

Abstract
Background:
Group 2 innate lymphoid cells (ILC2s) are involved in type 2 immune responses in mucosal organs and are associated with various allergic diseases in humans. Studies are needed to understand the molecules and pathways that control ILC2s.
Objective:
The aim of this study was to develop a mouse model that limits the innate type 2 immune response in the lung and to investigate the immunologic mechanisms involved in regulation of lung ILC2s.
Methods:
Naïve BALB/c mice were administered various toll-like receptor (TLR) agonists and exposed intranasally (i.n.) to the fungal allergen Alternaria alternata. The mechanisms were investigated using gene knockout mice and cultures of lung cells and isolated lung ILC2s.
Results:
Polyinosinic-polycytidylic acid [poly (I:C)] effectively inhibited innate type 2 response to A. alternata. Poly (I:C) promoted production of interferon (IFN)-α, -β, and -γ, and its inhibitory effects were dependent on the IFN-α/β receptor pathway. IFN-β was 100-times more potent than IFN-α at inhibiting type 2 cytokine production by lung ILC2s. Signal transducer and activator of transcription 5 (STAT5)-activating cytokines, including interleukin-2 (IL-2), IL-7, and thymic stromal lymphopoietin (TSLP), but not IL-33, promoted survival and proliferation of lung ILC2s in vitro, while IFN-β blocked these effects. Expression of the transcription factor GATA3, which is critical for differentiation and maintenance of ILC2s, was inhibited by IFN-β.
Conclusion:
IFN-β blocks the effects of STAT5-activating cytokines on lung ILC2s and inhibits their survival and effector functions. Administration of IFN-β may provide a new strategy to treat diseases involving ILC2s.
Keywords: Group 2 innate lymphoid cell, lung, poly (I:C), IL-5, IL-7, IL-13, IL-33, TSLP, IFN-β
Graphical Abstract
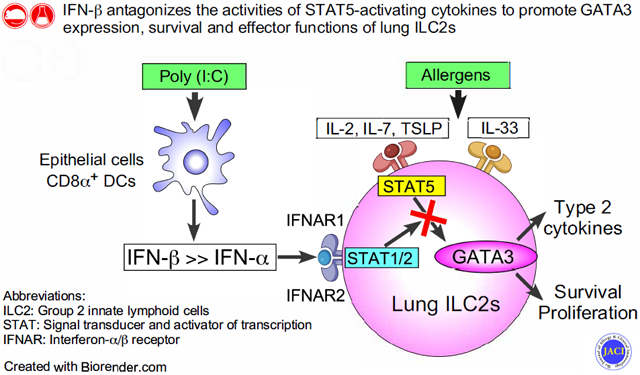
Capsule Summary
Using mouse models, Tei et al. demonstrate that IFN-β effectively inhibits proliferation and survival of lung ILC2s that are promoted by STAT5-activating cytokines and suppresses allergen-induced innate type 2 responses in the lung.
INTRODUCTION
The chronic inflammation of airways observed in allergic asthma is characterized by increased expression of type-2 cytokines, such as interleukin (IL)-5 and IL-13, and infiltration of eosinophils.1 Multiple cell types coordinate the inflammatory response, including CD4+ T cells, mast cells, eosinophils, and neutrophils.2 Group 2 innate lymphoid cells (ILC2s) recently were found to reside in mucosal organs, such as lungs, and contribute to type 2 immune responses and tissue inflammation.3, 4, 5 ILC2s do not express antigen-specific receptors; instead, they are directly activated by cytokines, such as IL-33, IL-25, and thymic stromal lymphopoietin (TSLP), derived from epithelial cells and other cell types, and rapidly produce large quantities of IL-5 and IL-13.6, 7 ILC2s also promote the development of antigen-specific type 2 helper (Th2) CD4+ T cells.8, 9 Increased numbers and activation of ILC2s are associated with asthma, allergic rhinitis, and chronic rhinosinusitis (CRS) in humans.7 More recently, glucocorticoid-resistant ILC2s have been found to be increased in airway tissues and peripheral blood in patients with asthma and correlated with the severity of disease.10 Furthermore, human asthma is associated with polymorphisms of genes related to ILC2s, including IL33, IL1RL1, IL7R, RORA, and IL2RB.11, 12, 13 These previous studies provide abundant and important information to explain how ILC2s are activated and how they might be involved in the pathophysiology of airway diseases associated with type 2 immunity. However, less is known about the molecular and immunologic mechanisms that regulate or suppress ILC2s.
Several molecules can suppress ILC2s, including cytokines, lipid mediators, and steroids.14 Interferons are the prototypic cytokines that inhibit ILC2s. Indeed, interferon (IFN)-β or IFN-γ suppressed type 2 cytokine production by ILC2s derived from bone marrow or fat-associated lymphoid clusters.15, 16 These cytokines also inhibited IL-33-mediated lung pathology when injected into the airways. However, our knowledge is limited regarding the relative contribution of these cytokines in regulating allergic airway inflammation induced by airborne allergens. Information is also scarce regarding the mechanisms to explain how these cytokines regulate lung ILC2s. In this regard, transcription factors have been known to play pivotal roles in promoting differentiation and functions of ILC2s.14 The RAR-related orphan receptor alpha (RORα) is necessary for development of ILC2s.17 Another transcription factor GATA-binding protein 3 (GATA3) is highly expressed by ILC2 precursors and mature ILC2s, and it is required for differentiation, maintenance and function of mouse and human ILC2s.18, 19 On other hand, little is known regarding how the expression of GATA3 is regulated in lung ILC2s.
Pattern recognition receptors (PRRs) recognize microorganisms and their components and promote anti-microbial immunity and Th1- or Th17-type adaptive immune responses, while potentially antagonizing the Th2-type immune responses associated with allergic asthma.20 An environment rich in microbes was associated with lower rates of asthma in humans21, and environmental endotoxin has been shown to protect against allergic immune responses.22 Toll-like receptors (TLRs) are PRRs and recognize a variety of microbial components, such as lipopolysaccharide (recognized by TLR4), lipopeptides (recognized by TLR2/1 and TLR2/6), flagellin (recognized by TLR5), unmethylated CpG motifs in DNA (recognized by TLR9), and RNA (recognized by TLRs 3, 7, and 8).23 More recently, TLR activation was found to suppress ILC2-mediated innate type 2 immune responses. For example, administration of TLR agonists, such as TLR7/8 agonist R848 and TLR9 agonist CpG, inhibited IL-33- or allergen-induced innate type 2 responses in mouse lungs.24, 25 Interferon (IFN)-α produced by plasmacytoid dendritic cells (pDCs) likely contributes to the inhibitory effects of TLR agonists by suppressing ILC2s directly24 or by promoting IFN-γ production by natural killer (NK) cells.25 These observations suggest that TLR agonists could provide an useful model to examine the immunological mechanisms controlling ILC2s.
The objective of this project was to identify the molecules and pathways that suppress lung ILC2s. Following in the footsteps of previous studies, we took a straightforward approach by administrating various TLR agonists into the airway of naive mice and examining the responses to the airborne allergen fungus Alternaria alternata. We found that IFN-β, which is induced by poly (I:C), inhibits the action of signal transducer and activator of transcription 5 (STAT5)-activating cytokines, such as IL-7 and TSLP, which promote survival and proliferation of lung ILC2s. The results provide new insight into how homeostasis and activation of ILC2s are controlled in the lung tissues and suggest potential strategies that could be used to regulate ILC2s in asthma and other allergic airway diseases.
METHODS
Mice
Wild-type (WT) BALB/c, C57BL/6, Ifngr1−/−, and Ifnar1−/− mice (C57BL/6 background) were purchased from the Jackson Laboratory (Bar Harbor, ME). The WT C57BL/6 mice and Ifngr1−/− or Ifnar1−/− mice were housed in the same room at least for 1 week before the start of experiments. The IL-5-reporter C. 129S4(B6)-Il5tm1Ktk (Il5Venus) mice26 were a gift from Dr. Kiyoshi Takatsu, Toyama University, Japan, and were maintained in the Mayo Clinic animal facility. All mice used in the experiments were female and in the age range of 6–12 weeks. All animal experiments and handling procedures were approved by the Mayo Clinic Institutional Animal Care and Use Committee and performed according to the Committee guidelines.
