

Abstract
Antibody fragments such as Fab’s require the formation of disulfide bonds to achieve a proper folding state. During their recombinant, periplasmic expression in Escherichia coli, oxidative folding is mediated by the DsbA/DsbB system in concert with ubiquinone. Thereby, overexpression of Fab’s is linked to the respiratory chain, which is not only immensely important for the cell’s energy household but also known as a major source of reactive oxygen species. However, the effects of an increased oxidative folding demand and the consequently required electron flux via ubiquinone on the host cell have not been characterized so far. Here, we show that Fab expression in E. coli BL21(DE3) interfered with the intracellular redox balance, thereby negatively impacting host cell performance. Production of four different model Fab’s in lab-scale fed-batch cultivations led to increased oxygen consumption rates and strong cell lysis. An RNA sequencing analysis revealed transcription activation of the oxidative stress-responsive soxS gene in the Fab-producing strains. We attributed this to the accumulation of intracellular superoxide, which was measured using flow cytometry. An exogenously supplemented ubiquinone analogue improved Fab yields up to 82%, indicating that partitioning of the quinone pool between aerobic respiration and oxidative folding limited ubiquinone availability and hence disulfide bond formation capacity. Combined, our results provide a more in-depth understanding of the profound effects that periplasmic Fab expression and in particular disulfide bond formation has on the host cell. Thereby, we show new possibilities to elaborate cell engineering and process strategies for improved host cell fitness and process outcome.
Keywords: E. coli, Fab, oxidative folding, periplasmic expression, reactive oxygen species, recombinant protein production, ubiquinone
Monoclonal antibodies and antibody-derived molecules are extensively used for various applications including therapeutics and diagnostics. With an increase in global annual sales from $84 billion to $163 billion between 2014 and 2019,1 they represent the fastest growing segment of the biopharmaceutical market.2 Due to cost-effective cultivation, microbial expression systems such as Escherichia coli represent a utile alternative for smaller-sized antibody fragments that do not rely on glycosylation for functionality.3 One format that retains its antigen-binding capacity (Fab) consists of the light chain (LC) and two domains of the heavy chain (HC) of an immunoglobulin G (IgG) molecule. Each chain is composed of a constant (CL, CH1) and a variable domain (VL, VH). Antigen binding is mediated by the complementary determining regions within the variable domains of the Fab. For correct folding, five disulfide bonds are required.4
Different approaches to express Fab’s in E. coli have been described. Efforts have been made to enable production in the cytoplasm, whereby investigated strategies aimed at expression as inclusion bodies (IBs) and subsequent refolding or at manipulating the prevalent reducing conditions, thereby allowing the formation of disulfide bonds.5,6 However, the method commonly used is fusion of the Fab’s HC and LC to a signal sequence for translocation across the inner membrane (IM) into the periplasmic space which is the only bacterial compartment that naturally provides the oxidizing conditions to enable the formation of disulfide bonds.7
In bacterial systems, various factors such as intracellular degradation or aggregation, and toxicity effects of the recombinant protein frequently lead to low product yields.8 In eukaryotic expression systems, oxidative stress has also been associated with the production of heterologous proteins. These studies implicate involvement of disulfide bond formation and breakage, secretion, and endoplasmic reticulum (ER) stress in eliciting the stress response.9−12 Bacteria lack a compartment equivalent to the ER. Instead, oxidative folding is mediated by the thiol:disulfide oxidoreductase DsbA and the thiol:quinone oxidoreductase DsbB within the periplasm. DsbA donates its disulfide bond to a nascent protein and gets re-oxidized by the IM protein DsbB in order to remain catalytic. DsbB in turn is regenerated by transferring the electrons to ubiquinone (UQ8). Oxidative folding in the bacterial periplasm is therefore directly linked to the respiratory chain.13,14
The respiratory chain of E. coli is extremely versatile, enabling the cell to optimize its energy household under various conditions. Multiple dehydrogenases and terminal oxidases for the utilization of different electron donors and acceptors are linked by the quinone pool. Combination of isozymes leads to different degrees of coupling between electron and proton transport.15 The resulting proton motive force (PMF) is fueling ATP formation through oxidative phosphorylation. During aerobic conditions, NADH is oxidized by NADH dehydrogenases I (NDH I, nuo operon) and II (NDH II, ndh), enabling varying flux distribution between them. NDH I recovers energy from NADH oxidation (2 H+/e–), while NDH II is noncoupling.16 Electron flow is directed mainly via UQ8 and to a lesser extent via menaquinone (MQ) and its precursor demethylmenaquinone (DMQ).17,18 The terminal oxidases cytochrome bo, bd I, and bd II transfer the electrons from the quinone pool to O2. At high O2 levels, mainly cytochrome bo is utilized.19,20
Partially reduced oxygen species are generated as inevitable byproducts of aerobic metabolism when O2 is reduced in single-electron reactions. The resulting products superoxide (O2•–), hydrogen peroxide (H2O2), and hydroxyl radical (OH•) generally referred to as reactive oxygen species (ROS) are therefore ubiquitous.21−23 Protective mechanisms to keep ROS at harmless levels have evolved in aerobes. Superoxide dismutases (SODs) and catalases/peroxidases prevent accumulation of endogenous O2•– and H2O2, respectively.24 Mutant strains deprived of the protective enzymes are poisoned by increased levels of O2•– and H2O2 when cultivated in the presence of O2.25−27 Basic defense mechanisms are quickly overcome when cells experience sudden elevated levels of ROS. The resulting imbalance of formation and elimination of ROS causes oxidative stress. Damage to proteins (through oxidation of flavin cofactors, metal centers, and amino acids), DNA, and phospholipids is the consequence. E. coli has a second, inducible line of defense to deal with oxidative stress. Diverse cellular antioxidant mechanisms are executed by redox stress sensors SoxR and OxyR.21,22,28 The two transcription factors are activated by oxidation of [2Fe–2S] clusters29,30 and cysteine residues,31,32 respectively. OxyR responds to H2O2;30 however, the signals sensed by SoxR are still a matter of debate.33 A known trigger of SoxR activation is accumulation of O2•– and nitric oxide.34−37 Recent research has also indicated other mechanisms of direct metal center oxidation and interference with SoxR inactivation (reduction) pathways as alternative activators.23,25,38 Oxidized SoxR in turn activates transcription of soxS, a gene coding for a secondary transcription factor. Targets of the SoxRS and OxyR regulons scavenge ROS, boost synthesis of reducing equivalents, repair oxidatively damaged proteins and DNA, and help to provide redox-resistant isozymes for sensitive enzymes.21−23,38,39
We hypothesized that overexpression of Fab’s in the periplasmic space requires higher oxidative folding activity to provide the disulfide bonds necessary for folding and consequently increased flux of electrons via UQ8 and through the respiratory chain. Since the respiratory chain is a known source of ROS,40 this might lead to disturbances of the redox balance and to metabolic changes. To address these interdependencies as a possible consequence of periplasmic Fab expression, we conducted lab-scale, fed-batch cultivations of a set of recombinant E. coli BL21(DE3) strains expressing four different Fab’s fused to the post-translational translocation signal sequence of the OmpA protein of E. coli (ompASS). We used genome-integrated expression systems to avoid plasmid-mediated metabolic load and other confounding factors as described elsewhere.41 Strong T7-based systems with a single Fab gene copy were chosen, in order to achieve a sufficiently strong cell response to Fab production, while reducing the metabolic load.42
In this study, we present evidence that perturbation of the host cell’s redox balance is indeed a consequence of expressing Fab’s in the periplasm of E. coli BL21(DE3) and that this interaction can be utilized for the improvement of production. We monitored increased oxygen consumption rates (qO2) and cell lysis as a consequence of Fab production in fed-batch cultivations. In fed-batch-like microtiter cultivations, we detected higher levels of intracellular O2•– in Fab-producing strains. Supplementing a UQ8 analogue to the growth medium led to increased Fab yields, indicating UQ8 deficiency during Fab expression. RNA sequencing (RNA-seq) revealed elevated transcript levels of the O2•–-inducible soxS gene at later stages of the fed-batch fermentations as well as changes in the gene expression behavior of NADH dehydrogenases.
Results and Discussion
For correct folding, Fab molecules require one inter- and four intrachain disulfide bonds. Twenty years ago, Bader et al. showed that oxidative folding in the periplasmic space is directly linked to the respiratory chain via the quinone pool.13 The main goal of this study was to investigate the interplay between increased oxidative folding demand in the periplasm, concomitant electron flux through the respiratory chain via UQ8, and effects thereof on host strain and process performance.
Increased Oxygen Consumption Rates of Fab-Producing Strains in Glucose-Limited Fed-Batch Cultivations
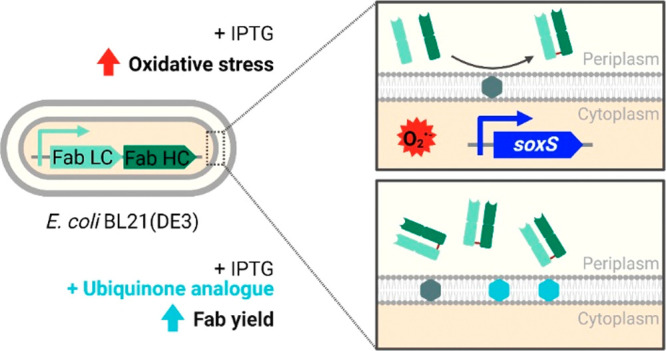
To observe the effects of periplasmic Fab expression on the host cells under relevant production conditions, we conducted lab-scale fed-batch cultivations of Fab-producing strains (strain abbreviations are listed in Table 1). Biomass accumulation and total specific soluble Fab titers including the intra- and extracellular fractions are shown in Figure 1A. The wildtype strain BL21(DE3) and BL21(DE3) expressing green fluorescent protein (GFP) as “easy-to-produce” protein43 without disulfide bonds were included as reference systems. Two variants of GFP were used: cytosolic GFPmut3.1 and periplasmic superfolder GFP (sfGFP) that were translocated by fusion to the signal sequence of the DsbA protein (dsbASS). All Fab-producing strains showed reduced accumulation of biomass compared to the wildtype strain, which reached a final biomass of 46.19 g of cell dry mass (CDM). Impact on growth was most pronounced in B⟨oFabx⟩ with 31.63 g of CDM, followed by B⟨oBIWA4⟩ with 39.12 g, B⟨oFTN2⟩ with 41.27 g, and B⟨oBIBH1⟩ with 42.00 g of final CDM. Cytosolic GFPmut3.1 production had no negative impact on cell growth and resulted in a final CDM of 48.39 g for B⟨GFPmut3.1⟩. Periplasmic expression of sfGFP led to slightly reduced biomass of 44.36 g for B⟨dsfGFP⟩. Intra- and extracellular Fab titers were analyzed from cell lysates and culture supernatant, respectively, using enzyme-linked immunosorbent assay (ELISA). GFP was quantified fluorometrically. Total specific Fab titer at the end of the production phase was the highest for FTN2 with 3.76 mg g–1 CDM, followed by BIWA4 with 2.88 mg g–1 CDM and BIBH1 with 1.46 mg g–1 CDM. The lowest titer was obtained for Fabx with 0.44 mg g–1 CDM. In addition to Fab molecules, also considerable amounts of unassembled LCs were detected. Different ratios of Fab to unassembled LCs in the soluble and IB fraction of cell lysates at the end of the production process are shown in LC-specific western blots (WBs) in Figure 1B. Unassembled HCs were hardly detectable in HC-specific WBs, presumably due to proteolysis. GFP was expressed at substantially higher levels compared to Fab’s. Cytosolic GFPmut3.1 reached a concentration of 293.67 mg g–1 CDM, while periplasmic expression levels of sfGFP were lower at 119.10 mg g–1 CDM.
Table 1. Used E. coli Strains and Genome-Integrated Expression Systems.
| strains and abbreviations | description | source |
|---|---|---|
| E. coli BL21(DE3) | F-ompT gal dcm lon hsdSB(rB–mB−) λ(DE3 [lacI lacUV5-T7p07 ind1 sam7 nin5]) | NEB |
| B⟨oFabx⟩ | BL21(DE3) expressing ompASS-Fabx | (41) |
| B⟨oBIBH1⟩ | BL21(DE3) expressing ompASS-BIBH1 | (41) |
| B⟨oBIWA4⟩ | BL21(DE3) expressing ompASS-BIWA4 | (41) |
| B⟨oFTN2⟩ | BL21(DE3) expressing ompASS-FTN2 | (41) |
| B⟨GFPmut3.1⟩ | BL21(DE3) expressing GFPmut3.1 | (92) |
| B⟨dsfGFP⟩ | BL21(DE3) expressing dsbASS-sfGFP | in house |
Figure 1.
