

Abstract
The gasdermin family is a newly identified class of pore-forming proteins that play as the executioners of pyroptosis, a lytic pro-inflammatory type of cell death triggered by sensing cytosolic infections and danger signals. Upon activation, the gasdermin N-terminal domain translocates to the cell membrane to form pores, which allow the release of proinflammatory cytokines and alarmins, and cause cell lysis. Many structural studies have been conducted in the past few years to investigate the mechanisms of gasdermin proteins in the activation and pore formation. Here, we review these high-resolution structures and highlight the mechanistic insights into the gasdermin activation and regulation that are provided.
Keywords: Gasdermin, GSDMD, GSDMA3, pyroptosis, pore-forming protein, inflammasome, caspase-1, caspase-4, caspase-5, caspase-11, IL-1β, IL-18
Graphical Abstract
Introduction
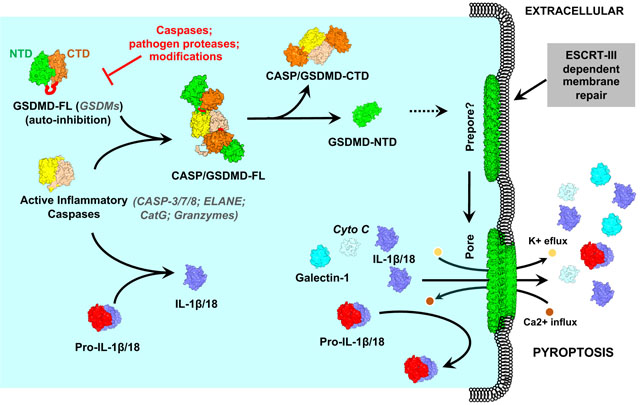
Pyroptosis, which features cell swelling, membrane rupture, and inflammatory intracellular content release, plays an important role in innate immune defense against microbial infections[1–3]. Pyroptosis was first defined as pro-inflammatory programmed cell death because of its dependence on caspase-1, but redefined as gasdermin-mediated programmed necrotic cell death since the recent discovery of gasdermin D (GSDMD) and the characterization of pore formation on the plasma membrane[4, 5].
Pyroptosis is often mediated by the proteolytic cleavage of GSDMD under the regulation by inflammasomes, which are stimulus-induced cytosolic multiprotein complexes in response to infections and damage, in canonical or non-canonical ways. In the canonical inflammasome pathways of pyroptosis, the signal is initiated by the oligomerization of cytosolic pattern recognition receptors (PRRs) such as those of nucleotide-binding domain (NBD) and leucine-rich repeat (LRR)-containing proteins (NLRs) or absent in melanoma 2 (AIM2) like receptors upon specifically sensing the pathogen-associated molecular patterns (PAMPs) and damage-associated molecular patterns (DAMPs)[6–11]. The assembled receptors then recruit the downstream effector pro-caspase-1 with or without the need of the adaptor such as apoptosis-associated, speck-like protein containing a caspase recruitment domain (ASC), resulting in the assembly of the inflammasome complexes[12–16]. The assembled inflammasomes ultimately cause the activation and self-processing of pro-caspase-1 molecules into their mature forms, which are responsible for the maturation of the pro-inflammatory precursor cytokines (pro-IL-1β and pro-IL-18)[17, 18], and the proteolytic cleavage of the pore-forming protein GSDMD[19]. The cleavage removes the inhibitory C-terminal domain (CTD) of GSDMD, resulting in the release of the active N-terminal domain (NTD)[19–22]. NTD fragments then translocate to the plasma membrane where they form membrane-embedded pores, which ultimately cause cell death and trigger a secondary activation of the canonical NLRP3 inflammasome for cytokine maturation and release[22–25].
In the non-canonical inflammasome pathway, non-canonical inflammatory caspases, including caspase-11 in mice and caspase-4 and caspase-5 in humans, function as both receptor and effector molecules. With the assistance of guanylate binding proteins (GBPs), caspase-4/-5/-11 directly recognize the lipid A moiety of intracellular lipopolysaccharides (LPS) from Gram-negative bacteria through their N-terminal caspase activation and recruitment domains (CARD)[26–29]. LPS binding then induces the oligomerization and consequently the activation of these caspases[4, 19–21, 30–33]. Unlike caspase-1, activated caspase-4/-5/-11 do not process pro-interleukins, but directly promote pyroptosis through the cleavage of GSDMD.
Pyroptosis can be mediated by other GSDM family members. Wang et al. and Rogers et al. showed that GSDME can be cleaved by caspase-3 and switches caspase-3-mediated apoptosis to pyroptosis in cells treated with chemotherapy agents or apoptosis stimulators[34, 35]. GSDMC can be cleaved by caspase-8, resulting in pyroptosis in cancer cells[36]. Moreover, two recent studies showed that pyroptosis can also be induced by granzyme A (Gzm A) and granzyme (Gzm B) through the cleavage of GSDMB and GSDME, respectively[37, 38].
Since GSDMs were identified as the executioners of pyroptosis, the GSDM family has become a hotspot in the field of immunology because of its critical roles in immune defense and autoinflammatory and other diseases. GSDMD deletion or inhibition has been shown to significantly decrease pyroptosis and remarkably protect mice from sepsis, which is a fatal condition with a great need for medical solutions[20, 21, 39]. Moreover, as the effector of inflammasome signaling, GSDMD is also involved in many common human diseases including gout, diabetes and neurodegenerative diseases[40, 41]. Genetic mutations in GSDME cause nonsyndromic hearing loss in humans[42, 43], while GSDMB polymorphisms increases the risk of asthma and inflammatory bowel disease (IBD)[44, 45]. GSDMA and GSDME are epigenetically silenced in gastric cancers and many cancer cell lines[46–49], while GSDMB is highly expressed in many cancers such as cervical, gastric, hepatic and breast cancers, suggesting that GSDMB might function as an oncogene[50–53]. The biological function of GSDMC is currently controversial, the expression of GSDMC is down-regulated in many esophageal and gastric cancer cells, but is up-regulated in metastatic melanoma cells[54, 55]. Given the crucial functions of GSDMs and their extensive associations with diseases, GSDMs have so far been considered an emerging therapeutic target.
In this review, we discuss our current understanding of GSDMs with a focus on the GSDM structures in both the inactive and active conformation, and the mechanistic insights they provide for GSDM-mediated pyroptosis.
Identification of GSDMD as an executioner of pyroptosis
GSDMD belongs to the conserved gasdermin family, which was named by its predominant expression in the upper gastrointestinal tract and skin. The GSDM family comprises six members in humans, including GSDMA, GSDMB, GSDMC, GSDMD, GSDME (DFNA5) and GSDMF (DFNB59 or PJVK). Mice lack the GSDMB but contain three isoforms of GSDMA (GSDMA1–3) and four of GSDMC (GSDMC1–4) (Table 1)[56, 57]. Most GSDMs share highly conserved NTD and CTD connected by a variable linker, while GSDMF possesses a truncated CTD. Several studies have shown that GSDMs are expressed in the epithelium with a distinct and restricted pattern and may have a differentiation-status specific role, however, their exact functions are poorly understood[54, 58].
Table 1.
Summary of GSDMs activation and regulation
| Human GSDMs | Mouse GSDMs | Pore-formation | Pore-size | Proteolytic Activation | Proteolytic Inhibition | Post-translational modifications | Ref. |
|---|---|---|---|---|---|---|---|
| GSDMA | GSDMA1–3 | Yes | 26–28 mer; 28 nm (outer) and 18 nm (inner) (27-mer)a |
Unknown | Unknown | Phosphorylation of Thr8 by PLK1 inhibits pore-forming activity. | [22, 99] |
| GSDMB | None | Yes | 13.5 – 15.6 nm (inner)b | Caspase-1/3/6/7; Granzyme A |
Unknown | Ubiquitination by IpaH7.8 mediates 26S proteasome degradation. | [22, 37, 79, 92] |
| GSDMC | GSDMC1–4 | Yes | Unknown | Caspase-8 | Unknown | Unknown | [22, 36] |
| GSDMD | GSDMD | Yes | 31–34 mer; 31.5 nm (outer) and 21.5 nm (inner) (33-mer)a |
Caspase-1/4/5/11, Caspase-8; neutrophil elastase; cathepsin G; |
Caspase-3/7, Pathogen proteases | Itaconation of Cys77 and succination of Cys56/191/268/309/467 inhibit pore-forming activity; Phosphorylation of Tyr37/158 and Ser185 with unknown function. |
[19–22, 72–76, 95–98, 100, 103] |
| GSDME (DFNA5) | GSDME (DFNA5) | Yes | Unknown | Caspase-3; Caspase-1/7c; Granzyme B |
Unknown | Phosphorylation of Thr6 inhibits pore-forming activity; Palmitoylation of Cys407 and Cys408 enhances pore-forming activity. |
[22, 34, 35, 38, 78, 99, 101] |
| GSDMF (DFNB59 or PJVK) | GSDMF (DFNB59 or PJVK) | Unknown | Unknown | Unknown | Unknown | Unknown |
The pore sizes are measured based on the cryo-EM structures; the outer and inner diameters are shown, respectively;
The pore size is based on the negative-stain EM measurement; the average of the inner diameter is shown.
The cleavage of GSDME by caspase-1 and -7 is currently observed only in teleosts.
In 2015, two independent groups simultaneously identified GSDMD as the executioner of pyroptosis by using a clustered regularly interspaced short palindromic repeats (CRISPR)-Cas9 screen in mouse Tlr4−/− bone marrow-derived macrophages, and a forward genetic screen with LPS-treated ethyl-N-nitrosourea (ENU) mutagenized mice, respectively[19, 20]. Both groups discovered that GSDMD is the direct substrate of inflammatory caspases and is required for pyroptosis. Mechanistically, GSDMD is specifically cleaved by activated caspase-1 and caspase-4/-5/-11 at the conserved “(F/L)L(S/T)D” motif in the NTD-CTD linker, generating a ~30kD N-terminal fragment that shows intrinsic pyroptotic activity[19, 20]. Meanwhile, another group identified GSDMD as a crucial component of NLRP3 inflammasomes based on the quantitative mass spectrometry-based proteomics, confirming that GSDMD serves as the executioner of pyroptosis[21].
