

Abstract
Implementation of precision medicine in lung cancer has benefited from intense research in the past years, developing subsequently an improved quality of life and increased overall survival of the patients. Targeted therapy has become one of the most important therapeutic innovations for the non-small cell lung cancer category with anaplastic lymphoma kinase (ALK) gene rearrangement. The aim of this review is to provide a thorough overview of the main molecules of ALK tyrosine kinase inhibitors (TKI) with their general and particular mechanisms of resistance, the main methods of ALK gene detection, each with advantages and limits, and the future perspectives currently under research which try to overcome the mechanisms of resistance. We have used two of the most reliable medical databases EMBASE and PubMed to properly select the latest and the most relevant articles for this topic. Encouraged by the promising results, the clinical practice was enriched by the approval of TKI molecules, three generations being developed, each one with more powerful agents than the previous ones. Unfortunately, the resistance to TKI eventually occurs and it may be induced by several mechanisms, either known or unknown. Crizotinib was the most intensely studied TKI, becoming the first molecule approved into clinical practice and although four other drugs have been broadly used (alectinib, ceritinib, brigatinib, and lorlatinib) it seems that even the most recently developed one remains imperfect due to the resistance mutations that developed. There are two types of resistance generally described for the entire class and for the particular drugs, but half of them remain unknown.
Keywords: Non-small cell lung cancer, anaplastic lymphoma kinase, anaplastic lymphoma kinase, tyrosine kinase inhibitors, resistance mechanisms
INTRODUCTION
Lung cancer has gained a top place in the cancer-related incidence and mortality worldwide. Non-small cell lung cancer (NSCLC) which represents almost 80-85% of the patients is considered a heterogeneous disease due to the wide spectrum of molecular targets identified which have benefited from personalized therapy in recent years [1,2]. To better select the patients for this type of treatment, a proper method of anaplastic lymphoma kinase (ALK) gene detection must be used according to the diagnosis standards and the main options applied in clinical practice are fluorescence in situ hybridization (FISH), immunohistochemistry (IHC), reverse transcriptase-polymerase chain reaction (RT-PCR), next-generation sequencing (NGS), liquid biopsy, and new potential biomarkers such as circulating tumor cells (CTCs), cell-free DNA, and exosomes are being investigated [3]. The therapeutic development in this domain has led to the implementation of three generations of ALK tyrosine kinase inhibitors (TKI) with five molecules approved into clinical practice and two more molecules, such as entrectinib and ensartinib, are waiting for clinical approval [4]. The development of the ALK TKI in NSCLC patients, beginning with crizotinib, the first molecule approved and followed by second and third-generation inhibitors (ceritinib, alectinib, brigatinib, and lorlatinib) has provided an increased progression-free survival (PFS) and overall survival (OS) in comparison with chemotherapy [5,6]. Unfortunately, despite the initial benefit of ALK TKI, resistance mechanisms have been identified upon progression [7,8]. From the available data, we have discovered that almost half of the mechanisms of resistance remain unknown at this moment, but we have tried to present the recognized mutations and the future perspectives, even though they are under research. To properly identify the mechanisms, we have encompassed the main detection methods implemented into clinical practice or in preclinical data. Besides the particular mutations for each molecule, we have also described the factors which determine secondary mutations for the entire class [9,10]. Regarding the challenges of overcoming resistance, promising results are represented by the combination between ALK and epidermal growth factor receptor (EGFR) TKI, metformin, MYC (avian myelocytomatosis viral oncogene homolog) – inhibitors, human EGFR (HER) family, mammalian target of rapamycin (mTOR) pathway, anti-angiogenesis factors, etc. Nevertheless, the most awaited results are for the combination between ALK TKI and immunotherapy, which represents the most innovative therapeutic option for NSCLC patients [11-14].
MATERIALS AND METHODS
In the present review, we have tried to encompass the most relevant and accurate available data from the literature regarding ALK TKI mechanisms of resistance in NSCLC patients. Our working hypothesis is based on describing the mechanisms of resistance for each molecule approved, presenting the advantages and disadvantages of the main detection methods currently used in clinical practice, and describing the future possibilities identified or still under research to provide therapeutic benefit. Our process of selection included two of the most reliable databases in the medical field, EMBASE, and PubMed, under specified criteria such as 10-years filter, English language, and Human species following A Preferred Reporting Items for Systematic Review and Meta-analysis statement as mentioned in Figure 1, but the final article selection remained subjective [15].
FIGURE 1.
Description of the literature identification process (A Preferred Reporting Items for Systematic Review and Meta-analysis flowchart) [15].
RESULTS
The role of ALK gene rearrangement in NSCLC
ALK gene has an incidence of 3-7% in patients with NSCLC and although more than 27 variants of ALK fusion proteins have been discovered, the most common partner is EML 4 (echinoderm microtubule-associated protein-like 4) [16,17]. The EML4-ALK fusion gene results from the paracentric inversion of chromosome 2 with at least 15 variants identified, variant 1 (v1) involving exon 13 being the most common, followed by the variant 3a/b (v3a/b) affecting exon 6 and variant 2 involving exon 20 [18,19]. This category of patients with ALK rearrangement has the following clinicopathological characteristics: Young age at diagnosis (a median of 50 years old), women gender, non-smokers/light smokers, histology of adenocarcinoma with particular morphologic patterns such as and cribriform and solid signet ring, expression of thyroid transcription factor 1, tendency to metastasize in pleura or pericardium, frequently with more metastatic sites than other molecular types, and predominant central nervous system (CNS) metastases [20-22].
Identifying the main mechanisms of resistance to ALK TKI
Despite the major therapeutic improvement of ALK TKI in NSCLC, disease progression after initial benefit has been described due to the development of resistance mechanisms with clinically progressive disease and a variable range of aggressiveness [23-25]. The mechanisms of resistance can be divided into “de novo” or acquired depending on the timeline of occurrence and according to the involvement of ALK are classified into ALK-dependent or ALK-independent mechanisms [26,27]. Regarding the ALK dependency, ALK-dependent “on-target” tumors are driven by ALK signaling, while ALK “off-target” means that the driver mutation and the tumor cells are based on a different mechanism such as by-pass signaling pathways activation, drug efflux mechanisms, or histological transition (such as small cell transformation) [28,29]. Secondary resistance can be divided into dominant (ALK intra-kinase domain mutation, increased copy number gain of ALK gene) and non-dominant such as tumor heterogeneity, bypass signaling pathways activation such as the EGFR, Kirsten rat sarcoma viral oncogene homolog (KRAS), v-kit Hardy-Zuckerman 4 feline sarcoma viral oncogene homolog (KIT), met proto-oncogene (MET), and insulin-like growth factor 1 receptor (IGF-1R), as presented in Figure 2 [30,31]. Studies have shown that multiple mechanisms of resistance can occur simultaneously by the identification of epithelial-to-mesenchymal transition (EMT) in ALK kinase domain mutations [31,32]. Furthermore, the presence of tumor protein 53 (TP53) mutations in ALK-rearranged NSCLC defines a category with the instability of the chromosomes, conferring a prognostic role and determines pathogenic aberrations which can co-occur [32-34]. Clinical data suggests that the baseline level of TP53 is correlated with worse PFS, a shorter OS, and a more aggressive disease [35-37]. Furthermore, other genes involved with low frequency were identified, such as: BRAF, FGFR2, MET, NRAS, and PIK3CA [38,39]. Most of the secondary resistance mutations from ALK tyrosine-kinase domain currently are mainly point mutations such as the gatekeeper mutation L1196M, G1269A, and G1202R [40,41]. Although in NSCLC patients, personalized therapy was developed using not only using ALK gene but also EGFR as molecular target, the mechanisms of resistance between these two genes are completely distinct due to different tumor biology including genomic instability and different oncogenic dependency. Furthermore, the TKI implemented has contrasting properties determined by the binding modalities and inhibiting potency of the molecules [42,43]. Among the by-pass signaling mechanisms which confer resistance to ALK inhibition, the HER family activation is one the most important identified [43,44]. The identification of EGFR in post-crizotinib resistance, even in the absence of mutations, suggests the activation of this pathway by the paracrine stimuli, such as neuregulin 1 (NRG-1) [44,45].
FIGURE 2.
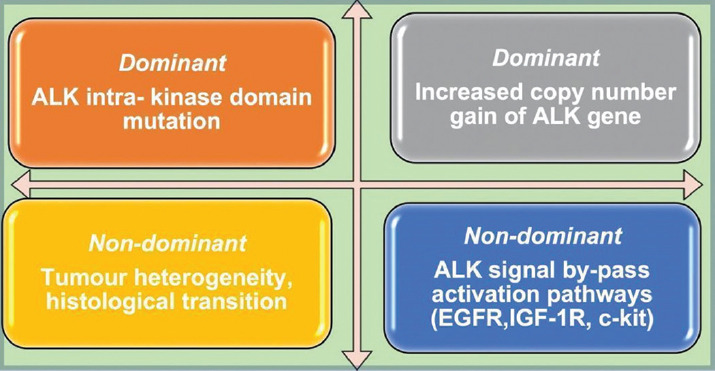
Describing the most common mechanisms of resistance to anaplastic lymphoma kinase tyrosine kinase inhibitors divided into dominant and non-dominant category.
Despite the development of TKI resistance mechanisms previously described, targeted therapy is considered the main therapeutic approach in ALK-rearranged NSCLC and literature data reports a median survival time of approximately 4 years in metastatic patients, significantly higher in comparison with chemotherapy [46,47].
Exploring ALK TKI and their particularities
The therapeutic implementation of the ALK TKI agents generates a clinical controversy based on the appropriate use, which should be guided by the potency and the CNS penetration, a frequent site of metastasis in patients with ALK rearrangement [48-51]. The main studies which have led to the approval of these molecules are summarized in Table 1. Another two molecules, ensartinib and entrectinib have been discovered and are currently into clinical trials, but currently, they have not gained FDA approval. Ensartinib is a potent ALK inhibitor with high CNS penetration and potential synergistic activity with the mTOR inhibitor rapamycin. Entrectinib is a new generation inhibitor of multiple targets, such as neurotrophic tyrosine kinase, receptor type 1, neurotrophic tyrosine kinase, receptor type 2, neurotrophic tyrosine kinase, receptor type 3, ROS proto-oncogene 1 (ROS1), and ALK which has been approved in NRK and ROS1 positive NSCLC [52-54]. Among the mechanisms of resistance, there are several point mutations identified for each molecule, which may be common in the same TKI generation or particularly, as it will be detailed in the following paragraphs and mentioned in Figure 3 [55-58]. Studies have shown that the development of ALK resistance mutations can be induced by the use of multiple sequential lines of TKI [58-60]. After exposure to crizotinib, ALK gene copy number gain has been observed, but it was not considered to have a clinical impact and therefore was not reported as resistance mechanism after more potent ALK TKI [61]. After developing resistance to second-generations ALK TKI, literature data have proven that half of the mutations are acquired, out of which G1202R is the most frequent [62,63]. The concept of progressive multistep genetic complexity was implemented due to the identification of two or more mutations in patients who benefited from ALK TKI molecules of the first- and second-generation [64].
