

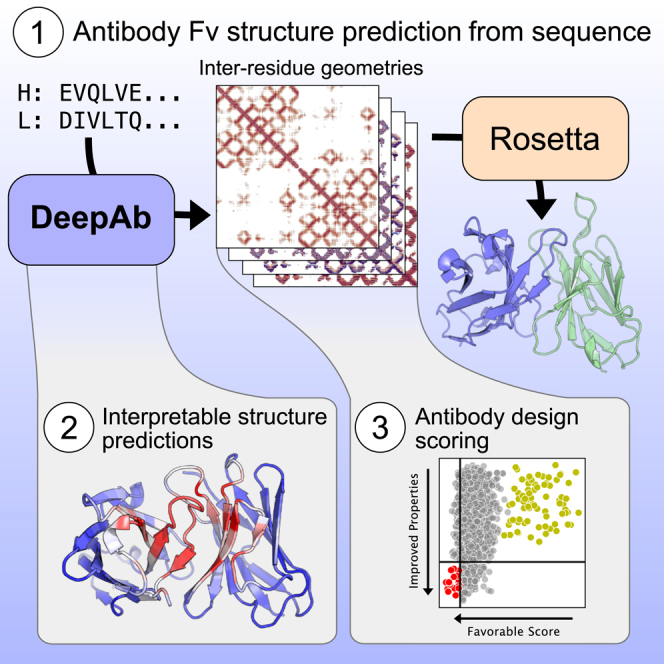
Summary
Therapeutic antibodies make up a rapidly growing segment of the biologics market. However, rational design of antibodies is hindered by reliance on experimental methods for determining antibody structures. Here, we present DeepAb, a deep learning method for predicting accurate antibody FV structures from sequence. We evaluate DeepAb on a set of structurally diverse, therapeutically relevant antibodies and find that our method consistently outperforms the leading alternatives. Previous deep learning methods have operated as “black boxes” and offered few insights into their predictions. By introducing a directly interpretable attention mechanism, we show our network attends to physically important residue pairs (e.g., proximal aromatics and key hydrogen bonding interactions). Finally, we present a novel mutant scoring metric derived from network confidence and show that for a particular antibody, all eight of the top-ranked mutations improve binding affinity. This model will be useful for a broad range of antibody prediction and design tasks.
Keywords: antibody design, deep learning, protein structure prediction, model interpretability
Graphical abstract
Highlights
-
•
DeepAb, a deep learning method for antibody structure, is presented
-
•
Structures from DeepAb are more accurate than alternatives
-
•
Outputs of DeepAb provide interpretable insights into structure predictions
-
•
DeepAb predictions should facilitate design of novel antibody therapeutics
The bigger picture
Accurate structure models are critical for understanding the properties of potential therapeutic antibodies. Conventional methods for protein structure determination require significant investments of time and resources and may fail. Although greatly improved, methods for general protein structure prediction still cannot consistently provide the accuracy necessary to understand or design antibodies. We present a deep learning method for antibody structure prediction and demonstrate improvement over alternatives on diverse, therapeutically relevant benchmarks. In addition to its improved accuracy, our method reveals interpretable outputs about specific amino acids and residue interactions that should facilitate design of novel therapeutic antibodies.
Accurate models of antibody structures are critical for the design of novel antibody therapeutics. We present DeepAb, a deep learning method for predicting antibody structure directly from amino acid sequence. When evaluated on benchmarks balanced for structural diversity and therapeutical relevance, DeepAb outperforms alternative methods. Finally, we dissect the interpretable elements of DeepAb to better understand the features contributing to its predictions and demonstrate how DeepAb could be applied to antibody design.
Introduction
The adaptive immune system of vertebrates is capable of mounting robust responses to a broad range of potential pathogens. Critical to this flexibility are antibodies, which are specialized to recognize a diverse set of molecular patterns with high affinity and specificity. This natural role in the defense against foreign particles makes antibodies an increasingly popular choice for therapeutic development.1,2 Presently, the design of therapeutic antibodies comes with significant barriers.1 For example, the rational design of antibody-antigen interactions often depends upon an accurate model of antibody structure. However, experimental methods for protein structure determination such as crystallography, NMR, and cryo-EM are low throughput and time consuming.
Antibody structure consists of two heavy and two light chains that assemble into a large Y-shaped complex. The crystallizable fragment (FC) region is involved in immune effector function and is highly conserved within isotypes. The variable fragment (FV) region is responsible for antigen binding through a set of six hypervariable loops that form a complementarity determining region (CDR). Structural modeling of the FV is critical for understanding the mechanism of antigen binding and for rational engineering of specific antibodies. Most methods for antibody FV structure prediction employ some form of grafting, by which pieces of previously solved FV structures with similar sequences are combined to form a predicted model.3, 4, 5, 6 Because much of the FV is structurally conserved, these techniques are typically able to produce models with an overall root-mean-square deviation (RMSD) less than 1 Å from the native structure. However, the length and conformational diversity of the third CDR loop of the heavy chain (CDR H3) make it difficult to identify high-quality templates. Further, the H3 loop’s position between the heavy and light chains makes it dependent on the chain orientation and multiple adjacent loops.7,8 Thus the CDR H3 loop presents a longstanding challenge for FV structure prediction methods.9
Machine learning methods have become increasing popular for protein structure prediction and design problems.10 Specific to antibodies11, machine learning has been applied to predict developability12, improve humanization13, generate sequence libraries14, and predict antigen interactions.15,16 In this work, we build on advances in general protein structure prediction17, 18, 19 to predict antibody FV structures. Our method consists of a deep neural network for predicting inter-residue distances and orientations and a Rosetta-based protocol for generating structures from network predictions. We show that deep learning approaches can predict more accurate structures than grafting-based alternatives, particularly for the challenging CDR H3 loop. The network used in this work is designed to be directly interpretable, providing insights that could assist in structural understanding or antibody engineering efforts. We conclude by demonstrating the ability of our network to distinguish mutational variants with improved binding using a prediction confidence metric. To facilitate further studies, all the code for this work, as well as pre-trained models, are provided.
Results
Overview of the method
Our method for antibody structure prediction, DeepAb, consists of two main stages (Figure 1). The first stage is a deep residual convolutional network that predicts FV structure, represented as relative distances and orientations between pairs of residues. The network requires only heavy and light chain sequences as input and is designed with interpretable components to provide insight into model predictions. The second stage is a fast Rosetta-based protocol for structure realization using the predictions from the network.
Figure 1.

Diagram of DeepAb method for antibody structure prediction
Starting from heavy and light chain sequences, the network predicts a set of inter-residue geometries describing the FV structure. Predictions are used for guided structure realization with Rosetta. Two interpretable components of the network are highlighted: a pre-trained antibody sequence model and output attention mechanisms.
Predicting inter-residue geometries from sequence
Due to the limited number of FV crystal structures available for supervised learning, we sought to make use of the abundant immunoglobin sequences from repertoire sequencing studies.20 We leveraged the power of unsupervised representation learning to embed general patterns from immunoglobin sequences that are not evident in the small subset with known structures into a latent representation. Although transformer models have become increasingly popular for unsupervised learning on protein sequences21, 22, 23, 24, we chose a recurrent neural network (RNN) model for ease of training on the limited data available. The fixed-size hidden state of RNNs forms an explicit information bottleneck ideal for representation learning. In the recent UniRep method, RNNs were demonstrated to learn rich feature representations from protein sequences when trained on next-amino-acid prediction.25 For our purposes, we developed an RNN encoder-decoder model26; the encoder is a bidirectional long short-term memory (biLSTM) and the decoder is a long short-term memory (LSTM).27 Briefly, the encoder learns to summarize an input sequence residue-by-residue into a fixed-size hidden state. This hidden state is transformed into a summary vector and passed to the decoder, which learns to reconstruct the original sequence one residue at a time. The model is trained using cross-entropy loss on a set of 118,386 paired heavy and light chain sequences from the Observed Antibody Space (OAS) database.28 After training the network, we generated embeddings for antibody sequences by concatenating the encoder hidden states for each residue. These embeddings are used as features for the structure prediction model described below.
