

Abstract
Various cellular insults and injury to renal epithelial cells stimulate repair mechanisms to adapt and restore the organ homeostasis. Renal tubular epithelial cells are endowed with regenerative capacity, which allows for a restoration of nephron function after acute kidney injury. However, recent evidence indicates that the repair is often incomplete, leading to maladaptive responses that promote the progression to chronic kidney disease. The dysregulated cell cycle and proliferation is also a key feature of renal tubular epithelial cells in polycystic kidney disease and HIV-associated nephropathy. Therefore, in this review, we provide an overview of cell cycle regulation and the consequences of dysregulated cell proliferation in acute kidney injury, polycystic kidney disease, and HIV-associated nephropathy. An increased understanding of these processes may help define better targets for kidney repair and combat chronic kidney disease progression.
Keywords: acute kidney injury, cell cycle, HIV-associated nephropathy, polycystic kidney disease, renal tubular epithelial cells
The ability to repair and regenerate damaged tissue is essential for the restoration of functional integrity of tissues and organs and ultimately the survival of complex organisms. However, complete recovery and regeneration of tissues can be observed during prenatal development in humans, but it is largely lost during adult life.1 In contrast, various nonmammalian vertebrates, such as certain salamanders (e.g., newts and axolotl), retain the capacity in adulthood to fully regenerate various organs and appendages and serve as model organisms to gain insights into near-perfect regeneration and scar-free healing.2,3 In the context of kidney injury and repair, it has long been held that a complete recovery was possible following an acute kidney injury (AKI), because proximal tubular cells, as the major target of AKI, are endowed with the capacity to proliferate and replenish the lost cells. However, recent evidence indicates that the repair is often incomplete, leading to maladaptive responses that promote the progression toward chronic kidney disease (CKD).4,5 Recent studies in experimental models of AKI have highlighted the cell cycle arrest of renal tubular epithelial cells (RTECs) as a key component in fibrosis development following AKI.6,7 A similar cell cycle arrest of RTECs has been observed in the setting of HIV-associated nephropathy (HIVAN) that features a prominent tubulointerstitial injury that accompanies the collapsing variant of glomerulosclerosis.8 In the contexts of polycystic kidney disease (PKD), such as autosomal dominant PKD (ADPKD), an unchecked tubular proliferation is a characteristic feature and a driver of disease pathogenesis. Thus, dysregulated cell cycle and proliferation of RTECs may underlie various modes of kidney injury and disease, whether in growth-arrested RTECs in AKI or hyperproliferative RTECs in ADPKD. Therefore, in this review, we provide a brief overview of the mammalian cell cycle and discuss the current understanding of the shared and nonoverlapping mechanisms of cell cycle dysregulation in RTECs in AKI, ADPKD, and HIVAN.
Canonical and noncanonical mitotic cell cycle
The mitotic cell cycle enables cell proliferation and division to occur with precise genome replication and duplication of its cellular components and machinery in the daughter cells necessary to repeat the process. The mammalian cell cycle consists of the S phase, in which genomic DNA is replicated; the M phase, in which the nuclear division (mitosis/karyokinesis) and cell division (cytokinesis) occur; and the 2 gap phases (G1 and G2) that separate the S and M phases. G1 is a particularly important regulatory period, as once the cell passes the G1 checkpoint and enters the S phase, it becomes irreversibly committed to progression through the cell division cycle. The fidelity and the coordination of each of these events are tightly regulated through the sequential activation of cyclin-dependent kinase (CDK) complexes, composed of cyclins that are expressed and degraded at specific cell cycle phases and CDKs whose activities are regulated by interactions with activators and inhibitors to ensure that the cell cycle progresses in one direction only.9,10 As outlined in Figure 1a, mammalian cells enter the cell cycle in response to cyclin D expression, which is required for CDK4 and CDK6 activation. Cyclin E accumulates in the late G1 phase and specifically activates CDK2. Together with cyclin D–CDK4/6 complex, cyclin E–CDK2 is responsible for passage through the late G1 and transitioning into the S phase, which involves the inhibitory hyperphosphorylation of retinoblastoma, allowing for the expression of genes necessary for DNA replication and further cell cycle progression. Among these genes is cyclin A that will complex with CDK2 and replace cyclin E that is degraded during the progression of S phase. The cyclin A–CDK2 remains active through S and early G2 phases. With the subsequent degradation of cyclin A during G2, the activation of a cyclin B–CDK1 complex drives the G2/M transition and commencement of mitosis. Concomitant with cyclin B/CDK1 degradation, mitotic exit and cytokinesis occur. In addition to these tightly coordinated activations of cyclin-CDK complexes, cell cycle progression is regulated by CDK inhibitors comprising the inhibitors of CDK4 (INK4) and CDK interacting protein/Kinase inhibitory protein (Cip/Kip) family members. The INK4 proteins (p21Cip1, p27Kip1, and p57Kip2) suppress the cyclin D–CDK4/6 complex during G1, whereas Cip/Kip family members (p21Cip1, p27Kip1, and p57Kip2) restrain the activities of a broader set of cyclin-CDK complexes11 (Figure 1a). These cell cycle regulators in the context of kidney cell homeostasis and injury have been widely studied and summarized in excellent reviews.12–14 Several quality-control checkpoints are also placed during the cell cycle to guard against damaged or incompletely replicated DNA (between G1/S, G2/M, and intra-S) and to monitor chromosome alignment for proper segregation of the sister chromatids (mitotic checkpoint, also called spindle assembly checkpoint). As cells exit the M phase at the end of the cell cycle, they commit to the next round of cell cycle progression or cell cycle exit (G0). G0 is a reversible quiescent nondividing state, which is nevertheless actively maintained by continual repression of genes necessary for cell division and activation of genes to block apoptosis and senescence.15–17 Terminal differentiation is coupled with a permanent exit from the cell cycle, where cells are refractory to proliferative signals and associated with low expression of cyclins and CDKs, hypophosphorylation of retinoblastoma, and upregulation of CDK inhibitors to ensure the inhibition G1 progression.18,19
Figure 1 |. Canonical and noncanonical cell cycle in eukaryotic cells.

(a) The schematics of the canonical mitotic cell cycle with specific expression patterns of cyclins and cyclin-dependent kinases (CDKs) are shown above the phases of the cell cycle with a dashed line. Cell cycle checkpoints are indicated below with arrows. (b) The schematics of endoreplication showing endomitosis (top) and endocycling (bottom). In endomitosis, cells undergo incomplete cytokinesis and generate a polyploid binucleate or polyploid mononucleate cell. In endocycling, cells increase their DNA by alternating between G and S phases without cell division. Polyploid cells can also reduce ploidy through multipolar spindle formation, a process known as ploidy reversal. During this ploidy reversal process, chromosome segregation errors are common, resulting in the formation of aneuploid daughter cells with a gain or loss of ≥1 individual chromosomes. C, total number of pairs of chromosomes in the cell, chromatin amount or DNA content as a multiple of haploid genome (e.g., 4C = chromatin amount of a diploid cell in the G2 phase of the cell cycle); CIP/KIP, CDK-interacting protein/Kinase inhibitory protein; CKI, cyclin-dependent kinase inhibitors; INK4, inhibitors of CDK4; n, number of sets of chromosomes as a multiple of the haploid genome (e.g., 2n = chromosomal content of a diploid cell); Rb, retinoblastoma.
The noncanonical mitotic cell cycle (i.e., endoreplication) is also observed in mammalian cells, including the mouse kidney RTECs.20,21 Although a strict consensus in the nomenclature is not yet found, endoreplication comprises endocycling, in which cycling cells with fully replicated DNA (from S or G2) bypasses the M phase entirely and re-enters the next G1; and endomitosis, in which cells that have entered M phase re-enter the G1 before completion of karyokinesis or cytokinesis22–24 (Figure 1b). As a result, endoreduplication results in polyploidization (duplicated chromosomes, e.g., 4C, 8C, or 16C). Although commonly observed in invertebrates and plants, the occurrence of endoreplication was considered to be rare in mammalian cells, with the exception of few specialized cell types, such as trophoblasts, megakaryocytes, and liver hepatocytes, as a part of development and differentiation programs.22 However, recent studies show more widespread occurrences of endoreplication in terminally differentiated mammalian cells. Endoreplication has been observed in epidermal differentiation,25 vascular smooth muscle cells in response to various hypertensive stimuli,26–28 mammary epithelial cells in lactation,29 and liver development30–32 and regeneration.33 The emerging theme is that within the correct timing and context, endoreplication in differentiated cells, particularly those with high metabolic activity, serves to promote mammalian tissue growth and repair.22–24 It is hypothesized that endoreplication, rather than mitotic cell division, would provide a homeostatic advantage in situations where energy sources are limiting and rapid growth is required to sustain mass production of proteins and high metabolic activity during tissue growth and repair. However, polyploidization and subsequent aneuploidy and genomic instability are also hallmarks of oncogenic tumors that drive cancer progression.34 Thus, it remains to be shown how the polyploid cells in tissue repair can remain mitotically active and accumulate genomic instability without becoming tumorigenic. In addition, molecular mechanisms coordinating proliferation and endoreplication in tissue adaptation, particularly in the kidney, remain to be better elucidated.
RTEC proliferation and cell cycle dysregulation
Acute kidney injury.