TABLE 1.
A summary description of the main studies and results of the ALK TKI in patients with ALK-positive NSCLC [70]
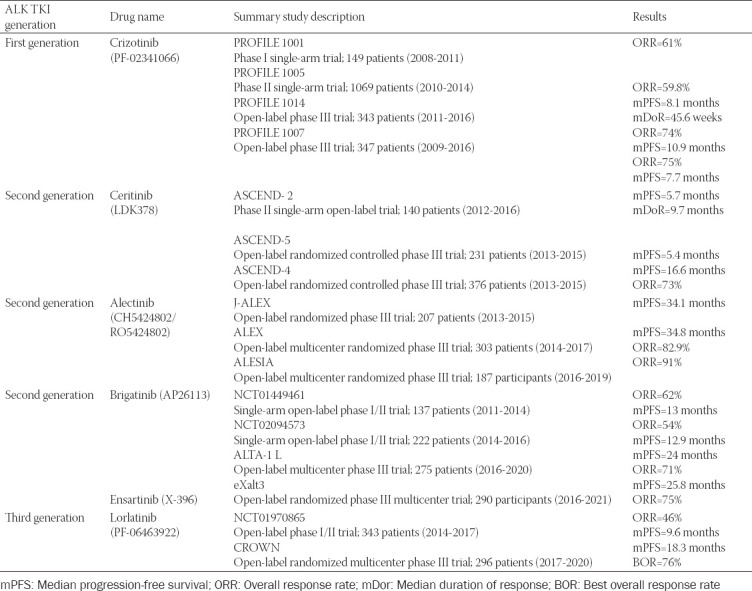
FIGURE 3.
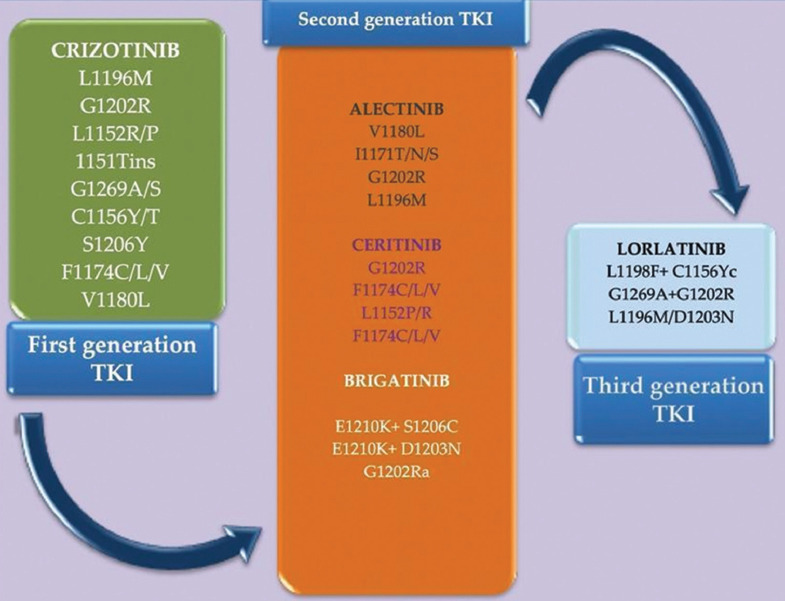
The most common anaplastic lymphoma kinase (ALK)- dependent resistance mutations associated with therapeutic resistance to ALK tyrosine kinase inhibitors (TKI). Each generation of ALK TKI is presented in the scheme with the approved drug and the main point mutations associated with resistance and different colors were used to highlight these point mutations.
Crizotinib is an oral TKI inhibitor of the first generation TKI with recommended oral dose is of 250 mg twice daily in a 28-day cycle until disease progression or no longer tolerated by the patient and its metabolism involves the CYP3A 4/5 mechanisms, the most common side effects include: Hepatotoxicity, interstitial lung disease (pneumonitis), prolongation of QT interval, bradycardia, visual toxicity (including visual loss), and gastrointestinal affection, which require either dose reduction or permanent interruption [65]. Ceritinib is a second-generation ALK TKI with a therapeutic dose of 450 mg orally once daily with a mainly hepatic metabolism through the enzyme complex CYP3A. Apart from the common adverse effects mentioned for the first-generation TKI, subsequently to the treatment with ceritinib the following toxicities have been identified: Hyperglycemia, pancreatitis, and embryo-fetal toxicity [66]. Alectinib is recommended in a twice-daily dose of 600 mg and its particular side effects include renal impairment, severe myalgia, and creatine phosphokinase elevation [67]. Brigatinib is approved with a dose of 90 mg once daily for the first 7 days of the treatment, increasing afterward at 180 mg once daily. It presents the common side effects of the entire class but also hypertension [68]. Lorlatinib is a third-generation TKI that is metabolized mainly by CYP3A4 and UGT1A4 pathways recommended in a dose of 100 mg once daily. The adverse effects of this class are different from the previous described and include CNS side effects (such as seizures, hallucinations, and changes in cognitive function), hyperlipidemia, and atrioventricular block [69].
Describing mechanisms of resistance in the first, second, and third generation of ALK TKI
Crizotinib was the first oral TKI molecule approved in 2011 by the FDA for metastatic NSCLC patients with ALK mutation, but almost a third of the patients had developed primary or secondary resistance within 1-2 years [71]. Crizotinib was first implemented to inhibit the c-MET pathway but has also proved important activity against ALK and ROS1 gene [72]. Furthermore, due to the promising results obtained from the clinical trials, crizotinib was approved in November 2013 with the indication of a second therapeutic option for patients with ALK mutation and progressive disease after platinum doublet treatment. About half of the patients developed metastasis in the CNS, considered the main or the exclusive site of disease progression because crizotinib has a decreased capacity to cross the blood-brain barrier [73]. One of the most frequent mutations identified in crizotinib resistance patients was L1196M gatekeeper mutation, analogous to EGFR T790M, which alters the gatekeeper residue at the bottom of the ATP-binding pocket and subsequently inhibits the binding with TKI. In addition, it has been discovered that patients who harbor L1196M mutation tend to have a shorter PFS [74]. The G1202R solvent front mutation has been identified in patients treated with first-generation ALK-TKI and determines the alteration of the binding activity of crizotinib [75,76]. Literature data have identified that the most common mutations in crizotinib-resistant patients are the following: L1152R, C1156Y, F1174C, L1196M, D1203N, C1156Y, and G1269A as well as EGFR activation working as the bypass pathway [77,78]. Other mechanisms of resistance to crizotinib have been identified: ALK gene amplification and copy number gain, new mutations such as 1151Tins and point mutation S1206Y in the solvent front of the kinase domain [79,80]. Intrinsic factors such as the concomitant KRAS (Kirsten rat sarcoma 2 viral oncogene homolog) mutations, MYC amplification, and the Bim deletion polymorphism could determine primary resistance to crizotinib [80,81]. In the acquired resistance, increased ErbB signaling through phosphorylation has been involved as well as the activation of the IGFR-1R pathway, EMT, and autophagy. The extended research in this domain has determined the identification of new mutation genes, such as CSMD3, CDKN2, MAGI1, CREBBP, DOT1L, PBX1, and PRKDC [82,83]. Potential mechanisms of resistance to crizotinib are represented by the activation of EGFR through the overexpression of the cKIT gene determined by its linkage to stem cell factor [83]. Activation of the PI3K/AKT/mTOR pathway has also been recognized as a resistance mechanism to crizotinib, possibly through increased autophagy of the ALK receptor. Synergy of crizotinib and a mTOR inhibitor in terms of inhibitory activity has been demonstrated in a cell line [84]. Therefore, due to the identification of crizotinib resistance, further ALK TKI molecules of the second and third generation have been implemented for the daily therapeutic approach [85].
Ceritinib was the initial ALK TKI molecule of the second-generation class approved to overcome resistance to crizotinib [86]. In 2014, ceritinib was indicated ALK-positive patients with disease progression on or intolerance to crizotinib and in 2017 was indicated as the first-line therapeutic setting. The mechanism of action is represented by the inhibition of the autophosphorylation of ALK gene and the molecular targets include IGF-1 R, InsR, and ROS1 and have an activity of 20 times higher against crizotinib–resistant tumor cell lines [87]. Ceritinib inhibits the most common ALK mutations, such as L1196 M, G1269A, I1171T, and S1206Y, which determine resistance to crizotinib [86,87]. In patients who progressed during ceritinib treatment, secondary mutations were detected such as G1202R, F1174 C/L, C1156Y, G1202del, and L1196M. The F1174L mutation which can be resistant to ceritinib but sensitive to alectinib and the I1171 mutation sensitive to ceritinib but resistant to alectinib has been discovered [88]. In addition, the F1174C/D1203N compound mutations found in patients treated with crizotinib and ceritinib have conferred resistance to these molecules and also to alectinib, brigatinib, and lorlatinib [89]. A combination between a MEK inhibitor and ALK TKI was developed due to ceritinib resistance identified by the MEK activating mutation (MAP2K1-K57N) [90,91].
Alectinib is a molecule from the second generation, which has proved antitumor effect on NSCLC patients with ALK rearrangement who have benefited previously from crizotinib which was approved in December 2015 and subsequently in November 2017 was indicated as first-line setting of patients with advanced ALK-positive NSCLC [92]. Due to its chemical structure, it has proved efficient for patients with crizotinib-resistant ALK mutations such as L1196M, F1174L C1156Y, G1269A, 1151Tins, and L1152R but not G1202R [91]. Potential mechanisms of resistance to alectinib include the activation of EGFR signaling pathway, increased activation of IGF1R, HER3 overexpression, and P2Y receptors (P2Y1 and P2Y2) which increase the levels of activated protein kinase C (PKC) [93]. At the relapse, among the acquired resistance mutations, G1202R, I1171T/S, and V1180L and L1196M were identified [94]. Another mechanism involved in the acquired resistance to alectinib was activation of the hepatocyte growth factor (HGF)/MET signaling pathway [95]. Recent studies have shown that in vitro inhibition of Src and MET restored the sensitivity to alectinib in patient-derived cell lines which have turned resistant to alectinib by the co-activation of these two mechanisms [96]. A potential resistance mechanism common for alectinib and crizotinib can be represented by the activation of HER3 pathway and overexpression of NRG1. [97]. Furthermore, human neuromedin U (NMU) gene is identified as a potential candidate which confers alectinib resistance in NSCLC patients, according to recent studies [98,99].
Brigatinib is a potent molecule from the second generation of ALK TKI with approval in 2017 for the indication of advanced NSCLC with ALK rearrangement with progressive disease on or intolerance to crizotinib. Furthermore, in 2020 it was approved as first-line treatment for ALK-positive metastatic NSCLC patients [100-102]. Brigatinib also inhibits ROS1 fusions and EGFR mutation (L858R) and has a remarkable activity on the CNS [102,103]. According to the clinical data, brigatinib is active against all the 17 known resistance mutations to the ALK gene, including G1202R and L1196M [104,105]. Among the mechanisms of resistance to brigatinib, gene mutations such as D1203, S1206, E1210K + S1206C, and E1210K + D1203N have been identified [106]. The molecular structure of brigatinib, which may lead to resistance development, is also influenced by the L1196M, G1269A, F1174L, and R1275Q mutations [107,108]. Furthermore, it has been discovered that the mutation G1202R occurs after previous exposure to ceritinib, alectinib, and brigatinib, meaning that lorlatinib is the only efficient therapeutic agent [108,109].