Subsequent studies further showed that GSDMD-NTD can oligomerize and form pores with an inner diameter of 12 to 20 nm on the membrane[22, 25, 59–61]. Extensive GSDMD pore formation on the plasma membrane triggers rapid pyroptotic cell death by disrupting osmotic pressure[62, 63]. In vitro lipid-binding assays demonstrated that GSDMD-NTD selectively binds acidic phospholipids such as phosphatidylinositol phosphates (PIPs), phosphatidylserine (PS), phosphatidic acid (PA) and cardiolipin[22, 25]. It is worth noting that PIPs and PS localize on the inner leaflet of the plasma membrane, while cardiolipin is a component of the mitochondrial and bacterial membranes. Such binding pattern suggests that GSDMD-NTD only causes pyroptosis through the cytosol, and has the potential to control the infection by killing the invasive bacteria[25].
Currently, NTDs of most GSDMs are able to induce pyroptotic cell death, therefore, pyroptosis has also been redefined as the GSDM-mediated programmed cell death[4, 22].
Structural auto-inhibition in the full-length GSDMs
Previous studies showed that it is the GSDMD-NTD but not full-length GSDMD or GSDMD-CTD that binds phospholipids and possesses pyroptotic activity[22, 25]. Overexpression of GSDMD-CTD significantly inhibited GSDMD-NTD induced pyroptotic cell death in LPS-stimulated Hela cells[22]. Crystal structures of full-length mouse GSDMA3 and human and mouse GSDMD revealed an auto-inhibited confirmation in full-length proteins with the CTD folding back on the NTD, preventing lipid binding and pore formation[22, 64]. In both GSDMD and GSDMA3 structures, the pore-forming NTD comprises a twisted β-sheet core surrounded by several α-helices, and a largely disordered transmembrane region, while the CTD contains a globin-like fold consisting of nine helices covered by a short anti-parallel three-stranded β-sheet, interacting extensively with the NTD[22, 64, 65] (Fig. 1A and B). In the crystal structure of full-length GSDMD, the CTD binds tightly to the globular region in the NTD (Interface I). In Interface I, the α1 helix and β1–β2 hairpin from the NTD form extensive charge-charge and hydrophobic interactions with the CTD, with the β1–β2 hairpin contributing the major interaction. Particularly, aromatic residues Phe49 and Trp50 (Phe48 and Trp49 in mouse GSDMA3) on the tip of the β1–β2 hairpin dock into a hydrophobic pocket in the CTD[64] (Fig. 1A and 2B). Interestingly, there is an additional interface (Interface II) in GSDMA3, which is formed where the α4 helix stretching out from the NTD inserts into another hydrophobic pocket on the CTD[22] (Fig. 1B). Amino acid sequence alignment shows that residues participating in the NTD-CTD interactions, especially in Interface I, are highly conserved among most GSDMs. Mutations abolishing NTD-CTD interactions cause constitutive auto-activation of full-length proteins, suggesting a conserved structural auto-inhibited mechanism in the GSDM family[22, 64].
Fig. 1. Structural auto-inhibition in full-length GSDMs.
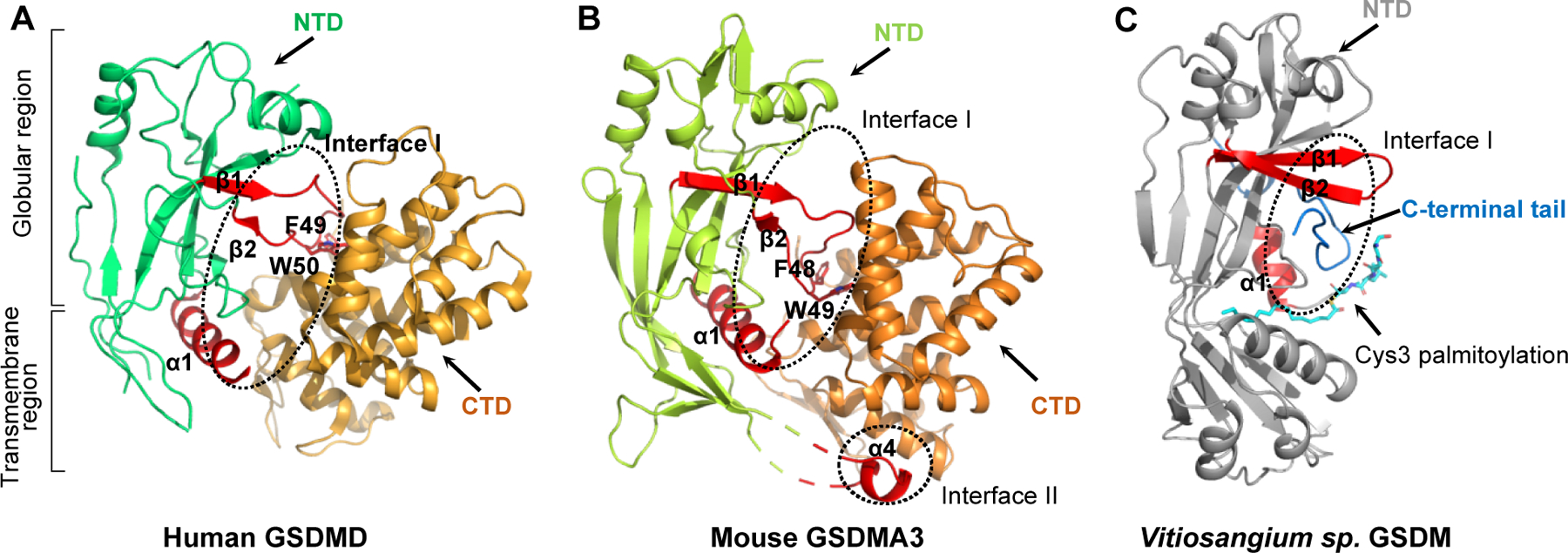
Crystal structures of full-length human GSDMD (A), mouse GSDMA3 (B) and a GSDM-like protein from Vitiosangium sp. (C). The N-terminal domains (NTD) and C-terminal domains (CTD) are colored as indicated by text labels, respectively. The NTD can be further divided into a globular region and a transmembrane region. The NTD-CTD interfaces are highlighted by black dotted ellipses. The secondary elements in the NTD that are involved in the autoinhibitory interactions are labeled and colored red. Mouse GSDMA3 contains an additional interface (Interface II) contributed by the α4 helix stretching out from the NTD. Vitiosangium sp. GSDM has an extremely short C-terminal tail containing about 30 amino acids. The N-terminal Cys3 in the Vitiosangium sp. GSDM-NTD is palmitoylated. The palmitoyl group is labeled and colored cyan.
Fig. 2. Proteolytic activation of GSDMD by inflammatory caspases.
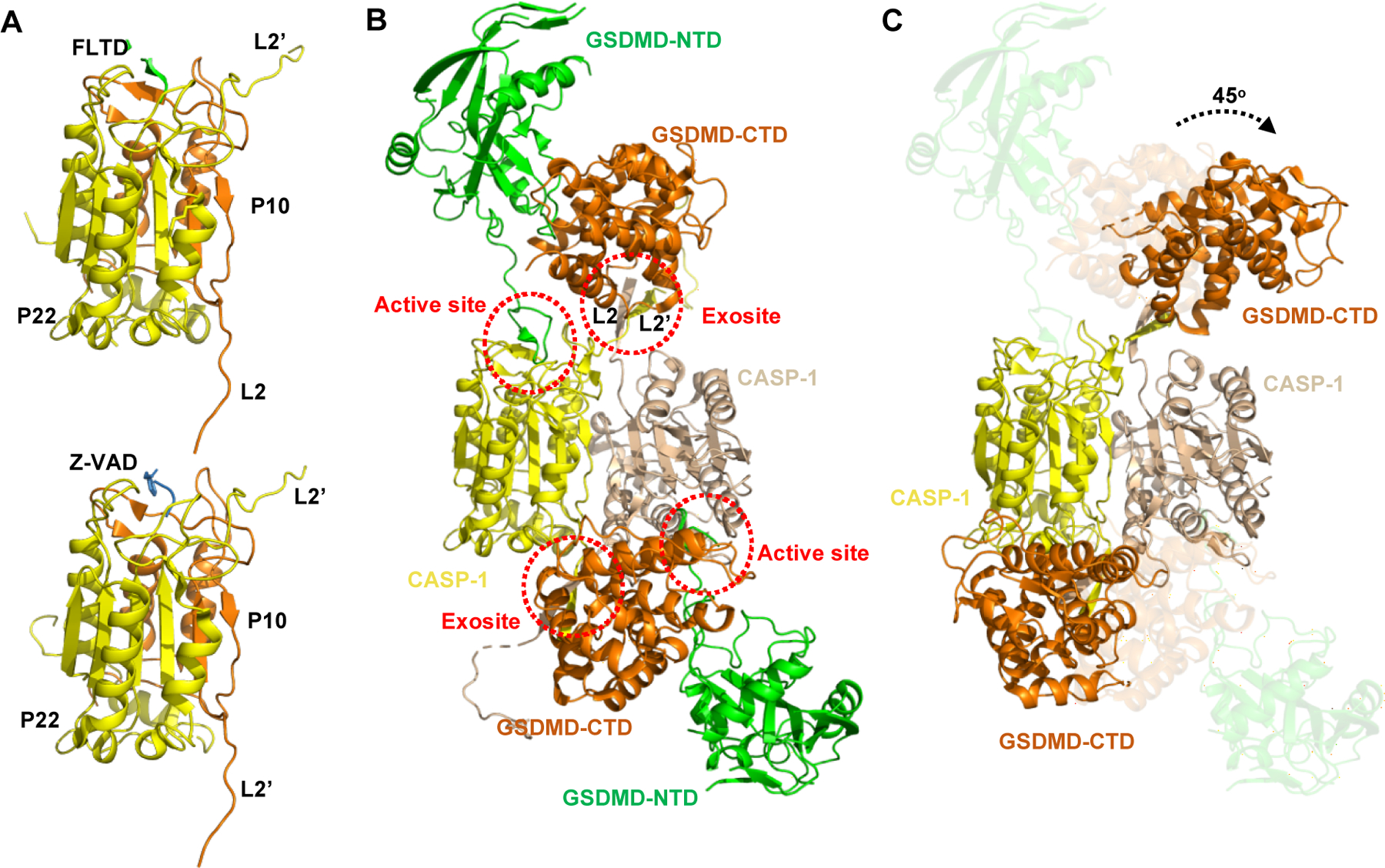
(A) Crystal structures of human CASP-1 catalytic domain in complexes with the human GSDMD derived peptide Ac-FLTD-CMK (Upper, PDB: 6BZ9) and the pan-caspase inhibitor z-VAD-FMK (Lower, PDB: 2HBQ). The p10 and p22 subdomains are colored orange and yellow, respectively. The Ac-FLTD-CMK and z-VAD-FMK are colored green and light blue, respectively. The p22 C-terminal L2’ and p10 N-terminal L2 loops involved in the GSDMD-CTD interactions are labeled. (B) Crystal structure of the human CASP-1 p22/p10(C285A) in complex with the full-length mouse GSDMD (PDB: 6VIE). The NTD and CTD of GSDMD are colored green and brown, respectively. The dimeric CASP-1 p22/p10 are colored yellow and wheat, respectively. The CASP-1/GSDMD interfaces are highlighted by red dotted ellipses. (C) Crystal structure of the human CASP-1 p22/p10(C285A) in complex with the human GSDMD-CTD (PDB: 6KN0). The molecules/domains are colored as in (B). Structural alignment shows a 45° degree rotation of the GSDMD-CTD after GSDMD-NTD is released from the complex.