Under the normal condition in the adult kidney, cells are mainly quiescent, with proliferation occurring in <1% of tubular cells.35,36 It is notable that a sizable portion of the proximal tubular cells, particularly in the S3 segment, expresses markers consistent with being in G1, rather than in G0, quiescence.37,38 This is thought to poise the S3 proximal tubular cells, most susceptible to acute kidney insult, to be primed for rapid cell division for efficiency in recovery, as cells entering from G0 have a significantly longer G1 phase than actively cycling ones.39 On AKI, such as those induced by toxic or ischemic insults, heavily injured cells are lost through apoptosis or necrosis, but surviving tubular cells rapidly re-enter the cell cycle and proliferate to restore the nephron function40–42 (Figure 2). Interestingly, concomitant with cell cycle entry, rapid induction of cell cycle inhibitor p21Cip1 (p21) has been observed in RTECs in various experimental models of AKI.43 The activation of p21 to put the brakes on the cell cycle progression seem counterproductive in RTECs in AKI, but p21 (and p27Kip1) has been shown to participate in the assembly and nuclear import of cyclin D/CDK4 complex during the early G1 progression, in addition to their cell cycle inhibitory roles.44,45 Thus, it is presumed that p21/p27’s participation in G1 progression while delaying the G1/S transition is an adaptive mechanism that allows sufficient time for proper DNA repair in injured RTECs. Such protective role of p21 in RTEC repair is in accordance with the observation that p21 knockout mouse kidneys displayed worsened tubular damage and increased mortality following cisplatin exposure or ischemia-reperfusion injury.46,47 Loss of p21 also mitigated the beneficial effects of renal ischemic preconditioning on ischemia-reperfusion injury in mice.48 Similarly, the delay of S phase entry by CDK4/6 inhibitors, palbociclib and ribociclib, reduced tubular cell apoptosis in experimental models of AKI.49,50 Because the inhibition of CDK4/6 by palbociclib and ribociclib is transient and reversible,49–51 it is thought that their actions sufficiently slow down the G1/S transition to mitigate cell death but allow subsequent cycling and division necessary for recovery, further supporting the notion that appropriately regulated G1 progression is essential in proper kidney repair after AKI. Paradoxically, in contrast to the protective role in the setting of AKI, p21’s actions are associated with the worsening of CKD progression.52 In the murine remnant kidney model, the genetic ablation of p21 attenuated the histologic lesions and prevented the progressive kidney decline in kidney function.52 These contrasting observations of p21 inhibition suggest that transient cell cycle arrest may be a protective mechanism in acute injury, whereas prolonged cell cycle arrest is deleterious. Moreover, well beyond the cell cycle regulatory role, p21 is now recognized as a multifunctional protein whose actions include regulation of apoptosis, differentiation, transcription, and migration.53–55 Thus, it is likely that the actions of p21 beyond the cell cycle regulation contribute to RTEC impairment and recovery after AKI.
Figure 2 |. Renal tubular epithelial cell (RTEC) repair following acute kidney injury (AKI).

On AKI, normally quiescent RTECs in G0 or G1 re-enter the cell cycle and proliferate. Slowing the G1/S transition aids in proper RTEC repair, but prolonged G2/M arrest leads to premature senescence and incomplete recovery. CDK, cyclin-dependent kinase.
The importance of proper cell cycle regulation in RTECs in kidneys recovering from AKI is further underscored by the work of Yang et al., which showed that the proliferating proximal tubular cells after AKI are often arrested in G2/M checkpoint and that the ensuing fibrosis development correlates with the extent of G2/M arrest.7 G2/M arrest typically triggered by DNA damage is a reversible process, following DNA repair and downregulation of p53. p53, as one of the key guardians of genome integrity, provides surveillance against various cellular insults, including missegregated chromosomes, by inducing cell cycle arrest and senescence.56 However, irreversible cell cycle arrest by p53 leads to premature cellular senescence.57,58 Consistent with prolonged and irreversibly arrested cells, G2/M-arrested proximal tubular cells in response to AKI assumed a senescent-associated secretory phenotype, thereby producing increased amounts of profibrotic factors.7 The heterogeneous composition of senescent-associated secretory phenotype molecules that are released into the surrounding microenvironment includes growth factors, inflammatory cytokines, and modulators of extracellular matrix, such as insulin-like growth factor 1, interleukin-6 and interleukin-8, plasminogen activator inhibitor-1, and transforming growth factor beta.59 Interestingly, in obstructed kidneys, enhanced transforming growth factor beta signaling in tubules also triggered p21-mediated G2/M arrest in RTECs,60,61 suggesting a vicious cycle between cell cycle arrest–mediated senescent-associated secretory phenotype and irreversible RTEC cell cycle arrest.
Although p53-mediated cell cycle arrest is an essential trigger of senescence induction, the activities of other pathways converge to make the process irreversible, such as those induced by p16INK4a/retinoblastoma, nuclear factor-κB, and mechanistic target of rapamycin.62–64 In oncogene-induced senescent cells, senescent-associated secretory phenotype production is further boosted by a reorganization of endomembrane compartments to coordinate protein synthesis and autophagy by mechanistic target of rapamycin, termed target of rapamycin–autophagy spatial coupling compartments.65 In line with these observations, Canaud et al. demonstrated the increased formation of target of rapamycin–autophagy spatial coupling compartments in G2/M-arrested proximal tubular cells during kidney fibrosis progression, in part mediated by p53-induced expression of atypical cyclin, cyclin G,6 such that genetic deletion of Raptor, a major component of mechanistic target of rapamycin complex 1, or pharmacologic inhibition of p53 reduced the target of rapamycin–autophagy spatial coupling compartment formation and ensuing renal fibrosis progression after AKI.6 However, analogous the opposing role of p21 in AKI and in CKD, although the inhibition of p53 has shown to promote better recovery after AKI recovery in rodent models,66–71 the long-term inhibition of p53 in kidneys after AKI is also associated with augmented renal inflammation and fibrosis.72–74 These observations are not entirely surprising, given the multifaceted role of p53 that can at times exert dichotomous effects on biological processes in that in contrast to its well-defined antiproliferative and proapoptotic functions, p53’s role is also implicated in promoting cell survival75 and reducing inflammatory responses and levels of intracellular reactive oxygen species.76–79 Thus, collectively, these studies underscore the complexities and importance of canonical cell cycle regulation in RTECs in the setting of AKI and CKD progression (Figure 2) and the need to consider the spatiotemporal inhibition of key cell cycle regulators in therapeutic approaches to AKI and CKD.
In addition to the canonical cell cycle entry following AKI, a recent study by Lazzeri et al. demonstrated that both the canonical (mitotic) cell cycle and noncanonical endoreplication (endocycling) occur in mouse RTECs following ischemia-reperfusion injury.21 Interestingly, only a small subset of cells that were of paired box (Pax)2+ progenitors proliferated via mitotic cell cycle, whereas the majority of tubular Pax8+ cells, particularly in the S1 and S2 segments of the proximal tubules, underwent endocycle-mediated hypertrophy.21 Although both Pax2 and Pax8 transcription factors are coexpressed at the onset of kidney development and together are critical regulators of nephric lineage specification,80–82 Pax2 expression becomes downregulated in most nephron segments in the adult kidneys except in the collecting duct and renal papilla, whereas Pax8 expression continues to be widespread in all epithelial cells. However, high expression of Pax2 has been noted in renal cancer83–85 and hyperproliferative cyst-lining cells in PKD.86–88 Congruent with the findings by Lazzeri et al., the study by Manolopoulou et al. also reported an increased number of polyploid proximal tubular cells following the aristolochic acid–mediated kidney injury.89 However, in another study examining the proliferating RTECs following ischemia-reperfusion injury in mice by lineage tracing of kidney injury molecule-1 (KIM1)–positive cells, the authors did not observe increased DNA content that would be consistent with endoreplication in this cell population.90 Moreover, their findings support the clonal expansion of injured KIM1–positive cells as mainly responsible for recovery after AKI, rather than proliferation of a subset of fixed progenitor cells.90 Thus, it remains to be further clarified whether the endoreplication of RTECs in addition to the canonical mitotic division and expansion occurs as a means of adaptive and reparative mechanism in response to AKI (Figure 2). If so, it also remains to be determined whether the polyploid RTECs in recovered kidneys will eventually undergo ploidy reversal to reduce the chromosome content, as observed in other organs, and what impact, if any, such reduction in ploidy would subsequently yield in RTECs and in the susceptibility of CKD progression.
Polycystic kidney disease.
PKD is a common genetic disorder characterized by progressive growth and expansion of fluid-filled cysts in the renal parenchyma and progressive development of tubulointerstitial fibrosis, leading to renal insufficiency.91 Notably, gene products whose mutations result in PKD are involved in the structure or function of the primary cilium. ADPKD, the most common form of PKD, is caused by a mutation in PKD1 and PKD2 genes that encode polycystin-1 (PC1) and polycystin-2 (PC2), respectively.92 It is believed that cyst initiation occurs when the level of functional polycystins falls below the “cystogenic threshold,” as a consequence of the germline mutation, somatic mutation (“second hit”), and/or genetic modifiers that affect PC1/PC2 expression93 (Figure 3). Nevertheless, the molecular mechanisms governing the cystogenic conversion as a consequence of lowered polycystin expression remain elusive, despite a large body of information that has been acquired over the past years on the genetics and cellular functions of cystic kidney cells. One of the hallmark features of ADPKD and a determinant of kidney failure is the hyperproliferation of RTECs with the continuous expansion of the cysts.94,95 Diverse signaling mechanisms are implicated in hyperproliferation and cell cycle dysregulation of cystic PKD cells.96 The first direct evidence linking PC1 and cell cycle regulation came from the observation that PC1 overexpression induces G1 arrest through the Janus kinase (JAK)–signal transducer and activator of transcription (STAT)–dependent expression of p21.97 Conversely, knockdown of Pkd1 lowered the fraction of cells in G1/S, suggesting that loss of PC1 may drive premature G1/S transition.98,99 PC2 overexpression similarly arrested RTECs in G1/S, which occurred through the cytosolic sequestration of inhibitor of DNA binding 2 (Id2),100 a member of the helix-loop-helix family of transcription regulators that enables cell cycle progression by repressing p21 and activation of various genes involved in G1/S transition by binding to retinoblastoma.101 Notably, the cytosolic sequestration of Id2 by PC2 was also shown to be PC1 dependent,100 suggesting that under basal conditions, both polycystins participate in keeping RTEC proliferation in check, in part through increased p21 expression. Indeed, increased nuclear Id2 expression was observed in cyst-lining epithelia in the Pkd1-null kidneys, and deletion of Id2 attenuated cyst formation in Pkd1-null mice.102 In line with cell cycle dysregulation and hyperproliferation as a major component driving PKD pathogenesis, roscovitine, a potent inhibitor of CDKs,103 was shown to effectively reduce the cystic volume and improve kidney function in jck and cpk PKD mice (characterized by the mutation of the Nek8 and Cystin genes, respectively),104,105 and more recently in collecting duct-specific cilia lacking Cep164-deficient PKD mice106 and in Pkd1-deficient mice.107
Figure 3 |. Renal tubular epithelial cell (RTEC) proliferation in autosomal dominant polycystic kidney disease.
