

Abstract
The germinal center (GC) response is critical for both effective adaptive immunity and establishing peripheral tolerance by limiting autoreactive B cells. Dysfunction in these processes can lead to defective immune responses to infection or contribute to autoimmune disease. To understand the gene regulatory principles underlying the GC response, we generated a single-cell transcriptomic and epigenomic atlas of the human tonsil, a widely studied and representative lymphoid tissue. We characterize diverse immune cell subsets and build a trajectory of dynamic gene expression and transcription factor activity during B cell activation, GC formation, and plasma cell differentiation. We subsequently leverage cell type-specific transcriptomic and epigenomic maps to interpret potential regulatory impact of genetic variants implicated in autoimmunity, revealing that many exhibit their greatest regulatory potential in GC-associated cellular populations. These included gene loci linked with known roles in GC biology (IL21, IL21R, IL4R, BCL6) and transcription factors regulating B cell differentiation (POU2AF1, HHEX). Together, these analyses provide a powerful new cell type-resolved resource for the interpretation of cellular and genetic causes underpinning autoimmune disease.
One sentence summary:
Single-cell ATAC sequencing maps the cell type-specific regulatory potential of transcription factors and autoimmune disease risk loci.
Introduction
Autoimmune diseases result from a loss of tolerance to otherwise harmless endogenous or exogenous antigens, in part as a consequence of dysregulation in the selection, differentiation, or function of immune cells. The propensity for such immune cell dysfunction can be potentiated by specific inherited genetic variants, as identified through genome wide association studies (GWAS). However, the majority of GWAS genetic variants reside in non-coding regions of the genome, and the identification of risk-associated genetic variants alone does not identify the cellular populations likely affected by the variant. Recent progress has been made linking autoimmune-associated genetic variants to immune cell type-specific gene regulation by examining functional epigenomic measures like chromatin accessibility, histone acetylation and/or chromatin topology, especially in activated immune cell states of immune subsets (1–3). However, such analysis remains incomplete due to limited mapping of important yet transient subpopulations of cells that exist in diverse immune organ contexts.
The development and commitment of different immune cell lineages occurs in primary lymphoid organs such as the bone marrow and thymus. Following lineage commitment and egress from these organs, adaptive immune cells can undergo additional maturation and differentiation in secondary lymphoid organs such as the spleen, lymph nodes and tonsils to generate T cell-mediated immunity and B cell-dependent antibody responses (4). The latter in particular is predominantly dependent on the formation of the germinal center (GC) response. This requires MHCII-dependent presentation of antigen-derived peptides by dendritic cells that can be recognized by naïve CD4+ T cells, leading to their differentiation into T follicular helper (Tfh) cells. Tfh are vital to support activated B cells to form GC reactions, undergo somatic hypermutation and affinity maturation of their antibody genes before differentiating into plasma cells or memory B cells.
Mechanisms that ensure immune tolerance to self-antigen target autoreactive B cell clones during early development in the bone marrow (central tolerance) and de novo generation in GC responses in secondary lymphoid organs (peripheral tolerance). Autoantibodies are a feature of many systemic autoimmune diseases, and numerous studies have found that autoantibodies can bear somatic hypermutation and class switch recombination signatures indicative of GC-derived B cell populations (5), pointing to defects in peripheral tolerance. Because these tissues and GC-associated immune cell populations are directly involved in establishing both peripheral tolerance and forming effective adaptive immune responses, mapping the regulatory potential of autoimmune-associated genetic variants in these dynamic populations will enable the interpretation of how these variants may contribute to autoimmunity.
Here we apply single-cell transcriptomics (scRNA-seq), surface-protein profiling (scADT-seq), and epigenomics (scATAC-seq) to map the cellular states and gene regulatory networks of immune cells from the human tonsil, a model secondary lymphoid organ. By integrating gene expression and chromatin accessibility across 37 immune cell populations spanning bone marrow, peripheral blood, and tonsils, we identify putative target genes of fine-mapped autoimmune-associated genetic variants and reveal extensive GC-specific regulatory potential, including at loci of major GC regulators such as IL21, IL21R/IL4R and BCL6, as well as two genes required for MBC fate commitment, POU2AF1 and HHEX. Our integrative analyses ultimately provide original insights into the cellular and genetic etiology of autoimmune-associated genetic variants and generate a framework to functionally dissect their potential in the maintenance of peripheral tolerance and the generation of adaptive immunity.
Results
Single-cell transcriptomics and epigenomics of a model human secondary lymphoid organ to define immune cell states.
To map the diverse immune cell states of the adaptive immune response in human secondary lymphoid organs, and the gene regulatory elements active in these different populations, we performed high throughput single-cell RNA sequencing (scRNA-seq) coupled with antibody-derived tags (scADT-seq) for twelve surface protein markers on tonsillar immune cells obtained from pediatric patients undergoing routine tonsillectomy for obstructive sleep apnea or recurrent tonsillitis (Fig1A–C, S1, Data file S1; n = 3). In parallel, we performed single-cell assay for transposase-accessible chromatin using sequencing (scATAC-seq) (6) to profile active chromatin regulatory elements in tonsillar immune cells (Fig1A–C, S1; Fig2 for more detailed analysis; n = 7). We first annotated 9 broad populations based on their surface protein and RNA levels of known markers (Fig1B) and observed good concurrence between RNA, surface protein expression, and chromatin accessibility of key marker genes and the frequency of different cell types (Fig1C, S1–2, Data file S1–2). We observed a relationship between patient age and the relative frequencies of B cells in our scRNA-seq datasets (FigS3A). CyTOF profiling of pediatric and adult tonsils revealed significantly fewer GC-specific B and T cell populations in older pediatric donors (>5 years old) and adults (FigS3B–D), consistent with reduced GC activity in older individuals (7). As the GC is a major site of many important cell fate decisions during adaptive immune responses, this demonstrates the need to profile pediatric and/or immunologically relevant (e.g. after vaccination or infection) lymphoid tissue, in contrast to peripheral blood-derived immune populations or lymphoid tissue from older individuals that lack these populations.
Figure 1. Single-cell mapping of immune cell subsets in human tonsils.
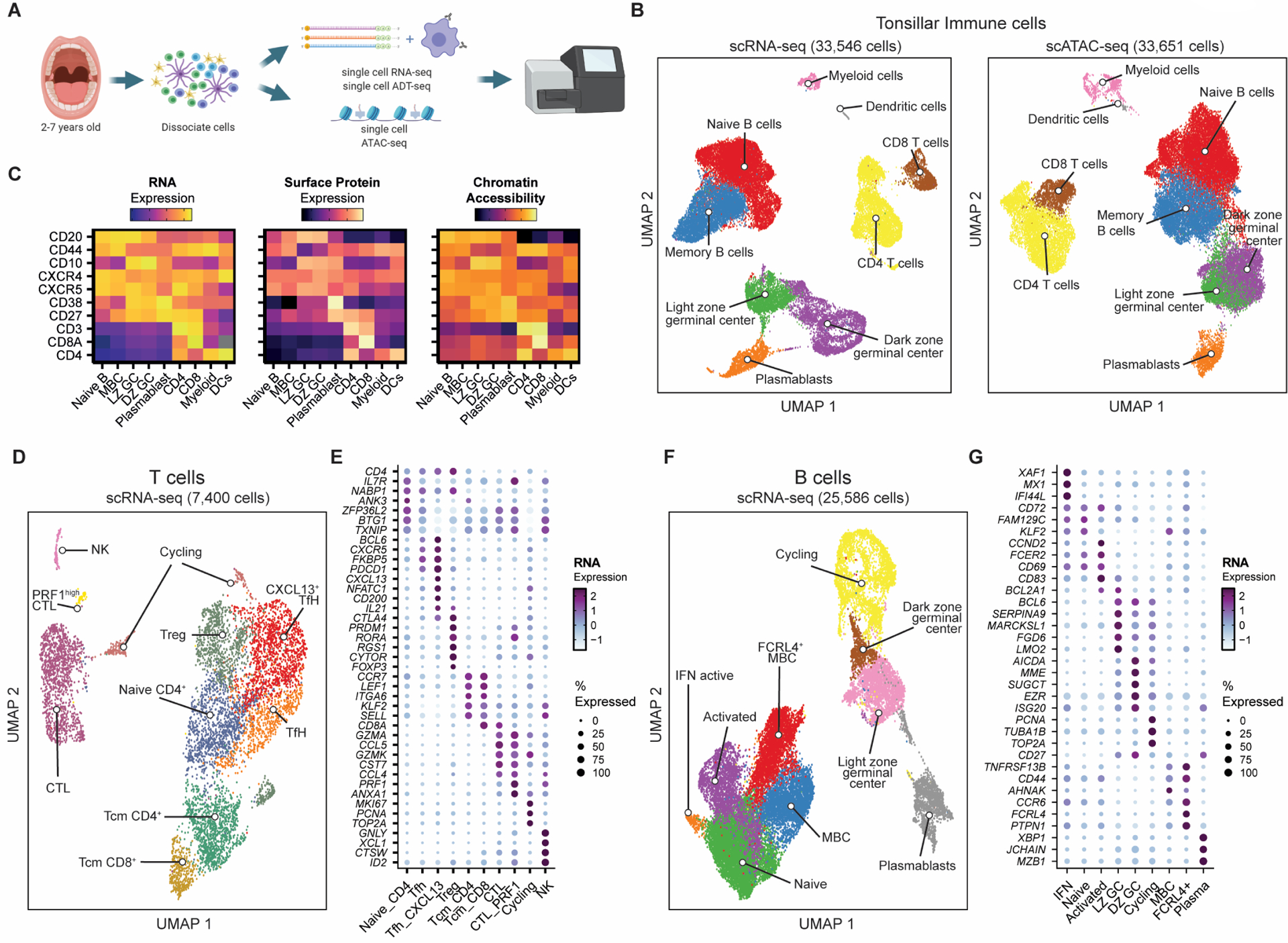
A) Experimental strategy for single-cell transcriptomics, surface marker expression, and chromatin accessibility of immune cells from pediatric tonsils.
B) UMAP of tonsillar immune scRNA-seq data (left; 3 donors) and scATAC-seq data (right; 7 donors).
C) Heatmap comparing gene expression, surface protein, and chromatin accessibility across immune cell types.
D) UMAP of T cell sub-populations in the tonsillar immune scRNA-seq data in B). NK = natural killer, CTL = cytotoxic lymphocyte, Treg = regulatory T cell, TfH = T follicular helper cell, Tcm = T central memory.
E) Mean expression of key marker genes for T cell sub-populations by scRNA-seq. Frequency of cells for which each gene is detected is denoted by size of the dots.
F) UMAP of B cell sub-populations in the tonsillar immune scRNA-seq data in B). MBC = memory B cell, LZ GC = light zone germinal center, DZ GC = dark zone germinal center; IFN = interferon.
G) Mean expression of key marker genes for B cell sub-populations by scRNA-seq. Frequency of cells for which each gene is detected is denoted by size of the dots.
Figure 2. Tonsillar immune cell type-specific transcription factor regulatory activity.
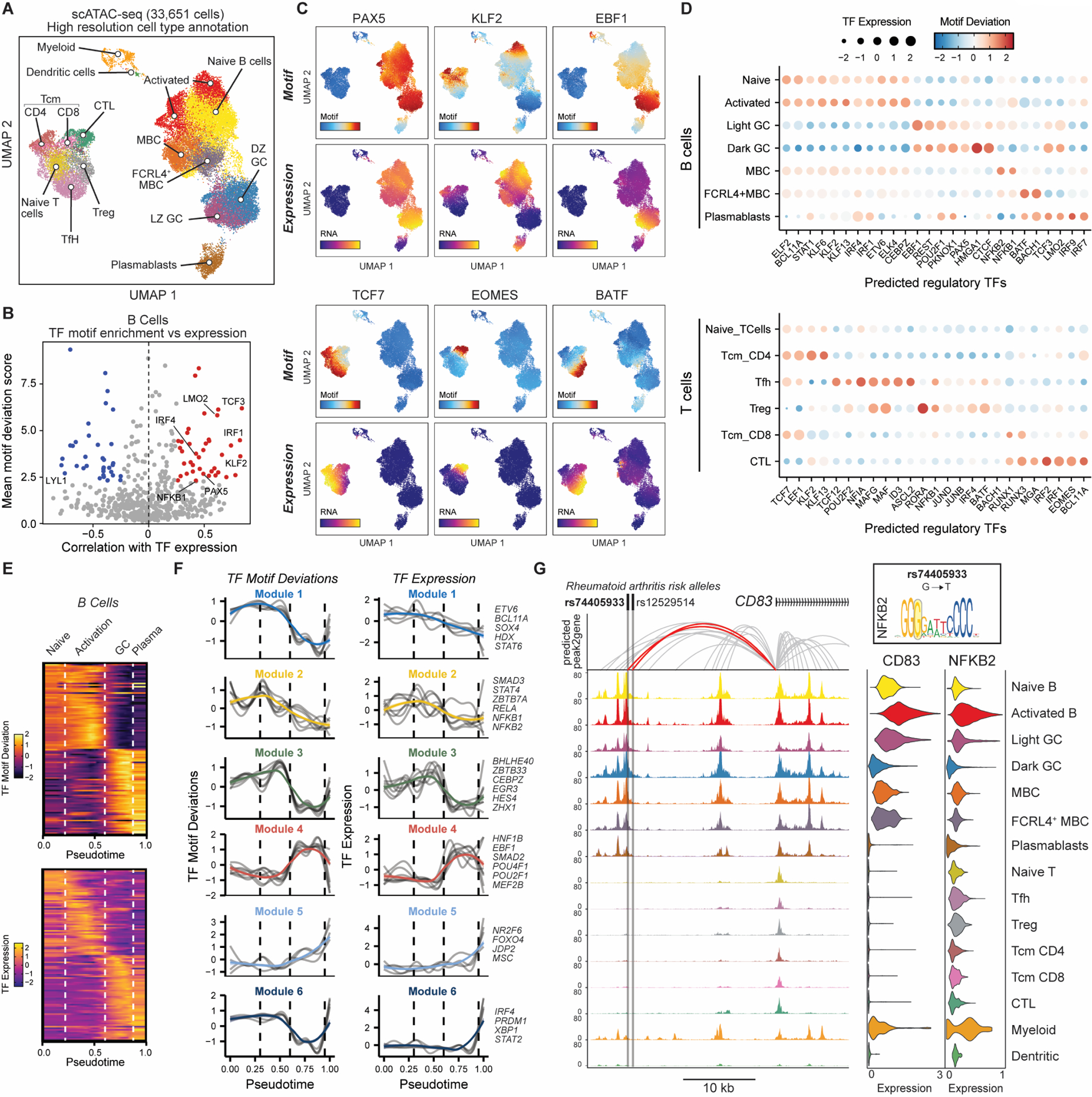
A) UMAP of tonsillar immune scATAC-seq with high resolution annotation of immune cell types.
