

Abstract

A series of brigatinib derivatives were designed and synthesized as new potent and selective EGFRT790M/C797S inhibitors. One of the most potent and selective compounds 18k strongly suppressed the EGFRL858R/T790M/C797S and EGFR19Del/T790M/C797S kinases with IC50 values of 0.7 and 3.6 nM, respectively, which were over 54-fold more potent than the lead compound. 18k also demonstrated promising EGFRT790M/C797S mutant selectivity, and was 94-fold less potent against the wild type EGFR. A cocrystal structure of EGFRT790M/C797S with a close derivative 18f was solved to provide insight on the inhibitor’s binding mode. Moreover, compound 18k was orally bioavailable and demonstrated highly desirable PK properties, making it a promising lead compound for further structural optimization.
Keywords: Epidermal Growth Factor Receptor (EGFR), Resistant Mutation, C797S, Antitumor
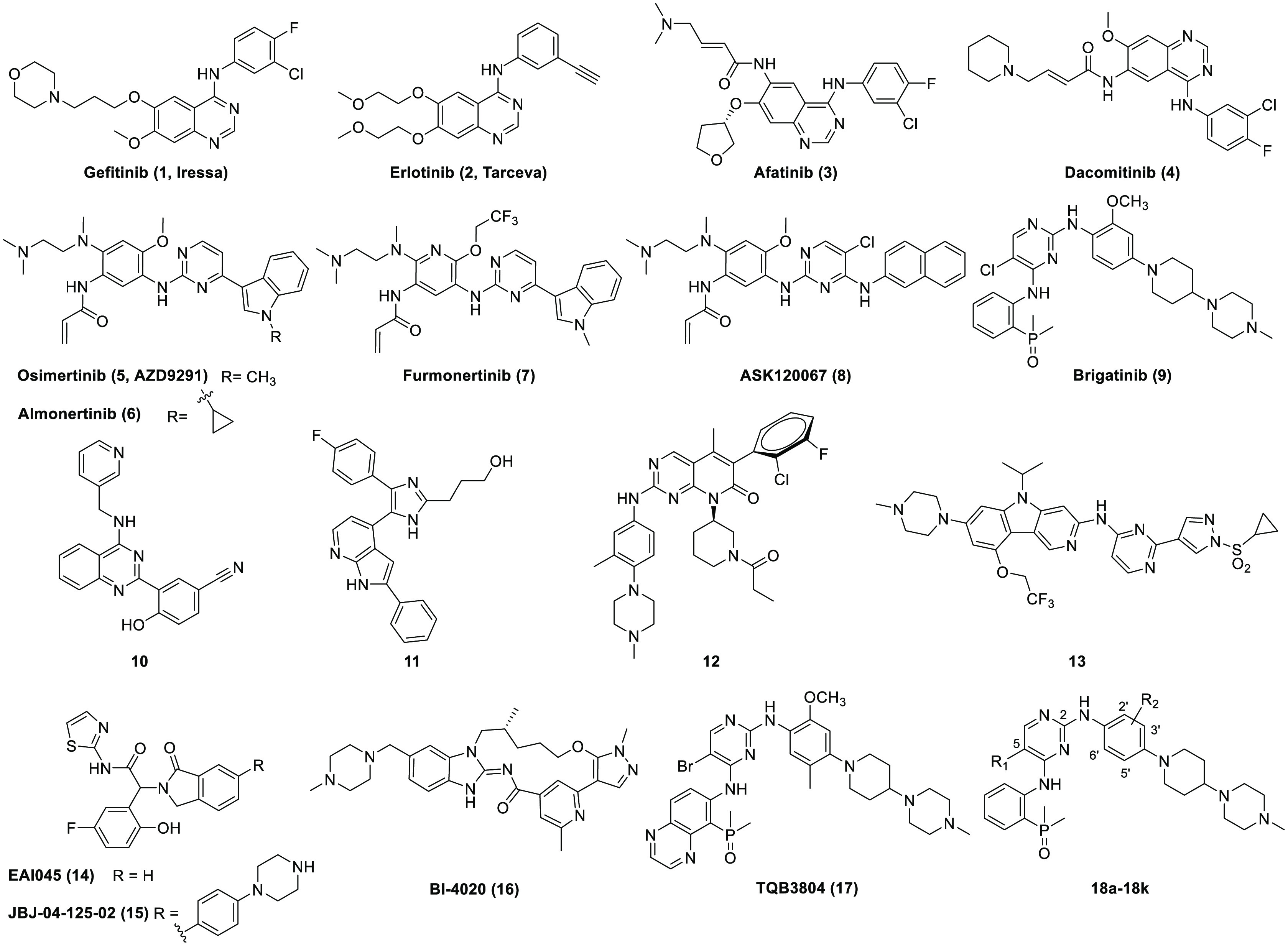
Epidermal growth factor receptor (EGFR) is a well validated target for non-small cell lung cancer (NSCLC) drug discovery.1 Five small molecular EGFR inhibitors (i.e., gefitinib (1),2 erlotinib (2),3 afatinib (3),4 dacomitinib (4),5 and osimertinib (AZD9291, 5),6,7Figure 1) have been approved by the U.S. Food and Drug Administration (FDA). The first generation drugs 1 and 2 clinically apply to the treatment of NSCLC patients with EGFR activating mutations (e.g., exon 19 deletion (19 Del) and a Leu858 → Arg858 point mutation (L858R)). Irreversible compounds 3 and 4 belong to the second generation EGFR inhibitor drugs which achieve significant clinical benefit for metastatic and advanced NSCLC patients harboring “gain of function” mutated EGFRs. Although drugs 3 and 4 exhibit strong suppression on the resistant Thr790 → Met790 (T790M) mutants in preclinical models, the low clinical maximal tolerated dose (MTD) attributing to the strong wild-type EGFR (EGFRWT) inhibition limited their application in resistant patients with the EGFRT790M mutant. Wild-type sparing EGFRT790M inhibitor 5 is the first FDA approved third generation drug for resistant NSCLC patients with EGFRT790M mutations, and it was later approved as a first-line treatment for metastatic NSCLC patients with positive EGFR mutations. Following the success of 5, two structurally close derivatives, i.e., almonertinib (6)8 and furmonertinib (7),9 were approved by National Medical Products Administration of China (NMPA) in 2020 and 2021, respectively. Several other third generation EGFR inhibitors, e.g., lazertinib, rezivertinib, and abivertinib, are in late stages of clinical investigation.10 We have also developed ASK120067 (8) as a highly potent and selective wild-type sparing EGFRT790M inhibitor. A New Drug Application (NDA) was filed recently in China based on its highly promising efficacy in advanced and multiple brain metastatic NSCLC patients harboring EGFRT790M mutations.11
Figure 1.
