

Summary
MED13L syndrome is a haploinsufficiency developmental disorder characterized by intellectual disability, heart malformation, and hypotonia. MED13L controls transcription by tethering the cyclin C-Cdk8 kinase module (CKM) to the Mediator complex. In addition, cyclin C has CKM-independent roles in the cytoplasm directing stress-induced mitochondrial fragmentation and regulated cell death. Unstressed MED13LS1497F/fs patient fibroblasts exhibited aberrant cytoplasmic cyclin C localization, mitochondrial fragmentation, and a 6-fold reduction in respiration. In addition, the fibroblasts exhibited reduced mtDNA copy number, reduction in mitochondrial membrane integrity, and hypersensitivity to oxidative stress. Finally, transcriptional analysis of MED13L mutant fibroblasts revealed reduced mRNA levels for several genes necessary for normal mitochondrial function. Pharmacological or genetic approaches preventing cyclin C-mitochondrial localization corrected the fragmented mitochondrial phenotype and partially restored organelle function. In conclusion, this study found that mitochondrial dysfunction is an underlying defect in cells harboring the MED13LS1497F/fs allele and identified cyclin C mis-localization as the likely cause. These results provide a new avenue for understanding this disorder.
Subject areas: Biological sciences, Biochemistry, Cell biology
Graphical Abstract
Highlights
-
•
MED13L haplo-insufficiency mutation causes aberrant cyclin C nuclear release
-
•
Cyclin C nuclear release fragments mitochondria and increases endogenous ROS
-
•
MED13L syndrome patient fibroblasts exhibit reduced mitochondrial function
-
•
MED13L syndrome patient fibroblasts exhibit reduced mtDNA copy number
Biological sciences; Biochemistry; Cell biology
Introduction
MED13L syndrome is an autosomal dominant spectrum disorder diagnosed in children that is denoted by several features including moderate intellectual disability (ID), speech impairment, dysmorphic facial features, heart malformation, and muscle hypotonia (Torring et al., 2019). Typical of these disorders, not all patients present with the same scope or severity of symptoms (Adegbola et al., 2015; Asadollahi et al., 2017; Snijders Blok et al., 2018; van Haelst et al., 2015). In the study of over 1000 patients with undiagnosed developmental disorders, 0.5%–1% exhibited de novo MED13L loss of function mutations (Deciphering Developmental Disorders, 2015). These findings suggest that MED13L mutation is a major factor in ID presentation. Although several MED13L mutations have been associated with this syndrome, a causative mechanism(s) for the multiple facets of this disorder has not been established.
CCNC (cyclin C), and one each of the CDK8/CDK19, MED12/MED12L, and MED13/MED13L paralogs, form the CDK8 kinase module (CKM), a highly conserved subcomplex of the RNA polymerase II Mediator (Bourbon, 2008). Transcriptome results revealed that cyclin C plays both a positive and negative role in transcription with many genes involved in the stress response and energetics (Li et al., 2014; Stieg et al., 2019). In addition to its transcriptional role, cyclin C also has a cytoplasmic function in stressed cells (Jezek et al., 2019b; Wang et al., 2015). Specifically, exposure to several stressors induces partial cyclin C nuclear release which in turn directly stimulates both stress-induced mitochondrial fission (Cooper et al., 2014; Ganesan et al., 2019) and intrinsic regulated cell death (iRCD) (Jezek et al., 2019a; Wang et al., 2015). Importantly, cytoplasmic cyclin C is sufficient to induce mitochondrial fission but not iRCD in the absence of additional stress (Jezek et al., 2019a; Wang et al., 2015) although it does make cells hypersensitive to oxidative damage (Jezek et al., 2019b).
Several results indicate that MED13 or MED13L anchors cyclin C in the nucleus. First, structural and genetic studies revealed that MED13 or MED13L tethers the CKM to the Mediator in mammalian cells (Hoeppner et al., 2005; Li et al., 2021). In addition, cyclin C-CDK8 promoter retention in unstressed cells is directly tied to MED13 turnover (Davis et al., 2013). Genetic analyses in yeast revealed that deleting MED13 results in partial cyclin C nuclear release (Khakhina et al., 2014). Moreover, preventing Med13 destruction in stressed yeast cells results in cyclin C nuclear retention (Stieg et al., 2018). Finally, constitutively cytoplasmic yeast cyclin C not only results in continuously fragmented mitochondria but also in a loss of mitochondrial function (Khakhina et al., 2014). Taken together, these studies point to a conserved role for MED13 and MED13L in anchoring cyclin C in the nucleus. In addition, these findings also suggest that long-term mitochondrial scission induced by constitutive cytoplasmic cyclin C is detrimental to organelle function. This report describes aberrant cyclin C subcellular location in MED13L syndrome patient fibroblasts and the impact this defect has on mitochondrial homeostasis and sensitivity to oxidative stress.
Results
Cyclin C exhibits aberrant cytoplasmic localization in unstressed MED13LS1497 F/fs fibroblasts
A six-year-old male patient presenting with heart malformation, ID, and hypotonia was further examined using whole exome sequencing of genomic DNA extracted from a peripheral blood sample. These results revealed a thymidine duplication NM_015335.5|:c.[5054dupT]; [(Ser1497Phe/fs)] in exon20 of MED13L. This duplication resulted in a frameshift mutation (MED13LS1497 F/fs) truncating the protein in exon21. Given the conserved role of MED13/MED13L in retaining cyclin C in the nucleus, immunofluorescence was used to monitor cyclin C localization and mitochondrial morphology in mortal skin fibroblasts isolated from the patient. As expected, cyclin C was primarily nuclear and the mitochondria exhibited long, interconnected tubules in the control WI-38 human lung fibroblasts (top row, Figure 1A). However, the MED13L+/S1497 F/fs (denoted MED13L+/fs for simplicity) cells exhibited elevated cyclin C cytoplasmic localization and extensive mitochondrial fragmentation (third row, Figure 1A). Quantification revealed over 90% of the MED13L+/fs cells exhibited fragmented mitochondria compared to 20% for the control (boxed numbers, Figure 1A). In addition, these studies revealed co-localization between cytoplasmic cyclin C and mitochondria (Figure 1A, arrows, third row in zoom panel). We previously found that treating cells with the cytoplasmic chaperone inhibitor pifithrin μ (PFT) (Strom et al., 2006) blocked cyclin C-mitochondrial localization (Wang et al., 2015). Treating cells with PFT (1 μM) partially restored mitochondrial fusion within 24 h (Figure 1A, bottom panels) arguing that cyclin C mis-localization is responsible for mitochondrial fragmentation in MED13L+/fs cells.
Figure 1.

MED13L+/fs cells exhibit cyclin C nuclear release and mitochondrial dysfunction
(A) The cultures indicated were monitored for nuclear (DAPI), cyclin C (immunohistochemistry), and mitochondrial morphology (MitoTracker). Numbers in mito panels indicate % of population exhibiting fragmented mitochondria (n = 3). Samples treated with PFT (1 μM, 24 h) are indicated. Arrows indicate cyclin C-mitochondrial co-localization. Bar = 20 μM, zoom = 6X magnification.
(B–D) (B) Seahorse analysis of the indicated cell lines quantifying basal respiration (B), ATP production (C) and maximum respiration (D) for vehicle and PFT treated (1 μM, 96 h).
