

Abstract
Activating mutations in Gαq/11 proteins are frequent in uveal melanoma, the most common eye cancer arising from the uveal tract. A small proportion of uveal melanomas have a D630Y mutation in phospholipase C β4 (PLCβ4), an effector of Gαq/11. Here, we found that the D630Y mutation in PLCβ4 results in a high level of constitutive PLCβ4 activity. Mutations at the corresponding position in other PLC isoforms also resulted in constitutive activity, revealing an unrecognized mechanism underlying PLC activation. In cultured human uveal melanoma cell lines, inhibition of PLC suppressed proliferation in Gαq/11–dependent cells. Furthermore, we found that PLCβ4(D630Y) mediated proliferation in cutaneous melanocytes and the growth of melanomas in mice. These results are consistent with PLCβ4(D630Y) driving oncogenic signaling downstream of Gαq/11.
Uveal melanoma (UM) is a form of intraocular cancer arising from melanocytes residing in the uvea. Primary uveal melanoma tumors can be effectively removed by radiation plaque therapy and enucleation. However, metastasis develops in approximately 50% of uveal melanoma cases and becomes deadly with overall survival of 12.8 months(1). Regarding the genetic landscape of uveal melanoma, 80 to ~90% of uveal melanoma tumors harbor activating mutations of Gαq/11 (1, 2). A smaller subset of uveal melanomas present activating mutations in the cysteinyl leukotriene receptor 2 (CYSLTR2) G protein-coupled receptor upstream of Gq/11 (3), or mutations in phospholipase C β4 (PLCβ4) downstream of Gq/11 (4). The constitutively activating mutation L129Q in CysLT2R drives the Gq/11 signaling pathway while poorly recruiting β-arrestin to the receptor (5). Activating mutations in Gαq/11 were shown to activate the Rho guanine nucleotide exchange factor (Rho-GEF) protein Trio and downstream focal adhesion kinase (FAK) and Yes-associated protein (YAP) (2, 6, 7) independently of the Gq/11–PLCβ pathway to promote cell proliferation in uveal melanoma. However, the existence of uveal melanoma-associated PLCβ4 mutations suggests that PLC activity may also play a role in Gq/11-driven uveal melanoma. In fact, these three genes have been proposed to form a mutually exclusive module of activating mutations in the CysLT2R–Gq/11–PLCβ4 signaling pathway that is characteristic of uveal melanoma tumors (3). However, it is unclear how uveal melanoma-associated PLCβ4 mutations affect catalytic activity, and whether PLCβ4 activation can drive cell proliferation and tumorigenesis.
PLC enzymes are critical regulators of cell physiology. PLCs hydrolyze phosphatidylinositol 4,5-bisphosphate (PIP2) at the plasma membrane to generate diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). DAG regulates the activity of protein kinase C (PKC), whereas IP3 mobilizes intracellular Ca2+, both of which initiate multiple signaling cascades to regulate a variety of cellular processes. Thus, PLC plays an essential role in cell physiology, and dysregulation of PLC function gives rise to pathological processes (8).
PLC enzymes are stimulated by activation of receptor or non-receptor protein kinases (9–13), small GTPases (14–17), and a wide range of Gq/11-, Gi/o- and G12/13-coupled receptors (18–26). The G protein-regulated PLCβ subfamily has been intensively studied at both physiological and structural levels and are regulated by Gαq/11, Gβγ or both, depending on the isoform (27). PLCβ4 is enriched in the retina and cerebellum (28), where it plays important roles in phototransduction (29–31) and motor function (32). PLCβ4 is activated by Gαq (33, 34) but not by Gβγ (34). The naturally occurring mutation D630Y in PLCβ4 that is found in UM tumors (4) might reveal other biological functions of this less well studied PLCβ isozyme. Here, we investigated the D630Y/V/N PLCβ4 mutants found in uveal melanoma.
Results
Asp630 mutation to Tyr leads to constitutive activation of PLCβ4
The PLC family contains a conserved core enzyme structure including a pleckstrin homology (PH) domain, four EF hands, a TIM barrel split into X and Y domains, and a C2 domain (24). The structure of PLCβ4 has not been solved, but Asp630 in PLCβ4 corresponds to Asp655 in the solved PLCβ3 structure (35) (Fig. 1, A and B; also in fig. S3). Based on the structure of PLCβ3, Asp655 (side chain shown in magenta) lies in a loop on the Y domain. Its position is distant from the catalytic site formed by Glu362, His332, His379, which in the basal state is capped by the X-Y linker (magenta and dashed line in Fig. 1B). To measure the enzymatic activity of WT and D630Y PLCβ4, we transfected COS-7 cells with cDNA containing plasmids expressing WT or PLCβ4(D630Y), with or without activators Gαq or Gβγ. Cells were labeled with [3H]-myo-inositol and PLC-dependent accumulation of total inositol phosphates (IP) was measured. Co-transfection of Gαq with PLCβ4(WT) increased total IP accumulation, whereas that of Gβγ did not (Fig. 1B), confirming results from a previous study (34). In the absence of activators, transfected PLCβ4(D630Y) resulted in markedly increased IP accumulation relative to PLCβ4(WT) at all DNA concentrations, with both proteins present at similar abundance (Fig. 1, D and E, and fig. S1B). Notably, co-transfection of Gαq with PLCβ4(D630Y) in COS-7 cells did not further increase total IP accumulation, whereas that of Gβγ reduced it (Fig. 1F). PLCβ4 protein levels did not change upon co-transfection with either Gαq or Gβγ (Fig. 1G). These results indicate that the D630Y mutation in PLCβ4 leads to constitutive activation of the enzyme that was resistant to further stimulation by Gαq and, thus, was likely maximally activated.
Figure 1: PLCβ4(D630Y) is constitutively active.
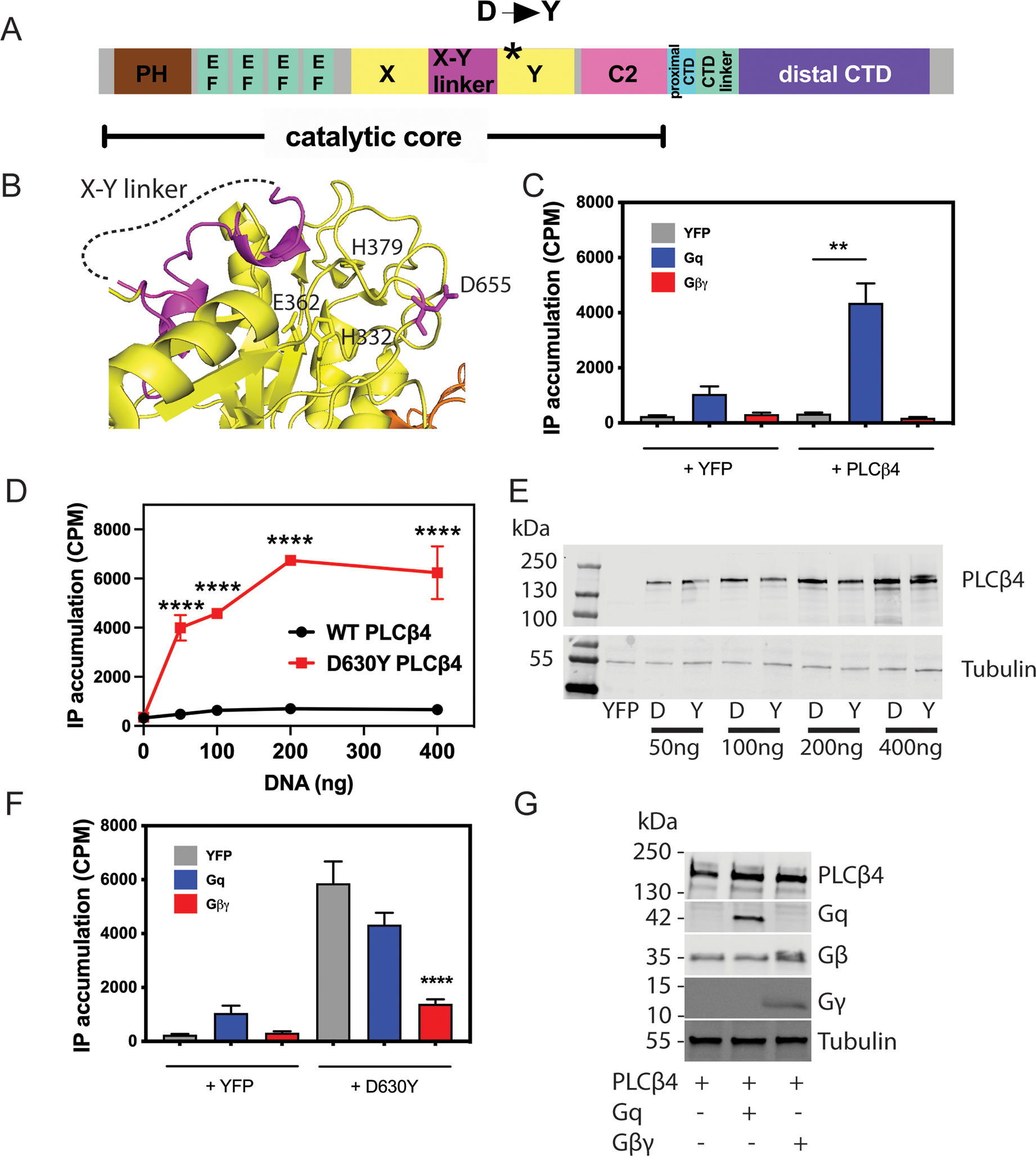
(A) PLCβ domain map. (B) Structure of PLCβ3 showing the position analogous to Asp630 mutation in PLCβ4 [Asp655 (D655) in PLCβ3; magenta, PDB 4GNK] relative to the catalytic site [labeled His (H) and Glu (E) residues] and the X-Y linker. (C) COS-7 cells were transfected with WT PLCβ4 or control vector (YFP, 200 ng) in the presence or absence of Gαq and Gβγ (200 ng). 24 hours after transfection, media was replaced with F-10 media containing 1.5 mCi/well myo[2-3H(N)] inositol. The next day LiCl was added for 1 hour followed by lysis and extraction of [3H]inositol phosphates ([3H]IPs). Total [3H]IPs accumulated were isolated using Dowex AGX8 anion exchange columns as described in the Methods. (D) COS-7 cells were transfected with control vector (YFP) and the indicated amount of WT or PLCβ4(D630Y), and total free [3H]inositol phosphate (IPs) were assayed and quantified as in (C). (E) Representative Western blots of PLCβ4 protein abundance in COS-7 cells transfected with the indicated concentrations of WT (“D” lanes) or PLCβ4(D630Y) (“Y” lanes) or the control vector (YFP). (F) COS-7 cells were transfected with PLCβ4(D630Y) or control vector (YFP) in the presence or absence of Gαq and Gβγ, and [3H]IP accumulation was assayed and quantified similarly to (C). (G) Representative Western blots showing the protein levels of transfected Gαq and Gβγ in cells described in (C). In all panels, data are either representative of or are mean ± SD pooled from at least three independent experiments. ** P < 0.01 and **** P < 0.0001 by one-way ANOVA with Bonferroni post-test.