Chemical structures of approved or literature reported EGFR inhibitors.
Structurally, all of the third generation EGFR inhibitors possess an acrylamide warhead to covalently react with the Cys797 residue to achieve improved competitiveness with the high concentration ATP (mM level) in cells. A tertiary C797S (Cys797 → Ser797) mutation would impair the covalent formation between Cys797 and the acrylamide warhead and induce the resistance against the third generation drugs in approximately 20% of the pretreated NSCLC patients.12 Although the patients with trans EGFRT790M/C797S mutations in which the T790M and C797S mutations are located on different alleles could be efficiently treated by combinational therapies of a first generation drug and 5, the cis- EGFRT790M/C797S mutation (mutations are on a same allele) mediated resistance remains an unmet clinical need.13,14 Therefore, development of the fourth generation EGFRT790M/C797S inhibitors is highly desirable for overcoming the new mutation mediated resistance.15
Several classes of EGFRT790M/C797S inhibitors have been reported. Examples include the traditional ATP competitive inhibitor brigatinib (9) and derivatives,16 4-hydroxy-3-(4-aminoquinazolin-2-yl)benzonitrile derivative 10,17 trisubstituted imidazole derivative 11,18 5-methylpyrimidopyridone derivative 12,19N-(pyrimidin-4-yl)-5H-pyrido[4,3-b]indol-3-amine analogue 13,20 allosteric inhibitors EAI045 (14)21 and JBJ-04-125-02 (15),22 and macrocyclic molecules BI4020 (16).23 To the best of our knowledge, only the brigatinib derivative TQB3804 (17) was advanced to clinical investigation in resistant NSCLC patients with EGFRT790M/C797S mutations (NCT04128085).24,25 Herein, we would like to report a straightforward structural optimization of compound 9. The effort eventually yielded the discovery of a new highly potent and selective EGFRT790M/C797S inhibitor 18k with approximatly 54-fold improved kinase inhibitory potency.
A cocrystal structure of EGFRWT with inhibitor 9 was recently reported (PDB 7AEM, Figure 2a in stick and line representation and Figure 2b in surface representation).26 The results showed that NH group of the aniline in 9 made a donor interaction with the backbone carbonyl of Met793, while the pyrimidine N accepted a hydrogen bond from the backbone NH of Leu792, in the hinge region. An oxygen atom of the dimethylphosphine oxide (DMPO) group formed acceptor interactions with the side chain NH3 of the conserved catalytic Lys745 and the side chain OH of Thr854, respectively, whereas the hydrophilic piperazinylpiperdine moiety was oriented to the solvent accessible surface, indicating it might not contribute much to the protein binding. A preliminary computational study suggested it might bind with EGFRT790M/C797S in a similar mode (Figure 2c). The structural information also suggested that the 2′-methoxyl group might cause potential steric clash with Pro794 although it could occupy a small hydrophobic cavity in EGFR to improve the target selectivity. This was consistent with the previous observation that removal of the corresponding methoxyl group resulted in improved EGFR kinase inhibitory potency for the third generation inhibitors.27,28
Figure 2.
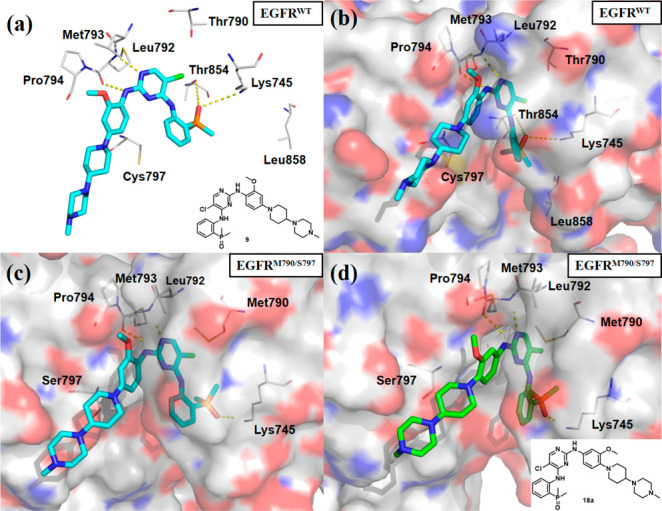
X-ray crystal structure of brigatinib (9) with EGFRThr790/Cys797(EGFRWT) kinase domain (PDB 7AEM) shown in cyan and gray in stick and line representation (a) or in surface representation (b). Preliminary computational docking complex of 9 (c) and 18a (d) with EGFRMet790/Ser797 (PDB 5ZTO). Hydrogen bonds are indicated by yellow hatched lines to key amino acids.