(A) CDM [g], total (intra- and extracellular) soluble Fab yields [mg g–1 CDM] and qO2 and qCO2 [mmol g–1 CDM h–1] during glucose-limited fed-batch cultivations. GFPmut3.1 and sfGFP yields were plotted in [mg g–1 CDM] × 101. Cultivations of the wildtype reference BL21(DE3) and the Fab-producing strains B⟨oFabx⟩, B⟨oBIBH1⟩, B⟨oBIWA4⟩, and B⟨oFTN2⟩ were performed in triplicate (mean + SEM, n = 3). Both GFP-producing strains were cultivated once. CDM and yield of cytoplasmic GFPmut3.1 of B⟨GFPmut3.1⟩ are shown by filled circles (●) and diamonds (gray ⧫), respectively, and CDM and yield of periplasmic sfGFP of B⟨dsfGFP⟩ are shown by empty circles (○) and diamonds (◊), respectively. Online data for qO2 and qCO2 are presented as moving average with a period of 60 data points (equals approx. 1 min) for a single representative experiment. For the GFP production strains, qO2 and qCO2 are only shown for B⟨GFPmut3.1⟩ since the measurements were very similar to B⟨dsfGFP⟩. Induction is indicated by the vertical dashed, gray line. The CDM of the BL21(DE3) wildtype strain is shown in the graphs of all recombinant strains for comparison. (B) Fab expression patterns of endpoint samples (after 16 h induction) analyzed by LC-specific WB. (1) Soluble and (2) IB fractions and (3) recombinant protein found extracellularly in the culture supernatant are shown. Fractions loaded in each lane were adjusted to the same biomass for comparability. Bands corresponding to Fab (approx. 50 kDa) and LC (approx. 25 kDa) are indicated by arrows.
Upon comparison of the online data measured during the different cultivations, it became obvious that Fab expression led to an increase in the qO2 compared to the BL21(DE3) wildtype and BL21(DE3) expressing either of the two GFP variants. The reference strains showed a constant qO2 of approx. 4 mmol g–1 h–1 throughout the process as expected44 (Figure 1A). The value is in accordance with numbers reported for glucose-limited growth at a rate of μ = 0.1 h–1.45 The strains expressing the four different Fab fragments exhibited a rather constant qO2 of approx. 4 mmol g–1 h–1 at the beginning of the process. However, concomitant with a deviation of the biomass from wildtype growth, the qO2 sharply increased up to approx. 8 mmol g–1 h–1 for B⟨oFabx⟩, B⟨oBIBH1⟩, and B⟨oBIWA4⟩ and approx. 10 mmol g–1 h–1 for B⟨oFTN2⟩. After reaching a peak, the qO2 slowly dropped again. The CO2 formation rate (qCO2) stayed rather constant for all strains. The surge in qO2 was accompanied by increasing levels of extracellular product (Figures 1B and S1) due to cell lysis (confirmed by measurement of increasing DNA levels in the culture supernatant using Hoechst dye; data not shown). Therefore, lower CDM yields of Fab-producing strains could also partly be attributed to loss of biomass by lysis.
There are multiple possible influence factors that could be responsible for the observed increase in qO2. Expression of Fab’s and the formation of disulfide bonds needed to reach their correct conformation lead to an increased oxidative folding demand, which in turn would require increased flux of electrons via UQ8. The respiratory chain is known as the major contributor to the formation of O2•–,40 which is scavenged by SODs.24 Single-electron transfer reactions to O2 are a prerequisite for the formation of O2•–; hence, increased O2•–formation would require higher O2 consumption. Assuming that all disulfide bonds are formed correctly and do not require breakage, re-formation leads to a theoretical consumption of 1 O2/LC and HC (4 e–/LC and HC) and 2.5 O2/Fab molecule (10 e–/Fab). Since considerable IB formation, production of unassembled LCs, and the formation of incorrect Fab derivatives as described by Schimek et al.46 were observed, it was not possible to quantify total recombinant protein production and calculate the respective amount of O2 needed as an electron acceptor. However, even at high recombinant protein titers, disulfide bond formation alone could not account for 100% (Fabx, BIBH1, and BIWA4) to 150% (FTN2) increase in qO2 when assuming stoichiometric O2 consumption. It has been discovered that the formation of disulfide bonds in secretory proteins is connected to the formation of ROS in eukaryotes.10,47 ER-resident proteins Ero1p and protein disulfide isomerases catalyze the formation of disulfides analogous to bacterial DsbB and DsbA.48 Ero1p regenerates by directly transferring electrons to O2 in a flavin-dependent reaction, thereby producing one molecule of H2O2 per disulfide bond.49 However, nonstoichiometric amounts of ROS produced by oxidative folding of overexpressed proteins have been determined experimentally.9,48 Incorrect disulfide bonds are broken and need to be reformed to reach their native state. Repeated breakage and re-formation of non-native disulfide bonds resulting in futile cycles have been proposed as a possible explanation for increased qO2 and ROS formation (e.g., when an uneven number of cysteines are present or folding is slow).9 In E. coli, disulfide bond isomerization is carried out by oxidoreductase DsbC in concert with the IM protein DsbD. Electrons are donated by the NADPH pool and transferred to DsbD by cytoplasmic thioredoxin.50 High levels of non-native disulfide bonds possibly lead to elevated O2 demand when they have to be re-formed. However, proteins with consecutive disulfide bonds such as Fab’s generally do not rely on DsbC and dsbC deletion has indeed been reported not to affect human Fab activity or yield when produced in E. coli.51 Additionally, supplementation of 10 mM glutathione which is described to aid reshuffling of disulfides and thereby improve titers of recombinant, disulfide bond-containing proteins52 led to decreased instead of increased Fab yields in both fed-batch-like microtiter and lab-scale fed-batch cultivations in our hands (data not shown). The effect on cell growth was not consistent and therefore not conclusive. Since the preliminary experiments did not show the anticipated improvements (as described by Kumar et al.53), we focused on other strategies to improve Fab production, even though the underlying mechanisms would be worth further investigation.
Campani et al. described that the metabolic burden exerted by recombinant protein production can impact qO2.54 However, in our case, constant qO2 during cultivation of the GFP-producing strains demonstrated that high-level expression of a recombinant protein and consequently increased ATP demand alone was not sufficient to increase qO2. Therefore, qO2 depended solely on the growth rate and the nature of the used carbon source in both GFP-producing strains. Furthermore, for periplasmic expression SecA-mediated, ATP- and PMF-driven translocation of the recombinant proteins across the IM is necessary.55 In contrast to cytosolic GFPmut3.1, expression of sfGFP and Fab’s required translocation and hence additional ATP, which could have influenced qO2. Nevertheless, expression of periplasmic sfGFP did not cause an increase in qO2; hence, energy consumption by translocation did not seem to have an impact on qO2.
Since rather high levels of cell lysis were occurring, starting at approx. 11 h of feed with up to 66% of the product found extracellularly at the end of the process (Figure S1), cellular components in the culture broth might also have influenced qO2. Cells are able to utilize nutrients liberated by lysed cells, which leads to higher qO2 as observed during the death phase and cryptic growth in the stationary phase.56
Finally, metabolic shifts and changes in respiration have been described upon perturbation of the respiratory chain and the PMF, which could be connected to increased oxidative folding activity. Manipulation of respiration has even been utilized for engineering metabolite distribution.57−60 Castan et al. also found increased levels of mixed acid fermentation metabolites upon use of O2-enriched process air.61
It is unclear to what extent each of the possibilities mentioned above impacted the observed increase in qO2. Probably, the surge and subsequent decline of qO2 were impacted by a combination of changes in metabolism, cell lysis, and oxidative folding. In any case, the need to use pure O2 in the in-gas stream to maintain dissolved oxygen (DO) at 30% demonstrated the pronounced effects of Fab production on the host cells. It needs to be mentioned that especially, cell lysis influenced total process performance, since it was associated with not only higher amounts of the extracellular product but also genomic DNA found in the fermentation broth. Loss of product and possibly product quality and decreased processability in downstream processing through higher viscosity due to extracellular DNA would be problematic consequences and need to be considered during process design.
Accumulation of Intracellular Superoxide in Fab-Producing Strains
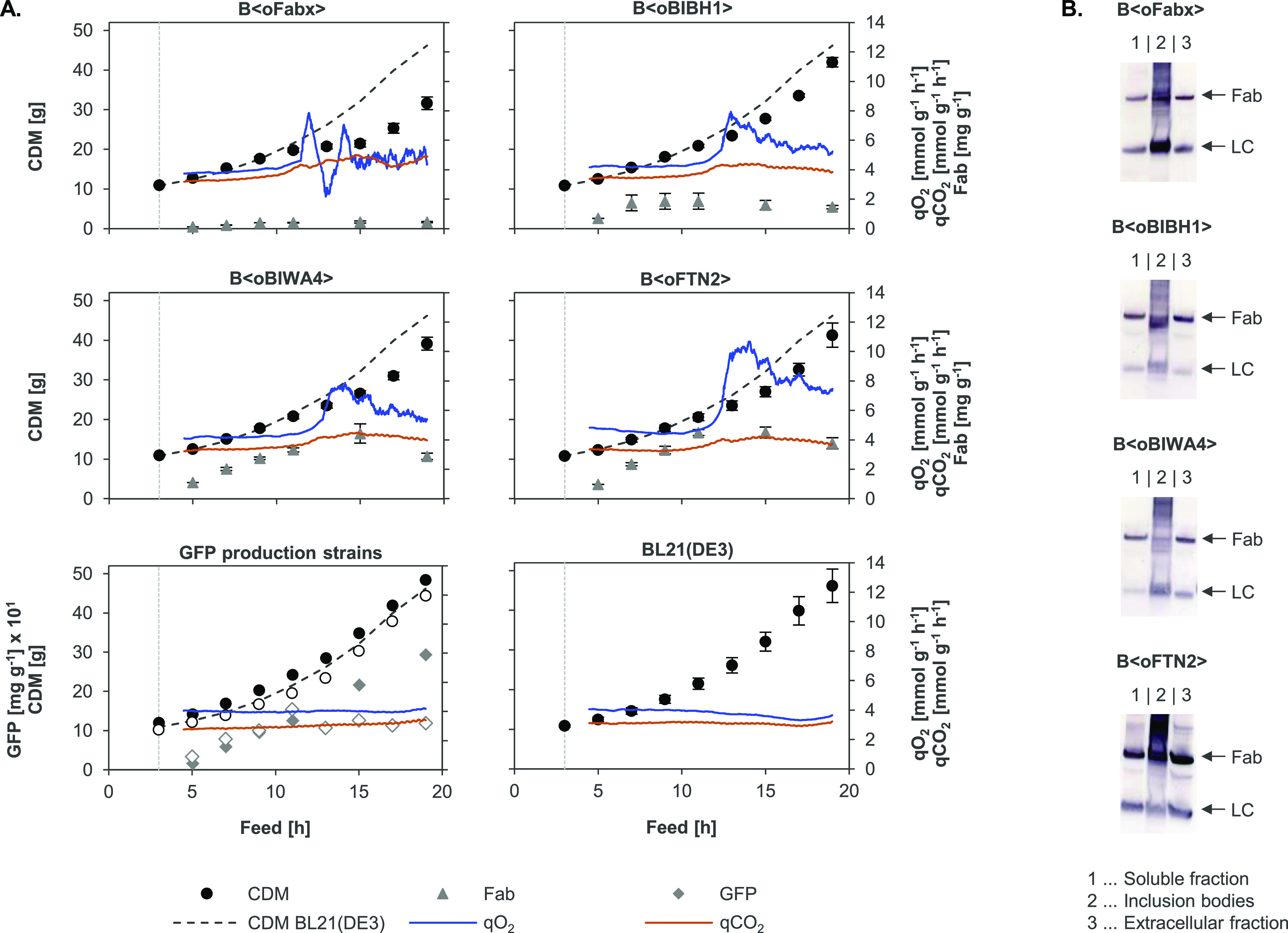
Even though the qO2 increase observed in Fab-producing strains during fed-batch cultivations was presumably not directly caused by disulfide bond formation, we assumed a connection to oxidative folding. This prompted us to test if Fab expression was indeed connected to the formation of ROS, more specifically O2•–. CellROX Green reagent was used to determine intracellular O2•– formation. This weakly fluorescent dye enters the cell and, when oxidized, becomes strongly fluorescent and binds to double-stranded DNA. According to the manufacturer, the dye is sensitive to oxidation by O2•– and OH•, but not H2O2, ONOO–, NO, and ClO–. It has also been demonstrated by McBee et al. that H2O2 treatment did not cause fluorescence increase in CellROX Green-stained E. coli cells.62 Fab-producing strains B⟨oFabx⟩, B⟨oBIBH1⟩, B⟨oBIWA4⟩, and B⟨oFTN2⟩ and the BL21(DE3) wildtype reference strain were grown in fed-batch-like cultivations in the microtiter format to achieve a higher amount of parallelization for including controls. CellROX Green-stained cells were analyzed flow cytometrically, and induced cultures were compared to noninduced ones 12 h after induction of Fab production. Cultures of induced BL21(DE3) wildtype were used as a reference. Wildtype BL21(DE3) treated with the redox cycling drug menadione (MD) that causes an increase in the CellROX Green signal due to the formation of O2•–,62 served as a positive control.