The D → Y mutants in PLCβ and ε isozymes have high enzymatic activity
The Asp630 residue of PLCβ4 that is mutated in uveal melanoma patients is conserved in all PLC isoforms across all 6 families (Fig. 2A), indicating that it plays an important role in protein function and/or regulation. We introduced equivalent Asp to Tyr (D → Y) mutations into PLCβ2, PLCβ3, and PLCε and measured total IP accumulation in COS-7 cells transfected with WT or D → Y PLC constructs. Similar to PLCβ4, substitution of the conserved Asp residue by a Tyr residue was sufficient to result in constitutively active PLCβ2, PLCβ3, and PLCε proteins (Fig. 2, B and C). This finding suggests that this amino acid plays a conserved regulatory function across PLC isoforms.
Figure 2: D-to-Y mutations at analogous residues in PLCβs and PLCε result in constitutively active enzymes.
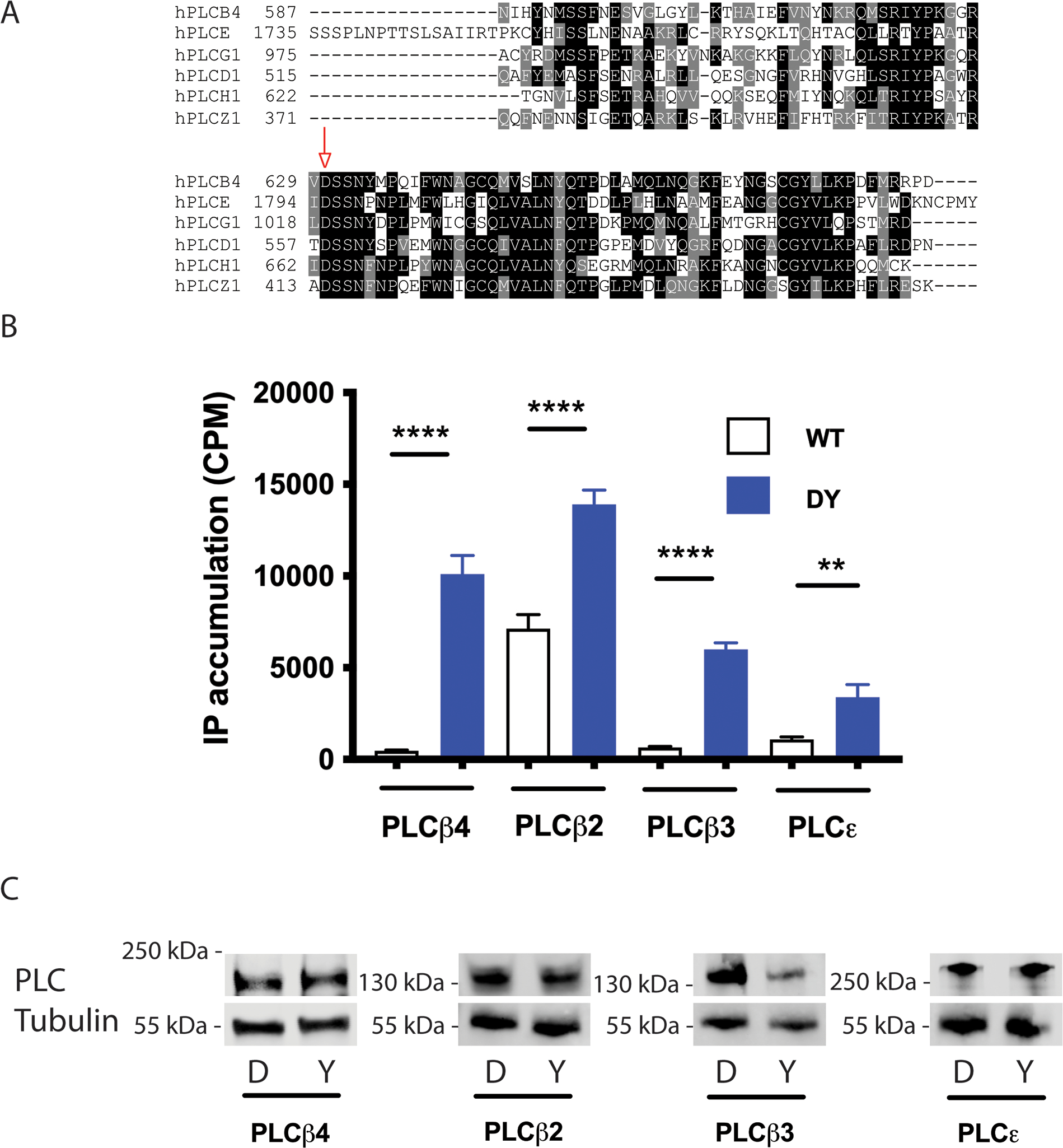
(A) Alignment of the protein segments encompassing the conserved aspartate (Asp, “D”) in 6 PLC subtypes. The red arrow indicates the position of the conserved D residue. Alignment was obtained using T-Coffee algorithm. (B) [3H]IP accumulation in COS-7 cells expressing the indicated D → Y mutant constructs of PLCβ and PLCε (blue bars) compared to corresponding WT constructs (white bars). Data are mean ± SD pooled from at least three independent experiments. ** P < 0.01 and **** P < 0.0001 by one-way ANOVA with Bonferroni post-test. (C) Representative blots of WT (“D”) or D→Y mutant (“Y”) PLCβ2, PLCβ3, PLCβ4, and PLCε in the cells described in (B).
Loss of the negative charge at Asp630 leads to high basal enzymatic activity and does not involve known mechanisms for PLC activation
The constitutive activity of PLCβ4(D630Y) inspired us to investigate the activity of other mutations in the PLCB4 gene found in uveal melanoma tumor samples. Three mutations in PLCB4 were found in uveal melanoma samples from the TCGA database and, notably, are at the same Asp residue: D630Y, D630N, and D630V). We also tested another 16 PLCB4 mutations from the Catalogue of Somatic Mutations in Cancer database (COSMIC, PLCB4_ENST00000278655), focusing on charge-altering mutations (Fig. 3A). Similar to the D630Y variant, the D630V and D630N variants of PLCβ4 showed strong gain-of-function (GOF) phenotypes (Fig. 3B). Other charge-altering mutants from the COSMIC database showed activities similar to that of WT PLCβ4 (Fig. 3B). Replacement of the negatively charged Asp residue with Tyr, Val, or Asn (which have different side chain characteristics—polar or hydrophobic) led to PLCβ4 GOF. That led us to ask whether it was the loss of the Asp residue or its replacement by certain amino acids at position 630 that altered PLCβ4 basal activity. We mutated Asp630 to each of a panel of amino acids (Fig. 3C), transfected the cDNA constructs into COS-7 cells, and measured total IP accumulation. The D630E mutant, which maintains the negative charge, showed basal activity equal to that of WT PLCβ4. Replacement of Asp630 with positively charged amino acids (D630K or D630R), polar amino acids (D630Y, D630N, or D630S), or hydrophobic amino acids (D630V, D630A, or D630F) all resulted in constitutively active mutant proteins. These data support a conclusion that a negatively charged amino acid at position 630 plays an important regulatory role for PLCβ4 to maintain its low basal activity.
Figure 3: Gain-of-function phenotype of UM-associated PLCβ4 mutations and the importance of the negatively charged Asp630 to PLCβ4 activity.
