

Abstract
Recent advances in immune therapy have fundamentally changed the landscape of cancer treatment by leveraging the specificity and selectivity of the adaptive immune system to kill cancer cells. These successes have ushered in a new wave of research aimed at understanding immune recognition with the hope of developing newer immunotherapies. The advent of CRISPR technologies and advancement of multi-omics modalities have greatly accelerated the discovery process. Here, we review the current literature surrounding CRISPR screens within the context of tumor immunology, provide essential components need to conduct immune-specific CRISPR screens, and present avenues for future research.
Introduction
Due to our continuous interaction with the external world, epithelial and mucosal barriers are constantly exposed to ultraviolet radiation and carcinogenic metabolites that can initiate the oncogenic process [1,2]. This is further exacerbated upon pathogenic infection whereby proinflammatory mediators like reactive oxygen and nitrogen species, which are used to eliminate pathogens, are also highly mutagenic [3]. Immune detection and elimination of metaplastic and dysplastic cells is critical in preventing tumor development. But as these cells continue to grow and proliferate, they can acquire additional mutations that enable them to evade immune detection, metastasize, and disrupt normal physiological processes [4].
Established research has shown that immune checkpoint blockade (ICB) neutralizes some of these evasion mechanisms, leading to increased survival rates in previously untreatable cancers [5]. Therefore, understanding how tumors develop over time in the setting of antitumor immunity is critical for the discovery of immune regulators and the development of new oncotherapeutics. Clustered regularly interspaced palindromic repeat (CRISPR) provides an unparalleled ability towards identifying novel immune mediators in a high-throughput fashion either through pooled CRISPR screens or multiplexed arrays to assess individual gene perturbations.
Here, we discuss the types of currently available CRISPR technologies and different methods of CRISPR library readouts to serve as a primer for understanding immune-related CRISPR screens (Graphic Abstract / Figure 1). In Table 1, we outline experimental parameters and readout for pivotal CRISPR screens performed in the context of tumor immunology and provide a supplemental table with all CRISPR screens discussed in this paper (see Table S1). We then place each CRISPR screen within its immunological context to highlight the importance of each screen. Finally, we provide some guiding principles for conducting CRISPR screens.
Figure 1.
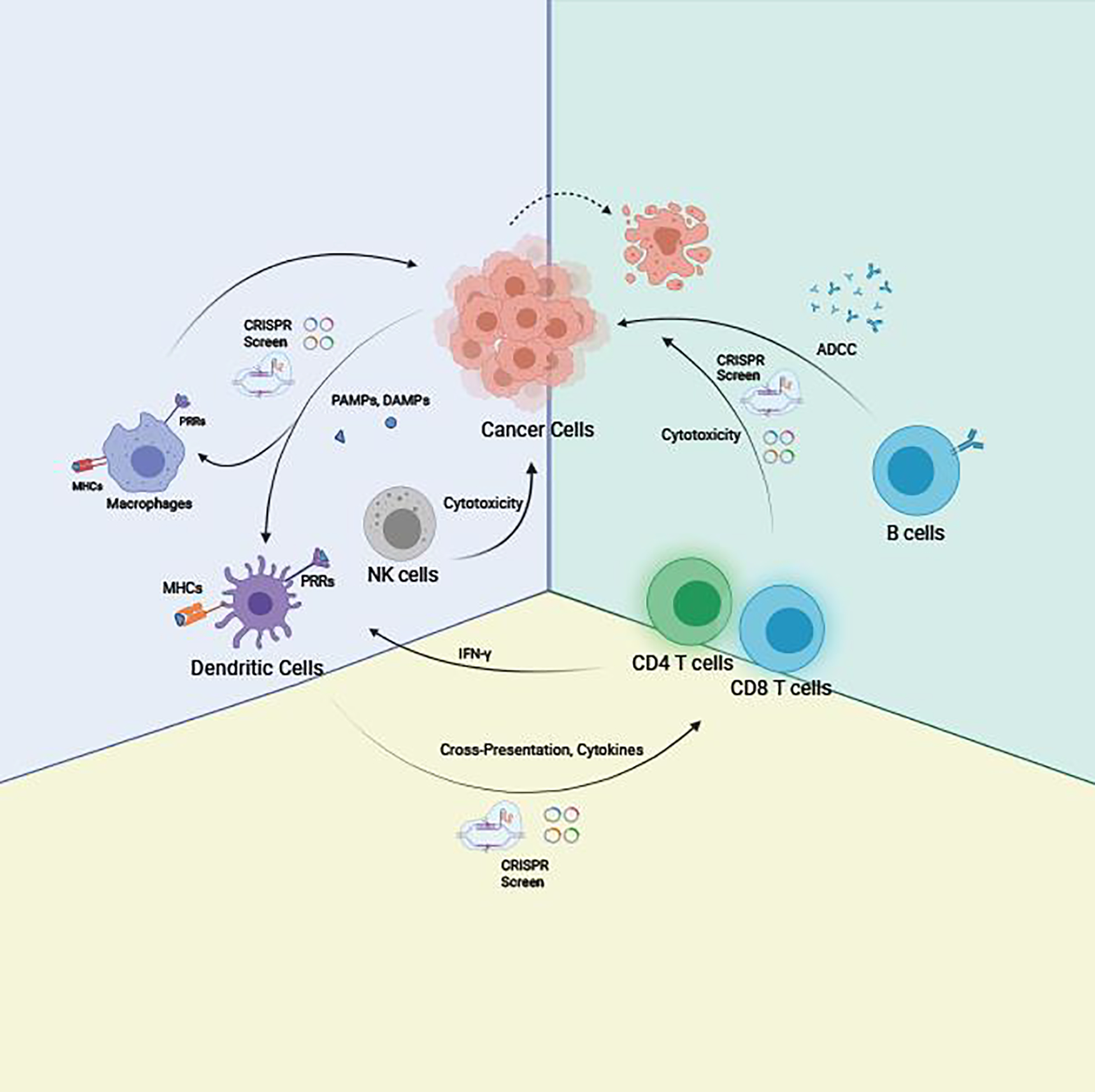
This schematic summarizes a simplified conceptual framework of immune cell interactions in cancer and the utility of CRISPR screen for identifying key regulators of tumor immunology. Macrophages and dendritic cells (DCs) recognize damage-associated molecular patterns (DAMPs) prior to the activation of the adaptive immune response via major histocompatibility complex (MHC) antigen presentation and cytokines expression. Neoantigens from tumor cells are presented by DCs to CD4 and CD8 T cells. CD8 T cells would get activated and mediated malignant cells killing. CD4 T helper cells release IFN-γ to mount innate immune response against cancer cells, like cytotoxicity mediated by natural killer cells (NK cells). B cells also get activated by tumor neoantigens and secret antibodies against the neoantigens, which would lead to antibody-dependent cellular cytotoxicity (ADCC) mediated by NK cells. As genetic regulations play crucial roles across all these immunological processes, CRISPR screens can in principle be applied in all these cell types for each specific process.
Table 1.
