

SUMMARY
A heterozygous missense mutation of the islet β cell-enriched MAFA transcription factor (p.Ser64Phe [S64F]) is found in patients with adult-onset β cell dysfunction (diabetes or insulinomatosis), with men more prone to diabetes than women. This mutation engenders increased stability to the unstable MAFA protein. Here, we develop a S64F MafA mouse model to determine how β cell function is affected and find sex-dependent phenotypes. Heterozygous mutant males (MafAS64F/+) display impaired glucose tolerance, while females are slightly hypoglycemic with improved blood glucose clearance. Only MafAS64F/+ males show transiently higher MafA protein levels preceding glucose intolerance and sex-dependent changes to genes involved in Ca2+ signaling, DNA damage, aging, and senescence. MAFAS64F production in male human β cells also accelerate cellular senescence and increase senescence-associated secretory proteins compared to cells expressing MAFAWT. These results implicate a conserved mechanism of accelerated islet aging and senescence in promoting diabetes in MAFAS64F carriers in a sex-biased manner.
Graphical abstract
In brief
Walker et al. show that mice harboring the missense mutation of the pancreatic islet-enriched transcription factor MAFA (S64F MAFA) demonstrate sex-dependent β cell dysfunction, similar to findings in human carriers. Only male S64F MAFA β cells show accelerated senescence and β cell dysfunction toward hyperglycemia.
INTRODUCTION
The large V-Maf avian musculoaponeurotic fibrosarcoma transcription factor MafA is a pancreatic β cell-enriched protein that is essential for activating rodent transcriptional programs to promote β cell maturation and function, as can be measured by glucose-stimulated insulin secretion (GSIS) (Artner et al., 2008, 2010; Hang and Stein, 2011; Hang et al., 2014; Zhang et al., 2005). The presence of MafA in only insulin+ β cells during islet cell development and postnatally distinguishes it from other important islet cell-enriched regulators (Golson and Kaestner, 2017; Hang and Stein, 2011; Pan and Wright, 2011; Shih et al., 2013). For example, transcription factors such as Pdx1 and Nkx6.1, which are expressed earlier and more broadly, also have a profound impact on pancreas organogenesis, islet β cell function, and whole-body glucose homeostasis in mice (Madsen et al., 1997; Pan and Wright, 2011). Pdx1 is present in both the exocrine and endocrine pancreas during development and then principally in islet β cells postnatally (Pan and Wright, 2011), while Nkx6.1 is found in endocrine progenitors and is later restricted to β cells (Golson and Kaestner, 2017; Hang and Stein, 2011; Pan and Wright, 2011; Shih et al., 2013).
In contrast, mice lacking MafA (i.e., MafA−/− (Zhang et al., 2005), MafAΔpanc (Artner et al., 2010; Hang et al., 2014), and MafAΔβ (Cyphert et al., 2019; Luan et al., 2019) have normal pancreas formation and a relatively subtle influence on postnatal physiology, primarily compromising GSIS in a sex-independent manner. However, MafA has been proposed to play a distinct late role in postnatal islet β cell maturation, in part supported by observations demonstrating that inducing MafA expression levels in normally non-glucose-responsive and MafALow neonatal rat islets increases GSIS (Aguayo-Mazzucato et al., 2011), while compromising levels reduce mouse (Artner et al., 2010; Cyphert et al., 2019; Hang et al., 2014; Luan et al., 2019; Zhang et al., 2005) and human (Guo et al., 2013) β cell activity. A key regulatory role is also implied by the ability of many islet-enriched transcription factors to coordinately stimulate MafA gene transcription, including Pdx1 and Nkx6.1 (Raum et al., 2006, 2010).
Expression of MafB, the other large Maf family member produced in the islet, differs from MafA in being produced in both murine α and β cells developmentally (Cyphert et al., 2019; Hang and Stein, 2011), then postnatally only in α cells and a subpopulation of maternal β cells during pregnancy (Banerjee et al., 2016; Cyphert et al., 2019). Moreover, genetic studies have demonstrated that MafB is dispensable in regulating adult murine islet β cell function, except during pregnancy (Banerjee et al., 2016; Cyphert et al., 2019). MafA expression during mouse β cell development compensates for the absence of MafB, although glucagon secretion from islet α cells is compromised (Conrad et al., 2016). In addition, misexpressing MafB in MafAΔpanc β cells cannot rescue MafA function (Cyphert et al., 2019). These results illustrate similarities and differences between MafA and MafB in expression and function in rodent islet cells.
While most islet-enriched transcription factors are produced in an analogous fashion in rodents and humans, there are unique differences in the temporal and islet cell type-specific expression pattern of the human MAFA and MAFB proteins. In humans, MAFA protein is not detected in the β cell until ~10 years of age, while MAFB is present throughout the lifespan of human insulin+ β cells (Cyphert et al., 2019; Dai et al., 2012). These observations imply that humans have at least two postnatal β cell populations (in terms of MAFA) capable of maintaining euglycemia, represented by the juvenile (i.e., <10 years old and MAFALow:MAFB+) and post-juvenile (≥10 years old and MAFAHigh:MAFB+) periods. In fact, independent studies have established distinct molecular and functional properties of these temporally produced human cell populations (Arda et al., 2016; Arrojo E Drigo et al., 2019; Camunas-Soler et al., 2020) and an association between MAFA levels and adult islet β cell functional heterogeneity (Chen et al., 2019). MAFA and MAFB levels are reduced in type 2 diabetic (T2D) islets (Guo et al., 2013), and (at least) MAFA is particularly sensitive to oxidative stress and glucotoxic conditions (Harmon et al., 2005). While the role of MAFA or MAFB has not been analyzed directly in intact human islets, both are required for GSIS in the human EndoC-βH1 β cell line (Scharfmann et al., 2014; Scoville et al., 2015). Moreover, we have recently shown that MAFB is essential to insulin production in human embryonic stem cell-derived β cells (Russell et al., 2020), which is in stark contrast to its dispensable role in rodents (Banerjee et al., 2016; Cyphert et al., 2019). These findings highlight species-specific differences in MAFA and MAFB production and islet cell distribution, likely implying that the homodimeric MAFB activator and the MAFA:MAFB heterodimeric activator provide unique functional characteristics to human islet β cells.
Individuals carrying a heterozygous mutation in the MAFA transactivation domain producing a substitution of the conserved serine at position 64 with a phenylalanine (p.Ser64Phe [S64F]) develop either diabetes mellitus or insulinomatosis, a non-syndromic condition of hyperinsulinemic hypoglycemia caused by multiple insulin-secreting neuroendocrine tumors. The mean age at diagnosis for both conditions is 38 years (Iacovazzo et al., 2018). These results demonstrate that MAFA is a causative gene for maturity-onset diabetes of the young (MODY), a collection of monogenic diseases predominantly driven by mutations in essential islet-enriched transcription factors (Barbetti and D’Annunzio, 2018). Interestingly, diabetes is prevalent in male MAFAS64F carriers (i.e., 3:1), while insulinomatosis is more common in female carriers (4:1) (Iacovazzo et al., 2018). In vitro analysis demonstrated that the S64F substitution prevents a key priming phosphorylation event at position S65 in MAFA, which profoundly increases the stability of the normally unstable MAFA protein by affecting processes necessary for ubiquitin-mediated degradation (Iacovazzo et al., 2018).
Due to the rare detection of MAFAS64F-mediated diseases and difficulty of performing mechanistic studies in a human context, we used CRISPR-Cas9-based mutagenesis to establish a mouse model (termed MafAS64F/+) harboring the same pathogenic single base pair substitution (C>T) as human carriers. Diabetes-related phenotypes were specifically associated with male MafAS64F/+ heterozygous mice by 5 weeks of age, which manifested glucose intolerance due in part to a reduced glucose-stimulated β cell Ca2+ response. These changes were preceded by an overt but transient increase in MafA protein levels in islet β cells at 4 weeks. In addition, the functional deficiencies in male MafAS64F/+ islet β cells accompanied the induction of markers of DNA damage, cell-cycle exit, senescence, and the senescence-associated secretory phenotype (SASP) at 5 weeks, consistent with accelerated cellular aging and senescence. In contrast, heterozygous mutant female mice not only had lower fasting blood glucose levels and improved glucose clearance but also did not display any of the overt aging and senescence signatures of littermate, heterozygous males. Expression of MAFAS64F in the male human β cell line, EndoC-βH2, showed increased senescent gene markers and secretion of human-specific SASP factors capable of inducing senescence in a cell non-autonomous manner. These results indicate that diabetes in human male MAFAS64F carriers is caused by the premature senescence and aging of islet β cells.
RESULTS
Male MafAS64F/+ mice are glucose intolerant due to impaired insulin secretion
Since MafA is a primary regulator of glucose clearance and insulin secretion in islet β cells (Hang and Stein, 2011), MafAS64F/+ and wild-type (WT) littermates were subjected to glucose tolerance testing (GTT) and fasting blood glucose measurements at various postnatal time points. While both male and female MafAS64F/+ animals had improved glucose clearance at 4 weeks, females also had modest fasting hypoglycemia (Figure 1A). Strikingly, male MafAS64F/+ animals developed persistent glucose intolerance beginning at 5 weeks of age, while females continued to have improved glucose tolerance and lower fasting blood glucose levels (Figure 1A). GTT results were stably maintained in male and female MafAS64F/+ mice (Figure S1). The sex-dependent male phenotype was even more penetrant in homozygous MafAS64F/S64F mutants, as males displayed overt diabetes with elevated fasting glucose levels that progressively worsened (Figures S2A and S2B). In contrast, the phenotype of homozygous females was much more variable and largely comparable to WT littermates (Figure S2C). Because of the generally poor survival of homozygous mutants postnatally (Figure S2D), possibly resulting from severe neurodevelopmental defects due to MafAS64F/S64F expression and its altered activity in the central nervous system (Lecoin et al., 2010; Niceta et al., 2015), all of the remaining experimentation was performed with male and female heterozygous MafAS64F/+ mice.
Figure 1. Male but not female MafAS64F/+ mice become glucose intolerant between 4 and 5 weeks of age.

(A) Fasted male and female animals underwent intraperitoneal glucose tolerance tests at 4 and 5 weeks of age. Male heterozygous (termed Het) MafAS64F/+ mice (red line) had improved glucose clearance at 4 weeks but become glucose intolerant by 5 weeks. Female MafAS64F/+ mice (teal line) had significantly lower fasting blood glucose levels and improved glucose clearance at both time points.
