

Abstract
Numerous molecular mechanisms are being examined in an attempt to discover disease-modifying drugs to slow down the underlying neurodegeneration in Alzheimer’s disease. Recent studies have shown the beneficial effects of epidermal growth factor receptor inhibitors on the enhancement of behavioral and pathological sequelae in Alzheimer’s disease. Despite the promising effects of epidermal growth factor receptor inhibitors in Alzheimer’s disease, there is no irrefutable neuroprotective evidence in well-established animal models using epidermal growth factor receptor inhibitors due to many un-explored downstream signaling pathways. This caused controversy about the potential involvement of epidermal growth factor receptor inhibitors in any prospective clinical trial. In this review, the mystery beyond the under-investigation of epidermal growth factor receptor in Alzheimer’s disease will be discussed. Furthermore, their molecular mechanisms in neurodegeneration will be explained. Also, we will shed light on SARS-COVID-19 induced neurological manifestations mediated by epidermal growth factor modulation. Finally, we will discuss future perspectives and under-examined epidermal growth factor receptor downstream signaling pathways that warrant more exploration. We conclude that epidermal growth factor receptor inhibitors are novel effective therapeutic approaches that require further research in attempts to be repositioned in the delay of Alzheimer’s disease progression.
Key Words: Alzheimer's disease, autophagy, drug re-positioning, epidermal growth factor receptor, human epidermal growth factor receptor-2, neurodegenerative diseases, neuroinflammation, oxidative stress, tyrosine kinase inhibitors
Introduction
Alzheimer’s disease (AD) is a multi-etiological disorder characterized by gradual cognitive decline, and it is the most prevalent type of dementia (Panpalli Ates et al., 2016). By 2050, the number of AD patients (65 years) in the United States will more than double, from 5.8 million to 13.8 million (Zhang et al., 2021). The most common hypotheses on the etiopathogenesis of AD are the amyloid cascade hypothesis and the hyperphosphorylated tau hypothesis. Amyloid precursor protein (APP) is a widely expressed transmembrane protein in the brain. It has a remarkable physiological role during brain maturation and memory. Normally, APP is cleaved by a proteolytic enzyme called α-secretase, generating soluble peptides (Müller et al., 2017). In AD, APP undergoes proteolytic cleavage by two amyloidogenic proteases, β- and γ- secretase forming insoluble amyloid-β (Aβ40–42). These insoluble proteins clump together, forming extracellular amyloid plaques that promote immune and inflammatory responses (Lichtenthaler and Haass, 2004; Müller et al., 2017; Figure 1).
Figure 1.
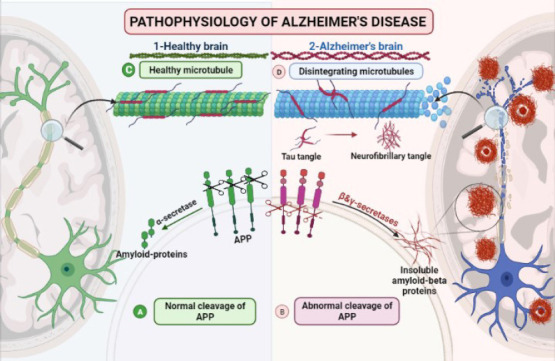
Comparison between a healthy brain and neuropathological changes in Alzheimer’s disease.
(A) Normal cleavage of APP by α-secretase. (B) Abnormal cleavage of the APP by β-secretase forming insoluble amyloid-β proteins. (C) Stabilized microtubules with unphosphorylated tau. (D) Disintegrating microtubule with hyperphosphorylated tau forming intracellular neurofibrillary tangles. APP: Amyloid precursor protein.
Tau proteins are microtubule-associated proteins. Microtubules’ stabilization and assembly depend on the phosphorylation status of tau proteins. The hyperphosphorylated tau proteins are transformed into paired helical aggregated fibers called intracellular neurofibrillary tangles. Change of the tau shape leads to disturbance in regulatory and cellular functions of the cytoskeleton, maintenance of normal morphology, and axonal transport (Malafaia et al., 2021; Figure 1).
The tau hypothesis suggests that the excessive hyperphosphorylation of tau seems to be crucial in the pathophysiology of AD. The phosphorylation state of tau is regulated by a balance between kinase(s) activity, and phosphatase(s) activity. Several studies have proven that hyperphosphorylation of tau might be due to increased tau kinase(s). Thus, inhibition of such kinases would be a promising avenue to suppress tau hyperphosphorylation (Ferguson and Gray, 2018).
Despite more than decades of extensive research, there is a lack of clarity on the etiopathogenesis of AD, and thus we still do not identify the best approaches for treating and mitigating this crippling and costly disorder. Furthermore, the prevalence of AD grows exponentially in the absence of truly successful therapies. Many epidemiological studies have shown that Alzheimer’s patients have a low risk of developing certain types of cancer (Rodriguez et al., 2021). Surprisingly, several studies have been reported over the last decade, demonstrating the valuable therapeutic effects of anti-cancer epidermal growth factor receptors (EGFR) inhibitors, on the enhancement of behavioral and neurological conditions in neurodegenerative disorders, including AD (Wang et al., 2012, 2017; Mansour et al., 2021). The potential commonalities between AD and cancer have triggered clinical trials in AD using approved anti-cancer drugs. A recent Phase III clinical study has revealed a cognitive ameliorating effect of the anti-cancer drug, masitinib as a tyrosine kinase inhibitor (TKI) (Folch et al., 2015). A meta-analysis demonstrated that the suppressed Aβ clearance in AD is associated with the genes encoding Fyn tyrosine kinase and EGFR, which are the key receptors in Aβ downstream signaling (Yuen et al., 2020). Several reports have examined whether the use of EGFR inhibitors protects against neurodegeneration in different AD models through multiple mechanisms, such as amelioration of p-tau and Aβ (Wang et al., 2012; Lee et al., 2021; Mansour et al., 2021), activation of autophagy (Wang et al., 2017), inhibition of reactive astrocytes (Chen et al., 2019; Lee et al., 2021; Mansour et al., 2021), and axonal regeneration via inhibition of glycogen synthase kinase-3β (GSK-3β) (Wakatsuki et al., 2015; Wakatsuki, 2019). Nevertheless, there are multiple caveats to engaging EGFR inhibitors in future clinical trials. For instance, some of the previously tested drugs, such as gefitinib and erlotinib, were intended for peripheral types of cancer and show modest effect in metastatic brain cancer, because they do not exhibit optimum blood-brain barrier (BBB) permeability (Ahluwalia et al., 2018; Kim et al., 2019). EGFR inhibitors displayed neuroprotective effects in spinal cord injuries. However, the ability of EGFR to exhibit antioxidant and anti-inflammatory effects in animal models of AD is still unexplored. In this review, we briefly described the mystery beyond EGFR under investigation in AD, and discussed potential therapeutic mechanisms of previously studied EGFR inhibitors, such as activation of autophagy (Wang et al., 2012, 2017), attenuation of reactive astrocyte (Chen et al., 2019), and inhibition of GSK-3β (Wakatsuki et al., 2015; Wakatsuki, 2019; Mansour et al., 2021). We also discussed some under-investigated nexus between EGFR and neuroinflammation, NADPH oxidase-1 (NOX-1)-mediated oxidative stress and glutamate-mediated excitotoxicity in AD. In addition, we shed light on the third generation of EGFR inhibitors of better BBB-permeability and dual EGFR inhibitors of possible additive effects to be repositioned in the treatment of AD.
Search Strategy and Selection Criteria
We performed a literature search on PubMed and Google Scholar until March 2021. The key words were: Alzheimer’s disease, EGFR, tyrosine kinase inhibitors, anti-cancer repositioning.