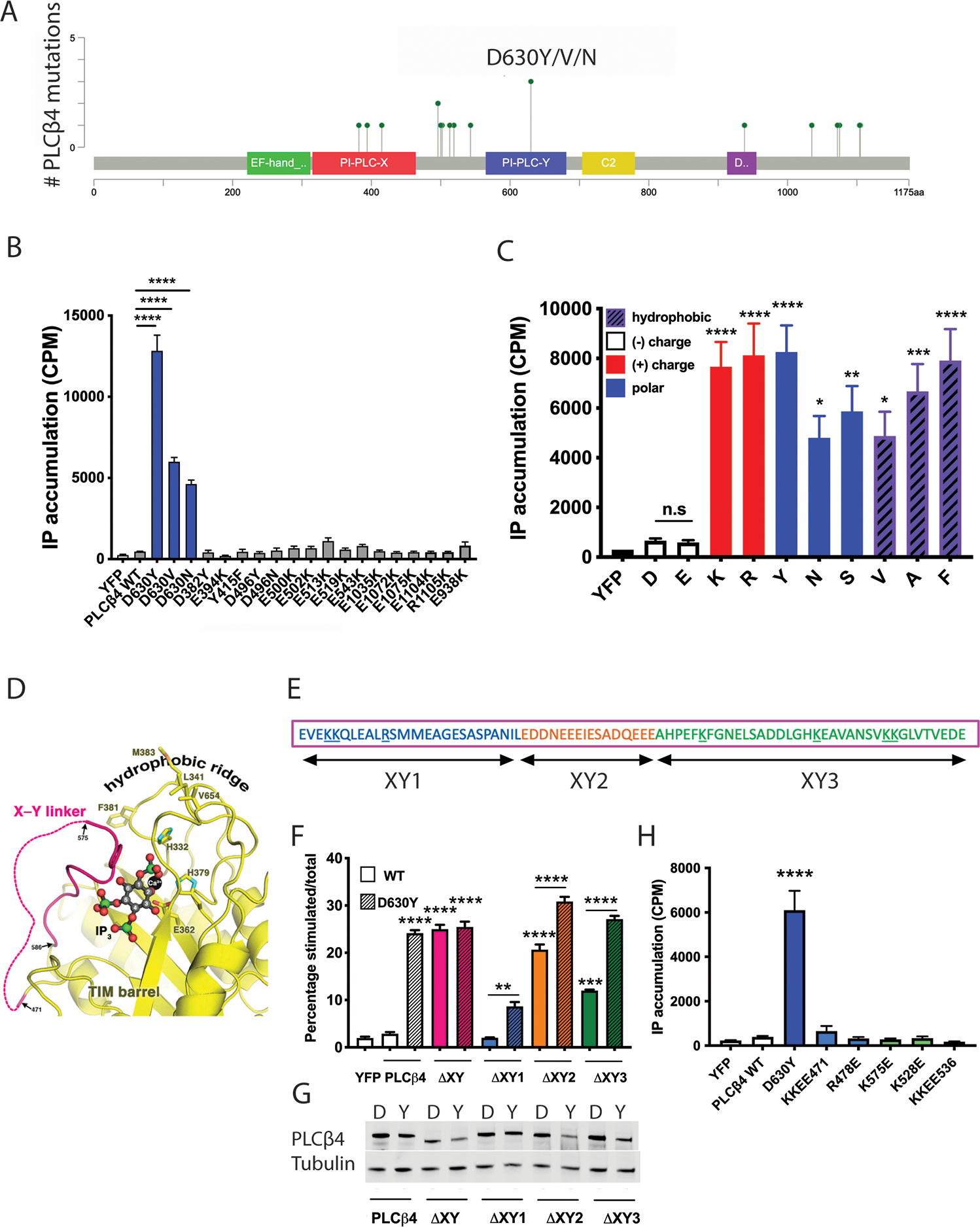
(A) A lollipop plot represents the positions of PLCβ4 mutations found in UM (from the TCGA database) or other cancers (COSMIC database). (B) Assessment of the activity of UM-associated PLCβ4 mutants D630Y/V/N and other charge-altering mutants by transfection of these mutant constructs to COS-7 cells. [3H]IP accumulation was assayed as in Fig. 1C. (C) Effect of replacement of the negative charge of Asp (or Gly) at residue 630 by other amino acids on PLCβ4 activity, assessed in COS-7 cells by measuring total [3H]IP accumulation. (D) Modeling of IP3 (derived from PDB ID 1DJX) bound to the PLCβ3 active site, wherein the X-Y linker forms a lid to occlude substrate from accessing the catalytic site. (E) Linear schematic of the X-Y linker in PLCβ, consisting of 3 sub-regions comprised of a disordered region at the amino terminal (blue; XY1), an acidic stretch region (orange; XY2), and a helix at the carboxyl terminal (green; XY3). Positively charged residues are underlined. (F) [3H]IP accumulation was measured in COS-7 cells transfected with mutants of PLCβ4, in which the whole or sub-regions of the X-Y linker were deleted in the WT (“D”) or D→Y mutant (“Y”) PLCβ4 background. (G) Cropped Western blotting for the abundance of the X-Y linker mutants in the WT (“D”) or D→Y mutant (“Y”) PLCβ4 background in (F). (H) ([3H]IP accumulation measurement in COS-7 cells transfected with PLCβ4 point-mutation constructs, in which positively charged residues in the X-Y linker were mutated to negatively charged glutamate residues in the WT (“D”) PLCβ4 background. In all panels, data are representative or are mean ± SD from at least three independent experiments. * P < 0.05, ** P < 0.01, *** P < 0.001, and **** P < 0.0001 by one-way ANOVA with Bonferroni post-test; where not otherwise indicated with brackets, comparison was with WT (“D”) PLCβ4.
Several structural elements in PLCβ are known to regulate its enzymatic activity (27, 35–38). The nearest regulatory element to Asp630 is the autoinhibitory X-Y linker. The X-Y linker regulates the basal activity of PLC by forming a lid that occludes the catalytic site inhibiting access to the substrate at the membrane (39) (Fig. 3D). Although no contacts between the aspartate residue corresponding to PLCβ4-Asp630 and the X-Y linker are observed in the crystal structures, much of the linker is disordered and may interact transiently with this aspartate. The X-Y linker itself can be further divided into sub-regions including a disordered amino terminal region, an acidic stretch and an ordered carboxy terminal region that sits above the catalytic site (27, 40) (Fig. 3, D and E). Given the vicinity of Asp630 to the X-Y linker, we performed a mutagenesis assay to test for an interaction between Asp630 and part of linker that may explain the constitutive activity of the D630Y mutant protein. We made WT or PLCβ4(D630Y) constructs that lack the entire X-Y linker, the disordered linker portion, the acidic stretch, or the “lid” region and transfected each into COS-7 cells to measure their activities. Whole X-Y linker deletion in either WT or PLCβ4(D630Y) resulted in comparably active PLCβ4 proteins, whereas deletion of the acidic stretch or the structured “lid” region, but not the disordered amino terminal region, led to high enzymatic activities (Fig. 3F). However, in all of the partial linker deletion constructs, PLCβ4(D630Y) showed higher activity than the deletion mutants of PLCβ4 with Asp at position 630, (Fig. 3, F and G), indicating that disruption of linker-Asp630 contacts is not responsible for D630Y-dependent activation of PLC. There are also several positively charged residues in the X-Y linker (underlined in Fig. 3E). To examine whether the Asp630 residue interacts with any of these positively charged amino acids, we individually mutated them to glutamate and measured their activities. None of these charge reversal mutants had higher basal activity (Fig. 3H). Altogether, these data suggest that the mechanism of activation of the PLCβ4(D630Y) does not involve disruption of autoinhibitory X-Y linker interactions with the catalytic site.
Constitutive activity of PLCβ4(D630Y) is independent of lipid membranes.
One possible mechanism for PLC regulation is through alteration in interaction with substrate containing lipid membranes. To test this and compare the enzymatic activities of WT and PLCβ4(D630Y) in vitro, the proteins were purified from insect cells and enzyme activity was measured using a water-soluble fluorescent substrate for mammalian PLCs, WH-15 (41). Using WH-15 as a substrate in a purified system enabled us to determine whether the heightened basal activity of PLCβ4(D630Y) was the result of its altered membrane association or was intrinsic to its catalytic activity. With WH-15 as a substrate, the enzymatic activity of PLCβ4(D630Y) was greater than that of WT PLCβ4 at different protein (Fig. 4, A and B), substrate (Fig. 4C) and Ca2+ concentrations (Fig. 4D). The D630Y mutation did not appear to change the substrate affinity or Ca2+ sensitivity of PLCβ4 but rather appeared to increase the intrinsic catalytic capacity of the enzyme in a membrane-independent fashion. We also examined the localization of PLCβ4 and PLCβ4(D630Y) in COS-7 cells and found that both were associated primarily with the plasma membrane (fig. S1A), supporting the idea that enhanced membrane binding is not responsible for the high activity of PLCβ4(D630Y).
Figure 4: Activity of purified WT and PLCβ4(D630Y) in solution.
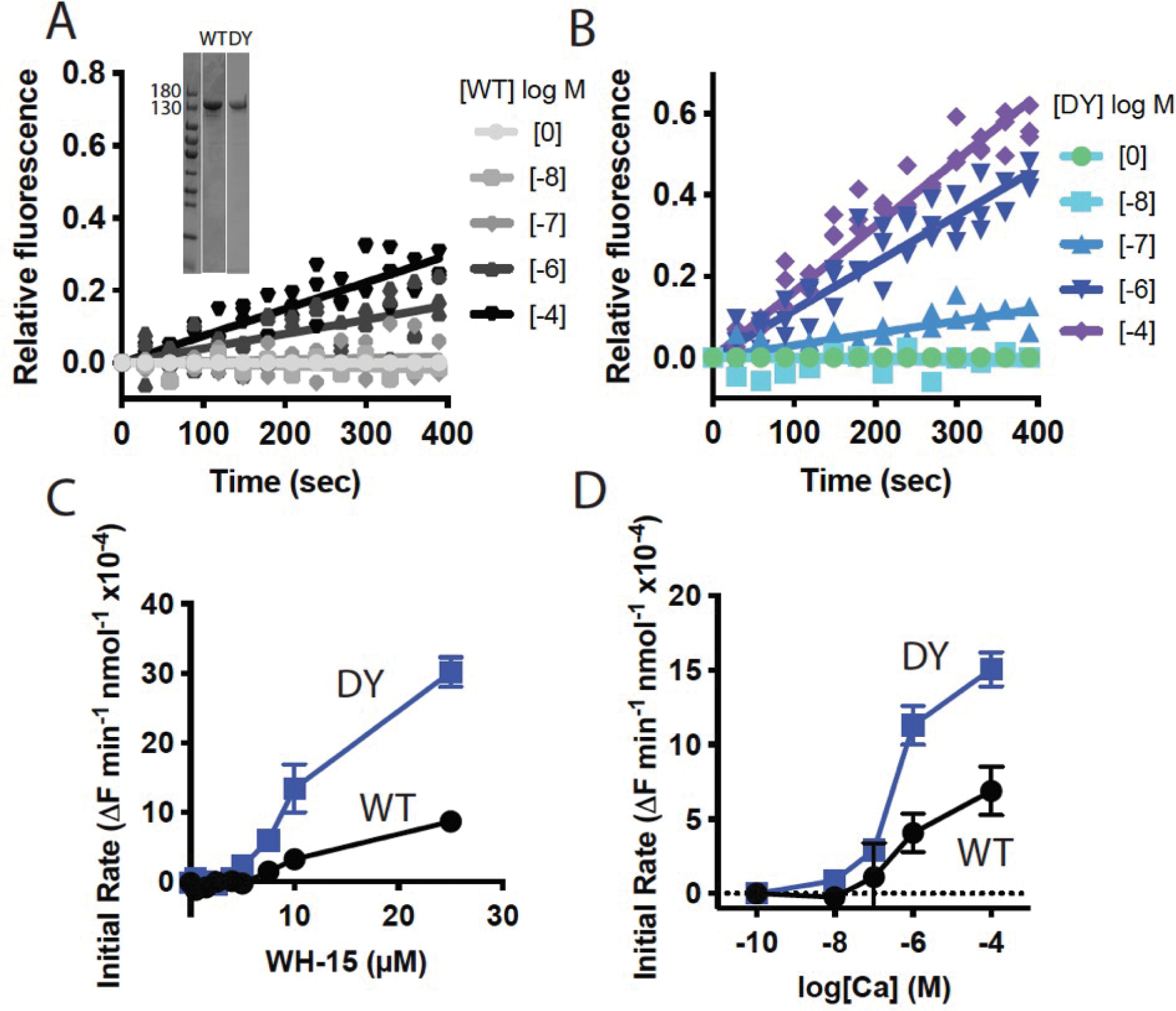
(A and B) In vitro analysis of activity of (A) WT PLCβ4 protein and (B) PLCβ4(D630Y) protein at the indicated concentrations, using soluble substrate WH-15 (5 μM). Equal amount of purified PLCβ4 proteins showed in inset, verified by SDS-PAGE followed by staining with Coomassie Blue. (C) Activity analysis of WT and PLCβ4(D630Y) in vitro with varying substrate WH-15 concentration. (D) Activity analysis of WT and PLCβ4(D630Y) in vitro at varying Ca2+ concentrations with 5 μM WH-15. Data are mean ± SD calculated from three independent experiments.
