

Abstract
The COVID-19 pandemic has shown itself to be an unprecedented challenge for vaccines which are widely recognized as the most important tool to exit this pandemic. We have witnessed vaccine scientists, developers, manufacturers, and stakeholders deliver several vaccines in just about a year. This is an unprecedented achievement in an environment that was not ready to manage such a global public health crisis. Indeed, the pandemic has highlighted some hurdles that need to be addressed in the system in order to streamline the regulatory processes and be in a situation where life-saving pharmaceutical solutions such as vaccines can be delivered quickly and equitably to people across the globe. More precisely, trade-offs had to be made between the need for regulatory flexibility in the requirements for manufacturing and controls to enable rapid availability of large volumes of vaccines vs the increased stringency and the lack of harmonization in the regulatory environment for vaccines globally. It is also characterized by a high heterogeneity in terms of review and approval processes, limiting equitable and timely access. We review and highlight the challenges relating to several topics, including process validation, comparability, stability, post-approval-changes, release testing, packaging, genetically modified organisms and variants. We see four areas for accelerating access to vaccines which provide solutions for the regulatory concerns, (1) science- and risk-based approaches, (2) global regulatory harmonization, (3) use of reliance, work-sharing, and recognition processes and (4) digitalization. These solutions are not new and have been previously highlighted. In recent months, we have seen some progress at the health authority level, but still much needs to be done. It is now time to reflect on the first lessons learnt from a devastating pandemic to ultimately ensure quick and wide access to medicines and vaccines for the citizens and patients.
Keywords: Regulatory convergence, Streamline, Vaccine, Reliance, Harmonization, Covid-19, Science-based, Risk-based, Digital
1. Introduction
History has shown that, after access to clean water, vaccines come next in improving the human life expectancy [1]. The importance of vaccines in leading a healthy life has been brought into the mainstream discussion by the current pandemic due to the SARS-CoV-2 virus.
Apart from the scientific challenge to develop safe and effective vaccines, the COVID-19 situation has exacerbated a number of hurdles which impact the timely supply of vaccines to all populations who need them. Vaccine developers and manufacturers, together with many stakeholders (regulatory bodies, academics, coalitions or consortiums, etc.) have engaged in discussions to find solutions to address those challenges.
This article is Part I of a two part discussion (see Fig. 1 ) and presents overarching solutions, focused on manufacturing and control, which can overcome a number of barriers to the timely access of vaccines.
Fig. 1.

How to accelerate the supply of vaccines to all populations worldwide – Part I and II.
2. Background and scope
Once a vaccine candidate has been identified and has proved to be safe and effective, vaccine manufacturers are rightfully expected to be able to manufacture it in sufficient quantities to supply all populations who need it in a timely and sustainable manner. This is particularly acute in the case of outbreaks which could become a pandemic. Over the past decades, the worldwide scientific, legal and regulatory environment has significantly evolved:
-
•
highly sophisticated scientific skills and tools have been developed, bringing huge improvements in the prediction and application of scientific knowledge, enabling therapeutic and preventive solutions to save millions of people from previously un-met medical needs
-
•
legislation across the world has increased considerably to provide people with more protection and prevent suffering from substandard products
-
•
the worldwide regulatory environment has become more stringent and demanding to constantly guarantee the Quality, Safety and Efficacy of ever more sophisticated drugs and vaccines
The current worldwide regulatory landscape is characterized by a high heterogeneity in terms of regulatory review, approval processes, release requirements, health authority batch testing, and approval timelines [2] which limits equitable and timely access to vaccines [3] . Much has also been advocated in the international arena to develop more regulatory harmonization, reliance and mutual recognition among agencies over the past decade. While some progress has been made with these aspects, harmonization is a very lofty goal, and we are still far from reaching worldwide convergence on requirements or reliance pathways which would enable all populations an equitable and timely access to all vaccines [4] .
In the last decades, prior to the COVID-19 pandemic, serious outbreaks have occurred, such as Ebola (1976–2020), SARS (2002), the H1N1 flu pandemic (2009), MERS (2012), Chikungunya (2014), Zika (2015), to name a few [5]. Those outbreaks triggered the scientific and regulatory communities to engage in discussions about how to be more prepared with vaccines to fight new outbreaks and from this the Coalition for Epidemic Preparedness Innovations (CEPI [6]) was started. In a related development, in 2018, an important joint EMA/FDA workshop was organized [7] in order to “discuss how regulators can better guide and support medicine developers in generating quality and manufacturing data packages in the context of development support programmes, such as the PRIority MEdicines scheme (PRIME) in the EU and Breakthrough Therapy designation program in the US“. The goal was to help patients to gain early access to therapies that target serious or life-threatening diseases or unmet medical needs.”
