

SUMMARY
Canonical CRISPR–Cas systems utilize RNA-guided nucleases for targeted cleavage of foreign nucleic acids, whereas some nuclease-deficient CRISPR–Cas complexes have been repurposed to direct the insertion of Tn7-like transposons. Here we established a bioinformatic and experimental pipeline to comprehensively explore the diversity of Type I-F CRISPR-associated transposons. We report DNA integration for 20 systems and identify a highly active subset that exhibit complete orthogonality in transposon DNA mobilization. We reveal the modular nature of CRISPR-associated transposons by exploring the horizontal acquisition of targeting modules and by characterizing a system that encodes both a programmable, RNA-dependent pathway, and a fixed, RNA-independent pathway. Finally, we analyzed transposon-encoded cargo genes and found the striking presence of anti-phage defense systems, suggesting a role in transmitting innate immunity between bacteria. Collectively, this study substantially advances our biological understanding of CRISPR-associated transposon function and expands the suite of RNA-guided transposases for programmable, large-scale genome engineering.
eTOC Blurb
Klompe et al. explore the natural diversity and function of CRISPR-associated transposons, which direct RNA-guided DNA integration. Further investigation of eighteen new systems reveals the modular nature of molecular machineries that perform DNA targeting and DNA insertion. A selection of high-efficiency, high-fidelity, and fully orthogonal systems is reported.
Graphical Abstract
INTRODUCTION
The past decade has revealed an astounding diversity of CRISPR–Cas systems that utilize RNA guides for sequence-specific nucleic acid targeting, thereby providing host organisms with adaptive immunity against invading mobile genetic elements (MGEs) (Hille et al., 2018; Klompe and Sternberg, 2018). CRISPR-Cas systems are currently grouped into two classes (1–2), six types (I-VI), and dozens of subtypes, depending on the signature and accessory genes that accompany the CRISPR array (Makarova et al., 2020). Although RNA-guided targeting typically leads to endonucleolytic cleavage of the bound substrate, recent studies have uncovered a range of noncanonical pathways in which CRISPR protein-RNA effector complexes have been naturally repurposed for alternative functions. For example, some Type I (Cascade) and Type II (Cas9) systems leverage dedicated guide RNAs to achieve potent transcriptional repression without cleavage (Li et al., 2021; Ratner et al., 2019; Workman et al., 2021), and other Type I (Cascade) and Type V (Cas12) systems are embedded within unusual Tn7-like transposons and lack nuclease components altogether (Faure et al., 2019; Peters et al., 2017). Indeed, rather than providing adaptive immunity, we and others showed that these CRISPR-Cas systems instead function together with transposases to direct RNA-guided DNA integration, thereby facilitating mobilization to new target molecules (Klompe et al., 2019; Strecker et al., 2019).
Tn7-like transposons are pervasive MGEs found in diverse bacterial and archaeal phyla (Parks and Peters, 2007). Unlike other well-known transposons that are mobilized by a single transposase gene product (e.g. Tn3 and Tn5), Tn7-like transposons are notable for their reliance on a heteromeric transposase and their ability to utilize multiple distinct transposition pathways (Haniford and Ellis, 2015; Nicolas et al., 2015; Peters, 2015). The well-studied Tn7 transposon from E. coli, for example, exploits three core transposition genes (tnsA, tnsB, tnsC) and one of two DNA targeting factors (tnsD, tnsE) to achieve a complex lifestyle that enables both specific transposition to a conserved genomic attachment site (tnsABC + tnsD) and promiscuous transposition to mobile plasmids (tnsABC + tnsE) (Kubo and Craig, 1990; Peters and Craig, 2001a). The target site is defined either by TnsD, a sequence-specific DNA binding protein that directs transposition downstream of the glmS gene (Mitra et al., 2010; Waddell and Craig, 1989), or TnsE, which directly binds components of the replication fork and biases transposition to conjugative plasmids (Peters and Craig, 2001b; Shi et al., 2015). Collectively, these alternative targeting pathways allow for both vertical and horizontal transmission of Tn7. TnsB is a DDE-family integrase that exhibits sequence-specific binding of both transposon ends and, together with TnsA, forms a heteromeric transposase that cleaves both ends of the transposon during excision and catalyzes transposon integration at the target (Sarnovsky et al., 1996; Tang et al., 1991). Importantly, the ATPase TnsC moderates between the transposase and both targeting modules through direct physical interactions (Choi et al., 2014; Stellwagen and Craig, 1998). Other Tn7-like transposons similarly encode the core transposition machinery in combination with distinct targeting factors, such as TniQ-family proteins that direct transposition to other conserved attachment sites (Peters, 2019; Peters et al., 2014), or CRISPR-Cas systems that allow for RNA-guided DNA-targeting (Klompe et al., 2019; Strecker et al., 2019).
We previously characterized a CRISPR-associated transposon (CRISPR-Tn, also referred to as CAST) derived from Vibrio cholerae (Tn6677), which employs a type I-F3 CRISPR-Cas effector complex for RNA-guided DNA integration (Figure 1A) (Klompe et al., 2019). This particular CRISPR-Cas subtype encodes an effector complex called Cascade, which consists of a CRISPR RNA (crRNA), Cas6, Cas7, and a Cas8-Cas5 fusion protein (hereafter Cas8), but lacks any adaptation or nuclease components required for adaptive immunity (Makarova et al., 2020). Intriguingly, we found that Cascade forms a co-complex with the transposition protein TniQ, providing a physical link between the otherwise independent CRISPR-Cas and transposition machineries (Halpin-Healy et al., 2020). Together, these CRISPR- and transposon-associated factors direct efficient and highly specific integration of the transposon ~50-bp downstream of the target site, which we refer to as INTEGRATE (INsertion of Transposable Elements by Guide RNA-Assisted TargEting) for genome engineering applications. Independent recruitment events have led to the evolution of CRISPR-Tn that utilize different CRISPR-Cas systems, including type V-K (Faure et al., 2019; Strecker et al., 2019) and type I-B (Peters et al., 2017; Saito et al., 2021), for RNA-guided targeting. CRISPR-Tn also encode specialized crRNAs that facilitate distinct RNA-guided pathways, as well as regulatory components that allow for the temporal control of transposition (Petassi et al., 2020). The plethora of interesting properties revealed for the CRISPR-Tn systems studied so far encouraged us to investigate the natural diversity of these systems more extensively, through both bioinformatic and experimental analysis.
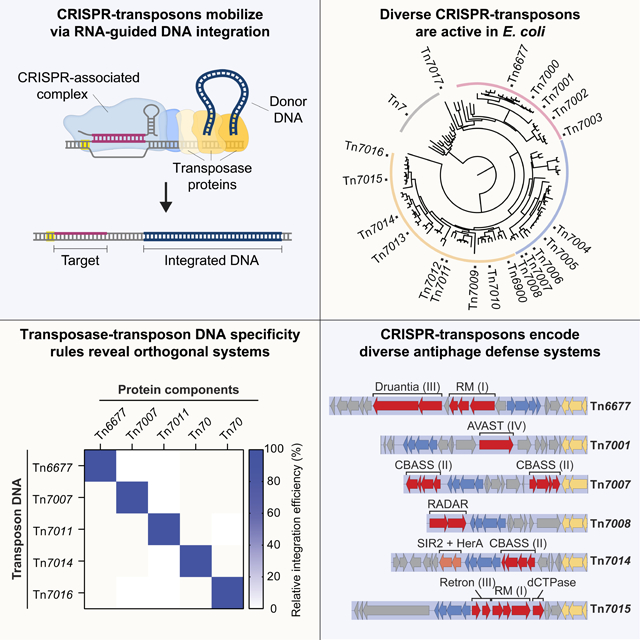
Figure 1. RNA-guided transposition activity for twenty diverse type I-F3 CRISPR-Tn.
(A) Genomic layout of Tn6677 (V. cholerae INTEGRATE, VchINT). The machinery required for transposon mobilization can be functionally divided into an excision and integration module that performs transposition chemistry, and a DNA targeting module that defines the integration site. Type I-F3 CRISPR-Tn use the RNA-guided DNA-binding complex TniQ-Cascade (crRNA1Cas81Cas76Cas61TniQ2) for target site determination. Other cargo genes are omitted for clarity. L, left end; R, right end. (B) Phylogenetic tree of TnsB proteins found in a similar genetic context as depicted in A. Tn numbers indicate the CRISPR-Tn systems tested in this study; TnsB from Tn7 is used as an outgroup. (C) Overview of selected Type I-F3 CRISPR-Tn homologs. Colors represent phylogenetic clustering of TnsB as in B. AB fusion, natural TnsA-TnsB fusion. Locus refers to the host gene found adjacent to the right end of transposon, which provides a target for the atypical crRNA homing pathway; no atypical homing crRNA was found for Tn7017/parE, marked with an *. (D) Schematic representation of a transposition assay in which a mini-Tn is targeted to a site in the E. coli genome and detected via junction PCR. (E) Integration efficiency for all homologs at 37 °C, measured by qPCR. ND, not detected. (F) Integration efficiency for select homologs at 37 °C and 25 °C, measured by qPCR. ND, not detected. Data in (E) and (F) are shown as mean ± s.d. for n = 3 biologically independent samples. See also Figure S1.
Here we explore the mechanistic diversity of type I-F3 CRISPR-Tn and describe activity for twenty diverse systems. CRISPR-Tn homologs are active at different temperatures, exhibit relaxed protospacer adjacent motif (PAM) requirements, and act orthogonally in both recognition and mobilization of the donor DNA. By examining the modular nature of the transposition and targeting components, we show that targeting modules can be exchanged horizontally and demonstrate activity for an alternate RNA-independent pathway directed to a conserved attachment site. Finally, we performed bioinformatic analysis of the cargo genes encoded by CRISPR-associated transposons and found enrichment in a diverse array of anti-phage defense systems, implicating these transposons in promoting rapid gain and loss of defense genes as part of the bacterial pan-immune system (Bernheim and Sorek, 2019).
RESULTS
Identification and characterization of twenty active Type I-F3 CRISPR-Tn systems
To explore the natural variation among CRISPR-associated transposons, we first established a bioinformatic pipeline to identify and prioritize a large set of Type I-F3 CRISPR-Tn systems for experimental analysis. Briefly, we used V. cholerae protein components from Tn6677 as a query and performed iterative rounds of psiBLAST to assemble large homolog sets, extracted genomic contigs encoding all protein components, and identified left and right transposon boundaries based on their characteristic structure (Methods). Enzymatic active sites and CRISPR arrays were manually inspected for a subset of candidate systems, and we ultimately selected twenty homologous systems from a range of gammaproteobacterial species whose TnsB transposase proteins are well distributed across a number of clearly distinguishable clades (Figure 1B). Newly characterized transposons were numbered Tn7000–7017, and together with V. cholerae Tn6677 (Klompe et al., 2019) and A. salmonicida Tn6900 (Petassi et al., 2020), our set of systems comprise variants that encode natural tnsA-tnsB fusions, possess multiple copies of tniQ, and are found next to distinct attachment sites (Figure 1C, Table S1).
For each system, we synthesized and cloned a donor plasmid (pDonor) encoding the mini-Tn, alongside an effector plasmid (pEffector) that encodes a crRNA and 6–8 protein components (Vo et al., 2021a). Transposition was assayed in E. coli BL21(DE3) cells using a crRNA targeting lacZ, and integration events in either of two possible orientations were quantified using qPCR (Figure 1D). The majority of systems were functional at 37 °C, albeit across a wide range of activities, with some catalyzing targeted integration at near 100% efficiency without selection for the insertion event (Figure 1E). Since many systems derive from species that grow at lower temperatures, we next repeated transposition assays at 25 °C and found that activity was greatly increased for multiple transposons, with Tn7012 and Tn7017 showing the most dramatic temperature-dependent improvement (Figure 1F and Figure S1A). We previously found that Tn6677 exhibits bidirectional integration but heavily favors one orientation product (Klompe et al., 2019), and we found that our transposon homologs were similarly biased, with some showing a >103:1 orientation bias (Figure S1B). In addition to their standard CRISPR arrays, both I-F3 and V-K CRISPR-Tn systems encode atypical CRISPR RNAs characterized by unusual repeats and spacers, which direct transposition to specific genomic attachment sites (Petassi et al., 2020; Saito et al., 2021). In some cases, these atypical crRNAs are differentially regulated or direct enhanced integration activity, when compared to typical crRNAs (Petassi et al., 2020). We identified the atypical CRISPR arrays for each of our CRISPR-Tn homologs and tested integration efficiency at the same target site using these atypical repeats with fully matching spacer sequences (Figure S1A, S1C, Table S1). Most systems functioned as well or slightly better with atypical crRNAs.
Collectively, these experiments demonstrate a robust bioinformatics and experimental pipeline for the prediction and characterization of active CRISPR-Tn systems that require co-expression of numerous interacting components. Furthermore, our ability to reconstitute activity in E. coli for 18 newly characterized systems that derive from a diverse range of bacterial species strongly suggests that these systems function autonomously and independently of specific host factors other than those needed for gap repair.
RNA-guided transposition with I-F3 systems exhibits flexible PAM requirements
Canonical DNA-targeting CRISPR-Cas systems rely on specific recognition of protospacer adjacent motifs (PAMs) for efficient binding and cleavage, and thereby avoid any accidental and lethal self-targeting of the CRISPR array (Leenay and Beisel, 2017; Mojica et al., 2009). However, we previously found that RNA-guided transposition with V. cholerae Tn6677 proceeded with a remarkably diverse set of PAMs (Klompe et al., 2019). Motivated by these findings, we sought to analyze the PAM requirements of our CRISPR-Tn homologs using a library approach, in which a fully randomized 5-bp sequence is cloned directly adjacent to the target site (Figure 2A). Junction PCR and deep sequencing then allows for selective amplification of successful integration products and comparison of enriched PAM motifs to the starting input library.