PLCβ contributes to Gαq/11 signaling in uveal melanoma
Gαq/11-driven uveal melanoma has been suggested to be the result of activation of the Trio pathway(2, 6, 7). PLCβ, although a canonical effector of Gαq/11, has not been examined in uveal melanoma. We treated 3 human uveal melanoma cell lines—OCM1A (harboring a V600E BRAF mutation), 92.1, and Mel202 (harboring Q209L Gαq mutations)—with the PLC inhibitor U-73122 or the negative-control compound U-73343 and measured cell numbers after 3 days. Addition of 1 μM U-73122 to cultures significantly reduced cell growth Mel202 cell line, whereas it had no effect on 92.1 and OCM1A lines, compared to cells treated with the control compound U-73343 or vehicle DMSO (Fig 5, A and B). At 5 μM, both U-73122 and U-73343 reduced cell proliferation and survival on all 3 cell lines compared to DMSO treatment (Fig. 5C), suggesting a general non-specific effect of these compounds. However, U-73122 had a significantly greater effect on cell proliferation than did U-73343 in Gαq(Q209L)-driven Mel202 and 92.1 cells, whereas it had no effect compared to the U-73343 control on proliferation of BRAF(V600E)-driven OCM1A cells (Fig. 5, C and D). Basal IP accumulation was greater in 92.1 and Mel202 cells compared to OCM1A cells (Fig. 5 B and D). U-73122 significantly inhibited IP accumulation in Mel202 cells, whereas U-73343 did not, and there was no significant effect on the low basal IP accumulation in OCM1A cells (Fig. 5B). Because the major difference in activity between U-73122 and U-73343 is the ability to inhibit PLC activity, we attribute the selective effect of U-73122 on inhibition of Gαq/11-mediated UM cell proliferation to its ability to inhibit PLC activity. Therefore, these data support the hypothesis that PLC activity downstream of Gαq/11 is important for Gαq/11 driven cell growth and survival of UM cells.
Figure 5: PLCβ is important for UM cell proliferation and survival.
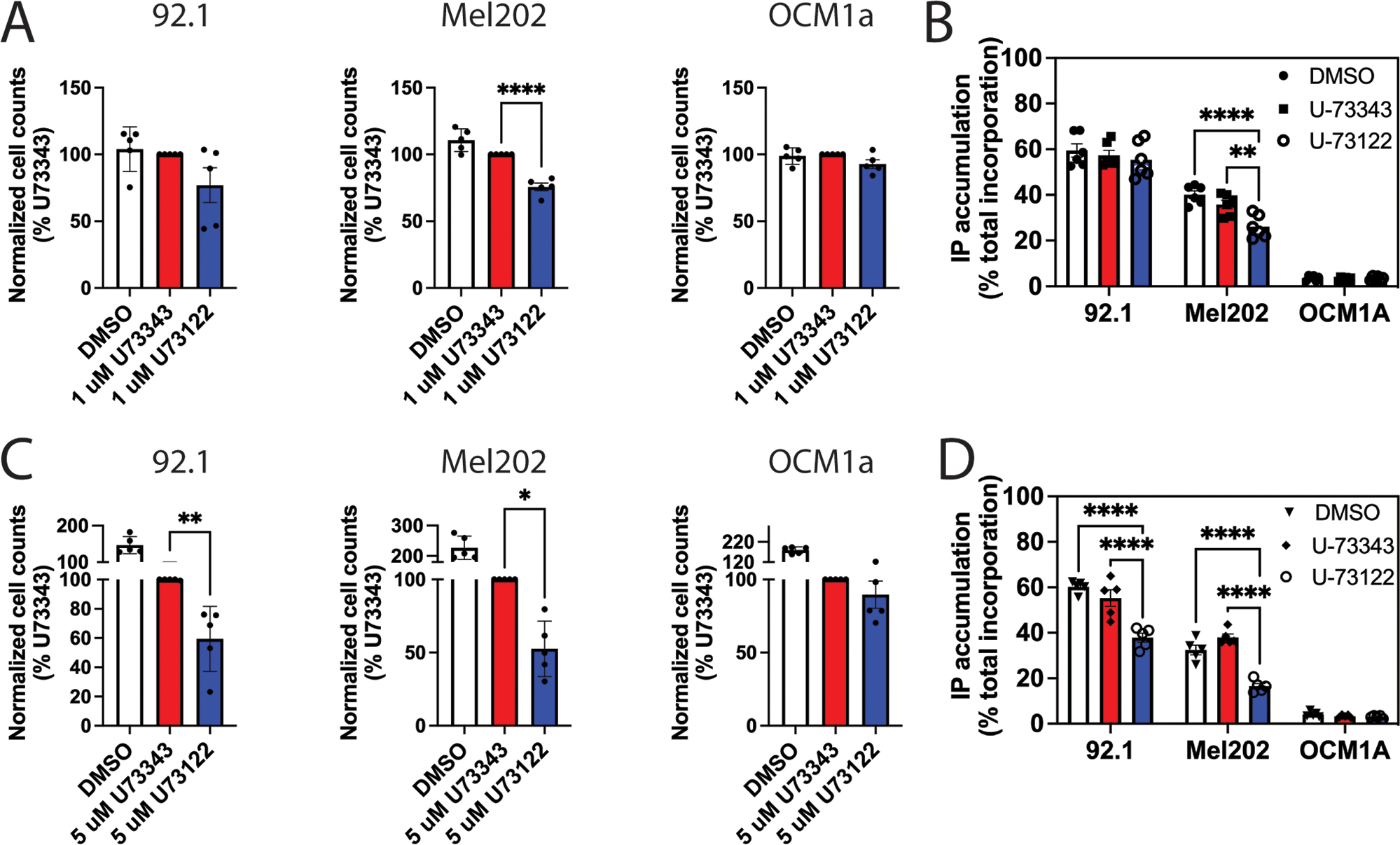
(A and B) Three human UM cell lines treated with 1 μM PLC inhibitor U-73122, the inactive control compound U-73343, or vehicle DMSO were assessed for (A) cell proliferation (3 days) by fixing and staining cells with DAPI, followed by cell counting using a Celigo Imaging cytometer and (B) [3H]IP accumulation as described in Fig. 1C. (C and D) As described in (A and B) in UM cells treated with 5 μM U-73122 or controls. Data in all panels are means ± SD pooled from at least three independent experiments. * P < 0.05, ** P < 0.01, and **** P < 0.0001 by one-way ANOVA with Dunnett post-test (A and C) or two-way ANOVA with Tukey post-test (B and D).
PLCβ4(D630Y) promotes phorbol ester-independent melanocyte proliferation in vitro and tumorigenesis in vivo
To test the ability of PLCβ4(D630Y) to promote melanocyte proliferation, we used a cultured mouse cutaneous melanocyte cell line that is similar to the Melan-a cell line in that it requires continuous PKC and Gs activation by addition of PMA and CTX for cell growth (42). We transduced the melanocyte cell line with lentiviral particles encoding empty vector control, WT PLCβ4, or PLCβ4(D630Y) then selected and pooled stable cell lines using puromycin. Like its GOF phenotype found in COS-7 cells through transient transfection, PLCβ4(D630Y) stably expressed in melanocytes had greater IP accumulation (fig. S2), whereas the protein level in mutant cells was similar to that of WT PLCβ4 (Fig. 6A). In the absence of PMA, cells expressing PLCβ4(D630Y) showed sustained proliferation, whereas melanocytes expressing empty vector or WT PLCβ4 failed to grow unless PMA was added to the medium (Fig. 6, B, C and D). To determine the oncogenic potential of mutant PLCβ4(D630Y) in melanoma tumor formation, we injected PLCβ4 WT- or PLCβ4(D630Y)-transduced melanocytes subcutaneously into the left and right hind flanks of NOD-scid-gamma (NSG) mice, respectively. The grafted PLCβ4(D630Y)-transduced melanocytes formed tumors that were larger—visibly by size and statistically significantly so by weight—than those formed by WT PLCβ4-transduced melanocytes (Fig. 6, E and F).
Figure 6: PLCβ4(D630Y) promotes PMA-independent cell proliferation in melanocytes and tumorigenesis in vivo.