(B) High glucose-stimulated insulin secretion in isolated islets was impaired in male MafAS64F/+ samples at 5 weeks. Islets were incubated with 4.6 mM (low, LG) or 16.8 mM (high, HG) glucose for 1 h. Secreted insulin was normalized to DNA content.
(C) Islet insulin content trended lower in MafAS64F/+ males and was significantly decreased in females. Levels were normalized to DNA content.
(D) Male (left panel) and female (right panel) islet β cell area was reduced at 7 weeks in MafAS64F/+ mice. The area was calculated by dividing the total insulin+ area by the total pancreatic area (eosin staining) in pancreas sections obtained every 50 μm multiplied by 100 to obtain percentage (%).
(E) Indicative of reduced islet area, the combined islet β and α cell area was significantly reduced in male and female MafAS64F/+ mice at 7 weeks.
(A–C) Two-tailed Student t test; *p < 0.05; **p < 0.01;
***p < 0.001.
(D and E) Two-way ANOVA; *p < 0.05; **p < 0.01; ***p < 0.005.
Insulin secretion by ex vivo GSIS was impaired in 5-week-old male MafAS64F/+ islets (Figure 1B), although insulin content trended lower in male MafAS64F/+ islets and was significantly reduced in female islets (Figure 1C). MafAS64F/+ female mice appear to have lower fasting blood glucose levels (Figure 1A) due to increased insulin secretion in both low (5.6) and high (16.7) glucose conditions as determined by dynamic perifusion assays (Figure S3A) and increased fasting serum insulin levels (Figure S3B). Thus, 8- to 10-week old female MafAS64F/+ islets had markedly improved 1st and 2nd phase GSIS properties compared to controls in the perifusion assays, while males MafAS64F/+ islets showed a mild improvement in this ex vivo system, but not nearly to the levels found in females (Figure S3C). Because of the counterregulatory effects of insulin on glucagon hormone secretion (Franklin et al., 2005), we considered that glucose-regulated glucagon levels may be altered in MafAS64F/+ mice. However, there was neither an obvious sex-dependent impact on glucose-stimulated glucagon secretion nor a change in glucagon content (Figures S3D and S3E). The islet α cell area was also unchanged in both male and female MafAS64F/+ islets, although this was variable among animals (Figure S3F).
The insulin+ β cell area was reduced in male and female MafAS64F/+ mice (Figure 1D), as were their combined β and α cell areas and β cell number per islet section (Figures 1E and S3H). The decrease in β cell area may be due to lower islet cell proliferation rates in MafAS64F/+ mice relative to WT littermates (Figure S3G). In addition, the changes in blood glucose levels were not attributable to differences in peripheral tissue insulin sensitivity or glucose uptake as insulin tolerance tests and glycogen storage were both unchanged (Figures S4A and S4B). Animal body weight was also not appreciably altered in MafAS64F/+ mice, except for a small decrease in female MafAS64F/+ mice at 5 weeks of age that disappeared by 6 weeks (Figure S4C). These combined results suggested a fundamental difference(s) in MafAS64F/+ β cell activity between the sexes, resulting in their distinguishing ability to control glucose homeostasis.
Because the sex hormones estradiol and testosterone affect islet cell function (Gannon et al., 2018), we questioned whether these hormones influenced the sex-biased phenotypes in MafAS64F/+ mice. Both sexes achieved puberty appropriately with fertility comparable to WT littermates (data not shown). Female MafAS64F/+ mice were ovariectomized to investigate whether estrogen was regulating mutant sex-biased activity. As expected, ovariectomy at 3 weeks of age produced glucose intolerance in WT female mice by 4 weeks of age (Figure S5A). In contrast, glucose tolerance was unaffected by ovariectomy in age-matched female MafAS64F/+ mice (data not shown; Figure S5A). These results indicate that the effects of the MafAS64F variant is dominant over the effects of estrogen deficiency on glucose intolerance. Furthermore, testosterone levels were not altered markedly in MafAS64F/+ males at both 4 and 5 weeks of age and were similar to age-matched females in these pre- and peripubertal mice (Figure S5B). The low circulating levels of testosterone produced during this phenotypic window lowers the likelihood of this hormone influencing MafAS64F actions.
Collectively, these results strongly suggest that MafAS64F regulates male and female islet β cells through common and distinct mechanisms; however, these processes are not principally influenced by the primary sex hormones.
MafA protein levels are transiently and profoundly upregulated before the changes in glucose clearance in male MafAS64F/+ islet β cells
The MAFAS64F variant impairs a key, priming phosphorylation event at position S65, which blocks subsequent glycogen synthase kinase 3 (GSK3)-mediated phosphorylation at positions S61, T51, T57, and S49 in the transactivation domain. These changes impede ubiquitin-mediated protein degradation of MAFAS64F, and its stability is dramatically increased when expressed in the human EndoC-βH1 β cell line (Han et al., 2007; Iacovazzo et al., 2018; Rocques et al., 2007). Consequently, we predicted that this mutation in the highly conserved region of the protein (Han et al., 2007; Iacovazzo et al., 2018; Rocques et al., 2007) would also increase MafA levels in MafAS64F/+ β cells. Surprisingly, we only found elevated MafA protein immunostaining intensity in male MafAS64F/+ islets at 4 weeks of age (Figure 2A), 1 week before the onset of glucose intolerance (Figure 1A). In contrast, MafA protein levels were not increased in female MafAS64F/+ β cells at 4 weeks or any other analyzed time point (Figure 2B). However, there was a roughly 3- to 5-fold decrease in MafA transcript levels at 4 and 5 weeks of age in both male and female MafAS64F/+ islets compared to WT littermates (Figure 2C). The reduction in MafA gene expression indicates that the more stable MafAS64F protein is acting in an autoregulatory manner, likely by binding at the conserved MafA binding site within the 5′-flanking sequences of the Region 3 transcription control domain (Raum et al., 2010).
Figure 2. MAFA protein levels were transiently upregulated at 4 weeks in male MafAS64F/+ islets.

(A) Immunostaining over the course of 3, 4, 5, 6, and 7 weeks revealed that MAFA protein staining intensity only increased at 4 weeks of age in male MafAS64F/+ (Het) islets.
(B) In contrast, MAFA staining intensity was unchanged between female MafAS64F/+ and WT islets.
(C) MafA mRNA levels were significantly reduced at 4 and 5 weeks in both male and female MafAS64F/+ islet samples. Fold change shown relative to male WT islets. Two-tailed Student t test; *p < 0.05; scale bars, 50 μm.
Glucose- and potassium chloride (KCl)-stimulated calcium handling were altered in both male and female MafAS64F/+ islets
To provide an unbiased and comprehensive perspective over how MafAS64F influences islet β cell gene expression, bulk RNA sequencing (RNA-seq) was performed on isolated islets from 5-week-old WT and MafAS64F/+ male and female mice when a clear delineation of phenotypes was observed. Female heterozygous islets had 736 differentially expressed genes (DEGs) (337 up and 399 down) compared to female WT islets, while males had 2,410 DEGs (2,031 up and 379 down) (Figure 3A). There were also 391 DEGs that were similarly regulated between the sexes, which were revealed by Gene Ontology analysis as having p < 0.05 and included factors associated with Ca2+ and K+ channels important in controlling β cell function (Figures 3B and 3C). In addition, other voltage-dependent Ca2+ channel genes as well as genes important to ion influx were also specifically upregulated in male MafAS64F/+ islets (Figure 3D), suggesting both sex-dependent and sex-independent changes in ion flux in MafAS64F/+ islets.
Figure 3. Male and female MafAS64F/+ islet cells regulate a common and distinct set of genes associated with Ca2+ and K+ channel activity.

(A) The Venn diagram illustrates the total number of RNA-seq-identified genes differentially up- or down-regulated between 5-week-old WT and MafAS64F/+ islets.
(B) Gene Ontology (GO): molecular function analysis (p < 0.05) of the 391 genes commonly up- or down-regulated in MafAS64F/+ (Het) islets revealed alterations in multiple ion channel activity pathways.
(C) Heatmaps showing channel gene expression changes common between male and female MafAS64F/+ islets.
(D) Heatmap of Ca2+ signaling pathway genes uniquely increased in male MafAS64F/+ islets identified by Kyoto Encyclopedia of Genes and Genomes (KEGG) analysis (also see Figure 5A). False discovery rate (FDR) < 0.05.
Since glucose-induced elevations of cytoplasmic Ca2+ mediate insulin release (Bergsten et al., 1994), WT and MafAS64F/+ islet Ca2+ handling was monitored in response to glucose stimulation and KCl-induced depolarization. As expected from the GSIS response of male MafAS64F/+ islets (Figures 1B and S3C), their Ca2+ entry was significantly blunted relative to the WT islets in response to 11 mM glucose stimulation (Figure 4A; represented by red trace in left panel and called a “non-responder”). The glucose-stimulated Ca2+ oscillatory pattern that is responsible for pulsatile insulin secretion was also significantly diminished in these MafAS64F/+ islets (Figure 4A). In contrast, there appeared to be two distinct Ca2+ responsive islet populations in female MafAS64F/+ animals. One was similar to male MafAS64F/+ islets in having a very limited initial glucose response and minimal oscillatory behavior (i.e., Figure 4A, dark green representative trace in right panel and labeled a non-responder). However, the remaining islets had more subtle decreases in their initial response to 11 mM glucose, and subsequently oscillate with higher frequency but equivalent amplitude to WT islets (Figure 4A, “responders,” teal representative trace in right panel). The average peak amplitude of the initial glucose-induced Ca2+ response was reduced in all male and female MafAS64F/+ islets (Figure 4B). This suggests that the MafAS64F/+ female islet responders population is able to compensate for the non-responders and maintain glucose tolerance in MafAS64F/+ female mice, which would be predicted because only a fraction (roughly 20%) of the β cell mass is thought to be required to maintain glucose tolerance (Gale, 2002).
Figure 4. Glucose-induced Ca2+ oscillations and KCl-induced Ca2+ responses are altered in MafAS64F/+ islets.

(A) Representative Fura2 traces show loss of the β cell glucose-induced Ca2+ oscillations (11 mM glucose [G]) in 5-week-old male MafAS64F/+ (Het) islets (red line, “non-responders”). Female MafAS64F/+ islets had 2 different functionally responsive islet populations (“responders,” teal line; non-responders, green line).
(B) Quantitation of male (left; 2-tailed Student t test; ***p < 0.001) and female (right; 1-way ANOVA; ****p < 0.0001) islets showed reduced cytoplasmic Ca2+ following stimulation with 11 mM glucose. The average peak amplitude was quantitated by dividing the first peak ΔCa2+ after 11 mM glucose application by the baseline response at 5 mM glucose.