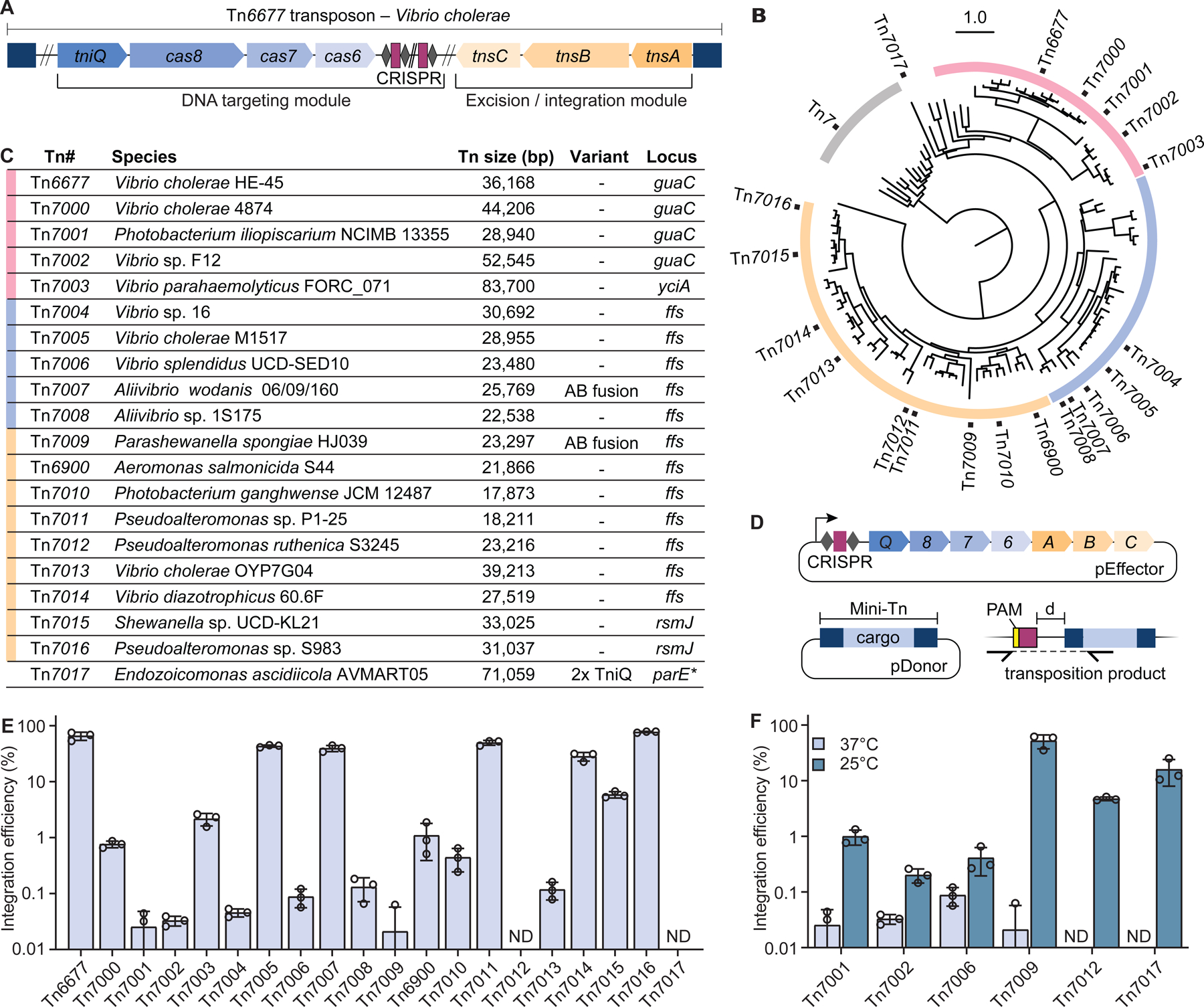
Figure 2. PAM requirements and integration site variation for CRISPR-Tn homologs.
(A) Schematic representation of a PAM library transposition assay, in which a pTarget plasmid encodes a 32-bp target sequence flanked by a 5-bp degenerate sequence. (B) Violin plots of PAM enrichment for five highly active Type I-F3 and a type V-K CRISPR-Tn (Tn6999, also referred to as S. hofmannii INTEGRATE, ShoINT). Lines represent 10-fold enrichment or depletion. *, PAM sequences not detected in the final library. (C) PAM preference is represented by WebLogos for the top 5% enriched PAM sequences, for CRISPR-Tn homologs indicated. PAM sequences are shown for the non-targeted strand, with the −1 position corresponding to the base immediately adjacent to the 5’ end of the target sequence as schematized in A. (D) Integration site distribution obtained from the PAM library data is shown for ‘CC’ PAMs (Tn6677, Tn7007, Tn7011, Tn7014, Tn7016) or ‘GTN’ PAMs (Tn6999) only. d, distance in bp from the 3’ end of the target to the integrated transposon. (E) Integration efficiencies for the CRISPR-Tn homologs and PAMs indicated, normalized to a ‘CC’ PAM. Data are shown as mean ± s.d. for n = 3 biologically independent samples. (F) Recognition of CC PAM by I-F1 Cascade from P. aeruginosa (left; PDB ID 6NE0) and I-F3 TniQ-Cascade (right; PDB ID 6VBW). The guanine base paired with C −1 forms hydrogen bonds with N250 (left) and N246 (right), alongside other subtype-specific contacts. Both systems exploit a positively charged residue (K247 and R243) as a ‘wedge’ to facilitate DNA unwinding. See also Figure S2.
Interestingly, we found that PAM enrichment values for most I-F3 CRISPR-Tn systems were narrowly distributed and often did not pass the 10-fold enrichment thresholds typically applied for PAM analysis of other CRISPR-Cas effectors (Figure 2B and Figure S2A) (Leenay et al., 2016). This was in sharp contrast to the data for a representative V-K CRISPR-Tn (Tn6999, also known as ShoINT) (Vo et al., 2021a), for which many nonfunctional PAMs dropped out altogether and a clear 5’-GTN-3’ PAM motif emerged from those that were enriched >10-fold. Therefore, we instead analyzed PAMs found within the top 5% of enriched sequences and detected a ‘CN’ preference for most I-F3 systems (Figure 2C and Figure S2B), consistent with PAM wheel visualizations and an empirically and bioinformatically deduced ‘CC’ PAM for related I-F1 CRISPR-Cas systems (Almendros et al., 2012; Rollins et al., 2019; Vercoe et al., 2013). Integration events for all homologs obeyed the rules previously described for Tn6677, occurring primarily 48–51 downstream of the target site for substrates bearing a ‘CC’ PAM (Figure 2D and Figure S2B). The presence of ‘self’ PAMs in the output library, albeit at lower abundance, suggests that transposition could occur downstream of the CRISPR array itself, consistent with our previous observation of self-targeting (Vo et al., 2021a). To validate these PAM library results, we measured the integration efficiency of Tn6677 and Tn7016 for individual ‘CN’ and ‘NC’ PAMs within the same target plasmid context (Figure 2E). These data revealed that plasmids with any ‘CN’ PAM could be targeted for transposition with indistinguishable efficiency, in excellent agreement with the library results. Tn7016 in particular exhibited nearly PAM-less activity, with only a modest 2-fold decrease in activity at the ‘AC’ PAM. This tolerance indicates that Type I-F3 CRISPR-Tn systems are intrinsically well suited for flexible genomic targeting without PAM restriction.
We were curious to interpret these data in light of our recent structures of V. cholerae TniQ-Cascade (QCascade) bound to a ‘CC’ target DNA, alongside similar structures from a I-F1 P. aeruginosa Cascade (aka Csy complex) (Halpin-Healy et al., 2020; Jia et al., 2020; Rollins et al., 2019). The −2 position, where C is preferred, is contacted by a highly conserved serine residue for the transposon-encoded Cascade complexes, whereas PaeCascade makes base-specific interactions using N111 (Figure 2F). Interestingly, both Vch and PaeCascade complexes employ an asparagine to make specific hydrogen bonds with the minor groove face of guanine within the C-G base pair at the −1 position; this interaction is predicted to be absent for Tn7016, which has alanine at the same position and may exhibit greater PAM tolerance as a result. The use of a positively charged residue as a ‘wedge’ to open up the DNA appears to be broadly conserved in both I-F1 and I-F3 complexes.
Distinct TniQ family proteins provide a protein-only transposon homing pathway
In our selection of CRISPR-Tn systems, we were intrigued by the unusual presence of two distinct tniQ family genes in Tn7017 from an Endozoicomonas ascidiicola (Eas) isolate (Figure 3A). We hypothesized that, as with canonical Tn7 pathway choice and I-B CRISPR-Tn systems (Saito et al. 2021), Tn7017 may encode two distinct transposition pathways that rely on alternative TniQ family proteins. We also noticed that Tn7017 was the only CRISPR-Tn system in our homolog set that lacked an identifiable CRISPR spacer that could explain the insertion of Tn7017 downstream of the highly conserved parE gene (Figure S1C), and reasoned that an alternative targeting pathway could potentially explain the presence of Tn7017 at this attachment site.
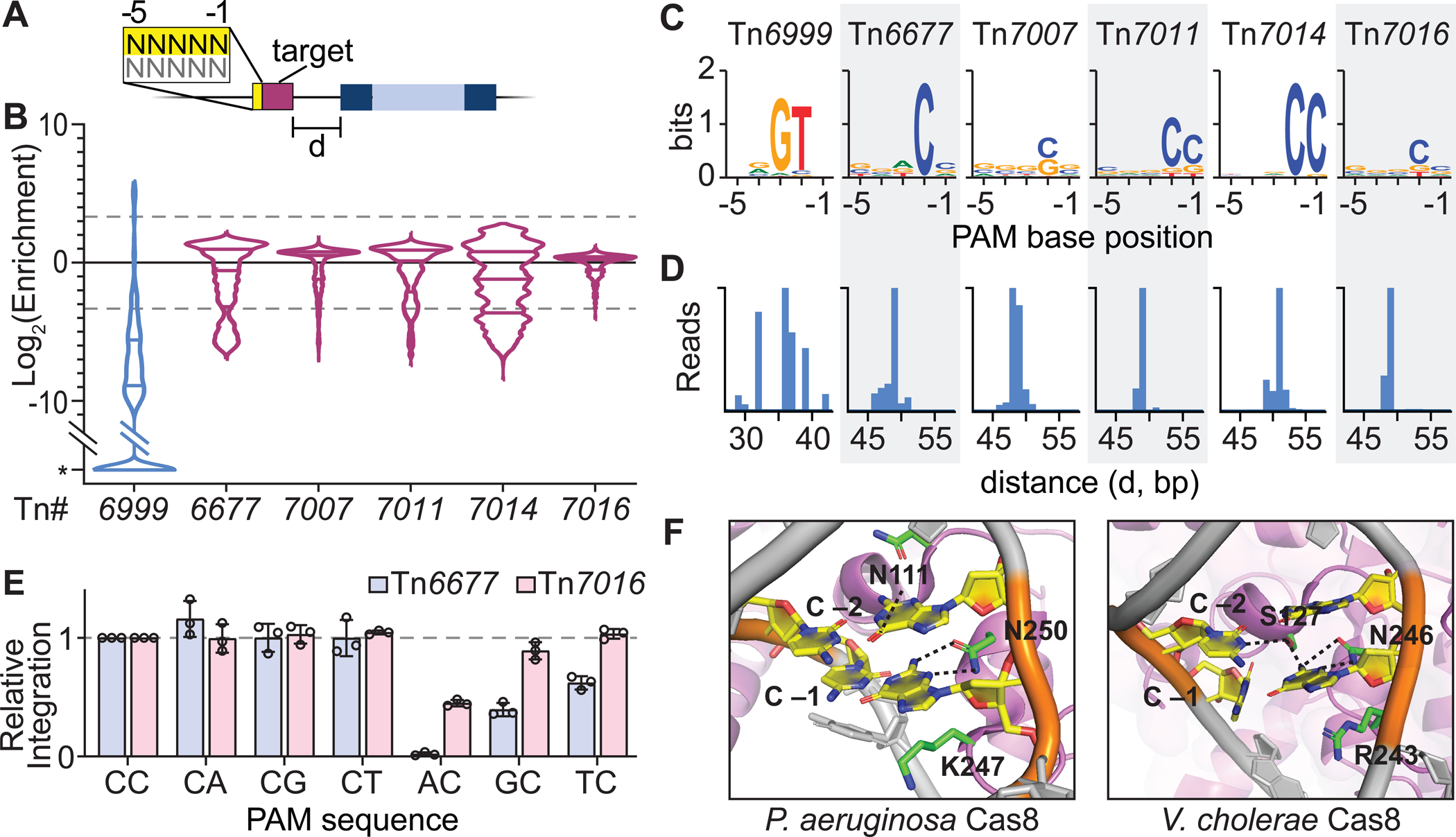
Figure 3. Tn7017 exploits distinct TniQ homologs for two different targeting pathways.
