

Abstract
Glycan arrays are indispensable for learning about the specificities of glycan-binding proteins. Despite the abundance of available data, the current analysis methods do not have the ability to interpret and use the variety of data types and to integrate information across datasets. Here we evaluated whether a novel, automated algorithm for glycan-array analysis could meet that need. We developed a regression-tree algorithm with simultaneous motif optimization and packaged it in software called MotifFinder. We applied the software to data from 8 different glycan-array platforms with widely divergent characteristics and observed an accurate analysis of each dataset. We then evaluated the feasibility and value of the combined analyses of multiple datasets. In an integrated analysis of datasets covering multiple lectin concentrations, the software determined approximate binding constants for distinct motifs and identified major differences between the motifs that were not apparent from single-concentration analyses. Furthermore, an integrated analysis of data sources with complementary sets of glycans produced broader views of lectin specificity than produced by the analysis of just one data source. MotifFinder therefore enables the optimal use of the expanding resource of glycan-array data and promises to advance the studies of protein-glycan interactions.
Introduction
Lectins are powerful tools for the study of glycans. All cells in every organism produce proteins that engage oligosaccharide ligands. Such proteins, known as lectins, can initiate, modulate, and carry out an array of functions, for example cell-cell communication, anti-microbial defense, cellular trafficking, or membrane organization1–2. The specificity of a lectin for specific arrangements of oligosaccharides is central to its biological function and to its value in research. If the specificity of a lectin or glycan-binding antibody is well understood, researchers can use the protein to localize and measure the corresponding glycans in biological specimens.
Characterizing the binding preferences of a lectin or glycan-binding antibody (collectively termed glycan-binding proteins, GBPs) is not simple. In the early studies, lectin binding was described in terms of monosaccharides, such as N-acetyl-galactosamine or galactose3. As chemists began to produce higher-order structures that better approximated the various features in biology, the studies showed a far higher degree of complexity than previously appreciated. It was revealed that most lectins bind not just one glycan substructure, but rather a family of substructures, all related in some way4. The strength of binding within the family depends on secondary features like the surrounding monosaccharides that are not directly in the binding pocket or the particular selection of monosaccharides that are tolerated within the pocket5. The ability to view this complexity depends on the experimental capabilities of synthesizing the complex structures and testing the binding to each one.
Fortunately, new synthesis strategies have resulted in the production of an increasing diversity of higher-complexity structures. The new structures include various types of sialylated structures6–8, human milk oligosaccharides9, asymmetrically-branched N-glycans10–11. In addition, microarrays have been developed to cover additional classes of glycans, including arrays of microbial glycans12, plant cell-wall glycans13, and fully-defined heparan sulfate glycans14. New experimental systems for glycan presentation and detection include a glycan bead array15, a Next-Generation Glycan Microarray (NGGM)16, and a Competitive Universal Proxy Receptor Assay (CUPRA)17. These new technologies offer the potential to identify complex interactions such as multivalent and hetero-multivalent interactions (binding to multiple identical or different ligands, respectively)18.
Bioinformatics tools are required to handle this increasing complexity and diversity of data. At the introduction of glycan microarrays, structure-alignment methods were developed for analyzing patterns in glycans19. Pattern analysis applied specifically to glycan-array data appeared with a motif-based approach20, followed by subtree mining methods21, a quantitative structure-activity relationship (QSAR) method22, an advanced structure alignment method for multiple carbohydrate alignment with weights (MCAW)23, and an advanced motif-based method that we introduced24. A recent, promising algorithm is carbohydrate classification accounting for restricted linkages (CCARL)25. Software to make the methods accessible to non-bioinformaticians include GlycoPattern26, Glycan Miner27, the MCAW database28, the Glycan Array Dashboard (GLAD)29, and the Glycan Microarray Database (GlyMDB)30.
While the above methods provided valuable advances in glycan-array analysis, they offer limited ability to fully explore the expanding depth and variety of data. In particular, the current software options are not easily adaptable to diverse glycan-array platforms, and they do not allow for the combined analysis of multiple datasets. Thus, we sought to develop the ability to use and integrate information from multiple data platforms. Further, we aimed at a system that users could directly run without intervention, which is necessary for increasing productivity, reducing bias, and broadening the range of researchers who could use the method.
Experimental Section
Software
The MotifFinder software was developed using Matlab R2017a and R Version 3.5.0. The analyses presented here used the latest beta branch of MotifFinder version 2.2.4. MotifFinder utilizes the DrawGlycan-SNFG tool for rendering glycan and motif structures31. Details on the use of MotifFinder, including screenshots and step-by-step instructions for all procedures used here, are available in the User’s Manual distributed with all release versions of MotifFinder. Slight adaptions were made to CCARL to run on a Window’s 10 operating system and to adapt to a continuous-fit model for the cross-validation analysis.
Data Preparation
Data were retrieved from various sources (Table S2). The input of MotifFinder is comma-delimited text (Excel/CSV) with columns for the ID, name, intensity, and, optionally, standard deviation associated with each glycan. The glycan name is the simplified IUPAC format used in CFG data. Data from non-CFG sources required modest reformatting. Data from the CUPRA array, which is provide as a depletion index (DI), was inverted (1 – DI) to fit MotifFinder’s expectation of a positive correlation between binding and the quantification metric. The structural-grafting data were given numerical values of 1 to indicate binders and 0 to indicate nonbinders. The results from the GlyMDB and MCAW-DB databases were retrieved from their respective websites.
Cross-Validation Analysis of Model Accuracy in 22 Datasets
The 22 datasets used for cross-validation analyses matched those in Coff et. al.25, with the addition of datasets for ECL and DSA. Each dataset was split into 5 folds (4/5 of the glycans for training and 1/5 for testing) using a built-in function in MotifFinder. The default parameters were used for the MotifFinder models. The CCARL models were converted from classification to continuous prediction by substituting lasso-regularized logistic regression with a regression-tree algorithm, using the Sklearn package in python. The regression trees used a cross-validation-selected tree depth without the added motif-optimization features of MotifFinder. The model from each training set was used to predict binding to each glycan in the test set (Supporting Methods), and for each prediction, R2 was calculated by comparing predicted to actual binding.
Graphics and Figure Preparation
Plots were made using Matlab R2017a and R 3.5.0 using the ggplot2 package. The figures were prepared using Deneba Canvas X and Canvas Draw 5.
Statistics
The accuracy of prediction fit was calculated as the , where prediction error = Σ(fitted value − observed value)2 and total error = Σ(mean value − observed value)2. Differences between groups of test-set fits were tested using a paired t-test with unequal variances.
Results
Identification of Complex Binding Determinants
We developed an algorithm to extract the complex specificities of glycan-binding proteins from glycan-array data. We previously reported the algorithm’s theoretical basis32, but we had not applied the method to the combined analysis of diverse glycan-array data. The algorithm uses a previously developed syntax for motifs24 that allows for variability in multiple components of the motif (Figure 1A). A new approach for using the motifs, developed for this study, is to represent the specificity of a GBP as two tiers of motifs (Figure 1B). The first tier is the primary motifs, which are the main determinants of binding, and the second tier is the fine specificities, which are modifications to the primary motifs that influence binding. Motif features outside of the glycan proper, such as density, linker composition, and polypeptide sequence, also could affect binding but were not covered in the current version of the syntax.
Figure 1. Automated Mining of Complex Specificities.