GSDMF is a more distantly related GSDM family member with a truncated non-homologous CTD. It is still unclear whether GSDMF possesses pore-forming activity or whether it is constitutively active because of its extremely short CTD. Recent discoveries of GSDM-like proteins in fungi and bacteria have expanded our understanding of CTD-mediated inhibition of GSDMs. Like GSDMF in mammals, GSDMs from both bacteria and fungi contain an N-terminal pore-forming domain and a short CTD containing tens to approximately one hundred amino acids [66–68]. Crystal structures of bacterial GSDMs (bGSDMs) from Bradyrhizobium tropiciagri and Vitiosangium sp reveal a complete absence of the α-helical CTD that appears in mammalian GSDMs[66]. However, full-length bGSDMs still adopt the same auto-inhibited confirmation as that in mammalian GSDMs, with the short CTD peptide wrapping around the twisted β-sheet core in the NTD (Fig. 1C). The short CTD peptide in bGSDMs directly interacts with the NTD helix α1 and β1–β2 hairpin, forming an interface equivalent to Interface I in the full-length mammalian GSDMs. bGSDMs also don’t have the Interface II, indicating that Interface I is sufficient to repress NTD activity (Fig. 1C). Truncation or proteolytic removal of the CTD peptide of bGSMDs by associated caspase-like proteases results in a cytotoxic active NTD, suggesting a direct role of CTD in maintaining the auto-inhibited conformation in bGSDMs[66].
It is interesting that the N-terminal Cys3 of bGSDMs is palmitoylated, and the palmitoylation is required for maintaining protein stability[66]. The 16-carbon palmitoyl is buried in an elongated hydrophobic tunnel formed by nonpolar residues on the transmembrane region (Fig. 1C). Removal of the palmitoylation of this cysteine with hydroxylamine-treatment or alanine mutagenesis destabilized the protein and significantly abolished the cellular toxicity[66]. The N-terminal cysteine does not exist in either human or mouse GSDMF, and whether a similar post-translation modification is required for mammalian GSDMF is not yet known. Taken together, the structures of both mammalian and bacterial GSDMs demonstrate that structural auto-inhibition is a conserved mechanism shared by the GSDM family from bacteria to animals.
Mechanism of GSDM pore formation
Proteolytic cleavage activation of GSDMs
The mechanism for GSDMD activation through proteolytic cleavage by inflammatory caspases has been revealed by many structures in the past few years. The crystal structure of human caspase-1 catalytic domain in complex with the peptide derived from the GSDMD cleavage site showed that the recognition of the “272FLTD275” tetrapeptide motif in human GSDMD by the caspase-1 catalytic pocket is similar to that in the previously reported structure of caspase-1 in complex with z-VAD-FMK (Fig. 2A), indicating a conserved mechanism for substrate engagement in GSDMD proteolytic processing[69].
However, subsequent structural and functional characterization demonstrated a more complicated model for GSDMD activation. In addition to auto-processing at Asp285, which corresponds to Asp316/Asp285 in caspase-1/-11, generating the p22/p10 heterodimer, caspase-4 can also be auto-processed at Asp270 to form p20/p12. Only p22/p10 is fully active, while p20/p12 is inactive and cannot bind GSDMD[70]. Crystal structures of caspase-1/-4/-11 catalytic domains in complex with GSDMD revealed a conserved dimeric structure of the p22/p10 heterodimer, which comprises a central six-stranded β sheet flanked by five helices. An antiparallel β sheet formed by the L2 (N-terminal of p10) and L2’ (C-terminal of p22’) loops in p22/p10 acts as a substrate-binding exosite that interacts with GSDMD-CTD through hydrophobic interactions and hydrogen bonds (Fig. 2B). The exosite interaction promotes complex formation and further recruits the GSDMD interdomain linker to enter the caspase catalytic pocket for cleavage. Mutations at the exosite-binding interface significantly reduced the cleavage of GSDMD by caspase-1/-4/-11 and compromised pyroptotic cell death, while the replacement of the “272FLTD275” tetrapeptide motif in the GSDMD interdomain linker with “AAAD” didn’t affect the caspase cleavage both in vitro and in cells, suggesting a sequence-independent proteolytic processing of GSDMD by caspases[70, 71]. It has to be mentioned that a ~45° rotation of the CTD domain is observed between the structures of caspase-1 in complex with full-length GSDMD and in complex with GSDMD-CTD, indicating a conformation change before and after GSDMD-NTD release after cleavage (Fig. 2C). Taken together, GSDMD-CTD not only functions as an inhibitory domain to maintain GSDMD in an autoinhibited state, but also provides the platform for activated inflammatory caspase recruitment and GSDMD activation.
In addition to the inflammatory caspases, GSDMD can also be activated by many other proteases (Table1). During Yersinia infection, host TAK1 or IκB kinase (IKK) inhibited by the Yersinia effector protein YopJ triggers RIPK1-caspase-8-dependent cleavage of GSDMD. Caspase-8 directly cleaves mouse GSDMD at Asp276 (human Asp275), generating the pore-forming active NTD that causes pyroptosis, which subsequently contributes to host defense against Yersinia infection[72, 73]. GSDMD can also be processed by neutrophil elastase (ELANE) and granule-associated protease cathepsin G (CatG) during “neutrophil extracellular trap”-osis (NETosis), a regulated form of neutrophil cell death induced by microbial infection[74–76]. Upon Gram-negative bacteria infection in neutrophils, GSDMD is proteolytically activated by ELANE which is released from granules. The liberated GSDMD-NTD then permeabilizes the primary granules and neutrophil plasma membrane to facilitate protease release and neutrophil extracellular trap extrusion, ultimately enhancing the NETosis in a feed-forward loop[75].
Recent studies have identified several upstream proteases for the activation of other GSDMs (Table 1). GSDME can be activated by caspase-3 by cleavage after Asp270 in the interdomain linker during classical apoptosis. The cleavage generates the pore-forming active NTD that converts noninflammatory apoptotic death into inflammatory pyroptotic death in cells that express GSDME[34, 35]. GSDME can also be cleaved and activated by GzmB, a death-inducing serine protease, that is released from natural killer (NK) cells and cytotoxic T lymphocytes (CTLs) into target cells, to induce pyroptosis during NK cell attack[38]. Moreover, GSDME is found to be activated in macrophages and neutrophils in a RIPK 1 kinase-dependent manner upon pathogenic Yersinia infection[77]. A recent study even demonstrated that GSDME is cleaved by caspase-1/-3/-7 and induces pyroptosis in teleosts[78]. GSDMB is cleaved by caspase-1/-3/-6 and -7, and by GzmA, another death-inducing serine protease, at Lys233 and Lys239, causing pyroptotic cell death in target cells[37, 79]. A recent study showed that GSDMC can be cleaved by caspase-8, switching TNF-α-induced apoptosis to pyroptosis[36].
By using a forward genetic screen, a study recently found that RagA in the Ragulator-Rag complex in the mTORC1 pathway is necessary for GSDMD pore formation but not plasma membrane localization[80]. However, another study demonstrated that Ragulator-Rag activates GSDMD through the RIPK1-caspase-8 signaling axis[81]. Whether RagA directly interacts with GSDMD-NTD to facilitate pore formation remains to be investigated.
Structural architecture of the GSDM Pore
Inflammatory caspase cleavage of GSDMD leads to the release of NTD, which then translocates to the plasma membrane and oligomerizes into pores. Negative-stain electron microscopy (EM) showed that GSDMD-NTD pores have inner diameters of 12–20 nm with approximately 16 symmetric protomers, while the molecular weight of a detergent extracted GSDMD-NTD pore from liposomes indicates an approximate symmetry of 24[22, 61]. These results were confirmed by high-resolution atomic force microscopy (AFM) in combination with cryo-EM, which revealed an average diameter of 20 nm with symmetries ranging from 15 to 45 of the GSDMD-NTD pores[59, 60]. The pores are sufficiently large to allow the passage of macromolecules smaller than their size, including the IL-1 family cytokines, and alarmins such as galectins[82–84].
A subsequent cryo-EM study revealed that human GSDMD-NTD forms a strikingly large, predominantly 33-subunit pore (range from 31 to 34). The 33-fold pore features a complete anti-parallel β-barrel with an inner diameter of around 21.5 nm and an outer diameter of around 31 nm (Fig. 3A), slightly larger than the previously reported 26–28-fold GSDMA3 pore[85, 86]. The membrane-inserted GSDMD-NTD subunit resembles a left hand with its cytosol globular domain as the palm, the N-terminal α1 helix as the thumb, and the four extended β-strands of the two membrane-inserted β-hairpins as fingers (Fig. 3B). The 132 β-strands define the central β-barrel about 40–50 Å high, which is sufficient to traverse a lipid bilayer composed of lipid acyl chains with a thickness of about 30–40 Å[86].
Fig. 3. Mechanism of GSDM pore formation.