Reactive Astrocyte, Oxidative Stress, and Impaired Autophagy Are the Main Culprits Implicated in Alzheimer’s Disease Pathogenesis following Epidermal Growth Factor Receptor Activation
EGFR activation with subsequent astrocyte reactivation and axonal degeneration
In adult healthy central nervous system (CNS), quiescent astrocytes are present with low levels of EGFR. Astrocytes are essential for neuronal functions and survival, but when they become activated, they lose their functions and initiate neurodegenerative disorders. Pathological insults occurred in neurodegenerative disorders, such as AD (Chen et al., 2019), Huntington’s disease (Gu et al., 2005; Faideau et al., 2010), Parkinson’s disease (Bandopadhyay et al., 2004; Neumann et al., 2004), and neural damage upregulate and stimulate EGFR in both astrocytes and neurons (Chen et al., 2019; Volonté et al., 2020). Here, we focus on the EGFR roles in AD. Further details regarding EGFR effects in other neurodegenerative disorders and spinal cord injuries can be found in a previous study (Tavassoly et al., 2020).
EGFR stimulation is accompanied by an increased level of proinflammatory molecules, such as inducible nitric oxide synthase, tumor necrosis factor-alpha, interleukin-1β, and cyclooxygenase-2, as well as elevated glial acidic fibrillary protein (GFAP) (Chen et al., 2019). Hyperphosphorylation of EGFR, astrocyte activation, and subsequent pro-inflammatory cytokine production result in demyelination, glial scar formation, and destruction of oligodendrocytes. A previous study revealed that EGFR inhibition using PD168393 reduced astrogliosis and production of cytokines in vitro, while suppressed demyelination, and neuronal death in vivo (Li et al., 2014).
In the CNS, neuronal degeneration and repair are under the influence of growth inhibitory proteins expressed in the CNS (McKerracher and Rosen 2015). Alterations in the cellular and molecular milieu of the CNS following insult or neurodegenerative disease lead to the production of an inhibitory environment that prohibits the regeneration of neurons (Leinster et al., 2013). A recent study showed that the inhibition of EGFR suppressed the activities of chondroitin sulfate proteoglycans and myelin inhibitors. Regeneration inhibitors stimulated EGFR phosphorylation. Consequently, EGFR inhibitors, such as erlotinib, enhanced the regeneration of optic nerve fibers, suggesting a promising therapeutic strategy for promoting axon regeneration after neurodegeneration (Koprivica et al., 2005). Another research demonstrated that administration of EGFR inhibitor PD168393 in injured spinal cord rats resulted in improvement of sensory, motor, and bladder emptying functions (Erschbamer et al., 2007). Taken together, EGFR inhibition is a potential target for enhancing axonal regeneration even in the presence of growth inhibitory molecules after injury in the CNS.
Oxidative stress and subsequent neuronal apoptosis mediated by EGFR activation
Oxidative stress has long been associated with neuronal death that characterizes certain neurodegenerative disorders, such as Parkinson’s disease, AD, and amyotrophic lateral sclerosis. Whether it is idiopathic or downstream signaling of neurodegeneration is still a controversial question (Wakatsuki and Araki, 2016). Oxidative stress is an eminent cause of axonal degeneration and neuronal apoptosis. A ubiquitin ligase enzyme called zinc and ring finger-1 (ZNRF1) is a ubiquitin ligase expressed in nearly all neurons in the CNS. Although ZNRF1 is expressed in every neuron, it does not work in steady conditions; however, in response to injury that triggers axonal dysfunction (Cao and Fang, 2015). Oxidative stress-activated ZNRF1 activity by activating EGFR-mediated phosphorylation at the 103rd tyrosine that in turn upregulated ZNRF1 by oxidative stress resulted in neuronal apoptosis and axonal degeneration (Wakatsuki et al., 2015; Araki and Wakatsuki, 2018).
Following axonal degeneration, ZNRF1 degraded AKT. As a result of Akt degradation, GSK3-β was stimulated, inhibiting collapsing response mediator protein 2 (CRMP2) that facilitated axonal growth via its interactions with the microtubules. Hence, degradation of CRMP2 resulted in the loss of axonal integrity (Wakatsuki et al., 2015). In addition, oxidative stress caused by treating neuronal cells with H2O2 or 6-hydroxydopamine or even in vivo by injury induction induced NOX in neuronal axons and activated the neuronal EGFR activity (Belambri et al., 2018). Phosphorylated EGFR activated ZNRF1 and consequently stimulated the ligase action of ZNRF1 (Araki and Wakatsuki, 2019). Interestingly, activation of the EGFR/ZNRF1/AKT/GSK3B/CRMP2 pathway in the neuronal body resulted in the stimulation of neuronal apoptosis (Cao and Fang, 2015). Accordingly, NOX is required for ZNRF1 stimulation by EGFR-dependent activation in response to axonal injury. Therefore, targeting EGFR-pluripotent oxidative stress warrants further research in neurodegenerative diseases, including AD (Figure 2c).
Figure 2.
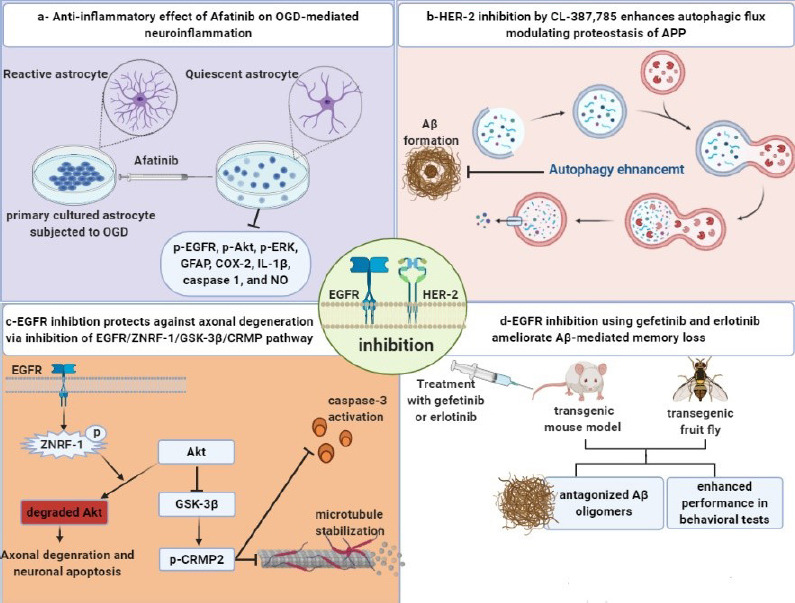
Graphical abstract recap the molecular mechanisms involved in the neuroprotective effects of EGFR inhibition.
(a) EGFR inhibition exerts an anti-inflammatory effect (Chen et al., 2019). (b) HER-2 inhibition augments autophagy (Wang et al., 2017a). (c) EGFR inhibition suppresses axonal degeneration and neuronal apoptosis (Wakatsuki et al., 2015a). (d) EGFR inhibition ameliorates Aβ-mediated memory loss (Wang et al., 2012a). APP: Amyloid precursor protein; Aβ: amyloid-beta; CRMP: collapsin response mediator protein; EGFR: epidermal growth factor receptor; ERK: extracellular regulated kinase; GFAP: glial acidic fibrillary protein; GSK-3β: glycogen synthase kinase 3-beta; HER-2: human epidermal growth factor 2; IL-1β: interleukin-1 beta; NO: nitric oxide; OGD: oxygen-glucose deprivation; ZNRF: zinc and ring finger. Unpublished data.
