RNA-sequencing (GSE141614). 555 bone genes were compiled from gene ontology bone gene sets. From this, OC and OB genes downregulated by DMAb were identified. Rank means of the OC and OB gene signatures, respectively, were used to assess correlation across the comprehensive bone gene set. Significant differential gene expression (DMAb vs placebo) or correlation with OC or OB gene signatures is defined as p<0.05. Significant genes were analyze by iPathway Analysis Software (Advaita) to identify pathways significantly affected by DMAb treatment (blue), or pathways correlating with OC (red) or OB (green) gene signatures. Significant pathways are defined as p<0.10. Pathways significantly associated with DMAb differential gene expression and OC and OB gene signatures may indicate pathways associated with the coupling of OC and OB.
In all three groups, ECM-receptor interaction was the most significantly correlated pathway. While more experiments are needed, these data support that altered ECM-receptor signaling may be crucial for coupling. It is possible that with DMAb treatment, the significant reduction in OCs and resorption leads to reduced exposure of ECM and surface topographies required for optimal OB differentiation and bone formation. However, it is important to note that patients with OC-rich osteopetrosis that also lack resorption pits still show bone formation[11]. Thus, the bone surface created within the resorption pit is not a sole requirement but part of a multifaceted regulatory system together with OC lineage-derived soluble and membrane factors to promote bone formation at sites of bone resorption.
4. Importance of energy metabolism for maintaining coupling
Bone formation, along with bone remodeling as a whole, is energy expensive. Altered energy availability and impaired nutrient sensing are linked to reduced bone mass and quality and increased fracture risk. Direct inhibition of these pathways in OBs lead to reduced OB number and bone formation[47,122,123]. Thus, OB cell energetics is a central to the ability of these cells to respond to OC and resorption-derived coupling signals.
Evaluation of bone cell energetics has provided insights into the importance of energy availability and nutrient sensing for normal bone formation responses. Esen, et al., showed that Wnt3a-induced OB differentiation was accompanied by a metabolic shift to aerobic glycolysis[124], or the “Warburg effect,” defined by preferential conversion of glucose to lactate even in the presence of oxygen[125]. Aerobic glycolysis has previously been shown to occur in bone explant studies[126,127], with anabolic PTH inducing lactate production, further supporting a role for aerobic glycolysis in bone formation[128–130]. More recent studies have indicated a role for oxidative phosphorylation in OB differentiation; however, differentiated OBs are more glycolytic than undifferentiated OBs[131]. Of interest, Wnt-induced bone formation was disrupted by knockdown of glycolytic enzymes[124]. Since Wnt signaling has been shown to contribute to coupling, this raises the question of how coupling is impacted by energy availability and nutrient sensing.
We and others have recently proposed RANKL-RANK signaling as a mechanism linking bone remodeling to energy metabolism. Bonnet, et al., show that RANKL drives peripheral insulin resistance in muscle, resulting in increased serum glucose; in contrast to muscle, RANKL increases bone glucose uptake. The effects of RANKL to drive peripheral insulin resistance and increased bone glucose uptake were prevented by DMAb neutralization of RANKL[132]. Consistent with this, pre-diabetic or diabetic patients treated with DMAb, but not calcium/vitamin D or BPs, showed improved glucose control[74]. Given that RANKL drives bone remodeling, this suggests that the peripheral insulin resistance increases glucose availability for the high energy demanded for bone remodeling. OCs were also positive for dipeptidyl-peptidase (DPP)-4, which degrade incretins leading to reduced insulin secretion and sensitivity[74]; this suggests an additional mechanism by which OCs promote peripheral insulin resistance to increase glucose availability.
Glucose is a major energy source for osteoblasts[133–135]. In addition to serving as an energy source, glycolysis provides metabolic intermediates for other metabolic pathways and the biosynthesis of proteins, lipids, and nucleotides. It is also possible that glycolysis provides intermediates for generation of bone matrix. Of interest, several of these energy and biosynthetic pathways are controlled via the Phosphoinositide 3-kinase (PI3K)-Akt signaling pathway.
In evaluating pathways correlating with OC and OB bone biopsy signatures and downregulated by DMAb, we also identified PI3K-Akt signaling pathway as a potential mediator of coupling (Figure 2). There is substantial evidence for PI3K-Akt signaling pathway in skeletal development and bone remodeling. Elevated PI3K-Akt signaling at specific stages of OB lineage leads to aberrant or increased OB differentiation[136,137] and/or increased bone formation[138]. The PI3K-Akt pathway is activated by and/or interacts with bone anabolic pathways including insulin, IGF-1 and Wnt and BMP signaling pathways[139]. Several of these factors have been identified as OC or resorption-derived coupling factors[47,48,51,56]. While OCs also exhibit a high energy demand during resorption, the impact of impaired OB nutrient sensing on bone formation along with the convergence of OC and resorption-derived coupling factors on the PI3K-Akt signaling pathway highlight the importance of OB cell energetics in the coupling of resorption to formation.
5. Impact of inflammation on coupling
Despite extensive mechanisms coordinating the coupling of resorption to bone formation, the rate of resorption exceeds bone formation with aging and certain disease conditions. Moreover, conditions such as rheumatoid arthritis (RA) and bone infections exhibit resorption independent of bone formation. Given the increase in inflammation with aging, and the prominent role for inflammation in RA and bone infections, inflammation likely contributes to imbalanced resorption and formation.