Fab expression patterns of the cultivations are shown in LC-specific WBs of the soluble and IB fractions in Figure S2. Histograms of the cell count plotted against the fluorescence intensity (FI) of BL21(DE3) and B⟨oFTN2⟩ are shown in Figure 2A as examples. B⟨oFabx⟩, B⟨oBIBH1⟩, and B⟨oBIWA4⟩ are shown in Figure S3A. Noninduced [0 mM β-d-1-thiogalactopyranoside (IPTG)] and induced cultures (0.5 mM IPTG) were measured with (+CG) and without (−CG) CellROX Green staining to detect changes in autofluorescence and avoid introduction of artifacts. Of the tested Fab-producing strains, only B⟨oFabx⟩ showed a slight increase in autofluorescence upon induction. Increased forward (FSC) and side scatter (SSC) signals indicated that altered autofluorescence was probably caused by changes in cell size and morphology (Figure S3B). For all strains, an increase in the SSC signal accompanied by a fluorescence shift could be seen for noninduced cultures upon staining with CellROX Green (Figure S3C). The geometric mean of the FI (GeoMean FI) was obtained for all samples. GeoMean FI values of the samples without CellROX Green staining were subtracted from stained cultures for every strain, for noninduced and induced cultures separately. The resulting values are plotted in Figure 2B. The CellROX Green-stained BL21(DE3) wildtype reference exhibited unchanged GeoMean FI regardless of IPTG addition. Fab-producing strains without the addition of IPTG exhibited GeoMean FI comparable to that of the wildtype reference. However, the induced, CellROX Green-stained Fab-producing strains all showed substantially higher GeoMean FI compared to the noninduced samples. Induction caused the strongest GeoMean FI increase in B⟨oBIBH1⟩, followed by B⟨oFabx⟩ and B⟨oFTN2⟩. Induced B⟨oBIWA4⟩ showed lower, but still clearly elevated GeoMean FI in the induced cultures compared to the noninduced ones. As expected, the addition of 350 μM MD to the BL21(DE3) wildtype led to an FI shift of nearly one log step in the positive control (Figure 2A) which equals an almost eightfold increase in the GeoMean FI (Figure 2B).
Figure 2.
Flow cytometric analysis of Fab-producing strains and the BL21(DE3) wildtype strain grown in fed-batch-like cultivations in the microtiter format after 12 h of cultivation/induction. Fluorescence at 488/525 nm of noninduced (0 mM IPTG) and induced cells (0.5 mM IPTG) was analyzed without (−CG) and with (+CG) staining with CellROX Green reagent. Wildtype BL21(DE3) treated with 350 μM MD served as a positive control. (A) Single representative measurements of BL21(DE3) and B⟨oFTN2⟩ are shown in histogram plots. The positive control in blue is shown in both diagrams. (B) GeoMeanFI × 103 of noninduced and induced samples with CellROX Green staining. All samples were analyzed in biological triplicate (n = 3, variance ⟨ 18%).
Hereby, we clearly demonstrated that induction of Fab expression in E. coli BL21(DE3) production strains caused increased oxidation of an O2•–-sensitive dye. The observed effects were somewhat lower than for the MD-treated positive control, indicating less-pronounced O2•– formation elicited by Fab production under the present conditions than by the action of the redox cycling drug. The intracellular site of O2•– formation in our experiment remains unclear. However, the respiratory chain has generally been identified as the major source of O2•– (but not H2O2) in the cell.40 It is tempting to speculate that at least a part of the detected O2•– was formed by an increasing number of single-electron transfer reactions to O2 within the respiratory chain, directly or indirectly caused by oxidative folding of the recombinant protein. One possibility is a higher rate of O2•– formation by the terminal oxidases simply due to higher flux of electrons from oxidative folding. Another possible site of O2•– formation is NDH II.18 NDH II is known to produce O2•– via autoxidation of its flavin cofactor21,40 and there are two explanations for increased autoxidation. Higher flux through NDH II instead of NDH I or a lack of downstream electron acceptors causes electrons to remain on the autoxidizable flavin.63 Electrons backed up on NDH II were identified as responsible for O2•– formation in membrane vesicles obtained from a UQ8-deficient mutant.64 Hence, partitioning of the UQ8 pool between NADH oxidation and DsbB regeneration during oxidative folding of the recombinant proteins might lead to a limitation of available UQ8 and in further consequence increased O2•– formation. Another explanation for higher O2•– formation, also assuming UQ8 deficiency, is the transfer of electrons to MQ. DsbB can use MQ as an alternative electron acceptor under aerobic and anaerobic conditions.13 Higher production of O2•– in ubiAC mutants that lack UQ8 and lower levels of O2•– in menA mutants with a deletion in the MQ synthesis pathway have been observed.18 The two quinones have different redox potentials (+0.113 V for UQ8 and −0.074 V for MQ), which is why MQ could also transfer electrons directly to O2 and thereby contribute to O2•– formation.
Exogenous Supplementation of Coenzyme Q1 (CoQ1) to Increase Fab Yields
Insufficient amounts of oxidized UQ8 within the cell might lead to increased formation of O2•– in the respiratory chain and to an insufficient oxidative folding capacity. Therefore, we tested if supplementation of the artificial UQ8 analogue CoQ1 to the growth medium could increase the yield of correctly folded, soluble Fab. CoQ1 has been used in other studies to control respiration in a ubiAC mutant.65 Compared to endogenous UQ8, the polyprenyl hydrophobic tail contains less isoprenyl units (1 instead of 8 in UQ8).66
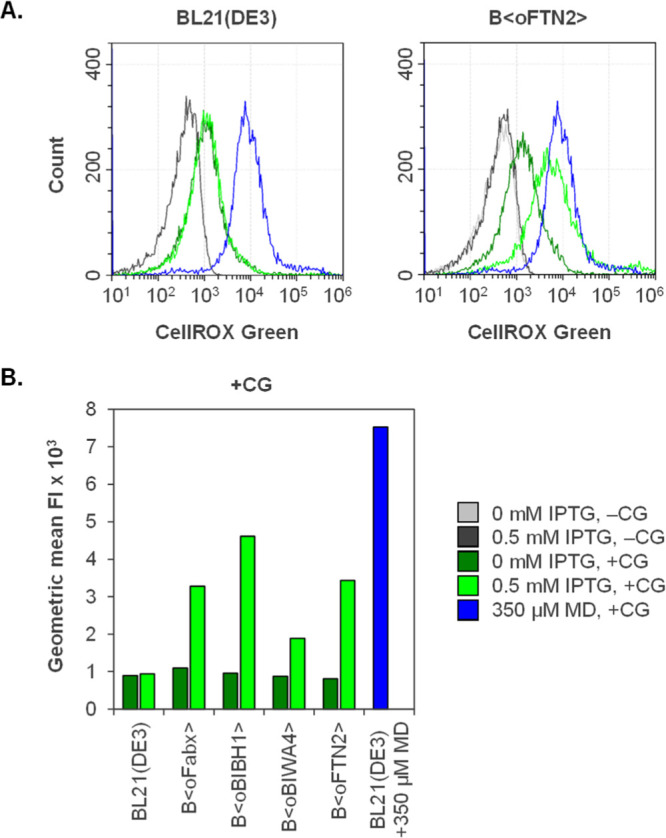
In a first approach, we tested the impact of supplementing 5 μM CoQ1 to an induced shake flask culture of B⟨oFTN2⟩, where the growth rate was not limited by glucose feeding. CoQ1 addition led to improved growth with a final OD600 of 3.2 compared to 2.0 of the control without CoQ1 after 4 h of Fab production. The soluble FTN2 band observed in LC-specific WB analysis was slightly increased when CoQ1 was added (Figure S4). Since fed-batch cultivation is more industrially relevant, batch experiments were not pursued further. Nevertheless, the experiment showed that boosting the available UQ8 pool seemed to positively impact ubiquinone availability for cell growth and oxidative folding under conditions without C-limitation.
The effect of supplementing different concentrations of CoQ1 was further analyzed using the strain B⟨oFTN2⟩ in fed-batch-like microtiter cultivations. The total UQ8 content of aerobically growing cells has been measured at approx. 1090 nmol g–1 CDM.67 In our setup (a final CDM of maximum 10 g L–1 in 800 μL working volume), this equals a concentration of approx. 11 μM CoQ1. Therefore, 0 μM, 5 μM (approx. 0.5× the endogenous intracellular UQ8 concentration), 10 μM (1×), 25 μM (2.5×), and 50 μM (5×) CoQ1 were supplemented to the cultivations at induction in addition to IPTG. Induction of recombinant protein production caused a slight decrease in final biomass compared to the noninduced cultures (9.8 g L–1). Induced cultures reached a final biomass of 7.9 g L–1 at all tested CoQ1 concentrations, including the reference without CoQ1 addition. Intracellular Fab yields were analyzed using ELISA. Extracellular amounts of Fab detected in LC-specific WB were negligible (data not shown) and were therefore not considered. A positive effect of all tested CoQ1 concentrations on FTN2 yield was observed. Fab yields obtained in cultivations with CoQ1 supplementation were normalized to the cultivation without CoQ1. An increase in FTN2 yield between 1.4- and 1.8-fold could be achieved (Figure 3A). In a follow-up cultivation, the remaining Fab-producing strains B⟨oFabx⟩, B⟨oBIBH1⟩, and B⟨oBIWA4⟩ were supplemented with 0 or 10 μM CoQ1 at induction (Figure 3B). In all tested Fab strains, CoQ1 improved Fab production to different degrees. Fabx yield was increased by 82%, BIBH1 by 39%, BIWA4 by 17%, and FTN2 as previously determined, by 50%.
Figure 3.
(A) FTN2 produced in fed-batch-like microtiter cultivations with different concentrations of the UQ8 analogue CoQ1 (0–50 μM). Biological duplicates were analyzed, and volumetric FTN2 yields were normalized to the cultivation without CoQ1 addition (n = 2). (B) Fab yields obtained in fed-batch-like microtiter cultivations of B⟨oFabx⟩, B⟨oBIBH1⟩, and B⟨oBIWA4⟩ with supplementation of 10 μM CoQ1. Volumetric Fab yields were normalized to yields obtained without CoQ1 addition for each respective Fab. One cultivation was analyzed for Fabx, BIBH1, and BIWA4 with analytical variance ⟨10%. (C) Log2FC of genes involved in the UQ8 synthesis pathway that were differentially expressed (α ≤ 0.05) after 12 h of induction in fed-batch cultivations relative to the sample drawn prior to induction as determined by RNA-seq (n = 3).
The fact that UQ8 plays a vital role in proper functioning of the respiratory chain (and hence energy household and growth)18,65,68,69 and oxidative folding13,70 has been well-established. A recent study found that disulfide bond formation was impaired in E. coli through growth on long-chain fatty acids, which causes increased levels of NADH.71 The authors reasoned that increased electron flux through the respiratory chain by NADH oxidation led to UQ8 deficiency and showed that providing UQ8 exogenously can restore disulfide bond formation. Comparably, increased oxidative folding demand exerted by periplasmic Fab expression seemed to exhaust the cells’ UQ8 pool, which could be counteracted by supplementation of CoQ1. Another study found that mutant E. coli forming only 20% of the normal amount of UQ8 showed decreased growth and decreased oxidase activity, even though UQ8 was still present in excess with respect to cytochrome bo.72 This highlights that sufficient availability of UQ8 is crucial to maintain both the respiratory chain and oxidative folding activity intact. By increasing Fab yields substantially upon supplementation of CoQ1, we showed that UQ8 indeed seemed to be a limiting factor during Fab expression.
Downregulation of ubi Genes during Glucose-Limited Fed-Batch Cultivations
To get a comprehensive view of the host cell response elicited upon Fab production stress on the transcription level, we investigated changes in gene expression over the course of the fed-batch cultivations using RNA-seq. We analyzed differential gene expression (DGE) in samples drawn 2, 12, and 16 h after induction relative to the noninduced sample after 3 h of feed. DGE profiles of the Fab-producing strains were compared to the BL21(DE3) wildtype strain to exclude changes in gene expression dependent on the process conditions or production of T7 RNA polymerase due to IPTG addition.