B) Correlation of TF motif deviation (enrichment) scores with TF expression (x axis) compared to TF motif deviation scores (y axis) to predict positive TF regulators across B cell populations.
C) Motif deviation scores (top panels) and RNA expression (bottom) for exemplar TFs.
D) Motif deviation scores for transcription factors (expressed in >25% cells in at least one cell type cluster). Mean gene expression is depicted by dot size.
E) Pseudotemporal reconstruction of B cell activation, GC entry and plasmablast differentiation using scATAC-seq. Dotted lines highlight major transition points between cell types. Top; TF motif deviations. Bottom; TF gene expression.
F) Grouped patterns of TF motif deviations (left) and TF gene expression (right) through B cell pseudotemporal reconstruction shown in (e). Colored line represents mean of all TFs per group (listed on right).
G) Genomic snapshot of tonsillar immune cell scATAC-seq tracks at CD83 locus, highlighting rheumatoid arthritis-associated SNPs rs74405933 and rs12529514 and correlated peak2gene linkages. rs74405933 falls within an NFKB2 predicted binding site (G→T). scRNA-seq expression of CD83 and NFKB2 are shown to the right.
We next annotated B or T lymphocyte sub-populations at a higher resolution using our scRNA-seq dataset (Fig1D–G, S4, Data file S3–4). Within the T cell lineage, we identified naïve and central memory T (Tcm) cells, cytotoxic lymphocytes (CTL), NK cells, regulatory T cells (Treg) and two populations of Tfh cells, with one population expressing high levels of CXCL13, CD200 and IL21, likely representing GC Tfh (8) (Fig1D–E). We also defined clusters with previously identified gene expression markers for many expected B cell populations, including naïve, activated, memory, tissue-resident FCRL4+ memory, GC (light zone and dark zone) B cells, as well as plasmablasts (Fig1F–G) (9). A large population of proliferating B cells were predominantly dark zone GC B cells, as expected (FigS4C). We also found a small cluster of B cells expressing markers of type I interferon response genes such as IFI44L, XAF1, and MX1 (Fig1F–G) that are known to be up-regulated after early stages of vaccination (10) and in patients with autoimmune diseases like lupus and Sjögren’s syndrome (FigS5) (11–13). Importantly, all cellular populations, including this rare IFN-responsive state, were identified at consistent frequencies across all patient donors (FigS4D–E), and these annotations broadly agreed with recent single-cell studies of lymphocytes in pediatric tonsils and adult lymph node (9, 14).
Mapping chromatin accessibility and transcription factor activity in tonsillar immune subsets.
Our high-resolution annotation of immune cell populations by scRNA-seq (Fig1) allowed us to more comprehensively annotate our scATAC datasets (Fig2A; see Materials and Methods for details) (15). We limited our annotations of the chromatin accessibility maps to 14 cell populations to maximize coverage and representation of cell type-specific peaks in subsequent analyses. We identified naïve, activated, memory, FCRL4+ memory and GC (light zone and dark zone) B cell subsets, as well as plasmablasts, Tfh, Treg, naïve, central memory and cytotoxic T cells, and two smaller clusters representing a combination of monocytes, macrophages and dendritic cells (Fig2A). We found a strong correspondence between cluster identities and cell type-specific markers used in both scATAC-seq and scRNA-seq annotation of our datasets (FigS1–2). Cells at different stages of the cell cycle, such as proliferating dark zone GC B cells, were difficult to distinguish based on their chromatin accessibility profiles, as we and others have observed few qualitative differences in chromatin accessibility profiles between mitotic and interphase cells (16, 17). As in our scRNA-seq analysis, most scATAC-seq clusters were identified reproducibly in all tissue donors (FigS6A–B), although we did observe higher frequencies of activated and DZ GC B cells in two recurrent tonsillitis patients compared to sleep apnea patients. However, previous studies, including scRNA-seq analysis, have found no or few differences in the cellular phenotypes of immune cells between these two patient groups (9, 18). Overall, we provide a comprehensive resource of cell type-specific gene regulatory elements across 14 tonsillar immune cell populations in this model secondary lymphoid organ (FigS7A–B, Data file S5–8), including at the immunoglobulin heavy chain locus (FigS7C–D). We also report putative peak-to-gene linkages by correlating peak chromatin accessibility with scRNA-seq expression in our integrated analysis pipeline (see Materials and Methods for details) (FigS7B, Data file S7–8) (15), which, when paired with cell type-specific accessibility and gene expression, can provide insights into potential gene regulatory landscapes across these different immune cell populations.
Lymphocyte activation, maturation, and differentiation are underpinned by transcriptional networks controlled by sequence-specific transcription factors (TFs). To understand the regulatory potential of different TFs in vivo we correlated the expression of TFs with the chromatin accessibility of their target motif sequences in B and T lymphocyte populations (Fig2B–D). Specifically, we sought to identify TFs whose enrichment of their motif sequences in accessible chromatin was significantly and positively correlated with expression of that TF within a given cell type (as shown for all B cells in Fig2B) as a means to predict TFs most likely to regulate gene expression in those cells. This successfully identified enrichment of TFs known to be important for gene regulation in B and T cell subset-specific states, such as PAX5, EBF1, TCF7 and BATF (Fig2C). Our analysis also revealed shared regulatory TF activities between similar cell states, such as those active in naïve, activated and memory B cells (KLF2, BCL11A, ELF2, ETV6, ELK4) or GC B cells (EBF1, REST, POU2F1, PKNOX1) (Fig2C–D). We also found highly cell type-specific activities, such as for EOMES, IRF1/2 and RUNX1/3 in cytotoxic lymphocytes, and ID3, ASCL2, NFIA and TCF12 in Tfh cells (Fig2C–D).
While these analyses of defined cell types and states revealed putative transcriptional regulators specific to different populations, TFs also play major roles in shaping dynamic cell fate decisions during activation or differentiation of immune cells. B cell activation and subsequent participation in the GC reaction is essential for high quality B cell-dependent immune responses, yet the dynamics of different gene regulatory networks involved in this key process are poorly understood. We therefore performed a pseudotemporal reconstruction of a single-cell trajectory encompassing B cell activation, the GC reaction and plasmablast differentiation and identified modules of TF regulatory activity that corresponded with different stages of this trajectory (Fig2E–F, S7E). Intriguingly, the pseudotemporal ordering of activated B cells identified two distinct peaks of dynamic TF expression and chromatin accessibility at corresponding motif sequences before commitment to the GC state (Fig2E–F; Modules 2 and 3). This included early expression of NFκB family members (Module 2; REL, RELA, NFKB1, NFKB2), which was highly correlated with chromatin accessibility at their predicted binding sites genome-wide. We identified a NFκB/RELA binding site predicted to be disrupted by a rheumatoid arthritis (RA)-associated SNP (rs74405933; G→T), for which chromatin accessibility is strongly correlated with CD83 expression (Fig2G), a key gene involved in B cell activation and maturation (19). In addition to this initial activation module, we identified a secondary activation state comprising several poorly understood TFs, including BHLHE40, CEBPE/Z, ZBTB33, and ZHX1 (Module 3). We also identified dynamic expression and chromatin activity in GC B cells, including one module that decreases through GC exit and plasma cell differentiation (Module 4; HNF1B, EBF1, SMAD2, POU2F1, MEF2B) and one module that is maintained or increases during commitment to the plasma fate (Module 5; NR2F6, FOXO4, JDP2, MSC). In contrast, a transcriptional regulatory module containing master plasma cell regulators such as IRF4, PRDM1 and XBP1 (Module 6) exhibited reduced accessibility at target sites within GC B cells compared to both naïve and plasma populations, suggesting that these sites may be actively repressed to prevent inappropriate or premature commitment to the plasma fate during affinity maturation in the GC. Unfortunately, we were not able to reconstruct a trajectory for the memory B cell fate, perhaps due to the presence of both GC-derived and extra-follicular sources of memory B cells in tonsil tissue, the proposed stochastic nature of this cell fate decision (20), or limited number of B cells within our scATAC datasets.
Integration of secondary lymphoid organ datasets with bone marrow and peripheral blood single-cell transcriptome and epigenome atlases.
Other scRNA-seq analyses have recently demonstrated that tonsils are a transferable model tissue to study secondary lymphoid organs and adaptive immune responses more generally (9, 14, 21). In contrast to circulating or bone-marrow resident lymphocyte populations, immune cells within secondary lymphoid organs exist in a range of activation and maturation states, including GC-associated populations, that may reflect varied tissue niches, cell-cell communication and cytokine signaling. To examine the potential relevance of tissue-specific gene expression and chromatin-based regulatory activities, we integrated our tonsillar scRNA-seq and scATAC-seq datasets with those from publicly available bone marrow and peripheral blood immune cell atlases (22) to generate an overview of leukopoiesis comprising data for 60,639 and 91,510 high quality cells for scRNA-seq and scATAC-seq respectively (Fig3A, S8–9, Data file S9–12). As expected, activated B cells, GC-associated lymphocytes (GC B and Tfh cells) and tissue-resident macrophages were strongly enriched in secondary lymphoid organs, while progenitor populations like common lymphoid progenitors (CLP) and granulocyte-monocyte progenitors (GMP), and circulating monocytes were enriched in the bone marrow and peripheral blood respectively (Fig3B). In addition to differences in the frequency of immune cell subsets, we also examined if there might be differences between circulating or tissue resident B cells. We found significant differences in both the chromatin accessibility and gene expression of naïve and memory B cells in the tonsil compared to matched populations in the periphery (Fig3C–D, S10). In particular, chromatin accessibility profiles of tonsillar B cells were enriched with POU2F2 (also known as OCT2) motif sequences (Fig3E), a TF known to be important in the regulation of humoral B cell responses (23). These tissue-specific phenotypes likely reflect differences in cytokine exposure and microenvironment of the tonsil compared to circulating blood and highlight that it is essential to examine immune cell populations across varied tissue contexts, even for a single cell type.
Figure 3. Integrated single-cell transcriptomics and epigenomics of human bone marrow, peripheral blood and tonsillar immune cell states.
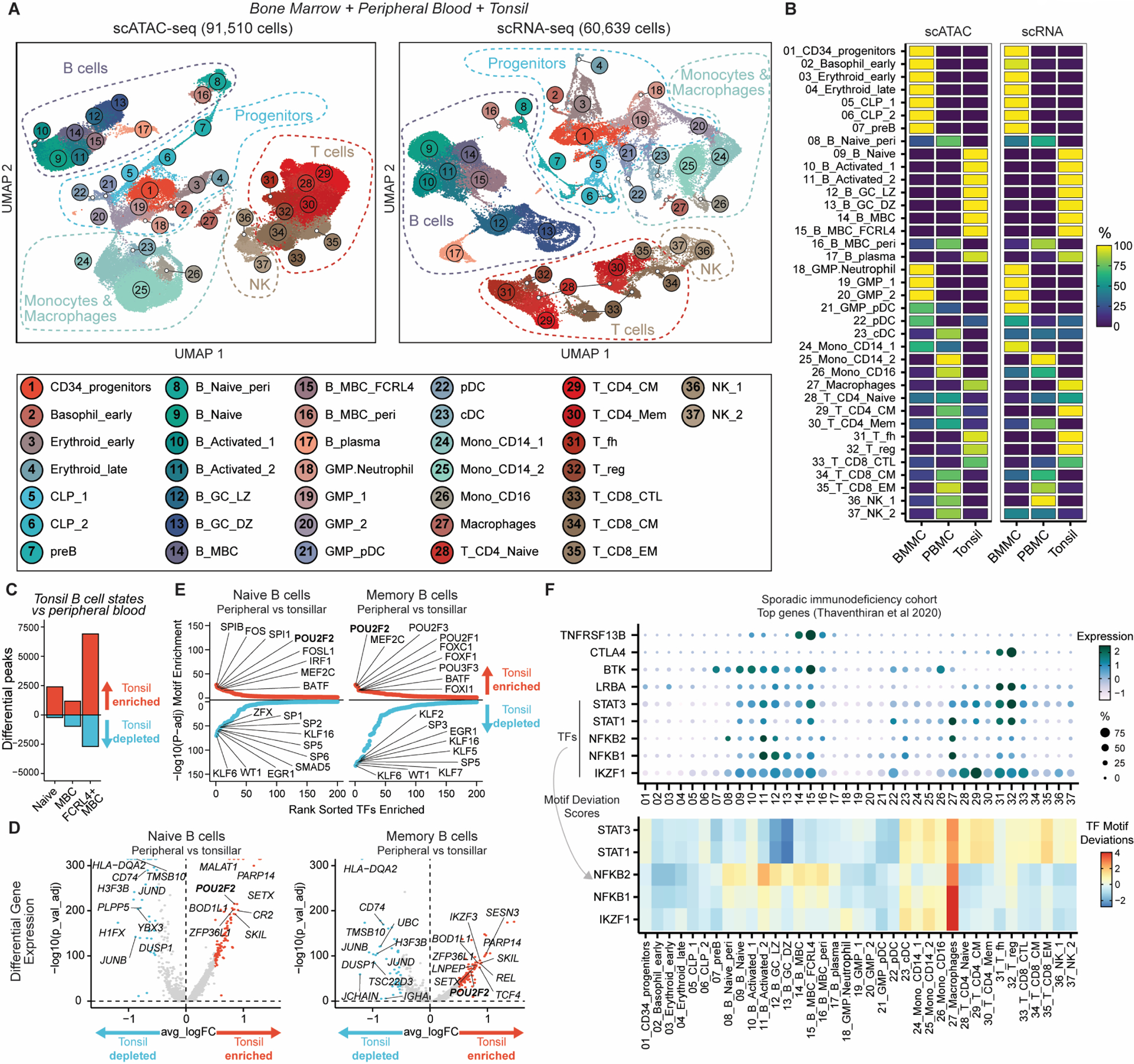
A) UMAP of integrated scATAC-seq and scRNA-seq for human bone marrow, peripheral blood and tonsils. CLP: common lymphoid progenitors. GMP: granulocyte-monocyte progenitors. CM: central memory. EM: effector memory. CTL: cytotoxic lymphocyte.
B) Relative frequency of cell type clusters in A) across different tissues.
C) Differential scATAC-seq peak analysis of tonsillar compared to peripheral blood/bone marrow-enriched naïve and memory B cell (MBC) clusters. FCRL4+ MBC cluster was compared to peripheral blood-enriched MBC cluster.
D) Differential gene expression analysis of tonsillar compared to peripheral blood/bone marrow-enriched naïve and MBC clusters in integrated scRNA-seq dataset. Selected genes are annotated.
E) Ranking of TF motif deviation enrichment within tissue-enriched (red, upper) or tissue-depleted (blue, lower) peaks naïve and MBCs.
F) Expression of top genes identified to be mutated by whole genome sequencing in a sporadic immunodeficiency cohort (24). For TFs, motif deviation scores are also provided.