Aging is associated with increased formation of aggressive, trench-forming OCs[140]. Separate studies revealed increased inflammatory OCs in autoimmune disease models[141,142], and a role for inflammasome signaling in driving resorption[143,144]. Trench forming OCs exhibit increased CTSK activity[145,146] and may itself be a population of inflammatory OCs. Given the increased acidification and CTSK activity exhibited by trench forming OCs[146], it is likely that the resulting resorption pits exhibit altered surface topographies, and potentially less optimal surfaces for OB differentiation. In addition, inflammatory OCs may exhibit altered secretomes. Thus, it seems likely that inflammatory and/or trench-forming OCs have an altered propensity to couple bone resorption to bone formation; however, this remains to be shown.
The BMU vasculature is a source of immune cells that can directly influence bone remodeling and the coupling of resorption and formation[38]. T cell involvement in bone remodeling has been well-established, and T cells can directly regulate OCs. T cells can express RANKL to induce OC differentiation and resorptive activity[147]. T cells isolated from RA patients were positive for RANKL, and other inflammatory factors, and stimulated OC differentiation[148]. In addition, interferon (IFN)-y, another T cell-derived cytokine, has inhibitory effects on OC. Disruption of T cell IFN-y signaling is associated with osteoporosis, osteomyelitis, and RA[149]. Not only can T cells induce OC activation and potentially activate aggressive resorption, T cells can also serve as a source of Wnt inhibitors to inhibit OB differentiation. Recently, T cell derived Dickkopf (DKK)-1 (Wnt inhibitor of Dickkopf-1) was shown to contribute to OVX-induced bone loss[150].
In addition to inflammatory immune populations, aging and certain diseases are associated with senescent cell accumulation that contribute to age-related pathologies[151]. Cellular senescence is defined as a terminal growth arrest in response to cell stress, such as DNA damage or oncogene activation. Senescent cells produce a senescence-associated secretory phenotype (SASP) composed of inflammatory factors such as IL-6 and TNF[151]. We identified senescent cells within the aging mouse skeleton[152], and showed that the SASP promotes OC progenitor survival and increased OC differentiation. Inhibition of the SASP or neutralization of SASP components prevented OC induction, and OCs were similarly reduced in a genetic model of senescent cell ablation. Of interest, senescent cell ablation reduced OCs, but increased bone formation[153]. This suggests that either SASP-induced OCs do not effectively couple resorption to bone formation (e.g. inflammatory subset of OCs) or alleviation of the SASP improves OB progenitor function. Related to altered OB progenitors, a recent report highlighted that aged skeletal stem cells have decreased bone forming potential, and increased propensity to drive inflammation and resorption[154]. Thus inflammation driven by aging and/or senescent cells alters OCs and OBs functions consistent with impacts on the altered coupling of resorption and formation.
5. Conclusions
OCs and bone resorption are coupled to subsequent bone formation during bone remodeling. Given the positive effects of OCs on bone formation even in the absence of resorptive activity, there is much interest in developing novel anti-resorptives to block bone degradation without reducing OCs. Odanacatib was pulled from development due to off-target risk for cardiovascular events[37]. However, if the CTSK inhibition could be localized to bone-resorbing OCs, it remains an effective strategy for reducing resorption without affecting OC number. In addition, targeting CTSK in animal models increases OC progenitors leading to increased OC-lineage derived coupling factors that protect bone formation[57,83]. As an alternative, pharmacologic agents that act on the OC lysosome, such as hydroxychloroquine[155], may also be effective to reduce CTSK protein maturation and acidification of the resorption lacunae.
The discovery of RANK positive EVs[100,104] offers the possibility of developing exosome-based therapies to promote bone formation. RANK positive exosomes or EVs could aid in the delivery of exogenous miRNAs, pharmacologic agents, or proteins specifically to RANKL positive OB progenitors. In such a case, it is theoretical that coupling activated bone formation may be possible even in the absence of OCs.
Further elucidation of the mechanisms by which OC couple resorption to bone formation, and how this is impacted by systemic energy metabolism and inflammation, may lead to novel therapeutic strategies to maintain bone mass or to stimulate bone accrual in conditions of severe bone loss. Conversely, these coupling mechanisms may also serve as targets to prevent increased bone mass in osteopetrosis.
Funding
The authors are supported by the National Institutes of Health [R01AR077538, MMW; K01AR070281, MMW; R61AR078073, MMW; P30AR069620, MMW; T32GM007315, MMD, RDA, MMW], the American Federation for Aging Research [MMW], and the University of Michigan Biointerfaces Institute. MMW has received honoraria from Amgen for lectures.
