

Abstract
Genetic alterations affecting transforming growth Factor–β (TGF-β) signaling are exceptionally common in diseases and cancers of the gastrointestinal system. As a regulator of tissue renewal, TGF-β signaling and the downstream SMAD-dependent transcriptional events play complex roles in the transition from a noncancerous disease state to cancer in the gastrointestinal tract, liver, and pancreas. Furthermore, this pathway also regulates the stromal cells and the immune system, which may contribute to evasion of the tumors from immune-mediated elimination. Here, we review the involvement of the TGF-β pathway mediated by the transcriptional regulators SMADs in disease progression to cancer in the digestive system. The review integrates human genomic studies with animal models that provide clues toward understanding and managing the complexity of the pathway in disease and cancer.
Keywords: Transforming Growth Factor–β, Digestive System, Cancers
Transforming growth Factor–β (TGF-β) signaling is crucial to homeostasis of epithelial cells, stromal compartments, and immune cells in the liver, pancreas, and gastrointestinal (GI) system. Altered activity contributes to the development of tissue-specific diseases and progression to malignancy. The importance of TGF-β signaling in digestive system health is epitomized by the high frequency of genomic alterations in genes encoding proteins in the TGF-β pathways in cancers of the digestive system: 40% of GI cancers have such alterations,1 38% of liver cancers,2 and 50% of pancreatic cancers.3
Multiple types of cells both produce and respond to TGF-β, creating a complex network that involves epithelial cells, tumor cells, immune cells, and stromal fibroblasts. This complex network both contributes to disease and changes over time, enabling TGF-β to have both tumor-suppressing and tumor-promoting or tumor-enabling roles. We focus on TGF-β signaling through SMAD proteins (Figure 1). Of the more than 30 members of the TGF-β superfamily, the TGF-β and bone morphogenetic protein (BMP) families are particularly relevant. The 12 serine/threonine kinase receptors form a heterotetrameric complex consisting of two type I receptors and two type II receptors; each receptor complex phosphorylates a specific set of SMADs. Phosphorylated SMADs form complexes, which translocate into the nucleus to regulate target gene expression. TGF-β1, TGF-β2, and TGF-β3, the TGF-β sub-family, signal through receptor complexes comprised of TGFBR1 and TGFBR2 to phosphorylate SMAD2 and SMAD3.
Figure 1.
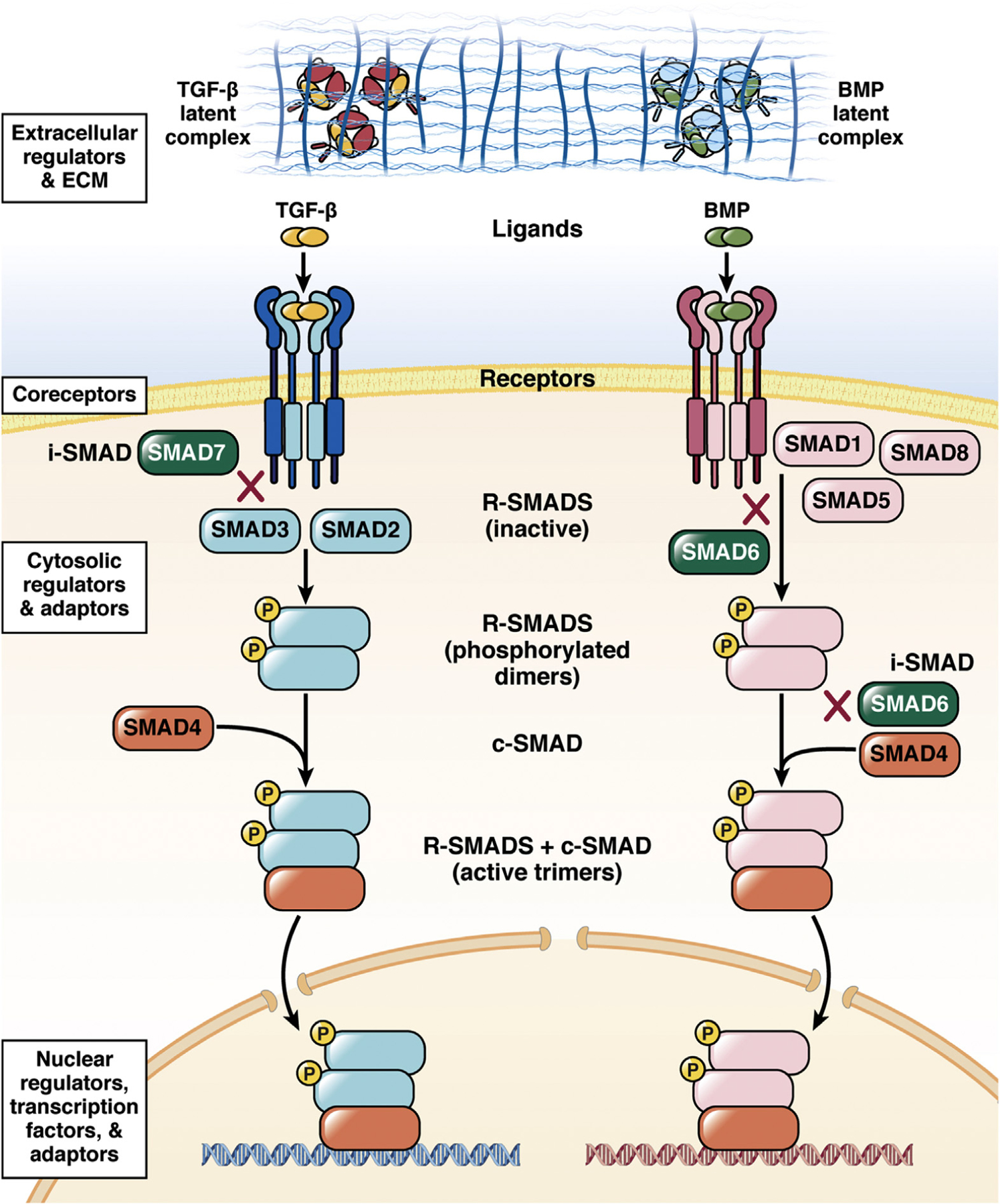
TGF-β signaling pathways. A simplified view of the core of the TGF-β and BMP pathways. X indicates an inhibitory interaction. The ligands are retained as inactive procomplexes, often associated with the ECM, before activation. The classes of various regulators are indicated at each level of the pathways in white boxes.
TGF-β proteins are secreted as inactive procomplexes consisting of the TGF-β dimer and latency-associated peptide. Release of active TGF-β can occur through either proteolytic mechanisms or allosteric mechanisms. Although the interaction of the procomplex with latent TGFb binding proteins results in deposition in the extracellular matrix (ECM);4,5 association with glycoprotein-A repetitions predominant protein enables the procomplexes to bind to the cell surface of various immune cells.6 Activation of the latent procomplexes can involve proteolysis of the ECM or allosteric interactions, such as those mediated by integrins.7
SMADs represent a regulatory family: 5 receptor-activated SMADs, SMAD1, SMAD2, SMAD3, SMAD5, and SMAD9 (also referred to as SMAD8); 1 co-mediator SMAD, SMAD4, which forms complexes with the receptor-activated SMAD dimers; and 2 inhibitory SMADs, which block SMAD activation or signaling. In addition to ligand-activated receptors, other proteins influence the activity of receptor-activated SMADs. SMAD-mediated transcriptional activity and target gene selectivity are regulated by various adaptors in both the cytoplasm and nucleus. Active SMAD complexes interact with other proteins to achieve target gene specificity and context-specific outcomes. TGF-β/SMAD signaling networks are complex, context-dependent, and critical for adult tissue homeostasis and immune function.
Transforming Growth Factor–β/SMAD Signaling in Diseases of the Digestive System and the Progression to Cancer
Throughout the digestive system, altered function of TGF-β is implicated in disease progression to cancer. The main text describes the roles of TGF-β1, TGF-β2, and TGF-β3; the Supplementary Material discusses BMP signals and celiac disease and includes a comprehensive mouse models table (Supplementary Table 1), and table of reviews (Supplementary Table 2).
Barrett’s Esophagus and Esophageal Cancer
Barrett’s esophagus (BE) results from the replacement of squamous epithelium by precancerous tissue (dysplasia). BE is often associated with gastroesophageal reflux disease, is more common in men, has an increasing incidence, and is associated with increased risk of developing esophageal adenocarcinoma (EAC).8 Impairment of TGF-β signaling is indicated by markedly reduced SMAD4 expression in all stages of BE from metaplasia to both low- and high-grade dysplasia, and the expressions of TGFBR2, TGFB1, and SMAD4 are lower in EAC compared to normal esophagus.9,10 In >80% of BE and EAC samples, signaling by TGF-β is impaired, and genomic alterations in members of the superfamily of TGF-β pathway components occur in 65% of EAC.1 Frequently altered in BE and EAC are TP53 (encoding the tumor suppressor p53) and CDKN2A (encoding the cell cycle inhibitor p16). The combination of such driver mutations with loss of function of members of the TGF-β pathways may predispose BE to transition to cancer (Figure 2).1
Figure 2.
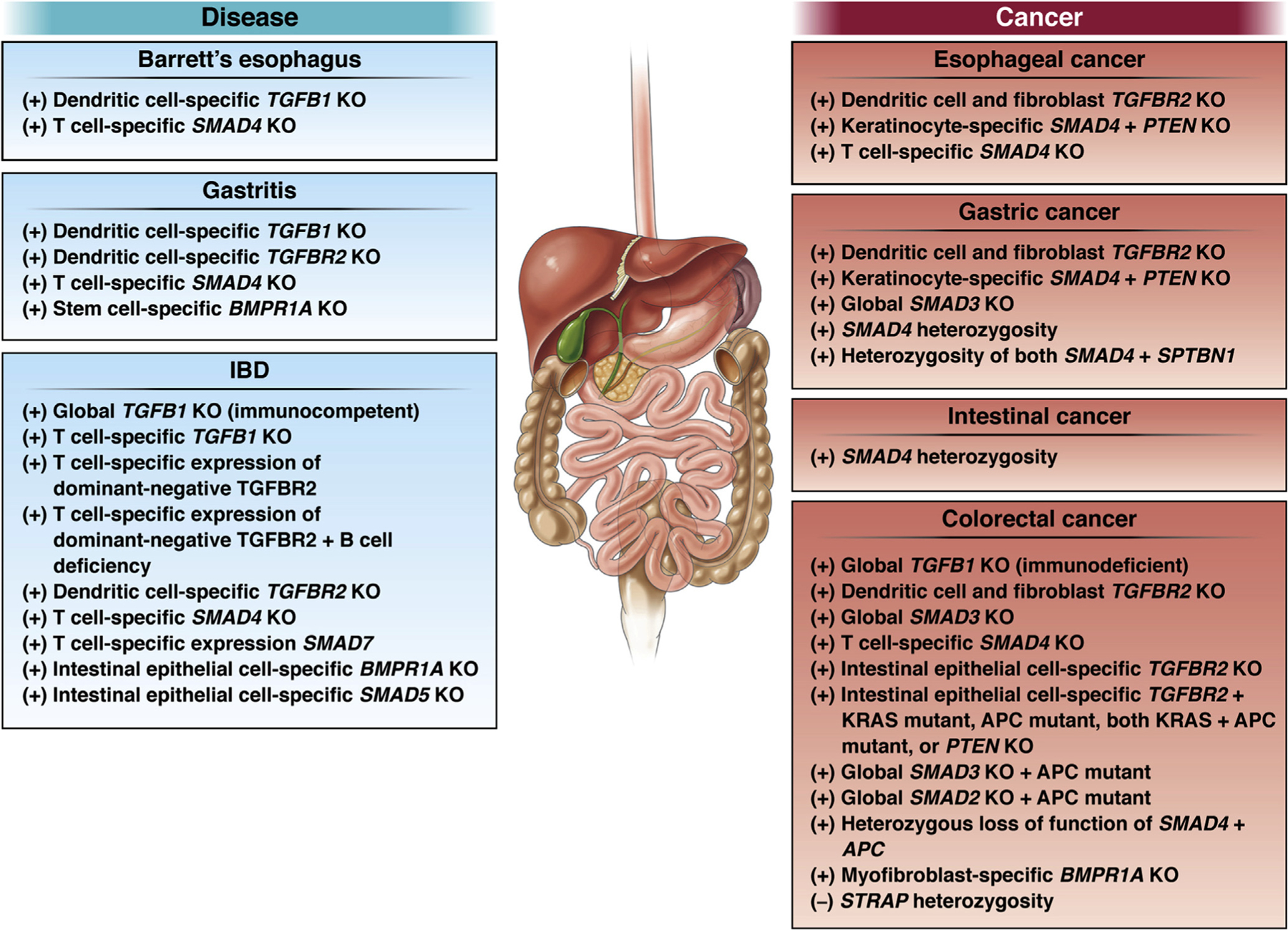
GI tract with relevant mouse models for investigating TGF-β signaling in disease or cancer. (+), increased susceptibility to disease or cancer; (–), decreased susceptibility.