Reagents
Fluorescently labeled antibodies (Abs) to CD3 (145-2C11), CD25 (PC51), CD44 (IM7), CD16/CD32 (2.4G2), CD14 (rmC5-3), CD45R/B220 (RA3-6B2), and IgG2a isotype control were purchased from BD Biosciences (San Jose, CA). Fluorescently labeled Abs to GATA3 (TWAJ) and IgG1 isotype control were purchased from eBioscience (San Diego, CA). Fluorescently labeled Annexin-V, IgG2b isotype control, and 7-AAD viability staining solution were purchased from BioLegend (San Diego, CA). Ghost Dye™ Red 780 was purchased from TONBO Biosciences (San Diego, CA). Recombinant mouse proteins, including IL-2, IL-7, IL-25, TSLP, and IFN-β were from R&D Systems (Minneapolis, MN). Mouse IL-33 and IFN-α2 were from eBioscience, and mouse IFN-γ was from PeproTech (Rocky Hill, NJ). High molecular weight poly (I:C), CpG A (ODN1585), and R848 were from InvivoGen (San Diego, CA). The culture filtrate extract of A. alternata was from Greer Laboratories (Lenoir, NC); the extract contained detectable, but minimal, amounts of endotoxin (i.e., 3 ng endotoxin/mg extract). Anti-IFNAR1 (MAR1-5A3) and isotype-matched control IgG for blocking experiments were purchased from BioXcell (West Lebanon, NH).
Mouse models of innate type 2 immune responses
Generally, naïve BALB/c or C57BL/6 mice were administered TLR agonists intranasally (i.n.) and then exposed i.n. to A. alternata extract. The timing and frequency for administration of TLR agonists and A. alternata were optimized for the purpose of each experiment. We collected bronchoalveolar lavage (BAL) fluids and lung tissues for immunologic analyses. The trachea was cannulated to collect BAL fluids, and lavage was performed in triplicate using Hank’s Balanced Salt Solution (HBSS; 0.5, 0.25, and 0.25 ml, respectively). Cell numbers were counted, and differentials were determined in cytospin preparations stained with Wright-Giemsa stain. More than 200 cells were counted using conventional morphologic criteria. The BAL fluid supernatants were stored at −20 °C for cytokine assays. The lungs were homogenized in 0.5 ml of PBS, and centrifuged at 10,000 × g at 4 °C for 15 min. The supernatants were analyzed for total protein concentration with the Pierce™ BCA Protein Assay kit (Thermo Fisher, Rockford, IL) and for cytokine levels (see below).
To examine the effects of TLR agonists on production of type 1 and type 2 IFNs in the lung, naïve BALB/c mice were administrated 25 μg of poly (I:C), R848, or CpG A intranasally (i.n.), and lungs were collected after 6 h or at times indicated. IFN-α, IFN-β, and IFN-γ levels in lung tissues were analyzed by ELISA. To examine the effects of TLR agonists on innate type 2 responses, naïve BALB/c mice or Il5Venus reporter mice were pretreated i.n. once with poly (I:C) or PBS (as a control) at 24 h prior to the administration of A. alternata. Mice were then administered 50 μg of A. alternata extract i.n., and BAL fluids and lungs were collected 4.5 h later. To examine the roles of IFN receptors, naïve BALB/c mice were administered anti-IFNAR1 or control IgG i.n. (50 μg) and intraperitoneally (i.p.) (250 μg) together with i.n. poly (I:C) (25 μg). After 24 h, mice were administered A. alternata extract i.n. and euthanized 4.5 h later. Alternatively, naïve WT C57BL/6 mice or Ifngr1−/− and Ifnar1−/− mice were administered poly (I:C) i.n. 24 h prior to administration of A. alternata extract. To examine the direct effects of IFN-β or IFN-γ, naïve BALB/c mice were also administered IFN-β or IFN-γ (1 μg/dose) or PBS i.n. for 3 consecutive days prior to administration of A. alternata extract. Finally, in some experiments, naïve BALB/c mice were administered poly (I:C) and A. alternata extract i.n. every 3 days for 6 days. Mice were euthanized 24 h after the last administration of A. alternata extract, and BAL fluids and lungs were collected for analyses.
Lung single cell culture
Lung single cell culture was used to examine ILC2 cytokine production in the presence of other immune cells in the lung. Lungs were collected from naïve BALB/c mice, and lung single-cell suspensions were obtained in buffer containing a cocktail of collagenases (Liberase TM, Roche Diagnostics, Indianapolis, IN) and Gentlemax Dissociator (Milteny Biotec) as described previously.27 Red blood cells were lysed with ammonium chloride/potassium lysing buffer (0.15 M NH4Cl, 10 mM KHCO3, and 0.1 mM Na2EDTA), and cells were resuspended in RPMI 1640 medium supplemented with 100 U/ml penicillin, 100 mg/ml streptomycin, and 10% fetal bovine serum (FBS) (RPMI 1640 medium). To examine the cytokine production, lung single cell suspensions were cultured at 1.0 × 106 cells/ml with 10 ng/ml IL-33 with or without serial dilutions or 100 ng/ml for each of IFN-α, IFN-β, IFN-γ, or IFN-λ in 48-well tissue culture plates for 4 days at 37 °C and 5% CO2. Supernatants were collected and analyzed for IL-5 and IL-13 by ELISA.
Lung ILC2 sorting and culture
To isolate lung ILC2s, naive BALB/c mice were injected i.p. with a cocktail of IL-25 and IL-33 (400 ng/dose each), once daily for 3 days. Twenty-four hours after the last injection, lungs were collected, and a lung single-cell suspension was generated as described above. In some experiments, lungs were collected from naïve BALB/c mice without prior treatment with cytokines. Cells was enriched for ILC2s using EasySep Mouse ILC2 Enrichment Kit (STEMCELL Technologies, Vancouver BC, Canada) according to manufacturer’s protocol. After staining with FITC-conjugated Abs to CD3, CD14, CD16/CD32, B220, PerCP Cy5.5-conjugated anti-CD44, and APC-conjugated anti-CD25, lung ILC2s were sorted by fluorescence-activated cell sorting (FACS) using BD FACSAria® as lineage-negative (Lin−)CD25+CD44high cells as described previously.27 Sorted lung ILC2s were cultured with 10 ng/ml or indicated concentration of cytokines, including IL-33, IL-7, IL-2, TSLP, IFN-α, IFN-β, or IFN-γ, at 5.0 × 104 to 1.0 × 105 cells/ml in RPMI 1640 medium in round-bottomed 96-well tissue culture plates for up to 4 days. For the cell proliferation assay, sorted lung ILC2s were labeled with 5 μM carboxyfluorescein succinimidyl ester (CFSE) before culture. After 4 days, CFSE dilution was analyzed using a BD FACSCanto® flow cytometer.
FACS analyses
Flow cytometry was used to analyze apoptosis and death of ILC2s as well as expression of GATA3 protein. After a 3-day culture or for another indicated period, lung ILC2s were stained with fluorescently labeled Annexin-V and 7-AAD viability-staining solution following the protocol recommended by the manufacture. For analysis of GATA3 protein expression, cells were stained with Ghost Dye™ Red 780 fixable cell viability dye and permeabilized with a Foxp3/transcription factor staining kit (eBioscience). Cells were then staining with PE-conjugated anti-GATA3 Ab or control IgG and analyzed with a BD FACSCanto® flow cytometer by gating separately on Ghost Dye™ Red-positive or -negative cells.
NanoString and quantitative polymerase chain reaction (PCR) gene expression assays
For Nano String™ gene expression analysis, isolated lung ILC2s were cultured with medium alone or with 10 ng/ml IL-7 for 16 h. Total RNA was purified from ILC2s with TRIzol and PureLink RNA Mini Kit columns (Thermo Fisher Scientific, Waltham, MA). mRNA was probed with the nCounter® analysis platform (NanoString Technologies, Seattle, WA) by using a Mouse Immunology Profiling Panel and following the protocol recommended by the manufacture. Data were analyzed with the nSolver™ Analysis Software package.
For quantitative PCR, total RNA was purified from lung ILC2s with TRIzol and PureLink RNA Mini Kit columns as described above, and cDNA was reverse transcribed with iScript (Bio-Rad Laboratories, Hercules, Calif). mRNA transcripts for Gata3 were quantified by real-time PCR using TaqMan Gene Expression Arrays and TaqMan Universal PCR Master Mix (Thermo Fisher Scientific) as per the manufacturer’s instructions. Data were normalized to the levels of gene expression in ILC2s cultured with medium alone.
ELISAs
The levels of IL-5, IL-13, IFN-α, IFN-β, IFN-γ, and IFN-λ were measured by Quantikine ELISA kits (R&D Systems). Cytokine concentrations in the cell supernatants were measured by DuoSet ELISA kits (R&D Systems) for IL-5 and IL-13. All ELISAs were performed as per manufacturer’s instructions.
Statistics
Data are presented as the mean ± standard error of the mean (SEM) for the numbers of mice or experiments as indicated. Statistics were performed using paired and unpaired Student t test, ANOVA, or repeated measures ANOVA as appropriate for each set of experimental conditions; p<0.05 was considered significant.