The UQ8 synthesis pathway in E. coli comprises multiple genes in different genomic locations (ubiCA, ubiD, ubiEJB, ubiHI, ubiX, ubiG, and ubiF).66 The genes encoding the enzymes dedicated to the first two steps in UQ8 synthesis, ubiC (chorismate pyruvate lyase) and ubiA (4-hydroxybenzoate octaprenyltransferse), were negatively affected by the process conditions in fed-batch cultivations after 12 h (Figure 3C) and 16 h of induction (Figure S8). Downregulation of the ubiCA operon was comparable in Fab-producing strains and the wildtype reference with a log2 fold change (log2FC) between 0.7 and 1.3 (see Table S1). Hence, Fab expression that exhausts the cells’ UQ8 pool did not trigger UQ8 synthesis. This is in accordance with Kwon et al.,73 who reported low expression levels of the operon during growth on fermentable carbon sources (such as glucose used in our study). Increased expression has been observed in cells provided with the oxidizable carbon source glycerol under aerobic conditions.73,74
Transcription Activation of soxS and marRAB upon Fab Expression
When ROS such as O2•– are formed at a higher rate than the cells’ basal defense mechanisms can disarm them, E. coli relies on two known lines of defense against oxidative stress, which are inducible on the transcription level: the SoxRS and the OxyR regulons. Since we observed elevated intracellular O2•– levels in Fab-producing strains in microtiter cultivations, we focused on genes that are activated either by SoxR/SoxS or OxyR. Log2FC of members of the SoxRS and OxyR regulons that were differentially expressed after 12 h of induction is shown in Figure 4A,B, respectively.
Figure 4.

DEGs (α ≤ 0.05) of members of the (A) SoxRS and (B) OxyR regulons as determined by RNA-seq. Log2FC after 12 h of induction relative to the sample drawn prior to induction are shown for the Fab-producing strains and the BL21(DE3) wildtype (n = 3).
We observed activation of soxS transcription in all Fab-producing strains 12 h after induction. Log2FC varied between 2.4 in B⟨oBIBH1⟩ and 4.2 in B⟨oFabx⟩ (see Table S1). Transcript levels of soxS were still elevated after 16 h of production albeit less-pronounced than earlier in the process (Figure S5). The only known activator of soxS transcription is SoxR, which triggers soxS transcription upon oxidation of its [2Fe2S] cluster.75,76 Hence, increased soxS transcript levels revealed activation of SoxR. Oxidation of SoxR is mediated, for example, by O2•–, which we showed accumulates in Fab-producing strains. No soxS upregulation was observed after 2 h of production. At the earlier stages of the production phase, the cells’ basal lines of defense apparently were sufficient to keep formed O2•–at harmless levels. After prolonged Fab production, basal defenses seemed to be overwhelmed, and the cells had to resort to inducible mechanisms to mitigate the formed O2•–. It was reported by Baez and Shiloach77 that the use of O2-enriched process air can lead to SoxRS activation in E. coli. A contribution thereof cannot be excluded; however, O2 levels in the in-gas stream did not necessarily coincide with soxS upregulation levels. Although the % O2 after 16 h induction was equal or higher compared to 12 h, soxS transcript levels decreased between the two time points (Figure S6A). This was confirmed by qPCR measurements of samples drawn from a B⟨oFTN2⟩ cultivation at additional time points. Increased soxS levels could be detected not only after 12 but also after 6 h of induction when no pure O2 had been added (Figure S6B). Additionally, soxS transcript levels were higher after 10 h than 12 h of induction, which does not coincide with the use of higher levels of O2, but with ceased productivity toward the end of the process (and parallelly decreasing soxS transcription upregulation). Furthermore, the increased levels of O2•– in Fab-producing strains cultivated in microtiter plates were independent of factors such as O2-enriched process air.
The list of genes activated by the secondary transcription factor SoxS is continuously being extended. Surprisingly, hardly any of the known SoxS target genes (e.g. zwf, nfo, fur, fldAB, ...23,39) were upregulated in Fab-producing strains, despite activated soxS transcription. B⟨oFabx⟩ showed slight upregulation (0.5 ⟨ log2FC ⟨ 0.8) of sodA, acrA, inaA, and rimK. Some SoxS-activated genes (fumC and acnA in all strains, and sodA in all strains, except B⟨oFabx⟩) were down- rather than upregulated. This can be attributed to process conditions since the same expression pattern was observed in wildtype BL21(DE3). The multiple-antibiotic-resistance operon marRAB was upregulated in all Fab-producing strains but not in the wildtype reference. Overlap between SoxRS and MarRAB operon and activation of marRAB transcription by SoxS have been reported and ascribed to the structural similarity between the transcription factors SoxS and MarA.39,75,78 Gene ybjC was described to be activated by MarA and SoxS39 and was also moderately upregulated in B⟨oFabx⟩ in our setup.
OxyR is activated when its cysteine residues are oxidized by H2O2.32 Targets include, for example, ahpCF, fur, trxA, and gor, most of which were not differentially expressed in our experiments. The small RNA OxyS was not captured due to size exclusion steps in the library preparation method. Genes katG, dps, the sufABCD operon, and mntH were downregulated in Fab-producing strains and the BL21(DE3) wildtype alike. Interestingly, all Fab-producing strains showed upregulated transcript levels of grxA (0.8 > log2FC > 1.5) as the sole OxyR target in response to Fab production.
RNA-seq provides a snapshot of global transcription but no information about protein abundances. SoxS expression is regulated not only on transcription but also on the translation level by the small RNA MgrR in an Hfq-binding manner79 and by proteolysis.80 Therefore, no clear answer can be derived from the transcriptome data, why almost no target genes of the SoxRS regulon were upregulated, while soxS transcription was activated. Possible explanations could be interferences with other regulatory pathways that affect expression either of SoxS or its target genes. For example, expression of MnSOD (sodA gene product) is regulated by four global transcription regulators in addition to SoxS (Fur, AcrA, Fnr, and IHF)81 and in a post-translational fashion.82
OxyR is activated by concentrations of H2O2 beyond 0.1 μM. However, high activity of peroxidase prevents accumulation of endogenously formed H2O2 exceeding 20 nM under nonstress conditions.21 Possibly, the concentration of H2O2 was not sufficient to saturate peroxidase activity and activate OxyR. Intriguingly, the gene product of the only upregulated OxyR target grxA (glutaredoxin-1) catalyzes reduction of activated OxyR via glutathione and therefore regulates OxyR in a negative feedback loop.83 Probably, other yet unknown transcription activators of grxA exist. Also, others have reported upregulation of the SoxRS but not the OxyR regulon under artificial oxidative stress conditions.39,77,84 Further investigations, for example, by means of proteomics or by measuring H2O2 levels, would be needed to shed more light on the observed results.
Gene ontology (GO) term enrichment analysis was performed to further explore the RNA-seq data. Among others (see Tables S2–S5), we identified the following enriched GO terms related to biological processes after 12 h of induction compared to the reference: “Cellular response to toxic substance” (GO: 0097237) in all Fab-producing strains, “Cellular response to oxidative stress” (GO: 0034599) in B⟨oBIBH1⟩ and B⟨oFTN2⟩, and “Response to oxidative stress” in BL21(DE3), B⟨oBIBH1⟩, and B⟨oFTN2⟩. Transcript levels of most genes within the three groups were downregulated, including the already described MnSOD (sodA). SodA is one of the three SODs in E. coli that contain different co-factors and are not functionally equivalent. Periplasmic CuZnSOD (sodC) which is expressed in an RpoS-dependent fashion in the stationary phase85 was downregulated as well in all strains including the BL21(DE3) wildtype (Figure S7). Within the enriched groups, sodB (coding for cytoplasmic FeSOD) was one of the few genes that showed expression upregulation. Transcript levels were increased in a Fab expression-dependent manner. Nevertheless, basal MnSOD and slightly increased FeSOD levels apparently were not sufficient to suppress SoxR activation during Fab expression.
Downregulation of nuo and Upregulation of ndh in Fab-Producing Strains
GO term enrichment analysis revealed an impact of Fab expression on the respiratory chain, especially on expression of NDH I. We found that transcription of the nuo operon coding for subunit proteins of NDH I was downregulated after 12 h (Figure 5) and 16 h of induction (Figure S8). Log2FC varied from −0.3 (nuoA in B⟨oFTN2⟩) to −1.7 (nuoH in B⟨oFabx⟩) depending on the gene and strain. Fabx expression caused the most pronounced downregulation. Additionally, ndh (NDH II) transcript levels were increased in B⟨oFabx⟩ (log2FC of 1.7 at 12 h and 1.5 at 16 h) and B⟨oBIWA4⟩ (log2FC of 0.6 at 12 h). The wildtype reference also showed slight upregulation at 16 h. We observed no DGE of NDH genes at 2 h of induction. Since no data points were analyzed between 2 and 12 h, possible dynamics of nuo downregulation and ndh upregulation between these two time points are unknown.
Figure 5.

Log2FC of genes coding for NDH I (nuo operon) and NDH II (ndh) after 12 h of induction relative to the sample drawn prior to induction as determined by RNA-seq (α ≤ 0.05, n = 3).
During glucose-limited growth, the electron flux is directed through both NADH dehydrogenases in the presence of O2.58 Although both dehydrogenases oxidize NADH to NAD+, only NDH I contributes to the generation of the PMF (2 H+/e–). Therefore, the degree of coupling between electron transfer and H+ translocation (and hence ATP generation) depends on the distribution of e– flux through NDH I and II (and between oxidases cytochrome bo and bd).58,86 Expression of nuo is influenced by growth conditions and ATP requirements.87,88 Growth impairment might have influenced downregulation of the operon in the strains producing the recombinant proteins. Since Fab productivity ceased toward the end of the process, ATP demand by recombinant protein production probably did as well. Downregulation of nuo and upregulation of ndh might indicate diverted electron flux from NDH I to NDH II. A change in usage of the two NADH dehydrogenases or rather increased activity of NDH II could have an implication for the formation of O2•–, since one of the sources of ROS is the autoxidizable flavin cofactor of NDH II.
Conclusions
Fab’s are proteins that are challenging to produce in microbes, owing to various reasons. During periplasmic expression, factors such as translocation, folding (especially the necessity for disulfide bonds), and balancing expression between the two chains increase complexity and impact expression.89,90 Here, we identified additional, previously uncharacterized implications of periplasmic Fab expression in E. coli: (1) accumulation of superoxide and transcription activation of the oxidative stress-responsive gene soxS and (2) an insufficient ubiquinone availability to meet oxidative folding demand. Ubiquinone is used for electron transport in oxidative folding and in the respiratory chain, which is a major site of O2•– formation; hence, the two observations are possibly connected and depend on process conditions. Oxidative stress and interference with the respiratory chain may have been involved in eliciting increased cell lysis and metabolic changes during Fab production in fed-batch processes, which impacted processability of the fermentation broth. A more detailed analysis, for example, of secreted metabolites would be desirable to further characterize shifts in energy metabolism. Within this study, we mainly focused on undesired O2•– formation and ubiquinone deficiency. Under production conditions, increased O2•– formation is an additional metabolic load and stress for the host cell. Oxidative stress was indicated by elevated transcript levels of soxS in all Fab production fed-batch processes which indicated activation of the SoxR transcription factor (presumably by O2•–). The source of O2•– in our experiments remains unidentified. There are multiple possible candidates (including NDH II); therefore, it might be difficult to pinpoint a single source as the one responsible for the observed increased O2•– levels. An RNA-seq analysis revealed that due to the glucose-limited growth conditions in our setup, different regulatory pathways were apparently counteracting (e.g., preventing increased expression of SoxS target sodA coding for ROS-scavenging MnSOD). Thereby, process conditions presumably impeded the cells’ capability to mitigate the harmful effects via the inducible oxidative stress response. This observation emphasizes the importance of applying relevant production conditions for comprehensive characterization of stress induced by recombinant protein expression (in our case, expression of Fab’s in a fed-batch process). Otherwise, conclusions drawn from small-scale batch experiments might not be valid under actual production conditions. The fact that supplementation of CoQ1 substantially improved Fab expression pointed toward ubiquinone shortage when the quinone pool is partitioned between dehydrogenases of the respiratory chain and oxidative folding. By artificially enhancing the available ubiquinone pool, for example, by supplementing UQ8 analogues, the cells’ disulfide bond formation capacity could be increased. The observation that the four studied Fab’s showed the same behavior albeit to a different extent can probably be explained by the heterodimeric nature of the model proteins. The HCs and LCs differ in sequence, which impacts expression and folding and dimerization of the chains and in further consequence accumulation of total recombinant protein. O2 consumption, O2•– formation, and the achievable improvement of Fab yield by CoQ1 addition are not solely influenced by the correctly folded, soluble (quantifiable) Fab’s. Instead, the mixture of Fab, free chains, possible dimers, or other derivatives can vary in composition46 and has a level of impact that is hard to assess. Additionally, the different tendencies of the different Fab’s and Fab-derived molecules to aggregate (possibly even before disulfide bonds are formed) presumably play a part as well. Therefore, some questions remain to be studied in more detail, for example, by using additional, monomeric model proteins with varying numbers of disulfide bonds. Another intriguing possibility is the comparison between periplasmic and cytoplasmic expression of disulfide-containing proteins regarding the interactions between recombinant protein expression and the redox balance (e.g., ROS formation). Currently, several systems for the cytoplasmic production of disulfide bond-containing proteins in E. coli are available (such as SHuffle, Origami, and CysDisCo). These systems rely either on disruption of reducing pathways or co-expression of sulfhydryl oxidases and disulfide bond isomerases to facilitate oxidative folding within the usually reducing environment of the cytoplasm.91 In the future, our data could aid in the development of additional strategies to obtain E. coli strains capable of counteracting some of the negative effects of Fab expression, thereby providing better yields and processability through improved cell fitness.