For SMAD4, loss of heterozygosity of region containing SMAD2 and SMAD4 on chromosome 18q is common in approximately 70% of EACs associated with BE.11,12 This loss of heterozygosity occurs in approximately 30%–70% of patients with premalignant BE, suggesting that this is an early event in neoplastic transformation.12 Methylation of the SMAD4 promoter is another mechanism of reducing SMAD4 expression in both BE and EAC.9 SMAD4 genomic alterations are more common in EAC (24%) than in esophageal squamous cell carcinoma (8%).
A switch in SMAD3-regulated genes in response to loss of SPTBN113 suggests that loss of SMAD4 could also induce a switch in target gene regulation to induce a neoplastic transcriptional profile. Consistent with a target gene-switching mechanism, analysis of transcript abundance biopsy tissue from EAC patients reveals enhanced expression of genes in either 1 or both of 2 pathways in approximately 70%–80% of the samples—JNK–JUN pathway and the TGF-β1–SMAD2/SMAD3 pathway.14 In cultured cells, TGF-β1–stimulated growth is SMAD4-independent and SMAD2/SMAD3-mediated signaling switches from growth-inhibiting to growth-promoting as the cells transition from dysplasia to neoplasia. Additional evidence supporting a switch from tumor-suppressing to tumor-promoting in TGF-β signaling comes from a study of the expression of genes in BE and EAC stromal tissue.15 Increased expression of inflammatory genes, such as those encoding interleukin (IL)-6, C/EBPb, and COX-2, and genes induced by TGF-β signaling, such as TSP1, POSTN, and TMEPAI, in BE fibroblasts occurs in the patients that progressed from metaplasia to cancer and is associated with poor outcomes.15 Induction of POSTN, which encodes a protein that promotes cell motility, stimulates invasive behavior of esophageal cancer cells in culture. However, knockout of TGFBR2 in fibroblasts and dendritic cells (DCs) using the FSP1 promoter triggers loss of parietal cells and development of squamous cell carcinoma in the forestomach (similar to human esophagus).16 Thus, either increased or decreased TGF-β signaling in the stromal fibroblasts can contribute to neoplasia.
Gastritis, Infection, and Gastric Cancer
The risk of developing stomach cancer is associated with inflammation. Infection with Helicobacter pylori or chronic atrophic gastritis results in inflammation and tissue injury, which are associated with epithelial cell metaplasia. Inflammation in Helicobacter-infected mice is partially controlled by TGF-β1 produced by DCs.17 Mice with DC-specific TGFB1 knockout develop severe gastritis and exhibit increased metaplasia, despite reduced colonization by the bacteria (Table 1). In patients with H pylori, interferon-gamma inhibits TGF-β signaling by increasing SMAD7, and exposing biopsy tissue to a SMAD7 antisense molecule restores signaling.18 Thus, TGF-β1 signaling is important for limiting metaplastic changes associated with H pylori–induced chronic gastritis (Figure 2). This is consistent with a tumor-suppressing role before neoplastic transformation.
Table 1.
Mouse Models Mentioned in the Text for Studying the Roles of Transforming Growth Factor–β Signaling in Digestive Diseases and Cancers
| Gene (and function) | Mice model | Disease relevance | Model design | Model phenotype | Reference no. |
|---|---|---|---|---|---|
| TGFB1 (ligand) |
Tgfb1+/− TGFB1 heterozygosity (C57B1/6 NCr) |
Liver cancer | Disruption of TGFB1 exon 1 and intron 1 with neomycin-resistance cassette | By 6 mo, mice have increased susceptibility to chemically induced liver cancer. | 96 |
|
Tgfb1−/− Immunocompetent TGFB1 knockout (C3H/HeN × C57BL/6J) |
Inflammation, IBD | Disruption of TGFB1 exon 6 with neomycin-resistance cassette | By 3 wk, mice die from a wasting syndrome with multifocal, mixed inflammatory cell response and tissue necrosis. | 32 | |
|
TGF-βΔDC DC-specific TGFB1 knockout (C57BL6) |
Gastritis | Disruption of TGFB1 exon 6 with cCD11c-Cre driving DC-specific knockout | After 6 mo of Helicobacter felis infection, mice develop severe gastritis, with a trend toward increased metaplasia. | 17 | |
|
CD4-Cre/Tgfb1fl/fl T cell–specific TGFB1 knockout (C57BL/6) |
IBD | LoxP sites flanking TGFB1 exon 1 with CD4-Cre driving T cell–specific knockout | By 4–12 mo, mice developed inflammatory disorder and severe colitis. | 36 | |
|
Tgfb1−/−Rag2−/− Immunodeficient TGFB1 knockout (129S6 × CF-1) |
CRC | Disruption of TGFB1 exon 6 with neomycin-resistance cassette | By 5 mo, all mice develop severe hyperplasia and nonmetastatic carcinoma in the cecum and colon. | 59 | |
| PF4CreTgfb1f/f Platelet-specific TGFB1 knockout (C57BL/6) |
Liver fibrosis | LoxP sites flanking TGFB1 exon 6, with PF4-Cre driving deletion in platelets | Mice develop less liver fibrosis in response to chemically induced liver damage. | 83 | |
|
Alb/TGF-Β1 Liver-specific expression of TGF-β1 (C57BL/6 × CBA/J) |
Liver cancer | Porcine TGFB1 expressed from albumin promoter | By 16–18 mo, ~60% of mice spontaneously develop HCC | 101 | |
|
myc/TGF-Β1 Liver-specific expression of MYC and TGF-β1 (C57BL/6 × CBA/J) |
Liver cancer | MYC and porcine TGFB1 expressed from albumin promoter | By 13 mo, 100% of mice develop multifocal tumors in different lobes of the liver. | 101 | |
|
Alb-TGF-β1/ LFABP-cyclin D1 Liver-specific expression of TGF-β1 and multi-tissue expression of cyclin D1 (B6CBA × C57BL/6) |
Liver cancer | Porcine TGFB1 expressed from albumin promoter and CCND1 expressed from LFABP promoter | By 12 mo, 69% of mice develop liver cancer or high-grade tumors. | 102 | |
| TGFBRII (Type II receptor subunit for TGFβ1, TGF-β2, TGF-β3) |
Tgfbr2fspKO TGFBR2 knockout specifically in cells positive for S100A4 (DBA× Balb/c× C57BL/6) |
Pancreatitis, esophageal and gastric cancer | LoxP sites flanking TGFBR2 exon 2, with FSP1-Cre driving deletion in multiple cells of mesenchymal origin, including DCs and stromal fibroblasts | By 6 wk, mice spontaneously develop autoimmune pancreatitis. By 7 wk, all mice spontaneously develop invasive squamous cell carcinoma of the forestomach. | 16 |
| CRP/ΔkTβRII Liver-specific expression of dominant-negative TGFBR2 |
Liver cancer | Dominant-negative TGFBR2 with CRP promoter driving liver-specific expression | Mice exhibit increased susceptibility to chemically induced multifocal preneoplastic lesions and liver cancer. | 97 | |
| CD4-dnTGFβRII T cell–specific expression of dominant-negative TGFBR2 (C57BL/6 × 6XC3H) |
IBD, hepatitis | Dominant-negative TGFBR2 with CD4 promoter driving T cell–specific expression | By 4 mo, mice spontaneously develop IBD; mice have increased susceptibility to chemically induced liver disease. | 37 | |
| DC-Tgfbr2 KO DC-specific TGFBR2 knockout (B6.129S6) |
Hepatitis, pancreatitis, colitis, gastritis | Flox sites flanking TGFBR exon2 with CD11c-Cre driving DC-specific deletion | By 15 wk, mice die of autoimmune inflammation in multiple organs of digestive tract. | 38 | |
|
Apc1638N/wtTgfbr2IEKO Intestinal epithelial cell–specific TGFBR2 knockout with heterozygous APC mutation (C57BL/6JIco × C57BL6) |
CRC | LoxP sites flanking TGFBR2 exon 2, with Villin-Cre driving intestinal epithelial cell–specific deletion in the context of heterozygous APC loss of function | By 12 mo, all mice spontaneously develop intestinal adenocarcinoma that progresses to invasive disease. | 67 | |
|
ApcΔ716
Tgfbr2ΔIEC Intestinal epithelial cell–specific TGFBR2 knockout with APC mutation |
CRC | LoxP sites flanking TGFBR2 exon 2, with Villin-Cre driving intestinal epithelial cell–specific deletion in the context of APC mutation | By 15 wk, mice spontaneously develop intestinal adenocarcinomas with submucosal invasion in large polyps. | 68 | |
|
LAKTP Lgr5eGFPCreERT2/ApcLoxp/KrasLSL-G12D/Tgfbr2Loxp/Trp53Loxp Intestinal stem cell-specific knockout of APC, TGFBR2, TRP53 with KRAS activation (C57BL/6J) |
CRC | LoxP sites flanking regions of APC, TGFBR2, and TRP53 with inducible with Lgr5eGFP-creERT2 driving intestinal stem cell-specific deletion and expression of activating KRAS mutant | Mice exhibit increased susceptibility of chemically induced metastatic colon cancer. | 71 | |
|
Ptf1acre/+LSL-KrasG12D/+Tgfbr2flox/flox Pancreatic epithelial cell–specific TGFBR2 knockout with KRAS activation (C57BL/6×DBA/2×129/SvJae) |
Pancreatic cancer | LoxP sites flanking TGFBR2 exon 2, with Ptf1a-Cre driving pancreatic epithelial cell–specific deletion and expression of activating KRAS mutant | By 2 mo, mice die of PDAC. | 122 | |
| SMAD3 (R-SMAD activated by TGF-β1, TGF-β2, TGF-β3, Activin A, Activin B, Nodal, GDF1, GDF2, GDF8, GDF9, GDF11) | Smad3−/− SMAD3 knockout (C57BL/6 × 129/S) | Gastric cancer, CRC | Disruption of SMAD3 exon 2 with IRES-LacZ and neomycin cassette | By 10 mo, mice spontaneously develop invasive gastric cancer arising in the forestomach–stomach junction. By 4–6 mo, mice (30%) spontaneously develop large polyps and aggressive, metastatic colon cancer that depends on Helicobacter. | 60 |
|
Smad3ex8/ex8 SMAD3 knockout (C57BL/6 × Black Swiss) |
IBD | Targeted deletion of Smad3 exon 8 by homologous recombination, resulting in disruption of the interaction between SMAD3 and the TGFB receptor | More than 6 mo, nearly 77% of the mice spontaneously developed chronic inflammation in the intestines. | 31 | |
|
Smad3−/− + Helicobacter SMAD3 knockout infected with Helicobacter (129/J) |
CRC | Disruption of SMAD3 exon 2 with IRES-LacZ and neomycin cassette | Mice exhibit increased susceptibility to infection-induced colon cancer. | 60, 61 | |
| ApcMin/+Smad3−/−SMAD3 knockout with heterozygous APC mutation (129/Sv) | CRC | Disruption of SMAD3 exon 2 with neomycin cassette and mutation of APC | By 2 mo, mice spontaneously develop tumors in the distal colon, resembling human familial adenomatous polyposis. | 70 | |
| SMAD4 (c-SMAD) |
Smad4co/co; Lck-cre Smad4co/co; CD4-cre T cell–specific SMAD4 knockout (C57BL/6 × SvEv129 × FVB) |
Gastrointestinal epithelial cancer | LoxP sites flanking SMAD4 exon 8, with Lck-Cre or CD4-Cre driving T cell–specific deletion | Mice spontaneously develop inflammation and epithelial cancers throughout the GI tract. | 65 |
|
Smad4+/E6sad SMAD4 heterozygosity (129Ola × C57BL/6JIco) |
Intestinal cancer | Single nucleotide deletion of SMAD4 exon 6 | By 9–18 mo, mice spontaneously develop adenomas and mixed polyposis of the upper GI tract. | 69 | |
|
Apc+/1638N/Smad4+/E6sad Heterozygous loss of function of both SMAD4 and APC in cis or trans (129Ola × C57BL/6JIco) |
CRC | Single nucleotide deletion of SMAD4 exon 6, and disruption of APC exon 15 | Both trans and cis mice spontaneously develop tumors of the GI tract, desmoids, and epidermal tumors. Trans mice spontaneously develop high numbers of tumors. Cis mice spontaneously develop rapidly progressing disease, dying within 6 wk. |
69 | |
| SMAD7 (I-SMAD for SMAD2 and SMAD3) |
Smad7Tg T cell–specific expression of SMAD7 (C57BL/6) |
IBD | SMAD7 with CD2 promoter/enhancer driving T cell–specific expression | Mice exhibit more susceptibility to chemically induced colitis but fewer colitis-induced tumors. | 77 |
|
S7tg Hepatocyte-specific expression of SMAD7 (C57BL/6) |
Liver fibrosis | Flag-tagged SMAD7 with CRP promoter driving hepatocyte-specific expression | Mice exhibit decreased susceptibility to chemically induced liver damage and fibrosis. | 108 | |
|
Smad7 KO SMAD7 knockout (C57BL/6) |
Liver cancer | Disruption of SMAD7 exon 1 with PGKneobpA cassette | Mice exhibit increased susceptibility to chemically induced HCC. | 110 | |
| TTR-Cre-SMAD7 KO, Hepatocyte-specific SMAD7 knockout (C57BL/6) |
Liver cancer | LoxP sites flanking SMAD7 exon 1 with TTR-Cre driving hepatocyte-specific deletion | Mice exhibit increased susceptibility to chemically induced HCC. | 109 | |
|
SMAD7Tg Pancreas-specific expression of SMAD7 (DBA2) |
Pancreatic cancer | Myc-tagged SMAD7 with elastase I promoter driving pancreas-specific expression | By 6 mo, mice develop PanIN. | 126 | |
| SPTBN1 (Adaptor for activated SMAD2 and SMAD3) |
SMAD7Tg Pancreas-specific expression of SMAD7 (DBA2) Smad4+/− Sptbn1+/−Heterozygous loss of function of both SMAD4 and SPTBN1 (129SvEv × C57BL/6) |
Gastric cancer, liver cancer | Disruption of SPTBN1 exon 25 and SMAD4 exon 8 with neomycin cassette | All mice spontaneously develop gastric polyps with a subset progressing to cancer; a subset of mice develop colon cancer. | 62, 64 |
| Sptbn1+/−SPTBN1 heterozygosity (129SvEv/Black Swiss) | Liver steatosis, fibrosis, liver cancer | Disruption of SPTBN1 exon 25 with neomycin cassette | By 15 mo, 40% of mice spontaneously develop liver diseases and cancer. | 99 | |
|
Sptbn1+/−Iitih4−/− Heterozygous loss of SPTBN1 and knockout of ITIH4 |
Liver cancer | Disruption of SPTBN1 exon 25, and ITIH4 exons 2 and 3 with neomycin cassette | Mice exhibit increased susceptibility to liver cancer. | 100 |
c-SMAD, common SMAD; I-SMAD, inhibitory SMAD; R-SMAD, receptor-activated SMAD.