Nexus between EGFR activation and autophagy impairment: PI3K/Akt/mTOR pathway
Autophagy plays an essential role in preserving neuronal integrity by clearing damaged organelles and misfolded proteins in AD to maintain cellular homeostasis (Wang et al., 2017). Upregulation of EGFR and human epidermal growth factor receptor-2 (HER-2) is tightly contributed to amyloid plaque formation in AD (Wang et al., 2012, 2017). p-EGFR was elevated in the hippocampus of the double transgenic mouse model of AD (Wang et al., 2012). Consistently, an elevated level of HER-2 was found in the hippocampus of postmortem AD patients. EGFR regulates autophagy, where p-EGFR binds to Beclin 1, the principal autophagy protein (Wang et al., 2017).
Furthermore, a recent study found that HER-2 effectively inhibited autophagy by physically separating Beclin-1 from the Vps34–Vps15 complex that forms the endosome required for autophagy (Backer, 2008; Wang et al., 2017). Inhibition of HER-2 by CL-387, 785 lowered the levels of APP c-terminal and Aβ in vitro, zebrafish, and transgenic mouse models of AD, via activation of autophagy. Moreover, administration of CL-387,785 improved the cognitive performance of APP/presenilin-1 (PS1) transgenic mice, unveiling the crucial role of HER-2 in modulating autophagy and recommended HER-2 as a therapeutic target for the treatment of AD (Wang et al., 2017).
SARS-COVID-19 associated neurological manifestations mediated by EGFR modulation
Retrospective case series have shown that a severe COVID-19 infection resulted in cerebrovascular problems as demonstrated by circulating virus nucleocapsid-proteins (ff-SARS-CoV-2-NC) in the cerebrovascular plasma (Mao et al., 2020). A recent study found that there are similarities between ff-SARS-CoV-2-NC and the EGF binding site on EGFR. Consequently, the EGF-EGFR binding can be hindered by ff-SARS-CoV-2-NC. Furthermore, SARS-Cov-19 S-spike glycoprotein may act as a competitive inhibitor with transforming growth factor-beta on EGFR receptors (Geurdes, 2020; Khan and Hatiboglu, 2020). These findings display a potential CNS-floating ff-SARS-CoV-2-NC involvement of AD. Surprisingly, the EGFR has been reported as being essential for the entrance of certain viruses, such as coronaviruses, and EGFR signaling is implicated in viral replication accompanied by lung fibrosis (Hondermarck et al., 2020). Surprisingly, EGFR inhibitor lapatinib has antiviral activity against SARS-COVID-19 (Raymonda et al., 2020). In conclusion, it appears insightful to monitor COVID-19 survivors taking into consideration the aftermath of SARS-COVID-19 from a neurodegenerative perspective. In addition, the incidence rate of COVID-19 was exceptionally low in chronic myeloid leukemia patients receiving TKIs (Breccia et al., 2020; Semih et al., 2020). Hence, a future retrospective study regarding neurological outcomes of COVID-19 in those patients taking TKIs compared with their control counterparts is highly warranted.
Underlying Causes of Under-Investigation of Epidermal Growth Factor Receptor Inhibitors in Neurodegenerative Disorders
The expression and activation of EGFR are critical for the maturation of neurons (Goldshmit et al., 2004; Liu and Neufeld, 2004; Niyomchan et al., 2015; da Rocha et al., 2020), astrocytes (Liu and Neufeld, 2007), and oligodendrocytes in the developing CNS (Romano and Bucci, 2020). For more details about the functions of EGFR in developmental CNS, see a detailed review by (Romano and Bucci, 2020). Surprisingly, EGFR is down-regulated in adult CNS that is accompanied by quiescent astrocytes (Poiana et al., 2020). Nevertheless, EGFR becomes upregulated again upon neurodegeneration or neuronal injuries associated with astrocyte reactivation (Lupo et al., 2019). In summary, in the last three decades, the myriad functions of EGFR in the developing CNS were investigated. However, the pathogenic mechanism elicited following EGFR activation in neurodegenerative disorders and the impact of EGFR inhibitors in these diseases has been poorly appreciated.
Molecular Mechanisms Involved in the Neuroprotective Effects of Epidermal Growth Factor Receptor Inhibition
Anti-inflammatory effect of EGFR inhibitors: suppression of reactive astrocyte
In 2019, Chen et al. examined the anti-inflammatory actions of EGFR inhibitor, afatinib using primary cultured astrocytes and CTX-TNA2 cells subjected to oxygen and glucose deprivation. They proved that oxygen and glucose deprivation mediated EGFR phosphorylation and stimulated downstream signaling pathways such as Akt and ERK. Furthermore, afatinib amended GFAP, cell proliferating biomarker; proliferating cell nuclear antigen levels, and hypoxia-mediated migratory capability. At the same time, afatinib suppressed cyclooxygenase-II, NO, inducible nitric oxide synthase, caspase-1, and interleukin-1β levels medium (Chen et al., 2019; Figure 2a). A recent study revealed that administration of ibrutinib, an EGFR inhibitor, ameliorated Alzheimer’s behavioral and pathological changes in transgenic mice. The neuroprotective actions were mediated via inhibition of Aβ, p-tau, cyclin-dependent kinase 5, and proinflammatory cytokines. Ibrutinib suppressed Aβ development in the primary and moderate phases of AD as demonstrated by decreased Aβ load in the hippocampus and cerebral cortex of 6-month-old transgenic AD mice (Lee et al., 2021).
EGFR and HER-2 are negative regulators of autophagy
Wang et al. (2017) have proved that HER-2 can control autophagy via its association with Beclin-1, which blocks the autophagy process. Although HER-2 expression becomes quiescent during adulthood, it is reactivated during AD, restricting the autophagy-induced clearance of the APP. As a result, the cumulative APP can be further manipulated by γ-secretase culminating in increased production of APP and amyloid-β. CL-387,785 inhibited HER-2, decreased P62 while increased the LC3II/LC3-I ratio, which resulted in autophagy augmentation with a concurrent clearance of Aβ plaques and successfully relieved cognitive dysfunction of APP/presenilin-1 transgenic AD mice (Wang et al., 2017), supposing that inhibition of HER-2 is a new target to stimulate autophagy.
CL-387,785 rescued memory decline in double transgenic mice at a low dose (5 mg/kg per day) as compared with gefitinib. The same authors used two cell cultures; HEK293-derived cell line and HEK293-derived cell line using different EGFR inhibitors such as CL-387,785 AG825, gefitinib, and lapatinib. Only CL-387,785 but not AG825, gefitinib, or lapatinib blocked the processing of APP in HEK293-derived cells but not in HEK293-derived cells (Wang et al., 2017), establishing the proof-of-principle base for the use of HER-2-targeted drugs for AD (Figure 2b). Tavassoly et al. (2021) found that administration of AZD3759, a highly selective BBB-penetrating EGFR inhibitor, for 3–10 weeks suppressed α-syncline pathology in the striatum and substantia nigra in C57BL6/C3H F1 Parkinson’s mouse model (Tavassoly et al., 2021).
Anti-amyloidogenic effect of EGFR inhibitors
For the first time, Chiang et al. (2010) reported the engagement of EGFR and its downstream PI3K/Akt pathway in AD pathogenesis. They demonstrated that fruit flies that overexpressed Aβ42 had activation of the PI3K/Akt pathway which was accompanied by severe memory impairment (Chiang et al., 2010). The same authors examined the upstream receptors of the PI3K/Akt pathway, insulin receptors, and EGFR through the assessment of behavioral alterations of EGFR-overexpressing fruit flies. Surprisingly, co-overexpression of EGFR and Aβ42 had a synergistic effect on memory loss, proving the pathogenic role of EGFR upregulation in AD (Chiang et al., 2010). Furthermore, inhibition of EGFR was responsible for ameliorating the Aβ-mediated memory decline in APP/PS1 double transgenic mice. Activation of both Aβ42 and p-EGFR was observed in 8-month old AD double transgenic mice. Also, administration of gefitinib in an AD mice model for 7 days resulted in memory retrieval in the Morris water maze test as compared to memantine for the same period (Scholtzowa et al., 2008; Martinez-Coria et al., 2010; Wang et al., 2012). Western blot analysis revealed an elevation of hippocampal expression of p-EGFRTyr1068, the domain for binding with Grb2 that resulted in activation of MAPK, in double transgenic mice. Accordingly, EGFR is the recommended target for the treatment of Aβ-induced cognitive impairment (Wang et al., 2012).
