

Summary
Upon tissue injury, cytokine expression reprogramming transiently remodels the inflammatory immune microenvironment to activate repair processes and subsequently return to homeostasis. However, chronic inflammation induces permanent changes in cytokine production which exacerbate tissue damage and may even favor tumor development. Here, we address the contribution of post-transcriptional regulation, by the RNA-binding protein CPEB4, to intestinal immune homeostasis and its role in inflammatory bowel diseases (IBD) and colorectal cancer (CRC) development. We found that intestinal damage induces CPEB4 expression in adaptive and innate immune cells, which is required for the translation of cytokine mRNA(s) such as the one encoding interleukin-22. Accordingly, CPEB4 is required for the development of gut-associated lymphoid tissues and to maintain intestinal immune homeostasis, mediating repair and remodeling after acute inflammatory tissue damage and promoting the resolution of intestinal inflammation. CPEB4 is chronically overexpressed in inflammatory cells in patients with IBD and in CRC, favoring tumor development.
Subject areas: Biological sciences, Molecular biology, Immunology
Graphical abstract
Highlights
-
•
CPEB4 is overexpressed in Th17 and ILC3 cells upon intestinal barrier damage
-
•
CPEB4 is required for Il-22 mRNA translation and IL-22 expression
-
•
CPEB4 promotes tissue repair in acute transient inflammation
-
•
In chronic inflammation CPEB4 exacerbates intestinal pathology and promotes tumor growth
Biological sciences; Molecular biology; Immunology
Introduction
The intestinal epithelium is a highly proliferative tissue which, in addition to the mechanical barrier and nutrient absorption properties of the epithelial cells, hosts innate and adaptive immune cells control host-microbial interactions and tissue homeostasis. Metabolic programs and host defense are integrated to ensure proper immune responses during stress. Mucosal barrier dysfunction contributes to the development of inflammatory bowel disease (IBD). IBD refers to a group of multifactorial, immunologically mediated chronic inflammatory diseases, of which Crohn’s disease (CD) and ulcerative colitis (UC) represent the two major forms of disease. Acute intestinal inflammation with immune cell activation and cytokine production restores tissue homeostasis and immune balance. However, when acute inflammatory responses are not properly shut down, the recovery of homeostasis is compromised and chronic inflammation can eventually take place (Garrett et al., 2010). In the context of intestine, the duration of the inflammatory response determines the equilibrium between the recovery of intestinal homeostasis or the development of IBD and eventually of colitis-associated colorectal cancer (CAC) or colorectal cancer (CRC) (Greten and Grivennikov, 2019; West et al., 2015). Genome-wide association studies (GWAS) have identified numerous susceptibility loci for CD and UC, encoding proteins with important roles in proteostasis (Coleman and Haller, 2019), including the RNA-binding protein and translational regulator CPEB4 (Franke et al., 2010; Jostins et al., 2012). In turn, how cancer-associated inflammation develops, interacts with, and modulates tumor immunity will determine its tumor immunosuppressive or immunosupportive role (Greten and Grivennikov, 2019; Shalapour and Karin, 2019).
Translational control of gene expression modulates the development and function of the immune system (Carpenter et al., 2014). By fine-tuning the strength and timing of innate and adaptive immune responses, post-transcriptional mechanisms remodel cell proteome to adapt the changes in the cell environment, without the need for de novo transcription (Piccirillo et al., 2014). Translational control is also required to shape the directionality of the tumoral immune responses (Truitt and Ruggero, 2016). A key mRNA translation/stability control mechanism of stored inactive mRNAs involves cytoplasmic changes in their poly(A) tail length, mediated by cytoplasmic polyadenylation element binding proteins (CPEBs). CPEB proteins bind the 3′-untranslated region (UTR) of mRNAs that contain cytoplasmic polyadenylation elements (CPE) (D'Ambrogio et al., 2013; Fernandez-Miranda and Mendez, 2012) and regulate their translation into proteins. CPEB4 mediates translational response to counteract chronic liver disease under ER-stress (Maillo et al., 2017) and regulates vascular endothelial growth factor (VEGF) translation, multifaceted inflammatory cytokine (Calderone et al., 2016). Mouse embryo fibroblasts lacking CPEB display mis-regulation of many mRNAs involved in inflammation (Alexandrov et al., 2012). CPEB4 has also been shown to be required for tumoral cell growth in a cell-autonomous manner in glioblastoma and melanoma (Ortiz-Zapater et al., 2011; Perez-Guijarro et al., 2016) xenografted tumors.
In this work, we aimed to determine the contribution of the translational regulator CPEB4 in maintaining intestinal homeostasis and during intestinal inflammation and development of colorectal cancer. We found that CPEB4 is required to maintain intestinal homeostasis, mediating repair, and remodeling after inflammatory tissue damage, by controlling T cell population. However, sustained accumulation of CPEB4 in the immune niche promotes tumor formation in colitis-associated cancer and in human CRC by modulating tumoral adaptive immune response. These functions are likely mediated by the temporal regulation of cytokine mRNA translation, such as Il22 mRNA.
Results
CPEB4 is required for the development of gut-associated lymphoid tissues and to maintain intestinal immune homeostasis
To elucidate the potential role of CPEB4 in intestinal barrier function, we used previously generated CPEB4 total KO mice (CPEB4KO) (Maillo et al., 2017) and determined whether CPEB4 depletion could compromise intestinal homeostasis. We found no differences in barrier integrity, as in intestinal permeability and proliferation (Figures S1A and S1B). We noticed alterations in the formation of gut-associated lymphoid tissues (GALT) in CPEB4KO mice, with reduction in the number and size of Peyer’s patches and cryptopatches, which maintain the intestinal immune barrier (Figures 1A–1C). Peyer’ s patches of CPEB4KO mice exhibited an aberrant composition of T and B lymphocytes, with decreased frequency of total CD3+ T lymphocytes (Figure 1D) due to reduced T-helper CD4+ and cytotoxic CD8+ T cells (Figures 1E and 1F), and increased frequency of CD19+ B lymphocytes (Figure 1G). In the colonic lamina propria, we also found a significant reduction in the frequency of CD3+ T-lymphocytes in CPEB4KO mice (Figure 1H), accompanied by increased CD19+ B lymphocyte frequency (Figure 1I). No changes in the myeloid population (CD11b+ cells) were detected (Figure S1C). These alterations were also observed in CPEB4KO thymus and lymph nodes, which resulted in less cellularity (Figures 1J, 1K, andS1D) caused by reduced number of CD4+ T lymphocytes in thymus (Figures 1L, S1E, S1F, and S1G), and CD4+ and CD8+ cells in lymph nodes (Figure 1K). Accordingly, other secondary lymphoid organs, such as blood and spleen, also displayed reduced frequency and number of CD3+ T lymphocytes in CPEB4KO mice (Figures 1M; S1H, and S1I). No changes in CD19+ B lymphocytes and CD11+ myeloid cells were detected (Figures 1M and S1H). In the blood, the decrease in CD3+ T cells was due to reduced CD4+ and CD8+ T cells (Figures 1N, S1J, and S1K), whereas in the spleen it was due to reduced CD8+ T cells (Figure S1L). These data indicate a function for CPEB4 in lymphoid organs homeostasis and suggested that CPEB4 regulates intestinal immune homeostasis by maintaining T cell populations.
Figure 1.

CPEB4 deficiency compromises the development of gut-associated lymphoid tissues and affects the composition of secondary lymphoid organs
(A) Peyer's patch (PP) number of WT and CPEB4KO mice (n = 16/group). Mean ±SEM ∗∗∗∗p <0.0001 (unpaired t-test).
(B andC) Cryptopatch (CP) number (B) and size (C) of WT and CPEB4KO mice (n = 10/group) were measured in H&E-stained colon sections. Representative H&E-stained CP images. Scale bar, 100 μm. CP number: ∗p = 0.042 (unpaired t-test); CP size: ∗p = 0.023 (Mann-Whitney test).
(D–G) Percentages of CD3+ (∗∗p = 0.0015), CD4+ (∗∗∗p = 0.0005), CD8+ (∗∗p = 0.0039) and CD19+ (∗∗p = 0.0039) lymphocytes were analyzed by FACS in PP of WT and CPEB4KO mice (Wilxocon test).
(H and I) WT and CPEB4KO lamina propria (LP) T lymphocytes (CD3+)(H) and B lymphocytes (CD19+)(I) were analyzed from CD45+ cells by flow cytometry analysis. Mean ±SEM ∗p = 0.0406 for CD3+; ∗p = 0.0317 for CD19+ (Mann-Whitney).
(J) Number of cells of WT (n = 12) and CPEB4KO (n = 10) thymus. Mean ±SEM ∗p = 0.0032 (Mann-Whitney).
(K) Inguinal lymph node analysis of total cells (∗∗∗p = 0.001), CD3+ (∗∗∗p = 0.0001), CD4+ (∗∗p = 0.0138) and CD8+ (∗∗∗∗p <0.0001) lymphocytes in WT (n = 13) and CPEB4KO (n = 11) mice. Data are shown as FC (mean ± SEM, two-way ANOVA test, multiple comparisons).
(L) Number of total CD4+ and CD8+ T lymphocytes from WT (n = 9) and CPEB4KO (n = 7) thymus. Mean ±SEM ∗p = 0.024 (two-way ANOVA test).
(M and N) Blood samples from unchallenged WT and CPEB4KO mice (n = 10/group) were analyzed by FACS for CD3+, CD19+, CD11b+, CD4+ and CD8+ lymphocytes. Data are shown as fold change (FC; mean ± SEM). CD3+: ∗∗∗p = 0.0006; CD4+: ∗∗p = 0.0012; CD8+: ∗∗p = 0.0031 (unpaired t-test). For all panels, data are pooled from a minimum of three biologically independent experiments
CPEB4 depletion exacerbates the susceptibility to DSS-induced acute colitis
Based on above described findings, we reasoned that CPEB4 might contribute to intestinal inflammation in response to epithelial damage. Thus, we induced acute colitis by disrupting the intestinal epithelial barrier with dextran sodium sulfate (DSS). CPEB4 protein expression was strongly increased following DSS treatment and maintained elevated during the recovery, when the epithelium is regenerated (Figure 2A). Notably, following DSS-treatment, CPEB4-deficient mice exhibited exacerbated disease, with more severe weight loss (Figure 2B), increased damage to the lamina propria, and extended crypt injury (Figures 2C, and 2D) compared with WT mice. Moreover, DSS-treated CPEB4KO mice displayed symptoms of persistent inflammation, with intense mixed inflammatory cells (Figures 2E and S2A) and increased mRNA expression of pro-inflammatory cytokines Il-6, Ccl2, Il-1a and I1-1b in the colons (Figure S2B). This phenotype was not reproduced in mice with selective deficiency of CPEB4 in the intestinal epithelium (Villin-CreERT2 and CPEB4lox/lox alleles, CPEB4KO-IEC) (Figures 2F, 2G, andS2C), implying that increased inflammation triggered by CPEB4 depletion originates in the mucosal microenvironment and not in the epithelial cell compartment. Indeed, when CPEB4 was selectively eliminated in T lymphocytes by crossing LCK-Cre (which depletes CD4 and CD8 T lineages) with CPEB4lox/lox mice (T cell-specific CPEB4KO mice, CPEB4-TKO), the repair of DSS-injured intestinal epithelia was equally affected compared with total CPEB4 depletion, with increased body weight loss and decreased survival compared with DSS-treated WT mice (Figures 2H, 2I, andS2D). Taken together, these findings indicate that in the absence of CPEB4, mice are more susceptible to DSS-induced acute colitis, suggesting a protective function of CPEB4 to resolve acute intestinal inflammation, most likely through a function of CPEB4 in the T lymphoid cell compartment.
Figure 2.

CPEB4 accumulates during colitis and its depletion exacerbates the susceptibility to DSS-induced colitis, by controlling T cell population
(A) Immunoblot analysis of CPEB4 in DSS-treated colons at indicated timepoints. Vinculin was used as loading control. ∗Marks specific band. Quantification is shown.
(B) Body weight of DSS-treated mice. 2.5% DSS was administered in drinking water for 5 days followed by regular drinking water. Body weight was plotted as percentage of initial weight (WT and CPEB4KO n = 8 until day 10, n = 4 up to day 22). Data are mean ± SEM; ∗∗p < 0.005, ∗p < 0.05 (two-way ANOVA test, multiple comparisons).