(E) RNA-seq analysis of CS mRNA levels in MEF cultures with the indicated genotypes. Values from three independent cultures were normalized to ActB. SEM is shown. ∗, p < 0.01
MED13L+/fs cells exhibit reduced mitochondrial activity
Previous studies (Hori et al., 2011; Scheibye-Knudsen et al., 2015), including our own in yeast (Khakhina et al., 2014), found that continuous mitochondrial fragmentation is associated with mitochondrial dysfunction. To test mitochondrial output in MED13L+/fs cells, oxygen consumption was measured via Seahorse metabolic flux analysis. Using WI-38 cultures as a control, we found that MED13L+/fs cells exhibited a 6-fold reduction in oxygen consumption rate (OCR, Figure 1B) and a 3-fold reduction in respiratory ATP production (Figure 1C). In addition, maximum respiratory activity was also reduced in MED13L+/fs cells (Figure 1D). These results suggest that mitochondrial respiration is reduced in MED13L+/fs cells. Support for this conclusion was obtained with the analysis of citric acid cycle and electron transport chain activities in a patient's skeletal muscle tissue biopsy. This study revealed reduced activity in components of both pathways (Table 1) indicating mitochondrial dysfunction in a different tissue type. As these studies were standardized to citrate synthase (CS) activity, one possible explanation is that CS mRNA levels are reduced in the MED13L+/fs cell line resulting in appearant lower overall activity for many of these assays. To test this possibility, we used RNAseq to measure CS mRNA levels in wild-type (Ccnc+/+) or deleted (Ccnc−/−) mouse embryonic fibroblasts. The rationale for this study was that deleting Ccnc should inactivate all CKM complexes providing a more robust signal. Feature counts of CS mRNA were normalized to ActB mRNA levels. These experiments revealed no change in CS transcription in Ccnc−/− cells (Figure 1E) indicating that CS levels were not altered and that MED13L+/fs cells exhibited reduced mitochondrial function.
Table 1.
Mitochondrial activities in muscle biopsy of patient with MED13LS1947 F/fs
| Test | Patient values mU CS | Normal range |
|---|---|---|
| [1-14C]pyruvate + malate | 2.83 | 3.61–7.48 |
| [1-14C]pyruvate + carnitine | 3.68 | 2.84–8.24 |
| [1-14C]pyruvate + malonate | 3.19 | 3.43–7.3 |
| [U-14C]malate + acetylcarnitine + malonate | 2.54 | 3.43–7.3 |
| [U-14C]malate + acetylcarnitine + arsenite | 1.24 | 2.05–3.85 |
| [1,4-14C]succinate + acetylcarnitine | 1.6 | 2.54–6.39 |
| Reduced ATP + Creatine-phosphate | 21 | 42–81 |
| CI NADH:Q1 Oxydereductase | 90 mU/U CS | 70–250 |
| CII Succinate Dehydrogenase | 77 mU/U CS | 67–177 |
| CIII cyt C Oxydoreductase | 1914 mU/U CS | 2200–6610 |
| CIV cyt C Oxydase | 1227 | 810–3120 |
| CII + CIII | 214 mU/U CS | 300–970 |
CS, citrate synthase.
Our results are consistent with a model that aberrant cyclin C nuclear release results in constitutive mitochondrial fragmentation and organelle dysfunction. To explore this possibility further, mitochondrial activity was measured in MED13L+/fs cells following PFT treatment (1 μM, 96 h). Although no significant change in activity was observed with WI-38 control, the MED13L+/fs cells exhibited increased OCR, respiratory ATP synthesis, and respiratory maximum (Figures 1B–1D). We were unable to extend these experiments for a longer duration as PFT treatment proved toxic after prolonged treatment. However, these results indicate that the mitochondrial hyper-fission phenotype, and to some extent mitochondrial dysfunction, is reversible by preventing cyclin C relocalization to the mitochondria.
Enhanced glycolysis and mitochondrial dysfunction were observed in MEF cells deleted for the fusion factors Mfn1 and Mfn2 (Chen et al., 2003, 2010). In addition, mitochondrial dysfunction has been associated with increased glycolytic activity in several pathological states including cancer and vascular disease (Hu et al., 2012). Therefore, we examined glycolytic activity in MED13L+/fs cells using Seahorse metabolic flux analysis by indirectly measuring lactate production using change in pH. Multiple experiments revealed either no change (trial 1) or increase (trial 2) in glycolytic activity for MED13L+/fs compared to control WI-38 cultures (Figure S1A). The addition of PFT for 96 h resulted in a slight reduction in glycolysis in for both cell lines. In addition, a modest elevation of serum lactate levels (2.4 mmol/L (normal range 0.5–2.2) was also noted in the patient. Finally, glycolytic activity was measured indirectly by quantifying lactate dehydrogenase A (LDHA) levels (Vander Heiden et al., 2009). Increased glycolysis stimulates LDHA production. Western blot analysis revealed a 1.6-fold increase in LDHA in the MED13L+/fs cells compared to control (Figure S1B, quantified in Figure S1C) but did not satisfy the significance cutoff. Taken together, these results indicate that mitochondrial function is reduced in MED13L+/fs cells. In addition, this reduced energy production may be compensated, at least in part, by increased glycolysis.
MED13L+/fs cells exhibit reduced mtDNA copy number
The reduction in mitochondrial output can be due to many factors including defects in nuclear transcription of mitochondria-destined proteins, loss of mtDNA integrity, or both. For mtDNA maintenance, mitochondrial dynamics play important roles as fusion is associated with elevated recombination and repair while constitutive mitochondrial fission contributes to nucleoid loss (Chen, 2013). We used a qPCR-based approach to measure mtDNA copy number (Phillips et al., 2014) by comparing the mtMinArc region (Figure S2A) to a genomic locus. These experiments revealed a 2-fold reduction in mtDNA levels in the mutant fibroblasts compared to control (Figure 2A). These findings suggest that reduced mtDNA copy number contributes to the mitochondrial dysfunction observed in MED13L+/fs cells.
Figure 2.

MED13L+/fs cells exhibit reduced mtDNA copy number, nucleoid retention, and mitochondrial maintenance gene transcription
(A) qPCR results from the minor arc with the indicated human fibroblasts. The -ΔCT was calculated with a nuclear gene (ß2M) used as the internal control. Error bars indicate SEM (n = 3 biological replicates performed in triplicate).
(B) Visualization of mtDNA nucleoids in WT and MED13L+/fs cells. Cells were fixed and co-stained with MitoTraker red and α-dsDNA antibodies. Boxed regions in merged images are enlarged in the right panels. Values presented represent the number of nucleoids per individual mitochondrion. Values for untreated WI-38 cells were not included as mitochondria were fused with multiple nucleoids. Asterisks indicate statistical differences from treated WI-38 value. Bar = 20 μM.
(C) RT-qPCR mRNA quantification of the indicated genes in unstressed MED13Lfs and WI-38 cells. Changes in mRNA levels are indicated (log2) compared to WI-38 control cells. SEM is shown. ∗ = p < 0.05; ∗∗ = p < 0.01; ∗∗∗ = p < 0.005; ∗∗∗∗ = p < 0.001.
The mtDNA is found in protein-DNA complexes termed nucleoids with each containing 1–15 copies of the mitochondrial genome (Satoh and Kuroiwa, 1991). Previous studies have identified several factors involved in nucleoid maintenance including mtDNA binding proteins (Chen and Butow, 2005) and factors controlling mitochondrial shape (Chan, 2020; Youngman et al., 2004). For example, causing constitutive mitochondrial fragmented by deleting the fusion factors Mfn1 and Mfn2 resulted in nucleoid loss in individual mitochondrion (Silva Ramos et al., 2019). Using antibodies directed at dsDNA, we calculated the number of nucleoids associated with individual mitochondrion in WI-38 and MED13L+/fs cells. Because WI-38 contains fused mitochondria with multiple nucleoids (Figure 2C), these cells were treated with H2O2 to induce fragmentation. This allowed the number of nucleoids per mitochondrion to be assessed. As expected, nearly all the mitochondria contained nucleoids in treated WI-38 cells (Figure 2C, second row). However, only 57% and 61% of the mitochondria contained nucleoids in untreated and treated MED13L+/fs cells, respectively. These results indicate that a significant portion of the mitochondria lack detectable nucleoid DNA which correlates with reduced mtDNA copy number and mitochondrial dysfunction.