Abbreviations
- AB
apoptotic body
- BMP
bone morphogenetic protein
- BMU
basic multicellular unit
- BP
bisphosphonate
- BRC
bone remodeling compartment
- CM
conditioned media
- CT
cardiotrophin
- CTSK
Cathepsin K
- DMAb
denosumab
- EV
extracellular vesicle
- IGF
insulin growth factor
- LIF
leukemia inhibitory factor
- OB
osteoblast
- OC
osteoclast
- OSM
oncostatin M
- OT
osteocyte
- PDGF
platelet derived growth factor
- PTH
parathyroid hormone
- RANK
Receptor Activator of NFkB
- RANKL
RANK ligand
- Sema
semaphorin
- TGF
Transforming growth factor
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Armas LA & Recker RR Pathophysiology of osteoporosis: new mechanistic insights. Endocrinol Metab Clin North Am 41, 475–486 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Farlay D, et al. Bone remodeling and bone matrix quality before and after menopause in healthy women. Bone 128, 115030 (2019). [DOI] [PubMed] [Google Scholar]
- 3.Hattner R, Epker BN & Frost HM Suggested sequential mode of control of changes in cell behaviour in adult bone remodelling. Nature 206, 489–490 (1965). [DOI] [PubMed] [Google Scholar]
- 4.Parfitt AM The mechanism of coupling: a role for the vasculature. Bone 26, 319–323 (2000). [DOI] [PubMed] [Google Scholar]
- 5.Andersen TL, et al. A physical mechanism for coupling bone resorption and formation in adult human bone. Am J Pathol 174, 239–247 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Kenkre JS & Bassett J The bone remodelling cycle. Ann Clin Biochem 55, 308–327 (2018). [DOI] [PubMed] [Google Scholar]
- 7.Sims NA & Martin TJ Osteoclasts Provide Coupling Signals to Osteoblast Lineage Cells Through Multiple Mechanisms. Annu Rev Physiol 82, 507–529 (2020). [DOI] [PubMed] [Google Scholar]
- 8.Parfitt AM The coupling of bone formation to bone resorption: a critical analysis of the concept and of its relevance to the pathogenesis of osteoporosis. Metab Bone Dis Relat Res 4, 1–6 (1982). [DOI] [PubMed] [Google Scholar]
- 9.Feng X & McDonald JM Disorders of bone remodeling. Annu Rev Pathol 6, 121–145 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Stark Z & Savarirayan R Osteopetrosis. Orphanet J Rare Dis 4, 5 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Sobacchi C, Schulz A, Coxon FP, Villa A & Helfrich MH Osteopetrosis: genetics, treatment and new insights into osteoclast function. Nat Rev Endocrinol 9, 522–536 (2013). [DOI] [PubMed] [Google Scholar]
- 12.Guerrini MM, et al. Human osteoclast-poor osteopetrosis with hypogammaglobulinemia due to TNFRSF11A (RANK) mutations. Am J Hum Genet 83, 64–76 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Del Fattore A, et al. Clinical, genetic, and cellular analysis of 49 osteopetrotic patients: implications for diagnosis and treatment. J Med Genet 43, 315–325 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Alatalo SL, et al. Osteoclast-derived serum tartrate-resistant acid phosphatase 5b in Albers-Schonberg disease (type II autosomal dominant osteopetrosis). Clin Chem 50, 883–890 (2004). [DOI] [PubMed] [Google Scholar]
- 15.Marks SC Jr. & Lane PW Osteopetrosis, a new recessive skeletal mutation on chromosome 12 of the mouse. J Hered 67, 11–18 (1976). [DOI] [PubMed] [Google Scholar]
- 16.Sakagami N, et al. Reduced osteoblastic population and defective mineralization in osteopetrotic (op/op) mice. Micron 36, 688–695 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Blin-Wakkach C, Wakkach A, Sexton PM, Rochet N & Carle GF Hematological defects in the oc/oc mouse, a model of infantile malignant osteopetrosis. Leukemia 18, 1505–1511 (2004). [DOI] [PubMed] [Google Scholar]
- 18.Thudium CS, et al. A Comparison of Osteoclast-Rich and Osteoclast-Poor Osteopetrosis in Adult Mice Sheds Light on the Role of the Osteoclast in Coupling Bone Resorption and Bone Formation. Calcified Tissue International 95, 83–93 (2014). [DOI] [PubMed] [Google Scholar]
- 19.Kim N, Odgren PR, Kim DK, Marks SC Jr. & Choi Y Diverse roles of the tumor necrosis factor family member TRANCE in skeletal physiology revealed by TRANCE deficiency and partial rescue by a lymphocyte-expressed TRANCE transgene. Proc Natl Acad Sci U S A 97, 10905–10910 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Neutzsky-Wulff AV, Karsdal MA & Henriksen K Characterization of the bone phenotype in ClC-7-deficient mice. Calcif Tissue Int 83, 425–437 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Khosla S & Hofbauer LC Osteoporosis treatment: recent developments and ongoing challenges. Lancet Diabetes Endocrinol 5, 898–907 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.McDonald MM, et al. Osteoclasts recycle via osteomorphs during RANKL-stimulated bone resorption. Cell 184, 1330–1347.e1313 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Drake MT, Clarke BL & Khosla S Bisphosphonates: mechanism of action and role in clinical practice. Mayo Clin Proc 83, 1032–1045 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Evans KD, et al. Long Term Cyclic Pamidronate Reduces Bone Growth by Inhibiting Osteoclast Mediated Cartilage-to-Bone Turnover in the Mouse. Open Orthop J 2, 121–125 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Weinstein RS, Roberson PK & Manolagas SC Giant Osteoclast Formation and Long-Term Oral Bisphosphonate Therapy. New England Journal of Medicine 360, 53–62 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sánchez A, et al. Effect of Denosumab on Bone Mineral Density and Markers of Bone Turnover among Postmenopausal Women with Osteoporosis. J Osteoporos 2016, 8738959 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Cummings SR, et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N Engl J Med 361, 756–765 (2009). [DOI] [PubMed] [Google Scholar]