Ensartinib is a novel ALK TKI molecule of the second generation with improved action on CNS metastasis and is a potential option for the first-line setting according to the preclinical data [110]. Compared to the other ALK TKI, ensartinib is active against the following mutations: G1123S, L1198F, F1174, C1156Y, L1196M, S1206R, and T1151, but less potent against G1202R and G1269A [111]. Upon progressive disease, two mutations have been identified: E1210K and S1206F. Apart from the ALK inhibition, ensartinib is also active against MET, Axl, ABL, EPHA2, Leukocyte Receptor Tyrosine Kinase, ROS1, and STE20 like kinase [112].
Entrectinib is another second-generation ALK inhibitor candidate, which is able to penetrate the blood-brain barrier exhibiting, therefore, consistent intracranial activity under clinical research. In addition, apart from the ALK inhibition, entrectinib is also active against Tropomyosin receptor kinase A, Tropomyosin receptor kinase B, Tropomyosin receptor kinase C, and ROS1. The results of the clinical data are awaited to conclude the efficacy of this molecule in ALK TKI setting [113].
Lorlatinib is a molecule of the third-generation approved in 2018 by the FDA in the first-line setting for metastatic NSCLC patients and ALK rearrangement with progressive disease on crizotinib and other ALK inhibitors [114,115]. Due to its activity, it targets mutations, which determine resistance to the other ALK TKI, including G1202R solvent-form mutation, and facilitates CNS penetration, subsequently determining an increased survival rate [116]. After analyzing the post-progression samples, the literature data suggest that half of the patients developed compound mutations such as ALK-G1202R/L1196M, ALK-E1210K/D1203N/G1269A, and ALK-I1171N/L1198F [117]. Although it proves efficacy against C1156Y gene, resistant to crizotinib or ceritinib, a potential mechanism of resistance is conferred by the co-occurrence of L1198Fmutation [118,119]. More clinical data are awaited because the previous mentioned mutations narrow the therapeutic options; therefore, this molecule is under active research.
The main methods of detecting ALK rearrangement and mechanisms of resistance in NSCLC
To provide the best therapeutic option, ALK gene rearrangement identification is essential. Several mechanisms of detection have been implemented, each with their advantages and limits, as described in Table 2. FISH testing with the ALK break-apart probe kit was the mandatory diagnosis test due to its use as a companion diagnostic tool in the clinical trials of crizotinib [120]. Although it has been considered the gold standard because it provides very good specificity, can be performed in small biopsy sample and it is used to validate and compare other ALK detection methods, FISH has disadvantages such as signal instability, expensiveness, difficulty in scoring, the long turnaround time, and the need for specific fluorescence microscopy and particular expertise to validate the results. Furthermore, FISH can only determine if there is a break in the ALK locus but does not have the ability to distinguish between the ALK fusion partners [121]. A modified FISH assay with filtration enrichment, filter-adapted FISH was implemented to identify ALK rearrangements on the CTC, but specific technological requirements are necessary for both CTC isolation and analysis, but due to the precise results obtained, it may become a non-invasive predictive biomarker [122].
TABLE 2.
The main methods used to identify ALK mutation and mechanism of resistance described according to their main advantages and limitations
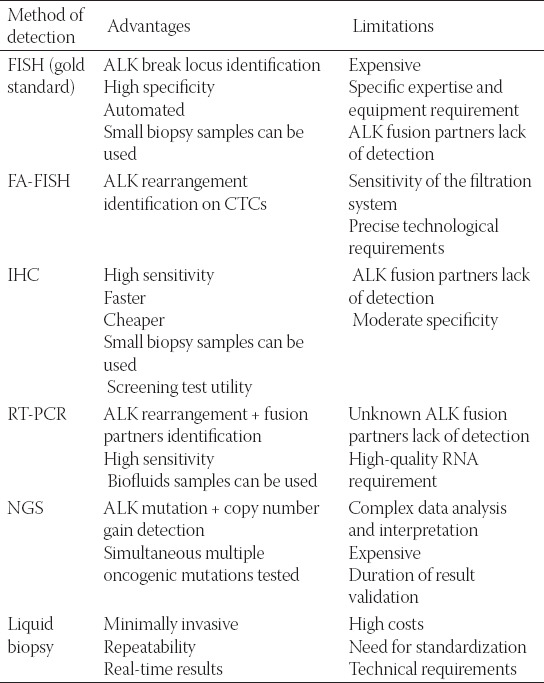
IHC has excellent sensitivity, with the advantage of being less expensive, following a simplified and rapid method than FISH with the utility as a screening test for diagnosis of NSCLC patients with ALK rearrangement, but it has poor sensitivity and it cannot identify the ALK fusion partner [123]. RT-PCR and NGS technologies allow the analysis of ALK gene rearrangement and its fusion partner. RT-PCR uses specific primers for identified ALK fusion partners, but a disadvantage is that several tests should be performed before identification of the ALK fusion partner variant and the unknown molecules could not be detected [124]. Furthermore, high-quality samples are needed which can determine either false-negative or positive results. RT-PCR was commonly used with screening purpose because of the high sensitivity and the applicability to biofluids, but it requires assay optimization for each new fusion partner and high-quality ribonucleic acid (RNA) [125]. NGS has the ability of identifying ALK mutations, copy number gain and provide identification of numerous oncogenic mutations at the same time in one assay with high sensitivity and reproducibility. At present, NGS has proved able to detect ALK fusion genes by sequencing the intron between exons 19 and 20, where they usually occur, but confirmation is required to gain approval for clinical application [126]. Tumor tissue samples used for the previous described testing methods are obtained from bronchoscopy or percutaneous lung biopsy and present several limitations, including the invasiveness acquisition, which may determine complications or may be insufficient and the tumor heterogeneity. To properly identify the mechanisms of resistance, liquid biopsy is considered of great significance due to its potential of detection against tumor heterogeneity and reflects the general particularities of the tumor. Its advantages include a less invasive procedure, which has a simple mechanism of interpretation providing real-time results and can be repeated at need [101,102]. CTC, circulating tumor DNA (ctDNA), and exosomes in body fluids are the main biomarkers which determine real-time resistance and can guide the follow-up treatment. The percentage of mutated CTCs, microRNAs, and proteins reflects the resistance mechanism developed by the activation of oncogenic drivers [124,125]. An important discovery was the detection of L1196M gene mutation on CTC at an early stage in patients who developed crizotinib secondary resistance [127]. Despite the low concentration in peripheral blood, CTC are ideal for detection, analysis, and overcoming tumor heterogeneity due to their distinct origin from the solid tumor or cancer sites. ctDNA is defined as tumor tissue-specific DNA fragment released in the bloodstream and its level is correlated with tumor progression, being the most frequent biomarker used in ALK-positive NSCLC resistance monitoring [123,124]. Studies have concluded that ctDNA NGS represents a non-invasive method of detecting targetable alterations and characterizing resistance mechanisms upon TKI progression [128]. Several detecting methods of TKI resistance are currently under research, such as exosome micro RNA because the concentration of exosomes in peripheral blood is greater than CTCs and it can also be identified in other body fluids besides serum and plasma [125,126]. In contrast, circulating ALK RNA analysis has proved low sensitivity due to the low blood stability [127]. A common method to detect resistance is the evaluation of tumor tissue before and after exposure to TKI by sequencing analysis. Furthermore, upon progression, an attempt to provide tissue sample or liquid biopsy is indicated [101,125]. On the other hand, studies reveal that plasma monitoring in ALK-rearranged NSCLC is feasible and could avoid re-biopsy from the tissue and monitoring plasmatic mutation levels could be used as a response parameter [128,129].
DISCUSSION
The actual research in this domain explores the options of overcoming TKI resistance by providing future therapeutic directions. Preclinical studies have identified that inhibitors against HSP90 (heat shock protein 90), the molecular protein responsible for ALK fusion stability, such as 17-AAG, 17-DMAG, and ganetespib have antitumor efficacy. Furthermore, HSP90 inhibitors have proved superiority against wild-type EML4-ALK mutation, L1196M, and F1174L gene mutations [130]. Ganetespib, which is also active on ROS1 rearrangements and RET kinases, has been evaluated individually and in combination with crizotinib and other ALK TKI to overcome crizotinib resistance in vitro and in vivo [131]. Literature data suggest the use of ALK-TKI ceritinib and the EGFR-TKI afatinib for patients who have acquired ALK TKI resistance through EGFR pathway activation because afatinib restored the sensitivity of H3122-CER cells and subsequently increased apoptosis and the antitumor activity [124,132]. Studies have identified that crizotinib is active not only against ALK but also against MET, AXL, and MST1R (Macrophage Stimulating 1 Protein Receptor) genes and it may target other TKI which are potential drivers of resistance to ALK inhibition in H3122 cells [133]. Another therapeutic option under research is represented by metformin, an oral antidiabetic drug which reverses resistance to crizotinib through the inhibition of the IGF-1R signaling pathway [124,131]. At patients with crizotinib resistance, EMT is associated with decreased expression of miR-200c and increased expression of ZEB1 (Zinc Finger E-Box Binding Homeobox 1); therefore, it determines cross-resistance to new-generation ALK inhibitors alectinib, ceritinib, and lorlatinib. Furthermore, in patients with coexistence of resistance mutations and EMT, pretreatment with histone deacetylase (HDAC) inhibitor quisinostat has proved efficacy [126,134]. ALK and vascular endothelial growth factor receptor (VEGF-R) share reciprocal downstream signaling; therefore, simultaneous inhibition of ALK gene and VEGFR by the linkage between alectinib with afatinib can provide overcoming of ALK resistance [135]. Another combination therapy undergoing preclinical studies is between alectinib and bevacizumab in patients with NSCLC and ALK rearrangement, which have CNS involvement with at least targetable lesion, because bevacizumab reshapes tumor vasculature and subsequently adjusts the systemic and intracranial drug activity [127,135]. In recent studies, MYC proto-oncogene amplification has been associated with developing primary resistance to crizotinib; therefore, a combination of MYC-directed inhibition treatment such as CDK4/6 inhibitors may provide an alternative option. In NSCLC patients with ALK rearrangement and TP-53 mutation, MYC overexpression determined a potential MYC-dependent resistance mechanism [136,137]. For patients with crizotinib resistance due to the presence of C1156Y mutation, methionine residue (M-1199) may represent a targetable approach [138]. In vitro studies have developed a structural similarity of Alectinib (JH-VIII-157-02) which is active against many resistance mutations, including G1202R and has important CNS activity [139]. Another study concluded that activated HER family signaling and mediated EGFR activation by amphiregulin protein are mechanisms which determine resistance to ALK inhibitors, suggesting that the resistance mechanism is potentially reversible [124,138]. Another potential mechanism of resistance to ALK inhibition is represented by the P2Y purinergic receptors, which co-stimulate and activate the EGFR/MAPK signaling pathway and increase the activation signal through the PKC (protein kinase C) activation [20]. In addition, IGF-1R has a synergistic effect on ALK signaling due to its protein Insulin receptor substrate 1 (IRS-1) which inhibits the IGF1R/IRS1 pathway and increases the sensibility of the tumor cells to ALK targeting [29]. Preclinical data exhibit the synergistic effect of saracatinib (a dual Src and Bcr-Abl inhibitor) on ALK inhibition through its mechanism of phosphorylation which determines resistance when SRC kinase is upregulated. Actually, the mechanism of phosphorylation of SRC substrates was expanded after both first and second-generation TKI [139,140]. Regarding the molecular changes, a tumor suppressor gene NF2 (neurofibromin 2), which activates the bypass signaling of PI3K-AKT-mTOR pathway, was identified in mutant forms at the progression with crizotinib and determined a sensibilization at the inhibitory effect of mTOR, providing possible clinical implications [117]. Another option for overcoming ALK-TKI resistance is represented by the Yes-associated protein, a downstream effector of the Hippo pathway, due to its overexpression, which inhibits the therapeutic response to alectinib as suggested by the preclinical data [141,142].