The choice of protein structure representation is critical for structure prediction methods.10 We represent the FV structure as a set of inter-residue distances and orientations, similar to previous methods for general protein structure prediction.18,19 Specifically, we predict inter-residue distances between three pairs of atoms (Cα—Cα, Cβ—Cβ, N—O) and the set of inter-residue dihedrals (ω: Cα—Cβ—Cβ—Cα, θ: N—Cα—Cβ—Cβ) and planar angles (φ: Cα—Cβ—Cβ) first described by Yang et al.18 and shown in their Figure 1. Each output geometry is discretized into 36 bins, with an additional bin indicating distant residue pairs . All distances are predicted in the range of 0–18 Å, with a bin width of 0.5 Å. Dihedral and planar angles are discretized uniformly into bins of 10° and 5°, respectively.
The general architecture of the structure prediction network is similar to our previous method for CDR H3 loop structure prediction29, with two notable additions: embeddings from the pre-trained language model and interpretable attention layers (Figure 1). The network takes as input the concatenated heavy and light chain sequences. The concatenated sequence is one-hot encoded and passed through two parallel branches: a 1D ResNet and the pre-trained language model. The outputs of the branches are combined and transformed into pairwise data. The pairwise data pass through a deep 2D ResNet that constitutes the main component of the predictive network. Following the 2D ResNet, the network separates into six output branches, corresponding to each type of geometric measurement. Each output branch includes a recurrent criss-cross attention module, allowing each residue pair in the output to aggregate information from all other residue pairs. The attention layers provide interpretability that is often missing from protein structure prediction models.
We opted to train with focal loss30 rather than cross-entropy loss to improve the calibration of model predictions, as models trained with cross-entropy loss have been demonstrated to overestimate the likelihood of their predicted labels.31 We pay special attention to model calibration as later in this work we attempt to distinguish between potential antibody variants on the basis of prediction confidence, which requires greater calibration. The model is trained on a nonredundant (at 99% sequence identity) set of 1,692 FV structures from the Structural Antibody Database (SAbDab).32 The pretrained language model, used as a feature extractor, is not updated while training the predictor network.
Structure realization
Similar to previous methods for general protein structure prediction17, 18, 19, we used constrained minimization to generate full 3D structures from network predictions. Unlike previous methods, which typically begin with some form of φ−ψ torsion sampling, we created initial models via multi-dimensional scaling (MDS). We opted to build initial structures through MDS, rather than torsion sampling, due to the high conservation of the framework structural regions. Through MDS, we can obtain accurate 3D coordinates for the conserved framework residues, thus bypassing costly sampling for much of the antibody structure.33 As a reminder, the relative positions of all backbone atoms are fully specified by the predicted L × L inter-residue , ω, θ, and φ geometries. Using the modal-predicted output bins for these four geometries, we construct a distance matrix between backbone atoms. From this distance matrix, MDS produces an initial set of 3D coordinates that are subsequently refined through constrained minimization.
Network predictions for each output geometry were converted to energetic potentials by negating the raw model logits (i.e., without softmax activation). These discrete potentials were converted to continuous constraints using a cubic spline function. Starting from the MDS model, the constraints are used to guide quasi-Newton minimization (L-BFGS) within Rosetta.34,35 First, the constraints are jointly optimized with a simplified Rosetta centroid energy function to produce a coarse-grained FV structure with the sidechains represented as a single atom. Next, constrained full-atom relaxation was used to introduce sidechains and remove clashes. After relaxation, the structure was minimized again with constraints and the Rosetta full-atom energy function (ref2015). This optimization procedure was repeated to produce 50 structures, and the lowest energy structure was selected as the final model. Although we opted to produce 50 candidate structures, five should be sufficient in practice due to the high convergence of the protocol (Figure S1). Five candidate structures can typically be predicted in 10 min on a standard CPU, making our method slower than grafting-only approaches (seconds to minutes per sequence), but significantly faster than extensive loop sampling (hours per sequence).
Benchmarking methods for FV structure prediction
To evaluate the performance of our method, we chose two independent test sets. The first is the RosettaAntibody benchmark set (47 targets), which has previously been used to evaluate antibody structure prediction methods.8,29,36 The second is a set of clinical-stage therapeutic antibodies (45 targets), which was previously assembled to study antibody developability.37 Taken together, these sets represent a structurally diverse, therapeutically relevant benchmark for comparing antibody FV structure prediction methods.
Deep learning outperforms grafting methods
Although our method bears resemblance to deep learning methods for general protein structure prediction, we opted to compare to antibody-specific methods as we have previously found general methods to not yet be capable of producing high-quality structures of the challenging CDR loops.29 Instead, we compared the performance of our method on the RosettaAntibody benchmark and therapeutic benchmark to three antibody-specific alternative methods: RosettaAntibody-G4,6, RepertoireBuilder5, and ABodyBuilder.3 Each of these methods is based on a grafting approach, by which complete FV structures are assembled from sequence-similar fragments of previously solved structures. To produce the fairest comparison, we excluded structures with greater than 99% sequence identity for the whole FV from use for grafting (similar to our training data set). We evaluated each method according to the backbone heavy-atom RMSD of the CDR loops and the framework regions of both chains. We also measured the orientational coordinate distance (OCD)8, a metric for heavy-light chain orientation accuracy. OCD is calculated as the sum of the deviations from native of four orientation coordinates (packing angle, interdomain distance, heavy-opening angle, light-opening angle) divided by the standard deviation of each coordinate.8 The results of the benchmark are summarized in Table 1 and fully detailed in Tables S1–S8.
Table 1.
Performance of FV structure prediction methods on benchmarks
| Method | OCD | H Fr (Å) | H1 (Å) | H2 (Å) | H3 (Å) | L Fr (Å) | L1 (Å) | L2 (Å) | L3 (Å) |
|---|---|---|---|---|---|---|---|---|---|
| RosettaAntibody benchmark | |||||||||
| RosettaAntibody-G | 5.19 | 0.57 | 1.22 | 1.14 | 3.48 | 0.67 | 0.80 | 0.87 | 1.06 |
| RepertoireBuilder | 5.26 | 0.58 | 0.86 | 1.00 | 2.94 | 0.51 | 0.63 | 0.52 | 1.03 |
| ABodyBuilder | 4.69 | 0.50 | 0.99 | 0.88 | 2.94 | 0.49 | 0.72 | 0.52 | 1.09 |
| DeepAb | 3.67 | 0.43 | 0.72 | 0.85 | 2.33 | 0.42 | 0.55 | 0.45 | 0.86 |
| Therapeutic benchmark | |||||||||
| RosettaAntibody-G | 5.43 | 0.63 | 1.42 | 1.05 | 3.77 | 0.55 | 0.89 | 0.83 | 1.48 |
| RepertoireBuilder | 4.37 | 0.62 | 0.91 | 0.96 | 3.13 | 0.47 | 0.71 | 0.52 | 1.08 |
| ABodyBuilder | 4.37 | 0.49 | 1.05 | 1.02 | 3.00 | 0.45 | 1.04 | 0.50 | 1.35 |
| DeepAb | 3.52 | 0.40 | 0.77 | 0.68 | 2.52 | 0.37 | 0.60 | 0.42 | 1.02 |
Orientational coordinate distance (OCD) is a unitless quantity calculated by measuring the deviation from native of four heavy-light chain coordinates.8 Heavy chain framework (H Fr) and light chain framework (L Fr) RMSDs are measured after superimposing the heavy and light chains, respectively. CDR loop RMSDs are measured using the Chothia loop definitions after superimposing the framework region of the corresponding chain. All RMSDs are measured over backbone heavy atoms.