- 28.Naylor KE, et al. Response of bone turnover markers to three oral bisphosphonate therapies in postmenopausal osteoporosis: the TRIO study. Osteoporosis International 27, 21–31 (2016). [DOI] [PubMed] [Google Scholar]
- 29.Jensen PR, Andersen TL, Chavassieux P, Roux J-P & Delaisse J-M Bisphosphonates impair the onset of bone formation at remodeling sites. Bone 145, 115850 (2021). [DOI] [PubMed] [Google Scholar]
- 30.Black DM, et al. The Effects of Parathyroid Hormone and Alendronate Alone or in Combination in Postmenopausal Osteoporosis. New England Journal of Medicine 349, 1207–1215 (2003). [DOI] [PubMed] [Google Scholar]
- 31.Finkelstein JS, et al. The effects of parathyroid hormone, alendronate, or both in men with osteoporosis. N Engl J Med 349, 1216–1226 (2003). [DOI] [PubMed] [Google Scholar]
- 32.Leder BZ, et al. Two years of Denosumab and teriparatide administration in postmenopausal women with osteoporosis (The DATA Extension Study): a randomized controlled trial. J Clin Endocrinol Metab 99, 1694–1700 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Delmas PD, et al. The anabolic effect of human PTH (1–34) on bone formation is blunted when bone resorption is inhibited by the bisphosphonate tiludronate—is activated resorption a prerequisite for the in vivo effect of PTH on formation in a remodeling system? Bone 16, 603–610 (1995). [DOI] [PubMed] [Google Scholar]
- 34.Ho N, et al. Mutations of CTSK Result in Pycnodysostosis via a Reduction in Cathepsin K Protein. Journal of Bone and Mineral Research 14, 1649–1653 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Recker R, et al. Effects of Odanacatib on Bone Structure and Quality in Postmenopausal Women With Osteoporosis: 5-Year Data From the Phase 3 Long-Term Odanacatib Fracture Trial (LOFT) and its Extension. J Bone Miner Res 35, 1289–1299 (2020). [DOI] [PubMed] [Google Scholar]
- 36.Langdahl B, et al. Odanacatib in the treatment of postmenopausal women with low bone mineral density: Five years of continued therapy in a phase 2 study. Journal of Bone and Mineral Research 27, 2251–2258 (2012). [DOI] [PubMed] [Google Scholar]
- 37.McClung MR, et al. Odanacatib for the treatment of postmenopausal osteoporosis: results of the LOFT multicentre, randomised, double-blind, placebo-controlled trial and LOFT Extension study. Lancet Diabetes Endocrinol 7, 899–911 (2019). [DOI] [PubMed] [Google Scholar]
- 38.Sims NA & Martin TJ Coupling the activities of bone formation and resorption: a multitude of signals within the basic multicellular unit. Bonekey Rep 3, 481 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Martin TJ & Sims NA Osteoclast-derived activity in the coupling of bone formation to resorption. Trends Mol Med 11, 76–81 (2005). [DOI] [PubMed] [Google Scholar]
- 40.Henriksen K, Karsdal MA & Martin TJ Osteoclast-derived coupling factors in bone remodeling. Calcif Tissue Int 94, 88–97 (2014). [DOI] [PubMed] [Google Scholar]
- 41.Howard GA, Bottemiller BL, Turner RT, Rader JI & Baylink DJ Parathyroid hormone stimulates bone formation and resorption in organ culture: evidence for a coupling mechanism. Proc Natl Acad Sci U S A 78, 3204–3208 (1981). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Bonewald LF & Mundy GR Role of transforming growth factor-beta in bone remodeling. Clin Orthop Relat Res, 261–276 (1990). [PubMed] [Google Scholar]
- 43.Janssens K, ten Dijke P, Janssens S & Van Hul W Transforming growth factor-beta1 to the bone. Endocr Rev 26, 743–774 (2005). [DOI] [PubMed] [Google Scholar]
- 44.Robertson IB, et al. Latent TGF-β-binding proteins. Matrix Biol 47, 44–53 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Oursler MJ Osteoclast synthesis and secretion and activation of latent transforming growth factor beta. J Bone Miner Res 9, 443–452 (1994). [DOI] [PubMed] [Google Scholar]
- 46.Tang Y, et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat Med 15, 757–765 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Xian L, et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat Med 18, 1095–1101 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ota K, et al. TGF-β induces Wnt10b in osteoclasts from female mice to enhance coupling to osteoblasts. Endocrinology 154, 3745–3752 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ota K, et al. Transforming growth factor beta 1 induces CXCL16 and leukemia inhibitory factor expression in osteoclasts to modulate migration of osteoblast progenitors. Bone 57, 68–75 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ruan M, Pederson L, Bradley EW, Bamberger AM & Oursler MJ Transforming growth factor-{beta} coordinately induces suppressor of cytokine signaling 3 and leukemia inhibitory factor to suppress osteoclast apoptosis. Endocrinology 151, 1713–1722 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Weivoda MM, et al. Osteoclast TGF-β Receptor Signaling Induces Wnt1 Secretion and Couples Bone Resorption to Bone Formation. J Bone Miner Res 31, 76–85 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Cornish J, Callon K, King A, Edgar S & Reid IR The effect of leukemia inhibitory factor on bone in vivo. Endocrinology 132, 1359–1366 (1993). [DOI] [PubMed] [Google Scholar]