COVID-19 is the most recent and unprecedented pandemic situation, impacting the entire globe and hence posing significant challenges for vaccines and therapeutics supply. In addition, due to the broad and fast spread of the disease, new virus variants are emerging, requiring even faster reaction and further increasing the need for vaccines [8], [9]. The scientific community has strongly responded, resulting in the development and authorization of multiple vaccines within a year. In just a few months after approval, over 400 million [10] have been vaccinated. Bringing online such huge manufacturing capacity in a short timeframe has logically come with considerable “growth pains” within the industry. There are also lessons that can be applied to improving the routine ways of working and better ensuring consistent supply of vaccines and vaccination infrastructure for all diseases.
There is no doubt that a new medicine or vaccine can only be approved with the provision of sufficient manufacturing and control information for the assurance of quality, safety and efficacy. However, there are ways of generating and providing information or updates outside of the standard approaches based on science and risk-based principles. The acceptance of these new approaches by regulatory agencies globally should be encouraged, to ensure the significant acceleration needed to provide sustainable vaccine supplies for patients, especially considering the expected high volume of post-approval changes for these vaccines. The current COVID-19 vaccines have been developed in record time without compromising product quality which means that much more data (e.g. additional process validation data, optimized control strategies and manufacturing processes, additional batch sizes, manufacturing and testing sites, long term stability data), that would normally be available would be provided after initial emergency use authorizations have been granted. This additional information would be included in updated and complete dossiers for full authorization submissions.
The objective of this article is to provide a first analysis of the Vaccines Europe/International Federation of Pharmaceutical Manufacturers and Associations (VE/IFPMA) Task Force activities, supported by the Developing Countries Vaccine Manufacturing Network (DCVMN), to inform what improvements could or should be considered moving forward for preparing for the next pandemic, as well as continuously improving the supply of vaccines outside of outbreak or pandemic situations.
3. Methods
With the emergence of SARS-CoV-2 virus, a specific international organization was put into place for COVID-19 vaccines called the COVAX Facility. COVAX is the vaccines pillar of the ACT Accelerator, co-led by the CEPI, Gavi, the Vaccine Alliance, and the World Health Organization (WHO). Its goal is to enable access to safe and effective vaccines to the most vulnerable in all participating economies [11]. Within the COVAX organization, a Regulatory Advisory Group (RAG) was established, made up of regulators from 10 nations, available to provide feedback and regulatory guidance on COVID-19 vaccine development and activities. COVAX has also established multiple SWAT (Support Work to Advance Teams) teams: groups of experts focused on resolving technical issues and challenges common across all COVID-19 vaccine development projects to promote and accelerate vaccine development. The Manufacturing SWAT, with representatives from the VE/IFPMA Task Force, focuses on the following [11]:
-
•
“Regulatory strategy and the identification of manufacturing capacity for initial manufacture and increase in the supply of vaccines.
-
•
Supply chain strategy to include securing raw materials, mutually agreed labelling and alignment with COVAX partners.
-
•
Support for batch release assays (including potency assay requirements); mutual recognition of the process for timely national batch release; and support for additional analytical capacity”
To inform the Manufacturing SWAT, VE and IFPMA set up a task force to share multi-national vaccine manufacturers concerns and views and develop solutions. Several working groups provided proposals to assist the COVAX Facility RAG in providing “guidance for regulatory science challenges related to SWAT team activities, towards harmonization and streamlined processes where feasible.” [11]
4. Results
Eight main regulatory and technical concerns were outlined in the Manufacturing SWAT team discussions. These are provided in Table 1 including a high-level summary of the challenges.
Table 1.
Regulatory Impact.