Our deep learning method showed improvement over all grafting-based methods on every metric considered. On both benchmarks, the structures predicted by our method achieved an average OCD less than 4, indicating that predicted structures were typically within one standard deviation of the native structure for each of the orientational coordinates. All of the methods predicted with sub-Angstrom accuracy on the heavy and light chain framework regions, which are highly conserved. Still, our method achieved average RMSD improvements of 14%–18% for the heavy chain framework and 16%–17% for light chain framework over the next best methods on the benchmarks. We also observed consistent improvement over grafting methods for CDR loop structure prediction.
Comparison of CDR H3 loop modeling accuracy
The most significant improvements by our method were observed for the CDR H3 loop (Figure 2A). On the RosettaAntibody benchmark, our method predicted H3 loop structures with an average RMSD of 2.33 Å (±1.32 Å), a 22% improvement over the next best method. On the therapeutic benchmark, our method had an average H3 loop RMSD of 2.52 Å (±1.50 Å), a 16% improvement over the next best method. The difficulty of predicting CDR H3 loop structures is due in part to the wide range of observed loop lengths. To understand the impact of H3 loop length on our method’s performance, we compared the average RMSD for each loop length across both benchmarks (Figure 2B). In general, all of the methods displayed degraded performance with increasing H3 loop length. However, DeepAb typically produced the most accurate models for each loop length.
Figure 2.

Comparison of CDR H3 loop structure prediction accuracy
(A) Average RMSD of H3 loops predicted by RosettaAntibody-G (RAb), RepertoireBuilder (RB), ABodyBuilder (ABB), and DeepAb on the two benchmarks. Error bars show standard deviations for each method on each benchmark.
(B) Average RMSD of H3 loops by length for all benchmark targets. Error bars show standard deviations for loop lengths corresponding to more than one target.
(C) Direct comparison of DeepAb and alternative methods H3 loop RMSDs, with diagonal band indicating predictions that were within ±0.25 Å.
(D) Comparison of native rituximab H3 loop structure (white, PDB: 3PP3) to predictions from DeepAb (green, 2.1 Å RMSD) and alternative methods (blue, 3.3–4.1 Å RMSD).
(E) Comparison of native sonepcizumab H3 loop structure (white, PDB: 3I9G) to predictions from DeepAb (green, 1.8 Å RMSD) and alternative methods (blue, 2.9–3.9 Å RMSD).
We also examined the performance of each method on individual benchmark targets. In Figure 2C, we plot the CDR H3 loop RMSD of our method versus that of the alternative methods. Predictions with an RMSD difference less than 0.25 Å (indicated by diagonal bands) were considered equivalent in quality. When compared to RosettaAntibody-G, RepertoireBuilder, and ABodyBuilder, our method predicted more/less accurate H3 loop structures for 64/17, 59/16, and 53/22 out of 92 targets, respectively. Remarkably, our method was able to predict nearly half of the H3 loop structures (42 of 92) to within 2 Å RMSD. RosettaAntibody-G, RepertoireBuilder, and ABodyBuilder achieved RMSDs of 2 Å or better on 26, 23, and 26 targets, respectively.
Accurate prediction of challenging, therapeutically relevant targets
To underscore and illustrate the improvements achieved by our method, we highlight two examples from the benchmark sets. The first is rituximab, an anti-CD20 antibody from the therapeutic benchmark (PDB: 3PP3).38 In Figure 2D, the native structure of the 12-residue rituximab H3 loop (white) is compared to our method’s prediction (green, 2.1 Å RMSD) and the predictions from the grafting methods (blue, 3.3–4.1 Å RMSD). The prediction from our method captures the general topology of the loop well, even placing many of the side chains near the native. The second example is sonepcizumab, an anti-sphingosine-1-phosphate antibody from the RosettaAntibody benchmark (PDB: 3I9G).39 In Figure 2E, the native structure of the 12-residue H3 loop (white) is compared to our method’s prediction (green, 1.8 Å) and the predictions from the grafting methods (blue, 2.9–3.9 Å). Again, our method captures the overall shape of the loop well, enabling accurate placement of several side chains. Interestingly, the primary source of error by our method in both cases is a tryptophan residue (around position H100) facing in the incorrect direction.
Impact of architecture on H3 loop modeling accuracy
The model presented in this work includes two primary additions over previous work for predicting H3 loop structures29: pre-trained LSTM sequence embeddings and criss-cross attention over output branches. To better understand the impact of each of these enhancements, we trained two additional model ensembles following the same procedure as described for the full model. The first model acts as a baseline, without LSTM features or criss-cross attention, and the second introduces the LSTM features. We made predictions for each of the 92 benchmark targets and compared the H3 loop modeling performance of these models to the full model (Figure S2A). The baseline model achieved an average H3 loop RMSD of 2.71 Å, outperforming grafting-based methods. Addition of the LSTM features yielded a moderate improvement in H3 accuracy (∼0.1 Å RMSD), while addition of criss-cross attention provided a slightly larger improvement (∼0.2 Å RMSD). We also analyzed the H3 loop lengths of each target while comparing the ablation models (Figure S2B) and found that improvements were relatively consistent across lengths.
Interpretability of model predictions
Despite the popularity of deep learning approaches for protein structure prediction, little attention has been paid to model interpretability. Interpretable models offer utility beyond their primary predictive task.40,41 The network used in this work was designed to be directly interpretable and should be useful for structural understanding and antibody engineering.
Output attention tracks model focus
Each output branch in the network includes a criss-cross attention module42, similar to the axial attention used in other protein applications.24,43,44 We have selected the criss-cross attention in order to efficiently aggregate information over a 2D grid (e.g., pairwise distance and orientation matrices). The criss-cross attention operation allows the network to attend across output rows and columns when predicting for each residue pair (as illustrated in Figure 3A). Through the attention layer, we create a matrix (where L is the total number of residues in the heavy and light chain Fv domains) containing the total attention between each pair of residues (see experimental procedures). To illustrate the interpretative power of network attention, we considered an anti-peptide antibody (PDB: 4H0H) from the RosettaAntibody benchmark set. Our method performed well on this example (H3 RMSD = 1.2 Å), so we expected it would provide insights into the types of interactions that the network captures well. We collected the attention matrix for predictions and averaged over the residues belonging to each CDR loop to determine which residues the network focuses on while predicting each loop’s structure (Figure 3B). As expected, the network primarily attends to residues surrounding each loop of interest. For the CDR1-2 loops, the network attends to the residues in the neighborhood of the loop, with little attention paid to the opposite chain. For the CDR3 loops, the network attends more broadly across the heavy-light chain interface, reflecting the interdependence between the loop conformations and the overall orientation of the chains.
Figure 3.

Interpretability of model components
(A) Diagram of attention mechanism (with attention matrix A and value matrix V) and example H3 loop attention matrix, with attention on other loops indicated. Attention values increase from blue to red.
(B) Model attention over FV structure while predicting each CDR loop for an anti-peptide antibody (PDB: 4H0H).
(C) Key interactions for H3 loop structure prediction identified by attention. The top five non-H3 attended residues (H32-Y, L32-Y, L49-Y, L55-F, and L91-S) are labeled, as well as an H3 residue participating in a hydrogen bond (H100-S).
(D) Two-dimensional t-SNE projection of sequence-averaged LSTM embeddings labeled by source species.
(E) Two-dimensional t-SNE projects of LSTM embeddings averaged over CDR1 loop residues labeled by loop structural clusters.
To better understand what types of interactions the network considers, we examined the residues assigned high attention while predicting the H3 loop structure (Figure 3C). Within the H3 loop, we found that the highest attention was on the residues forming the C-terminal kink. This structural feature has previously been hypothesized to contribute to H3 loop conformational diversity45, and it is likely critical for correctly predicting the overall loop structure. Of the five non-H3 residues with the highest attention, we found that one was a phenylalanine and three were tyrosines. The coordination of these bulky side chains appears to play a significant role in the predicted H3 loop conformation. The fifth residue was a serine from the L3 loop (residue L91) that forms a hydrogen bond with a serine of the H3 loop (residue H100), suggesting some consideration by the model of biophysical interactions between neighboring residues. To understand how the model attention varies across different H3 loops and neighboring residues, we performed a similar analysis for the 47 targets of the RosettaAntibody benchmark (Figure S3). Although some neighboring residues were consistently attended to, we observed noticeable changes in attention patterns across the targets (Figure S4), demonstrating the sensitivity of the attention mechanism for identifying key interactions for a broad range of structures.