Several phase I and II studies are currently studying the potential benefit and tolerability of the combination between ALK TKI and immune checkpoint inhibitors in lung cancer, such as ceritinib with nivolumab, alectinib with cobimetinib, lorlatinib with crizotinib and binimetinib, ceritinib with trametinib, alectinib with cobimetinib, and brigatinib with binimetinib and the results are highly awaited [47,143]. Research data have revealed that the expression of PD-L1 is 5 times higher in patients with ALK gene rearrangement; therefore, promising results could highlight the efficacy of anti-PD-1/PD-L1 antibodies in this category of patients. Silibinin treatment inhibited the upregulation of the programmed death-ligand 1(PD-L1) and EMT regulators in crizotinib-resistance cells, suggesting a potential improvement of ALK TKI resistant NSCLC patients with silibinin-based drugs [145]. Furthermore, combination therapies such as inhibitors of ALK and MAPK signaling pathways, ceritinib + CDK4/6 inhibitor, ceritinib + mTOR inhibitor, and alectinib + anti-angiogenesis inhibitor have potential mechanisms of overcoming resistance [47,143-146].
CONCLUSION
We consider that identifying and overcoming mechanisms of resistance to ALK-rearranged TKI represents a promising research domain, which still requires continuous efforts to discover the remaining questions. In our opinion, this topic represents one of the most challenging of oncological research with unmet clinical needs so far.
Footnotes
Conflict of interest statement: Authors declare no conflict of interest
Funding: The authors received no financial support for the research and authorship
REFERENCES
- 1.Gridelli S, Peters S, Sgambato A, Casaluce F, Adjei AA, Ciardello F. ALK inhibitors in the treatment of advanced NSCLC. Cancer Treat Rev. 2014;40(2):300–6. doi: 10.1016/j.ctrv.2013.07.002. https://doi.org/10.1016/j.ctrv.2013.07.002. [DOI] [PubMed] [Google Scholar]
- 2.Russo A, Franchina T, Ricciardi GR, Ferraro G, Scimone A, Bronte G, et al. Central nervous system involvement in ALK-rearranged NSCLC:Promising strategies to overcome crizotinib resistance. Expert Rev Anticancer Ther. 2016;16(6):615–23. doi: 10.1080/14737140.2016.1182427. https://doi.org/10.1080/14737140.2016.1182427. [DOI] [PubMed] [Google Scholar]
- 3.Cameron L, Solomon B. Treatment of ALK-rearranged non-small cell lung cancer:Recent progress and future directions. Drugs. 2015;75(10):1059–70. doi: 10.1007/s40265-015-0415-9. https://doi.org/10.1007/s40265-015-0415-9. [DOI] [PubMed] [Google Scholar]
- 4.Sun JM, Lira M, Pandya K, Choi YL, Ahn JS, Mao M, et al. Clinical characteristics associated with ALK rearrangements in never-smokers with pulmonary adenocarcinoma. Lung Cancer. 2014;83(2):259–64. doi: 10.1016/j.lungcan.2013.11.009. https://doi.org/10.1016/j.lungcan.2013.11.009. [DOI] [PubMed] [Google Scholar]
- 5.Hallberg B, Palmer RH. Mechanistic insight into ALK receptor tyrosine kinase in human lung cancer biology. Nat Rev Cancer. 2013;13(10):685–700. doi: 10.1038/nrc3580. https://doi.org/10.1038/nrc3580. [DOI] [PubMed] [Google Scholar]
- 6.Katayama R, Lovly CM, Shaw AT. Therapeutic targeting of anaplastic lymphoma kinase in lung cancer:A paradigm for precision medicine. Clin Cancer Res. 2015;21(10):2227–35. doi: 10.1158/1078-0432.CCR-14-2791. https://doi.org/10.1158/1078-0432.ccr-14-2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Camidge DR, Doebele RC. Treating ALK-positive lung cancer-early successes and future challenges. Nat Rev Clin Oncol. 2012;9(5):268–77. doi: 10.1038/nrclinonc.2012.43. https://doi.org/10.1038/nrclinonc.2012.43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Passaro A, Lazzari C, Karachaliou N, Spitaleri G, Pochesci A, Catania C, et al. Personalized treatment in advanced ALK-positive non-small cell lung cancer:From bench to clinical practice. Onco Targets Ther. 2016;9:6361–76. doi: 10.2147/OTT.S98347. https://doi.org/10.2147/ott.s98347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu C, Zhuang W, Chen L, Yang W, Ou WB. Frontiers of ctDNA, targeted therapies, and immunotherapy in non-small-cell lung cancer. Transl Lung Cancer Res. 2020;9(1):111–38. doi: 10.21037/tlcr.2020.01.09. https://doi.org/10.21037/tlcr.2020.01.09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Vavalà T, Novello S. Alectinib in the treatment of ALK-positive non-small cell lung cancer:An update on its properties, efficacy, safety and place in therapy. Ther Adv Med Oncol. 2018;10:1758835918789364. doi: 10.1177/1758835918789364. https://doi.org/10.1177/1758835918789364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Iacono D, Chiari R, Metro G, Bennati C, Bellezza G, Cenci M, et al. Future options for ALK-positive non-small cell lung cancer. Lung Cancer. 2015;87(3):211–19. doi: 10.1016/j.lungcan.2014.12.017. https://doi.org/10.1016/j.lungcan.2014.12.017. [DOI] [PubMed] [Google Scholar]
- 12.Ziogas DC, Tsiara A, Tsironis G, Lykka M, Liontos M, Bamias A, et al. Treating ALK-positive non-small cell lung cancer. Ann Transl Med. 2018;6(8):141. doi: 10.21037/atm.2017.11.34. https://doi.org/10.21037/atm.2017.11.34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Addeo A, Tabbò F, Robinson T, Buffoni L, Novello S. Precision medicine in ALK rearranged NSCLC:A rapidly evolving scenario. Crit Rev Oncol Hematol. 2018;122:150–6. doi: 10.1016/j.critrevonc.2017.12.015. https://doi.org/10.1016/j.critrevonc.2017.12.015. [DOI] [PubMed] [Google Scholar]
- 14.Crystal AS, Shaw AT. New targets in advanced NSCLC:EML4-ALK. Clin Adv Hematol Oncol. 2011;9(3):207–14. [PubMed] [Google Scholar]
- 15.Moher D, Liberati A, Tetzlaff J, Altman DG. The PRISMA group preferred reporting items for systematic reviews and meta-analyses:The PRISMA statement. PLoS Med. 2009;6(7):e1000097. doi: 10.1371/journal.pmed.1000097. https://doi.org/10.1371/journal.pmed.1000097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Shaw AT, Solomon B. Targeting anaplastic lymphoma kinase in lung cancer. Clin Cancer Res. 2011;17(8):2081–6. doi: 10.1158/1078-0432.CCR-10-1591. https://doi.org/10.1158/1078-0432.ccr-10-1591. [DOI] [PubMed] [Google Scholar]
- 17.Lovly CM, Pao W. Escaping ALK inhibition:Mechanisms of and strategies to overcome resistance. Sci Transl Med. 2012;4(120):120ps2. doi: 10.1126/scitranslmed.3003728. https://doi.org/10.1126/scitranslmed.3003728. [DOI] [PubMed] [Google Scholar]
- 18.Kwak EL, Bang YJ, Camidge DR, Shaw AT, Solomon B, Maki RG, et al. Anaplastic lymphoma kinase inhibition in non-small cell lung cancer. N Engl J Med. 2010;363(18):1693–703. doi: 10.1056/NEJMoa1006448. https://doi.org/10.1517/14728222.2011.550880. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Steuer CE, Ramalingam SS. ALK-positive non-small cell lung cancer:Mechanisms of resistance and emerging treatment options. Cancer. 2014;120(16):2392–402. doi: 10.1002/cncr.28597. https://doi.org/10.1002/cncr.2↕. [DOI] [PubMed] [Google Scholar]
- 20.Wilson FH, Johannessen CM, Piccioni F, Tamayo P, Kim JW, van Allen AM, et al. A functional landscape of resistance to ALK inhibition in lung cancer. Cancer Cell. 2015;27(3):397–408. doi: 10.1016/j.ccell.2015.02.005. https://doi.org/10.1016/j.ccell.2015.02.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yu Y, Ou Q, Wu X, Bao H, Ding Y, Shao YW, et al. Concomitant resistance mechanisms to multiple tyrosine kinase inhibitors in ALK-positive non-small cell lung cancer. Lung Cancer. 2019;127:19–24. doi: 10.1016/j.lungcan.2018.11.024. https://doi.org/10.1016/j.lungcan.2018.11.024. [DOI] [PubMed] [Google Scholar]
- 22.Ulivi P. Non-invasive methods to monitor mechanisms of resistance to tyrosine kinase inhibitors in non-small-cell lung cancer:Where do we stand? Int J Mol Sci. 2016;17(7):1186. doi: 10.3390/ijms17071186. https://doi.org/10.3390/ijms17071186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Choi YL, Soda M, Yamashita T, Ueno T, Takashima J, Nakajima T, et al. EML4-ALK mutations in lung cancer that confer resistance to ALK inhibitors. N Engl J Med. 2010;363(18):1734–9. doi: 10.1056/NEJMoa1007478. https://doi.org/10.1056/nejmoa1007478. [DOI] [PubMed] [Google Scholar]
- 24.Dagogo-Jack I, Shaw AT. Tumour heterogeneity and resistance to cancer therapies. Nat Rev Clin Oncol. 2018;15(2):81–94. doi: 10.1038/nrclinonc.2017.166. https://doi.org/10.1038/nrclinonc.2017.166. [DOI] [PubMed] [Google Scholar]
- 25.Schram AM, Chang MT, Jonsson P, Drilon A. Fusions in solid tumours:Diagnostic strategies, targeted therapy, and acquired resistance. Nat Rev Clin Oncol. 2017;14(12):735–48. doi: 10.1038/nrclinonc.2017.127. https://doi.org/10.1038/nrclinonc.2017.127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Du X, Shao Y, Qin HF, Tai YH, Gao HJ. ALK-rearrangement in non-small-cell lung cancer (NSCLC) Thorac Cancer. 2018;9(4):423–30. doi: 10.1111/1759-7714.12613. https://doi.org/10.1111/1759-7714.12613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Yoda S, Lin JJ, Lawrence MS, Burke BJ, Friboulet L, Langenbucher A, et al. Sequential ALK inhibitors can select for lorlatinib-resistant compound ALK mutations in ALK-positive lung cancer. Cancer Discov. 2018;8(6):714–29. doi: 10.1158/2159-8290.CD-17-1256. https://doi.org/10.1158/2159-8290.cd-17-1256. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Mc Cusker MG, Russo A, Scilla KA, Mehra R, Rolfo C. How I treat ALK-positive non-small cell lung cancer. ESMO Open. 2019;4(Suppl 2):e000524. doi: 10.1136/esmoopen-2019-000524. https://doi.org/10.1136/esmoopen-2019-000524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lovly CM, McDonald NT, Chen H, Ortiz-Curan S, Heukamp LC, Yan Y, et al. Rationale for co-targeting IGF-1R and ALK in ALK fusion-positive lung cancer. Nat Med. 2014;20(9):1027–34. doi: 10.1038/nm.3667. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Qin A, Gadgeel S. The current landscape of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer:Emerging treatment paradigms and future directions. Target Oncol. 2017;12(6):709–18. doi: 10.1007/s11523-017-0526-1. https://doi.org/10.1007/s11523-017-0526-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Drizou M, Kotteas EA, Syrigos N. Treating patients with ALK-rearranged non-small-cell lung cancer:Mechanisms of resistance and strategies to overcome it. Clin Transl Oncol. 2017;19(6):658–66. doi: 10.1007/s12094-016-1605-y. https://doi.org/10.1007/s12094-016-1605-y. [DOI] [PubMed] [Google Scholar]