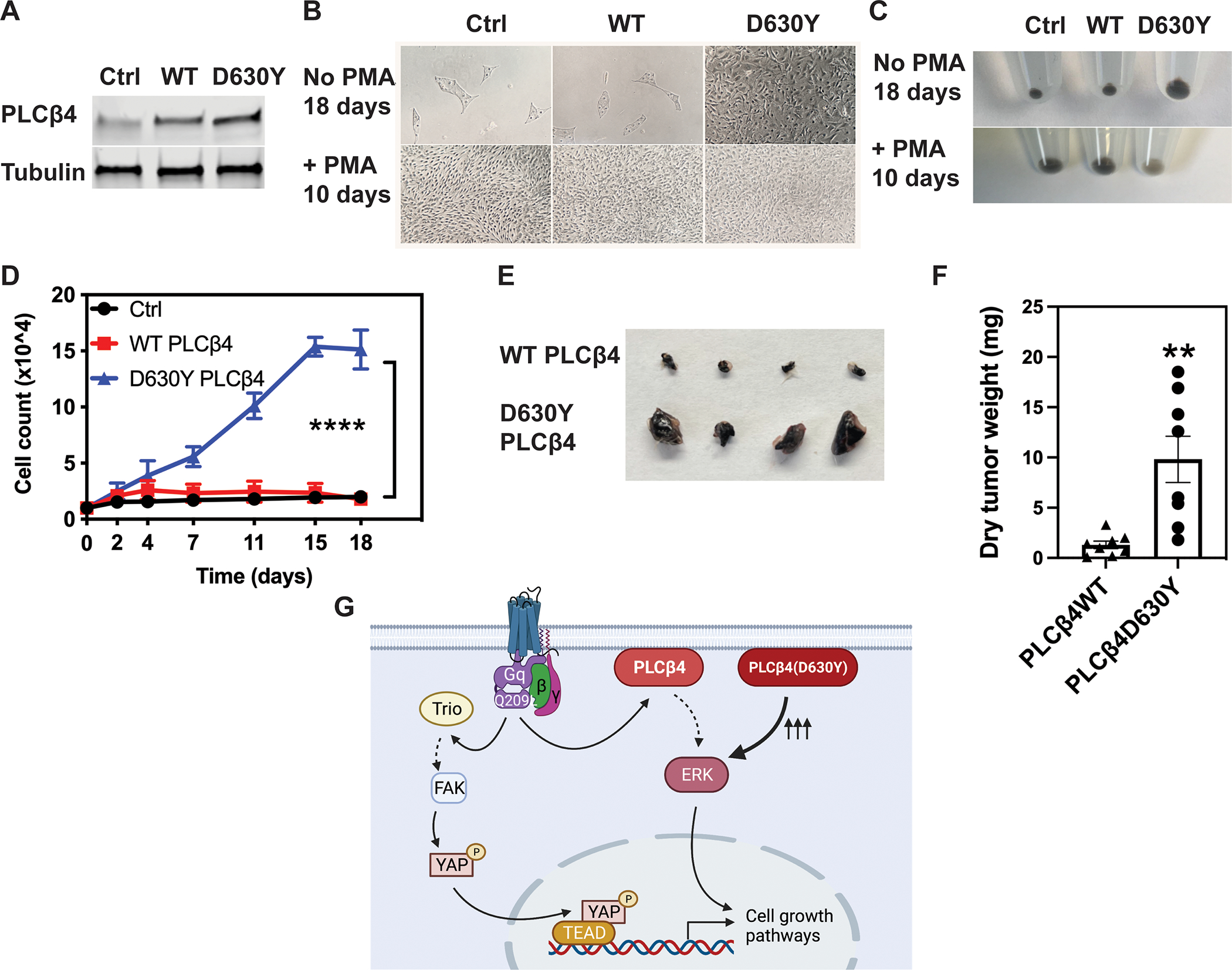
(A) Cropped immunoblots of PLCβ4 in cutaneous melanocytes expressing WT and D630Y PLCβ4. Control (Ctrl) refers to cells transfected with empty vector. Blots are representative of 3 experiments. (B and C) Bright field images (B) and cell pellet photos (C) of melanocytes expressing control vector, WT and D630Y PLCβ4 in in melanocyte media supplemented with or without PMA for 10 or 18 days, respectively. Results are representative of 3 experiments. (D) Cell proliferation of melanocytes expressing empty vector, WT and PLCβ4(D630Y) cultured without PMA for 18 days. At each time point, cells were fixed in formaldehyde 4%, stained with DAPI, and counted using a Celigo imaging cytometer. Data are mean ± SD from three independent experiments. **** P < 0.0001 by two-way ANOVA with Tukey post-test. (E and F) Photographs of representative melanocyte xenograft tumors explanted at 12 weeks after injection (E) and dry tumor weights (F) from 8 mice per group. ** P < 0.01 by paired Student t-test. (G) Cartoon depiction of the two parallel pathways that mediate cell proliferation downstream of active Gαq/11. PLCβ4 containing a Tyr at residue 630 (D630Y mutation) drives cell growth by itself, irrespective of upstream stimuli from either receptor or Gαq/11.
Discussion
In this study, we report the constitutive activity of the PLCβ4 D630Y/V/N mutations found in uveal melanoma and demonstrate the oncogenic-driver potential of the PLCβ4(D630Y) mutation, regardless of upstream stimuli (Fig. 6G). The D-to-Y mutation resulted in constitutive activation of all PLCβ and PLCε enzymes that were tested. This residue likely plays a role in regulating activity in other PLCs as well, given the conserved nature of this residue. In a genome-wide N-ethyl-N-nitrosourea (ENU) mutagenesis screen in mice, mutations at this aspartate residue have been found and characterized in PLCγ1 and PLCγ2 (43). Here, the basal activities of D1019G/A/L/N PLCγ1 or D993G PLCγ2 (positions analogous to Asp630 in PLCβ4) were similar to those of WT PLCγ1 or PLCγ2 (43, 44) and showed the GOF phenotype upon receptor-dependent activation. Another mutation of this aspartate residue in PLCγ1, D1019K has constitutively high basal activity in cells (45). Overall, this aspartate residue plays an important role in maintaining basal activity of PLCs, at least in the case of PLC isoforms β, γ, and ε.
The atomic resolution structures of PLCβ3 (35, 46), PLCδ1 (47), PLCγ1 (48) (shown for reference in fig. S3) and PLCε (49) reveal that the aspartate residue is located adjacent to a hydrophobic ridge, which anchors the enzyme catalytic site to the acyl-chains of the inner leaflet of the plasma membrane to facilitate phospholipid substrate hydrolysis (27, 50). The GOF mutation D1019L (in PLCγ1) or D993G (in PLCγ2) was proposed to confer GOF by facilitating PLCγ interaction with the plasma membrane through this hydrophobic ridge region (43, 44). Our data using a soluble substrate showed that the increase in PLCβ4 activity conferred by the D630Y mutation is membrane independent, indicating that the mutation must result in a conformational change in the enzyme, resulting in increased activity.
In another report, it was proposed that Asp1019 in PLCγ1 played an autoinhibitory role, possibly by interfacing the positively charged surface of the cSH2 domain (45) located in the X-Y linker domain. In the high-resolution crystal structures of PLCβ2, PLCβ3 and PLCε, the amino acid corresponding to D630 is surface exposed and does not interact with other domains, including the autoinhibitory X-Y linker. The X-Y linker of PLCβ is different from that of PLCγ (51) and does not contain SH2 domains. Nevertheless, there are several positively charged residues in the X-Y linker of PLCβ4 that could possibly be involved in transient ionic interactions with Asp630 that were not stabilized under the conditions for crystallization. However, substitution of these residues to negatively charged amino acids had no effect on the basal enzymatic activity of PLCβ4, suggesting that the mechanism of D630-mutant GOF phenotypes does not involve interaction with these residues. Furthermore, PLCβ4(D630Y) retained the GOF phenotype with deletion of either the unstructured amino terminus, the acidic stretch or the structured carboxyl terminal X-Y linker (38, 52). Therefore, the constitutive activity of PLCβ4(D630Y) does not seem to involve regulation of the autoinhibitory X-Y linker.
These data indicates that the constitutive activity of the D630Y mutant relies on a hitherto unknown regulatory mechanism intrinsic to PLC enzymes. The Asp residue does not appear to make contact with other parts of the protein in static crystal structures, however, it has become clear that PLCβ can adopt different conformational states that may not be represented in the crystal structures (53). The fact that mutation of this aspartate to a tyrosine leads to constitutive activity of PLCβ4 that is resistant to further Gαq activation suggests that it might be involved in the mechanism of regulation by Gαq. PLCβ4(D630Y), therefore, may serve as a model for structural determination of a fully active PLC enzyme.
Regardless of the mechanism for activation of PLCβ4 by these mutations, its ability to promote melanocyte proliferation is important with respect to uveal melanoma development. Our results agree with a relatively recent report that demonstrated the central role of PLCβ4 in mediating Gαq/11 oncogenic signaling through activation of MAPK and FAK (54). PLCβs have been implicated in the regulation of cellular signaling in cancer mechanisms(55); however, the ability of enhanced PLCβ activity to promote cell proliferation and tumor formation has not been previously demonstrated.