(C) The Ca2+ response to 30 mM KCl is reduced in male MafAS64F/+ islets but increased in female MafAS64F/+ islets. Representative traces for this experiment are shown in Figure S8. Two-tailed Student t test; **p < 0.01; ***p < 0.001.
Male and female MafAS64F/+ islets also had disparate Ca2+ responses to KCl-mediated depolarization. The KCl-induced Ca2+ influx (ΔCa2+ after KCl application divided by baseline Ca2+) was significantly reduced in male MafAS64F/+ islets when compared to WT male islets (Figures 4C and S5C). Interestingly, however, the female MafAS64F/+ islets showed greater KCl-induced Ca2+ influx when compared to WT female islets, which was observed for both glucose-stimulated Ca2+ responders and non-responders (Figures 4C, S3A, and S5C). Importantly, female MafAS64F/+ islets also showed greater baseline (5 mM glucose) Ca2+ levels than WT female islets (Figure S5D), which is consistent with the fasting hypoglycemia and increased fasting insulin levels observed in female MafAS64F/+ mice (Figures 1A and S3B).
Only male MafAS64F/+ islets express markers of accelerated cellular senescence and aging
Because 5-week-old male MafAS64F/+ islet β cells were not only defined by a predominant, Ca2+ non-responsive population (Figures 4A and 4B) but also by glucose intolerance (Figure 1A), we focused our attention on identifying molecular determinants to their poor function. In addition to the previously illustrated gene expression alterations involved in Ca2+ signaling (index ranking 6, Figure 5A), 5-week-old male MafAS64F/+ RNA-seq data revealed the upregulation of many metabolic pathway genes implicated in cellular aging (e.g., index ranking pathways 4, 6, and 8) and senescence (e.g., index ranking pathways 1, 2, 3, 5, 6, 7, and 8 in Figure 5A) (Basisty et al., 2018; Campisi and d’Adda di Fagagna, 2007; Coppé et al., 2010; Franceschi et al., 2018; Greenhill, 2019; Kabir et al., 2016; Mancini et al., 2012; Martin and Bernard, 2018). Furthermore, the preponderance of upregulated genes detected by RNA-seq implied that MafAS64F is acting as a dominant activator in (at least) male MafAS64F/+ islets (Figure 3A).
Figure 5. Male MafAS64F/+ islets display increased expression of aging, DDR, SASP, and cytokine pathway gene signatures.

(A) KEGG analysis of the 1,842 genes specifically upregulated in 5-week-old male MafAS64F/+ (Het) islets.
(B–E) Heatmaps reveal male MafAS64F/+ islets have increased expression of pathway genes associated with (B) aging, (C) DDR, (D) SASP, and (E) cytokine-cytokine receptor interactions. FDR < 0.05.
(F) qRT-PCR confirmation of pathway gene changes in 5-week-old male MafAS64F/+ islets (solid bars). Gene expression was unchanged at 4-week-old male MafAS64F/+ islets, with the exception of Cdkn1a (p21) upregulation (hash-marked bars). Two-tailed Student t test; *p < 0.05; **p < 0.01.
Cellular senescence is a durable, cell-cycle arrest response that regulates cell fate specification, tissue patterning, and function during development, cell maturation, and organismal aging (Campisi, 2014; Gorgoulis et al., 2019; Helman et al., 2016; Herranz and Gil, 2018). It is also induced in a variety of disease states and primarily serves as a stress response to many internal and external insults (Gorgoulis et al., 2019; Herranz and Gil, 2018). The importance of premature aging and senescence in driving male MafAS64F/+ β cell dysfunction was supported by the many genes associated with these responses, including those controlling Ca2+ signaling (Figure 3D), aging (Figure 5B), the DNA damage response (DDR; Figure 5C), SASP (Figure 5D), and cytokine-cytokine receptor interactions (Figure 5E). As expected, the expression of candidate genes linked to aging and senescence were increased in 5-week-old male MafAS64F/+ islets (Figure 5F), although not before manifesting glucose intolerance in 4-week-old males (Figures 1A and 5F). Importantly, these genes were largely unaltered in MafAS64F/+ female islets (Figure S6A), although many other female candidate DEGs were validated by qPCR (Figures 3D, 5B-5F, S6B, and S6C).
Senescent cells undergo a progression of changes after an initial insult: early initiation of cellular arrest by activation of cell-cycle inhibitors (e.g., p53, p21, and/or p16) and DDR responses in an attempt to regain homeostasis (Gorgoulis et al., 2019; Herranz and Gil, 2018). DDR can be characterized by markers such as phosphorylated histone H2AX (gH2AX) and 53BP1 recruitment to chromatin. We identified increased immunostaining for the gH2AX marker for DNA double-strand breaks, the P53 binding protein-1 (53BP1) DNA damage marker (Schultz et al., 2000), and the p21 cell-cycle arrest and senescence marker (Fang et al., 1999) in 5-week-old MafAS64F/+ male, but not female islets (Figures 6A-6C and S7A-S7C). While only 3% of the male WT islets showed >10% p21+ β cells, this proportion increased to 37% (including 15% with >20% p21+ β cells) in male MafAS64F/+ islets (Figure 6C, right panel).
Figure 6. DNA damage and senescence markers are increased in dysfunctional male MafAS64F/+ islets.

(A–C) gH2AX staining (A) and 53BP1 (B), markers of DNA double-strand breaks, and p21, a cell-cycle inhibitor (C), were present in male MafAS64F/+ (Het) islets. Male MafAS64F/+ islets had a significant increase in the proportion of islets with >10% p21+ cells.
(D) Male MafAS64F/+ islets showed reduced LaminB1 protein (left) and mRNA (right).
(E and F) SA-β-gal was not produced in 4-week-old male MafAS64F/+ islets, but was detected in MafAS64F/+ males by 7 weeks. (E) The SA-β-gal in 7-week-old MafAS64F/+ islets was of similar intensity to 10- to 12-month-old WT male mouse islets. (F) The proportion of SA-β-gal+ islets was quantitated for each sample (n = 3–4). Two-tailed Student t test; **p < 0.01; scale bar, 50 μm.
Apoptosis was not induced in response to DNA damage in either male or female MafAS64F/+ islets, as determined by the inability to detect islet TUNEL+ cells between 4 and 7 weeks of age (data not shown; Figure S7F). Induction of anti-apoptosis genes (e.g., the BCL2 family members; Figure 5F) and resistance to apoptosis are consistent with the senescence phenotype (Herranz and Gil, 2018; Thompson et al., 2019). In fact, key markers of senescence, such as decreases in LaminB1 mRNA and protein (Figure 6D) (Freund et al., 2012) and increased endogenous senescence-associated β-galactosidase (SA-β-gal) staining was detected in male MafAS64F/+ islets at levels comparable to mice aged 10–12 months (Figures 6E and 6F). Notably, SA-β-gal was undetectable at 4 weeks in male MafAS64F/+ islets, a time point before β cell dysfunction or ever in female MafAS64F/+ islets, nor was LaminB1 expression decreased in female MafAS64F/+ islets (Figures 6D-6F, and S7D and S7E). These results illustrate a novel sex-dependent pathophysiology of accelerated β cell aging and senescence that contributes to glucose intolerance in MafAS64F/+ males.
Human EndoC-βH2 β cells expressing MAFAS64F show increased markers of cellular senescence and a functional senescence-associated secretory phenotype
Human EndoC-βH2 cells were transduced with human MAFAWT or MAFAS64F expression constructs using lentivirus to investigate whether cellular senescence could be prematurely induced in human β cells. Immunoblot analysis of protein extracts from these EndoC-βH2 cells confirmed the faster migrating properties of the phosphorylation-impaired MAFAS64F (Figure S8A) (Iacovazzo et al., 2018). As in our MafAS64F/+ mouse model, MAFAS64F-producing cells had significantly increased SA-β-gal, p21, and 53BP1 expression and decreased LaminB1 expression compared to those expressing the human MAFAWT protein (Figures 7A-7D and S8B). Gene expression analysis of MAFAS64F-expressing β cells showed changes consistent with senescence markers identified in male MafAS64F/+ mice (i.e., increased p21, increased BCL2, and decreased LMNB1) and decreased expression of MAFB (Figure 7C), a transcription factor not produced in rodent islet β cells postnatally (Conrad et al., 2016). As found in male MafAS64F/+ mutant mouse islets, over-expressing MAFAS64F in human EndoC-βH2 β cells caused misexpression of a number of critical calcium channel genes, including CACNA1A, CACNA1C, CACNA1E, and CACNA1G without altering insulin secretion machinery gene expression (Figure S8C).
Figure 7. Human β cells expressing MAFAS64F demonstrate accelerated senescence and a species-specific SASP signature.

(A) SA-β-gal staining on EndoC-βH2 cells transduced to express WT MAFA or MAFAS64F and stained for MAFA (red). Scale bar, 100 μm.
(B) Inset areas outlined in boxes in (A) are magnified in the left panel. Scale bar, 25 μm. β-gal+ area was significantly increased in MAFAS64F cells (right panel).
(C) Human expression of β cell-enriched proteins and senescence associated proteins identified in male MafAS64F/+ mice. Two-tailed Student t test; *p < 0.05; **p < 0.01.
(D) Immunostaining for senescence markers p21 (green) was increased and LaminB1 (red) decreased in MAFAS64F-expressing EndoC-βH2 cells.
(E) CM from WT MAFA or MAFAS64F-expressing EndoC-βH2 cells were collected, purified and added to EndoC-βH2 cells cultured for 72 h. SASP genes upregulated in male MafAS64F/+ mice were not identified in EndoC-βH2 cells. However, novel, species-specific secretory senescence-associated factors were identified in this context. N.D., not detectable. One-way ANOVA; *p < 0.05; **p < 0.01; ****p < 0.0001.