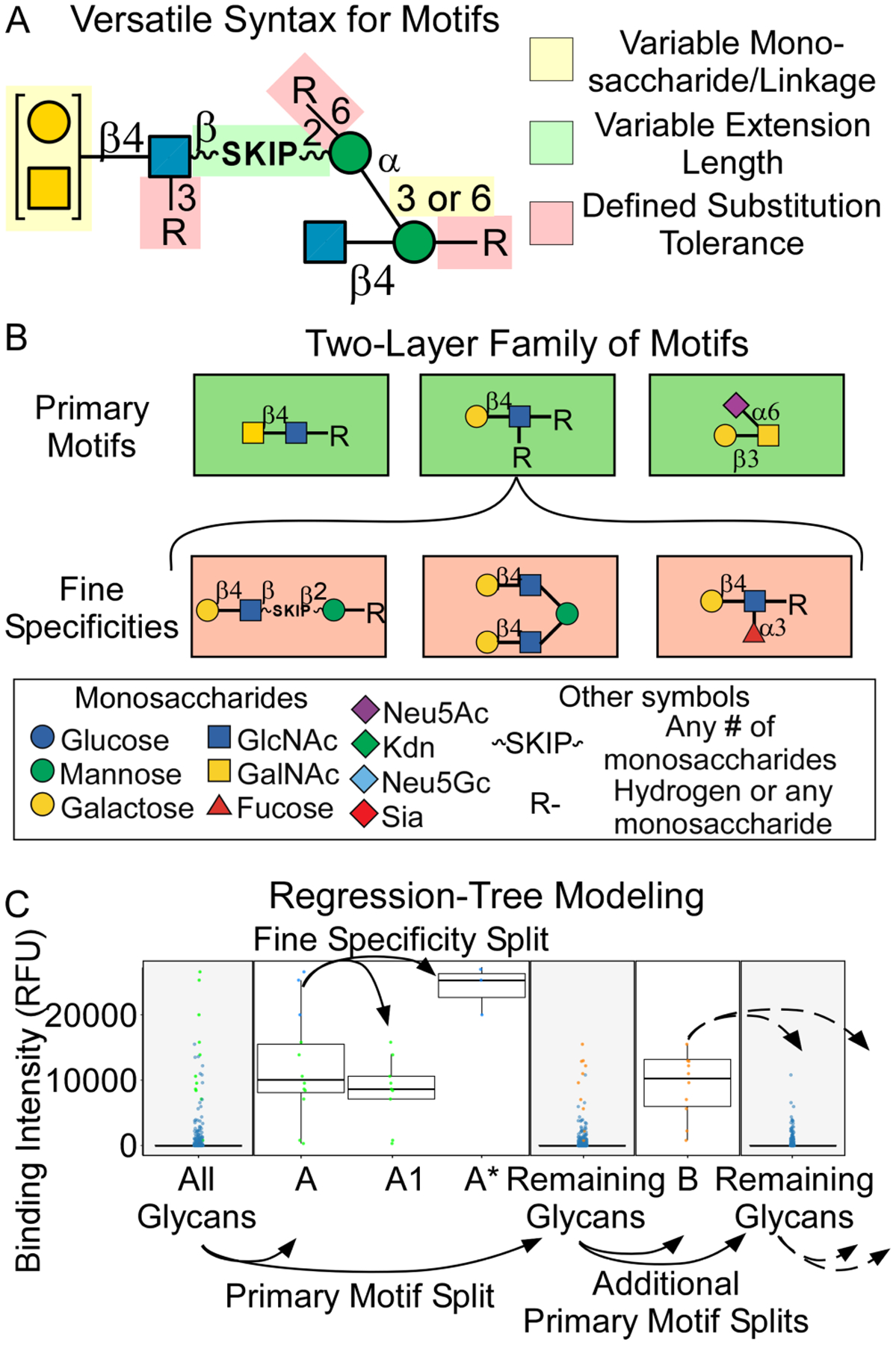
A. Versatile syntax for individual motifs. The syntax allows for the explicit definition of variability in multiple aspects of the structure, which enables the description of motifs of almost any complexity. B. Two-layer family of motifs. The specificity of a glycan-binding protein is represented as a family of motifs. The first layer is the primary motifs, and the second layer is the fine specificities. C. Regression-tree modeling. MotifFinder uses repeated splitting of the data to identify the primary motifs (A and B) and the fine specificities (A1 and A*).
The algorithm uses regression-tree modeling to uncover the two-tiered family of motifs representing a GBP’s specificity. The algorithm splits the data repeatedly to optimize the separation of the data into groups with distinct binding (Figure 1C), and it concurrently produces new motifs to further optimize the accuracy of the splits. The optimization of the motifs is performed by testing all versions and incrementally growing the ones with the best ability to improve the accuracy of the splits. In this way, the initial splits give the primary motifs (motifs A and B, Figure 1C), and the secondary splits give the fine specificities (motif A1, with the remainder of a primary motif denoted as A*).
In a test set of lectins that covers a wide range of protein folds, source organisms, and primary binding determinants, the analyses demonstrated useful insights into the nuances of lectin specificities (Figure S1). For example, a comparison between the jacalin lectin and PNA, which have similar primary motifs but different fine specificities, correctly found the main primary motif of terminal Galβ1–3GalNAc for both lectins as well as fine specificities that agree with previous research. For example, jacalin had been found to bind Galβ1–3GalNAc when attached directly to a polypeptide backbone or as a disaccharide33 and PNA was found to permit binding when the reducing end is extended3. For DSA, the results exactly replicated previous findings34 by identifying ranked preferences for LacNAc attached 6’ to Man, short chains of β1–4 linked GlcNAc (chitin), and poly-LacNAc, and they added information by identifying a preference for LacNAc in the context of a tetra-antennary N-glycan over LacNAc only on the 6’ branch. The unambiguous syntax also provided greater clarity than qualitative descriptions. For example, MotifFinder gave precise definitions of the canonical specificities of MAL-II as di-sialylated core 1 O-GalNAc and its sulfated variants (motif A1 and the fine specificities of motif B), motifs that are frequently misinterpreted due to confused definitions of specificity35. Further, the algorithm added information by identifying binding to the mono-sialylated forms of core 1 O-GalNAc and differences between the variants of the 3’ sulfated epitopes.
Analysis of Data from Divergent Platforms
A prerequisite for the integration of diverse data was the seamless processing of data from any glycan array. To this end, we used a parser for the glycan names that allows the input of any data in which the glycan names are in text. We required only an association of each glycan with a quantity that is indicative of binding. We tested data from platforms with differences between them in types of glycans, methods of data acquisition, and modes of quantification. For each dataset, we asked whether the program returned results that concorded with the known specificities and manual analyses, and whether it provided insights into the fine specificities.
A source with similarities to the CFG in the types of glycans was the glycan bead array15. MotifFinder analyzed these data without any modification. In the analysis of PNA (Figure 2A), the algorithm found Galβ1–3GalNAc (Motif A) as the top determinant, in agreement with previous information. Relative to the CFG array (Figure S1), the bead-array analysis gave fewer fine specificities, but it identified evidence of weak binding to Lewis A epitopes (motif B) and uncommon glycans with a terminal alpha-linkage (Motif C). A source with a completely different set of glycans is a microbial-glycan array12. This array includes non-mammalian monosaccharides such as xylose, arabinose, or N-acetyl-fucosamine, as well as oligosaccharide sequences not seen in mammalian glycans. MotifFinder parsed the majority of the glycans (126/141) in a dataset for the bacterial Burkholderia cenocepacia lectin (BC2L-A), and it correctly identified binding to glycans containing L-glycero D-manno-heptose (LDmanHep)36 (Motifs B-B2, Figure 2B). The MotifFinder analysis of this lectin on the CFG array identified only high-mannose glycans (Figure S2) owing to the absence of LDmanHep-containing glycans on the CFG array.