A preliminary docking study also suggested that a change of the 2′-methoxyl group to 3′- position would increase the EGFR inhibitory activity by avoiding the potential steric clash (Figure 2d). Therefore, a series of brigatinib derivatives with substituents at the 3′- position of 2-aniline group were synthesized by following the reported procedures (Scheme 1).29 Beginning with the commercially available 2-iodoaniline (19) and dimethylphosphine oxide, intermediate 20 was obtained via a palladium-catalyzed coupling reaction. Then a two-step, sequential SNAr reaction was conducted to give the target compounds 18a–18k. Biological evaluation showed that compound 18a indeed exhibited a 16-fold improved EGFRL858R/T790M/C797S (EGFRTM1) inhibitory potency with an IC50 value of 2.4 nM compared with 9 (Table 1). In addition , the switch of the substituent position also resulted in EGFRWT and EGFRL858R/T790M (EGFRDM) potency improvement by factors of 5.1 and 21.2, respectively. Notably, compound 18a exhibited an improved wild-type sparing selectivity with an EGFRWT/EGFRTM1 ratio of 21.3, while the corresponding value for 9 was 6.8. Further investigation also revealed that the 3′-position could be substituted by a variety of substituents, e.g., −CH3 (18b), −OCF3 (18c), −CF3 (18d), −CF2H (18e), and −Cl (18f), to demonstrate wild-type sparing strong inhibition against the resistant EGFRTM1 and EGFRDM kinases. Particularly, compounds 18b and 18f exhibited the strongest EGFRTM1 inhibition with IC50 values of 0.7 and 0.6 nM, respectively. The compounds could also potently suppress EGFRDM kinase with IC50 values of 2.7 and 1.8 nM, but their activities against EGFRWT were 23- and 21-fold less potent, respectively. Additionally, results showed that a replacement of 5-Cl of the pyrimidine motif in 18f by a Br group (18g) barely led to an obvious effect on the activity and selectivity, while the 5-CF3 substituted analogue (18h) totally abolished the EGFR inhibitory potency. The investigation also suggested the aniline group at the pyrimidine 2-position could be replaced by a pyridinyl moiety (18i) without causing a significant potency change to the EGFR triple mutated kinase (IC50 = 2.1 nM). Further investigation revealed that 3′,5′-dicholor analogue 18j was 55-fold less potent than 18f against EGFRTM1 triple mutant, while the 2′,5′-dicholor compound 18k exhibited identical EGFRTM1 inhibition to that of 18f, with an IC50 value of 0.7 nM. Further evaluation suggested that 18k potently suppressed the EGFR19Del/T790M/C797S mutated kinase, which is a clinically more common resistant mutation, with an IC50 value of 3.6 (±1.3) nM. In addition, 18k was also highly potent against EGFRL858R/T790M with an IC50 value of 0.7 nM, but its potency against EGFRWT was 94.1-fold less potent.
Scheme 1. Synthesis of Compounds 18a–18k.

Reagents and conditions: (a) dimethylphosphine oxide, Pd(OAc)2, xantphos, K3PO4, DMF, 120 °C, overnight, 82%; (b) R1 substituted pyrimidine, K2CO3, n-Bu4NHSO4, DMF, 65 °C, overnight, 63–84%; (c) R2 substituted aniline, 2.5 M HCl in EtOH, 2-methoxyethanol, 120 °C, overnight, 37–75%.
Table 1. In Vitro EGFR Inhibition and Antiproliferation Data of Compounds 18a–18ka.

| substituents |
kinase
activity IC50 (nM)b |
|||||
|---|---|---|---|---|---|---|
| compd | R1 | R2 | EGFRWT | EGFRDM | EGFRTM1 | WT/TM1 ratioc |
| 18a | Cl | 2′-H, 3′-OCH3 | 51.2 ± 21.2 | 2.4 ± 0.4 | 2.4 ± 1.0 | 21.3 |
| 18b | Cl | 2′-H, 3′-CH3 | 62.3 ± 11.5 | 2.7 ± 0.7 | 0.7 ± 0.3 | 89.0 |
| 18c | Cl | 2′-H, 3′-OCF3 | 157.0 ± 26.4 | 9.4 ± 3.1 | 12.9 ± 3.2 | 12.2 |
| 18d | Cl | 2′-H, 3′-CF3 | 538.1 ± 62.7 | 32.8 ± 3.6 | 3.5 ± 0.9 | 153.7 |
| 18e | Cl | 2′-H, 3′-CF2H | 198.7 ± 41.2 | 13.7 ± 3.3 | 23.1 ± 1.6 | 8.6 |
| 18f | Cl | 2′-H, 3′-Cl | 38.3 ± 5.6 | 1.8 ± 0.4 | 0.6 ± 0.3 | 63.8 |
| 18g | Br | 2′-H, 3′-Cl | 113.1 ± 24.2 | 1.8 ± 0.5 | 3.3 ± 0.4 | 34.3 |
| 18h | CF3 | 2′-H, 3′-Cl | >1000 | >1000 | >1000 | |
| 18i | Cl | 2′-H, 3′-CH3, 5′ = N | 337.6 ± 141.6 | 25.3 ± 6.1 | 2.1 ± 0.7 | 160.8 |
| 18j | Cl | 3′-Cl, 5′-Cl | 299.8 ± 169.7 | 29.4 ± 8.0 | 33.5 ± 3.5 | 8.9 |
| 18k | Cl | 2′-Cl, 5′-Cl | 65.9 ± 10.7 | 0.7 ± 0.1 | 0.7 ± 0.3 | 94.1 |
| 9 | Cl | 2′-OCH3, 3′-H | 261.3 ± 78.7 | 50.9 ± 12.8 | 38.3 ± 7.0 | 6.8 |
| 5 | 207.5 ± 93.6 | 2.0 ± 0.1 | 236.6 ± 96.0 | 0.88 | ||
EGFRWT, EGFRDM and EGFRTM1 kinase inhibition was tested by ELISA assay. The data are mean values ± SD from at least three independent experiments.