Repertoire sequence model learns evolutionary and structural representations
To better understand what properties of antibodies are accessible through unsupervised learning, we interrogated the representation learned by the LSTM encoder, which was trained only on sequences. First, we passed the entire set of paired heavy and light chain sequences from the OAS database through the network to generate embeddings like those used for the structure prediction model. The variable-length embedding for each sequence was averaged over its length to generate a fixed-size vector describing the entire sequence. We projected the vector embedding for each sequence into two dimensions via t-distributed stochastic neighbor embedding (t-SNE)46 and found that the sequences were naturally clustered by species (Figure 3D). Because the structural data set is predominately composed of human and murine antibodies, the unsupervised features are likely providing evolutionary context that is otherwise unavailable.
The five non-H3 CDR loops typically adopt one of several canonical conformations.47,48 Previous studies have identified distinct structural clusters for these loops and described each cluster by a characteristic sequence signature.49 We hypothesized that our unsupervised learning model should detect these sequence signatures and thus encode information about the corresponding structural clusters. Similar to before, we created fixed-size embedding vectors for the five non-H3 loops by averaging the whole-sequence embedding over the residues of each loop (according to Chothia definitions47). In Figure 3E, we show t-SNE embeddings for the CDR1 loops labeled by their structural clusters from PyIgClassify.49 These loops are highlighted because they have the most uniform class balance among structural clusters; similar plots for the remaining loops are provided in Figure S5. We observed clustering of labels for both CDR1 loops, indicating that the unsupervised model has captured some structural features of antibodies through sequence alone.
Applicability to antibody design
Moving toward the goal of antibody design, we sought to test our method’s ability to distinguish between beneficial and disruptive mutations. First, we gathered a previously published deep mutational scanning (DMS) data set for an anti-lysozyme antibody.50 Anti-lysozyme was an ideal subject for evaluating our network’s design capabilities, as it was part of the benchmark set and thus already excluded from training. In the DMS data set, anti-lysozyme was subjected to mutational scanning at 135 positions across the FV, including the CDR loops and the heavy-light chain interface. Each variant was transformed into yeast and measured for binding enrichment over the wild type.
Prediction confidence is indicative of mutation tolerability
We explored two strategies for evaluating mutations with our network. First, we measured the change in the network’s structure prediction confidence for a variant sequence relative to the wild type (visualized in Figure 4A) as a change in categorical cross-entropy:
where seqWt and seqvar are the wild type and variant sequences, respectively, and the conditional probability term describes the probability of a particular geometric output given seqWt or seqvar. Only residue pairs ij with predicted were used in the calculation. Second, we used the LSTM decoder described previously to calculate the negative log likelihood of a particular point mutation given the wild type sequence, termed dLSTM:
where seqvar is a variant sequence with a point mutation to aa at position i, and zWt is the biLSTM encoder summary vector for the wild type sequence. To evaluate the discriminative power of the two metrics, we calculated ΔCCE and dLSTM for each variant in the anti-lysozyme data set. We additionally calculated a combined metric as ΔCCE + 0.01 × dLSTM, roughly equating the magnitudes of both terms, and compared to the experimental binding data (Figure 4B). Despite having no explicit knowledge of the antigen, the network was moderately predictive of experimental binding enrichment (Figure 4C). The most successful predictions (true positives in Figure 4B) were primarily for mutations in CDR loop residues (Figure 4D). This is not surprising, given that our network has observed the most diversity in these hypervariable regions and is likely less calibrated to variance among framework residues. Nevertheless, if the ΔCCE + 0.01 × dLSTM were for ranking, all the top-8 and 22 of the top-100 single-point mutants identified would have experimental binding enrichments above the wild type (Figure 4E).
Figure 4.

Prediction of mutational effects with DeepAb model
(A) Diagram of ΔCCE calculation for model output predictions for an arbitrary residue pair. Plots show the change in probability density of the predicted geometries for the residue pair after making a mutation.
(B) Plot of the combined network metric against experimental binding enrichment over wild type, with negative values corresponding to beneficial mutations for both axes. True positive predictions (red) and mutations to wild type cysteines (yellow) are highlighted.
(C) Receiver operating characteristic for predicting experimental binding enrichment over wild type with the combined network metric and each component metric. Area under the curve (AUC) values are provided for each metric.
(D) Position of true positive predictions on anti-lysozyme FV structure.
(E) Positive predictive value for mutants ranked by the combined metric.
(F) Comparison of ΔCCE for a designed eight-point variant (D44.1des, red) to sequences with random mutations at the same positions.
Network distinguishes stability-enhanced designs
The anti-lysozyme DMS data set was originally assembled to identify residues for design of multi-point variants.50 The authors designed an anti-lysozyme variant with eight mutations, called D44.1des, that displayed improved thermal stability and nearly 10-fold increase in affinity. To determine whether our network could recognize the cumulative benefits of multiple mutations, we created a set of variants with random mutations at the same positions. We calculated ΔCCE for D44.1des and the random variants and found that the model successfully distinguished the design (Figure 4F). We found similar success at distinguishing enhanced multi-point variants for other targets from the same publication (Figure S6), suggesting that our approach will be a useful screening step for a broad range of antibody design tasks. Despite being trained only for structure prediction, these results suggest that our model may be a useful tool for screening or ranking candidates in antibody design pipelines.
Discussion
The results presented in this work build on advances in general protein structure prediction to effectively predict antibody FV structures. We found that our deep learning method consistently produced more accurate structures than grafting-based alternatives on benchmarks of challenging, therapeutically relevant targets. Although we focused on prediction of FV structures, our method is also capable of modeling single-chain nanobodies (Figure S7). In these limited cases, the framework RMSD and several of the CDR1 and CDR2 loops are predicted with sub-Angstrom accuracy. However, we observe that the CDR3 predictions tend to resemble antibody FV CDR H3 loops, indicating that there may be value in training models specifically for nanobody structure prediction.
As deep learning methods continue to improve, model interpretability will become increasingly important to ensure practitioners can gain insights beyond the primary predictive results. In addition to producing accurate structures, our method also provides interpretable insights into its predictions. Through the attention mechanism, we can track the network’s focus while predicting FV structures. We demonstrated interpretation of predictions for a CDR H3 loop and identified several interactions with neighboring residues that the model deemed important for structure. In the future, similar insights could be used within antibody engineering workflows to prevent disruption of key interactions, reducing the need for time-consuming human analysis and focusing antibody library design.
As part of this work, we developed an unsupervised representation model for antibody sequences. We found that critical features of antibody structure, including non-H3 loop clusters, were accessible through a simple LSTM encoder-decoder model. While we limited training to known pairs of heavy and light chains, several orders of magnitude more unpaired immunoglobins have been identified through next-generation repertoire sequencing experiments.28 We anticipate that a more advanced language model trained on this larger sequence space will enable further advances across all areas of antibody bioinformatics research.
While this work was under review, improved deep learning methods for general protein structure prediction were published.43,44 These methods make extensive use of attention for the end-to-end prediction of protein structures. Both methods additionally separate pairwise residue information from evolutionary information in the form of multiple sequence alignments, with RoseTTAFold going further and learning a nascent structural representation in a third track. While these methods were designed for single-chain predictions, we anticipate that similar methods may yield advances in protein complex prediction (including antibody FV structures). Further improvements still may come from directly incorporating the antigen into predictions, as antigen binding can lead to significant conformational changes.51 DMPfold52, a similar method for general proteins, has been shown to contain flexibility information within inter-residue distance distributions.53 In principle, DeepAb might provide similar insights into CDR loop flexibility, but further investigation is necessary.