- 53.Cornish J, Callon KE, Edgar SG & Reid IR Leukemia inhibitory factor is mitogenic to osteoblasts. Bone 21, 243–247 (1997). [DOI] [PubMed] [Google Scholar]
- 54.Fahiminiya S, et al. Mutations in WNT1 are a cause of osteogenesis imperfecta. J Med Genet 50, 345–348 (2013). [DOI] [PubMed] [Google Scholar]
- 55.Keupp K, et al. Mutations in WNT1 cause different forms of bone fragility. Am J Hum Genet 92, 565–574 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Pederson L, Ruan M, Westendorf JJ, Khosla S & Oursler MJ Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc Natl Acad Sci U S A 105, 20764–20769 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Lotinun S, et al. Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J Clin Invest 123, 666–681 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Joeng KS, et al. The swaying mouse as a model of osteogenesis imperfecta caused by WNT1 mutations. Hum Mol Genet 23, 4035–4042 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Joeng KS, et al. Osteocyte-specific WNT1 regulates osteoblast function during bone homeostasis. J Clin Invest 127, 2678–2688 (2017) [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Palomo T, et al. Skeletal characteristics associated with homozygous and heterozygous WNT1 mutations. Bone 67, 63–70 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Walker EC, et al. Oncostatin M promotes bone formation independently of resorption when signaling through leukemia inhibitory factor receptor in mice. J Clin Invest 120, 582–592 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Ryu J, et al. Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. Embo j 25, 5840–5851 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bennett CN, et al. Wnt10b Increases Postnatal Bone Formation by Enhancing Osteoblast Differentiation. Journal of Bone and Mineral Research 22, 1924–1932 (2007). [DOI] [PubMed] [Google Scholar]
- 64.Goumans MJ & Mummery C Functional analysis of the TGFbeta receptor/Smad pathway through gene ablation in mice. Int J Dev Biol 44, 253–265 (2000). [PubMed] [Google Scholar]
- 65.Tasca A, et al. SMAD1/5 signaling in osteoclasts regulates bone formation via coupling factors. PLoS One 13, e0203404 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Marzia M, et al. Decreased c-Src expression enhances osteoblast differentiation and bone formation. J Cell Biol 151, 311–320 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Henriksen K, et al. Dissociation of bone resorption and bone formation in adult mice with a non-functional VATPase in osteoclasts leads to increased bone strength. PLoS One 6, e27482 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Thudium CS, et al. A comparison of osteoclast-rich and osteoclast-poor osteopetrosis in adult mice sheds light on the role of the osteoclast in coupling bone resorption and bone formation. Calcif Tissue Int 95, 83–93 (2014). [DOI] [PubMed] [Google Scholar]
- 69.Karsdal MA, Neutzsky-Wulff AV, Dziegiel MH, Christiansen C & Henriksen K Osteoclasts secrete non-bone derived signals that induce bone formation. Biochem Biophys Res Commun 366, 483–488 (2008). [DOI] [PubMed] [Google Scholar]
- 70.Henriksen K, et al. A specific subtype of osteoclasts secretes factors inducing nodule formation by osteoblasts. Bone 51, 353–361 (2012). [DOI] [PubMed] [Google Scholar]
- 71.Walker EC, et al. Cardiotrophin-1 is an osteoclast-derived stimulus of bone formation required for normal bone remodeling. J Bone Miner Res 23, 2025–2032 (2008). [DOI] [PubMed] [Google Scholar]
- 72.Fernandes TJ, et al. Cord Blood-Derived Macrophage-Lineage Cells Rapidly Stimulate Osteoblastic Maturation in Mesenchymal Stem Cells in a Glycoprotein-130 Dependent Manner. PLOS ONE 8, e73266 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Poulton IJ, McGregor NE, Pompolo S, Walker EC & Sims NA Contrasting roles of leukemia inhibitory factor in murine bone development and remodeling involve region-specific changes in vascularization. J Bone Miner Res 27, 586–595 (2012). [DOI] [PubMed] [Google Scholar]
- 74.Weivoda MM, et al. Identification of osteoclast-osteoblast coupling factors in humans reveals links between bone and energy metabolism. Nat Commun 11, 87 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Koide M, et al. Bone Formation Is Coupled to Resorption Via Suppression of Sclerostin Expression by Osteoclasts. Journal of Bone and Mineral Research 32, 2074–2086 (2017). [DOI] [PubMed] [Google Scholar]