Figure 2. Analysis of Data from Divergent Platforms.
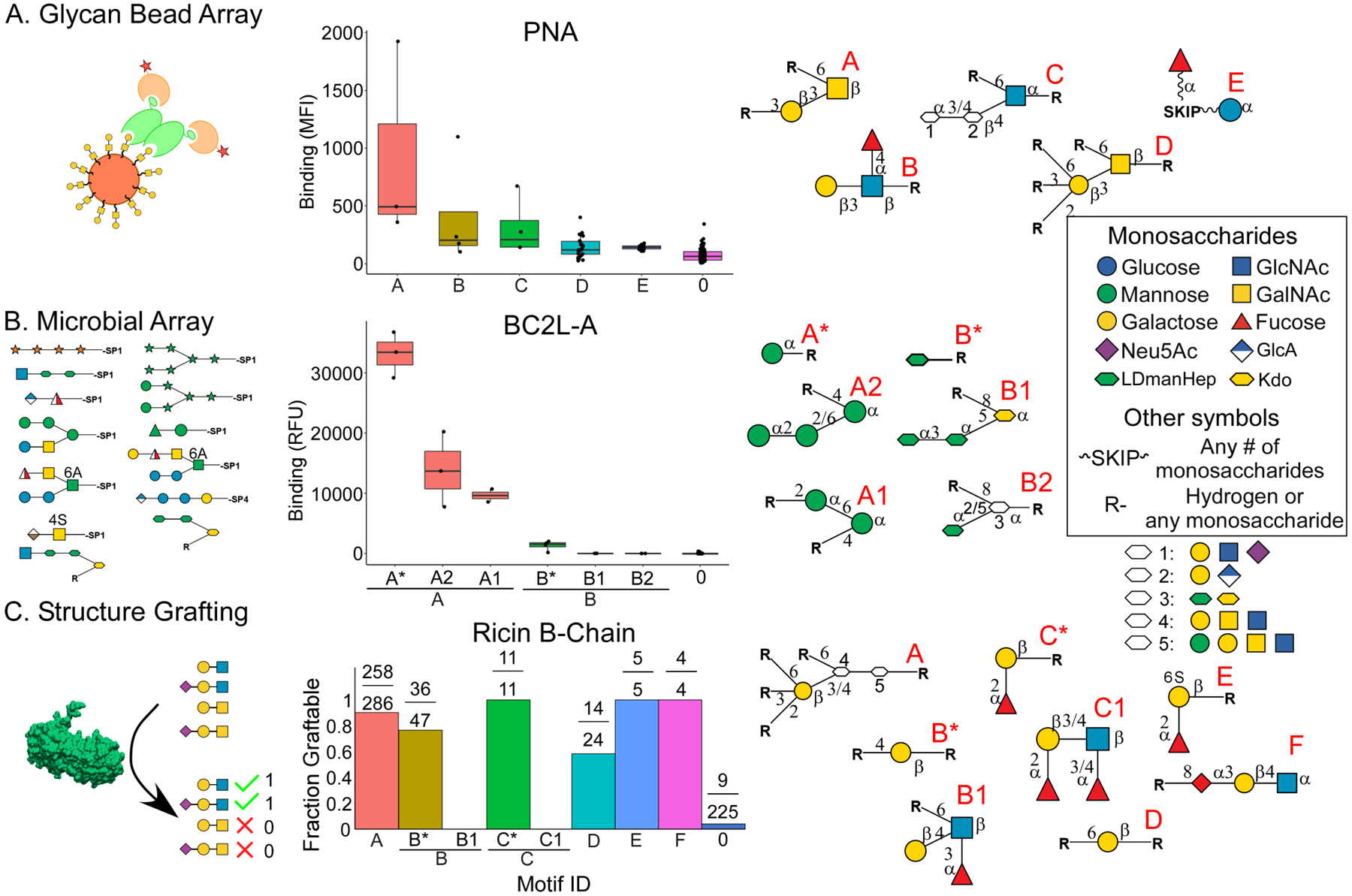
The graphs show the intensities of binding to the glycans grouped according to their motifs, which are presented graphically at right. The units of binding vary between the methods. A. The bead array uses mean fluorescence intensity (MFI). B. The microbial array uses relative fluorescence units (RFU). C. Structure grafting denotes compatible and incompatible glycans respectively with a 1 or 0.
Finally, we tested the method on semi-quantitative data. The glycan-grafting method developed by Grant et al. integrates protein structure into glycan binding consideration37 and categorically reports whether a glycan could or could not be grafted. We assigned glycans a value of 1 or 0 to indicate success or failure of grafting, respectively. The MotifFinder analysis of Ricin B-Chain (PDB 2AAI) indicated tolerance for a wide variety of galactose-containing glycans, including certain 2’ fucosylated or 3’ and 6’ sialylated structures (motif A, Figure 2C). This broad tolerance contrasts with the stricter specificity of RCA-I, which has high sequence similarity to Ricin B-Chain. In any case, the result demonstrates a method to objectively analyze grafting data and to compare it with results from glycan arrays.
As further testing on diverse platforms, we applied MotifFinder to the CUPRA array, the NGGM array, the neoglycoprotein array, a plant cell-wall array, and an asymmetric N-glycan array (Figure S3). In each case, the results matched the manually determined specificities and were supported by analyses of the same lectins on the CFG array (Figure S2). Thus, MotifFinder readily analyzed data from a wide diversity of platforms.
Integrated Analysis of Datasets with Varying Lectin Concentrations
The automated capabilities of this system opened the potential for an integrated analysis of lectin binding over multiple concentrations. To explore this possibility, we developed a function to analyze logistic regression fits of motif binding across protein concentrations (Supporting Methods and Figure 3A). We evaluated whether the analysis could provide information about binding preferences that could not be derived using single-concentration analyses.
Figure 3. Combined Analysis of Datasets with Varied Protein Concentrations.