(A) Schematic representation of Tn7017 from E. ascidiicola, showing the presence of two distinct TniQ-family genes, labeled tniQ and tnsD. ‘TniQ’ and ‘TnsD’ are used to describe TniQ-family proteins involved in the RNA-dependent and RNA-independent targeting pathways, respectively. (B) Pruned phylogenetic tree of TniQ/TnsD family proteins based on tniQ domain (PF06527) alignment. Clades containing known Tn7-like transposons and CRISPR-Tn systems (I-B1, I-B2, V-K, and I-F3) are indicated. Black text indicates a TniQ protein, red text indicates a TnsD protein. A red square indicates that a TnsD domain (PF15978) was identified in the full-length protein. Also shown are TniQ and TnsD proteins from two Parashewanella species (Ptr and Pcu) described in Petassi et al. (2020), which are also predicted to facilitate RNA-guided transposition and RNA-independent parE homing, respectively. (C) Transposition assay design for simultaneous detection of DNA integration at a genomic target site (RNA-dependent) and a putative, plasmid-borne attachment site (RNA-independent). Arrows indicate the predicted integration sites relative to the target sites. (D) Integration efficiency for pTarget and the genomic target site, as measured by qPCR, under different gene deletion conditions. Data are shown as mean ± s.d. for n = 3 biologically independent samples. See also Figure S3.
One of the homologous genes, EasTniQ, is found within the same operon as cas8-cas7-cas6 and encodes a similarly sized protein as other I-F3 CRISPR-Tn systems (397 aa), whereas the second variant, denoted EasTnsD, is encoded on its own operon downstream of the CRISPR array and is much larger (630 aa) (Figure S3A). Phylogenetic analysis further revealed that EasTniQ was more closely related to TniQ proteins involved with RNA-guided transposition (Figure 3B, S3B), while EasTnsD showed little sequence homology to other TniQ homologs from our selection (Figure S3C). We therefore suspected that Tn7017 mobilizes through an RNA-dependent pathway that exploits EasQCascade to promote horizontal transmission, as well as an RNA-independent pathway that exploits EasTnsD for sequence-specific targeting of the attachment site parE to promote vertical transmission, in a process termed ‘homing’.
To test this hypothesis, we generated a target plasmid (pTarget) with the 3’ end of the E. ascidiicola parE gene, which contains the anticipated EasTnsD binding site, and monitored simultaneous transposition to pTarget (RNA-independent) and a genomic target site (RNA-dependent) (Figure 3C). Transposition was indeed directed to both target sites, with the insertion site downstream of parE recapitulating the native genomic location of Tn7017 (Figure 3D, S3D). Next, we performed gene deletions and showed that integration into pTarget required EasTnsD but proceeded independently of Cascade, demonstrating that TnsABCD constitutes an independent targeting pathway directed at the parE attachment. In contrast, EasTniQ was necessary for the RNA-guided transposition pathway but functioned only when combined with Cascade, in agreement with our previous studies elucidating the structure and function of the QCascade co-complex (Halpin-Healy et al., 2020; Klompe et al., 2019). Interestingly, RNA-guided transposition efficiency at the genomic target increased drastically when EasTnsD was omitted, whether or not pTarget was present (Figure 3D, S3E), suggesting that EasTnsD may somehow inhibit QCascade formation or compete for binding downstream transposase components. Notably, similar trends were also seen for gene deletion experiments with a Type I-B CRISPR-Tn system (AvCAST) (Saito et al., 2021).
Collectively, these data provide the first experimental evidence of a type I-F3 CRISPR-Tn system that leverages two TniQ-family proteins for distinct targeting pathways, as initially proposed by Peters and colleagues (Petassi et al., 2020). Future structural studies will be needed to reveal how two divergent TniQ homologs with less than 25% sequence identity are still able to functionally recruit the same core TnsABC transposase machinery, and whether they do so using a common or divergent architectural scaffold.
Horizontal acquisition of TniQ-Cascade operons reveals functional modularity
When we performed sequence analyses of all protein components involved in RNA-guided transposition (Figure S4A), we found key discrepancies in sequence conservation between protein components derived from the transposition (TnsABC) and targeting (QCascade) operons. We were especially intrigued to find extensive heterogeneity in the degree of synteny between two CRISPR-associated transposons, Tn7005 and Tn7013 (Figure 4A). Both of these transposons are integrated downstream of the ffs gene in related V. cholerae strains and are nearly identical in their tniQ-cas8-cas7-cas6 operons (97% identity at the DNA level), while encoding diverged tnsA-tnsB-tnsC operons (48% identity at the DNA level, and ~35% identity at the protein level) (Figure 4B). This specific case illustrates the existence of distinct evolutionary trajectories governing the two operons within these transposons, as has been previously described (McDonald et al., 2019).
Figure 4. Modularity of transposition and DNA targeting modules.
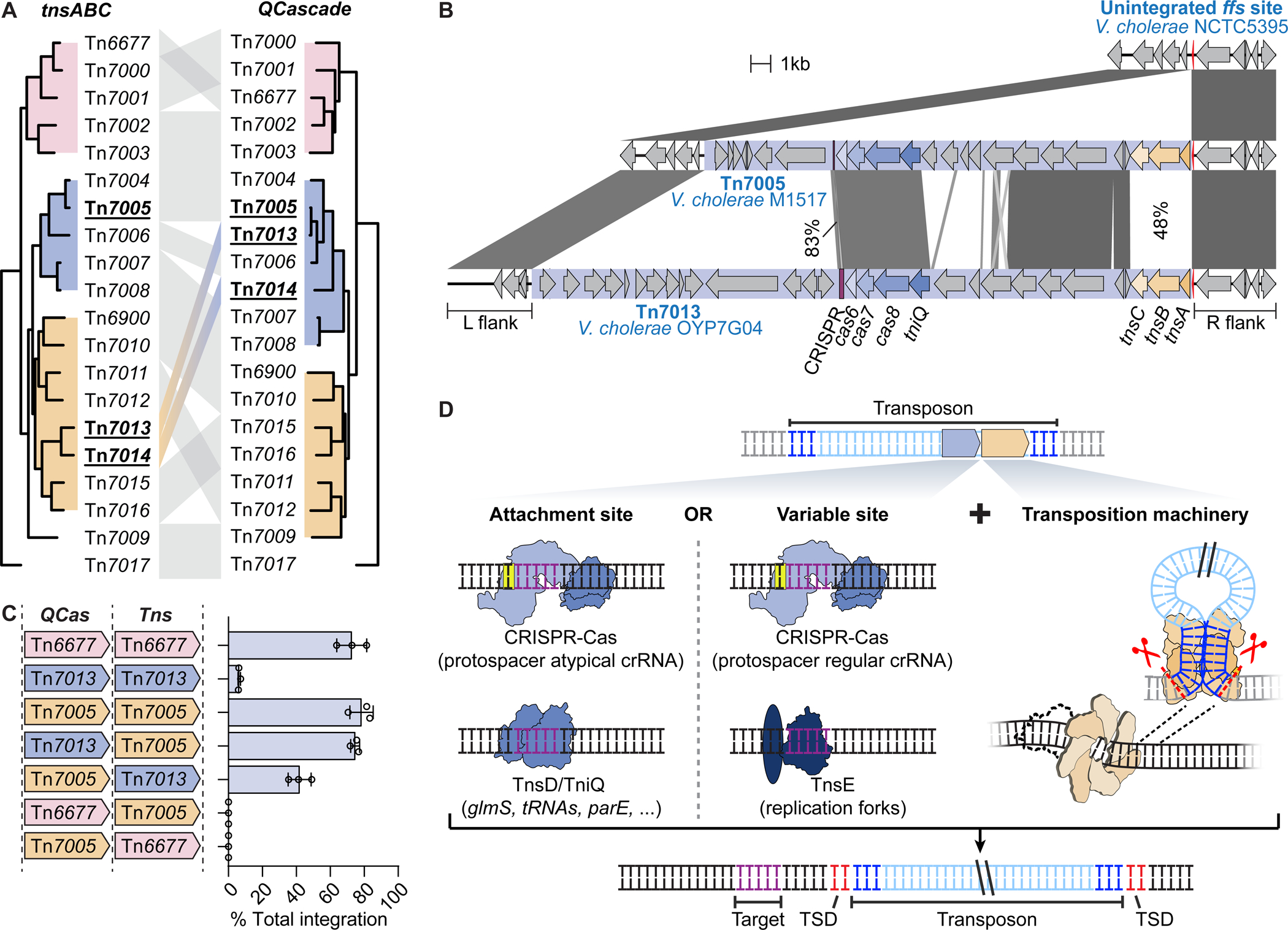
(A) Phylogenetic comparison of the tnsABC and tniQ-cas876 operons of CRISPR-Tn tested in this study. Tn7013 and Tn7014 show evidence of module exchange, in which a recombination event may have disrupted co-evolution of the transposition and DNA targeting modules. (B) Schematic representation of the sequence similarity between Tn7005 and Tn7013, both of which are found downstream of the ffs gene in separate V. cholerae strains. DNA sequence similarity is indicated for homologous regions. (C) Integration efficiency for different combinations of tniQ-cas876 (QCas) and tnsABC (Tns) operons from Tn6677, Tn7005, and Tn7013, as measured by qPCR. Data are shown as mean ± s.d. for n = 3 biologically independent samples. (D) Tn7-like transposons are modular in nature and encode homing pathways alongside flexible pathways that target mobile genetic elements. TSD, target site duplication. See also Figure S4.
The observation that nearly identical targeting modules can function with diverged transposition modules implies flexible communication between the QCascade and TnsABC machineries. To test this assumption, we cloned a series of pEffector variants in which various genes/operons between distinct CRISPR-Tn systems were combined, and then assayed transposition activity (Figure 4C, S4B). When we exchanged operons between Tn7005 and Tn7013, we found that transposition was equally active (mini-Tn7005) or even more active (mini-Tn7013), consistent with the natural synteny of these systems. In contrast, pEffector variants with module exchange between Tn7005 and Tn6677, whose tnsA-tnsB-tnsC operons show similarly low sequence identity as those from Tn7005 and Tn7013, were entirely nonfunctional (Figure 4C), indicating that the ability for QCascade and TnsABC to cross-react is not a generalizable property of CRISPR-Tn systems. Furthermore, any exchange of individual genes within the tnsA-tnsB-tnsC operon between Tn7005 and Tn7013 also ablated transposition (Figure S4B), highlighting the critical role of cognate protein-protein interactions between transposase components for productive assembly of an enzymatically active transposition complex.
In the case of Tn7005 and Tn7013, the divergence between the transposition genes and transposon end sequences suggests that two evolutionarily distinct transposon lineages captured the same CRISPR-Cas module independently, and in both cases retained the ability to be efficiently programmed by the same RNA-guided targeting machinery. When taken together with our Tn7017 data, these results suggest the TnsABC module is remarkably adaptable to novel targeting factors, including TnsD, TnsE, TniQ-Cas12k, TniQ-Cascade (Figure 4D), and possibly additional candidate proteins that have yet to be experimentally investigated (Peters, 2019). In the case of E. coli Tn7, Craig and colleagues have shown that TnsC functions as a key regulator between the targeting factors (TnsD or TnsE) and the heteromeric TnsAB transposase (Gamas and Craig, 1992). Given that TnsAB interactions have been mapped to the C-terminus of TnsC, whereas TnsD interactions have been mapped to the N-terminus — with the core AAA+ ATPase domain sandwiched in between (Choi et al., 2014; Mitra et al., 2010; Ronning et al., 2004) — we speculate that one ‘face’ of this protein (or protein complex) directly engages the targeting factors, with the remainder of the structural scaffold maintaining conserved interactions with the transposase components. Recent cryo-EM and crystal structures of EcoTnsC indeed confirm that one side of the TnsC ring is constituted by the N-terminal regions that interact with the targeting factors, while the other side of the ring structure consists of C-terminal tails that are expected to interact with TnsA and TnsB (Shen et al., 2021). This multi-component architecture likely enabled Tn7-like transposons to coopt alternative targeting mechanisms throughout their evolution, without disrupting the core enzymatic machinery..
Type I-F3 CRISPR-Tn systems encode diverse bacterial defense systems as cargo
Transposase (tnsABC) and CRISPR-Cas (tniQ-cas8-cas7-cas6) operons are often separated by tens of kilobases within their native transposons, and can be flanked by numerous additional genes. This large physical distance at the primary sequence level could offer substantial opportunities for recombination and thereby facilitate targeting module exchange in Tn7-like transposons, as observed with Tn7005 and Tn7013. Indeed, a comparison of closely related Tn7005-like transposons located at the ffs locus in distinct V. cholerae strains (Figure S4C) revealed rapid loss and gain of many transposon-encoded genes, including the CRISPR-Cas system itself.
We previously highlighted the presence of defense systems in V. cholerae Tn6677 and Type V-K CRISPR-associations transposons, including restriction-modification (RM) systems (Klompe et al., 2019), and were intrigued to further analyze the cargo genes mobilized by other Type I-F3 CRISPR-associated transposons. Analysis of all genes embedded within the transposons from this study using DefenseFinder (Tesson et al., 2021) revealed a striking and diverse array of innate immune systems. These antiviral defense systems included Druantia (Doron et al., 2018), AVAST (Gao et al., 2020), CBASS (Cohen et al., 2019), retrons (Gao et al., 2020; Millman et al., 2020) and even a recently described dCTPase (Severin et al., 2021; Tal et al., 2021) (Figure 5, S5). Although more comprehensive analyses will be necessary, our observations strongly suggest that these transposons may provide a fitness benefit to host cells by mobilizing defense systems to confer broad protection against phage predation. Remarkably, other classes of mobile genetic elements, including the SXT integrative and conjugative elements (ICEs), similarly encode and mobilize phage defense systems within Vibrio cholerae (LeGault et al., 2020), highlighting the critical role played by diverse horizontal gene transfer events in sculpting bacterial immune system repertoires. Given the rate of cargo gene exchange between closely related CRISPR-associated transposons, one can envision these mobile genetic elements experiencing many opportunities to diversify both their specific genetic payload capacity, as well as the machinery dictating their specific mobilization mechanism.