TM1, L858R/T790M/C797S; DM, L858R/T790M.
WT/TM1 ratio was calculated by IC50(EGFRWT)/IC50(EGFRTM1), which reflected the selectivity of the compounds to EGFRWT and EGFRTM1.
The X-ray cocrystal structure of EGFRT790M/C797S with 18f was solved (PDB 7ER2, Figure 3a). Similar to our prediction, the results showed that the molecule anchored in the hinge region of the ATP-binding pocket with bidentate hydrogen bond interactions between Met793 and 2-aminopyrimidine motif. Interestingly, structural alignment of EGFR-18f complex and EGFR-brigatinib complex (PDB 7AEM) showed that the backbone amido linkages between Thr854 and Asp855 pointed in opposite directions (Figure 3b). Therefore, instead of forming a hydrogen bond directly with side chain OH of Thr854 as exhibited in Figure 2a, the oxygen atom on the DMPO group of 18f fostered water-molecule-mediated hydrogen bond interactions with the backbone carbonyl of Thr854 and additionally the side chain NH3 of Lys745, inducing a stable conformation and improving binding affinity toward the protein. In addition, the 3′-Cl in 18f located in an appropriate cavity with no steric clash with any residues, providing a rational explanation for the improved EGFR inhibitory potency. Additionally, the C5-chlorine atom on the pyrimidine scaffold was directed toward the “gatekeeper” residue Met790 with a distance of 3.2 Å, indicating that this position was unable to accommodate large hydrophobic groups such as CF3, which explains the poor potency of 18h.
Figure 3.
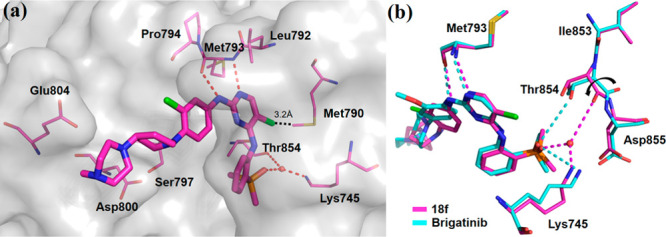
(a) X-ray crystal structure of EGFRT790M/C797S in complex with 18f (PDB 7ER2). Key residues and 18f are shown in magenta by the element in lines and in sticks, respectively. Hydrogen bond interactions are showed as red dotted lines, and the water molecule is indicated by a red dot. (b) Superposition of 18f (PDB 7ER2; compound and protein shown as magenta sticks and lines, respectively) and brigatinib complex (PDB 7AEM; compound and protein shown as cyan sticks and lines, respectively).
Having demonstrated the selectivity of 18k for its direct target EGFR, we next examined its inhibitory activities against a small kinome panel at a concentration of 100 nM. The results in Figure 4 showed that 18k exhibited >30× selectivity in all 28 tyrosine kinases examined (see the Supporting Information for raw inhibition rate). Strikingly, 18k markedly attenuated the inhibitory effect on ALK, the original target of brigatinib, indicating its improved kinase selectivity.
Figure 4.

Kinase inhibitory activity of 18k against 28 different kinases at a concentration of 100 nM. Kinase inhibition was tested by ELISA assay. Inhibition rate are shown as mean ± SD.
Furthermore, the antiproliferative activity of 18k was evaluated in BaF3 cells stably expressing EGFRL858R/T790M/C797S (BaF3-EGFRTM1) and EGFR19Del/T790M/C797S (BaF3-EGFRTM2) mutants (Table 2). It was shown that 18k exhibited moderate growth inhibition against the cell models with IC50 values of 0.258 and 0.141 μM, respectively, which were comparable to those of drug 9, whereas its cytotoxic effect on BaF3 parental cells was a bit more potent with an IC50 value of 1.69 μM. Compound 18k also exhibited a 0.60 μM IC50 value against the proliferation of self-constructed osimertinib-resistant PC9 NSCLC cells harboring EGFR19Del/T790M/C797S mutation (PC9-EGFRTM2). Selectivity was observed against PC9 (EGFR19Del) and H1299 (EGFRWT) cell lines, which was within the kinase selectivity margin. Further immunoblotting analysis suggested that the compound dose-dependently suppressed the phosphorylation of EGFR in BaF3-EGFRTM1, BaF3-EGFRTM2, as well as PC9-EGFRTM2 cells, supporting its cellular inhibitory potency on the resistance related EGFR mutations (Figure 5).