Once the gastric epithelia become neoplastic, TGF-β signaling becomes hyperactivated, suggesting a switch from tumor-suppressing to tumor-promoting or tumor-supporting. Activation of a TGF-β signaling transcriptional profile is higher in gastric cancer (GC) compared with intestinal metaplasia of the stomach.15 Furthermore, biopsy samples reveal high abundance of TGF-β1 and TGF-β3 in GC compared with normal tissue.19 However, a mouse model with FSP1-mediated knockout of TGFBR2 shows that loss of stromal TGF-β signaling can also lead to tumorigenesis of specific epithelial populations, including those of the stomach. Consistent with this finding in mice, patients with SMAD7-positive tumors had a worse survival rate than those with SMAD7-negative tumors, suggesting that impaired TGF-β signaling is also associated with poor outcomes in some patients, and underscoring the complexity in considering whether TGF-β signaling is tumor suppressive or tumor enabling.20
Alterations in TGF-β signaling are associated with metastasis of GC. Increased or decreased signaling occurs in multiple cells of the tumor microenvironment. A patient with GC that metastasized to the ovaries had a biallelic loss-of-function genetic alteration in TGFBR2.21 Subsequent studies with mouse organoids in vitro and using the cells in a mouse xenograft model show that loss of TGFBR2 in the context of genetic knockout of CDH1 (encoding E-cadherin) and TP53 results in metastatic phenotypes. GCs are molecularly diverse, with each subset having different connections to TGF-β signaling22: 9% are positive for Epstein-Barr virus (EBV), 22% are associated with microsatellite instability (MSI), 20% are genomically stable, and 50% exhibit chromosomal instability. EBV-positive GCs have high DNA hypermethylation, especially of the CDKN2A promoter, and mutations in PIK3CA, encoding the catalytic subunit of PI3K.22 Although TGF-β1 stimulates apoptosis of GC cell lines, introduction of the EBV oncoprotein LMP2A blocks this apoptotic effect by stimulating PI3K-mediated activation of AKT.23 EBV-infected gastric epithelial cell lines produce TGF-β1 and respond to this signal by activating EBV gene expression.24 Thus, ineffective TGF-β1–mediated apoptosis and ongoing inflammation due to viral reactivation may contribute to progression of EBV-positive GC. In MSI GC, mutations are common in genes that are associated with increased susceptibility to altered TGF-β signaling, or are core to the TGF-β pathway—TP53 (50%), PIK3CA (12%), CDH1 (11%), SMAD4 (8%), and SMAD2 (2%).22 Mutations in TGFBR2 occur in GCs associated with either MSI or replication errors.25 Reduced expression of TGFBR2 is associated with a high frequency of MSI at this gene (approximately 52%) in GC.26 Like the other subtypes of GC, genes associated with increased susceptibility to altered TGF-β signaling were common in the genomically stable and the chromosomal instability types. In the genomically stable tumors, 37% had mutations in CDH1; in the chromosomal instability tumors, 71% had mutations in TP53. Thus, even though these subtypes were not associated with mutations in genes of the TGF-β pathway, these cancers can arise through impairment of TGF-β–mediated tumor-suppressor signaling.22
Inflammatory Bowel Disease, Hereditary Colon Cancer Syndromes, and Colorectal Cancer
Inflammatory bowel disease (IBD), including Crohn’s disease and ulcerative colitis (UC), is characterized by chronic or recurring inflammation of the GI tract. IBD is associated with disrupted immune homeostasis that involves T cells and innate lymphoid cells. Through genome-wide association studies, more than 200 risk loci are associated with IBD or the subtypes of Crohn’s disease and UC.27,28 Of particular relevance here is the identification of loci containing SMAD2, SMAD3, SMAD4, and SMAD7 (Figure 2). A SMAD3 locus was found to be associated with IBD.28 Single-nucleotide polymorphisms in SMAD2, SMAD3, SMAD4, and SMAD7 were identified specifically in patients with UC, not those with Crohn’s disease.29
Biallelic loss-of-function mutations in TGFB1 lead to severe infantile IBD and central nervous system defects.30 Consistent with a loss of immune homeostasis, the lamina propria of 1 patient had decreased frequency of multiple subpopulations of T cells, including regulatory T cells (Tregs), T helper (Th) type 1 cells, and Th17 cells. These findings in children support the translational validity of the observations that mice with global SMAD3 knockout31 spontaneously develop IBD and mice with a global TGFB1 knockout develop severe multiorgan inflammatory disease that includes the intestine.32 Mice in which TGF-β is induced by oral administration of haptens to induce “tolerance” or before induction of experimental colitis with the same molecules, results in a reduction in inflammation and colitis-associated symptoms.33
Intestinal epithelial cells release proinflammatory signals in response to IL-22, which was transcriptionally up-regulated in biopsies from patients with active Crohn’s disease.34 In the presence of IL-6 and TGF-β, Th17 cells produce IL-17 and transcriptionally suppress IL22.35 Thus, context-dependent TGF-β signaling can provide an anti-inflammatory input and loss of this signal can contribute to intestinal inflammation. Mice with either a T cell–specific deletion of TGFB1 36 or introduction of dominant-negative TGBR2 specifically in T cells37 develop IBD. DC-specific TGFBR2 knockout also results in multiorgan inflammatory disease, which, in the colon, involves increased production of pro-inflammatory cytokines, reduced function of Tregs,38 an altered microbiota, goblet cell loss, and a reduction in the mucous layer.39 Together these mouse studies indicate that TGF-β1 signaling in DCs is important not only for maintaining T cell homeostasis, but also for enabling the differentiation of mucous-producing goblet cells that are critical for maintaining a healthy epithelium that is continuously exposed to enteric microbes.
Analysis of tissue and experiments with explanted tissue reveals increased abundance of SMAD7, anti-SMAD, and strongly reduced phosphorylated SMAD3 in the Crohn’s disease tissue and a moderate reduction in UC tissue.40 Exposing immune cells isolated from the lamina propria of these tissues to SMAD7-specific antisense oligonucleotides increases TGF-β1–stimulated SMAD3 phosphorylation. Furthermore, these antisense oligonucleotides reduce interferon-gamma and tumor necrosis factor–α in the explanted tissue from patients with Crohn’s disease, consistent with a shift from pro-inflammatory T cells (Th1 and Th17 cells) to Tregs in response to increasing TGF-β1 signaling.41 Studies with mice support the increase in SMAD7 in IBD and the effectiveness of SMAD7-targeted antisense oligonucleotides in enabling TGF-β1–mediated SMAD3 activation and reducing inflammation.42 These studies resulted in clinical trials of an orally available SMAD7 antisense oligonucleotide (mongersen) for active Crohn’s disease, which yielded positive results in phase II43,44 but failed in phase III.45
Small intestinal fibrosis and stricture formation often occurs in Crohn’s disease. TGF-β signaling regulates many genes involved in fibrosis, such as those encoding collagens and matrix metalloproteinases (MMPs) and their tissue inhibitors (TIMPs). TGF-β also stimulates fibroblasts to become myofibroblasts, which both produce proteins that alter the ECM and have contractile properties. Tissue injury and the activity of MMPs, such as occurs in chronic inflammatory conditions like IBD, release latent TGF-β from the ECM, and integrins mediate local activation of latent TGF-β. Tissues from both patients with UC46 and patients with Crohn’s disease47 exhibit changes in TGF-β signaling and pro-fibrotic pathways, including MMPS, TIMPs, and integrins.48
Myofibroblasts contribute to fibrosis and stenosis. The phenotypes of the myofibroblasts associated with Crohn’s disease or UC are different. Myofibroblasts from fibrotic Crohn’s disease tissue exhibit increased expression of TIMP1 and release higher amounts of TIMP1 into the medium compared with myofibroblasts cultured from normal tissue or from patients with UC.49 Intestinal myofibroblasts from patients with Crohn’s disease or UC exhibit differential production of TGF-β isoforms; those from Crohn’s disease tissue produce all 3 isoforms (TGF-β1, TGF-β2, TGF-β3), those from UC tissue produce primarily TGF-β1 and TGF-β3, and those from normal tissue produce mostly TGF-β3.50 Despite producing all 3 isoforms, only the myofibroblasts from the Crohn’s disease tissue are independent of the autocrine effects of TGF-β for proliferation in culture. In culture, TGF-β3 inhibits proliferation the least and TGF-β2 is the least effective in promoting cell migration.50
Differences in TGF-β signaling exist in the mucosal tissue and myofibroblasts from strictured and nonstrictured regions of patients with fibrostenosing Crohn’s disease, from inflamed tissue from patients with nonfibrostenosing disease, and from normal intestine.51 Strictured areas have the highest activity of TGF-β signaling and the highest amounts of TIMP1 and collagen, whereas inflamed areas from patients with nonfibrostenosing disease have the highest amount of SMAD7, MMP12, and MMP3. These differences relate to the myofibroblasts in the tissues and suggest that myofibroblast SMAD7 abundance may regulate TGF-β signaling at different stages of the disease process or contribute to the type of disease present.