As we hinted earlier, there is an association between p-EGFR and amyloidogenesis. COS-7 cultured cells were co-transfected with genes encoding human EGFRwt and Aβ42. The finding indicated that Aβ42 was drawn down with EGFR, proving the binding of EGFR with amyloid proteins (Wang et al., 2012). Also, 10-day-old male fruit flies treated with erlotinib or gefitinib for one week demonstrated significantly improved memory in a dose-dependent manner for gefitinib. Furthermore, it has been established that EGFR inhibitors, JKF-006, JKF-011, and JKF-027 reversed Aβ42-mediated activation of human EGFR expressing COS-7 cells (Wang et al., 2012).
Wang et al. (2013) demonstrated that the pathogenic role of EGFR is age-dependent. In 10-day-old flies expressing human pan-Aβ42, EGFR expression was elevated, and Aβ42 stimulated EGFR/PI3K, distorted the synaptic plasticity, and eventually resulted in memory impairment. Despite a decrease in total EGFR and p-EGFR in APP/PS1 8-month-old double transgenic mice, the ratio of p-EGFR/EGFR increased relative to wild-type mice (Wang et al., 2013). The previous convergent outcomes endorsed the theory that EGFR serves as a cellular receptor of amyloid peptides, as well as the Aβ-mediated stimulation of EGFR, which plays a crucial role in amyloidogenesis and memory loss. Taken together, the behavioral examination of different EGFR inhibitors warrants more studies to make us conclude that EGFR inhibition is a favorable target for slowing down Aβ-mediated memory decline (Figure 2d). TKIs studies in different AD models are summarized in Additional Table 1 (119.8KB, pdf) .
Under-Explored Downstream Cellular Mechanisms Associated with Epidermal Growth Factor Receptor Inhibition and Neuroprotection
The possible neuroprotective effect induced via inhibition of the EGFR/MAPK pathway
P38 mitogen-activated protein kinase (P38 MAPK) is a member of the MAPK family that responds to stressful stimuli like ROS and inflammatory cytokines. P38-MPAK is highly expressed in the CNS, especially in regions that are pivotal for memory and learning. Hence, dysfunction in p38-MAPK pathways is associated with the pathophysiology of AD (Lee et al., 2017; Figure 3).
Figure 3.
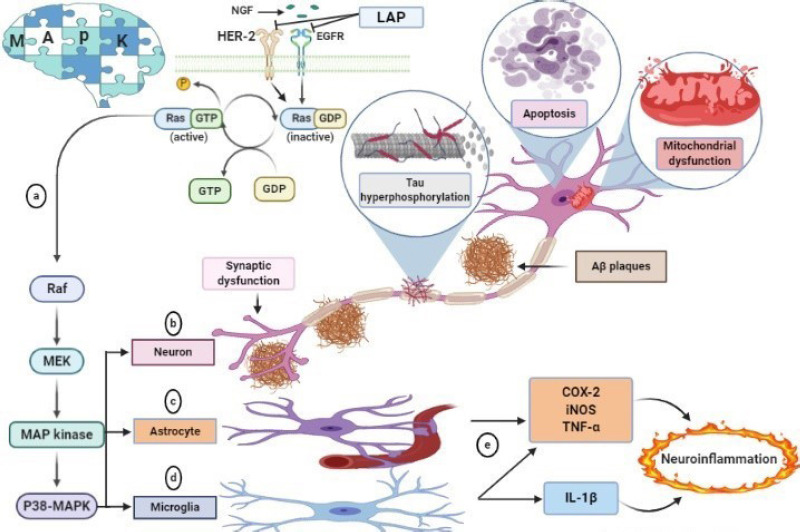
Different pathological roles of P38 MAPK in AD.
(a) The activated EGFR and HER-2 increase the phosphorylation of p38 MAPK. (b) Activation of P38 MAPK in neurons evokes neuronal damages, including tau hyperphosphorylation, synaptic dysfunction, apoptosis, and mitochondrial dysfunction. (c) Activation of p38 MAPK in astrocytes is induced by amyloid plaques. This activation produces TNFα, resulting in excitotoxicity. (d) Activation of P38 MAPK in microglia-mediated by amyloid plaques releases TNFα and IL-1β. (e) Astrocyte and microglial activation promotes neuroinflammation by releasing pro-inflammatory mediators. COX-2: Cyclooxygenase-2; EGFR: epidermal growth factor receptor; HER-2: human epidermal growth factor receptor; IL-1β: interleukin-1 beta; iNOS: inducible nitric oxide synthase; MAPK: mitogen-activated protein; NGF: nerve growth factor; TNF-α: tumor necrosis factor-alpha.
P38 MAPK is a common underlying cause for the initiation of AD. Aβ toxicity (Colié et al., 2017), tau hyperphosphorylation, the elevation of RAGE expression (Fang et al., 2018), deregulated autophagy, and induction of apoptosis are partly tangled and being triggered by stimulation of P38 MAPK. Also, p38 MAPK causes NF-κB activation, disruption of synaptic plasticity, and glutamate excitotoxicity, which justifies that p38 MAPK might be a possible central avenue to break the vicious cycle of Aβ toxicity (Kheiri et al., 2018).
Growing evidence shows that reactive microglia-mediated inflammatory reactions cause secondary damage following traumatic spinal cord injury. EGFR activation caused microgliosis, while EGFR inhibition in both microglial cell culture and spinal cord injury rat model, using AG1478 or C225 for 7 days, suppressed astrocyte and microglial activation and production of tumor necrosis factor alpha (TNFα), interleukin-1β, and GFAP with subsequent inhibition of P38 MAPK, p-EGFR, and ERK (Qu et al., 2012).
Eight-month-old Tg (APPswe.PSEN1dE9) double transgenic mice showed an escalation of hippocampal expression of p-EGFRTyr1068, which resulted in activation of MAPK. Oral administration of gefitinib brought p-EGFRTyr1068 back to normal level (Wang et al., 2012). However, the neuroinflammatory effect exerted by gefitinib that occurred via MAPK inhibition was not studied. Other parameters should be measured such as P38-MAPK, and cytokines such as TNF-α and interleukin-1β with immunofluorescent images of Ib-a and GFAP as markers of microgliosis and astrogliosis including quantification levels. Another study demonstrated that oral administration of lapatinib ditosylate, a dual EGFR inhibitor, in the D-galactose/ovariectomized Alzheimer’s rat model improved Alzheimer’s hallmarks, including Aβ and p-tau and enhanced cognitive performance as proved by Morris water maze and novel object recognition tests. The previous neuroprotective effects were shown to be partly mediated through activation of the pro-survival pathway; PI3K/AKT/GSK-3β along with suppression of neuroinflammatory P38 MAPK, TNF-α, and reactive astrocyte marker; GFAP (Mansour et al., 2021). In conclusion, EGFR signaling is necessary for cytokine production and microglia activation, making it a potential target for the treatment of inflammatory-mediated CNS disorders. Only few studies demonstrate the nexus between EGFR and neuroinflammation in AD (Lee et al., 2021; Mansour et al., 2021). Whether or not curbing neuroinflammation by EGFR inhibition would have neuroprotective effects warrants further studies.