Figure 5. Type I-F3 CRISPR-Tn systems carry diverse bacterial defense systems as cargo.
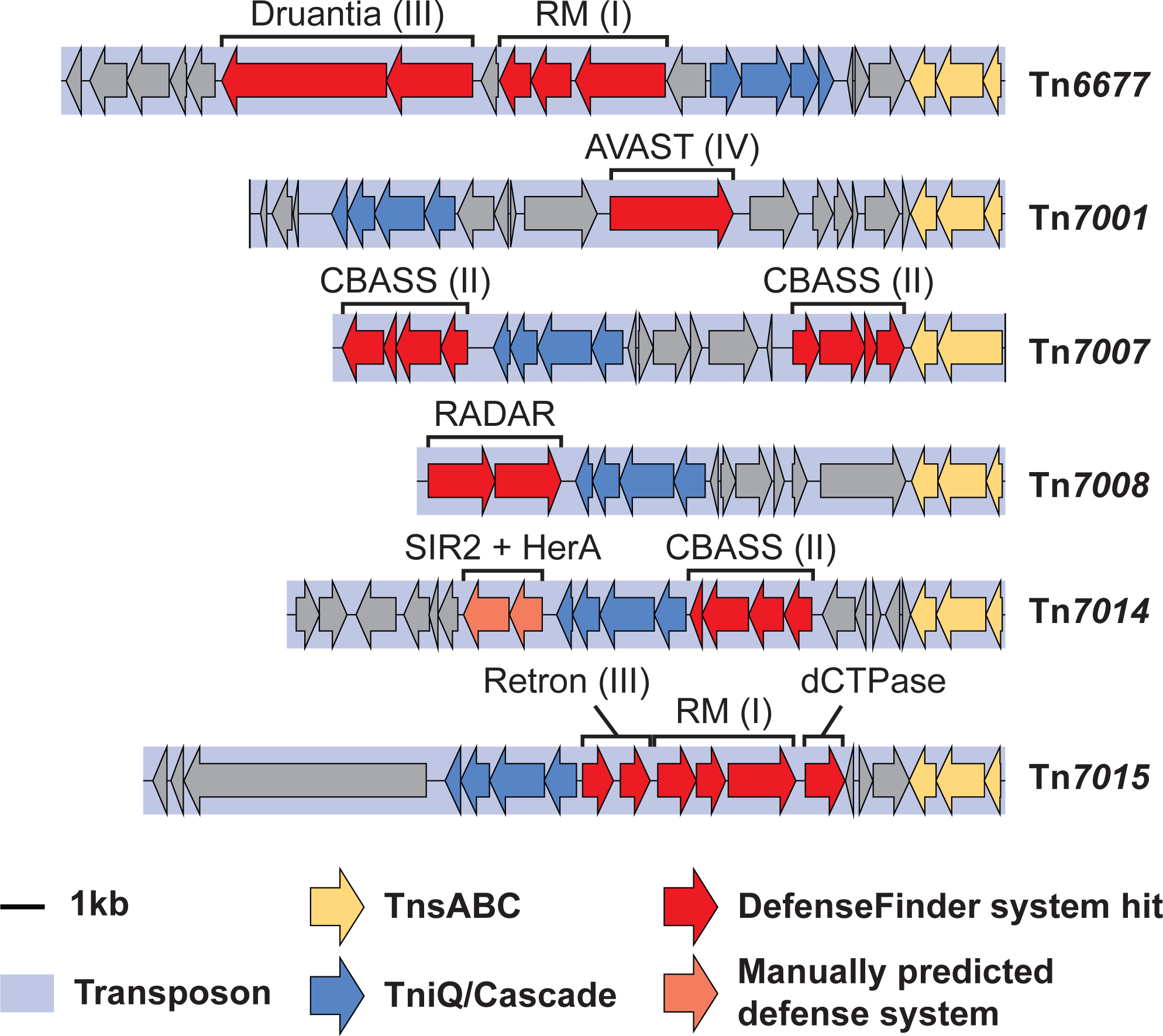
The native genomic layout of a subset of CRISPR-Tn homologs is shown. Transposons are highlighted with a light blue box and shown with the right (R) end on the right side of the figure. Transposition and DNA targeting modules are shown in yellow and blue, respectively. Putative defense systems are highlighted in red when the complete gene set was found. Roman numerals in brackets indicate the predicted subtype of the defense system. A large diversity of defense systems are found, suggesting that CRISPR-Tn systems provide a benefit to bacterial hosts by providing innate immunity from phage predation. See also Figure S5.
Diverse CRISPR-associated transposons exhibit orthogonal, high-accuracy integration
Our analysis of CRISPR-Tn phylogeny, and the general observation of modularity in the targeting and transposase machinery, led us to next investigate the extent of transposase-transposon co-evolution and DNA sequence specificity during transposition. Tn7-like transposons comprise left and right ends that each harbor 2–4 partially repetitive sequences that are ~20-bp in length (Figure S6A), and experiments with E. coli Tn7 have demonstrated that these sequences are specifically bound by TnsB (Arciszewska and Craig, 1991). However, the degree of TnsB substrate specificity has not been carefully investigated, especially for related transposons within this larger Tn7 family, and we were curious to determine how flexibly the transposase machinery from our homologous CRISPR-Tn systems would recognize each other’s donor DNAs. To test this, we performed pooled library transposition assays, in which pEffector plasmids were reacted with all twenty pDonor substrates in a single transformation step (Figure 6A). Successful integration products were then deep sequenced, and comparison to the starting library yielded enrichment scores describing the relative activity between each pDonor and the protein components from a given CRISPR-Tn system (Methods).
Figure 6. Orthogonality of type I-F3 CRISPR-Tn through specific sequence determinants.
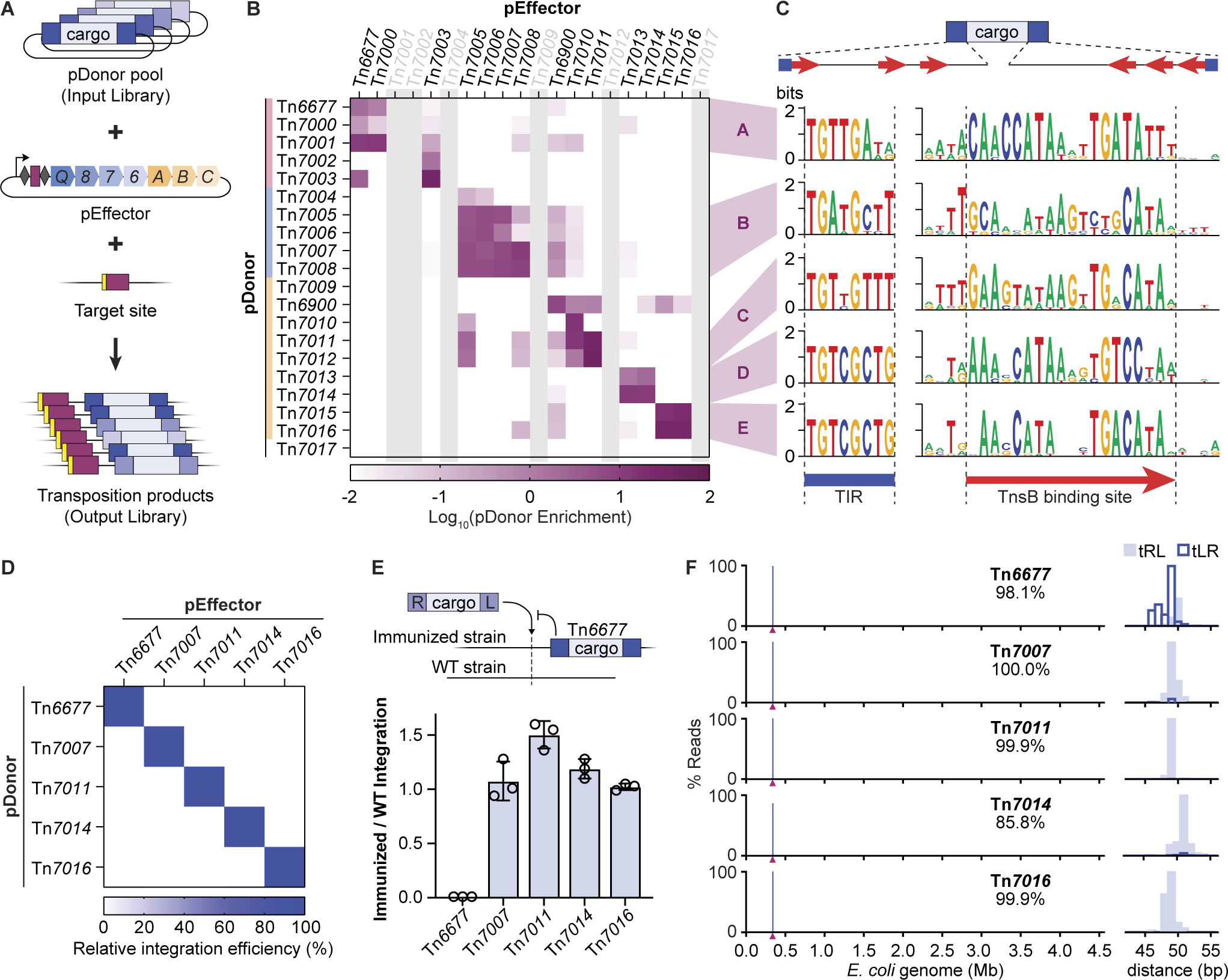
(A) Schematic of the pooled library approach to determine cross-reactivity between protein-RNA machinery (pEffector) and the mini-transposon DNA (pDonor). (B) Integration efficiency for pairwise pEffector (x-axis) and pDonor (y-axis) combinations from the library experiment shown in (A), plotted as the log enrichment of each pDonor per pEffector variant as compared to the input library. Mini-Tn with similar compatibility patterns are indicated with letters A-E. Systems with wild-type integration activity below 0.5%, when incubated at 37 °C, are greyed out. (C) Schematic representation of sequence motifs within the transposon ends (top). WebLogos of the terminal inverted repeat (TIR) sequences (left) and TnsB binding site sequences (right) for each of the predicted CRISPR-Tn compatibility groups. The red arrow represents the 19-bp TnsB binding site sequence, and high sequence conservation outside or low sequence conservation inside of this region indicates that TnsB binding site length might differ between CRISPR-Tn. (D) Integration efficiencies for each individual pEffector + pDonor combination shown, as measured by qPCR, to validate the assignment of compatibility groups shown in B. Data for each mini-Tn were plotted relative to the cognate pEffector/pDonor combination. (E) Relative integration efficiency for each CRISPR-Tn system shown, tested in a strain with or without a pre-existing mini-Tn6677, measured by qPCR. Data are shown as mean ± s.d. for n = 3 biologically independent samples. These data demonstrate that orthogonal CRISPR-Tn systems can be used for high-efficiency tandem insertions of genetic payloads. (F) Genome-wide specificity data for highly active, orthogonal CRISPR-Tn systems using Tn-seq (left), and the accompanying base-pair resolution view of the integration site (right). The maroon triangle on the x-axis indicates the target site for RNA-guided integration. Distance refers to the distance in bp from the 3’ end of the target to the integrated transposon. On-target percentages are shown. See also Figure S6.
Pooled library transposition results revealed hotspots of integration activity, with most pDonor substrates acted upon only by a narrow range of effectors (Figure 6B). Intriguingly, some substrates (Tn7009 and Tn7017) could not be acted upon by any pEffector in our collection, aside from their cognate pairing, which corresponds well with their low TnsB sequence identity compared to the other systems tested (Figure S4A). The greatest pDonor enrichment was found along the diagonal, suggesting that, as expected, the RNA-guided transposase machinery is most active on its own cognate transposon ends. Some of the larger clusters of active transposition (e.g. Tn7005–Tn7008) indicated a greater degree of cross-reactivity, and the clear clustering between evolutionarily related transposons indicated the presence of CRISPR-Tn ‘compatibility groups,’ which we classified into groups A–E (Figure 6B-C). We hypothesized that transposons within each group would have conserved sequence motifs in the predicted TnsB binding sites, explaining why their donor DNA substrates could be acted upon by RNA-guided transposases occupying the same group. We performed a detailed analysis of the transposon ends from each homologous CRISPR-Tn system, identified the location and sequence of each TnsB binding site in the left and right ends, and then generated consensus sequences for each individual CRISPR-Tn, as well as the transposons within each compatibility group (Figure S6B, 6C). Although there were highly conserved nucleotides across all members of the Type I-F3 CRISPR-Tn systems, each compatibility group exhibited subtype-specific motifs.
To validate the anticipated cross-reactivity between compatibility groups, as well as our pooled library results, we selected one system from each group and individually assayed transposition activity for all pEffector-pDonor combinations. All five systems directed high-efficiency and completely orthogonal RNA-guided DNA integration, with exquisite specificity for only its cognate donor DNA substrate (Figure 6D). In light of these data, we hypothesized that orthogonal CRISPR-Tn systems would allow for genomic sites to be efficiently retargeted for the generation of tandem DNA insertions, without any repressive effect from target immunity. E. coli Tn7 has been shown to prevent multiple insertions at the same target site through the action of TnsB and TnsC (Stellwagen and Craig, 1997), and we recently showed that the same immunity mechanism exists for V. cholerae Tn6677 (Vo et al., 2021a). However, if a genomically integrated transposon cannot be efficiently used as a donor DNA substrate by the machinery from an orthogonal CRISPR-Tn system, then presumably the mechanism of blocking multiple insertions would also be lost. We compared the integration efficiency of orthogonal CRISPR-Tn systems in E. coli strains that either lacked a pre-existing transposon, or contained a mini-transposon derived from Tn6677 downstream of the same site being targeted by the orthogonal system. Unlike the target immunity data with Tn6677, where the efficiency of a second insertion was close to 0%, orthogonal CRISPR-Tn systems could generate a second insertion with the same efficiency, regardless of the presence of mini-Tn6677 (Figure 6E, S6C). Furthermore, when we performed Tn-seq analysis, we found that each of the homologs exhibited highly specific genome-wide integration (Figure 6F).