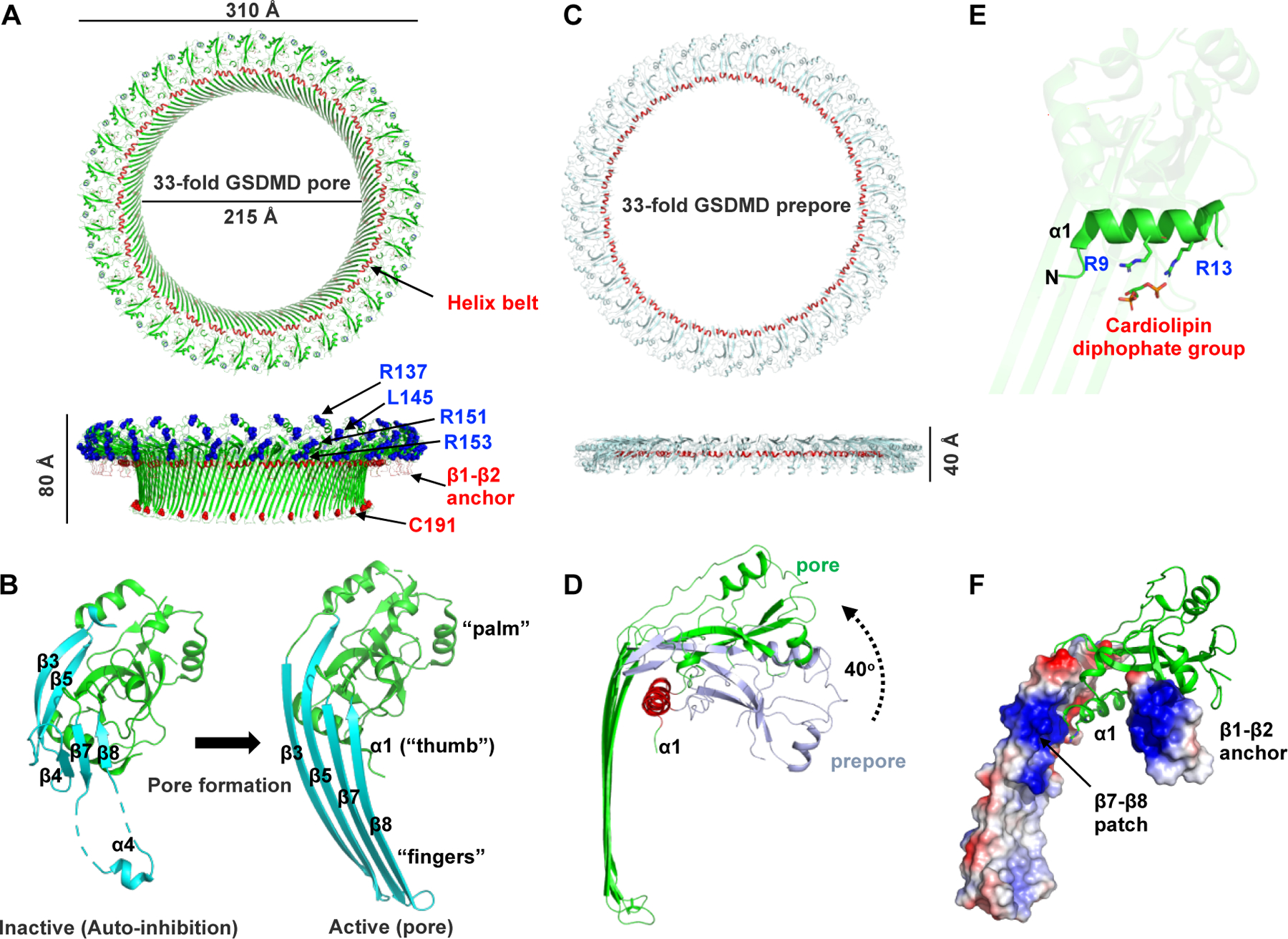
(A) Cryo-EM structure of the 33-subunit human GSDMD pore (PDB: 6VFE). The pore features a large transmembrane β-barrel and a globular domain on the cytosolic side. The “helix belt” and the β1–β2 anchor that binds to the plasma membrane are labeled and colored red. A previously reported “four-basic-residue” patch is also labeled and colored blue. Cys191 residue that are modified by disulfiram (DSF) are labeled. (B) Conformation change of GSDMD subunit upon pore formation. The pore forms a structure that resembles a human left hand, with the globular domain as the ‘palm’, the α1 helix as the ‘thumb’, and the membrane-inserted β-hairpins as the ‘fingers’. (C) Cryo-EM structure of the 33-subunit human GSDMD prepore. (D) Prepore-to-pore transition of a GSDMD subunit. The globular domain rotates away from the membrane by approximately 40° upon membrane insertion. (E) The two conserved basic residues in the α1 helix interact with the cardiolipin diphosphate group. (F) GSDMD pore subunit with the β1–β2 anchor and the additional β7–β8 patch is shown in the local electrostatic surface.
Comparison of the membrane-inserted NTD with the auto-inhibited conformation in the full-length structures reveals drastic conformational changes in the transmembrane region. The entire β3-β4-β5 region extends to form the first transmembrane β-hairpin, and β7, β8, as well as the disordered region in between, form the second transmembrane β-hairpin (Fig. 3B). The polar and nonpolar residues on the transmembrane β-strands are alternately arranged, with the nonpolar residues oriented outside to form a hydrophobic β-barrel exterior that contacts the surrounding lipids[22, 64, 85, 86].
The oligomerization of the pore is mediated by both the cytosolic globular domain and the transmembrane region through extensive hydrophobic and charge-charge interactions and hydrogen-bond network, respectively. Particularly, the N-terminal α1 thumb helix interacts with the neighboring one in a head-to-tail mode, forming a helical belt on the membrane to stabilize the entire pore (Fig. 3A)[85, 86]. Loss-of-function mutations of the residues either in the globular oligomerization interface, such as E14K in GDMDA3, or in the transmembrane region, such as L184D in GSMDA3 and I104N/I105N in human and mouse GSDMD compromised the pore-forming activity[20, 22].
Prepores in both GSDMA3 and GSDMD
Previous studies on pore-forming proteins/toxins demonstrated that pore formation usually involves several steps with multiple conformational changes, including an intermediate step with a stable ring-like oligomer without membrane insertion, which is called a ‘prepore’ [87–91]. Strikingly, a prepore conformation was identified in both GSDMA3 and GSDMD cryo-EM samples (Fig. 3C)[85, 86]. 2-dimensional (2D) and 3D classifications of both pores revealed a population of particles containing two ring structures, in which one ring is the pore, and the other ring represents the prepore with the membrane insertion region disordered. Structural comparison of GSDMD prepore and pore showed that the prepore is similar in the inner and outer diameters to the pore, but about 40 Å shorter in height, which is due to a rigid-body rotation of the globular domain away from the membrane during pore formation[86] (Fig. 3C and D). Although the globular domain in the prepore resembles the auto-inhibited conformation, the ordered transmembrane region adopts a conformation more like that in the pore, indicating the transition of prepore to pore (Fig. 1A and Fig. 3D)[85, 86]. Therefore, the existence of the prepore in both GSDMD and GSDMA3 suggests a conserved, simplest model that GSDMs used for membrane insertion–the concerted, all-or-none prepore-to-pore transition.
However, this concerted model is in contradiction with the observation of the ring-, arc- and slit-shaped GSDMD pore assemblies on lipid bilayers by AFM[59, 60]. Measuring the height of the oligomer on the supported lipid membrane by AFM topography indicated a nearly full insertion of the oligomers into the membrane, even for the slit-shaped ones[59]. These data suggested a sequential, rather than concerted, model of pore formation that the NTDs insert individually into the membrane, and oligomerize in the membrane to form a partial conductive pore, to which more NTDs are recruited to enlarge the pore. Experiments like patch-clamp electrophysiology on a single GSDM pore or the direct observation of the prepore on the membrane will help to address the pore formation process of GSDMs.
Lipid binding and membrane contacts
Previous studies showed that GSDMA3 and GSDMD NTDs selectively bind acidic phospholipids including PIPs, PS and PA, and kill cells by targeting plasma membrane from cytosol but do not harm neighboring cells[22, 25]. The NTDs also bind cardiolipin, a component of the mitochondrial and bacterial membranes, and GSDMB-NTD can even interacts with the lipid A moiety of LPS at a relatively lower affinity[92]. Both GSDMD and GSDMB are able to kill bacteria by targeting bacterial membrane cardiolipin, suggesting their potential role in host to limit bacterial infection[25, 92]. GSDMD, GSDME, and GSDMA3 are shown to permeabilize the mitochondrial outer membrane and disrupt mitochondrial function, and finally activate caspase-3 and cause apoptosis[93, 94]. GSDMB-NTD also binds sulfatide, a sphingolipid highly enriched in myelin in the central nervous system (CNS), however, with the function unknown[79].
By noting that all those phospholipids contain a negatively charged head group, a “four-basic-residue” binding mode was proposed for GSDMD recognizing specific lipids based on the amino sequence alignment and secondary structure prediction[25]. Four conserved basic residues Arg137, Lys145, Arg151, and Arg153 in human GSDMD (Arg138, Lys146, Arg152, and Arg154 in mouse) on the α2 and α3 helices in the NTD globular domain are thought to be responsible for the lipid-binding (Fig. 3A). Double, triple, or quadruple mutation of these residues to alanine completely abolished the ability of the protein to bind lipids, oligomerize and induce pyroptosis, without altering the secondary structure[25]. However, subsequent cryo-EM structures of GSDMA3 and human GSDMD pores showed that only Arg151 and Arg153 directly contact the negatively charged membrane surface, while Lys145 is involved in the oligomerization (Fig. 3A) [85, 86].
Structural analysis of the GSDMA3 pore identified the N-terminal helix α1 as a critical element for the recognition of acidic lipids. In the cryo-EM structure of the GSDMA3 pore, two highly conserved basic residues Arg9 and Arg13 with their positively charged sidechains pointing to the double phosphate head group of the cardiolipin form the first membrane contact site for the lipid binding of GSMDs[85] (Fig. 3E). The previously known inhibition mediated by Thr8 and Thr6 phosphorylation in GSDMA and GSDME is probably through interfering with the interaction between the α1 helix and the lipid[93]. In both GSDMA3 and human GSDMD pores, the β1–β2 loop that features a hydrophobic tip flanked by basic residues forms another membrane contact site. Aromatic residues, Tyr48, Phe49, and Tyr50 in human GSDMD (Leu48, Phe48 and Tyr49 in GSDMA3) insert into the lipid bilayer acting as an anchor, while the surrounding basic residues on the β1, β2 strands and α3 helix form a basic patch that contacts the negatively charged membrane surface[85, 86] (Fig. 3A and F). Interestingly, an additional basic patch formed by the β7–β8 transmembrane hairpin was found in the GSDMD pore (Fig. 3F). Mutation of Arg174 on β7 and Lys204 on β8 significantly compromised pore formation[86]. However, amino sequence alignment shows that this additional patch is less conserved in GSDMs. More structural studies will be needed to elucidate whether this additional patch is a common feature in GSDMs or only specific in GSDMD.