MED13L+/fs cells exhibit reduced transcription of nuclear-encoded mitochondrial proteins
Of the ∼1500 proteins that compose the mitochondria, the vast majority are encoded by the nuclear genome (Diaz and Moraes, 2008). We previously described the cyclin C-dependent transcriptome in mice focusing on the oxidative stress response (Stieg et al., 2019). Interrogation of these datasets revealed several genes exhibiting reduced transcription in the Ccnc−/− mouse embryonic fibroblast (MEF) cell line that had mitochondrion GO terms (Figure S2B). We tested five genes and found that two genes, TFB1M (a mitochondrial transcription specificity factor) and MDH1 (malate dehydrogenase) exhibited modest reductions in mRNA levels in MED13L+/fs cells compared to WI-38 controls (Figure 2D). Transcript levels of SDHB (Succinate Dehydrogenase Complex Iron Sulfur Subunit B) were slightly more suppressed while HSPA9 (mitochondrial chaperone) and NDUFV3 (NADH: Ubiquinone Oxidoreductase Subunit V3) were more severely affected. Consistent with this latter finding, ubiquinone oxioreductase activity was at the lower end of the normal range in the muscle biopsy (Table 1). Therefore, despite the presence of two wild-type copies of MED13 and one copy of MED13L, transcriptional defects in mitochondrial-directed genes were detected in MED13L+/fs cells. These results suggest that the mitochondrial dysfunction observed in MED13L+/fs cells are due to both nuclear and mitochondrial-specific defects. The exact contribution of each arm of the mitochondrial maintenance pathway is yet to be established.
MED13L+/fs cells exhibit elevated ROS and sensitivity to oxidative stress
Previous studies have reported that cells undergoing extensive mitochondrial fragmentation also exhibit elevated endogenous reactive oxygen species (ROS) levels perhaps due to disrupting the ETC complexes (Hung et al., 2018). To examine this question, MED13L+/fs cells were treated with a mitochondrial-specific redox sensor (MitoSOX) followed by fluorescence-activated cell analyses. We found that MED13L+/fs cells exhibited a significant elevation in MitoSOX oxidation compared to the WI-38 control (Figure 3A). To further explore the health of the mitochondria, WI-38 and MED13L+/fs cells were treated with TMRM, a dye that fluoresces in the oxidative environment of intact mitochondria. These studies found statistically significant reduction in membrane potential (Δψ) in MED13L+/fs cells (Figure 3B). These findings indicate that the MED13L+/fs mutation reduces mitochondrial function and health. Repeating these experiments with PFT treatment for 96 h did not produce a statistically significant recovery of mitochondrial membrane potential although the results were trending in that direction.
Figure 3.

MED13L+/fs cells exhibit loss of mitochondrial integrity and enhanced ROS sensitivity
(A) Endogenous mitochondrial ROS was measured in the cells indicated using mitochondrial-specific ROS activated fluorescent dye MitoSOX. H2O2-treated WI-38 cells served as a positive control for MitoSox oxidation. N = 3 for all samples. Means ± SEM are shown.
(B) Mitochondrial membrane potential for the indicated cell lines with or without PFT added (1 μM, 96 h) was calculated using TMRM staining as described in STAR methods.
(C) WI-38 and MED13L+/fs cells were treated with H2O2 (0.4 mM, 4 h) as indicated and mitochondrial morphology (MitoTracker) and the subcellular localization of the nucleus (DAPI staining) and cyclin C (immunohistochemistry) were determined. The percentages of the population exhibiting fragmented mitochondria are shown on the right.
(D) Intrinsic regulated cell death (iRCD) was measured in the indicated cell lines with or without H2O2 addition (0.4 mM, 16 h). The percentages of the populations that were Annexin V positive, propidium iodide negative as determined by FAC analysis are shown. N = 3 for all experiments, means ± SEM are shown. ∗ = p < 0.05; ∗∗ = p < 0.01. Bar = 20 μM.
(E) Mitochondrial morphology and cyclin C subcellular localization were determined for MED13L+/fs cells with and without NAC treatment as indicated. The percent of cells exhibiting fragmented mitochondria or cytoplasmic cyclin C (>50 cells counted) are indicated in the MitoRed and α-cyclin C panels, respectively. Bar = 10 μM
Many cell types are sensitive to elevated endogenous ROS including neurons (Calabrese et al., 2005; Li et al., 2013). To test whether the MED13fs allele sensitized the fibroblasts to reactive oxygen, these cells were treated with H2O2 and cyclin C subcellular localization, mitochondrial morphology and intrinsic regulated cell death (iRCD, a.k.a. apoptosis) efficiency were monitored. In WI-38 control cells, H2O2 treatment induced the anticipated cyclin C release and mitochondrial fragmentation (Figure 3C). In the MED13L+/fs cells, cyclin C levels in the cytoplasm were elevated over untreated controls but no quantitative difference in percentage of cells exhibiting mitochondrial fragmentation was observed following H2O2 treatment (Figure 3C).
Our previous work indicated that cytoplasmic cyclin C stimulated mitochondrial localization of Bax, a Bcl-2 family member that triggers iRCD (Jezek et al., 2019a). Although cytoplasmic cyclin C did not stimulate cell death on its own, it did make cells hypersensitive to oxidative stress. To test whether the cytoplasmic cyclin C in MED13L+/fs cells altered sensitivity to H2O2-induced iRCD, two cell death signals, Annexin V reactivity and propidium iodide (PI) permeability were monitored. In these assays, only Annexin V-positive, PI-negative cells were counted to eliminate the contribution from necrotic cells. In the absence of any exogenous stress, the presence of the MED13Lfs allele did not enhance iRCD execution (Figure 3D). However, treatment with low-dose H2O2 stimulated a significant increase in iRCD in the MED13Lfs cells compared to the control. These findings indicate that the MED13Lfs allele does not induce cell death on its own but does sensitize fibroblasts to oxidative insults. As cyclin C relocalizes to the cytoplasm following oxidative stress (Wang et al., 2015), one possible cause of aberrant cyclin C localization in MED13L+/fs cells is elevated ROS and not disruption of binding to the Mediator. To test this possibility, N-acetyl cysteine (NAC) was added to MED13L+/fs cells for 2 h; then, cyclin C localization and mitochondrial morphology were accessed. Two hours was chosen as this was sufficient time to blunt the impact of H2O2 treatment on cyclin C function (Jezek et al., 2019a). These studies revealed no change in cyclin C localization or mitochondrial fragmentation following NAC treatment (Figure 3E). Taken together, these results indicate that cyclin C cytoplasmic localization is due to the MED13Lfs mutation and that the presence of cyclin C at the mitochondria sensitizes cells to oxidative stress.
Correcting the MED13Lfs mutation restores mitochondrial fusion
As described earlier, PFT treatment partially reversed MED13Lfs-induced mitochondrial hyper-fragmentation indicating that aberrant cyclin C localization to the mitochondria was the basis for organelle fragmentation (Figure 1A). However, as a chaperone inhibitor, it is still possible that the impact of PFT treatment is not solely mediated through cyclin C localization. As described earlier, the MED13Lfs allele possesses an extra thymidine in exon 20 terminating the protein in exon 21 (Figure 4A). To correct this mutation, we used a new CRISPR approach termed in situ cut-and-paste (iCAP) that utilizes the Cas9 or Cas12a genome editing enzymes to generate precise double-strand breaks (Figure 4B). Placing the identical nuclease target sites flanking the wild-type sequence on the replacement template (RT) provides a substrate for seamless replacement of the mutant allele. In addition, the RT harbors a puromycin resistance gene to identify transfectants (see Figure 4B for Cas12a example). Following transfection, two antibiotic resistant pools obtained from the Cas9 and Cas12a iCAP procedures (4-1-2 and 5-1-2, respectively) were selected for further study. PCR genotyping and sequencing of iCAP replacement insertions revealed that the mutation in exon 20 was corrected. To determine if correcting the frameshift mutation altered mitochondrial morphology and cyclin C localization, fluorescence microscopy was employed. Compared to the MED13L+/fs cell line, 4-1-2 and 5-1-2 cell pools exhibited reduced mitochondrial fragmentation and predominantly nuclear cyclin C similar to that observed in wild-type fibroblasts (representative images shown in Figure 4C, quantified in Figure 4D). These results confirmed that the aberrant cytoplasmic cyclin C and mitochondrial fragmentation was a result of the MED13Lfs frameshift allele and not due to a secondary mutation. Attempts to measure mitochondrial activity in the 4-1-2 or 5-1-2 corrected cell pools were confounded by reduced growth kinetics of these mortal cell lines due to senescence. Therefore, the extent to which mitochondrial function can be restored by inhibiting aberrant cyclin C-dependent fission needs further study (see Discussion).