Table 2. Antiproliferative Activity of 18k in BaF3, PC9, PC9-EGFRTM2, and H1299 Cell Linesa.
| BaF3 |
||||||
|---|---|---|---|---|---|---|
| IC50 (μM) | EGFRTM1 | EGFRTM2 | parental cells | PC9 | PC9-EGFRTM2 | H1299b |
| 18k | 0.258 ± 0.167 | 0.141 ± 0.081 | 1.691 ± 0.253 | 1.192 ± 0.727 | 0.603 ± 0.294 | 2.115 ± 0.302 |
| brigatinib (9) | 0.286 ± 0.176 | 0.155 ± 0.046 | 4.191 ± 1.614 | 0.829 ± 0.148 | 1.110 ± 0.028 | 1.049 ± 0.160 |
| AZD9291 (5) | 2.893 ± 0.601 | 2.885 ± 0.689 | 4.427 ± 1.162 | 0.011 ± 0.005 | 3.210 ± 0.853 | >10.0 |
BaF3-EGFRTM1, BaF3-EGFRTM2, and PC9-EGFRTM2 cell lines were built by Jian Ding’s laboratory, Shanghai Institute of Materia Medica, Chinese Academy of Sciences. PC9-EGFRTM2 cell line was constructed by introducing the T790M/C797S mutant into PC9 (EGFR19Del). Cellular assay was determined using the CCK8 assay.
H1299 (EGFRWT) cellular assay was determined using sulforhodamine B (SRB) assay. The data were shown as mean ± SD from at least three independent experiments. TM1, L858R/T790M/C797S; TM2, 19Del/T790M/C797S.
Figure 5.

Target inhibition potency of 18k in EGFRC797S mutant BaF3 cells and PC9 cells. Cells were treated with indicated compounds for 2 h, and the activation of EGFR and downstream signaling molecule were evaluated using Western Blot analysis.
The pharmacokinetic (PK) parameters of 18f and 18k were also assessed in Sprague–Dawley (SD) rats (Table 3). Both compounds exhibited good PK properties with high oral bioavailability, decent exposure, and acceptable half-life. For instance, 18k exhibited an oral bioavailability value of 81.7%, an AUC (area under curve) value of 5219 h·ng/mL, and a T1/2 value of 2.67 h, which were optimal for oral administration for the following in vivo efficacy investigation.
Table 3. Mean PK Parameters of 18f and 18k after po or iv Administration in SD Ratsa.
|
18f |
18k |
|||
|---|---|---|---|---|
| parameters | pob | ivc | pob | ivc |
| T1/2 (h) | 2.84 ± 0.11 | 3.73 ± 1.36 | 2.16 ± 0.2 | 2.67 ± 0.45 |
| Tmax (h) | 1.50 ± 0.87 | 2.67 ± 1.15 | ||
| Cmax (ng/mL) | 470 ± 155 | 345 ± 52 | 868 ± 181 | 288 ± 16 |
| AUC(0-t) (h·ng/mL) | 2553 ± 704 | 862 ± 88 | 4552 ± 1154 | 929 ± 91 |
| AUC(0-∞) (h·ng/mL) | 2563 ± 709 | 898 ± 57 | 5219 ± 1536 | 1060 ± 144 |
| CL (mL/min/kg) | 9.30 ± 0.57 | 7.97 ± 1.17 | ||
| MRT(0-∞) (h) | 4.79 ± 0.49 | 4.08 ± 1.24 | 4.37 ± 0.62 | 3.65 ± 0.56 |
| Vss (mL/kg) | 2275 ± 710 | 1720 ± 40 | ||
| F (%) | 49.3 ± 11.8 | 81.7 ± 20.7 | ||
n = Three animals per study.
Dosed at 3 mg/kg.
Dosed at 0.5 mg/kg.
In summary, a straightforward structure optimization of brigatinib was conducted to yield compound 18k as a new potent and selective EGFRT790M/C797S inhibitor. The compound strongly suppressed the EGFRL858R/T790M/C797S and EGFR19Del/T790M/C797S kinases with sub-nanomolar IC50 values but was approximately 94-fold less potent against the wild type EGFR. A cocrystal structure of EGFRT790M/C797S with a close derivative 18f was also solved to provide understanding of the binding mode of the inhibitor. Moreover, compound 18k exhibited cellular activities in all three cell models, BaF3-EGFRTM1, BaF3-EGFRTM2, and PC9-EGFRTM2, comparable to that of brigatinib. Compound 18k is orally bioavailable and demonstrates highly desirable PK properties, making it a promising lead compound for further structural optimization.
Acknowledgments
The authors appreciate the financial support from National Natural Science Foundation of China (21807045, 82173650, 81820108029, 81874284, 22037003, 22077050, and 81903638), Guangdong Province (2021A1515011239 and 2018B030337001), the National Science & Technology Major Project “Key New Drug Creation and Manufacturing Program”, China (2019ZX09301157-004), and the ″Personalized Medicines-Molecular Signature-based Drug Discovery and Development″, Strategic Priority Research Program of the Chinese Academy of Sciences (XDA12020112).
Glossary
Abbreviations
- EGFR
epidermal growth factor receptor
- NSCLC
nonsmall cell lung cancer
- 19Del
Exon 19 deletion
- MTD
maximal tolerated dose
- WT
wild type
- NMPA
National Medical Products Administration of China
- FDA
U.S. Food and Drug Administration
- NDA
New Drug Application
- DMPO
dimethylphosphine oxide group
- TM1
L858R/T790M/C797S
- TM2
19Del/T790M/C797S
- DM
L858R/T790M
- PK
pharmacokinetic
- AUC
area under curve
- SD rats
Sprague–Dawley rats
- ALK
anaplastic lymphoma kinase
- CCK8
cell counting kit 8
- SRB
sulforhodamine.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsmedchemlett.1c00555.