Intestinal microbial populations are altered in patients with IBD.52–54 Of particular relevance to TGF-β signaling are the reductions in the butyrate-producing Clostridium populations and Bacteroides populations and in the increase in Enterobacteriaceae. Studies with mice suggest that the reduction in butyrate-producing species impairs intestinal epithelial cell TGF-β1 production,55 as well as reduces the responsiveness of T cells to Treg-inducing TGF-β1 signals by affecting histone acetylation.56 The consequent change in FOX3P expression shifts the T-cell balance away from the inflammation-suppressing Treg, contributing to colitis. Combined with the high abundance of SMAD7 in the immune cells from inflamed IBD patient tissue,40 this dysbiosis likely contributes to the hyperinflammatory conditions by further impairing immuno-suppressive TGF-β signaling. Mouse studies suggest that impaired TGF-β signaling in DCs could contribute to the increase in Enterobacteriaceae.39 Other studies in mice show that dysfunction of the TGF-β pathway in immune cells57 is associated with IBD susceptibility in response to commensal microbes. An attractive concept for treating IBD is a therapy that restores a microbial community to enable TGF-β responsiveness, promote Treg differentiation, and limit inflammation.58
Inflammation and microbial dysbiosis are risk factors for colorectal cancer (CRC), which includes both colon adenocarcinoma and rectal adenocarcinoma. Furthermore, mutations in any of 43 genes encoding components in the superfamily of TGF-β pathways are present in 65% of colon adenocarcinomas and 50% of rectal adenocarcinomas.1 Various studies in mice provide mechanistic connections between changes in the intestinal microbiome or susceptibility to microbial infection, gut inflammation, and cancer development (Figure 2). Mice with global TGFB1 knockout that are also genetically immunodeficient (Tgfb1−/−Rag2−/−) require gut microbes for inflammation-induced adenocarcinoma and progression from colitis-associated inflammation to cancer.59 SMAD3 global knockout mice housed under normal conditions, but not those lacking Helicobacter in the GI tract, develop metastatic CRC.60 The introduction of Helicobacter induced inflammation that progressed to mucinous adenocarcinoma.61 Mice heterozygous for SMAD4 and for the gene encoding the SMAD3 adaptor protein SPTBN1 spontaneously develop CRC, and these mice exhibit an altered gut microbiota that is enriched in microbial populations associated with human CRC.62 The CRC-associated mutants exhibit enhanced ability to inhibit TGF-β signaling in cultured cells.62,63
The high frequency of alterations in genes in the components of the superfamily of TGF-β pathways in CRC suggests that these pathways prevent these cancers or that altered activity enables these cancers to progress in the presence of gut microbiota. SMAD3−/−, SMAD4+/–, SPTBN1+/– double heterozygous mice develop CRC, and T cell–specific, not epithelial cell–specific, SMAD4 knockout causes CRC.60,62,64,65 The T cell–specific SMAD4 knockout models phenocopy familial juvenile polyposis syndrome, some cases of which are associated with germline mutations in BMPR1A or SMAD466 and increased risk of CRC. Furthermore, disruption of TGFBR2, specifically in intestinal epithelial cells; SMAD4 heterozygosity; or SMAD3 knockout accelerate the development of invasive, inflammation-associated colorectal adenocarcinomas in mice with APC mutations (Supplementary Table 1).67–70 Thus, in the context of APC mutations, suppression of TGF-β signaling either in the intestinal cells or the T cell population contributes to CRC development.
APC mutations cause aberrant Wnt signaling by constitutively activating b-catenin (encoded by CTNNB). Various classifications exist for CRC, one of which defines 4 main subtypes: 13% exhibit epithelial differentiation with metabolic dysregulation (CMS3); 14% are associated with MSI and immune cells with Th1 and cytotoxic phenotypes (CMS1); 23% are associated with a mesenchymal phenotype that includes high activity of TGF-β signaling, stromal invasion, increased abundance of ECM proteins, and angiogenesis (CMS4); and 37% exhibit epithelial differentiation with increased Wnt and MYC signaling, frequent amplifications in oncogenes, and losses in tumor suppressor genes (CMS2). CMS4 is also associated with late-stage, metastatic tumors, suggesting that by stimulating epithelial-to-mesenchymal transition (EMT), TGF-β signaling functions as a tumor promoter in this subtype.
Severe, invasive CRC develops in mice with intestinal stem cell–targeted deletion of genes encoding APC, TGFBR2, and TRP53, and introduction of an activated KRAS mutant.71 Although the tumors could not respond to TGF-β signals, TGF-β signaling in the stroma was high at the invasive margins of the tumors, which correlated with the immune “cold” character of these tumors, suggesting that high TGF-β signaling may interfere with T cell infiltration of the tumor. Transcriptional profiling of tumors transplanted into the ceca of syngeneic mice, but not those grown in culture, revealed that these tumors resemble the CMS4 subtype. Single-cell analysis of patient-derived CRC reveals enrichment in stromal cells, especially cancer-associated fibroblasts (CAFs), in the subset of patients with poor prognosis.72,73 High expression of TGFB1 and TGFB3, along with induction of a TGF-β response transcriptional profile in fibroblasts, is also associated with poor prognosis. Multiple mouse models reveal how the stroma contributes to the protumor effects of TGF-β signaling in CRC: Mice with tumors derived from a patient with inactivating mutations in both TGFBR2 alleles,73 mouse tumor organoids with intestinal cell-targeted mutations,71 or patient-derived tumor organoids with high expression of TGFB1.72 In these models, blocking stromal TGF-β signaling with the selective TGFBR1 inhibitor Galunisertib (LY2157299) reduces metastatic burden.71–73 Thus, independently from effects on the cancer cells, TGF-β signaling in the tumor microenvironment plays a key role in both tumor development and progression to highly invasive CRC.
SMAD7 has dual roles in development and progression of CRC, reflecting the cell-specific effects of TGF-β signaling in cancer. Genome-wide association studies reveal that SMAD7 variants are associated with increased risk of developing CRC.74 Overexpression of SMAD7 in non-tumorigenic CRC-derived cell line responsive to TGF-β signaling results in tumorigenicity and liver metastasis in nude mice, along with antagonism of TGF-β–mediated growth inhibition and apoptosis.75,76 These studies indicate that SMAD7 interferes with a tumor-suppressive role for TGF-β signaling within the tumor cells. In contrast, mice with T cell–specific overexpression of SMAD7 develop fewer colitis-associated tumors, despite exhibiting increased susceptibility to chemically induced colitis.77 These mice are also resistant to tumor development by subcutaneously transplanted syngeneic CRC cells.78 The protective role of SMAD7 against CRC in these models involves interferon-gamma, tumor necrosis factor–α, and infiltration of T cells into the tumor microenvironment. These studies indicate that TGF-β signaling within T cells provides a tumor-enabling, immune-suppressive microenvironment.
Nonalcoholic Fatty Liver Disease, Nonalcoholic Steatohepatitis, and Hepatocellular Carcinoma
Nonalcoholic fatty liver disease (NAFLD) ranges from benign steatosis to aggressive nonalcoholic steatohepatitis (NASH). NAFLD often progresses to fibrosis, cirrhosis, liver failure, or hepatocellular carcinoma (HCC). The amount of fibrosis is the key clinical indicator of outcomes of NAFLD, independent of histologic evidence of NASH.79,80 In NALFD, there is an infiltration of immune cells and activation of Kuppfer cells, all of which produce TGF-β81–83 and also respond to TGF-β. Although TGF-β signaling is critical for liver repair, in NAFLD the initiating condition does not resolve appropriately. Thus, the carefully orchestrated TGF-β response fails to move from the initial phase to the resolution phase.
In the presence of injury or inflammation, like that in NAFLD or NASH, TGF-β1 induces apoptosis and inhibits proliferation of hepatocytes84,85 and activates hepatic stellate cells.86–88 Studies with cultured cells and mice indicate that TGF-β1 also promotes the differentiation of mesothelial cells into hepatic stellate cells and myofibroblasts during chemically induced liver fibrosis.89 The activation of hepatic stem cells and formation of myofibroblasts are pro-fibrotic consequences of TGF-β1 signaling: Both activated hepatic stellate cells and myofibroblasts produce pro-inflammatory signals, remodel the ECM, and deposit collagen.86
This remodeling of the ECM can release TGF-β latent complexes. This activated TGF-β from the ECM can synergize with inflammatory mediators, such as IL-22, to activate hepatic stellate cells.90 Both IL-22 and IL-17 are associated with fibrotic liver disease and enhance responsiveness to TGF-β signaling in hepatic stellate cells,90,91 connecting inflammation and TGF-β signaling to progression of fibrotic liver disease. The chronic inflammation and fibrotic tissue damage associated with NAFLD and NASH are risk factors for HCC. As treatments become more available for viral-associated HCC,92,93 the frequency of NAFLD- and NASH-associated HCC will increase. Fibrosis results in a stiffer ECM, which enhances TGF-β1 autocrine signaling and triggers EMT in HCC cells in culture and enhances HCC metastasis in a rat model.94 Furthermore, a stiffer matrix also induces HCC cells in culture to express markers of cancer stem cells.95
The role of TGF-β in the progression from inflammation to fibrosis to HCC is complex; TGF-β1 signaling has different effects on multiple cells at various stages of progression to neoplasia (Figure 3).93 Studies with mice show that TGF-β1 functions as tumor suppressor, limiting hepatocyte proliferation under normal conditions, but enabling chemically induced HCC in the heterozygous state96 or upon expression of a dominant-negative TGFBR2.97 Impaired TGF-β signaling is linked to the development of cancer stem cells in HCC, consistent with an early tumor-suppressing role for TGF-β. Studies with cultured hepatocyte progenitor cells reveal suppression of SMAD3-mediated gene expression downstream of TLR4, a receptor involved in innate immune responses, contributes to the induction of cancer stem cell properties.98 Altered SMAD3 function that occurs in mice heterozygous for the SMAD3 adaptor protein SPTBN1 results in spontaneous development of fibrosis and HCC, which is associated with hyperproliferative endothelial cells that result in aberrant angiogenesis.99 Crossing these SPTBN+/– with mice lacking ITIH4, which is stimulated by IL-6 and encodes a serine protease, suppressed cancer stem cell generation and HCC.100 Tissue from living donor liver–transplanted specimens contains small groups of cells that are positive for stem cell markers and for SPTBN1 and TGFBRII,100 indicating that healthy tissue contains a subset of cells poised to become cancer stem cells should they lose TGF-β–mediated inhibition.
Figure 3.
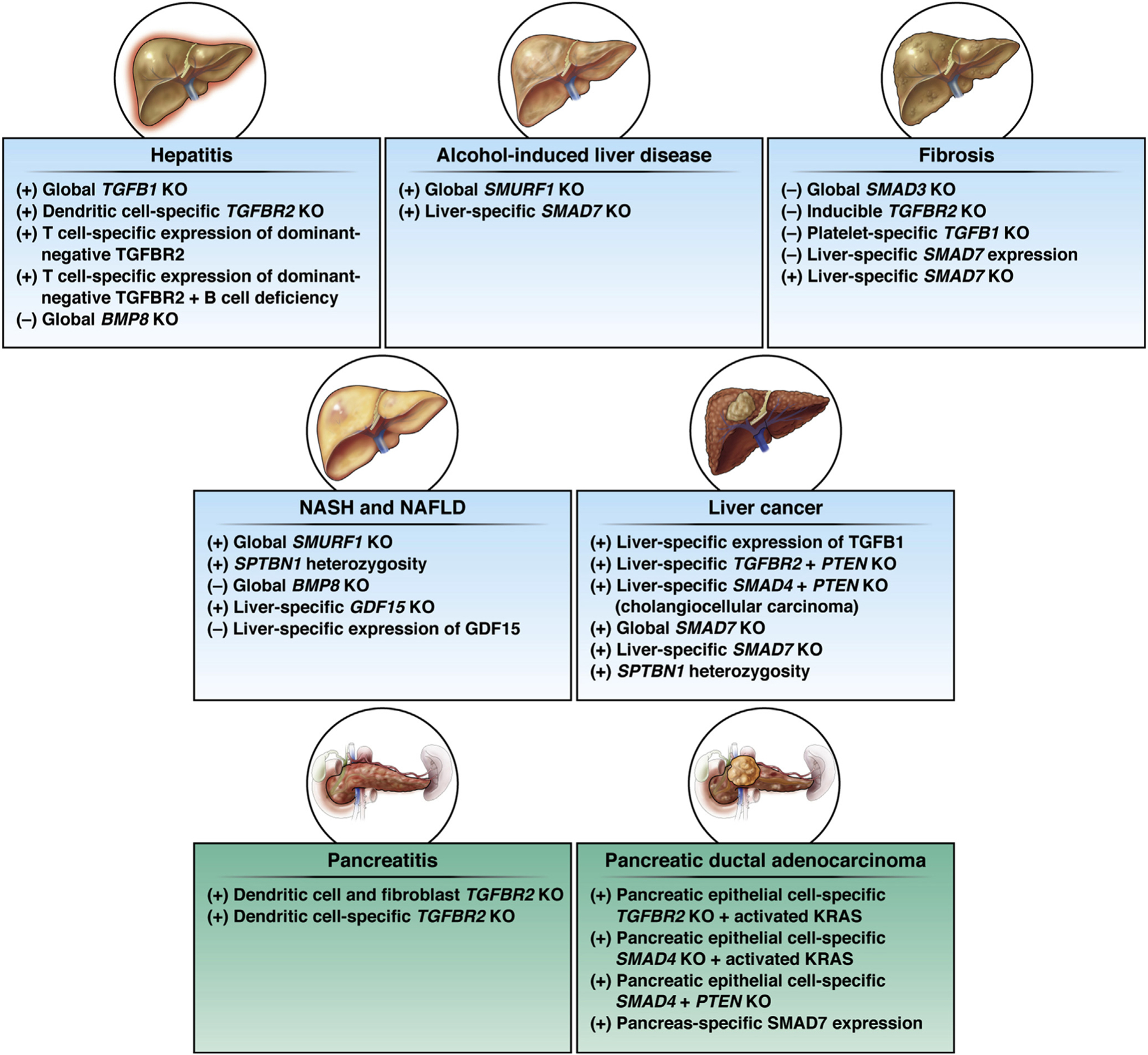
Pancreas and liver with relevant mouse models for investigating TGF-β signaling in disease or cancer. (+), increased susceptibility to disease or cancer; (–), decreased susceptibility.