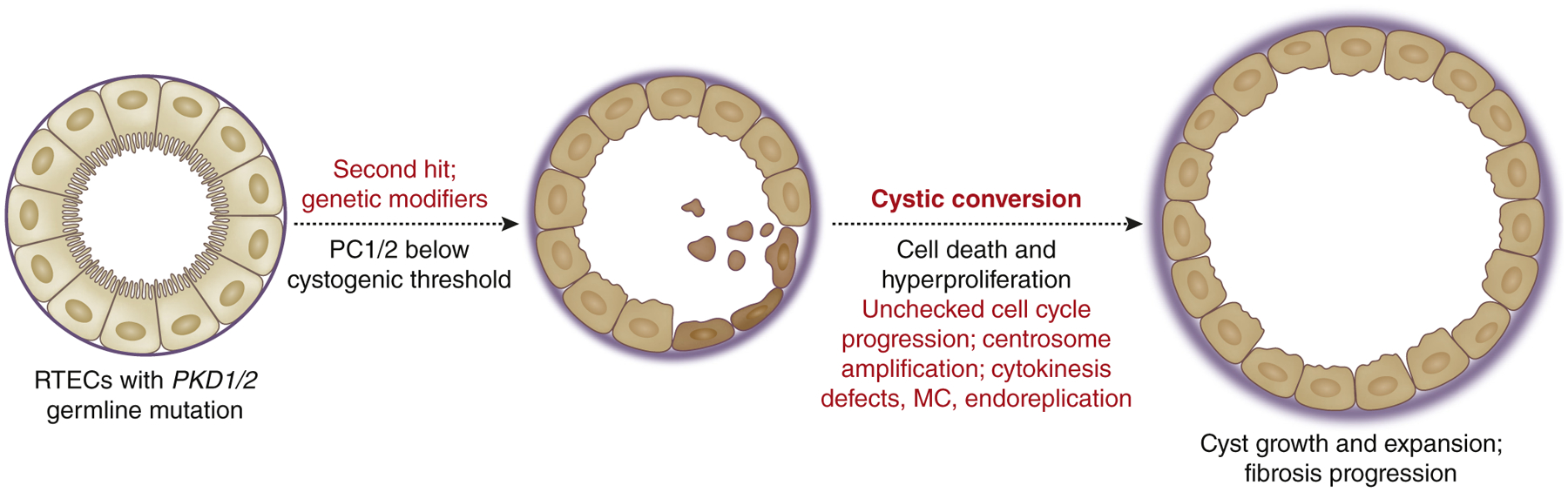
Loss of polycystin-1 (PC1) or polycystin-2 (PC2) proteins beyond the “cystogenic threshold” in RTECs with the PKD1 or PKD2 germline mutation through the genetic mutation alone, an acquired somatic mutation (“second hit”), and/or genetic modifiers results in cystogenesis and hyperproliferative RTECs marked with cell cycle anomalies. MC, mitotic catastrophe.
Associated with the hyperproliferative phenotype of PKD RTECs is the reactivation of developmentally regulated genes, such as Cux1 and Pax2.80–82,108 The homeodomain protein Cux1, which promotes cell cycle progression by suppressing the transcription of the G1-checkpoint CDK inhibitors p21 and p27,109,110 was shown to be highly expressed in PKD kidneys.111,112 The loss of Cux1 in a murine model with conditional inactivation of Pkd1 in collecting ducts increased the expression of p27, reduced the severity of the cystic disease, and prolonged survival.113 Persistent Pax2 expression is also observed in proliferating cystic RTECs of human juvenile cystic and ADPKD kidneys, and in various murine models of PKD.86,87,114 Reduction in Pax2 gene expression attenuated the cystic severity in the cpk mouse model (that resembles autosomal recessive PKD)86 and in the mouse model of ADPKD.87 The interference with growth-promoting pathways has shown to be similarly effective in reducing the cystic burden in various experimental PKD models. Although signaling by cyclic adenosine monophosphate in RTECs tends to have an inhibitory effect on proliferation, its high accumulation in cystic cells was shown to promote proliferation in a B-Raf and mitogen-activated protein kinase dependent manner.115,116 Cyclic adenosine monophosphate also contributes to cystic growth by stimulating chloride and fluid secretion.117 Pharmacologic antagonism of the vasopressin V2 receptor that attenuates cyclic adenosine monophosphate signaling was shown to be effective in various PKD animal models.118,119 Tolvaptan, a select V2 receptor antagonist, was shown to slow the kidney function decline in ADPKD patients,120 although its effect on estimated glomerular filtration rate was moderate, and a considerable proportion of patients reported polyuria and polydipsia among the adverse events. It is the first Food and Drug Administration–approved drug for slow kidney function decline in adults at risk of rapidly progressing ADPKD.121
Interestingly, many of the cellular pathways dysregulated in PKD cells bear much resemblance with those in cancer cells, including cell cycle dysregulation, even though PKD does not lead to the formation of spontaneous renal tumors.122 Similar to tumorigenesis, renal cysts are thought to arise from clonal populations that have undergone additional “hits” beyond the genetic mutation.123–130 Other intriguing parallel features noted between malignant lesions and renal cystic cells are the genomic instability and the presence of supernumerary centrosomes as a result of aberrant cell division and endomitosis. Battini et al. showed that the knockdown of Pkd1 expression in cultured collecting duct cells induced centrosome amplification, leading to multipolar spindle formation and defective cytokinesis that subsequently led to mitotic catastrophe (MC).131 MC is distinct from programmed cell death as a result of prolonged arrest at G2/M checkpoint.34 But it is functionally defined, as widely studied in the context of neoplastic transformation, as “an oncosuppressive cascade that precedes but operates through apoptosis, necrosis, or senescence for the avoidance of genomic instability in cells sensing mitotic failure.”34,132 Although the precise mechanisms that trigger MC in cells are not well defined, it is triggered during the prolonged mitotic arrest and results in gross nuclear changes, such as multinucleation, macronucleation, or micronucleation.132 Interestingly, extensive studies in cancer cells with antimitotic agents have shown that during a prolonged mitotic arrest, a subset of cells can escape the demise of MC through endomitosis and exiting the cell cycle prematurely by cyclin B degradation.133,134 Similarly, although PKD1 loss resulted in MC in cultured collecting duct cells, a subset of hyperproliferative cells with abnormal ploidy was also observed.131 Interestingly, the surviving polyploid cells over time underwent ploidy reversal, converging toward a parental ploidy and establishing a relatively stable cell population, although cytologic abnormalities, such as micronucleation and aneuploidy, remained common in the proliferating population of cells.131 Similar observations of defective cytokinesis and polyploidy were made in endothelial cells with Pkd1 or Pkd2 loss135 and in capillary endothelial cells from ADPKD patients.136 A common aberration in cells with abnormal ploidy is centrosome amplification in both cancer and in renal cysts,137–139 as duplication of centrosomes is tightly coordinated with cell cycle and division. Mahjoub et al. demonstrated that the overduplication of centrosomes leads to disrupted primary cilium signaling in epithelial cells,140 and Dionne et al. further demonstrated that centrosome amplification in vivo predisposed kidneys in adult mice to cystic development following ischemic kidney injury, without alteration in established cystic genes.141 These studies highlight the role of centrosome and ciliary signaling in cell cycle aberration in cystic cells142 and suggest that it may be a common denominator in renal cystic diseases (Figure 3). Together, these observations indicate that although proliferation is not per se sufficient to cause cystogenesis, targeting the hyperproliferation and aberrant cell cycle and division may be a promising approach to slow down the cystic growth and expansion, and thereby the disease progression.
HIV-1–associated nephropathy.