| Topics of Concern | Challenges |
|---|---|
|
A traditional process validation approach for a new vaccine adds several additional months or even years to global technical development timelines. |
|
To make billions of doses, post-launch supply requires the use of multiple manufacturing sites (& concurrent expansion), and the need for many post-approval changes. Given the challenges associated with the COVID-19 emergency, comparability assessment of vaccines may be on the critical path for supply. Building strong, quality risk-based comparability strategies is key to support fast access to vaccines and sustainable lifecycle management. |
|
Stability is frequently on the critical path for drug substance and drug product development and commercial vaccine supply. Additionally, the rigid application of ICH Q5C [12] indications, like the core stability data package and requirements for real-time data, is not compatible with the accelerated vaccine development and industrial plan needed for urgent global supply of COVID-19 vaccines. |
|
Due to the accelerated development in combination with the overall complexity of vaccines, a significant number (i.e. dozens of changes times 150+ countries; resulting in thousands of variation dossiers) of post-approval changes are needed within a short period of time to reflect the maintenance and optimization of the manufacturing processes as well as expiry extensions, additional raw material sourcing, manufacturing and testing capacity to expand supply. As change is inevitable, innovative solutions such as mutual recognition and reliance are needed for this global problem. |
|
A normal vaccine batch is tested by the original releasing authority in the country where the vaccine is manufactured, then tested again in the receiving country or multiple receiving countries in independent laboratories not always using the same test method and applying the same specifications. Initial COVID-19 vaccine batches have limited expiry date. Therefore, redundant local testing in addition to the reference National Control Laboratory (NCL) reduces the remaining shelf life for a vaccine, potentially resulting in insufficient time to reach the patient or discarding expired doses. |
|
All aspects of packaging materials are regulated by National Regulatory Authorities (NRAs) as required by their laws. In this pandemic, two key imperatives are impacted: speed of introduction of new vaccines, and flexibility of world vaccine supply chains. Access is delayed due to packaging materials’ approval occurring after initial registration but prior to distribution in that country. Country-specific language and packaging requirements severely limits interchangeability of global vaccine supplies, resulting in small allotments that can only be used in one country. With vaccines being developed so quickly, it is likely that labeling and leaflet information will be subject to many changes as additional information becomes available (e.g. clinical data, stability, etc.). Regulatory change management will be challenging in the current system, further fracturing supply chains. |
|
In national authorities that require GMO dossiers, different positions between countries are often observed, particularly with regard to the BioSafety Level (BSL). This has an impact on conducting clinical trials, the transport of material between countries and on manufacturing decisions. |
|
The fast emergence of multiple variants to the parent strain leads to the need to react very quickly by developing, manufacturing, controlling and obtaining regulatory authorizations of the vaccines against variants as appropriate in a dramatically accelerated way. Most of the above challenges are also applicable for vaccines against the variants. |
Note: Further details are provided in the Part II article.
We generally see four over-arching areas for accelerating access to vaccines which can provide solutions for the regulatory concerns:
-
1.
Science- and Risk- based approaches
-
2.
Global regulatory convergence and harmonization
-
3.
Use of reliance, work-sharing, and recognition processes
-
4.
Digitalization
These can aid in streamlining the regulatory processes in future pandemics but could equally contribute to the increased efficiency of regulatory activities for vaccines in a regular setting. And indeed, familiarization with these processes in routine regulatory processes globally can only aid in the preparation for emergencies to come.
4.1. Science- and risk-based approaches
When a candidate vaccine targets an unmet medical need with the aim to protect patients as early as possible, for example in a pandemic situation, some flexibility on the provision and type of Chemistry, Manufacturing & Control (CMC) data packages in the context of regulatory submissions would clearly be beneficial. This would take into consideration the overall benefit/risk of the product, while never compromising on the quality of vaccine.
ICH Q9 [12] provides risk-based approaches to validation and NRA’s have developed their own requirements for the types of data required and timing for availability of such data. A common strategy across all NRA’s that recognizes appropriate and harmonized risk-based approaches would help ensure fast and equitable access to vaccines. Allowing risk recognition to define appropriate levels of validation for equipment, process, and analytical methods at the time of submission can allow vaccine manufacturers to manage some aspects within their pharmaceutical quality system [13] during vaccine development and accelerate access to patients. It would also allow the NRAs to receive data as post-approval commitments, for example concurrent validation, with drug product validation being a post-approval commitment, as suggested in recent (draft) guidance documents [14], [15].
A risk-based validation approach should also take prior knowledge into account [15]. This can be key in accelerated scenarios where there is limited manufacturing and available drug substance lots. For example:
-
•
Accelerate process validation using platform technology, use of lots manufactured prior to validation, and small scale lots
-
•
Use of Continuous Process Verification (CPV) to validate the process
-
•
Eliminate re-validation of aseptic and cleaning processes
-
•
Decouple drug substance and drug product validations
An illustration of a need for more flexibility, is the unprecedented scale at which vaccines for COVID-19 must be manufactured: many developers have had to use multiple drug substance (often two or more) and drug product (often three or more) sites simultaneously. These sites can be located around the globe, representing manufacturing on many continents and in numerous countries, highlighting the need for a common approach across different regulatory agencies and regional authorities overseeing these sites. Additionally, some sites may have been recently renovated to accommodate new unit operations or elements of the manufacturing process for the newly introduced COVID-19 vaccine. These changes impact both validation (discussed above) and comparability.
Risk-based analytical comparability assessment of manufacturing changes includes, for instance, focus on only critical quality attributes impacted by the change; matrixing and bracketing; degradation studies; and linking to pivotal clinical lots. In addition, product-specific kinetic models supported by prior knowledge should be considered. In this context “good modeling practices” should be ensured, combining advanced kinetics and statistical analysis of stability data obtained under accelerated and recommended storage conditions [16].