Mouse studies also support a tumor-promoting role for TGF-β1 in HCC. Overexpression of TGF-β1 alone or in combination with MYC, both specifically in hepatocytes, results in HCC and accelerates chemically induced HCC in mice.101 Combined hepatocyte-targeted overexpression of TGF-β1 and cyclin D1 in mice results in initial inhibition of hepatocyte proliferation, followed by development of highly invasive HCC, suggesting that TGF-β1 signaling switches from tumor-suppressing to tumor-promoting (or enabling) in this noninflammatory HCC model.102 Analysis of HCC cell lines reveals diverse outcomes in response to TGF-β1. Some cells respond with EMT or partial EMT, and cells that respond with partial EMT have increased expression of genes indicative of stem cells.103 Furthermore, knocking down TGFBRI reduces these invasive malignant properties of the cells. Another study shows that some HCC cell lines respond with apoptosis, some with proliferation, and some with inhibition of proliferation.104 Furthermore, by altering the immune environment to one that is immunosuppressive, TGF-β1 also promotes progression of HCC and immune evasion that may be exacerbated by metabolic changes associated with NAFLD.105,106
Genetic and transcriptomic data reveal profiles consistent with an activated or an inactivated TGF-β signaling system in patient HCC tissue.2 Patients with an inactivated TGF-β signature have the worst prognosis. Consistent with the TGF-β signal predominantly affecting stromal cells, expression of genes associated with activated fibroblasts was increased in HCC with the activated TGF-β signature. Like CRC, the TGF-β inactivated signature was associated with HCC with mutations in genes associated with DNA repair. HCC patients with TGF-β–positive gene signature (one dependent on TGFBR2) can be divided into 2 groups based on the temporal profile of the response to TGF-β: one with an “early” TGF-β signature and the other with a “late” TGF-β signature.107 HCC patients with a late TGF-β signature display worse prognosis and overall survival, consistent with TGF-β signaling having different outcomes at different stages of cancer progression.
Despite tumor-suppressing roles for TGF-β, interest exists for inhibiting TGF-β signaling in chronic liver disease to prevent fibrosis and progression to invasive HCC. This concept is supported by mouse studies. Mice with hepatocyte-specific overexpression of SMAD7 exhibit protection from chemically induced fibrosis and liver damage.108 Mice with hepatocyte-specific deletion of SMAD7 have increased susceptibility to chemically induced HCC.109,110 However, context is important: Mice with SMAD7, MYC, and MCL overexpression or SMAD7 overexpression and activated AKT have accelerated development of liver cancer.111
HCC patients treated with sorafenib, an inhibitor of multiple tyrosine kinases, and who have high circulating TGF-β1 have worse prognosis and shorter survival than patients with low circulating TGF-β1.2,112 Various preclinical studies show beneficial effects of LY2109761, a TGFBR1/TGFBR2 inhibitor,113 in blocking tumor growth and tumor angiogenesis in xenograft models of HCC114 and blocking HCC cell invasiveness.115,116 HCC tissues exposed to galunisertib, alone or in combination with sorafenib, exhibit reduced proliferation markers and increased apoptotic markers.117 Galunisertib reduces stem cell markers in HCC cell lines and patient tissue samples.118
Pancreatic Cancer
Pancreatic ductal adenocarcinoma (PDAC), the most common and deadly form of pancreatic cancer, typically follows a predictable morphologic and genetic course that converts normal epithelial cells to noninvasive pancreatic intraepithelial neoplasia (PanIN) that progresses to PDAC. The median survival of patients with PDAC is 6 months, and the 5-year survival rate is approximately 6%.119 The relative risk of developing PDAC is high in individuals with germline mutations in STK11 (also known as LKB1, encoding a serine/threonine kinase), PRSS1 (encoding a serine protease associated with hereditary pancreatitis), and CDKN2A. Inactivating TP53 and SMAD4 mutations occur in approximately 50% of these cancers and activating KRAS mutations in >90%.3,120 Loss of SMAD4 occurs in later stages in the progression of PanIN to PDAC.121
The TGF-β pathway likely plays a role in the progression to malignant cancer through loss of the tumor-suppressing activity, such as occurs with combined activation of KRAS and inactivation of SMAD4 (Figure 3). Mice with pancreas-specific expression of KRASG12D and pancreas-specific heterozygous or homozygous knockout of TGFBR2 rapidly develop invasive PDAC.122 Furthermore, the tumors also contained CDNK2A genomic alterations,122 consistent with progressive acquisition of enabling mutations resulting in the progressive phenotypic switch from noninvasive PanIN to invasive PDAC.123 Cell culture and mouse xenograft studies indicate that loss of SMAD4 activity or acquired resistance to TGF-β does not alter proliferation of human pancreatic ductal epithelial cell line expressing an activated KRAS mutant. However, loss of TGF-β responsiveness enhances invasive and metastatic phenotypes, consistent with loss of TGF-β responsiveness contributing to the later stages of progression to malignant, aggressive cancer.124 Thus, aberrant or loss of TGF-β signaling within the cancer cells appears to serve a cancer-enabling or cancer-progressing function rather than a tumor-initiating function in the context of PDAC. Comparison of PanIN triggered by chemically induced pancreatitis in mice with pancreas-specific expression of an activating KRAS mutant in the presence or absence of SMAD4 knockout shows that TGF-β signaling induces apoptosis in signaling-competent cells through induction of a lethal EMT program.125 The failure of PanIN to progress to PDAC in the context of functional TGF-β–SMAD signaling provides an explanation for the high-frequency SMAD4 mutations in PDAC patients.125 Consistent with a tumor-suppressing role of TGF-β signaling early in PDAC development, pancreas-specific expression of SMAD7 in mice disrupts TGF-β signaling and promotes PanIN.126
However, many PDAC tumors exhibit high amounts of TGF-β1, TGF-β2, or TGF-β3, or a combination thereof, with high amounts of TGF-β2 particularly associated with advanced tumor stage and high amounts of each correlated with worse outcome.127 Reduced expression of the inhibitory SMAD7 gene in patients is associated with poor prognosis.128 Patients with higher serum TGF-β before treatment tend to have better treatment outcomes, and a concentration that is higher after treatment than at diagnosis correlates with patients that progress.129 Thus, TGF-β signaling may enable disease progression through effects on the tumor microenvironment.
Later stages of PanIN or PDAC are associated with a transcriptional profile of inactivated T cells and populations of immune-suppressing innate immune cells, consistent with the established immune-suppressing effects of TGF-β signaling.130,131 PDAC has a desmoplastic stroma, consistent with TGF-β–induced myofibroblasts or activated pancreatic stellate cells.130,132 At least 2 populations of CAFs are identified by transcriptional profiling,131,132 an inflammatory type and a myofibroblast type. Studies with mouse PDAC organoids and pancreatic stellate cells shows that TGF-β signaling drives the myofibroblast phenotype and suppresses the inflammatory phenotype.133 Tissue samples reveal differences between the immune cell populations in PDAC with different stromal properties, consistent with communication between the CAFs and the immune cells.134
Studies of pancreatic cancer cells and xenografts in mice found a reduction in tumor growth in response to kinase inhibitors of the TGFBR1/TGFBR2 complex, and this reduction is associated with less fibrosis and increased immune cell infiltration.135,136 Mouse PDAC models show that combination treatment with galunisertib to inhibit the TGF-β receptor and lapatinib, an inhibitor of epidermal growth factor receptors (EGFR and HER2), reduces tumor growth and metastasis by inhibiting lymphangiogenesis and angiogenesis.137 These results are consistent with finding a pro-angiogenic transcriptional profile in patient samples, angiogenesis in patient-derived xenografts, and angiogenesis in a PDAC mouse model.138 Studies with cultured cells and mouse xenografts show that LY2109761, an inhibitor of the kinase activity of both TGFBR1 and TGFBR2, has synergistic anti-tumor effects with gemcitabine and reduces metastasis.113 Targeting the fibrotic stiff stroma of tumors is another possibility. Even in tumors with cancer cells deficient in SMAD4 function, the epithelial cells contribute to stromal thickening through activation of other pathways, such as through JAK-mediated STAT3 signaling.139 Mouse studies and patient tissues show that PDAC deficient in TGF-β signaling is more aggressive, in part because of EMT driven by a mechanically activated tumor microenvironment involving both TGF-β–responsive stromal cells and nonresponsive epithelial cells.139
Future Directions
Given the burden that these diseases and cancer have on the health care system (approximately $135.9 billion in 2015 for the United States) and the high mortality rates of many of these conditions, the ability to treat existing disease and prevent progression to severe disease or cancer is urgently needed.119 Preclinical studies tested approaches to inhibiting TGF-β signaling: neutralizing antibodies against the TGF-β subfamily or the receptor, ligand traps, small molecule inhibitors of the kinase activity of the receptor or integrin-mediated activation of the ligand latent complexes or antisense oligonucleotides targeting TGF-β2. However, neutralizing antibodies and some TGF-β receptor inhibitors have resulted in cardiac toxicity in nonhuman primates, dogs, and rodents140–142 and skin toxicity, including skin cancer in patients.143 Although the TGFBR1 inhibitor galunisertib is in clinical trials for CRC, HCC, and PDAC,144,145 toxicities may preclude systemic targeting of TGF-β.
Targeted blocking of specific TGF-β signals could be beneficial. Inspired by high TGF-β signaling in CAFs of patients with urothelial cancer that failed to respond to immune checkpoint inhibition (the PD-L1 antibody atezolizumab), blocking stromal TGF-β signaling improves the response to a PD-L1–targeted antibody in a mouse model.146 In a melanoma model, specifically blocking the TGF-β present on the surface of Tregs restores anti-tumor cytotoxicity mediated by effector T cells ex vivo.147 Thus, targeted disruption of tumor-enabling signaling within the tumor microenvironment or combining TGF-β signaling inhibitors with immune-modulating therapies may be therapeutic options that overcome toxicities associated with systemic TGF-β inhibition.