(C) Representative H&E-stained colon sections from mice treated with DSS, analyzed at day 10. The regenerating region is highlighted by a red line. Scale bars, 1 mm. Larger magnification, 100 μm.
(D and E) Colitis severity of WT and CPEB4KO mice (n = 4/group) was measured by disease score (D) and inflammation score (E) at day 10 after DSS treatment. ∗p = 0.0286 (Mann-Whitney).
(F) Body weight record as percentage of initial weight of WT IEC and CPEB4KO IEC DSS-treated mice (n = 12 until day 7/genotype; n = 6 up to day 13/genotype).
(G) Disease score at day 13 of DSS treatment (n = 5/genotype).
(H) Percentage of body weight loss of 3% DSS-treated WT (n = 15) and T cell-specific CPEB4 TKO (n = 13) mice. ∗∗p < 0.05, ∗∗∗p = 0.0005, ∗∗∗∗p < 0.0001 (mean ± SEM, two-way ANOVA test, multiple comparisons).
(I) Survival rate of DSS-treated WT and CPEB4 TKO mice. ∗∗∗∗p < 0.001. Data are representative of three (B,C), two (A,D,E,F,G) and one (H,I) biologically independent experiments.
CPEB4 is upregulated in patients with inflammatory bowel diseases and correlates with Th17 lineage
To determine if CPEB4 could also impact chronic inflammatory condition(s), we first analyzed the expression of CPEB4 mRNA in mucosal biopsies from adult patients with IBD. We found that CPEB4 mRNA levels were upregulated in IBD patients, both UC and CD patients, compared to healthy controls (Figure 3A). In non-inflamed colitis patients who do not show macroscopic signs of inflammation, CPEB4 mRNA levels were similar to healthy controls (Figure 3A). CPEB4 mRNA accumulation was also confirmed in pediatric patients with UC and CD (Figure 3B). These observations point to a potential role of CPEB4 in the IBD-associated chronic inflammatory response.
Figure 3.

CPEB4 increases in patients with IBD and correlates with Th17 lineage
(A and B) CPEB4 mRNA expression (arbitrary units, AU) was measured in intestinal mucosa of adult (A) and pediatric (B) healthy human controls, and in biopsies from patients with noninflamed ulcerative colitis (UC), inflamed UC and Crohn's disease (CD), combining GSE13367, GSE16879, GSE59071, GSE9452, GEO public adult datasets (A) or using GSE10616 pediatric dataset (B); ∗∗∗∗p < 0.0001, ∗∗p < 0.05.
(C and D) Pearson correlation between CPEB4 expression and immune signatures (used in (Guinney et al., 2015)) in intestinal mucosa from adult healthy individuals (n = 41), and adult patients with non-inflamed ulcerative colitis (UC; n = 54), inflamed UC (n = 122) and Crohn’s disease (CD; n = 27), combining GSE13367, GSE16879, GSE59071, GSE9452, GEO public datasets and from healthy (n = 11), UC (n = 10), CD Colon (n = 14) and CD Colon-ileo (n = 18) pediatric individuals, GEO dataset GSE10616.
To assess whether this CPEB4 function was related to immune responses, we performed correlation studies between CPEB4 and immune response signatures in human intestinal mucosa obtained from IBD patients. CPEB4 positively correlated with immune response signature in both adult and pediatric UC and CD samples compared with healthy mucosa (Figures 3C and 3D). Notably, CPEB4 expression positively correlated with T-helper 17 (Th17) activation signature in adult UC samples (Figure 3C) and also in pediatric UC and CD samples (Figure 3D), whereas no consistent correlation with T-regulatory (T-reg) activation was found (Figures 3C, 3D, andS2E). Th17 cells are CD4+ T lymphocytes of the adaptive immunity and are essential for mucosal inflammation, regeneration and clearance of extracellular bacteria by producing a set of cytokines that attract neutrophils and induce the release of anti-microbial peptides from epithelial cells (Kempski et al., 2017). Collectively, these results suggest a putative role of CPEB4 inducing sustained and inappropriate immune responses in chronic intestinal inflammation and associate CPEB4 to Th17 response in IBD patients.
CPEB4 posttranscriptionally regulates IL-22 signaling
Because the main functions of Th17 cells are the cytokine-mediated mucosal regeneration, inflammation, and the clearance of extracellular bacteria, we next measured the effect of CPEB4 depletion in Th17 cytokine mRNA levels in colons of adult untreated WT and CPEB4KO mice. Interleukin-22 (Il-22) mRNA strongly accumulated in colons of CPEB4KO mice, while other Th17 cytokine mRNAs, such as Il-17a, Il-17f, and Il-1a, Il-1b, Tnf, Infγ did not change (Figure 4A). This effect was not observed in CPEB4KO-IEC animals (Figure S3A). The main cell types responsible for the production of intestinal IL-22, in addition to Th17 (Zheng et al., 2007), are the recently discovered Th22 - a distinct CD4+ T cell subset which produces IL-22 but not IL-17 (Trifari et al., 2009)-, as well as ILC3 cells of the innate immune system, including lymphoid tissue-inducer (LTi) cells and natural killer cell cytotoxicity receptor-positive (NCR+) cells (Bando and Colonna, 2016). All these cells also express Cpeb4 mRNA (Figure S3B). Il-22 mRNA levels were significantly increased in sorted CD4+ T and ILC3 cells from the lamina propria of CPEB4KO mice (Figures 4B and 4C). Of note, the percentage of CD4+ T cells was decreased in CPEB4KO mice compared to WT, without changes in ILC3 frequency (Figures 4D and 4E). These results indicated that the observed Il-22 mRNA increase in adaptive CD4+ T lymphocytes and innate ILC3 cells from CPEB4KO mice was not because of increased number of IL-22-expressing cells. Because IL-22 production by Th17 cells is induced by IL-23 from intestinal dendritic cells and macrophages, which are activated by microbial products reaching the lamina propria (Sugimoto et al., 2008), we next tested whether Il-22 mRNA overexpression was caused by increased IL-23 signaling. We found no changes in Il-23r mRNA or in Il-23 mRNA in the lamina propria of CPEB4KO mice (Figures S3C–S3E).
Figure 4.

CPEB4 controls IL-22 protein production, by binding Il-22 mRNA
(A) Gene expression of Il-22, Il-17a, Il-17f, Il-1a, Il-1b, Tnfa, Ifnγ mRNA in colon extracts from untreated WT (n = 8) and CPEB4KO (n = 8) mice, relative to Gapdh. Mean ±SEM ∗∗∗∗p <0.0001 (two-way ANOVA test, multiple comparisons).
(B and C) Il-22 mRNA expression in CD4+ T cells (B) and ILC3 (LTi and NCR+) (C) cells from the colon lamina propria of WT and CPEB4KO mice (n = 3/group). Mean ±SEM ∗∗p = 0.0037; ∗LTi p = 0.0222; ∗NCR+p = 0.0472 (unpaired t-test).
(D and E) Frequency of CD4+ T cells(D) (WT, n = 11; CPEB4KO, n = 13) and ILC3 cells (E) (WT, n = 8; CPEB4KO, n = 9) in colon lamina propria. ∗∗p = 0.0042 (unpaired t-test).
(F) ELISA of IL-22 in CD4+ naive T cells isolated from WT (n = 7) and CPEB4KO (n = 7) mice treated for 72 h with IL-23 (50 ng/mL). Mean ±SEM ∗∗∗∗p < 0.0001 (unpaired t-test).
(G) ELISA of IL-22 from four 0.5-cm segments of colon from WT and CPEB4KO mice cultured ex vivo for 24 h. Mean ± SEM of six mice/genotype. ∗p = 0.026 (Mann-Whitney test).
(H) RT-qPCR of Saa1/2 and Ang4, mRNA from isolated intestinal epithelial cells from WT (n = 8) and CPEB4KO (n = 6) mice. Mean ±SEM ∗∗p = 0.0023, ∗p = 0.008 (Mann-Whitney test).
(I) Binding of CPEB4 to Il-22, Il-17a, Il-17f and Il-23r mRNA was measured by RNA-immunoprecipitation (RIP)-qPCR. Cpeb4 and Rpl0 mRNAs were used as positive and negative CPEB4 binding controls, respectively. IgG was used as IP control. Data are pooled from three biologically independent experiments for panels D, E, I or two for F, G, H.
To determine whether the observed increase in Il-22 mRNA translated into higher IL-22 protein levels and signaling, or rather reflected the accumulation of silenced mRNA, we purified naive T cells from spleen and lymph nodes and measured IL-22 production after IL-23 stimulation. We found a clear reduction of IL-22 protein level (Figure 4F). This observation was further confirmed by measuring IL-22 protein production in colon cultures from WT and CPEB4KO mice (Figure 4G). The decrease in IL-22 protein was accompanied by downregulation of IL-22 signaling in intestinal epithelial cells, the only cell type that responds to IL-22 in colonic mucosa (Sonnenberg and Artis, 2015), as demonstrated by the significant downregulation of the IL-22 downstream targets, serum amyloid A (Saa1/2) and angiogenin-4 (Ang4) mRNA, observed in CPEB4KO mice (Figure 4H). Taken together, these results indicate that CD4+ T and ILC3 cells store Il-22 mRNA, which cannot be translated into protein in the absence of CPEB4.
To determine if this regulation was directly mediated by the binding of CPEB4 to Il-22 mRNA, we used CPEB4-RNA immunoprecipitation (RIP) followed by Il-22 qPCR. We were able to immunoprecipitate Il-22 mRNA, demonstrating that it is a direct target of CPEB4 in Th17 cells (Figures 4I andS3F). Other mRNAs relevant for Th17 function, such as Il-17a, Il-17f or Il-23r, were also co-immunoprecipitated with CPEB4 (Figure 4I). However, we did not find differences in rorc mRNA and RORyt protein levels between WT and CPEB4KO Th17 cells, suggesting that CPEB4 does not control Th17 differentiation (Figures S3G and S3H).
CPEB4 depletion reduces the incidence of colitis-associated colorectal cancer in mice
Besides supporting the integrity of intestinal barrier and resolution of acute inflammation, Th17 cells massively infiltrate the inflamed intestine of IBD patients (Fujino et al., 2003), where they amplify an overactive inflammatory process that promotes pathogenicity and eventually induces tumor formation (Kempski et al., 2017). In fact, Th17 cells negatively influence the prognosis of CRC patients (Tosolini et al., 2011). Th17 cells associate with advanced stages of CRC and produce cytokines, which mainly exert pro-tumorigenic effects (Martin et al., 2015; Sharp et al., 2017; Wu et al., 2009). Continued activation of the IL-22 pathway increases the risk of CRC in patients with an extended history of IBD (Mizoguchi et al., 2018) and favors tumorigenesis in mouse models of CAC (Huber et al., 2012). Thus, we next tested whether CPEB4 depletion could also affect the development of colorectal tumors induced by injection of the carcinogen azoxymethane (AOM) followed by DSS-induced chronic colitis. We found that the number of macroscopic colon tumors was significantly reduced in AOM/DSS-treated CPEB4KO animals compared with WT control mice (Figures 5A, 5B; S4A, S4B, and S4C). Despite the lower tumor incidence, the tumors that were formed in the presence or absence of CPEB4 were indistinguishable in size (Figure 5C), proportion of polypoid and flat adenomas and adenocarcinomas (Figure S4D), cell proliferation (Ki67 immunohistochemistry) (Figure S4E) or cell apoptosis (caspase-3 immunohistochemistry) (Figure S4F). These results suggest that CPEB4 plays a role in the pre-tumoral niche, but does not affect the tumoral cell properties. Accordingly, we found no statistically significant differences in average colon tumor number, load or size between AOM/DSS-treated CPEB4KO-IEC and WT-IEC mice with intestinal epithelium-specific deletion of CPEB4 (Figures 5D and 5E). These data imply that, as in DSS-colitis, the role of CPEB4 in CAC results from a tumor microenvironment-dependent effect rather than an intestinal epithelial cell autonomous effect and it is consistent with a possible regulation of CPEB4 on Th17-IL22 axis.
Figure 5.

CPEB4 deficiency reduces CAC development and shapes adaptive tumoral immune response, controlling IL-22 production
(A) Schematic representation of the two-stage murine model of CAC. Mice were intraperitoneally injected with one dose of azoxymethane (AOM, tumor-inducing agent), followed by three rounds of 2.5% dextran sodium sulfate (DSS, tumor-promoting agent) in drinking water. Representative tumor images in WT and CPEB4KO mice. Scale bar, 5 mm.