Landmark cancer-immune screens
| Cell Type | Species; Conditions | CRISPR Library | Brief Description | Read-out | References |
|---|---|---|---|---|---|
| Ramos: Burkitt’s lymphoma cell line | Human; in vitro | Whole genome CRISPR-ko library; whole genome CRISPRa library | To identify intrinsic regulators of cancer cell phagocytosis, Cas9-Ramos cells were incubated with anti-CD20 (rituximab) antibody and J774 macrophages in the presence or absence of anti-CD47 antibodies. | crRNA libraries were prepared from suviving cells and compared to untreated Ramos cell controls. For validate gene, authors specifically looked at genes depleted in CRISPR-ko cells to identify mutated genes that sensitize cancer cells to macrophage-mediated phagocytosis | [45] |
| RAW 264.7 monocytic cell line | Murine; in vitro | Whole genome CRISPR-ko library | To identify genes required for NLRP1B-mediated pyroptosis, Cas9-RAW 264.7 cells were treated with either nonselective-NLRP1B inflammasome inducer (DPP8/9 inhibitor) or selective-NLRP1B inflammasome inducer (anthrax lethal factor) | Genes enriched in response to both treatments were broadly involved with inflammasome activation. Gene enriched in lethal factor treatment only group identified genes involved selectively with NLRP1B-mediated pyroptosis | [48] |
| THP-1 monocytic cell line | Human; in vitro | Whole genome CRISPRi library | Cyclic dinucleotides (CDN) are potent inducers of the cGas-STING pathway. To identify genes involved with CDN uptake and metabolism, Cas9-THP-1 were transduced with a CDN-inducible reporter and cocultured with various CDNs | Genes involved with CDN uptake/metabolism were enriched in reporter low cells compared to reporter high cells. | [28] |
| U937 monocytic cell line | Human; in vitro | Whole genome CRISPR-ko library | To identify modulators of phagocytosis, Cas9-U937 were treated with different antigens conjugated to different iron reagents and sorted through magnetic separation | Genes enriched in the unbound magnetic fraction were determined to be positive regulators of phagocytosis and genes enriched in the magnetically bound fraction were consider to be negative regulators of phagocytosis | [63] |
| E0771 Triple Negative Breast Cancer cell line, Pan02 PDA cell line, B16F10 melanoma cell line | Mouse; in vivo | Whole genome CRISPRa library | Demonstrating the utility of gene activation via CRISPRa to elicit antitumor immunity | Tumor growth curves to test efficacy of gene activation | [69] |
| Primary conventional dendritic cells | Mouse; in vitro | Pathway specific, transcriptome-based focused CRISPR-ko library | To identify regulators of cross presentation, sorted cDC2s were individually infected with crRNAs and then cocultured with antigen-specific CD4 T cells. | Regulators of cross presentation were identified by crRNAs that attenuated T cell proliferation in the presence of antigen via CFSE cell proliferation assay. | [72] |
| Jurkat: T acute lymphoblastic leukemia cell line | Human; in vitro | Whole genome CRISPR-ko library | To identify genes involved with proximal T cell antigen receptor signaling, Cas9-Jurkat T cells were stimulated with anti-TCR antibody to induce TCR crosslinking and subsequent upregulation of CD69. | crRNAs enriched in CD69 high cells respresent negative regulators of TCR signaling whereas crRNAs enriched in CD69 low cells respresent positive regulator of TCR signaling | [80] |
| Primary CD8 T cells | Human; in vitro | Whole genome CRISPR-ko library | To identify genes involved with T cell proliferation, Cas9-CD8 T cells were stained with the cytoplasmic CSFE proliferation dye prior to being stimulated with anti-CD3/CD28 bead to induce T cell activation and proliferation. Nonproliferating cells were sorted on CFSE-hi expression and Proliferating cells were sorted on CFSE-lo expression | crRNAs enriched in nonproliferating T cells represent positive regulators of TCR signaling. crRNAs enriched in proliferating T cells represent regulatory genes that suppresses TCR signaling and proliferation. | [82] |
| Primary CD8 T cells | Mouse; in vitro and in vivo | Whole genome CRISPR-ko library | To identify genes involved with tumor killing, Cas9-CD8 T cells were coincubated with cancer cells expressing their cognate antigen and stained with anti-CD107a antibody to capture TCR-induced degranulation/killing. To identify genes involved with tumor infiltration, Cas9-CD8 T cells were injected into antigen-expressing tumor-bearing mice and after several days tumors were extracted to identify T cells that were enriched in tumors | crRNAs enriched in CD107a-high cells represent negative regulators of T cell degranulation/killing. crRNAs enriched in tumors represent negative regulators of T cell infiltration | [83] |
| Primary CD8 T cells | Mouse; in vitro | CRISPR-ko library targeting kinases | Multiple flow cytometric assays were designed to identify genes involved with cell proliferation (CFSE), memory formation (CD62L), oxidative stress (DCFDA), and genomic stability (gH2AX). | Favorable crRNAs were enriched in cell that induced high cell expansion and increased memory formation while limiting oxidative stress and genome instability. These crRNAs represent genes that normally inhibit these functions due to the ability of the crRNAs to induce silencing frameshift mutations | [84] |
| B16F10 melanoma cell line | Mouse; in vivo | Whole genome CRISPR-ko library | Cas9-B16F10 melanoma cell lines were subcutaneously transplanted into Tcra−/− mice, C57BL6 mice treated with GM-CSF + irradiated B16F10 (GVAX), or C57BL6 mice treated with GVAX + anti-PD-1 | Surviving cells from different treatment groups were sequenced. Enriched crRNAs in GVAX or GVAX + anti-PD-1 treatment compared to Tcra−/− represent mutated genes that confer immune escape. | [101] |
| B16F10 melanoma cell line; 4T1 and EMT6 breast cancer cell lines; CT26 and MC38 colon adenocarcinoma cell line; RenCa renal cell carcinoma cell line |
Mouse; in vitro | Whole genome CRISPR-ko library | To identify genes involved with T cell evasion, Cas9-expressing cell lines were treated with OT-1 T cells | Sensitizer genes are mutated genes that were depleted under T cell selection; resistor genes are mutated genes that were enriched under T cell selection | [102] |
| B16F10 melanoma cell line | Mouse; in vitro | Whole genome CRISPR-ko library | To identify genes involved with T cell evasion, Cas9-B16F10-OVA cell lines were treated with OT-1 T cells | Sensitive cells bear mutated genes that were depleted under T cell selection; Resistant cells possess mutated genes that were enriched under T cell selection | [104] |
Brief Primer of CRISPR technologies and Multi-omic Advancements
CRISPR encodes an adaptive immune response that protects bacteria against prior bacteriophage infections [6]. This system is comprised of Cas9 nuclease, trans-activating CRISPR RNA (tracrRNA) scaffold, and CRISPR RNA (crRNA) spacer sequence that function in tandem to selectively mutagenize DNA sequences complementary to the crRNA [7]. Fusion of crRNA with tracrRNA to generate single guide RNAs enables highly efficient gene-editing in mammalian cells [8,9] (Figure 2). The specificity and efficiency of CRISPR-Cas9, along with the generation of crRNA libraries targeting every gene in the genome, create a robust platform that enables genomic screening in mammalian cells [10–16]. On top of single-gene perturbation, combinatorial CRISPR-screens were developed to dissect gene interaction networks [16,17] (Figure 2). Beyond loss-of-function screen, fusion of catalytically inactive Cas9 to different epigenetic modifiers (such as histone acetylation by p300 or DNA demethylation by TET dioxygenases) enables CRISPR activation (CRISPRa) screens [18–23] or CRISPR interference (CRISPRi) screens [24], which can be used in cancer immunology (later in text) (Figure 3).
Figure 2.
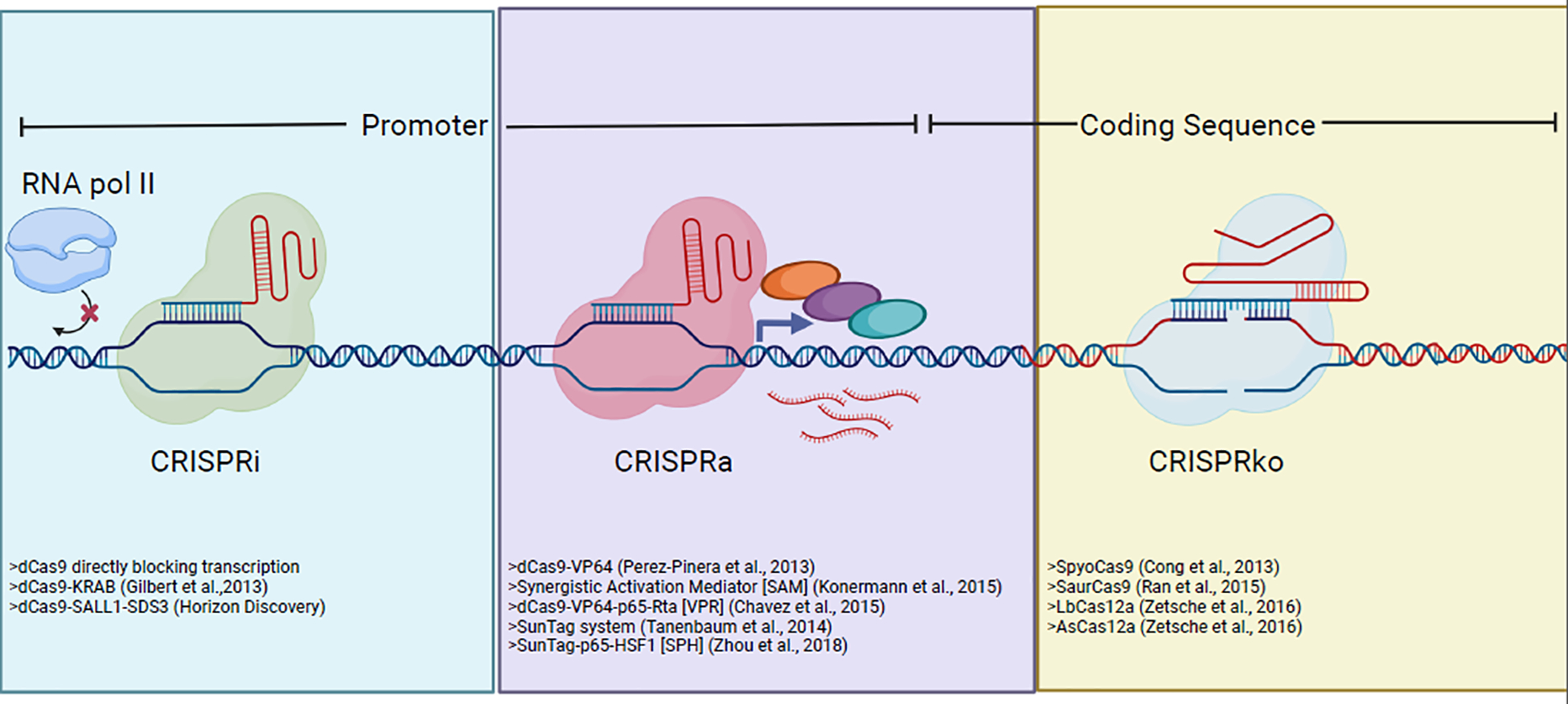
This schematic summarizes the three main modes of CRISPR gene editing or gene expression perturbations used in mammalian cell biology including tumor immunology.
Figure 3.