Classic HIVAN, caused by HIV-1 infection of renal epithelial cells, including the podocytes,143–145 is characterized histologically by the collapsing form of glomerulosclerosis, tubular microcystic dilatation and degeneration, interstitial inflammation, and fibrosis8,146 (for recent comprehensive reviews on HIVAN, see these studies147–149). HIVAN patients develop nephrotic-range proteinuria with renal insufficiency, which often rapidly progresses to end-stage kidney disease. Characterization of HIVAN pathology in experimental mouse models has suggested that the glomerular injury precedes and likely disposes the tubular epithelial cells to subsequent inflammation and apoptosis.150,151 Nevertheless, early histologic findings of HIVAN kidneys have noted a significant tubular injury that appeared disproportionate to the glomerular pathology,8 suggesting that some tubular injury may arise directly through HIV infection. Indeed, HIV-1 gene expression and ensuing microcyst development were observed in multiple segments of the nephron.144,152
The pathogenic mechanisms of HIVAN have been significantly aided by the use of transgenic mouse models, which have shown that the renal expression of HIV genes, particularly the genes that encode accessory proteins Negative Regulatory Factor (Nef) and Viral Protein R (Vpr), are directly responsible for HIVAN pathogenesis.150,153–155 HIV-1 encodes 4 accessory proteins, namely Nef, Vpr, Viral Infectivity Factor (Vif), and Viral Protein U (Vpu), that are dispensable for replication in some cell types in vitro, but are essential for viral pathogenesis.156 Although the full function of the accessory proteins is not entirely understood, the emerging theme from various studies is that the accessory proteins serve to counteract the host antiviral defense mechanisms through the sequestration and recruitment of multiple host proteins for the cellular degradation machinery.156,157 Interestingly, a recent study using unbiased quantitative proteomics showed that almost all proteomic changes as a result of the HIV accessory protein-mediated degradation in HIV-1–infected T cells were mediated by Vpr.158 Vpr is a multifunctional protein whose role includes the transactivation of the viral promoter, nuclear import of HIV preintegration complexes, activation of DNA damage response, cell cycle arrest, and apoptosis.159 In HIVAN, various studies have also demonstrated Vpr as a major culprit in the demise of RTECs. Vpr expression in human RTECs (HK-2 cells) induced DNA damage response160 and apoptosis that was dependent on sustained extracellular signal-regulated kinase activity and caspase-8–and caspase-9–mediated apoptosis.161 In addition to Vpr’s well-established role of inducing G2/M arrest, cell cycle anomaly by Vpr extends to aberrant mitosis due to centrosome amplification, aberrant mitotic spindles, and cytokinesis failure, which result in multinucleated cells and/or mitotic cell death in various cells in vitro.162–164 In vivo, hypertrophic multinucleated tubular cells were observed in kidneys of Tg26 HIVAN transgenic mice, and increased chromosome content in human HIVAN kidney biopsies by in situ hybridization.164 One mechanism by which Vpr induces RTEC apoptosis and polyploidy was shown to be mediated through HLA-F–adjacent transcript 10 (FAT10), a ubiquitin-like modifier that targets proteins for proteasomal degradation. FAT10 was shown to be one of the most highly upregulated genes in HIV-infected RTECs,165 and the suppression of FAT10 expression in cultured RTECs prevented Vpr-induced apoptosis and polyploidy.166 Although the exact mechanism of FAT10 in Vpr-induced RTEC cell cycle regulation requires further analysis, these observations in RTECs are consistent with the recent findings showing the involvement of FAT10 in the regulation of mitotic progression.167 Interestingly, a recent study has implicated common variants in UBD, encoding FAT10, as a genetic modifier of Apolipoprotein-L1 (APOL1)–mediated CKD risk, and showed the protective effects of FAT10 in cells overexpressing APOL1 risk variants in cultured cells.168 Given that APOL1 risk variants are strongly associated with HIVAN,169,170 whether FAT10 is also involved in Vpr-mediated cell cycle deregulation and injury in glomerular epithelial cells (i.e., podocytes) remains to be explored.
More recently, work by Payne et al. has unraveled the molecular mechanism of RTEC cell cycle deregulation by Vpr.171 They demonstrated that although the Vpr expression in RTECs initially results in G2 arrest, many cells eventually enter mitosis and the majority of the cells succumb to MC and death as a result of supernumerary centrosomes and multipolar spindle formation.171 Notably, a smaller fraction of Vpr-expressing RTECs overcame MC and death by exiting mitosis via endomitosis and polyploidization.171 The induction of Vpr expression in RTECs under the control of Pax8 promoter-driven reverse tetra-cycline transactivator transgene in mice (Pax8-Vpr mice) resulted in overt tubulointerstitial injury, cystic development, and renal failure (Chen Y, D’Agati V, Lee K, He JC, manuscript in preparation, personal communication, 2021). Consistent with in vitro observations of cell cycle arrest, MC, and polyploid RTEC fates by Vpr, multinucleated RTECs were readily visible in Pax8-Vpr kidneys as well as atrophic tubules. In addition, Pax8-Vpr mice manifested overtly cystic kidneys over time with increased proliferation in cyst-lining cells. The eventual fate of Vpr-induced polyploid RTECs that escape MC in vivo remains to be determined, and at present, it is unclear what molecular pathways dictate the fates of Vpr-expressing RTECs to cell death and tubular atrophy versus cystic conversion and hyperproliferation. Intriguingly, the observations of centrosomal amplification, MC, and endoreplication observed in the subset of Vpr-expressing RTECs are redolent of cystic cells in PKD and raise the speculation that RTECs that escape the MC and death in Pax8-Vpr kidneys may acquire changes toward cystic conversion that is associated with PKD (Figure 4). Future investigations to explore different cell fates after Vpr-induced RTEC injury may shed additional insights into the initial molecular signals initiating cystic conversion in PKD that remains yet to be defined.
Figure 4 |. Renal tubular epithelial cell (RTEC) proliferation and injury in HIV-associated nephropathy (HIVAN).
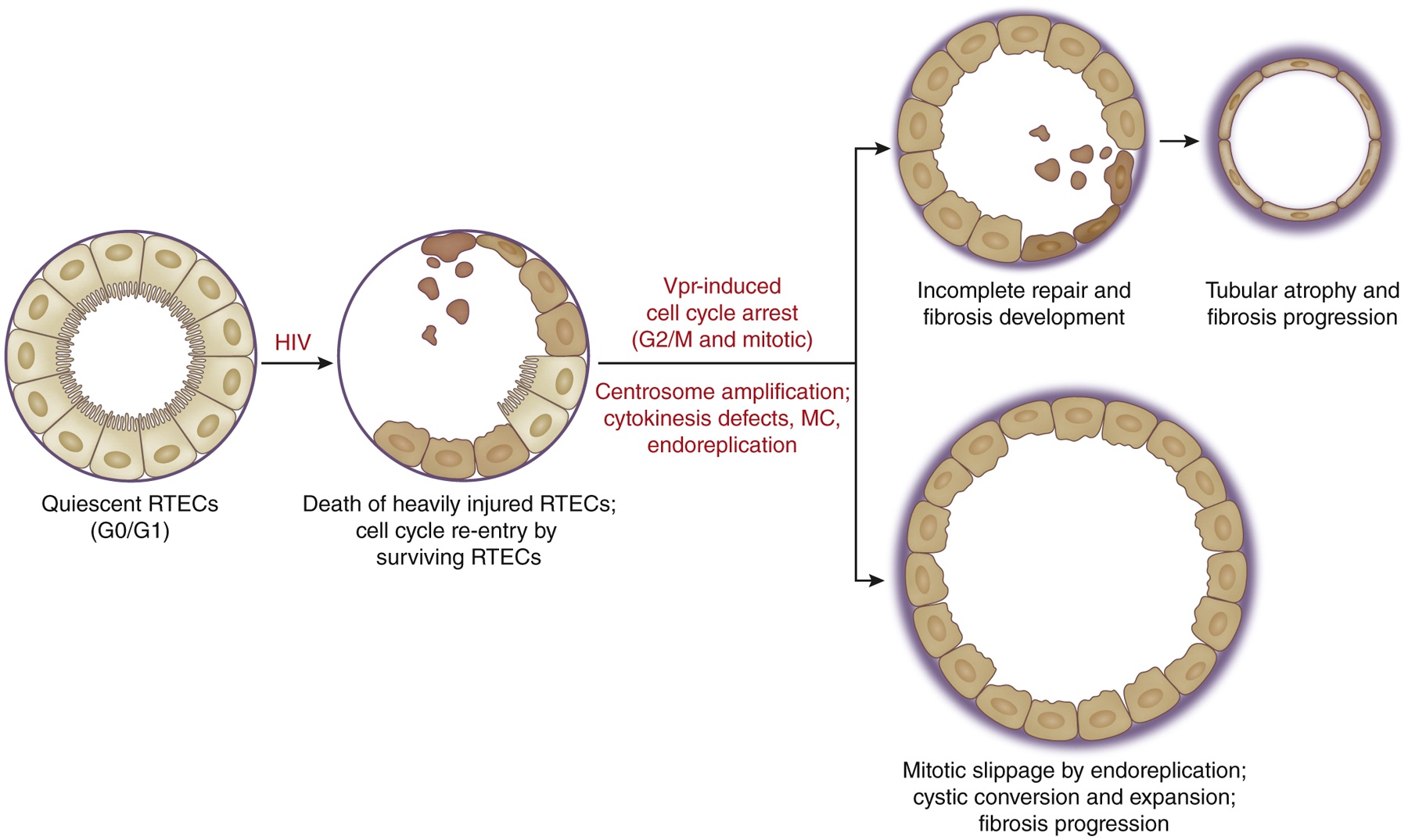
HIV infection of RTECs (and Vpr expression) results in ongoing RTEC injury, marked by cell cycle arrest, tubular atrophy, cystic development, and fibrosis. The 2 distinct fates of RTEC injury due to Vpr injury in HIVAN are hypothesized from in vitro and in vivo findings. MC, mitotic catastrophe.
Concluding remarks
Irrespective of the etiology of RTEC injury and whether the injury is acute or persistent, it is clear that cell cycle dysregulation underlies the fate of RTECs in progression to CKD (Table 16,7,46–50,60,61,86,87,89,97–100,108,111,112,114,131,141,160–166,171–173). Rapid progression through phases of the cell cycle with improper checkpoints, as well as prolonged cell cycle arrest, are both deleterious in RTECs. Although considerable insights have been gained in the cell cycle dysregulation in the respective renal epithelial cells, much remains to be explored for a broader molecular insight in the complete cell cycle–associated pathways (canonical and noncanonical) for appropriate therapeutic interventions (accounting for both duration and intensity) in the treatment of renal diseases.
Table 1 |.
