

Abstract
O-GlcNAc is a common post-translational modification of nuclear, mitochondrial, and cytoplasmic proteins that regulates normal physiology and the cell stress response. Dysregulation of O-GlcNAc cycling is implicated in the etiology of type II diabetes, heart failure, hypertension, and Alzheimer’s disease, as well as cardioprotection. These protocols cover simple and comprehensive techniques for detecting proteins modified by O-GlcNAc and studying the enzymes that add or remove O-GlcNAc.
Basic Protocol 1: Increasing the Stoichiometry of O-GlcNAc on Proteins Before Analysis
Basic Protocol 2: Detection of Proteins Modified by O-GlcNAc Using Antibodies
Basic Protocol 3: Detection of Proteins Modified by O-GlcNAc Using the Lectin sWGA
Support Protocol 1: Control for O-Linked Glycosylation
Basic Protocol 4: Detection and Enrichment of Proteins Using WGA-Agarose
Support Protocol 2: Digestion of Proteins with Hexosaminidase
Alternate Protocol 1: Detection of Proteins Modified by O-GlcNAc Using Galactosyltransferase
Support Protocol 3: Autogalactosylation of Galactosyltransferase
Support Protocol 4: Assay of Galactosyltransferase Activity
Basic Protocol 5: Characterization of Labeled Glycans by β-Elimination and Chromatography
Basic Protocol 6: Detection of O-GlcNAc in 96 well plates
Basic Protocol 7: Assay for OGT Activity
Support Protocol 5: Desalting the O-GlcNAc Transferase
Basic Protocol 8: Assay for O-GlcNAcase Activity
Keywords: signal transduction, glycosylation, O-GlcNAc, detection, analysis, galactosyltransferase, O-linked
INTRODUCTION
The modification of Ser/Thr residues of nuclear, cytosolic, and mitochondrial proteins by O-linked β-N-acetylglucosamine (O-GlcNAc) is a dynamic post-translational modification of metazoans. While O-GlcNAc has been identified in all metazoans studied, recent data suggest that plants use both O-fucose and O-GlcNAc (Xu et al., 2012; Zentella et al., 2017), protozoans use either O-GlcNAc or O-fucose (Banerjee et al., 2009), and that while filamentous yeast use O-GlcNAc (Schindler et al., 1987), Saccharomyces cerevisiae may use mannose (Halim et al., 2015).
O-GlcNAc is cycled on and off proteins by two dedicated enzymes; the O-GlcNAc transferase (OGT) that catalyzes the addition of O-GlcNAc and the O-GlcNAcase (OGA) that catalyzes the removal of O-GlcNAc. The reversible nature of the O-GlcNAc modification allows for multiple rounds of regulation/alteration of function over the lifetime of proteins. Indeed, protein O-GlcNAcylation has been demonstrated to impact protein function by altering structure and stability (Elbaum and Zondlo, 2014; Brister et al., 2014; Yuzwa et al., 2012; Levine et al., 2019), enzymatic activity (Han et al., 2017; Rao et al., 2015; Yi et al., 2012), protein-protein interactions (Toleman et al., 2018), and post-translational modifications (Comer and Hart, 2001; Chou et al., 1995). O-GlcNAcylation is responsive to stimuli/signaling including nutrient availability, cellular stress, insulin stimulation, or cellular stages such as the cell cycle suggesting that cells use O-GlcNAc to remodel cellular pathways (Hart et al., 2011).
Proteomic studies suggest that more than 4000 protein of diverse function are modified by O-GlcNAc. Proteins modified by O-GlcNAc include cytoskeletal proteins, nuclear pore proteins, RNA polymerase II (RNA Pol II), transcription factors, proto-oncogene products, tumor suppressors, hormone receptors, phosphatases, and kinases (Nandi et al., 2006; Vosseller et al., 2006; Chalkley et al., 2009; Khidekel et al., 2007; Trinidad et al., 2012). Not surprisingly, O-GlcNAc has been implicated in regulating key proteins within cellular pathways, including epigenetics, transcription, translation, protein homeostasis, autophagy, nutrient sensing and metabolism, immune signaling, and the cell cycle. Moreover, perturbations in the metabolism of UDP-GlcNAc, which alter the regulation of many O-GlcNAc-modified proteins, have been implicated in Alzheimer’s disease, diabetes, and cancer (Hart et al., 2011; Zhu et al., 2014; Ma and Vosseller, 2014).