Figure 4.

Correcting the MED13Lfs mutation using iCAP genome editing restores mitochondrial fusion and cyclin C nuclear localization
(A) Nucleotide sequence of MED13Lfs allele with the extra thymidine and aberrant protein sequence indicated.
(B) iCAP strategy to introduce MED13L correction replacement template.
(C) Representative images displaying cyclin C localization and mitochondrial morphology for the 4-1-2 and 5-1-2 corrected cell pools. Bar = 20 μM.
(D) Percent of the population displaying fragmented mitochondria are quantified in the indicated cell lines (n = 3). SEM is shown, ∗∗, p ≤0.01.
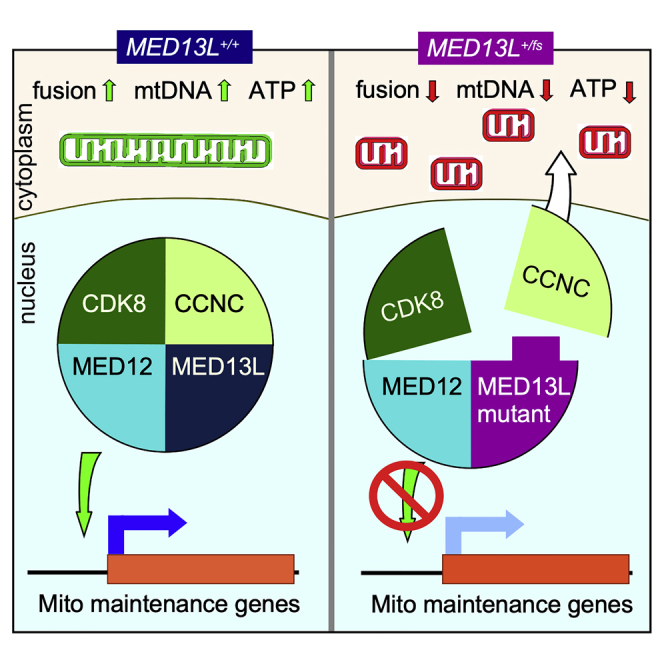
(E) In normal cells, the CKM supports mitochondrial health by stimulating transcription of mitochondrial maintenance genes (MMG) and retaining cyclin C in the nucleus resulting in increased mitochondrial fusion, ATP production, stabilized mitochondrial membranes, and enhanced mtDNA integrity.
(F) MED13Lfs mutation allows partial cyclin C nuclear release resulting in reduced MMG transcription and constitutive fragmentation leading to mitochondrial dysfunction.
Discussion
Typical of spectrum disorders, patients with MED13L syndrome present with different subsets of afflictions that vary in their severity. However, the underlying cause(s) of the individual phenotypes, as well as their differences in intensity, is unknown. This study reports that MED13L+/S1497F/fs patient fibroblasts exhibit reduced mitochondrial function in both fibroblasts and skeletal muscle. Mitochondrial dysfunction is at the heart of many disorders (Srivastava, 2017) including neuronal development/function (Nunnari and Suomalainen, 2012) and muscle strength (Chan, 2020; Zane et al., 2017). The proper balance between mitochondrial fission/fusion (Scheibye-Knudsen et al., 2015), and the continued supply of nuclear-encoded proteins (Jazwinski, 2013), is required for normal mitochondrial function. Importantly, loss of fusion activity through mutating the mitofusions Mfn1 and Mfn2 results in a similar response (Chen et al., 2010). With continuously fragmented mitochondria, there is a reduction in mtDNA content as well as overall activity. The underlying mechanism for this observation is not completely understood. However, defects in the distribution of mtDNA replication machinery have been observed in these fragmented organelles (Silva Ramos et al., 2019). Our findings phenocopy the fusion defective cells arguing that cyclin C-induced fragmentation is at least partially responsible for mitochondrial dysfunction observed in MED13L+/fs cells. For example, in wild-type cells, the intact CKM sequesters cyclin C in the nucleus both stimulating the transcription of nuclear encoded mitochondrial genes and preventing continuous mitochondrial fission (Figure 4E). The MED13Lfs allele allows aberrant cyclin C nuclear release that diminishes both the morphology and transcriptional arms of the mitochondrial maintenance system (Figure 4F). Finally, we demonstrated that MED13L+/fs cells exhibit elevated endogenous ROS levels and that these cells are hypersensitivity to H2O2. We have previously demonstrated that forced cyclin C relocalization to the mitochondria enhances cell sensitivity to oxidative stress-induced cell death in cancer cells (Jezek et al., 2019a). In addition, neurons and muscle can be highly sensitive to elevated oxidative stress (Cheung et al., 2007; Choi et al., 2016; Kirkland et al., 2007; Reddy et al., 2011; Santos and Cardoso, 2012). Taken together, our findings provide a potential explanation underlying at least a subset of the symptoms associated with MED13L syndrome.
MED13/MED13L is not unique in that mutations in almost all CKM components are associated with similar developmental syndromes (reviewed in (Poot, 2020)). For example, MED13 mutations cause heart defects and ID (Minerath et al., 2019; Snijders Blok et al., 2018). Similarly, haplo-insufficient MED12L or MED12 mutations are also associated with ID, speech impairment, and facial dysmorphology (Caro-Llopis et al., 2016; Nizon et al., 2019; Srivastava et al., 2019). Finally, several patients presenting with hypotonia, autism-like spectrum disorders, and congenital heart abnormalities were found to harbor CDK8 (Calpena et al., 2019; Uehara et al., 2020) or CDK19 (Chung et al., 2020; Zarate et al., 2021) mutations. Taken together, these syndromes present with overlapping disorders and have been placed into a general category of transcriptomopathies (Caro-Llopis et al., 2016; Yuan et al., 2015). The lone exception to this pattern is CCNC, which has not been associated with any syndromic disorders. This would not be expected if transcriptional defects were the only driving force behind these syndromes. This is especially true for CCNC as a paralog has not been identified unlike the other CKM components. One simple explanation is that the CCNC locus is resistant to mutagenesis. Another possibility is that mutations in other CKM components also require cyclin C to express their pathology. In this model, a combination of transcription mis-regulation and aberrant cytoplasmic cyclin C localization are required for expression of this disorder. Currently, we do not know if cyclin C subcellular localization is altered in these different patients, but the possibility is supported by the multiple contacts cyclin C makes with the other CKM components (Li et al., 2021). Mutating any component could impact CKM integrity and stimulate cyclin C nuclear release. Consistent with these results, a Ccnc mutation that disrupted its interaction with Med13 or Med13L resulted in aberrant cytoplasmic localization, increased mitochondrial fission, and dysfunction in developing mouse cardiac tissue (Ponce et al., 2020). Elaborating cyclin C subcellular localization in patients with differing CKM mutations would provide insight into this question.