Materials and Methods
Bacterial Strains
All used strains originated from E. coli BL21(DE3) (New England Biolabs (NEB), USA) and are listed in Table 1. Enzymes and kits for generation of the strains were purchased from NEB (USA). All constructs were confirmed by Sanger sequencing (Microsynth AG, Switzerland).
The design of the four different model Fab’s (Fabx, BIBH1, BIWA4, and FTN2), the construction of the integration cassettes, and the genome integration procedure of the constructs are described in detail in our previous work.41 Briefly, the Fab LCs and HCs were both fused to ompASS for post-translational translocation to the periplasm. LC and HC were expressed as bicistronic constructs, and each chain was equipped with its own ribosome-binding site. We used production systems with a single copy of the gene of interest integrated into the bacterial chromosome at the attTn7 site. Construction of the reference strain BL21(DE3) expressing GFPmut3.1 from a single copy integrated into the genome is described elsewhere.92 Since sfGFP exhibits fluorescence regardless of cellular localization, it was used as periplasmic reference protein. The co-translational dsbASS was used to mitigate cytosolic fluorescence. The dsbASS-sfGFP construct was amplified from an in-house pET30a plasmid according to the same procedure as the Fab’s41 and integrated into the genome of BL21(DE3).93
Media and Cultivation Conditions
Cell Banks
Master cell banks (MCBs) were prepared from cells grown in M9ZB medium. Baffled shake flasks were inoculated with a single colony and incubated at 37 °C and 180 rpm. Exponentially growing cultures were mixed 1:2 with 80% glycerol (Merck, Germany) when OD600 had reached 3.5 and aliquots were frozen at −80 °C. Working cell banks (WCBs) were inoculated from MCBs and grown in baffled shake flasks in semisynthetic medium (SSM). WCBs were cultured at 37 °C and 180 rpm. Like for the MCBs, cell aliquots were frozen in 40% glycerol at −80 °C after cultures had reached an OD600 of 3.5.
Shake Flask Cultivation
Shake flasks were grown in 25 mL of SSM in 250 mL baffled shake flasks. The cultures were inoculated from overnight cultures at on OD600 of 0.1 and grown at 37 °C and 180 rpm on an orbital shaker. Fab production was induced at an OD600 of 1.0 with 0.5 mM IPTG and the cultures were transferred to 30 °C. Cells were harvested after 4 h of production, and pellets equivalent to 1 mg CDM were frozen at −20 °C.
Fed-Batch-like Cultivation in a Microbioreactor System
Microscale cultivations were performed in the BioLector system (m2p-labs GmbH, Germany) as described by ref (41) with some modifications. Feed-in-time (FIT) medium containing 1 g L–1 glucose and 33 g L–1 EnPump200 dextran (Enpresso GmbH, Germany) was used in all experiments. Enzyme-mediated release of glucose (Carl Roth, Germany) was achieved by the addition of 0.6% (v v–1) EnzMix (Enpresso GmbH, Germany). FIT medium was supplemented with (1) 148.3 mM MOPS, (2) 1.7 μM CoCl2·6H2O, (3) 56.1 mM (NH4)2SO4, (4) 12.8 mM K2HPO4, (5) 7.6 mM Na3Citrate·2H2O, (6) 3.1 M MgSO4·7H2O, (7) 1.4 μM ZnSO4·7H2O, (8) 114.4 μM FeCl3·6H2O, (9) 10.4 mM Na2SO4, (10) 22.0 μM Thiamine·HCl, (11) 1.5 μM CuSO4·5H2O, (12) 1.3 μM MnSO4·H2O, (13) 66.5 μM Titriplex III, (14) 13.9 mM NH4Cl, and (15) 10.1 μM CaCl2·2H2O. (1) and (2) were purchased from Sigma-Aldrich, USA, (3–8) from Carl Roth, Germany, (9–13) from Merck, Germany, and (14) and (15) from Applichem, Germany. 48-well flower plates (m2p-labs GmbH, Germany) with a working volume of 800 μL were used. The cultures were inoculated from WCBs with an initial OD600 of 0.3. Temperature was maintained at 30 °C, the shaking frequency at 1400 rpm, and the humidity at >85%. Biomass accumulation was analyzed as described by ref (41). Additionally, biomass of endpoint samples was determined gravimetrically. Fab production was induced with 0.5 mM IPTG (GERBU Biotechnik, Germany) after 9 h of cultivation. Endpoint samples were drawn 12 h after induction.
CoQ1 Supplementation
CoQ1 was supplemented to shake flask cultivations or selected wells of microtiter cultivations at induction. CoQ1 was obtained from Sigma-Aldrich, USA, and a 200 mM stock solution was prepared in acetone. The stock was further diluted to 100× the final concentrations in 50% ethanol and the respective amount added to the cultivations.
Fed-Batch Cultivation
Fed-batch cultivations were conducted in a DASGIP Parallel Bioreactor System (Eppendorf AG, Germany) as described by ref 9494 with 0.6 L batch volume and 1.25 L final working volume. Reactors were equipped with standard control units and a GA4X-module (Eppendorf AG, Germany) for off-gas analysis. Temperature was maintained at 37 ± 0.5 °C during the batch phase and was decreased to 30 ± 0.5 °C at the beginning of the feeding phase. The pH was kept constant at 7.00 ± 0.05 by the addition of 12.5% ammonia solution (w w–1). DO was set to 30% and maintained by adjusting the stirrer speed, aeration rate, and in-gas composition. The batch was inoculated from precultures according to ref (94). Cells were grown to 6 g in the batch phase after which the exponential carbon-limited feed was started. The growth rate was controlled at μ = 0.1 h–1. Recombinant protein production was induced after 3 h of feed with 2 μmol IPTG g–1 CDM. Production was continued for 16 h (approx. 2 generations) resulting in a theoretical biomass of 40 g. Components for batch and fed-batch media were obtained from Carl Roth, Germany. Media were prepared gravimetrically according to final biomass. Glucose was added to batch and fed-batch media according to the theoretical final biomass based on a yield coefficient of YX/S = 0.3 g/g. Compositions of batch and fed-batch media are described elsewhere.94 To prevent the formation of foam, PPG2000 antifoam (BASF, Germany) was added on demand. Cultivations used for RNA-seq were conducted in triplicate.
Offline Analytics
Biomass Quantification
CDM from fed-batch cultivations was determined gravimetrically as described by ref (94).
Sampling and Cell Disruption
To analyze recombinant protein production, cell aliquots corresponding to 1 mg of CDM were sampled. Endpoint samples were drawn from shake flask and microscale cultivations. Samples from fed-batch fermentations were drawn every 2 h. The aliquots were pelleted (10 min, 13,000g) and frozen at −20 °C. Total protein was extracted enzymatically as described by ref (41).
Samples for RNA-seq and qPCR were drawn from fed-batch cultivation preinduction and after 2, 12, and 16 h of induction. Sampled cells were immediately transferred into 0.5× the volume of a 5% phenol in ethanol solution on ice. 3 mg CDM aliquots were spun down at 4 °C and 13,000g for 2 min and stored at −80 °C.
Analysis of Recombinant Protein by WB
Expression of Fab and LCs in the soluble and IB fraction of cell lysates and in the culture supernatant were analyzed by LC-specific WB analysis using anti-human κ-LC (bound and free) goat antibody, conjugated to alkaline phosphatase (A3813; Sigma-Aldrich, USA) as described by ref (41).
Quantification of Fab by ELISA
Fab in the soluble fraction of cell extracts and the culture supernatant was quantified using a sandwich ELISA as described by ref (41).
Quantification of GFP by Fluorometry Analysis
GFP was quantified using a Tecan analyzer infinite 200Pro (485/520 nm) and a calibration curve constructed with in-house-purified GFP as previously described.92
Flow Cytometric Superoxide Measurement
Intracellular superoxide levels were measured flow cytometrically using CellROX Green reagent (Invitrogen, USA). Cells were sampled from the respective cultivations and diluted to a final OD600 of 0.035 in 1× PBS. CellROX Green reagent was thawed, diluted in 1× PBS (flow cytometry grade), and added to the cells at a final concentration of 4 μM. The optimal ratio of cell to dye concentration (114× CellROX Green) had been determined in preliminary experiments and was defined as the concentration at which a maximal signal was obtained, without using excessive amounts of dye. The cells were incubated at 37 °C for 30 min with gentle mixing for staining. Once thawed, the CellROX Green reagent aliquots were protected from light and used within 2 h. Cells treated with 350 μM MD served as a positive control. MD treatment was included during CellROX Green staining. Samples were measured on a CytoFLEX S flow cytometer (Beckman Coulter, USA). CellROX Green reagent was excited by a 488 nm laser and emission detected with a 525/40 nm band pass filter. The flow rate was set to 60 μL min–1, and 15,000 events were collected for each sample. Samples were analyzed in biological triplicate. The obtained data were analyzed using the CytExpert Software (Beckman Coulter, USA). E. coli-sized particles were gated in FSC/SSC plots to remove small particles and cell debris (Figure S9).
Gene Expression Analysis
RNA Extraction
Cell pellets were thawed, and cells were disrupted for 10 min with 10 mg mL–1 lysozyme in TE-buffer (1 mM EDTA, 10 mM Tris–HCl) while vortexing. Then, TRIzol reagent (Invitrogen, USA) was added to a final cell count of approx. 1.5 × 108 and the samples were incubated for another 5 min on the vortex. RNA was extracted using a Direct-zol RNA Miniprep Kit (Zymo Research, USA) according to the manufacturer’s instructions. An equal volume of ethanol was added to the samples in TRIzol. The samples were transferred to a Zymo-Spin Mini column, spun down at 13,400g for 30 s, and washed. To remove DNA, the samples were treated with DNase I for 30 min according to the kit manual. After three washing steps, the RNA was eluted in 25 μL of nuclease-free water. Quantification of total RNA and assessment of protein and phenol contamination was performed using a NanoDrop ND-1000 (Thermo Fisher, USA). RNA integrity and the absence of genomic DNA were analyzed with an Agilent 2100 Bioanalyzer using the RNA 6000 Nano Kit (Agilent Technologies, USA). Only samples with an RNA integrity number (RIN) > 8 were used. RNA extracts were stored at −80 °C.
RNA-Seq Library Preparation and Sequencing
Ribosomal RNA removal was performed with the Ribo-Zero rRNA Removal Kit Bacteria (Illumina, USA). Sequencing libraries of the rRNA depleted samples were prepared using the NEBNext Ultra II Directional RNA Library Prep Kit for Illumina (New England Biolabs, USA). Libraries were sequenced in single-read mode on a HiSeq 2500 system (Illumina, USA). rRNA depletion, library preparation, and sequencing were performed by the Next Generation Sequencing Facility at Vienna BioCenter Core Facilities (VBCF), member of the Vienna BioCenter (VBC), Austria.
Sequencing Read Preprocessing and Mapping
The quality and the adapter content of the raw RNA-seq reads were analyzed with FastQC.95 Then, the raw reads were trimmed with Trimmomatic v0.3896 to remove adapters and low-quality reads. Reads with a Phred quality score ≥ 25 and a length ≥ 35 bp after trimming were kept for further analysis. The trimming was conducted providing the NEBNext adapter sequence as a template to assess and remove any adapter content. The quality and the adapter content of the trimmed reads were then re-assessed with FastQC and a summary was obtained with MultiQC.97 The quality-trimmed RNA-seq reads from each sample were mapped onto the corresponding reference genome. The reference genome of E. coli BL21(DE3) used for mapping of the RNA-seq reads had been previously determined in-house by whole genome sequencing.92 The mapping was conducted with HISAT298 using the following adapted parameters: --score-min L,0.0,-0.2 --no-spliced-alignment --no-softclip. The remaining parameters of the program were left as default. The BAM files resulting from the mapping were filtered with samtools99 removing unmapped reads and secondary alignments (-F0x4-F0x0100). The filtered BAM files were sorted and indexed with samtools.