These protocols concentrate on techniques for the detection and analysis of proteins modified by O-GlcNAc, as well as methods for the analysis of enzymes responsible for the addition and removal of O-GlcNAc. The major focus is on methods that require standard laboratory equipment. First, a protocol for increasing the stoichiometry of O-GlcNAc on proteins is given (see Basic Protocol 1). This is followed by simple techniques for the detection and screening of O-GlcNAc modified proteins either by immunoblotting or lectin affinity chromatography (see Basic Protocols 2 to 5). Separate protocols are used to verify that the glycan is O-linked GlcNAc (see Support Protocols 1 and 2). These methods are followed by protocols for a more comprehensive analysis of O-GlcNAc-modified proteins, including labeling of O-GlcNAc residues with [3H]Gal, and subsequent product analysis (see Alternate Protocol 1, see Basic Protocol 5, and see Support Protocols 3 and 4). There is also a protocol that enables the detection of O-GlcNAc in 96-well plate format (see Basic Protocol 6). The final two protocols assay for O-GlcNAc transferase and O-GlcNAcase activity, respectively (see Basic Protocols 7 and 8 and Support Protocol 5).
BASIC PROTOCOL 1 Increasing the Stoichiometry of O-GlcNAc on Proteins Before Analysis
In cultured mammalian cells, as well as tissue slices and tissues in vivo, the stoichiometry of O-GlcNAc moieties per protein molecule can be increased by treating cells/tissues/animals with inhibitors of O-GlcNAcase. Several inhibitors exist (Macauley et al., 2005; Dennis et al., 2006; Kim et al., 2006; Shanmugasundaram et al., 2006; Yuzwa et al., 2008), although only PUGNAc (Toronto Research Chemicals; (Haltiwanger, 1998)) and Thiamet-G (Cayman Chemicals; (Yuzwa et al., 2008)) are commercially available. Unlike Thiamet-G, PUGNAc also inhibits lysosomal hexosaminidases. Several OGT inhibitors have been developed (Gross et al., 2005). While these initial inhibitors were demonstrated to work in neonatal cardiomyocytes (1 to 5 μM (Ngoh et al., 2008)) and breast cancer cells (500 μM (Caldwell et al., 2010)), recent data suggests that these inhibitors react with the protein backbone limiting their utility (Jiang et al., 2011). Recently, high-throughput screening led to the development of a small-molecule OGT inhibitor, OSMI-1, which is cell-permeable (Ortiz-Meoz et al., 2015). Further structure-based evolution of OSMI-1 yielded three additional compounds OSMI −2, −3 and −4 with higher affinity for OGT (Martin et al., 2018). One alternative strategy has been to use cell-permeable compounds that are metabolized by the cell to generate non-hydrolyzable analogs of UDP-GlcNAc. Examples include Ac45SGlcNAc (effective in cells (Gloster et al., 2011)) or 5SGlcNHex (effective in animals (Liu et al., 2018)). However, it is important to note that OGT inhibitors that mimic UDP-GlcNAc are likely to impact other enzymes utilizing UDP-GlcNAc as a substrate. Streptozotocin (STZ; (Roos et al., 1998)), glucosamine (Han et al., 2000), and the glutamine fructose-6-phosphate amidotransferase (GFAT) inhibitors 6-diazo-5-oxonorleucine (DON) and Azaserine have also been used to alter the stoichiometry of O-GlcNAc on proteins. However, STZ has been shown to induce poly-(ADP-ribose) polymerase–mediated apoptosis in Min6 cells (Gao et al., 2000) and should be used with caution. STZ and glucosamine treatments are most effective in cells that express the glucose transporter GLUT-2 (Schnedl et al., 1994). Notably, inhibitors of GFAT can have off-target effects on other glutamine utilizing enzymes. Benzyl-α-GalNAc and benzyl-β-GlcNAc, which have been reported to inhibit OGT, should be avoided, as these have been characterized as inhibitors of prototypical O-GalNAc-type glycosylation. Care should be taken when using inhibitors of OGT and OGA, as treatment at too high a dose or for too long can impact the maturation of the mRNAs encoding OGT and OGA and thus protein expression (Park et al., 2017; Tan et al., 2020).
Materials
Cells of interest growing in monolayer culture, and appropriate culture medium
1 mM Thiamet-G (Millipore-SIGMA; SML0244) stock in 1 M HEPES, pH 7.4 (filter sterilize and store in aliquots up to 6 to 12 months at –80°C)
100 mM PUGNAc (Millipore-SIGMA; A7229) stock in PBS, pH 7.5 (filter sterilize and store in aliquots up to 6 to 12 months at –80°C)
500 mM glucosamine stock in 500 mM HEPES, pH 7.5 (make just prior to use; filter sterilize)
Ice
100-mm tissue culture dishes
Humidified water-jacket CO2 incubator
Additional reagents and equipment for SDS-PAGE and electroblotting (Gallagher, 2001).
Grow cells in monolayer culture in a sufficient number of 100-mm dishes.