Correcting the MED13Lfs mutation or prohibiting cyclin C-to-mitochondrial transport resulted in rapid restoration of mitochondrial tubules. Although an indication that mitochondrial function was also returning, the activity remained well below control values. Unfortunately, these experiments were cut short when the corrected mortal MED13L+/fs cell line senesced. However, these results beg the question of whether mitochondrial function can be restored by simply preventing cyclin C nuclear exit. The reduction in mtDNA copy number in MED13L+/fs cells is well below the normal range of hundreds to thousands depending on the cell type. There are a few examples in which low mtDNA copy number is routinely observed. For example, sperm contains ∼100 mtDNA copies and reducing this number to only a third does not significantly impede sperm cell function (Wai et al., 2010).Therefore, although the overall copy number is reduced, the range is higher than that of mitochondriopathies such as Kearns-Sayre syndrome or certain cancers (Bai and Wong, 2005; Lee et al., 2010) that exhibit more severe copy number changes. Taken together, these observations raise the possibility that mitochondrial function can be restored at least to some extent in MED13L syndrome cells. To what extent mitochondrial function returns and the time frame required remains an open question that will require more study.
Limitations of the study
The most important caveat to these conclusions is that only one MED13L syndrome cell line was examined. Concerted effort was made to secure another cell line to test but they were unsuccessful. Only through analysis of additional MED13L syndrome, as well as cell lines harboring mutations in other CKM components, will the generality of the findings described here be ascertained. Second, the use of mortal cell lines placed constraints on our analysis of the iCAP-corrected derivatives. The concern with immortalizing these corrected cell lines was the potential impact this would have on metabolic activity. However, in the absence of a pool of patient samples, a MED13L mutant-immortalized cell platform may be required to address questions concerning the rate and extent at which mitochondrial function can be restored by correcting the mutation or through pharmacological inhibition of cyclin C-dependent mitochondrial fission.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| cyclin C | ThermoScientific | PA5-16227; RRID:AB_10982613 |
| Alexa Fluor 488-conjugated secondary antibody | ThermoScientific | A11008; RRID:AB_143165 |
| alkaline phosphatase-conjugated rabbit secondary antibody | Abcam | ab97061; RRID:AB_10680575 |
| LDHA | Santa Cruz Biotechnology | SC-137243; RRID:AB_2137192 |
| β-actin | Merck | A1978; RRID:AB_476692 |
| dsDNA | Dev. Studies Hybridoma Bank | RRID:AB_10805293 |
| Chemicals, peptides, and recombinant proteins | ||
| H2O2 | ACS Chemical | UN2014 |
| Pifithrin μ | TGI Chemica | P2048 |
| carbonyl cyanide p-trifluoromethoxyphenylhydrazone | Cayman Chemical | 15218 |
| Oligomycin | Sigma | 49455 |
| Rotenone | Sigma | R8875 |
| Antimycin A | Sigma | R8875 |
| MitoTempo | Sigma | SML0737 |
| Actinomycin D | Cayman Chemical | 11421 |
| Tetramethylrhodamine (TMRM) | Invitrogen | I34361 |
| Critical commercial assays | ||
| Annexin V Cell Death Assay | BD Biosciences | 556419 |
| Bradford Protein | Bio-Rad | 5000001 |
| Experimental models: Cell lines | ||
| MED13LS1497F/+ human dermal fibroblast | Center for Molecular Medicine UMC Utrecht Netherlands | N/A |
| MED13L+/+ human dermal fibroblast corrected allele | This study | N/A |
| WI-38 | ATCC | CCL-75 |
| Experimental models: Organisms/strains | ||
| XL-1 Blue Escherichia coli | Agilent | 200249 |
| Oligonucleotides | ||
|
MED13L exon 20, Forward AGCCTAGTCCAAGTTTTAGAGAG |
This study | N/A |
|
MED13L exon 20, Reverse AAACTGCCCAGAACACCAAACTGG |
This study | N/A |
| M13 Sequencing Primer Forward TGTAAAACGACGGCCAGT |
This study | N/A |
| M13 Sequencing Primer, Reverse CAGGAAACAGCTATGAC |
This study | N/A |
|
MED13L intron19-20, Forward CGATCAGCATACTCACTGCTTCAG |
This study | N/A |
|
MED13L intron19-20, Reverse GTCTCCTTTCAGACTGATTCCATG |
This study | N/A |
| mtMinArc Forward CTAAATAGCCCACACGTTCCC | mt 16,528 – 16,548 | (Phillips et al., 2014) |
| mtMinArc Reverse AGAGCTCCCGTGAGTGGTTA | mt 23-42 | (Phillips et al., 2014) |
| Fβ2M Forward GCTGGGTAGCTCTAAACAATGTATTCA | Chr15 15,798,932 – 15,798,95 | (Phillips et al., 2014) |
| Fβ2M Reverse CCATGTACTAACAAAT GTCTAAAATGGT |
Chr15 15,798,999 – 15,799,02 | (Phillips et al., 2014) |
| Recombinant DNA | ||
| pGL3-U6-sgRNA-PGK-puromycin | Addgene | 51133 |
| pTE4398 | Addgene | 74042 |
| pX330S-2 | Addgene | 58778 |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to, and will be fulfilled by, the lead contact, Randy Strich (strichra@rowan.edu)
Materials availability
The MED13L+/fs mutant and corrected cell lines described in this study will be made available upon request and proper consent.
Experimental model and subject details
Dermal fibroblasts from a MED13L syndrome 6 year old male patient were obtained from a skin biopsy, cultured and stored. Cells were obtained with written consent of the patient’s representative. WI-38 cells were obtained from the ATCC. The human fibroblast cell line (MED13LS1497 F/fs) was obtained following informed consent with the identification of the donor blinded from the participants in this study. WI-38 embryonic lung fibroblasts were obtained from the ATCC.
Method details
Cell culture
The cells were cultured in DMEM, 10% fetal bovine serum (FBS) and 1% penicillin/streptomycin (PS). Cells were passaged by brief treatment (2 min) with trypsin/EDTA with gentle tapping of the dish. Collected cells were counted and replated to provide approximately 20% confluency for WI-38, 25% for MED13Lfs cells. All incubations were conducted in this medium except for H2O2 treatment, which was performed in FBS- and PS-free medium. Pifithrin μ (PFT) was dissolved in DMSO and added to cells at a final concentration of 1 μM for the times indicated. The cell culture medium was changed daily and additional PFT was daily for multi-day experiments.
Metabolic flux analysis
The oxygen consumption rate (OCR) and extracellular acidification rate (ECAR) were measured by the Seahorse XF96 Analyzer (Agilent Technologies, Santa Clara, CA). WI-38 or MED13L+/fs cells were grown in XF96 microplates then incubated in a 37°C, non-CO2 incubator for one hour with the designated medium. The assay medium for the mitochondrial stress assay was Seahorse XF DMEM medium, pH 7.4 containing 10 mM glucose, 2 mM glutamine and 1 mM pyruvate. The assay medium for glycolytic stress assays was Seahorse XF DMEM medium, pH 7.4 containing 2 mM glutamine. Metabolic data were normalized to cell number after the assay. Each experiment was conducted in 14 separate wells with the results averaged ± standard error of the mean (SEM). Statistical significance was determined by the Students T test.
Western blot analysis
Whole-cell extracts (WCE) were prepared from cells harvested following trypsin-EDTA treatment. The cells were washed with 4 °C PBS and incubated with CHAPS lysis buffer (CLB,150 mM NaCl, 10 mM HEPES, 1% CHAPS, pH 7.4) containing 1% protease inhibitor cocktail (PIC) for 30 min at 4°C. WCEs were centrifuged at 14,000 × g for 15 min at 4 °C to separate soluble proteins from cell debris. Soluble protein concentrations were determined by Bradford assay (Bio-Rad, Hercules, CA). Western blots were visualized and quantified by phosphorimaging (iBright 1500, Thermo Scientific) using alkaline phosphatase-conjugated rabbit secondary antibody and CDP-Star as substrate. Blots were initially probed with LDHA antibodies then stripped and reprobed with β-actin for a loading control.