RESULTS
TLR3 agonist poly (I:C) effectively inhibits innate type 2 immune response
Previous studies showed that the fungus A. alternata induces innate type 2 immune responses in the lungs of mice.27 Naïve BALB/c and C57BL/6 mice exposed to A. alternata extract quickly produced large quantities of IL-5 and IL-13 dependent on the IL-33/ST2 pathway and lung ILC2s.27 TLR agonists, such as R848 (TLR7/8 agonist) and CpG (TLR9 agonist), were shown recently to inhibit IL-33- or allergen-induced innate type 2 responses in mice.24, 25 To establish a mouse model to investigate the molecules and pathways that suppress ILC2s in the lung in vivo, we pretreated naïve BALB/c mice i.n. with two different doses (5 μg and 25 μg) of poly (I:C) (TLR3 agonist), R848, and CpG A. Twenty-four hours later, mice were exposed to A. alternata extract, and the lungs were analyzed for type 2 cytokines 4.5 h later (Fig. 1A). Administration of A. alternata increased lung levels of IL-5 and IL-13 (Fig. 1B), which is consistent with previous observations.27, 28 These cytokine responses were significantly inhibited in mice pretreated with 5 μg of poly (I:C) (p<0.05) and further reduced in those treated with 25 μg of poly (I:C) (p<0.05). In this model, R848 showed modest effects, and CpG A inhibited the response only at the 25-μg dose (p<0.05).
Figure 1.

TLR3 agonist poly (I:C) inhibits Alternaria alternata-induced innate type 2 response. (A) Experimental model for Panel B. (B) Lung levels of type 2 cytokines were analyzed by ELISA. *p<0.05 compared with mice pretreated with PBS and exposed i.n. to A. alternata. Data are presented as the mean ± SEM (n = 2 in each group). (C) IL-5venus mice were pretreated with poly (I:C) or PBS i.n., and exposed 24 h later to A. alternata or PBS. At 6 h, lung single-cell suspensions were analyzed by gating on Lin−C25+CD44high lung ILC2s. Representative scattergrams are shown. (D) Total number of ILC2s, proportion of IL-5venus-positive ILC2s and mean fluorescence intensity (MFI) of IL-5venus in ILC2s are presented. **p<0.01 between the groups is indicated by horizontal lines. Data are presented as the mean ± SEM (n = 2-3 in each group) and are a pool of two experiments. (E) Experimental model for Panels F and G. (F) Cell numbers and differentials in BAL fluids were analyzed. (G) Lung levels of type 2 cytokines were analyzed by ELISA. *p<0.05, **p<0.01 between the groups indicated by horizontal lines. Data are presented as the mean ± SEM (n = 4 in each group).
To examine whether the inhibition by poly (I:C) is mediated by ILC2s, we examined IL-5venus reporter mice. Naïve Il5Venus reporter mice were pretreated with poly (I:C) i.n. and then exposed i.n. to A. alternata extract. After 4.5 h, the expression of IL-5venus was analyzed as an indicator of Il5 transcription by gating on the lung ILC2 population (Lin−CD25+CD44high), as previously described.27, 29 When mice were exposed to A. alternata, the proportion of IL-5-expressing ILC2s increased (Fig. 1C); however, IL-5-expressing ILC2s significantly decreased in mice pretreated with poly (I:C). The proportion of IL-5-positive ILC2s and mean fluorescence intensity (MFI) for IL-5venus verified that IL-5 production in ILC2s was significantly increased by A. alternata exposure, and this effect was suppressed by pretreatment with poly (I:C) (Fig. 1D, p<0.01). In contrast, no significant change was observed in the total number of ILC2s in this acute model.
We next examined whether poly (I:C) affects allergen-induced airway inflammation. Naïve BALB/c mice were exposed to poly (I:C) (50 μg/dose) and A. alternata (50 μg/dose) three times over a period of 6 days (Fig. 1E); poly (I:C) was administrated 1-day prior to each administration of A. alternata. When exposed to PBS or poly (I:C) alone, no eosinophils or neutrophils were detectable in BAL fluids (Fig. 1F); however, the number of eosinophils in the BAL fluid significantly increased in mice exposed to A. alternata, and the airway eosinophilia was nearly abolished in mice treated with poly (I:C) (p<0.01). A slight increase in neutrophils was also observed in mice administered A. alternata with poly (I:C) compared to those administered A. alternata alone. Similarly, the levels of IL-5 and PL-13 in lungs increased in mice exposed to A. alternata and were inhibited by administration of poly (I:C) (Fig. 1G, p<0.05 and p<0.01). Taken together, these findings suggest that poly (I:C) effectively inhibits allergen-induced ILC2 production of type 2 cytokines and eosinophilic airway inflammation.
Type 1 interferons mediate the inhibitory effect of poly (I:C) in vivo
Given that poly (I:C) inhibits the ILC2-mediated innate type 2 immune response to A. alternata in the lung, we next investigated the mechanism. Previous studies showed that i.n. administration of R848 or CpG promotes production of IFN-α and IFN-γ in the lung24, 25, so we first measured lung levels of type 1 and type 2 IFNs at 6 h after i.n. administration of poly (I:C), R848, or CpG A (25 μg for each). Roughly equal amounts of IFN-γ were produced by all three TLR agonists tested (Fig. 2A). Poly (I:C) also robustly induced both IFN-α and IFN-β, whereas R848 induced modest levels of these cytokines. In contrast, CpG A failed to induce IFNα or IFN-β. A kinetic study showed that when poly (I:C) was administered, the lung levels of IFN-α, IFN-β, and IFN-γ reached a plateau at 6 h and IFN-α and IFN-β remained elevated for at least 24 h (Fig. 2B, p<0.01).
Figure 2.
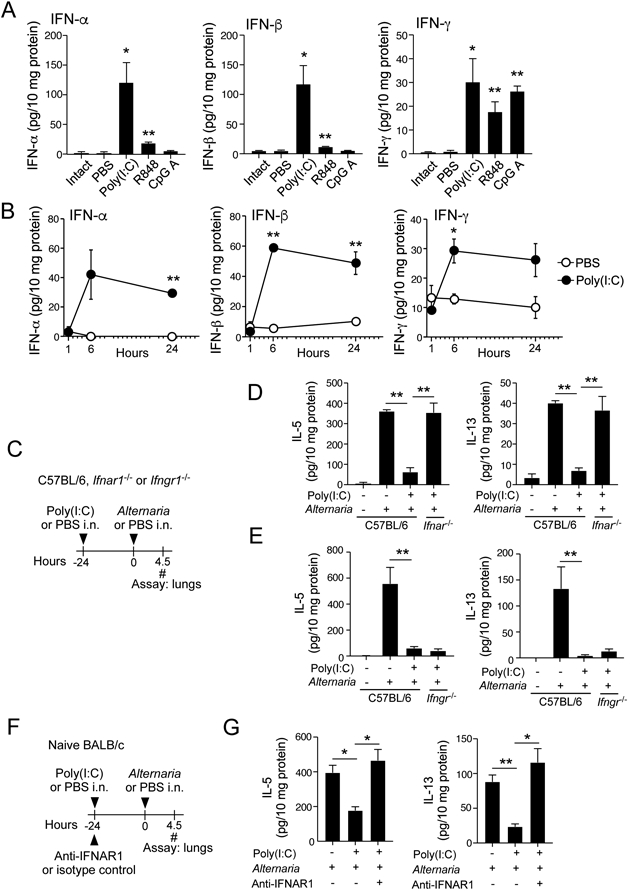
The inhibitory effects of poly (I:C) is mediated by type 1 IFNs. (A) Naïve BALB/c mice were administrated poly (I:C), R848, or CpG A i.n.. After 6 h, lung levels of interferons were analyzed by ELISA. The intact group indicates naïve mice without any manipulations. *p<0.05, **p<0.01 compared to mice administered PBS. Data are presented as the mean ± SEM (n = 3 in each group). (B) Naïve BALB/c mice were administrated poly (I:C) or PBS. Kinetic changes in the lung levels of interferons were analyzed. *p<0.05, **p<0.01 compared to mice administered PBS. Data are presented as the mean ± SEM (n = 3 in each group). (C) Experimental model for Panels D and E. (D, E) The results with Ifnar1−/− and Ifngr1−/− mice are presented in Panel D and Panel E, respectively. Data are presented as the mean ± SEM (n = 5 in each group) and are representative of two experiments. **p<0.01 between the groups indicated by horizontal lines. (F) Experimental model for Panel G. (G) Lung levels of type 2 cytokines are presented. *p<0.05, **p<0.01 between the groups indicated by horizontal lines. Data are presented as the mean ± SEM (n = 3 in each group) and are representative of three experiments.
To evaluate which IFNs are involved in poly (I:C)-mediated suppression of ILC2s in vivo, we used Ifnar1−/− and Ifngr1−/− mice that are deficient in receptors for IFN α/β and IFN-γ, respectively (Fig. 2C). In WT C57BL/6 mice, lung levels of type 2 cytokines increased after i.n. exposure to A. alternata, and poly (I:C) significantly reduced these cytokines (p<0.01). Importantly, Ifnar1−/− mice reversed the inhibitory effect of poly (I:C) (Fig. 2D), suggesting a critical role for the IFNAR1 pathway to inhibit the ILC2 response. In contrast, Ifngr1−/− mice failed to reverse the inhibitory effects of poly (I:C) (Fig. 2E).