- Add (or replace) growth medium with fresh medium containing 0.1 to 1 μM Thiamet-G (added from 1 mM stock) or 10 to 100 μM PUGNAc (added from 100mM stock) or 5 mM glucosamine (added from 500 mM stock). Incubate the cells in a humidified water-jacket CO2 incubator at 37°C for 4 to 18 hr.Treatments should be optimized for the desired cell type for 4–18h. Longer treatments alter the expression of OGT and OGA, as well as leading to significant toxicity (Slawson et al., 2005).The vehicle is either 1 M HEPES, PBS, or 0.5 M HEPES, pH 7.5.When using glucosamine, mannitol is often added to the controls at the same concentration. The use of mannitol controls for changes in osmolarity due to the additional sugar in the medium (Heart et al., 2000).
- At the end of treatment, take the dishes out of the incubator and place on ice. Extract as desired. Separate proteins by SDS-PAGE and electroblot onto an appropriate membrane (Gallagher, 2001).Alternatively, extract proteins and proceed with protein purification or immunoprecipitation (Immunoprecipitation., 2001).
BASIC PROTOCOL 2 Detection of Proteins Modified by O-GlcNAc Using Antibodies
Several antibodies have been developed that recognize terminal GlcNAc residues or the O-GlcNAc modification (Table 1). Additionally, site-specific O-GlcNAc antibodies have been raised against glycosylation sites on proteins including c-Myc (Kamemura et al., 2002), Tau (Cameron et al., 2013; Yuzwa et al., 2011), SirT1 (Shan et al., 2018), CRMP2 (Muha et al., 2019), as well as IRS2 and a subset of histones (H2A, H2B, H3, H4; GlycoScientific). These tools allow researchers to probe the O-GlcNAc modification state of key proteins without purification of the protein first. Site-specific antibodies are not discussed here since they are not widely available. When using O-GlcNAc pan-specific antibodies (antibodies that recognize O-GlcNAc linked to Ser/Thr residues) or GlcNAc pan-specific antibodies (antibodies that recognize any terminal beta GlcNAc residue, including those linked to longer glycan structures), it is important to run appropriate controls, as some antibodies cross-react with peptide sequences that mimic GlcNAc residues (Shikhman et al., 1994), while others have some dependence on the peptide backbone (Holt et al., 1987; Snow et al., 1987). Appropriate controls include: (1) elevating O-GlcNAc levels in tissue culture using inhibitors of the O-GlcNAcase or glucosamine (see Basic Protocol 1); (2) lowering O-GlcNAcylation in tissue culture cells using inhibitors of OGT or the hexosamine biosynthetic pathway; (3) removing O-GlcNAc from samples in vitro using hexosaminidase (see Support Protocol 3); (4) elevating the levels of O-GlcNAc in tissue culture by overexpressing OGT or performing RNAi of O-GlcNAcase; (5) lowering O-GlcNAcylation by overexpressing O-GlcNAcase or performing RNAi of OGT; (6) treating lysates with PNGase F to remove N-linked glycans; (7) using appropriate purified control proteins, such as ovalbumin, which bears N-linked glycans with terminal GlcNAc residues, or synthetic neoglycoconjugates, such as BSA-aminophenyl-GlcNAc (BSA-AP-GlcNAc), or (8) competing away antibody binding with free GlcNAc which controls for non-specific binding and cross-reactivity with endogenous immunoglobulin.
Table 1.
Antibodies and Lectins that Recognize O-GlcNAc and GlcNAc
| Name | Antibody isotype | Recognizes | Commercially available | Positive control | Negative control | Reference |
|---|---|---|---|---|---|---|
| CTD110.6 | IgM | βGlcNAc | Covance, Pierce, Millipore SIGMA, Cell Signaling, Santa Cruz Biotechnology | BSA-AP-GlcNAc | BSA-APa or ovalbumin | (Comer et al., 2001) |
| RL2 | IgG | O-GlcNAc | Abcam, Affinity Bioreagents, Santa Cruz Biotechnology | BSA-AP-GlcNAc | BSA-AP or ovalbumin | (Snow et al., 1987) |
| MY95 | IgG | O-GlcNAc | No | BSA-AP-GlcNAc | BSA-AP or ovalbumin | (Matsuoka et al., 2002) |
| 18B10.C7(3) | IgG | O-GlcNAc | Millipore | BSA-AP-GlcNAc | BSA-AP or ovalbumin | (Teo et al., 2010) |
| 9D1.E4(10) | IgG | βGlcNAc | Millipore | BSA-AP-GlcNAc | BSA-AP or ovalbumin | (Teo et al., 2010) |
| 1F5.D6(14) | IgG | O-GlcNAc | Millipore | BSA-AP-GlcNAc | BSA-AP or ovalbumin | (Teo et al., 2010) |
| HGAC 85 | IgG | βGlcNAc | Novus Biologicals, Abcam, Pierce, Enzo life sciences, Affinity Bioreagents | BSA-AP-GlcNAc | BSA-AP or BSA | (Turner et al., 1990) |
| HGAC 49 | IgM | βGlcNAc | No | BSA-AP-GlcNAc | BSA-AP or BSA | (Turner et al., 1990) |
| HGAC 39 | IgG | βGlcNAc | No | BSA-AP-GlcNAc | BSA-AP or BSA | (Turner et al., 1990) |
| Multi-mAbb | IgG | O-GlcNAc | Cell Signaling Technology | BSA | N/A |
BSA-AP, BSA-aminophenol.