The possible neuroprotective effect induced via interrupting the vicious cycle between EGFR and NADPH oxidase-1
NADPH oxidase-1 (NOX-1) is a multi-subunit membrane-bound protein whose function is to transfer electrons from the plasma membrane to oxygen, which generates superoxide anion and ROS such as hydroxyl radicals and hydrogen peroxide. Over the last few years, NOX-1-derived reactive oxygen species such as H2O2 and superoxide mutase have been thoroughly studied as factors of pathophysiological processes in AD (Fragoso-Morales et al., 2021). NOX-1 activation in the hippocampus is linked to cognitive decline following cerebral hypoperfusion in the cerebral ischemia rat model, which has been shown to activate astrocytes and microglia generating high levels of ROS through NOX (Raz et al., 2010). NOX activity is elevated by Aβ peptides. There was a direct relationship between NOX activity in the AD mouse model in an age-dependent manner (Bruce-Keller et al., 2011). The fact that AD has a direct interrelationship with NOX makes NOX exceptionally up-and-coming biomarkers and therapeutic molecular targets when developing new therapies.
NADPH-oxidase 1 (NOX-1) is implicated in the production of ROS in response to various stimuli including growth factors like transforming growth factor-beta, TNF-α, interleukin-1, angiotensin 2, platelet-derived growth factor, and epidermal growth factor (Ishii and Warabi, 2019). Accordingly, EGFR stimulation, by any of the aforementioned growth factors, induces ROS production through stimulation of the PI3K pathway that results in the activation of the Ras-Rac1 cascade which, in turn, activates NOX. ROS inactivates protein tyrosine phosphatase, a negative regulator of RTK, allowing for sustained activation of RTK. The ROS produced from NOX sustainably activates RTK signaling pathways, creating a continuous niche of oxidative stress and inflammation. In 2021, Mansour et al. showed that oral administration of lapatinib ditosylate, an EGFR/HER-2 inhibitor, in D-gal/OVX Alzheimer’s rat model curbed the expression of hippocampal NOX-1. However, the exact cellular nexus between EGFR inhibition and NOX-1 remains unclear. These data raised the question of how EGFR stimulates oxidative stress in AD? Another question is, along with the previously mentioned activation of the ZNRF1–AKT–GSK3β–CRMP2 cascade (Wakatsuki et al., 2015c), are any other EGFR downstream sig¬naling pathways associated with NOX-1 activation also activated? Worth mentioning, it was claimed that AKT inhibits GSK3β, which, in turn, increased CRMP2 and stabilized microtubules to ameliorate axonal degeneration (Wakatsuki et al., 2015; Araki and Wakatsuki, 2018). Further studies of the nexus between EGFR signaling and NOX-mediated oxidative stress are needed to extend an improved understanding of the oxidative stress regulatory mechanisms in AD.
Possible neuroprotective effect of EGFR induced via inhibition of glutamate-mediated excitotoxicity
Undeniably, glutamate receptors are vital for neuronal functions. However, their excessive stimulation is accused of neuronal death which occurs in AD (Olajide et al., 2021). In 2020, Jia et al. found that oral administration of 100 mg/kg lapatinib inhibited kainic acid-induced epilepsy in mice. This neuroprotective effect was mediated via an increase of glutathione peroxidase 4 and inhibition of oxidative stress, and ferroptosis-mediated cell death. Moreover, lapatinib suppressed glutamate-mediated excitotoxicity in the ferroptotic cell death mice model (Jia et al., 2020). The last result is consistent with a previous study that reported inhibition of GluR-II following oral lapatinib in D-gal/OVX Alzheimer’s rat model (Mansour et al., 2021). Ferroptosis has been a recently discovered form of controlled necrosis distinguished by the accumulation of iron and a massive increase of lipid peroxidative (Dixon et al., 2012). Although many studies have proven that ferroptosis is related to many neurodegenerative disorders such as AD (Yan and Zhang, 2020), further studies are needed to examine the under-investigated possible modulatory effect of EGFR on ferroptosis and glutamate-mediated excitotoxicity in AD.
Future Perspective and Research Implications
Although some EGFR inhibitors exert neuroprotection against neuronal cell death in AD models, via diverse attractive mechanisms (Wang et al., 2012, 2017; Araki and Wakatsuki, 2019; Lee et al., 2021; Mansour et al., 2021), several critical questions are still to be answered before clinical trials begin:
(1) What are the exact mechanisms that link EGFR inhibition with its downstream autophagic and anti-inflammatory effects? Defining the complete scope of downstream signaling pathways that differ in response to EGFR inhibition, might shed further light on the mechanisms by which EGFR inhibition protects neurons in AD models.
(2) Does HER-2 inhibition protect against Aβ toxicity? Only two studies have revealed that enhanced autophagy via using HER-2 inhibitors can reduce amyloid-β burden (Wang et al., 2017; Mansour et al., 2021). Hence, stopping the over-activation of mTOR via EGFR inhibition might protect against Aβ accumulation.
(3) Despite the hopeful ameliorative effects of EGFR inhibitors in AD, there are only two recently published reports that used the BBB crossing RTK-EGFR inhibitors, lapatinib, and ibrutinib (Lee et al., 2021; Mansour et al., 2021). This lack of research caused an issue about the potential repositioning of EGFR inhibitors in AD. So, BBB-crossing EGFR inhibitors are needed to target AD, raising an important question about the use of the third generation of EGFR inhibitors, which are characterized by good CNS-penetrating capacities, such as dacomitinib and AZD3759 (Mok et al., 2017).
(4) EGFR or HER-2 inhibition had a marvelous neuroprotective effect in different AD models (Wang et al., 2012, 2017; Lee et al., 2021), so the use of dual tyrosine kinase inhibitors such as lapatinib has an additive effect upon mono-tyrosine kinase inhibitor (Mansour et al., 2021). So, the use of other dual TKIs is highly recommended.
(5) EGFR has been reported as being essential for certain viruses’ entrance and replication (Hondermarck et al., 2020). Also, EGFR inhibitors can have antiviral activity (Lupberger et al., 2011). These data ignited old un-answered questions; do viruses have a role in the pathophysiology of AD? And what is the nexus between EGFR inhibition and viral entry suppression in AD?
(6) It would be exciting to screen for any possible EGFR inhibition-mediated epigenetic changes in AD models. In Particular, a previous study has shown a nexus between polymorphism in three EGFR nucleotide polymorphisms and AD (Chen et al., 2018). So, such epigenetic alterations could be biomarkers to target in a future clinical trial.
Conclusion
In the current review, we discussed different pathological effects elicited by EGFR activation. Several studies demonstrated that EGFR inhibition amended astrogliosis after injuries (Chen et al., 2019), enhanced autophagy (Wang et al., 2017), ameliorated Aβ toxicity (Wang et al., 2012; Mansour et al., 2021), neuroinflammation (Lee et al., 2021; Mansour et al., 2021) and regenerated axonal degradation (Wakatsuki et al., 2015; Wakatsuki, 2019). Many aspects, however, appear to be understudied, such as the link between EGFR and NOX-1-mediated oxidative stress, and the neuroprotective effect of EGFR inhibition via inhibition of glutamate-mediated excitotoxicity. In conclusion, changes in EGFR signaling pathways are linked with the onset and progression of AD authenticating that inhibition of EGFR expression or signaling could be valuable to counteract neurodegeneration. We recommend that clinical trials should be started to address the potential of EGFR inhibitors as disease-modifying therapies in AD. Consistently, the mechanism of action research, answering the previously outlined questions may connect the dots and elucidate how modulating downstream signaling pathways by EGFR inhibition might endorse neuroprotection.
Additional file:
Additional Table 1 (119.8KB, pdf) : Summary of the effect of epidermal growth factor receptor (EGFR) modulation in different in vivo and in vitro models of Alzheimer’s disease.
Summary of the effect of epidermal growth factor receptor (EGFR) modulation in different in vivo and in vitro models of Alzheimer's disease
Footnotes
Conflicts of interest: The authors declare no conflicts of interest.