A possible role for PLCβ as an oncogene in uveal melanoma also illuminates possibilities for targeted therapeutics for this form of cancer. A majority of uveal melanoma tumors harbor activating mutations of CysLT2R and Gαq/11(54). These mutations strongly promote Gαq/11 oncogenic signaling. One way to inhibit the signaling driven by these mutation is to directly target the mutated proteins. Inhibition of Gαq/11 by using YM-254890 (54) or FR900359 (56) has shown to be effective to inhibit UM cell line growth and the progression of xenografted tumors carrying Gαq/11 mutations. Given that the activating CysLT2R mutation selectively activates Gαq/11 without recruiting β-arrestin, Gαq/11 inhibitors might be effective at inhibiting downstream signaling elicited by the receptor(57). However, inhibition of upstream CysLT2R or Gαq/11 proteins will not have any effect on tumors or cell lines carrying the D630Y/N/V PLCβ4 mutations. This is supported by data showing that YM-254890 had no effect on IP1 accumulation mediated by PLCβ4(D630Y)(54). Another treatment strategy would be to target the downstream signaling pathways elicited by these oncogenes with several clinical trials being concluded or underway(1). At the molecular level, there are two vital parallel pathways that mediate signaling initiated by uveal melanoma oncogenes. One is the Gαq/11–Trio–FAK–YAP pathway(2, 6, 7), and the other is the Gαq/11–PLCβ–PKC–MEK pathway(54) (Fig. 6G). Inhibition of only one signaling branch might not be sufficient to maintain therapeutic efficacy because this inhibition will potentially shunt signaling to the other branch, allowing compensatory feedback for a persistent mitogenic response. A combination of inhibitors of both pathways should be considered for better management of uveal melanoma. In fact, co-targeting MEK downstream of PLCβ and FAK downstream of Trio exhibits robust cancer growth inhibition (58). Ultimately, our study’s results are consistent with PLCβ4 being a driver oncogene in UM and reinforces the essential role of the Gαq/11–PLCβ pathway in this tumor type.
Materials and Methods
Cloning and mutagenesis
The Gateway entry vector encoding PLCβ4 was purchased from Genecopoeia (Cat No GC-Y5168-CF-GS). A stop codon was introduced and PLCβ4 mutations were made using the QuikChange II XL Site-Directed Mutagenesis Kit (Agilent) following the manufacturer’s protocol. Gateway™ pDEST™10 vector was purchased from ThermoFisher Scientific (Cat No 11806015). The destination vector pEZYegfp was a gift from Yu-Zhu Zhang (Addgene plasmid # 18671), pLenti CMV Puro DEST (w118-1) was a gift from Eric Campeau and Paul Kaufman (Addgene plasmid # 17452). The complete PLCβ4 ORF was transferred from the entry vector to pEZYegfp, Gateway™ pDEST™10 or pLenti CMV Puro DEST vectors using Gateway™ LR Clonase™ II Enzyme Mix (Invitrogen, Cat No 11791-020), following the manufacturer’s protocol, resulting in a mammalian expression vector encoding N-terminally tagged EGFP PLCβ4 which was used for all Cos cell based experiments, a baculovirus vector encoding N-terminally tagged 6xHis PLCβ4 and a lentiviral vector expressing PLCβ4. All mutagenesis was performed with the Agilent QuickChange II XL Site-Directed Mutagenesis Kit according to the manufacturers protocol. Mutagenesis primer sequences were generated using Quickchange Primer Design Software (Agilent) and were purchased from IDT. All mutations were verified by Sanger Sequencing.
[3H]-IPx accumulation assay
COS-7 monkey kidney fibroblast cells (ATCC CRL-1651) were maintained in DMEM supplemented with 10% FBS and 1% Pen/Strep at 37 °C with 5% CO2. Reverse transfection using lipofectamine 2000 (Thermo Scientific) was adapted from the manufacturer’s protocol. The maximal amount of DNA/well/24 well plate was 450 ng at a DNA:lipofectamine 2000 ratio of 1:3. COS-7 cells in antibiotic free DMEM supplemented with 10% FBS were mixed with the DNA:lipofectamine 2000 in a 24-well plate at 100,000 cells per well. Twenty-four hours after transfection, the media was replaced with Ham’s F10 media supplemented with 1.5 mCi/well myo[2-3H(N)] inositol (Perkin Elmer) and incubated overnight. 10 mM lithium chloride (LiCl) was then added to the cells and incubated for 1–1.5 hours to inhibit the activity of inositol phosphatases. The media was aspirated, and cells were washed with ice-cold phosphate-buffered saline (PBS), followed by the addition of 300 μl ice cold 50 mM formic acid per well for 1 hour for extraction of [3H]-inositol phosphates. The extract in each well was transferred to Dowex AGX8 anion exchange columns in a 96-well vacuum manifold to isolate the inositol phosphates. Columns were washed 6 times with 50 mM formic acid, 3 times with 100 mM formic acid, and then the inositol phosphates were eluted into a 96 well plate using buffer containing 1.2 M ammonium formate and 0.1 M formic acid. The eluates were transferred to scintillation vials, 4 ml of EcoLume Scintillation Cocktail (MP Biomedicals) was added to each vial and counted. All experiments were performed at least three times.
For uveal melanoma cell line measurements, PLC inhibitor (U73122), its inactive analog (U73343), or DMSO vehicle were added immediately after LiCl and incubated for 1.5 hours. [3H]IP accumulation was calculated as a percentage of the total myo[2-3H(N)] inositol incorporated into membrane lipids to account for any cell loss.
His6-PLCβ4 WT and PLCβ4 D630Y protein purification
Sf9 (Themofisher 12659017) and HighFive (Thermofisher B85502) insect cells were were authenticated by the suppliers. Both cell lines were used up to a maximum of 20 passages. Insect cells were maintained in Sf-900™ II serum free medium. Bacmids and baculoviruses were prepared following the Bac-to-Bac baculovirus expression system protocol (ThermoFisher Scientific). HighFive insect cells were infected with baculovirus at a density between 1.5×106 – 2×106 cells/ml at an MOI of 1. After 48h cells were harvested by centrifugation, snap frozen in liquid N2 and stored at −80°C. Frozen insect cell pellets expressing His6 PLCβ4 WT or D630Y were lysed in 15 ml lysis buffer (per liter of insect cell culture) containing 20 mM HEPES, pH 8, 50 mM NaCl, 10 mM β-mercaptoethanol, 0.1 mM EDTA, 0.1 M EGTA, 0.1 mM DTT, protease inhibitors including 133 μM PMSF, 21 μg/ml TLCK and TPCK, 0.5 μg/ml, Aprotonin, 0.2 μg/ml Leupeptin, 1 μg/ml Pepstatin A, 42 μg/ml TAME, 10 μg/ml SBTI by subjecting the cell suspension to 4 cycles of thawing in a 37 °C water bath and snap freezing in liquid N2. The lysate was diluted with 45 ml cold lysis buffer with addition of NaCl to a final concentration of 1 M and centrifuged at 40,000 rpm using a Ti60 rotor. The supernatant was collected and diluted 5x with buffer containing 10 mM HEPES, pH 8, 10 mM β-mercaptoethanol, 0.1 mM EDTA, 0.1 M EGTA, 0.5% Polyoxyethylene (10) lauryl ether (C12E10), and protease inhibitors. The diluted supernatant was then centrifuged at 100,000 × g and the supernatant was loaded onto a Ni-NTA column pre-equilibrated with buffer A (20 mM HEPES, pH 8, 100 mM NaCl, 10 mM β-mercaptoethanol, 0.1 mM EDTA, and 0.1 M EGTA). The column was washed with 3 column volumes (CVs) of buffer A, followed by 3 CVs of buffer A supplemented with 300 mM NaCl and 10 mM imidazole. The protein was eluted from the column with 3–10 CVs of buffer A, supplemented with 200 mM imidazole. Proteins were concentrated and loaded onto a gel filtration Superdex column equilibrated with buffer containing 20 mM HEPES pH 8, 200 mM NaCl, 2 mM DTT, 0.1 mM EGTA and 0.1 mM EDTA. Fractions of His6 PLCβ4 at greater than 95% purity were confirmed by SDS-PAGE and Coomassie staining, pooled, concentrated, and snap frozen in liquid nitrogen. Protein concentrations were determined by Nanodrop absorbance at 280 nm and confirmed by a BCA protein assay.
Fluorescence-based assay for quantitation of PLC enzymatic activity
WH-15 (KXTbio, Durham, NC) at indicated concentration was dissolved in assay buffer containing 50 mM HEPES (pH 7.2), 70 mM KCl, 2.9 mM CaCl2 (or varying concentration to determine Ca2+ sensitivity), 3 mM EGTA, 2 mM DTT, and 50 μg/ml fatty acid-free BSA. Assays were initiated by adding recombinant PLCβ4 proteins at indicated concentration. To assess [Ca2+] sensitivity, The free Ca2+ concentration in the assay was controlled with an EGTA-Ca2+ buffer system and calculated using Maxchelator program (http://maxchelator.stanford.edu) to result in indicated final free [Ca2+]. Excitation and emission intensities were recorded for 2 hours at 30 second intervals at 25°C, using a Varioskan LUX Multimode Microplate Reader (Thermo Fisher).
Melanocyte culture, lentiviral infection, PLC inhibition, and proliferation assays
An immortalized murine cutaneous melanocyte cell line derived from a C57Bl6J mouse with floxed Ric-8A alleles was developed previously using methods reproduced from those of Bennett (1987) to produce Melan-a cells (59) and extensively characterized as similar to Melan-a cells (42). Melanocytes were cultured in RPMI 1640 supplemented with 10% FBS, 2 mM L-Glutamine, 100 U/ml penicillin, 100 μg/ml streptomycin, 200 nM phorbol 12-myristate 13-acetate (PMA), 200 pM cholera toxin (CTX) and 200 μM phenylthiourea(42). Lentiviral particles were produced from pLenti CMV Puro DEST vectors encoding WT and PLCβ4(D630Y) by University of Michigan Biomedical Research vector core. Melanocytes were transduced with viral particles and medium was replaced after one day. Cells expressing PLCβ4 were selected with puromycin. To assess cell proliferation, melanocytes were grown in 6-well plates in melanocyte medium with or without PMA and were fixed with 4% w/v paraformaldehyde and stained with DAPI nuclear stain at various times as indicated in the figure.
Human uveal melanoma (UM) cell lines were kind gifts from Dr. Kendall Blumer (60). UM cells including OCM1A, 92.1 and Mel202, were maintained in RPMI 1640 media supplemented with 10% FBS, 100 U/ml penicillin, 100 μg/ml streptomycin. UM cells were plated in 6-well plates overnight and treated with PLC inhibitor U-73122 or inactive compound U-73343 (MedChemExpress) as control. Media containing indicated concentration of compounds or DMSO were changed every day for 3 days. UM cells were fixed with 4% w/v paraformaldehyde and stained with DAPI nuclear stain. DAPI-stained images of fixed cells were captured and counted using Celigo Image cytometer (Nexcelom Bioscience).