These data confirm that transposase-transposon DNA sequence specificity dictates both transposition activity and target immunity effects, and provide a straightforward opportunity to leverage multiple orthogonal CRISPR-Tn systems for high-efficiency genomic DNA integration in a given bacterial strain, without spatial restriction. Importantly though, such efforts must also consider similarities between CRISPR repeat sequences, which could lead to crRNA cross-reactivity when orthogonal systems are co-expressed.
DISCUSSION
CRISPR-associated transposons are an elegant example of the guns-for-hire principle (Koonin et al., 2019), as Tn7-like transposons co-opted and repurposed targeting modules from the microbial pangenome, but also because they help sustain their lifestyle by mobilizing cargo genes that might provide a selective advantage to their host. In this study we present a comprehensive experimental survey of evolutionarily diverse, Type I-F3 CRISPR-associated transposons (also known as CAST). We report activity for twenty CRISPR-Tn homologs that mediate RNA-guided transposition, and provide insights into the modular architecture that enables communication between DNA targeting and DNA integration machineries. Additional analysis of the broader genetic context reveals the likely role of CRISPR-Tn systems in mobilizing antiviral defense systems and other biological pathways between bacteria via horizontal gene transfer.
CRISPR-Tn systems as generators of defense islands
CRISPR-associated transposons can carry large cargoes in addition to their transposition modules. Our bioinformatic analysis of this content revealed a surprisingly high degree of gene diversity between closely related transposons and illustrates high cargo turnover rates. These cargo genes frequently encode operons predicted to encode antiviral defense systems. Bacterial defense mechanisms are often predicted through a guilt-by-association approach, in which the recurring proximity of a candidate gene or gene cluster to other characterized defense systems increases its likelihood of also representing a defense system (Doron et al., 2018; Makarova et al., 2011). These clusters of defense genes are referred to as ‘defense islands,’ yet a plausible mechanism that explains how these islands came to be remains unclear. More recently, it has become clear that mobile genetic elements can serve as hotspots for anti-phage defense systems (LeGault et al., 2020; Rousset et al., 2021), and that these systems exhibit exceedingly rapid evolutionary turnover, leading to extremely high diversity in the degree of phage sensitivity/resistance between otherwise clonal strains (Hussain et al., 2021; Piel et al., 2021). These findings are in excellent agreement with our CRISPR-Tn observations, in which transposons with highly diverse cargo genes are targeted to the same conserved locus in different bacterial strains. Collectively, these lines of evidence support the pan-immune system model that posits the critical role of HGT in enabling rapid exchange and turnover of diverse defense mechanisms between closely related strains (Bernheim and Sorek, 2019).
Targetable space of RNA-guided transposition
Canonical CRISPR-Cas systems are protected from self-targeting of the CRISPR array through the presence of a protospacer adjacent motif (PAM), which provides the only distinction between an invading nucleic acid and the copy of that intruder that is retained in the CRISPR array. However, in this work we show that the PAM requirements of most Type I-F3 CRISPR-associated transposons is quite relaxed. While self-targeting of the CRISPR array by canonical CRISPR-Cas systems is detrimental to the host (Stern et al., 2010), self-targeted transposition does not create DNA breaks and would lead to downstream integration, thus leaving the spacer-repeat sequence intact. It is therefore likely that the cost of a flexible PAM requirement, and thereby potential self-targeting, is not as high for CRISPR-Tn as it is for canonical CRISPR-Cas systems. Additionally, self-targeting may be inhibited by a phenomenon called target immunity, in which the presence of transposon ends near the target site prevents transposition to that location (Stellwagen and Craig, 1997; Vo et al., 2021a). Stringent PAM recognition is also thought to accelerate the target search process (Collias and Beisel, 2021), as is required to successfully counter phage infections during adaptive immunity. Transposases, however, generally evolve to be suboptimal for transposition in order to balance the cost and benefits of mobilization (Bourque et al., 2018; Lampe et al., 1999), and may hence prefer relaxed PAM requirements. On top of that, CRISPR-Tn are restricted in their targetable space when it comes to RNA-guided homing pathways. These dedicated atypical guides require targeting of a highly conserved sequence without interrupting this likely essential region by integration of the transposon. Relaxed PAM requirements might provide CRISPR-Tn systems access to these important regions, thereby supporting the vertical spread of the transposon.
Transposition lifestyle of Type I-F3 CRISPR-associated transposons
All of the Type I-F3 systems in this study encode a heteromeric TnsA-TnsB transposase that is expected to completely excise the transposon from the donor prior to target insertion, and thereby generate simple insertion products. In contrast, Type V-K CRISPR-Tn lack TnsA altogether and transpose through the formation of a cointegrate structure (Rice et al., 2020; Strecker et al., 2020; Vo et al., 2021b). Interestingly, two systems we investigated encode natural TnsA-TnsB fusions and demonstrated highly efficient transposition. This, and the fact that similar fusions were found for Type I-B2 CRISPR-associated transposons (Saito et al., 2021), suggest there might be a benefit to linking TnsA-TnsB activities or expression levels. A fusion product could potentially remove a rate-limiting step in the form of TnsA recruitment, or by ensuring concerted cleavage reactions between TnsA and TnsB during the excision step of transposition. The latter could also play a role in regulating the relative frequency of simple insertions versus cointegrate products, as we recently described (Vo et al., 2021b). It would be interesting to see if certain environments favor one transposition lifestyle (by simple insertion or through cointegrate formation) over the other, and what the driving forces would be behind this.
Concluding remarks
The suite of homologous RNA-guided transposons presented in this study has not only revealed natural mechanistic differences, but also inspires future screening and engineering efforts. The apparent modularity of targeting modules, and the knowledge that transposition and targeting operons need not be physically proximal, encourage future efforts to mine Tn7-like transposons for novel targeting pathways. The activity of natural protein fusions, and the increase in transposition efficiency with limited changes in the targeting module, inspire the pursuit of protein engineering. Furthermore, CRISPR-Tn provide an exciting path for generating large programmed DNA insertions without generating double-strand breaks in the target DNA, requiring homology arms in the donor DNA, or the need for host DNA recombination machinery. Therefore, having a large array of diverse homologous systems will facilitate ongoing efforts to harness programmable, targeted DNA integration for bacterial genome engineering, and also considerably expand the available toolbox for heterologous reconstitution in other cell types.
Limitations of the Study
Although our study surveyed a broad sampling of Type I-F3 CRISPR-associated transposons, non-canonical systems that exhibit unexpected genetic architectures or highly diverged genes may have escaped detection by our bioinformatics approach. Future studies will be necessary to further mine sequenced genomes and catalog the full diversity of Tn7-like transposons and their associated targeting modules. Furthermore, while cargo gene analyses provided compelling insights into the likely selective advantages that CRISPR-Tn systems confer on their hosts, this approach should be complemented by future microbiology and/or metagenomics experiments to investigate RNA-guided transposition within native ecological samples, ideally while monitoring any effects on strain fitness and phage sensitivity. These approaches will be critical to reveal the native biological roles played by CRISPR-associated transposons.
STAR METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Samuel H. Sternberg (shsternberg@gmail.com).
Materials availability
Reagents generated in this study are available from the lead contact upon reasonable request. A subset of the plasmids generated in this study will also be made available on Addgene.
Data and code availability
Deep sequencing data have been deposited at the sequence read archive (SRA) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original agarose gel images have been deposited at Mendeley and are publicly available as of the date of publication. DOIs are listed in the key resources table.
All original code has been deposited at Mendeley and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli NEB® Turbo | NEB | Cat# C2984 |
| Escherichia coli BL21(DE3) | NEB | Cat# C2527 |
| Escherichia coli 10-beta electrocompetent | NEB | Cat# C3020K |
| Escherichia coli BL21(DE3) electrocompetent | Millipore Sigma | Cat# CMC0016 |
| Chemicals, peptides, and recombinant proteins | ||
| LB Broth | Fisher | Cat# BP1426 |
| LB Agar | Fisher | DF0445-07-6 |
| Spectinomycin | Gold Bio | Cat# S-140 |
| Kanamycin | Gold Bio | Cat# K-120 |
| Carbenicillin | Gold Bio | Cat# C-103 |
| IPTG | Gold Bio | Cat# I2481C |
| Klenow Fragment | NEB | Cat# M0212L |
| Critical commercial assays | ||
| SsoAdvanced Universal SYBR Green Supermix | BioRad | Cat# 1725275 |
| Promega Wizard Genomic DNA purification kit | Promega | Cat# A1120 |
| NEBNext Library Quant Kit | NEB | Cat# E7630L |
| Deposited data | ||
| Raw agarose gel image | This paper, Mendeley Data | doi:10.17632/89p4wdryt3.1 |
| Illumina sequencing data | This paper, SRA | SRA: PRJNA769066 |
| Custom python scripts | This paper, Mendeley Data | doi:10.17632/89p4wdryt3.1 |
| Oligonucleotides | ||
| All oligonucleotides used in this study. | This paper. | Table S3 |
| Recombinant DNA | ||
| All plasmids used in this study. | This paper. | Table S2 |
| Software and algorithms | ||
| PSI-BLAST (2.10.0+) | NCBI | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| NCBI Efetch | NCBI | https://www.ncbi.nlm.nih.gov/books/NBK25499/ |
| Entrez Programming Utilities | NCBI | https://www.ncbi.nlm.nih.gov/books/NBK25501/ |
| CRISPRCasFinder (4.2.2) | Couvin et al., 2018 | https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index |
| Krona | Ondov et al., 2011 | https://github.com/marbl/Krona |
| WebLogo (2.8.2) | Crooks et al., 2004 | https://weblogo.berkeley.edu/ |
| Illumina pipeline script | Vo et al., 2020 | https://github.com/sternberglab/Illumina-pipeline |
| CD-HIT | Li et al., 2001, 2002 | https://github.com/weizhongli/cdhit |
| MUSCLE (v3.8.1551) | Edgar, 2004 | N/A |
| FastTree (version 2.1.10) | Price et al., 2010 | http://www.microbesonline.org/fasttree/ |
| iTOL (6.4) | Letunic and Bork, 2021 | https://itol.embl.de/itol.cgi |
| HMMER 3.3.2 | Eddy, 1995 | http://hmmer.org/ |
| Geneious Prime® 2019.2.1 | Biomatters | https://www.geneious.com/ |
| Prokka (version 1.14.6) | Seemann, 2014 | https://github.com/tseemann/prokka |
| DefenseFinder (v0.9, model v0.0.2) | Tesson et al., 2021 | https://defense-finder.mdmparis-lab.com/ |
| EasyFig | Sullivan et al., 2011 | https://mjsull.github.io/Easyfig/ |
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Bacterial strains
E. coli strains were routinely grown in Luria Broth (LB) at 37°C with shaking or on LB agar plates at 37°C overnight. When required, antibiotics were added at the following concentrations: carbenicillin (100 μg/ml), kanamycin (50 μg/ml), or spectinomycin (50 μg/ml). For protein expression, 0.1 mM isopropyl β-d-1-thiogalactopyranoside (IPTG) was added.
METHOD DETAILS
Bioinformatics pipeline for system detection
Protein sequences corresponding to Vibrio cholerae TnsA, TnsB, TnsC, TniQ, Cas8, Cas7, and Cas6 from the Tn6677 transposon were obtained from Klompe et al. (2019). These sequences were used as queries for PSI-BLAST (ncbi-blast-2.10.0+ release) against the nr database (version 3/27/20) using the parameters: -evalue 0.005 -num_alignments 9999999 -num_iterations 15. Unique protein IDs were extracted from each PSI-BLAST result file and used for further analysis. The genomic accession ID corresponding to each protein ID was retrieved using NCBI Efetch, and genomic IDs with hits for TniQ, Cas8, Cas7, Cas6, TnsA, TnsB, and TnsC, referred to as the Minimal Gene Set (MGS), form our initial set of potential homologs. A genomic accession ID was scored as containing a Type I-F3 CRISPR-Tn system if it contained PSI-BLAST hits in the following order (with no restriction on the linear distance between each PSI-BLAST hit): [TnsA,TnsB,TnsC,TniQ,Cas8,Cas7,Cas6], [TnsA,TnsB,TnsC,Cas6,Cas7,Cas8,TniQ], [TnsC,TnsB,TnsA,Cas6,Cas7,Cas8,TniQ], [Cas6,Cas7,Cas8,TniQ,TnsC,TnsB,TnsA], [TniQ,Cas8,Cas7,Cas6,TnsC,TnsB,TnsA], [Cas6,Cas7,Cas8,TniQ,TnsA,TnsB,TnsC], [TniQ,Cas8,Cas7,Cas6,TnsA,TnsB,TnsC], [TnsB,TnsA,TnsC,TniQ,Cas8,Cas7,Cas6], [Cas6,Cas7,Cas8,TniQ,TnsC,TnsA,TnsB], [TnsA,TnsB,TnsB,TnsC,TniQ,Cas8,Cas7,Cas6], or [Cas6,Cas7,Cas8,TniQ,TnsC,TnsB,TnsB,TnsA].