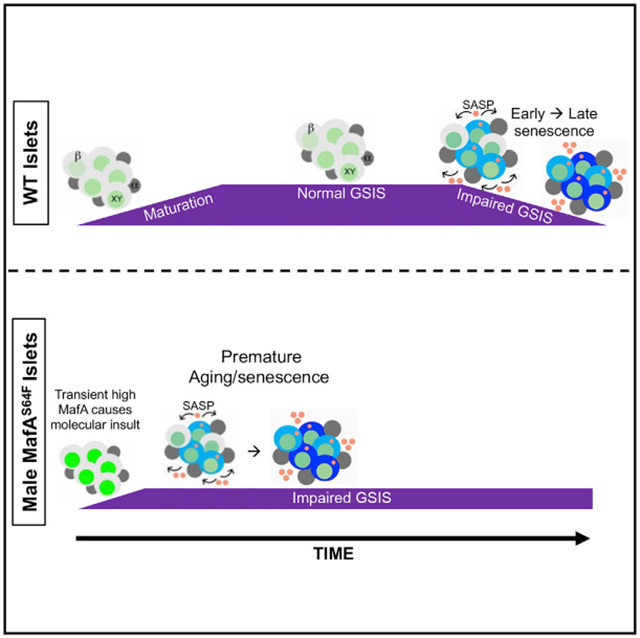
(F) Schematic of temporal senescence and aging responses in MafAS64F-expressing male β cells. In male MafAS64F/+, transiently high MafA protein triggers molecular insults to induce premature senescence marked by cell-cycle arrest, senescence staining (blue cytoplasmic shade), and initiation of a senescence-associated secretory phenotype (SASP, pink secretory factors), resulting in impaired GSIS. Late (progressive) senescence propagates this phenotype with SASP amplification and diversification.
Progression of senescence includes the development of SASP factors, which upon secretion induce senescence by reprogramming neighboring cells in a cell non-autonomous manner (Gorgoulis et al., 2019; Herranz and Gil, 2018). To determine whether SASP factors are released by human cells expressing MAFAS64F, control medium or conditioned media (CM) collected from EndoC-βH2 cells expressing either MAFAWT or MAFAS64F was applied to untransduced EndoC-βH2 cells (Figure 7E). Notably, SASP genes identified in male MafAS64F/+ mice were not increased in EndoC-βH2 cells in response to MAFAWT or MAFAS64F CM; however, human-specific SASP markers in gene families related to those identified in MafAS64F/+ male mice were specifically increased by MAFAS64F CM (Figure 7E). These results illustrate the ability of MAFAS64F to promote cellular senescence in human β cells and to generate a functional, human β cell SASP via species-specific mediators.
DISCUSSION
Post-translational modifications of the islet-enriched MAFA protein are critical for regulating its activity, stability, and cellular localization for appropriate β cell function (Guo et al., 2009, 2010; Han et al., 2007; Iacovazzo et al., 2018; Rocques et al., 2007). Here, we have developed a mouse model of the human MAFAS64F variant to understand mechanistically the sex-biased pathophysiological outcomes of MODY or insulinomatosis in affected heterozygous human carriers (Iacovazzo et al., 2018). Significantly, the physiological outcomes of MafAS64F/+ mice appear to mimic the outcomes expected in human subjects, with glucose intolerance in males and improved glucose clearance and hypoglycemia in females.
This mutation blocks a key priming phosphorylation event at S65, which normally directs post-translational modifications affecting MAFA protein stability (Guo et al., 2009; Han et al., 2007; Rocques et al., 2007), transactivation (Han et al., 2007; Rocques et al., 2007), oncogenesis (Rocques et al., 2007), and DNA binding (Guo et al., 2010). These modifications are coupled to two antagonistic regulatory processes: increased transactivation activity and ubiquitin-mediated degradation (Rocques et al., 2007), illustrating that impeccable regulation of this protein is linked to islet β cell health. Earlier in vitro analysis demonstrated that the MAFAS64F variant converted this normally unstable protein (t1/2 ~30 min) to a very stable form (t1/2 ≥4 h) (Iacovazzo et al., 2018). However, both male and female MafAS64F/+ mice had similar improvements in glucose tolerance at 4 weeks, before overt and transient elevation in protein levels in only males (Figures 1 and 2). Notably, this increase in MafAS64F protein levels preceded glucose intolerance seen by 5 weeks of age in males, while females continued to be modestly hypoglycemic, with improved glucose clearance. The changes in male and female MafAS64F/+ β cell activity were maintained throughout the period of analysis despite the presence of WT-like protein levels after 5 weeks. However, we propose that MafAS64F is more abundantly and persistently produced than MAFAWT throughout the lifetime of the β cell despite similar protein levels because of 3- to 5-fold lower MafA mRNA levels in male and female MafAS64F/+ islets in relation to the WT islets (Figure 2C). In line with this observation, human MAFA protein levels were largely unchanged by immunohistochemistry between the normal and insulinoma β cells of MAFAS64F patients (Iacovazzo et al., 2018).
Our examination of ovariectomized female MafAS64F/+ mice suggested that the estrogen sex hormone did not have a direct regulatory role, so it is presently unclear what factors are controlling the sexually dimorphic phenotypes of MafAS64F mutant mice. Since MAFAS64F-induced disease is not observed until ~38 years of age in both sexes without reports of affected pubertal development (Iacovazzo et al., 2018), we believe that neither estrogen nor testosterone have an impact on the divergent disease processes in humans. Instead, we propose that future efforts focus on determining whether sex chromosome-linked genes are influenced by MAFAS64F, such as the X chromosome-linked candidate genes found differentially expressed in 5-week-old MafAS64F/+ male islets (Figure S9A).
Bulk RNA-seq analysis of 5-week-old male and female MafAS64F/+ mouse islets illustrated similar dysregulation of genes involved in β cell identity and ion channel function (Figures 3, S9B, and S9C). For example, the expression of several markers of β cell maturation were diminished in both MafAS64F/+ male and female islets (Figures S9B and S9C), including the Mnx1 (Pan et al., 2015) and Pdx1 transcription factors as well as the Ucn3 neuropeptide (van der Meulen et al., 2015). In addition, the expression of both sex-dependent and sex-independent genes involved in Ca2+ responses were identified (Figure 3). Accordingly, functional analyses after glucose stimulation revealed dysfunctional Ca2+ responses. While all male 5-week-old MafAS64F/+ islets appear to have severely blunted glucose-induced Ca2+ responses (called non-responders in Figure 4), females contained both non-responder and unique responder islets with higher frequency oscillations in response to glucose. Presumably, responder islets mediate the high basal glucose-induced insulin secretion properties that are characteristic of female MafAS64F/+ islets (Figures 4A and S3), while downstream effectors of Mnx1 and Pdx1 activation that are involved in cell signaling (e.g., Gcgr, Glp1r), secretion (Syt2, Ucn3, Ins1, Ins2), and ion channel activity contribute to β cell non-responder dysfunction (Blum et al., 2014; Gilbert and Blum, 2018; Jacobson and Shyng, 2020; Kalwat and Cobb, 2017).
Because of the heterogeneity of the Ca2+ signaling changes in female MafAS64F/+ islets, we narrowed our focus to determine possible mechanisms contributing to the MafAS64F-induced diabetic phenotype in males. Future single-cell sequencing efforts could reveal the factors regulating the interplay of distinct responder and non-responder islet β cell populations of MafAS64F/+ females as well as their relative homogeneity in male non-responder islets. Importantly, our results clearly show that the many gene products associated with male MafAS64F/+ β cell inactivity are not made in female heterozygotes (Figures 5 and S6). As divergent regulation of metabolism between the sexes in aging and disease is increasingly recognized (Sampathkumar et al., 2020), MAFAS64F may provide a penetrant model to study sex-dependent effects on β cell health.
As expected, dynamic perifusion studies showed a marked reduction in baseline and stimulated insulin secretion in male compared to female MafAS64F/+ islets (Figure S3). However, the mildly elevated insulin secretion levels of 10- to 12-week-old male MafAS64F/+ islets appears at odds with the impairment in static insulin secretion at 5 weeks and temporally stable glucose intolerance levels (Figure 1). We propose that the age of the mutant mice may account for this discrepancy. Potentially, the older and more active male β cells “compensate” for the less active 5-week-old cells that were affected by the SASP factors secreted at the onset of glucose intolerance. Future studies will be focused on directly determining whether functionally and molecularly distinct β cell populations are progressively produced upon MAFAS64F exposure.
Since Ca2+ release in MafAS64F/+ male islets appeared to contribute to β cell inactivity, we focused on understanding how this process was affected in this context. Our results have demonstrated that MafAS64F produces premature aging and senescence signatures in male murine β cells and human EndoC-βH2 cells (Figures 5, 6, and 7). In contrast, neither an aging nor senescence signature was observed in female murine β cells expressing MafAS64F (Figures S6 and S7). Future studies will also investigate whether changes in Ca2+ handling occurs before or after MafAS64F/+ male islets transition to a senescent phenotype as Ca2+ is also implicated in cellular senescence (Martin and Bernard, 2018).
Unlike the temporary cell-cycle arrest of quiescence, senescence is thought to be irreversible and refractory to mitogenic stimuli. Senescent cells undergo a progression of changes after early insult, including cell-cycle arrest by the activation of inhibitors (i.e., p53, p21, and/or p16, among others; Figures 5, 6, and 7) and DDR responses (Gorgoulis et al., 2019; Herranz and Gil, 2018). Progression to senescence involves chromatin remodeling to influence gene expression, metabolism, autophagy, and SASP, with the release of a heterogeneous mix of SASP effector proteins influencing neighboring cells in a cell non-autonomous manner (Gorgoulis et al., 2019; Herranz and Gil, 2018). Terminal senescence from persistent damage involves autocrine and paracrine SASP amplification, loss of nuclear integrity, and diversification of the SASP phenotype (Gorgoulis et al., 2019). Notably, senescence-associated SA-β-gal staining was not induced in 4-week-old MafAS64F/+ β cells, just 1 week before the detection of glucose intolerance (Figures 1 and 6).
Ultimately, senescent cells become resistant to apoptosis by the upregulation of anti-apoptotic proteins, such as those in the BCL2 family, and are often cleared by immune cells (Gorgoulis et al., 2019; Herranz and Gil, 2018). Interestingly, effective clearance of β cells is not apparent in MafAS64F/+ male islets as we see accumulation of senescent cells, and there was no evidence of β cell death or immune cell infiltration (data not shown; Figures 6 and S7F). Senescent cells can play a causal role in aging-related pathology (van Deursen, 2014), and targeted removal of senescent cells can improve health span and reduce the incidence of aging-related diseases (Baker et al., 2011, 2016; Chang et al., 2016; Childs et al., 2016). Independent studies have shown that removal of the rare, senescent β cells in mouse models of T1D and T2D helps restore β cell function and glucose homeostasis in vivo (Aguayo-Mazzucato et al., 2019; Thompson et al., 2019). These senescent β cells showed distinct SASP signatures depending on modeling context (T1D versus T2D) (Aguayo-Mazzucato et al., 2019; Thompson et al., 2019), and these signatures were also species dependent such that only a subset of SASP mediators identified in respective mouse models were detected in human, diabetic islets (Aguayo-Mazzucato et al., 2019; Thompson et al., 2019).