Availability of data and materials: All data generated or analyzed during this study are included in this published article and its supplementary information files.
References
- 1.Ahluwalia MS, Becker K, Levy BP. Epidermal growth factor receptor tyrosine kinase inhibitors for central nervous system metastases from non-small cell lung cancer. Oncologist. 2018;23:1199–1209. doi: 10.1634/theoncologist.2017-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Araki T, Wakatsuki S. Regulation of neuronal/axonal degeneration by ZNRF1 ubiquitin ligase. Neurosci Res. 2019;139:21–25. doi: 10.1016/j.neures.2018.07.008. [DOI] [PubMed] [Google Scholar]
- 3.Backer JM. The regulation and function of class III PI3Ks: novel roles for Vps34. Biochem J. 2008;410:1–17. doi: 10.1042/BJ20071427. [DOI] [PubMed] [Google Scholar]
- 4.Bandopadhyay R, Kingsbury AE, Cookson MR, Reid AR, Evans IM, Hope AD, Pittman AM, Lashley T, Canet-Aviles R, Miller DW, McLendon C, Strand C, Leonard AJ, Abou-Sleiman PM, Healy DG, Ariga H, Wood NW, de Silva R, Revesz T, Hardy JA, et al. The expression of DJ-1 (PARK7) in normal human CNS and idiopathic Parkinson's disease. Brain. 2004;127:420–430. doi: 10.1093/brain/awh054. [DOI] [PubMed] [Google Scholar]
- 5.Belambri SA, Rolas L, Raad H, Hurtado-Nedelec M, Dang PM, El-Benna J. NADPH oxidase activation in neutrophils: role of the phosphorylation of its subunits. Eur J Clin Invest. 2018;48(Suppl 2):e12951. doi: 10.1111/eci.12951. [DOI] [PubMed] [Google Scholar]
- 6.Breccia M, Abruzzese E, Bocchia M, Bonifacio M, Castagnetti F, Fava C, Galimberti S, Gozzini A, Gugliotta G, Iurlo A, Latagliata R, Luciano L, Pregno P, Rege-Cambrin G, Rosti G, Stagno F, Tiribelli M, Foà R, Saglio G, Campus CML working group Chronic myeloid leukemia management at the time of the COVID-19 pandemic in Italy. A campus CML survey. Leukemia. 2020;34:2260–2261. doi: 10.1038/s41375-020-0904-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Bruce-Keller AJ, Gupta S, Knight AG, Beckett TL, McMullen JM, Davis PR, Murphy MP, Van Eldik LJ, St Clair D, Keller JN. Cognitive impairment in humanized APP×PS1 mice is linked to Aβ 1-42 and NOX activation. Neurobiol Dis. 2011;44:317–326. doi: 10.1016/j.nbd.2011.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cao X, Fang Y. Transducing oxidative stress to death signals in neurons. J Cell Biol. 2015;211:741–743. doi: 10.1083/jcb.201510105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chen X, Wang C, Zhou S, Li X, Wu L. The impact of EGFR gene polymorphisms on the risk of Alzheimer’s disease in a Chinese Han population: a case-controlled study. Med Sci Monit. 2018;24:5035–5040. doi: 10.12659/MSM.907809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chen YJ, Hsu CC, Shiao YJ, Wang HT, Lo YL, Lin AMY. Anti-inflammatory effect of afatinib (an EGFR-TKI) on OGD-induced neuroinflammation. Sci Rep. 2019;9:2516. doi: 10.1038/s41598-019-38676-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chiang HC, Wang L, Xie Z, Yau A, Zhong Y. PI3 kinase signaling is involved in Aβ-induced memory loss in drosophila. Proc Natl Acad Sci U S A. 2010;107:7060–7065. doi: 10.1073/pnas.0909314107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Colié S, Sarroca S, Palenzuela R, Garcia I, Matheu A, Corpas R, Dotti CG, Esteban JA, Sanfeliu C, Nebreda AR. Neuronal P38α mediates synaptic and cognitive dysfunction in an Alzheimer’s mouse model by controlling β-amyloid production. Sci Rep. 2017;7:45306. doi: 10.1038/srep45306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.da Rocha JF, Bastos L, Domingues SC, Bento AR, Konietzko U, da Cruz E Silva OAB, Vieira SI. APP binds to the EGFR ligands HB-EGF and EGF, acting synergistically with EGF to promote ERK signaling and neuritogenesis. BioRxiv. 2020 doi: 10.1007/s12035-020-02139-2. doi:10.1101/2020.06.12.149062. [DOI] [PubMed] [Google Scholar]
- 14.Dixon SJ, Lemberg KM, Lamprecht MR, Skouta R, Zaitsev EM, Gleason CE, Patel DN, Bauer AJ, Cantley AM, Yang WS, Morrison B 3rd, Stockwell BR. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell. 2012;149:1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Erschbamer M, Pernold K, Olson L. Inhibiting epidermal growth factor receptor improves structural locomotor sensory and bladder recovery from experimental spinal cord injury. J Neurosci. 2007;27:6428–6435. doi: 10.1523/JNEUROSCI.1037-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Faideau M, Kim J, Cormier K, Gilmore R, Welch M, Auregan G, Dufour N, Guillermier M, Brouillet E, Hantraye P, Déglon N, Ferrante RJ, Bonvento G. In vivo expression of polyglutamine-expanded huntingtin by mouse striatal astrocytes impairs glutamate transport: a correlation with Huntington’s disease subjects. Hum Mol Genet. 2010;19:3053–3067. doi: 10.1093/hmg/ddq212. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Fang F, Yu Q, Arancio O, Chen D, Gore SS, Yan SS, Yan SF. RAGE mediates aβ accumulation in a mouse model of Alzheimers disease via modulation of β- and γ-secretase activity. Hum Mol Genet. 2018;27:1002–1014. doi: 10.1093/hmg/ddy017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ferguson FM, Gray NS. Kinase inhibitors: the road ahead. Nat Rev Drug Discov. 2018;17:353–377. doi: 10.1038/nrd.2018.21. [DOI] [PubMed] [Google Scholar]
- 19.Folch J, Petrov D, Ettcheto M, Pedrós I, Abad S, Beas-Zarate C, Lazarowski A, Marin M, Olloquequi J, Auladell C, Camins A. Masitinib for the treatment of mild to moderate Alzheimer’s disease. Expert Rev Neurother. 2015;15:587–596. doi: 10.1586/14737175.2015.1045419. [DOI] [PubMed] [Google Scholar]
- 20.Fragoso-Morales LG, Correa-Basurto J, Rosales-Hernández MC. Implication of nicotinamide adenine dinucleotide phosphate (nadph) oxidase and its inhibitors in Alzheimer’s disease murine models. Antioxidants. 2021;10:218. doi: 10.3390/antiox10020218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Geurdes H. Can free floating SARS-CoV-2 viral material diminish the amount of epidermal growth factor and promote Alzheimer disease in women with COVID-19 in menopause transition? SSRN. 2020 doi:10.2139/ssrn.3670114. [Google Scholar]
- 22.Goldshmit Y, Walters CE, Scott HJ, Greenhalgh CJ, Turnley AM. SOCS2 induces neurite outgrowth by regulation of epidermal growth factor receptor activation. J Biol Chem. 2004;279:16349–16355. doi: 10.1074/jbc.M312873200. [DOI] [PubMed] [Google Scholar]
- 23.Gu X, Li C, Wei W, Lo V, Gong S, Li SH, Iwasato T, Itohara S, Li XJ, Mody I, Heintz N, Yang XW. Pathological cell-cell interactions elicited by a neuropathogenic form of mutant huntingtin contribute to cortical pathogenesis in HD mice. Neuron. 2005;46:433–444. doi: 10.1016/j.neuron.2005.03.025. [DOI] [PubMed] [Google Scholar]