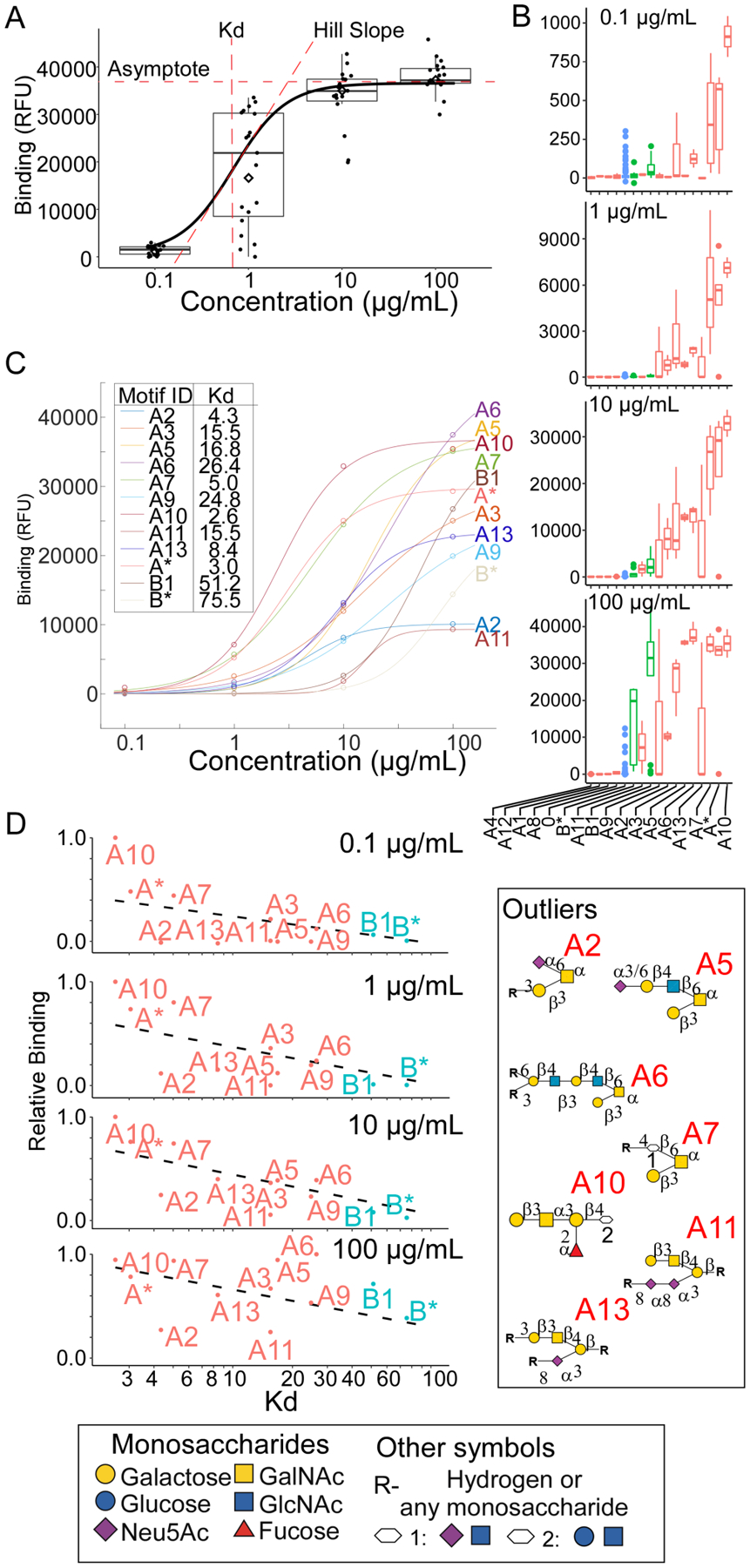
A. The fit of the logistic curve across concentrations provides the asymptote, slope, and half-maximum binding for each motif, the latter approximating the Kd. B. The simultaneous analysis of data for PNA over multiple concentrations gave the average binding to each motif at each concentration. C. The logistic curves fit to the average binding for each motif revealed distinct binding profiles across motifs. D. The relative binding (motif binding normalized to the highest motif binding) was plotted with respect to the curve-derived Kd values.
We used CFG data for PNA across four lectin concentrations (0.1, 1, 10, and 100 μg/mL). The analysis gave estimates of motif binding at each concentration (Figure 3B) and produced logistic curves with a wide range of binding parameters across the motifs (Figure 3C). These calculations enabled a comparison of the apparent Kd values derived across all concentrations with the relative-binding values derived from each single concentration (Figure 3D). For some motifs, the rank order was significantly different. For example, motifs A2 and A11 had relatively weak binding at all concentrations yet had strong (numerically low) apparent Kd values. Other motifs such as A5, A6, and B1 showed strong relative binding at higher concentrations but weak apparent Kd values, making them likely to have overestimated relative binding in higher-concentration datasets.
The divergence between apparent Kd and relative binding could stem from experimental sources, such as differences between the glycans in their densities on the surface or their accessibility to lectins when immobilized. Alternatively, the divergence could stem from fundamental kinetics, such as differences between the motifs in their association and disassociation rates. Motifs A2 and A11, which have lower apparent Kd values, have sialic acid near the canonical epitope of Galβ1,3GalNAc, while Motifs A5 and A6, which have much higher apparent Kd values, have an extended branch neighboring the shorter Galβ1,3GalNAc. The extended branch could potentially weaken the interaction (increasing the dissociation rate). The kinetics cannot be directly measured with conventional microarray technology, but within the limits of the experiment, this analysis demonstrates the importance of an integrated analysis across lectin concentrations. The derivation of approximate binding constants could provide more accurate identifications of the strong and weak motifs than would be possible at any single concentration.
Integrating Data from Diverse Arrays for a Single Lectin
Another application opened up by this system is the combined analysis of arrays with complementary glycans. Because the universe of glycans is so vast, and because each array contains only a particular subset of glycans, no single data source could provide a complete picture of specificity. We explored the value of joining information across arrays using data for the Ricinus communis agglutinin I (RCA-I) collected over 7 different arrays (Table S2). To place the datasets in similar ranges and scales, we used post-model mapping and motif curve fitting (Supporting Methods and Figure S4). Based on the assumption that independent datasets should show basic agreement to build a coherent model, we selected datasets that optimized the generalization of a model from a training set (4/5 of the sets) to a test set (1/5 of the sets) (Supporting Methods). This process selected the CFG array, the NCFG data (the latest CFG array content and an ASN-linked N-glycan array), an array of asymmetrically extended N-glycans5, and an array of neoglycoproteins38. Datasets excluded from training a combined analysis model were reserved for model validation (Figure S4).
We compared the results obtained using only the CFG data (Figure 4A) to the results obtained using multiple data sources (Figure 4B). Both analyses found that terminal LacNAc (Galβ1–4GlcNAc) was required, and that 6’ substitutions on the Gal are tolerated. Whereas the single-source model found biantennary LacNAc as the top motif (Motif A3, Figure 4A), the mixed-source model found terminal poly-LacNAc as the strongest motif (Motifs A8 and A9, Figure 4B). The mixed-source model also found that bi-antennary, terminal LacNAc binds markedly better than mono-antennary LacNAc (Motifs A and B, Figure 4B). The additional information was provided mainly from the monoantennary glycans in the asymmetric N-glycan array and the neoglycoprotein array.
Figure 4. Integration of Data from Mixed Datasets for RCA-I.