Regarding stability, it is acknowledged that post marketing commitments to provide full shelf-life data may be acceptable with appropriate justification. Yet, it is not clear to what extent the vaccine manufacturers will be allowed to leverage prior knowledge and scientific approaches to model or set the vaccine expiry date for the initial authorization and to submit confirmatory stability data generated on commercial batches as post approval commitments.
The WHO Good Regulatory Practices [17] supports that “regulatory oversight and regulatory decisions should be proportional to risk and the regulator’s capacity to implement and enforce” and that “the regulatory system should provide the flexibility to apply good judgement.” This is aligned with the stated objective of the ICH Q12 [12] guideline “A harmonized approach regarding technical and regulatory considerations for lifecycle management will benefit patients, industry, and regulatory agencies by promoting innovation and continual improvement in the biopharmaceutical sector, strengthening quality assurance and improving supply of medicinal products.”
The extent of operational and regulatory flexibility is subject to product and process understanding (ICH Q8 and Q11) [12], application of quality risk management principles (ICH Q9) [12], and an effective Pharmaceutical Quality System (PQS) (ICH Q10) [12], all enabled by appropriate knowledge management. The Parenteral Drug Association One Voice of Quality publication [18] outlines a standard approach for the steps necessary to establish and demonstrate an effective quality system to fully leverage the risk-based approach to post-approval changes. This can reduce regulatory requirements and corresponding administrative workload for low risk changes that can be handled by an effective PQS.
The benefits of a risk-based approach are focusing regulatory, quality and manufacturing resources on activities that may have a potential to impact product quality as it relates to safety and efficacy and eliminating delays of regulatory approvals. From a patient point of view, this would in fact represent a “benefit-based approach” by being able to combine high quality standards with timely supply.
4.2. Global regulatory convergence and harmonization
There is an urgent and critical need to develop a global regulatory framework based on the lessons learned from COVID-19 for future pandemics; a global regulatory framework that allows for the use of an exceptional approach to the development and provision of CMC data in support of the initial Marketing Authorization as well as the post-approval CMC changes (including comparability).
We need to ensure that such a global framework could be agreed upon through early and frequent dialogue with regulators globally. This framework will ensure that appropriate technical action can be taken in a timely manner by vaccine developers, allowing important vaccines get to patients as soon as possible. It is hoped that regulators globally can accept, further develop, and work with industry on the proposals from the EMA/FDA 2018 PRIME/BT Quality workshop, the principles of the EFPIA MAPPs CMC [19], and AAPS Pharm Sci CMC acceleration papers [20] for all potential Covid-19 vaccines. While this approach is applauded for scientific risk-based CMC and GMP approaches, the application of this thinking must be expanded in a truly global context to ensure early access for all global patients.
In addition, specific consideration needs to be given to the challenges posed by accelerated development for life cycle management contrasted with the known lack of a homogeneous process and assessment globally (which can lead to four years or longer for a global change to be approved). This will be critical for sustainable supply of vaccines in relation to the timely addition of manufacturing and testing capacity. Harmonized international regulatory requirements on PACs would avoid numerous country specific submissions which can cause inconsistencies and create delays in product availability.
There is a high risk that vaccines required to combat global pandemics, approved via an accelerated approval process, could experience shortages or discontinuities in supply to patients, as a result of difficulties in meeting standard regulatory life cycle management expectations. This global harmonization concept should include post approval changes that are needed as stability data becomes available, supply chains and processes evolve, to maintain availability, increase supply, and improve delivery of vaccines globally.
A further unforeseen complication that has arisen from manufacturing scale-up, to a volume required in a pandemic, has been the need to secure alternatives to basic manufacturing supplies. These include items such as reagents, single use devices, vials, and stoppers necessary to meet the manufacturing demand.
Alignment is needed for the technical dossier content as well as on reporting categories of PACs and mechanisms, data requirements, review and approval timelines, allowance for multiple sites to be registered at all stages, expectations with regards to implementation, and the use of regulatory tools such as Post-Approval Change Management Protocols (PACMP). The WHO Good Regulatory Practices [17] aims to provide ”a set of principles and practices that are applied to the development, implementation and maintenance of regulatory instruments – including laws, regulations and guidelines – in order to achieve a public health policy objective in the most efficient way”.
Current worldwide heterogeneity with regards to CMC requirements, pharmacopoeias, and regulatory and inspection processes represents the limiting factor in terms of having the maximum industrial flexibility to answer the demand. Lack of alignment consumes both significant resources and time to meet all specificities, without any added value for patients. Vaccine manufacturers have experienced this situation which resulted in a large number of questions asked by National Regulatory Authorities while the initial dossier and subsequent PACs have been scrutinized, reviewed and approved by Stringent Regulatory Authorities.