Cell cycle dysregulation in RTECs and podocytes in kidney injury and disease
| Cell cycle regulation/impairment | Mediators | Results | References |
|---|---|---|---|
| RTECs in AKI-CKD | |||
| G1/S transition delay | ↑p53/p21 (and p27) and ↓CDK2/cyclin E activity | Improved RTEC recovery after AKI | 46–48 |
| Transient CDK4/6 inhibition | Improved RTEC recovery after AKI | 49, 50 | |
| G2/M checkpoint arrest | Cyclin → ↑p53/p21 | SASP, TASCC formation, and fibrosis | 6, 7 |
| TGF-β → ↑p21 | Tubular damage, EMT, fibrosis | 60, 61 | |
| Endocycling (Pax8+ RTECs) | Not identified | Polyploidy and hypertrophy | 89 |
| PKD (ADPKD) | |||
| Unchecked cell cycle progression | Loss of PC1 → ↓p53/p21 | RTEC hyperproliferation | 97, 172, 173 |
| Loss of PC1 → ↑Cux1, ↑Pax2 |
108, 111, 112 86, 87, 114 |
||
| Premature G1/S transition | Loss of PC1 → ↓STAT1 activation → ↓Yp21 | Cell cycle progression and hyperproliferation | 97 |
| Loss of PC1 → ↑cyclin A | 98, 99 | ||
| Loss of PC2 → ↑Id2 nuclear translocation and gene expression | 100 | ||
| Cytokinesis defects | Loss of PC1 | Centrosomal amplification, MC | 131, 141 |
| Mitotic slippage/endomitosis, aberrant ploidy | |||
| RTECs in HIVAN | |||
| G2/M checkpoint arrest | Vpr → DNA damage response (ATM/ATR) | Caspase-mediated cell death | 160, 161, 171 |
| Cytokinesis defects | Vpr→ FAT10 | Centrosome amplification, MC | 162–166, 171 |
| Vpr → DNA-PK | Mitotic slippage/endomitosis, aberrant ploidy |
ADPKD, autosomal dominant polycystic kidney disease; AKI, acute kidney injury; ATM, Ataxia telangiectasia mutated; ATR, ATM and RAD3-related; CDK, cyclin-dependent kinase; CKD, chronic kidney disease; Cux, Cut-like homeobox; DNA-PK, DNA-dependent protein kinase; EMT, epithelial-to-mesenchymal transition; FAT10, HLA-F–adjacent transcript 10; HIVAN, HIV-associated nephropathy; MC, mitotic catastrophe; Pax, paired box; PC1, polycystin-1; PC2, polycystin-2; PKD, polycystic kidney disease; RTEC, renal tubular epithelial cell; SASP, senescence-associated secretory phenotype; STAT, signal transducers and activators of transcription; TASCC, target of rapamycin–autophagy spatial coupling compartment; TGF-β, transforming growth factor-β; Vpr, Viral Protein R.
ACKNOWLEDGEMENTS
KL is supported by a National Institutes of Health (NIH) grant R01DK117913. GLG is supported by NIH grant R01DK106035. JCH is supported by NIH grants R01DK078897, 1R01DK088541, and P01DK56492 and Veterans Affairs Merit Award IBX000345C.
Footnotes
DISCLOSURE
All the authors declared no competing interests.
REFERENCES
- 1.Gurtner GC, Werner S, Barrandon Y, et al. Wound repair and regeneration. Nature. 2008;453:314–321. [DOI] [PubMed] [Google Scholar]
- 2.Godwin J The promise of perfect adult tissue repair and regeneration in mammals: learning from regenerative amphibians and fish. Bioessays. 2014;36:861–871. [DOI] [PubMed] [Google Scholar]
- 3.Brockes JP, Kumar A. Appendage regeneration in adult vertebrates and implications for regenerative medicine. Science. 2005;310:1919–1923. [DOI] [PubMed] [Google Scholar]
- 4.Basile DP, Bonventre JV, Mehta R, et al. Progression after AKI: understanding maladaptive repair processes to predict and identify therapeutic treatments. J Am Soc Nephrol. 2016;27:687–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Venkatachalam MA, Weinberg JM, Kriz W, et al. Failed tubule recovery, AKI-CKD transition, and kidney disease progression. J Am Soc Nephrol. 2015;26:1765–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Canaud G, Brooks CR, Kishi S, et al. Cyclin G1 and TASCC regulate kidney epithelial cell G2-M arrest and fibrotic maladaptive repair. Sci Transl Med. 2019;11:eaav4754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Yang L, Besschetnova TY, Brooks CR, et al. Epithelial cell cycle arrest in G2/M mediates kidney fibrosis after injury. Nat Med. 2010;16:535–543, 1p following 143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.D’Agati V, Suh JI, Carbone L, et al. Pathology of HIV-associated nephropathy: a detailed morphologic and comparative study. Kidney Int. 1989;35:1358–1370. [DOI] [PubMed] [Google Scholar]
- 9.Malumbres M, Barbacid M. Mammalian cyclin-dependent kinases. Trends Biochem Sci. 2005;30:630–641. [DOI] [PubMed] [Google Scholar]
- 10.Harashima H, Dissmeyer N, Schnittger A. Cell cycle control across the eukaryotic kingdom. Trends Cell Biol. 2013;23:345–356. [DOI] [PubMed] [Google Scholar]
- 11.Sherr CJ, Roberts JM. CDK inhibitors: positive and negative regulators of G1-phase progression. Genes Dev. 1999;13:1501–1512. [DOI] [PubMed] [Google Scholar]
- 12.Shankland SJ. Cell-cycle control and renal disease. Kidney Int. 1997;52: 294–308. [DOI] [PubMed] [Google Scholar]
- 13.Shankland SJ, Wolf G. Cell cycle regulatory proteins in renal disease: role in hypertrophy, proliferation, and apoptosis. Am J Physiol Renal Physiol. 2000;278:F515–F529. [DOI] [PubMed] [Google Scholar]
- 14.Thomasova D, Anders HJ. Cell cycle control in the kidney. Nephrol Dial Transplant. 2015;30:1622–1630. [DOI] [PubMed] [Google Scholar]
- 15.Coller HA, Sang L, Roberts JM. A new description of cellular quiescence. PLoS Biol. 2006;4:e83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Litovchick L, Sadasivam S, Florens L, et al. Evolutionarily conserved multisubunit RBL2/p130 and E2F4 protein complex represses human cell cycle-dependent genes in quiescence. Mol Cell. 2007;26:539–551. [DOI] [PubMed] [Google Scholar]
- 17.Sang L, Coller HA, Roberts JM. Control of the reversibility of cellular quiescence by the transcriptional repressor HES1. Science. 2008;321: 1095–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Buttitta LA, Edgar BA. Mechanisms controlling cell cycle exit upon terminal differentiation. Curr Opin Cell Biol. 2007;19:697–704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Pack LR, Daigh LH, Meyer T. Putting the brakes on the cell cycle: mechanisms of cellular growth arrest. Curr Opin Cell Biol. 2019;60:106–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Lazzeri E, Angelotti ML, Conte C, et al. Surviving acute organ failure: cell polyploidization and progenitor proliferation. Trends Mol Med. 2019;25: 366–381. [DOI] [PubMed] [Google Scholar]
- 21.Lazzeri E, Angelotti ML, Peired A, et al. Endocycle-related tubular cell hypertrophy and progenitor proliferation recover renal function after acute kidney injury. Nat Commun. 2018;9:1344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gjelsvik KJ, Besen-McNally R, Losick VP. Solving the polyploid mystery in health and disease. Trends Genet. 2019;35:6–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Ovrebo JI, Edgar BA. Polyploidy in tissue homeostasis and regeneration. Development. 2018;145:dev156034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pandit SK, Westendorp B, de Bruin A. Physiological significance of polyploidization in mammalian cells. Trends Cell Biol. 2013;23:556–566. [DOI] [PubMed] [Google Scholar]
- 25.Zanet J, Freije A, Ruiz M, et al. A mitosis block links active cell cycle with human epidermal differentiation and results in endoreplication. PLoS One. 2010;5:e15701. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Barrett TB, Sampson P, Owens GK, et al. Polyploid nuclei in human artery wall smooth muscle cells. Proc Natl Acad Sci U S A. 1983;80:882–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Dominiczak AF, Devlin AM, Lee WK, et al. Vascular smooth muscle polyploidy and cardiac hypertrophy in genetic hypertension. Hypertension. 1996;27:752–759. [DOI] [PubMed] [Google Scholar]
- 28.Hixon ML, Muro-Cacho C, Wagner MW, et al. Akt1/PKB upregulation leads to vascular smooth muscle cell hypertrophy and polyploidization. J Clin Invest. 2000;106:1011–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rios AC, Fu NY, Jamieson PR, et al. Essential role for a novel population of binucleated mammary epithelial cells in lactation. Nat Commun. 2016;7:11400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Epstein CJ. Cell size, nuclear content, and the development of polyploidy in the mammalian liver. Proc Natl Acad Sci U S A. 1967;57: 327–334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kudryavtsev BN, Kudryavtseva MV, Sakuta GA, et al. Human hepatocyte polyploidization kinetics in the course of life cycle. Virchows Arch B Cell Pathol Incl Mol Pathol. 1993;64:387–393. [DOI] [PubMed] [Google Scholar]
- 32.Margall-Ducos G, Celton-Morizur S, Couton D, et al. Liver tetraploidization is controlled by a new process of incomplete cytokinesis. J Cell Sci. 2007;120:3633–3639. [DOI] [PubMed] [Google Scholar]
- 33.Miyaoka Y, Ebato K, Kato H, et al. Hypertrophy and unconventional cell division of hepatocytes underlie liver regeneration. Curr Biol. 2012;22: 1166–1175. [DOI] [PubMed] [Google Scholar]
- 34.Vitale I, Galluzzi L, Castedo M, et al. Mitotic catastrophe: a mechanism for avoiding genomic instability. Nat Rev Mol Cell Biol. 2011;12:385–392. [DOI] [PubMed] [Google Scholar]
- 35.Schmitt R, Cantley LG. The impact of aging on kidney repair. Am J Physiol Renal Physiol. 2008;294:F1265–F1272. [DOI] [PubMed] [Google Scholar]
- 36.Nadasdy T, Laszik Z, Blick KE, et al. Proliferative activity of intrinsic cell populations in the normal human kidney. J Am Soc Nephrol. 1994;4: 2032–2039. [DOI] [PubMed] [Google Scholar]