To determine the transposon ends of potential homolog systems, a user-defined length of genomic sequence (default = 100,000 bp) upstream and downstream of the MGS was extracted using Entrez Programming Utilities. Genomic “flanks” upstream and downstream of the MGS were then used for target site duplication (TSD) + terminal inverted repeat (TIR) detection in intergenic regions. Thereafter, we first predicted all open reading frames (ORFs) within the genomic flanks using EMBOSS getorf (minsize = 200; table = 11). All genomic sequences within predicted ORFs were excluded from the TSD+TIR search. A 5’ sliding window searched between the ORFs downstream of the transposon MGS for a 5 bp TSD candidate. For every TSD candidate, a 3’ sliding window searched upstream of the transposon MGS for a matching TSD candidate. Once a pair of 5’ and 3’ TSDs was found, the 2 bp upstream and downstream of the respective TSD were checked to match a TG/AC dinucleotide motif.
To predict TnsB binding sites within putative transposon ends, a sliding window of length 18 bp was defined downstream of a putative 5’ TSD. In order to determine repeats on the same end, a second window iterated from the first window position until the 5’ MGS coordinate was reached (or up to 500 bp). After each iteration, the hamming distance (defined as the number of mismatches) was calculated between the first and second windows. A match was registered if the sequences had a Hamming distance <= 3. All positions of the second sliding window that produced matches were recorded, along with the position of the first window. Subsequently, a third sliding window iterated from the 3’ TSD until the 3’ MGS coordinate was reached (or up to 500 bp). The first sliding window was compared to the reverse complement of the third sliding window and registered a match if the sequences had a Hamming distance <=3. The reverse complement was taken because TnsB binding sites in each transposon end are oriented in opposite directions. All positions of the third sliding window that produced matches were recorded, along with the position of the first window. This analysis yielded the hamming distance between all possible pairs of 18-mers within 500 bp from each transposon end, and the most plausible transposon ends were selected manually.
CRISPR arrays were predicted using CRISPRCasFinder 4.2.2 (Couvin et al., 2018) (Standard settings: no Cas gene detection), and were checked for the presence of a CUGCC-like stem-loop in CRISPR repeats. Conservation of active site residues in TnsA, TnsB, TnsC, TniQ, and Cas6 were checked manually (delineated in Klompe et al., Supplementary Figures 2–6). Atypical repeats were predicted manually based on similarity to the typical repeats and stem loop-forming sequences.
Experimental pipeline for system characterization
Vector design:
Expression vectors (pEffector) were designed according to Vo et al. (2020), where a single T7 promoter drives the expression of a CRISPR array (repeat-spacer-repeat), the native tniQ-cas8-cas7-cas6 operon, and the native tnsA-tnsB-tnsC operon from a pCDF-Duet-1 backbone. The accompanying pDonor vectors were designed to encode 250 bp encompassing the Left and Right transposon end sequences, on either end of a chloramphenicol resistance gene, generating a mini-Tn of 1307-bp in size, on a pUC19 backbone. Single-plasmid versions (pSPIN) were generated by amplifying the mini-Tn from pDonor through PCR, and inserting it into digested pEffector plasmids following the design described in Vo et al. (2020).
Transposition assays:
All transposition experiments were performed in E. coli BL21(DE3) cells (NEB). For experiments including pDonor and pEffector, chemically competent cells harboring one of the plasmids were transformed with the second plasmid, and transformants were isolated by selective plating on double antibiotic LB-agar plates containing IPTG. For experiments with pSPIN vectors, transformants were plated on LB-agar plates containing spectinomycin and IPTG. Transformations were done via heat shock at 42 °C for 30 sec, and after recovering cells in fresh LB medium at 37 °C for 1 h, cells were plated on LB-agar plates containing the appropriate antibiotics and inducer (100 μg ml−1 carbenicillin, 50 μg ml−1 spectinomycin, 0.1 mM IPTG). After overnight growth at 37 °C for 18 h, hundreds of colonies were scraped from the plates, resuspended in LB medium, and prepared for subsequent analysis. Experiments performed at 25 °C were incubated for 62 h instead. Cell lysates were then prepared as described in Klompe et al. (2019). We note that Tn7017 did not actually yield any colonies at 37 °C, suggesting that a lower incubation temperature may also be affecting integration efficiency by mitigating toxicity issues. We note that 32-bp spacer sequences were used regardless of the length of the predicted spacer targeting the attachment site.
qPCR analysis:
Pairs of transposon- and target DNA-specific primers were designed to amplify fragments resulting from RNA-guided DNA integration at the expected loci in either orientation. A separate pair of genome-specific primers was designed to amplify an E. coli reference gene (rssA) for normalization purposes. qPCR reactions (10 μl) contained 5 μl of SsoAdvanced Universal SYBR Green Supermix (BioRad), 1 μl H2O, 2 μl of 2.5 μM primers, and 2 μl of tenfold-diluted lysate prepared from scraped colonies, as described for the PCR analysis above. Reactions were prepared in 384-well clear/white PCR plates (BioRad), and measurements were performed on a CFX384 Real-Time PCR Detection System (BioRad) using the following thermal cycling parameters: polymerase activation and DNA denaturation (98 °C for 2.5 min), 35 cycles of amplification (98 °C for 10 s, 62 °C for 1 min), and terminal melt-curve analysis (65–95 °C in 0.5 °C per 5 s increments). Each biological sample was analyzed in three parallel reactions: one reaction contained a primer pair for the E. coli reference gene, a second reaction contained a primer pair for one of the two possible integration orientations, and a third reaction contained a primer pair for the other possible integration orientation (Table S3). Transposition efficiency for each orientation was then calculated as 2ΔCq, in which ΔCq is the Cq difference between the experimental reaction and the control reaction. Total transposition efficiency for a given experiment was calculated as the sum of transposition efficiencies for both orientations. All measurements presented in the text and figures were determined from three independent biological replicates.
NGS approaches
PAM libraries:
To clone the ‘NNNNN’ PAM library, two partially overlapping oligos (oSL3352 and oSL3353) were annealed (95 °C for 2 min, then cooled to room temperature), subjected to Klenow Fragment (37 °C for 30 min) to produce double-stranded DNA without overhangs, and purified using a Qiagen PCR Clean-Up kit. This double-stranded insert as well as the vector backbone (pETDuet-1) were then digested with BstEII and BamHI (37 °C for 1.5 h), gel purified, and ligated following the NEB T4 DNA ligase protocol. The ligation reaction was then used for four individual electroporation reactions with 10-beta electrocompetent E. coli cells (NEB) following the manufacturer’s protocol and plated on large pre-warmed bioassay dishes (LB-agar with 50 μg/ml kanamycin). After overnight growth at 37 °C, the bioassay dishes were inspected, and the plate with the fewest colonies was used to estimate the total number of colonies (>>65,000 colonies). The bioassay dishes were scraped, and the PAM library was extracted from the resuspended colonies using a Qiagen MidiPrep kit.
Transposition assays for the PAM library were performed as described above, except the transformations were performed via electroporation of BL21(DE3) electrocompetent cells with pSPIN and pTarget, and cells were plated on large bioassay dishes (LB agar with 50 μg/ml kanamycin, 50 μg/ml spectinomycin, and 0.1mM IPTG).
To determine the PAM preference for RNA-guided DNA-integration, the following steps were performed using custom Python scripts. First, reads were filtered based on the requirement that they contain 10 bp of perfectly matching transposon end sequence (in the case of the output library), as well as a perfect 32-bp target site. The five bases immediately upstream of the target site were then extracted, and enrichment values were calculated as: ((reads PAM output)/(total output reads)) / ((reads PAM input)/(total input reads)). These enrichment values were used for PAM wheel generation using the Krona template (Ondov et al., 2011) and to determine the top 5% of enriched PAM sequences for WebLogo generation (Crooks et al., 2004).
To determine the integration site preference from the same PAM library dataset, we extracted reads from the output library specifically for targets that contained a ‘CC’ PAM sequence. These reads were then subjected to the Illumina pipeline script described previously (Vo et al. (2020)), which extracts a 17-bp fingerprint from the integration site, maps it back to the targeted sequence, and plots the number of reads found per base position relative to the 3’ end of the target site.
pDonor libraries:
A pDonor library containing twenty different mini-Tn constructs, derived from our I-F3 CRISPR-Tn homologs, was prepared by combining the individual pDonor plasmids. Chemically competent E. coli BL21(DE3) cells containing a pEffector were transformed with ~165 ng of the pDonor library and plated on LB agar containing 100 μg ml−1 carbenicillin, 50 μg ml−1 spectinomycin, and 0.1mM IPTG. After 18 hours, cells were scraped and resuspended in 500 ul of LB. An equivalent of 500 ul of OD7.0 was aliquoted for each sample, and the gDNA was purified using a Promega Wizard Genomic DNA purification kit and used for NGS sample preparation, as described below.
Reads from the output libraries (the amplicons that result from RNA-guided, genomic integration) were filtered based on a perfect 20 bp sequence match to the target locus, and the presence of specific 15-bp mini-Tn ends was tallied. This was done for tRL integration only, but for both the left- and right-end boundaries. Reads for the input libraries (the amplicons resulting from the pDonor pooled library) were filtered based on a 45 bp sequence (25 bp transposon-end + 20 bp flanking sequence) or 25 bp sequence (20 bp flanking + 5 bp TSD) for the left- and right-end amplicons respectively, and the number of occurrences for each mini-Tn homolog were tallied. Enrichment values were then calculated as:
Tn-seq:
genome-wide specificity was determined, as described previously (Klompe et al. (2019)). Briefly, one of the transposon ends was mutated to contain an MmeI recognition site (Table S2) that allows for specific fragmentation of the genome at integration sites after transposition has occurred. These mutations did not drastically affect transposition efficiency (Figure S6D). An adapter with an ‘NN’ overhang was then ligated and used in combination with a transposon-specific primer to enrich transposon-target boundaries from the host genome. Deep sequencing then allowed for identification of both on-target and potential off-target integration events simultaneously.
Product amplification:
PCR products were generated with Q5 Hot Start High-Fidelity DNA Polymerase (NEB) from extracted genomic DNA (as described by the Wizard® Genomic DNA Purification Kit), miniprepped plasmid samples, or 20-fold diluted PCR1 samples. Reactions contained 200 μM dNTPs and 0.5 μM primers, and were generally subjected to 20 or 10 thermal cycles (PCR1 and PCR2, respectively) with an annealing temperature of 65 °C. Primer pairs contained one target-specific primer and one transposon-specific primer (output library), two pTarget-specific primers (PAM input library), one pDonor backbone-specific primer and one transposon-specific primer (pDonor input library), or one transposon-specific and one adapter-specific primer (Tn-seq samples). PCR amplicons were resolved by 1–2% agarose gel electrophoresis and visualized by staining with SYBR Safe (Thermo Scientific), DNA was isolated by Gel Extraction Kit (Qiagen), and NGS libraries were quantified by qPCR using the NEBNext Library Quant Kit (NEB). Illumina sequencing was performed using a NextSeq mid or high output kit with 150-cycle reads and automated demultiplexing and adaptor trimming (Illumina). Next-generation sequencing data are summarized in Table S4 and are available in the National Center for Biotechnology Information Sequence Read Archive (BioProject Accession: PRJNA769066).
Sequence and Phylogenetic Analyses
TnsB diversity and system selection:
To ensure we explored a diverse set of CRISPR-associated transposons, we selected systems with more diverged TnsB proteins. The bioinformatics pipeline (described above) revealed 304 unique TnsB protein IDs that were found in genomic sequences together with all other required CRISPR-Tn protein components. To reduce redundancy, this set was filtered for <90% sequence identity using CD-HIT with default settings (Li et al., 2001, 2002). To create an outgroup, TnsB sequences from Tn7-like elements described in the literature (found in CP000681, CP000606.1, AE017340, CP000155, CP000142, CP002738.1, CP002431.1, CP000644.1, NZ_CP044399.1, and CP025573.1) were included in the phylogenetic analysis (Parks and Peters, 2007; Peters et al., 2014). The TnsB protein sequences were aligned using MUSCLE (v3.8.1551), the resulting alignment was manually inspected, truncated sequences that were the result of nonsense mutations were discarded, and the remaining sequences were aligned again (Edgar, 2004). The resulting alignment was subjected to FastTree (version 2.1.10 Double precision (No SSE3)) to generate a phylogenetic tree (Price et al., 2010). iTOL was used for downstream visualization purposes (Letunic and Bork, 2021). The EcoTnsB-derived sequences indeed formed a distinct clade and were used to root the tree. Colored clades have support values >0.97 (Figure 1B).