Regulation of GSDMs
Proteolytic cleavage inhibition of GSDMs
The activities of GSDMs can be negatively regulated by proteolytic cleavage at noncanonical sites in the NTD (Table 1). A recent study showed that GSDMD-mediated pyroptosis is inhibited by apoptotic caspase-3 and caspase-7[95]. In serine peptidases DPP8/9 inhibitor-induced pyroptosis in monocytes and macrophages, caspase-1 activates both caspase-3 and caspase-7, which subsequently cleave GSDMD at Asp87 within the pore-forming NTD and abolish its ability to form pores, blocking pyroptosis[95]. GSDMD can also be cleaved at Gln193-Gly194 by 3C proteases from Enterovirus 71 (EV71), and Coronaviruses (CoV) including SARS-CoV-2, MERS-CoV, and PD-CoV, to generate a truncated N-terminal fragment that is unable to trigger pyroptosis, suggesting an important strategy that pathogens use to escape from GSDMD-mediated immune response[96, 97].
Post-translational modification of GSDMs
Different types of post-translational modifications have been identified on most GSDMs (Table 1). The modifications can either negatively regulate or enhance the activities of GSDMs. A recent study showed that GSDMD can be itaconated at Cys77 by endogenous accumulated itaconate in macrophages after sustained LPS stimulation. This modification blocked caspase-1-dependent GSDMD cleavage and inhibited pyroptosis[98]. However, Cys77 is not involved in GSDMD/caspase-1 interaction, the mechanism of how Cys77 itaconation suppresses caspase-1 cleavage needs further investigation. GSMDB can be ubiquitinated by Shigella flexneri type 3 effector IpaH7.8. IpaH7.8 specifically recognizes GSDMB-NTD and catalyzes K48-ubiquitination on multiple lysines on GSDMB. The ubiquitinated GSDMB is then targeted for 26S proteasomal degradation, therefore preventing the GSDMB-mediated immune response[92]. Moreover, GSDMA and GSDME have been reported to be phosphorylated at Thr8 and Thr6, respectively[93, 99]. Thr8 phosphorylation in GSDMA is mediated by serine-threonine kinase Polo-like kinase 1 (PLK1)[99], while it remains unknown in GSDME. Both phosphorylations inhibit pore formation and pyroptosis. Phosphorylation has also been identified on Tyr37, Tyr158, and Ser185 on GSDMD in cancer cells[100], however, whether these phosphorylations affect the pyroptotic activity of GSDMD remains to be uncovered.
Post-translational modifications can also enhance the activation of GSDMs. Palmitoylations at Cys407 and Cys408 on GSDME by the palmitoyltransferases ZDHHC 2, 7, 11, and 15 have been reported to induce chemotherapy-induced cancer cell pyroptosis in a recent study[101]. Cys407 and Cys408 are not directly involved in the NTD-CTD interaction based on the full-length GSDM structures. The modification probably causes conformation changes of GSDME-CTD that weaken the interactions between NTD and CTD, which facilitates the release of the cleaved GSDME-NTD. It is also possible that palmitoylation alters the behaviors of GSDME by enhancing its ability for membrane association, or recruiting a partner protein for membrane localization. The Cys407 and Cys408 are not conserved in other GSDMs, but highly conserved in GSDME from zebrafish to humans. Further studies would be required to address the mechanism of palmitoylation in GSDME activation.
Inhibition of GSDMs by small molecules
Several small-molecule inhibitors have been reported to inhibit GSDMD activity. Disulfiram (DSF), an FDA-approved drug for treating chronic alcoholism, and necrosulfonamide (NSA), a drug previously known to inhibit MLKL in necroptotic cell death, were shown to inhibit GSDMD pore formation[39, 102]. Dimethyl fumarate (DMF), an FDA-approved drug for the treatment of multiple sclerosis (MS), was reported to prevent GSDMD from interacting with caspases, limit its processing and oligomerization, and block pyroptosis[103]. All three drugs are cysteine-reactive and capable of modifying cysteines on GSDMD. Among these, DSF and NSA specifically modify Cys191, while DMF succinates multiple cysteines, including Cys56, Cys191, Cys268, Cys309 and Cys467. Cys191 is an exposed cysteine on the α4 helix in the transmembrane region (Fig. 3A), modification of which would probably interfere with the formation of transmembrane β-hairpins[86]. Modifications of the other cysteines would probably block the pore oligomerization, like Cys56[86], or limit the cleavage by interfering with the interactions between GSDMD and the inflammatory caspases, like Cys268 and Cys309[70, 71].
LDC7559, a pyrazolo-oxazepine-based compound, and punicalagin, an antioxidant polyphenol from pomegranates, are the other two small molecules that are reported to inhibit pyroptosis[75, 104]. Although GSDMD can be specifically enriched from cell lysates by pull-down using LDC7559, LDC7559 is not able to inhibit GSDMD pore formation on liposomes[75]. Similarly, direct interaction between punicalagin and GSDMD has not been confirmed. Further studies are required to elucidate the mechanism of how LDC7559 and punicalagin inhibit GSDMD.
Selectivity of GSDM pores
It has previously been proposed that the GSDM pore uses a “size-exclusion” mechanism to allow the passage of molecules smaller than a certain size, such as ions, nucleotides, and intracellular proteins such as IL-1β/18 (mature form of 17/18 kDa) and galectin-1 (14 kDa), but not larger ones such as tetramerized LDH (~140 kDa)[82, 84]. Protein high mobility group box 1 (HMGB1) was previously thought to be released through GSDM pores, however, a recent study showed that the HMGB1 tetramer (~150 kDa) is only released after cellular rupture[105–107]. Liposome leakage assays using charge-neutral dextrans revealed a cutoff molecular weight of 40 kDa for passing through the GSDMD pores[22]. Surprisingly, liposome leakage assay using 40 kDa dextrans carrying different charges indicated a charge-based effect of GSDMD pore-mediated cargo release. While the positively charged diethylaminoethyl (DEAE)-dextran showed a similar or slightly enhanced release compared with the neutral ones, negatively charged carboxymethyl (CM)-dextran was barely released, suggesting that the GSDMD pore repels acidic cargoes but favors basic or neutral cargoes[86]. Surface electrostatic analysis of GSDMA3 and GSDMD pores revealed that selectivity is contributed by the negative potentials originating from the globular domain and β-barrel and decaying towards the center of both (Fig. 4A)[86].
Fig 4. Selectivity of the GSDMD pore.
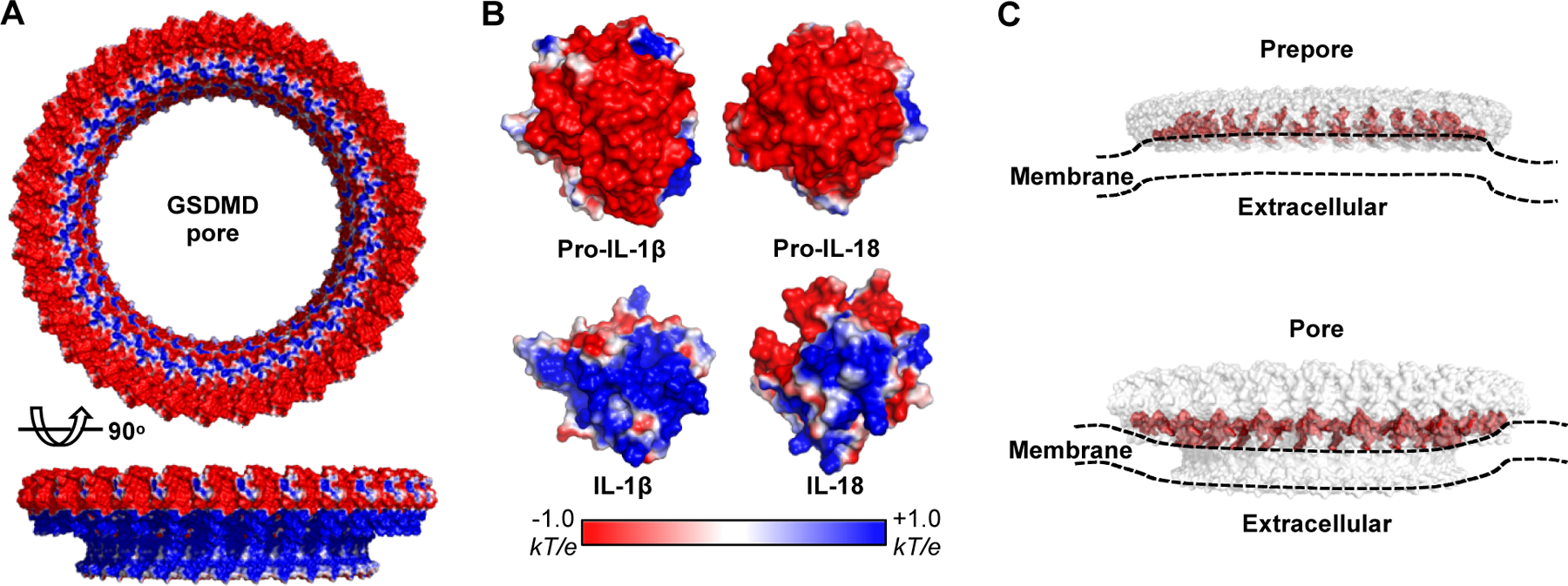
(A) Electrostatic surface (−1 to +1 kT/e) of the GSDMD pore. (B) Electrostatic surface (−1 to +1 kT/e) of the pro- and mature IL-1 β and IL-18. The molecules are basified upon maturation. (C) Potential membrane distortion caused upon GSDMD prepore and pore attachment.
It is interesting that the precursor and mature IL-1β and IL-18 are similar in size but different in their surface potentials. Both cytokines are largely basified after proteolytic maturation (Fig. 4B). Liposome release assays indicated that mature IL-1β and IL-18 are secreted considerably faster than their precursors, suggesting that the GSDMD pore acts as a ‘filter’ to repel the precursors. Mutations in GSDMD that diminish the negative potential of the pore or mutations in the mature IL-1β and IL-18 that diminish the positive potential remarkably limited the charge-based filtration effect on secretion[86]. It is worth noting that previously reported proteins that are released in a GSDMD-dependent manner, including Rac-1, cytochrome c, and galectin-1, are all non-acidic[83, 84, 93]. Collectively, the GSDM pore functions as an unconventional secretion channel using both “size-exclusion” and charge-based “filtration” to guide the secretion of neutral or basic intracellular cargoes, such as inflammatory cytokines.