Chemistry, biological assay, X-ray crystallography data, and copies of NMR spectra and HPLC purity data (PDF)
Author Contributions
∇ S.L., T.Z., and S.-J.Z. contributed equally to this work.
The authors declare no competing financial interest.
Supplementary Material
References
- Hynes N. E.; Lane H. A. ERBB receptors and cancer: the complexity of targeted inhibitors. Nat. Rev. Cancer 2005, 5 (5), 341–354. 10.1038/nrc1609. [DOI] [PubMed] [Google Scholar]
- Paez J. G.; Janne P. A.; Lee J. C.; Tracy S.; Greulich H.; Gabriel S.; Herman P.; Kaye F. J.; Lindeman N.; Boggon T. J.; Naoki K.; Sasaki H.; Fujii Y.; Eck M. J.; Sellers W. R.; Johnson B. E.; Meyerson M. EGFR mutations in lung cancer: correlation with clinical response to gefitinib therapy. Science 2004, 304 (5676), 1497–1500. 10.1126/science.1099314. [DOI] [PubMed] [Google Scholar]
- Dowell J.; Minna J. D.; Kirkpatrick P. Erlotinib hydrochloride. Nat. Rev. Drug Discov 2005, 4 (1), 13–14. 10.1038/nrd1612. [DOI] [PubMed] [Google Scholar]
- Dungo R. T.; Keating G. M. Afatinib: first global approval. Drugs 2013, 73 (13), 1503–1515. 10.1007/s40265-013-0111-6. [DOI] [PubMed] [Google Scholar]
- Wu Y. L.; Cheng Y.; Zhou X.; Lee K. H.; Nakagawa K.; Niho S.; Tsuji F.; Linke R.; Rosell R.; Corral J.; Migliorino M. R.; Pluzanski A.; Sbar E. I.; Wang T.; White J. L.; Nadanaciva S.; Sandin R.; Mok T. S. Dacomitinib versus gefitinib as first-line treatment for patients with EGFR-mutation-positive non-small-cell lung cancer (ARCHER 1050): a randomised, open-label, phase 3 trial. Lancet Oncol 2017, 18 (11), 1454–1466. 10.1016/S1470-2045(17)30608-3. [DOI] [PubMed] [Google Scholar]
- Finlay M. R.; Anderton M.; Ashton S.; Ballard P.; Bethel P. A.; Box M. R.; Bradbury R. H.; Brown S. J.; Butterworth S.; Campbell A.; Chorley C.; Colclough N.; Cross D. A.; Currie G. S.; Grist M.; Hassall L.; Hill G. B.; James D.; James M.; Kemmitt P.; Klinowska T.; Lamont G.; Lamont S. G.; Martin N.; McFarland H. L.; Mellor M. J.; Orme J. P.; Perkins D.; Perkins P.; Richmond G.; Smith P.; Ward R. A.; Waring M. J.; Whittaker D.; Wells S.; Wrigley G. L. Discovery of a potent and selective EGFR inhibitor (AZD9291) of both sensitizing and T790M resistance mutations that spares the wild type form of the receptor. J. Med. Chem. 2014, 57 (20), 8249–8267. 10.1021/jm500973a. [DOI] [PubMed] [Google Scholar]
- Cross D. A.; Ashton S. E.; Ghiorghiu S.; Eberlein C.; Nebhan C. A.; Spitzler P. J.; Orme J. P.; Finlay M. R.; Ward R. A.; Mellor M. J.; Hughes G.; Rahi A.; Jacobs V. N.; Red Brewer M.; Ichihara E.; Sun J.; Jin H.; Ballard P.; Al-Kadhimi K.; Rowlinson R.; Klinowska T.; Richmond G. H.; Cantarini M.; Kim D. W.; Ranson M. R.; Pao W. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discovery 2014, 4 (9), 1046–1061. 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J. C.; Camidge D. R.; Yang C. T.; Zhou J.; Guo R.; Chiu C. H.; Chang G. C.; Shiah H. S.; Chen Y.; Wang C. C.; Berz D.; Su W. C.; Yang N.; Wang Z.; Fang J.; Chen J.; Nikolinakos P.; Lu Y.; Pan H.; Maniam A.; Bazhenova L.; Shirai K.; Jahanzeb M.; Willis M.; Masood N.; Chowhan N.; Hsia T. C.; Jian H.; Lu S. Safety, Efficacy, and Pharmacokinetics of Almonertinib (HS-10296) in Pretreated Patients With EGFR-Mutated Advanced NSCLC: A Multicenter, Open-label, Phase 1 Trial. J. Thorac Oncol 2020, 15 (12), 1907–1918. 10.1016/j.jtho.2020.09.001. [DOI] [PubMed] [Google Scholar]
- Shi Y.; Hu X.; Zhang S.; Lv D.; Wu L.; Yu Q.; Zhang Y.; Liu L.; Wang X.; Cheng Y.; Ma Z.; Niu H.; Wang D.; Feng J.; Huang C.; Liu C.; Zhao H.; Li J.; Zhang X.; Jiang Y.; Gu C. Efficacy, safety, and genetic analysis of furmonertinib (AST2818) in patients with EGFR T790M mutated non-small-cell lung cancer: a phase 2b, multicentre, single-arm, open-label study. Lancet Respir Med. 2021, 9 (8), 829–839. 10.1016/S2213-2600(20)30455-0. [DOI] [PubMed] [Google Scholar]