Finally, we examined the cell type-specific expression of nine genes recently identified to be most commonly mutated within a sporadic primary immunodeficiency cohort (Fig3F) (24). Two genes, TNFRSF13B and CTLA4 were relatively cell type-specific in their expression pattern. TNFRSF13B (encoding TACI) was most highly expressed in memory B cells, particularly tonsillar FCRL4+ memory B cells. Patients with immunodeficiency and TNFRSF13B mutations have fewer memory B cells expressing class-switched antibodies, although the mechanisms and penetrance of different coding TNFRSF13B mutations remain unclear given the prevalence of coding variants in healthy individuals (25, 26). CTLA4 expression peaked in Tfh and Treg populations as expected. In contrast, BTK, LRBA, and the TF genes STAT1, STAT3, NFKB1, NFKB2 and IZKF1 were broadly expressed across varied subsets. We used our scATAC-seq data to examine the enrichment of their motif sequences in accessible chromatin to determine which cell type might be most sensitive to altered activity of these TF genes. This revealed that tonsillar myeloid cells (labelled here primarily as macrophages) had the highest activity of these immunodeficiency-associated TFs (Fig3F), although we observed enrichment of NFKB2 in activated B cells (Fig2F–G, 3F) and STAT1/STAT3 in circulating monocyte and T cells (Fig3F).
Identification of fine-mapped autoimmune GWAS variants in cell type-specific chromatin.
Our integrated scRNA-seq and scATAC-seq atlas of immune cell populations in bone marrow, peripheral blood and tonsils provided a unique opportunity to understand the regulatory potential and cell type-specificity of autoimmune-associated genetic variants across a broad diversity of immune cell types. By examining 12,902 statistically fine-mapped SNPs, of which 9,493 were significantly associated with disorders of the immune system (1, 27), we found that our single-cell accessibility profiles of immune cells were broadly enriched in immune-related genetic variants compared to non-immune related traits and background genetic variation (Fig4A, S11A–B). We found specific enrichment of disease-specific genetic variants in different immune cell lineages or subsets (Fig4B, S11C–D). For example, we found a strong enrichment of genetic variants associated with Kawasaki disease and systemic lupus erythematosus in chromatin accessibility maps of the B cell lineage, particularly tonsillar naïve and memory B cells, as well as enrichment of genetic variants associated with alopecia, autoimmune thyroiditis, systemic sclerosis and Behçets disease in cytotoxic lymphocyte regulatory elements (Fig4B, S11C–D). In contrast, genetic variants associated with multiple sclerosis were enriched in both B and T cell-specific chromatin, perhaps reflecting the multigenic nature and complex etiology of this disease (Fig4B, S11C–D).
Figure 4. Autoimmune-associated genetic variants enriched in immune cell chromatin accessibility maps.
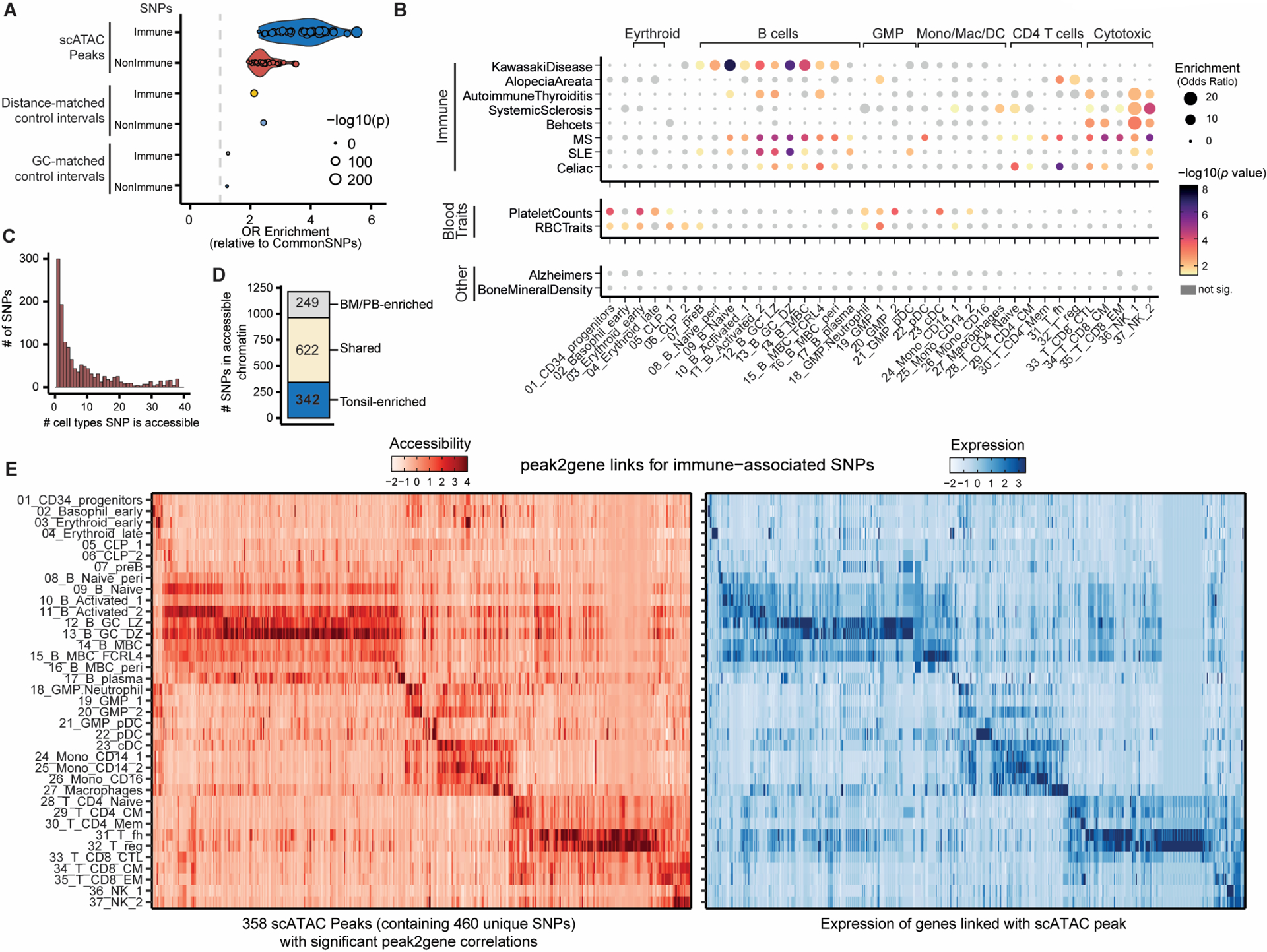
A) Fisher enrichment test of immune-associated fine mapped genetic variants, compared to common genetic variants, for chromatin accessibility scATAC peaks across 37 immune cell populations. Results for non-immune traits and background control peaks are shown. Dot size conveys significance (−log10(p value)).
B) Fisher enrichment test for trait-specific SNPs, compared to the complete fine-mapped SNP set, within cell type-specific chromatin accessibility peaks. Dot size conveys enrichment (odds ratio) and color denotes significance of enrichment.
C) Frequency histogram of immune-associated SNPs that fall within chromatin accessibility peaks across 37 immune cell types.
D) Tissue-specificity of chromatin accessibility peaks overlapping autoimmune SNPs.
E) Chromatin accessibility of peaks containing >1 immune-associated SNP (scATAC; left) for which at least one significant peak2gene correlation is identified. Expression of linked genes (scRNA; right) is also plotted. Accessibility or expression counts are scaled by peak or gene respectively.
Of the 1213 immune-related SNPs that overlapped with accessible chromatin peaks in our atlas (Data file S13), many were localized in cell type- or lineage-specific chromatin (Fig4C). Importantly, 342 (28.2%) of these SNPs fell within accessible chromatin only identified in tonsil-enriched immune subsets (Fig4D), demonstrating the value of our tonsillar immune cell atlas for interpretation of GWAS genetic variants. We next predicted the putative gene targets of these genetic variants by using our integrated scRNA-seq and scATAC-seq to identify highly correlated accessibility at chromatin regions to nearby gene expression (15, 22). This enabled us to examine 358 chromatin accessible regions (containing 460 unique immune-linked SNPs) for which we identified significant peak-to-gene linkage correlations (Fig4E). These linkages revealed cell type-specific patterns of both the chromatin accessibility at autoimmune genetic variants and correlated expression of putative gene targets, providing a powerful resource to explore the potential regulatory mechanisms of these genetic variants and their relationship to autoimmune disease.
Chromatin regulatory activity at immune-associated genetic variants predicts importance of GC activity in autoimmunity.
Many studies examining the relationship between immune-associated genetic variants and their regulatory activity with functional genomics methods such as ATAC-seq or ChIP-seq have been limited to studying peripheral immune cell populations. This limitation is likely significant, given our knowledge that many lymphocyte maturation and antibody-based selection events occur in secondary lymphoid organs, and that GC-derived autoantibody production is a feature of many autoimmune diseases. Although we found examples of genetic variants in cell type-specific chromatin across diverse immune subsets (Fig4E, S12–S13; e.g. GZMB/GZMH, NKX2–3, COTL1/KLHL36, KSR1/LGALS9, TNFRSF1A/LTBR), we observed a striking enrichment of fine-mapped autoimmune variants in chromatin accessibility regions specific to GC-associated B and T populations, such as GC B cells and Tfh cells (Fig4E), including the IL21, IL21R/IL4R, BCL6/LPP, CD80, PRAG1, SLC38A9, VAV3/SLC25A24, DLEU1/DLEU1/TRIM13 loci (Fig5–6, S14–S15).
Figure 5. Chromatin regulatory landscapes of GC-specific autoimmune risk variants.

A) Genomic snapshot of fine-mapped autoimmune-associated GWAS variants that localize to accessible chromatin in the integrated human bone marrow, peripheral blood and tonsil scATAC-seq atlas. High resolution of individual SNP loci and larger view of the IL21 locus are shown, with significantly correlated peak2gene linkages colored in red and significant links between SNPs and gene promoters (SNP2gene) in blue and bold. Significant associations between individual SNPs and autoimmune diseases are shown in black boxes and gene expression is shown as violin plots for matched populations in the scATAC tracks. AID: autoimmune disease. IBD: inflammatory bowel disease. Juv Idio Arthritis: Juvenile idiopathic arthritis. Scl cholangitis: Primary sclerosing cholangitis.
B) Same as A), at the IL4R/IL21R locus.
C) Same as A), at the BCL6/LPP locus. A germinal center (GC)-specific locus control region (LCR) is highlighted in green. MS: multiple sclerosis.
Figure 6. Autoimmune risk variants at transcription regulator genes POU2AF1 and HHEX.

A) Genomic snapshot of fine-mapped autoimmune-associated GWAS variants at the POU2AF1 locus that localize to accessible chromatin in the integrated human bone marrow, peripheral blood and tonsil scATAC-seq atlas. Significantly correlated peak2gene linkages colored in red and significant links between SNPs and gene promoters (SNP2gene) in blue and bold. Significant associations between individual SNPs and autoimmune diseases are shown in black boxes and gene expression is shown as violin plots for matched populations in scATAC tracks. PBC: primary biliary cirrhosis.
B) Same as A), at the HHEX locus. MS: multiple sclerosis.
We identified GC-specific regulatory elements at the IL21 locus and the locus of its receptor IL21R (Fig5A–B, FigS16). Cytokine signaling by IL-21, primarily secreted by Tfh cells, is essential for B cells to form and participate in normal GC reactions. B cells respond to IL-21 through the IL-21 receptor (IL-21R). We identified several fine-mapped SNPs at the IL21 locus highly correlated with both chromatin accessibility and gene expression at the IL21 promoter (Fig5A). These SNPs exhibited Tfh-specific chromatin accessibility, although one SNP, rs13140464, was also highly accessible in several progenitor populations. These fine-mapped SNPs at IL21 have been associated with alopecia (1), juvenile idiopathic arthritis or autoimmunity more generally (27), and some of these same SNPs are also significantly associated with celiac disease (rs7682241, rs6840978) (28), inflammatory bowel disease (rs7662182) (29), primary sclerosing cholangitis (rs13140464) (30) and lupus (rs13140464) (31). Conversely, we found two fine-mapped SNPs in strong linkage disequilibrium (rs6498021, rs6498019) located in close proximity to IL21R in B cell-specific chromatin accessibility regions that have been linked with allergy (1) and/or asthma (32) (Fig5B, S16). As well as significant correlations with IL21R expression, the chromatin accessibility of these two SNPs were also highly correlated with the nearby IL4R gene, encoding the IL-4 receptor (IL-4R), which, similar to IL21R, was most highly expressed in GC B cells and is vital for T cell-dependent maturation of B cells.
Autoimmune risk variants within a GC-specific locus control region.
Our analysis of genetic variants linked with autoimmunity identified a concentration of recently fine-mapped autoimmune-associated SNPs from the UKBB databank (27) in a GC-specific locus control region (LCR) (33) located between BCL6 and LPP (Fig5C, S16). Of the genetic variants that fell within accessible chromatin across this locus, there were associations with celiac disease (rs11709472 (34), rs7628982 (UKBB), rs9834159 (35), rs4686484 (1)), allergy (rs56046601 and rs12639588 (1)), multiple sclerosis (rs4686953 (formerly rs66756607) (36, 37)), asthma (rs7640550 and rs7628982 (38)) and vitiligo (rs7628982 (39)). Many of these SNPs were present in chromatin accessible regions specific to GC B or Tfh cells, in which BCL6, LPP and the long non-coding RNA at the LCR (LINC01991) are most highly expressed (Fig5C). We report significant correlations in chromatin accessibility between many of these SNPs (and the LCR in general) with the expression of both BCL6 and LPP, consistent with chromosome conformation interactions detected in GC B cells between this LCR and the BCL6 promoter (33). Importantly, deletion of this LCR has been shown in mouse models to lead to defects in GC B cell formation (33), presumably through its transcriptional regulation of BCL6, one of the master regulatory TFs required for both GC B cells and Tfh cells. These observations suggest that association of this locus with autoimmunity is primarily driven through GC B and Tfh defects. However, some genetic variants (rs142486803, rs76288334, rs78146088) were accessible across many different immune lineages, as well as rs4686484 that was previously proposed to be located in a B cell-specific enhancer (35), revealing an additional layer of complexity to this autoimmune regulatory locus.
Autoimmune risk variants at the loci of transcriptional regulators POU2AF1 and HHEX.