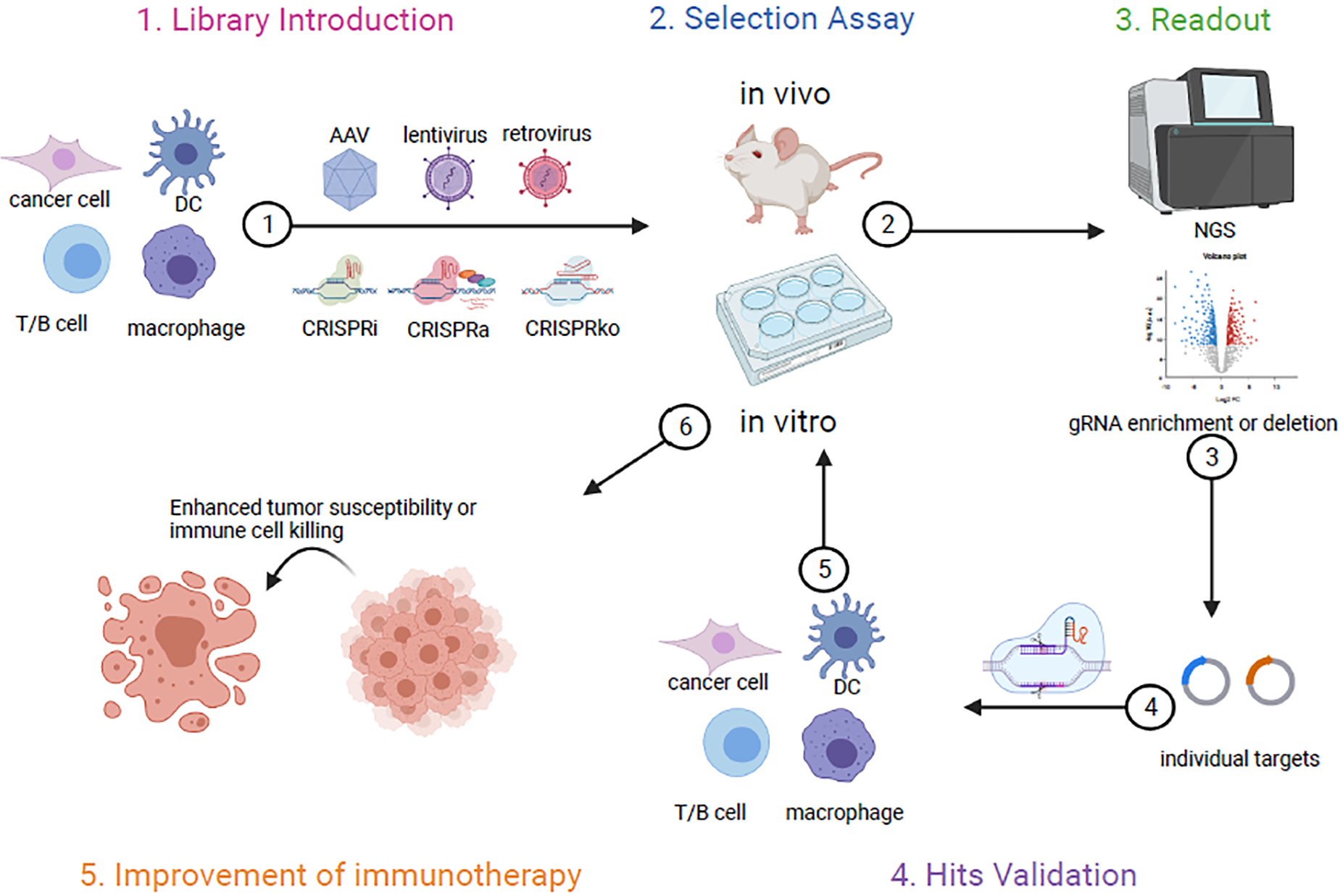
This schematic outlines the general workflow of CRISPR screen in various settings. 1) CRISPRi, CRISPRa or CRISRPko components (guide RNA library included) can be introduced by viral vectors like AAV, lentivirus or retrovirus into different cell types. 2) Depending on the phenotypes of interest, either in vitro or vivo based assays can be adopted for guide RNA selection. 3) Next generation sequencing (NGS) is then performed to check guide RNA enrichment or deletion. 4) Top hits are then validated by cloning guide RNAs individually to target gene of interest, followed by similar in vitro or in vivo assays used for the screen initially. 5) The final goal of these screens is to identify targets that are able to increase tumor cell susceptibility or to improve immune cells mediated anti-tumor responses.
In order to identify relevant genes, pooled CRISPR screens require well-characterized phenotypic assays to differentiate genetic mutants from wild-type and/or nontargeting crRNA controls. Examples of such techniques include survival assays [25–27], the upregulation of critical effector molecules [28], or engineered promoters and transcription factor fusion proteins [29–32]. Often, these types of experiments require library readouts of enriched gene-specific crRNAs, followed by individual gene validation.
To increase throughput, CRISPR screens have been applied in conjunction with multi-omics modalities to facilitate simultaneous phenotypic assessment for a given gene perturbation. For example, Pro-code is a technology that enables simultaneous proteomic analyses with CRISPR screening via mass spectrometry [33]. This method demarcates the causative crRNA with a unique epitope signature within a library of combinatorically concatenated epitope tags. Conversely, unique barcodes placed immediately downstream of lentiviral/retroviral reporters [34] and poly-A tails attached to the crRNAs themselves [35] make CRISPR screening compatible with single cell RNA sequencing (scRNAseq). (EC)CITE seq permits simultaneous analyses of the proteome, transcriptome, and gene perturbation from an individual cell via scRNAseq by covalently binding unique DNA barcodes to commercially available antibodies and utilizing scRNAseq-compatible crRNAs [36]. Meanwhile, CRISPR screens targeting nuclear and epigenetic factors can be read out using scRNAseq that captures the crRNA, proteome, and epigenetic landscape of an individual cell [37]. The limitation of these studies is the total number of cells that can be analyzed. Recent developments have increased scRNAseq throughput 100-fold by first indexing methanol-fixed cells with a first set of primers prior to overloading scRNAseq chip [38].
Application of these technologies in tumor cell lines and immune cells has been shown to successfully identify known mediators and novel regulators, which highlights the overwhelming success of these technologies.
Contextualization of Cancer Immune-Related CRISPR screens
Innate immune detection, activation, and antigen presentation
Underlying the epithelium and mucosa are innate and adaptive immune cells. Of note, tissue resident macrophages and dendritic cells (DCs) play a central role detecting pathogens and tissue damage, initiating the adaptive immune response, and clearing cellular debris [39,40]. In the context of cancer, tumor apoptosis and necrosis triggers the release of different chemotactic agents that attract resident macrophages and DCs [41,42]. Upon their arrival, these cells clear cellular debris using various phagocytic receptors and survey the composition of the debris using pattern recognition receptors (PRRs) that can detect common molecular patterns associated with microbial pathogens (PAMPs) and tissue damage (DAMPs). PRRs then initiate a signaling cascade to upregulate various inflammatory mediators, cytokines, chemokines, and effector molecules that triggers DCs to process antigens, present antigenic epitopes via major histocompatibility complexes (MHC) and activate T cells.
Malignant cells adapt a multitude of ways to evade innate immune recognition. One mechanism is by modifying glycosylation patterns to enhance lectin receptor binding, which often inhibits immune activation. Siglecs are a major class of lectin receptors that are preferentially expressed on immune cells. To find genes that enhance Siglec-binding, researchers have stained library-infected K562 cells with recombinant Siglec-7-Fc and sorted on Siglec-7-Fc-negative cells, reasoning that gene perturbations to glycan modifying genes would prevent Siglec-7-binding [43]. Readout of this study identified CD43. Subsequent knockout and antibody-neutralization studies of CD43 in K562 cells displayed enhanced NK cell-mediated tumor lysis [43]. To further elucidate the role of glycan-modifying enzymes in tumor evasion, computational analyses of five different CRISPR screens found that crRNAs targeting the glycan-modifying enzyme, Man2a1, were depleted under immune selection [44]. Validation studies performed in mice showed that genetic perturbation of Man2a1 resulted in robust antitumor responses [44]. While these studies identified the importance of glycosylation patterning and secondary modifications to the surface proteome for innate immune evasion, these signals are balanced by coexisting immunostimulatory cues in the tumor microenvironment derived from tissue damage and apoptotic debris [42].
Tumor cells undergo or necrosis if they outgrow their nutritional or oxygen supply. Resident macrophages express an array of scavenger receptors, phosphatidyl serine receptors, integrins, and complement receptors that aid in debris detection and clearance. Recent CRISPR-knockout and activation screens performed in tumor cells have identified genes that enhance and resist phagocytic clearance by macrophages [45]. This study highlighted the role of mutated adipocyte plasma membrane-associated protein (APMAP) gene and its ability to synergize with pro-phagocytic therapies, which augmented cancer cell uptake by macrophages [45]. Avoiding macrophage- and dendritic cell-mediated phagocytosis prevent malignant cell antigens from being displayed on antigen presenting cells, resulting in an inability to initiate the adaptive T cell response.
Cellular debris that is taken up can be detected by membrane-bound Toll-like receptors (TLRs) and cytosolic Rig-like receptors, cyclic GMP-AMP synthase (cGAS), and NOD-like receptors in macrophages and DCs [42]. These PRRs activate critical transcriptional programs that initiate the localized immune response through the upregulation of chemokines, cytokines, proinflammatory mediators, and antigen processing machinery. Pooled knockout screens have identified genes that modulate the expression of PRRs [46] as well as their downstream mediators [47–50], by enriching for either: a) genetic perturbations in negative regulators with heightened PRR expression, or, b) positive regulator perturbations with attenuated PRR expression. TLR signaling transduced either by MyD88 or TRIF, ultimately converges on NF-kB and IRF signaling [51]. Screens have identified how the NF-kB pathway is regulated at the genetic, protein, and post-translational level [29,32,52–54].
Although PRR screens in myeloid cells have largely centered around TLR signaling pathways, innate immune detection of apoptotic tumor cells has been attributed to the cGAS-STING pathway [55–57]. In order to elucidate how the cGAS-STING pathway is initiated in the tumor microenvironment, CRISPR screens were performed in human macrophage cell lines in the presence of cGAMP, a cGAS agonist, which identified SLC19A1 [28,58]. Further studies are needed to determine the range of dinucleotide transporters expressed in other myeloid populations that can trigger cGAS-STING activation. Of note, CD8 conventional dendritic cells (DCs) are purportedly critical for eliciting anti-tumor CD8 T cell responses [59,60]. Therefore, knockout studies performed specifically in CD8 DCs will be critical at identifying both upstream and downstream mediators of cGAS-STING pathways in tumor models. These studies can provide mechanistic insight as to how antitumor innate immune responses are initiated in an immunosuppressive tumor microenvironment.