Tumor formation assay
NOD.Cg-Prkdcscid Il2rgtm1Wjl/SzJ (NSG) mice were purchased from Jackson laboratory. Melanocytes expressing WT PLCβ4 were grown in PMA-containing melanocyte medium. Melanocytes expressing PLCβ4(D630Y) were grown in PMA-free medium. Cells were harvested during logarithmic growth phase by trypsinization. Live cell numbers were determined by trypan blue exclusion. 150 μl cell suspension containing 5 × 106 live cells (WT or D630Y PLCβ4-transduced cells) in RPMI media were injected subcutaneously on each hind flank of an NSG mouse. Tumor formation was monitored, and mice were sacrificed at ~12 weeks after cell grafting. Tumor size and weight were measured and compared using a paired Student’s t-test.
SDS-PAGE and immunoblotting
Gel electrophoresis and Western blotting were performed as previously described with minor modifications(61). After transfer, the membrane was incubated with TBS buffer supplemented with 0.1% Tween 20 (TBS-T) and 5% BSA at room temperature for 30 minutes on a shaker, then probed with primary antibodies (anti-PLCβ4 (Sigma Aldrich HPA007951), in-house anti-Gq, anti Gβ, anti-PLCβ2, anti-PLCβ3 and PLCε antiserum, anti-tubulin (invitrogen PA5-16863), anti-Gγ2 (Santa Cruz sc-166419)) diluted 1:1000 in TBS buffer supplemented with 0.1% Tween 20 and 3% BSA at 4 °C overnight. Membrane was washed with TBS-T four times and probed with secondary antibody (goat anti-rabbit IgG, DyLight 800 (invitrogen SA535571) at room temperature for 1 hour. After another four washes with TBS-T, protein bands on membrane were visualized using Li-Cor Odyssey CLx and analyzed using StudioLite software.
Supplementary Material
Acknowledgments:
We thank Kendall Blumer for gifting us the uveal melanoma cell lines, and Yu-Zhu Zhang, Eric Campeau and Paul Kaufman for providing the noted vectors through Addgene.
Funding:
This work was funded by a grant from the National Institutes of Health (R35GM127303) to A.V.S. We thank Dr. Kendall Blumer for sharing the uveal melanoma cell lines.
Footnotes
Competing interests: All authors declare that they have no competing interests.
Supplementary Materials
Figures S1 to S3
Data and materials availability:
All data needed to evaluate the conclusions are present in the paper or the supplementary materials. All newly created materials are available upon request.
References and Notes
- 1.Li Y, Shi J, Yang J, Ge S, Zhang J, Jia R, Fan X, Uveal melanoma: progress in molecular biology and therapeutics. Ther Adv Med Oncol 12, 1758835920965852 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Feng X, Degese MS, Iglesias-Bartolome R, Vaque JP, Molinolo AA, Rodrigues M, Zaidi MR, Ksander BR, Merlino G, Sodhi A, Chen Q, Gutkind JS, Hippo-independent activation of YAP by the GNAQ uveal melanoma oncogene through a trio-regulated rho GTPase signaling circuitry. Cancer Cell 25, 831–845 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Moore AR, Ceraudo E, Sher JJ, Guan Y, Shoushtari AN, Chang MT, Zhang JQ, Walczak EG, Kazmi MA, Taylor BS, Huber T, Chi P, Sakmar TP, Chen Y, Recurrent activating mutations of G-protein-coupled receptor CYSLTR2 in uveal melanoma. Nat Genet 48, 675–680 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Johansson P, Aoude LG, Wadt K, Glasson WJ, Warrier SK, Hewitt AW, Kiilgaard JF, Heegaard S, Isaacs T, Franchina M, Ingvar C, Vermeulen T, Whitehead KJ, Schmidt CW, Palmer JM, Symmons J, Gerdes AM, Jonsson G, Hayward NK, Deep sequencing of uveal melanoma identifies a recurrent mutation in PLCB4. Oncotarget 7, 4624–4631 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Ceraudo E, Horioka M, Mattheisen JM, Hitchman TD, Moore AR, Kazmi MA, Chi P, Chen Y, Sakmar TP, Huber T, Direct evidence that the GPCR CysLTR2 mutant causative of uveal melanoma is constitutively active with highly biased signaling. J Biol Chem 296, 100163 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Vaque JP, Dorsam RT, Feng X, Iglesias-Bartolome R, Forsthoefel DJ, Chen Q, Debant A, Seeger MA, Ksander BR, Teramoto H, Gutkind JS, A genome-wide RNAi screen reveals a Trio-regulated Rho GTPase circuitry transducing mitogenic signals initiated by G protein-coupled receptors. Mol Cell 49, 94–108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Feng X, Arang N, Rigiracciolo DC, Lee JS, Yeerna H, Wang Z, Lubrano S, Kishore A, Pachter JA, Konig GM, Maggiolini M, Kostenis E, Schlaepfer DD, Tamayo P, Chen Q, Ruppin E, Gutkind JS, A Platform of Synthetic Lethal Gene Interaction Networks Reveals that the GNAQ Uveal Melanoma Oncogene Controls the Hippo Pathway through FAK. Cancer Cell 35, 457–472 e455 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Cocco L, Follo MY, Manzoli L, Suh PG, Phosphoinositide-specific phospholipase C in health and disease. J Lipid Res 56, 1853–1860 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kim HK, Kim JW, Zilberstein A, Margolis B, Kim JG, Schlessinger J, Rhee SG, PDGF stimulation of inositol phospholipid hydrolysis requires PLC-gamma 1 phosphorylation on tyrosine residues 783 and 1254. Cell 65, 435–441 (1991). [DOI] [PubMed] [Google Scholar]
- 10.Kim UH, Fink D Jr., Kim HS, Park DJ, Contreras ML, Guroff G, Rhee SG, Nerve growth factor stimulates phosphorylation of phospholipase C-gamma in PC12 cells. J Biol Chem 266, 1359–1362 (1991). [PubMed] [Google Scholar]
- 11.Park DJ, Rho HW, Rhee SG, CD3 stimulation causes phosphorylation of phospholipase C-gamma 1 on serine and tyrosine residues in a human T-cell line. Proc Natl Acad Sci U S A 88, 5453–5456 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Carter RH, Park DJ, Rhee SG, Fearon DT, Tyrosine phosphorylation of phospholipase C induced by membrane immunoglobulin in B lymphocytes. Proc Natl Acad Sci U S A 88, 2745–2749 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Park DJ, Min HK, Rhee SG, IgE-induced tyrosine phosphorylation of phospholipase C-gamma 1 in rat basophilic leukemia cells. J Biol Chem 266, 24237–24240 (1991). [PubMed] [Google Scholar]
- 14.Jezyk MR, Snyder JT, Gershberg S, Worthylake DK, Harden TK, Sondek J, Crystal structure of Rac1 bound to its effector phospholipase C-beta2. Nat Struct Mol Biol 13, 1135–1140 (2006). [DOI] [PubMed] [Google Scholar]
- 15.Song C, Hu CD, Masago M, Kariyai K, Yamawaki-Kataoka Y, Shibatohge M, Wu D, Satoh T, Kataoka T, Regulation of a novel human phospholipase C, PLCepsilon, through membrane targeting by Ras. J Biol Chem 276, 2752–2757 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Kelley GG, Reks SE, Ondrako JM, Smrcka AV, Phospholipase C(epsilon): a novel Ras effector. EMBO J 20, 743–754 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wing MR, Snyder JT, Sondek J, Harden TK, Direct activation of phospholipase C-epsilon by Rho. J Biol Chem 278, 41253–41258 (2003). [DOI] [PubMed] [Google Scholar]
- 18.Taylor SJ, Chae HZ, Rhee SG, Exton JH, Activation of the beta 1 isozyme of phospholipase C by alpha subunits of the Gq class of G proteins. Nature 350, 516–518 (1991). [DOI] [PubMed] [Google Scholar]
- 19.Blank JL, Ross AH, Exton JH, Purification and characterization of two G-proteins that activate the beta 1 isozyme of phosphoinositide-specific phospholipase C. Identification as members of the Gq class. J Biol Chem 266, 18206–18216 (1991). [PubMed] [Google Scholar]
- 20.Smrcka AV, Hepler JR, Brown KO, Sternweis PC, Regulation of polyphosphoinositide-specific phospholipase C activity by purified Gq. Science 251, 804–807 (1991). [DOI] [PubMed] [Google Scholar]
- 21.Katz A, Wu D, Simon MI, Subunits beta gamma of heterotrimeric G protein activate beta 2 isoform of phospholipase C. Nature 360, 686–689 (1992). [DOI] [PubMed] [Google Scholar]
- 22.Camps M, Carozzi A, Schnabel P, Scheer A, Parker PJ, Gierschik P, Isozyme-selective stimulation of phospholipase C-beta 2 by G protein beta gamma-subunits. Nature 360, 684–686 (1992). [DOI] [PubMed] [Google Scholar]
- 23.Smrcka AV, Sternweis PC, Regulation of purified subtypes of phosphatidylinositol-specific phospholipase C beta by G protein alpha and beta gamma subunits. J Biol Chem 268, 9667–9674 (1993). [PubMed] [Google Scholar]
- 24.Kadamur G, Ross EM, Mammalian phospholipase C. Annu Rev Physiol 75, 127–154 (2013). [DOI] [PubMed] [Google Scholar]
- 25.Hains MD, Wing MR, Maddileti S, Siderovski DP, Harden TK, Galpha12/13- and rho-dependent activation of phospholipase C-epsilon by lysophosphatidic acid and thrombin receptors. Mol Pharmacol 69, 2068–2075 (2006). [DOI] [PubMed] [Google Scholar]
- 26.Kelley GG, Reks SE, Smrcka AV, Hormonal regulation of phospholipase Cepsilon through distinct and overlapping pathways involving G12 and Ras family G-proteins. Biochem J 378, 129–139 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Lyon AM, Tesmer JJ, Structural insights into phospholipase C-beta function. Mol Pharmacol 84, 488–500 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adamski FM, Timms KM, Shieh BH, A unique isoform of phospholipase Cbeta4 highly expressed in the cerebellum and eye. Biochim Biophys Acta 1444, 55–60 (1999). [DOI] [PubMed] [Google Scholar]