We verified the roles for IFNAR by using a blocking Ab. The WT BALB/c mice were treated with anti-IFNAR1 Ab or control IgG at the same time as i.n. administration of poly (I:C) (Fig. 2F) and then exposed to A. alternata extract 24 h later. A. alternata-induced type 2 cytokine responses were inhibited by poly (I:C) in mice given isotype control IgG; however, the anti-IFNAR1 blocking Ab reversed the inhibitory effects (Fig. 2G). Together, these results suggest that type 1 IFNs, but not IFN-γ, mediate the inhibitory effects of poly (TC) in vivo. Furthermore, the involvement of type 1 IFNs was observed in both BALB/c and C57BL/6 mice, suggesting that their effects are not limited to a certain mouse strain.
IFN-β potently inhibits innate type 2 responses in the lung
We next examined the effects of ligands for IFNAR, namely IFN-α and -β, on type 2 immune responses in the lungs by using in vitro and in vivo models. First, we obtained single-cell suspensions of lungs from naïve BALB/c mice, which maintained the composition of various cell types in the lung. Cells were cultured with IL-33, which promotes a ILC2-mediated type 2 cytokine response27, with serial dilutions of IFNs for 4 days. Without IL-33, IL-5 and IL-13 were undetectable in culture supernatants (data not shown). When cultured with IL-33 at 1 ng/ml, large quantities of IL-5 and IL-13 were detectable, and the levels were reduced by IFN-β, with an IC50 of approximately 100 pg/ml for both IL-5 and IL-13 (Fig. 3A). IFN-β nearly abolished type 2 cytokines at 1 ng/ml. IL-33-induced cytokine production was also inhibited partially by IFN-α at 100 ng/ml, whereas lower concentrations of IFN-α appear to enhance type 2 cytokine response. When lung single cell cultures were treated with IL-33 at 10 ng/ml, even higher concentrations of IL-5 and IL-13 were detectable in the culture supernatants. IFN-β inhibited these cytokine responses with IC50 of <100 pg/ml. IFN-α also partially inhibited type 2 cytokine production in a concentration-dependent manner, but IFN-β was approximately 100-fold more potent than IFN-α.
Figure 3.
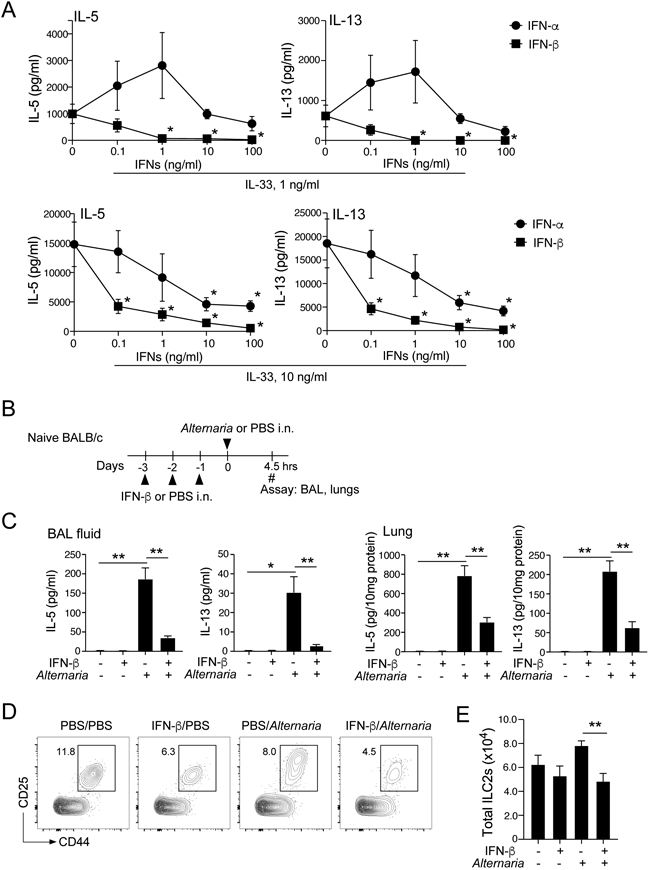
IFN-β effectively inhibits IL-33- or allergen-induced innate type 2 response in the lung. (A) Lung single-cell suspensions were cultured with indicated concentrations of IL-33 with serial dilution of IFN-α or IFN-β for 96 h. The levels of IL-5 and IL-13 in the supernatants were determined by ELISA. Data are presented as the mean ± SEM (n = 3 in each group) and are representative of three experiments. *p<0.05, compared with cells cultured without IFNs. (B) Experimental model for Panels B, C, D and E. (C) BAL and lung levels of IL-5 and IL-13 were analyzed by ELISA. Data are presented as the mean ± SEM (n = 4 in each group). *p<0.05, **p<0.01 between the groups indicated by horizontal lines. (D) ILC2s in the lung were analyzed by gating on the Lin−CD25+CD44high population. Representative scattergrams are shown. (E) The number of ILC2 cells are presented. Data are presented as the mean ± SEM (n = 4 in each group). **p<0.01 between the groups indicated by horizontal lines.
We next examined the role of IFN-β in an innate type 2 response in an in vivo model. We administrated IFN-β i.n. for 3 consecutive days, and mice were then exposed i.n. to A. alternata extract (Fig. 3B). A. alternata induced robust production of type 2 cytokines at 4.5 h in both BAL fluids and lung tissues, and IFN-β significantly reduced the cytokine levels (Fig. 3C, p<0.05 and p<0.01). We also found that the number of ILC2s in the lung was significantly reduced in mice treated with IFN-β and exposed to A. alternata as compared to those treated with PBS and then exposed to A. alternata (Figs. 3D and 3E, p<0.01). We also examined the effects of IFN-γ by using the same model. As shown in Supplemental Fig. E1, administration of IFN-γ significantly reduced the levels of IL-5 and IL-13 (p<0.01) whereas IFN-γ did not affect the number of ILC2s. These findings suggest that exogenous IFN-β inhibits type 2 cytokine production by lung ILC2s in vitro and in vivo, and that IFN-β is likely to be the key cytokine that mediates inhibition in mice treated with poly (I:C), as shown by its potency compared to IFN-α. Furthermore, IFN-β and IFN-γ might act differently on lung ILC2s as judged by their effects on ILC2 numbers.
IFN-β inhibits ILC2 proliferation induced by STATS-activating cytokines
To address the mechanisms involved in regulation of ILC2s by IFN-β, we moved to in vitro models using lung ILC2s purified by FACS. IL-7 is indispensable for development of ILCs6, 18, 19 and often used in combination with IL-33 to stimulate cytokine production by ILC2s in culture.15, 16 Therefore, we isolated ILC2s from the lungs of BALB/c mice and stimulated them with IL-33 alone or IL-33 plus IL-7 in the presence of IFN-β for 4 days; IFN-γ was used as a control. Although the amounts of type 2 cytokines produced by ILC2s stimulated with IL-33 alone or IL-7 alone were modest, IL-33 and IL-7 together induced large quantities of IL-5 and IL-13 (Supplemental Fig. E2). When ILC2s were stimulated with IL-33 alone, production of IL-5 and IL-13 was partially inhibited by IFN-γ, but not by IFN-β (Fig. 4A, p<0.01). In contrast, when ILC2s were stimulated with IL-33 plus IL-7, IFN-β significantly inhibited type 2 cytokine production in a concentration-dependent manner (Fig. 4B, p<0.05 and p<0.01). In this condition, IFN-β and IFN-γ at 100 ng/ml showed roughly comparable inhibitory effects.
Figure 4.

IFN-β inhibits type 2 cytokine production and proliferation of lung ILC2s. (A and B). Isolated lung ILC2s were cultured with medium alone, IL-33 (10 ng/ml), or IL-33 plus IL-7 (10 ng/ml) (Panel B) for 96 h with serial dilutions of IFN-β or 100 ng/ml IFN-γ. The levels of IL-5 and IL-13 in the supernatants were determined by ELISA. Data are presented as the mean ± SEM (n = 3) and are representative of three experiments. *p<0.05, **p<0.01 compared with cells cultured without IFNs. (C) CFSE-labeled lung ILC2s were cultured with medium alone, IL-7, IL-2, and TSLP with or without IL-33 for 96 h. Dilutions of CFSE were analyzed by flow cytometry. Representative histograms are shown. (D) CFSE-labeled lung ILC2s were cultured as described above with or without IFN-β and IFN-γ. Dilutions of CFSE were analyzed by flow cytometry. Representative histograms are shown. (E) MFI of CFSE dilution is presented. Data are presented as the mean ± SEM (n = 2) and are representative of three experiments. *p<0.05, **p<0.01 between the groups indicated by horizontal lines.