Membrane repair and beyond
The GSDMD-perforated plasma membranes can be repaired. Many studies have reported that the GSDMD pore causes calcium influx[108, 109]. Calcium influx then triggers the assembly of the Endosomal Sorting Complexes Required for Transport III (ESCRT III) machinery at the plasma membrane where it removes the GSDMD pores by shedding them into vesicles, thus preventing GSDMD-mediated pyroptosis[110]. It is interesting that GSDMD pore formation may cause membrane curvature[86]. Based on the structures, the plasma membrane would be convex (curved towards the cytosol) upon prepore attachment, and concave (curved towards the extracellular space) upon pore attachment (Fig. 4C). This GSDMD pore-mediated membrane curvature is probably a signal for initiating ESCRT-III-dependent membrane repair[111].
Osmotic pressure caused by the GSDM pore has been considered the cause of the cell losing its membrane integrity and rupture[62, 63]. Surprisingly, a recent discovery of NINJ1 as the plasma membrane rupture mediator overturns this idea. Cells deficient in NINJ1 do not rupture after cell death stimulation despite the formation of GSDMD pores. NINJ1 is a 16 kDa cell surface protein containing two transmembrane helices and extracellular N- and C- termini. The N-terminal extracellular helix appears to be responsible for the cytotoxicity[112]. However, the exact mechanism of how NINJ1 is activated and mediates membrane rupture is still unknown.
Conclusions and future perspective
Over the past few years, significant progress has been made in understanding the mechanism by which GSDMD, and more generally the gasdermin family members, execute pyroptosis. Current studies have described a clear picture for GSDM activation and regulation. Briefly, proteases including caspases, granule-associated proteases, and granzymes cleave the interdomain linker to liberate NTD from inhibition by the CTD. The NTD then translocates to the membrane and forms pores through recognition and binding of specific lipids. The activity of GSDMs is tightly regulated by posttranslational modifications and cleavage by other cellular proteases to maintain an appropriate level of the immune response. Pathogens use their own mechanisms through posttranslational modification or cleavage within the NTD to inhibit GSDMs and enhance their survival in the host.
However, our current understanding of the activation and pore formation of GSDMs is mostly based on GSDMD, and whether those mechanisms can be applied to other GSDMs is still unknown. Despite protein sequence similarities, GSDMB shows different lipid-binding behaviors from GSDMD, such as full-length GSDMB binding to phospholipids[79, 92], suggesting distinct activation mechanism and biological roles of GSDMB. The GSDMD pore contains a predominantly acidic conduit with an electrostatic preference for the non-acidic cargo release[86]. Whether this electrostatic preferential release exists in other GSDM pores, and whether the “secretomes” mediated by different GSDM pores are unique to each other remain to be uncovered. There are also more questions raised when we discuss the cellular consequences after GSDM pore formation. As the plasma membrane can be repaired through the rapid removal of the GSDM pores by the ESCRT-III machinery[110], then what is the fate of those GSDM pores? Will they be active in the extracellular space as antibacterial cytolysins or are they degraded? And what is the fate of the CTD once it is released from full-length GSDM? Will it be involved in other cellular events? We anticipate the answers to these questions in the near future, and we believe that through these investigation, GSDMs will be linked to more physiological and pathological processes.
Highlights.
Gasdermins are a family of proteins that execute pyroptosis.
Full-length gasdermin adopts an auto-inhibited confirmation.
Gasdermin N-terminal domain forms membrane pores upon release from its inhibitory C terminal domain.
The gasdermin pore mediates the secretion of proinflammatory cytokines and alarmins.
Gasdermin activity can be regulated in many ways.
Acknowledgments
This work was supported by UConn Health Startup fund and NIH R01AI158435 to J.R. The authors apologize for the incomplete citations due to space limitations.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of interest
The authors declare no conflicts of interest related to this study.
CRediT author statement
Chengliang Wang: Writing-Original draft preparation. Jianbin Ruan: Writing-Reviewing and Editing.
Reference
- [1].Zhang Y, Chen X, Gueydan C, Han J. Plasma membrane changes during programmed cell deaths. Cell Res. 2018;28:9–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol. 2009;7:99–109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tan Y, Chen Q, Li X, Zeng Z, Xiong W, Li G, et al. Pyroptosis: a new paradigm of cell death for fighting against cancer. J Exp Clin Cancer Res. 2021;40:153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Shi J, Gao W, Shao F. Pyroptosis: Gasdermin-Mediated Programmed Necrotic Cell Death. Trends Biochem Sci. 2017;42:245–54. [DOI] [PubMed] [Google Scholar]
- [5].Galluzzi L, Vitale I, Aaronson SA, Abrams JM, Adam D, Agostinis P, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [6].Kagan JC, Magupalli VG, Wu H. SMOCs: supramolecular organizing centres that control innate immunity. Nat Rev Immunol. 2014;14:821–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Sharma D, Kanneganti TD. The cell biology of inflammasomes: Mechanisms of inflammasome activation and regulation. J Cell Biol. 2016;213:617–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Broz P, Dixit VM. Inflammasomes: mechanism of assembly, regulation and signalling. Nat Rev Immunol. 2016;16:407–20. [DOI] [PubMed] [Google Scholar]
- [9].Wang B, Bhattacharya M, Roy S, Tian Y, Yin Q. Immunobiology and structural biology of AIM2 inflammasome. Mol Aspects Med. 2020;76:100869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Wang L, Hauenstein AV. The NLRP3 inflammasome: Mechanism of action, role in disease and therapies. Mol Aspects Med. 2020;76:100889. [DOI] [PubMed] [Google Scholar]
- [11].Tuladhar S, Kanneganti TD. NLRP12 in innate immunity and inflammation. Mol Aspects Med. 2020;76:100887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Lu A, Magupalli VG, Ruan J, Yin Q, Atianand MK, Vos MR, et al. Unified polymerization mechanism for the assembly of ASC-dependent inflammasomes. Cell. 2014;156:1193–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Zhang L, Chen S, Ruan J, Wu J, Tong AB, Yin Q, et al. Cryo-EM structure of the activated NAIP2-NLRC4 inflammasome reveals nucleated polymerization. Science. 2015;350:404–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Hollingsworth LR, Sharif H, Griswold AR, Fontana P, Mintseris J, Dagbay KB, et al. DPP9 sequesters the C terminus of NLRP1 to repress inflammasome activation. Nature. 2021;592:778–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Robert Hollingsworth L, David L, Li Y, Griswold AR, Ruan J, Sharif H, et al. Mechanism of filament formation in UPA-promoted CARD8 and NLRP1 inflammasomes. Nat Commun. 2021;12:189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Gong Q, Robinson K, Xu C, Huynh PT, Chong KHC, Tan EYJ, et al. Structural basis for distinct inflammasome complex assembly by human NLRP1 and CARD8. Nat Commun. 2021;12:188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Howard AD, Kostura MJ, Thornberry N, Ding GJ, Limjuco G, Weidner J, et al. IL-1-converting enzyme requires aspartic acid residues for processing of the IL-1 beta precursor at two distinct sites and does not cleave 31-kDa IL-1 alpha. J Immunol. 1991;147:2964–9. [PubMed] [Google Scholar]
- [18].Thornberry NA, Bull HG, Calaycay JR, Chapman KT, Howard AD, Kostura MJ, et al. A novel heterodimeric cysteine protease is required for interleukin-1 beta processing in monocytes. Nature. 1992;356:768–74. [DOI] [PubMed] [Google Scholar]
- [19].Shi J, Zhao Y, Wang K, Shi X, Wang Y, Huang H, et al. Cleavage of GSDMD by inflammatory caspases determines pyroptotic cell death. Nature. 2015;526:660–5. [DOI] [PubMed] [Google Scholar]
- [20].Kayagaki N, Stowe IB, Lee BL, O’Rourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526:666–71. [DOI] [PubMed] [Google Scholar]
- [21].He WT, Wan H, Hu L, Chen P, Wang X, Huang Z, et al. Gasdermin D is an executor of pyroptosis and required for interleukin-1beta secretion. Cell Res. 2015;25:1285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Ding J, Wang K, Liu W, She Y, Sun Q, Shi J, et al. Pore-forming activity and structural autoinhibition of the gasdermin family. Nature. 2016;535:111–6. [DOI] [PubMed] [Google Scholar]
- [23].Rathinam VA, Vanaja SK, Waggoner L, Sokolovska A, Becker C, Stuart LM, et al. TRIF licenses caspase-11-dependent NLRP3 inflammasome activation by gram-negative bacteria. Cell. 2012;150:606–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Yi YS. Functional crosstalk between non-canonical caspase-11 and canonical NLRP3 inflammasomes during infection-mediated inflammation. Immunology. 2020;159:142–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Liu X, Zhang Z, Ruan J, Pan Y, Magupalli VG, Wu H, et al. Inflammasome-activated gasdermin D causes pyroptosis by forming membrane pores. Nature. 2016;535:153–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Santos JC, Boucher D, Schneider LK, Demarco B, Dilucca M, Shkarina K, et al. Human GBP1 binds LPS to initiate assembly of a caspase-4 activating platform on cytosolic bacteria. Nat Commun. 2020;11:3276. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Pilla DM, Hagar JA, Haldar AK, Mason AK, Degrandi D, Pfeffer K, et al. Guanylate binding proteins promote caspase-11-dependent pyroptosis in response to cytoplasmic LPS. Proc Natl Acad Sci USA. 2014;111:6046–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Santos JC, Dick MS, Lagrange B, Degrandi D, Pfeffer K, Yamamoto M, et al. LPS targets host guanylate-binding proteins to the bacterial outer membrane for non-canonical inflammasome activation. EMBO J. 2018;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Cerqueira DM, Gomes MTR, Silva ALN, Rungue M, Assis NRG, Guimaraes ES, et al. Guanylate-binding protein 5 licenses caspase-11 for Gasdermin-D mediated host resistance to Brucella abortus infection. PLoS Pathog. 2018;14:e1007519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Hagar JA, Powell DA, Aachoui Y, Ernst RK, Miao EA. Cytoplasmic LPS activates caspase-11: implications in TLR4-independent endotoxic shock. Science. 2013;341:1250–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Shi J, Zhao Y, Wang Y, Gao W, Ding J, Li P, et al. Inflammatory caspases are innate immune receptors for intracellular LPS. Nature. 2014;514:187–92. [DOI] [PubMed] [Google Scholar]