Deep learning models for antibody structure prediction present several promising avenues toward antibody design. In this work, we demonstrated how our network could be used to suggest or screen point mutations. Even with no explicit knowledge of the antigen, this approach was already moderately predictive of mutational tolerability. Further, because our approach relies only on the model outputs for a given sequence, it is capable of screening designs for any antibody. Inclusion of antigen structural context through extended deep learning models or traditional approaches like Rosetta should only improve these results. Other quantities of interest such as stability or developability metrics could be predicted by using the DeepAb network for transfer learning or feature engineering.12 Furthermore, comparable networks for general protein structure prediction have recently been re-purposed for design through direct sequence optimization.54, 55, 56 With minimal modification, our network should enable similar methods for antibody design.
Experimental procedures
Resource availability
Lead contact
Further information and requests for resources should be directed to and will be fulfilled by the lead contact, Jeffrey J. Gray (jgray@jhu.edu).
Materials availability
This study did not generate any unique reagents.
Independent test sets
To evaluate the performance of our method, we considered two independent test sets. The first is the RosettaAntibody benchmark set of 49 structures, which was previously assembled to evaluate methods over a broad range of CDR H3 loop lengths (ranging 7–17 residues).8,36 Each structure in this set has greater than 2.5 Å resolution, a maximum R value of 0.2, and a maximum B factor of 80 Å2. The second comes from a set of 56 clinical-stage antibody therapeutics with solved crystal structures, which was previously assembled to study antibody developability.37 We removed five of the therapeutic antibodies that were missing one or more CDR loops (PDB: 3B2U, 3C08, 3HMW, 3S34, and 4EDW) to create a therapeutic benchmark set. The two sets shared two common antibodies (PDB: 3EO9 and 3GIZ) that we removed from the therapeutic benchmark set.
While benchmarking alternative methods, we found that some methods were unable to produce structures for every target. Specifically, RosettaAntibody failed to produce predictions for four targets (PDB: 1X9Q, 3IFL, 4D9Q, and 4K3J) and both RepertoireBuilder and ABodyBuilder failed to produce predictions for two targets (PDB: 4O02 and 5VVK). To compare consistently across all methods, we report values for only the targets that all methods succeeded in modeling. However, we note that DeepAb was capable of producing structures for all of the targets attempted. From the RosettaAntibody benchmark set, we omit PDB: 1X9Q and 3IFL. From the therapeutic benchmark set, we omit PDB: 4D9Q, 4K3J, 4O02, and 5VKK. We additionally omit the long L3 loop of target 3MLR, which not all alternative methods were able to model. In total, metrics are reported for 92 targets: 47 from the RosettaAntibody benchmark and 45 from the therapeutic benchmark. We use the Chothia CDR loop definitions to measure RMSD throughout this work.47
Representation learning on repertoire sequences
Training data set
To train the sequence model, paired FV heavy and light chain sequences were collected from the OAS database28, a set of immunoglobin sequences from next-generation sequencing experiments of immune repertoires. Each sequence in the database had previously been parsed with ANARCI57 to annotate sequences and detect potentially erroneous entries. For this work, we extract only the FV region of the sequences, as identified by ANARCI. Sequences indicated to have failed ANARCI parsing were discarded from the training data set. We additionally remove any redundant sequences. These steps resulted in a set of 118,386 sequences from five studies58, 59, 60, 61, 62 for model training.
Model and training details
To learn representations of immunoglobin sequences, we adopted an RNN encoder-decoder model26 consisting of two LSTMs.27 In an encoder-decoder model, the encoder learns to summarize the input sequence into a fixed-dimension summary vector, from which the decoder learns to reconstruct the original sequence. For the encoder model, we used a bidirectional two-layer stacked LSTM with a hidden state size of 64. The model input was created by concatenation of paired heavy and light chain sequences to form a single sequence. Three additional tokens were added to the sequence to mark the beginning of the heavy chain, the end of the heavy chain, and the end of the light chain. The concatenated sequence was one-hot encoded, resulting in an input of dimension (L + 3) × 23, where L is the combined heavy and light chain length. The summary vector is generated by stacking the final hidden states from the forward and backward encoder LSTMs, followed by a linear transformation from 128 to 64 dimensions and tanh activation. For the decoder model, we used a two-layer stacked LSTM with a hidden state size of 64. The decoder takes as input the summary vector and the previously decoded amino acid to sequentially predict the original amino acid sequence.
The model was trained using cross-entropy loss and the Adam optimizer63 with a learning rate of 0.01, with learning rate reduced upon plateauing of validation loss. A teacher forcing rate of 0.5 was used to stabilize training. The model was trained on one NVIDIA K80 GPU, requiring ∼4 h for 5 epochs over the entire data set. We used a batch size of 128, maximized to fit into GPU memory.
Predicting inter-residue geometries from antibody sequence
Training data set
To train the structure prediction model, we collected a set of FV structures from the SAbDab32, a curated set of antibody structures from the PDB.64 We removed structures with less than 4-Å resolution and applied a 99% sequence identity threshold to remove redundant sequences. We chose this high sequence similarity due to the high conservation characteristic of antibody sequences, as well as the over-representation of many identical therapeutic antibodies in structural databases. Additionally, we hoped to expose the model to examples of small mutations that lead to differences in structures. This is particularly important for the challenging CDR H3 loop, which has been observed to occupy an immense diversity of conformations even at the level of four-level fragments.65 Finally, any targets from the benchmark sets, or structures with 99% sequence similarity to a target, were removed from the training data set. These steps resulted in a set of 1,692 Fv structures, a mixture of antigen bound and unbound, for model training.
Model and training details
The structure prediction model takes as input concatenated heavy and light chain sequences. The sequences are one-hot encoded and passed through two parallel branches: a 1D ResNet and the biLSTM encoder described above. For the 1D ResNet, we add an additional delimiter channel to mark the end of the heavy chain, resulting in a dimension of L × 21, where L is the combined heavy and light chain length. The 1D ResNet begins with a 1D convolution that projects the input features up to dimension L × 64, followed by three 1D ResNet blocks (two 1D convolutions with kernel size 17) that maintain dimensionality. The second branch consists of the pre-trained biLSTM encoder. Before passing the one-hot encoded sequence to the biLSTM, we add the three delimiters described previously, resulting in dimension (L +3) × 23. From the biLSTM, we concatenate the hidden states from the forward and backward LSTMs after encoding each residue, resulting in dimension L × 128. The outputs of the 1D ResNet and the biLSTM are stacked to form a final sequential tensor of dimension L × 160. We transform the sequential tensor to pairwise data by concatenating row- and column-wise expansions. The pairwise data, dimension L × L × 320, is passed to the 2D ResNet. The 2D ResNet begins with a 2D convolution that reduces dimensionality to L × L × 64, followed by 25 2D ResNet blocks (two 2D convolutions with kernel size 5 × 5) that maintain dimensionality. The 2D ResNet blocks cycle through convolution dilation values of 1, 2, 4, 8, and 16 (five cycles in total). After the 2D ResNet, the network branches into six separate paths. Each output branch consists of a 2D convolution that projects down to dimension L × L × 37, followed by a recurrent criss-cross attention (RCCA) module.42 The RCCA modules use two criss-cross attention operations that share weights, allowing each residue pair to gather information across the entire spatial dimension. Attention queries and keys are projected to dimension L × L × 1 (one attention head). Symmetry is enforced for , , and ω predictions by averaging the final outputs with their transposes. All convolutions in the network are followed by ReLU activation. In total, the model contains about 6.4 million trainable parameters.
We trained five models on random 90/10% training/validation splits and averaged over model logits to make predictions, following previous methods.8,19 Models were trained using focal loss30 and the Adam optimizer63 with a learning rate of 0.01, with learning rate reduced upon plateauing of validation loss. Learning rate was reduced upon plateauing of the validation loss. Each model was trained on one NVIDIA K80 GPU, requiring ∼60 h for 60 epochs over the entire data set.