DGE Analysis
The filtered, sorted, and indexed BAM files were used together with the GFF annotation of the reference genome to count read occurrences at each gene with HTSeq-count.100 The following parameters were used: --format bam --order pos --stranded reverse --minaqual 20 --type exon --mode union --secondary-alignments ignore --supplementary-alignments ignore --idattr Name. Read counts per gene produced by HTSeq for each sample were merged into a single table of counts with a custom python script. The table of counts was used as input to calculate differentially expressed genes (DEGs) with DESeq2.101 Genes with an average of less than 10 counts across all replicates were excluded from the analysis. A normalized distribution of counts was obtained with samples within the function DESeq(sfType = “poscounts”). DEGs were computed within the same strains at different time points, comparing 2, 12, and 16 h postinduction samples against their corresponding noninduced (0 h) samples. DEGs were calculated with the function res(alpha = 0.05, altHypothesis = “greaterAbs”, lfcThreshold = 0.1). Computed log2FoldChanges were reduced using the function lfcShrink(type = “ashr”). Genes showing a log2FC ≥ 0.1 (in absolute value) and a p-value ≤ 0.05 were considered differentially expressed and statistically significant.
GO Term Analysis
The potential enrichment of any GO term102 in the DEGs was assessed in a custom R script with ClusterProfiler.103 In each sample, the enrichment of the GO terms associated to each DEG (i.e., “Selection”) was compared to their abundance in the complete E. coli gene set (i.e., “Universe”, source: ecocyc). The library “org.EcK12.eg.db” was used to convert gene aliases to Entrez Gene IDs using the function “org.EcK12.egALIAS2EG”. Enriched GO Terms were extracted using the function enrichGO(), targeting biological process (“BP”), molecular function (“MF”), and cellular component (“CC”) terms in three separate runs. The complete function arguments were declared as follows: enrichGO(Selection, org.EcK12.eg.db, keyType = “ENTREZID”, ont = “BP”, pvalueCutoff = 0.05, pAdjustMethod = “BH”, Universe, qvalueCutoff = 0.05, minGSSize = 15, maxGSSize = 500, readable = TRUE, pool = FALSE). The “ont” argument was changed to MF and CC according to the type of GO term assessed. Resulting enriched GO terms were simplified merging similar GO terms using the simplify() function, with a similarity cutoff of 0.8. Simplified enriched GO terms were then filtered using the function gofilter(level = ...). For each of the three runs (BP, MF, and CC), four independent filtering runs were generated, each at a different GO term level (3, 4, 5, and 6), representing increasing depths of GO term characterization.
Reverse Transcription
Approx. 1.5 μg of total RNA was reverse-transcribed to cDNA using Superscript III Reverse Transcriptase (Invitrogen, USA) in 20 μL reaction volume according to the manufacturer’s instructions. 230 ng of random hexamer primers was used; hence, the reaction mixtures were incubated for 10 min at room temperature. Reverse transcription was performed at 50 °C for 50 min. 40 U of RNasin Ribonuclease Inhibitor (Promega, USA) were added to the reaction mixtures. No reverse transcriptase controls were prepared for all samples by replacing Superscript III Reverse Transcriptase with nuclease-free water. Reverse-transcribed samples were treated with 5 U RNase H (NEB, USA) for 20 min at 37 °C to remove the RNA template. A Qubit 4 fluorometer was used to quantify cDNA with a Qubit dsDNA HS Assay Kit (Invitrogen, USA) according to the manufacturer’s instructions. Samples were stored at −20 °C.
qPCR
Efficiencies and melting curves of selected primer pairs (Table S6, purchased from Sigma-Aldrich, USA) binding to transcripts of the target soxS and the reference gene cysG were evaluated using fivefold dilution series of the pooled samples as a template (Figure S10). Stability of cysG transcript levels and similar expression between all tested strains had been previously determined by RNA-seq (Figure S11). 2× iQ SYBR Green Supermix (Bio-Rad, USA) was used according to manufacturer’s instructions. Primers were diluted to a final concentration of 300 nM. Approx. 1 ng of cDNA samples was used in a reaction volume of 20 μL. No template controls were included. The PCRs were carried out in white 48-well PCR plates (Bio-Rad, USA) in a MiniOpticon Real-Time PCR System (Bio-Rad, USA). Thermocycling included an initial denaturation and enzyme activation step (95 °C, 3 min), 39 cycles of denaturing (95 °C, 10 s), annealing (62 °C, 30 s), and extension (72 °C, 30 s), and determination of the melt curve (55–95 °C in 0.5 °C increments with 20 s holding time). All cDNA samples were measured in triplicate. CFX Manager Software (Bio-Rad, USA) was used for data analysis. Quantification cycles (Cq) were determined in the single threshold mode (automatic). Expression of the target gene was normalized to the reference gene according to the ΔΔCq method. The noninduced sample was used as a reference.
Acknowledgments
The financial support provided by the Austrian Federal Ministry for Digital and Economic Affairs, the National Foundation for Research, Technology, and Development, and the Christian Doppler Research Association is gratefully acknowledged. Furthermore, we gratefully acknowledge the financial support received from our industry partner, Boehringer Ingelheim RCV GmbH & Co KG. We thank the colleagues of BI RCV for the scientific input and their continuous scientific support. We thank Christopher Tauer, Florian Simon, Mathias Fink, and Alexander Doleschal for providing help and knowledge regarding generation of the strains and fermentations and the student assistants who helped with protein analytics. The TOC figure was created with BioRender.com.
Glossary
Abbreviations
- CDM
cell dry mass
- CoQ1
coenzyme Q1
- DEGs
differentially expressed genes
- DGE
differential gene expression
- DMQ
demethylmenaquinone
- DO
dissolved oxygen
- E. coli
Escherichia coli
- ELISA
enzyme-linked immunosorbent assay
- ER
endoplasmic reticulum
- Fab
fragment antigen binding
- FIT
feed-in-time
- FSC
forward scatter
- (GeoMean) FI
(geometric mean of) fluorescence intensity
- GFP
green fluorescent protein
- GO
gene ontology
- HC
heavy chain
- IB
inclusion body
- IM
inner membrane
- IPTG
β-d-1-thiogalactopyranoside
- LC
light chain
- log2FC
log2 fold change
- MD
menadione
- MQ
menaquinone
- NDH I/II
NADH dehydrogenase I/II
- OD
optical density
- ompASS
ompA signal sequence
- PMF
proton motive force
- qCO2
CO2 formation rate
- qO2
O2 consumption rate
- RNA-seq
RNA sequencing
- ROS
reactive oxygen species
- SSC
side scatter
- SSM
semisynthetic medium
- UQ8
ubiquinone
- WB
western blot
- WCB
working cell bank
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acssynbio.1c00502.
% of total soluble Fab found extracellularly in the culture supernatant in fed-batch fermentations; Fab fragment expression patterns (LC-specific WB) obtained by fed-batch-like microtiter cultivations that were used for flow cytometric measurement of O2•–; histograms and FSC/SSC plots of flow cytometric measurements of B⟨oFabx⟩, B⟨oBIBH1⟩ and B⟨oBIWA4⟩ cells obtained from fed-batch-like microtiter cultivations; LC-specific WB of B⟨oFTN2⟩ shake flask cultivations without and with 5 μM CoQ1 supplementation; log2FC of differentially expressed members of the SoxRS and OxyR regulons 16 h postinduction as determined by RNA-seq; O2 [%] in the in-gas stream of fed-batch cultivations and log2FC of soxS transcript levels; log2FC of sodB and sodC in fed-batch cultivations; log2FC of the nuo operon, ndh and the ubi genes 16 h postinduction; gating of the E. coli population in flow cytometry data; efficiencies and melting peaks of primers used for soxS qPCR; and cysG transcript levels in fed-batch cultivations obtained by RNA-seq (PDF)
Transcriptomic data are available under the SRA BioProject PRJNA750301 (PDF)
Author Contributions
S.V. contributed to conception and design of the experiments, data analysis, and interpretation, performed the practical work, and drafted the manuscript; M.S. conducted the sequencing data analysis; M.W. contributed to data interpretation and paper draft revision; M.C.-P. contributed to study design, data interpretation, and paper draft revision; G.S. designed the study, revised the paper draft, and secured funding together with S.V.; and M.C.-P. prepared the manuscript. All authors approved the final manuscript.
The authors declare no competing financial interest.
Supplementary Material
References
- Ecker D. M.; Crawford T. J.; Seymour P.. The Therapeutic Monoclonal Antibody Product Market. BioProcess International, 2020, No. October 2020 Featured Report.
- Grilo A. L.; Mantalaris A. The Increasingly Human and Profitable Monoclonal Antibody Market. Trends Biotechnol. 2019, 37, 9–16. 10.1016/j.tibtech.2018.05.014. [DOI] [PubMed] [Google Scholar]
- Spadiut O.; Capone S.; Krainer F.; Glieder A.; Herwig C. Microbials for the Production of Monoclonal Antibodies and Antibody Fragments. Trends Biotechnol. 2014, 32, 54–60. 10.1016/j.tibtech.2013.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feige M. J.; Hendershot L. M.; Buchner J. How Antibodies Fold. Trends Biochem. Sci. 2010, 35, 189–198. 10.1016/j.tibs.2009.11.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gupta S. K.; Shukla P. Microbial Platform Technology for Recombinant Antibody Fragment Production: A Review. Crit. Rev. Microbiol. 2017, 43, 31–42. 10.3109/1040841X.2016.1150959. [DOI] [PubMed] [Google Scholar]
- Lobstein J.; Emrich C. A.; Jeans C.; Faulkner M.; Riggs P.; Berkmen M. SHuffle, a Novel Escherichia Coli Protein Expression Strain Capable of Correctly Folding Disulfide Bonded Proteins in Its Cytoplasm. Microb. Cell Fact. 2012, 11, 753. 10.1186/1475-2859-11-56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- de Marco A. Recombinant Antibody Production Evolves into Multiple Options Aimed at Yielding Reagents Suitable for Application-Specific Needs. Microb. Cell Fact. 2015, 14, 125. 10.1186/s12934-015-0320-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rosano G. L.; Ceccarelli E. A. Recombinant Protein Expression in Escherichia Coli: Advances and Challenges. Front. Microbiol. 2014, 5, 172. 10.3389/fmicb.2014.00172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tyo K. E.; Liu Z.; Petranovic D.; Nielsen J. Imbalance of Heterologous Protein Folding and Disulfide Bond Formation Rates Yields Runaway Oxidative Stress. BMC Biol. 2012, 10, 16. 10.1186/1741-7007-10-16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez J. L.; Meza E.; Petranovic D.; Nielsen J. The impact of respiration and oxidative stress response on recombinant α-amylase production by Saccharomyces cerevisiae. Metab. Eng. Commun. 2016, 3, 205–210. 10.1016/j.meteno.2016.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ali A. S.; Raju R.; Kshirsagar R.; Ivanov A. R.; Gilbert A.; Zang L.; Karger B. L. Multi-Omics Study on the Impact of Cysteine Feed Level on Cell Viability and mAb Production in a CHO Bioprocess. Biotechnol. J. 2019, 14, 1800352. 10.1002/biot.201800352. [DOI] [PubMed] [Google Scholar]
- Chevallier V.; Andersen M. R.; Malphettes L. Oxidative stress-alleviating strategies to improve recombinant protein production in CHO cells. Biotechnol. Bioeng. 2020, 117, 1172–1186. 10.1002/bit.27247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bader M.; Muse W.; Ballou D. P.; Gassner C.; Bardwell J. C. A. Oxidative Protein Folding Is Driven by the Electron Transport System. Cell 1999, 98, 217–227. 10.1016/S0092-8674(00)81016-8. [DOI] [PubMed] [Google Scholar]