Although engagement of PRRs can initiate transcription of downstream effectors, full activation of macrophage and DCs also require concomitant proinflammatory cytokine signaling [42]. Once activated, these innate immune cells increase their cytokine production, phagolysosome capacity, and proinflammatory metabolite production. To identify genes involved with phagolysosome maturation, pH-sensitive and pH-insensitive reporters were used to delineate different stages of phagolysosome maturation and identify genes that serve important roles in each process [61,62]. These studies showed that crRNAs targeting SLC4A7 were depleted in cells with mature phagolysosomes [62]. Follow-up studies revealed that SLC4A7 was involved with bicarbonate transport and required for phagosome acidification [62]. In a parallel study, researchers used antigens conjugated to ferrous nanoparticles to enrich for cells that were either able to or unable take up antigen when placed in a magnetic field [63]. This study identified a number of genes involved with cytoskeletal rearrangement, lipid metabolism, and protein recycling pathways [63]. Once phagocytosed by macrophages, these antigens are thoroughly digested using several acidic hydrolases for nutrient recycling [64]. The ability of macrophages to phagocytose senescent and pathogenic cells and digest these cells into their principle components makes these cells an important target for immunotherapy. Previous studies have shown that CD40-mediated activation of infiltrating macrophages can eliminate pancreatic ductal adenocarcinoma cells in a T cell-independent manner [65]. Understanding which macrophage effector function(s) contributes to this tumoricidal activity is crucial for the expansion of macrophage-directed immunotherapy.
Initiation of the adaptive T cell response
Tumor-specific adaptive immune responses require antigens that are uniquely expressed by tumors. These neoantigens can be derived from missense mutations and translocations that generate new functional proteins, viral oncogenes, re-expression of neonatal antigens and/or enhanced expression of tissue-specific antigens driven by global hypomethylation [66]. Due to the plethora of FDA-approved HDAC inhibitors, CRISPR screens specifically targeting epigenetic modifiers have been designed to identify epigenetic modulators that inhibit neoantigen expression in animal models of lung adenocarcinoma and pancreatic ductal carcinoma [67,68]. Subsequent knockdown experiments demonstrated enhanced T cell tumor infiltration and attenuated tumor growth in response to immune checkpoint blockade (ICB) therapy when compared to control, highlighting the therapeutic potential of using epigenetic modifiers in conjunction with ICBs. Further studies and clinical trials are needed to confirm these findings in patients. Additionally, whole-genome and focused library-based CRISPR activation (CRISPRa) studies have been performed in mouse models of triple-negative breast cancer, pancreatic cancer, and melanoma, which resulted in robust antitumor immunity [69]. Although the suggested mechanism of these studies collectively points towards enhanced T cells responses driven by the augmented neoantigen presentation, the exact mechanism underlying the heightened T cell activity is yet to be determined.
To elicit antitumor adaptive immune responses, PRR-activated DCs upregulate antigen presentation machinery and lymph node-homing receptors, so that they can efficiently present antigenic peptides on major histocompatibility complex class I (MHCI) and class II (MHCII) to naïve CD8 and CD4 T cells, respectively [41,42]. More specifically, activated DCs incompletely digest phagocyted neoantigens into short peptides that can then be loaded on MHCII to activate CD4 T cells in draining lymph nodes. In order to identify genes involved with MHCII antigen presentation, antigen presenting cells were cultured with SteD, a bacterial effector that negatively regulates MHCII expression, and enriched for cells that were still able to express MHCII [70]. TMEM127 and WWP2 were both found to be required by SteD to ubiquitinate MHCII, resulting in attenuated MHCII expression [70]. In contrast, MHCI antigen presentation commonly requires processed antigens to be expressed in the cytosol of infected cells where they can be processed by the proteosome. The resulting peptides are then shunted into the ER via TAP-mediated transport and loaded on to MHCI before it can be presented to CD8 T cells. CD8 DCs possess the unique ability to present exogenous antigen on MHCI through a process called cross presentation [71]. However, it is not entirely known how exogenous antigens are translocated into the cytosol to be processed by the proteosome. To address this issue, a select library of genes uniquely expressed in cross-presenting DCs was created and cloned in a 96-well format in order to study single-gene perturbations in DCs using multiplexed arrays [72]. Mutant DCs were then cultured with cell-associated antigen to facilitate antigenic cross presentation, and subsequently cultured with antigen-specific CD8 T cells. Perturbations to genes involved with cross presentation impaired CD8 T cell proliferation. These assays helped identify Wdfy4, a BEACH domain-containing protein that is involved in surface membrane and endosomal trafficking [72].
The purpose of dividing antigen presentation into two different classes of MHC is to direct critical resources to necessary immune cells. For cancer-specific responses, CD8 T cells mount crucial responses against cytosolic antigens presented on MHCI, which is expressed on most nucleated cells. CD4 T cells, on the other hand, secrete out IFN-gamma to enhance local NK cell and CD8 T cell activity as well as increase the phagocytic capacity of macrophages. This is mediated by T cell antigen receptor (TCR) recognition of neoepitopes presented on MHCII by resident DCs. In other immune contexts, the composition of signals generated by DCs varies to generate different types of CD4 and CD8 T cell responses, which are out of scope for this review.
Initiation of B cell responses
Neoantigens can be transported to the lymph node via two pathways – active transport to the lymph node by DCs or passive diffusion through lymphatic channels. Once in the lymph node, soluble antigens can either be taken up by subcapsular macrophages and transferred to B cells, or they can be taken up directly by B cells from the conduits of the lymph node [73]. Upon binding to their cognate antigen, B cells initiate a signaling cascade that ultimately results in its activation, proliferation, and differentiation. This response is augmented if B cells receive T cell help, which is partially mediated by CD40-CD40L costimulation. CRISPR screening has unraveled the role of post-translational RNA modification in negatively regulating CD40 expression as well as the role of the ubiquitin ligase FBX011 at heightening CD40 expression by attenuating regulators of CD40 [74]. Having received both the antigenic and costimulatory signals, B cells are able to generate highly specific antibodies directed against these neoantigens.
Researchers have developed therapeutic antibodies that target antigens like CD20 (B cell lymphomas, multiple sclerosis, and rheumatoid arthritis), CD52 (chronic lymphocytic leukemia), EGFR (colon cancer, head and neck cancers), and HER2 (breast and gastric cancers). These antibodies promote tumor cell death through antibody-dependent cell cytotoxicity by NK cells, as well as by direct tumor lysis through complement-mediated formation of the membrane attack complex. Engineered neoantigen-specific antibodies can also serve as a protein carrier for potent chemotherapeutic drugs. To determine the mechanisms by which these therapeutic antibodies work, enrichment screens were conducted to confirm that genes involved in complement activation sensitize cancer cells to therapy, and depletion screens confirmed that regulators of the endolysosome protect cancer cells from therapy [75,76]. These neoantigen-specific antibodies can also be bioengineered to have multiple targets, the most common example being bispecific antibodies targeting the neoantigen and CD3zeta chain. These bispecific antibodies bypass the specificity of the T cell receptor by having the neoantigen directly engage the TCR signaling component (CD3zeta), which has been shown to elicit T cell-mediated killing. In order to identify genes that confer resistance to CD20xCD3 bispecific antibody killing, CRISPRa survival screens have been performed in human mantle lymphoma cells treated with CD20xCD3 bispecific antibody under CD8 T cell selection pressure. These studies identified genes involved with protein glycosylation [77] and fucosylation [78] can reduce the therapeutic effectiveness of bispecific antibodies.
Antitumor humoral responses presently are a highly under-investigated area in cancer immunology. Understanding how B cell maturation is affected by immunosuppressive environments, such as by comparing B cell populations in the tumor draining lymph nodes to non-draining lymph nodes, can offer some insight. Characterizing the antibody repertoire in these environments may provide the best insight as to how the tumors shape the humoral immune response.
Cell-mediated Antitumor Responses
Once activated, CD8 T cells migrate to the tumor site where they selectively target tumor cells expressing their cognate peptide on MHCI. This interaction initiates the formation of the immunological synapse, which concentrates and directs the cytotoxic payload towards the targeted tumor cell [79]. Early CRISPR screens used human T cell lines to reidentify genetic pathways involved with T cell activation [80] and PD-1 expression [81]. These screens were followed by similar screens performed in primary human CD8 T cells [82]. Although these human CRISPR screens identified important modulators of CD8 T cell signaling and PD-1 expression, these studies were performed in the absence of an experimental tumor model, which may influence the types of genes identified. To address this issue, subsequent CD8 T cell screens were performed using primary murine CD8 T cells that were tumor-specific [83–86]. The Cas9-transgenic mice [12] was utilized and crossed it to the OT-I TCR which recognizes the SIINFEKL peptide of chicken ovalbumin [87], in order to generate OT-I;Cas9 double transgenic animals [83]. These animals allowed rapid isolation of OT-I;Cas9 T cells, which was transduced with a whole genome library [83]. Dong et al. performed converging in vivo and in vitro screens to identify genes that enhanced the ability of the T cells to infiltrate into the tumors and augment antigen-induced degranulation [83]. This study identified Dhx37 and Odc1 as new regulators of T cell activity with Dhx37-knockout CD8 T cells demonstrating the most robust antitumor efficacy [83].
Subsequent screens have been designed to identify: metabolic regulators that negatively impact CD8 T cell persistence [85]; kinases that modulate CD8 T cell memory, cytotoxicity, and expansion [84]; and genes that positively or negatively impact T cell responses to chemotherapy [88,89]. Although CD8 T cells are directly able to kill tumor cells, successful antitumor responses still require CD4 T cell-help [90]. Performing CRISPR screens in CD4 T cells remains difficult because their effects are mediated by other cell types. Identifying which cytokine(s) and effector molecule(s) that are important for these effects may serve as the first step towards designing a CRISPR screening assay.