This antibody was raised against glycoproteins. Modulating O-GlcNAc levels is an appropriate control.
Using the Millipore Sigma Immobilon Western Enhanced Chemiluminescent (ECL) Horse radish peroxidase (HRP) Substrate system the authors have found that 10 μg of nuclear or total cell extract is sufficient. However, 20 to 30 μg of cell extract provides the highest quality data. For the detection of O-GlcNAc from tissue samples, the authors have established that 25 to 30 μg of total tissue lysate is required for detection using CTD110.6. For detection using the RL2 antibody, 15 to 20 μg of tissue lysate is sufficient to be within the linear range of detection using the aforementioned ECL system. For purified proteins, Comer and co-workers found that 25 to 50 ng of a neoglycoconjugate was sufficient (Comer et al., 2001). The protocol given is for CTD110.6 (Comer et al., 2001), which appears to have the least peptide dependence and recognize the greatest number of O-GlcNAc modified proteins. A table summarizing conditions for a subset of commercial antibodies is included and use a similar protocol to CTD110.6 (Table 2).
Table 2.
Conditions for Some Common Commercial Antibodies
| Antibody | block * | Primary Concentration | Primary Buffer | Secondary | Secondary Buffer* | GlcNAc Competition |
|---|---|---|---|---|---|---|
| CTD110.6 | 3% Milk | 1μg/ml | 3% BSA | Mouse Anti-IgM | 3% BSA | 100mM |
| RL2 | 3% Milk | 1μg/ml | 3% BSA | Mouse Anti-IgG | 3% BSA | 500mM |
| HGAC 85 | 3% BSA | 1μg/ml | 3% BSA | Mouse Anti-IgG | 3% BSA | 250mM |
| HGAC 49 | 3% BSA | 1μg/ml | 3% BSA | Mouse Anti-IgM | 3% BSA | 250mM |
| HGAC 39 | 3% BSA | 1μg/ml | 3% BSA | Mouse Anti-IgG | 3% BSA | 250mM |
| Multi-mAB (CST) | 3% Milk | 1μg/ml | 3% BSA | Rabbit Anti-IgG | 3% BSA | 250mM |
Milk and BSA are w/v and are dissolved in TBST
Materials
Purified or crude protein (e.g., Basic Protocol 1) separated by SDS-PAGE and electroblotted to polyvinylidene difluoride (PVDF; (Gallagher, 2001)) or nitrocellulose (preferred to PVDF) (duplicate blots are required)
Blocking buffer (3% milk w/v in TBST)
Antibody: CTD 110.6 (Millipore SIGMA, MABS1254) diluted to 1 μg/mL in antibody dilution buffer (3% BSA w/v in TBST; see recipe for TBST in Reagents and Solutions)
N-acetylglucosamine (GlcNAc; Millipore SIGMA, A4106)
TBST (see recipe in Reagents and Solutions)
HRP-conjugated anti–mouse IgM (Millipore SIGMA, A8786) diluted 1/5000 in antibody dilution buffer (3% BSA w/v in TBST)
TBS (see recipe in Reagents and Solutions)
Immobilon Western Chemiluminescence HRP Substrate (Millipore SIGMA, WBKLS0500)
Additional reagents and equipment for visualization with chromogenic and luminescent substrates (Gallagher, 2001).
IMPORTANT NOTE: Volumes are given for 9 × 14–cm membranes washed in 10.5 × 15.5–cm boxes. Volumes can be scaled down or up, but membranes should be covered.
Block the blots by incubating with 25 ml blocking buffer for 60 min at room temperature.
Wash the blots with 25 ml TBST three times, each time for 10 min at room temperature.
- Incubate the blots with CTD 110.6 (diluted to 1 μg/mL in antibody dilution buffer), in duplicate, with and without 100 mM GlcNAc, overnight at 4°C.An antibody that has been antigen purified using a GlcNAc-agarose column yields higher quality blots.Commercially available CTD110.6 is purified by ammonium sulfate precipitation. Thus, it may be necessary to titrate antibody concentration when using a new lot number. Typically, we find that 1 μg/mL is a good place to start. For the detection of O-GlcNAc in tissue lysates, we find that higher concentrations of CTD110.6 are necessary,To control for specificity, it is important to perform a control blot. Here, the antibody is pre-incubated with 100 mM GlcNAc for ~5 min on ice before being applied to the control blot. This control is most important when probing immunoprecipitations and tissue lysates that may contain immunoglobulin. Table 2 reports the concentration of free GlcNAc to use for other O-GlcNAc antibodies.