Global pharmacopeia harmonization is admittedly a heavy lift, though we have seen that agencies have been willing to be flexible on this in the initial vaccine reviews. In emergency situations, the use and recognition of one pharmacopoeia can allow for a harmonized approach for compendial standards. This could take the approach of utilizing one of the globally recognized pharmacopoeias used to develop the product and applying to all countries, thus not having to meet the needs of 40+ different pharmacopoeias. Such an action would benefit independent batch testing and facilitate reliance for vaccine Health Authority batch release.
The need to better harmonize regulatory requirements and processes is not a new concept and many discussions refer to the patient benefits of further harmonization (one such reference is the One Voice of Quality paper [18]). Worldwide harmonization is seen to be key to success of early and sustained availability of vaccines globally.
4.3. Use of reliance, work-sharing, and recognition processes
The implementation of reliance or recognition of approval from a Stringent Regulatory Authority (SRA) would benefit global patients by addressing supply constraints and potential vaccine shortages, particularly when emphasis is put on working towards accelerated approval timelines. There is a substantial opportunity for WHO and ICMRA to drive for a global reliance system, where there is an agreed reference agency with specific platform expertise.
Definition of Reliance (WHO Good Reliance Practices[21]): The act whereby the NRA in one jurisdiction may take into account and give significant weight to assessments performed by another NRA or trusted institution, or to any other authoritative information in reaching its own decision. The relying authority remains independent, responsible and accountable regarding the decisions taken, even when it relies on the decisions and information of others.
The potential exists for the global recognition of data for vaccines with the current legislative framework, by making use of the many reliance or recognition processes that already exist worldwide (including countries such as: Australia, Egypt, Jordan, Saudi Arabia, South Africa, and at least 14 countries in the LATAM region e.g. Brazil, Mexico, Argentina, Peru, as well as ACCESS [22] and EAEU recognition pathways and similar to the ORBIS workshare applied for Oncology products [23]. Another process that could be considered for applicability is the OPEN initiative with the EMA [24]. As applied to the review of COVID-19 vaccine, it could lead to alignment in understanding of the critical quality areas of the vaccine. This would allow the same data and product to be received by National Regulatory Authorities i.e. the ‘sameness’ of product being understood: one science, one product, one dossier.
For COVID-19 vaccines, our proposal is to utilize existing and/or creating new reliance procedures for regulatory approvals and post-approval changes, predicated on the acceptance of an approval from a reference authority. This includes agreeing to a set short timing (e.g. 15–30 working days) for approval.
It is key that manufacture and distribution of vaccines takes place under adequate Good Manufacturing and inspection practices. While it is recognized there may be specific country or regional differences, again applying the principle of ‘one science’, we must drive for rationalization of the assessment processes, which can often be rate determining for vaccine approval and therefore availability to patients. Acknowledgement and application of one assessment could be applied, for example via the PIC/S organization or mutual recognition of existing GMP inspections, so waiving the need for each (and many) individual country inspection [25].
We realize that worldwide reliance is a considerable challenge, in particular with regards to large extent of local regulations. However, we see that many agencies were able to quickly introduce emergency approval procedures in the COVID-19 pandemic, thus demonstrating there can be rapid agreement to new legislation to help bring vaccines to patients. Despite legal challenges, developing reliance mechanisms by leveraging the WHO “Good Reliance Practices in the regulation of medical products” [21], offers many advantages. It can lead to a more efficient way to streamline the initial licensing process and the subsequent PAC approvals, making the best use of scarce resources, preventing vaccine shortages and, most importantly, meeting the needs of global patients waiting for a vaccine. Even where legal processes make harmonization difficult, it must still be acknowledged that already considerable improvements can be made around the timeframes applied to approval and lifecycle management.
The point of sovereignty of review is often noted as a reason that global reliance cannot be achieved. But it must be noted that other industries, like aviation, have found a way through this in the interest of global citizens [4].
The need to apply reliance type procedures was agreed in Regulatory forums such as the CEPI/WHO RAG group, which includes key regulatory agencies. However, the reality is that many major agencies, with full access to the same data as approved by a reference regulatory authority, have generally chosen to also perform their own reviews due to many reasons (e.g. legal, sovereignty, population). This is observed by the many thousands of questions on CMC (Module 3 of the marketing authorization dossier) that are typically received, all relating to the same product and exact same data package and therefore time taken to gain approval and access to products in some countries. Even markets involved in the EMA OPEN initiative, while spring boarding off the review that included the questions and answers, still felt a need to ask further clarification.