These observations together with those in Fig. 3E and Supplemental Fig. E1D led us to speculate that the mechanisms of ILC2 suppression by IFN-β and IFN-γ are different and that IFN-β may control the effects of IL-7, rather than the effects of IL-33, on ILC2s. To address this question directly, we examined the proliferation of ILC2s by isolating lung ILC2s, labeling them with CFSE, and culturing the cells for 4 days with IL-7 or IL-33. As compared to medium alone, IL-7 induced proliferation of ILC2s, resulting in robust dilution of CFSE (Fig. 4C) over 4 days. Other STAT5-activating cytokines, including IL-2 and TSLP30, also induced ILC2 proliferation. In contrast, IL-33 alone induced minimal proliferation of ILC2s, and the combination of IL-33 and STAT5-activating cytokines showed comparable effects as STAT5-activating cytokines alone.
Given the robust activities of IL-7 and other STAT5-activating cytokines on ILC2 proliferation, we examined the effects of IFN-β and IFN-γ. CFSE-labeled ILC2s were cultured with each cytokine in the presence of interferons, and CFSE dilution was measured as an indicator of ILC2 proliferation. Both IL-7 and IL-2 induced robust proliferation of ILC2s (Fig. 4D and 4E), which was inhibited by IFN-β nearly to the baseline level (i.e., medium alone, P<0.01). Although IFN-γ also inhibited IL-2- and IL-7-induced proliferation of ILC2s, its effects were weaker than those by IFN-β, especially when ILC2s were cultured with IL-2. TSLP induced modest proliferation of ILC2s, which was inhibited both by IFN-β or IFN-γ. When ILC2s were cultured with IL-33 plus STAT5-activated cytokines, they proliferated vigorously (Supplemental Fig. E3A), and this proliferation was inhibited strongly by IFN-β but weakly by IFN-γ (Supplemental Fig. E3A and E3B). When we analyzed the cytokine levels in the cell-free supernatants of these cell proliferation experiments, both IFN-β and IFN-γ significantly decreased the levels of IL-5 and IL-13 in lung ILC2s cultured with STAT5-activated cytokines alone (Supplemental Fig. E4A) or STAT5-activating cytokines plus IL-33 (Supplemental Fig. E4B) (p<0.05). Altogether, these findings suggest that STAT5-activating cytokines, but not IL-33, promote proliferation of lung ILC2s in vitro and that IFN-β inhibits the effects of these cytokines. In contrast, the inhibitory effects of IFN-γ might be independent of cell proliferation.
IFN-β suppresses IL-7-induced survival of lung ILC2s
To explore the effects of IFN-β on ILC2s further, we examined their viability by flow cytometry. Isolated lung ILC2s were cultured with medium alone, IL-33, or IL-7 with or without IFN-β for 72 h and stained with Annexin V and 7-aminoactinomycin D (7-AAD). In culture with medium alone without any growth factors, approximately 70% of ILC2s became apoptotic and then necrotic, as indicated by cellular membranes that were permeable to 7-AAD (Fig. 5A) and flow cytometry scatter plots. IL-33 showed minimal effects on the distribution of these apoptotic and necrotic cells. In contrast, IL-7 significantly inhibited apoptosis of ILC2s, resulting in 80% viable cells over a period of 72 h of culture (Fig. 5A and 5B). Importantly, in the presence of IFN-β, a large proportion of cells became apoptotic and necrotic even in the presence of IL-7. In contrast, IFN-β showed minimal effects on apoptosis when ILC2s were cultured with medium alone or with IL-33, suggesting that IFN-β disrupted the molecular pathway activated by IL-7 to maintain survival of ILC2s in vitro.
Figure 5.

IFN-β induces apoptosis of lung ILC2s. (A) Isolated lung ILC2s were cultured with medium alone, IL-33, or IL-7 with or without IFN-β for 72 h. Cell viability was analyzed by staining with Annexin-V and 7-AAD. (A) Representative scattergrams are shown. (B) Proportions of live (Annexin V−7-AAD−), apoptotic (Annexin V+7-AAD−), and necrotic (Annexin V+7-AAD+) cells are shown. Data are presented as the mean ± SEM (n = 2) and are representative of three experiments. **p<0.01 between the groups indicated by horizonal lines.
To examine the effects of IL-7 on lung ILC2s at molecular levels, we analyzed their gene expression using a NanoString® assay. Isolated lung ILC2s were cultured with medium alone or with IL-7 for 16 h. Using unsupervised heat map analysis, IL-7 was shown to promote expression of a number of genes while it inhibited fewer genes (Fig. 6A). According to dot plots, upregulated genes included Il5, Il13, Icos, Il2ra, Il2rb, and Gata3 (Fig. 6B). Increased expression of Gata3 was also verified by quantitative RT-PCR (Fig. 6C). It has been reported previously that GATA3 is indispensable for differentiation and maintenance of ILC2s.18, 19, 31 Therefore, we examined GATA3 protein expression in ILC2s by flow cytometry by gating separately on alive (i.e., negative for Ghost Dye Red 780 staining) and dead cells. When cultured with medium alone for 72 h, approximately 50% of live cells lost their expression of GATA3 (Fig. 6D). In contrast, when cultured with IL-7, a majority of live ILC2s expressed GATA3. Dead cells, detected as the Ghost-positive population, did not express GATA3 regardless of whether they were cultured with medium alone or IL-7. Furthermore, the levels of GATA3 in the Ghost-negative cells (i.e. alive cells) were higher in ILC2s cultured with IL-7 as compared to those with medium alone. Collectively, IL-7 enhanced survival of isolated ILC2s in vitro and the effects were inhibited significantly by IFN-β. IL-7 promoted expression of GATA3, which may explain the supportive effects of IL-7 on lung ILC2s.
Figure 6.

IL-7 induces GATA3 mRNA and protein expression in lung ILC2s. (A) Isolated lung ILC2s were cultured with medium alone or with IL-7 for 16 h. mRNA was analyzed by the Nanostring® assay. (A) The results of unsupervised heat map analysis are shown. Sample numbers indicate a paired and separate experiment. (B) Scatter plots of all the analyzed genes are shown. Red dots indicate notable genes. (C) Isolated lung ILC2s were cultured with medium alone or with IL-7 for 16 h. mRNA was analyzed by real-time RT-PCR, and the results were normalized to the cells cultured with medium alone. (D) Isolated lung ILC2s were cultured with medium alone or with IL-7 for 72 h. Cells were stained with Ghost Dye Red 780, followed by staining for GATA3 protein. Cells were analyzed by gating separately on live cells (Ghost Dye Red 780-negative) and dead cells (Ghost Dye Red 780-positive). Representative scatter grams and histograms are shown.
GATA3 expression in lung ILC2s is regulated reciprocally by STAT5-activating cytokines and IFN-β
Finally, to examine the molecular mechanisms involved in IFN-β-mediated suppression of ILC2s, we examined the effects of various cytokines on GATA3 protein expression using the same approach as described above; GATA3 protein was analyzed by gating on viable cells. IL-7 enhanced expression of GATA3 within the live ILC2 population (Fig. 7A), resulting in increased MFI of GATA3 staining (Fig. 7B) and a smaller proportion of GATA3-negative cells (Fig. 7C) compared to ILC2s cultured with medium alone. Similarly, IL-2 and TSLP enhanced GATA3 expression. The ranked order of STAT5-activating cytokines which promoted GATA3 expression was IL-7>IL-2>TSLP, as indicated by GATA3 MFI. In contrast, IL-33 showed no effects on GATA3 expression. A kinetic study showed that lung ILC2s lose their expression of GATA3 over 72 hours when they were cultured in vitro with medium alone (Supplemental Fig. E5). In contrast, IL-7 enhanced GATA3 expression, resulting in increased GATA3 MFI in the same time period.
Figure 7.

IFN-β inhibits GATA3 expression induced by STAT5-activating cytokines in lung ILC2s. (A) Isolated lung ILC2s were cultured with medium alone, IL-7, IL-2, TSLP or IL-33 for 72 h, and GATA3 protein expression in live ILC2s (i.e., Ghost Dye Red 780-negative) was analyzed. Representative histograms and scattergrams are shown. (B) MFI for GATA3 protein. (C) Proportion of GATA3-negative cells among the live cells. Data are presented as the mean ± SEM (n = 2). *p<0.05, **p<0.01 compared with cells cultured with medium alone. (D) Isolated lung ILC2s were cultured with medium alone or IL-7 with or without IFN-β or IFN-γ for 72 h, and GATA3 protein expression in live ILC2s (i.e., Ghost Dye Red 780-negative) was analyzed. (E) MFI for GATA3 protein. (F) Proportion of GATA3-negative cells among the live cells. Data are presented as the mean ± SEM (n = 3) and are representative of two experiments. *p<0.05, **p<0.01 between the groups indicated by horizontal lines.