As tumors grow and infiltrate into adjacent tissues, they acquire mutations that enable them to evade immune detection [91]. Critical effectors have been identified through forward genetic approaches [92–94]. The discovery of these evasion mechanisms has greatly accelerated with CRISPR screening, which identified pathways involved in the upregulation of checkpoint inhibition [36,95,96], downregulation of MHC processing [70,95,97] altered cytokine signaling [98–101], and autophagy [102]. In their landmark paper, Manguso and colleagues performed a series of in vivo CRISPR screens in Tcra−/− and C57BL6 mice treated with either irradiated tumor vaccine (GVAX) or GVAX + anti-PD-1 to determine which mutants contribute to immune escape [101]. They identified a number of genes involved in interferon signaling that when genetically perturbed aid in resistance to T cell responses [101]. Conversely, depletion screens performed using similar tumor models revealed that mutations in chromosomal modifiers and cytosolic regulators of antiviral sensing can sensitize tumors cells to immunotherapy [103,104]. These observations were later confirmed using multiple tumor cell lines derived from different organs [102].
Tumor cells commonly mutate genes involved with antigen presentation to evade CD8 T cell detection. To guard against this, our immune system deploys NK cells, which express a panoply of inhibitory [105] and activation receptors that enable these cells to target cells that downregulate MHCI expression [106]. CRISPR screens have been performed to identify mutated genes that confer NK cell resistance [107] as well as enhanced sensitization to NK cell killing [108]. Individual CRISPR screens using either NK cell selection or T cell selection have been compared to identify cytotoxic resistance genes [109] but screens applying simultaneous and/or sequential NK cell- and T cell-mediated selection are needed to determine how tumors evolve over time under different selection conditions. Although these cells have developed mechanisms to evade cell-mediated immunity, additional studies could determine if these selection mechanisms also confer resistance to other tumoricidal cells, such as macrophages and NKT cells.
Adoptive T cell therapies
Bioengineering of CD8 T cells have been of tremendous interest to the scientific community due to their unique ability to selectively target tumor cells. The most successful therapy to-date are chimeric antigen receptors (CARs), which integrate vital components of the TCR signaling complex with the antigenic specificity of well-characterized antibodies. This has enabled clinicians to direct the cytotoxicity of CD8 T cells to surface-bound neoantigens that are selectively expressed on pathogenic tumor cells. CAR-T cells have achieved tremendous success in patients with hematopoietic malignancies, but poor to moderate success in patients with solid tumors [110]. In order to enhance the efficacy of these therapies, researchers have employed CAR-T cell screens have identified that genetic perturbations to TLE4 and IKZF2 enhanced CAR-T cell killing while suppressing exhaustion programs [111]. Activation screens in CAR-T cells will need to be performed in different tumor models to determine which genes are important for stabilizing the immunological synapse in both liquid and solid tumors. Additional activation and knockout screens are needed to determine which genetic circuits are necessary to generate durable and effective CAR-T cell responses.
Due to the success of CAR-T cell therapy, many CARs have been developed to either target different antigens on the same cell type or to modify the composition of the cytoplasmic signaling domains. To compare the efficacy of the various CAR-T cells head-to-head, pooled knock-in CAR screens have utilized CRISPR to determine the therapeutic efficacy of each CAR construct simultaneously [112]. Newer CAR therapies have been focused on identifying TCRs that are neoantigen-specific with minimal responsiveness to autologous (self) and allogenic (non-self) MHC alleles to create off-the-shelf-CAR therapies. In a cleverly designed study, Crowther and colleagues isolated a primary human T clone (MC.7.G5) that could expand when cocultured with multiple human cell lines that was independent of allogenic MHC recognition [113]. They then employed CRISPR screens in HEK293T cells, which identified the MHC class I-like protein MR1 as the ligand for MC.7.G5 T cells, demonstrating that CRISPR can serve as a potent tool to identify ligands for orphan receptors and antibody targets [113].
Immunotherapeutic Screening Design
Continued investigation into CRISPR technologies is necessary to expand our knowledge of tumor immunology as well as identifying additional regulators for therapeutic intervention. As such, we provide four essential considerations, along with recommendations for publicly available resources, below to help facilitate future immunotherapeutic screens.
Genome-Integrating Pseudovirus
The first component is a genome-integrating pseudovirus, like integrase-expressing lentivirus or retrovirus, which enable the causative genes to be identified in pooled CRISPR sequences via next generation sequencing (NGS). Each crRNA contains a unique 20 base pair sequence that targets Cas9 to complementary gene segments to facilitate gene editing. Therefore, the identity of the gene is intricately linked to the sgRNA or crRNA sequence. Integrating pseudoviral systems propagates the sgRNA or crRNA sequence with each cellular division, enabling the identification of genetic perturbations. In order to expand the pseudoviral systems that enable CRISPR-based screening, Ye et al. has generated a transposon-based sleeping beauty system that was adapted into the nonintegrating adeno-associated virus (AAV) system in order to enable genomic integration and readout enriched sgRNAs / crRNAs in a screen setting [86].
Robust Reporter Gene
The second component is being able to identify cells that have been genetically perturbed. This is mediated through an antibiotic resistance gene, a fluorescent reporter, or a congenic marker that can be detected and enriched using commercially available antibodies and kits. These reporters enable researchers to accurately calculate library representation, which is particularly important due the variability in targeting efficiency between guides [14,16]. A successfully pooled genetic screen should aim for sufficient library coverage (e.g. 300x) of each crRNA across replicates to ensure consistent representation of each mutant within the cellular pool. Unlike antibiotic resistance-based reporters, fluorescent-based reporters and congenic markers give the added benefit of being able to calculate relative protein expression through flow cytometric analysis. Between the two, congenic-based reporters are superior to fluorescent-based reporters due to the flexibility provided by available antibodies, as well as the ability to enrich for infected cells via bead-based purification.
crRNA Library Construction and Pre-Screen Considerations
The third component is the crRNA library. Whole genome crRNA libraries for both mice and humans are available on Addgene [16,114]. For more tailored libraries, the Broad Institute has created CRISPick, a publicly available online tool that provides crRNA sequences for genes of interest [114,115]. crRNA libraries can be constructed based on signaling pathways [80], enzymatic families [84], metabolic function [85], and cellular localization [86]. For simple gene lists, gene ontology lists can be utilized to identify relevant genes of interest. Additionally, more rigorous computational approaches can be employed to design more stringent gene lists [44,116]. Such approaches will include expressed and non-expressed genes. If the goal is, instead, to identify the contributions of expressed genes only, then consideration should be given to designing crRNA libraries based on carefully curated RNA sequencing data [117]. Most importantly, when designing customized libraries, it is important to ensure that ~10% of all crRNAs within the library are non-targeting controls (NTCs) or guides targeting unexpressed proteins. These controls serve as internal computational benchmarks to see if an crRNA is enriched or not [16,115]. Prior to performing a screen, it is also important to confirm that the cells of interest respond uniformly to selection pressure and appropriately across a range of dose responses. During the screen, there should be a discernable difference between library infected cells compared to vector control, since reliable detection of infected cells increases the likelihood of identifying causative genes.
Cell-type of Interest
The fourth component is the cell type of interest. Adaptive immune cells contain a vast array of antigenic receptors due to somatically recombining their antigen receptor loci, which enables these cells to mount pathogen-specific responses. Therefore, for adaptive immune screens, CARs, TCR-constructs (e.g. NY-ESO), and mouse transgenic lines (OT-I, OT-II, PMEL, MD4) should be used to fix the TCR and BCR repertoires to ensure that the observed phenotype is due to gene perturbation as opposed to the specificity of the TCR or BCR. Unlike the adaptive immune system, the innate immune system expresses genetically encoded PRRs that detect PAMPs and DAMPs. Consequently, CRISPR screening of innate immune cells does not require transgenic systems. However, there are inborn errors of innate immune signaling that may confound innate immune screens [118]. Outbreeding of C567BL6-Cas9 transgenic mice can help address these issues.
Concluding Remarks
Tumor immunology encompasses more than just the responses mounted by macrophages, DCs, T cells, B cells, and NK cells. Largely, screens have been centered around these cell types because of their prevalence in circulation, their direct relevance towards tumor immunotherapy, and established protocols that allow these cells to be cultured ex vivo. However, there are other immune cell types with shorter half-lives, as well as other tissue-resident cell types that may have a comparable affect to antitumor responses. To study the effect of different genes in short lived immune cells, small library screens can be performed in HSCs to give rise to most hematopoietic lineages, though these efforts have primarily looked at T cell responses [119]. Due to the potential for gene toxicity at different hematopoietic developmental stages, temporal regulation of Cas9-mediated gene editing can facilitate more refined immunogenetic studies [120]. Applications of these approaches to the study of neutrophils and MDSCs may be of tremendous interest.
The study of tissue resident immune cells as well as complex immune cell networks presents a more difficult challenge for CRISPR screening due issues of scarcity and pleiotropic effects, respectively. The advent of CRISPR activation (CRISPRa) and interference (CRISPRi) screens may aid these studies when used in conjunction with complex genetic mouse models that are able to evaluate cell-cell interaction [121]. CRISPR-ko, CRISPRa, CRISPRi, as well as various combinatorial screens may be used for immunotherapy gene and genetic network discovery in the future (Figure 3).
Another limitation present in the current body of knowledge are that they are largely performed using orthotopic tumor cells to which the recipient mice are immunologically naïve. Therefore, immunotherapeutic screens should be performed using genetic models that have already undergone immune editing, to make the tumor models more physiologic. This may display different results from published screens.