Structure realization
Multi-dimensional scaling
From the network predictions, we create real-value matrices for the , ω, θ, and φ outputs by taking the midpoint value of the modal probability bin for each residue pair. From these real-valued distances and orientations, we create an initial backbone atom (N, Cα, and C) distance matrix. For residue pairs predicted to have , we approximate the distances between atoms using the Floyd-Warshall shortest path algorithm.66 From this distance matrix, we use MDS67 to produce an initial set of 3D coordinates. The initial structures from MDS typically contained atom clashes and non-ideal geometries that required further refinement.
Energy minimization refinement
Initial structures from MDS were refined by constrained energy minimization in Rosetta. For each pair of residues, the predicted distributions for each output were converted to energy potentials by negating the raw model logits (i.e., without softmax activation) and dividing by the squared prediction.
The discrete potentials were converted to continuous functions using the built-in Rosetta spline function. We disregarded potentials for residue pairs with predicted , as well as those with a modal bin probability below 10%. For potentials, we also discarded with predicted or modal bin probability below 30% to create a local backbone hydrogen-bonding potential. The remaining potentials are applied to the MDS structure as inter-residue constraints in Rosetta.
Modeling in Rosetta begins with a coarse-grained representation, in which the side-chain atoms are represented as a single artificial atom (centroid). The centroid model is optimized by gradient-based energy minimization (MinMover) using the L-BFGS algorithm.34,35 The centroid energy function includes the following score terms in addition to learned constraints: vdw (clashes), cen_hb (hydrogen bonds), and rama and omega (backbone torsion angles). After centroid optimization, we add side-chain atoms and relax the structure to reduce steric clashes (FastRelax). Finally, we repeat the gradient-based energy minimization step in the full-atom representation to produce a final model. We repeat this procedure to produce 50 decoy models and select the structure with the lowest energy as the final prediction. Only the relaxation step in the protocol is non-deterministic, leading to high convergence among decoys. In practice, we expect 5–10 decoys will be sufficient for most applications.
Predicting structures with other recent methods
To contextualize the performance of our method, we benchmarked three recent methods for antibody FV structure prediction: RosettaAntibody-G6, RepertoireBuilder5, and ABodyBuilder.3 RosettaAntibody-G predictions were generated using the command-line arguments recommended by Jeliazkov et al.6 (Appendix S1). We note that we only used the RosettaAntibody grafting protocol (antibody), omitting the extensive but time-consuming H3 loop sampling (antibody_H3).4,6 RepertoireBuilder and ABodyBuilder predictions were generated using their respective web servers. For each target in the benchmarks, we excluded structures with sequence similarity greater than 99% from use for predictions, to mirror the conditions of our training set. We note that this sequence cutoff does not prevent methods from grafting identical loops from slightly different sequences.
Attention matrix calculation
During the criss-cross attention operation42, we create an attention matrix , where for each residue pair in the L × L spatial dimension, we have 2L−1 entries corresponding to the attention values over other residue pairs in the same row and column (including the residue pair itself). To interpret the total attention between pairs of residues, we simplify the attention matrix to , where for each residue i in the sequence, we only consider the attention values in the i-th row and column. In A′, for each residue i there are two attention values for each other residue j, corresponding to the row- and column-wise attention between i and j. We further simplify by summing these row- and column-wise attention values, resulting in an attention matrix , containing the total attention between pairs of residues. In the main text, we refer to A″ as A for simplicity.
Acknowledgments
We thank Dr. Sai Pooja Mahajan for helpful discussions and advice. This work was supported by National Institutes of Health grants R01-GM078221 and T32-GM008403 (J.A.R.) and AstraZeneca (J.A.R.). Computational resources were provided by the Maryland Advanced Research Computing Cluster (MARCC).
Author contributions
J.A.R. and J.J.G. conceptualized the project. All authors developed the methodology. J.A.R. developed the software and conducted the investigation. J.S. and J.J.G. supervised the project. All authors wrote the manuscript.
Declaration of interests
J.J.G. is an unpaid board member of the Rosetta Commons. Under institutional participation agreements between the University of Washington, acting on behalf of the Rosetta Commons, Johns Hopkins University may be entitled to a portion of revenue received on licensing Rosetta software including methods discussed/developed in this study. As a member of the Scientific Advisory Board, J.J.G. has a financial interest in Cyrus Biotechnology. Cyrus Biotechnology distributes the Rosetta software, which may include methods developed in this study. These arrangements have been reviewed and approved by the Johns Hopkins University in accordance with its conflict-of-interest policies.
Published: December 9, 2021
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.patter.2021.100406.
Supplemental information
Data and code availability
Structure prediction data generated in this study have been deposited to Zenodo: 10.5281/zenodo.5525257 and are publicly available as of the date of publication. All original code is available at https://github.com/RosettaCommons/DeepAb and deposited to Zenodo: 10.5281/zenodo.5683647. Any additional information required to reanalyze the data reported in this paper is available from the lead author upon request.
References
- 1.Lu R.-M., Hwang Y.-C., Liu I.-J., Lee C.-C., Tsai H.-Z., Li H.-J., Wu H.-C. Development of therapeutic antibodies for the treatment of diseases. J. Biomed. Sci. 2020;27:1. doi: 10.1186/s12929-019-0592-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Kaplon H., Reichert J.M. Antibodies to watch in 2021. MAbs. 2021;13:1860476. doi: 10.1080/19420862.2020.1860476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Dunbar J., Krawczyk K., Leem J., Marks C., Nowak J., Regep C., Georges G., Kelm S., Popovic B., Deane C.M. SAbPred: a structure-based antibody prediction server. Nucleic Acids Res. 2016;44:W474–W478. doi: 10.1093/nar/gkw361. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Weitzner B.D., Jeliazkov J.R., Lyskov S., Marze N., Kuroda D., Frick R., Adolf-Bryfogle J., Biswas N., Dunbrack R.L., Gray J.J. Modeling and docking of antibody structures with Rosetta. Nat. Protoc. 2017;12:401–416. doi: 10.1038/nprot.2016.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Schritt D., Li S., Rozewicki J., Katoh K., Yamashita K., Volkmuth W., Cavet G., Standley D.M. Repertoire Builder: high-throughput structural modeling of B and T cell receptors. Mol. Syst. Des. Eng. 2019;4:761–768. doi: 10.1039/C9ME00020H. [DOI] [Google Scholar]
- 6.Jeliazkov J.R., Frick R., Zhou J., Gray J.J. Robustification of RosettaAntibody and Rosetta SnugDock. PLoS One. 2021;16:e0234282. doi: 10.1371/journal.pone.0234282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Dunbar J., Fuchs A., Shi J., Deane C.M. ABangle: characterising the VH-VL orientation in antibodies. Protein Eng. Des. Sel. 2013;26:611–620. doi: 10.1093/protein/gzt020. [DOI] [PubMed] [Google Scholar]