Cyclin C immunofluorescence and mitochondrial fragmentation assay
Cells were cultured on poly-l-lysine-coated coverslips for 2 d, stained with 100 nM MitoTracker Red CMXRos for 30 min at 37°C, fixed with 4% paraformaldehyde for 10 min, permeabilized with 0.2% Triton X-100 for 10 min, blocked with 2% BSA for 30 min, and incubated with 4 mg/mL cyclin C antibody at 4°C overnight. Slides were washed in PBS and 1 mg/mL Alexa Fluor 488-conjugated secondary antibody for 1 h. Fixed cells were mounted with 4′,6-diamidino-2-phenylindole (DAPI)-containing medium to stain the nuclei. Images were acquired with an Eclipse 90i microscope (Nikon, Tokyo, Japan) using the 100× oil objective (NA 1.25). Mitochondrial DNA immunofluorescence was accomplished as just described using α-dsDNA antibodies as primary. All images of a particular stain were collected with identical exposure times. Imaging processing was limited to adjusting exposures to permit visualization of all components in merged images. All merged images were adjusted identically. Mitochondria were scored as fragmented when cells contained >15 puncta with <10 mitochondria with a length of 10 μM or more were considered fragmented (Wang et al., 2015). NIS-Elements software (Nikon) was used for image deconvolution and analysis.
Mitochondrial functional assays
Cells were stained with 4 μM MitoSOX Red for 15 min at 37°C, washed twice with FluoroBrite DMEM, and signals quantified using an Accuri C6 flow cytometer (BD Biosciences, San Jose, CA). Tetramethylrhodamine methyl ester (TMRM) staining was conducted as suggested by the manufacturer. Changes in mitochondrial membrane potential (ΔΨm) were calculated using TMRM fluorescence levels as described (Li et al., 2009) using the Nernst equation (Nicholls, 2006). Cells were seeded in 12 well plates and treated with or without PFT (1 μM). For the assay, cells were treated with 80 μM CCCP for 5 min and then treated with 200 nM TMRM at 37°C for 30 min. Cells were harvested and then analyzed by flow cytometry (Attune NXT, ThermoFisher). To calculate membrane potential, CCCP + TMRM intensities for WI-38 and MED13Lfs were used as the unstimulated (or background) values. TMRM stimulated = fluorescent intensity (TMRM with or without PFT added) – background values.
Cell death assays
Cells were grown in 12-well plates (VWR, Radnor, PA) for 2 days and treated with H2O2 (0.4 mM for 16 h) as indicated in the text. Annexin V assays were conducted as described by the manufacturer on an Accuri C6 flow cytometer (BD Biosciences). Only Annexin V+ and propidium iodide (PI)– cells were plotted as they were considered early apoptotic without necrotic cell contribution to the signal.
In situcut-and-paste (iCAP) CRISPR correction of MED13LS1497 F/fs
Briefly, genomic DNA extracted from frozen cell culture samples of human WI-38 and MED13L+/fs fibroblasts was subjected to PCR amplification of exon 20. The fragments were subcloned into pGL3-U6-sgRNA-PGK-puromycin using XL1-Blue E. coli for amplification prior to DNA sequence analysis. The analysis revealed a mutation in MED13L exon 20 NM_015335.5|:c.[5054dupT];[(Ser1497Phefs)] (Chr12(GRCh38):t.[116252045dup]). Briefly, iCAP editing was accomplished by excising the endogenous mutant exon 20 from the MED13L allele by cleavages induced by Cas9 (pX330S-2) or Cas12a/Cpf1 (pTE4398) at two gRNA target sites, one in intron 19-20 and the other in intron 20-21. Intronic sequences flanking exon 20 are identical in the wild-type and mutant MED13L alleles. Potential Cas9 or Cas12a (Cpf1) gRNA target sequences were identified using the online tool "Benchling" (https://www.benchling.com/crispr/, San Francisco, CA). The mutant allele was replaced by pasting the wild-type exon 20 containing a puromycin selection marker gene placed in intron 19-20 (P.J. and R.S., personal communication). The edited isogenic fragment was pre-constructed in a DNA replacement template (dRT) in vitro and was excised from the dRT by CRISPR/Cas12a cleavages which also simultaneously occurred to the endogenous MED13L gene in situ.
Quantification and statistical analysis
The student’s t test was applied to calculate statistical differences with p values < 0.05 considered significantly different. p values are discriminated for each experiment as described in the figure legends. Standard error of the mean (S.E.M.) is presented in each figure.
Acknowledgments
We thank Erin Seifert and the MitoCircle facility (Thomas Jefferson University, Philadelphia USA) for assistance in the Seahorse analysis. We thank Katrina Cooper and Vidyaramanan Ganesan for helpful comments on manuscript preparation. We thank Stephen Willis for assistance in CS mRNA determinations. This work was supported by a grant from the National Institutes of Health (GM113052) and the New Jersey Health Foundation to R. S.
Author contributions
K-T.C., J.J., and A.N.C. conducted experiments, prepared figures, and edited the manuscript. Z.A.K. analyzed LDHA levels. K.K. and P.J. performed CRISPR correction experiments. H-O.L. and W.D.K performed Seahorse experiments. R.S. and P.M.vH. conceived the experiments. R.S. wrote the manuscript and prepared figures.
Declaration of interests
The authors declare no competing interests in this study.
Published: February 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103823.
Supplemental information
Data and code availability
Data generated in this study will be provided upon request. The Ccnc−/− transcriptome dataset has been deposited previously (GEO accession is GSE126450). Methods to interpret the data are detailed in the STAR method section of this manuscript. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adegbola A., Musante L., Callewaert B., Maciel P., Hu H., Isidor B., Picker-Minh S., Le Caignec C., Delle Chiaie B., Vanakker O., et al. Redefining the MED13L syndrome. Eur. J. Hum. Genet. 2015;23:1308–1317. doi: 10.1038/ejhg.2015.26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Asadollahi R., Zweier M., Gogoll L., Schiffmann R., Sticht H., Steindl K., Rauch A. Genotype-phenotype evaluation of MED13L defects in the light of a novel truncating and a recurrent missense mutation. Eur. J. Med. Genet. 2017;60:451–464. doi: 10.1016/j.ejmg.2017.06.004. [DOI] [PubMed] [Google Scholar]
- Bai R.K., Wong L.J. Simultaneous detection and quantification of mitochondrial DNA deletion(s), depletion, and over-replication in patients with mitochondrial disease. J. Mol. Diagn. 2005;7:613–622. doi: 10.1016/S1525-1578(10)60595-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourbon H.M. Comparative genomics supports a deep evolutionary origin for the large, four-module transcriptional mediator complex. Nucleic Acids Res. 2008;36:3993–4008. doi: 10.1093/nar/gkn349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabrese V., Lodi R., Tonon C., D'Agata V., Sapienza M., Scapagnini G., Mangiameli A., Pennisi G., Stella A.M., Butterfield D.A. Oxidative stress, mitochondrial dysfunction and cellular stress response in Friedreich's ataxia. J. Neurol. Sci. 2005;233:145–162. doi: 10.1016/j.jns.2005.03.012. [DOI] [PubMed] [Google Scholar]
- Calpena E., Hervieu A., Kaserer T., Swagemakers S.M.A., Goos J.A.C., Popoola O., Ortiz-Ruiz M.J., Barbaro-Dieber T., Bownass L., Brilstra E.H., et al. De novo missense substitutions in the gene encoding CDK8, a regulator of the mediator complex, cause a syndromic developmental disorder. Am. J. Hum. Genet. 2019;104:709–720. doi: 10.1016/j.ajhg.2019.02.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caro-Llopis A., Rosello M., Orellana C., Oltra S., Monfort S., Mayo S., Martinez F. De novo mutations in genes of mediator complex causing syndromic intellectual disability: mediatorpathy or transcriptomopathy? Pediatr. Res. 2016;80:809–815. doi: 10.1038/pr.2016.162. [DOI] [PubMed] [Google Scholar]