We then examined the effects of IFN-β on GATA3 expression in lung ILC2s by culturing them with IL-7, as a representative of STAT5-activating cytokines. Again, IL-7 enhanced expression of GATA3 in ILC2s compared with those cultured with medium alone (Fig. 7D-F), and IFN-β significantly inhibited GATA3 expression to levels roughly comparable to ILC2s cultured without IL-7 (p<0.01). IFN-γ also partially inhibited GATA3 expression, but not as strongly as IFN-β. The inhibitory effects of IFN-β on IL-7-induced GATA3 expression were reproduced in a kinetic study (Supplemental Fig. E5). Altogether, these results suggest that STAT5-activating cytokines enhance expression of GATA3 by lung ILC2s, whereas IFN-β blocks the effects of these cytokines.
Finally, we investigated the effects of IFN-β on GATA3 expression in vivo. Naïve BALB/c mice were administrated PBS or IFN-β i.n. for 3 consecutive days, and lungs were analyzed 24 h after the last administration of IFN-β by gating on the Lin−CD25+CD44high ILC2 population (Fig. 8A, 8B). We found that GATA3 protein levels in ILC2s were significantly lower in mice administered IFN-β than those administered PBS (Fig. 8C, p<0.01), consistent with decreased cytokine production by those lung ILC2s when they are exposed to Alternaria in vivo (Fig. 3). On the other hand, the number of ILC2s was not significantly affected by IFN-β (Fig. 8D)
Figure 8.

IFN-β inhibits GATA3 expression in lung ILC2s in vivo. (A) Experimental model. Naïve BALB/c were administered i.n. IFN-β or PBS for 3 consecutive days. GATA3 protein expression in lung ILC2s was analyzed by gating on the Lin−CD25+CD44high population. (B) Representative scattergrams and histograms are shown. (C) MFI for GATA3 protein and proportion of GATA3-negative cells within the lung ILC2 population. (D) Proportion of lung ILC2s within the Lin− cell population is shown (n.s., not significant). Data are presented as the mean ± SEM (n = 3-4 in each group) and are representative of two experiments. **p<0.01 between the groups indicated by horizontal lines.
DISCUSSION
The objective of this project was to identify a strategy to suppress innate type 2 immunity, which could potentially be used to treat patients with allergic airway diseases. We found that activation of TLR3 by poly (I:C) induces IFN-α, -β, and -γ in the lung of naïve mice and suppresses ILC2-mediated allergic airway inflammation. Previous reports showed that activation of TLR7/8 and TLR9 by R848 and CpG A, respectively, suppressed ILC2-driven airway inflammation by producing IFN-α or IFN-γ.24, 25 Our findings are consistent with a previous report showing that poly (I:C) ameliorates Aspergillus flavus- or IL-33-induced type 2 immune response in the lung.32 Our observations add to this knowledge by demonstrating that poly (FC)-mediated IFN-β plays a major role in suppressing lung ILC2s and allergic airway inflammation. Indeed, we compared the effects of poly (I:C), R848, and CpG A in parallel in an acute innate type 2 response model and found that the poly (I:C) was the most effective at suppressing A. alternata-induced production of type 2 cytokines (Fig. 1). The results of Ifnar1-deficient mice and blocking Abs support the roles for IFNAR and its ligands IFN-α and -β while involvement of the IFNAR specifically expressed on lung ILC2s needs to be verified in future with conditional knockout mice once they become available. Results of in vitro culture experiments show that IFN-β was more than 100x more potent than IFN-α. Altogether, these results lead us to conclude that the IFN-β that activates the IFNAR-pathway likely play a major role in inhibiting the innate type 2 response to A. alternata exposure in vivo.
The observations in this study are consistent previous observations, which indicated that R848 and CpG A inhibit allergen-induced innate type 2 response through the IFN-α and IFN-γ pathways.24, 25 Rather, our findings demonstrate the diversity of IFN responses depending on the nature of TLRs involved. Agonists for TLR7/8 and TLR9 activate pDCs, which results in their production of IFN-α.24, 25 On the other hand, pDCs do not express the poly (I:C) receptor TLR3.33 Although the identification of the targets for poly (I:C) was not the primary goal of this project, we speculate that either airway epithelial cells or CD8α+ conventional DCs might be involved. IFN-β is expressed by bronchial epithelial cells in response to rhinovirus infection.34 The CD8α+ population of conventional DCs express TLR3, but not TLR7.33, 35 We also found previously that bone marrow-derived conventional DCs produce IFN-β when they are stimulated with poly (I:C) in vitro.36 Thus, microbes may activate distinct sets of IFNs, depending on their nature (i.e., dsRNA virus, ssRNA virus, or bacteria), TLRs, and innate immune cells. Such redundant mechanisms may represent fine-tuning of anti-microbial immune responses and regulation of allergic immune responses.
The findings in Fig. 2 demonstrating that the inhibitory effects of poly (I:C) are dependent on type 1 interferons (i.e. IFN-α and -β) but not on type 2 interferon (i.e. IFN-γ) are rather unexpected. Nonetheless, our findings are consistent with those of other investigators who reported that the inhibitory effects of poly (I:C) on IL-33-induced type 2 response are fully reversed in mice deficient in IFNAR1.32 Earlier studies15, 16 and observations in this manuscript clearly show that both IFN-β or IFN-γ are capable of inhibiting IL-33-induced type 2 cytokine production by ILC2s in vitro and in vivo. Furthermore, all IFN-α, -β, and -γ were detected in the lungs of mice exposed to poly (I:C) (Fig. 2B). This discrepancy may reflect complexity of the lung immunity and we can speculate several reasons: First, in vitro experiments often use relatively high concentrations of IFN-β and IFN-γ, which may not be achievable in an in vivo setting. Second, more IFN-β than IFN-γ is produced for a prolonged period in the lungs of mice exposed to poly (I:C) (Fig. 2B). Finally, in vitro culture may not precisely reflect the spacious distribution of IFN-β- or IFN-γ-producing cells and ILC2s, which may be pivotal for effective inhibitory actions of these cytokines on lung ILC2s. Thus, the experimental designs that engage endogenous sources of cytokines can complement those using exogenous cytokines and likely help to elucidate the complexity of the biological system.
By comparing IFN-α, -β and -γ using several in vitro approaches, we also found unique features and a potent capacity of IFN-β to suppress lung ILC2s. IFN-β failed to inhibit type 2 cytokine response when ILC2s were stimulated with IL-33 alone, but it did inhibit ILC2s when they were stimulated with IL-33 and IL-7 (Fig. 4A and 4B). IFN-β was effective at suppressing the responses of ILC2s to IL-7 and other STAT5-activating cytokines, including proliferation, survival, and GATA3 expression (Fig. 4, Fig. 5 and Fig. 7), but IFN-γ showed only modest effects. ILC2s are known to express receptors for all three interferons16, but the downstream signaling pathways are likely distinct. For example, type 1 IFNs generally promote the IFN-stimulated gene factor 3, consisting of STAT1, STAT2, and IRF9, which binds IFN-stimulated response (ISRE) elements in DNA.37 On the other hand, type 2 IFN or IFN-γ predominantly signal through a STAT1 homodimer, which binds IFN-γ-activated site (GAS) elements.37 Although IFN-α and IFN-β share the IFNAR1 and IFNAR2 receptor complex, they are known to generate distinct biological outcomes. This is likely due to variable affinities to the receptor complex.38, 39 Indeed, IFN-α binds IFNAR1 and IFNAR2 at affinities of 0.5 to 5 μM and 0.4 to 5 nM, respectively; IFN-β binds these receptors with affinities of 0.1 μM and 0.1 nM, respectively.40 In addition, cell lines with low IFNAR1 expression respond to IFN-β but not to IFN-α.41 INF-β inhibited differentiation of human osteoclasts 100-fold more potently than IFN-α42, consistent with our findings with mouse lung ILC2s. Finally, distinct differences in the effects of IFN-α and IFN-β have also been reported in other cell types.43, 44 On the other hand, a possibility remains that the observed differences between IFN-α and IFN-β in Fig. 3A could be due to the quality of recombinant proteins used in the experiments. Further studies will be necessary to dissect the differential responses of ILC2s to these cytokines.
Although the molecular targets of IFN-β on ILC2s have not been fully understood, we speculate that GATA3 might be involved. GATA3 has been shown to be indispensable for differentiation, maintenance, and function of ILC2s in both in mice and humans.18, 19 We found that IL-7 and other STAT5-activating cytokines, but not IL-33, enhanced expression of GATA3 protein by lung ILC2s in vitro, and IFN-β inhibited GATA3 expression both in vitro and in vivo. Our observations are consistent with a previous report in human peripheral blood ILC2s showing that TSLP, but not IL-33, increases GATA3 expression.19 In CD4+ T cells, GATA3 expression is induced by the TCR signals and IL-4.45, 46 However, the mechanisms involved in the expression of GATA3 in mature ILC2s have remained an enigma. Our findings suggest that GATA3 expression in lung ILC2s is controlled by the balance between the activities of two key transcription factors, STAT5 and STAT1/2 (as a downstream of IFN-β). This model is consistent with the observation in CD4+ T cells that STAT5 activators promote expression of GATA3 in differentiated Th2 cells.30 IFN-β, but not IFN-γ, suppressed GATA3 during Th2 cell development and in fully committed Th2 cells.47 In addition, the level of STAT5 activation likely plays a central role in homeostasis and the functions of ILCs in general.48 Further studies in the molecular mechanisms involved in GATA3 expression in ILC2s in mucosal organs and roles of STAT protein(s) in the process will likely provide important information to understand the immunobiology of ILC2s and to find ways to control them.