(B and C) Tumor number per animal (B) (WT, n = 15; CPEB4KO, n = 16; mean ± SEM; Mann-Whitney test, ∗∗∗p = 0.0006) and percentage of tumor sizes (C) in mm.
(D and E) Average tumor number (D) and size (E) of Villin-CreERT2 Cpeb4+/+(IEC WT, n = 10) and Villin-CreERT2 Cpeb4lox/lox (IEC CPEB4KO, n = 9) mice. Data represent mean ±SEM.
(F) Representative stainings of myeloperoxidase (MPO, an enzyme produced by neutrophil granulocytes to potentiate inflammation and apoptotic oxidative damage), T lymphocytes (CD3) and granzyme B (GrzB) in AOM/DSS colon tumor sections from WT and CPEB4KO mice (n = 8/genotype). Related quantifications are shown in the histograms. Data represent means ±SEM ∗p = 0.0111; ∗∗∗p = 0.0003; ∗p = 0.0207 (unpaired t-test for MPO; Mann-Whitney test for CD3 and GrzB).
(G) Granzyme B mRNA levels, relative to Gapdh, in colon tumors from WT (n = 14) and CPEB4KO (n = 13) mice. ∗∗p = 0.0023 (Mann-Whitney test).
(H) Flow cytometry analysis of CD3+ T lymphocytes (gated from CD45+ cells) from WT and CPEB4KO mice. Data are mean ± SEM (n = 7/genotype). ∗p = 0.0148 (Mann-Whitney test).
(I) Flow cytometry analysis of CD4 and CD8 T lymphocyte populations of colon tumors from WT (n = 7) and CPEB4KO (n = 8) mice. ∗∗∗p = 0.0001 (two-way ANOVA test, multiple comparisons).
(J) Ifnγ mRNA levels, relative to Gapdh, in CD4 tumor sorted T cells from WT (n = 5) and CPEB4KO (n = 7) mice. ∗p = 0.048 (Mann-Whitney test).
(K) IL-22 ELISA in adjacent mucosa and AOM-DSS tumors of WT (n = 4) and CPEB4KO (n = 5) mice. ∗p = 0.0241 (unpaired t-test).
(L) Relative Il-22 mRNA levels in WT (n = 12) and CPEB4KO (n = 13) tumors analyzed by RT-qPCR ∗p = 0.0109 (unpaired t-test). Data are pooled from two (B,C,G,L) or representative of two (D,E) biologically independent experiments
CPEB4 shapes the adaptive immunity and modulates IL-22 production in CAC
To further define the contribution of CPEB4 to CAC, we analyzed the immune response that characterizes CPEB4-deficient tumors. Immunohistochemistry of AOM/DSS-treated CPEB4KO mice showed an increased tumor infiltration by myeloperoxidase (MPO+) producing-neutrophils and total T lymphocytes (CD3+), compared with WT mice (Figure 5F). This was associated with a concomitant boost of secreted granzyme B and accumulation of its mRNA (Figures 5F and 5G), which plays a major role in eliminating cancer cells (Trapani and Smyth, 2002). Flow cytometry confirmed the increase of T lymphocytes in CPEB4KO tumors (Figure 5H), without changes in the amount of immune (CD45+) cells (Figure S4G). Tumor infiltrating CD3+ lymphocytes derived mainly from activated T-helper lymphocytes (CD4+) and not from cytotoxic T lymphocytes (CD8+) (Figure 5I). Moreover, CD4+ T cells from CPEB4KO tumors and total lysates from CPEB4KO tumors showed increased mRNA levels of interferon gamma (Infγ) (Figures 5J andS4H), a conventional cytokine associated with anti-tumoral responses and protective adaptive immunity (West et al., 2015). Collectively, these results support the idea that CPEB4 impacts on cancer occurrence by influencing the immune landscape of the CAC tumors.
The findings that CPEB4 depletion decreases CAC incidence but also aggravates acute intestinal inflammation could be linked with Th17 cells and the observed regulation of IL-22, which is key to both immune protection and immune pathology (Keir et al., 2020). Indeed, IL-22 promotes epithelial homeostasis and host defense and resolves acute colitis (Sugimoto et al., 2008), but it also supports tumor growth (Huber et al., 2012; Kryczek et al., 2014). Thus, we measured the levels of IL-22 and IL-17a in colon tumors from AOM/DSS-treated CPEB4KO mice. There were no differences in IL-17a protein levels and mRNA levels were only slightly increased (Figures S4I and S4J). However, IL-22 protein expression was reduced in absence of CPEB4 (Figure 5K), despite a robust increase in Il-22 mRNA (Figure 5L), suggesting that CPEB4 is necessary for the translation of Il-22 mRNA also in cancer, and that a translation impairment was buffered, or was compensated by increased mRNA levels. Taken together, these data indicate that the pathogenic role of CPEB4 in colon tumorigenesis is at least in part mediated by activation of Th17 response and production of interleukins such as IL-22.
CPEB4 correlates with Th17 activation in CRC and is specifically overexpressed in the tumoral immune compartment
To determine the impact of CPEB4 to CRC prognosis, we interrogated a metacohort of 1705 patients with CRC for association between CPEB4 mRNA levels and disease-free survival. We consistently observed that the expression of CPEB4 was negatively correlated with disease-free survival of CRC patients (Figure 6A). This result is consistent with previous findings showing that CPEB4 mRNA is overexpressed in whole CRC tumor samples (Zhong et al., 2015), where associates with poor prognosis (Cortes-Guiral et al., 2017; He et al., 2017). To determine the source, tumoral cell or immune niche, of this increased CPEB4 mRNA levels, we searched immune cell expression signatures (Guinney et al., 2015) that correlated with CPEB4 in healthy tissue, tumor adjacent mucosa and tumor mucosa from the human CRC samples. We found that CPEB4 positively correlated with immune responses and activation of Th17 cells in tumor mucosa, compared with healthy or tumor adjacent mucosa (Figure 6B). Interestingly, CPEB4 protein and mRNA were mainly expressed by differentiated epithelial cells in healthy mouse colons, but their expression strongly increased in tumoral stroma in AOM/DSS induced tumors from WT mice, where CPEB4 especially accumulated in immune cells (Figure 6C, S5A, and S5B). Accordingly, when we analyzed the expression of CPEB4 in samples specific for stroma or epithelium collected from human colorectal cancer and normal tissues (GSE35602) (Nishida et al., 2012), we found that CPEB4 expression was increased in tumor stroma compared with normal stroma, whereas it was strongly reduced in tumor epithelia compared with normal epithelia (Figure 6D). Consistently, the analysis of a transcriptomic dataset of sorted cells from human CRC samples (GSE39397) (Calon et al., 2012) revealed that CPEB4 mRNA levels were higher in the immune cell population (CD45+), compared to epithelial cells (Epcam+), endothelial cells (CD31+) and cancer-associated fibroblasts (FAP+) (Figure 6E). Therefore, CPEB4 differentially accumulated in the immune tumor microenvironment, suggesting a major role for CPEB4 in immune tumor-infiltrating population. Thus, we next analyzed the correlation between CPEB4 expression and the genes of Th17 signature in human immune sorted CRC cells and we found that CPEB4 strongly correlates with many Th17-specific mRNAs, including IL-17A and IL-22 (Figure 6F).
Figure 6.

High CPEB4 expression correlates with poor prognosis of CRC patients, accumulates in tumoral immune population where associates with Th17 signature
(A) Association plot between CPEB4 expression and disease-free survival (DFS) of a colorectal patient metacohort, combining Gene Expression Omnibus (GEO) datasets GSE33113, GSE14333, GSE39582 and GSE37892. LRT-pv = 0.039364.
(B) Pearson correlation between CPEB4 expression and immune signatures (used in (Guinney et al., 2015)) in 10 CRC datasets (see STAR Methods).
(C) Representative stainings of CPEB4 in healthy colons and AOM/DSS-induced tumors from WT mice. CPEB4KO tissues were used as control. NE: normal epithelia, NS: normal stroma, TE: tumor epithelia, TS: tumor stroma. Scale bars, 100 μm.
(D) CPEB4 mRNA expression (arbitrary units, AU) of normal and tumoral epithelia versus normal and tumoral stroma in human samples (GSE35602). ∗∗∗p < 0.001.
(E) CPEB4 mRNA expression of epithelial cells (Epcam), immune cells (CD45), endothelial cells (CD31) and fibroblasts (FAP) in tumors of human samples (GSE39397). ∗∗∗∗p < 0.0001; ∗p =0.007; #p =0.069.
(F) Spearman correlation between CPEB4 expression and Th17 signature in tumoral immune population (used in M23). ∗p < 0.05; ∗∗p < 0.05.
Discussion
Intestinal immune homeostasis and intestinal inflammation resolution requires a tight temporal regulation of gene expression adaptive responses to stress. We found that the stress-induced expression of CPEB4 in immune cells is indispensable to preserve intestinal immune homeostasis under normal conditions and to restore homeostasis following acute intestinal inflammatory processes. The targets of CPEB4 in these intestinal immune cells include a set of cytokines produced by Th17 cells of adaptive immunity and ILC3 cells of innate immunity. Of those, IL-17a, IL-17f, and IL-22 are cytokines with beneficial roles when generated at moderate levels and under tight temporal control, but whose under- or overexpression cause unbalanced immune responses that affect lymphoid tissue homeostasis and promote further inflammation (Kempski et al., 2017). In particular, IL-22 production is required for organogenesis and functions of the gut-associated lymphoid tissue, which controls intestinal epithelia homeostasis and preserves appropriate host-microbial interactions (Dempsey, 2017; Hernandez et al., 2018; Mizoguchi et al., 2018; Pearson et al., 2012). IL-22 is also required for intestinal epithelial regeneration and resolution of intestinal inflammation (Aden et al., 2016; Sonnenberg and Artis, 2015; Sugimoto et al., 2008). Accordingly, we show that increased Il-22 mRNA levels cannot be translated into IL-22 protein in the absence of CPEB4, leading to reduced IL-22 signaling in intestinal epithelial cells and exacerbation of acute intestinal inflammation. In fact, CPEB4-deficient mice share phenotypic characteristics with reduced IL-22 models. Thus, mice deficient in IL-22 show worse DSS-induced colitis (Sonnenberg and Artis, 2015).
Consistently with these results, we found increased levels of CPEB4 in chronic intestinal inflammation, such as in UC and CD, where it accumulates in inflammatory cells and associates with Th17 adaptive response. CPEB4 has been identified as one of the IBD and Crohn’s risk/susceptibility loci (Franke et al., 2010; Jostins et al., 2012; Momozawa et al., 2018). Although the functional role of IL-22 in chronic inflammation is controversial, targeting IL-22 alleviates chronic colitis by modulating ER stress (Powell et al., 2020), a function also shared with CPEB4 (Maillo et al., 2017).
In many cases, inflammation predisposes to the development of cancer and promotes the different stages of tumorigenesis. Thus, patients suffering from longstanding IBD have increased risk for developing CRC (Lasry et al., 2016). However, only around 2% of CRCs are preceded by intestinal inflammation, which can also suppress tumorigenesis and promote anti-tumoral effects (Ullman and Itzkowitz, 2011). How cancer-associated inflammation develops, interacts with, and modulates tumor microenvironment will determine its tumor immunosupportive or immunosuppressive role (Greten and Grivennikov, 2019; Shalapour and Karin, 2019). CPEB4 KO mice have reduced CAC incidence despite showing defects to repair the damaged intestinal epithelia and more sustained inflammation. CPEB4 has been found to be overexpressed in samples from whole (including tumoral niche) CRC (Zhong et al., 2015) and associated with poor prognosis (Cortes-Guiral et al., 2017; He et al., 2017). Because pancreatic ductal adenocarcinoma, glioblastoma, and melanoma require high levels of CPEB4 in a tumoral cell autonomous manner to proliferate when xenografted in immune-incompentent mice (Ortiz-Zapater et al., 2011; Perez-Guijarro et al., 2016), these previous observations were interpreted as a pro-tumoral function for CPEB4 in CRC. However, here we show that the increased levels of CPEB4 in CRC take place in the immune niche and not in the tumoral epithelia. Accordingly, epithelial-specific deficiency of CPEB4 does not compromise CAC development or resolution of colitis. As was the case in inflammation, the phenotype of CPEB4 depletion in CAC is again consistent with the impaired translation of IL-22. Thus, Th17 cells and Th17 cytokines, such as IL-17A, IL-21 and IL-22, have been described to mainly be pro-tumorigenic in the context of CRC (Doulabi et al., 2018; Kryczek et al., 2014; Perez et al., 2020). Consistently, continued production of IL-22 by Th17 cells increases the risk of CRC in patients with an extended history of IBD (Mizoguchi et al., 2018) and in mouse models of CAC (Huber et al., 2012). The dual functions of Th17 and ILC3 produced cytokines, IL-17A, IL-17F and IL-22, appear to be defined by the temporal regulation of their expression. Thus, short term IL-22 production protects against genotoxic stress (Gronke et al., 2019), whereas uncontrolled IL-22 activity promotes tumor growth (Huber et al., 2012). A deregulation of this temporal control in the absence or upon chronic expression of CPEB4 would be consistent with the phenotypes of CPEB4 KO mice in intestinal homeostasis, inflammation, and tumorigenesis. Thus, we found that global CPEB4 deficiency exacerbates acute colitis but reduces tumorigenesis, suggesting that CPEB4 supports a tumor-promoting adaptive immunity. This, at first sight, counterintuitive phenotype can be explained by the regulation that we found of Th17-IL-22 response by CPEB4.