This has been discussed at the CEPI RAG as well as at the joint ICMRA (International Coalition of Medicines Regulatory Authorities) – Industry workshop in July 2021 [26], and at the “Extraordinary ICDRA” (International Conference of Drug Regulatory Authorities) conference in September 2021 [27]. Both ICMRA and ICDRA conferences highlighted the pre-requisites needed for ensuring appropriate regulatory flexibility and the need to better develop reliance mechanisms respectively.
4.4. Digital levers
The COVID-19 pandemic situation has exacerbated the need for rapid exchange of information amongst all stakeholders, in particular between vaccine manufacturers and regulators, and between regulators themselves. Digital tools are a key enabler for this. A lot can still be explored and leveraged in that space, and initiatives such as the “Accumulus Synergy” are welcome[28]. “Accumulus Synergy” is “a global information exchange platform to transform how drug innovators and health regulators interact to bring safe and effective medicines to patients faster and more efficiently”. “The single-platform approach aims to improve speed, transparencies and efficiencies in the regulatory process by leveraging advanced technology and tools for data exchange”. While “Accumulus Synergy” is still in its early development phase, this provides great hope for the future development of global regulatory reliance mechanisms whereby all agencies could access at the exact same time the information and data provided by vaccine manufacturers, run regulatory worksharing activities, parallel review of dossiers, reliance- or recognition-based assessments, under a single and global management system.
A key component for global access is the use of e-labelling. The use and acceptance of a digital link to label information provides up to date information in multiple languages on additional clinical data, safety information, and extending expiry. This can allow manufacturers to distribute finished products swiftly to any market and avoid discards due to outdated information.
Digital tools also provide other huge possibilities such as, but not limited to, enabling quicker development of medicinal products, greater predictabilities, increased knowledge of products and processes, faster decision-making processes, better supply chain management, and regulatory oversight. The use of digital technologies should then be factored into the preparation of a next pandemic as well as in the “new normal” situations.
5. Discussion
A key lever to accelerate access to patients is by embracing the weight of clinical benefit versus CMC risk. Certain regulatory agencies have already recognized the approval of a Stringent Regulatory Authority (SRA), for example through the WHO’s Emergency use Listing procedure, Pre-qualification process or recognition. Furthermore, use of the collaborative approach [29] based on the SRA approval for review of initial authorizations and PACs, means that life-saving vaccines can get to patients in those countries sooner. If this approach is carried through to post approval setting, this acceleration will be sustained. It is also recognized that some agencies now see lack of a global approach as a real issue where we need to find a solution for patients. The EMA for example recognize this in their goals [30] and the FDA have a draft reflection paper on Enhancing Regulator and Manufacturer Agility through proposed harmonization of some key information Standards [31] which would enable reliance.
There are many opportunities to accelerate the regulatory process and provide quicker access to a global population. The themes of science/risk-based approaches, global harmonization, and reliance are widely applicable to the regulatory concerns for rapid access. These are highlighted in Table 2 .
Table 2.
Identification of Opportunities for Acceleration.
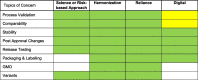 |
Note: Green – Direct Impact; Yellow – Indirect Impact.
As seen in the WHO Technical Brief: Regulation of COVID-19 Vaccines [13] there is general agreement on the principles outlined in the proposals above. However, there is concern that in many instances case-by-case discussions are still required between manufacturers and agencies [32].
As with other facets of global pandemic preparedness, a lack of unified global regulatory pathways adds unnecessary complexity and delays in product supply, thus preventing maximally agile supply chains and minimization of vaccine distribution delays. Regulations supporting agility and speed for pandemic vaccines should prioritize regulatory concerns prior to the next pandemic.
Although several safe, efficacious vaccines are now available in many countries, the need for further regulatory work has not gone away. We know there will be further rapid development and commercialization of new vaccines, for example, to meet the need of COVID-19 mutations, and likely that other future pandemic situations will arise. The complex, heterogeneous regulatory processes and pathways, with undefined common approval times still exist. Lack of global alignment and acceleration of approvals will continue to prevent sustainable global supply of COVID-19 vaccine for patients, as could be seen in the very real issue of component and excipient supply constraints for vaccines. The need therefore for further collaboration and agreement between Regulatory Agencies and also vitally partnering with Industry is key to ensure these future patient needs are met, through collaborative, pragmatic science and acceptance of ‘sameness’ of product: one patient centric science, one product, one review. An illustration of how agencies can move towards these solutions can be seen in Fig. 2 [21].
Fig. 2.

Progression of inter-agency collaboration.
These solutions should also be applied to the non-pandemic vaccines which also provide a necessary lifesaving function and would greatly benefit from a more streamlined global regulatory process.