We identified cell type-specific chromatin accessibility at autoimmune risk variants across loci for many regulatory TFs or transcriptional regulators including POU2AF1, HHEX, ETS1, STAT4, IKZF3, NKX2–3 and IRF8 (Fig6, S12, S17–18), in addition to the GC master regulator BCL6 (Fig5C). Of particular interest were POU2AF1 and HHEX, which have recently been proposed to control memory B cell fate selection in the GC (40, 41). POU2AF1, also known as OCT binding factor 1 (OBF1), is a largely B cell-specific transcriptional coactivator with no intrinsic DNA binding activity that interacts with TFs POU2F1 (OCT1) and POU2F2 (OCT2). It is indispensable for formation of GCs and GC-dependent B cell maturation (42). We found two genetic variants associated with primary biliary cirrhosis/cholangitis (PBC) (rs4938541 and rs4393359 (1, 43)) within B cell-specific accessible chromatin and observed that POU2AF1 expression peaks in GC B cells (Fig6A). Our analysis of B cell activation dynamics predicted POU2F1/POU2F2 as regulators in GC B cells (Fig2) and POU2F2 is more highly expressed in tonsillar B cells compared to those circulating in peripheral blood (Fig3), suggesting that B cells within lymphoid tissues are likely to be most sensitive to altered POU2AF1 levels.
HHEX has recently been reported to be an essential regulator of the memory B cell fate decision by GC B cells (41), although its potential mechanistic involvement in autoimmune disease is not known. Our integrated epigenomic and transcriptomic analyses identified three fine-mapped SNPs at the HHEX locus that fell within B cell specific-accessible chromatin, were implicated in the regulation of HHEX through peak-to-gene correlation analysis, and were associated with multiple sclerosis (MS) (rs11187144, rs4933736, rs10882106) (Fig6B). We also identified correlated peak-to-gene linkages between these SNPs and neighboring genes KIF11 and EXOC6 (FigS19). We note that rs4933736 falls within a predicted KLF TF binding site (Fig6B), providing a potential mechanism for disruption of HHEX expression.
Discussion
Here, we generated paired transcriptome and epigenome atlases of immune cell subsets in the human tonsil, a model system to study the GC reaction which is a major site for developing adaptive immunity to respond to infection and establishing peripheral tolerance to prevent autoimmunity. We defined gene expression and gene regulatory elements across dynamic immune cell states and examined the regulatory potential of transcription factors in these populations. We subsequently leveraged our single-cell resource to profile the cell type-specific chromatin accessibility at fine-mapped GWAS variants linked with autoimmune disorders to reveal that the chromatin of many such variants is most accessible in GC-associated cell types and this accessibility is highly correlated with cell type-specific expression of genes required for normal cytokine signaling or transcriptional regulation in the GC response.
Our single-cell transcriptomic analysis identified a rare B cell population that expresses high levels of IFN-induced gene expression (Fig1). Unfortunately, we were unable to identify this rare B cell population in our scATAC profiling to explore how it may be linked to different autoimmune traits at the chromatin level. One of the genes most highly expressed by the IFN-responsive B cells was IFI44L. Splice and missense genetic variants at the IFI44L locus (rs1333973 and rs273259) have previously been linked with neutralizing antibody titers to the measles vaccine (44), and type I interferon-positive B cells have previously been implicated in the development of autoreactive B cells (45). Many of the genes uniquely expressed by this B cell state are also upregulated in the peripheral blood B cells of patients with lupus (FigS5) (13). These observations suggest this rare and poorly characterized B cell state may be involved in B cell-mediated antibody responses to vaccines and/or processes linked with autoimmunity.
The joint analysis of gene expression with chromatin accessibility landscapes allowed us to predict putative TF regulators in both steady state and dynamic immune cell populations, including temporally dynamic TFs during B cell activation and their participation in the GC reaction. As part of a dominant B cell activation, maturation and plasma cell differentiation trajectory, we identified a secondary B cell activation state, after an initial NFκB-associated activation presumably linked with strong BCR activation and/or T cell help. One particularly interesting TF identified was BHLHE40, which has previously been shown to be required for the transition from an activated state prior to entry into the GC (46, 47) and is capable of binding key regulatory elements at the immunoglobulin heavy chain locus (9). Recent spatial epigenomic mapping of the human tonsil found BHLHE40 regulatory activity outside of the GC reaction, consistent with our pseudotemporal analyses (48). How this, and other putative regulators we identify in this secondary activation state (such as CEBPE/Z, ZBTB33, and ZHX1) may contribute to the transition from the activated B cell state to a GC-associated gene expression program will be an important question for future mechanistic studies. However, as the human tonsil represents a highly polyclonal source of B cells, which may arise from many different antigen sources, sub-tissue locations or clonal expansion events, it remains challenging to resolve potentially more complex B cell fate trajectories, such as whether the chromatin accessibility and transcription factor network dynamics in antigen-naïve or –experienced (memory) B cells vary during activation and the GC response.
The molecular mechanisms by which many GWAS-identified genetic polymorphisms contribute to autoimmune disease remain poorly understood. To address this, we and others have examined the relationships between non-coding SNPs and lineage- or cell type-specific expression of putative gene targets to predict the potential functional relevance of genetic variants (reviewed in 49). For immune-associated GWAS variants, many resources have focused on gene expression or epigenomic profiles of cell types circulating in the peripheral blood or bone marrow (1, 50), although there is an emerging prioritization of activation or tissue-specific immune cell states (2, 3). Our analysis of chromatin accessibility and gene expression at GWAS loci in tonsillar immune cell states highlights the importance of examining cellular populations in secondary lymphoid tissues, especially of pediatric patients with highly active GC responses, to understand how regulatory activity at non-coding genetic variants in dynamic and tissue-specific populations might contribute to autoimmune disease. Specifically, we found that many autoimmune disease-associated genetic variants are localized within chromatin most accessible in GC B and T cell populations, including at the loci of genes with well-established roles in B cell activation (CD83, CD80), survival and participation in the GC (IL21, IL21R, IL4R, BCL6) and fate selection (POU2AF1, HHEX, IRF8). While our findings do not exclude dysregulation of autoimmune-associated loci in stromal cell populations which we did not profile here, or potential pleiotropic genetic effects from variants that are accessible across multiple immune cell lineages or tissues, they strongly implicate lymphocyte-intrinsic dysfunctional GC responses as a major feature in the genetic etiology of autoimmune disease.
Our integrated scRNA-seq and scATAC-seq resource maps the cell type-specific chromatin accessibility of autoimmune variant loci genome-wide and identifies highly correlated peak accessibility-gene expression relationships to identify gene targets that may be affected by those SNPs (15). Chromosome conformation capture methods such as Hi-C have also been used to predict putative gene targets of autoimmune GWAS variants in GC-associated cell populations (33, 51), but these experimental approaches can be limited in their ability to detect short range interactions (e.g. <10kb) and are challenging to perform at scale across many cell types at once or at single-cell resolution. While the inferred peak-to-gene relationships we report here do not provide direct evidence of physical interactions and will require experimental follow up in future studies, our integrated approach to predict gene targets has advantages over other co-accessibility models that link distal regulatory elements to promoters without taking account changes in gene expression, and our approach has successfully linked GWAS variants with putative targets in previous studies (22, 52).
To explain how individual non-coding genetic variants may contribute towards the development or pathology of autoimmune disease, it will be necessary to further understand their precise regulatory impact on gene expression. Our analyses do not predict whether specific polymorphisms might positively or negatively regulate gene expression of their putative gene targets. Expression quantitative trait loci (eQTL) analyses can be used to infer whether genetic variants are associated with loss or gain of gene expression (53). However, current eQTL databases have profiled either circulating immune cell subsets or whole tissues (e.g. spleen) from adult donors (GTEX median donor age is 50–59 years old). In both cases, these resources lack adequate representation of GC-associated gene expression to confidently dissect the directionality of many SNP-to-gene relationships we predict in our analyses. New advances in neural-network derived methods may prove useful to quantitatively model effects on gene expression in cell type-resolved chromatin accessibility maps (17, 54).
While at some loci we identified variants that appear likely to disrupt predicted TF binding sites, the highly context-dependent activating or repressive gene regulatory functions for many TFs remain poorly understood. This therefore makes it difficult to confidently predict whether the downstream gene targets are more likely to be activated or repressed. Inferring downstream targets of TFs without cell type-specific ChIP-seq datasets is likewise challenging, making prediction of the phenotypic impact of potentially altered TF expression at several loci we predict (BCL6, HHEX, POU2AF1, ETS1, IKZF3, STAT4, IRF8) difficult. Functional genomics, single-cell multi-omics and eQTL analyses in varied healthy and diseased immune organs and model systems will be essential to provide further mechanistic insights, as studies of healthy individuals lacking specific variants may miss gain-of-function mutations that create disease-specific regulatory elements de novo (55). Although functional (epi)genomic editing of primary human immune cells remains challenging, high throughput screening strategies are emerging as powerful new tools (56) to assign loss- or gain-of-function to GWAS variants linked with autoimmune disease. However, whichever method is employed to dissect mechanism of non-coding polymorphisms, the fact that many variants associated with disease are in linkage disequilibrium poses a significant challenge to confidently identify causal variants for any given locus.
While we are unable to confidently predict whether expression of a specific gene is enhanced or disrupted by autoimmune-associated genetic variants, either defective or enhanced GC phenotypes could contribute to the development of autoimmune disease by providing an opportunity for the expansion of self-reactive B cells that are normally inhibited in the periphery of healthy individuals (57). As a model example to illustrate this principle, we discuss here how altered signaling by IL-21 through IL-21R, for which we identified several autoimmune-associated genetic variants in Tfh- or GC B cell-enriched gene regulatory elements, could lead to altered cellular and immunological phenotypes that might contribute to autoimmunity. If at these loci, any of the genetic variants we characterize result in decreased IL21 or IL21R expression, and subsequently reduced IL-21 signaling, even subtly, this could result in reduced B cell survival within the GC, and enhanced cell death would lead to high concentrations of nuclear autoantigens that might promote autoreactive B cells and loss of tolerance. Conversely, if IL21 or IL21R gene expression was enhanced by genetic variation at distal regulatory elements, elevated autocrine IL-21 signaling by Tfh cells could result in Tfh expansion and proliferation that limit competition amongst GC B cells and lead to the survival of self-reactive B cells (58, 59). Indeed, B cell-specific depletion of IL-21R in a mouse model of lupus prevents the development of autoantibodies and disease (60), demonstrating that this pathway can play a major role in autoimmunity. While many of the precise molecular and immunological pathways involved in autoimmunity remain unclear, our genetic analyses provide a powerful resource to dissect the transcriptional and epigenetic landscapes of immune cells in secondary lymphoid organs of healthy individuals.
Finally, the development of transient GC-like lymphoid follicles in non-lymphoid tissue (termed ectopic GCs) has been associated with site-specific inflammation in autoimmune diseases and may contribute to loss of tolerance by promoting maturation of self-reactive B cell clones (61). Analysis of B cells from ectopic GCs in several autoimmune diseases provide evidence of site-specific clonal expansion and somatic hypermutation of antibody genes, and an absence of normal GC regulation (62–64). Single-cell analyses of “defective” and “ectopic” immune structures in different autoimmune diseases will be essential to understand how the regulatory and gene expression dysfunction we predict in the normal immune cell landscape may drive autoimmunity through altered GC response dynamics.
Materials and Methods
Study design
In this study we aimed to define the gene expression and accessible DNA landscapes of different immune cell populations found in the human tonsil, a model secondary lymphoid organ to study adaptive immune responses. This study used tonsil samples from pediatric patients undergoing routine tonsillectomy, numbers of samples per experiment are reported in Data file S1. We first looked at patients covering a wide range of ages and chose to focus for this study on patients ranging from age 3–7 where germinal center population were most abundant for subsequent analysis by scRNA-seq coupled with CITE-seq, and scATAC-seq, performed at Stanford University (n=3). During initial analysis, four additional tonsillar scATAC-seq datasets that had been generated with an identical protocol at Queen Mary University of London were integrated into the data analysis pipeline and used in all subsequent analyses. We used known gene expression markers to define different cell populations in the human tonsil scRNA-seq resource, before using this fine-scaled definition to annotate clusters in matched scATAC-seq datasets. Pseudotemporal ordering of single-cell chromatin accessibility profiles was performed to examine the dynamics of transcription factor activities between different B cell maturation stages. To understand cell type-specific regulatory potential of autoimmune genetic variants, we intersected published statistically fine-mapped GWAS variants with regions of cell-type-specific chromatin accessibility and examined the chromatin accessibility and gene expression of exemplar autoimmune gene loci.
Human ethics, tissue collection and preparation
Tonsil samples were collected from children and adults undergoing routine tonsillectomy. All participants provided written informed consent and the protocols were approved by Stanford University’s Institutional Review Board (protocol numbers 30837 and 47690). Whole tonsils were collected in saline and processed within four hours of receipt. Tissues were treated with penicillin, streptomycin, and normocin for 30 minutes on ice and heavily clotted or cauterized areas of the tissue were removed. Tonsils were then dissected into small pieces (roughly 5–8 pieces per tonsil) before mechanical dissociation through a 100 μm cell strainer using a syringe plunger. Mononuclear cells were isolated by Ficoll density gradient centrifugation (GE Healthcare) and the buffy coats were collected. Cells were cryopreserved in 90% fetal bovine serum, 10% DMSO until use. Four additional cryopreserved tonsil samples at Queen Mary University of London included for scATAC-seq analyses were prepared as described previously (9) under approval from North West/Greater Manchester East Research Ethics Committee (17/NW/0664).
CyTOF staining and analysis
Cryopreserved samples were thawed in pre-warmed cell culture medium (RPMI1640 with 10 % FBS, non-essential amino acids, sodium pyruvate, antibiotics), washed, and rested for 1 hour at 37°C in culture medium supplemented with DNase (25 U/ml). Cells were then washed and resuspended in FACS buffer (PBS with 0.1% w/v bovine serum albumin, 2 mM EDTA, 0.05% v/v sodium azide). Individual donor samples were barcoded using a combination of metal-tagged CD45 antibodies, combined into barcoded pools, stained for surface antibody markers (Table S1), and treated with cisplatin for viability staining as described (65). Samples were then fixed overnight with 2% paraformaldehyde diluted in PBS. The next day, cells were permeabilized using a permeabilization buffer (eBioscience), stained with a DNA intercalator for 30 minutes, and washed. Just prior to CyTOF data collection, samples were washed three times with PBS, then three times with MilliQ water. Barcoded pools were run on a CyTOF2 instrument (Fluidigm) and fcs files were exported for analysis in FlowJo software. Live intact singlets were gated and samples were manually debarcoded using combinations of CD45 channels (5-choose-2 scheme) and individual donor samples were exported as separate fcs files before dimensionality reduction analyses.
Single-cell library preparation, sequencing and alignment.