IFN-β response is compromised in bronchial epithelial cells from patients with asthma.34 In this study, we found that exogenous IFN-β, when administered i.n., suppresses ILC2 expression of GATA3 (Fig. 8) and allergen-induced innate type 2 responses in mouse models (Fig. 3). Human asthma is associated with polymorphisms of genes associated with ILC2s, including IL33, IL1RL1, IL7R, RORA, and IL2RB.11, 12, 13 Therefore, IFN-β can be considered an option to treat patients with asthma. Indeed, a clinical trial was performed to examine the efficacy of inhaled IFN-β in asthma patients who are accompanied by cold symptoms as IFN-β might promote anti-viral immune responses.49 The study found that inhaled IFN-β was safe, reduced medication use to treat cold-induced asthma exacerbations and improved clinical symptoms and lung function in patients with difficult-to-treat asthma. In a recent clinical trial, inhaled IFN-β was safe and promoted recovery in patients with SARS-CoV-2 infection.50 Furthermore, systemic treatment with IFN-β has been well tolerated and is currently used to treat patients with relapsing multiple sclerosis.51, 52, 53 Therefore, more clinical trials with IFN-β are warranted to examine the clinical efficacy of IFN-β in asthma, to optimize the regimens, and to develop biomarkers to identify patients who are most benefitted from the treatment. Treatment with inhaled poly (I:C) could also be another option as it can promote production of both type 1 and type 2 interferons (Fig. 2). Nonetheless, potential risks associated with increased production of TSLP53 and other pro-inflammatory mediators54 by inhaled poly (I:C) may need to be taken into consideration.
In conclusion, we found that TLR3-driven IFN-β inhibits allergen-induced ILC2-mediated airway inflammation and that IFN-β blocks GATA3 expression, proliferation, and survival in lung ILC2s that are promoted by STAT5-activating cytokines. The results of our study also suggest that IFN-β suppresses ILC2s likely through a distinct mechanism(s) from IFN-γ. Further studies to understand the inhibitory mechanisms of ILC2s will likely provide renewed understanding of the immunobiology of this unique cell type and novel therapeutic options for asthma and allergic airway diseases.
Supplementary Material
Key Messages.
Poly (I:C) inhibits allergen-induced innate type 2 responses in the lung that are mediated by ILC2s.
The inhibitory effects of poly (I:C) are dependent on the IFN-α/β receptor pathway.
IFN-β inhibits proliferation and survival of lung ILC2s induced by STAT5-activating cytokines.
The ability of IL-7 to promote GATA3 expression in ILC2s is blocked by IFN-β.
ACKNOWLEDGEMENTS
This work was supported by grants from the National Institutes of Health (R37AI71106, R01HL117823), the Mayo Graduate School of Biomedical Sciences, and the Mayo Foundation. We thank Ms. Tammy Brehm-Gibson and Ms. Carole Viso at the Flow Cytometry Core, Mayo Clinic Arizona, for their excellent technical assistance in ILC2 cell sorting.
Abbreviations
- 7-AAD
7-aminoactinomycin D
- Ab
antibody
- BAL
bronchoalveolar lavage
- CFSE
carboxyfluorescein succinimidyl
- FACS
fluorescence-activated cell sorting
- FBS
fetal bovine serum
- GAS
IFN-γ-activated site
- GATA3
GATA-binding protein 3
- IFNAR
interferon-α/β receptor
- IFNGR
interferon-γ receptor
- ILC2
group 2 innate lymphoid cell
- IL
interleukin
- i.n.
intranasally
- i.p.
intraperitoneally
- ISRE
IFN-stimulated response
- Lin−
lineage-negative
- NK
natural killer
- PBS
phosphate-buffered saline
- pDCs
plasmacytoid dendritic cells
- poly (I:C)
polyinosinic-polycytidylic acid
- RORα
RAR-related orphan receptor alpha
- PRRs
pattern recognition receptors
- RT
room temperature
- SEM
standard error of the mean
- STAT
signal transducer and activator of transcription
- Th2
type 2 helper T
- TLRs
toll-like receptors
- TSLP
thymic stromal lymphopoietin
- WT
wild-type
Footnotes
All authors acknowledge no conflict of interest related to this manuscript.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
REFERENCES
- 1.Busse WW, Lemanske RF Jr. Asthma. N Engl J Med 2001; 344:350–62. [DOI] [PubMed] [Google Scholar]
- 2.Boonpiyathad T, Sözener ZC, Satitsuksanoa P, Akdis CA. Immunologic mechanisms in asthma. Semin Immunol 2019; 46:101333. [DOI] [PubMed] [Google Scholar]
- 3.Moro K, Yamada T, Tanabe M, Takeuchi T, Ikawa T, Kawamoto H, et al. Innate production of T(H)2 cytokines by adipose tissue-associated c-Kit(+)Sca-1(+) lymphoid cells. Nature 2010; 463:540–4. [DOI] [PubMed] [Google Scholar]
- 4.Neill DR, Wong SH, Bellosi A, Flynn RJ, Daly M, Langford TK, et al. Nuocytes represent a new innate effector leukocyte that mediates type-2 immunity. Nature 2010; 464:1367–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Price AE, Liang HE, Sullivan BM, Reinhardt RL, Eisley CJ, Erie DJ, et al. Systemically dispersed innate IL-13-expressing cells in type 2 immunity. Proc Natl Acad Sci U S A 2010; 107:11489–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Walker JA, Barlow JL, McKenzie AN. Innate lymphoid cells--how did we miss them? Nat Rev Immunol 2013; 13:75–87. [DOI] [PubMed] [Google Scholar]
- 7.Morita H, Moro K, Koyasu S. Innate lymphoid cells in allergic and nonallergic inflammation. J Allergy Clin Immunol 2016; 138:1253–64. [DOI] [PubMed] [Google Scholar]
- 8.Halim TY, Steer CA, Mathä L, Gold MJ, Martinez-Gonzalez I, McNagny KM, et al. Group 2 innate lymphoid cells are critical for the initiation of adaptive T helper 2 cell-mediated allergic lung inflammation. Immunity 2014; 40:425–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Halim TY, Hwang YY, Scanlon ST, Zaghouani H, Garbi N, Fallon PG, et al. Group 2 innate lymphoid cells license dendritic cells to potentiate memory TH2 cell responses. Nat Immunol 2016; 17:57–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.van der Ploeg EK, Golebski K, van Nimwegen M, Fergusson JR, Heesters BA, Martinez-Gonzalez I, et al. Steroid-resistant human inflammatory ILC2s are marked by CD45RO and elevated in type 2 respiratory diseases. Sci Immunol 2021; 6. [DOI] [PubMed] [Google Scholar]
- 11.Gudbjartsson DF, Bjornsdottir US, Halapi E, Helgadottir A, Sulem P, Jonsdottir GM, et al. Sequence variants affecting eosinophil numbers associate with asthma and myocardial infarction. Nat Genet 2009; 41:342–7. [DOI] [PubMed] [Google Scholar]
- 12.Moffatt MF, Gut IG, Demenais F, Strachan DP, Bouzigon E, Heath S, et al. A large-scale, consortium-based genomewide association study of asthma. N Engl J Med 2010; 363:1211–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kurz T, Hoffjan S, Hayes MG, Schneider D, Nicolae R, Heinzmann A, et al. Fine mapping and positional candidate studies on chromosome 5p13 identify multiple asthma susceptibility loci. J Allergy Clin Immunol 2006; 118:396–402. [DOI] [PubMed] [Google Scholar]
- 14.Bartemes KR, Kita H. Roles of innate lymphoid cells (ILCs) in allergic diseases: The 10-year annivesary for ILC2s. J Allergy Clin Immunol 2021; 147:1531–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Duerr CU, McCarthy CD, Mindt BC, Rubio M, Meli AP, Pothlichet J, et al. Type I interferon restricts type 2 immunopathology through the regulation of group 2 innate lymphoid cells. Nat Immunol 2016; 17:65–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Moro K, Kabata H, Tanabe M, Koga S, Takeno N, Mochizuki M, et al. Interferon and IL-27 antagonize the function of group 2 innate lymphoid cells and type 2 innate immune responses. Nat Immunol 2016; 17:76–86. [DOI] [PubMed] [Google Scholar]
- 17.Wong SH, Walker JA, John HE, Drynan LF, Hams E, Camelo A, et al. Transcription factor RORα is critical for nuocyte development. Nat Immunol 2012; 13:229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoyler T, Klose CS, Souabni A, Turqueti-Neves A, Pfeifer D, Rawlins EL, et al. The transcription factor GATA-3 controls cell fate and maintenance of type 2 innate lymphoid cells. Immunity 2012; 37:634–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Mjösberg J, Bernink J, Golebski K, Karrich JJ, Peters CP, Blom B, et al. The transcription factor GATA3 is essential for the function of human type 2 innate lymphoid cells. Immunity 2012; 37:649–59. [DOI] [PubMed] [Google Scholar]