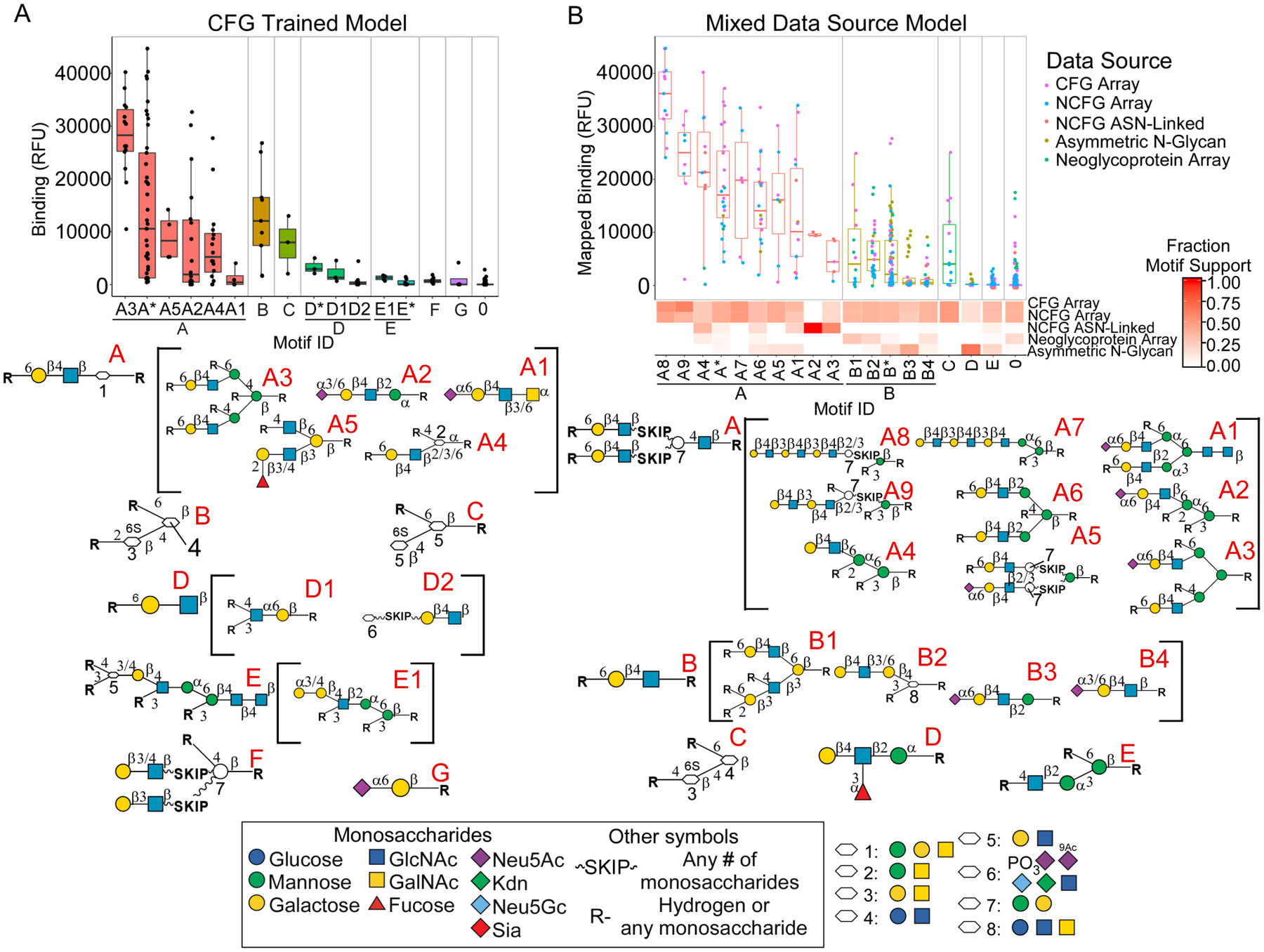
A. The single-source analysis of datasets from the CFG gave six primary motifs and 15 fine specificities. B. The integrated analysis of datasets from five different platforms revealed features not found in the single-source analysis, such as terminal poly-LacNAc (motifs A8, A9, and A7).
As a further evaluation, we compared the abilities of the models to predict the levels of lectin binding to independent sets glycans (Supporting Methods and Figure S4). In the application of a model trained only on CFG data (single source) and a model trained on multiple sources (mixed source) to the glycans in the CFG data, the R2 fits between predicted and actual binding were similar. But when applied to glycans from all 5 sources used in the mixed-source model or from 5 additional sources used in neither model, the R2 fits were significantly higher using the mixed-source model. This result supports the conclusion of increased accuracy in defining lectin specificity using integrated information from diverse sources.
Comparisons of Output Between Analysis Tools
We next assessed the output of MotifFinder by comparing it to the output from other programs. We used CFG-array data for the Erythrina cristagalli lectin (ECL) for this purpose. Two databases, GlyMDB and MCAW-DB, had pre-analyzed results for the data, and the programs MotifFinder and CCARL provided fresh analyses. These analysis tools have appeared in publications in the last 5 years and are still supported by their developers. We did not include older tools that are no longer supported by the developers. To enable objective comparisons of the methods, we examined the overall fit of the glycans between predicted and actual binding (Supporting Methods).
ECL binds type-2 LacNAc (Galβ1–4GlcNAc), especially in multivalent presentations39. It lacks tolerance for 6’ galactose substitutions but allows 2’ galactose substitutions such as found in H-antigen (blood group O)40–41. The motifs given by the two databases were able to correctly identify many high binding glycans but GlyMDB lacked specificity while MCAW-DB lacked sensitivity. GlyMDB (Figure 5A) uses subtree motifs that lack the ability to specify substitution intolerance (carbons that must be unsubstituted for a glycan to contain the motif). MCAW-DB (Figure 5B) uses an alignment-based method to identify the structure in common for the highest binding glycans but gives little information regarding less strongly bound glycans. CCARL (Fig. 5C) is similar to GlyMDB in the use of subtree modeling, but on account of the use of “restricted nodes” that define substitution intolerance, it gave a superior R2 of 0.32. MotifFinder, in contrast, identified features associated with each degree of binding. Binding was highest to Galβ1–4GlcNAc on two separate branches (Motif A), less to the same feature on a single branch (Motif B), and weak to GalNAcβ1–4GlcNAc (Motif C) or Fucα1–2Galβ1–4GlcNAc (Motif D). Accordingly, the R2 of 0.75 was over double that of the next highest, MCAW-DB. Thus, all of methods found the main motif of Galβ1–4GlcNAc, but only MotifFinder identified all of the canonical binding motifs (Figure S5) and their associations with gradations in binding.
Figure 5. Comparisons of Analysis Tools.
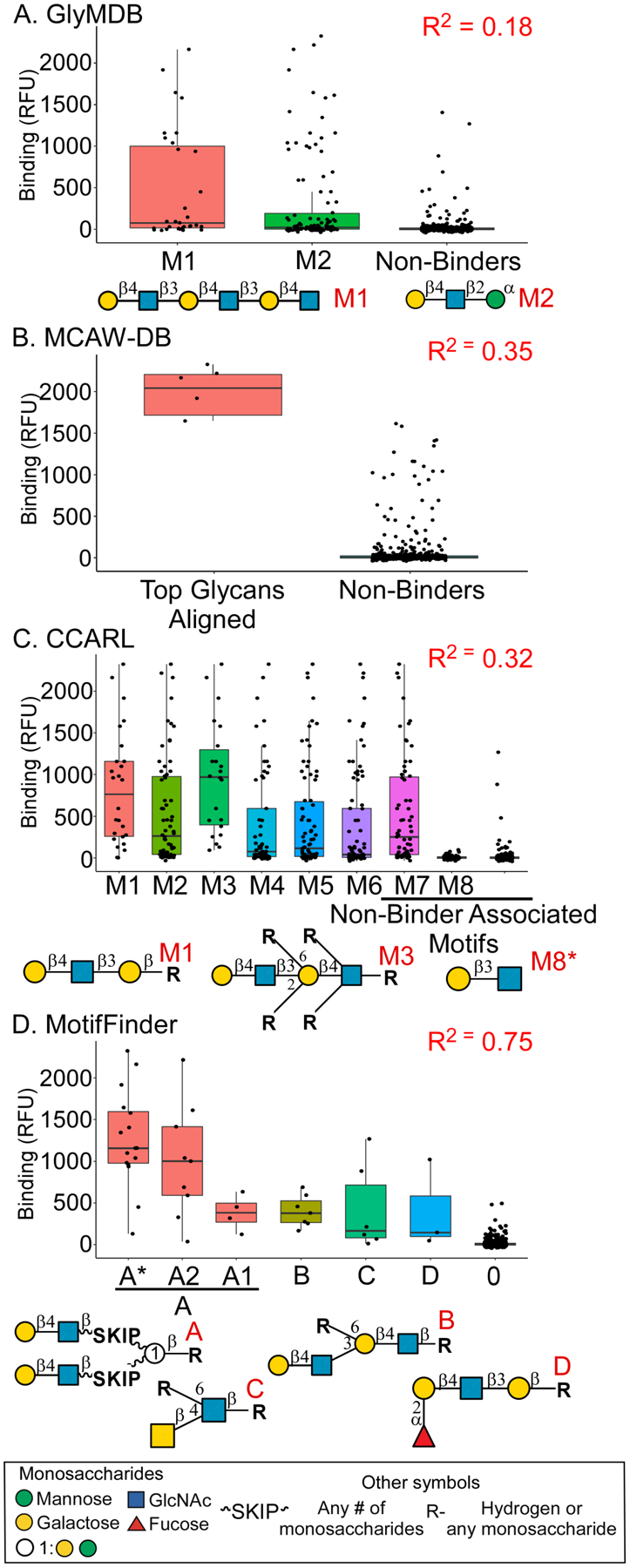
Each tool was evaluated using a CFG dataset for the lectin ECL. The plots present the ECL binding to the glycans organized by the motifs found by each method. A. GlyMDB. B. MCAW-DB. C. CCARL. D. MotifFinder. The R2 values were calculated using the observed binding compared to predicted binding.