Tonsillar immune cells were loaded on to the 10X Genomics Chromium according to the manufacturer’s protocol using either the single-cell 3’ kit (v3) or the single-cell ATAC kit (v1). Cell surface labelling for scADT-seq libraries was performed with 12 oligo-labelled TotalSeq™ antibodies (BioLegend; Table S2). Library preparation was performed according to the manufacturer’s protocol prior to sequencing on either the Illumina NovaSeq 6000 or NextSeq 500 platforms. scRNA-seq libraries were sequenced with 28/10/10/90 bp cycles while scATAC-seq libraries were sequenced with 70/8/16/70 bp read configurations. BaseCall files were used to generate FASTQ files with either cellranger mkfastq (v3; 10X Genomics) or cellranger-atac (v1; 10X Genomics) prior to running cellranger count with the cellranger-GRCh38–3.0.0 reference or cellranger-atac count with the cellranger-atac-GRCh38–1.1.0 reference for scRNA-seq and scATAC-seq libraries respectively.
Quality control, integration and cell type annotation of tonsillar scRNA-seq
Gene expression count matrices from cellranger were processed with Seurat (v3.0.2) (66, 67) for genes detected in greater than 3 cells. Cell barcodes were filtered based on the number of genes per cell (between 200–7500), percentage of mitochondrial reads per cell (0–20 %) and the number of ADTs (less than 4000). Initial data quality control was performed separately on each biological sample. Data from technical replicate libraries were combined, normalized with SCTransform (68) before highly variable gene identification and PCA dimensionality reduction. Jackstraw plots were visually assessed to determine the number of principal components (PCs) for subsequent analysis: Tonsil1 = 11, Tonsil2 = 13, Tonsil3 = 12. Preliminary clusters were identified (FindClusters; res = 0.8) before computing UMAP dimensionality reduction and identifying putative doublets with DoubletFinder (69) (sct=TRUE, expected_doublets=3.9%). Pre-processed Seurat objects were then merged, with SCTransform normalization and PCA computation repeated using all variable features (except for IGKC, IGLC, IGLV, HLA, and IGH genes). Batch correction was performed with harmony (70). UMAP dimensionality reduction and cluster identification were performed (27 PCs, res = 0.8). Broad cell type cluster frequencies (as in Fig1B) from an independent scRNA-seq analysis of human tonsils (9) were obtained to compare cell type frequencies between patients of different ages. For higher resolution analysis of B cells and T cells, data from B or T cells only were processed separately, with repeated variable gene identification (removing IGKC, IGLC, IGLV, HLA, and IGH) before repeated PCA, batch correction with Harmony, UMAP reduction and cluster identification (30 PCs, res = 0.6 for B cells; 20 PCs, res = 0.6 for T cells). Gene expression markers for clusters were identified (FindAllMarkers; log fold change > 1, adjusted p value < 0.05). Imputation of gene expression counts (for plotting only) was performed with MAGIC (71). Mean gene expression values per cell type per donor were used to calculate Spearman correlation coefficients between donors. Top 50 marker genes for the IFN_active B cell cluster were analyzed with the “Gene Set Query” function in the Autoimmune Disease Explorer (https://adex.genyo.es/) (11).
scATAC-seq quality control, batch correction and integration with scRNA-seq datasets
Mapped Tn5 insertion sites (fragments.tsv files) from cellranger were read into the ArchR (v0.9.4) package (15) retaining cell barcodes with at least 1000 fragments per cell and a TSS enrichment score > 4. Doublets were identified and filtered (addDoubletScores and filterDoublets, filter ratio = 1.4) before iterative LSI dimensionality reduction was computed (iterations = 2, res = 0.2, variable features = 25000, dim = 30). Sample batch correction was performed with harmony (70). Clustering was then performed on the harmony-corrected data (addClusters, res = 0.8) before UMAP dimensionality reduction (nNeighbors = 30, metric = cosine, minDist = 0.4). One cluster enriched for high doublet scores (cluster 7) was removed. A preliminary cell type annotation was performed using gene accessibility scores of known cell type markers. Tonsillar scRNA-seq gene expression and metadata were integrated with tonsillar scATAC data with ArchR as previously described (15). To improve cell type assignment of closely related cell types, we performed this step as a constrained integration, grouping GC B cell clusters, other B cell clusters and non-B cell clusters together during addGeneIntegrationMatrix. The most common predicted cell type from the integration with RNA expression in each previously identified ATAC-seq cluster was used to annotate scATAC cluster identity. The quality of mapping between the RNA and ATAC was confirmed by identifying marker gene scores in scATAC clusters using getMarkerFeatures. Additionally, cluster annotations derived from scATAC-only analysis were compared with annotations derived from scRNA-seq integration.
For high resolution clustering of B and T cell subsets (Fig2), scATAC clusters identified as B cells or T cells following scATAC/scRNA integration were subset, and use to recompute iterative LSI dimensionality reduction as described above, except 30 dimensions were used for B cell analysis. Batch correction, cluster identification and UMAP reduction were also performed as above, except that minDist = 0.1 (T cells) or 0.3 (B cells). Integration of B cell and T cell scATAC-seq datasets with gene expression and high resolution cluster annotations was performed using the T cell- or B cell-specific scRNA-seq Seurat objects as previously described with addGeneIntegrationMatrix in ArchR. Integration between assays were constrained with the following broad groups: B cell subgroups; plasmablasts, memory, naïve/activated and GC B cell clusters, T cells; CD8+/cytotoxic T cells and remaining T cell clusters. Mean peak accessibility scores per cell type per donor were used to calculate Spearman correlation coefficients between donors.
Peak calling and inference of transcription factor activity in scATAC-seq datasets.
Single-cell chromatin accessibility data were used to generate pseudobulk group coverages based on high resolution cluster identities of scATAC-seq datasets before peak calling with macs2 (72) using addReproduciblePeakSet in ArchR. A background peak set controlling for total accessibility and GC-content was generated using addBgdPeaks and used for TF motif enrichment analyses. Chromvar (73) was run with addDeviationsMatrix using the cisbp motif set to calculate enrichment of chromatin accessibility at different TF motif sequences in single cells. To identify correlations between the gene expression and transcription factor activity, RNA-expression projected into the ATAC subspace (GeneIntegrationMatrix) and the Chromvar deviations (MotifMatix) were correlated using correlateMatrices. A correlation of greater than 0.25 was used to determine if TF expression and activity were positively correlated, and the list of correlated TFs was further subset by only including TFs that were expressed in at least 25 percent of cells in one or more cell type cluster. To analyze transcription factor activity during B cell activation, GC entry and plasma differentiation, the harmony-corrected B cell ArchR object was subjected to “addTrajectory” from ArchR using the following user-defined trajectory as a guide: Naive→Activated→LZ GC→DZ GC→Plasmablasts. Gene expression and Chromvar deviation scores were correlated throughout pseudotime using correlateTrajectories (corCutOff = 0.25, varCutOff1 = 0.25, varCutOff2 = 0.25) and visualized using plotTrajectoryHeatmap. “Peak-to-gene links” were calculated using correlations between peak accessibility and integrated scRNA-seq expression data using addPeak2GeneLinks.
Integration of tonsil scATAC-seq and scRNA-seq with bone marrow and peripheral blood datasets
Published bone marrow and peripheral blood scRNA-seq and scATAC-seq (22) were aligned to the hg38 genome as described above. Additional hg38-aligned PBMC scATAC-seq datasets were downloaded from 10X Genomics (https://support.10xgenomics.com/single-cell-atac/datasets).
scRNA-seq
Cellranger gene expression matrices were used to sum and quantify mitochondrial gene expression before mitochondrial genes were removed from the gene expression matrices. Similarly, V, D and J gene counts from T cell and immunoglobulin receptors were summed and removed from matrices. Closely related IgH constant region genes were also summed and removed (IgG1–4, IgA1–2). Cell barcodes expressing >200 genes and genes detected in >3 cells were then processed in Seurat (66, 67), with doublet prediction using default settings with scrublet (74) (expected doublet frequency 8×10−6 × 1000 cells). Predicted doublets were removed, and cell barcodes with <750 or >30000 UMIs, <500 or >6000 genes detected, or >20% mitochondrial gene expression were also removed. Individual datasets were then merged together, before normalization and batch correction with SCTransform (3000 variable features) and scoring of cell cycle phase with Seurat. “IGLsum”, “IGKsum”, “IGHG”, “IGHA”, “IGHM”, and “IGHD” were subsequently removed from highly variable gene list so they would not contribute to downstream dimensionality reductions. PCA was then computed before UMAP reduction (n.neighbors = 20, min.dist = 0.35, dims = 1:50), nearest neighbor identification (FindNeighbours; dims = 1:50) and cluster identification (FindClusters; res = 1.75). Some additional subclustering was performed to better match cell type annotations from previous tonsil analysis (this study) and peripheral blood/bone marrow analysis (22). In general, previous annotations were closely adhered to and confirmed by examination of known cell type-specific gene expression markers. Differential gene expression between clusters was performed with FindAllMarkers or FindMarkers, with padj < 0.05 and avg_logFC > 0.5. Imputation of gene expression counts (for plotting only) was performed with MAGIC (71).
scATAC-seq
Cellranger-derived fragments.tsv files of tonsil, peripheral blood and bone marrow samples were processed with ArchR (15) (createArrowFiles; filterTSS = 6, filterFrags = 1000, minFrags = 500, maxFrags = 1e+05). Doublets were identified (addDoubletScores; k=10) and removed with a filterRatio = 1.4, before additional filtering of cell barcodes to remove those with TSSEnrichment < 6, < 103.25 or > 105 fragments per barcode, nucleosome ratio of > 2.5, ReadsInBlacklist > 800, or BlacklistRatio > 0.009. Preliminary LSI reduction was performed with addIterativeLSI (corCutOff = 0.25, varFeatures = 30000, dimsToUse = 1:40, selectionMethod = “var”, LSIMethod = 1, iterations = 6, filterBias = FALSE, clusterParams = list(resolution = c(0.1,0.2,0.4,0.6,0.8,1), sampleCells = 10000, n.start = 10). To account for differences in sequencing coverage, Harmony batch correction (corCutOff = 0.25, lambda = 0.75, sigma = 0.2) was performed using library ID for tonsil samples, public 10X Genomics PBMC datasets and sample BMMC_D6T1, while remaining samples from Granja et al were treated as a single batch. Preliminary identification of clusters (addClusters; res = 1.5) identified two poor quality clusters enriched with doublets (C38, C7). These were removed from subsequent analysis. Quality controlled datasets were then subjected to new LSI dimensionality reduction and Harmony batch correction with the same settings, before computing UMAP (RunUMAP; nNeighbors = 80, minDist = 0.45, seed = 1) and identifying cell type clusters with at least 80 cells (addClusters (method = “Seurat”, res = 1.1 or 1.5, nOutlier = 80). Broad lineages were first annotated to help with integration and transfer of scRNA expression. Normalized, non-corrected scRNA expression counts and annotated cell types were transferred to nearest neighbor scATAC cells using addGeneIntegrationMatrix (sampleCellsATAC = 10000, nGenes (RNA) = 4000, sampleCellsRNA = 10000) with a constrained integration to the following groups: CD4T_cells, CD8T_cells, GC_PB, MBC_B_cells, Myeloid_cells, NaiveAct_B_cells, NK, Peripheral_B_cells, Progenitors. Accessibility gene scores and transferred RNA expression counts were imputed with addImputeWeights(corCutOff = 0.25). Cell type clusters were carefully annotated with a combination of pre-existing annotations from Granja et al. (22) and tonsil immune cell scATAC data (this study), transferred cell annotations from scRNA-seq and examination of known subset markers.
Pseudobulk group coverages of cell type clusters were calculated with addGroupCoverages and used for peak calling using macs2 (addReproduciblePeakSet in ArchR). A background peak set controlling for total accessibility and GC-content was generated using addBgdPeaks for TF enrichment analyses. Cell type-specific marker peaks were identified with getMarkerFeatures with the wilcoxon test and controlled for TSSEnrichment and fragment count. Peak accessibility was deemed significantly different between clusters if FDR < 0.05 and log2fc > 0.56. “Peak-to-gene links” were calculated using correlations between peak accessibility and integrated scRNA-seq expression data using addPeak2GeneLinks. Motif annotations and enrichment were calculated as described above with addMotifAnnotations and addDeviationsMatrix.
Analysis of fine-mapped GWAS variants
The results of two independent GWAS statistical fine-mapping studies (1, 27) (https://www.finucanelab.org/data) were combined. PICS SNPs from both immune and non-immune traits were included in analyses (1), while only SNPs from the study mapping the UK BioBank resource that were associated with a combined autoimmune disease trait (AID; labelled as AID_UKBB) were included (27). This provided a total of 12,902 non-redundant SNPs, of which 9,493 were significantly associated with disorders of the immune system. Fisher’s exact test was used to calculate enrichment of immune trait-associated SNPs and non-immune trait-associated SNPs, against a background of common genetic variants (Common dbSnp153), in cell type-resolved peak sets or control background genomic intervals (either matched for GC content or distance to nearest TSS). Trait-specific enrichment analysis was performed using cell type-specific marker peaks (FDR < 0.05, log2FC > 0.25), with a background SNP set comprising all fine-mapped SNPs across all traits. Cell type- and tissue-specificity of accessibility at SNPs was determined by presence or absence of a scATAC peak in each cell type, with cell type clusters regrouped based on enrichment in tonsils, peripheral blood or bone marrow. Of the immune-related SNPs that overlapped with accessible chromatin peaks (1213, 12.8%), we subsequently identified 460 unique immune-linked SNPs that fell within 358 chromatin accessible regions for which a significant Peak2Gene link had been identified to at least one gene (P2G_Correlation > 0.4; FDR < 0.01). Mean normalized chromatin accessibility counts (scATAC) and RNA expression counts for linked genes (scRNA) for each cell type cluster were calculated and used for heatmap visualization while pyGenomeTracks was used to visualize grouped scATAC pseudobulk tracks (75). Linkage disequilibrium scores of top candidate SNPs were calculated using LDlink across all populations (76).
Supplementary Material
Data file S1. Tonsillectomy patient donor details, experimental study design and cell type frequencies across donors.
Data file S2. Broad resolution of tonsil immune cell subset scRNA-seq gene expression markers.
Data file S3. High resolution B cell subset scRNA-seq gene expression markers.
Data file S4. High resolution T cell subset scRNA-seq gene expression markers.
Data file S5. Differential chromatin accessibility peaks from high resolution annotation of tonsil immune cell scATAC-seq.
Data file S6. Differential chromatin accessibility gene scores from high resolution annotation of tonsil immune cell scATAC-seq.
Data file S7. Peak2gene predictions for tonsil scATAC-seq and scRNA-seq analysis.
Data file S8. Loop coordinates to visualize predicted tonsil immune peak2gene interactions.
Data file S9. Gene expression markers for integrated tonsil, peripheral blood and bone marrow immune cell populations from scRNA-seq.
Data file S10. Differential chromatin accessibility peak markers for integrated tonsil, peripheral blood and bone marrow immune cell populations from scATAC-seq.
Data file S11. Peak2gene predictions for integrated bone marrow, blood and tonsil scATAC-seq and scRNA-seq analysis.