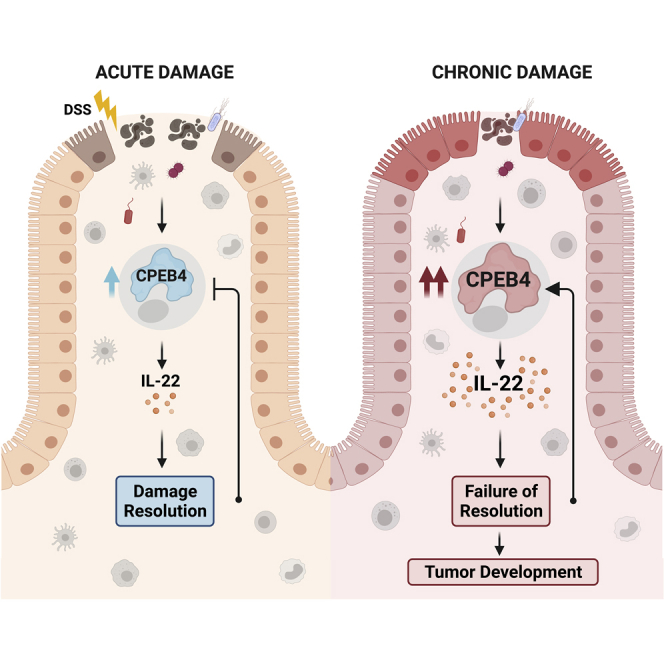
Taken together, these findings demonstrate that the CPEB4-Th17/ILC3 cells-IL-22 circuit regulates intestinal inflammatory responses and colorectal cancer development. Our results are consistent with a model where CPEB4 provides temporal control of IL-22 expression. Thus, CPEB4 would be transiently overexpressed in Th17 and ILC3 cells upon intestinal barrier damage, upregulating IL-22 expression and promoting tissue repair and acute transient inflammation. Chronic damage, as in Crohn’s disease and UC, would force continuous overexpression of CPEB4 and IL-22 synthesis, exacerbating intestinal pathology and promoting tumor growth (Kamanaka et al., 2011).
Limitations of the study
Despite the fact that CPEB4 depletion phenocopies IL22 dysregulation, suggesting that in colon mucosa homeostasis Il-22 mRNA is the most relevant target, CPEB4 potentially regulates hundreds of transcripts. Defining how much of the CPEB4 KO phenotype is mediated by lack of IL22 synthesis will require a rescue of the CPEB4 KO with ectopic IL22 expression. However, this rescue will need to consider the temporal window of expression of IL22, presumably defined by CPEB4, as constitutive interleukin expression is often as deleterious as its lack of expression.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-F4/80 | Thermo Fisher Scientific | Cat#14-4801; RRID:AB_2314387 |
| Rabbit polyclonal anti-MPO | Dako | Cat#A0398; RRID:AB_2335676 |
| Rabbit polyclonal anti-cleaved caspase-3 | Cell Signaling | Cat#9661; RRID:AB_2341188 |
| Flex Rabbit polyclonal Anti-CD3 | Dako-Agilent | Cat#IR503; RRID:AB_273200 |
| Rabbit polyclonal anti-Granzyme B | Abcam | Cat#ab4059; RRID:AB_304251 |
| Mouse anti-Ki67 | Novacastra, NCL | Cat#ACK02; N/A |
| Mouse monoclonal anti-CPEB4 149C/D10 | Homemade | N/A |
| Rabbit polyclonal anti-CPEB4 | Abcam | |
| Mouse monoclonal anti-vinculin | AbCam | Cat#ab130007; RRID:AB_11156698 |
| Mouse monoclonal anti-b-Actin | AbCam | Cat#ab8224; RRID:AB_449644 |
| HRP conjugated anti-rabbit | ImmunoLogic | Cat#DPVR110 |
| Goat Anti-Mouse Immunoglobulins/HRP antibody | Dako-Agilent | Cat#P0447; RRID:AB_2617137 |
| Polyclonal Rabbit Anti- Rat Immunoglobulins antibody | Dako-Agilent | Cat#P0450; RRID:AB_2630354 |
| TruStain FcX(TM) (anti-mouse CD16/32) antibody | Biolegend, Palex | Cat#101320; RRID:AB_1574975 |
| CD326 (EpCAM)-FITC, mouse (clone: caa7-9G8) | Miltenyi Biotec | Cat#130-102-995;RRID:AB_2657514 |
| PE/Cyanine5 anti-mouse CD45 (clone 30-F11) | Biolegend | Cat#103110;RRID:AB_312975 |
| CD31-APC, mouse (clone: 390) | Miltenyi Biotec | Cat#130-102-978; RRID:AB_2657310 |
| CD140a-PE, mouse (clone: REA637) | Miltenyi Biotec | Cat#130-109-736; RRID:AB_2655071 |
| CD3e Monoclonal (145-2C11) | ThermoFisher | Cat#MA5-17655;RRID:AB_2539045 |
| CD28 Monoclonal Antibody (CD28.6) | eBioscience | Cat#16-0288-85, RRID:AB_468925 |
| CD3ε-PE, mouse (clone: 145-2C11) | Miltenyi Biotec | Cat#130-102-792; RRID:AB_2660393 |
| PerCP/Cy5.5 anti-mouse CD3ε (clone 145-2C11) | Biolegend/palex | Cat#100327;RRID:AB_893320 |
| CD4 Monoclonal (clone GK1.5), APC-eFluor 780 | ThermoFisher | Cat#47-0041-80; RRID:AB_11219883 |
| CD4-FITC, mouse (clone GK1.5) | Miltenyi Biotec | Cat#130-102-779; RRID:AB_2659901 |
| CD8a-APC, mouse (clone: REA601) | Miltenyi Biotec | Cat#130-109-326; RRID:AB_2659493 |
| CD19 VioBlue, mouse Clone: 6D5 | Miltenyi Biotec | Cat#130-103-139;RRID:AB_2661115 |
| CD19-FITC, mouse (clone: 6D5) | Miltenyi Biotec | Cat#130-102-822;RRID:AB_2661107 |
| PE/Cy7 anti-mouse/human CD11b, (clone: REA592) | Biolegend/palex | Cat#101215; RRID:AB_312798 |
| CD11b-PE, mouse (clone: REA592) | Miltenyi Biotec | Cat#130-109-363; RRID:AB_2654644 |
| CD11b-APC-Vio770, human and mouse, (clone: REA592) | Miltenyi Biotec | Cat#130-098-089; RRID:AB_2660134 |
| FITC Rat Anti-Mouse CD90.2 Clone 53-2.1(RUO) | BD biosciences | Cat#553003;RRID:AB_394542 |
| CD127 Monoclonal Antibody (A7R34), PE-Cyanine5 | ThermoFisher | Cat#15-1271-81; RRID:AB_468792 |
| CD49b (DX5)-PE Mouse | Miltenyi Biotec | Cat#130-123-702; RRID:AB_2811545 |
| APC anti-mouse CD335 (NKp46) Clone 29A1.4 | Biolegend/palex | Cat#137607;RID:AB_10612749 |
| V450 Mouse Lineage Antibody Cocktail, with Isotype Control | BDBiosciences | Cat#561301;RRID:AB_10611731 |
| Anti-Gr-1 VioBlue, mouse Clone: RB6-8C5 | Miltenyi Biotec | Cat#130-102-830;RRID:AB_2659864 |
| Anti-TCRγ/δ VioBlue, mouse Clone: GL3 | Miltenyi Biotec | Cat#130-104-046; RRID:AB_2654081 |
| CD19-VioBlue, mouse Clone: 6D5 | Miltenyi Biotec | Cat#130-103-139, RRID:AB_2661115 |
| CD8a-VioBlue, mouse Clone: 53-6.7 | Miltenyi Biotec | Cat#130-102-804, RRID:AB_2659888 |
| CD44-APC, mouse Clone IM7 | Miltenyi Biotec | Cat#130-110-118, RRID:AB_2658154 |
| CD25-PE-Vio770, mouse (clone: REA568) | Miltenyi Biotec | Cat#130-112-836, RRID:AB_2658858 |
| CD62L-PE, mouse (clone: REA828) | Miltenyi Biotec | Cat#130-112-836, RRID:AB_2658858 |
| Chemicals, peptides, and recombinant proteins | ||
| Dextran Sodium Sulfate (MW: 40,000) | Cymitquimica | Cat#02-J63606 |
| Azoxymethane | Sigma-Aldrich | Cat#A2853; CAS 25843-45-2 |
| Percoll®GE Healthcare | Vwr | Cat#17-0891-01 |
| TRIzol® Reagent | Life technologies | Cat#15596026 |
| Trisure | Ecogen | Cat#BIO-38033 |
| 4′,6-diamidino-2-phenylindole (DAPI) | Sigma-Aldrich | Cat#D9542; CAS 28718-90-3 |
| Recombinant Human IL-23 | Peprotech | Cat#200-23 |
| Recombinant Human IL-6 | Peprotech | Cat#200-06 |
| Recombinant Murine IL-1β | Peprotech | Cat#211-11B |
| 4-Hydroxytamoxifen (4-OHT) | Sigma-Aldrich | Cat#H6278; CAS 68047-06-3 |
| Critical commercial assays | ||
| Tumor Dissociation Kit, mouse | Miltenyi Biotec | Cat#130-096-730 |
| Lamina Propria Dissociation Kit | Miltenyi Biotec | Cat#130-097-410 |
| Dynabeads-Flowcomp mouse CD4 kit | Invitrogen | Cat#11461D |
| Murine IL-22 Mini ABTS ELISA Development Kit | Peprotech | Cat#900-M257 |
| Dynabeads Protein A | Invitrogen | Cat#10001D |
| Turbo DNA-free Kit | Ambion | Cat#AM1907 |
| SYBR green master mix | ThermoFisher | Cat#A25778 |
| SuperScript® IV First-Strand Synthesis System | Invitrogen | Cat#18091200 |
| Experimental models: Cell lines | ||
| Naïve CD4+ T cells | This paper | N/A |
| Total CD4+ T cells | This paper | N/A |
| Experimental models: Organisms/strains | ||
| Mouse: CPEB4 total body knockout (C57Bl6JxSv129, mostly in C57BL/6 background) | This paper | N/A |
| Mouse: CPEB4lox/lox mice | Maillo et al. (2017) | N/A |
| Mouse: Villin-Cre ERT2 | Angel Nebreda, IRB Barcelona | JAX: 020282 |
| Mouse: LCK-Cre | Balbino Alarcón, CBMSO-Madrid | JAX: 003802 |
| Oligonucleotides | ||
| Primers for XX, see Table S1 | This paper | N/A |
| Software and algorithms | ||
| BD-FACSDIVA | BD Biosciences | https://www.bdbiosciences.com/en-eu |
| FlowJo | N/A | https://www.flowjo.com/solutions/flowjo; RRID:SCR_008520 |
| Image J | N/A | https://imagej.net/; RRID:SCR_003070 |
| QuPath | N/A | https://qupath.github.io/; RRID:SCR_018257 |
| T Marker | Schüffler et al. (2013) | http://thomasfuchslab.org/tmarker |
| NDP Scan | Hamamatsu | https://www.hamamatsu.com/eu/en/product/type/U12388-01/index.html |
| GraphPad Prism software, version 7.0c | N/A | https://www.graphpad.com; RRID:SCR_002798 |
| Other | ||
| Accession number of existing, publicly available data analyzed, see Table S2 | This paper | N/A |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Raul Mendez (raul.mendez@irbbarcelona.org).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Mice
Weight- and gender-matched CPEB4 total body knockout mice (genetic background C57Bl6JxSv129, mostly in C57BL/6 background) were used at an age of 8–14 weeks for all experiments. Intestinal-specific Cpeb4 knockout mice (CPEB4KO IEC) were obtained by crossing Cpeb4lox/lox mice (described in (Maillo et al., 2017)) with Villin-Cre ERT2 transgenic animals (Angel Nebreda, IRB-Barcelona). To induce CPEB4 depletion, the animals were intraperitoneally injected with 4-hydroxy tamoxifen (10 mg/ml) for 5 days. CPEB4 depletion was confirmed by western blotting. T cell-specific Cpeb4 knockout mice (CPEB4 TKO) were generated by crossing LCK-CreT/+ transgenic animals (Balbino Alarcón, CBMSO-Madrid) with Cpeb4lox/lox animals. Mice were given free access to food and water and maintained in individually ventilated cages under specific-pathogen-free conditions. All experimental protocols were approved by the Animal Ethics Committee at the Parc Cientific deBarcelona.