- 32.Qian M, Zhu B, Wang X, Liebman M. Drug resistance in ALK-positive non-small cell lung cancer patients. Semin Cell Dev Biol. 2017;64:150–7. doi: 10.1016/j.semcdb.2016.09.016. https://doi.org/10.1016/j.semcdb.2016.09.016. [DOI] [PubMed] [Google Scholar]
- 33.Golding B, Luu A, Jones R, Viloria-Petit AM. The function and therapeutic targeting of anaplastic lymphoma kinase (ALK) in non-small cell lung cancer (NSCLC) Mol Cancer. 2018;17(1):52. doi: 10.1186/s12943-018-0810-4. https://doi.org/10.1186/s12943-018-0810-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Gainor JF, Dardaei L, Yoda S, Friboulet L, Leshchiner I, Katayama R, et al. Molecular mechanisms of resistance to first-and second-generation ALK inhibitors in ALK-rearranged lung cancer. Cancer Discov. 2016;6(10):1118–33. doi: 10.1158/2159-8290.CD-16-0596. https://doi.org/10.1016/s0959-8049(16)33009-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Kron A, Alidousty C, Scheffler M, Merkelbach-Bruse S, Seidel D, Riedel R, et al. Impact of TP53 mutation status on systemic treatment outcome in ALK-rearranged non-small-cell lung cancer. Ann Oncol. 2018;29(10):2068–75. doi: 10.1093/annonc/mdy333. https://doi.org/10.1093/annonc/mdy333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Christopoulos P, Kirchner M, Bozorgmehr F, Endris V, Elsayed M, Budczies J, et al. Identification of a highly lethal V3+TP53+subset in ALK+lung adenocarcinoma. Int J Cancer. 2019;144(1):190–9. doi: 10.1002/ijc.31893. https://doi.org/10.1002/ijc.31893. [DOI] [PubMed] [Google Scholar]
- 37.Christopoulos P, Dietz S, Kirchner M, Volckmar AL, Endris V, Neumann O, et al. Detection of TP53 mutations in tissue or liquid rebiopsies at progression identifies ALK+lung cancer patients with poor survival. Cancers (Basel) 2019;11(1):124. doi: 10.3390/cancers11010124. https://doi.org/10.3390/cancers11010124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Levacq D, D'Haene N, de Wind R, Remmelink M, Berghmans T. Histological transformation of ALK rearranged adenocarcinoma into small cell lung cancer:A new mechanism of resistance to ALK inhibitors. Lung Cancer. 2016;102:38–41. doi: 10.1016/j.lungcan.2016.10.012. https://doi.org/10.1016/j.lungcan.2016.10.012. [DOI] [PubMed] [Google Scholar]
- 39.Miyamoto S, Ikushima S, Ono R, Awano N, Kondo K, Furuhata Y, et al. Transformation to small-cell lung cancer as a mechanism of acquired resistance to crizotinib and alectinib. Jpn J Clin Oncol. 2016;46(2):170–3. doi: 10.1093/jjco/hyv173. https://doi.org/10.1093/jjco/hyv173. [DOI] [PubMed] [Google Scholar]
- 40.Pinto JA, Raez LE, Domingo G. Clinical consequences of resistance to ALK inhibitors in non-small cell lung cancer. Expert Rev Respir Med. 2020;14(4):385–90. doi: 10.1080/17476348.2020.1721285. https://doi.org/10.1080/17476348.2020.1721285. [DOI] [PubMed] [Google Scholar]
- 41.Itchins M, Chia PL, Hayes SA, Howell VM, Gill AJ, Cooper WA, et al. Treatment of ALK-rearranged non-small cell lung cancer:A review of the landscape and approach to emerging patterns of treatment resistance in the Australian context. Asia Pac J Clin Oncol. 2017;13(Suppl 3):3–13. doi: 10.1111/ajco.12754. https://doi.org/10.1111/ajco.12754. [DOI] [PubMed] [Google Scholar]
- 42.Lobello C, Bikos V, Janikova A, Pospisilova S. The role of oncogenic tyrosine kinase NPM-ALK in genomic instability. Cancers (Basel) 2018;10(3):64. doi: 10.3390/cancers10030064. https://doi.org/10.3390/cancers10030064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kougioumtzi A, Ntellas P, Papadopoulou E, Nasioulas G, Kampletsas E, Pentheroudakis G. Molecular findings reveal possible resistance mechanisms in a patient with ALK-rearranged lung cancer:A case report and literature review. ESMO Open. 2019;4(5):e000561. doi: 10.1136/esmoopen-2019-000561. https://doi.org/10.1136/esmoopen-2019-000561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Sasaki T, Koivunen J, Ogino A, Yanagita M, Nikiforow S, Zheng W, et al. A novel ALK secondary mutation and EGFR signaling cause resistance to ALK kinase inhibitors. Cancer Res. 2011;71(18):6051–60. doi: 10.1158/0008-5472.CAN-11-1340. https://doi.org/10.1158/0008-5472.can-11-1340. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yamada T, Takeuchi S, Nakade J, Kita K, Nakagawa T, Nanjo S, et al. Paracrine receptor activation by microenvironment triggers bypass survival signals and ALK inhibitor resistance in EML4-ALK lung cancer cells. Clin Cancer Res. 2012;18(13):3592–602. doi: 10.1158/1078-0432.CCR-11-2972. https://doi.org/10.1158/1078-0432.ccr-11-2972. [DOI] [PubMed] [Google Scholar]
- 46.Lin JJ, Zhu VW, Yoda S, Yeap BY, Schrock AB, Dagogo-Jack I, et al. Impact of EML4-ALK variant on resistance mechanisms and clinical outcomes in ALK-positive lung cancer. J Clin Oncol. 2018;36(12):1199–206. doi: 10.1200/JCO.2017.76.2294. https://doi.org/10.1016/j.jtho.2017.09.508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Lovly CM. Combating acquired resistance to tyrosine kinase inhibitors in lung cancer. Am Soc Clin Oncol Educ Book. 2015:e165–73. doi: 10.14694/EdBook_AM.2015.35.e165. https://doi.org/10.14694/edbook_am.2015.35.e165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Iragavarapu C, Mustafa M, Akinleye A, Furqan M, Vittal M, Cang S, et al. Novel ALK inhibitors in clinical use and development. J Hematol Oncol. 2015;8:17. doi: 10.1186/s13045-015-0122-8. https://doi.org/10.1186/s13045-015-0122-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hallberg B, Palmer RH. Crizotinib-latest champion in the cancer wars? N Engl J Med. 2010;363:1760–2. doi: 10.1056/NEJMe1010404. https://doi.org/10.1056/nejme1010404. [DOI] [PubMed] [Google Scholar]
- 50.Shaw AT, Kim DW, Nakagawa K, Seto T, Crino L, Ahn MJ, et al. Crizotinib versus chemotherapy in advanced ALK-positive lung cancer. N Engl J Med. 2013;368(25):2385–94. doi: 10.1056/NEJMoa1214886. [DOI] [PubMed] [Google Scholar]
- 51.Solomon BJ, Mok T, Kim DW, Wu YL, Nakagawa K, Mekhail T, et al. First-line crizotinib versus chemotherapy in ALK-positive lung cancer. N Engl J Med. 2014;371(23):2167–177. doi: 10.1056/NEJMoa1408440. https://doi.org/10.1056/nejmoa1408440. [DOI] [PubMed] [Google Scholar]
- 52.Katayama R. Drug resistance in anaplastic lymphoma kinase-rearranged lung cancer. Cancer Sci. 2018;109(3):572–80. doi: 10.1111/cas.13504. https://doi.org/10.1111/cas.13504. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Shaw AT, Engelman JA. ALK in lung cancer:Past, present, and future. J Clin Oncol. 2013;31(8):1105–11. doi: 10.1200/JCO.2012.44.5353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Matikas A, Kentepozidis N, Georgoulias V, Kotsakis A. Management of resistance to crizotinib in anaplastic lymphoma kinase-positive non-small-cell lung cancer. Clin Lung Cancer. 2016;17(6):474–82. doi: 10.1016/j.cllc.2016.05.006. https://doi.org/10.1016/j.cllc.2016.05.006. [DOI] [PubMed] [Google Scholar]
- 55.Hatcher JM, Bahcall M, Choi HG, Gao Y, Sim T, George R, et al. Discovery of inhibitors that overcome the G1202R anaplastic lymphoma kinase resistance mutation. J Med Chem. 2015;58(23):9296–308. doi: 10.1021/acs.jmedchem.5b01136. https://doi.org/10.1021/acs.jmedchem.5b01136. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shackelford RE, Ansari JM, Wei EX, Alexander JS, Cotelingam J. Anaplastic lymphoma kinase rearrangements in non-small-cell lung cancer:Novel applications in diagnostics and treatment. Pharmacogenomics. 2017;18(12):1179–92. doi: 10.2217/pgs-2017-0098. https://doi.org/10.2217/pgs-2017-0098. [DOI] [PubMed] [Google Scholar]
- 57.Lin JJ, Riely GJ, Shaw AT. Targeting ALK:Precision medicine takes on drug resistance. Cancer Discov. 2017;7(2):137–55. doi: 10.1158/2159-8290.CD-16-1123. https://doi.org/10.1158/2159-8290.cd-16-1123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Rosas G, Ruiz R, Araujo JM, Pinto JA, Mas L. ALK rearrangements:Biology, detection and opportunities of therapy in non-small cell lung cancer. Crit Rev Oncol Hematol. 2019;136:48–55. doi: 10.1016/j.critrevonc.2019.02.006. https://doi.org/10.1016/j.critrevonc.2019.02.006. [DOI] [PubMed] [Google Scholar]
- 59.Thai AA, Solomon BJ. Treatment of ALK-positive non-small cell lung cancer:Recent advances. Curr Opin Oncol. 2018;30(2):84–91. doi: 10.1097/CCO.0000000000000431. [DOI] [PubMed] [Google Scholar]
- 60.Kay M, Dehghanian F. Exploring the crizotinib resistance mechanism of NSCLC with the L1196M mutation using molecular dynamics simulation. J Mol Model. 2017;23(11):323. doi: 10.1007/s00894-017-3495-5. https://doi.org/10.1007/s00894-017-3495-5. [DOI] [PubMed] [Google Scholar]
- 61.Ma D, Zhang Y, Xing P, Hao X, Wang M, Wang Y, et al. Clinical features and outcomes of ALK rearranged non-small cell lung cancer with primary resistance to crizotinib. Thorac Cancer. 2019;10(5):1213–9. doi: 10.1111/1759-7714.13071. https://doi.org/10.1111/1759-7714.13071. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.van der Wekken AJ, Saber A, Hiltermann TJ, Kok K, van den Berg A, Groen HJ, et al. Resistance mechanisms after tyrosine kinase inhibitors afatinib and crizotinib in non-small cell lung cancer, a review of the literature. Crit Rev Oncol Hematol. 2016;100:107–16. doi: 10.1016/j.critrevonc.2016.01.024. https://doi.org/10.1016/j.critrevonc.2016.01.024. [DOI] [PubMed] [Google Scholar]
- 63.Kim S, Kim TM, Kim DW, Go H, Keam B, Lee SH, et al. Heterogeneity of genetic changes associated with acquired crizotinib resistance in ALK-rearranged lung cancer. J Thorac Oncol. 2013;8(4):415–22. doi: 10.1097/JTO.0b013e318283dcc0. https://doi.org/10.1097/jto.0b013e318283dcc0. [DOI] [PubMed] [Google Scholar]
- 64.Tabbò F, Reale ML, Bironzo P, Scagliotti GV. Resistance to anaplastic lymphoma kinase inhibitors:Knowing the enemy is half the battle won. Transl Lung Cancer Res. 2020;9(6):2545–56. doi: 10.21037/tlcr-20-372. https://doi.org/10.21037/tlcr-20-372. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65. Available from: https:// www.accessdata.fda.gov/drugsatfda _docs/label/2012/202570s002lbl.pdf .