Other options for limiting toxicity of TGF-β–targeted therapies include stratifying patients to treat only those with high signatures of activated TGF-β signaling. For example, patients with activated TGF-β signaling signatures that correlate with poor prognosis have been identified among patients with HCC,107 PDAC,148 and CRC.149 Using treatment holidays to limit toxicity or combination with other therapies to reduce the effective dose are also possibilities. Targeting pathways that are aberrantly activated by disruption of TGF-β signaling or targeting regulators of TGF-β signaling, such as adaptors or E3 ubiquitin ligases, could yield safer alternatives. For example, targeting the TGF-β–regulated protein vascular endothelial growth factor and immune checkpoint blockade in HCC.150
Conclusions
Efforts ranging from analysis of patient tissue to development of mouse models to studies of cells in culture have yielded progress in understanding the complex roles of TGF-β signaling. Mouse models have been critical (Table 1, Supplementary Table 1). Given the complexity in cells that produce and respond to TGF-β signals, single-cell approaches will help determine the sources of TGF-β and the responding cells and provide insight regarding how specific cell types respond to these signals or adapt to the absence of the ability to respond to these signals and clarify the roles of TGF-β signaling in various stages of disease. Such information may pave the way for combination therapies and identify TGF-β pathway biomarkers heralding the switch from dysplasia to cancer, identifying high-risk individuals.
Supplementary Material
Acknowledgments
The authors thank Shuyun Rao and Xiaochun Yang for technical support.
Funding
This research work was supported by National Institutes of Health (NIH) grants R01AA023146 (Lopa Mishra), NIH R01CA236591 (Lopa Mishra), NIH U01 CA230690-01 (Lopa Mishra), and Elaine H. Snyder Cancer Research Award (Lopa Mishra).
Abbreviations used in this paper:
- BE
Barrett’s esophagus
- BMP
bone morphogenetic protein
- CAF
cancer-associated fibroblast
- CRC
colorectal cancer
- DC
dendritic cell
- EAC
esophageal adenocarcinoma
- EBV
Epstein-Barr virus
- ECM
extracellular matrix
- EMT
epithelial-to-mesenchymal transition
- GC
gastric cancer
- GI
gastrointestinal
- IBD
inflammatory bowel disease
- IL
interleukin
- MMP
matrix metalloproteinase
- MSI
microsatellite instability
- NAFLD
nonalcoholic fatty liver disease
- NASH
nonalcoholic steatohepatitis
- PanIN
pancreatic intraepithelial neoplasia
- PDAC
pancreatic ductal adenocarcinoma
- TGF-β
transforming growth factor β
- Th
T helper cell
- TIMP
tissue inhibitor of metalloproteinases
- Treg
regulatory T cells
- UC
ulcerative colitis
Footnotes
Supplementary Material
To access the supplementary material accompanying this article, visit the online version of Gastroenterology at www.gastrojournal.org, and at http://dxdoi.org/10.1053/j.gastro.2021.04.064
CRediT Authorship Contributions
Nancy Gough, PhD (Visualization: Lead; Writing – original draft: Lead; Writing – review & editing: Lead).
Xiyan Xiang, PhD (Investigation: Lead; Visualization: Supporting; Writing – original draft: Supporting; Writing – review & editing: Supporting).
Lopa Mishra, MD (Conceptualization: Lead; Funding acquisition: Lead; Supervision: Lead; Validation: Lead; Writing – original draft: Equal; Writing – review & editing: Equal).
Conflicts of interest
The authors disclose no conflicts.
References
- 1.Korkut A, Zaidi S, Kanchi RS, et al. A pan-cancer analysis reveals high-frequency genetic alterations in mediators of signaling by the TGF-β superfamily. Cell Syst 2018;7:422–437.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Chen J, Zaidi S, Rao S, et al. Analysis of genomes and transcriptomes of hepatocellular carcinomas identifies mutations and gene expression changes in the transforming growth factor-β pathway. Gastroenterology 2018;154:195–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Waddell N, Pajic M, Patch A-M, et al. Whole genomes redefine the mutational landscape of pancreatic cancer. Nature 2015;518:495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Yang Z, Mu Z, Dabovic B, et al. Absence of integrin-mediated TGFbeta1 activation in vivo recapitulates the phenotype of TGFbeta1-null mice. J Cell Biol 2007; 176:787–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Aluwihare P, Mu Z, Zhao Z, et al. Mice that lack activity of alphavbeta6- and alphavbeta8-integrins reproduce the abnormalities of Tgfb1- and Tgfb3-null mice. J Cell Sci 2009;122:227–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Tran DQ, Andersson J, Wang R, et al. GARP (LRRC32) is essential for the surface expression of latent TGF-βeta on platelets and activated FOXP3+ regulatory T cells. Proc Natl Acad Sci U S A 2009;106:13445–13450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi M, Zhu J, Wang R, et al. Latent TGF-β structure and activation. Nature 2011;474:343–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Dong J, Buas MF, Gharahkhani P, et al. Determining risk of Barrett’s esophagus and esophageal adenocarcinoma based on epidemiologic factors and genetic variants. Gastroenterology 2018;154:1273–1281.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Onwuegbusi BA, Aitchison A, Chin S-F, et al. Impaired transforming growth factor β signalling in Barrett’s carcinogenesis due to frequent SMAD4 inactivation. Gut 2006;55:764–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lee S-W, Lien H-C, Lin C-C, et al. Low expression of transforming growth factor β in the epithelium of Barrett’s esophagus. Gastroenterol Res 2018;11:189–194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Barrett MT, Schutte M, Kern SE, et al. Allelic loss and mutational analysis of the DPC4 gene in esophageal adenocarcinoma. Cancer Res 1996;56:4351–4353. [PubMed] [Google Scholar]
- 12.Wu T-T, Watanabe T, Heitmiller R, et al. Genetic alterations in Barrett esophagus and adenocarcinomas of the esophagus and esophagogastric junction region. Am J Pathol 1998;153:287–294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Song S, Maru DM, Ajani JA, et al. Loss of TGF-β adaptor b2SP activates Notch signaling and SOX9 expression in esophageal adenocarcinoma. Cancer Res 2013; 73:2159–2169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Blum AE, Venkitachalam S, Ravillah D, et al. Systems biology analyses show hyperactivation of transforming growth factor-β and JNK signaling pathways in esophageal cancer. Gastroenterology 2019;156:1761–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Saadi A, Shannon NB, Lao-Sirieix P, et al. Stromal genes discriminate preinvasive from invasive disease, predict outcome, and highlight inflammatory pathways in digestive cancers. Proc Natl Acad Sci 2010;107:2177–2182. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Bhowmick NA, Chytil A, Plieth D, et al. TGF-ß signaling in fibroblasts modulates the oncogenic potential of adjacent epithelia. Science 2004;303:848–851. [DOI] [PubMed] [Google Scholar]
- 17.Owyang SY, Zhang M, El-Zaatari M, et al. Dendritic cell-derived TGF-β mediates the induction of mucosal regulatory T-cell response to Helicobacter infection essential for maintenance of immune tolerance in mice. Helicobacter 2020;25:e12763. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Monteleone G, Del Vecchio Blanco G, Palmieri G, et al. Induction and regulation of Smad7 in the gastric mucosa of patients with Helicobacter pylori infection. Gastroenterology 2004;126:674–682. [DOI] [PubMed] [Google Scholar]
- 19.Naef M, Ishiwata T, Friess H, et al. Differential localization of transforming growth factor-β isoforms in human gastric mucosa and overexpression in gastric carcinoma. Int J Cancer 1997;71:131–137. [DOI] [PubMed] [Google Scholar]
- 20.Kim YH, Lee HS, Lee HJ, et al. Prognostic significance of the expression of Smad4 and Smad7 in human gastric carcinomas. Ann Oncol 2004;15:574–580. [DOI] [PubMed] [Google Scholar]
- 21.Nadauld LD, Garcia S, Natsoulis G, et al. Metastatic tumor evolution and organoid modeling implicate TGFBR2 as a cancer driver in diffuse gastric cancer. Genome Biol 2014;15:428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.The Cancer Genome Atlas Research Network. Comprehensive molecular characterization of gastric adenocarcinoma. Nature 2014;513:202–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fukuda M, Longnecker R. Latent membrane protein 2A inhibits transforming growth factor-beta 1-induced apoptosis through the phosphatidylinositol 3-kinase/Akt pathway. J Virol 2004;78:1697–1705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Fukuda M, Ikuta K, Yanagihara K, et al. Effect of transforming growth factor-beta1 on the cell growth and Epstein-Barr virus reactivation in EBV-infected epithelial cell lines. Virology 2001;288:109–118. [DOI] [PubMed] [Google Scholar]
- 25.Myeroff LL, Parsons R, Kim SJ, et al. A transforming growth factor beta receptor type II gene mutation common in colon and gastric but rare in endometrial cancers with microsatellite instability. Cancer Res 1995;55:5545–5547. [PubMed] [Google Scholar]
- 26.Verma R, Agarwal AK, Sakhuja P, et al. Microsatellite instability in mismatch repair and tumor suppressor genes and their expression profiling provide important targets for the development of biomarkers in gastric cancer. Gene 2019;710:48–58. [DOI] [PubMed] [Google Scholar]
- 27.Liu JZ, van Sommeren S, Huang H, et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat Genet 2015;47:979–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Jostins L, Ripke S, Weersma RK, et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature 2012;491:119–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Yamashita A, Inamine T, Suzuki S, et al. Genetic variants of SMAD2/3/4/7 are associated with susceptibility to ulcerative colitis in a Japanese genetic background. Immunol Lett 2019;207:64–72. [DOI] [PubMed] [Google Scholar]
- 30.Kotlarz D, Marquardt B, Barøy T, et al. Human TGF-β1 deficiency causes severe inflammatory bowel disease and encephalopathy. Nat Genet 2018;50:344–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Yang X, Letterio JJ, Lechleider RJ, et al. Targeted disruption of SMAD3 results in impaired mucosal immunity and diminished T cell responsiveness to TGF-β. EMBO J 1999;18:1280–1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Shull MM, Ormsby I, Kier AB, et al. Targeted disruption of the mouse transforming growth factor-β1 gene results in multifocal inflammatory disease. Nature 1992; 359:693–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Neurath MF, Fuss I, Kelsall BL, et al. Experimental granulomatous colitis in mice is abrogated by induction of TGF-βeta-mediated oral tolerance. J Exp Med 1996; 183:2605–2616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Brand S, Beigel F, Olszak T, et al. IL-22 is increased in active Crohn’s disease and promotes proinflammatory gene expression and intestinal epithelial cell migration. Am J Physiol Gastrointest Liver Physiol 2006;290:G827–G838. [DOI] [PubMed] [Google Scholar]
- 35.Rutz S, Noubade R, Eidenschenk C, et al. Transcription factor c-Maf mediates the TGF-β-dependent suppression of IL-22 production in T(H)17 cells. Nat Immunol 2011;12:1238–1245. [DOI] [PubMed] [Google Scholar]
- 36.Li MO, Wan YY, Flavell RA. T cell-produced transforming growth factor-β1 controls T cell tolerance and regulates Th1- and Th17-cell differentiation. Immunity 2007; 26:579–591. [DOI] [PubMed] [Google Scholar]
- 37.Gorelik L, Flavell RA. Abrogation of TGFβ signaling in T cells leads to spontaneous T cell differentiation and autoimmune disease. Immunity 2000;12:171–181. [DOI] [PubMed] [Google Scholar]
- 38.Ramalingam R, Larmonier CB, Thurston RD, et al. Dendritic cell-specific disruption of TGF-β receptor II leads to altered regulatory T cell phenotype and spontaneous multiorgan autoimmunity. J Immunol 2012;189:3878–3893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ihara S, Hirata Y, Serizawa T, et al. TGF-β signaling in dendritic cells governs colonic homeostasis by controlling epithelial differentiation and the luminal microbiota. J Immunol 2016;196:4603–4613. [DOI] [PubMed] [Google Scholar]