We further evaluated MotifFinder and CCARL by cross-validation. Using datasets from 22 lectins, we derived a specificity model from 4/5 of the glycans in each dataset and used the model to predict binding to the remaining 1/5 of the glycans, with iteration over the five splits. The average R2 over all splits and lectins was 0.4 and 0.37 using multiple or single lectin concentrations with MotifFinder, respectively, and 0.27 using CCARL (Table S2). The values using MotifFinder were higher in 19/22 datasets.
Discussion
The potential value of glycan-array data is increasing. More and more groups produce arrays, and the arrays have greater variety in the glycans. In order to make full use of this resource, researchers need software that can process information from a wide mix of array types and that can integrate information in the combined analysis of multiple datasets. In this work, we present a system to address this need. We used a powerful glycan-name parser with a versatile syntax for motifs to input data from any source, and we developed a decision-tree algorithm with built-in motif optimization to derive the specificities of glycan-binding proteins. We demonstrated the ability to 1) seamlessly analyze data from any glycan-array platform; and 2) perform integrated analyses of multiple datasets. These capabilities are without precedent among methods to analyze glycan-array data.
A useful application of the integrated analysis is to identify concentration-dependent changes in apparent lectin binding. Previous analyses were manual and focused on individual glycans rather than motifs. Here we demonstrate a software tool that calculates apparent binding parameters for individual motifs. By evaluating binding to the motifs that are common between the glycans, the approach reflects the way in which lectins actually bind. The analysis presented here revealed that certain features in the motifs bound by PNA could link to differences between the motifs in apparent Kd. Future research could test the hypotheses that neighboring sialic acids reduce dissociation rates and that extended branches increase dissociation rates of PNA binding. The limitations to the analysis are that the separate concentrations must be run under highly controlled, consistent conditions, and that the concentrations should provide enough range and data to enable accurate curves.
The second application of integrated analysis, to combine information from disparate arrays, showed the potential for obtaining more complete views of specificity. It is clear that no single array can provide enough content to give full information about specificity. Data from complementary arrays could help to meet this need, but without software to combine the analyses, researchers could gather only limited insights. In the present demonstration, the integration of arrays containing motifs not in the CFG array led to useful insights into differential binding between branched glycans, extended monoantennary glycans, and short monoantennary glycans. A limitation of the approach is that at least a subset of the strong-to-moderately bound glycans should overlap between the datasets. In the present analysis, we excluded the microbial array because of poor overlap with the other arrays. Another limitation is that the datasets should have similar lectin concentrations in order to minimize concentration-dependent differences. Finally, the platforms should not have many inherent differences in the types of glycans that are bound. Such differences are not predictable but occasionally occur42. Here, we excluded the glycan bead array and the NGGM datasets from training owing to low agreement with the rest of the datasets. The apparent difference in specificity may be due to the novel microarray formats used for those datasets.
An important implication of this work relates to the use of lectins and glycan-binding antibodies as probes for their targets in biological samples. In such studies, the interpretation is typically based on simplified information about the GBP specificities. But given the complexities of the specificities, simple interpretations may not be accurate. To provide higher accuracy, an algorithm that makes use of the output from glycan-array analyses could prove useful. We previously introduced an algorithm for this purpose43, in which we convert the measurements of lectin binding into estimates of the motifs in the sample. Using a similar algorithm, one could predict the binding of a lectin to a previously untested glycan, which could be useful for the design of synthetic glycans.
The current study also suggested important areas for further development. Going forward, analyses could address the fact that other features beyond the motifs used here could influence lectin binding. For example, the microbial glycan array contains glycans with atypical substitutions that are not monosaccharides or common chemical modifiers, such as pyruvates or phosphoamino-pentanol. In other cases, the linker attaching the glycan to the substrate could have significant effects on binding38, as in single amino acids resulting in weaker binding due to steric hindrance44. Other factors include the sequence of the polypeptide backbone and the density of glycosylation, which could be studied using polypeptide arrays45 and density-variant arrays46–47. New analyses also could accommodate glycans that do not have fully defined sequences. An array with such glycans is the natural (shotgun) microarray, derived from cell or tissue sources5, 48. The framework of the motif syntax used here would support buildout to accommodate these features.
Conclusions
Among the various tools currently available for analyzing glycan-array data, the ability to integrate information across arrays and platforms is unique to MotifFinder. The need for such a tool is ever greater, given the expansion and diversification of glycan-array platforms. The ability to run the analysis without intervention by the user and the readily interpretable output make the method accessible to researchers who are not experts in glycobiology or bioinformatics. Thus, an increased number of researchers could analyze data from diverse arrays and leverage the unique advantages of each individual array.
Supplementary Material
Acknowledgements
We thank the National Institute of General Medical Sciences (R44GM131430 and R42GM112750) and the National Cancer Institute (Early Detection Research Network, U01CA152653; Alliance of Glycobiologists for Cancer Research, U01CA226158) for support of this research. We thank Dr. Jian Zhang and colleagues at Z Biotech for helpful suggestions on the interface and functionality of MotifFinder. Special thanks to Dr. Andrew Guy of the RMIT University in Melbourne, Australia, for advice regarding subtree mining methods and recommendations for the adaptation of CCARL for continuous prediction.
Abbreviations
- CFG
Consortium for Functional Glycomics
- NCFG
National Center for Functional Glycomics
- NGGM
Next Generation Glycan Microarray
- CUPRA
Competitive Universal Proxy Receptor Assay
- KCAM
KEGG Carbohydrate Matcher
- GlyMDB
Glycan Microarray Database
- CCARL
Carbohydrate Classification Accounting for Restricted Linkages
- DI
Depletion Index
- PNA
Peanut Agglutinin
- MAL-II
Maackia Amurensis Lectin 2
- DSA
Datura Stramonium Agglutinin
- HPA
Helix Pomatia Agglutinin
- BC2L-A
Burkholderia Cenocepacia Lectin 2 A
- RCA-I
Ricinus Communis Agglutinin
- ECL
Erythrina Crista-galli Lectin
- GSL-I-B4
Griffonia (Bandeiraea) Simplicifolia Lectin 1 Isolectin B4
- SNA
Sambucus Nigra Agglutinin
- ConA
Canavalia Ensiformis Agglutinin
Footnotes
Supporting Information
Methods for curve fitting, mapping array data onto comparable scales, and predicting lectin binding to glycans; results from MotifFinder analysis of common lectins and additional glycan-array platforms; additional analysis of the comparisons between analysis programs.