- 66. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2019/211225s000lbl.pdf .
- 67. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2017/208434s 003lbl.pdf .
- 68. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2020/20≄s008lbl.pdf .
- 69. Available from: https://www.accessdata.fda.gov/drugsatfda_docs/label/2018/210868s000lbl.pdf .
- 70.Gristina V, La Mantia M, Iacono F, Galvano A, Russo A, Bazan V. The emerging therapeutic landscape of ALK inhibitors in non-small cell lung cancer. Pharmaceuticals (Basel) 2020;13(12):474. doi: 10.3390/ph13120474. https://doi.org/10.3390/ph13120474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yanagitani N, Uchibori K, Koike S, Tsukahara M, Kitazono S, Yoshizawa T, et al. Drug resistance mechanisms in Japanese anaplastic lymphoma kinase-positive non-small cell lung cancer and the clinical responses based on the resistant mechanisms. Cancer Sci. 2020;111(3):932–9. doi: 10.1111/cas.14314. https://doi.org/10.1111/cas.14314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Sun HY, Ji FQ. A molecular dynamics investigation on the crizotinib resistance mechanism of C1156Y mutation in ALK. Biochem Biophys Res Commun. 2012;432(2):319–24. doi: 10.1016/j.bbrc.2012.05.120. https://doi.org/10.1016/j.bbrc.2012.05.120. [DOI] [PubMed] [Google Scholar]
- 73.Casaluce F, Sgambato A, Sacco PC, Palazzolo G, Maione P, Rossi A, et al. Resistance to crizotinib in advanced non-small cell lung cancer (NSCLC) with ALK rearrangement:Mechanisms, treatment strategies and new targeted therapies. Curr Clin Pharmacol. 2016;11(2):77–87. doi: 10.2174/1574884711666160502124134. https://doi.org/10.2174/1574884711666160502124134. [DOI] [PubMed] [Google Scholar]
- 74.Doebele RC, Pilling AB, Aisner DL, Kutateladze TG, Le AT, Weickhardt AJ, et al. Mechanisms of resistance to crizotinib in patients with ALK gene rearranged non-small cell lung cancer. Clin Cancer Res. 2012;18(5):1472–82. doi: 10.1158/1078-0432.CCR-11-2906. https://doi.org/10.1158/1078-0432.ccr-11-2906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dagogo-Jack I, Shaw AT. Crizotinib resistance:Implications for therapeutic strategies. Ann Oncol. 2016;27(Suppl 3):42–50. doi: 10.1093/annonc/mdw305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Katayama R. Therapeutic strategies and mechanisms of drug resistance in anaplastic lymphoma kinase (ALK)-rearranged lung cancer. Pharmacol Ther. 2017;177:1–8. doi: 10.1016/j.pharmthera.2017.02.015. https://doi.org/10.1016/j.pharmthera.2017.02.015. [DOI] [PubMed] [Google Scholar]
- 77.Taniguchi H, Takeuchi S, Fukuda K, Nakagawa T, Arai S, Nanjo S, et al. Amphiregulin triggered epidermal growth factor -receptor activation confers in vivo crizotinib-resistance of EML4-ALK lung cancer and circumvention by epidermal growth factor receptor inhibitors. Cancer Sci. 2017;108(1):53–60. doi: 10.1111/cas.13111. https://doi.org/10.1111/cas.13111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Yamaguchi N, Lucena-Araujo AR, Nakayama S, de Figueiredo-Pontes LL, Gonzalez DA, Yasuda H, et al. Dual ALK and EGFR inhibition targets a mechanism of acquired resistance to the tyrosine kinase inhibitor crizotinib in ALK rearranged lung cancer. Lung Cancer. 2014;83(1):37–43. doi: 10.1016/j.lungcan.2013.09.019. https://doi.org/10.1016/j.lungcan.2013.09.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kang J, Chen HJ, Zhang XC, Su J, Zhou Q, Tu HY, et al. Heterogeneous responses and resistant mechanisms to crizotinib in ALK-positive advanced non-small cell lung cancer. Thorac Cancer. 2018;9(9):1093–103. doi: 10.1111/1759-7714.12791. https://doi.org/10.1111/1759-7714.12791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Soria JC, Tan DS, Chiari R, Wu YL, Paz-Ares L, Wolf J, et al. First-line ceritinib versus platinum-based chemotherapy in advanced ALK-rearranged non small-cell lung cancer (ASCEND-4):A randomised, open-label, phase 3 study. Lancet. 2017;389(10072):917–29. doi: 10.1016/S0140-6736(17)30123-X. https://doi.org/10.1016/s0140-6736(17)30123-x. [DOI] [PubMed] [Google Scholar]
- 81.Forde PM, Rudin CM. Crizotinib in the treatment of non-small-cell lung cancer. Expert Opin Pharmacother. 2012;13(8):1195–201. doi: 10.1517/14656566.2012.688029. https://doi.org/10.1517/14656566.2012.688029. [DOI] [PubMed] [Google Scholar]
- 82.Ricciuti B, de Giglio A, Mecca C, Arcuri C, Marini S, Metro G, et al. Precision medicine against ALK-positive non-small cell lung cancer:Beyond crizotinib. Med Oncol. 2018;35(5):72. doi: 10.1007/s12032-018-1133-4. https://doi.org/10.1007/s12032-018-1133-4. [DOI] [PubMed] [Google Scholar]
- 83.Li L, Wang Y, Peng T, Zhang K, Lin C, Han R, et al. Metformin restores crizotinib sensitivity in crizotinib-resistant human lung cancer cells through inhibition of IGF1-R signaling pathway. Oncotarget. 2016;7(23):34442–52. doi: 10.18632/oncotarget.9120. https://doi.org/10.18632/oncotarget.9120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rihawi K, Alfieri R, Fiorentino M, Fontana F, Capizzi E, Cavazzoni A, et al. MYC amplification as a potential mechanism of primary resistance to crizotinib in ALK-rearranged non-small cell lung cancer:A brief report. Translational Oncol. 2019;12(1):116–21. doi: 10.1016/j.tranon.2018.09.013. https://doi.org/10.1016/j.tranon.2018.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Gadgeel SM. Sequencing of ALK inhibitors in ALK+non-small cell lung cancer. Curr Treat Options Oncol. 2017;18(6):36. doi: 10.1007/s11864-017-0479-8. https://doi.org/10.1007/s11864-017-0479-8. [DOI] [PubMed] [Google Scholar]
- 86.Amin AD, Li L, Rajan SS, Gokhale V, Groysmann MJ, Pongtornpipat P, et al. TKI sensitivity patterns of novel kinase-domain mutations suggest therapeutic opportunities for patients with resistant ALK+tumors. Oncotarget. 2016;7(17):23715–29. doi: 10.18632/oncotarget.8173. https://doi.org/10.18632/oncotarget.8173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Friboulet L, Li N, Katayama R, Lee CC, Gainor JF, Crystal AS, et al. The ALK inhibitor ceritinib overcomes crizotinib resistance in non-small cell lung cancer. Cancer Discov. 2014;4(6):662–73. doi: 10.1158/2159-8290.CD-13-0846. https://doi.org/10.1158/2159-8290.cd-13-0846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Facchinetti F, Caramella C, Auger N, Planchard D, Adam J, Lacroix L, et al. Crizotinib primary resistance overcome by ceritinib in a patient with ALK-rearranged non-small cell lung cancer. Tumori. 2016;102(Suppl 2):S46–9. doi: 10.5301/tj.5000520. https://doi.org/10.5301/tj.5000520. [DOI] [PubMed] [Google Scholar]
- 89.Massarelli E, Papadimitrakopoulou V. Ceritinib for the treatment of late-stage (metastatic) non-small cell lung cancer. Clin Cancer Res. 2015;21(4):670–4. doi: 10.1158/1078-0432.CCR-14-1291. https://doi.org/10.1158/1078-0432.ccr-14-1291. [DOI] [PubMed] [Google Scholar]
- 90.Dong X, Fernandez-Salas E, Li E, Wang S. Elucidation of resistance mechanisms to second-generation ALK inhibitors alectinib and ceritinib in non-small cell lung cancer cells. Neoplasia. 2016;18(3):162–71. doi: 10.1016/j.neo.2016.02.001. https://doi.org/10.1016/j.neo.2016.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Toyokawa G, Seto T. Updated evidence on the mechanisms of resistance to ALK inhibitors and strategies to overcome such resistance:Clinical and preclinical data. Oncol Res Treat. 2015;38(6):291–8. doi: 10.1159/000430852. https://doi.org/10.1159/000430852. [DOI] [PubMed] [Google Scholar]
- 92.Peters S, Camidge DR, Shaw AT, Gadgeel S, Ahn JS, Kim DW, et al. Alectinib versus crizotinib in untreated ALK-positive non-small cell lung cancer. New Engl J Med. 2017;377(9):829–38. doi: 10.1056/NEJMoa1704795. https://doi.org/10.1056/nejmoa1704795. [DOI] [PubMed] [Google Scholar]
- 93.He M, Li W, Zheng Q, Zhang H. A molecular dynamics investigation into the mechanisms of alectinib resistance of three ALK mutants. J Cell Biochem. 2018;119(7):5332–42. doi: 10.1002/jcb.26666. https://doi.org/10.1002/jcb.26666. [DOI] [PubMed] [Google Scholar]
- 94.Jamme P, Descarpentries C, Gervais R, Dansin E, Wislez M, Gregoire V, et al. Relevance of detection of mechanisms of resistance to ALK inhibitors in ALK-rearranged NSCLC in routine practice. Clin Lung Cancer. 2019;20(4):297–304. doi: 10.1016/j.cllc.2019.02.013. https://doi.org/10.1016/j.cllc.2019.02.013. [DOI] [PubMed] [Google Scholar]