- 37.Vogetseder A, Picard N, Gaspert A, et al. Proliferation capacity of the renal proximal tubule involves the bulk of differentiated epithelial cells. Am J Physiol Cell Physiol. 2008;294:C22–C28. [DOI] [PubMed] [Google Scholar]
- 38.Iwakura T, Fujigaki Y, Fujikura T, et al. A high ratio of G1 to G0 phase cells and an accumulation of G1 phase cells before S phase progression after injurious stimuli in the proximal tubule. Physiol Rep. 2014;2: e12173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Coller HA. What’s taking so long? S-phase entry from quiescence versus proliferation. Nat Rev Mol Cell Biol. 2007;8:667–670. [DOI] [PubMed] [Google Scholar]
- 40.Lin F, Moran A, Igarashi P. Intrarenal cells, not bone marrow-derived cells, are the major source for regeneration in postischemic kidney. J Clin Invest. 2005;115:1756–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Humphreys BD, Valerius MT, Kobayashi A, et al. Intrinsic epithelial cells repair the kidney after injury. Cell Stem Cell. 2008;2:284–291. [DOI] [PubMed] [Google Scholar]
- 42.Humphreys BD, Czerniak S, DiRocco DP, et al. Repair of injured proximal tubule does not involve specialized progenitors. Proc Natl Acad Sci U S A. 2011;108:9226–9231. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Megyesi J, Udvarhelyi N, Safirstein RL, et al. The p53-independent activation of transcription of p21 WAF1/CIP1/SDI1 after acute renal failure. Am J Physiol. 1996;271:F1211–F1216. [DOI] [PubMed] [Google Scholar]
- 44.Cheng M, Olivier P, Diehl JA, et al. The p21(Cip1) and p27(Kip1) CDK “inhibitors” are essential activators of cyclin D-dependent kinases in murine fibroblasts. EMBO J. 1999;18:1571–1583. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.LaBaer J, Garrett MD, Stevenson LF, et al. New functional activities for the p21 family of CDK inhibitors. Genes Dev. 1997;11:847–862. [DOI] [PubMed] [Google Scholar]
- 46.Megyesi J, Safirstein RL, Price PM. Induction of p21WAF1/CIP1/SDI1 in kidney tubule cells affects the course of cisplatin-induced acute renal failure. J Clin Invest. 1998;101:777–782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Megyesi J, Andrade L, Vieira JM Jr, et al. Positive effect of the induction of p21WAF1/CIP1 on the course of ischemic acute renal failure. Kidney Int. 2001;60:2164–2172. [DOI] [PubMed] [Google Scholar]
- 48.Nishioka S, Nakano D, Kitada K, et al. The cyclin-dependent kinase inhibitor p21 is essential for the beneficial effects of renal ischemic preconditioning on renal ischemia/reperfusion injury in mice. Kidney Int. 2014;85:871–879. [DOI] [PubMed] [Google Scholar]
- 49.DiRocco DP, Bisi J, Roberts P, et al. CDK4/6 inhibition induces epithelial cell cycle arrest and ameliorates acute kidney injury. Am J Physiol Renal Physiol. 2014;306:F379–F388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pabla N, Gibson AA, Buege M, et al. Mitigation of acute kidney injury by cell-cycle inhibitors that suppress both CDK4/6 and OCT2 functions. Proc Natl Acad Sci U S A. 2015;112:5231–5236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Roberts PJ, Bisi JE, Strum JC, et al. Multiple roles of cyclin-dependent kinase 4/6 inhibitors in cancer therapy. J Natl Cancer Inst. 2012;104:476–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Megyesi J, Price PM, Tamayo E, et al. The lack of a functional p21WAF1/CIP1 gene ameliorates progression to chronic renal failure. Proc Natl Acad Sci U S A. 1999;96:10830–10835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Besson A, Dowdy SF, Roberts JM. CDK inhibitors: cell cycle regulators and beyond. Dev Cell. 2008;14:159–169. [DOI] [PubMed] [Google Scholar]
- 54.Abbas T, Dutta A. p21 in cancer: intricate networks and multiple activities. Nat Rev Cancer. 2009;9:400–414. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Hydbring P, Malumbres M, Sicinski P. Non-canonical functions of cell cycle cyclins and cyclin-dependent kinases. Nat Rev Mol Cell Biol. 2016;17:280–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Aylon Y, Oren M. p53: guardian of ploidy. Mol Oncol. 2011;5:315–323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Campisi J Senescent cells, tumor suppression, and organismal aging: good citizens, bad neighbors. Cell. 2005;120:513–522. [DOI] [PubMed] [Google Scholar]
- 58.Serrano M, Lin AW, McCurrach ME, et al. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16INK4a. Cell. 1997;88:593–602. [DOI] [PubMed] [Google Scholar]
- 59.Kuilman T, Peeper DS. Senescence-messaging secretome: SMS-ing cellular stress. Nat Rev Cancer. 2009;9:81–94. [DOI] [PubMed] [Google Scholar]
- 60.Lovisa S, LeBleu VS, Tampe B, et al. Epithelial-to-mesenchymal transition induces cell cycle arrest and parenchymal damage in renal fibrosis. Nat Med. 2015;21:998–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Wu CF, Chiang WC, Lai CF, et al. Transforming growth factor beta-1 stimulates profibrotic epithelial signaling to activate pericyte-myofibroblast transition in obstructive kidney fibrosis. Am J Pathol. 2013;182:118–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chien Y, Scuoppo C, Wang X, et al. Control of the senescence-associated secretory phenotype by NF-kappaB promotes senescence and enhances chemosensitivity. Genes Dev. 2011;25:2125–2136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Beausejour CM, Krtolica A, Galimi F, et al. Reversal of human cellular senescence: roles of the p53 and p16 pathways. EMBO J. 2003;22:4212–4222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Demidenko ZN, Zubova SG, Bukreeva EI, et al. Rapamycin decelerates cellular senescence. Cell Cycle. 2009;8:1888–1895. [DOI] [PubMed] [Google Scholar]
- 65.Narita M, Young AR, Arakawa S, et al. Spatial coupling of mTOR and autophagy augments secretory phenotypes. Science. 2011;332:966–970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Kelly KJ, Plotkin Z, Vulgamott SL, et al. P53 mediates the apoptotic response to GTP depletion after renal ischemia-reperfusion: protective role of a p53 inhibitor. J Am Soc Nephrol. 2003;14:128–138. [DOI] [PubMed] [Google Scholar]
- 67.Wei Q, Dong G, Yang T, et al. Activation and involvement of p53 in cisplatin-induced nephrotoxicity. Am J Physiol Renal Physiol. 2007;293: F1282–F1291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Molitoris BA, Dagher PC, Sandoval RM, et al. siRNA targeted to p53 attenuates ischemic and cisplatin-induced acute kidney injury. J Am Soc Nephrol. 2009;20:1754–1764. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhou L, Fu P, Huang XR, et al. Activation of p53 promotes renal injury in acute aristolochic acid nephropathy. J Am Soc Nephrol. 2010;21:31–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zhang D, Liu Y, Wei Q, et al. Tubular p53 regulates multiple genes to mediate AKI. J Am Soc Nephrol. 2014;25:2278–2289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Ying Y, Kim J, Westphal SN, et al. Targeted deletion of p53 in the proximal tubule prevents ischemic renal injury. J Am Soc Nephrol. 2014;25:2707–2716. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Dagher PC, Mai EM, Hato T, et al. The p53 inhibitor pifithrin-alpha can stimulate fibrosis in a rat model of ischemic acute kidney injury. Am J Physiol Renal Physiol. 2012;302:F284–F291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Sutton TA, Hato T, Mai E, et al. p53 Is renoprotective after ischemic kidney injury by reducing inflammation. J Am Soc Nephrol. 2013;24:113–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mulay SR, Thomasova D, Ryu M, et al. MDM2 (murine double minute-2) links inflammation and tubular cell healing during acute kidney injury in mice. Kidney Int. 2012;81:1199–1211. [DOI] [PubMed] [Google Scholar]
- 75.Jänicke RU, Sohn D, Schulze-Osthoff K. The dark side of a tumor suppressor: anti-apoptotic p53. Cell Death Differ. 2008;15:959–976. [DOI] [PubMed] [Google Scholar]
- 76.Komarova EA, Krivokrysenko V, Wang K, et al. p53 Is a suppressor of inflammatory response in mice. FASEB J. 2005;19:1030–1032. [DOI] [PubMed] [Google Scholar]
- 77.Muñoz-Fontela C, Mandinova A, Aaronson SA, et al. Emerging roles of p53 and other tumour-suppressor genes in immune regulation. Nat Rev Immunol. 2016;16:741–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Sablina AA, Budanov AV, Ilyinskaya GV, et al. The antioxidant function of the p53 tumor suppressor. Nat Med. 2005;11:1306–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Liu B, Chen Y, St. Clair DK. ROS and p53: a versatile partnership. Free Radic Biol Med. 2008;44:1529–1535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Torres M, Gomez-Pardo E, Dressler GR, et al. Pax-2 controls multiple steps of urogenital development. Development. 1995;121:4057–4065. [DOI] [PubMed] [Google Scholar]
- 81.Bouchard M, Souabni A, Mandler M, et al. Nephric lineage specification by Pax2 and Pax8. Genes Dev. 2002;16:2958–2970. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Dressler GR, Douglass EC. Pax-2 is a DNA-binding protein expressed in embryonic kidney and Wilms tumor. Proc Natl Acad Sci U S A. 1992;89: 1179–1183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Tong GX, Melamed J, Mansukhani M, et al. PAX2: a reliable marker for nephrogenic adenoma. Mod Pathol. 2006;19:356–363. [DOI] [PubMed] [Google Scholar]