- 76.van Lierop AH, Appelman-Dijkstra NM & Papapoulos SE Sclerostin deficiency in humans. Bone 96, 51–62 (2017). [DOI] [PubMed] [Google Scholar]
- 77.Spiegel S & Milstien S Sphingosine-1-phosphate: an enigmatic signalling lipid. Nat Rev Mol Cell Biol 4, 397–407 (2003). [DOI] [PubMed] [Google Scholar]
- 78.Tian J, et al. Sphingosine 1-phosphate and osteoporosis: pathophysiology and therapeutic aspects-a narrative review. Ann Palliat Med 10, 4799–4805 (2021). [DOI] [PubMed] [Google Scholar]
- 79.Ishii M, et al. Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature 458, 524–528 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Weske S, et al. Targeting sphingosine-1-phosphate lyase as an anabolic therapy for bone loss. Nat Med 24, 667–678 (2018). [DOI] [PubMed] [Google Scholar]
- 81.Kusumbe AP, Ramasamy SK & Adams RH Coupling of angiogenesis and osteogenesis by a specific vessel subtype in bone. Nature 507, 323–328 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Ramasamy SK, Kusumbe AP, Wang L & Adams RH Endothelial Notch activity promotes angiogenesis and osteogenesis in bone. Nature 507, 376–380 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Xie H, et al. PDGF-BB secreted by preosteoclasts induces angiogenesis during coupling with osteogenesis. Nat Med 20, 1270–1278 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Sanchez-Fernandez MA, Gallois A, Riedl T, Jurdic P & Hoflack B Osteoclasts Control Osteoblast Chemotaxis via PDGF-BB/PDGF Receptor Beta Signaling. PLOS ONE 3, e3537 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Kreja L, et al. Non-resorbing osteoclasts induce migration and osteogenic differentiation of mesenchymal stem cells. J Cell Biochem 109, 347–355 (2010). [DOI] [PubMed] [Google Scholar]
- 86.Brun J, et al. PDGF Receptor Signaling in Osteoblast Lineage Cells Controls Bone Resorption Through Upregulation of Csf1 Expression. J Bone Miner Res 35, 2458–2469 (2020). [DOI] [PubMed] [Google Scholar]
- 87.Xu T, et al. GIT1 is critical for formation of the CD31(hi)Emcn(hi) vessel subtype in coupling osteogenesis with angiogenesis via modulating preosteoclasts secretion of PDGF-BB. Bone 122, 218–230 (2019). [DOI] [PubMed] [Google Scholar]
- 88.Bruna A, et al. High TGFbeta-Smad activity confers poor prognosis in glioma patients and promotes cell proliferation depending on the methylation of the PDGF-B gene. Cancer Cell 11, 147–10 (2007). [DOI] [PubMed] [Google Scholar]
- 89.Garimella R, et al. Expression and synthesis of bone morphogenetic proteins by osteoclasts: a possible path to anabolic bone remodeling. J Histochem Cytochem 56, 569–577 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Takeshita S, et al. Osteoclast-secreted CTHRC1 in the coupling of bone resorption to formation. J Clin Invest 123, 3914–3924 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Matsuoka K, Park KA, Ito M, Ikeda K & Takeshita S Osteoclast-derived complement component 3a stimulates osteoblast differentiation. J Bone Miner Res 29, 1522–1530 (2014). [DOI] [PubMed] [Google Scholar]
- 92.Kim B-J, et al. Osteoclast-secreted SLIT3 coordinates bone resorption and formation. The Journal of Clinical Investigation 128, 1429–1441 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Zhao C, et al. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab 4, 111–121 (2006). [DOI] [PubMed] [Google Scholar]
- 94.Tonna S, et al. EphrinB2 signaling in osteoblasts promotes bone mineralization by preventing apoptosis. Faseb j 28, 4482–4496 (2014). [DOI] [PubMed] [Google Scholar]
- 95.Allan EH, et al. EphrinB2 regulation by PTH and PTHrP revealed by molecular profiling in differentiating osteoblasts. J Bone Miner Res 23, 1170–1181 (2008). [DOI] [PubMed] [Google Scholar]
- 96.Negishi-Koga T, et al. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat Med 17, 1473–1480 (2011) [DOI] [PubMed] [Google Scholar]
- 97.Zhang Y, et al. Serum Sema4D levels are associated with lumbar spine bone mineral density and bone turnover markers in patients with postmenopausal osteoporosis. Int J Clin Exp Med 8, 16352–16357 (2015). [PMC free article] [PubMed] [Google Scholar]
- 98.Furuya Y, et al. Stimulation of bone formation in cortical bone of mice treated with a receptor activator of nuclear factor-κB ligand (RANKL)-binding peptide that possesses osteoclastogenesis inhibitory activity. J Biol Chem 288, 5562–5571 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Zhang S, et al. Osteoclast regulation of osteoblasts via RANK-RANKL reverse signal transduction in vitro. Mol Med Rep 16, 3994–4000 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Ikebuchi Y, et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature 561, 195–200 (2018). [DOI] [PubMed] [Google Scholar]
- 101.Huynh N, et al. Characterization of Regulatory Extracellular Vesicles from Osteoclasts. J Dent Res 95, 673–679 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Holliday LS, Patel SS & Rody WJ Jr. RANKL and RANK in extracellular vesicles: surprising new players in bone remodeling. Extracell Vesicles Circ Nucl Acids 2, 18–28 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.El Andaloussi S, Mäger I, Breakefield XO & Wood MJA Extracellular vesicles: biology and emerging therapeutic opportunities. Nature Reviews Drug Discovery 12, 347–357 (2013). [DOI] [PubMed] [Google Scholar]