In summary, we briefly outline current CRISPR technologies and multi-omic advancements. These advancements opened many areas of research as well as many outstanding questions (for example, see Outstanding Questions Box). We place the currently published peer-revied cancer immune-related CRISPR screens within its proper immunological context. Through our experiment with CRISPR screens, we provide four essential components for any immune-specific screens. Finally, we discuss potential avenues of researcher for immune-related CRISPR screens. Through this review, we hope to provide the reader with a cancer immunology primer and essential experimental parameters for CRISPR-immune screens to conduct their own CRISPR screen.
Outstanding Questions Box.
How would CRISPR-ko screen’s performance differ from CRISPRi screen’s in cancer immunology.
How can CRISPRa screen identify gain-of-function immune effectors in cancer.
How to harness CRISPR activation (CRISPRa) and interference (CRISPRi) screens to complement the discoveries that are challenging in CRISPR knockout screens.
How can combinatorial screens be used for the discovery of immunotherapy genetic network and genetic interactions.
What types of other CRISPR screen technologies can come into play of tumor immunology.
How can multi-omics open the new dimensions of CRISPR screening in cancer immunology.
What other cell types can be studied using CRISPR screening in the tumor microenvironment.
What other regulators will be identified from genetic screening in macrophages, DCs, T cells, B cells, or NK cells in the context of cancer immunity.
Supplementary Material
Supplemental Table 1. Exhaustive list of cancer-immune CRISPR screens.
Highlights.
CRISPR screens have recently been extensively utilized for cancer immunotherapy gene discovery
Multiple types of CRISPR screen technologies, including CRISPR-ko, CRISPRa and CRISPRi screens are available for different modes of gene identification
CRISPR screens allows identification of targets in both cancer cells and immune cells, such as T cells
In vivo CRISPR screens enable the discovery of genetic and cellular regulators in the tumor microenvironment
Acknowledgments
We thank all members in Chen laboratory for various discussions on gene editing and CRISPR screens.
S.C. is supported by Yale SBI/Genetics Startup Fund, NIH/NCI/NIDA (DP2CA238295, R01CA231112, U54CA209992-8697, R33CA225498, RF1DA048811), DoD (W81XWH-17-1-0235, W81XWH-20-1-0072, W81XWH-21-10514, W81XWH-21-1-0019), Damon Runyon Dale Frey Award (DFS-13-15), Melanoma Research Alliance (412806, 16-003524), St-Baldrick’s Foundation (426685), Breast Cancer Alliance, Cancer Research Institute (CLIP), AACR (499395, 17-20-01-CHEN), The Mary Kay Foundation (017-81), The V Foundation (V2017-022), Alliance for Cancer Gene Therapy, Sontag Foundation (DSA), Pershing Square Sohn Cancer Research Alliance, Yale Cancer Center Pilot Award, Dexter Lu Gift, Ludwig Family Foundation, Blavatnik Family Foundation, and Chenevert Family Foundation.
MBD is supported by the Yale MSTP training grant from NIH (T32GM007205). KT is supported by Yale PhD training grant from NIH (T32GM007499). JJZ is supported by Yale College Fellowships.
Footnotes
Declaration of Interest
No competing interest related to this study. SC is a scientific founder of EvolveImmune Therapeutics, unrelated to the study.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Reference:
- 1.Bernard JJ et al. (2019) Photoimmunology: how ultraviolet radiation affects the immune system. Nature Reviews Immunology 19, 688–701. 10.1038/s41577-019-0185-9 [DOI] [PubMed] [Google Scholar]
- 2.O’Keefe SJD (2016) Diet, microorganisms and their metabolites, and colon cancer. Nature Reviews. Gastroenterology & Hepatology 13, 691–706. 10.1038/nrgastro.2016.165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Liou G-Y and Storz P (2010) Reactive oxygen species in cancer. Free radical research 44, 10.3109/10715761003667554 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.O’Donnell JS et al. (2019) Cancer immunoediting and resistance to T cell-based immunotherapy. Nature Reviews Clinical Oncology 16, 151–167. 10.1038/s41571-018-0142-8 [DOI] [PubMed] [Google Scholar]
- 5.Hargadon KM et al. (2018) Immune checkpoint blockade therapy for cancer: An overview of FDA-approved immune checkpoint inhibitors. International Immunopharmacology 62, 29–39. 10.1016/j.intimp.2018.06.001 [DOI] [PubMed] [Google Scholar]
- 6.Lander ES (2016) The heroes of CRISPR. Cell 164, 18–28 [DOI] [PubMed] [Google Scholar]
- 7.Jinek M et al. (2012) A programmable dual-RNA–guided DNA endonuclease in adaptive bacterial immunity. science 337, 816–821 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cong L et al. (2013) Multiplex Genome Engineering Using CRISPR/Cas Systems. Science 339, 819–823. 10.1126/science.1231143 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Mali P et al. (2013) CAS9 transcriptional activators for target specificity screening and paired nickases for cooperative genome engineering. Nature Biotechnology 31, 833–838. 10.1038/nbt.2675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mali P et al. (2013) RNA-Guided Human Genome Engineering via Cas9. Science 339, 823–826. 10.1126/science.1232033 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koike-Yusa H et al. (2014) Genome-wide recessive genetic screening in mammalian cells with a lentiviral CRISPR-guide RNA library. Nature Biotechnology 32, 267–273. 10.1038/nbt.2800 [DOI] [PubMed] [Google Scholar]
- 12.Platt RJ et al. (2014) CRISPR-Cas9 knockin mice for genome editing and cancer modeling. Cell 159, 440–455. 10.1016/j.cell.2014.09.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chen S et al. (2015) Genome-wide CRISPR screen in a mouse model of tumor growth and metastasis. Cell 160, 1246–1260. 10.1016/j.cell.2015.02.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Hart T et al. (2015) High-Resolution CRISPR Screens Reveal Fitness Genes and Genotype-Specific Cancer Liabilities. Cell 163, 1515–1526. 10.1016/j.cell.2015.11.015 [DOI] [PubMed] [Google Scholar]
- 15.Parnas O et al. (2015) A Genome-wide CRISPR Screen in Primary Immune Cells to Dissect Regulatory Networks. Cell 162, 675–686. 10.1016/j.cell.2015.06.059 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Sanjana NE et al. (2014) Improved vectors and genome-wide libraries for CRISPR screening. Nature Methods 11, 783–784. 10.1038/nmeth.3047 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao D et al. (2018) Combinatorial CRISPR-Cas9 Metabolic Screens Reveal Critical Redox Control Points Dependent on the KEAP1-NRF2 Regulatory Axis. Molecular Cell 69, 699–708.e697. 10.1016/j.molcel.2018.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Perez-Pinera P et al. (2013) RNA-guided gene activation by CRISPR-Cas9-based transcription factors. Nat Methods 10, 973–976. 10.1038/nmeth.2600 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Perez-Pinera P et al. (2013) Synergistic and tunable human gene activation by combinations of synthetic transcription factors. Nat Methods 10, 239–242. 10.1038/nmeth.2361 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chavez A et al. (2015) Highly efficient Cas9-mediated transcriptional programming. Nat Methods 12, 326–328. 10.1038/nmeth.3312 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Tanenbaum ME et al. (2014) A protein-tagging system for signal amplification in gene expression and fluorescence imaging. Cell 159, 635–646. 10.1016/j.cell.2014.09.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Zhou H et al. (2018) In vivo simultaneous transcriptional activation of multiple genes in the brain using CRISPR-dCas9-activator transgenic mice. Nat Neurosci 21, 440–446. 10.1038/s41593-017-0060-6 [DOI] [PubMed] [Google Scholar]
- 23.Konermann S et al. (2013) Optical control of mammalian endogenous transcription and epigenetic states. Nature 500, 472–476. 10.1038/nature12466 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gilbert LA et al. (2013) CRISPR-mediated modular RNA-guided regulation of transcription in eukaryotes. Cell 154, 442–451. 10.1016/j.cell.2013.06.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Napier BA et al. (2016) Complement pathway amplifies caspase-11–dependent cell death and endotoxin-induced sepsis severity. Journal of Experimental Medicine 213, 2365–2382. 10.1084/jem.20160027 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Nedeva C et al. (2020) TREML4 receptor regulates inflammation and innate immune cell death during polymicrobial sepsis. Nature Immunology 21, 1585–1596. 10.1038/s41590-020-0789-z [DOI] [PubMed] [Google Scholar]
- 27.Orvedahl A et al. (2019) Autophagy genes in myeloid cells counteract IFNγ-induced TNF-mediated cell death and fatal TNF-induced shock. Proceedings of the National Academy of Sciences 116, 16497–16506. 10.1073/pnas.1822157116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Luteijn RD et al. (2019) SLC19A1 transports immunoreactive cyclic dinucleotides. Nature 573, 434–438. 10.1038/s41586-019-1553-0 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Covarrubias S et al. (2020) High-Throughput CRISPR Screening Identifies Genes Involved in Macrophage Viability and Inflammatory Pathways. Cell Reports 33, 108541. 10.1016/j.celrep.2020.108541 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Elster D et al. (2018) TRPS1 shapes YAP/TEAD-dependent transcription in breast cancer cells. Nature Communications 9, 3115. 10.1038/s41467-018-05370-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Feldman D et al. (2019) Optical Pooled Screens in Human Cells. Cell 179, 787–799.e717. 10.1016/j.cell.2019.09.016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Sato R et al. (2017) Requirement of glycosylation machinery in TLR responses revealed by CRISPR/Cas9 screening. International Immunology 29, 347–355. 10.1093/intimm/dxx044 [DOI] [PubMed] [Google Scholar]