- [32].Kayagaki N, Wong MT, Stowe IB, Ramani SR, Gonzalez LC, Akashi-Takamura S, et al. Noncanonical inflammasome activation by intracellular LPS independent of TLR4. Science. 2013;341:1246–9. [DOI] [PubMed] [Google Scholar]
- [33].Downs KP, Nguyen H, Dorfleutner A, Stehlik C. An overview of the non-canonical inflammasome. Mol Aspects Med. 2020;76:100924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Rogers C, Fernandes-Alnemri T, Mayes L, Alnemri D, Cingolani G, Alnemri ES. Cleavage of DFNA5 by caspase-3 during apoptosis mediates progression to secondary necrotic/pyroptotic cell death. Nat Commun. 2017;8:14128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Wang Y, Gao W, Shi X, Ding J, Liu W, He H, et al. Chemotherapy drugs induce pyroptosis through caspase-3 cleavage of a gasdermin. Nature. 2017;547:99–103. [DOI] [PubMed] [Google Scholar]
- [36].Hou J, Zhao R, Xia W, Chang CW, You Y, Hsu JM, et al. PD-L1-mediated gasdermin C expression switches apoptosis to pyroptosis in cancer cells and facilitates tumour necrosis. Nat Cell Biol. 2020;22:1264–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Zhou Z, He H, Wang K, Shi X, Wang Y, Su Y, et al. Granzyme A from cytotoxic lymphocytes cleaves GSDMB to trigger pyroptosis in target cells. Science. 2020;368. [DOI] [PubMed] [Google Scholar]
- [38].Zhang Z, Zhang Y, Xia S, Kong Q, Li S, Liu X, et al. Gasdermin E suppresses tumour growth by activating anti-tumour immunity. Nature. 2020;579:415–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Hu JJ, Liu X, Xia S, Zhang Z, Zhang Y, Zhao J, et al. FDA-approved disulfiram inhibits pyroptosis by blocking gasdermin D pore formation. Nat Immunol. 2020;21:736–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Rashidi M, Simpson DS, Hempel A, Frank D, Petrie E, Vince A, et al. The Pyroptotic Cell Death Effector Gasdermin D Is Activated by Gout-Associated Uric Acid Crystals but Is Dispensable for Cell Death and IL-1beta Release. J Immunol 2019;203:736–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Guo H, Callaway JB, Ting JP. Inflammasomes: mechanism of action, role in disease, and therapeutics. Nat Med. 2015;21:677–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Op de Beeck K, Van Camp G, Thys S, Cools N, Callebaut I, Vrijens K, et al. The DFNA5 gene, responsible for hearing loss and involved in cancer, encodes a novel apoptosis-inducing protein. Eur J Hum Genet. 2011;19:965–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Van Laer L, Huizing EH, Verstreken M, van Zuijlen D, Wauters JG, Bossuyt PJ, et al. Nonsyndromic hearing impairment is associated with a mutation in DFNA5. Nat Genet. 1998;20:194–7. [DOI] [PubMed] [Google Scholar]
- [44].Acevedo N, Reinius LE, Greco D, Gref A, Orsmark-Pietras C, Persson H, et al. Risk of childhood asthma is associated with CpG-site polymorphisms, regional DNA methylation and mRNA levels at the GSDMB/ORMDL3 locus. Hum Mol Genet. 2015;24:875–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Soderman J, Berglind L, Almer S. Gene Expression-Genotype Analysis Implicates GSDMA, GSDMB, and LRRC3C as Contributors to Inflammatory Bowel Disease Susceptibility. Biomed Res Int. 2015;2015:834805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Croes L, Beyens M, Fransen E, Ibrahim J, Vanden Berghe W, Suls A, et al. Large-scale analysis of DFNA5 methylation reveals its potential as biomarker for breast cancer. Clin Epigenetics. 2018;10:51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Moussette S, Al Tuwaijri A, Kohan-Ghadr HR, Elzein S, Farias R, Berube J, et al. Role of DNA methylation in expression control of the IKZF3-GSDMA region in human epithelial cells. PLoS One. 2017;12:e0172707. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Kim MS, Chang X, Yamashita K, Nagpal JK, Baek JH, Wu G, et al. Aberrant promoter methylation and tumor suppressive activity of the DFNA5 gene in colorectal carcinoma. Oncogene. 2008;27:3624–34. [DOI] [PubMed] [Google Scholar]
- [49].Akino K, Toyota M, Suzuki H, Imai T, Maruyama R, Kusano M, et al. Identification of DFNA5 as a target of epigenetic inactivation in gastric cancer. Cancer Sci. 2007;98:88–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Carl-McGrath S, Schneider-Stock R, Ebert M, Rocken C. Differential expression and localisation of gasdermin-like (GSDML), a novel member of the cancer-associated GSDMDC protein family, in neoplastic and non-neoplastic gastric, hepatic, and colon tissues. Pathology. 2008;40:13–24. [DOI] [PubMed] [Google Scholar]
- [51].Hergueta-Redondo M, Sarrio D, Molina-Crespo A, Megias D, Mota A, Rojo-Sebastian A, et al. Gasdermin-B promotes invasion and metastasis in breast cancer cells. PLoS One. 2014;9:e90099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Sun Q, Yang J, Xing G, Sun Q, Zhang L, He F. Expression of GSDML Associates with Tumor Progression in Uterine Cervix Cancer. Transl Oncol. 2008;1:73–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Hergueta-Redondo M, Sarrio D, Molina-Crespo A, Vicario R, Bernado-Morales C, Martinez L, et al. Gasdermin B expression predicts poor clinical outcome in HER2-positive breast cancer. Oncotarget. 2016;7:56295–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Saeki N, Usui T, Aoyagi K, Kim DH, Sato M, Mabuchi T, et al. Distinctive expression and function of four GSDM family genes (GSDMA-D) in normal and malignant upper gastrointestinal epithelium. Genes Chromosomes Cancer. 2009;48:261–71. [DOI] [PubMed] [Google Scholar]
- [55].Watabe K, Ito A, Asada H, Endo Y, Kobayashi T, Nakamoto K, et al. Structure, expression and chromosome mapping of MLZE, a novel gene which is preferentially expressed in metastatic melanoma cells. Jpn J Cancer Res. 2001;92:140–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Tamura M, Tanaka S, Fujii T, Aoki A, Komiyama H, Ezawa K, et al. Members of a novel gene family, Gsdm, are expressed exclusively in the epithelium of the skin and gastrointestinal tract in a highly tissue-specific manner. Genomics. 2007;89:618–29. [DOI] [PubMed] [Google Scholar]
- [57].Ruan J Structural Insight of Gasdermin Family Driving Pyroptotic Cell Death. Adv Exp Med Biol. 2019;1172:189–205. [DOI] [PubMed] [Google Scholar]
- [58].Saeki N, Kuwahara Y, Sasaki H, Satoh H, Shiroishi T. Gasdermin (Gsdm) localizing to mouse Chromosome 11 is predominantly expressed in upper gastrointestinal tract but significantly suppressed in human gastric cancer cells. Mamm Genome. 2000;11:718–24. [DOI] [PubMed] [Google Scholar]
- [59].Mulvihill E, Sborgi L, Mari SA, Pfreundschuh M, Hiller S, Muller DJ. Mechanism of membrane pore formation by human gasdermin-D. EMBO J. 2018;37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [60].Sborgi L, Ruhl S, Mulvihill E, Pipercevic J, Heilig R, Stahlberg H, et al. GSDMD membrane pore formation constitutes the mechanism of pyroptotic cell death. EMBO J. 2016;35:1766–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Aglietti RA, Estevez A, Gupta A, Ramirez MG, Liu PS, Kayagaki N, et al. GsdmD p30 elicited by caspase-11 during pyroptosis forms pores in membranes. Proc Natl Acad Sci USA. 2016;113:7858–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [62].de Vasconcelos NM, Van Opdenbosch N, Van Gorp H, Parthoens E, Lamkanfi M. Single-cell analysis of pyroptosis dynamics reveals conserved GSDMD-mediated subcellular events that precede plasma membrane rupture. Cell Death Differ. 2019;26:146–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [63].Zheng J, Wang D, Chen Q, Liu Q, Lin Z, Hu Q, et al. Hypertonic saccharide solution delays pyroptosis in murine macrophages regardless of the membrane binding of gasdermin D N-terminal. Eur J Immunol. 2020;50:464–7. [DOI] [PubMed] [Google Scholar]
- [64].Liu Z, Wang C, Yang J, Zhou B, Yang R, Ramachandran R, et al. Crystal Structures of the Full-Length Murine and Human Gasdermin D Reveal Mechanisms of Autoinhibition, Lipid Binding, and Oligomerization. Immunity. 2019;51:43–9 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Kuang S, Zheng J, Yang H, Li S, Duan S, Shen Y, et al. Structure insight of GSDMD reveals the basis of GSDMD autoinhibition in cell pyroptosis. Proc Natl Acad Sci USA. 2017;114:10642–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [66].Johnson AG, Wein T, Mayer ML, Duncan-Lowey B, Yirmiya E, Oppenheimer-Shaanan Y, et al. Bacterial gasdermins reveal an ancient mechanism of cell death. bioRxiv. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Clave C, Dyrka W, Granger-Farbos A, Pinson B, Saupe SJ, Daskalov A. Fungal gasdermin-like proteins are controlled by proteolytic cleavage. bioRxiv. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].De Schutter E, Roelandt R, Riquet FB, Van Camp G, Wullaert A, Vandenabeele P. Punching Holes in Cellular Membranes: Biology and Evolution of Gasdermins. Trends Cell Biol. 2021;31:500–13. [DOI] [PubMed] [Google Scholar]