- 24.Hondermarck H, Bartlett NW, Nurcombe V. The role of growth factor receptors in viral infections: an opportunity for drug repurposing against emerging viral diseases such as COVID-19. FASEB Bioadv. 2020;2:296–303. doi: 10.1096/fba.2020-00015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ishii T, Warabi E. Mechanism of rapid nuclear factor-E2-related factor 2 (Nrf2) activation via membrane-associated estrogen receptors: roles of NADPH oxidase 1, neutral sphingomyelinase 2 and epidermal growth factor receptor (EGFR) Antioxidants. 2019;8:69. doi: 10.3390/antiox8030069. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jia JN, Yin XX, Li Q, Guan QW, Yang N, Chen KN, Zhou HH, Mao XY. Neuroprotective effects of the anti-cancer drug lapatinib against epileptic seizures via suppressing glutathione peroxidase 4-dependent ferroptosis. Front Pharmacol. 2020;11:601572. doi: 10.3389/fphar.2020.601572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Khan I, Hatiboglu MA. Can COVID-19 induce glioma tumorogenesis through binding cell receptors. Med Hypotheses. 2020;144:110009. doi: 10.1016/j.mehy.2020.110009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kheiri G, Dolatshahi M, Rahmani F, Rezaei N. Role of P38/MAPKs in Alzheimer’s disease: implications for amyloid beta toxicity targeted therapy. Rev Neurosci. 2018;30:9–30. doi: 10.1515/revneuro-2018-0008. [DOI] [PubMed] [Google Scholar]
- 29.Kim M, Laramy JK, Mohammad AS, Talele S, Fisher J, Sarkaria JN, Elmquist WF. Brain distribution of a panel of epidermal growth factor receptor inhibitors using cassette dosing in wild-type and ABCB1/ABCG2-deficient mice. Drug Metab Dispos. 2019;47:393–404. doi: 10.1124/dmd.118.084210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Koprivica V, Cho KS, Park JB, Yiu G, Atwal J, Gore B, Kim JA, Lin E, Tessier-Lavigne M, Chen DF, He Z. Neuroscience: EGFR activation mediates inhibition of axon regeneration by myelin and chondroitin sulfate proteoglycans. Science. 2005;310:106–110. doi: 10.1126/science.1115462. [DOI] [PubMed] [Google Scholar]
- 31.Lee HJ, Jeon SG, Kim J, Kang RJ, Kim SM, Han KM, Park H, Kim KT, Sung YM, Nam HY, Koh YH, Song M, Suk K, Hoe HS. Ibrutinib modulates Aβ/Tau pathology, neuroinflammation, and cognitive function in mouse models of Alzheimer's disease. Aging Cell. 2021;20:e13332. doi: 10.1111/acel.13332. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lee JK, Kim NJ. Recent advances in the inhibition of P38 MAPK as a potential strategy for the treatment of Alzheimer’s disease. Molecules. 2017;22:1287. doi: 10.3390/molecules22081287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Leinster VH, Joy MT, Vuononvirta RE, Bolsover SR, Anderson PN. ErbB1 epidermal growth factor receptor is a valid target for reducing the effects of multiple inhibitors of axonal regeneration. Exp Neurol. 2013;239:82–90. doi: 10.1016/j.expneurol.2012.09.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Li ZW, Li JJ, Wang L, Zhang JP, Wu JJ, Mao XQ, Shi GF, Wang Q, Wang F, Zou J. Epidermal growth factor receptor inhibitor ameliorates excessive astrogliosis and improves the regeneration microenvironment and functional recovery in adult rats following spinal cord injury. J Neuroinflammation. 2014;11:71. doi: 10.1186/1742-2094-11-71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Lichtenthaler SF, Haass C. Amyloid at the cutting edge: activation of α-secretase prevents amyloidogenesis in an alzheimer disease mouse model. J Clin Invest. 2004;113:1384–1387. doi: 10.1172/JCI21746. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Liu B, Neufeld AH. Activation of epidermal growth factor receptors directs astrocytes to organize in a network surrounding axons in the developing rat optic nerve. Dev Biol. 2004;273:297–307. doi: 10.1016/j.ydbio.2004.06.006. [DOI] [PubMed] [Google Scholar]
- 37.Lupberger J, Zeisel MB, Xiao F, Thumann C, Fofana I, Zona L, Davis C, Mee CJ, Turek M, Gorke S, Royer C, Fischer B, Zahid MN, Lavillette D, Fresquet J, Cosset FL, Rothenberg SM, Pietschmann T, Patel AH, Pessaux P, et al. EGFR and EphA2 are host factors for hepatitis c virus entry and possible targets for antiviral therapy. Nat Med. 2011;17:589–595. doi: 10.1038/nm.2341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lupo G, Gioia R, Nisi PS, Biagioni S, Cacci E. Molecular mechanisms of neurogenic aging in the adult mouse subventricular zone. J Exp Neurosci. 2019;13:117906951982904. doi: 10.1177/1179069519829040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Malafaia D, Albuquerque HMT, Silva AMS. Amyloid-β and Tau aggregation dual-inhibitors: a synthetic and structure-activity relationship focused review. Eur J Med Chem. 2021;214:113209. doi: 10.1016/j.ejmech.2021.113209. [DOI] [PubMed] [Google Scholar]
- 40.Mansour HM, Fawzy HM, El-Khatib AS, Khattab MM. Lapatinib ditosylate rescues memory impairment in D-galactose/ovariectomized rats: potential repositioning of an anti-cancer drug for the treatment of Alzheimer’s disease. Exp Neurol. 2021;341:113697. doi: 10.1016/j.expneurol.2021.113697. [DOI] [PubMed] [Google Scholar]
- 41.Mao L, Wang M, Chen S, He Q, Chang J, Hong C, Zhou Y, Wang D, Li Y, Jin H, Hu B. China: a retrospective case series study. MedRxiv; 2020. Neurological manifestations of hospitalized patients with COVID-19 in Wuhan. doi:10.1101/2020.02.22.20026500. [Google Scholar]
- 42.Martinez-Coria H, Green KN, Billings LM, Kitazawa M, Albrecht M, Rammes G, Parsons CG, Gupta S, Banerjee P, LaFerla FM. Memantine improves cognition and reduces Alzheimer’s-like neuropathology in transgenic mice. Am J Pathol. 2010;176:870–880. doi: 10.2353/ajpath.2010.090452. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.McKerracher L, Rosen KM. MAG, myelin and overcoming growth inhibition in the CNS. Frontiers in Molecular Neuroscience. Front Mol Neurosci. 2015;8:51. doi: 10.3389/fnmol.2015.00051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Mok TS, Wu YL, Ahn MJ, Garassino MC, Kim HR, Ramalingam SS, Shepherd FA, He Y, Akamatsu H, Theelen WS, Lee CK, Sebastian M, Templeton A, Mann H, Marotti M, Ghiorghiu S, Papadimitrakopoulou VA; AURA3 Investigators. Osimertinib or platinum–pemetrexed in EGFR T790M–positive lung cancer. N Engl J Med. 2017;376:629–640. doi: 10.1056/NEJMoa1612674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Müller UC, Deller T, Korte M. Not just amyloid: physiological functions of the amyloid precursor protein family. Nat Rev Neurosci. 2017;18:281–298. doi: 10.1038/nrn.2017.29. [DOI] [PubMed] [Google Scholar]
- 46.Neumann M, Müller V, Görner K, Kretzschmar HA, Haass C, Kahle PJ. Pathological properties of the Parkinson’s disease-associated protein DJ-1 in α-synucleinopathies and tauopathies: relevance for multiple system atrophy and Pick’s disease. Acta Neuropathol. 2004;107:489–496. doi: 10.1007/s00401-004-0834-2. [DOI] [PubMed] [Google Scholar]