- 33.Wroblewska A et al. (2018) Protein Barcodes enable high-dimensional single cell CRISPR screens. Cell 175, 1141–1155.e1116. 10.1016/j.cell.2018.09.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Jaitin DA et al. (2016) Dissecting Immune Circuits by Linking CRISPR-Pooled Screens with Single-Cell RNA-Seq. Cell 167, 1883–1896.e1815. 10.1016/j.cell.2016.11.039 [DOI] [PubMed] [Google Scholar]
- 35.Dixit A et al. (2016) Perturb-Seq: Dissecting Molecular Circuits with Scalable Single-Cell RNA Profiling of Pooled Genetic Screens. Cell 167, 1853–1866.e1817. 10.1016/j.cell.2016.11.038 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Papalexi E et al. (2021) Characterizing the molecular regulation of inhibitory immune checkpoints with multimodal single-cell screens. Nature Genetics 53, 322–331. 10.1038/s41588-021-00778-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Rubin AJ et al. (2019) Coupled Single-Cell CRISPR Screening and Epigenomic Profiling Reveals Causal Gene Regulatory Networks. Cell 176, 361–376.e317. 10.1016/j.cell.2018.11.022 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Datlinger P et al. (2021) Ultra-high-throughput single-cell RNA sequencing and perturbation screening with combinatorial fluidic indexing. Nature Methods 18, 635–642. 10.1038/s41592-021-01153-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Kabashima K et al. (2019) The immunological anatomy of the skin. Nature Reviews. Immunology 19, 19–30. 10.1038/s41577-018-0084-5 [DOI] [PubMed] [Google Scholar]
- 40.Kayama H et al. (2020) Interaction Between the Microbiota, Epithelia, and Immune Cells in the Intestine. Annual Review of Immunology 38, 23–48. 10.1146/annurev-immunol-070119-115104 [DOI] [PubMed] [Google Scholar]
- 41.Demaria O et al. (2019) Harnessing innate immunity in cancer therapy. Nature 574, 45–56. 10.1038/s41586-019-1593-5 [DOI] [PubMed] [Google Scholar]
- 42.Gajewski TF et al. (2013) Innate and adaptive immune cells in the tumor microenvironment. Nature Immunology 14, 1014–1022. 10.1038/ni.2703 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Wisnovsky S et al. (2021) Genome-wide CRISPR screens reveal a specific ligand for the glycan-binding immune checkpoint receptor Siglec-7. Proceedings of the National Academy of Sciences 118. 10.1073/pnas.2015024118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi S et al. (2020) Inhibition of MAN2A1 Enhances the Immune Response to Anti-PD-L1 in Human Tumors. Clinical Cancer Research: An Official Journal of the American Association for Cancer Research 26, 5990–6002. 10.1158/1078-0432.CCR-20-0778 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kamber RA et al. (2021) Inter-cellular CRISPR screens reveal regulators of cancer cell phagocytosis. Nature 597, 549–554. 10.1038/s41586-021-03879-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Jimenez-Duran G et al. (2020) Pharmacological validation of targets regulating CD14 during macrophage differentiation. EBioMedicine 61. 10.1016/j.ebiom.2020.103039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Benaoudia S et al. (2019) A genome-wide screen identifies IRF2 as a key regulator of caspase-4 in human cells. EMBO reports 20, e48235. 10.15252/embr.201948235 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Chui AJ et al. (2019) N-terminal degradation activates the NLRP1B inflammasome. Science 364, 82–85. 10.1126/science.aau1208 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lee BL et al. (2018) ASC- and caspase-8-dependent apoptotic pathway diverges from the NLRC4 inflammasome in macrophages. Scientific Reports 8, 3788. 10.1038/s41598-018-21998-3 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Xu H et al. (2019) The N-end rule ubiquitin ligase UBR2 mediates NLRP1B inflammasome activation by anthrax lethal toxin. The EMBO journal 38, e101996. 10.15252/embj.2019101996 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kawasaki T and Kawai T (2014) Toll-Like Receptor Signaling Pathways. Frontiers in Immunology 5, 461. 10.3389/fimmu.2014.00461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Covarrubias S et al. (2017) CRISPR/Cas-based screening of long non-coding RNAs (lncRNAs) in macrophages with an NF-κB reporter. The Journal of Biological Chemistry 292, 20911–20920. 10.1074/jbc.M117.799155 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Wang Q et al. (2018) Condensin Smc4 promotes inflammatory innate immune response by epigenetically enhancing NEMO transcription. Journal of Autoimmunity 92, 67–76. 10.1016/j.jaut.2018.05.004 [DOI] [PubMed] [Google Scholar]
- 54.Zablocki-Thomas L et al. (2020) A genome-wide CRISPR screen identifies regulation factors of the TLR3 signalling pathway. Innate Immunity 26, 459–472. 10.1177/1753425920915507 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Deng L et al. (2014) STING-Dependent Cytosolic DNA Sensing Promotes Radiation-Induced Type I Interferon-Dependent Antitumor Immunity in Immunogenic Tumors. Immunity 41, 843–852. 10.1016/j.immuni.2014.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Klarquist J et al. (2014) STING-mediated DNA sensing promotes antitumor and autoimmune responses to dying cells. Journal of Immunology (Baltimore, Md.: 1950) 193, 6124–6134. 10.4049/jimmunol.1401869 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Woo S-R et al. (2014) STING-Dependent Cytosolic DNA Sensing Mediates Innate Immune Recognition of Immunogenic Tumors. Immunity 41, 830–842. 10.1016/j.immuni.2014.10.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Ritchie C et al. (2019) SLC19A1 Is an Importer of the Immunotransmitter cGAMP. Molecular Cell 75, 372–381.e375. 10.1016/j.molcel.2019.05.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Fuertes MB et al. (2011) Host type I IFN signals are required for antitumor CD8+ T cell responses through CD8{alpha}+ dendritic cells. The Journal of Experimental Medicine 208, 2005–2016. 10.1084/jem.20101159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Hildner K et al. (2008) Batf3 Deficiency Reveals a Critical Role for CD8α+ Dendritic Cells in Cytotoxic T Cell Immunity. Science 322, 1097–1100. 10.1126/science.1164206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Lindner B et al. (2021) A genome-wide CRISPR/Cas9 screen to identify phagocytosis modulators in monocytic THP-1 cells. Scientific Reports 11, 12973. 10.1038/s41598-021-92332-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Sedlyarov V et al. (2018) The Bicarbonate Transporter SLC4A7 Plays a Key Role in Macrophage Phagosome Acidification. Cell Host & Microbe 23, 766–774.e765. 10.1016/j.chom.2018.04.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Haney MS et al. (2018) Identification of phagocytosis regulators using magnetic genome-wide CRISPR screens. Nature Genetics 50, 1716–1727. 10.1038/s41588-018-0254-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Gordon S (2016) Phagocytosis: An Immunobiologic Process. Immunity 44, 463–475. 10.1016/j.immuni.2016.02.026 [DOI] [PubMed] [Google Scholar]
- 65.Beatty GL et al. (2011) CD40 Agonists Alter Tumor Stroma and Show Efficacy Against Pancreatic Carcinoma in Mice and Humans. Science (New York, N.y.) 331, 1612–1616. 10.1126/science.1198443 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Coulie PG et al. (2014) Tumour antigens recognized by T lymphocytes: at the core of cancer immunotherapy. Nature Reviews. Cancer 14, 135–146. 10.1038/nrc3670 [DOI] [PubMed] [Google Scholar]
- 67.Li F et al. (2020) In Vivo Epigenetic CRISPR Screen Identifies Asf1a as an Immunotherapeutic Target in Kras-Mutant Lung Adenocarcinoma. Cancer Discovery 10, 270–287. 10.1158/2159-8290.CD-19-0780 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Li J et al. (2021) Epigenetic and Transcriptional Control of the Epidermal Growth Factor Receptor Regulates the Tumor Immune Microenvironment in Pancreatic Cancer. Cancer Discovery 11, 736–753. 10.1158/2159-8290.CD-20-0519 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Wang G et al. (2019) Multiplexed activation of endogenous genes by CRISPRa elicits potent antitumor immunity. Nature Immunology 20, 1494–1505. 10.1038/s41590-019-0500-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Alix E et al. (2020) The Tumour Suppressor TMEM127 Is a Nedd4-Family E3 Ligase Adaptor Required by Salmonella SteD to Ubiquitinate and Degrade MHC Class II Molecules. Cell Host & Microbe 28, 54–68.e57. 10.1016/j.chom.2020.04.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Dudziak D et al. (2007) Differential Antigen Processing by Dendritic Cell Subsets in Vivo. Science 315, 107–111. 10.1126/science.1136080 [DOI] [PubMed] [Google Scholar]
- 72.Theisen DJ et al. (2018) WDFY4 is required for cross-presentation in response to viral and tumor antigens. Science 362, 694–699. 10.1126/science.aat5030 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Thomas SN et al. (2016) Implications of Lymphatic Transport to Lymph Nodes in Immunity and Immunotherapy. Annu Rev Biomed Eng 18, 207–233. 10.1146/annurev-bioeng-101515-014413 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jiang C et al. (2019) CRISPR/Cas9 Screens Reveal Multiple Layers of B cell CD40 Regulation. Cell Reports 28, 1307–1322.e1308. 10.1016/j.celrep.2019.06.079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Thomsen EA et al. (2020) Identification of BLNK and BTK as mediators of rituximab-induced programmed cell death by CRISPR screens in GCB-subtype diffuse large B-cell lymphoma. Molecular Oncology 14, 1978–1997. 10.1002/1878-0261.12753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Tsui CK et al. (2019) CRISPR-Cas9 screens identify regulators of antibody–drug conjugate toxicity. Nature Chemical Biology 15, 949–958. 10.1038/s41589-019-0342-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Decker CE et al. (2019) Genome-scale CRISPR activation screen uncovers tumor-intrinsic modulators of CD3 bispecific antibody efficacy. Scientific Reports 9, 20068. 10.1038/s41598-019-56670-x [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Liu S-Q et al. (2021) A CRISPR Screen Reveals Resistance Mechanisms to CD3-Bispecific Antibody Therapy. Cancer Immunology Research 9, 34–49. 10.1158/2326-6066.CIR-20-0080 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Voskoboinik I et al. (2006) Perforin-mediated target-cell death and immune homeostasis. Nature Reviews Immunology 6, 940–952. 10.1038/nri1983 [DOI] [PubMed] [Google Scholar]