- Nagasaka M.; Zhu V. W.; Lim S. M.; Greco M.; Wu F.; Ou S. I. Beyond Osimertinib: The Development of Third-Generation EGFR Tyrosine Kinase Inhibitors For Advanced EGFR+ NSCLC. J. Thorac Oncol 2021, 16 (5), 740–763. 10.1016/j.jtho.2020.11.028. [DOI] [PubMed] [Google Scholar]
- Zhang T.; Qu R.; Chan S.; Lai M.; Tong L.; Feng F.; Chen H.; Song T.; Song P.; Bai G.; Liu Y.; Wang Y.; Li Y.; Su Y.; Shen Y.; Sun Y.; Chen Y.; Geng M.; Ding K.; Ding J.; Xie H. Discovery of a novel third-generation EGFR inhibitor and identification of a potential combination strategy to overcome resistance. Mol. Cancer 2020, 19 (1), 90. 10.1186/s12943-020-01202-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thress K. S.; Paweletz C. P.; Felip E.; Cho B. C.; Stetson D.; Dougherty B.; Lai Z.; Markovets A.; Vivancos A.; Kuang Y.; Ercan D.; Matthews S. E.; Cantarini M.; Barrett J. C.; Janne P. A.; Oxnard G. R. Acquired EGFR C797S mutation mediates resistance to AZD9291 in non-small cell lung cancer harboring EGFR T790M. Nat. Med. 2015, 21 (6), 560–562. 10.1038/nm.3854. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H. N.; Jung K. S.; Yoo K. H.; Cho J.; Lee J. Y.; Lim S. H.; Kim H. S.; Sun J. M.; Lee S. H.; Ahn J. S.; Park K.; Choi Y. L.; Park W.; Ahn M. J. Acquired C797S Mutation upon Treatment with a T790M-Specific Third-Generation EGFR Inhibitor (HM61713) in Non-Small Cell Lung Cancer. J. Thorac. Oncol. 2016, 11 (4), E45–E47. 10.1016/j.jtho.2015.12.093. [DOI] [PubMed] [Google Scholar]
- Niederst M. J.; Hu H.; Mulvey H. E.; Lockerman E. L.; Garcia A. R.; Piotrowska Z.; Sequist L. V.; Engelman J. A. The Allelic Context of the C797S Mutation Acquired upon Treatment with Third-Generation EGFR Inhibitors Impacts Sensitivity to Subsequent Treatment Strategies. Clin. Cancer Res. 2015, 21 (17), 3924–3933. 10.1158/1078-0432.CCR-15-0560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ayeni D.; Politi K.; Goldberg S. B. Emerging Agents and New Mutations in EGFR-Mutant Lung Cancer. Clin. Cancer Res. 2015, 21 (17), 3818–3820. 10.1158/1078-0432.CCR-15-1211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Uchibori K.; Inase N.; Araki M.; Kamada M.; Sato S.; Okuno Y.; Fujita N.; Katayama R. Brigatinib combined with anti-EGFR antibody overcomes osimertinib resistance in EGFR-mutated non-small-cell lung cancer. Nat. Commun. 2017, 8, 14768. 10.1038/ncomms14768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park H.; Jung H. Y.; Mah S.; Hong S. Discovery of EGF Receptor Inhibitors That Are Selective for the d746–750/T790M/C797S Mutant through Structure-Based de Novo Design. Angew. Chem. Int. Edit 2017, 56 (26), 7634–7638. 10.1002/anie.201703389. [DOI] [PubMed] [Google Scholar]
- Gunther M.; Juchum M.; Kelter G.; Fiebig H.; Laufer S. Lung Cancer: EGFR Inhibitors with Low Nanomolar Activity against a Therapy-Resistant L858R/T790M/C797S Mutant. Angew. Chem. Int. Edit 2016, 55 (36), 10890–10894. 10.1002/anie.201603736. [DOI] [PubMed] [Google Scholar]
- Shen J.; Zhang T.; Zhu S. J.; Sun M.; Tong L.; Lai M.; Zhang R.; Xu W.; Wu R.; Ding J.; Yun C. H.; Xie H.; Lu X.; Ding K. Structure-Based Design of 5-Methylpyrimidopyridone Derivatives as New Wild-Type Sparing Inhibitors of the Epidermal Growth Factor Receptor Triple Mutant (EGFR(L858R/T790M/C797S)). J. Med. Chem. 2019, 62 (15), 7302–7308. 10.1021/acs.jmedchem.9b00576. [DOI] [PubMed] [Google Scholar]
- Kashima K.; Kawauchi H.; Tanimura H.; Tachibana Y.; Chiba T.; Torizawa T.; Sakamoto H. CH7233163 overcomes osimertinib resistant EGFR-Del19/T790M/C797S mutation. Mol. Cancer Ther 2020, 19 (11), 2288–2297. 10.1158/1535-7163.MCT-20-0229. [DOI] [PubMed] [Google Scholar]