Importantly, up to 50%–60% of human T2D β cells were shown to be senescent (Aguayo-Mazzucato et al., 2019), compared to their rare occurrence in previously described mouse models (Aguayo-Mazzucato et al., 2019; Thompson et al., 2019). This rate of β cell senescence is comparable to that found in MafAS64F/+ male islets, suggesting that this variant models the widespread β cell senescence in human T2D (Figure 6). In fact, MAFAS64F expression produced senescence in human EndoC-βH2 β cells with the development of a functional SASP. Treatment with CM from MAFAS64F-expressing EndoC-βH2 cells induced β cell senescence and SASP factor expression differently from those identified in MafAS64F/+ male mouse islets, albeit from similar molecular families. In summary, these studies implicate MAFAS64F-induced senescence factors in causing β cell dysfunction and open doors to the identification of β cell senescence signatures across etiologies of diabetes (Figure 7F).
Human and rodent islets differ substantially in architecture, cell composition, proliferative capacity, islet amyloid formation, antioxidant enzyme levels, and, most significantly for the results described herein, MAFA and MAFB transcription factor expression (Bosco et al., 2010; Brissova et al., 2005; Butler et al., 2007; Cabrera et al., 2006; Dai et al., 2012; Fiaschi-Taesch et al., 2010; Henquin et al., 2006; Tyrberg et al., 2001). Because MafA is expressed at the onset of rodent β cell formation during embryogenesis, but not until 10 years of age in humans (Cyphert et al., 2019; Dai et al., 2012; Hang and Stein, 2011), we appreciate that some of the observations made in MafAS64F/+ mice will not be directly relevant to the human disease. For example, since substantial β cell proliferation ceases in humans before MAFA expression (Cyphert et al., 2019; Dai et al., 2012; Hang and Stein, 2011), the decreased islet cell proliferation observed in male and female MafAS64F/+ mice may not be directly relevant in humans. We also did not expect to find that MafAS64F/+ male mice would manifest the overt fasting hyperglycemia seen in affected humans (Iacovazzo et al., 2018), since only 1 of the 7 human MODY transcription factors has a phenotype in heterozygous mice comparable to human carriers (i.e., Pdx1 (Ahlgren et al., 1998)). As found in other studies of MODY transcription factor function in mice, only the homozygous (male) MafAS64F/S64F mice manifested an explicit elevation in fasting blood glucose levels and glucose intolerance, which worsened with age (Figure S2). MafAS64F presumably represents another example of how gene dosage of critical regulatory gene variants affects islet cell health in a species-specific manner.
It is also likely that the unique requirement of MAFB in human (but not rodent) β cells influences the effects of MAFAS64F in human carriers. The human MAFAS64F:MAFB heterodimeric activator could impart a unique influence on β cells compared to the mouse MafAS64F:MafA homodimeric activator (Cyphert et al., 2019; Hang and Stein, 2011), which may explain why neuroendocrine tumors and overt hypoglycemia was not observed in aged MafAS64F/+ female mice (data not shown). Notably, MAFB was recently shown to be essential for the formation of human embryonic-derived β cells and insulin production (Russell et al., 2020), whereas there is no phenotype associated with the loss of MafB in mouse islet β cells except during pregnancy (Cyphert et al., 2019). Thus, we believe that it will be important to extend the analysis of MAFAS64F control to human islets—acutely in vitro and chronically after the transplantation of human pseudoislets into immunocompromised mice to directly determine its effect in vivo. Such studies should generate keen insight into unique, species-specific, age-dependent, and sex-biased molecular and genetic mechanisms controlling human islet β cell activity.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Roland Stein (roland.stein@vanderbilt.edu).
Materials availability
All unique/stable reagents generated in this study are available from the lead contact with a completed materials transfer agreement.
Data and code availability
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplemental information. Bulk RNA-seq data have been deposited to GEO and are publicly available at the date of publication. Accession number is listed in the Key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Guinea pig anti-Insulin | Thermo Fisher | Cat#PA1 26938; RRID:AB_794668 |
| Mouse anti-Glucagon | Sigma | Cat#G2654; RRID:AB_259852 |
| Mouse anti-Ki67 | BD Biosciences | Cat#550609; RRID:AB_393778 |
| DAPI Fluoromount-G mounting medium | SouthernBiotech | Cat#0100-20 |
| Rabbit anti-MAFA | Cell Signaling | Cat#79737; RRID:AB_2799938 |
| Goat anti-PDX-1 | Chris Wright, Vanderbilt | N/A |
| Rabbit anti-UCN3 | Phoenix | Cat#H-019-028; RRID:AB_2889826 |
| Goat anti-P21 (for cells) | Santa Cruz | Cat#sc-397G; RRID:AB_632127 |
| Rat anti-P21 (for tissue sections) | Abcam | Cat#ab107099; RRID:AB_10891759 |
| Rabbit anti-53BP1 | Bethyl | Cat#A300-272A; RRID:AB_185520 |
| Rabbit anti-gH2AX | Abcam | Cat#ab81299; RRID:AB_1640564 |
| Rabbit anti-LaminB1 | Abcam | Cat#16048; RRID:AB_443298 |
| Bacterial and virus strains | ||
| Lentivirus: MAFAWT-pCDH-EF1-MCS-IRES-GopGFP | Modified for this paper from System Biosciences | Cat#CD521A-1 |
| Lentivirus: MAFAS64F-pCDH-EF1-MCS-IRES-GopGFP | Modified for this paper from System Biosciences | Cat#CD521A-1 |
| Chemicals, peptides, and recombinant proteins | ||
| Fura-2 AM | Life Technologies | Cat#F1221 |
| Critical commercial assays | ||
| Cell Death Detection Kit | Roche | Cat#11684795910 |
| Senescence β-Galactosidase Staining Kit | Cell Signaling | Cat#9860 |
| Glycogen Assay Kit | Cayman Chemicals | Cat#700480 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO:GSE183561 |
| Experimental models: Cell lines | ||
| Human: EndoC-βH2 Cells | Scharfmann et al., 2014 | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: MafAS64F/+ | This paper | N/A |
| Oligonucleotides | ||
| Primers for mouse and human see Table S1 | This paper | N/A |
| Software and algorithms | ||
| Genialis visual informatics platform: BBDuk, START aligner, featureCounts | https://www.genialis.com | N/A |
| DESeq2 | Love et al., 2014 | N/A |
| ImageJ | https://imagej.nih.gov/ij/ | N/A |
| ImageScope Software | Aperio Technologies | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Animals
S64F MafA-expressing mice were generated using CRISPR/Cas9 targeting by the University of Michigan Transgenic Core in the C57BL/6J mouse strain. Viable animals and progeny were screened for the appropriate mutation by DNA sequencing (Molecular Resource Center, University of Tennessee Health Science Center). Wild-type (WT) littermates were used as controls. MafAS64F/+ were born at normal Mendelian ratios while homozygous MafAS64F/S64F variants were not (see Figure S2). Mice of both sexes were used in this study; details can be found in the results section and figure legends. All animal studies were reviewed and approved by the Vanderbilt University Institutional Animal Care and Use Committee. Mice were housed and cared for according to the Vanderbilt Department of Animal Care and the Institutional Animal Care and Use Committee of Animal Welfare Assurance Standards and Guidelines.
Human EndoC-βH2 cells
Human EndoC-βH2 cells were grown in DMEM containing 5.6mM glucose, 2% BSA, 50 μM 2-mercaptoethanol, 10mM nicotinamide, 5.5 μg/mL transferrin, 6.7 ng/mL selenite, 100 units/mL penicillin, and 100 units/mL streptomycin (Scharfmann et al., 2014). For gene transfection, cells were incubated with lentiviral particles (150ng for cells cultured in a 6cm dish) containing either MAFAWT-expressing or MAFAS64F-expressing sequences one day after plating. Assays were performed one week following infection unless specified otherwise.
METHOD DETAILS
Intraperitoneal glucose and insulin tolerance testing
Glucose tolerance testing was performed on WT and MafAS64F/+ mice (n = 4-16) given an intraperitoneal injection of D-glucose (2 mg/g body weight) prepared in sterile PBS (20% w/v) after a 6-hour fast. Insulin tolerance tests were conducted by intraperitoneal injection of 0.5 IU/kg body weight insulin (Novolin, regular human insulin, recombinant DNA origin) into mice (n = 3-5) fasted for 6 hours. Blood glucose was measured using a FreeStyle glucometer (Abbott Diabetes Care) before (0 minutes) and at 15, 30, 60, and 120 minutes following injection. Serum insulin was measured by radioimmunoassay at the Vanderbilt Hormone Assay and Analytical Services Core using blood collected following the 6-hour fast. Serum testosterone was measured by the University of Virginia Center for Research in Reproduction Ligand Assay and Analysis Core.
Glucose-stimulated hormone secretion
WT and MafAS64F/+ mouse (n = 3-6) islets were isolated using standard islet isolation conditions, and hormone secretion was assessed as described previously (Cyphert et al., 2019). The outcome was presented as secreted insulin or glucagon relative to the total islet DNA content (ng/mg DNA (insulin) or pg/μg DNA (glucagon)). Islet hormone content was presented as the concentration of insulin or glucagon per DNA content (Quant-iT PicoGreen, Invitrogen) in each reaction (ng/ng DNA).
For perifusion analysis, islets from 8-10 week old female and male WT and MafAS64F/+ mice were studied in a dynamic cell perifusion system at a perifusate flow rate of 1 mL/min (Walker et al., 2020) in the Vanderbilt Islet Procurement and Analysis Core. The effluent was collected at 3-minute intervals using an automatic fraction collector. The insulin concentration in each perifusion fraction and islet extract was measured by radioimmunoassay (Millipore).
Tissue and cell preparation and immunostaining
For immunostaining, WT and MafAS64F/+ pancreata were fixed in 4% (v/v) paraformaldehyde, embedded in either Tissue-Plus O.C.T. (Thermo Fisher Scientific) or paraffin wax, and sectioned to 6 mm thickness. Immunofluorescent images were obtained using a Zeiss Axio Imager M2 widefield microscope with ApoTome. Immunofluorescence staining was performed as previously described with the antibodies listed in Key resources table. Islet β- and α- cell areas were determined as described previously (Cyphert et al., 2019). Briefly, pancreatic sections taken every 50 μm (n = 3-4 animals per genotype) were scanned using a ScanScope CS scanner (Aperio Technologies, Vista, CA). Images from each experiment were processed with ImageScope Software (Aperio Technologies, Vista, CA). Islet β- and α-cell areas were calculated as the ratio of the insulin- or glucagon-positive area to total pancreas area (eosin stained). The TUNEL assay was performed using an in situ cell death detection kit (Roche, #11684795910). In EndoC-βH2 cells, immunostaining for LaminB1, 53BP1, and p21 were also performed (see Key resources table).