- 20.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010; 140:805–20. [DOI] [PubMed] [Google Scholar]
- 21.Stein MM, Hrusch CL, Gozdz J, Igartua C, Pivniouk V, Murray SE, et al. Innate Immunity and Asthma Risk in Amish and Hutterite Farm Children. N Engl J Med 2016; 375:411–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Schuijs MJ, Willart MA, Vergote K, Gras D, Deswarte K, Ege MJ, et al. Farm dust and endotoxin protect against allergy through A20 induction in lung epithelial cells. Science 2015; 349:1106–10. [DOI] [PubMed] [Google Scholar]
- 23.Arpaia N, Barton GM. Toll-like receptors: key players in antiviral immunity. Curr Opin Virol 2011; 1:447–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maazi H, Banie H, Aleman Muench GR, Patel N, Wang B, Sankaranarayanan I, et al. Activated plasmacytoid dendritic cells regulate type 2 innate lymphoid cell-mediated airway hyperreactivity. J Allergy Clin Immunol 2018; 141:893–905.e6. [DOI] [PubMed] [Google Scholar]
- 25.Thio CL, Lai AC, Chi PY, Webster G, Chang YJ. Toll-like receptor 9-dependent interferon production prevents group 2 innate lymphoid cell-driven airway hyperreactivity. J Allergy Clin Immunol 2019; 144:682–97.e9. [DOI] [PubMed] [Google Scholar]
- 26.Ikutani M, Yanagibashi T, Ogasawara M, Tsuneyama K, Yamamoto S, Hattori Y, et al. Identification of innate IL-5-producing cells and their role in lung eosinophil regulation and antitumor immunity. J Immunol 2012; 188:703–13. [DOI] [PubMed] [Google Scholar]
- 27.Bartemes KR, Iijima K, Kobayashi T, Kephart GM, McKenzie AN, Kita H. IL-33-responsive lineage- CD25+ CD44(hi) lymphoid cells mediate innate type 2 immunity and allergic inflammation in the lungs. J Immunol 2012; 188:1503–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Doherty TA, Khorram N, Chang JE, Kim HK, Rosenthal P, Croft M, et al. STAT6 regulates natural helper cell proliferation during lung inflammation initiated by Alternaria. Am J Physiol Lung Cell Mol Physiol 2012; 303:L577–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Bartemes K, Chen CC, Iijima K, Drake L, Kita H. IL-33-responsive group 2 innate lymphoid cells are regulated by female sex hormones in the uterus. J Immunol 2018; 200:229–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Guo L, Wei G, Zhu J, Liao W, Leonard WJ, Zhao K, et al. IL-1 family members and STAT activators induce cytokine production by Th2, Th17, and Th1 cells. Proc Natl Acad Sci U S A 2009; 106:13463–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Furusawa J, Moro K, Motomura Y, Okamoto K, Zhu J, Takayanagi H, et al. Critical role of p38 and GATA3 in natural helper cell function. J Immunol 2013; 191:1818–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.She L, Alanazi HH, Yan L, Brooks EG, Dube PH, Xiang Y, et al. Sensing and signaling of immunogenic extracellular RNAs restrain group 2 innate lymphoid cell-driven acute lung inflammation and airway hyperresponsiveness. PLoS One 2020; 15:e0236744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Edwards AD, Diebold SS, Slack EM, Tomizawa H, Hemmi H, Kaisho T, et al. Toll-like receptor expression in murine DC subsets: lack of TLR7 expression by CD8 alpha+ DC correlates with unresponsiveness to imidazoquinolines. Eur J Immunol 2003; 33:827–33. [DOI] [PubMed] [Google Scholar]
- 34.Wark PA, Johnston SL, Bucchieri F, Powell R, Puddicombe S, Laza-Stanca V, et al. Asthmatic bronchial epithelial cells have a deficient innate immune response to infection with rhinovirus. J Exp Med 2005; 201:937–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Széles L, Meissner F, Dunand-Sauthier I, Thelemann C, Hersch M, Singovski S, et al. TLR3-Mediated CD8+ dendritic cell activation is coupled with establishment of a cell-intrinsic antiviral state. J Immunol 2015; 195:1025–33. [DOI] [PubMed] [Google Scholar]
- 36.Wada K, Kobayashi T, Matsuwaki Y, Moriyama H, Kita H. Altemaria inhibits double-stranded RNA-induced cytokine production through Toll-like receptor 3. Int Arch Allergy Immunol 2013; 161 Suppl 2:75–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Platanias LC. Mechanisms of type-I- and type-II-interferon-mediated signalling. Nat Rev Immunol 2005; 5:375–86. [DOI] [PubMed] [Google Scholar]
- 38.van Boxel-Dezaire AH, Rani MR, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 2006; 25:361–72. [DOI] [PubMed] [Google Scholar]
- 39.de Weerd NA, Vivian JP, Nguyen TK, Mangan NE, Gould JA, Braniff SJ, et al. Structural basis of a unique interferon-β signaling axis mediated via the receptor IFNAR1. Nat Immunol 2013; 14:901–7. [DOI] [PubMed] [Google Scholar]
- 40.Piehler J, Thomas C, Garcia KC, Schreiber G. Structural and dynamic determinants of type I interferon receptor assembly and their functional interpretation. Immunol Rev 2012; 250:317–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Uzé G, Schreiber G, Piehler J, Pellegrini S. The receptor of the type I interferon family. Curr Top Microbiol Immunol 2007; 316:71–95. [DOI] [PubMed] [Google Scholar]
- 42.Leomil Coelho LF, Almeida GMF, Mennechet FJD, Blangy A, Uze G. Interferon-α and -β differentially rgulate osteoclastogenesis: role of differential induction of chemokine CXCL11 expression. Proc Natl Acad Sci USA 2005; 11917–11922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.van Boxel-Dezaire AHH, Rani MRS, Stark GR. Complex modulation of cell type-specific signaling in response to type I interferons. Immunity 2006; 25:361–72. [DOI] [PubMed] [Google Scholar]
- 44.Kaur S, Platanias LC. IFN-β-specific signaling via a unique IFNAR1 interaction. Nat Immunol 2013; 14:884–885. [DOI] [PubMed] [Google Scholar]
- 45.Scheinman EJ, Avni O. Transcriptional regulation of GATA3 in T helper cells by the integrated activities of transcription factors downstream of the interleukin-4 receptor and T cell receptor. J Biol Chem 2009; 284:3037–48. [DOI] [PubMed] [Google Scholar]
- 46.Paul WE, Zhu J. How are T(H)2-type immune responses initiated and amplified? Nat Rev Immunol 2010; 10:225–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Huber JP, Ramos HJ, Gill MA, Farrar JD. Cutting edge: Type IIFN reverses human Th2 commitment and stability by suppressing GATA3. J Immunol 2010; 185:813–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Villarino AV, Sciumè G, Davis FP, Iwata S, Zitti B, Robinson GW, et al. Subset- and tissue-defined STAT5 thresholds control homeostasis and function of innate lymphoid cells. J Exp Med 2017; 214:2999–3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Djukanović R, Harrison T, Johnston SL, Gabbay F, Wark P, Thomson NC, et al. The effect of inhaled IFN-β on worsening of asthma symptoms caused by viral infections. A randomized trial. Am J Respir Crit Care Med 2014; 190:145–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Monk PD, Marsden RJ, Tear VJ, Brookes J, Batten TN, Mankowski M, et al. Safety and efficacy of inhaled nebulized interferon beta -1a (SNG001) for treatment of SARS-CoV-2 infection: a randomized, double-blind, placebo-controlled, phase 2 trial. Lancet Respir Med 2021; 9:196–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Jakimovski D, Kolb C, Ramanathan M, Zivadinov R, Weinstock-Guttman B. Interferon β for Multiple Sclerosis. Cold Spring Harb Perspect Med 2018; 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Reder AT, Feng X. How type I interferons work in multiple sclerosis and other diseases: some unexpected mechanisms. J Interferon Cytokine Res 2014; 34:589–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Markowitz CE. Interferon-beta: mechanism of action and dosing issues. Neurology 2007; 68:S8–11. [DOI] [PubMed] [Google Scholar]
- 54.Kato A, Favoreto S Jr, Avila PC, Schleimer RP. TLR3- and Th2 cytokine dependent production of thymic stromal lymphopoietin in human airway epithelial cells. J Immunol 2007; 179:1080–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Ammi R, De Waele J, Willemen Y, Van Brussel I, Schrijvers DM, Lion E, et al. Poly (I:C) as cancer vaccine adjuvant: knocking on the door of medical breakthroughs. Pharmacol Ther 2015; 146:120–31. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.