Human dataset analysis
Association and correlation studies were performed with GEO public datasets of colorectal cancer and inflammatory bowel disease samples, as described below.
Method details
Dextran sodium sulfate (DSS)-induced colitis mouse model
3Mice were given 2.5% or 3% dextran sodium sulfate (as specified in Figure legends) (DSS, molecular weight: 40,000; Cymit) in their drinking water for 5 days, followed by regular drinking water. The mice were weighed every day and sacrificed at the indicated time points. Colons were removed, flushed with cold phosphate-buffered saline (PBS), opened longitudinally, fixed as ‘‘swiss-rolls’’ in 10% formalin solution or frozen for further analysis. Sections from the ascending colon of each mouse were analyzed using hematoxylin and eosin (H&E) staining.
Induction of colitis-associated colorectal cancer (CAC)
To induce colorectal tumors, we used a combination of the carcinogen azoxymethane (AOM) with repeated administration of DSS in the drinking water, which causes colitis (Gupta et al., 2014). Mice (8–10 weeks old) were injected intraperitoneally with a single dose of AOM (10 mg/kg; Sigma, #A2853). After 5 days, 2.5% DSS was given in the drinking water for 5 days, followed by 14 days of regular drinking water. DSS treatment was repeated for two additional cycles, and mice were sacrificed 100 days after AOM injection. Colons were removed, flushed with cold PBS, and opened longitudinally. Colon tumors were counted macroscopically. Then, the colons were fixed and prepared as ‘‘swiss-rolls’’ in 10% formalin solution for histopathological analysis or collected and flash-frozen for further RNA and protein analysis. When indicated, the tumors were processed for FACS analysis.
Histopathological analysis
Colitis
A qualitative and quantitative histological assessment of H&E-stained colon slides was performed. Colitis severity was assessed by a combined score of colon cellular infiltration (0–4, according to the number and localization of inflammatory cells), tissue disruption (0–4, according to the severity of mucosal and crypt damage), depth of injury (0–4) and crypt damage (0–4) as described previously (Bai et al., 2010). Histological scoring was performed in a blinded fashion. Final scores were obtained by summing individual lesions multiplied by the tissue disruption score.
Colitis-associated colorectal cancer (CAC).
Tumor grades were scored using a two-tiered system based on up-to-date WHO guidelines for tumors of the digestive system. Briefly, adenomas with low-grade dysplasia consist of stratified dysplastic epithelium retaining its columnar shape, with the nuclei in the basal part of epithelium, whereas adenomas with high-grade dysplasia have lost their columnar shape and the nuclei are located mainly in the surface of the crypts. The Mitotic Index was scored (low-medium-high) as a measurement of the proliferation rate, and inflammation was classified from none to very intense (score 0-4). Infiltration score of immunohistochemistry stainings of AOM-DSS tumors was semiquantitative and considered intensity (0, undetectable; 1, very low; 2, low; 3, moderate; 4, intense) and ratio of positive stained /unstained cells (0, 0-10%; 1, 11-30%; 2, 31-60%; 3, 61-100%).
Immunohistochemical analysis
Immunohistochemical analysis of H&E was conducted on formalin-fixed paraffin-embedded tissues by an IRB Barcelona pathologist in blinded fashion for tumor grade, epithelial damage, and inflammation using the scoring systems described above. Paraffin-embedded colon sections were stained with the following antibodies against Ki67 (Novacastra, NCL; 1:500, 1hat RT), F4/80 (eBiosciences #14-4801; 1:50, 2hat RT), MPO (Dako #A0398; 1:1000, 30 min at RT), cleaved caspase-3 (Cell Signaling #9661; 1:200, 1hat RT), CD3 ((IR503, Dako-Agilent; 1:10, 2hat RT), Granzyme B ((ab4059, Abcam; 1:1000, 2hat RT) and CPEB4 (149C/D10, monoclonal homemade). The secondary antibodies were HRP conjugated anti-rabbit (ImmunoLogic #DPVR110 HRP, 45 min at RT), anti-mouse (Dako #P0447; 1:100, 30 min at RT) and anti-Rat (Dako #P0450; 1:75, 30 min at RT). Measurements were performed in slides scanned using a Hamamatsu Nanozoomer SQ digital slide scanner running NDP Scan software. The quantification was performed by TMarker software, using a color deconvolution plugin counting positive cell nuclei of total nuclei number, and by QuPath software, quantitative pathology & bioimage analysis.
Preparation of cell suspension
Thymus, lymph nodes and spleen were isolated and mechanically dissociated through 70-μm filters. To remove erythrocytes or dead cells, blood, thymus, lymph nodes and spleen were incubated in Red Blood Cell Lysis Solution for 10 min at room temperature, followed by centrifugation for 10 min at 400g. Cell pellets were counted by TC20 Automated Cell Counter (Biorad) and used for further analysis. Tumor cell suspensions were prepared using Tumor Dissociation Kit, mouse (Miltenyi Biotec #130-096-730). The tumors were cut into small pieces of 2–4 mm. 2.5 ml of digestion media was added to each tube, and digestion to single cell suspension was performed using GentleMACS Dissociator on program ‘37C_m_TDK_2’. Colons were isolated, opened longitudinally, washed with 10 mM HEPES in HBSS solution and cut into pieces of approximately 0.5 cm in length. These pieces were incubated in 20 ml of predigestion solution containing 10 mM HEPES, 5 mM EDTA, 5% fetal bovine serum (FBS), and 1 mM DTT in HBSS under continuous rotation at 37°C for 20 min. They were then passed through a 100-μm filter and treated with a second round of predigestion solution. The flow-throughs were combined and centrifuged at 400g for 10 min to isolate intestinal epithelial cells (IECs). Remaining lamina propria tissues were digested using the Lamina Propria Dissociation Kit (Miltenyi Biotec #130-097-410), following the manufacturer’s protocol. 2.5 ml of media containing digestion enzymes was added to each tube and digestion to single cell suspension was achieved using GentleMACS Dissociator on program ‘37C_m_LPDK_1’. Digested tissues were then passed through a 70-μm cell strainer. Specifically, lamina propria lymphocytes were isolated by resuspending the cell suspension in 8 ml of 30% Percoll, underlined with 5 ml of 80% Percoll, and centrifuged at 900g for 25 min with no brake. Lymphocytes were isolated from the interface and resuspended in HBSS containing 0.5% FBS for further analyses.
Flow cytometry and cell sorting
Flow cytometry analysis and cell sorting were performed using a FACS Aria flow cytometer (BD-Biosciences).Data analysis was performed using BD-DIVA Software. Cell suspensions were incubated with purified anti-CD16/32 antibody (TruStain fcX™, Palex #101320) for 10 min on ice to block Fc receptors and then incubated with specific antibodies for 15 min on ice. Live-dead labelling with 4′,6-diamidino-2-phenylindole (DAPI) (Sigma) was performed, and only live cells were analyzed.
Flow cytometry stainings and cell sorting strategies
Tumor cell suspensions were stained with Epcam (CD326), anti-CD45 (30-F11), anti-CD31 (390), anti-CD140a, anti-CD3ε (145-2C11), anti-CD4 (GK1.5), and anti-CD8α (REA601). Lamina propria cell suspensions were stained with anti-CD45 (30-F11), anti-CD3ε (145-2C11), anti-CD4 (GK1.5), anti-CD8α (REA601), anti-CD19 (6D5), and anti-CD11b (M1/70). Blood, spleen and lymph nodes cells suspensions were stained with anti-CD45 (30-F11), anti-CD3ε (145-2C11), anti-CD4 (GK1.5), anti-CD8α (REA601), and anti-CD19 (6D5). Lamina propria innate lymphoid cells (ILC3) were stained with anti-CD45 (30-F11), anti-CD90.2 (53-2.1), anti-CD127 (A7R34), anti-CD49b (DX5), and anti-CD335 (29A1.4). Lin. Mouse Lineage Antibody Cocktail (Lin)(561301, BD Biosciences) was composed of the following: anti-CD3e (clone 500A2); anti-CD11b (M1/70); anti-CD45R/B220 (RA3-6B2); anti-Ly-76 (Ter119); and anti-Ly6G/Ly6C (RB6-8C5). Additional markers, namely anti-Gr1 (RB6-8C5), anti-CD19 (6D5), anti-CD8α (REA601), and anti-TCR©™ (GL3), were used in the mix lineage. ILC3 were sorted as lymphoid tissue initiator cells Lti (CD45+Thy1.2+Lin-CD127+CD4-DX5-NKP46-) and natural cytotoxicity receptor NCR+ (CD45+Thy1.2+Lin-CD127+CD4-DX5-NKP46+). Naïve CD4 T cells were stained with anti-CD44 (IM7), anti-CD4 (GK1.5), anti-CD25 (PC61.5), and anti-CD62L (MEL-14). Antibodies were purchased from Biolegend, BD Biosciences, and Miltenyi Biotec.
T-cell differentiation in vitro
Naïve CD4+T cells were isolated from spleen and lymph node suspensions from 8-week-old mice by sorting as CD4+CD25-CD44-CD62L+. They were stimulated with plate-bound antibody against CD3 (145-2C11, 2 μg/ml) and soluble anti-CD28 antibody (CD28.6, 0.5 μg/ml) in the presence of IL-23 (50 ng/ml; 200-23 Peprotech) for 72 h. For RIP and Th17 differentiation experiments, total CD4+T cells were isolated from the spleen and lymph nodes by dynabeads-Flowcomp mouse CD4 kit (11461D, Invitrogen). Th17 differentiation was achieved by stimulating the cells with anti-CD3 (2 μg/ml) and anti-CD28 (0.5 μg/ml), IL-23 (50 ng/ml), IL-6 (20 ng/ml), and IL-1b (10 ng/ml) for 96 h. RPMI-based medium was supplemented with sodium pyruvate (1 mM), L-glutamine (1 mM), penicillin (100 U/ml), streptomycin (100 U/ml), 10% heat-inactivated FBS (BioWhittaker, Inc.) and 50 μM of 2-mercaptoethanol.
Colon culture
Colons were washed in cold PBS supplemented with penicillin and streptomycin (Gibco) and cut longitudinally. Four segments of 0.5 cm length were cultured in 24-well flat-bottom culture plates in RPMI-1640 medium (Gibco) for 24hat 37°C. Supernatants were analyzed for IL-22 concentration using an IL-22 ELISA kit and following the manufacturer’s protocol (PeproTech).
Cytokine analysis
Secreted IL-22 was measured in tumor extracts, DSS-treated mucosa lysates, supernatants of IL-23-stimulated CD4+T cells and supernatants of colon culture samples by using the Murine IL-22 Mini ABTS ELISA Development kit (900-M257, Peprotech), following the manufacturer’s protocol.