- Chan D.C. Mitochondrial dynamics and its involvement in disease. Annu. Rev. Pathol. 2020;15:235–259. doi: 10.1146/annurev-pathmechdis-012419-032711. [DOI] [PubMed] [Google Scholar]
- Chen H., Detmer S.A., Ewald A.J., Griffin E.E., Fraser S.E., Chan D.C. Mitofusins Mfn1 and Mfn2 coordinately regulate mitochondrial fusion and are essential for embryonic development. J. Cell Biol. 2003;160:189–200. doi: 10.1083/jcb.200211046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen H., Vermulst M., Wang Y.E., Chomyn A., Prolla T.A., McCaffery J.M., Chan D.C. Mitochondrial fusion is required for mtDNA stability in skeletal muscle and tolerance of mtDNA mutations. Cell. 2010;141:280–289. doi: 10.1016/j.cell.2010.02.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.J. Mechanism of homologous recombination and implications for aging-related deletions in mitochondrial DNA. Microbiol. Mol. Biol. Rev. 2013;77:476–496. doi: 10.1128/MMBR.00007-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen X.J., Butow R.A. The organization and inheritance of the mitochondrial genome. Nat. Rev. Genet. 2005;6:815–825. doi: 10.1038/nrg1708. [DOI] [PubMed] [Google Scholar]
- Cheung E.C., McBride H.M., Slack R.S. Mitochondrial dynamics in the regulation of neuronal cell death. Apoptosis. 2007;12:979–992. doi: 10.1007/s10495-007-0745-5. [DOI] [PubMed] [Google Scholar]
- Choi M.H., Ow J.R., Yang N.D., Taneja R. Oxidative stress-mediated skeletal muscle degeneration: molecules, mechanisms, and therapies. Oxid Med. Cell Longev. 2016;2016:6842568. doi: 10.1155/2016/6842568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chung H.L., Mao X., Wang H., Park Y.J., Marcogliese P.C., Rosenfeld J.A., Burrage L.C., Liu P., Murdock D.R., Yamamoto S., et al. De novo variants in CDK19 are associated with a syndrome involving intellectual disability and epileptic encephalopathy. Am. J. Hum. Genet. 2020;106:717–725. doi: 10.1016/j.ajhg.2020.04.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cooper K.F., Khakhina S., Kim S.K., Strich R. Stress-induced nuclear-to-cytoplasmic translocation of cyclin C promotes mitochondrial fission in yeast. Dev. Cell. 2014;28:161–173. doi: 10.1016/j.devcel.2013.12.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis M.A., Larimore E.A., Fissel B.M., Swanger J., Taatjes D.J., Clurman B.E. The SCF-Fbw7 ubiquitin ligase degrades MED13 and MED13L and regulates CDK8 module association with Mediator. Genes Dev. 2013;27:151–156. doi: 10.1101/gad.207720.112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deciphering Developmental Disorders Study Large-scale discovery of novel genetic causes of developmental disorders. Nature. 2015;519:223–228. doi: 10.1038/nature14135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diaz F., Moraes C.T. Mitochondrial biogenesis and turnover. Cell Calcium. 2008;44:24–35. doi: 10.1016/j.ceca.2007.12.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ganesan V., Willis S.D., Chang K.T., Beluch S., Cooper K.F., Strich R. Cyclin C directly stimulates Drp1 GTP affinity to mediate stress-induced mitochondrial hyperfission. Mol. Biol. Cell. 2019;30:302–311. doi: 10.1091/mbc.E18-07-0463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoeppner S., Baumli S., Cramer P. Structure of the mediator subunit cyclin C and its implications for CDK8 function. J. Mol. Biol. 2005;350:833–842. doi: 10.1016/j.jmb.2005.05.041. [DOI] [PubMed] [Google Scholar]
- Hori A., Yoshida M., Ling F. Mitochondrial fusion increases the mitochondrial DNA copy number in budding yeast. Genes Cells. 2011;16:527–544. doi: 10.1111/j.1365-2443.2011.01504.x. [DOI] [PubMed] [Google Scholar]
- Hu Y., Lu W., Chen G., Wang P., Chen Z., Zhou Y., Ogasawara M., Trachootham D., Feng L., Pelicano H., et al. K-ras(G12V) transformation leads to mitochondrial dysfunction and a metabolic switch from oxidative phosphorylation to glycolysis. Cell Res. 2012;22:399–412. doi: 10.1038/cr.2011.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hung C.H., Cheng S.S., Cheung Y.T., Wuwongse S., Zhang N.Q., Ho Y.S., Lee S.M., Chang R.C. A reciprocal relationship between reactive oxygen species and mitochondrial dynamics in neurodegeneration. Redox Biol. 2018;14:7–19. doi: 10.1016/j.redox.2017.08.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jazwinski S.M. The retrograde response: when mitochondrial quality control is not enough. Biochim. Biophys. Acta. 2013;1833:400–409. doi: 10.1016/j.bbamcr.2012.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jezek J., Chang K.T., Joshi A.M., Strich R. Mitochondrial translocation of cyclin C stimulates intrinsic apoptosis through Bax recruitment. EMBO Rep. 2019;20:e47425. doi: 10.15252/embr.201847425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jezek J., Smethurst D.G.J., Stieg D.C., Kiss Z.A.C., Hanley S.E., Ganesan V., Chang K.T., Cooper K.F., Strich R. Cyclin C: the story of a non-cycling cyclin. Biology (Basel) 2019;8 doi: 10.3390/biology8010003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khakhina S., Cooper K.F., Strich R. Med13p prevents mitochondrial fission and programmed cell death in yeast through nuclear retention of cyclin C. Mol. Biol. Cell. 2014;25:2807–2816. doi: 10.1091/mbc.E14-05-0953. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kirkland R.A., Saavedra G.M., Franklin J.L. Rapid activation of antioxidant defenses by nerve growth factor suppresses reactive oxygen species during neuronal apoptosis: evidence for a role in cytochrome c redistribution. J. Neurosci. 2007;27:11315–11326. doi: 10.1523/JNEUROSCI.3590-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee H.C., Chang C.M., Chi C.W. Somatic mutations of mitochondrial DNA in aging and cancer progression. Ageing Res. Rev. 2010;9:S47–S58. doi: 10.1016/j.arr.2010.08.009. [DOI] [PubMed] [Google Scholar]
- Li J., O W., Li W., Jiang Z.G., Ghanbari H.A. Oxidative stress and neurodegenerative disorders. Int. J. Mol. Sci. 2013;14:24438–24475. doi: 10.3390/ijms141224438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li N., Fassl A., Chick J., Inuzuka H., Li X., Mansour M.R., Liu L., Wang H., King B., Shaik S., et al. Cyclin C is a haploinsufficient tumour suppressor. Nat. Cell Biol. 2014;16:1080–1091. doi: 10.1038/ncb3046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li R., Beebe T., Cui J., Rouhanizadeh M., Ai L., Wang P., Gundersen M., Takabe W., Hsiai T.K. Pulsatile shear stress increased mitochondrial membrane potential: implication of Mn-SOD. Biochem. Biophys. Res. Commun. 2009;388:406–412. doi: 10.1016/j.bbrc.2009.08.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li Y.C., Chao T.C., Kim H.J., Cholko T., Chen S.F., Li G., Snyder L., Nakanishi K., Chang C.E., Murakami K., et al. Structure and noncanonical Cdk8 activation mechanism within an Argonaute-containing Mediator kinase module. Sci. Adv. 2021;7:eabd4484. doi: 10.1126/sciadv.abd4484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Minerath R.A., Dewey C.M., Hall D.D., Grueter C.E. Regulation of cardiac transcription by thyroid hormone and Med13. J. Mol. Cell Cardiol. 2019;129:27–38. doi: 10.1016/j.yjmcc.2019.01.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicholls D.G. Simultaneous monitoring of ionophore- and inhibitor-mediated plasma and mitochondrial membrane potential changes in cultured neurons. J. Biol. Chem. 2006;281:14864–14874. doi: 10.1074/jbc.M510916200. [DOI] [PubMed] [Google Scholar]