Data file S12. Loop coordinates to visualize predicted integrated bone marrow, blood and tonsil immune peak2gene interactions.
Data file S13. Peak2gene linkage annotation of fine-mapped SNPs found in chromatin accessibility peaks from integrated tonsil, peripheral blood and bone marrow scATAC-seq datasets.
Data file S14. Raw Data file.
Figure S1. Single-cell library metadata, integration, batch correction and quality control.
Figure S2. Comparison of RNA expression, cell surface protein expression and chromatin accessibility of key marker genes.
Figure S3. Age-related changes in tonsillar immune cell populations by scRNA-seq and CyTOF.
Figure S4. Tonsil scRNA-seq marker gene heatmaps, annotation of GC B cells and reproducibility of cell types across donors.
Figure S5. Differential expression in autoimmune disease of top scRNA-seq marker genes for the IFN_active B cell cluster.
Figure S6. Reproducibility of scATAC-seq cluster frequencies and correlation of peak accessibilities between donors.
Figure S7. Differential peak analysis of scATAC-seq clusters, peak-to-gene predictions and alternative pseudotime analysis.
Figure S8. Batch correction and quality control of integrated bone marrow, blood and tonsil immune scRNA-seq and scATAC-seq.
Figure S9. Integrated bone marrow, blood and tonsil scRNA-seq and scATAC-seq markers.
Figure S10. Tonsil B cell-enriched gene expression markers compared to peripheral blood B cells.
Figure S11. Enrichment of fine-mapped autoimmune variants in immune cell subsets.
Figure S12. Genome snapshots of fine-mapped autoimmune variants at GZMB/GZMH, NKX2–3 and COTL1/KHLH36 loci.
Figure S13. Genome snapshots of fine-mapped autoimmune variants at KSR1/LGALS9 and TNFRSF1A/LTBR loci.
Figure S14. Genome snapshots of germinal center-associated cell type-specific regulatory activity at fine-mapped autoimmune variants at CD80, PRAG1 and SLC38A9/DDX4 loci.
Figure S15. Genome snapshots of germinal center-associated cell type-specific regulatory activity at fine-mapped autoimmune variants at VAV3 and DLEU2 loci.
Figure S16. Linkage disequilibrium scores for variants at IL21, IL21R and BCL6 loci.
Figure S17. Genome snapshots of fine-mapped autoimmune variants at ETS1 and IKZF3 loci.
Figure S18. Genome snapshots of fine-mapped autoimmune variants at STAT4 and IRF8 loci.
Figure S19. Genomic landscape at HHEX and expression of KLF family transcription factors.
Table S1. CyTOF phenotyping antibody panel.
Table S2. CITE-seq antibody details.
Acknowledgements
We thank all members of the Greenleaf and James laboratories for helpful comments and advice. We thank the QMUL Genome Centre for sequencing support and A. Orantes for administrative support.
Funding
This work was supported by funding from the Rita Allen Foundation (W.J.G.), the Human Frontiers Science (RGY006S) (W.J.G) and the Wellcome Trust (213555/Z/18/Z) (H.W.K). Z.S is supported by EMBO Long-Term Fellowship (EMBO ALTF 1119-2016) and by Human Frontier Science Program Long-Term Fellowship (HFSP LT 000835/2017-L). K.L.W is supported by a National Science Foundation GRFP award (DGE-1656518). W.J.G. is a Chan Zuckerberg Biohub investigator and acknowledges grants 2017-174468 and 2018-182817 from the Chan Zuckerberg Initiative.
Footnotes
Competing interests
WJG is a consultant for 10x Genomics and Guardant Health, and is named as an inventor on patents describing ATAC-seq methods.
Data and materials availability
Raw and processed data for this study are available at Gene Expression Omnibus under accession GSE165860. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. All code and scripts necessary to repeat analysis in this manuscript is available upon request.
References
- 1.Farh KK-H, Marson A, Zhu J, Kleinewietfeld M, Housley WJ, Beik S, Shoresh N, Whitton H, Ryan RJH, Shishkin AA, Hatan M, Carrasco-Alfonso MJ, Mayer D, Luckey CJ, Patsopoulos NA, De Jager PL, Kuchroo VK, Epstein CB, Daly MJ, Hafler DA, Bernstein BE, Genetic and epigenetic fine mapping of causal autoimmune disease variants. Nature 518, 337–343 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Calderon D, Nguyen MLT, Mezger A, Kathiria A, Müller F, Nguyen V, Lescano N, Wu B, Trombetta J, Ribado JV, Knowles DA, Gao Z, Blaeschke F, Parent AV, Burt TD, Anderson MS, Criswell LA, Greenleaf WJ, Marson A, Pritchard JK, Landscape of stimulation-responsive chromatin across diverse human immune cells. Nat Genet 51, 1494–1505 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Soskic B, Cano-Gamez E, Smyth DJ, Rowan WC, Nakic N, Esparza-Gordillo J, Bossini-Castillo L, Tough DF, Larminie CGC, Bronson PG, Willé D, Trynka G, Chromatin activity at GWAS loci identifies T cell states driving complex immune diseases. Nat Genet 51, 1486–1493 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ruddle NH, Akirav EM, Secondary lymphoid organs: responding to genetic and environmental cues in ontogeny and the immune response. J Immunol 183, 2205–2212 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Suurmond J, Diamond B, Autoantibodies in systemic autoimmune diseases: specificity and pathogenicity. J Clin Invest 125, 2194–2202 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Buenrostro JD, Giresi PG, Zaba LC, Chang HY, Greenleaf WJ, Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat Methods 10, 1213–1218 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lee J, Chang D-Y, Kim S-W, Choi YS, Jeon S-Y, Racanelli V, Kim DW, Shin E-C, Age-related differences in human palatine tonsillar B cell subsets and immunoglobulin isotypes. Clin Exp Med 16, 81–87 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Crotty S, T follicular helper cell differentiation, function, and roles in disease. Immunity 41, 529–542 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.King HW, Orban N, Riches JC, Clear AJ, Warnes G, Teichmann SA, James LK, Single-cell analysis of human B cell maturation predicts how antibody class switching shapes selection dynamics. Sci Immunol 6, (2021). [DOI] [PubMed] [Google Scholar]
- 10.Zhang J, Shao J, Wu X, Mao Q, Wang Y, Gao F, Kong W, Liang Z, Type I interferon related genes are common genes on the early stage after vaccination by meta-analysis of microarray data. Hum Vaccin Immunother 11, 739–745 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Martorell-Marugán J, López-Domínguez R, García-Moreno A, Toro-Domínguez D, Villatoro-García JA, Barturen G, Martín-Gómez A, Troule K, Gómez-López G, Al-Shahrour F, González-Rumayor V, Peña-Chilet M, Dopazo J, Sáez-Rodríguez J, Alarcón-Riquelme ME, Carmona-Sáez P, A comprehensive database for integrated analysis of omics data in autoimmune diseases. BMC Bioinformatics 22, 343 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Hutcheson J, Scatizzi JC, Siddiqui AM, Haines GK III, Wu T, Li Q-Z, Davis LS, Mohan C, Perlman H, Combined Deficiency of Proapoptotic Regulators Bim and Fas Results in the Early Onset of Systemic Autoimmunity. Immunity 28, 206–217 (2008). [DOI] [PubMed] [Google Scholar]
- 13.Becker AM, Dao KH, Han BK, Kornu R, Lakhanpal S, Mobley AB, Li Q-Z, Lian Y, Wu T, Reimold AM, Olsen NJ, Karp DR, Chowdhury FZ, Farrar JD, Satterthwaite AB, Mohan C, Lipsky PE, Wakeland EK, Davis LS, SLE Peripheral Blood B Cell, T Cell and Myeloid Cell Transcriptomes Display Unique Profiles and Each Subset Contributes to the Interferon Signature. PLoS ONE 8, e67003 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Kleshchevnikov V, Shmatko A, Dann E, Aivazidis A, King HW, Li T, Lomakin A, Kedlian V, Jain MS, Park JS, Ramona L, Tuck E, Arutyunyan A, Vento-Tormo R, Gerstung M, James L, Stegle O, Bayraktar OA, Comprehensive mapping of tissue cell architecture via integrated single cell and spatial transcriptomics. bioRxiv, 10.1101/2020.1111.1115.378125 (2020). [DOI] [Google Scholar]
- 15.Granja JM, Corces MR, Pierce SE, Bagdatli ST, Choudhry H, Chang HY, Greenleaf WJ, ArchR is a scalable software package for integrative single-cell chromatin accessibility analysis. Nat Genet, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Hsiung CCS, Morrissey CS, Udugama M, Frank CL, Keller CA, Baek S, Giardine B, Crawford GE, Sung M-H, Hardison RC, Blobel GA, Genome accessibility is widely preserved and locally modulated during mitosis. Genome Res 25, 213–225 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Trevino AE, Muller F, Andersen J, Sundaram L, Kathiria A, Shcherbina A, Farh K, Chang HY, Pasca AM, Kundaje A, Pasca SP, Greenleaf WJ, Chromatin and gene-regulatory dynamics of the developing human cerebral cortex at single-cell resolution. bioRxiv, 10.1101/2020.1112.1129.424636 (2020). [DOI] [PubMed] [Google Scholar]
- 18.Dan JM, Havenar-Daughton C, Kendric K, Al-Kolla R, Kaushik K, Rosales SL, Anderson EL, LaRock CN, Vijayanand P, Seumois G, Layfield D, Cutress RI, Ottensmeier CH, Lindestam Arlehamn CS, Sette A, Nizet V, Bothwell M, Brigger M, Crotty S, Recurrent group A Streptococcus tonsillitis is an immunosusceptibility disease involving antibody deficiency and aberrant TFH cells. Sci Transl Med 11, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Breloer M, Fleischer B, CD83 regulates lymphocyte maturation, activation and homeostasis. Trends Immunol 29, 186–194 (2008). [DOI] [PubMed] [Google Scholar]
- 20.Duffy KR, Wellard CJ, Markham JF, Zhou JHS, Holmberg R, Hawkins ED, Hasbold J, Dowling MR, Hodgkin PD, Activation-induced B cell fates are selected by intracellular stochastic competition. Science 335, 338–341 (2012). [DOI] [PubMed] [Google Scholar]
- 21.Wagar LE, Salahudeen A, Constantz CM, Wendel BS, Lyons MM, Mallajosyula V, Jatt LP, Adamska JZ, Blum LK, Gupta N, Jackson KJL, Yang F, Röltgen K, Roskin KM, Blaine KM, Meister KD, Ahmad IN, Cortese M, Dora EG, Tucker SN, Sperling AI, Jain A, Davies DH, Felgner PL, Hammer GB, Kim PS, Robinson WH, Boyd SD, Kuo CJ, Davis MM, Modeling human adaptive immune responses with tonsil organoids. Nat Med 27, 125–135 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Granja JM, Klemm S, McGinnis LM, Kathiria AS, Mezger A, Corces MR, Parks B, Gars E, Liedtke M, Zheng GXY, Chang HY, Majeti R, Greenleaf WJ, Single-cell multiomic analysis identifies regulatory programs in mixed-phenotype acute leukemia. Nat Biotechnol 37, 1458–1465 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corcoran L, Emslie D, Kratina T, Shi W, Hirsch S, Taubenheim N, Chevrier S, Oct2 and Obf1 as Facilitators of B:T Cell Collaboration during a Humoral Immune Response. Front Immunol 5, 108 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thaventhiran JED, Lango Allen H, Burren OS, Rae W, Greene D, Staples E, Zhang Z, Farmery JHR, Simeoni I, Rivers E, Maimaris J, Penkett CJ, Stephens J, Deevi SVV, Sanchis-Juan A, Gleadall NS, Thomas MJ, Sargur RB, Gordins P, Baxendale HE, Brown M, Tuijnenburg P, Worth A, Hanson S, Linger RJ, Buckland MS, Rayner-Matthews PJ, Gilmour KC, Samarghitean C, Seneviratne SL, Sansom DM, Lynch AG, Megy K, Ellinghaus E, Ellinghaus D, Jorgensen SF, Karlsen TH, Stirrups KE, Cutler AJ, Kumararatne DS, Chandra A, Edgar JDM, Herwadkar A, Cooper N, Grigoriadou S, Huissoon AP, Goddard S, Jolles S, Schuetz C, Boschann F, Primary NB Immunodeficiency Consortium for the, Lyons PA, Hurles ME, Savic S, Burns SO, Kuijpers TW, Turro E, Ouwehand WH, Thrasher AJ, Smith KGC, Whole-genome sequencing of a sporadic primary immunodeficiency cohort. Nature 583, 90–95 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Salzer U, Chapel HM, Webster ADB, Pan-Hammarström Q, Schmitt-Graeff A, Schlesier M, Peter HH, Rockstroh JK, Schneider P, Schäffer AA, Hammarström L, Grimbacher B, Mutations in TNFRSF13B encoding TACI are associated with common variable immunodeficiency in humans. Nat Genet 37, 820–828 (2005). [DOI] [PubMed] [Google Scholar]