- 29.Peng YW, Rhee SG, Yu WP, Ho YK, Schoen T, Chader GJ, Yau KW, Identification of components of a phosphoinositide signaling pathway in retinal rod outer segments. Proc Natl Acad Sci U S A 94, 1995–2000 (1997). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Jiang H, Lyubarsky A, Dodd R, Vardi N, Pugh E, Baylor D, Simon MI, Wu D, Phospholipase C beta 4 is involved in modulating the visual response in mice. Proc Natl Acad Sci U S A 93, 14598–14601 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jiang Z, Yue WWS, Chen L, Sheng Y, Yau KW, Cyclic-Nucleotide- and HCN-Channel-Mediated Phototransduction in Intrinsically Photosensitive Retinal Ganglion Cells. Cell 175, 652–664 e612 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Nakamura M, Sato K, Fukaya M, Araishi K, Aiba A, Kano M, Watanabe M, Signaling complex formation of phospholipase Cbeta4 with metabotropic glutamate receptor type 1alpha and 1,4,5-trisphosphate receptor at the perisynapse and endoplasmic reticulum in the mouse brain. Eur J Neurosci 20, 2929–2944 (2004). [DOI] [PubMed] [Google Scholar]
- 33.Lee CW, Lee KH, Lee SB, Park D, Rhee SG, Regulation of phospholipase C-beta 4 by ribonucleotides and the alpha subunit of Gq. J Biol Chem 269, 25335–25338 (1994). [PubMed] [Google Scholar]
- 34.Jiang H, Wu D, Simon MI, Activation of phospholipase C beta 4 by heterotrimeric GTP-binding proteins. J Biol Chem 269, 7593–7596 (1994). [PubMed] [Google Scholar]
- 35.Lyon AM, Dutta S, Boguth CA, Skiniotis G, Tesmer JJ, Full-length Galpha(q)-phospholipase C-beta3 structure reveals interfaces of the C-terminal coiled-coil domain. Nat Struct Mol Biol 20, 355–362 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Lyon AM, Tesmer VM, Dhamsania VD, Thal DM, Gutierrez J, Chowdhury S, Suddala KC, Northup JK, Tesmer JJ, An autoinhibitory helix in the C-terminal region of phospholipase C-beta mediates Galphaq activation. Nat Struct Mol Biol 18, 999–1005 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Charpentier TH, Waldo GL, Barrett MO, Huang W, Zhang Q, Harden TK, Sondek J, Membrane-induced allosteric control of phospholipase C-beta isozymes. J Biol Chem 289, 29545–29557 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Lyon AM, Begley JA, Manett TD, Tesmer JJG, Molecular mechanisms of phospholipase C beta3 autoinhibition. Structure 22, 1844–1854 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Hicks SN, Jezyk MR, Gershburg S, Seifert JP, Harden TK, Sondek J, General and versatile autoinhibition of PLC isozymes. Mol Cell 31, 383–394 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Esquina CM, Garland-Kuntz EE, Goldfarb D, McDonald EK, Hudson BN, Lyon AM, Intramolecular electrostatic interactions contribute to phospholipase Cbeta3 autoinhibition. Cell Signal 62, 109349 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Huang W, Hicks SN, Sondek J, Zhang Q, A fluorogenic, small molecule reporter for mammalian phospholipase C isozymes. ACS Chem Biol 6, 223–228 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Patel BR, Tall GG, Ric-8A gene deletion or phorbol ester suppresses tumorigenesis in a mouse model of GNAQ(Q209L)-driven melanoma. Oncogenesis 5, e236 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Everett KL, Bunney TD, Yoon Y, Rodrigues-Lima F, Harris R, Driscoll PC, Abe K, Fuchs H, de Angelis MH, Yu P, Cho W, Katan M, Characterization of phospholipase C gamma enzymes with gain-of-function mutations. J Biol Chem 284, 23083–23093 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Yu P, Constien R, Dear N, Katan M, Hanke P, Bunney TD, Kunder S, Quintanilla-Martinez L, Huffstadt U, Schroder A, Jones NP, Peters T, Fuchs H, de Angelis MH, Nehls M, Grosse J, Wabnitz P, Meyer TP, Yasuda K, Schiemann M, Schneider-Fresenius C, Jagla W, Russ A, Popp A, Josephs M, Marquardt A, Laufs J, Schmittwolf C, Wagner H, Pfeffer K, Mudde GC, Autoimmunity and inflammation due to a gain-of-function mutation in phospholipase C gamma 2 that specifically increases external Ca2+ entry. Immunity 22, 451–465 (2005). [DOI] [PubMed] [Google Scholar]
- 45.Hajicek N, Charpentier TH, Rush JR, Harden TK, Sondek J, Autoinhibition and phosphorylation-induced activation of phospholipase C-gamma isozymes. Biochemistry 52, 4810–4819 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Waldo GL, Ricks TK, Hicks SN, Cheever ML, Kawano T, Tsuboi K, Wang X, Montell C, Kozasa T, Sondek J, Harden TK, Kinetic scaffolding mediated by a phospholipase C-beta and Gq signaling complex. Science 330, 974–980 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Essen LO, Perisic O, Katan M, Wu Y, Roberts MF, Williams RL, Structural mapping of the catalytic mechanism for a mammalian phosphoinositide-specific phospholipase C. Biochemistry 36, 1704–1718 (1997). [DOI] [PubMed] [Google Scholar]
- 48.Hajicek N, Keith NC, Siraliev-Perez E, Temple BR, Huang W, Zhang Q, Harden TK, Sondek J, Structural basis for the activation of PLC-gamma isozymes by phosphorylation and cancer-associated mutations. Elife 8:e51700, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Rugema NY, Garland-Kuntz EE, Sieng M, Muralidharan K, Van Camp MM, O’Neill H, Mbongo W, Selvia AF, Marti AT, Everly A, McKenzie E, Lyon AM, Structure of phospholipase Cepsilon reveals an integrated RA1 domain and previously unidentified regulatory elements. Commun Biol 3, 445 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Ellis MV, James SR, Perisic O, Downes CP, Williams RL, Katan M, Catalytic domain of phosphoinositide-specific phospholipase C (PLC). Mutational analysis of residues within the active site and hydrophobic ridge of plcdelta1. J Biol Chem 273, 11650–11659 (1998). [DOI] [PubMed] [Google Scholar]
- 51.Rhee SG, Regulation of phosphoinositide-specific phospholipase C. Annu Rev Biochem 70, 281–312 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hudson BN, Jessup RE, Prahalad KK, Lyon AM, Galphaq and the Phospholipase Cbeta3 X-Y Linker Regulate Adsorption and Activity on Compressed Lipid Monolayers. Biochemistry 58, 3454–3467 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Muralidharan K, Van Camp MM, Lyon AM, Structure and regulation of phospholipase Cbeta and epsilon at the membrane. Chem Phys Lipids 235, 105050 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ma J, Weng L, Bastian BC, Chen X, Functional characterization of uveal melanoma oncogenes. Oncogene 40, 806–820 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Owusu Obeng E, Rusciano I, Marvi MV, Fazio A, Ratti S, Follo MY, Xian J, Manzoli L, Billi AM, Mongiorgi S, Ramazzotti G, Cocco L, Phosphoinositide-Dependent Signaling in Cancer: A Focus on Phospholipase C Isozymes. Int J Mol Sci 21, 2581 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Onken MD, Makepeace CM, Kaltenbronn KM, Choi J, Hernandez-Aya L, Weilbaecher KN, Piggott KD, Rao PK, Yuede CM, Dixon AJ, Osei-Owusu P, Cooper JA, Blumer KJ, Targeting primary and metastatic uveal melanoma with a G protein inhibitor. J Biol Chem 296, 100403 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Hitchman TD, Bayshtok G, Ceraudo E, Moore AR, Lee C, Jia R, Wang N, Pachai MR, Shoushtari AN, Francis JH, Guan Y, Chen J, Chang MT, Taylor BS, Sakmar TP, Huber T, Chi P, Chen Y, Combined Inhibition of Galphaq and MEK Enhances Therapeutic Efficacy in Uveal Melanoma. Clin Cancer Res 27, 1476–1490 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Paradis JS, Acosta M, Saddawi-Konefka R, Kishore A, Gomes F, Arang N, Tiago M, Coma S, Lubrano S, Wu X, Ford K, Day CP, Merlino G, Mali P, Pachter JA, Sato T, Aplin AE, Gutkind JS, Synthetic Lethal Screens Reveal Cotargeting FAK and MEK as a Multimodal Precision Therapy for GNAQ-Driven Uveal Melanoma. Clin Cancer Res 27, 3190–3200 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Bennett DC, Cooper PJ, Hart IR, A line of non-tumorigenic mouse melanocytes, syngeneic with the B16 melanoma and requiring a tumour promoter for growth. Int J Cancer 39, 414–418 (1987). [DOI] [PubMed] [Google Scholar]
- 60.Onken MD, Makepeace CM, Kaltenbronn KM, Kanai SM, Todd TD, Wang S, Broekelmann TJ, Rao PK, Cooper JA, Blumer KJ, Targeting nucleotide exchange to inhibit constitutively active G protein alpha subunits in cancer cells. Sci Signal 11, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Phan HTN, Sjogren B, Neubig RR, Human Missense Mutations in Regulator of G Protein Signaling 2 Affect the Protein Function Through Multiple Mechanisms. Mol Pharmacol 92, 451–458 (2017). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data needed to evaluate the conclusions are present in the paper or the supplementary materials. All newly created materials are available upon request.