- 47.Niyomchan A, Watcharasit P, Visitnonthachai D, Homkajorn B, Thiantanawat A, Satayavivad J. Insulin attenuates arsenic-induced neurite outgrowth impairments by activating the PI3K/Akt/SIRT1 signaling pathway. Toxicol Lett. 2015;236:138–144. doi: 10.1016/j.toxlet.2015.05.008. [DOI] [PubMed] [Google Scholar]
- 48.Olajide OJ, Gbadamosi IT, Yawson EO, Arogundade T, Lewu FS, Ogunrinola KY, Adigun OO, Bamisi O, Lambe E, Arietarhire LO, Oluyomi OO, Idowu OK, Kareem R, Asogwa NT, Adeniyi PA. Hippocampal degeneration and behavioral impairment during Alzheimer-like pathogenesis involves glutamate excitotoxicity. J Mol Neurosci. 2021;71:1205–1220. doi: 10.1007/s12031-020-01747-w. [DOI] [PubMed] [Google Scholar]
- 49.Panpalli Ates M, Karaman Y, Guntekin S, Ergun MA. Analysis of genetics and risk factors of Alzheimer’s disease. Neuroscience. 2016;325:124–131. doi: 10.1016/j.neuroscience.2016.03.051. [DOI] [PubMed] [Google Scholar]
- 50.Poiana G, Gioia R, Sineri S, Cardarelli S, Lupo G, Cacci E. Transcriptional regulation of adult neural stem/ progenitor cells: tales from the subventricular zone. Neural Regen Res. 2020;15:1773–1783. doi: 10.4103/1673-5374.280301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Qu WS, Tian DS, Guo ZB, Fang J, Zhang Q, Yu ZY, Xie MJ, Zhang HQ, Lü JG, Wang W. Inhibition of EGFR/MAPK signaling reduces microglial inflammatory response and the associated secondary damage in rats after spinal cord injury. J Neuroinflammation. 2012;9:178. doi: 10.1186/1742-2094-9-178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Raymonda MH, Ciesla JH, Monaghan M, Leach J, Asantewaa G, Smorodintsev-Schiller LA, Lutz MM, Schafer XL, Takimoto T, Dewhurst S, Munger J, Harris IS. Pharmacologic profiling reveals lapatinib as a novel antiviral against SARS-CoV-2 in vitro. BioRxiv. 2020 doi: 10.1016/j.virol.2021.11.008. doi:10.1101/2020.11.25.398859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Raz L, Zhang QG, Zhou CF, Han D, Gulati P, Yang LC, Yang F, Wang RM, Brann DW. Role of Rac1 GTPase in NADPH oxidase activation and cognitive impairment following cerebral ischemia in the rat. PLoS One. 2010;5:e12606. doi: 10.1371/journal.pone.0012606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Rodriguez S, Hug C, Todorov P, Moret N, Boswell SA, Evans K, Zhou G, Johnson NT, Hyman BT, Sorger PK, Albers MW, Sokolov A. Machine learning identifies candidates for drug repurposing in Alzheimer’s disease. Nat Commun. 2021;12:1033. doi: 10.1038/s41467-021-21330-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Romano R, Bucci C. Role of EGFR in the nervous system. Cells. 2020;9:1887. doi: 10.3390/cells9081887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Scholtzova H, Wadghiri YZ, Douadi M, Sigurdsson EM, Li YS, Quartermain D, Banerjee P, Wisniewski T. Memantine leads to behavioral improvement and amyloid reduction in Alzheimer’s-disease-model transgenic mice shown as by micromagnetic resonance imaging. J Neurosci Res. 2008;86:2784–2791. doi: 10.1002/jnr.21713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Semih B, Tuğçe Nur Y, Mehmet Sinan D, Serdal K, Burhan T, Fevzi A. Tyrosine kinase inhibitors and COVID-19. J Oncol Pharm Pract. 2020;26:2072–2073. doi: 10.1177/1078155220967081. [DOI] [PubMed] [Google Scholar]
- 58.Tavassoly O, Del Cid Pellitero E, Larroquette F, Cai E, Thomas RA, Soubannier V, Luo W, Durcan TM, Fon EA. Pharmacological inhibition of brain EGFR activation by a BBB-penetrating inhibitor, AZD3759, attenuates a-synuclein pathology in a mouse model of α-synuclein propagation. Neurotherapeutics. 2021 doi: 10.1007/s13311-021-01017-6. doi:10.1007/s13311-021-01017-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Tavassoly O, Sato T, Tavassoly I. Inhibition of brain epidermal growth factor receptor activation: a novel target in neurodegenerative diseases and brain injuries. Mol Pharmacol. 2020;98:13–22. doi: 10.1124/mol.120.119909. [DOI] [PubMed] [Google Scholar]
- 60.Volonté C, Morello G, Spampinato AG, Amadio S, Apolloni S, D’Agata V, Cavallaro S. Omics-based exploration and functional validation of neurotrophic factors and histamine as therapeutic targets in ALS. Ageing Res Rev. 2020;62:101121. doi: 10.1016/j.arr.2020.101121. [DOI] [PubMed] [Google Scholar]
- 61.Wakatsuki S. Regulation of neuronal/axonal degeneration by ZNRF1 ubiquitin ligase. Neurosci Res. 2019;139:21–25. doi: 10.1016/j.neures.2018.07.008. [DOI] [PubMed] [Google Scholar]
- 62.Wakatsuki S, Araki T. NADPH oxidases promote apoptosis by activating ZNRF1 ubiquitin ligase in neurons treated with an exogenously applied oxidant. Commun Integr Biol. 2016;9:e1143575. doi: 10.1080/19420889.2016.1143575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Wakatsuki S, Furuno A, Ohshima M, Araki T. Oxidative stress-dependent phosphorylation activates ZNRF1 to induce neuronal/axonal degeneration. J Cell Biol. 2015;211:881–896. doi: 10.1083/jcb.201506102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wang BJ, Her GM, Hu MK, Chen YW, Tung YT, Wu PY, Hsu WM, Lee H, Jin LW, Hwang SL, Chen RP, Huang CJ, Liao YF. ErbB2 regulates autophagic flux to modulate the proteostasis of APP-CTFs in Alzheimer’s disease. Proc Natl Acad Sci U S A. 2017;114:E3129–3138. doi: 10.1073/pnas.1618804114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Wang L, Chiang HC, Wu W, Liang B, Xie Z, Yao X, Ma W, Du S, Zhong Y. Epidermal growth factor receptor is a preferred target for treating amyloid-β-induced memory loss. Proc Natl Acad Sci U S A. 2012;109:16743–16748. doi: 10.1073/pnas.1208011109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wang L, Liang B, Zhong Y. Reduced EGFR level potentially mediates the aβ42-induced neuronal loss in transgenic fruit fly and mouse. Protein Cell. 2013;4:647–649. doi: 10.1007/s13238-013-3043-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Yan N, Zhang J. Iron metabolism ferroptosis and the links with Alzheimer’s disease. Front Neurosci. 2020;13:1443. doi: 10.3389/fnins.2019.01443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Yuen SC, Zhu H, Leung SW. A systematic bioinformatics workflow with meta-analytics identified potential pathogenic factors of Alzheimer’s disease. Front Neurosci. 2020;14:209. doi: 10.3389/fnins.2020.00209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang XX, Tian Y, Wang ZT, Ma YH, Tan L, Yu JT. The epidemiology of Alzheimer’s disease modifiable risk factors and prevention. J Prev Alzheimers Dis. 2021;8:313–321. doi: 10.14283/jpad.2021.15. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Summary of the effect of epidermal growth factor receptor (EGFR) modulation in different in vivo and in vitro models of Alzheimer's disease