- Jia Y.; Yun C. H.; Park E.; Ercan D.; Manuia M.; Juarez J.; Xu C.; Rhee K.; Chen T.; Zhang H.; Palakurthi S.; Jang J.; Lelais G.; DiDonato M.; Bursulaya B.; Michellys P. Y.; Epple R.; Marsilje T. H.; McNeill M.; Lu W.; Harris J.; Bender S.; Wong K. K.; Janne P. A.; Eck M. J. Overcoming EGFR(T790M) and EGFR(C797S) resistance with mutant-selective allosteric inhibitors. Nature 2016, 534 (7605), 129–132. 10.1038/nature17960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To C.; Jang J.; Chen T.; Park E.; Mushajiang M.; De Clercq D. J. H.; Xu M.; Wang S.; Cameron M. D.; Heppner D. E.; Shin B. H.; Gero T. W.; Yang A.; Dahlberg S. E.; Wong K. K.; Eck M. J.; Gray N. S.; Janne P. A. Single and Dual Targeting of Mutant EGFR with an Allosteric Inhibitor. Cancer Discov 2019, 9 (7), 926–943. 10.1158/2159-8290.CD-18-0903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt H.; Bose D.; Petronczki M.; Scharn D.; Bader G.; Baum A.; Bergner A.; Chong E.; Dobel S.; Egger G.; Engelhardt C.; Ettmayer P.; Fuchs J. E.; Gerstberger T.; Gonnella N.; Grimm A.; Grondal E.; Haddad N.; Hopfgartner B.; Kousek R.; Krawiec M.; Kriz M.; Lamarre L.; Leung J.; Mayer M.; Patel N. D.; Simov B. P.; Reeves J. T.; Schnitzer R.; Schrenk A.; Sharps B.; Solca F.; Stadtmuller H.; Tan Z.; Wunberg T.; Zoephel A.; McConnell D. B. Start Selective and Rigidify: The Discovery Path toward a Next Generation of EGFR Tyrosine Kinase Inhibitors. J. Med. Chem. 2019, 62 (22), 10272–10293. 10.1021/acs.jmedchem.9b01169. [DOI] [PubMed] [Google Scholar]
- Liu X. L.; Zhang X. Q.; Yang L.; Tian X.; Dong T. T.; Ding C. Z.; Hu L. H.; Wu L. Y.; Zhao L. L.; Mao J.; Ji Q. S.; Yan S. Y.; Zhu Z. Z.; Xia Y. F.; Chan C. C.; Chen S. H. Preclinical evaluation of TQB3804, a potent EGFR C797S inhibitor. Cancer Res. 2019, 79 (13), 1320. 10.1158/1538-7445.AM2019-1320. [DOI] [Google Scholar]
- Dong R. F.; Zhu M. L.; Liu M. M.; Xu Y. T.; Yuan L. L.; Bian J.; Xia Y. Z.; Kong L. Y. EGFR mutation mediates resistance to EGFR tyrosine kinase inhibitors in NSCLC: From molecular mechanisms to clinical research. Pharmacol. Res. 2021, 167, 105583. 10.1016/j.phrs.2021.105583. [DOI] [PubMed] [Google Scholar]
- Finlay M. R. V.; Barton P.; Bickerton S.; Bista M.; Colclough N.; Cross D. A. E.; Evans L.; Floc’h N.; Gregson C.; Guerot C. M.; Hargreaves D.; Kang X.; Lenz E. M.; Li X.; Liu Y.; Lorthioir O.; Martin M. J.; McKerrecher D.; McWhirter C.; O’Neill D.; Orme J. P.; Mosallanejad A.; Rahi A.; Smith P. D.; Talbot V.; Ward R. A.; Wrigley G.; Wylot M.; Xue L.; Yao T.; Ye Y.; Zhao X. Potent and Selective Inhibitors of the Epidermal Growth Factor Receptor to Overcome C797S-Mediated Resistance. J. Med. Chem. 2021, 64 (18), 13704–13718. 10.1021/acs.jmedchem.1c01055. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Ercan D.; Chen L.; Yun C. H.; Li D.; Capelletti M.; Cortot A. B.; Chirieac L.; Iacob R. E.; Padera R.; Engen J. R.; Wong K. K.; Eck M. J.; Gray N. S.; Janne P. A. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462 (7276), 1070–1074. 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S.; Xu T.; Zhang L.; Zhang Z.; Luo J.; Liu Y.; Lu X.; Tu Z.; Ren X.; Ding K. Design, synthesis, and biological evaluation of 2-oxo-3,4-dihydropyrimido[4,5-d]pyrimidinyl derivatives as new irreversible epidermal growth factor receptor inhibitors with improved pharmacokinetic properties. J. Med. Chem. 2013, 56 (21), 8803–8813. 10.1021/jm4012388. [DOI] [PubMed] [Google Scholar]
- Huang W. S.; Liu S.; Zou D.; Thomas M.; Wang Y.; Zhou T.; Romero J.; Kohlmann A.; Li F.; Qi J.; Cai L.; Dwight T. A.; Xu Y.; Xu R.; Dodd R.; Toms A.; Parillon L.; Lu X.; Anjum R.; Zhang S.; Wang F.; Keats J.; Wardwell S. D.; Ning Y.; Xu Q.; Moran L. E.; Mohemmad Q. K.; Jang H. G.; Clackson T.; Narasimhan N. I.; Rivera V. M.; Zhu X.; Dalgarno D.; Shakespeare W. C. Discovery of Brigatinib (AP26113), a Phosphine Oxide-Containing, Potent, Orally Active Inhibitor of Anaplastic Lymphoma Kinase. J. Med. Chem. 2016, 59 (10), 4948–4964. 10.1021/acs.jmedchem.6b00306. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.