- 104.Ma Q, et al. Mature osteoclast-derived apoptotic bodies promote osteogenic differentiation via RANKL-mediated reverse signaling. J Biol Chem 294, 11240–11247 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Liang M, et al. Osteoclast-derived small extracellular vesicles induce osteogenic differentiation via inhibiting ARHGAP1. Mol Ther Nucleic Acids 23, 1191–1203 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Li D, et al. Osteoclast-derived exosomal miR-214–3p inhibits osteoblastic bone formation. Nature Communications 7, 10872 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Sun W, et al. Osteoclast-derived microRNA-containing exosomes selectively inhibit osteoblast activity. Cell Discovery 2, 16015 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Lin X, Patil S, Gao Y-G & Qian A The Bone Extracellular Matrix in Bone Formation and Regeneration. Frontiers in Pharmacology 11(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Engler AJ, Sen S, Sweeney HL & Discher DE Matrix Elasticity Directs Stem Cell Lineage Specification. Cell 126, 677–689 (2006). [DOI] [PubMed] [Google Scholar]
- 110.Hwang MP, et al. Approximating bone ECM: Crosslinking directs individual and coupled osteoblast/osteoclast behavior. Biomaterials 103, 22–32 (2016). [DOI] [PubMed] [Google Scholar]
- 111.Hefti T, Frischherz M, Spencer ND, Hall H & Schlottig F A comparison of osteoclast resorption pits on bone with titanium and zirconia surfaces. Biomaterials 31, 7321–7331 (2010). [DOI] [PubMed] [Google Scholar]
- 112.Boyan BD, et al. Pretreatment of bone with osteoclasts affects phenotypic expression of osteoblast-like cells. J Orthop Res 21, 638–647 (2003). [DOI] [PubMed] [Google Scholar]
- 113.Gray C, Boyde A & Jones SJ Topographically induced bone formation in vitro: Implications for bone implants and bone grafts. Bone 18, 115–123 (1996). [DOI] [PubMed] [Google Scholar]
- 114.Martin JY, et al. Effect of titanium surface roughness on proliferation, differentiation, and protein synthesis of human osteoblast-like cells (MG63). J Biomed Mater Res 29, 389–401 (1995). [DOI] [PubMed] [Google Scholar]
- 115.Zhao G, Raines AL, Wieland M, Schwartz Z & Boyan BD Requirement for both micron- and submicron scale structure for synergistic responses of osteoblasts to substrate surface energy and topography. Biomaterials 28, 2821–2829 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Boyan BD, et al. Titanium surface roughness alters responsiveness of MG63 osteoblast-like cells to 1 alpha,25-(OH)2D3. J Biomed Mater Res 39, 77–85 (1998). [DOI] [PubMed] [Google Scholar]
- 117.Schwartz Z, et al. Implant surface characteristics modulate differentiation behavior of cells in the osteoblastic lineage. Adv Dent Res 13, 38–48 (1999). [DOI] [PubMed] [Google Scholar]
- 118.Klokkevold PR, Nishimura RD, Adachi M & Caputo A Osseointegration enhanced by chemical etching of the titanium surface. A torque removal study in the rabbit. Clin Oral Implants Res 8, 442–447 (1997). [DOI] [PubMed] [Google Scholar]
- 119.Khang W, Feldman S, Hawley CE & Gunsolley J A multi-center study comparing dual acid-etched and machined-surfaced implants in various bone qualities. J Periodontol 72, 1384–1390 (2001). [DOI] [PubMed] [Google Scholar]
- 120.Yang GL, He FM, Yang XF, Wang XX & Zhao SF Bone responses to titanium implants surface-roughened by sandblasted and double etched treatments in a rabbit model. Oral Surg Oral Med Oral Pathol Oral Radiol Endod 106, 516–524 (2008). [DOI] [PubMed] [Google Scholar]
- 121.Davies JE Bone bonding at natural and biomaterial surfaces. Biomaterials 28, 5058–5067 (2007). [DOI] [PubMed] [Google Scholar]
- 122.Ferron M, et al. Insulin signaling in osteoblasts integrates bone remodeling and energy metabolism. Cell 142, 296–308 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Riddle RC, et al. Tsc2 is a molecular checkpoint controlling osteoblast development and glucose homeostasis. Mol Cell Biol 34, 1850–1862 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Esen E, et al. WNT-LRP5 signaling induces Warburg effect through mTORC2 activation during osteoblast differentiation. Cell Metab 17, 745–755 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Warburg O On the origin of cancer cells. Science 123, 309–314 (1956). [DOI] [PubMed] [Google Scholar]
- 126.Borle AB, Nichols N & Nichols G Metabolic Studies of Bone in Vitro: I. NORMAL BONE. Journal of Biological Chemistry 235, 1206–1210 (1960). [PubMed] [Google Scholar]
- 127.Cohn DV & Forscher BK Aerobic Metabolism of Glucose by Bone. Journal of Biological Chemistry 237, 615–618 (1962). [PubMed] [Google Scholar]
- 128.Borle AB, Nichols N & Nichols G Jr. Metabolic studies of bone in vitro. II. The metabolic patterns of accretion and resorption. J Biol Chem 235, 1211–1214 (1960). [PubMed] [Google Scholar]
- 129.Neuman WF, Neuman MW & Brommage R Aerobic glycolysis in bone: lactate production and gradients in calvaria. Am J Physiol 234, C41–50 (1978). [DOI] [PubMed] [Google Scholar]