- 40.Monteleone G, Kumberova A, Croft NM, et al. Blocking Smad7 restores TGF-β1 signaling in chronic inflammatory bowel disease. J Clin Inves 2001;108:601–609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Benahmed M, Meresse B, Arnulf B, et al. Inhibition of TGF-β signaling by IL-15: a new role for IL-15 in the loss of immune homeostasis in celiac disease. Gastroenterology 2007;132:994–1008. [DOI] [PubMed] [Google Scholar]
- 42.Boirivant M, Pallone F, Di Giacinto C, et al. Inhibition of Smad7 with a specific antisense oligonucleotide facilitates TGF-βeta1-mediated suppression of colitis. Gastroenterology 2006;131:1786–1798. [DOI] [PubMed] [Google Scholar]
- 43.Monteleone G, Neurath MF, Ardizzone S, et al. Mongersen, an oral SMAD7 antisense oligonucleotide, and Crohn’s disease. N Engl J Med 2015;372:1104–1113. [DOI] [PubMed] [Google Scholar]
- 44.Monteleone G, Fantini MC, Onali S, et al. Phase I clinical trial of Smad7 knockdown using antisense oligonucleotide in patients with active Crohn’s disease. Mol Ther 2012;20:870–876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Sands BE, Feagan BG, Sandborn WJ, et al. Mongersen (GED-0301) for active Crohn’s disease: results of a phase 3 study. Am J Gastroenterol 2020; 115:738–745. [DOI] [PubMed] [Google Scholar]
- 46.Gundersen MD, Goll R, Fenton CG, et al. Fibrosis mediators in the colonic mucosa of acute and healed ulcerative colitis. Clin Transl Gastroenterol 2019; 10:e00082. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kirkegaard T, Hansen A, Bruun E, et al. Expression and localisation of matrix metalloproteinases and their natural inhibitors in fistulae of patients with Crohn’s disease. Gut 2004;53:701–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Bailey CJ, Hembry RM, Alexander A, et al. Distribution of the matrix metalloproteinases stromelysin, gelatinases A and B, and collagenase in Crohn’s disease and normal intestine. J Clin Pathol 1994;47:113–116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.McKaig BC, McWilliams D, Watson SA, et al. Expression and regulation of tissue inhibitor of metalloproteinase-1 and matrix metalloproteinases by intestinal myofibroblasts in inflammatory bowel disease. Am J Pathol 2003; 162:1355–1360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.McKaig BC, Hughes K, Tighe PJ, et al. Differential expression of TGF-β isoforms by normal and inflammatory bowel disease intestinal myofibroblasts. Am J Physiol Cell Physiol 2002;282:C172–C182. [DOI] [PubMed] [Google Scholar]
- 51.Sabatino AD, Jackson CL, Pickard KM, et al. Transforming growth factor β signalling and matrix metalloproteinases in the mucosa overlying Crohn’s disease strictures. Gut 2009;58:777–789. [DOI] [PubMed] [Google Scholar]
- 52.Frank DN, Amand ALS, Feldman RA, et al. Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc Natl Acad Sci U S A 2007;104:13780–13785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sokol H, Seksik P, Furet JP, et al. Low counts of Faecalibacterium prausnitzii in colitis microbiota. Inflamm Bowel Dis 2009;15:1183–1189. [DOI] [PubMed] [Google Scholar]
- 54.Gevers D, Kugathasan S, Denson Lee A, et al. The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 2014;15:382–392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Atarashi K, Tanoue T, Oshima K, et al. T reg induction by a rationally selected mixture of Clostridia strains from the human microbiota. Nature 2013;500:232–236. [DOI] [PubMed] [Google Scholar]
- 56.Furusawa Y, Obata Y, Fukuda S, et al. Commensal microbe-derived butyrate induces the differentiation of colonic regulatory T cells. Nature 2013;504:446–450. [DOI] [PubMed] [Google Scholar]
- 57.Bloom Seth M, Bijanki Vinieth N, Nava Gerardo M, et al. Commensal bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe 2011;9:390–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Mishima Y, Sartor RB. Manipulating resident microbiota to enhance regulatory immune function to treat inflammatory bowel diseases. J Gastroenterol 2020; 55:4–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Engle SJ, Ormsby I, Pawlowski S, et al. Elimination of colon cancer in germ-free transforming growth factor beta 1-deficient mice. Cancer Res 2002;62:6362–6366. [PubMed] [Google Scholar]
- 60.Zhu Y, Richardson JA, Parada LF, et al. Smad3 mutant mice develop metastatic colorectal cancer. Cell 1998; 94:703–714. [DOI] [PubMed] [Google Scholar]
- 61.Maggio-Price L, Treuting P, Zeng W, et al. Helicobacter infection is required for inflammation and colon cancer in SMAD3-deficient mice. Cancer Res 2006;66:828–838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Gu S, Zaidi S, Hassan MI, et al. Mutated CEACAMs disrupt transforming growth factor beta signaling and alter the intestinal microbiome to promote colorectal carcinogenesis. Gastroenterology 2020;158:238–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Chen J, Raju GS, Jogunoori W, et al. Mutational profiles reveal an aberrant TGF-β-CEA regulated pathway in colon adenomas. PLoS One 2016;11:e0153933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Tang Y, Katuri V, Srinivasan R, et al. Transforming growth factor-β suppresses nonmetastatic colon cancer through Smad4 and adaptor protein ELF at an early stage of tumorigenesis. Cancer Res 2005;65:4228–4237. [DOI] [PubMed] [Google Scholar]
- 65.Kim BG, Li C, Qiao W, et al. Smad4 signalling in T cells is required for suppression of gastrointestinal cancer. Nature 2006;441:1015–1019. [DOI] [PubMed] [Google Scholar]
- 66.Calva-Cerqueira D, Chinnathambi S, Pechman B, et al. The rate of germline mutations and large deletions of SMAD4 and BMPR1A in juvenile polyposis. Clin Genet 2009;75:79–85. [DOI] [PubMed] [Google Scholar]
- 67.Muñoz NM, Upton M, Rojas A, et al. Transforming growth factor b receptor type II inactivation induces the malignant transformation of intestinal neoplasms initiated by apc mutation. Cancer Res 2006;66:9837–9844. [DOI] [PubMed] [Google Scholar]
- 68.Oshima H, Nakayama M, Han TS, et al. Suppressing TGFβ signaling in regenerating epithelia in an inflammatory microenvironment is sufficient to cause invasive intestinal cancer. Cancer Res 2015;75:766–776. [DOI] [PubMed] [Google Scholar]
- 69.Alberici P, Jagmohan-Changur S, De Pater E, et al. Smad4 haploinsufficiency in mouse models for intestinal cancer. Oncogene 2006;25:1841–1851. [DOI] [PubMed] [Google Scholar]
- 70.Sodir NM, Chen X, Park R, et al. Smad3 deficiency promotes tumorigenesis in the distal colon of ApcMin/+mice. Cancer Res 2006;66:8430–8438. [DOI] [PubMed] [Google Scholar]
- 71.Tauriello DVF, Palomo-Ponce S, Stork D, et al. TGFβ drives immune evasion in genetically reconstituted colon cancer metastasis. Nature 2018;554:538–543. [DOI] [PubMed] [Google Scholar]
- 72.Calon A, Lonardo E, Berenguer-Llergo A, et al. Stromal gene expression defines poor-prognosis subtypes in colorectal cancer. Nat Genet 2015;47:320–329. [DOI] [PubMed] [Google Scholar]
- 73.Calon A, Espinet E, Palomo-Ponce S, et al. Dependency of colorectal cancer on a TGF-β-driven program in stromal cells for metastasis initiation. Cancer Cell 2012; 22:571–584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Broderick P, Carvajal-Carmona L, Pittman AM, et al. A genome-wide association study shows that common alleles of SMAD7 influence colorectal cancer risk. Nat Genet 2007;39:1315–1317. [DOI] [PubMed] [Google Scholar]
- 75.Halder SK, Beauchamp RD, Datta PK. Smad7 induces tumorigenicity by blocking TGF-βeta-induced growth inhibition and apoptosis. Exp Cell Res 2005;307:231–246. [DOI] [PubMed] [Google Scholar]
- 76.Halder SK, Rachakonda G, Deane NG, et al. Smad7 induces hepatic metastasis in colorectal cancer. Br J Cancer 2008;99:957–965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Rizzo A, Waldner MJ, Stolfi C, et al. Smad7 expression in T cells prevents colitis-associated cancer. Cancer Res 2011;71:7423–7432. [DOI] [PubMed] [Google Scholar]
- 78.Rizzo A, De Mare V, Rocchi C, et al. Smad7 induces plasticity in tumor-infiltrating Th17 cells and enables TNF-alpha-mediated killing of colorectal cancer cells. Carcinogenesis 2014;35:1536–1546. [DOI] [PubMed] [Google Scholar]
- 79.Angulo P, Kleiner DE, Dam-Larsen S, et al. Liver fibrosis, but no other histologic features, is associated with long-term outcomes of patients with nonalcoholic fatty liver disease. Gastroenterology 2015;149:389–397.e10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hagström H, Nasr P, Ekstedt M, et al. Fibrosis stage but not NASH predicts mortality and time to development of severe liver disease in biopsy-proven NAFLD. J Hepatol 2017;67:1265–1273. [DOI] [PubMed] [Google Scholar]
- 81.Karlmark KR, Weiskirchen R, Zimmermann HW, et al. Hepatic recruitment of the inflammatory Gr1+ monocyte subset upon liver injury promotes hepatic fibrosis. Hepatology 2009;50:261–274. [DOI] [PubMed] [Google Scholar]
- 82.Meijer C, Wiezer MJ, Diehl AM, et al. Kupffer cell depletion by CI2MDP-liposomes alters hepatic cytokine expression and delays liver regeneration after partial hepatectomy. Liver 2000;20:66–77. [DOI] [PubMed] [Google Scholar]
- 83.Ghafoory S, Varshney R, Robison T, et al. Platelet TGF-β1 deficiency decreases liver fibrosis in a mouse model of liver injury. Blood Adv 2018;2:470–480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Herrera B, Alvarez AM, Beltrán J, et al. Resistance to TGF-βeta-induced apoptosis in regenerating hepatocytes. J Cell Physiol 2004;201:385–392. [DOI] [PubMed] [Google Scholar]
- 85.Bissell DM, Wang SS, Jarnagin WR, et al. Cell-specific expression of transforming growth factor-beta in rat liver. Evidence for autocrine regulation of hepatocyte proliferation. J Clin Invest 1995;96:447–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Hellerbrand C, Stefanovic B, Giordano F, et al. The role of TGFβ1 in initiating hepatic stellate cell activation in vivo. J Hepatol 1999;30:77–87. [DOI] [PubMed] [Google Scholar]
- 87.Kanzler S, Lohse AW, Keil A, et al. TGF-βeta1 in liver fibrosis: an inducible transgenic mouse model to study liver fibrogenesis. Am J Physiol 1999;276:G1059–G1068. [DOI] [PubMed] [Google Scholar]
- 88.Dooley S, Hamzavi J, Breitkopf K, et al. Smad7 prevents activation of hepatic stellate cells and liver fibrosis in rats. Gastroenterology 2003;125:178–191. [DOI] [PubMed] [Google Scholar]
- 89.Li Y, Wang J, Asahina K. Mesothelial cells give rise to hepatic stellate cells and myofibroblasts via mesothelial–mesenchymal transition in liver injury. Proc Natl Acad of Sci U S A 2013;110:2324–2329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Fabre T, Molina MF, Soucy G, et al. Type 3 cytokines IL-17A and IL-22 drive TGF-β-dependent liver fibrosis. Sci Immunol 2018;3(28):eaar7754. [DOI] [PubMed] [Google Scholar]
- 91.Fabre T, Kared H, Friedman SL, et al. IL-17A enhances the expression of profibrotic genes through upregulation of the TGF-β receptor on hepatic stellate cells in a JNK-dependent manner. J Immunol 2014;193:3925–3933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ringelhan M, McKeating JA, Protzer U. Viral hepatitis and liver cancer. Philos Trans R Soc Lond B Biol Sci 2017;372(1732):20160274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Matsuzaki K, Seki T, Okazaki K. TGF-β signal shifting between tumor suppression and fibro-carcinogenesis in human chronic liver diseases. J Gastroenterol 2014; 49:971–981. [DOI] [PubMed] [Google Scholar]
- 94.Dong Y, Zheng Q, Wang Z, et al. Higher matrix stiffness as an independent initiator triggers epithelial-mesenchymal transition and facilitates HCC metastasis. J Hematol Oncol 2019;12:112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Schrader J, Gordon-Walker TT, Aucott RL, et al. Matrix stiffness modulates proliferation, chemotherapeutic response, and dormancy in hepatocellular carcinoma cells. Hepatology 2011;53:1192–1205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Tang B, Böttinger EP, Jakowlew SB, et al. Transforming growth factor-β1 is a new form of tumor suppressor with true haploid insufficiency. Nat Med 1998;4:802–807. [DOI] [PubMed] [Google Scholar]
- 97.Kanzler S, Meyer E, Lohse AW, et al. Hepatocellular expression of a dominant-negative mutant TGF-β type II receptor accelerates chemically induced hepatocarcinogenesis. Oncogene 2001;20:5015–5024. [DOI] [PubMed] [Google Scholar]