- 95.Isozaki H, Ichihara E, Takigawa N, Ohashi K, Ochi N, Yasugi M, et al. Non-small cell lung cancer cells acquire resistance to the ALK inhibitor alectinib by activating alternative receptor tyrosine kinases. Cancer Res. 2016;76(6):1506–16. doi: 10.1158/0008-5472.CAN-15-1010. https://doi.org/10.1158/0008-5472.can-15-1010. [DOI] [PubMed] [Google Scholar]
- 96.Katayama R, Friboulet L, Koike S, Lockerman EL, Khan TM, Gainor JF, et al. Two novel ALK mutations mediate acquired resistance to the next-generation ALK inhibitor alectinib. Clin Cancer Res. 2014;22:5686–96. doi: 10.1158/1078-0432.CCR-14-1511. https://doi.org/10.1158/1078-0432.ccr-14-1511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Ou SH, Klempner SJ, Greenbowe JR, Azada M, Schrock AB, Ali SM, et al. Identification of a novel HIP1-ALK fusion variant in non-small-cell lung cancer (NSCLC) and discovery of ALK I1171 (I1171N/S) mutations in two ALK-rearranged NSCLC patients with resistance to alectinib. J Thorac Oncol. 2014;9(12):1821–5. doi: 10.1097/JTO.0000000000000368. https://doi.org/10.1097/jto.0000000000000368. [DOI] [PubMed] [Google Scholar]
- 98.Shuangjie Y, Gao L. Identification of NMU as a potential gene conferring alectinib resistance in non-small cell lung cancer based on bioinformatics analyses. Gene. 2018;678:137–42. doi: 10.1016/j.gene.2018.08.032. https://doi.org/10.1016/j.gene.2018.08.032. [DOI] [PubMed] [Google Scholar]
- 99.Rossi A. Alectinib for ALK-positive non-small-cell lung cancer. Expert Rev Clin Pharmacol. 2016;9(8):1005–13. doi: 10.1080/17512433.2016.1195262. [DOI] [PubMed] [Google Scholar]
- 100.Genova C, Rijavec E, Biello F, Rossi G, Barletta G, Dal Bello MG, et al. New systemic strategies for overcoming resistance to targeted therapies in non-small cell lung cancer. Expert Opin Pharmacother. 2017;18(1):19–33. doi: 10.1080/14656566.2016.1261109. https://doi.org/10.1080/14656566.2016.1261109. [DOI] [PubMed] [Google Scholar]
- 101.Passiglia F, Pilotto S, Facchinetti F, Bertolaccini L, Del Re M, Ferrara R, et al. Treatment of advanced non-small-cell lung cancer:The 2019 AIOM (Italian Association of Medical Oncology) clinical practice guidelines. Crit Rev Oncol Hematol. 2020;146:102858. doi: 10.1016/j.critrevonc.2019.102858. https://doi.org/10.1016/j.critrevonc.2019.102858. [DOI] [PubMed] [Google Scholar]
- 102.Planchard D, Popat S, Kerr K, Novello S, Smit EF, Faivre-Finn C, et al. Metastatic non-small cell lung cancer:ESMO clinical practice guidelines for diagnosis, treatment and follow-up. Ann Oncol. 2018;29(Suppl 4):iv192–237. doi: 10.1093/annonc/mdy275. https://doi.org/10.1093/annonc/mdy275. [DOI] [PubMed] [Google Scholar]
- 103.Tu J, Song LT, Liu RR, Zhai HL, Wang J, Zhang XY. Molecular inhibitory mechanism study on the potent inhibitor brigatinib against four crizotinib-resistant ALK mutations. J Cell Biochem. 2019;120(1):562–74. doi: 10.1002/jcb.27412. https://doi.org/10.1002/jcb.27412. [DOI] [PubMed] [Google Scholar]
- 104.Passaro A, Prelaj A, Pochesci A, Spitaleri G, Rossi G, Del Signore E, et al. Brigatinib for the treatment of ALK-positive advanced non-small cell lung cancer patients. Drugs Today (Barc) 2017;53(8):435–46. doi: 10.1358/dot.2017.53.8.2676119. https://doi.org/10.1358/dot.2017.53.8.2676119. [DOI] [PubMed] [Google Scholar]
- 105.Ali R, Arshad J, Palacio S, Mudad R. Brigatinib for ALK-positive metastatic non-small-cell lung cancer:Design, development and place in therapy. Drug Des Devel Ther. 2019;13:569–80. doi: 10.2147/DDDT.S147499. https://doi.org/10.2147/dddt.s147499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Sabari JK, Santini FC, Schram AM, Bergagnini I, Chen R, Mrad C, et al. The activity, safety, and evolving role of brigatinib in patients with ALK-rearranged non-small cell lung cancers. Onco Targets Ther. 2017;10:1983–92. doi: 10.2147/OTT.S109295. https://doi.org/10.2147/ott.s109295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Hamilton G, Hochmair MJ. An evaluation of brigatinib as a promising treatment option for non-small cell lung cancer. Expert Opin Pharmacother. 2019;20(13):1551–61. doi: 10.1080/14656566.2019.1643839. https://doi.org/10.1080/14656566.2019.1643839. [DOI] [PubMed] [Google Scholar]
- 108.Umbela S, Ghacha S, Matuknauth R, Gause S, Joshee S, Deshmukh RR. Brigatinib:New-generation ALK inhibitor for non small cell lung cancer. Curr Probl Cancer. 2019;43(6):100477. doi: 10.1016/j.currproblcancer.2019.03.005. https://doi.org/10.1016/j.currproblcancer.2019.03.005. [DOI] [PubMed] [Google Scholar]
- 109.Bedi S, Khan SA, AbuKhader MM, Alam P, Siddiqui NA, Husain A. A comprehensive review on Brigatinib-a wonder drug for targeted cancer therapy in non-small cell lung cancer. Saudi Pharm J. 2018;26(6):755–63. doi: 10.1016/j.jsps.2018.04.010. https://doi.org/10.1016/j.jsps.2018.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Horn L, Infante JR, Reckamp KL, Blumenschein GR, Leal TA, Waqar SN, et al. Ensartinib (X-396) in ALK-positive non-small cell lung cancer:Results from a first-inhumanPhaseI/II, multicenter study. Clin Cancer Res. 2018;24(12):2771–9. doi: 10.1158/1078-0432.CCR-17-2398. https://doi.org/10.1158/1078-0432.ccr-17-2398. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Yang Y, Zhou J, Zhou J, Feng J, Zhuang W, Chen J, et al. Efficacy, safety, and biomarker analysis of ensartinib in crizotinib-resistant, ALK-positive non-small-cell-lung cancer:A multicentre, phase 2 trial. Lancet Respir Med. 2020;8:45–53. doi: 10.1016/S2213-2600(19)30252-8. [DOI] [PubMed] [Google Scholar]
- 112.Selvaggi G, Wakelee HA, Mok T, Wu YL, Reck M, Chiappori A, et al. ID:1882 phase III randomized study of ensartinib vs crizotinib in anaplastic lymphoma kinase (ALK) positive NSCLC patients:eXalt3. J Thorac Oncol. 2020;15:E41–2. https://doi.org/10.1016/j.jtho.2020.08.003. [Google Scholar]
- 113.Drilon A, Siena S, Ou SH, Patel M, Ahn MJ, Lee J, et al. Safety and antitumor activity of the multitargeted pan-TRK, ROS1, and ALK inhibitor entrectinib:Combined results from two phase I trials (ALKA-372-001 and STARTRK-1) Cancer Discov. 2017;7(4):400–9. doi: 10.1158/2159-8290.CD-16-1237. https://doi.org/10.3410/f.727294853.793541382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Shaw AT, Friboulet L, Leshchiner I, Gainor JF, Bergqvist S, Brooun A, et al. Resensitization to crizotinib by the lorlatinib ALK resistance mutation L1198F. N Engl J Med. 2016;374(1):54–61. doi: 10.1056/NEJMoa1508887. https://doi.org/10.1056/nejmoa1508887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Redaelli S, Ceccon M, Zappa M, Gainor JF, Bergqvist S, Brooun A, et al. Lorlatinib treatment elicits multiple on-and off-target mechanisms of resistance in ALK-driven cancer. Cancer Res. 2018;8(24):6866–80. doi: 10.1158/0008-5472.CAN-18-1867. https://doi.org/10.1158/0008-5472.can-18-1867. [DOI] [PubMed] [Google Scholar]
- 116.Shaw AT, Solomon BJ, Besse B, Bauer TM, Lin CC, Soo RA, et al. ALK resistance mutations and efficacy of lorlatinib in advanced anaplastic lymphoma kinase-positive non-small-cell lung cancer. J Clin Oncol. 2019;16:1370–9. doi: 10.1200/JCO.18.02236. https://doi.org/10.1200/jco.18.02236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Recondo G, Mezquita L, Facchinetti F, Planchard D, Gazzah A, Bigot L, et al. Diverse resistance mechanisms to the third-generation ALK inhibitor lorlatinib in ALK-rearranged lung cancer. Clin Cancer Res. 2020;1:242–55. doi: 10.1158/1078-0432.CCR-19-1104. https://doi.org/10.1158/1078-0432.ccr-19-1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Akamine T, Toyokawa G, Tagawam T, Seto T. Spotlight on lorlatinib and its potential in the treatment of NSCLC:The evidence to date. Onco Targets Ther. 2018;11:5093–101. doi: 10.2147/OTT.S165511. https://doi.org/10.2147/ott.s165511. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.El Darsa H, Abdel-Rahman O, Sangha R. Pharmacological and clinical properties of lorlatinib in the treatment of ALK-rearranged advanced non-small cell lung cancer. Expert Opin Pharmacother. 2020;21(13):1547–54. doi: 10.1080/14656566.2020.1774552. https://doi.org/10.1080/14656566.2020.1774552. [DOI] [PubMed] [Google Scholar]