Wash the blots with 25 ml TBST three times, each time for 10 min at room temperature.
- Incubate the blots with HRP-conjugated anti–mouse IgM (1/5000 dilution in antibody dilution buffer) for 50 min at room temperature.The concentration of secondary antibody varies from lot to lot and should be optimized each time with each new preparation.
Wash the blots with 25 ml TBST four times, each time for 10 min at room temperature.
Wash the blots with 25 ml TBS once, for 10 min at room temperature.
- Develop the HRP reaction using, e.g., the ECL system(Gallagher, 2001).CTD110.6 should bind BSA-AP-GlcNAc, but not BSA-AP. A number of bands in total cell lysate should be detected predominantly over 40 kDa. Treatment with Thiamet-G should elevate the levels of O-GlcNAc 2- to 3-fold on numerous proteins in total cell lysate. Reactivity toward both BSA-GlcNAc and total cell lysate should be completely competed away by free GlcNAc. Note that the antibody often cross-reacts with pre-stained markers.
BASIC PROTOCOL 3 Detection of Proteins Modified by O-GlcNAc Using the Lectin sWGA
Many lectins are reportedly specific for β-GlcNAc residues. The authors have typically used succinylated wheat germ agglutinin (sWGA), which is widely available and is derivatized with a number of useful functional groups including HRP. Before succinylation, WGA will recognize both sialic acid and GlcNAc (Monsigny et al., 1979). For additional information concerning lectin chromatography, see Lectin Affinity Chromatography (Freeze, 2001).
The amount of “test” protein used is dependent on the technique(s) used to develop the HRP reaction. Using the Millipore Sigma Immobilon Western Enhanced Chemiluminescent HRP Substrate system, the authors find that 15 μg of cytoplasmic or nuclear extract is sufficient, but 20 to 30 μg of protein produces the best data.
It is important to include an appropriate positive (100 ng ovalbumin) and negative (100 ng of BSA) control. As a control, a portion of the sample should also be treated with hexosaminidase (see Support Protocol 2), to show that reactivity is toward GlcNAc. Alternatively, the levels of O-GlcNAc can be manipulated in cell culture (see Basic Protocol 1). An additional control is to subject the blot to mild reductive β-elimination to verify that lectin/antibody reactivity is towards O-linked glycans (see Support Protocol 1), rather than N-linked glycans.
Materials
Purified or crude protein separated by SDS-PAGE and electroblotted (Gallagher, 2001) to polyvinylidene difluoride (PVDF) or nitrocellulose (duplicate blots are needed)
5% (w/v) BSA in TBST (see recipe for TBST)
TBST (see recipe in Reagents and Solutions)
0.1 μg/ml HRP-conjugated sWGA (EY Labs; H-2102) in TBST (see recipe for TBST): the lectin can be stored at 1 mg/ml in 0.01 M PBS, pH 7.4 (appendix 2e), at –20°C for at least 1 year
N-acetylglucosamine (GlcNAc; Millipore Sigma; A4106)
High-salt TBST (HS-TBST): TBST (see recipe in Reagents and Solutions) containing 1 M NaCl
Tris-buffered saline (TBS; see recipe in Reagents and Solutions)
ECL kit (Millipore Sigma Immobilon Western Chemiluminescent HRP Substrate, WBKLS0500)
Additional reagents and equipment for visualization with chromogenic and luminescent substrates (Gallagher, 2001)
IMPORTANT NOTE: Volumes are given for 9 × 14–cm membranes washed in 10.5 × 15.5–cm boxes. Volumes can be scaled down or up, but membranes should be covered.
Wash the duplicate blots for 10 min in 25 ml TBST at room temperature.
- Block by incubating the blots in 25 ml 5% (w/v) BSA/TBST for at least 60 min at room temperature.IMPORTANT NOTE: Milk cannot be used as the blocking agent since many of the proteins in milk are modified by glycans that react with sWGA.
Wash the blots three times, each time for 10 min with 25 ml TBST at room temperature.
- Incubate the blots in 0.1 μg/ml sWGA-HRP in HS-TBST, in duplicate, with and without 1 M GlcNAc, overnight at 4°C.To control for lectin specificity, it is important to perform a control blot. Here, the lectin is pre-incubated with 1 M GlcNAc for ~5 min on ice before being applied to the control blot.
Wash the blots six times, each time for 10 min, with 25 ml HS-TBST at room temperature.
Wash the blots once with 25 ml TBS for 10 min at room temperature.