TniQ diversity:
Protein sequences corresponding to TnsD/TniQ from Tn7, Tn6677, and Tn7017 (WP_001243518.1, WP_000479715.1, and WP_067516660.1 and WP_157673483.1, respectively) were used as queries for PSI-BLAST (ncbi-blast-2.10.0+ release) against the nr database (version 02/04/2021) using the parameters: -evalue 0.005 -num_alignments 9999999 -num_iterations 10. Unique protein IDs were extracted, combined, and filtered for <50% sequence identity using CD-HIT with default settings. This list of 5,278 sequences was complemented with TniQ/TnsD sequences from characterized Type I-B1, I-B2, and V-K CRISPR-Tn (CAST) systems (WP_011320206.1, WP_011320212.1, WP_094348676.1, WP_094348672.1, WP_015212837.1, WP_029636334.1, and WP_148662752.1) (Saito et al., 2021; Strecker et al., 2019; Vo et al., 2021a), as well as from predicted Type I-F3 CRISPR-Tn (CAST) systems (WP_121838250.1, WP_121838257.1, WP_133408060.1, and WP_170308330.1) (Petassi et al., 2020), Type I-F3 CRISPR-Tn from this study (see Table S1), and TnsD sequences from Tn7 to create an outgroup as described above. The combined protein list was subjected to hmmsearch (HMMER 3.3.2, Nov 2020) for detection of the TniQ domain (TniQ, PF06527) with a p-value of 0.001 (Eddy, 1995). The list of sequences with TniQ domain hits was filtered to retain entries <900 amino acids in length and >85% query coverage of the TniQ domain query, before being subjected to the same hmmsearch settings for both TniQ (PF06527) and TnsD (PF15978) domain detection. The resulting alignment file for the TniQ domain was used to generate a phylogenetic tree with the default settings of FastTree (version 2.1.10 Double precision (No SSE3)). The tree was subsequently visualized in iTOL (Figure S3B), where TnsD domain annotations were added and a pruned view version of the tree was generated (Figure 3B).
Smaller scale analysis was performed for the full length characterized TnsD/TniQ proteins mentioned above. Sequences were aligned with MUSCLE (v3.8.1551) with default settings and allowing for N iterations. FastTree (version 2.1.10 Double precision (No SSE3)) was then used with default settings to generate a phylogenetic tree that was visualized using iTOL. Tn7 sequences were used as an outgroup to root the tree (Figure S3C).
Protein and operon comparison:
Protein sequences of the individual protein components were extracted from the native transposon. Multiple sequence alignments were generated in Geneious using the MUSCLE plugin with default settings and allowing for 8 iterations. Protein identity matrices were extracted and visualized in Prism (Figure S4A). DNA sequences spanning the start codon of TnsA to the stop codon of TnsC (TnsABC operon) and the start codon of TniQ to the stop codon of Cas6 (QCascade operon) were extracted from the native transposon sequences. Multiple sequence alignments were generated in Geneious using the MUSCLE alignment plugin with default settings allowing for 8 iterations. Phylogenetic trees were subsequently generated using the FastTree plugin with default settings. Trees were visualized in iTOL, and Tn7017 was used as an outgroup (based on the high divergence of individual protein components of Tn7017 as compared to the other Type I-F3 CRISPR-Tn systems tested in this study).
DNA sequence motifs:
Transposon end sequences were predicted as part of the computational pipeline described above, and provided a starting point for manual analysis. As we expect the first TnsB binding site to be located at the edge of the transposon, we used the 19-bp sequence directly adjacent to the predicted 8-bp TIR as a query to detect similar sequences within 250 bp of the predicted transposon end sequences. The location of the sequence hits revealed a consistent pattern across the different CRISPR-Tn systems (Figure S6) and indicated that spacing, and not just sequence identity, of the TnsB binding sites is important. Repeat sequences were extracted together with 4 bp upstream and downstream, resulting in multiple 27-bp sequence hits per transposon end. Sequence conservation of the predicted binding sites was then visualized using WebLogo (Version 2.8.2, 2005–09-08), providing a specific motif for TnsB binding sites for compatibility groups.
Cargo analysis:
To explore the presence of anti-phage systems within the CRISPR-associated transposons described in this study, we extracted the entire sequence between the predicted left and right transposon ends and annotated predicted genes using Prokka (version 1.14.6) with default settings (Seemann, 2014). We then applied DefenseFinder (v0.9, model v0.0.2) to detect all anti-phage systems and genes involved in anti-phage systems (Tesson et al., 2021). Predicted defense systems and defense associated genes were manually confirmed using BLASTp against known defense proteins (Cohen et al., 2019; Doron et al., 2018; Gao et al., 2020; Millman et al., 2020; Severin et al., 2021; Tal et al., 2021) before being annotated (Figure 5, S5).
ffs locus occupancy:
To analyze transposons present at the ffs site between distinct V. cholerae strains, 5 kb upstream and downstream of the Tn7005 and Tn7013 integration sites were used for BLASTn with default settings against the Vibrio cholerae (ID:666) set of the ‘nr’ database. The majority of hits were from naive genomes without transposons at this location, but eleven sequences hinted at the presence of a transposon by having an interrupted ffs locus. tBLASTn, using the TnsA sequences of Tn7005 and Tn7013, revealed that six out of eleven genomes encoded a TnsA protein 99% similar to Tn7005 in the interrupted ffs locus (CP053808.1, CP053822, CP053816.1, CP053820.1, CP036499.1, and CP046742.1). Five ffs loci with unique interrupting sequences were aligned to a naive V. cholerae genome (CP013317) and to a locus containing Tn7005 using EasyFig (Sullivan et al., 2011). These alignments revealed rapid turnover of cargo genes (Figure S4C). The same approach was used to compare the native locations of Tn7005 and Tn7013 to an un-integrated ffs locus of V. cholerae (Figure 4B).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were plotted using GraphPad Prism 9.2.0 (332). Statistical details for each experiment are provided in the figure legends. Generally, data are shown as mean ± s.d. for n = 3 biologically independent samples.
Supplementary Material
Table S1: Overview of Type I-F3 CRISPR-Tn systems in this study, related to Figure 1.
Table S2: Description and sequence of plasmids used in this study, related to STAR Methods.
Table S3: Oligonucleotides used for cloning, qPCR, and NGS experiments in this study, related to STAR Methods.
Highlights.
A diverse suite of 20 CRISPR-associated transposons function robustly in E. coli
Some systems exhibit unique modularity and possess multiple DNA-targeting pathways
RNA-guided transposons encode diverse antiphage defense systems
A selection of efficient, orthogonal transposases provides new genome editing tools
ACKNOWLEDGEMENTS
We thank A. Millman for discussions on phylogenetic analyses, the JP Sulzberger Columbia Genome Center for NGS support, and L. F. Landweber for qPCR instrument access. This research was supported by NIH grant DP2HG011650-01, a Pew Biomedical Scholarship and Sloan Research Fellowship, and a generous start-up package from the Columbia University Irving Medical Center Dean’s Office and the Vagelos Precision Medicine Fund (S.H.S.).
Footnotes
DECLARATION OF INTERESTS
Columbia University has filed a patent application related to this work for which S.E.K. and S.H.S. are inventors. S.E.K. and S.H.S. are inventors on other patents and patent applications related to CRISPR–Cas systems and uses thereof. S.H.S. is a co-founder and scientific advisor to Dahlia Biosciences, a scientific advisor to Prime Medicine and CrisprBits, and an equity holder in Dahlia Biosciences and Caribou Biosciences.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- Almendros C, Guzmán NM, Díez-Villaseñor C, García-Martínez J, and Mojica FJM (2012). Target Motifs Affecting Natural Immunity by a Constitutive CRISPR-Cas System in Escherichia coli. Plos One 7, e50797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arciszewska LK, and Craig NL (1991). Interaction of the Tn7-encoded transposition protein TnsB with the ends of the transposon. Nucleic Acids Res 19, 5021–5029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernheim A, and Sorek R (2019). The pan-immune system of bacteria: antiviral defence as a community resource. Nat Rev Microbiol 1–7. [DOI] [PubMed] [Google Scholar]
- Bourque G, Burns KH, Gehring M, Gorbunova V, Seluanov A, Hammell M, Imbeault M, Izsvák Z, Levin HL, Macfarlan TS, et al. (2018). Ten things you should know about transposable elements. Genome Biology 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Choi KY, Spencer JM, and Craig NL (2014). The Tn7 transposition regulator TnsC interacts with the transposase subunit TnsB and target selector TnsD. Proc National Acad Sci 111, E2858–E2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cohen D, Melamed S, Millman A, Shulman G, Oppenheimer-Shaanan Y, Kacen A, Doron S, Amitai G, and Sorek R (2019). Cyclic GMP–AMP signalling protects bacteria against viral infection. Nature 574, 691–695. [DOI] [PubMed] [Google Scholar]
- Collias D, and Beisel CL (2021). CRISPR technologies and the search for the PAM-free nuclease. Nat Commun 12, 555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Couvin D, Bernheim A, Toffano-Nioche C, Touchon M, Michalik J, Néron B, Rocha EPC, Vergnaud G, Gautheret D, and Pourcel C (2018). CRISPRCasFinder, an update of CRISRFinder, includes a portable version, enhanced performance and integrates search for Cas proteins. Nucleic Acids Res 46, W246–W251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crooks GE, Hon G, Chandonia J-M, and Brenner SE (2004). WebLogo: A Sequence Logo Generator. Genome Res 14, 1188–1190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doron S, Melamed S, Ofir G, Leavitt A, Lopatina A, Keren M, Amitai G, and Sorek R (2018). Systematic discovery of antiphage defense systems in the microbial pangenome. Science 4120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eddy SR (1995). Multiple alignment using hidden Markov models. Proc Int Conf Intelligent Syst Mol Biology Ismb Int Conf Intelligent Syst Mol Biology 3, 114–120. [PubMed] [Google Scholar]
- Edgar RC (2004). MUSCLE: multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res 32, 1792–1797. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faure G, Shmakov SA, Yan WX, Cheng DR, Scott DA, Peters JE, Makarova KS, and Koonin EV (2019). CRISPR–Cas in mobile genetic elements: counter-defence and beyond. Nature Reviews Microbiology 1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamas P, and Craig NL (1992). Purification and characterization of TnsC, a Tn7 transposition protein that binds ATP and DNA. Nucleic Acids Res 20, 2525–2532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao L, Altae-Tran H, Böhning F, Makarova KS, Segel M, Schmid-Burgk JL, Koob J, Wolf YI, Koonin EV, and Zhang F (2020). Diverse enzymatic activities mediate antiviral immunity in prokaryotes. Science 369, 1077–1084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halpin-Healy TS, Klompe SE, Sternberg SH, and Fernández IS (2020). Structural basis of DNA targeting by a transposon-encoded CRISPR–Cas system. Nature 577, 271–274. [DOI] [PubMed] [Google Scholar]
- Haniford DB, and Ellis MJ (2015). Mobile DNA III. Microbiol Spectr 3, 631–645. [DOI] [PubMed] [Google Scholar]
- Hille F, Richter H, Wong SP, Bratovič M, Ressel S, and Charpentier E (2018). The Biology of CRISPR-Cas: Backward and Forward. Cell 172, 1239–1259. [DOI] [PubMed] [Google Scholar]
- Hussain FA, Dubert J, Elsherbini J, Murphy M, VanInsberghe D, Arevalo P, Kauffman K, Rodino-Janeiro BK, and Polz M (2021). Rapid evolutionary turnover of mobile genetic elements drives microbial resistance to viruses. Biorxiv 2021.03.26.437281. [DOI] [PubMed] [Google Scholar]
- Jia N, Xie W, Cruz M.J. de la, Eng ET, and Patel DJ (2020). Structure–function insights into the initial step of DNA integration by a CRISPR–Cas–Transposon complex. Cell Res 1–3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klompe SE, and Sternberg SH (2018). Harnessing “A Billion Years of Experimentation”: The Ongoing Exploration and Exploitation of CRISPR–Cas Immune Systems. Crispr J 1, 141–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klompe SE, Vo PLH, Halpin-Healy TS, and Sternberg SH (2019). Transposon-encoded CRISPR–Cas systems direct RNA-guided DNA integration. Nature 571, 219–225. [DOI] [PubMed] [Google Scholar]
- Koonin EV, Makarova KS, Wolf YI, and Krupovic M (2019). Evolutionary entanglement of mobile genetic elements and host defence systems: guns for hire. Nat Rev Genet 1–13. [DOI] [PubMed] [Google Scholar]
- Kubo KM, and Craig NL (1990). Bacterial transposon Tn7 utilizes two different classes of target sites. J Bacteriol 172, 2774–2778. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lampe DJ, Akerley BJ, Rubin EJ, Mekalanos JJ, and Robertson HM (1999). Hyperactive transposase mutants of the Himar1 mariner transposon. Proc National Acad Sci 96, 11428–11433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenay RT, and Beisel CL (2017). Deciphering, Communicating, and Engineering the CRISPR PAM. J Mol Biol 429, 177–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leenay RT, Maksimchuk KR, Slotkowski RA, Agrawal RN, Gomaa AA, Briner AE, Barrangou R, and Beisel CL (2016). Identifying and Visualizing Functional PAM Diversity across CRISPR-Cas Systems. Mol Cell 62, 137–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LeGault KN, Hays SG, Angermeyer A, McKitterick AC, Johura F, Sultana M, Alam M, and Seed KD (2020). Temporal Shifts in Antibiotic Resistance Elements Govern Virus-Pathogen Conflicts. Biorxiv 2020.12.16.423150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Letunic I, and Bork P (2021). Interactive Tree Of Life (iTOL) v5: an online tool for phylogenetic tree display and annotation. Nucleic Acids Res 49, gkab301-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li M, Gong L, Cheng F, Yu H, Zhao D, Wang R, Wang T, Zhang S, Zhou J, Shmakov SA, et al. (2021). Toxin-antitoxin RNA pairs safeguard CRISPR-Cas systems. Science 372, eabe5601. [DOI] [PubMed] [Google Scholar]