- Inaba K.; Ito K. Structure and mechanisms of the DsbB-DsbA disulfide bond generation machine. Biochim. Biophys. Acta, Mol. Cell Res. 2008, 1783, 520–529. 10.1016/j.bbamcr.2007.11.006. [DOI] [PubMed] [Google Scholar]
- Unden G.; Bongaerts J. Alternative Respiratory Pathways of Escherichia Coli: Energetics and Transcriptional Regulation in Response to Electron Acceptors. Biochim. Biophys. Acta, Bioenerg. 1997, 1320, 217–234. 10.1016/S0005-2728(97)00034-0. [DOI] [PubMed] [Google Scholar]
- Noda S.; Takezawa Y.; Mizutani T.; Asakura T.; Nishiumi E.; Onoe K.; Wada M.; Tomita F.; Matsushita K.; Yokota A. Alterations of Cellular Physiology in Escherichia coli in Response to Oxidative Phosphorylation Impaired by Defective F 1 -ATPase. J. Bacteriol. 2006, 188, 6869–6876. 10.1128/JB.00452-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sharma P.; Teixeira de Mattos M. J.; Hellingwerf K. J.; Bekker M. On the Function of the Various Quinone Species in Escherichia Coli: The Role of DMK in the Electron Transfer Chains of E. Coli. FEBS J. 2012, 279, 3364–3373. 10.1111/j.1742-4658.2012.08608.x. [DOI] [PubMed] [Google Scholar]
- Korshunov S.; Imlay J. A. Detection and Quantification of Superoxide Formed within the Periplasm of Escherichia Coli. J. Bacteriol. 2006, 188, 6326–6334. 10.1128/JB.00554-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinsiek S.; Stagge S.; Bettenbrock K. Analysis of Escherichia Coli Mutants with a Linear Respiratory Chain. PLoS One 2014, 9, e87307 10.1371/journal.pone.0087307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borisov V. B.; Murali R.; Verkhovskaya M. L.; Bloch D. A.; Han H.; Gennis R. B.; Verkhovsky M. I. Aerobic Respiratory Chain of Escherichia Coli Is Not Allowed to Work in Fully Uncoupled Mode. Proc. Natl. Acad. Sci. U.S.A. 2011, 108, 17320–17324. 10.1073/pnas.1108217108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay J. A. Cellular Defenses against Superoxide and Hydrogen Peroxide. Annu. Rev. Biochem. 2008, 77, 755–776. 10.1146/annurev.biochem.77.061606.161055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sheng Y.; Abreu I. A.; Cabelli D. E.; Maroney M. J.; Miller A.-F.; Teixeira M.; Valentine J. S. Superoxide Dismutases and Superoxide Reductases. Chem. Rev. 2014, 114, 3854–3918. 10.1021/cr4005296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Touati D. Sensing and Protecting against Superoxide Stress in Escherichia Coli – How Many Ways Are There to Trigger SoxRS Response?. Redox Rep. 2000, 5, 287–293. 10.1179/135100000101535825. [DOI] [PubMed] [Google Scholar]
- Arts I. S.; Gennaris A.; Collet J.-F. Reducing Systems Protecting the Bacterial Cell Envelope from Oxidative Damage. FEBS Lett. 2015, 589, 1559–1568. 10.1016/j.febslet.2015.04.057. [DOI] [PubMed] [Google Scholar]
- Gu M.; Imlay J. A. The SoxRS Response of Escherichia Coli Is Directly Activated by Redox-Cycling Drugs Rather than by Superoxide: Redox-Cycling Drugs Directly Activate SoxR. Mol. Microbiol. 2011, 79, 1136–1150. 10.1111/j.1365-2958.2010.07520.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carlioz A.; Touati D. Isolation of Superoxide Dismutase Mutants in Escherichia Coli: Is Superoxide Dismutase Necessary for Aerobic Life?. EMBO J. 1986, 5, 623–630. 10.1002/j.1460-2075.1986.tb04256.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang S.; Imlay J. A. Micromolar Intracellular Hydrogen Peroxide Disrupts Metabolism by Damaging Iron-Sulfur Enzymes. J. Biol. Chem. 2007, 282, 929–937. 10.1074/jbc.M607646200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezraty B.; Gennaris A.; Barras F.; Collet J.-F. Oxidative Stress, Protein Damage and Repair in Bacteria. Nat. Rev. Microbiol. 2017, 15, 385–396. 10.1038/nrmicro.2017.26. [DOI] [PubMed] [Google Scholar]
- Ding H.; Hidalgo E.; Demple B. The Redox State of the [2Fe-2S] Clusters in SoxR Protein Regulates Its Activity as a Transcription Factor. J. Biol. Chem. 1996, 271, 33173–33175. 10.1074/jbc.271.52.33173. [DOI] [PubMed] [Google Scholar]
- Green J.; Paget M. S. Bacterial Redox Sensors. Nat. Rev. Microbiol. 2004, 2, 954–966. 10.1038/nrmicro1022. [DOI] [PubMed] [Google Scholar]
- Zheng M.; Åslund F.; Storz G. Activation of the OxyR Transcription Factor by Reversible Disulfide Bond Formation. Science 1998, 279, 1718–1722. 10.1126/science.279.5357.1718. [DOI] [PubMed] [Google Scholar]
- Aslund F.; Zheng M.; Beckwith J.; Storz G. Regulation of the OxyR Transcription Factor by Hydrogen Peroxide and the Cellular Thiol--Disulfide Status. Proc. Natl. Acad. Sci. U.S.A. 1999, 96, 6161–6165. 10.1073/pnas.96.11.6161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singh A. K.; Shin J.-H.; Lee K.-L.; Imlay J. A.; Roe J.-H. Comparative Study of SoxR Activation by Redox-Active Compounds: SoxR Activation by Redox-Active Compounds. Mol. Microbiol. 2013, 90, 983–996. 10.1111/mmi.12410. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liochev S. I.; Benov L.; Touati D.; Fridovich I. Induction of the SoxRS Regulon of Escherichia Coli by Superoxide. J. Biol. Chem. 1999, 274, 9479–9481. 10.1074/jbc.274.14.9479. [DOI] [PubMed] [Google Scholar]
- Fujikawa M.; Kobayashi K.; Kozawa T. Direct Oxidation of the [2Fe-2S] Cluster in SoxR Protein by Superoxide. J. Biol. Chem. 2012, 287, 35702–35708. 10.1074/jbc.M112.395079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greenberg J. T.; Monach P.; Chou J. H.; Josephy P. D.; Demple B. Positive Control of a Global Antioxidant Defense Regulon Activated by Superoxide-Generating Agents in Escherichia Coli. Proc. Natl. Acad. Sci. U.S.A. 1990, 87, 6181–6185. 10.1073/pnas.87.16.6181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsaneva I. R.; Weiss B. SoxR, a Locus Governing a Superoxide Response Regulon in Escherichia Coli K-12. J. Bacteriol. 1990, 172, 4197–4205. 10.1128/JB.172.8.4197-4205.1990. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krapp A. R.; Humbert M. V.; Carrillo N. The SoxRS Response of Escherichia Coli Can Be Induced in the Absence of Oxidative Stress and Oxygen by Modulation of NADPH Content. Microbiology 2011, 157, 957–965. 10.1099/mic.0.039461-0. [DOI] [PubMed] [Google Scholar]
- Pomposiello P. J.; Bennik M. H. J.; Demple B. Genome-Wide Transcriptional Profiling of TheEscherichia Coli Responses to Superoxide Stress and Sodium Salicylate. J. Bacteriol. 2001, 183, 3890–3902. 10.1128/JB.183.13.3890-3902.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Imlay J. A.; Fridovich I. Assay of Metabolic Superoxide Production in Escherichia Coli. J. Biol. Chem. 1991, 266, 6957–6965. 10.1016/s0021-9258(20)89596-9. [DOI] [PubMed] [Google Scholar]
- Fink M.; Vazulka S.; Egger E.; Jarmer J.; Grabherr R.; Cserjan-Puschmann M.; Striedner G. Microbioreactor Cultivations of Fab-ProducingEscherichia coliReveal Genome-Integrated Systems as Suitable for Prospective Studies on Direct Fab Expression Effects. Biotechnol. J. 2019, 14, 1800637. 10.1002/biot.201800637. [DOI] [PubMed] [Google Scholar]
- Mairhofer J.; Scharl T.; Marisch K.; Cserjan-Puschmann M.; Striedner G. Comparative Transcription Profiling and In-Depth Characterization of Plasmid-Based and Plasmid-Free Escherichia Coli Expression Systems under Production Conditions. Appl. Environ. Microbiol. 2013, 79, 3802–3812. 10.1128/AEM.00365-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cormack B. P.; Valdivia R. H.; Falkow S. FACS-Optimized Mutants of the Green Fluorescent Protein (GFP). Gene 1996, 173, 33–38. 10.1016/0378-1119(95)00685-0. [DOI] [PubMed] [Google Scholar]
- Garcia-Ochoa F.; Gomez E.; Santos V. E.; Merchuk J. C. Oxygen Uptake Rate in Microbial Processes: An Overview. Biochem. Eng. J. 2010, 49, 289–307. 10.1016/j.bej.2010.01.011. [DOI] [Google Scholar]
- Marr A. G. Growth Rate of Escherichia Coli. Microbiol. Rev. 1991, 55, 316–333. 10.1128/mr.55.2.316-333.1991. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schimek C.; Kubek M.; Scheich D.; Fink M.; Brocard C.; Striedner G.; Cserjan-Puschmann M.; Hahn R. Three-Dimensional Chromatography for Purification and Characterization of Antibody Fragments and Related Impurities from Escherichia Coli Crude Extracts. J. Chromatogr. A 2021, 1638, 461702. 10.1016/j.chroma.2020.461702. [DOI] [PubMed] [Google Scholar]
- Tu B. P.; Weissman J. S. Oxidative Protein Folding in Eukaryotes: Mechanisms and Consequences. J. Cell Biol. 2004, 164, 341–346. 10.1083/jcb.200311055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tu B. P.; Weissman J. S. The FAD- and O2-Dependent Reaction Cycle of Ero1-Mediated Oxidative Protein Folding in the Endoplasmic Reticulum. Mol. Cell 2002, 10, 983–994. 10.1016/S1097-2765(02)00696-2. [DOI] [PubMed] [Google Scholar]
- Sevier C. S.; Kaiser C. A. Ero1 and Redox Homeostasis in the Endoplasmic Reticulum. Biochim. Biophys. Acta, Mol. Cell Res. 2008, 1783, 549–556. 10.1016/j.bbamcr.2007.12.011. [DOI] [PubMed] [Google Scholar]
- Berkmen M. Production of Disulfide-Bonded Proteins in Escherichia Coli. Protein Expression Purif. 2012, 82, 240–251. 10.1016/j.pep.2011.10.009. [DOI] [PubMed] [Google Scholar]
- Joly J. C.; Swartz J. R. In Vitro and in Vivo Redox States of the Escherichia Coli Periplasmic Oxidoreductases DsbA and DsbC. Biochemistry 1997, 36, 10067–10072. 10.1021/bi9707739. [DOI] [PubMed] [Google Scholar]
- Schäffner J.; Winter J.; Rudolph R.; Schwarz E. Cosecretion of Chaperones and Low-Molecular-Size Medium Additives Increases the Yield of Recombinant Disulfide-Bridged Proteins. Appl. Environ. Microbiol. 2001, 67, 3994–4000. 10.1128/AEM.67.9.3994-4000.2001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar D.; Batra J.; Komives C.; Rathore A. QbD Based Media Development for the Production of Fab Fragments in E. Coli. Bioengineering 2019, 6, 29. 10.3390/bioengineering6020029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campani G.; Gonçalves da Silva G.; Zangirolami T. C.; Perencin de Arruda Ribeiro M. Recombinant Escherichia Coli Cultivation in a Pressurized Airlift Bioreactor: Assessment of the Influence of Temperature on Oxygen Transfer and Uptake Rates. Bioprocess Biosyst. Eng. 2017, 40, 1621–1633. 10.1007/s00449-017-1818-7. [DOI] [PubMed] [Google Scholar]
- Knyazev D. G.; Kuttner R.; Zimmermann M.; Sobakinskaya E.; Pohl P. Driving Forces of Translocation Through Bacterial Translocon SecYEG. J. Membr. Biol. 2018, 251, 329–343. 10.1007/s00232-017-0012-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riedel T. E.; Berelson W. M.; Nealson K. H.; Finkel S. E. Oxygen Consumption Rates of Bacteria under Nutrient-Limited Conditions. Appl. Environ. Microbiol. 2013, 79, 4921–4931. 10.1128/AEM.00756-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalnenieks U.; Balodite E.; Rutkis R. Metabolic Engineering of Bacterial Respiration: High vs. Low P/O and the Case of Zymomonas Mobilis. Front. Bioeng. Biotechnol. 2019, 7, 327. 10.3389/fbioe.2019.00327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun M. W.; Oden K. L.; Gennis R. B.; de Mattos M. J.; Neijssel O. M. Energetic Efficiency of Escherichia Coli: Effects of Mutations in Components of the Aerobic Respiratory Chain. J. Bacteriol. 1993, 175, 3020–3025. 10.1128/JB.175.10.3020-3025.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- San K.-Y.; Bennett G. N.; Berríos-Rivera S. J.; Vadali R. V.; Yang Y.-T.; Horton E.; Rudolph F. B.; Sariyar B.; Blackwood K. Metabolic Engineering through Cofactor Manipulation and Its Effects on Metabolic Flux Redistribution in Escherichia Coli. Metab. Eng. 2002, 4, 182–192. 10.1006/mben.2001.0220. [DOI] [PubMed] [Google Scholar]