- 98.Chen C-L, Tsukamoto H, Liu J-C, et al. Reciprocal regulation by TLR4 and TGF-β in tumor-initiating stem-like cells. J Clin Invest 2013;123:2832–2849. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 99.Baek HJ, Lim SC, Kitisin K, et al. Hepatocellular cancer arises from loss of transforming growth factor beta signaling adaptor protein embryonic liver fodrin through abnormal angiogenesis. Hepatology 2008;48:1128–1137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Tang Y, Kitisin K, Jogunoori W, et al. Progenitor/stem cells give rise to liver cancer due to aberrant TGF-β and IL-6 signaling. Proc Natl Acad Sci 2008;105:2445–2450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Factor VM, Kao C-Y, Santoni-Rugiu E, et al. Constitutive expression of mature transforming growth factor β1 in the liver accelerates hepatocarcinogenesis in transgenic mice. Cancer Res 1997;57:2089–2095. [PubMed] [Google Scholar]
- 102.Deane NG, Lee H, Hamaamen J, et al. Enhanced tumor formation in cyclin D1 × transforming growth factor b1 double transgenic mice with characterization by magnetic resonance imaging. Cancer Res 2004;64:1315–1322. [DOI] [PubMed] [Google Scholar]
- 103.Malfettone A, Soukupova J, Bertran E, et al. Transforming growth factor-β-induced plasticity causes a migratory stemness phenotype in hepatocellular carcinoma. Cancer Lett 2017;392:39–50. [DOI] [PubMed] [Google Scholar]
- 104.Dzieran J, Fabian J, Feng T, et al. Comparative analysis of TGF-β/Smad signaling dependent cytostasis in human hepatocellular carcinoma cell lines. PLoS One 2013;8:e72252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ma C, Kesarwala AH, Eggert T, et al. NAFLD causes selective CD4(+) T lymphocyte loss and promotes hepatocarcinogenesis. Nature 2016;531:253–257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Shen Y, Wei Y, Wang Z, et al. TGF-β regulates hepatocellular carcinoma progression by inducing Treg cell polarization. Cell Physiol Biochem 2015;35:1623–1632. [DOI] [PubMed] [Google Scholar]
- 107.Coulouarn C, Factor VM, Thorgeirsson SS. Transforming growth factor-beta gene expression signature in mouse hepatocytes predicts clinical outcome in human cancer. Hepatology 2008;47:2059–2067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Dooley S, Hamzavi J, Ciuclan L, et al. Hepatocyte-specific Smad7 expression attenuates TGF-βeta-mediated fibrogenesis and protects against liver damage. Gastroenterology 2008;135:642–659. [DOI] [PubMed] [Google Scholar]
- 109.Feng T, Dzieran J, Yuan X, et al. Hepatocyte-specific Smad7 deletion accelerates DEN-induced HCC via activation of STAT3 signaling in mice. Oncogenesis 2017;6:e294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Wang J, Zhao J, Chu ES, et al. Inhibitory role of Smad7 in hepatocarcinogenesis in mice and in vitro. J Pathol 2013;230:441–452. [DOI] [PubMed] [Google Scholar]
- 111.Wang H, Song X, Liao H, et al. Overexpression of SMAD7 activates the YAP/NOTCH cascade and promotes liver carcinogenesis in mice and humans [published online ahead of print December 24, 2020]. Hepatology 10.1002/hep.31692. [DOI] [Google Scholar]
- 112.Lin T-H, Shao Y-Y, Chan S-Y, et al. High serum transforming growth factor- 1 levels predict outcome in hepatocellular carcinoma patients treated with sorafenib. Clin Cancer Res 2015;21:3678–3684. [DOI] [PubMed] [Google Scholar]
- 113.Melisi D, Ishiyama S, Sclabas GM, et al. LY2109761, a novel transforming growth factor beta receptor type I and type II dual inhibitor, as a therapeutic approach to suppressing pancreatic cancer metastasis. Mol Cancer Ther 2008;7:829–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Mazzocca A, Fransvea E, Lavezzari G, et al. Inhibition of transforming growth factor β receptor I kinase blocks hepatocellular carcinoma growth through neo-angiogenesis regulation. Hepatology 2009;50:1140–1151. [DOI] [PubMed] [Google Scholar]
- 115.Fransvea E, Angelotti U, Antonaci S, et al. Blocking transforming growth factor–beta up-regulates E-cadherin and reduces migration and invasion of hepatocellular carcinoma cells. Hepatology 2008;47:1557–1566. [DOI] [PubMed] [Google Scholar]
- 116.Fransvea E, Mazzocca A, Antonaci S, et al. Targeting transforming growth factor (TGF)-βRI inhibits activation of β1 integrin and blocks vascular invasion in hepatocellular carcinoma. Hepatology 2009;49:839–850. [DOI] [PubMed] [Google Scholar]
- 117.Serova M, Tijeras-Raballand A, Santos CD, et al. Effects of TGF-βeta signalling inhibition with galunisertib (LY2157299) in hepatocellular carcinoma models and in ex vivo whole tumor tissue samples from patients. Oncotarget 2015;6:21614–21627. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Rani B, Malfettone A, Dituri F, et al. Galunisertib suppresses the staminal phenotype in hepatocellular carcinoma by modulating CD44 expression. Cell Death Dis 2018;9:1–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Peery AF, Crockett SD, Murphy CC, et al. Burden and cost of gastrointestinal, liver, and pancreatic diseases in the United States: update 2018. Gastroenterology 2019; 156:254–272.e11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jones S, Zhang X, Parsons DW, et al. Core Signaling pathways in human pancreatic cancers revealed by global genomic analyses. Science 2008;321:1801–1806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Wilentz RE, Iacobuzio-Donahue CA, Argani P, et al. Loss of expression of Dpc4 in pancreatic intraepithelial neoplasia: evidence that DPC4 Inactivation occurs late in neoplastic progression. Cancer Res 2000;60:2002–2006. [PubMed] [Google Scholar]
- 122.Ijichi H, Chytil A, Gorska AE, et al. Aggressive pancreatic ductal adenocarcinoma in mice caused by pancreas-specific blockade of transforming growth factor-beta signaling in cooperation with active Kras expression. Genes Dev 2006;20:3147–3160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Mueller S, Engleitner T, Maresch R, et al. Evolutionary routes and KRAS dosage define pancreatic cancer phenotypes. Nature 2018;554:62–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Leung L, Radulovich N, Zhu C-Q, et al. Loss of canonical Smad4 signaling promotes KRAS driven malignant transformation of human pancreatic duct epithelial cells and metastasis. PLOS One 2013;8:e84366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.David Charles J, Huang Y-H, Chen M, et al. TGF-β tumor suppression through a lethal EMT. Cell 2016;164:1015–1030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kuang C, Xiao Y, Liu X, et al. In vivo disruption of TGF-beta signaling by Smad7 leads to premalignant ductal lesions in the pancreas. Proc Natl Acad Sci U S A 2006; 103:1858–1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Friess H, Yamanaka Y, Büchler M, et al. Enhanced expression of transforming growth factor beta isoforms in pancreatic cancer correlates with decreased survival. Gastroenterology 1993;105:1846–1856. [DOI] [PubMed] [Google Scholar]
- 128.Wang P, Fan J, Chen Z, et al. Low-level expression of Smad7 correlates with lymph node metastasis and poor prognosis in patients with pancreatic cancer. Ann Surg Oncol 2009;16:826–835. [DOI] [PubMed] [Google Scholar]
- 129.Park H, Bang J-H, Nam A-R, et al. The prognostic role of soluble TGF-βeta and its dynamics in unresectable pancreatic cancer treated with chemotherapy. Cancer Med 2020;9:43–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Peng J, Sun B-F, Chen C-Y, et al. Single-cell RNA-seq highlights intra-tumoral heterogeneity and malignant progression in pancreatic ductal adenocarcinoma. Cell Res 2019;29:725–738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Bernard V, Semaan A, Huang J, et al. Single-cell transcriptomics of pancreatic cancer precursors demonstrates epithelial and microenvironmental heterogeneity as an early event in neoplastic progression. Clin Cancer Res 2019;25:2194–2205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Moffitt RA, Marayati R, Flate EL, et al. Virtual microdissection identifies distinct tumor- and stroma-specific subtypes of pancreatic ductal adenocarcinoma. Nat Genet 2015;47:1168–1178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Biffi G, Oni TE, Spielman B, et al. IL1-induced JAK/STAT signaling is antagonized by TGFβ to shape CAF heterogeneity in pancreatic ductal adenocarcinoma. Cancer Discov 2019;9:282–301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Mahajan UM, Langhoff E, Goni E, et al. Immune cell and stromal signature associated with progression-free survival of patients with resected pancreatic ductal adenocarcinoma. Gastroenterology 2018;155:1625–1639.e2. [DOI] [PubMed] [Google Scholar]
- 135.Medicherla S, Li L, Ma JY, et al. Antitumor activity of TGF-β, inhibitor is dependent on the microenvironment. Anticancer Res 2007;27:4149–4157. [PubMed] [Google Scholar]
- 136.Murakami T, Hiroshima Y, Miyake K, et al. Color-coded intravital imaging demonstrates a transforming growth factor-β (TGF-β) antagonist selectively targets stromal cells in a human pancreatic-cancer orthotopic mouse model. Cell Cycle 2017;16:1008–1014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Gore J, Imasuen-Williams IE, Conteh AM, et al. Combined targeting of TGF-β, EGFR and HER2 suppresses lymphangiogenesis and metastasis in a pancreatic cancer model. Cancer Lett 2016;379:143–153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Gore J, Craven KE, Wilson JL, et al. TCGA data and patient-derived orthotopic xenografts highlight pancreatic cancer-associated angiogenesis. Oncotarget 2015; 6:7504–7521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Laklai H, Miroshnikova YA, Pickup MW, et al. Genotype tunes pancreatic ductal adenocarcinoma tissue tension to induce matricellular fibrosis and tumor progression. Nat Med 2016;22:497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Anderton MJ, Mellor HR, Bell A, et al. Induction of heart valve lesions by small-molecule ALK5 inhibitors. Toxicol Pathol 2011;39:916–924. [DOI] [PubMed] [Google Scholar]
- 141.Mitra MS, Lancaster K, Adedeji AO, et al. A potent pan-TGFβ neutralizing monoclonal antibody elicits cardiovascular toxicity in mice and cynomolgus monkeys. Toxicol Sci 2020;175:24–34. [DOI] [PubMed] [Google Scholar]
- 142.Rak GD, White MR, Augustine-Rauch K, et al. Intermittent dosing of the transforming growth factor beta receptor 1 inhibitor, BMS-986260, mitigates class-based cardiovascular toxicity in dogs but not rats. J Appl Toxicol 2020;40:931–946. [DOI] [PubMed] [Google Scholar]
- 143.Lacouture ME, Morris JC, Lawrence DP, et al. Cutaneous keratoacanthomas/squamous cell carcinomas associated with neutralization of transforming growth factor β by the monoclonal antibody fresolimumab (GC1008). Cancer Immunol Immunother 2015;64:437–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Kelley RK, Gane E, Assenat E, et al. A phase 2 study of galunisertib (TGF-β1 receptor type I inhibitor) and sorafenib in patients with advanced hepatocellular carcinoma. Clin Transl Gastroenterol 2019;10:e00056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Melisi D, Garcia-Carbonero R, Macarulla T, et al. Galunisertib plus gemcitabine vs. gemcitabine for first-line treatment of patients with unresectable pancreatic cancer. Br J Cancer 2018;119:1208–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Mariathasan S, Turley SJ, Nickles D, et al. TGFβ attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554:544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Budhu S, Schaer DA, Li Y, et al. Blockade of surface-bound TGF-β on regulatory T cells abrogates suppression of effector T cell function in the tumor microenvironment. Sci Signal 2017:10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Collisson EA, Sadanandam A, Olson P, et al. Subtypes of pancreatic ductal adenocarcinoma and their differing responses to therapy. Nat Med 2011;17:500–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Fessler E, Drost J, van Hooff SR, et al. TGFβ signaling directs serrated adenomas to the mesenchymal colorectal cancer subtype. EMBO Mol Med 2016;8:745–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lee WS, Yang H, Chon HJ, et al. Combination of anti-angiogenic therapy and immune checkpoint blockade normalizes vascular-immune crosstalk to potentiate cancer immunity. Exp Mol Med 2020;52:1475–1485. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.