- Li W, Jaroszewski L, and Godzik A (2001). Clustering of highly homologous sequences to reduce the size of large protein databases. Bioinformatics 17, 282–283. [DOI] [PubMed] [Google Scholar]
- Li W, Jaroszewski L, and Godzik A (2002). Tolerating some redundancy significantly speeds up clustering of large protein databases. Bioinformatics 18, 77–82. [DOI] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Snir S, and Koonin EV (2011). Defense Islands in Bacterial and Archaeal Genomes and Prediction of Novel Defense Systems ‡. J Bacteriol 193, 6039–6056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Makarova KS, Wolf YI, Iranzo J, Shmakov SA, Alkhnbashi OS, Brouns SJJ, Charpentier E, Cheng D, Haft DH, Horvath P, et al. (2020). Evolutionary classification of CRISPR–Cas systems: a burst of class 2 and derived variants. Nat Rev Microbiol 18, 67–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McDonald ND, Regmi A, Morreale DP, Borowski JD, and Boyd FE (2019). CRISPR-Cas systems are present predominantly on mobile genetic elements in Vibrio species. BMC Genomics 20, 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Millman A, Bernheim A, Stokar-Avihail A, Fedorenko T, Voichek M, Leavitt A, Oppenheimer-Shaanan Y, and Sorek R (2020). Bacterial Retrons Function In Anti-Phage Defense. Cell 183, 1551–1561.e12. [DOI] [PubMed] [Google Scholar]
- Mitra R, McKenzie GJ, Yi L, Lee CA, and Craig NL (2010). Characterization of the TnsD-attTn7 complex that promotes site-specific insertion of Tn7. Mobile DNA 1, 18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mojica FJM, Díez-Villaseñor C, García-Martínez J, and Almendros C (2009). Short motif sequences determine the targets of the prokaryotic CRISPR defence system. Microbiology+ 155, 733–740. [DOI] [PubMed] [Google Scholar]
- Nicolas E, Lambin M, Dandoy D, Galloy C, Nguyen N, Oger CA, and Hallet B (2015). Mobile DNA III. Microbiol Spectr 3, 693–726. [DOI] [PubMed] [Google Scholar]
- Ondov BD, Bergman NH, and Phillippy AM (2011). Interactive metagenomic visualization in a Web browser. Bmc Bioinformatics 12, 385–385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parks AR, and Peters JE (2007). Transposon Tn7 Is Widespread in Diverse Bacteria and Forms Genomic Islands▿†. J Bacteriol 189, 2170–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petassi MT, Hsieh S-C, and Peters JE (2020). Guide RNA Categorization Enables Target Site Choice in Tn7-CRISPR-Cas Transposons. Cell 183, 1757–1771.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE (2015). Mobile DNA III. pp. 647–667. [Google Scholar]
- Peters JE (2019). Targeted transposition with Tn7 elements: safe sites, mobile plasmids, CRISPR/Cas and beyond. Mol Microbiol. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE, and Craig NL (2001a). Tn7: smarter than we thought. Nature Reviews Molecular Cell Biology 2, 806–814. [DOI] [PubMed] [Google Scholar]
- Peters JE, and Craig NL (2001b). Tn7 recognizes transposition target structures associated with DNA replication using the DNA-binding protein TnsE. Genes & Development 15, 737–747. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters JE, Fricker AD, Kapili BJ, and Petassi MT (2014). Heteromeric transposase elements: generators of genomic islands across diverse bacteria. Mol Microbiol 93, 1084–1092. [DOI] [PubMed] [Google Scholar]
- Peters JE, Makarova KS, Shmakov S, and Koonin EV (2017). Recruitment of CRISPR-Cas systems by Tn7-like transposons. Proc National Acad Sci 114, E7358–E7366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piel D, Bruto M, Labreuche Y, Blanquart F, Chenivesse S, Lepanse S, James A, Barcia-Cruz R, Dubert J, Petton B, et al. (2021). Genetic determinism of phage-bacteria coevolution in natural populations. Biorxiv 2021.05.05.442762. [Google Scholar]
- Price MN, Dehal PS, and Arkin AP (2010). FastTree 2 – Approximately Maximum-Likelihood Trees for Large Alignments. Plos One 5, e9490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ratner HK, Escalera-Maurer A, Rhun AL, Jaggavarapu S, Wozniak JE, Crispell EK, Charpentier E, and Weiss DS (2019). Catalytically Active Cas9 Mediates Transcriptional Interference to Facilitate Bacterial Virulence. Mol Cell 75, 498–510.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rice PA, Craig NL, and Dyda F (2020). Comment on “RNA-guided DNA insertion with CRISPR-associated transposases.” Science 368, eabb2022. [DOI] [PubMed] [Google Scholar]
- Rollins MF, Chowdhury S, Carter J, Golden SM, Miettinen HM, Santiago-Frangos A, Faith D, Lawrence CM, Lander GC, and Wiedenheft B (2019). Structure Reveals a Mechanism of CRISPR-RNA-Guided Nuclease Recruitment and Anti-CRISPR Viral Mimicry. Mol Cell 74, 132–142.e5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ronning DR, Li Y, Perez ZN, Ross PD, Hickman AB, Craig NL, and Dyda F (2004). The carboxy- terminal portion of TnsC activates the Tn7 transposase through a specific interaction with TnsA. The EMBO Journal 23, 2972–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousset F, Dowding J, Bernheim A, Rocha EPC, and Bikard D (2021). Prophage-encoded hotspots of bacterial immune systems. Biorxiv 2021.01.21.427644. [Google Scholar]
- Saito M, Ladha A, Strecker J, Faure G, Neumann E, Altae-Tran H, Macrae RK, and Zhang F (2021). Dual modes of CRISPR-associated transposon homing. Cell. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarnovsky R, May E, and journal C-N (1996). The Tn7 transposase is a heteromeric complex in which DNA breakage and joining activities are distributed between different gene products. [PMC free article] [PubMed] [Google Scholar]
- Seemann T (2014). Prokka: rapid prokaryotic genome annotation. Bioinformatics 30, 2068–2069. [DOI] [PubMed] [Google Scholar]
- Severin GB, Hsueh BY, Elg CA, Dover JA, Rhoades CR, Wessel AJ, Ridenhour BJ, Top EM, Ravi J, Parent KN, et al. (2021). A Broadly Conserved Deoxycytidine Deaminase Protects Bacteria from Phage Infection. Biorxiv 2021.03.31.437871. [Google Scholar]
- Shen Y, Gomez-Blanco J, Petassi MT, Peters JE, Ortega J, and Guarne A (2021). Structural basis for DNA targeting by the Tn7 transposon. BioRxiv. [DOI] [PubMed] [Google Scholar]
- Shi Q, Straus MR, Caron JJ, Wang H, Chung Y, Guarné A, and Peters JE (2015). Conformational toggling controls target site choice for the heteromeric transposase element Tn7. Nucleic Acids Research 43, 10734–10745. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen AE, and Craig NL (1997). Avoiding self: two Tn7- encoded proteins mediate target immunity in Tn7 transposition. Embo J 16, 6823–6834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stellwagen AE, and Craig NL (1998). Mobile DNA elements: controlling transposition with ATP-dependent molecular switches. Trends in Biochemical Sciences 23, 486–490. [DOI] [PubMed] [Google Scholar]
- Stern A, Keren L, Wurtzel O, Amitai G, and Sorek R (2010). Self-targeting by CRISPR: gene regulation or autoimmunity? Trends Genet 26, 335–340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker J, Ladha A, Gardner Z, Schmid-Burgk JL, Makarova KS, Koonin EV, and Zhang F (2019). RNA-guided DNA insertion with CRISPR-associated transposases. Science eaax9181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Strecker J, Ladha A, Makarova KS, Koonin EV, and Zhang F (2020). Response to Comment on “RNA-guided DNA insertion with CRISPR-associated transposases.” Science 368, eabb2920. [DOI] [PubMed] [Google Scholar]
- Sullivan MJ, Petty NK, and Beatson SA (2011). Easyfig: a genome comparison visualizer. Bioinformatics 27, 1009–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tal N, Millman A, Stokar-Avihail A, Fedorenko T, Leavitt A, Melamed S, Yirmiya E, Avraham C, Amitai G, and Sorek R (2021). Antiviral defense via nucleotide depletion in bacteria. Biorxiv 2021.04.26.441389. [Google Scholar]
- Tang Y, Lichtenstein C, and Cotterill S (1991). Purification and characterisation of the TnsB protein of Tn7: a transposition protein that binds to the ends of Tn7. Nucleic Acids Res 19, 3395–3402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tesson F, Hervé A, Touchon M, d’Humières C, Cury J, and Bernheim A (2021). Systematic and quantitative view of the antiviral arsenal of prokaryotes. Biorxiv 2021.09.02.458658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vercoe RB, Chang JT, Dy RL, Taylor C, Gristwood T, Clulow JS, Richter C, Przybilski R, Pitman AR, and Fineran PC (2013). Cytotoxic Chromosomal Targeting by CRISPR/Cas Systems Can Reshape Bacterial Genomes and Expel or Remodel Pathogenicity Islands. Plos Genet 9, e1003454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo PLH, Ronda C, Klompe SE, Chen EE, Acree C, Wang HH, and Sternberg SH (2021a). CRISPR RNA-guided integrases for high-efficiency, multiplexed bacterial genome engineering. Nat Biotechnol 39, 480–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vo PLH, Acree C, Smith ML, and Sternberg SH (2021b). Unbiased profiling of CRISPR RNA-guided transposition products by long-read sequencing. Mobile Dna-Uk 12, 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Waddell CS, and Craig NL (1989). Tn7 transposition: recognition of the attTn7 target sequence. Proc National Acad Sci 86, 3958–3962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Workman RE, Pammi T, Nguyen BTK, Graeff LW, Smith E, Sebald SM, Stoltzfus MJ, Euler CW, and Modell JW (2021). A natural single-guide RNA repurposes Cas9 to autoregulate CRISPR-Cas expression. Cell 184, 675–688.e19. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Table S1: Overview of Type I-F3 CRISPR-Tn systems in this study, related to Figure 1.
Table S2: Description and sequence of plasmids used in this study, related to STAR Methods.
Table S3: Oligonucleotides used for cloning, qPCR, and NGS experiments in this study, related to STAR Methods.
Data Availability Statement
Deep sequencing data have been deposited at the sequence read archive (SRA) and are publicly available as of the date of publication. Accession numbers are listed in the key resources table. Original agarose gel images have been deposited at Mendeley and are publicly available as of the date of publication. DOIs are listed in the key resources table.
All original code has been deposited at Mendeley and is publicly available as of the date of publication. DOIs are listed in the key resources table.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and virus strains | ||
| Escherichia coli NEB® Turbo | NEB | Cat# C2984 |
| Escherichia coli BL21(DE3) | NEB | Cat# C2527 |
| Escherichia coli 10-beta electrocompetent | NEB | Cat# C3020K |
| Escherichia coli BL21(DE3) electrocompetent | Millipore Sigma | Cat# CMC0016 |
| Chemicals, peptides, and recombinant proteins | ||
| LB Broth | Fisher | Cat# BP1426 |
| LB Agar | Fisher | DF0445-07-6 |
| Spectinomycin | Gold Bio | Cat# S-140 |
| Kanamycin | Gold Bio | Cat# K-120 |
| Carbenicillin | Gold Bio | Cat# C-103 |
| IPTG | Gold Bio | Cat# I2481C |
| Klenow Fragment | NEB | Cat# M0212L |
| Critical commercial assays | ||
| SsoAdvanced Universal SYBR Green Supermix | BioRad | Cat# 1725275 |
| Promega Wizard Genomic DNA purification kit | Promega | Cat# A1120 |
| NEBNext Library Quant Kit | NEB | Cat# E7630L |
| Deposited data | ||
| Raw agarose gel image | This paper, Mendeley Data | doi:10.17632/89p4wdryt3.1 |
| Illumina sequencing data | This paper, SRA | SRA: PRJNA769066 |
| Custom python scripts | This paper, Mendeley Data | doi:10.17632/89p4wdryt3.1 |
| Oligonucleotides | ||
| All oligonucleotides used in this study. | This paper. | Table S3 |
| Recombinant DNA | ||
| All plasmids used in this study. | This paper. | Table S2 |
| Software and algorithms | ||
| PSI-BLAST (2.10.0+) | NCBI | https://blast.ncbi.nlm.nih.gov/Blast.cgi |
| NCBI Efetch | NCBI | https://www.ncbi.nlm.nih.gov/books/NBK25499/ |
| Entrez Programming Utilities | NCBI | https://www.ncbi.nlm.nih.gov/books/NBK25501/ |
| CRISPRCasFinder (4.2.2) | Couvin et al., 2018 | https://crisprcas.i2bc.paris-saclay.fr/CrisprCasFinder/Index |
| Krona | Ondov et al., 2011 | https://github.com/marbl/Krona |
| WebLogo (2.8.2) | Crooks et al., 2004 | https://weblogo.berkeley.edu/ |
| Illumina pipeline script | Vo et al., 2020 | https://github.com/sternberglab/Illumina-pipeline |
| CD-HIT | Li et al., 2001, 2002 | https://github.com/weizhongli/cdhit |
| MUSCLE (v3.8.1551) | Edgar, 2004 | N/A |
| FastTree (version 2.1.10) | Price et al., 2010 | http://www.microbesonline.org/fasttree/ |
| iTOL (6.4) | Letunic and Bork, 2021 | https://itol.embl.de/itol.cgi |
| HMMER 3.3.2 | Eddy, 1995 | http://hmmer.org/ |
| Geneious Prime® 2019.2.1 | Biomatters | https://www.geneious.com/ |
| Prokka (version 1.14.6) | Seemann, 2014 | https://github.com/tseemann/prokka |
| DefenseFinder (v0.9, model v0.0.2) | Tesson et al., 2021 | https://defense-finder.mdmparis-lab.com/ |
| EasyFig | Sullivan et al., 2011 | https://mjsull.github.io/Easyfig/ |