Data Availability
The input data (Table S2) were obtained through publicly accessible sources. The output files that were presented in the current study are available in the Supporting Data. The CCARL source code was downloaded from its source: https://github.com/andrewguy/CCARL. The results for GlyMDB and MCAW-DB were accessed from their respective websites: http://www.glycanstructure.org/glymdb/ and https://mcawdb.glycoinfo.org/. The MotifFinder software is available for download for Windows, MacOS, and Linux at our laboratory website: https://haablab.vai.org/tools/. The download package includes a User’s Manual and tutorials.
References
- 1.Rudiger H; Gabius HJ Plant Lectins: Occurrence, Biochemistry, Functions and Applications Glycoconj J 2001, 8, 589–613. [DOI] [PubMed] [Google Scholar]
- 2.Reily C; Stewart TJ; Renfrow MB; Novak J Glycosylation in Health and Disease Nat Rev Nephrol 2019, 6, 346–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pereira ME; Kabat EA; Lotan R; Sharon N Immunochemical Studies on the Specificity of the Peanut (Arachis Hypogaea) Agglutinin Carbohydr Res 1976, 1, 107–118. [DOI] [PubMed] [Google Scholar]
- 4.Taylor ME; Drickamer K Structural Insights into What Glycan Arrays Tell Us About How Glycan-Binding Proteins Interact with Their Ligands Glycobiology 2009, 11, 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li L; Guan W; Zhang G; Wu Z; Yu H; Chen X; Wang PG Microarray Analyses of Closely Related Glycoforms Reveal Different Accessibilities of Glycan Determinants on N-Glycan Branches Glycobiology 2020, 5, 334–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Nycholat CM; McBride R; Ekiert DC; Xu R; Rangarajan J; Peng W; Razi N; Gilbert M; Wakarchuk W; Wilson IA; Paulson JC Recognition of Sialylated Poly-N-Acetyllactosamine Chains on N- and O-Linked Glycans by Human and Avian Influenza a Virus Hemagglutinins Angew Chem Int Ed Engl 2012, 4860–4863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Song X; Yu H; Chen X; Lasanajak Y; Tappert MM; Air GM; Tiwari VK; Cao H; Chokhawala HA; Zheng H; Cummings RD; Smith DF A Sialylated Glycan Microarray Reveals Novel Interactions of Modified Sialic Acids with Proteins and Viruses J Biol Chem 2011, 31610–31622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Padler-Karavani V; Song X; Yu H; Hurtado-Ziola N; Huang S; Muthana S; Chokhawala HA; Cheng J; Verhagen A; Langereis MA; Kleene R; Schachner M; de Groot RJ; Lasanajak Y; Matsuda H; Schwab R; Chen X; Smith DF; Cummings RD; Varki A Cross-Comparison of Protein Recognition of Sialic Acid Diversity on Two Novel Sialoglycan Microarrays J Biol Chem 2012, 27, 22593–22608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Prudden AR; Liu L; Capicciotti CJ; Wolfert MA; Wang S; Gao Z; Meng L; Moremen KW; Boons GJ Synthesis of Asymmetrical Multiantennary Human Milk Oligosaccharides Proc Natl Acad Sci U S A 2017, 27, 6954–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Wang Z; Chinoy ZS; Ambre SG; Peng W; McBride R; de Vries RP; Glushka J; Paulson JC; Boons GJ A General Strategy for the Chemoenzymatic Synthesis of Asymmetrically Branched N-Glycans Science 2013, 6144, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Wu Z; Liu Y; Li L; Wan XF; Zhu H; Guo Y; Wei M; Guan W; Wang PG Decoding Glycan Protein Interactions by a New Class of Asymmetric N-Glycans Org Biomol Chem 2017, 42, 8946–8951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Geissner A; Reinhardt A; Rademacher C; Johannssen T; Monteiro J; Lepenies B; Thepaut M; Fieschi F; Mrazkova J; Wimmerova M; Schuhmacher F; Gotze S; Grunstein D; Guo X; Hahm HS; Kandasamy J; Leonori D; Martin CE; Parameswarappa SG; Pasari S; Schlegel MK; Tanaka H; Xiao G; Yang Y; Pereira CL; Anish C; Seeberger PH Microbe-Focused Glycan Array Screening Platform Proc Natl Acad Sci U S A 2019, 6, 1958–1967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Ruprecht C; Bartetzko MP; Senf D; Dallabernadina P; Boos I; Andersen MCF; Kotake T; Knox JP; Hahn MG; Clausen MH; Pfrengle F A Synthetic Glycan Microarray Enables Epitope Mapping of Plant Cell Wall Glycan-Directed Antibodies Plant Physiol 2017, 3, 1094–1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Horton M; Su G; Yi L; Wang Z; Xu Y; Pagadala V; Zhang F; Zaharoff DA; Pearce K; Linhardt RJ; Liu J Construction of Heparan Sulfate Microarray for Investigating the Binding of Specific Saccharide Sequences to Proteins Glycobiology 2020, 3, 188–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Purohit S; Li T; Guan W; Song X; Song J; Tian Y; Li L; Sharma A; Dun B; Mysona D; Ghamande S; Rungruang B; Cummings RD; Wang PG; She JX Multiplex Glycan Bead Array for High Throughput and High Content Analyses of Glycan Binding Proteins Nat Commun 2018, 1, 258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Yan M; Zhu Y; Liu X; Lasanajak Y; Xiong J; Lu J; Lin X; Ashline D; Reinhold V; Smith DF; Song X Next-Generation Glycan Microarray Enabled by DNA-Coded Glycan Library and Next-Generation Sequencing Technology Anal Chem 2019, 14, 9221–9228. [DOI] [PubMed] [Google Scholar]
- 17.Kitov PI; Kitova EN; Han L; Li Z; Jung J; Rodrigues E; Hunter CD; Cairo CW; Macauley MS; Klassen JS A Quantitative, High-Throughput Method Identifies Protein-Glycan Interactions Via Mass Spectrometry Commun Biol 2019, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Han L; Kitov PI; Li J; Kitova EN; Klassen JS Probing Heteromultivalent Protein-Glycosphingolipid Interactions Using Native Mass Spectrometry and Nanodiscs Anal Chem 2020, 5, 3923–3931. [DOI] [PubMed] [Google Scholar]
- 19.Aoki KF; Yamaguchi A; Ueda N; Akutsu T; Mamitsuka H; Goto S; Kanehisa M Kcam (Kegg Carbohydrate Matcher): A Software Tool for Analyzing the Structures of Carbohydrate Sugar Chains Nucleic Acids Res 2004, Web Server issue, W267–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Porter A; Yue T; Heeringa L; Day S; Suh E; Haab BB A Motif-Based Analysis of Glycan Array Data to Determine the Specificities of Glycan-Binding Proteins Glycobiology 2010, 3, 369–380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Cholleti SR; Agravat S; Morris T; Saltz JH; Song X; Cummings RD; Smith DF Automated Motif Discovery from Glycan Array Data Omics : a journal of integrative biology 2012, 10, 497–512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Xuan P; Zhang Y; Tzeng TR; Wan XF; Luo F A Quantitative Structure-Activity Relationship (Qsar) Study on Glycan Array Data to Determine the Specificities of Glycan-Binding Proteins Glycobiology 2011, 4, 552–560. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Hosoda M; Akune Y; Aoki-Kinoshita KF Development and Application of an Algorithm to Compute Weighted Multiple Glycan Alignments Bioinformatics 2017, 9, 1317–1323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Klamer Z; Staal B; Prudden AR; Liu L; Smith DF; Boons GJ; Haab BB Mining High-Complexity Motifs in Glycans: A New Language to Uncover the Fine-Specificities of Lectins and Glycosidases Analytical Chemistry 2017, 22, 12342–12350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Coff L; Chan J; Ramsland PA; Guy AJ Identifying Glycan Motifs Using a Novel Subtree Mining Approach BMC Bioinformatics 2020, 1, 42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Agravat SB; Saltz JH; Cummings RD; Smith DF Glycopattern: A Web Platform for Glycan Array Mining Bioinformatics 2014, 23, 3417–3418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Akune Y; Hosoda M; Kaiya S; Shinmachi D; Aoki-Kinoshita KF The Rings Resource for Glycome Informatics Analysis and Data Mining on the Web Omics 2010, 4, 475–486. [DOI] [PubMed] [Google Scholar]