- 84.Daniel L, Lechevallier E, Giorgi R, et al. Pax-2 expression in adult renal tumors. Hum Pathol. 2001;32:282–287. [DOI] [PubMed] [Google Scholar]
- 85.Gnarra JR, Dressler GR. Expression of Pax-2 in human renal cell carcinoma and growth inhibition by antisense oligonucleotides. Cancer Res. 1995;55:4092–4098. [PubMed] [Google Scholar]
- 86.Ostrom L, Tang MJ, Gruss P, et al. Reduced Pax2 gene dosage increases apoptosis and slows the progression of renal cystic disease. Dev Biol. 2000;219:250–258. [DOI] [PubMed] [Google Scholar]
- 87.Stayner C, Iglesias DM, Goodyer PR, et al. Pax2 gene dosage influences cystogenesis in autosomal dominant polycystic kidney disease. Hum Mol Genet. 2006;15:3520–3528. [DOI] [PubMed] [Google Scholar]
- 88.Winyard PJ, Risdon RA, Sams VR, et al. The PAX2 transcription factor is expressed in cystic and hyperproliferative dysplastic epithelia in human kidney malformations. J Clin Invest. 1996;98:451–459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Manolopoulou M, Matlock BK, Nlandu-Khodo S, et al. Novel kidney dissociation protocol and image-based flow cytometry facilitate improved analysis of injured proximal tubules. Am J Physiol Renal Physiol. 2019;316:F847–F855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Chang-Panesso M, Kadyrov FF, Lalli M, et al. FOXM1 drives proximal tubule proliferation during repair from acute ischemic kidney injury. J Clin Invest. 2019;129:5501–5517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Bergmann C, Guay-Woodford LM, Harris PC, et al. Polycystic kidney disease. Nat Rev Dis Primers. 2018;4:50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Harris PC, Torres VE. Polycystic kidney disease. Annu Rev Med. 2009;60: 321–337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Ong AC, Harris PC. A polycystin-centric view of cyst formation and disease: the polycystins revisited. Kidney Int. 2015;88:699–710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Grantham JJ, Torres VE, Chapman AB, et al. Volume progression in polycystic kidney disease. N Engl J Med. 2006;354:2122–2130. [DOI] [PubMed] [Google Scholar]
- 95.Grantham JJ, Chapman AB, Torres VE. Volume progression in autosomal dominant polycystic kidney disease: the major factor determining clinical outcomes. Clin J Am Soc Nephrol. 2006;1:148–157. [DOI] [PubMed] [Google Scholar]
- 96.Takiar V, Caplan MJ. Polycystic kidney disease: pathogenesis and potential therapies. Biochim Biophys Acta. 2011;1812:1337–1343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Bhunia AK, Piontek K, Boletta A, et al. PKD1 induces p21(waf1) and regulation of the cell cycle via direct activation of the JAK-STAT signaling pathway in a process requiring PKD2. Cell. 2002;109:157–168. [DOI] [PubMed] [Google Scholar]
- 98.Kim H, Bae Y, Jeong W, et al. Depletion of PKD1 by an antisense oligodeoxynucleotide induces premature G1/S-phase transition. Eur J Hum Genet. 2004;12:433–440. [DOI] [PubMed] [Google Scholar]
- 99.Battini L, Fedorova E, Macip S, et al. Stable knockdown of polycystin-1 confers integrin-alpha2beta1-mediated anoikis resistance. J Am Soc Nephrol. 2006;17:3049–3058. [DOI] [PubMed] [Google Scholar]
- 100.Li X, Luo Y, Starremans PG, et al. Polycystin-1 and polycystin-2 regulate the cell cycle through the helix-loop-helix inhibitor Id2. Nat Cell Biol. 2005;7:1202–1212. [DOI] [PubMed] [Google Scholar]
- 101.Sikder HA, Devlin MK, Dunlap S, et al. Id proteins in cell growth and tumorigenesis. Cancer Cell. 2003;3:525–530. [DOI] [PubMed] [Google Scholar]
- 102.Fan LX, Li X, Magenheimer B, et al. Inhibition of histone deacetylases targets the transcription regulator Id2 to attenuate cystic epithelial cell proliferation. Kidney Int. 2012;81:76–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Meijer L, Borgne A, Mulner O, et al. Biochemical and cellular effects of roscovitine, a potent and selective inhibitor of the cyclin-dependent kinases cdc2, cdk2 and cdk5. Eur J Biochem. 1997;243:527–536. [DOI] [PubMed] [Google Scholar]
- 104.Bukanov NO, Smith LA, Klinger KW, et al. Long-lasting arrest of murine polycystic kidney disease with CDK inhibitor roscovitine. Nature. 2006;444:949–952. [DOI] [PubMed] [Google Scholar]
- 105.Husson H, Moreno S, Smith LA, et al. Reduction of ciliary length through pharmacologic or genetic inhibition of CDK5 attenuates polycystic kidney disease in a model of nephronophthisis. Hum Mol Genet. 2016;25:2245–2255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Airik R, Airik M, Schueler M, et al. Roscovitine blocks collecting duct cyst growth in Cep164-deficient kidneys. Kidney Int. 2019;96:320–326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Zhang C, Balbo B, Ma M, et al. Cyclin-dependent kinase 1 activity is a driver of cyst growth in polycystic kidney disease. J Am Soc Nephrol. 2021;32:41–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Vanden Heuvel GB, Bodmer R, McConnell KR, et al. Expression of a cut-related homeobox gene in developing and polycystic mouse kidney. Kidney Int. 1996;50:453–461. [DOI] [PubMed] [Google Scholar]
- 109.Coqueret O, Berube G, Nepveu A. The mammalian Cut homeodomain protein functions as a cell-cycle-dependent transcriptional repressor which downmodulates p21WAF1/CIP1/SDI1 in S phase. EMBO J. 1998;17:4680–4694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Livingston S, Carlton C, Sharma M, et al. Cux1 regulation of the cyclin kinase inhibitor p27(kip1) in polycystic kidney disease is attenuated by HDAC inhibitors. Gene X. 2019;2:100007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Sharma M, Brantley JG, Alcalay NI, et al. Differential expression of Cux-1 and p21 in polycystic kidneys from Pkd1 null and cpk mice. Kidney Int. 2005;67:432–442. [DOI] [PubMed] [Google Scholar]
- 112.Paul BM, Vassmer D, Taylor A, et al. Ectopic expression of Cux1 is associated with reduced p27 expression and increased apoptosis during late stage cyst progression upon inactivation of Pkd1 in collecting ducts. Dev Dyn. 2011;240:1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Porath B, Livingston S, Andres EL, et al. Cux1 promotes cell proliferation and polycystic kidney disease progression in an ADPKD mouse model. Am J Physiol Renal Physiol. 2017;313:F1050–F1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Qin S, Taglienti M, Cai L, et al. c-Met and NF-kappaB-dependent overexpression of Wnt7a and −7b and Pax2 promotes cystogenesis in polycystic kidney disease. J Am Soc Nephrol. 2012;23:1309–1318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Yamaguchi T, Nagao S, Wallace DP, et al. Cyclic AMP activates B-Raf and ERK in cyst epithelial cells from autosomal-dominant polycystic kidneys. Kidney Int. 2003;63:1983–1994. [DOI] [PubMed] [Google Scholar]
- 116.Hanaoka K, Guggino WB. cAMP regulates cell proliferation and cyst formation in autosomal polycystic kidney disease cells. J Am Soc Nephrol. 2000;11:1179–1187. [DOI] [PubMed] [Google Scholar]
- 117.Terryn S, Ho A, Beauwens R, et al. Fluid transport and cystogenesis in autosomal dominant polycystic kidney disease. Biochim Biophys Acta. 2011;1812:1314–1321. [DOI] [PubMed] [Google Scholar]
- 118.Gattone VH 2nd, Wang X, Harris PC, et al. Inhibition of renal cystic disease development and progression by a vasopressin V2 receptor antagonist. Nat Med. 2003;9:1323–1326. [DOI] [PubMed] [Google Scholar]
- 119.Wang X, Gattone V 2nd, Harris PC, et al. Effectiveness of vasopressin V2 receptor antagonists OPC-31260 and OPC-41061 on polycystic kidney disease development in the PCK rat. J Am Soc Nephrol. 2005;16:846–851. [DOI] [PubMed] [Google Scholar]
- 120.Torres VE, Chapman AB, Devuyst O, et al. Tolvaptan in patients with autosomal dominant polycystic kidney disease. N Engl J Med. 2012;367: 2407–2418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Chebib FT, Perrone RD, Chapman AB, et al. A practical guide for treatment of rapidly progressive ADPKD with tolvaptan. J Am Soc Nephrol. 2018;29:2458–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Seeger-Nukpezah T, Geynisman DM, Nikonova AS, et al. The hallmarks of cancer: relevance to the pathogenesis of polycystic kidney disease. Nat Rev Nephrol. 2015;11:515–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Qian F, Watnick TJ, Onuchic LF, et al. The molecular basis of focal cyst formation in human autosomal dominant polycystic kidney disease type I. Cell. 1996;87:979–987. [DOI] [PubMed] [Google Scholar]
- 124.Brasier JL, Henske EP. Loss of the polycystic kidney disease (PKD1) region of chromosome 16p13 in renal cyst cells supports a loss-of-function model for cyst pathogenesis. J Clin Invest. 1997;99:194–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Torra R, Badenas C, San Millan JL, et al. A loss-of-function model for cystogenesis in human autosomal dominant polycystic kidney disease type 2. Am J Hum Genet. 1999;65:345–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Pei Y, Watnick T, He N, et al. Somatic PKD2 mutations in individual kidney and liver cysts support a “two-hit” model of cystogenesis in type 2 autosomal dominant polycystic kidney disease. J Am Soc Nephrol. 1999;10:1524–1529. [DOI] [PubMed] [Google Scholar]
- 127.Watnick TJ, Torres VE, Gandolph MA, et al. Somatic mutation in individual liver cysts supports a two-hit model of cystogenesis in autosomal dominant polycystic kidney disease. Mol Cell. 1998;2:247–251. [DOI] [PubMed] [Google Scholar]