RNA sequencing, analysis, and validation by quantitative real-time PCR
RNA was isolated from WT and MafAS64F/+ islets (n = 4 each for males, n = 5 each for females) using the RNAqueous total RNA isolation kit (Ambion; Thermo Fisher), and then analyzed on an Agilent 2100 Bioanalyzer. Only samples with an RNA Integrity Number >8.0 were used for library preparation. cDNA libraries were constructed and paired-end sequencing of 4-5 replicates was performed on an Illumina NovaSeq6000 (150 nucleotide reads). The generated FASTQ files were processed and interpreted using the Genialis visual informatics platform (https://www.genialis.com). Reads were preprocessed by BBDuk which removes adapters, trims reads for quality from the 3′ end, and discards reads that are too short after trimming. Preprocessed reads were aligned by START aligner and quantification was done using featureCounts. FastQC reports, alignment statistics and rRNA/globin depletion rate QC information is automatically summarized by the MultiQC tool. Differential gene expression analyses were performed with DESeq2 (Love et al., 2014) with a log2(fold change) threshold of 1 and FDR <0.05 using the genome build GRCm38. Tab-delimited text files include FPKM values for each sample.
Quantitative real-time PCR expression analysis of selected candidates were performed on independently isolated mouse islets and EndoC-βH2 cells. Trizol reagent (Life Technologies) was used to collect RNA. The cDNAs produced from the iScript cDNA Synthesis Kit (Bio-Rad) were processed in a LightCycler 480 II system (Roche), and analyzed by the ΔΔCT method with primers provided in Key resources table. Significance was calculated by comparing the ΔCT values.
Islet cytosolic calcium imaging
Approximately 60 WT and MafAS64F/+ islets from at least 6 female and male mice were analyzed with the ratiometric calcium indicator fura-2-acetoxymethylester (Fura-2 AM) (Life Technologies). Islets were maintained in 5 mM glucose culture media for 30 min prior to being loaded with 2 μM Fura-2 AM for 20 min, washed, transferred to a hand-made agarose gel small-volume chamber in a glass bottom dish filled with regular HBSS (5 mM glucose, Thermo Fisher). The 11 mM glucose-induced calcium oscillations and depolarization-activated calcium influx at 30 mM KCl were measured. Images were taken using a Nikon Eclipse TE2000-U microscope equipped with an epifluorescence illuminator (SUTTER, Inc), a CCD camera (HQ2; Photometrics, Inc), and Nikon Elements software (NIKON, Inc) as described before (Dadi et al., 2015).
Senescence-associated β-galactosidase (SA-β-gal) staining
WT and MafAS64F/+ pancreata were snap frozen in O.C.T. and cryosections were prepared at 16 μm thickness. SA-β-gal activity staining was performed at pH 6.0 (Kurz et al., 2000) using a commercial kit (Cell Signaling, #9860). To compare the intensity of SA-β-gal staining, sections from different genotypes and ages were processed on the same slide. Staining reactions were developed for 18 hours at 37°C, then stopped by 3x PBS washes (pH 7.4). Slides were then subject to immunostaining for insulin by fixing in 4% paraformaldehyde for 45 minutes, permeabilized with Tris-buffered saline with 0.2% Triton X-100 for 15 minutes, blocked in 2% normal donkey serum/1% BSA in PBS and incubated overnight with guinea pig anti-insulin (1:500, Abcam) at 4°C. HRP-conjugated secondary antibodies were incubated on slides for 1 hour and detected with the DAB+ chromogen kit (DAKO). After washing, slides were mounted and imaged by brightfield microscopy. The total number of SA-β-gal+ islets per pancreas section were analyzed on ImageJ.
The same kit and condition were also used to evaluate the SA-β-gal activity in EndoC-βH2 cells cultured on chamber slides from different conditions. Staining reactions were stopped after developing for 2 to 3 hours at 37°C with 3x PBS washes (pH 7.4). Slides were then subject to immunostaining for MAFA by fixing in 4% paraformaldehyde for 12 minutes, permeabilized with Tris-buffered saline with 0.5% Triton X-100 for 5 minutes, blocked in 2% normal donkey serum/1% BSA in PBS and incubated overnight with Rabbit α-MAFA (1:500, Novus (NBP1-00121) at 4°C. After washing, slides were mounted and imaged on a Zeiss Axio Imager M2 widefield microscope with ApoTome. Average SA-β-gal+ area/cell was analyzed on ImageJ.
Ovariectomy methods
WT and MafAS64F/+ female mice underwent ovariectomy at 3 weeks to remove the contribution of endogenous ovarian hormones and prevent completion of sexual maturity (Martinez et al., 2012). Mice were anesthetized with 1%–5% inhaled isoflurane, and placed prone on a heating pad to maintain their body temperature at 37°C. The mice received subcutaneous ketofen at 5-10 mg/kg prior to surgery and post-operatively for two days for pain relief. Intraperitoneal ceftriaxone at 20-40 mg/kg was given once intraoperatively for infection prophylaxis. After adequate anesthesia and analgesia, a midline 1.5 cm incision was made along the shaved mid-dorsal surface of the mouse. Within the retroperitoneal cavity, the ovaries were located and ligated. The incision was closed using surgical staples, which were removed on post-operative day three. Mice were housed singly during recovery and regrouped post-operatively.
Glycogen storage assay
Liver and gastrocnemius muscle from WT and MafAS64F/+ mice (n = 5 per genotype and sex) were collected and flash frozen in liquid nitrogen. Tissue was homogenized with a Dounce homogenizer prior to determining glycogen content using a glycogen assay kit (Cayman Chemical) according to the manufacturer’s protocol.
Paracrine conditioned media assay
Conditioned media (CM) produced after 24 hours of culture from MAFAS64F- and MAFAWT-expressing human EndoC-βH2 cells were clarified by centrifugation at 500xg for 5 minutes and then at 3000xg for 5 minutes. CM was stored at 4°C for up to 3 weeks prior to use. EndoC-βH2 cells plated on 12-well plate were cultured with a 1:1 mix of EndoC-βH2 islet media (4% BSA) and albumin-free CM collected from MAFAWT or MAFAS64F expressing cells (resulting in 2% BSA in the final medium). For controls, EndoC-βH2 cells from the same passage were cultured in a 1:1 mix of regular EndoC-βH2 cells media and albumin-free media alone (not conditioned by cells), also resulting in 2% BSA in the final medium. After 72 hours, EndoC-βH2 cells were harvested for analysis.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical significance was determined using the two-tailed Student t test, one-way ANOVA or two-way ANOVA and Tukey post hoc test as indicated in the figure legends. Data are presented as the mean ± SEM. A threshold of at least p < 0.05 was used to declare significance.
Supplementary Material
Highlights.
MafAS64F/+ mice show sex-dependent β cell dysfunction (diabetes or hypoglycemia)
MafAS64F/+ male and female mice show aberrant islet Ca2+ signaling
Only MafAS64F/+ males show markers of islet aging and senescence
MAFAS64F expression in male human EndoC-βH2 cells accelerates cellular senescence
ACKNOWLEDGMENTS
This research was performed using resources and/or funding provided by NIH grants to R.S. (DK090570), E.M.W. (F32 DK109577), J.C. (T32 DK007061), F.M.-J. (DK074970 and DK107444), J.S. (DK109102, HL144846), and D.A.J. (DK097392) and by the Vanderbilt Diabetes Research and Training Center (DK20593). X.T. was supported by a JDRF Fellowship (3-PDF-2019-738-AN); J.C. by a Doris Duke Charitable Foundation Physician Scientist Fellowship (2020063), and VUMC Harrison Society funds; D.I. by a George Alberti Research Training Fellowship funded by Diabetes UK (16/0005395); and F.M.-J. by a US Department of Veterans Affairs Merit Review Award (BX003725). S.E.F. is supported by a Sir Henry Dale Fellowship jointly sponsored by the Wellcome Trust and the Royal Society (105636/Z/14/Z). We also thank Drs. Raphaël Scharfmann and Phillippe Ravassard for generously providing EndoC-βH2 cells. Imaging was performed with NIH support from the Vanderbilt University Medical Center Cell Imaging Shared Resource (NCI grant CA68485; NIDDK grants DK20593, DK58404, and DK59637; NICHD grant HD15052; and NEI grant EY08126). Islet hormone analysis was performed in the Vanderbilt University Medical Center Islet Procurement and Analysis Core (NIH DK20593) and the Vanderbilt University Neurochemistry Core (NIH U54 HD083211). MafAS64F-expressing mice were produced in the Diabetes Research Center Animal Studies Core at the University of Michigan, which is supported by NIH P30 DK020572.
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.109813.
INCLUSION AND DIVERSITY
We worked to ensure sex balance in the selection of non-human subjects. We worked to ensure diversity in experimental samples through the selection of the cell lines. We worked to ensure diversity in experimental samples through the selection of the genomic datasets. While citing references scientifically relevant for this work, we also actively worked to promote gender balance in our reference list. The author list of this paper includes contributors from the location where the research was conducted who participated in the data collection, design, analysis, and/or interpretation of the work.