RNA isolation and RT-qPCR
Total RNA was extracted from frozen and pulverize colon segments and tumors, or sorted cells, using TRIzol (Invitrogen) or TRisure (Bioline, Ecogene), following the manufacturer’s protocol. For sorted ILC3 cells, total RNA was extracted by PicoProfiling. RNA concentration was determined using Nanodrop Spectrophotometer (Nanodrop Technologies). Total RNA (0.5-5 μg) was reverse transcribed using a Super script IV Reverse Transcriptase (Invitrogen #18091200). qPCR was performed in triplicates using from 2.5 to 10 ng of cDNA and SYBR green master mix (ThermoFisher #A25778) in a total volume of 10 μl on a Bio-Rad C1000 thermal cycler machine. Relative quantities (Δ cycle threshold values) were obtained by normalizing against gapdh, hprt or tbp housekeeping genes, depending on the experiment. The primers used are listed in the Table S1.
RNA immunoprecipitation-qPCR
Th17 WT T lymphocytes stimulated for 96 h, as described above, were cross-linked with PBS 0.5% formaldehyde for 5minat room temperature under constant gentle agitation. The crosslinking reaction was quenched by glycine to a final concentration of 0.25 M for 5 min. Cells were washed twice with 10 ml cold PBS, lysed with RIPA buffer (25 mM Tris-Cl pH7.6, 1% Nonidet P-40, 1% sodium deoxycholate, 0.1% SDS, 100 mM EDTA, 150 mM NaCl, protease inhibitor cocktail, RNase inhibitors), and sonicated for 5minat low intensity with Standard Bioruptor Diagenode. After centrifugation (5 min, max speed, 4°C) supernatants were collected, precleared, and immunoprecipitated (ON, 4°C on rotation) with 10 μg of anti-CPEB4 monoclonal antibody (clone 149C/D10, homemade) or normal mouse IgG polyclonal antibody (12-371 Sigma), previously conjugated with 50 μl of Dynabeads Protein A (Invitrogen). Beads were washed 4 times with cold RIPA buffer supplemented with Protease inhibitors (Sigma-Aldrich). They were then resuspended in 100 μl Proteinase-K buffer with 70 ug Proteinase-K (Roche), and incubated for 60minat 65°C. RNA was extracted by standard phenol-chloroform extraction, followed by Turbo DNA-free Kit (AM1907, Ambion) treatment. All recovered RNA was retrotranscribed and qPCR was performed.
Western blotting
Colon tissues or colon tumors were extracted, frozen and pulverize using a liquid nitrogen-cooled mortar. The same amount of powder samples was lysed in buffer containing 1% NP40, 150 mM NaCl, 50 mM Tris HCl pH 7.5, 2 mM EDTA, 2 mM EGTA, 20 mM sodium fluoride, 2 mM PMSF, 2 mM sodium orthovanadate, 1 mM DTT and 1x EDTA-free complete protease inhibitor cocktail (Roche, #11873580001), and sonicated at high intensity for 5 min with Standard Bioruptor Diagenode. Protein content was quantified using DC Protein Assay (Biorad), and 30 μg of total protein lysate in Laemmli buffer was separated on SDS-PAGE and transferred to nitrocellulose membrane (GE10600001, Sigma). After blocking (5% non-fat milk and 1% BSA in PBS, 1hat room temperature) membranes were incubated at 4°C overnight with anti-CPEB4 (Abcam) or anti-CPEB4 monoclonal (149C/D10, homemade) primary antibodies or at room temperature for 1 h with anti-vinculin (ab130007, AbCam) and anti-b-Actin (ab8224, AbCam) primary antibodies. After incubation with secondary antibodies for 1hat room temperature, the membranes were incubated with Amersham ECL™ Western Blotting Detection Reagents (GERPN2106, Sigma).
Human dataset analysis
CRC transcriptomic metacohort –dataset integration
Public CRC transcriptomic datasets were downloaded from GEO (Barrett and Edgar, 2006) and NCI GDC commons (Grossman et al., 2016), and were pre-processed and integrated in order to create a unique metacohort for analysis. This metacohort included 1705 tumor samples from the following 6 datasets: TCGA (Cancer Genome Atlas, 2012); GSE33113 (Kemper et al., 2012); GSE14333 (Jorissen et al., 2009); GSE39582 (Marisa et al., 2013); GSE38832 (Tripathi et al., 2014); andGSE44076 (Sanz-Pamplona et al., 2014). Importantly, three GEO datasets and the colon TCGA data (TCGA-COAD) include disease-free survival information with a median follow-up ranging from 2.3 to 5.4 years. Following the TCGA PanCanAtlas Clinical Data Resource (TCGA-CDR) guidelines (Liu et al., 2018), TCGA-COAD data were included in the disease-free survival analyses despite their short available follow-up. Rectum TCGA samples (TCGA-READ) were excluded from these analyses following these same guidelines. Other clinical information commonly available across datasets were gender, age, stage, and location of primary tumor. When not available, Microsatellite Instability (MSI) status was imputed using the transcriptomic signature reported by Jorissen et al. (2008) and the methodology described below (section MSI imputation in transcriptomic CRC samples). Datasets were processed separately (see sections CRC transcriptomic metacohort – Processing of TCGA data and CRC transcriptomic metacohort – Pre-processing of microarray datasets) and merged after gene-wise standardization (median and median absolute deviation) to the GSE39582 dataset, according to their distribution of gender, age, tumor site, MSI and stage, when available. Such standardization was carried out by inverse probability weighting using a logistic regression model. To this end and for each dataset D, a model was fitted on D and GSE39582, which included the aforementioned clinical parameters as predictors of the dataset affiliation. Clinical parameters were considered in the model as they were informative according to the Akaike information criterion (AIC) after running a forward step-wise algorithm for variables selection. Next, conditional probabilities of membership to datasets D and GSE39582 (Pd and Pr, respectively) were retrieved from the model. Weights were then computed as the ratio of Pd/Pr and finally truncated to value “five”, in order to avoid the overrepresentation of any one sample in the standardization process. Expression values were truncated to the maximum and minimum values observed in this reference dataset.
CRC transcriptomic metacohort – processing of TCGA data
RNA-Seq (version 2) data from the The Cancer Genome Atlas (TCGA) project were retrieved from the legacy version of the GDC commons repository (Grossman et al., 2016). Clinical and follow-up information was retrieved from the TCGA-CDR (Liu et al., 2018). Expression measures were expressed in RSEM (Li and Dewey, 2011) in this data version, which were log2-transformed and quantile normalized. Data from different tumor locations (colon and rectum) and platforms (Genome Analyzer and HiSeq) were processed separately. Non-primary tumor samples were filtered out, and duplicated samples measured in the different platforms were excluded from the HiSeq subset. Only primary tumors from patients with no previous cancer diagnosis were kept for analyses. An exploratory analysis was carried out using Principal Component Analyses in order to identify samples with abnormal expression values in each dataset. Samples TCGA-A6-2679-01A and TCGA-AA-A004-01A were excluded as their gene expression showed an anomalous distribution compared to the rest of samples in their dataset, even after quantile normalization. Finally, each dataset was corrected gene-wise by technical variation using a mixed-effect linear model as implemented in the lme4 R package, in which the center of origin and plate identifiers of each sample were modeled as a fixed and a random effect, respectively. Coefficients from the imputations of the fixed effects and random effects provided by the models were then used to correct these technical effects.
CRC transcriptomic metacohort – pre-processing of microarray datasets
Microarray datasets were processed separately using R packages affy and affyPLM from Bioconductor (Huber et al., 2015). Raw cel files data were normalized using RMA (Irizarry et al., 2003) and annotated with information available on the Affymetrix web page. Standard quality controls were performed to identify abnormal samples regarding: a) spatial artifacts in the hybridization process (scan images and pseudo-images from probe level models); b) intensity dependences of differences between chips (MvA plots); c) RNA quality (RNA digest plot); d) global intensity levels (boxplot of perfect match log-intensity distributions before and after normalization and RLE plots); and e) anomalous intensity profile compared to the rest of samples (NUSE plots, Principal Component Analyses). Technical information concerning sample processing and hybridization was retrieved from the original CEL files: scanning dates were collected in order to define scan batches in each dataset separately. The technical metrics described by Eklund AC and Szallasi Z (Eklund and Szallasi, 2008) were computed and recorded as additional features for each sample. Microarray expression values were summarized at the gene level (entrez) using the first principal component of the probesets mapping to the same gene. These components were centered and scaled to the weighted means of the means and standard deviations of their probesets using the corresponding contribution to the component as weight. The sign of this gene-based expression summary was imputed so that it was congruent to the sign of the probeset contributing the most to the original first component. Before gene-level summarization, each microarray dataset was corrected a priori by Eklund-Szallasi metrics (Eklund and Szallasi, 2008) effects, as estimated from a standard linear model. In addition, mixed-effect linear models were used to correct microarray expression by the sample’s center of origin (if more than one) and date of scanning. Expression values were corrected using the coefficients from the fixed effects and the imputations of the random effects provided by the models.
CRC transcriptomic metacohort – MSI imputation
When not available in the published clinical information, MSI status was imputed in each dataset separately on the basis of the expression of genes included in a published transcriptomic signature (Jorissen et al., 2008). To this end, Pearson’s correlation coefficients were computed between the profile of each sample and an artificial MSI profile consisting of “one” values for genes included in the signature that were overexpressed in MSI samples and “zero” values for those overexpressed in MSS compared to MSI tumors. Assignation to MSI or MSS was performed according to results of a cluster analysis based on non-parametric density estimation (DeBin and Risso, 2011) applied to these correlation coefficients. This imputation procedure was performed separately for each dataset at the gene level using the entrez identifier and Affymetrix annotation before correction by technical effects. To this end, not-corrected microarray datasets were summarized at the gene level by the most variable probeset mapping to the same gene, as measured by the median absolute deviation. In each case, the same number of top MSI and MSS overexpressed genes were used in order to avoid biases towards the group that included more genes (MSI). Accuracy of this imputation was evaluated in dataset GSE39582 and the TCGA colon dataset, which included annotation of MicroSatellite-Stable (MSS) and-Instable (MSI) samples in their clinical information (GSE39582: 97% and 77% accuracy for MSS and MSI samples, respectively; TCGA colon: 97% and 92% accuracy for MSS and MSI, respectively).
Transcriptomic datasets of tumoral cell populations
Three GEO (Barrett and Edgar, 2006) datasets, namely GSE39395, GSE39396 (Calon et al., 2012) and GSE35602 (Nishida et al., 2012), were used to examine the contribution of the tumor microenvironment to the expression of CPEB4 in CRC. In GSE39395 and GSE39396, FACS was used to separate the following populations from 14 fresh human CRC samples: CD45+EpCAM−CD31−FAP−, CD45−EpCAM+CD31−FAP−, CD45−EpCAM−CD31+FAP− and CD45−EpCAM− CD31−FAP+. GSE35602 contains transcriptomic human data for epithelial and stromal cells microdissected from 13 CRC tissue samples and 4 adjacent morphologically normal colorectal mucosae (>5 cm from the tumor). GSE39395 and GSE39396 were processed using RMA (Irizarry et al., 2003) as described above (see section Pre-processing of microarray datasets) and integrated in a single gene expression matrix using a gene-wise linear model in which the sample’s cell population was included as covariate. Regarding GSE35602, the series matrix originally provided by their authors was used in the analyses.
Th17 gene signature
The activation status of T helper 17 (Th17) cells was measured using a gene expression signature published by Keerthivasan S et al. (Keerthivasan et al., 2014). Genes included in the signature were retrieved from the Synapse repository (Derry et al., 2012) (Synapse identifier syn2321865). To obtain a measure of the gene signatures’ expression activity in a transcriptomic dataset, z-scores were computed for each gene and each sample in the dataset, which were then averaged across all genes included in that signature. The resulting score was in turn centered and scaled across samples.
Association analyses
Association analyses with prognosis in human transcriptomic datasets were assessed using a Cox proportional hazards model. For the rest of the analyses of association with gene expression levels, linear models were used. When suitable (CRC transcriptomic metacohort, GSE39395 and GSE39396), the sample’s dataset or origin was included in the models for statistical adjustment. In addition, and for evaluation of the Th17 gene expression profile, a global signature was computed using all the genes in the expression matrix and used as adjusting variable in the models. This strategy has proven useful to avoid systematic biases caused by the gene-correlation structure present in the data and to adjust for the expectation under gene randomization, i.e., the association expected for a signature whose genes have been chosen at random. Statistical significance was assessed by means of Log-likelihood Ratio Test (LRT, for Cox models) and F-test (linear models), while Wald tests were used for pair-wise comparisons. Sample groups of low, medium, and high expression levels were defined using terciles as cut-off points, which were computed across all the samples in the dataset. Hazard Ratios (HR) and partial Pearson’s correlation coefficients were computed as measures of association. For visualization purposes, Kaplan-Meier (KM) survival curves and boxplots were computed, while scatter plots were used to display correlation with gene expression levels. KM curves were adjusted by the sample’s dataset or origin using inverse probability weighting by means of a logistic regression model, in an analogous way to that described above for the integration of transcriptomic data (see section CRC transcriptomic metacohort – Datasets integration).