- 120.Shackelford RE, Vora M, Mayhall K, Cotelingam J. ALK-rearrangements and testing methods in non-small cell lung cancer:A review. Genes Cancer. 2014;5(1-2):1–14. doi: 10.18632/genesandcancer.3. https://doi.org/10.18632/genesandcancer.3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Qiao H, Lovly CM. Cracking the code of resistance across multiple lines of ALK inhibitor therapy in lung cancer. Cancer Discov. 2016;6(10):1084–6. doi: 10.1158/2159-8290.CD-16-0910. https://doi.org/10.1158/2159-8290.cd-16-0910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.McCoach CE, Blakely CM, Banks KC, Levy B, Chue BM, Raymond VM, et al. Clinical utility of cell-free DNA for the detection of ALK fusions and genomic mechanisms of ALK inhibitor resistance in non-small cell lung cancer. Clin Cancer Res. 2018;12:2758–70. doi: 10.1158/1078-0432.CCR-17-2588. https://doi.org/10.1158/1078-0432.ccr-17-2588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chen Y, Guo W, Fan J, Chen Y, Zhang X, Chen X, et al. The applications of liquid biopsy in resistance surveillance of anaplastic lymphoma kinase inhibitor. Cancer Manag Res. 2017;9:801–11. doi: 10.2147/CMAR.S151235. https://doi.org/10.2147/cmar.s151235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Sharma GG, Mota I, Mologni L, Patrucco E, Gambacorti-Passerini C, Chiarle R. Tumor resistance against ALK targeted therapy-where it comes from and where it goes. Cancers (Basel) 2018;10(3):62. doi: 10.3390/cancers10030062. https://doi.org/10.3390/cancers10030062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Horn L, Whisenant JG, Wakelee H, Reckamp KL, Qiao H, Leal TA, et al. Monitoring therapeutic response and resistance:Analysis of circulating tumor DNA in patients with ALK+lung cancer. J Thorac Oncol. 2019;14(11):1901–11. doi: 10.1016/j.jtho.2019.08.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Santarpia M, Gil N, Rosell R. Strategies to overcome resistance to tyrosine kinase inhibitors in non-small-cell lung cancer. Expert Rev Clin Pharmacol. 2015;8(4):461–77. doi: 10.1586/17512433.2015.1055252. https://doi.org/10.1586/17512433.2015.1055252. [DOI] [PubMed] [Google Scholar]
- 127.Simionato F, Frizziero M, Carbone C, Tortora G, Melisi D. Current strategies to overcome resistance to ALK-inhibitor agents. Curr Drug Metab. 2015;16(7):585–96. doi: 10.2174/1389200216666150812142059. https://doi.org/10.2174/1389200216666150812142059. [DOI] [PubMed] [Google Scholar]
- 128.Friedlaender A, Banna G, Patel S, Adeo A. Diagnosis and treatment of ALK aberrations in metastatic NSCLC. Curr Treat Options Oncol. 2019;20(10):79. doi: 10.1007/s11864-019-0675-9. https://doi.org/10.1007/s11864-019-0675-9. [DOI] [PubMed] [Google Scholar]
- 129.Wynes MW, Sholl LM, Dietel M, Schuuring E, Tsao MS, Yatabe Y, et al. An international interpretation study using the ALK IHC antibody D5F3 and a sensitive detection kit demonstrates high concordance between ALK IHC and ALK FISH and between evaluators. J Thorac Oncol. 2014;9(5):631–8. doi: 10.1097/JTO.0000000000000115. https://doi.org/10.1097/jto.0000000000000115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Sang J, Acquaviva J, Friedland JC, Smith DL, Sequeira M, Zhang C, et al. Targeted inhibition of the molecular chaperone Hsp90 overcomes ALK inhibitor resistance in non-small cell lung cancer. Cancer Discov. 2013;3(4):430–43. doi: 10.1158/2159-8290.CD-12-0440. https://doi.org/10.1158/2159-8290.cd-12-0440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Huang D, Kim DW, Kotsakis A, Deng S, Lira P, Ho SN, et al. Multiplexed deep sequencing analysis of ALK kinase domain identifies resistance mutations in relapsed patients following crizotinib treatment. Genomics. 2013;102(3):157–62. doi: 10.1016/j.ygeno.2013.02.006. https://doi.org/10.1016/j.ygeno.2013.02.006. [DOI] [PubMed] [Google Scholar]
- 132.Miyawaki M, Yasuda H, Tani T, Hamamoto J, Arai D, Ishioka K, et al. Overcoming EGFR bypass signal-induced acquired resistance to ALK tyrosine kinase inhibitors in ALK-translocated lung cancer. Mol Cancer Res. 2017;15(1):106–14. doi: 10.1158/1541-7786.MCR-16-0211. https://doi.org/10.1158/1541-7786.mcr-16-0211. [DOI] [PubMed] [Google Scholar]
- 133.Bordi P, Tiseo M, Rofi E, Petrini I, Restante G, Danesi R, et al. Detection of ALK and KRAS mutations in circulating tumor DNA of patients with advanced ALK-positive NSCLC with disease progression during crizotinib treatment. Clin Lung Cancer. 2017;18(6):692–7. doi: 10.1016/j.cllc.2017.04.013. https://doi.org/10.1016/j.cllc.2017.04.013. [DOI] [PubMed] [Google Scholar]
- 134.Muller IB, De Langen AJ, Honeywell RJ, Giovannetti E, Peters GJ. Overcoming crizotinib resistance in ALK-rearranged NSCLC with the second-generation ALK-inhibitor ceritinib. Exp Rev Anticancer Ther. 2016;16(2):147–57. doi: 10.1586/14737140.2016.1131612. https://doi.org/10.1586/14737140.2016.1131612. [DOI] [PubMed] [Google Scholar]
- 135.Katayama R, Shaw AT, Khan TM, Mino-Kenudson M, Solomon BJ, Halmos B, et al. Mechanisms of acquired crizotinib resistance in ALK-rearranged lung Cancers. Sci Transl Med. 2012;4(120):120ra17. doi: 10.1126/scitranslmed.3003316. https://doi.org/10.1158/1538-7445.am2012-5593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Gower A, Hsu WH, Hsu ST, Wang Y, Giaccone G. EMT is associated with, but does not drive resistance to ALK inhibitors among EML4-ALK non-small cell lung cancer. Mol Oncol. 2016;10(4):601–9. doi: 10.1016/j.molonc.2015.11.007. https://doi.org/10.1016/j.molonc.2015.11.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Alidousty C, Baar T, Martelotto LG, Heydt C, Wagener S, Fassunke J, et al. Genetic instability and recurrent MYC amplification in ALK-translocated NSCLC:A central role of TP53 mutations. J Pathol. 2018;246(1):67–76. doi: 10.1002/path.5110. https://doi.org/10.26226/morressier.5b4709866f4cb30010951ee6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Tanizaki J, Okamoto I, Okabe T, Sakai K, Tanaka K, Hayashi H, et al. Activation of HER family signaling as a mechanism of acquired resistance to ALK inhibitors in EML4-ALK-positive non-small cell lung cancer. Clin Cancer Res. 2012;18(22):6219–26. doi: 10.1158/1078-0432.CCR-12-0392. https://doi.org/10.1158/1078-0432.ccr-12-0392. [DOI] [PubMed] [Google Scholar]
- 139.Crystal AS, Shaw AT, Sequist LV, Friboulet L, Niederst MJ, Lockerman EL, et al. Patient-derived models of acquired resistance can identify effective drug combinations for cancer. Science. 2014;346(6216):1480–6. doi: 10.1126/science.1254721. https://doi.org/10.1126/science.1254721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tsuji T, Ozasa H, Aoki W, Aburaya S, Funazo T, Furugaki K, et al. Alectinib resistance in ALK-rearranged lung cancer by dual salvage signaling in a clinically paired resistance model. Mol Cancer Res. 2019;17(1):212–24. doi: 10.1158/1541-7786.MCR-18-0325. https://doi.org/10.1158/1541-7786.mcr-18-0325. [DOI] [PubMed] [Google Scholar]
- 141.Tsuji T, Ozasa H, Aoki W, Aburaya S, Funazo TK, Furugaki K, et al. YAP1 mediates survival of ALK-rearranged lung cancer cells treated with alectinib via pro-apoptotic protein regulation. Nat Commun. 2020;11(1):74. doi: 10.1038/s41467-019-13771-5. https://doi.org/10.1038/s41467-019-13771-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Yun MR, Choi HM, Lee YW, Joo HS, Park CW, Choi JW, et al. Targeting YAP to overcome acquired resistance to ALK inhibitors in ALK-rearranged lung cancer. EMBO Mol Med. 2019;11(12):e10581. doi: 10.15252/emmm.201910581. https://doi.org/10.15252/emmm.201910581. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Ota K, Azuma K, Kawahara A, Hattori S, Iwama E, Tanizaki J, et al. Induction of PD-L1 expression by the EML4-ALK oncoprotein and downstream signalling pathways in non-small cell lung cancer. Clin Cancer Res. 2015;21(17):4014–21. doi: 10.1158/1078-0432.CCR-15-0016. https://doi.org/10.1158/1078-0432.ccr-15-0016. [DOI] [PubMed] [Google Scholar]
- 144.Felip E, de Braud FG, Maur M, Long HH, Shaw AT, Vansteenkiste JF, et al. Ceritinib plus nivolumab in patients with advanced ALK-rearranged non-small cell lung cancer:Results of an open-label, multicenter, phase 1B study. J Thorac Oncol. 2020;15(3):392–403. doi: 10.1016/j.jtho.2019.10.006. https://doi.org/10.1016/j.jtho.2019.10.006. [DOI] [PubMed] [Google Scholar]
- 145.Cuyàs E, Pérez-Sánchez A, Micol V, Menendez JA, Bosch-Barrera J. STAT3-targeted treatment with silibinin overcomes the acquired resistance to crizotinib in ALK-rearranged lung cancer. Cell Cycle. 2016;15(24):3413–8. doi: 10.1080/15384101.2016.1245249. https://doi.org/10.1080/15384101.2016.1245249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Tashiro H, Shimokawa H, Sadamatu K, Yamamoto K. Prognostic significance of plasma concentrations of transforming growth factor-beta in patients with coronary artery disease. Coron Artery Dis. 2002;13(3):139–43. doi: 10.1097/00019501-200205000-00001. https://doi.org/10.1097/00019501-200205000-00001. [DOI] [PubMed] [Google Scholar]