- 26.Pan-Hammarström Q, Salzer U, Du L, Björkander J, Cunningham-Rundles C, Nelson DL, Bacchelli C, Gaspar HB, Offer S, Behrens TW, Grimbacher B, Hammarström L, Reexamining the role of TACI coding variants in common variable immunodeficiency and selective IgA deficiency. Nat Genet 39, 429–430 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Weeks EM, Ulirsch JC, Cheng NY, Trippe BL, Fine RS, Miao J, Patwardhan TA, Kanai M, Nasser J, Fulco CP, Tashman KC, Aguet F, Li T, Ordovas-Montanes J, Smillie CS, Biton M, Shalek AK, Ananthakrishnan AN, Xavier RJ, Regev A, Gupta RM, Lage K, Ardlie KG, Hirschhorn JN, Lander ES, Engreitz JM, Finucane HK, Leveraging polygenic enrichments of gene features to predict genes underlying complex traits and diseases. medRxiv, 10.1101/2020.1109.1108.20190561 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.van Heel DA, Franke L, Hunt KA, Gwilliam R, Zhernakova A, Inouye M, Wapenaar MC, Barnardo MCNM, Bethel G, Holmes GKT, Feighery C, Jewell D, Kelleher D, Kumar P, Travis S, Walters JRF, Sanders DS, Howdle P, Swift J, Playford RJ, McLaren WM, Mearin ML, Mulder CJ, McManus R, McGinnis R, Cardon LR, Deloukas P, Wijmenga C, A genome-wide association study for celiac disease identifies risk variants in the region harboring IL2 and IL21. Nat Genet 39, 827–829 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robertson CC, Inshaw JRJ, Onengut-Gumuscu S, Chen W-M, Santa Cruz DF, Yang H, Cutler AJ, Crouch DJM, Farber E, Bridges SL, Edberg JC, Kimberly RP, Buckner JH, Deloukas P, Divers J, Dabelea D, Lawrence JM, Marcovina S, Shah AS, Greenbaum CJ, Atkinson MA, Gregersen PK, Oksenberg JR, Pociot F, Rewers MJ, Steck AK, Dunger DB, Wicker LS, Concannon P, Todd JA, Rich SS, Type C 1 Diabetes Genetics, Fine-mapping, trans-ancestral and genomic analyses identify causal variants, cells, genes and drug targets for type 1 diabetes. Nat Genet, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Liu JZ, Hov JR, Folseraas T, Ellinghaus E, Rushbrook SM, Doncheva NT, Andreassen OA, Weersma RK, Weismüller TJ, Eksteen B, Invernizzi P, Hirschfield GM, Gotthardt DN, Pares A, Ellinghaus D, Shah T, Juran BD, Milkiewicz P, Rust C, Schramm C, Müller T, Srivastava B, Dalekos G, Nöthen MM, Herms S, Winkelmann J, Mitrovic M, Braun F, Ponsioen CY, Croucher PJP, Sterneck M, Teufel A, Mason AL, Saarela J, Leppa V, Dorfman R, Alvaro D, Floreani A, Onengut-Gumuscu S, Rich SS, Thompson WK, Schork AJ, Næss S, Thomsen I, Mayr G, König IR, Hveem K, Cleynen I, Gutierrez-Achury J, Ricaño-Ponce I, van Heel D, Björnsson E, Sandford RN, Durie PR, Melum E, Vatn MH, Silverberg MS, Duerr RH, Padyukov L, Brand S, Sans M, Annese V, Achkar J-P, Boberg KM, Marschall H-U, Chazouillères O, Bowlus CL, Wijmenga C, Schrumpf E, Vermeire S, Albrecht M, Consortium U-P, Rioux JD, Alexander G, Bergquist A, Cho J, Schreiber S, Manns MP, Färkkilä M, Dale AM, Chapman RW, Lazaridis KN, Group IPS, Franke A, Anderson CA, Karlsen TH, Consortium IIG, Dense genotyping of immune-related disease regions identifies nine new risk loci for primary sclerosing cholangitis. Nat Genet 45, 670–675 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Hughes T, Kim-Howard X, Kelly JA, Kaufman KM, Langefeld CD, Ziegler J, Sanchez E, Kimberly RP, Edberg JC, Ramsey-Goldman R, Petri M, Reveille JD, Martín J, Brown EE, Vilá LM, Alarcón GS, James JA, Gilkeson GS, Moser KL, Gaffney PM, Merrill JT, Vyse TJ, Alarcón-Riquelme ME, Network B, Nath SK, Harley JB, Sawalha AH, Fine-mapping and transethnic genotyping establish IL2/IL21 genetic association with lupus and localize this genetic effect to IL21. Arthritis Rheum 63, 1689–1697 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Olafsdottir TA, Theodors F, Bjarnadottir K, Bjornsdottir US, Agustsdottir AB, Stefansson OA, Ivarsdottir EV, Sigurdsson JK, Benonisdottir S, Eyjolfsson GI, Gislason D, Gislason T, Guðmundsdóttir S, Gylfason A, Halldorsson BV, Halldorsson GH, Juliusdottir T, Kristinsdottir AM, Ludviksdottir D, Ludviksson BR, Masson G, Norland K, Onundarson PT, Olafsson I, Sigurdardottir O, Stefansdottir L, Sveinbjornsson G, Tragante V, Gudbjartsson DF, Thorleifsson G, Sulem P, Thorsteinsdottir U, Norddahl GL, Jonsdottir I, Stefansson K, Eighty-eight variants highlight the role of T cell regulation and airway remodeling in asthma pathogenesis. Nat Commun 11, 393 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bunting KL, Soong TD, Singh R, Jiang Y, Béguelin W, Poloway DW, Swed BL, Hatzi K, Reisacher W, Teater M, Elemento O, Melnick AM, Multi-tiered Reorganization of the Genome during B Cell Affinity Maturation Anchored by a Germinal Center-Specific Locus Control Region. Immunity 45, 497–512 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sharma A, Liu X, Hadley D, Hagopian W, Liu E, Chen W-M, Onengut-Gumuscu S, Simell V, Rewers M, Ziegler A-G, Lernmark Å, Simell O, Toppari J, Krischer JP, Akolkar B, Rich SS, Agardh D, She J-X, Group TS, Identification of Non-HLA Genes Associated with Celiac Disease and Country-Specific Differences in a Large, International Pediatric Cohort. PLoS ONE 11, e0152476 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Almeida R, Ricaño-Ponce I, Kumar V, Deelen P, Szperl A, Trynka G, Gutierrez-Achury J, Kanterakis A, Westra H-J, Franke L, Swertz MA, Platteel M, Bilbao JR, Barisani D, Greco L, Mearin L, Wolters VM, Mulder C, Mazzilli MC, Sood A, Cukrowska B, Núñez C, Pratesi R, Withoff S, Wijmenga C, Fine mapping of the celiac disease-associated LPP locus reveals a potential functional variant. Hum Mol Genet 23, 2481–2489 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lill CM, Luessi F, Alcina A, Sokolova EA, Ugidos N, de la Hera B, Guillot-Noël L, Malhotra S, Reinthaler E, Schjeide B-MM, Mescheriakova JY, Mashychev A, Wohlers I, Akkad DA, Aktas O, Alloza I, Antigüedad A, Arroyo R, Astobiza I, Blaschke P, Boyko AN, Buttmann M, Chan A, Dörner T, Epplen JT, Favorova OO, Fedetz M, Fernández O, García-Martínez A, Gerdes L-A, Graetz C, Hartung H-P, Hoffjan S, Izquierdo G, Korobko DS, Kroner A, Kubisch C, Kümpfel T, Leyva L, Lohse P, Malkova NA, Montalban X, Popova EV, Rieckmann P, Rozhdestvenskii AS, Schmied C, Smagina IV, Tsareva EY, Winkelmann A, Zettl UK, Binder H, Cournu-Rebeix I, Hintzen R, Zimprich A, Comabella M, Fontaine B, Urcelay E, Vandenbroeck K, Filipenko M, Matesanz F, Zipp F, Bertram L, Genome-wide significant association with seven novel multiple sclerosis risk loci. J Med Genet 52, 848–855 (2015). [DOI] [PubMed] [Google Scholar]
- 37.C. International Multiple Sclerosis Genetics, Beecham AH, Patsopoulos NA, Xifara DK, Davis MF, Kemppinen A, Cotsapas C, Shah TS, Spencer C, Booth D, Goris A, Oturai A, Saarela J, Fontaine B, Hemmer B, Martin C, Zipp F, D’Alfonso S, Martinelli-Boneschi F, Taylor B, Harbo HF, Kockum I, Hillert J, Olsson T, Ban M, Oksenberg JR, Hintzen R, Barcellos LF, C. Wellcome Trust Case Control, I. B. D. G. C. International, Agliardi C, Alfredsson L, Alizadeh M, Anderson C, Andrews R, Søndergaard HB, Baker A, Band G, Baranzini SE, Barizzone N, Barrett J, Bellenguez C, Bergamaschi L, Bernardinelli L, Berthele A, Biberacher V, Binder TMC, Blackburn H, Bomfim IL, Brambilla P, Broadley S, Brochet B, Brundin L, Buck D, Butzkueven H, Caillier SJ, Camu W, Carpentier W, Cavalla P, Celius EG, Coman I, Comi G, Corrado L, Cosemans L, Cournu-Rebeix I, Cree BAC, Cusi D, Damotte V, Defer G, Delgado SR, Deloukas P, di Sapio A, Dilthey AT, Donnelly P, Dubois B, Duddy M, Edkins S, Elovaara I, Esposito F, Evangelou N, Fiddes B, Field J, Franke A, Freeman C, Frohlich IY, Galimberti D, Gieger C, Gourraud P-A, Graetz C, Graham A, Grummel V, Guaschino C, Hadjixenofontos A, Hakonarson H, Halfpenny C, Hall G, Hall P, Hamsten A, Harley J, Harrower T, Hawkins C, Hellenthal G, Hillier C, Hobart J, Hoshi M, Hunt SE, Jagodic M, Jelčić I, Jochim A, Kendall B, Kermode A, Kilpatrick T, Koivisto K, Konidari I, Korn T, Kronsbein H, Langford C, Larsson M, Lathrop M, Lebrun-Frenay C, Lechner-Scott J, Lee MH, Leone MA, Leppä V, Liberatore G, Lie BA, Lill CM, Lindén M, Link J, Luessi F, Lycke J, Macciardi F, Männistö S, Manrique CP, Martin R, Martinelli V, Mason D, Mazibrada G, McCabe C, Mero I-L, Mescheriakova J, Moutsianas L, Myhr K-M, Nagels G, Nicholas R, Nilsson P, Piehl F, Pirinen M, Price SE, Quach H, Reunanen M, Robberecht W, Robertson NP, Rodegher M, Rog D, Salvetti M, Schnetz-Boutaud NC, Sellebjerg F, Selter RC, Schaefer C, Shaunak S, Shen L, Shields S, Siffrin V, Slee M, Sorensen PS, Sorosina M, Sospedra M, Spurkland A, Strange A, Sundqvist E, Thijs V, Thorpe J, Ticca A, Tienari P, van Duijn C, Visser EM, Vucic S, Westerlind H, Wiley JS, Wilkins A, Wilson JF, Winkelmann J, Zajicek J, Zindler E, Haines JL, Pericak-Vance MA, Ivinson AJ, Stewart G, Hafler D, Hauser SL, Compston A, McVean G, De Jager P, Sawcer SJ, McCauley JL, Analysis of immune-related loci identifies 48 new susceptibility variants for multiple sclerosis. Nat Genet 45, 1353–1360 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Johansson Å, Rask-Andersen M, Karlsson T, Ek WE, Genome-wide association analysis of 350 000 Caucasians from the UK Biobank identifies novel loci for asthma, hay fever and eczema. Hum Mol Genet 28, 4022–4041 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Singh PK, van den Berg PR, Long MD, Vreugdenhil A, Grieshober L, Ochs-Balcom HM, Wang J, Delcambre S, Heikkinen S, Carlberg C, Campbell MJ, Sucheston-Campbell LE, Integration of VDR genome wide binding and GWAS genetic variation data reveals co-occurrence of VDR and NF-κB binding that is linked to immune phenotypes. BMC Genomics 18, 132 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Levels MJ, Fehres CM, van Baarsen LGM, van Uden NOP, Germar K, O’Toole TG, Blijdorp ICJ, Semmelink JF, Doorenspleet ME, Bakker AQ, Krasavin M, Tomilin A, Brouard S, Spits H, Baeten DLP, Yeremenko NG, BOB.1 controls memory B-cell fate in the germinal center reaction. J Autoimmun 101, 131–144 (2019). [DOI] [PubMed] [Google Scholar]
- 41.Laidlaw BJ, Duan L, Xu Y, Vazquez SE, Cyster JG, The transcription factor Hhex cooperates with the corepressor Tle3 to promote memory B cell development. Nat Immunol 21, 1082–1093 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Schubart DB, Rolink A, Kosco-Vilbois MH, Botteri F, Matthias P, B-cell-specific coactivator OBF-1/OCA-B/Bob1 required for immune response and germinal centre formation. Nature 383, 538–542 (1996). [DOI] [PubMed] [Google Scholar]
- 43.Nakamura M, Nishida N, Kawashima M, Aiba Y, Tanaka A, Yasunami M, Nakamura H, Komori A, Nakamuta M, Zeniya M, Hashimoto E, Ohira H, Yamamoto K, Onji M, Kaneko S, Honda M, Yamagiwa S, Nakao K, Ichida T, Takikawa H, Seike M, Umemura T, Ueno Y, Sakisaka S, Kikuchi K, Ebinuma H, Yamashiki N, Tamura S, Sugawara Y, Mori A, Yagi S, Shirabe K, Taketomi A, Arai K, Monoe K, Ichikawa T, Taniai M, Miyake Y, Kumagi T, Abe M, Yoshizawa K, Joshita S, Shimoda S, Honda K, Takahashi H, Hirano K, Takeyama Y, Harada K, Migita K, Ito M, Yatsuhashi H, Fukushima N, Ota H, Komatsu T, Saoshiro T, Ishida J, Kouno H, Kouno H, Yagura M, Kobayashi M, Muro T, Masaki N, Hirata K, Watanabe Y, Nakamura Y, Shimada M, Hirashima N, Komeda T, Sugi K, Koga M, Ario K, Takesaki E, Maehara Y, Uemoto S, Kokudo N, Tsubouchi H, Mizokami M, Nakanuma Y, Tokunaga K, Ishibashi H, Genome-wide association study identifies TNFSF15 and POU2AF1 as susceptibility loci for primary biliary cirrhosis in the Japanese population. Am J Hum Genet 91, 721–728 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Haralambieva IH, Ovsyannikova IG, Kennedy RB, Larrabee BR, Zimmermann MT, Grill DE, Schaid DJ, Poland GA, Genome-wide associations of CD46 and IFI44L genetic variants with neutralizing antibody response to measles vaccine. Hum Genet 136, 421–435 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Domeier PP, Chodisetti SB, Schell SL, Kawasawa YI, Fasnacht MJ, Soni C, Rahman ZSM, B-Cell-Intrinsic Type 1 Interferon Signaling Is Crucial for Loss of Tolerance and the Development of Autoreactive B Cells. Cell Rep 24, 406–418 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Camponeschi A, Todi L, Cristofoletti C, Lazzeri C, Carbonari M, Mitrevski M, Marrapodi R, Del Padre M, Fiorilli M, Casato M, Visentini M, DEC1/STRA13 is a key negative regulator of activation-induced proliferation of human B cells highly expressed in anergic cells. Immunol Lett 198, 7–11 (2018). [DOI] [PubMed] [Google Scholar]
- 47.Rauschmeier R, Reinhardt A, Gustafsson C, Glaros V, Artemov AV, Taneja R, Adameyko I, Månsson R, Busslinger M, Kreslavsky T, Cell-intrinsic functions of the transcription factor Bhlhe40 in activated B cells and T follicular helper cells restrain the germinal center reaction and prevent lymphomagenesis. bioRxiv, 2021.2003.2012.435122 (2021). [Google Scholar]
- 48.Deng Y, Bartosovic M, Ma S, Zhang D, Liu Y, Qin X, Su G, Xu ML, Halene S, Craft JE, Castelo-Branco G, Fan R, Spatial-ATAC-seq: spatially resolved chromatin accessibility profiling of tissues at genome scale and cellular level. bioRxiv, 2021.2006.2006.447244 (2021). [Google Scholar]