- 8.Marze N.A., Lyskov S., Gray J.J. Improved prediction of antibody V L –V H orientation. Protein Eng. Des. Sel. 2016;29:409–418. doi: 10.1093/protein/gzw013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Almagro J.C., Teplyakov A., Luo J., Sweet R.W., Kodangattil S., Hernandez-Guzman F., Gilliland G.L. Second antibody modeling assessment (AMA-II) Proteins Struct. Funct. Bioinform. 2014;82:1553–1562. doi: 10.1002/prot.24567. [DOI] [PubMed] [Google Scholar]
- 10.Gao W., Mahajan S.P., Sulam J., Gray J.J. Deep learning in protein structural modeling and design. Patterns. 2020;1:100142. doi: 10.1016/j.patter.2020.100142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Graves J., Byerly J., Priego E., Makkapati N., Parish S., Medellin B., Berrondo M. A review of deep learning methods for antibodies. Antibodies. 2020;9:12. doi: 10.3390/antib9020012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chen X., Dougherty T., Hong C., Schibler R., Cong Zhao Y., Sadeghi R., Matasci N., Wu Y.C., Kerman I. Predicting antibody developability from sequence using machine learning. bioRxiv. 2020 doi: 10.1101/2020.06.18.159798. [DOI] [Google Scholar]
- 13.Marks C., Hummer A.M., Chin M., Deane C.M. Humanization of antibodies using a machine learning approach on large-scale repertoire data. Bioinformatics. 2021:1–7. doi: 10.1093/bioinformatics/btab434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shin J.-E., Riesselman A.J., Kollasch A.W., McMahon C., Simon E., Sander C., Manglik A., Kruse A.C., Marks D.S. Protein design and variant prediction using autoregressive generative models. Nat. Commun. 2021;12:2403. doi: 10.1038/s41467-021-22732-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Pittala S., Bailey-Kellogg C. Learning context-aware structural representations to predict antigen and antibody binding interfaces. Bioinformatics. 2020;36:3996–4003. doi: 10.1093/bioinformatics/btaa263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Akbar R., Robert P.A., Pavlović M., Jeliazkov J.R., Snapkov I., Slabodkin A., Weber C.R., Scheffer L., Miho E., Haff I.H., et al. A compact vocabulary of paratope–epitope interactions enables predictability of antibody-antigen binding. Cell Rep. 2021;34:108856. doi: 10.1016/j.celrep.2021.108856. [DOI] [PubMed] [Google Scholar]
- 17.Senior A.W., Evans R., Jumper J., Kirkpatrick J., Sifre L., Green T., Qin C., Žídek A., Nelson A.W.R., Bridgland A., et al. Improved protein structure prediction using potentials from deep learning. Nature. 2020;577:706–710. doi: 10.1038/s41586-019-1923-7. [DOI] [PubMed] [Google Scholar]
- 18.Yang J., Anishchenko I., Park H., Peng Z., Ovchinnikov S., Baker D. Improved protein structure prediction using predicted interresidue orientations. Proc. Natl. Acad. Sci. U S A. 2020;117:1496–1503. doi: 10.1073/pnas.1914677117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Xu J., McPartlon M., Li J. Improved protein structure prediction by deep learning irrespective of co-evolution information. Nat. Mach. Intell. 2021;3:601–609. doi: 10.1038/s42256-021-00348-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Georgiou G., Ippolito G.C., Beausang J., Busse C.E., Wardemann H., Quake S.R. The promise and challenge of high-throughput sequencing of the antibody repertoire. Nat. Biotechnol. 2014;32:158–168. doi: 10.1038/nbt.2782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Rives A., Meier J., Sercu T., Goyal S., Lin Z., Liu J., Guo D., Ott M., Zitnick C.L., Ma J., et al. Biological structure and function emerge from scaling unsupervised learning to 250 million protein sequences. Proc. Natl. Acad. Sci. U S A. 2021;118 doi: 10.1073/pnas.2016239118. e2016239118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Vig J., Madani A., Varshney L.R., Xiong C., Socher R., Rajani N.F. BERTology meets biology: interpreting attention in protein language models. bioRxiv. 2020:1–24. doi: 10.1101/2020.06.26.174417. [DOI] [Google Scholar]
- 23.Rao R., Meier J., Sercu T., Ovchinnikov S., Rives A. Transformer protein language models are unsupervised structure learners. bioRxiv. 2020:1–24. doi: 10.1101/2020.12.15.422761. [DOI] [Google Scholar]
- 24.Rao R., Liu J., Verkuil R., Meier J., Canny J.F., Abbeel P., et al. MSA transformer. bioRxiv. 2021:1–16. doi: 10.1101/2021.02.12.430858. [DOI] [Google Scholar]
- 25.Alley E.C., Khimulya G., Biswas S., AlQuraishi M., Church G.M. Unified rational protein engineering with sequence-based deep representation learning. Nat. Methods. 2019;16:1315–1322. doi: 10.1038/s41592-019-0598-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Cho K., van Merrienboer B., Gulcehre C., Bahdanau D., Bougares F., Schwenk H., Bengio Y. Proceedings of the 2014 Conference on Empirical Methods in Natural Language Processing (EMNLP) Association for Computational Linguistics; 2014. Learning phrase representations using RNN encoder–decoder for statistical machine translation; pp. 1724–1734. [DOI] [Google Scholar]
- 27.Gers F.A., Schmidhuber J., Cummins F. Learning to forget: continual prediction with LSTM. Neural Comput. 2000;12:2451–2471. doi: 10.1162/089976600300015015. [DOI] [PubMed] [Google Scholar]
- 28.Kovaltsuk A., Leem J., Kelm S., Snowden J., Deane C.M., Krawczyk K. Observed antibody space: a resource for data mining next-generation sequencing of antibody repertoires. J. Immunol. 2018;201:2502–2509. doi: 10.4049/jimmunol.1800708. [DOI] [PubMed] [Google Scholar]
- 29.Ruffolo J.A., Guerra C., Mahajan S.P., Sulam J., Gray J.J. Geometric potentials from deep learning improve prediction of CDR H3 loop structures. Bioinformatics. 2020;36:i268–i275. doi: 10.1093/bioinformatics/btaa457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lin T.-Y., Goyal P., Girshick R., He K., Dollar P. 2017 IEEE International Conference on Computer Vision (ICCV) IEEE; 2017. Focal loss for dense object detection; pp. 2999–3007. [DOI] [Google Scholar]
- 31.Mukhoti J., Kulharia V., Sanyal A., Golodetz S., Torr P.H.S., Dokania P.K. Calibrating deep neural networks using focal loss. Advances in Neural Information Processing Systems. 2020;33:1–12. [Google Scholar]
- 32.Dunbar J., Krawczyk K., Leem J., Baker T., Fuchs A., Georges G., Shi J., Deane C.M. SAbDab: the structural antibody database. Nucleic Acids Res. 2014;42:D1140–D1146. doi: 10.1093/nar/gkt1043. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zhang J., Wang Q., Barz B., He Z., Kosztin L., Shang Y., Xu D. MUFOLD: a new solution for protein 3D structure prediction. Proteins Struct. Funct. Bioinform. 2010;78:1137–1152. doi: 10.1002/prot.22634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Leaver-Fay A., Tyka M., Lewis S.M., Lange O.F., Thompson J., Jacak R., Kaufman K., Renfrew P.D., Smith C.A., Sheffler W., et al. Rosetta3: an object-oriented software suite for the simulation and design of macromolecules. Methods Enzymol. 2011;487:545–574. doi: 10.1016/B978-0-12-381270-4.00019-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Leman J.K., Weitzner B.D., Lewis S.M., Adolf-Bryfogle J., Alam N., Alford R.F., Aprahamian M., Baker D., Barlow K.A., Barth P., et al. Macromolecular modeling and design in Rosetta: recent methods and frameworks. Nat. Methods. 2020;17:665–680. doi: 10.1038/s41592-020-0848-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Weitzner B.D., Gray J.J. Accurate structure prediction of CDR H3 loops enabled by a novel structure-based C-terminal constraint. J. Immunol. 2017;198:505–515. doi: 10.4049/jimmunol.1601137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Raybould M.I.J., Marks C., Krawczyk K., Taddese B., Nowak J., Lewis A.P., Bujotzek A., Shi J., Deane C.M. Five computational developability guidelines for therapeutic antibody profiling. Proc. Natl. Acad. Sci. U S A. 2019;116:4025–4030. doi: 10.1073/pnas.1810576116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Niederfellner G., Lammens A., Mundigl O., Georges G.J., Schaefer W., Schwaiger M., Franke A., Wiechmann K., Jenewein S., Slootstra J.W., et al. Epitope characterization and crystal structure of GA101 provide insights into the molecular basis for type I/II distinction of CD20 antibodies. Blood. 2011;118:358–367. doi: 10.1182/blood-2010-09-305847. [DOI] [PubMed] [Google Scholar]