CPEB4 expression in patients with IBD
CPEB4 mRNA expression (arbitrary unit, au) was measured in mucosa of healthy human controls, and of patients with Crohn’s disease, noninflamed ulcerative colitis and inflamed ulcerative colitis, combining transcriptomic data from GEO public adult datasets with accession numbers: GSE13367, GSE16879, GSE59071, GSE9452. GSE10616 dataset was used for pediatric patients.
Quantification and statistical analysis
Data are expressed as mean ± standard error of the mean (SEM) unless otherwise specified. Data set statistics were analyzed using the GraphPad Prism software, version 7.0c. The significance of experimental differences was evaluated by an unpaired (2-tailed) Student’s t-test or a Mann Whitney test, when comparing two groups. Comparisons between more than groups were carried out with one-way and two-way ANOVA. Differences under p < 0.05 were considered statistically significant.
Acknowledgments
This work was supported by grants from the SpanishMinistry of Science, Innovation and Universities (BFU2017-83561-P to RM; SAF2017-87988-R to MF), and the Spanish Association Against Cancer (AECC) (GCB15152955MÉND), Worldwide Cancer Research Foundation (20-0284), World Cancer Research Fund International (2020_021), BBVA Foundation (BBVABIOMED/18), “la Caixa” Foundation (LCF/PR/HR19/52160020) and Fundacio Marato TV3 (201926-30-31) to RM and MF. Cpeb4 mouse model generation (2008–2012) was supported by the EU-funded Infrafrontier project.IRB and IDIBAPS are supported by the CERCA Program (Catalan Government). IRB is the recipient of a Severo Ochoa Award of Excellence from the Spanish Government. We thank the Biostatistics/Bioinformatics, Histopathology (especially the pathophysiologist Neus Prats and Monica Aguilera), Mouse Mutant, and Functional Genomics facilities at IRB Barcelona. The Flow Cytometry Facility of the UB/PCB and the CRG Genomic Unit are also acknowledged. We thank members of Dr. Raul Mendez’s laboratory for useful discussions.
Author contributions
AS performed all the studies and contributed to experimental design, data analysis and interpretation, wrote the manuscript and assembled the figures. CS performed part of the studies, contributed to all experimental design, data analysis and interpretation and helped with manuscript preparation. RM conceived, directed, and discussed the study, wrote the manuscript and obtained funding. MFA performed the experiments of lymphoid organs characterization. JM contributed to in vivo mouse experiments. AB performed all gene expression analysis of human cancer datasets. AC performed the analysis of IBD patients. VC performed the tissue immunohistochemistry. AMR, GFM, and MF discussed the results and contributed to the presented idea. EB and MF contributed to the final manuscript.
Declaration of interests
The authors declare that they have no conflict of interest
Published: February 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103790.
Supplemental information
Data and code availability
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Aden K., Rehman A., Falk-Paulsen M., Secher T., Kuiper J., Tran F., Pfeuffer S., Sheibani-Tezerji R., Breuer A., Luzius A., et al. Epithelial IL-23r signaling licenses protective IL-22 responses in intestinal inflammation. Cell Rep. 2016;16:2208–2218. doi: 10.1016/j.celrep.2016.07.054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexandrov I.M., Ivshina M., Jung D.Y., Friedline R., Ko H.J., Xu M., O'Sullivan-Murphy B., Bortell R., Huang Y.T., Urano F., et al. Cytoplasmic polyadenylation element binding protein deficiency stimulates PTEN and Stat3 mRNA translation and induces hepatic insulin resistance. PLoS Genet. 2012;8 doi: 10.1371/journal.pgen.1002457. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bai A., Yong M., Ma A.G., Ma Y., Weiss C.R., Guan Q., Bernstein C.N., Peng Z. Novel anti-inflammatory action of 5-aminoimidazole-4-carboxamide ribonucleoside with protective effect in dextran sulfate sodium-induced acute and chronic colitis. J. Pharmacol. Exp. Ther. 2010;333:717–725. doi: 10.1124/jpet.109.164954. [DOI] [PubMed] [Google Scholar]
- Bando J.K., Colonna M. Innate lymphoid cell function in the context of adaptive immunity. Nat. Immunol. 2016;17:783–789. doi: 10.1038/ni.3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett T., Edgar R. Gene expression omnibus: microarray data storage, submission, retrieval, and analysis. Methods Enzymol. 2006;411:352–369. doi: 10.1016/S0076-6879(06)11019-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calderone V., Gallego J., Fernandez-Miranda G., Garcia-Pras E., Maillo C., Berzigotti A., Mejias M., Bava F.A., Angulo-Urarte A., Graupera M., et al. Sequential functions of CPEB1 and CPEB4 regulate pathologic expression of vascular endothelial growth factor and angiogenesis in chronic liver disease. Gastroenterology. 2016;150:982–997 e30. doi: 10.1053/j.gastro.2015.11.038. [DOI] [PubMed] [Google Scholar]
- Calon A., Espinet E., Palomo-Ponce S., Tauriello D.V., Iglesias M., Cespedes M.V., Sevillano M., Nadal C., Jung P., Zhang X.H., et al. Dependency of colorectal cancer on a TGF-beta-driven program in stromal cells for metastasis initiation. Cancer Cell. 2012;22:571–584. doi: 10.1016/j.ccr.2012.08.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas N. Comprehensive molecular characterization of human colon and rectal cancer. Nature. 2012;487:330–337. doi: 10.1038/nature11252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carpenter S., Ricci E.P., Mercier B.C., Moore M.J., Fitzgerald K.A. Post-transcriptional regulation of gene expression in innate immunity. Nat. Rev. Immunol. 2014;14:361–376. doi: 10.1038/nri3682. [DOI] [PubMed] [Google Scholar]
- Coleman O.I., Haller D. ER stress and the UPR in shaping intestinal tissue homeostasis and immunity. Front.Immunol. 2019;10:2825. doi: 10.3389/fimmu.2019.02825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cortes-Guiral D., Pastor-Iodate C., Diaz Del Arco C., Del Puerto-Nevado L., Fernandez-Acenero M.J. CPEB4 immunohistochemical expression is associated to prognosis in stage IV colorectal carcinoma. Pathol. Res. Pract. 2017;213:639–642. doi: 10.1016/j.prp.2017.04.020. [DOI] [PubMed] [Google Scholar]
- D'Ambrogio A., Nagaoka K., Richter J.D. Translational control of cell growth and malignancy by the CPEBs. Nat. Rev. Cancer. 2013;13:283–290. doi: 10.1038/nrc3485. [DOI] [PubMed] [Google Scholar]
- De Bin R., Risso D. A novel approach to the clustering of microarray data via nonparametric density estimation. BMC Bioinformatics. 2011;12:49. doi: 10.1186/1471-2105-12-49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dempsey L.A. IL-22 in Peyer's patches. Nat. Immunol. 2017;18:715. doi: 10.1038/ni.3786. [DOI] [PubMed] [Google Scholar]
- Derry J.M., Mangravite L.M., Suver C., Furia M.D., Henderson D., Schildwachter X., Bot B., Izant J., Sieberts S.K., Kellen M.R., et al. Developing predictive molecular maps of human disease through community-based modeling. Nat. Genet. 2012;44:127–130. doi: 10.1038/ng.1089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doulabi H., Rastin M., Shabahangh H., Maddah G., Abdollahi A., Nosratabadi R., Esmaeili S.A., Mahmoudi M. Analysis of Th22, Th17 and CD4(+)cells co-producing IL-17/IL-22 at different stages of human colon cancer. Biomed.Pharmacother. 2018;103:1101–1106. doi: 10.1016/j.biopha.2018.04.147. [DOI] [PubMed] [Google Scholar]
- Eklund A.C., Szallasi Z. Correction of technical bias in clinical microarray data improves concordance with known biological information. Genome Biol. 2008;9:R26. doi: 10.1186/gb-2008-9-2-r26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandez-Miranda G., Mendez R. The CPEB-family of proteins, translational control in senescence and cancer. Ageing Res. Rev. 2012;11:460–472. doi: 10.1016/j.arr.2012.03.004. [DOI] [PubMed] [Google Scholar]
- Franke A., McGovern D.P., Barrett J.C., Wang K., Radford-Smith G.L., Ahmad T., Lees C.W., Balschun T., Lee J., Roberts R., et al. Genome-wide meta-analysis increases to 71 the number of confirmed Crohn's disease susceptibility loci. Nat. Genet. 2010;42:1118–1125. doi: 10.1038/ng.717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fujino S., Andoh A., Bamba S., Ogawa A., Hata K., Araki Y., Bamba T., Fujiyama Y. Increased expression of interleukin 17 in inflammatory bowel disease. Gut. 2003;52:65–70. doi: 10.1136/gut.52.1.65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrett W.S., Gordon J.I., Glimcher L.H. Homeostasis and inflammation in the intestine. Cell. 2010;140:859–870. doi: 10.1016/j.cell.2010.01.023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greten F.R., Grivennikov S.I. Inflammation and cancer: triggers, mechanisms, and consequences. Immunity. 2019;51:27–41. doi: 10.1016/j.immuni.2019.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gronke K., Hernandez P.P., Zimmermann J., Klose C.S.N., Kofoed-Branzk M., Guendel F., Witkowski M., Tizian C., Amann L., Schumacher F., et al. Interleukin-22 protects intestinal stem cells against genotoxic stress. Nature. 2019;566:249–253. doi: 10.1038/s41586-019-0899-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grossman R.L., Heath A.P., Ferretti V., Varmus H.E., Lowy D.R., Kibbe W.A., Staudt L.M. Toward a shared vision for cancer genomic data. New Engl. J. Med. 2016;375:1109–1112. doi: 10.1056/NEJMp1607591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Guinney J., Dienstmann R., Wang X., de Reyniès A., Schlicker A., Soneson C., Marisa L., Roepman P., Nyamundanda G., Angelino P., et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015;21:1350–1356. doi: 10.1038/nm.3967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta J., del Barco Barrantes I., Igea A., Sakellariou S., Pateras I.S., Gorgoulis V.G., Nebreda A.R. Dual function of p38alpha MAPK in colon cancer: suppression of colitis-associated tumor initiation but requirement for cancer cell survival. Cancer Cell. 2014;25:484–500. doi: 10.1016/j.ccr.2014.02.019. [DOI] [PubMed] [Google Scholar]
- He X., Lin X., Cai M., Fan D., Chen X., Wang L., Wu X., Lan P., Wang J. High expression of cytoplasmic polyadenylation element-binding protein 4 correlates with poor prognosis of patients with colorectal cancer. Virchows Arch. 2017;470:37–45. doi: 10.1007/s00428-016-2037-3. [DOI] [PubMed] [Google Scholar]
- Hernandez P., Gronke K., Diefenbach A. A catch-22: interleukin-22 and cancer. Eur. J. Immunol. 2018;48:15–31. doi: 10.1002/eji.201747183. [DOI] [PubMed] [Google Scholar]
- Huber S., Gagliani N., Zenewicz L.A., Huber F.J., Bosurgi L., Hu B., Hedl M., Zhang W., O'Connor W., Jr., Murphy A.J., et al. IL-22BP is regulated by the inflammasome and modulates tumorigenesis in the intestine. Nature. 2012;491:259–263. doi: 10.1038/nature11535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huber W., Carey V.J., Gentleman R., Anders S., Carlson M., Carvalho B.S., Bravo H.C., Davis S., Gatto L., Girke T., et al. Orchestrating high-throughput genomic analysis with Bioconductor. Nat. Methods. 2015;12:115–121. doi: 10.1038/nmeth.3252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Irizarry R.A., Hobbs B., Collin F., Beazer-Barclay Y.D., Antonellis K.J., Scherf U., Speed T.P. Exploration, normalization, and summaries of high density oligonucleotide array probe level data. Biostatistics. 2003;4:249–264. doi: 10.1093/biostatistics/4.2.249. [DOI] [PubMed] [Google Scholar]