- 28.Hosoda M; Takahashi Y; Shiota M; Shinmachi D; Inomoto R; Higashimoto S; Aoki-Kinoshita KF Mcaw-Db: A Glycan Profile Database Capturing the Ambiguity of Glycan Recognition Patterns Carbohydr Res 2018, 44–56. [DOI] [PubMed] [Google Scholar]
- 29.Mehta AY; Cummings RD Glad: Glycan Array Dashboard, a Visual Analytics Tool for Glycan Microarrays Bioinformatics 2019, 18, 3536–3537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Cao Y; Park SJ; Mehta AY; Cummings RD; Im W Glymdb: Glycan Microarray Database and Analysis Toolset Bioinformatics 2019, 8, 2438–2442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cheng K; Zhou Y; Neelamegham S Drawglycan-Snfg: A Robust Tool to Render Glycans and Glycopeptides with Fragmentation Information Glycobiology 2017, 3, 200–205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Klamer Z; Haab B Automated Identification of Lectin Fine Specificities from Glycan-Array Data; Glycan-Based Cellular Communication: Techniques for Carbohydrate-Protein Interactions American Chemical Society 2020, 67–82. [Google Scholar]
- 33.Sastry MV; Banarjee P; Patanjali SR; Swamy MJ; Swarnalatha GV; Surolia A Analysis of Saccharide Binding to Artocarpus Integrifolia Lectin Reveals Specific Recognition of T-Antigen (Beta-D-Gal(1----3)D-Galnac) J Biol Chem 1986, 25, 11726–11733. [PubMed] [Google Scholar]
- 34.Crowley JF; Goldstein IJ; Arnarp J; Lönngren J Carbohydrate Binding Studies on the Lectin from Datura Stramonium Seeds Arch Biochem Biophys 1984, 2, 524–533. [DOI] [PubMed] [Google Scholar]
- 35.Geisler C; Jarvis DL Effective Glycoanalysis with Maackia Amurensis Lectins Requires a Clear Understanding of Their Binding Specificities Glycobiology 2011, 8, 988–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Marchetti R; Malinovska L; Lameignère E; Adamova L; de Castro C; Cioci G; Stanetty C; Kosma P; Molinaro A; Wimmerova M; Imberty A; Silipo A Burkholderia Cenocepacia Lectin a Binding to Heptoses from the Bacterial Lipopolysaccharide Glycobiology 2012, 10, 1387–1398. [DOI] [PubMed] [Google Scholar]
- 37.Grant OC; Tessier MB; Meche L; Mahal LK; Foley BL; Woods RJ Combining 3d Structure with Glycan Array Data Provides Insight into the Origin of Glycan Specificity Glycobiology 2016, 7, 772–783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Temme JS; Campbell CT; Gildersleeve JC Factors Contributing to Variability of Glycan Microarray Binding Profiles Faraday Discuss 2019, 0, 90–111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu AM; Wu JH; Tsai MS; Yang Z; Sharon N; Herp A Differential Affinities of Erythrina Cristagalli Lectin (Ecl) toward Monosaccharides and Polyvalent Mammalian Structural Units Glycoconj J 2007, 9, 591–604. [DOI] [PubMed] [Google Scholar]
- 40.Itakura Y; Nakamura-Tsuruta S; Kominami J; Sharon N; Kasai K; Hirabayashi J Systematic Comparison of Oligosaccharide Specificity of Ricinus Communis Agglutinin I and Erythrina Lectins: A Search by Frontal Affinity Chromatography J Biochem 2007, 4, 459–469. [DOI] [PubMed] [Google Scholar]
- 41.Wang Y; Yu G; Han Z; Yang B; Hu Y; Zhao X; Wu J; Lv Y; Chai W Specificities of Ricinus Communis Agglutinin 120 Interaction with Sulfated Galactose FEBS Lett 2011, 24, 3927–3934. [DOI] [PubMed] [Google Scholar]
- 42.Wang L; Cummings RD; Smith DF; Huflejt M; Campbell CT; Gildersleeve JC; Gerlach JQ; Kilcoyne M; Joshi L; Serna S; Reichardt NC; Parera Pera N; Pieters RJ; Eng W; Mahal LK Cross-Platform Comparison of Glycan Microarray Formats Glycobiology 2014, 6, 507–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Klamer Z; Hsueh P; Ayala-Talavera D; Haab B Deciphering Protein Glycosylation by Computational Integration of on-Chip Profiling, Glycan-Array Data, and Mass Spectrometry Mol Cell Proteomics 2019, 1, 29–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Grant OC; Smith HM; Firsova D; Fadda E; Woods RJ Presentation, Presentation, Presentation! Molecular-Level Insight into Linker Effects on Glycan Array Screening Data Glycobiology 2014, 1, 17–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wandall HH; Blixt O; Tarp MA; Pedersen JW; Bennett EP; Mandel U; Ragupathi G; Livingston PO; Hollingsworth MA; Taylor-Papadimitriou J; Burchell J; Clausen H Cancer Biomarkers Defined by Autoantibody Signatures to Aberrant O-Glycopeptide Epitopes Cancer Res 2010, 4, 1306–1313. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Oyelaran O; Li Q; Farnsworth DW; Gildersleeve JC Microarrays with Varying Carbohydrate Density Reveal Distinct Subpopulations of Serum Antibodies J Proteome Res 2009, 3529–3538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Godula K; Bertozzi CR Density Variant Glycan Microarray for Evaluating CrossLinking of Mucin-Like Glycoconjugates by Lectins J Am Chem Soc 2012, 38, 15732–15742. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Song X; Lasanajak Y; Xia B; Heimburg-Molinaro J; Rhea JM; Ju H; Zhao C; Molinaro RJ; Cummings RD; Smith DF Shotgun Glycomics: A Microarray Strategy for Functional Glycomics Nat Methods 2011, 1, 85–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The input data (Table S2) were obtained through publicly accessible sources. The output files that were presented in the current study are available in the Supporting Data. The CCARL source code was downloaded from its source: https://github.com/andrewguy/CCARL. The results for GlyMDB and MCAW-DB were accessed from their respective websites: http://www.glycanstructure.org/glymdb/ and https://mcawdb.glycoinfo.org/. The MotifFinder software is available for download for Windows, MacOS, and Linux at our laboratory website: https://haablab.vai.org/tools/. The download package includes a User’s Manual and tutorials.