- Develop the HRP-reaction using, e.g., the ECL system (Gallagher, 2001).sWGA should bind BSA-AP-GlcNAc and ovalbumin, but not BSA-AP. A number of bands, predominantly over 40 kDa, in total cell lysate should be detected. Treatment with Thiamet-G should elevate the levels of O-GlcNAc 2- to 3-fold on numerous proteins in total cell lysate. Reactivity toward BSA-GlcNAc, ovalbumin, and total cell lysate should be completely competed away by free GlcNAc. Using the ECL system described, 100 ng of ovalbumin should be visualized in 5 to 15 sec.
SUPPORT PROTOCOL 1 CONTROL FOR O-LINKED GLYCOSYLATION
Traditionally, mild alkaline reduction (reductive β-elimination) has been used to release O-linked carbohydrates from proteins (Amano and Kobata, 1989). This method has been adapted for blots to show that lectin/antibody reactivity is toward O-linked rather than N-linked glycans (Duk et al., 1997). Proteins blotted to PVDF are treated with 55 mM NaOH overnight (releasing O-linked sugars) and then probed using lectins or antibodies (Reeves et al., 2014). The authors note that this protocol will not differentiate between O-GlcNAc and other forms of O-linked glycosylation.
There are a number of reasons why lectin/antibody reactivity could be lost after NaOH treatment, e.g., the sugars were destroyed instead of being released, or the protein was degraded. To control for these, it is important to have control proteins with N- and O-linked sugars, and to stain one blot for protein after treatment, preferably with an antibody. The authors suggest a control blot of bovine asialofetuin (Millipore Sigma), which contains both N- and O-linked sugars terminating in GlcNAc, treated and not treated with PNGase F (Powell, 2001).
Materials
Protein samples and controls blotted to PVDF (triplicate blots are needed; nitrocellulose is not suitable as it dissolves in 55 mM NaOH)
Tris-buffered saline (TBS; see recipe in Reagents and Solutions)
55 mM NaOH
Milli-Q water
TBST (see recipe in Reagents and Solutions)
3% (w/v) BSA in TBST (see recipe for TBST in Reagents and Solutions)
40°C water bath
Additional reagents and equipment for probing protein blots with protein-specific antibodies (see Basic Protocol 2) or lectins (see Basic Protocol 3)
IMPORTANT NOTE: Volumes are given for 9 × 14–cm membranes washed in 10.5 × 15.5–cm boxes. Volumes can be scaled down or up, but membranes should be covered.
Wash the blots once with 25 ml TBS for 10 min at room temperature.
- Incubate the two blots in 25 ml 55 mM NaOH at 40°C overnight; incubate the control blot in 25 ml Milli-Q water at 40°C overnight.The blots treated with NaOH will yellow slightly.
Wash the blots three times, each time for 10 min at room temperature, with 25 ml TBST.
Block by incubating the blots in 25 ml of 3% (w/v) BSA/TBST for 60 min at room temperature.
- Probe the blots (one treated and one untreated) with carbohydrate-specific lectins (see Basic Protocol 3) or antibodies (see Basic Protocol 2). Probe the second NaOH-treated blot with a protein-specific antibody (see Basic Protocol 2).On the untreated blot, asialofetuin ± PNGase F should react with sWGA, as both the N- and O-linked sugars contain terminal GlcNAc residues. On the treated blot, only the asialofetuin – PNGase F should react with sWGA.
BASIC PROTOCOL 4 DETECTION AND ENRICHMENT OF PROTEINS USING WGA-AGAROSE
WGA lectin affinity chromatography provides a convenient method for enriching and detecting O-GlcNAc modified proteins. This procedure has been adapted for detecting proteins that are difficult to purify or are present in low copy numbers, such as transcription factors. In this protocol, the protein of interest is synthesized in a rabbit reticulocyte lysate (RRL) in vitro transcription-translation (TNT) system (Promega) and labeled with either [35S]Met, [35S]Cys, or [14C]Leu. After desalting, the proteins are tested for their ability to bind WGA-agarose in a GlcNAc-specific manner (Roquemore et al., 1994). This protocol is readily adapted to purifying proteins from cell extracts, but, as WGA binds proteins with both terminal GlcNAc and sialic acid residues, typically, one would purify proteins from nuclear and cytoplasmic extracts to avoid co-purifying proteins with prototypical glycans.
Alternatively, the lectin Ricinus communis agglutinin 1 (RCA1) has been used to select for O-GlcNAc proteins that have previously been labeled by galactosyltransferase (see Alternate Protocol 1). Proteins modified by terminal Gal are specifically retained on an RCA1 affinity column. Labeled O-GlcNAc-modified proteins are released under mild conditions, while those containing N-linked structures require lactose addition to the buffer before elution results (Hayes et al., 1995; Greis and Hart, 1998). The method described in this protocol can be adapted for RCA1 affinity chromatography by substituting RCA1-agarose (Vector Laboratories) for WGA-agarose and changing the order of the Gal and GlcNAc elution buffer.