- 80.Shang W et al. (2018) Genome-wide CRISPR screen identifies FAM49B as a key regulator of actin dynamics and T cell activation. Proceedings of the National Academy of Sciences 115, E4051–E4060. 10.1073/pnas.1801340115 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Okada M et al. (2017) Blockage of Core Fucosylation Reduces Cell-Surface Expression of PD-1 and Promotes Anti-tumor Immune Responses of T Cells. Cell Reports 20, 1017–1028. 10.1016/j.celrep.2017.07.027 [DOI] [PubMed] [Google Scholar]
- 82.Shifrut E et al. (2018) Genome-wide CRISPR Screens in Primary Human T Cells Reveal Key Regulators of Immune Function. Cell 175, 1958–1971 e1915. 10.1016/j.cell.2018.10.024 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Dong MB et al. (2019) Systematic Immunotherapy Target Discovery Using Genome-Scale In Vivo CRISPR Screens in CD8 T Cells. Cell 178, 1189–1204.e1123. 10.1016/j.cell.2019.07.044 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Gurusamy D et al. (2020) Multi-phenotype CRISPR-Cas9 Screen Identifies p38 Kinase as a Target for Adoptive Immunotherapies. Cancer Cell 37, 818–833.e819. 10.1016/j.ccell.2020.05.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Wei J et al. (2019) Targeting REGNASE-1 programs long-lived effector T cells for cancer therapy. Nature 576, 471–476. 10.1038/s41586-019-1821-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Ye L et al. (2019) In vivo CRISPR screening in CD8 T cells with AAV–Sleeping Beauty hybrid vectors identifies membrane targets for improving immunotherapy for glioblastoma. Nature Biotechnology 37, 1302–1313. 10.1038/s41587-019-0246-4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Hogquist KA et al. (1994) T cell receptor antagonist peptides induce positive selection. Cell 76, 17–27 [DOI] [PubMed] [Google Scholar]
- 88.Dufva O et al. (2020) Integrated drug profiling and CRISPR screening identify essential pathways for CAR T-cell cytotoxicity. Blood 135, 597–609. 10.1182/blood.2019002121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lizotte PH et al. (2018) A High-Throughput Immune-Oncology Screen Identifies EGFR Inhibitors as Potent Enhancers of Antigen-Specific Cytotoxic T-lymphocyte Tumor Cell Killing. Cancer Immunology Research 6, 1511–1523. 10.1158/2326-6066.CIR-18-0193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Alspach E et al. (2019) MHC-II neoantigens shape tumour immunity and response to immunotherapy. Nature 574, 696–701. 10.1038/s41586-019-1671-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schreiber RD et al. (2011) Cancer Immunoediting: Integrating Immunity’s Roles in Cancer Suppression and Promotion. Science 331, 1565–1570. 10.1126/science.1203486 [DOI] [PubMed] [Google Scholar]
- 92.Klampfl T et al. (2013) Somatic Mutations of Calreticulin in Myeloproliferative Neoplasms. New England Journal of Medicine 369, 2379–2390. 10.1056/NEJMoa1311347 [DOI] [PubMed] [Google Scholar]
- 93.Ortmann CA et al. (2015) Effect of Mutation Order on Myeloproliferative Neoplasms. New England Journal of Medicine 372, 601–612. 10.1056/NEJMoa1412098 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Zaretsky JM et al. (2016) Mutations Associated with Acquired Resistance to PD-1 Blockade in Melanoma. New England Journal of Medicine 375, 819–829. 10.1056/NEJMoa1604958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Gu SS et al. (2021) Therapeutically Increasing MHC-I Expression Potentiates Immune Checkpoint Blockade. Cancer Discovery 11, 1524–1541. 10.1158/2159-8290.CD-20-0812 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Zhang J-P et al. (2019) A novel model of controlling PD-L1 expression in ALK+ anaplastic large cell lymphoma revealed by CRISPR screening. Blood 134, 171–185. 10.1182/blood.2019001043 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Spel L et al. (2018) Nedd4-Binding Protein 1 and TNFAIP3-Interacting Protein 1 Control MHC-1 Display in Neuroblastoma. Cancer Research 78, 6621–6631. 10.1158/0008-5472.CAN-18-0545 [DOI] [PubMed] [Google Scholar]
- 98.Codina A et al. (2019) Convergent Identification and Interrogation of Tumor-Intrinsic Factors that Modulate Cancer Immunity In Vivo. Cell Systems 8, 136–151.e137. 10.1016/j.cels.2019.01.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Han P et al. (2019) Genome-Wide CRISPR Screening Identifies JAK1 Deficiency as a Mechanism of T-Cell Resistance. Frontiers in Immunology 10, 251. 10.3389/fimmu.2019.00251 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Kearney CJ et al. (2018) Tumor immune evasion arises through loss of TNF sensitivity. Science Immunology 3. 10.1126/sciimmunol.aar3451 [DOI] [PubMed] [Google Scholar]
- 101.Manguso RT et al. (2017) In vivo CRISPR screening identifies Ptpn2 as a cancer immunotherapy target. Nature 547, 413–418. 10.1038/nature23270 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Lawson KA et al. (2020) Functional genomic landscape of cancer-intrinsic evasion of killing by T cells. Nature 586, 120–126. 10.1038/s41586-020-2746-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Ishizuka JJ et al. (2019) Loss of ADAR1 in tumours overcomes resistance to immune checkpoint blockade. Nature 565, 43–48. 10.1038/s41586-018-0768-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pan D et al. (2018) A major chromatin regulator determines resistance of tumor cells to T cell-mediated killing. Science (New York, N.Y.) 359, 770–775. 10.1126/science.aao1710 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Zhang X et al. (2019) Synergized regulation of NK cell education by NKG2A and specific Ly49 family members. Nature Communications 10, 5010. 10.1038/s41467-019-13032-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Huang R-S et al. (2020) Enhanced NK-92 Cytotoxicity by CRISPR Genome Engineering Using Cas9 Ribonucleoproteins. Frontiers in Immunology 0. 10.3389/fimmu.2020.01008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhuang X et al. (2019) Genome-Wide CRISPR Screen Reveals Cancer Cell Resistance to NK Cells Induced by NK-Derived IFN-γ. Front Immunol 10, 2879. 10.3389/fimmu.2019.02879 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Pech MF et al. (2019) Systematic identification of cancer cell vulnerabilities to natural killer cell-mediated immune surveillance. eLife 8, e47362. 10.7554/eLife.47362 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Freeman AJ et al. (2019) Natural Killer Cells Suppress T Cell-Associated Tumor Immune Evasion. Cell Reports 28, 2784–2794.e2785. 10.1016/j.celrep.2019.08.017 [DOI] [PubMed] [Google Scholar]
- 110.Pettitt D et al. (2018) CAR-T Cells: A Systematic Review and Mixed Methods Analysis of the Clinical Trial Landscape. Molecular Therapy: The Journal of the American Society of Gene Therapy 26, 342–353. 10.1016/j.ymthe.2017.10.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wang D et al. (2021) CRISPR Screening of CAR T Cells and Cancer Stem Cells Reveals Critical Dependencies for Cell-Based Therapies. Cancer Discovery 11, 1192–1211. 10.1158/2159-8290.CD-20-1243 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Roth TL et al. (2020) Pooled Knockin Targeting for Genome Engineering of Cellular Immunotherapies. Cell 181, 728–744.e721. 10.1016/j.cell.2020.03.039 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Crowther MD et al. (2020) Genome-wide CRISPR–Cas9 screening reveals ubiquitous T cell cancer targeting via the monomorphic MHC class I-related protein MR1. Nature Immunology 21, 178–185. 10.1038/s41590-019-0578-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Doench JG et al. (2016) Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nature Biotechnology 34, 184–191. 10.1038/nbt.3437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Sanson KR et al. (2018) Optimized libraries for CRISPR-Cas9 genetic screens with multiple modalities. Nature Communications 9, 5416. 10.1038/s41467-018-07901-8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Fu J et al. (2020) Large-scale public data reuse to model immunotherapy response and resistance. Genome Medicine 12, 21. 10.1186/s13073-020-0721-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Zhang Y et al. (2020) Histone Loaders CAF1 and HIRA Restrict Epstein-Barr Virus B-Cell Lytic Reactivation. mBio 11. 10.1128/mBio.01063-20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Sellers RS et al. (2012) Immunological Variation Between Inbred Laboratory Mouse Strains: Points to Consider in Phenotyping Genetically Immunomodified Mice. Veterinary Pathology 49, 32–43. 10.1177/0300985811429314 [DOI] [PubMed] [Google Scholar]
- 119.LaFleur MW et al. (2019) A CRISPR-Cas9 delivery system for in vivo screening of genes in the immune system. Nature Communications 10, 1668. 10.1038/s41467-019-09656-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Laidlaw BJ et al. (2020) The transcription factor Hhex cooperates with the corepressor Tle3 to promote memory B cell development. Nature Immunology 21, 1082–1093. 10.1038/s41590-020-0713-6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Pickar-Oliver A and Gersbach CA (2019) The next generation of CRISPR–Cas technologies and applications. Nature Reviews Molecular Cell Biology 20, 490–507. 10.1038/s41580-019-0131-5 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplemental Table 1. Exhaustive list of cancer-immune CRISPR screens.