- 49.Cano-Gamez E, Trynka G, From GWAS to Function: Using Functional Genomics to Identify the Mechanisms Underlying Complex Diseases. Front Genet 11, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ulirsch JC, Lareau CA, Bao EL, Ludwig LS, Guo MH, Benner C, Satpathy AT, Kartha VK, Salem RM, Hirschhorn JN, Finucane HK, Aryee MJ, Buenrostro JD, Sankaran VG, Interrogation of human hematopoiesis at single-cell and single-variant resolution. Nat Genet 51, 683–693 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Su C, Johnson ME, Torres A, Thomas RM, Manduchi E, Sharma P, Mehra P, Le Coz C, Leonard ME, Lu S, Hodge KM, Chesi A, Pippin J, Romberg N, Grant SFA, Wells AD, Mapping effector genes at lupus GWAS loci using promoter Capture-C in follicular helper T cells. Nat Commun 11, 3294 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Corces MR, Granja JM, Shams S, Louie BH, Seoane JA, Zhou W, Silva TC, Groeneveld C, Wong CK, Cho SW, Satpathy AT, Mumbach MR, Hoadley KA, Robertson AG, Sheffield NC, Felau I, Castro MAA, Berman BP, Staudt LM, Zenklusen JC, Laird PW, Curtis C, Greenleaf WJ, Chang HY, The chromatin accessibility landscape of primary human cancers. Science 362, eaav1898 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Aguet F, Brown AA, Castel SE, Davis JR, He Y, Jo B, Mohammadi P, Park Y, Parsana P, Segrè AV, Strober BJ, Zappala Z, Cummings BB, Gelfand ET, Hadley K, Huang KH, Lek M, Li X, Nedzel JL, Nguyen DY, Noble MS, Sullivan TJ, Tukiainen T, MacArthur DG, Getz G, Addington A, Guan P, Koester S, Little AR, Lockhart NC, Moore HM, Rao A, Struewing JP, Volpi S, Brigham LE, Hasz R, Hunter M, Johns C, Johnson M, Kopen G, Leinweber WF, Lonsdale JT, McDonald A, Mestichelli B, Myer K, Roe B, Salvatore M, Shad S, Thomas JA, Walters G, Washington M, Wheeler J, Bridge J, Foster BA, Gillard BM, Karasik E, Kumar R, Miklos M, Moser MT, Jewell SD, Montroy RG, Rohrer DC, Valley D, Mash DC, Davis DA, Sobin L, Barcus ME, Branton PA, Abell NS, Balliu B, Delaneau O, Frésard L, Gamazon ER, Garrido-Martín D, Gewirtz ADH, Gliner G, Gloudemans MJ, Han B, He AZ, Hormozdiari F, Li X, Liu B, Kang EY, McDowell IC, Ongen H, Palowitch JJ, Peterson CB, Quon G, Ripke S, Saha A, Shabalin AA, Shimko TC, Sul JH, Teran NA, Tsang EK, Zhang H, Zhou Y-H, Bustamante CD, Cox NJ, Guigó R, Kellis M, McCarthy MI, Conrad DF, Eskin E, Li G, Nobel AB, Sabatti C, Stranger BE, Wen X, Wright FA, Ardlie KG, Dermitzakis ET, Lappalainen T, Aguet F, Ardlie KG, Cummings BB, Gelfand ET, Getz G, Hadley K, Handsaker RE, Huang KH, Kashin S, Karczewski KJ, Lek M, Li X, MacArthur DG, Nedzel JL, Nguyen DT, Noble MS, Segrè AV, Trowbridge CA, Tukiainen T, Abell NS, Balliu B, Barshir R, Basha O, Battle A, Bogu GK, Brown A, Brown CD, Castel SE, Chen LS, Chiang C, Conrad DF, Cox NJ, Damani FN, Davis JR, Delaneau O, Dermitzakis ET, Engelhardt BE, Eskin E, Ferreira PG, Frésard L, Gamazon ER, Garrido-Martín D, Gewirtz ADH, Gliner G, Gloudemans MJ, Guigo R, Hall IM, Han B, He Y, Hormozdiari F, Howald C, Kyung Im H, Jo B, Yong Kang E, Kim Y, Kim-Hellmuth S, Lappalainen T, Li G, Li X, Liu B, Mangul S, McCarthy MI, McDowell IC, Mohammadi P, Monlong J, Montgomery SB, Muñoz-Aguirre M, Ndungu AW, Nicolae DL, Nobel AB, Oliva M, Ongen H, Palowitch JJ, Panousis N, Papasaikas P, Park Y, Parsana P, Payne AJ, Peterson CB, Quan J, Reverter F, Sabatti C, Saha A, Sammeth M, Scott AJ, Shabalin AA, Sodaei R, Stephens M, Stranger BE, Strober BJ, Sul JH, Tsang EK, Urbut S, van de Bunt M, Wang G, Wen X, Wright FA, Xi HS, Yeger-Lotem E, Zappala Z, Zaugg JB, Zhou Y-H, Akey JM, Bates D, Chan J, Chen LS, Claussnitzer M, Demanelis K, Diegel M, Doherty JA, Feinberg AP, Fernando MS, Halow J, Hansen KD, Haugen E, Hickey PF, Hou L, Jasmine F, Jian R, Jiang L, Johnson A, Kaul R, Kellis M, Kibriya MG, Lee K, Billy Li J, Li Q, Li X, Lin J, Lin S, Linder S, Linke C, Liu Y, Maurano MT, Molinie B, Montgomery SB, Nelson J, Neri FJ, Oliva M, Park Y, Pierce BL, Rinaldi NJ, Rizzardi LF, Sandstrom R, Skol A, Smith KS, Snyder MP, Stamatoyannopoulos J, Stranger BE, Tang H, Tsang EK, Wang L, Wang M, Van Wittenberghe N, Wu F, Zhang R, Nierras CR, Branton PA, Carithers LJ, Guan P, Moore HM, Rao A, Vaught JB, Gould SE, Lockart NC, Martin C, Struewing JP, Volpi S, Addington AM, Koester SE, Little AR, Consortium G, Genetic effects on gene expression across human tissues. Nature 550, 204–213 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Avsec Ž, Weilert M, Shrikumar A, Krueger S, Alexandari A, Dalal K, Fropf R, McAnany C, Gagneur J, Kundaje A, Zeitlinger J, Base-resolution models of transcription-factor binding reveal soft motif syntax. Nat Genet 53, 354–366 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mu Z, Wei W, Fair B, Miao J, Zhu P, Li YI, The impact of cell type and context-dependent regulatory variants on human immune traits. Genome Biol 22, 122 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Freimer JW, Shaked O, Naqvi S, Sinnott-Armstrong N, Kathiria A, Chen AF, Cortez JT, Greenleaf WJ, Pritchard JK, Marson A, Systematic discovery and perturbation of regulatory genes in human T cells reveals the architecture of immune networks. bioRxiv, 2021.2004.2018.440363 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Vinuesa CG, Sanz I, Cook MC, Dysregulation of germinal centres in autoimmune disease. Nat Rev Immunol 9, 845–857 (2009). [DOI] [PubMed] [Google Scholar]
- 58.Pratama A, Vinuesa CG, Control of TFH cell numbers: why and how? Immunol Cell Biol 92, 40–48 (2014). [DOI] [PubMed] [Google Scholar]
- 59.Linterman MA, Beaton L, Yu D, Ramiscal RR, Srivastava M, Hogan JJ, Verma NK, Smyth MJ, Rigby RJ, Vinuesa CG, IL-21 acts directly on B cells to regulate Bcl-6 expression and germinal center responses. J Exp Med 207, 353–363 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.McPhee CG, Bubier JA, Sproule TJ, Park G, Steinbuck MP, Schott WH, Christianson GJ, Morse HC, Roopenian DC, IL-21 is a double-edged sword in the systemic lupus erythematosus-like disease of BXSB.Yaa mice. J Immunol 191, 4581–4588 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Domeier PP, Schell SL, Rahman ZSM, Spontaneous germinal centers and autoimmunity. Autoimmunity 50, 4–18 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Qin Y, Duquette P, Zhang Y, Talbot P, Poole R, Antel J, Clonal expansion and somatic hypermutation of V(H) genes of B cells from cerebrospinal fluid in multiple sclerosis. J Clin Invest 102, 1045–1050 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Kim HJ, Krenn V, Steinhauser G, Berek C, Plasma cell development in synovial germinal centers in patients with rheumatoid and reactive arthritis. J Immunol 162, 3053–3062 (1999). [PubMed] [Google Scholar]
- 64.Amft N, Curnow SJ, Scheel-Toellner D, Devadas A, Oates J, Crocker J, Hamburger J, Ainsworth J, Mathews J, Salmon M, Bowman SJ, Buckley CD, Ectopic expression of the B cell-attracting chemokine BCA-1 (CXCL13) on endothelial cells and within lymphoid follicles contributes to the establishment of germinal center-like structures in Sjögren’s syndrome. Arthritis Rheum 44, 2633–2641 (2001). [DOI] [PubMed] [Google Scholar]
- 65.Wagar LE, Live cell barcoding for efficient analysis of small samples by mass cytometry. Methods Mol Biol 1989, 125–135 (2019). [DOI] [PubMed] [Google Scholar]
- 66.Butler A, Hoffman P, Smibert P, Papalexi E, Satija R, Integrating single-cell transcriptomic data across different conditions, technologies, and species. Nat Biotechnol 36, 411–420 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902.e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Hafemeister C, Satija R, Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol 20, 296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.McGinnis CS, Murrow LM, Gartner ZJ, DoubletFinder: Doublet Detection in Single-Cell RNA Sequencing Data Using Artificial Nearest Neighbors. Cell Syst 8, 329–337.e324 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, Baglaenko Y, Brenner M, Loh P-R, Raychaudhuri S, Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods 16, 1289–1296 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Dijk D, Sharma R, Nainys J, Yim K, Kathail P, Carr AJ, Burdziak C, Moon KR, Chaffer CL, Pattabiraman D, Bierie B, Mazutis L, Wolf G, Krishnaswamy S, Pe’er D, Recovering Gene Interactions from Single-Cell Data Using Data Diffusion. Cell 174, 716–729.e727 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, Liu XS, Model-based analysis of ChIP-Seq (MACS). Genome Biol 9, R137 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schep AN, Wu B, Buenrostro JD, Greenleaf WJ, chromVAR: inferring transcription-factor-associated accessibility from single-cell epigenomic data. Nat Methods 14, 975–978 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Wolock SL, Lopez R, Klein AM, Scrublet: Computational Identification of Cell Doublets in Single-Cell Transcriptomic Data. Cell Syst 8, 281–291.e289 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Lopez-Delisle L, Rabbani L, Wolff J, Bhardwaj V, Backofen R, Grüning B, Ramírez F, Manke T, pyGenomeTracks: reproducible plots for multivariate genomic data sets. Bioinformatics, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Machiela MJ, Chanock SJ, LDlink: a web-based application for exploring population-specific haplotype structure and linking correlated alleles of possible functional variants. Bioinformatics 31, 3555–3557 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S1. Tonsillectomy patient donor details, experimental study design and cell type frequencies across donors.
Data file S2. Broad resolution of tonsil immune cell subset scRNA-seq gene expression markers.
Data file S3. High resolution B cell subset scRNA-seq gene expression markers.
Data file S4. High resolution T cell subset scRNA-seq gene expression markers.
Data file S5. Differential chromatin accessibility peaks from high resolution annotation of tonsil immune cell scATAC-seq.
Data file S6. Differential chromatin accessibility gene scores from high resolution annotation of tonsil immune cell scATAC-seq.
Data file S7. Peak2gene predictions for tonsil scATAC-seq and scRNA-seq analysis.
Data file S8. Loop coordinates to visualize predicted tonsil immune peak2gene interactions.
Data file S9. Gene expression markers for integrated tonsil, peripheral blood and bone marrow immune cell populations from scRNA-seq.
Data file S10. Differential chromatin accessibility peak markers for integrated tonsil, peripheral blood and bone marrow immune cell populations from scATAC-seq.
Data file S11. Peak2gene predictions for integrated bone marrow, blood and tonsil scATAC-seq and scRNA-seq analysis.
Data file S12. Loop coordinates to visualize predicted integrated bone marrow, blood and tonsil immune peak2gene interactions.
Data file S13. Peak2gene linkage annotation of fine-mapped SNPs found in chromatin accessibility peaks from integrated tonsil, peripheral blood and bone marrow scATAC-seq datasets.
Data file S14. Raw Data file.
Figure S1. Single-cell library metadata, integration, batch correction and quality control.
Figure S2. Comparison of RNA expression, cell surface protein expression and chromatin accessibility of key marker genes.
Figure S3. Age-related changes in tonsillar immune cell populations by scRNA-seq and CyTOF.
Figure S4. Tonsil scRNA-seq marker gene heatmaps, annotation of GC B cells and reproducibility of cell types across donors.
Figure S5. Differential expression in autoimmune disease of top scRNA-seq marker genes for the IFN_active B cell cluster.
Figure S6. Reproducibility of scATAC-seq cluster frequencies and correlation of peak accessibilities between donors.
Figure S7. Differential peak analysis of scATAC-seq clusters, peak-to-gene predictions and alternative pseudotime analysis.
Figure S8. Batch correction and quality control of integrated bone marrow, blood and tonsil immune scRNA-seq and scATAC-seq.
Figure S9. Integrated bone marrow, blood and tonsil scRNA-seq and scATAC-seq markers.
Figure S10. Tonsil B cell-enriched gene expression markers compared to peripheral blood B cells.
Figure S11. Enrichment of fine-mapped autoimmune variants in immune cell subsets.
Figure S12. Genome snapshots of fine-mapped autoimmune variants at GZMB/GZMH, NKX2–3 and COTL1/KHLH36 loci.
Figure S13. Genome snapshots of fine-mapped autoimmune variants at KSR1/LGALS9 and TNFRSF1A/LTBR loci.
Figure S14. Genome snapshots of germinal center-associated cell type-specific regulatory activity at fine-mapped autoimmune variants at CD80, PRAG1 and SLC38A9/DDX4 loci.
Figure S15. Genome snapshots of germinal center-associated cell type-specific regulatory activity at fine-mapped autoimmune variants at VAV3 and DLEU2 loci.
Figure S16. Linkage disequilibrium scores for variants at IL21, IL21R and BCL6 loci.
Figure S17. Genome snapshots of fine-mapped autoimmune variants at ETS1 and IKZF3 loci.
Figure S18. Genome snapshots of fine-mapped autoimmune variants at STAT4 and IRF8 loci.
Figure S19. Genomic landscape at HHEX and expression of KLF family transcription factors.
Table S1. CyTOF phenotyping antibody panel.
Table S2. CITE-seq antibody details.
Data Availability Statement
Raw and processed data for this study are available at Gene Expression Omnibus under accession GSE165860. All other data needed to evaluate the conclusions in the paper are present in the paper or the Supplementary Materials. All code and scripts necessary to repeat analysis in this manuscript is available upon request.