Materials
cDNA subcloned into an expression vector with an SP6 or T7 promoter (~0.5 to 1 μg/μl)
TNT ® Lysate systems kit system kit (Promega)
Label: [35S]Met or [35S]Cys, or [14C]Leu
Ensure that the label used is well represented in your protein of interest. Although [35S]Met is usually the most cost-effective and sensitive choice.
WGA-agarose (Vector Laboratories; AL1023) WGA wash buffer: PBS (appendix 2e) containing 0.2% (v/v) NP-40
WGA Gal elution buffer (see recipe in Reagents and Solutions)
WGA GlcNAc elution buffer (see recipe in Reagents and Solutions)
TCA or methanol
1-ml tuberculin syringe with glass wool plug at the bottom to support chromatography matrix or Bio-Rad Bio-Spin disposable chromatography column
Liquid scintillation counter
Additional reagents and equipment for digesting proteins with hexosaminidase (see Support Protocol 2), desalting (see Support Protocol 5), SDS-PAGE, and autoradiography (Gallagher, 2001).
Prepare the proteins
-
1
Synthesize the proteins to incorporate the desired label ([35S]Met, [35S]Cys, or [14C]Leu) using the TNT ® system according to the manufacturer’s instructions. Include the protein of interest, a positive control for WGA binding (e.g., the nuclear pore protein p62), a negative control (luciferase, supplied with kit), and a no-DNA control.
-
2
Treat half of each sample with hexosaminidase (see Support Protocol 2).
-
3
Desalt the samples using spin filtration (e.g., Cytiva Life Sciences Microspin G-50 columns) or a 1-ml G-50 desalting column (as for desalting O-GlcNAc transferase; see Support Protocol 5).
Apply the protein samples to chromatography columns
The following procedure is carried out at 4°C.
-
4Equilibrate WGA-agarose and pack column as follows:
- If the resin is supplied as 50% slurry (i.e., 50% resin/50% storage solution), remove 300 μl (double the volume required) and pipet into a 1-ml tuberculin syringe or disposable chromatography column.WGA is used here rather than sWGA as it has a higher affinity for GlcNAc and O-GlcNAc
- Let the storage solution drain from resin.
- Equilibrate resin by washing column four times, each time with 1 ml of WGA wash buffer. Cap the column.
The volumes given are appropriate for a sample derived from an TNT. For enrichment of other protein samples, the volume of WGA should be optimized for the protein sample applied. The authors find that 50 mg of cell extract requires 5 ml of WGA-agarose, assuming that 1% to 2% of the total cell extract is modified by O-GlcNAc. -
5Apply the sample (~30 μl of an TNT reaction) to the column and let stand at 4°C for 30 min, or cap and incubate at 4°C for 30 min with rotating or rocking.Save a small amount (1%) of the starting material as a control to count and to run on a gel.
Wash the column and elute GlcNAc
-
6
At the end of the 30 min incubation, uncap the column and allow the sample to “run through” the resin. Collect this as the “run through” fraction. Wash the column with 15 ml WGA wash buffer at 10 ml/hr, collecting 0.5 ml fractions.
-
7
Load the column with 300 μl WGA Gal elution buffer and let stand at 4°C for 20 min.
-
8
Wash the column with 5 ml WGA Gal elution buffer, collecting 0.5 ml fractions.
-
9
Repeat steps 6 to 8 using GlcNAc elution buffer.
-
10Count 25 μl of each fraction using a liquid scintillation counter.Depending on the stoichiometry, or the number of O-GlcNAc residues per molecule of protein, 1% to 10% of the labeled protein should bind the WGA.
-
11Pool positive fractions that elute in the presence of GlcNAc and precipitate using TCA or methanol.To precipitate proteins with methanol, mix 1 vol of sample with 10 vol of ice-cold methanol. Incubate overnight at –20°C. Recover protein by centrifuging for 10 min at 16,000 × g, 4°C, in a microcentrifuge tube (which is the most efficient procedure) or in 15-ml conical centrifuge tubes for 10 min at 3000 × g, 4°C. Resuspend samples in SDS-PAGE sample buffer (Gallagher, 2001).As many proteins in rabbit reticulocyte lysate contain O-GlcNAc and bind WGA, the authors do not recommend the addition of carrier proteins at this point. Typically, a fraction of the GlcNAc elution containing a total of 1000 to 2000 dpm [35S]Met is precipitated and analyzed by SDS-PAGE and autoradiography (Gallagher, 2001).The use of acetone to precipitate proteins is not recommended, as free GlcNAc will also precipitate.
-
12Analyze the pellet by SDS-PAGE and autoradiography (Gallagher, 2001).Expect a band at the approximate molecular weight of the protein of interest.