6. Conclusion
The COVID-19 pandemic has shown itself to be an unprecedented situation for vaccines. Clearly the availability of vaccines is widely recognized as the most important element to exit this pandemic. At the same time, as often is the case with these kinds of catastrophic events, it has highlighted some hurdles that need to be addressed in the system and more precisely in the regulatory flow for manufacturing and controls. The collaborative review of the VE/IFPMA Task Force has identified potential solutions and regulatory flexibilities that can overcome these barriers and provide significant improvement for the benefit of all. The regulatory flexibilities described in this paper provides sustainable solutions to be better prepared to improve the supply of vaccines when the next pandemic occurs. These solutions and tools are not new, and several have been highlighted many times by the pharmaceutical industry before this pandemic started. Some of the lessons learned can also improve the ways of working in order to provide a consistent supply of vaccines under a routine mode. Quick and global access of medicines and vaccines to patients remains an important goal even outside of a pandemic.
Although we have seen some great progress in adopting some of the solutions, there is still work that must be done to provide the needed accelerated processes to continuously improve the timely supply of vaccines to all populations who need them. Taking incremental approaches as short term solutions should not undermine developing thoughts for more disruptive and longer term solutions that would enable equitable and timely access of vaccines to the worldwide population.
Declaration of Competing Interest
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Footnotes
Important note to the readers: This report summarizes and reflects the views of an international group of professionals, as discussed and elaborated in a given time point and context and does not necessarily represent the decisions or the stated policy of any institution or corporation. This work is supported by IFPMA, Vaccines Europe and DCVMN. There was no funding provided to support this work.
References
- 1.Plotkin S.L., Plotkin S.A. In: Vaccines. Plotkin S.A., Orenstein W.A., editors. WB Saunders; Philadelphia: 2004. A short history of vaccination; pp. 1–15. https://www.pnas.org/content/111/34/12283. [Google Scholar]
- 2.Nora Dellepiane, Sonia Pagliusi et al. Alignment in post-approval changes (PAC) guidelines in emerging countries may increase timely access to vaccines: An illustrative assessment by manufacturers. https://www.sciencedirect.com/science/article/pii/S259013622030022X?via%3Dihub [DOI] [PMC free article] [PubMed]
- 3.Nora Dellepiane, Sonia Pagliusi et al. Challenges for registration and opportunities to increase alignment of requirements in emerging countries, Vaccine 2018; 36 (24): 3389-3396. https://www.sciencedirect.com/science/article/pii/S0264410X18304055 [DOI] [PMC free article] [PubMed]
- 4.Gastineau T. What can vaccines learn from aviation. Vaccine. 2020;38(2020):5082–5084. doi: 10.1016/j.vaccine.2020.06.027. https://www.sciencedirect.com/science/article/pii/S0264410X20308008?via%3Dihub [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Excler J.-L., Saville M., Berkley S., Kim J.H. Vaccine development for emerging infectious diseases. Nat Med. 2021;27(4):591–600. doi: 10.1038/s41591-021-01301-0. [DOI] [PubMed] [Google Scholar]
- 6.Gouglas D., Christodoulou M., Plotkin S.A., Hatchett R. CEPI: driving progress toward epidemic preparedness and response. Epidemiol. Rev. 2019;41:28–33. doi: 10.1093/epirev/mxz012. https://pubmed.ncbi.nlm.nih.gov/31673694/ [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Stakeholder workshop on support to quality development in early access approaches, such as PRIME and Breakthrough Therapies; EMA 26/11/2018 https://www.ema.europa.eu/en/events/stakeholder-workshop-support-quality-development-early-access-approaches-such-prime-breakthrough
- 8.Callaway E., Ledford H. How to redesign COVID vaccines so they protect against variants. Nature. 2021;590(7844):15–16. doi: 10.1038/d41586-021-00241-6. [DOI] [PubMed] [Google Scholar]
- 9.COVAX workshop - Multivalent COVID vaccines to help address emergence of Variants: CMC and Clinical Implications; April 14, 2021 https://media.tghn.org/medialibrary/2021/04/20210414_Workshop_Clinical__Manufacturing_FINAL.pdf
- 10.Coronavirus (COVID-19) Vaccinations: Our World in Data, Project of the Global Change Data Lab (sited 9-Jun-2021). https://ourworldindata.org/covid-vaccinations