- [69].Yang J, Liu Z, Wang C, Yang R, Rathkey JK, Pinkard OW, et al. Mechanism of gasdermin D recognition by inflammatory caspases and their inhibition by a gasdermin D-derived peptide inhibitor. Proc Natl Acad Sci USA. 2018;115:6792–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Wang K, Sun Q, Zhong X, Zeng M, Zeng H, Shi X, et al. Structural Mechanism for GSDMD Targeting by Autoprocessed Caspases in Pyroptosis. Cell. 2020;180:941–55 e20. [DOI] [PubMed] [Google Scholar]
- [71].Liu Z, Wang C, Yang J, Chen Y, Zhou B, Abbott DW, et al. Caspase-1 Engages Full-Length Gasdermin D through Two Distinct Interfaces That Mediate Caspase Recruitment and Substrate Cleavage. Immunity. 2020;53:106–14 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Demarco B, Grayczyk JP, Bjanes E, Le Roy D, Tonnus W, Assenmacher CA, et al. Caspase-8-dependent gasdermin D cleavage promotes antimicrobial defense but confers susceptibility to TNF-induced lethality. Sci Adv. 2020;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Orning P, Weng D, Starheim K, Ratner D, Best Z, Lee B, et al. Pathogen blockade of TAK1 triggers caspase-8-dependent cleavage of gasdermin D and cell death. Science. 2018;362:1064–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Kambara H, Liu F, Zhang X, Liu P, Bajrami B, Teng Y, et al. Gasdermin D Exerts Anti-inflammatory Effects by Promoting Neutrophil Death. Cell Rep. 2018;22:2924–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Sollberger G, Choidas A, Burn GL, Habenberger P, Di Lucrezia R, Kordes S, et al. Gasdermin D plays a vital role in the generation of neutrophil extracellular traps. Sci Immunol. 2018;3. [DOI] [PubMed] [Google Scholar]
- [76].Burgener SS, Leborgne NGF, Snipas SJ, Salvesen GS, Bird PI, Benarafa C. Cathepsin G Inhibition by Serpinb1 and Serpinb6 Prevents Programmed Necrosis in Neutrophils and Monocytes and Reduces GSDMD-Driven Inflammation. Cell Rep. 2019;27:3646–56 e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Chen KW, Demarco B, Ramos S, Heilig R, Goris M, Grayczyk JP, et al. RIPK1 activates distinct gasdermins in macrophages and neutrophils upon pathogen blockade of innate immune signaling. Proc Natl Acad Sci USA. 2021;118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [78].Jiang S, Gu H, Zhao Y, Sun L. Teleost Gasdermin E Is Cleaved by Caspase 1, 3, and 7 and Induces Pyroptosis. J Immunol. 2019;203:1369–82. [DOI] [PubMed] [Google Scholar]
- [79].Chao KL, Kulakova L, Herzberg O. Gene polymorphism linked to increased asthma and IBD risk alters gasdermin-B structure, a sulfatide and phosphoinositide binding protein. Proc Natl Acad Sci USA. 2017;114:E1128–E37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Evavold CL, Hafner-Bratkovic I, Kagan JC. Downstream of gasdermin D cleavage, a Ragulator-Rag-mTORC1 pathway promotes pore formation and pyroptosis. bioRxiv. 2020. [Google Scholar]
- [81].Zheng Z, Deng W, Bai Y, Miao R, Mei S, Zhang Z, et al. The lysosomal Rag-Ragulator complex licenses RIPK1–and caspase-8–mediated pyroptosis by Yersinia. Science. 2021;372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [82].Evavold CL, Ruan J, Tan Y, Xia S, Wu H, Kagan JC. The Pore-Forming Protein Gasdermin D Regulates Interleukin-1 Secretion from Living Macrophages. Immunity. 2018;48:35–44 e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [83].Heilig R, Dick MS, Sborgi L, Meunier E, Hiller S, Broz P. The Gasdermin-D pore acts as a conduit for IL-1beta secretion in mice. Eur J Immunol. 2018;48:584–92. [DOI] [PubMed] [Google Scholar]
- [84].Russo AJ, Vasudevan SO, Mendez-Huergo SP, Kumari P, Menoret A, Duduskar S, et al. Intracellular immune sensing promotes inflammation via gasdermin D-driven release of a lectin alarmin. Nat Immunol. 2021;22:154–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ruan J, Xia S, Liu X, Lieberman J, Wu H. Cryo-EM structure of the gasdermin A3 membrane pore. Nature. 2018;557:62–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Xia S, Zhang Z, Magupalli VG, Pablo JL, Dong Y, Vora SM, et al. Gasdermin D pore structure reveals preferential release of mature interleukin-1. Nature. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Dal Peraro M, van der Goot FG. Pore-forming toxins: ancient, but never really out of fashion. Nat Rev Microbiol. 2016;14:77–92. [DOI] [PubMed] [Google Scholar]
- [88].Wade KR, Hotze EM, Kuiper MJ, Morton CJ, Parker MW, Tweten RK. An intermolecular electrostatic interaction controls the prepore-to-pore transition in a cholesterol-dependent cytolysin. Proc Natl Acad Sci USA. 2015;112:2204–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [89].Yamashita D, Sugawara T, Takeshita M, Kaneko J, Kamio Y, Tanaka I, et al. Molecular basis of transmembrane beta-barrel formation of staphylococcal pore-forming toxins. Nature Communications. 2014;5. [DOI] [PubMed] [Google Scholar]
- [90].Degiacomi MT, Lacovache I, Pernot L, Chami M, Kudryashev M, Stahlberg H, et al. Molecular assembly of the aerolysin pore reveals a swirling membrane-insertion mechanism. Nat Chem Biol. 2013;9:623–9. [DOI] [PubMed] [Google Scholar]
- [91].van Pee K, Neuhaus A, D’Imprima E, Mills DJ, Kuhlbrandt W, Yildiz O. CryoEM structures of membrane pore and prepore complex reveal cytolytic mechanism of Pneumolysin. Elife. 2017;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Hansen JM, de Jong MF, Wu Q, Zhang LS, Heisler DB, Alto LT, et al. Pathogenic ubiquitination of GSDMB inhibits NK cell bactericidal functions. Cell. 2021;184:3178–91 e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Rogers C, Erkes DA, Nardone A, Aplin AE, Fernandes-Alnemri T, Alnemri ES. Gasdermin pores permeabilize mitochondria to augment caspase-3 activation during apoptosis and inflammasome activation. Nat Commun. 2019;10:1689. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Lin PH, Lin HY, Kuo CC, Yang LT. N-terminal functional domain of Gasdermin A3 regulates mitochondrial homeostasis via mitochondrial targeting. J Biomed Sci. 2015;22:44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Taabazuing CY, Okondo MC, Bachovchin DA. Pyroptosis and Apoptosis Pathways Engage in Bidirectional Crosstalk in Monocytes and Macrophages. Cell Chem Biol. 2017;24:507–14 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Shi F, Lv Q, Wang T, Xu J, Xu W, Shi Y, et al. The nonstructural protein 5 of coronaviruses antagonizes GSDMD-mediated pyroptosis by cleaving and inactivating its pore-forming p30 fragment. bioRxiv. 2021. [Google Scholar]
- [97].Lei X, Zhang Z, Xiao X, Qi J, He B, Wang J. Enterovirus 71 Inhibits Pyroptosis through Cleavage of Gasdermin D. J Virol. 2017;91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Bambouskova M, Potuckova L, Paulenda T, Kerndl M, Mogilenko DA, Lizotte K, et al. Itaconate confers tolerance to late NLRP3 inflammasome activation. Cell Rep. 2021;34:108756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Santamaria A, Wang B, Elowe S, Malik R, Zhang F, Bauer M, et al. The Plk1-dependent phosphoproteome of the early mitotic spindle. Mol Cell Proteomics. 2011;10:M110 004457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Rush J, Moritz A, Lee KA, Guo A, Goss VL, Spek EJ, et al. Immunoaffinity profiling of tyrosine phosphorylation in cancer cells. Nat Biotechnol. 2005;23:94–101. [DOI] [PubMed] [Google Scholar]
- [101].Hu L, Chen M, Chen X, Zhao C, Fang Z, Wang H, et al. Chemotherapy-induced pyroptosis is mediated by BAK/BAX-caspase-3-GSDME pathway and inhibited by 2-bromopalmitate. Cell Death Dis. 2020;11:281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Rathkey JK, Zhao J, Liu Z, Chen Y, Yang J, Kondolf HC, et al. Chemical disruption of the pyroptotic pore-forming protein gasdermin D inhibits inflammatory cell death and sepsis. Sci Immunol. 2018;3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Humphries F, Shmuel-Galia L, Ketelut-Carneiro N, Li S, Wang B, Nemmara VV, et al. Succination inactivates gasdermin D and blocks pyroptosis. Science. 2020;369:1633–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Martin-Sanchez F, Diamond C, Zeitler M, Gomez AI, Baroja-Mazo A, Bagnall J, et al. Inflammasome-dependent IL-1beta release depends upon membrane permeabilisation. Cell Death Differ. 2016;23:1219–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Volchuk A, Ye A, Chi L, Steinberg BE, Goldenberg NM. Indirect regulation of HMGB1 release by gasdermin D. Nat Commun. 2020;11:4561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Tan G, Huang C, Chen J, Zhi F. HMGB1 released from GSDME-mediated pyroptotic epithelial cells participates in the tumorigenesis of colitis-associated colorectal cancer through the ERK1/2 pathway. J Hematol Oncol. 2020;13:149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Li W, Deng M, Loughran PA, Yang M, Lin M, Yang C, et al. LPS Induces Active HMGB1 Release From Hepatocytes Into Exosomes Through the Coordinated Activities of TLR4 and Caspase-11/GSDMD Signaling. Front Immunol. 2020;11:229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Russo HM, Rathkey J, Boyd-Tressler A, Katsnelson MA, Abbott DW, Dubyak GR. Active Caspase-1 Induces Plasma Membrane Pores That Precede Pyroptotic Lysis and Are Blocked by Lanthanides. J Immunol. 2016;197:1353–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Yang X, Cheng X, Tang Y, Qiu X, Wang Y, Kang H, et al. Bacterial Endotoxin Activates the Coagulation Cascade through Gasdermin D-Dependent Phosphatidylserine Exposure. Immunity. 2019;51:983–96 e6. [DOI] [PubMed] [Google Scholar]
- [110].Ruhl S, Shkarina K, Demarco B, Heilig R, Santos JC, Broz P. ESCRT-dependent membrane repair negatively regulates pyroptosis downstream of GSDMD activation. Science. 2018;362:956–60. [DOI] [PubMed] [Google Scholar]
- [111].Lee IH, Kai H, Carlson LA, Groves JT, Hurley JH. Negative membrane curvature catalyzes nucleation of endosomal sorting complex required for transport (ESCRT)-III assembly. Proc Natl Acad Sci USA. 2015;112:15892–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [112].Kayagaki N, Kornfeld OS, Lee BL, Stowe IB, O’Rourke K, Li Q, et al. NINJ1 mediates plasma membrane rupture during lytic cell death. Nature. 2021;591:131–6. [DOI] [PubMed] [Google Scholar]