- 39.Wojciak J.M., Zhu N., Schuerenberg K.T., Moreno K., Shestowsky W.S., Hiraiwa M., Sabbadini R., Huxford T. The crystal structure of sphingosine-1-phosphate in complex with a Fab fragment reveals metal bridging of an antibody and its antigen. Proc. Natl. Acad. Sci. U S A. 2009;106:17717–17722. doi: 10.1073/pnas.0906153106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Doshi-Velez F., Kim B. Towards a rigorous science of interpretable machine learning. arXiv. 2017:1–13. [Google Scholar]
- 41.Lipton Z.C. The Mythos of model interpretability. Queue. 2018;16:31–57. doi: 10.1145/3236386.3241340. [DOI] [Google Scholar]
- 42.Huang Z., Wang X., Wei Y., Huang L., Shi H., Liu W., Huang T.S. CCNet: criss-cross attention for semantic segmentation. IEEE Trans. Pattern Anal. Mach. Intell. 2020;14:1. doi: 10.1109/TPAMI.2020.3007032. [DOI] [PubMed] [Google Scholar]
- 43.Baek M., DiMaio F., Anishchenko I., Dauparas J., Ovchinnikov S., Lee G.R., Wang J., Cong Q., Kinch L.N., Schaeffer R.D., et al. Accurate prediction of protein structures and interactions using a three-track neural network. Science. 2021;373:871–876. doi: 10.1126/science.abj8754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jumper J., Evans R., Pritzel A., Green T., Figurnov M., Ronneberger O., Tunyasuvunakool K., Bates R., Žídek A., Potapenko A., et al. Highly accurate protein structure prediction with AlphaFold. Nature. 2021;596:583–589. doi: 10.1038/s41586-021-03819-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Weitzner B.D., Dunbrack R.L., Gray J.J. The origin of CDR H3 structural diversity. Structure. 2015;23:302–311. doi: 10.1016/j.str.2014.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Van der Maaten L., Hinton G. Visualizing data using t-SNE. J. Mach. Learn. Res. 2008;9:2579–2605. [Google Scholar]
- 47.Chothia C., Lesk A.M. Canonical structures for the hypervariable regions of immunoglobulins. J. Mol. Biol. 1987;196:901–917. doi: 10.1016/0022-2836(87)90412-8. [DOI] [PubMed] [Google Scholar]
- 48.North B., Lehmann A., Dunbrack R.L. A new clustering of antibody CDR loop conformations. J. Mol. Biol. 2011;406:228–256. doi: 10.1016/j.jmb.2010.10.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Adolf-Bryfogle J., Xu Q., North B., Lehmann A., Dunbrack R.L. PyIgClassify: a database of antibody CDR structural classifications. Nucleic Acids Res. 2015;43:D432–D438. doi: 10.1093/nar/gku1106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Warszawski S., Borenstein Katz A., Lipsh R., Khmelnitsky L., Ben Nissan G., Javitt G., Dym O., Unger T., Knop O., Albeck S., et al. Optimizing antibody affinity and stability by the automated design of the variable light-heavy chain interfaces. PLoS Comput. Biol. 2019;15:e1007207. doi: 10.1371/journal.pcbi.1007207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Fernández-Quintero M.L., Kraml J., Georges G., Liedl K.R. CDR-H3 loop ensemble in solution–conformational selection upon antibody binding. MAbs. 2019;11:1077–1088. doi: 10.1080/19420862.2019.1618676. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Greener J.G., Kandathil S.M., Jones D.T. Deep learning extends de novo protein modelling coverage of genomes using iteratively predicted structural constraints. Nat. Commun. 2019;10:3977. doi: 10.1038/s41467-019-11994-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schwarz D., Georges G., Kelm S., Shi J., Vangone A., Deane C.M. Co-evolutionary distance predictions contain flexibility information. Bioinformatics. 2021:1–8. doi: 10.1093/bioinformatics/btab562. [DOI] [PubMed] [Google Scholar]
- 54.Linder J., Seelig G. Fast differentiable DNA and protein sequence optimization for molecular design. arXiv. 2020 2005.11275v2. [Google Scholar]
- 55.Anishchenko I., Chidyausiku T.M., Ovchinnikov S., Pellock S.J., Baker D. De novo protein design by deep network hallucination. bioRxiv. 2020 doi: 10.1101/2020.07.22.211482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Norn C., Wicky B.I.M., Juergens D., Liu S., Kim D., Tischer D., Koepnick B., Anishchenko I., Baker D., Ovchinnikov S. Protein sequence design by conformational landscape optimization. Proc. Natl. Acad. Sci. U S A. 2021;118 doi: 10.1073/pnas.2017228118. e2017228118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dunbar J., Deane C.M. ANARCI: antigen receptor numbering and receptor classification. Bioinformatics. 2015;32:btv552. doi: 10.1093/bioinformatics/btv552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Goldstein L.D., Chen Y.-J.J., Wu J., Chaudhuri S., Hsiao Y.-C., Schneider K., Hoi K.H., Lin Z., Guerrero S., Jaiswal B.S., et al. Massively parallel single-cell B-cell receptor sequencing enables rapid discovery of diverse antigen-reactive antibodies. Commun. Biol. 2019;2:304. doi: 10.1038/s42003-019-0551-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Setliff I., Shiakolas A.R., Pilewski K.A., Murji A.A., Mapengo R.E., Janowska K., Richardson S., Oosthuysen C., Raju N., Ronsard L., et al. High-throughput mapping of B cell receptor sequences to antigen specificity. Cell. 2019;179:1636–1646.e15. doi: 10.1016/j.cell.2019.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Eccles J.D., Turner R.B., Kirk N.A., Muehling L.M., Borish L., Steinke J.W., Payne S.C., Wright P.W., Thacker D., Lahtinen S.J., et al. T-bet+ memory B cells link to local cross-reactive IgG upon human rhinovirus infection. Cell Rep. 2020;30:351–366.e7. doi: 10.1016/j.celrep.2019.12.027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Alsoussi W.B., Turner J.S., Case J.B., Zhao H., Schmitz A.J., Zhou J.Q., Chen R.E., Lei T., Rizk A.A., McIntire K.M., et al. A potently neutralizing antibody protects mice against SARS-CoV-2 infection. J. Immunol. 2020;205:915–922. doi: 10.4049/jimmunol.2000583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.King H.W., Orban N., Riches J.C., Clear A.J., Warnes G., Teichmann S.A., James L.K. Single-cell analysis of human B cell maturation predicts how antibody class switching shapes selection dynamics. Sci. Immunol. 2021;6:eabe6291. doi: 10.1126/sciimmunol.abe6291. [DOI] [PubMed] [Google Scholar]
- 63.Kingma D.P., Ba J. 3rd Int. Conf. Learn. Represent. ICLR 2015 - Conf. Track Proc. 2014. Adam: a method for stochastic optimization; pp. 1–15. [Google Scholar]
- 64.Berman H.M., Battistuz T., Bhat T.N., Bluhm W.F., Bourne P.E., Burkhardt K., Feng Z., Gilliland G.L., Iype L., Jain S., et al. The protein data bank. Acta Crystallogr. Sect. D Biol. Crystallogr. 2002;58:899–907. doi: 10.1107/S0907444902003451. [DOI] [PubMed] [Google Scholar]
- 65.Regep C., Georges G., Shi J., Popovic B., Deane C.M. The H3 loop of antibodies shows unique structural characteristics. Proteins Struct. Funct. Bioinform. 2017;85:1311–1318. doi: 10.1002/prot.25291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Floyd R.W. Algorithm 97: shortest path. Commun. ACM. 1962;5:345. doi: 10.1145/367766.368168. [DOI] [Google Scholar]
- 67.Borg I., Groenen P.J. Springer; 2005. Modern Multidimensional Scaling. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Structure prediction data generated in this study have been deposited to Zenodo: 10.5281/zenodo.5525257 and are publicly available as of the date of publication. All original code is available at https://github.com/RosettaCommons/DeepAb and deposited to Zenodo: 10.5281/zenodo.5683647. Any additional information required to reanalyze the data reported in this paper is available from the lead author upon request.