REFERENCES
- Aguayo-Mazzucato C, Koh A, El Khattabi I, Li WC, Toschi E, Jermendy A, Juhl K, Mao K, Weir GC, Sharma A, and Bonner-Weir S (2011). Mafa expression enhances glucose-responsive insulin secretion in neonatal rat β cells. Diabetologia 54, 583–593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aguayo-Mazzucato C, Andle J, Lee TB Jr., Midha A, Talemal L, Chipashvili V, Hollister-Lock J, van Deursen J, Weir G, and Bonner-Weir S (2019). Acceleration of β cell aging determines diabetes and senolysis improves disease outcomes. Cell Metab. 30, 129–142.e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ahlgren U, Jonsson J, Jonsson L, Simu K, and Edlund H (1998). β-cell-specific inactivation of the mouse Ipf1/Pdx1 gene results in loss of the β-cell phenotype and maturity onset diabetes. Genes Dev. 12, 1763–1768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arda HE, Li L, Tsai J, Torre EA, Rosli Y, Peiris H, Spitale RC, Dai C, Gu X, Qu K, et al. (2016). Age-Dependent Pancreatic Gene Regulation Reveals Mechanisms Governing Human β Cell Function. Cell Metab. 23, 909–920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrojo E Drigo R, Lev-Ram V, Tyagi S, Ramachandra R, Deerinck T, Bushong E, Phan S, Orphan V, Lechene C, Ellisman MH, and Hetzer MW (2019). Age mosaicism across multiple scales in adult tissues. Cell Metab. 30, 343–351.e3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artner I, Hang Y, Guo M, Gu G, and Stein R (2008). MafA is a dedicated activator of the insulin gene in vivo. J. Endocrinol 198, 271–279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artner I, Hang Y, Mazur M, Yamamoto T, Guo M, Lindner J, Magnuson MA, and Stein R (2010). MafA and MafB regulate genes critical to β-cells in a unique temporal manner. Diabetes 59, 2530–2539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Wijshake T, Tchkonia T, LeBrasseur NK, Childs BG, van de Sluis B, Kirkland JL, and van Deursen JM (2011). Clearance of p16Ink4a-positive senescent cells delays ageing-associated disorders. Nature 479, 232–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baker DJ, Childs BG, Durik M, Wijers ME, Sieben CJ, Zhong J, Saltness RA, Jeganathan KB, Verzosa GC, Pezeshki A, et al. (2016). Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banerjee RR, Cyphert HA, Walker EM, Chakravarthy H, Peiris H, Gu X, Liu Y, Conrad E, Goodrich L, Stein RW, and Kim SK (2016). Gestational Diabetes Mellitus From Inactivation of Prolactin Receptor and MafB in Islet β-Cells. Diabetes 65, 2331–2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barbetti F, and D’Annunzio G (2018). Genetic causes and treatment of neonatal diabetes and early childhood diabetes. Best Pract. Res. Clin. Endocrinol. Metab 32, 575–591. [DOI] [PubMed] [Google Scholar]
- Basisty N, Meyer JG, and Schilling B (2018). Protein Turnover in Aging and Longevity. Proteomics 18, e1700108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bergsten P, Grapengiesser E, Gylfe E, Tengholm A, and Hellman B (1994). Synchronous oscillations of cytoplasmic Ca2+ and insulin release in glucose-stimulated pancreatic islets. J. Biol. Chem 269, 8749–8753. [PubMed] [Google Scholar]
- Blum B, Roose AN, Barrandon O, Maehr R, Arvanites AC, Davidow LS, Davis JC, Peterson QP, Rubin LL, and Melton DA (2014). Reversal of β cell de-differentiation by a small molecule inhibitor of the TGFβ pathway. eLife 3, e02809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosco D, Armanet M, Morel P, Niclauss N, Sgroi A, Muller YD, Giovannoni L, Parnaud G, and Berney T (2010). Unique arrangement of α- and β-cells in human islets of Langerhans. Diabetes 59, 1202–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brissova M, Fowler MJ, Nicholson WE, Chu A, Hirshberg B, Harlan DM, and Powers AC (2005). Assessment of human pancreatic islet architecture and composition by laser scanning confocal microscopy. J. Histochem. Cytochem 53, 1087–1097. [DOI] [PubMed] [Google Scholar]
- Butler PC, Meier JJ, Butler AE, and Bhushan A (2007). The replication of β cells in normal physiology, in disease and for therapy. Nat. Clin. Pract. Endocrinol. Metab 3, 758–768. [DOI] [PubMed] [Google Scholar]
- Cabrera O, Berman DM, Kenyon NS, Ricordi C, Berggren PO, and Caicedo A (2006). The unique cytoarchitecture of human pancreatic islets has implications for islet cell function. Proc. Natl. Acad. Sci. USA 103, 2334–2339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J (2014). Cell biology: the beginning of the end. Nature 505, 35–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campisi J, and d’Adda di Fagagna F (2007). Cellular senescence: when bad things happen to good cells. Nat. Rev. Mol. Cell Biol 8, 729–740. [DOI] [PubMed] [Google Scholar]
- Camunas-Soler J, Dai X, Hang Y, Bautista A, Lyon J, Suzuki K, Kim S, Quake S, and MacDonald P (2020). Patch-Seq links single-cell transcriptomes to human islet dysfunction in diabets. Cell Metab. 5, 1017–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang J, Wang Y, Shao L, Laberge RM, Demaria M, Campisi J, Janakiraman K, Sharpless NE, Ding S, Feng W, et al. (2016). Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med 22, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen C, Shiota C, Agostinelli G, Ridley D, Jiang Y, Ma J, Prasadan K, Xiao X, and Gittes GK (2019). Evidence of a developmental origin for β-cell heterogeneity using a dual lineage-tracing technology. Development 146, dev164913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Childs BG, Baker DJ, Wijshake T, Conover CA, Campisi J, and van Deursen JM (2016). Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354, 472–477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Conrad E, Dai C, Spaeth J, Guo M, Cyphert HA, Scoville D, Carroll J, Yu WM, Goodrich LV, Harlan DM, et al. (2016). The MAFB transcription factor impacts islet α-cell function in rodents and represents a unique signature of primate islet β-cells. Am. J. Physiol. Endocrinol. Metab 310, E91–E102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coppé JP, Desprez PY, Krtolica A, and Campisi J (2010). The senescence-associated secretory phenotype: the dark side of tumor suppression. Annu. Rev. Pathol 5, 99–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cyphert HA, Walker EM, Hang Y, Dhawan S, Haliyur R, Bonatakis L, Avrahami D, Brissova M, Kaestner KH, Bhushan A, et al. (2019). Examining how the MAFB transcription factor affects islet β cell function postnatally. Diabetes 68, 337–348. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dadi PK, Luo B, Vierra NC, and Jacobson DA (2015). TASK-1 Potassium Channels Limit Pancreatic α-Cell Calcium Influx and Glucagon Secretion. Mol. Endocrinol 29, 777–787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dai C, Brissova M, Hang Y, Thompson C, Poffenberger G, Shostak A, Chen Z, Stein R, and Powers AC (2012). Islet-enriched gene expression and glucose-induced insulin secretion in human and mouse islets. Diabetologia 55, 707–718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fang L, Igarashi M, Leung J, Sugrue MM, Lee SW, and Aaronson SA (1999). p21Waf1/Cip1/Sdi1 induces permanent growth arrest with markers of replicative senescence in human tumor cells lacking functional p53. Oncogene 18, 2789–2797. [DOI] [PubMed] [Google Scholar]
- Fiaschi-Taesch NM, Salim F, Kleinberger J, Troxell R, Cozar-Castellano I, Selk K, Cherok E, Takane KK, Scott DK, and Stewart AF (2010). Induction of human β-cell proliferation and engraftment using a single G1/S regulatory molecule, cdk6. Diabetes 59, 1926–1936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Franceschi C, Garagnani P, Parini P, Giuliani C, and Santoro A (2018). Inflammaging: a new immune-metabolic viewpoint for age-related diseases. Nat. Rev. Endocrinol 14, 576–590. [DOI] [PubMed] [Google Scholar]
- Franklin I, Gromada J, Gjinovci A, Theander S, and Wollheim CB (2005). Beta-cell secretory products activate alpha-cell ATP-dependent potassium channels to inhibit glucagon release. Diabetes 54, 1808–1815. [DOI] [PubMed] [Google Scholar]
- Freund A, Laberge RM, Demaria M, and Campisi J (2012). Lamin B1 loss is a senescence-associated biomarker. Mol. Biol. Cell 23, 2066–2075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gale EA (2002). Can we change the course of beta-cell destruction in type 1 diabetes? N. Engl. J. Med 346, 1740–1742. [DOI] [PubMed] [Google Scholar]
- Gannon M, Kulkarni RN, Tse HM, and Mauvais-Jarvis F (2018). Sex differences underlying pancreatic islet biology and its dysfunction. Mol. Metab 15, 82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert JM, and Blum B (2018). Synaptotagmins Tweak Functional β Cell Maturation. Dev. Cell 45, 284–286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Golson ML, and Kaestner KH (2017). Epigenetics in formation, function, and failure of the endocrine pancreas. Mol. Metab 6, 1066–1076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorgoulis V, Adams PD, Alimonti A, Bennett DC, Bischof O, Bishop C, Campisi J, Collado M, Evangelou K, Ferbeyre G, et al. (2019). Cellular Senescence: Defining a Path Forward. Cell 179, 813–827. [DOI] [PubMed] [Google Scholar]
- Greenhill C (2019). Regulating metabolism and ageing - the role of PI3K. Nat. Rev. Endocrinol 15, 376–377. [DOI] [PubMed] [Google Scholar]
- Guo S, Burnette R, Zhao L, Vanderford NL, Poitout V, Hagman DK, Henderson E, Ozcan S, Wadzinski BE, and Stein R (2009). The stability and transactivation potential of the mammalian MafA transcription factor are regulated by serine 65 phosphorylation. J. Biol. Chem 284, 759–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Vanderford NL, and Stein R (2010). Phosphorylation within the MafA N terminus regulates C-terminal dimerization and DNA binding. J. Biol. Chem 285, 12655–12661. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guo S, Dai C, Guo M, Taylor B, Harmon JS, Sander M, Robertson RP, Powers AC, and Stein R (2013). Inactivation of specific β cell transcription factors in type 2 diabetes. J. Clin. Invest 123, 3305–3316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han SI, Aramata S, Yasuda K, and Kataoka K (2007). MafA stability in pancreatic β cells is regulated by glucose and is dependent on its constitutive phosphorylation at multiple sites by glycogen synthase kinase 3. Mol. Cell. Biol 27, 6593–6605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang Y, and Stein R (2011). MafA and MafB activity in pancreatic β cells. Trends Endocrinol. Metab 22, 364–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hang Y, Yamamoto T, Benninger RK, Brissova M, Guo M, Bush W, Piston DW, Powers AC, Magnuson M, Thurmond DC, and Stein R (2014). The MafA transcription factor becomes essential to islet β-cells soon after birth. Diabetes 63, 1994–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Harmon JS, Stein R, and Robertson RP (2005). Oxidative stress-mediated, post-translational loss of MafA protein as a contributing mechanism to loss of insulin gene expression in glucotoxic beta cells. J. Biol. Chem 280, 11107–11113. [DOI] [PubMed] [Google Scholar]