- 11.The COVAX Pillar; WHO Website (sited 15-Apr-2021) COVAX: The vaccines pillar of the access to COVID-19 tools (ACT) Accelerator (who.int)
- 12.ICH Quality Guidances; Website (sited 15-Apr-2021) https://www.ich.org/page/quality-guidelines
- 13.WHO Technical Brief: Regulation of COVID-19 Vaccines – Synopsis from the August to November 2020 COVAX RAG meetings, February 2021 https://www.who.int/publications/m/item/annex-1st-technical-brief-regulation-of-covid-19-vaccines
- 14.FDA Guidance for Industry – Development and Licensure of Vaccines to Prevent COVID-19, June 2020. https://www.fda.gov/regulatory-information/search-fda-guidance-documents/development-and-licensure-vaccines-prevent-covid-19
- 15.EMA Draft toolbox guidance on scientific elements and regulatory tools to support quality data packages for PRIME marketing authorisation applications, February 2021. https://www.ema.europa.eu/en/documents/scientific-guideline/draft-toolbox-guidance-scientific-elements-regulatory-tools-support-quality-data-packages-prime_en.pdf
- 16.Roque C, Ausar SF, Raham N, Clénet D. Chapter, PDA book “Quality by Design - An Indispensable Approach to Accelerate Biopharmaceutical Product Development”, ISBN number: 978-1-945584-22-0, 2021 Stability Modeling in QbD: Accelerating Formulation Development and Predicting Shelf Life of Products
- 17.Good regulatory practices in the regulation of medical products (Annex 11); WHO; pages 255-285 https://apps.who.int/iris/bitstream/handle/10665/340323/9789240020900-eng.pdf
- 18.PDA, Effective Management of Post-Approval Changes in the Pharmaceutical Quality System (PQS) - Through Enhanced Science and Risk-Based Approaches Industry One-Voice-of-Quality (1VQ) Solutions, 2020: https://journal.pda.org/content/early/2020/05/28/pdajpst.2020.011734. [DOI] [PubMed]
- 19.EFPIA-EBE White Paper on Expedited CMC Development: Accelerated Access for Medicines of Unmet Medical Need – CMC Challenges and Opportunities (Final Version – December 2017) https://www.efpia.eu/media/288657/efpia-ebe-white-paper-expedited-cmc-development-accelerated-access-for-medicines-of-unmet-medical-need-december-2017.pdf
- 20.Examining Manufacturing Readiness for Breakthrough Drug Development; AAPS. https://www.focr.org/sites/default/files/Final%20-%20Manufacturing%20Readiness%20Paper%20-%20AAPS.pdf [DOI] [PubMed]
- 21.Good reliance practices in the regulation of medical products: high-level principles and considerations (Annex 10); WHO; pages 287-322. TRS 1033 - 55th report of the WHO Expert Committee on Specifications for Pharmaceutical Preparations
- 22.Access Consortium, SwissMedic Website (Sited 15-Apr-2021). https://www.swissmedic.ch/swissmedic/en/home/about-us/international-collaboration/multilateral-co-operation-with-international-organisations---ini/multilateral-co-operation-with-international-organisations---ini.html
- 23.Project ORBIS, FDA; Website (sited 15-Apr-2021) https://www.fda.gov/about-fda/oncology-center-excellence/project-orbis
- 24.Questions and Answers on the Pilot Project 'OPEN'; EMA; 03-Feb-20201 Questions and Answers on the Pilot Project 'OPEN' (europa.eu)
- 25.IFPMA; 2020; Points to Consider for Virtual GMP Inspections – An Industry perspective; https://www.ifpma.org/wp-content/uploads/2020/07/Best-Practices-for-Virtual-Inspections_vF.pdf
- 26.ICMRA-Industry Virtual Workshop Report on Enabling Manufacturing Capacity in the COVID-19 Pandemic; 8 to 9 July 2021 (sited 10-Nov-21) https://icmra.info/drupal/sites/default/files/2021-10/covid-19_manufacturing_capacity_ws_report.pdf
- 27.Extraordinary International Conference of Drug Regulatory Authorities (ICDRA); 20 to 24 September 2021 (sited 10-Nov-21) https://www.who.int/teams/regulation-prequalification/regulation-and-safety/regulatory-convergence-networks/icdra
- 28.Accumulus Synergy; Website (sited 15-Apr-2021) https://www.accumulus.org/
- 29.WHO Annex 6, Good practices of national regulatory authorities in implementing the collaborative registration procedures for medical products https://www.who.int/medicines/areas/quality_safety/quality_assurance/WHO_TRS_1019_Annex6.pdf?ua=1
- 30.EMA Regulatory Science to 2025 – Strategic Reflection https://www.ema.europa.eu/en/documents/regulatory-procedural-guideline/ema-regulatory-science-2025-strategic-reflection_en.pdf
- 31.FDA Draft Reflection paper to ICH (Dec. 8, 2020) on “Enhancing Regulator and Manufacturer Agility by Harmonizing Key Identifiers and Other Information Standards Enabling Reliance”
- 32.WHO Regulatory Update on Covid-19, WHO Website; Regulation and Prequalification (Sited 15-Apr-2021). https://www.who.int/teams/regulation-prequalification/overview