- Berríos-Rivera S. Metabolic Engineering of Escherichia Coli: Increase of NADH Availability by Overexpressing an NAD+-Dependent Formate Dehydrogenase. Metab. Eng. 2002, 4, 217–229. 10.1006/mben.2002.0227. [DOI] [PubMed] [Google Scholar]
- Castan A.; Näsman A.; Enfors S.-O. Oxygen Enriched Air Supply in Escherichia Coli Processes: Production of Biomass and Recombinant Human Growth Hormone. Enzyme Microb. Technol. 2002, 30, 847–854. 10.1016/S0141-0229(01)00490-2. [DOI] [Google Scholar]
- McBee M. E.; Chionh Y. H.; Sharaf M. L.; Ho P.; Cai M. W. L.; Dedon P. C. Production of Superoxide in Bacteria Is Stress- and Cell State-Dependent: A Gating-Optimized Flow Cytometry Method That Minimizes ROS Measurement Artifacts with Fluorescent Dyes. Front. Microbiol. 2017, 8, 459. 10.3389/fmicb.2017.00459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seaver L. C.; Imlay J. A. Are Respiratory Enzymes the Primary Sources of Intracellular Hydrogen Peroxide?. J. Biol. Chem. 2004, 279, 48742–48750. 10.1074/jbc.M408754200. [DOI] [PubMed] [Google Scholar]
- Messner K. R.; Imlay J. A. The Identification of Primary Sites of Superoxide and Hydrogen Peroxide Formation in the Aerobic Respiratory Chain and Sulfite Reductase Complex of Escherichia Coli. J. Biol. Chem. 1999, 274, 10119–10128. 10.1074/jbc.274.15.10119. [DOI] [PubMed] [Google Scholar]
- Zhu J.; Sánchez A.; Bennett G. N.; San K.-Y. Manipulating Respiratory Levels in Escherichia Coli for Aerobic Formation of Reduced Chemical Products. Metab. Eng. 2011, 13, 704–712. 10.1016/j.ymben.2011.09.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aussel L.; Pierrel F.; Loiseau L.; Lombard M.; Fontecave M.; Barras F. Biosynthesis and Physiology of Coenzyme Q in Bacteria. Biochim. Biophys. Acta, Bioenerg. 2014, 1837, 1004–1011. 10.1016/j.bbabio.2014.01.015. [DOI] [PubMed] [Google Scholar]
- Nitzschke A.; Bettenbrock K. All Three Quinone Species Play Distinct Roles in Ensuring Optimal Growth under Aerobic and Fermentative Conditions in E. Coli K12. PLoS One 2018, 13, e0194699 10.1371/journal.pone.0194699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wallace B. J.; Young I. G. Role of Quinones in Electron Transport to Oxygen and Nitrate in Escherichia Coli. Studies with a UbiA– MenA– Double Quinone Mutant. Biochim. Biophys. Acta, Bioenerg. 1977, 461, 84–100. 10.1016/0005-2728(77)90071-8. [DOI] [PubMed] [Google Scholar]
- Cox G. B.; Newton N. A.; Gibson F.; Snoswell A. M.; Hamilton J. A. The Function of Ubiquinone in Escherichia Coli. Biochem. J. 1970, 117, 551–562. 10.1042/bj1170551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobayashi T.; Kishigami S.; Sone M.; Inokuchi H.; Mogi T.; Ito K. Respiratory Chain Is Required to Maintain Oxidized States of the DsbA-DsbB Disulfide Bond Formation System in Aerobically Growing Escherichia Coli Cells. Proc. Natl. Acad. Sci. U.S.A. 1997, 94, 11857–11862. 10.1073/pnas.94.22.11857. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jaswal K.; Shrivastava M.; Roy D.; Agrawal S.; Chaba R. Metabolism of Long-Chain Fatty Acids Affects Disulfide Bond Formation in Escherichia Coli and Activates Envelope Stress Response Pathways as a Combat Strategy. PLoS Genet. 2020, 16, e1009081 10.1371/journal.pgen.1009081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Newton N. A.; Cox G. B.; Gibson F. Function of Ubiquinone in Escherichia Coli: A Mutant Strain Forming a Low Level of Ubiquinone. J. Bacteriol. 1972, 109, 69–73. 10.1128/JB.109.1.69-73.1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kwon O.; Druce-Hoffman M.; Meganathan R. Regulation of the Ubiquinone (Coenzyme Q) Biosynthetic Genes UbiCA in Escherichia Coli. Curr. Microbiol. 2005, 50, 180–189. 10.1007/s00284-004-4417-1. [DOI] [PubMed] [Google Scholar]
- Martínez I.; Zelada P.; Guevara F.; Andler R.; Urtuvia V.; Pacheco-Leyva I.; Díaz-Barrera A. Coenzyme Q Production by Metabolic Engineered Escherichia Coli Strains in Defined Medium. Bioprocess Biosyst. Eng. 2019, 42, 1143–1149. 10.1007/s00449-019-02111-y. [DOI] [PubMed] [Google Scholar]
- Blanchard J. L.; Wholey W.-Y.; Conlon E. M.; Pomposiello P. J. Rapid Changes in Gene Expression Dynamics in Response to Superoxide Reveal SoxRS-Dependent and Independent Transcriptional Networks. PLoS One 2007, 2, e1186 10.1371/journal.pone.0001186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pomposiello P. J.; Demple B. Redox-Operated Genetic Switches: The SoxR and OxyR Transcription Factors. Trends Biotechnol. 2001, 19, 109–114. 10.1016/s0167-7799(00)01542-0. [DOI] [PubMed] [Google Scholar]
- Baez A.; Shiloach J. Escherichia Coli Avoids High Dissolved Oxygen Stress by Activation of SoxRS and Manganese-Superoxide Dismutase. Microb. Cell Fact. 2013, 12, 23. 10.1186/1475-2859-12-23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin R. G.; Gillette W. K.; Rhee S.; Rosner J. L. Structural Requirements for Marbox Function in Transcriptional Activation of Mar/Sox/Rob Regulon Promoters in Escherichia Coli: Sequence, Orientation and Spatial Relationship to the Core Promoter. Mol. Microbiol. 1999, 34, 431–441. 10.1046/j.1365-2958.1999.01599.x. [DOI] [PubMed] [Google Scholar]
- Lee H.-J.; Gottesman S. SRNA Roles in Regulating Transcriptional Regulators: Lrp and SoxS Regulation by SRNAs. Nucleic Acids Res. 2016, 44, 6907–6923. 10.1093/nar/gkw358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffith K. L.; Shah I. M.; E. Wolf R. Proteolytic degradation of Escherichia coli transcription activators SoxS and MarA as the mechanism for reversing the induction of the superoxide (SoxRS) and multiple antibiotic resistance (Mar) regulons. Mol. Microbiol. 2004, 51, 1801–1816. 10.1046/j.1365-2958.2003.03952.x. [DOI] [PubMed] [Google Scholar]
- Compan I.; Touati D. Interaction of Six Global Transcription Regulators in Expression of Manganese Superoxide Dismutase in Escherichia Coli K-12. J. Bacteriol. 1993, 175, 1687–1696. 10.1128/JB.175.6.1687-1696.1993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang Y.; Quail M. A.; Artymiuk P. J.; Guest J. R.; Green J. Escherichia Coli Aconitases and Oxidative Stress: Post-Transcriptional Regulation of SodA Expression. Microbiology 2002, 148, 1027–1037. 10.1099/00221287-148-4-1027. [DOI] [PubMed] [Google Scholar]
- Dubbs J. M.; Mongkolsuk S. Peroxide-Sensing Transcriptional Regulators in Bacteria. J. Bacteriol. 2012, 194, 5495–5503. 10.1128/JB.00304-12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edelmann D.; Berghoff B. A. Type I Toxin-Dependent Generation of Superoxide Affects the Persister Life Cycle of Escherichia Coli. Sci. Rep. 2019, 9, 14256. 10.1038/s41598-019-50668-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gort A. S.; Ferber D. M.; Imlay J. A. The Regulation and Role of the Periplasmic Copper, Zinc Superoxide Dismutase of Escherichia Coli. Mol. Microbiol. 1999, 32, 179–191. 10.1046/j.1365-2958.1999.01343.x. [DOI] [PubMed] [Google Scholar]
- Fischer S.; Gräber P.; Turina P. The Activity of the ATP Synthase from Escherichia Coli Is Regulated by the Transmembrane Proton Motive Force. J. Biol. Chem. 2000, 275, 30157–30162. 10.1074/jbc.275.39.30157. [DOI] [PubMed] [Google Scholar]
- Hua Q.; Yang C.; Oshima T.; Mori H.; Shimizu K. Analysis of Gene Expression in Escherichia Coli in Response to Changes of Growth-Limiting Nutrient in Chemostat Cultures. Appl. Environ. Microbiol. 2004, 70, 2354–2366. 10.1128/aem.70.4.2354-2366.2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wackwitz B.; Bongaerts J.; Goodman S. D.; Unden G. Growth Phase-Dependent Regulation of NuoA-N Expression in Escherichia Coli K-12 by the Fis Protein: Upstream Binding Sites and Bioenergetic Significance. Mol. Gen. Genet. 1999, 262, 876–883. 10.1007/s004380051153. [DOI] [PubMed] [Google Scholar]
- Baneyx F.; Mujacic M. Recombinant Protein Folding and Misfolding in Escherichia Coli. Nat. Biotechnol. 2004, 22, 1399–1408. 10.1038/nbt1029. [DOI] [PubMed] [Google Scholar]
- Humphreys D. P.; Carrington B.; Bowering L. C.; Ganesh R.; Sehdev M.; Smith B. J.; King L. M.; Reeks D. G.; Lawson A.; Popplewell A. G. A plasmid system for optimization of Fab′ production in Escherichia coli: importance of balance of heavy chain and light chain synthesis. Protein Expression Purif. 2002, 26, 309–320. 10.1016/S1046-5928(02)00543-0. [DOI] [PubMed] [Google Scholar]
- Saaranen M. J.; Ruddock L. W. Applications of Catalyzed Cytoplasmic Disulfide Bond Formation. Biochem. Soc. Trans. 2019, 47, 1223–1231. 10.1042/BST20190088. [DOI] [PubMed] [Google Scholar]
- Schuller A.; Cserjan-Puschmann M.; Köppl C.; Grabherr R.; Wagenknecht M.; Schiavinato M.; Dohm J. C.; Himmelbauer H.; Striedner G. Adaptive Evolution in Producing Microtiter Cultivations Generates Genetically Stable Escherichia Coli Production Hosts for Continuous Bioprocessing. Biotechnol. J. 2020, 16, 2000376. 10.1002/biot.202000376. [DOI] [PubMed] [Google Scholar]
- Egger E.; Tauer C.; Cserjan-Puschmann M.; Grabherr R.; Striedner G. Fast and Antibiotic Free Genome Integration into Escherichia Coli Chromosome. Sci. Rep. 2020, 10, 16510. 10.1038/s41598-020-73348-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fink M.; Cserjan-Puschmann M.; Reinisch D.; Striedner G. High-Throughput Microbioreactor Provides a Capable Tool for Early Stage Bioprocess Development. Sci. Rep. 2021, 11, 2056. 10.1038/s41598-021-81633-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wingett S. W.; Andrews S. FastQ Screen: A Tool for Multi-Genome Mapping and Quality Control. F1000Research 2018, 7, 1338. 10.12688/f1000research.15931.2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger A. M.; Lohse M.; Usadel B. Trimmomatic: A Flexible Trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ewels P.; Magnusson M.; Lundin S.; Käller M. MultiQC: Summarize Analysis Results for Multiple Tools and Samples in a Single Report. Bioinformatics 2016, 32, 3047–3048. 10.1093/bioinformatics/btw354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D.; Paggi J. M.; Park C.; Bennett C.; Salzberg S. L. Graph-Based Genome Alignment and Genotyping with HISAT2 and HISAT-Genotype. Nat. Biotechnol. 2019, 37, 907–915. 10.1038/s41587-019-0201-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H.; Handsaker B.; Wysoker A.; Fennell T.; Ruan J.; Homer N.; Marth G.; Abecasis G.; Durbin R. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. 10.1093/bioinformatics/btp352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Anders S.; Pyl P. T.; Huber W. HTSeq--a Python Framework to Work with High-Throughput Sequencing Data. Bioinformatics 2015, 31, 166–169. 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love M. I.; Anders S.; Kim V.; Huber W. RNA-Seq Workflow: Gene-Level Exploratory Analysis and Differential Expression. F1000Research 2015, 4, 1070. 10.12688/f1000research.7035.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashburner M.; Ball C. A.; Blake J. A.; Botstein D.; Butler H.; Cherry J. M.; Davis A. P.; Dolinski K.; Dwight S. S.; Eppig J. T.; Harris M. A.; Hill D. P.; Issel-Tarver L.; Kasarskis A.; Lewis S.; Matese J. C.; Richardson J. E.; Ringwald M.; Rubin G. M.; Sherlock G. Gene Ontology: Tool for the Unification of Biology. Nat. Genet. 2000, 25, 25–29. 10.1038/75556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu G.; Wang L.-G.; Han Y.; He Q.-Y. ClusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS: J. Integr. Biol. 2012, 16, 284–287. 10.1089/omi.2011.0118. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.