- Nizon M., Laugel V., Flanigan K.M., Pastore M., Waldrop M.A., Rosenfeld J.A., Marom R., Xiao R., Gerard A., Pichon O., et al. Variants in MED12L, encoding a subunit of the mediator kinase module, are responsible for intellectual disability associated with transcriptional defect. Genet. Med. 2019;21:2713–2722. doi: 10.1038/s41436-019-0557-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nunnari J., Suomalainen A. Mitochondria: in sickness and in health. Cell. 2012;148:1145–1159. doi: 10.1016/j.cell.2012.02.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phillips N.R., Sprouse M.L., Roby R.K. Simultaneous quantification of mitochondrial DNA copy number and deletion ratio: a multiplex real-time PCR assay. Sci. Rep. 2014;4:3887. doi: 10.1038/srep03887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ponce J.M., Coen G., Spitler K.M., Dragisic N., Martins I., Hinton A., Jr., Mungai M., Tadinada S.M., Zhang H., Oudit G.Y., et al. Stress-induced cyclin C translocation regulates cardiac mitochondrial dynamics. J. Am. Heart Assoc. 2020;9:e014366. doi: 10.1161/JAHA.119.014366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poot M. Mutations in mediator complex genes CDK8, MED12, MED13, and MEDL13 mediate overlapping developmental syndromes. Mol. Syndromol. 2020;10:239–242. doi: 10.1159/000502346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reddy T.P., Manczak M., Calkins M.J., Mao P., Reddy A.P., Shirendeb U., Park B., Reddy P.H. Toxicity of neurons treated with herbicides and neuroprotection by mitochondria-targeted antioxidant SS31. Int. J. Environ. Res. Public Health. 2011;8:203–221. doi: 10.3390/ijerph8010203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Santos D., Cardoso S.M. Mitochondrial dynamics and neuronal fate in Parkinson's disease. Mitochondrion. 2012;12:428–437. doi: 10.1016/j.mito.2012.05.002. [DOI] [PubMed] [Google Scholar]
- Satoh M., Kuroiwa T. Organization of multiple nucleoids and DNA molecules in mitochondria of a human cell. Exp. Cell Res. 1991;196:137–140. doi: 10.1016/0014-4827(91)90467-9. [DOI] [PubMed] [Google Scholar]
- Scheibye-Knudsen M., Fang E.F., Croteau D.L., Wilson D.M., 3rd, Bohr V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2015;25:158–170. doi: 10.1016/j.tcb.2014.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silva Ramos E., Motori E., Bruser C., Kuhl I., Yeroslaviz A., Ruzzenente B., Kauppila J.H.K., Busch J.D., Hultenby K., Habermann B.H., et al. Mitochondrial fusion is required for regulation of mitochondrial DNA replication. PLoS Genet. 2019;15:e1008085. doi: 10.1371/journal.pgen.1008085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Snijders Blok L., Hiatt S.M., Bowling K.M., Prokop J.W., Engel K.L., Cochran J.N., Bebin E.M., Bijlsma E.K., Ruivenkamp C.A.L., Terhal P., et al. De novo mutations in MED13, a component of the Mediator complex, are associated with a novel neurodevelopmental disorder. Hum. Genet. 2018;137:375–388. doi: 10.1007/s00439-018-1887-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S. The mitochondrial basis of aging and age-related disorders. Genes (Basel) 2017;8:398. doi: 10.3390/genes8120398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Srivastava S., Niranjan T., May M.M., Tarpey P., Allen W., Hackett A., Jouk P.S., Raymond L., Briault S., Skinner C., et al. Dysregulations of sonic hedgehog signaling in MED12-related X-linked intellectual disability disorders. Mol. Genet. Genomic Med. 2019;7:e00569. doi: 10.1002/mgg3.569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieg D.C., Chang K.T., Cooper K.F., Strich R. Cyclin C regulated oxidative stress responsive transcriptome in Mus musculus embryonic fibroblasts. G3 (Bethesda) 2019;9:1901–1908. doi: 10.1534/g3.119.400077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stieg D.C., Willis S.D., Ganesan V., Ong K.L., Scuorzo J., Song M., Grose J., Strich R., Cooper K.F. A complex molecular switch directs stress-induced cyclin C nuclear release through SCF(Grr1)-mediated degradation of Med13. Mol. Biol. Cell. 2018;29:363–375. doi: 10.1091/mbc.E17-08-0493. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strom E., Sathe S., Komarov P.G., Chernova O.B., Pavlovska I., Shyshynova I., Bosykh D.A., Burdelya L.G., Macklis R.M., Skaliter R., et al. Small-molecule inhibitor of p53 binding to mitochondria protects mice from gamma radiation. Nat. Chem. Biol. 2006;2:474–479. doi: 10.1038/nchembio809. [DOI] [PubMed] [Google Scholar]
- Torring P.M., Larsen M.J., Brasch-Andersen C., Krogh L.N., Kibaek M., Laulund L., Illum N., Dunkhase-Heinl U., Wiesener A., Popp B., et al. Is MED13L-related intellectual disability a recognizable syndrome? Eur. J. Med. Genet. 2019;62:129–136. doi: 10.1016/j.ejmg.2018.06.014. [DOI] [PubMed] [Google Scholar]
- Uehara T., Abe K., Oginuma M., Ishitani S., Yoshihashi H., Okamoto N., Takenouchi T., Kosaki K., Ishitani T. Pathogenesis of CDK8-associated disorder: two patients with novel CDK8 variants and in vitro and in vivo functional analyses of the variants. Sci. Rep. 2020;10:17575. doi: 10.1038/s41598-020-74642-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Haelst M.M., Monroe G.R., Duran K., van Binsbergen E., Breur J.M., Giltay J.C., van Haaften G. Further confirmation of the MED13L haploinsufficiency syndrome. Eur. J. Hum. Genet. 2015;23:135–138. doi: 10.1038/ejhg.2014.69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wai T., Ao A., Zhang X., Cyr D., Dufort D., Shoubridge E.A. The role of mitochondrial DNA copy number in mammalian fertility. Biol. Reprod. 2010;83:52–62. doi: 10.1095/biolreprod.109.080887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang K., Yan R., Cooper K.F., Strich R. Cyclin C mediates stress-induced mitochondrial fission and apoptosis. Mol. Biol. Cell. 2015;26:1030–1043. doi: 10.1091/mbc.E14-08-1315. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youngman M.J., Hobbs A.E., Burgess S.M., Srinivasan M., Jensen R.E. Mmm2p, a mitochondrial outer membrane protein required for yeast mitochondrial shape and maintenance of mtDNA nucleoids. J. Cell Biol. 2004;164:677–688. doi: 10.1083/jcb.200308012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuan B., Pehlivan D., Karaca E., Patel N., Charng W.L., Gambin T., Gonzaga-Jauregui C., Sutton V.R., Yesil G., Bozdogan S.T., et al. Global transcriptional disturbances underlie Cornelia de Lange syndrome and related phenotypes. J. Clin. Invest. 2015;125:636–651. doi: 10.1172/JCI77435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zane A.C., Reiter D.A., Shardell M., Cameron D., Simonsick E.M., Fishbein K.W., Studenski S.A., Spencer R.G., Ferrucci L. Muscle strength mediates the relationship between mitochondrial energetics and walking performance. Aging Cell. 2017;16:461–468. doi: 10.1111/acel.12568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zarate Y.A., Uehara T., Abe K., Oginuma M., Harako S., Ishitani S., Lehesjoki A.E., Bierhals T., Kloth K., Ehmke N., et al. CDK19-related disorder results from both loss-of-function and gain-of-function de novo missense variants. Genet. Med. 2021;23:1050–1057. doi: 10.1038/s41436-020-01091-9. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data generated in this study will be provided upon request. The Ccnc−/− transcriptome dataset has been deposited previously (GEO accession is GSE126450). Methods to interpret the data are detailed in the STAR method section of this manuscript. This paper does not report original code. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.