- 130.Rodan GA, Rodan SB & Marks SC Jr. Parathyroid hormone stimulation of adenylate cyclase activity and lactic acid accumulation in calvaria of osteopetrotic (ia) rats. Endocrinology 102, 1501–1505 (1978). [DOI] [PubMed] [Google Scholar]
- 131.Guntur AR, Le PT, Farber CR & Rosen CJ Bioenergetics during calvarial osteoblast differentiation reflect strain differences in bone mass. Endocrinology 155, 1589–1595 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bonnet N, Bourgoin L, Biver E, Douni E & Ferrari S RANKL inhibition improves muscle strength and insulin sensitivity and restores bone mass. J Clin Invest 129, 3214–3223 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Zoch ML, Abou DS, Clemens TL, Thorek DLJ & Riddle RC In vivo radiometric analysis of glucose uptake and distribution in mouse bone. Bone Res 4, 16004–16004 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Wei J, et al. Glucose Uptake and Runx2 Synergize to Orchestrate Osteoblast Differentiation and Bone Formation. Cell 161, 1576–1591 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Lee WC, Ji X, Nissim I & Long F Malic Enzyme Couples Mitochondria with Aerobic Glycolysis in Osteoblasts. Cell Rep 32, 108108 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Ford-Hutchinson AF, et al. Inactivation of Pten in osteo-chondroprogenitor cells leads to epiphyseal growth plate abnormalities and skeletal overgrowth. J Bone Miner Res 22, 1245–1259 (2007). [DOI] [PubMed] [Google Scholar]
- 137.Guntur AR, Reinhold MI, Cuellar J Jr. & Naski MC Conditional ablation of Pten in osteoprogenitors stimulates FGF signaling. Development 138, 1433–1444 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Liu X, et al. Lifelong accumulation of bone in mice lacking Pten in osteoblasts. Proc Natl Acad Sci U S A 104, 2259–2264 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.McGonnell IM, Grigoriadis AE, Lam EW, Price JS & Sunters A A specific role for phosphoinositide 3-kinase and AKT in osteoblasts? Front Endocrinol (Lausanne) 3, 88 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Møller AMJ, et al. Aging and menopause reprogram osteoclast precursors for aggressive bone resorption. Bone Res 8, 27 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Madel MB, et al. Dissecting the phenotypic and functional heterogeneity of mouse inflammatory osteoclasts by the expression of Cx3cr1. Elife 9(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Ibáñez L, et al. Inflammatory Osteoclasts Prime TNFα-Producing CD4(+) T Cells and Express CX(3) CR1. J Bone Miner Res 31, 1899–1908 (2016). [DOI] [PubMed] [Google Scholar]
- 143.Alippe Y, et al. Bone matrix components activate the NLRP3 inflammasome and promote osteoclast differentiation. Sci Rep 7, 6630 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Chen Y, et al. NLRP3 regulates alveolar bone loss in ligature-induced periodontitis by promoting osteoclastic differentiation. Cell Prolif 54, e12973 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Borggaard XG, Pirapaharan DC, Delaissé JM & Søe K Osteoclasts’ Ability to Generate Trenches Rather Than Pits Depends on High Levels of Active Cathepsin K and Efficient Clearance of Resorption Products. Int J Mol Sci 21(2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Merrild DM, et al. Erratum: Pit- and trench-forming osteoclasts: a distinction that matters. Bone Res 4, 16006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Takayanagi H, et al. T-cell-mediated regulation of osteoclastogenesis by signalling cross-talk between RANKL and IFN-gamma. Nature 408, 600–605 (2000). [DOI] [PubMed] [Google Scholar]
- 148.Miranda-Carús ME, et al. Peripheral blood T lymphocytes from patients with early rheumatoid arthritis express RANKL and interleukin-15 on the cell surface and promote osteoclastogenesis in autologous monocytes. Arthritis Rheum 54, 1151–1164 (2006). [DOI] [PubMed] [Google Scholar]
- 149.Komatsu N & Takayanagi H Immune-bone interplay in the structural damage in rheumatoid arthritis. Clinical & Experimental Immunology 194, 1–8 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lehmann J, et al. Mice lacking DKK1 in T cells exhibit high bone mass and are protected from estrogen-deficiency-induced bone loss. iScience 24, 102224 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Tchkonia T, Zhu Y, van Deursen J, Campisi J & Kirkland JL Cellular senescence and the senescent secretory phenotype: therapeutic opportunities. J Clin Invest 123, 966–972 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Farr JN, et al. Identification of Senescent Cells in the Bone Microenvironment. J Bone Miner Res 31, 1920–1929 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Farr JN, et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med 23, 1072–1079 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Ambrosi TH, et al. Aged skeletal stem cells generate an inflammatory degenerative niche. Nature (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Both T, et al. Hydroxychloroquine affects bone resorption both in vitro and in vivo. J Cell Physiol 233, 1424–1433 (2018). [DOI] [PubMed] [Google Scholar]