- 128.Wu G, D’Agati V, Cai Y, et al. Somatic inactivation of Pkd2 results in polycystic kidney disease. Cell. 1998;93:177–188. [DOI] [PubMed] [Google Scholar]
- 129.Takakura A, Contrino L, Zhou X, et al. Renal injury is a third hit promoting rapid development of adult polycystic kidney disease. Hum Mol Genet. 2009;18:2523–2531. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Weimbs T Third-hit signaling in renal cyst formation. J Am Soc Nephrol. 2011;22:793–795. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Battini L, Macip S, Fedorova E, et al. Loss of polycystin-1 causes centrosome amplification and genomic instability. Hum Mol Genet. 2008;17:2819–2833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Galluzzi L, Vitale I, Aaronson SA, et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death Differ. 2018;25:486–541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Brito DA, Rieder CL. Mitotic checkpoint slippage in humans occurs via cyclin B destruction in the presence of an active checkpoint. Curr Biol. 2006;16:1194–1200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Gascoigne KE, Taylor SS. Cancer cells display profound intra- and interline variation following prolonged exposure to antimitotic drugs. Cancer Cell. 2008;14:111–122. [DOI] [PubMed] [Google Scholar]
- 135.AbouAlaiwi WA, Ratnam S, Booth RL, et al. Endothelial cells from humans and mice with polycystic kidney disease are characterized by polyploidy and chromosome segregation defects through survivin down-regulation. Hum Mol Genet. 2011;20:354–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kathem SH, AbouAlaiwi WA, Zi X, et al. Capillary endothelia from two ADPKD patients are polyploidy. Ann Clin Cytol Pathol. 2016;2:1022. [PMC free article] [PubMed] [Google Scholar]
- 137.Grantham JJ, Mulamalla S, Grantham CJ, et al. Detected renal cysts are tips of the iceberg in adults with ADPKD. Clin J Am Soc Nephrol. 2012;7: 1087–1093. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Bae KT, Zhou W, Shen C, et al. Growth pattern of kidney cyst number and volume in autosomal dominant polycystic kidney disease. Clin J Am Soc Nephrol. 2019;14:823–833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Burtey S, Riera M, Ribe E, et al. Centrosome overduplication and mitotic instability in PKD2 transgenic lines. Cell Biol Int. 2008;32:1193–1198. [DOI] [PubMed] [Google Scholar]
- 140.Mahjoub MR, Stearns T. Supernumerary centrosomes nucleate extra cilia and compromise primary cilium signaling. Curr Biol. 2012;22:1628–1634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Dionne LK, Shim K, Hoshi M, et al. Centrosome amplification disrupts renal development and causes cystogenesis. J Cell Biol. 2018;217:2485–2501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Pan J, Seeger-Nukpezah T, Golemis EA. The role of the cilium in normal and abnormal cell cycles: emphasis on renal cystic pathologies. Cell Mol Life Sci. 2013;70:1849–1874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Bruggeman LA, Dikman S, Meng C, et al. Nephropathy in human immunodeficiency virus-1 transgenic mice is due to renal transgene expression. J Clin Invest. 1997;100:84–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Ross MJ, Bruggeman LA, Wilson PD, et al. Microcyst formation and HIV-1 gene expression occur in multiple nephron segments in HIV-associated nephropathy. J Am Soc Nephrol. 2001;12:2645–2651. [DOI] [PubMed] [Google Scholar]
- 145.Barisoni L, Kriz W, Mundel P, et al. The dysregulated podocyte phenotype: a novel concept in the pathogenesis of collapsing idiopathic focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 1999;10:51–61. [DOI] [PubMed] [Google Scholar]
- 146.Cohen AH, Nast CC. HIV-associated nephropathy: a unique combined glomerular, tubular, and interstitial lesion. Mod Pathol. 1988;1:87–97. [PubMed] [Google Scholar]
- 147.Rednor SJ, Ross MJ. Molecular mechanisms of injury in HIV-associated nephropathy. Front Med (Lausanne). 2018;5:177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Rosenberg AZ, Naicker S, Winkler CA, et al. HIV-associated nephropathies: epidemiology, pathology, mechanisms and treatment. Nat Rev Nephrol. 2015;11:150–160. [DOI] [PubMed] [Google Scholar]
- 149.Mallipattu SK, Salem F, Wyatt CM. The changing epidemiology of HIV-related chronic kidney disease in the era of antiretroviral therapy. Kidney Int. 2014;86:259–265. [DOI] [PubMed] [Google Scholar]
- 150.Zhong J, Zuo Y, Ma J, et al. Expression of HIV-1 genes in podocytes alone can lead to the full spectrum of HIV-1-associated nephropathy. Kidney Int. 2005;68:1048–1060. [DOI] [PubMed] [Google Scholar]
- 151.Gu L, Dai Y, Xu J, et al. Deletion of podocyte STAT3 mitigates the entire spectrum of HIV-1-associated nephropathy. AIDS. 2013;27:1091–1098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Bruggeman LA, Ross MD, Tanji N, et al. Renal epithelium is a previously unrecognized site of HIV-1 infection. J Am Soc Nephrol. 2000;11:2079–2087. [DOI] [PubMed] [Google Scholar]
- 153.Lu TC, He JC, Klotman P. Animal models of HIV-associated nephropathy. Curr Opin Nephrol Hypertens. 2006;15:233–237. [DOI] [PubMed] [Google Scholar]
- 154.Dickie P, Roberts A, Uwiera R, et al. Focal glomerulosclerosis in proviral and c-fms transgenic mice links Vpr expression to HIV-associated nephropathy. Virology. 2004;322:69–81. [DOI] [PubMed] [Google Scholar]
- 155.Zuo Y, Matsusaka T, Zhong J, et al. HIV-1 genes vpr and nef synergistically damage podocytes, leading to glomerulosclerosis. J Am Soc Nephrol. 2006;17:2832–2843. [DOI] [PubMed] [Google Scholar]
- 156.Strebel K HIV accessory proteins versus host restriction factors. Curr Opin Virol. 2013;3:692–699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Malim MH, Emerman M. HIV-1 accessory proteins–ensuring viral survival in a hostile environment. Cell Host Microbe. 2008;3:388–398. [DOI] [PubMed] [Google Scholar]
- 158.Greenwood EJD, Williamson JC, Sienkiewicz A, et al. Promiscuous targeting of cellular proteins by Vpr drives systems-level proteomic remodeling in HIV-1 infection. Cell Rep. 2019;27:1579–1596.e1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Fabryova H, Strebel K. Vpr and its cellular interaction partners: R we there yet? Cells. 2019;8:1310. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Rosenstiel PE, Chan J, Snyder A, et al. HIV-1 Vpr activates the DNA damage response in renal tubule epithelial cells. AIDS. 2009;23:2054–2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Snyder A, Alsauskas ZC, Leventhal JS, et al. HIV-1 viral protein r induces ERK and caspase-8-dependent apoptosis in renal tubular epithelial cells. AIDS. 2010;24:1107–1119. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Chang F, Re F, Sebastian S, et al. HIV-1 Vpr induces defects in mitosis, cytokinesis, nuclear structure, and centrosomes. Mol Biol Cell. 2004;15: 1793–1801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Watanabe N, Yamaguchi T, Akimoto Y, et al. Induction of M-phase arrest and apoptosis after HIV-1 Vpr expression through uncoupling of nuclear and centrosomal cycle in HeLa cells. Exp Cell Res. 2000;258:261–269. [DOI] [PubMed] [Google Scholar]
- 164.Rosenstiel PE, Gruosso T, Letourneau AM, et al. HIV-1 Vpr inhibits cytokinesis in human proximal tubule cells. Kidney Int. 2008;74:1049–1058. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Ross MJ, Wosnitzer MS, Ross MD, et al. Role of ubiquitin-like protein FAT10 in epithelial apoptosis in renal disease. J Am Soc Nephrol. 2006;17:996–1004. [DOI] [PubMed] [Google Scholar]
- 166.Snyder A, Alsauskas Z, Gong P, et al. FAT10: a novel mediator of Vpr-induced apoptosis in human immunodeficiency virus-associated nephropathy. J Virol. 2009;83:11983–11988. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Merbl Y, Refour P, Patel H, et al. Profiling of ubiquitin-like modifications reveals features of mitotic control. Cell. 2013;152:1160–1172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhang JY, Wang M, Tian L, et al. UBD modifies APOL1-induced kidney disease risk. Proc Natl Acad Sci U S A. 2018;115:3446–3451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Kopp JB, Nelson GW, Sampath K, et al. APOL1 genetic variants in focal segmental glomerulosclerosis and HIV-associated nephropathy. J Am Soc Nephrol. 2011;22:2129–2137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Papeta N, Kiryluk K, Patel A, et al. APOL1 variants increase risk for FSGS and HIVAN but not IgA nephropathy. J Am Soc Nephrol. 2011;22:1991–1996. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Payne EH, Ramalingam D, Fox DT, et al. Polyploidy and mitotic cell death are two distinct HIV-1 Vpr-driven outcomes in renal tubule epithelial cells. J Virol. 2018;92. e01718–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Park JY, Schutzer WE, Lindsley JN, et al. p21 Is decreased in polycystic kidney disease and leads to increased epithelial cell cycle progression: roscovitine augments p21 levels. BMC Nephrol. 2007;8:12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Nishio S, Hatano M, Nagata M, et al. Pkd1 regulates immortalized proliferation of renal tubular epithelial cells through p53 induction and JNK activation. J Clin Invest. 2005;115:910–918. [DOI] [PMC free article] [PubMed] [Google Scholar]