- Jorissen R.N., Gibbs P., Christie M., Prakash S., Lipton L., Desai J., Kerr D., Aaltonen L.A., Arango D., Kruhoffer M., et al. Metastasis-associated gene expression changes predict poor outcomes in patients with dukes stage B and C colorectal cancer. Clin. Cancer Res. 2009;15:7642–7651. doi: 10.1158/1078-0432.CCR-09-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jorissen R.N., Lipton L., Gibbs P., Chapman M., Desai J., Jones I.T., Yeatman T.J., East P., Tomlinson I.P., Verspaget H.W., et al. DNA copy-number alterations underlie gene expression differences between microsatellite stable and unstable colorectal cancers. Clin. Cancer Res. : official J. Am. Assoc. Cancer Res. 2008;14:8061–8069. doi: 10.1158/1078-0432.CCR-08-1431. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jostins L., Ripke S., Weersma R.K., Duerr R.H., McGovern D.P., Hui K.Y., Lee J.C., Schumm L.P., Sharma Y., Anderson C.A., et al. Host-microbe interactions have shaped the genetic architecture of inflammatory bowel disease. Nature. 2012;491:119–124. doi: 10.1038/nature11582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamanaka M., Huber S., Zenewicz L.A., Gagliani N., Rathinam C., O'Connor W., Jr., Wan Y.Y., Nakae S., Iwakura Y., Hao L., et al. Memory/effector (CD45RB(lo)) CD4 T cells are controlled directly by IL-10 and cause IL-22-dependent intestinal pathology. J. Exp. Med. 2011;208:1027–1040. doi: 10.1084/jem.20102149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keerthivasan S., Aghajani K., Dose M., Molinero L., Khan M.W., Venkateswaran V., Weber C., Emmanuel A.O., Sun T., Bentrem D.J., et al. beta-Catenin promotes colitis and colon cancer through imprinting of proinflammatory properties in T cells. Sci. Transl Med. 2014;6:225ra228. doi: 10.1126/scitranslmed.3007607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Keir M., Yi Y., Lu T., Ghilardi N. The role of IL-22 in intestinal health and disease. J. Exp. Med. 2020;217 doi: 10.1084/jem.20192195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kemper K., Versloot M., Cameron K., Colak S., de Sousa e Melo F., de Jong J.H., Bleackley J., Vermeulen L., Versteeg R., Koster J., et al. Mutations in the Ras-Raf Axis underlie the prognostic value of CD133 in colorectal cancer. Clin. Cancer Res. 2012;18:3132–3141. doi: 10.1158/1078-0432.CCR-11-3066. [DOI] [PubMed] [Google Scholar]
- Kempski J., Brockmann L., Gagliani N., Huber S. TH17 cell and epithelial cell crosstalk during inflammatory bowel disease and carcinogenesis. Front.Immunol. 2017;8:1373. doi: 10.3389/fimmu.2017.01373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kryczek I., Lin Y., Nagarsheth N., Peng D., Zhao L., Zhao E., Vatan L., Szeliga W., Dou Y., Owens S., et al. IL-22(+)CD4(+) T cells promote colorectal cancer stemness via STAT3 transcription factor activation and induction of the methyltransferase DOT1L. Immunity. 2014;40:772–784. doi: 10.1016/j.immuni.2014.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lasry A., Zinger A., Ben-Neriah Y. Inflammatory networks underlying colorectal cancer. Nat. Immunol. 2016;17:230–240. doi: 10.1038/ni.3384. [DOI] [PubMed] [Google Scholar]
- Li B., Dewey C.N. RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinformatics. 2011;12:323. doi: 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu J., Lichtenberg T., Hoadley K.A., Poisson L.M., Lazar A.J., Cherniack A.D., Kovatich A.J., Benz C.C., Levine D.A., Lee A.V., et al. An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell. 2018;173:400–416 e411. doi: 10.1016/j.cell.2018.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maillo C., Martin J., Sebastian D., Hernandez-Alvarez M., Garcia-Rocha M., Reina O., Zorzano A., Fernandez M., Mendez R. Circadian- and UPR-dependent control of CPEB4 mediates a translational response to counteract hepatic steatosis under ER stress. Nat. Cel. Biol. 2017;19:94–105. doi: 10.1038/ncb3461. [DOI] [PubMed] [Google Scholar]
- Marisa L., de Reynies A., Duval A., Selves J., Gaub M.P., Vescovo L., Etienne-Grimaldi M.C., Schiappa R., Guenot D., Ayadi M., et al. Gene expression classification of colon cancer into molecular subtypes: characterization, validation, and prognostic value. PLoS Med. 2013;10 doi: 10.1371/journal.pmed.1001453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin M., Kesselring R.K., Saidou B., Brunner S.M., Schiechl G., Mouris V.F., Wege A.K., Rummele P., Schlitt H.J., Geissler E.K., et al. RORgammat(+) hematopoietic cells are necessary for tumor cell proliferation during colitis-associated tumorigenesis in mice. Eur. J. Immunol. 2015;45:1667–1679. doi: 10.1002/eji.201444915. [DOI] [PubMed] [Google Scholar]
- Mizoguchi A., Yano A., Himuro H., Ezaki Y., Sadanaga T., Mizoguchi E. Clinical importance of IL-22 cascade in IBD. J. Gastroenterol. 2018;53:465–474. doi: 10.1007/s00535-017-1401-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Momozawa Y., Dmitrieva J., Théâtre E., Deffontaine V., Rahmouni S., Charloteaux B., Crins F., Docampo E., Elansary M., Gori A.S., et al. IBD risk loci are enriched in multigenic regulatory modules encompassing putative causative genes. Nat. Commun. 2018;9:2427. doi: 10.1038/s41467-018-04365-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida N., Nagahara M., Sato T., Mimori K., Sudo T., Tanaka F., Shibata K., Ishii H., Sugihara K., Doki Y., et al. Microarray analysis of colorectal cancer stromal tissue reveals upregulation of two oncogenic miRNA clusters. Clin. Cancer Res. 2012;18:3054–3070. doi: 10.1158/1078-0432.CCR-11-1078. [DOI] [PubMed] [Google Scholar]
- Ortiz-Zapater E., Pineda D., Martinez-Bosch N., Fernandez-Miranda G., Iglesias M., Alameda F., Moreno M., Eliscovich C., Eyras E., Real F.X., et al. Key contribution of CPEB4-mediated translational control to cancer progression. Nat. Med. 2011;18:83–90. doi: 10.1038/nm.2540. [DOI] [PubMed] [Google Scholar]
- Pearson C., Uhlig H.H., Powrie F. Lymphoid microenvironments and innate lymphoid cells in the gut. Trends Immunol. 2012;33:289–296. doi: 10.1016/j.it.2012.04.004. [DOI] [PubMed] [Google Scholar]
- Perez L.G., Kempski J., McGee H.M., Pelzcar P., Agalioti T., Giannou A., Konczalla L., Brockmann L., Wahib R., Xu H., et al. TGF-beta signaling in Th17 cells promotes IL-22 production and colitis-associated colon cancer. Nat. Commun. 2020;11:2608. doi: 10.1038/s41467-020-16363-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perez-Guijarro E., Karras P., Cifdaloz M., Martinez-Herranz R., Canon E., Grana O., Horcajada-Reales C., Alonso-Curbelo D., Calvo T.G., Gomez-Lopez G., et al. Lineage-specific roles of the cytoplasmic polyadenylation factor CPEB4 in the regulation of melanoma drivers. Nat. Commun. 2016;7:13418. doi: 10.1038/ncomms13418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccirillo C.A., Bjur E., Topisirovic I., Sonenberg N., Larsson O. Translational control of immune responses: from transcripts to translatomes. Nat. Immunol. 2014;15:503–511. doi: 10.1038/ni.2891. [DOI] [PubMed] [Google Scholar]
- Powell N., Pantazi E., Pavlidis P., Tsakmaki A., Li K., Yang F., Parker A., Pin C., Cozzetto D., Minns D., et al. Interleukin-22 orchestrates a pathological endoplasmic reticulum stress response transcriptional programme in colonic epithelial cells. Gut. 2020;69:578–590. doi: 10.1136/gutjnl-2019-318483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanz-Pamplona R., Berenguer A., Cordero D., Mollevi D.G., Crous-Bou M., Sole X., Pare-Brunet L., Guino E., Salazar R., Santos C., et al. Aberrant gene expression in mucosa adjacent to tumor reveals a molecular crosstalk in colon cancer. Mol. Cancer. 2014;13:46. doi: 10.1186/1476-4598-13-46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schüffler P.J., Fuchs T.J., Ong C.S., Wild P.J., Rupp N.J., Buhmann J.M. TMARKER: a free software toolkit for histopathological cell counting and staining estimation. J. Pathol. Inform. 2013;4:2. doi: 10.4103/2153-3539.109804. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shalapour S., Karin M. Pas de Deux: control of anti-tumor immunity by cancer-associated inflammation. Immunity. 2019;51:15–26. doi: 10.1016/j.immuni.2019.06.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharp S.P., Avram D., Stain S.C., Lee E.C. Local and systemic Th17 immune response associated with advanced stage colon cancer. J. Surg. Res. 2017;208:180–186. doi: 10.1016/j.jss.2016.09.038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sonnenberg G.F., Artis D. Innate lymphoid cells in the initiation, regulation and resolution of inflammation. Nat. Med. 2015;21:698–708. doi: 10.1038/nm.3892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sugimoto K., Ogawa A., Mizoguchi E., Shimomura Y., Andoh A., Bhan A.K., Blumberg R.S., Xavier R.J., Mizoguchi A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Invest. 2008;118:534–544. doi: 10.1172/JCI33194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tosolini M., Kirilovsky A., Mlecnik B., Fredriksen T., Mauger S., Bindea G., Berger A., Bruneval P., Fridman W.H., Pages F., et al. Clinical impact of different classes of infiltrating T cytotoxic and helper cells (Th1, th2, treg, th17) in patients with colorectal cancer. Cancer Res. 2011;71:1263–1271. doi: 10.1158/0008-5472.CAN-10-2907. [DOI] [PubMed] [Google Scholar]
- Trapani J.A., Smyth M.J. Functional significance of the perforin/granzyme cell death pathway. Nat. Rev. Immunol. 2002;2:735–747. doi: 10.1038/nri911. [DOI] [PubMed] [Google Scholar]
- Trifari S., Kaplan C.D., Tran E.H., Crellin N.K., Spits H. Identification of a human helper T cell population that has abundant production of interleukin 22 and is distinct from T(H)-17, T(H)1 and T(H)2 cells. Nat. Immunol. 2009;10:864–871. doi: 10.1038/ni.1770. [DOI] [PubMed] [Google Scholar]
- Tripathi M.K., Deane N.G., Zhu J., An H., Mima S., Wang X., Padmanabhan S., Shi Z., Prodduturi N., Ciombor K.K., et al. Nuclear factor of activated T-cell activity is associated with metastatic capacity in colon cancer. Cancer Res. 2014;74:6947–6957. doi: 10.1158/0008-5472.CAN-14-1592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Truitt M.L., Ruggero D. New frontiers in translational control of the cancer genome. Nat. Rev. Cancer. 2016;16:288–304. doi: 10.1038/nrc.2016.27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ullman T.A., Itzkowitz S.H. Intestinal inflammation and cancer. Gastroenterology. 2011;140:1807–1816. doi: 10.1053/j.gastro.2011.01.057. [DOI] [PubMed] [Google Scholar]
- West N.R., McCuaig S., Franchini F., Powrie F. Emerging cytokine networks in colorectal cancer. Nat. Rev. Immunol. 2015;15:615–629. doi: 10.1038/nri3896. [DOI] [PubMed] [Google Scholar]
- Wu S., Rhee K.J., Albesiano E., Rabizadeh S., Wu X., Yen H.R., Huso D.L., Brancati F.L., Wick E., McAllister F., et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009;15:1016–1022. doi: 10.1038/nm.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y., Danilenko D.M., Valdez P., Kasman I., Eastham-Anderson J., Wu J., Ouyang W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature. 2007;445:648–651. doi: 10.1038/nature05505. [DOI] [PubMed] [Google Scholar]
- Zhong X., Xiao Y., Chen C., Wei X., Hu C., Ling X., Liu X. MicroRNA-203-mediated posttranscriptional deregulation of CPEB4 contributes to colorectal cancer progression. Biochem.Biophys. Res. Commun. 2015;466:206–213. doi: 10.1016/j.bbrc.2015.09.008. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.