- Helman A, Klochendler A, Azazmeh N, Gabai Y, Horwitz E, Anzi S, Swisa A, Condiotti R, Granit RZ, Nevo Y, et al. (2016). p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat. Med 22, 412–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Henquin JC, Dufrane D, and Nenquin M (2006). Nutrient control of insulin secretion in isolated normal human islets. Diabetes 55, 3470–3477. [DOI] [PubMed] [Google Scholar]
- Herranz N, and Gil J (2018). Mechanisms and functions of cellular senescence. J. Clin. Invest 128, 1238–1246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iacovazzo D, Flanagan SE, Walker E, Quezado R, de Sousa Barros FA, Caswell R, Johnson MB, Wakeling M, Brändle M, Guo M, et al. (2018). MAFA missense mutation causes familial insulinomatosis and diabetes mellitus. Proc. Natl. Acad. Sci. USA 115, 1027–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobson DA, and Shyng SL (2020). Ion Channels of the Islets in Type 2 Diabetes. J. Mol. Biol 432, 1326–1346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kabir TD, Leigh RJ, Tasena H, Mellone M, Coletta RD, Parkinson EK, Prime SS, Thomas GJ, Paterson IC, Zhou D, et al. (2016). A miR-335/COX-2/PTEN axis regulates the secretory phenotype of senescent cancer-associated fibroblasts. Aging (Albany NY) 8, 1608–1635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalwat MA, and Cobb MH (2017). Mechanisms of the amplifying pathway of insulin secretion in the β cell. Pharmacol. Ther 179, 17–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurz DJ, Decary S, Hong Y, and Erusalimsky JD (2000). Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J. Cell Sci 113, 3613–3622. [DOI] [PubMed] [Google Scholar]
- Lecoin L, Rocques N, El-Yakoubi W, Ben Achour S, Larcher M, Pouponnot C, and Eychène A (2010). MafA transcription factor identifies the early ret-expressing sensory neurons. Dev. Neurobiol 70, 485–497. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luan C, Ye Y, Singh T, Barghouth M, Eliasson L, Artner I, Zhang E, and Renström E (2019). The calcium channel subunit gamma-4 is regulated by MafA and necessary for pancreatic beta-cell specification. Commun. Biol 2, 106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madsen OD, Jensen J, Petersen HV, Pedersen EE, Oster A, Andersen FG, Jørgensen MC, Jensen PB, Larsson LI, and Serup P (1997). Transcription factors contributing to the pancreatic beta-cell phenotype. Horm. Metab. Res 29, 265–270. [DOI] [PubMed] [Google Scholar]
- Mancini M, Saintigny G, Mahé C, Annicchiarico-Petruzzelli M, Melino G, and Candi E (2012). MicroRNA-152 and -181a participate in human dermal fibroblasts senescence acting on cell adhesion and remodeling of the extracellular matrix. Aging (Albany NY) 4, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin N, and Bernard D (2018). Calcium signaling and cellular senescence. Cell Calcium 70, 16–23. [DOI] [PubMed] [Google Scholar]
- Martinez MN, Emfinger CH, Overton M, Hill S, Ramaswamy TS, Cappel DA, Wu K, Fazio S, McDonald WH, Hachey DL, et al. (2012). Obesity and altered glucose metabolism impact HDL composition in CETP transgenic mice: a role for ovarian hormones. J. Lipid Res 53, 379–389. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Niceta M, Stellacci E, Gripp KW, Zampino G, Kousi M, Anselmi M, Traversa A, Ciolfi A, Stabley D, Bruselles A, et al. (2015). Mutations Impairing GSK3-Mediated MAF Phosphorylation Cause Cataract, Deafness, Intellectual Disability, Seizures, and a Down Syndrome-like Facies. Am. J. Hum. Genet 96, 816–825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan FC, and Wright C (2011). Pancreas organogenesis: from bud to plexus to gland. Dev. Dyn 240, 530–565. [DOI] [PubMed] [Google Scholar]
- Pan FC, Brissova M, Powers AC, Pfaff S, and Wright CV (2015). Inactivating the permanent neonatal diabetes gene Mnx1 switches insulin-producing β-cells to a δ-like fate and reveals a facultative proliferative capacity in aged β-cells. Development 142, 3637–3648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raum JC, Gerrish K, Artner I, Henderson E, Guo M, Sussel L, Schisler JC, Newgard CB, and Stein R (2006). FoxA2, Nkx2.2, and PDX-1 regulate islet beta-cell-specific mafA expression through conserved sequences located between base pairs -8118 and -7750 upstream from the transcription start site. Mol. Cell. Biol 26, 5735–5743. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raum JC, Hunter CS, Artner I, Henderson E, Guo M, Elghazi L, Sosa-Pineda B, Ogihara T, Mirmira RG, Sussel L, and Stein R (2010). Islet beta-cell-specific MafA transcription requires the 5′-flanking conserved region 3 control domain. Mol. Cell. Biol 30, 4234–4244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rocques N, Abou Zeid N, Sii-Felice K, Lecoin L, Felder-Schmittbuhl MP, Eychène A, and Pouponnot C (2007). GSK-3-mediated phosphorylation enhances Maf-transforming activity. Mol. Cell 28, 584–597. [DOI] [PubMed] [Google Scholar]
- Russell R, Carnese PP, Hennings TG, Walker EM, Russ HA, Liu JS, Giacometti S, Stein R, and Hebrok M (2020). Loss of the transcription factor MAFB limits β-cell derivation from human PSCs. Nat. Commun 11, 2742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sampathkumar NK, Bravo JI, Chen Y, Danthi PS, Donahue EK, Lai RW, Lu R, Randall LT, Vinson N, and Benayoun BA (2020). Widespread sex dimorphism in aging and age-related diseases. Hum. Genet 139, 333–356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scharfmann R, Pechberty S, Hazhouz Y, von Bülow M, Bricout-Neveu E, Grenier-Godard M, Guez F, Rachdi L, Lohmann M, Czernichow P, and Ravassard P (2014). Development of a conditionally immortalized human pancreatic β cell line. J. Clin. Invest 124, 2087–2098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schultz LB, Chehab NH, Malikzay A, and Halazonetis TD (2000). p53 binding protein 1 (53BP1) is an early participant in the cellular response to DNA double-strand breaks. J. Cell Biol 151, 1381–1390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scoville DW, Cyphert HA, Liao L, Xu J, Reynolds A, Guo S, and Stein R (2015). MLL3 and MLL4 methyltransferases bind to the MAFA and MAFB transcription factors to regulate islet β cell function. Diabetes 64, 3772–3783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shih HP, Wang A, and Sander M (2013). Pancreas organogenesis: from lineage determination to morphogenesis. Annu. Rev. Cell Dev. Biol 29, 81–105. [DOI] [PubMed] [Google Scholar]
- Thompson PJ, Shah A, Ntranos V, Van Gool F, Atkinson M, and Bhushan A (2019). Targeted elimination of senescent beta cells prevents type 1 diabetes. Cell Metab. 29, 1045–1060.e10. [DOI] [PubMed] [Google Scholar]
- Tyrberg B, Andersson A, and Borg LA (2001). Species differences in susceptibility of transplanted and cultured pancreatic islets to the beta-cell toxin alloxan. Gen. Comp. Endocrinol 122, 238–251. [DOI] [PubMed] [Google Scholar]
- van der Meulen T, Donaldson CJ, Cáceres E, Hunter AE, Cowing-Zitron C, Pound LD, Adams MW, Zembrzycki A, Grove KL, and Huising MO (2015). Urocortin3 mediates somatostatin-dependent negative feedback control of insulin secretion. Nat. Med 21, 769–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van Deursen JM (2014). The role of senescent cells in ageing. Nature 509, 439–446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Walker JT, Haliyur R, Nelson HA, Ishahak M, Poffenberger G, Aramandla R, Reihsmann C, Luchsinger JR, Saunders DC, Wang P, et al. (2020). Integrated human pseudoislet system and microfluidic platform demonstrate differences in GPCR signaling in islet cells. JCI Insight 5, e137017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C, Moriguchi T, Kajihara M, Esaki R, Harada A, Shimohata H, Oishi H, Hamada M, Morito N, Hasegawa K, et al. (2005). MafA is a key regulator of glucose-stimulated insulin secretion. Mol. Cell. Biol 25, 4969–4976. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplemental information. Bulk RNA-seq data have been deposited to GEO and are publicly available at the date of publication. Accession number is listed in the Key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this work paper is available from the Lead Contact upon request.
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Guinea pig anti-Insulin | Thermo Fisher | Cat#PA1 26938; RRID:AB_794668 |
| Mouse anti-Glucagon | Sigma | Cat#G2654; RRID:AB_259852 |
| Mouse anti-Ki67 | BD Biosciences | Cat#550609; RRID:AB_393778 |
| DAPI Fluoromount-G mounting medium | SouthernBiotech | Cat#0100-20 |
| Rabbit anti-MAFA | Cell Signaling | Cat#79737; RRID:AB_2799938 |
| Goat anti-PDX-1 | Chris Wright, Vanderbilt | N/A |
| Rabbit anti-UCN3 | Phoenix | Cat#H-019-028; RRID:AB_2889826 |
| Goat anti-P21 (for cells) | Santa Cruz | Cat#sc-397G; RRID:AB_632127 |
| Rat anti-P21 (for tissue sections) | Abcam | Cat#ab107099; RRID:AB_10891759 |
| Rabbit anti-53BP1 | Bethyl | Cat#A300-272A; RRID:AB_185520 |
| Rabbit anti-gH2AX | Abcam | Cat#ab81299; RRID:AB_1640564 |
| Rabbit anti-LaminB1 | Abcam | Cat#16048; RRID:AB_443298 |
| Bacterial and virus strains | ||
| Lentivirus: MAFAWT-pCDH-EF1-MCS-IRES-GopGFP | Modified for this paper from System Biosciences | Cat#CD521A-1 |
| Lentivirus: MAFAS64F-pCDH-EF1-MCS-IRES-GopGFP | Modified for this paper from System Biosciences | Cat#CD521A-1 |
| Chemicals, peptides, and recombinant proteins | ||
| Fura-2 AM | Life Technologies | Cat#F1221 |
| Critical commercial assays | ||
| Cell Death Detection Kit | Roche | Cat#11684795910 |
| Senescence β-Galactosidase Staining Kit | Cell Signaling | Cat#9860 |
| Glycogen Assay Kit | Cayman Chemicals | Cat#700480 |
| Deposited data | ||
| Raw and analyzed data | This paper | GEO:GSE183561 |
| Experimental models: Cell lines | ||
| Human: EndoC-βH2 Cells | Scharfmann et al., 2014 | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: MafAS64F/+ | This paper | N/A |
| Oligonucleotides | ||
| Primers for mouse and human see Table S1 | This paper | N/A |
| Software and algorithms | ||
| Genialis visual informatics platform: BBDuk, START aligner, featureCounts | https://www.genialis.com | N/A |
| DESeq2 | Love et al., 2014 | N/A |
| ImageJ | https://imagej.nih.gov/ij/ | N/A |
| ImageScope Software | Aperio Technologies | N/A |