SUPPORT PROTOCOL 2 DIGESTION OF PROTEINS WITH HEXOSAMINIDASE
Terminal GlcNAc and O-GlcNAc can be removed from proteins using commercially available hexosaminidases; these enzymes will also cleave terminal GalNAc residues. Unlike O-GlcNAcase, commercial hexosaminidases have low pH optima, typically pH 4.0 to 5.0.
Materials
Protein sample for digestion (include a positive control, e.g., ovalbumin)
2% (w/v) SDS (see appendix 2e for 20% stock)
2× hexosaminidase reaction mixture (see recipe in Reagents and Solutions)
Additional reagents and equipment for SDS-PAGE and electroblotting (Gallagher, 2001)
Mix the sample 1:1 with 2% SDS and boil for 5 min.
Mix the sample 1:1 with 2× hexosaminidase reaction mixture and incubate at 37°C for 4 to 24 hr.
- To assess the completeness of the digestion, separate an aliquot of the reaction by SDS-PAGE and electroblot (Gallagher, 2001) onto an appropriate membrane. Probe blots with carbohydrate-specific lectins or antibodies.Ovalbumin should move several kilodaltons on a 10% to 12% gel, and reactivity toward WGA should be ablated. Reactivity of O-GlcNAc modified proteins to WGA and CTD110.6 should be reduced significantly.
ALTERNATE PROTOCOL 1 Detection of Proteins Modified by O-GlcNAc Using Galactosyltransferase
The enzyme β−1,4-galactosyltransferase (from bovine milk) will label any terminal GlcNAc residue with Gal, using uridine diphospho-d-Gal (UDP-Gal) as a donor substrate (Brew et al., 1968). Hart and colleagues have exploited this property, using the enzyme to label terminal GlcNAc residues on proteins with [6-3H]Gal, forming a [3H]-βGal1–4βGlcNAc (Torres and Hart, 1984; Roquemore et al., 1994; Greis et al., 1996). The labeled sugar can be chemically released (via β-elimination) and analyzed by size-exclusion chromatography on a BioGel-P4 column, using the 3H radiolabel to detect the fraction of interest (Roquemore et al., 1994). Labeling the O-GlcNAc residue allows for the subsequent detection of proteins and peptides of interest during SDS-PAGE, HPLC, protease digestion, and Edman degradation steps. Researchers have been able to identify glycosylation sites on as little as 10 pmol using these methods (Greis et al., 1996). Recently, this technique has been adapted to allow the incorporation of unnatural sugar analogs that can be derivatized to facilitate either the purification or detection of O-GlcNAc-modified proteins and peptides (Kim, 2018), a technique that is marketed by Invitrogen under the “Click-it” brand (Invitrogen).
To achieve efficient labeling of some proteins, it is necessary to denature samples, for example, by boiling in the presence of 10 mM DTT and 0.5% (w/v) SDS. Galactosyltransferase has been shown to be active in solutions containing 5 mM DTT, 0.5 M NaCl, up to 2% (v/v) Triton X-100, up to 2% (v/v) NP-40, and 1 M urea. Up to 0.5% (w/v) SDS can be used if it is titrated with a 10-fold molar excess of either Triton X-100 or NP-40 in the final reaction mixture. Digitonin, which is commonly used to solubilize cells, should be used with caution, as it is a substrate for galactosyltransferase. The total ionic strength should be less than 0.2 M.
Galactosyltransferase requires 1 to 5 mM Mn2+ for activity but is inhibited by Mg2+ and concentrations of Mn2+ >20 mM. EDTA (or analogs) should be avoided unless titrated with appropriate levels of Mn2+. Note that 1 mole of EDTA binds 2 moles of Mn2+.
Free UDP is also an inhibitor of galactosyltransferase. For studies where complete labeling of the GlcNAc is preferable, such as site mapping, calf intestinal alkaline phosphatase is included in the reaction, as it degrades UDP (Unverzagt et al., 1990). While this increases the efficiency of the reaction, it is important to add this to the control as some preparations of alkaline phosphatase contain proteins that will label with galactosyltransferase (R.N. Cole, pers. commun.).
NOTE: Protease inhibitors, such as PIC 1, PIC 2, and PMSF (see the recipe for 1000× protease inhibitors in Reagents and Solutions), can be included (final concentrations, 1×), but GlcNAc and 1-amino GlcNAc should be removed prior to labeling by spin filtration or another method of desalting.
Materials
Protein sample(s)
Dithiothreitol (DTT)
Sodium dodecyl sulfate (SDS; see appendix 2e for 20% stock solution)
Label: 1.0 mCi/ml UDP-[3H]Gal, (17.6 Ci/mmol; American Radiolabeled Chemicals) in 70% (v/v) ethanol
Nitrogen source
25 mM 5′-adenosine monophosphate (5′-AMP) in Milli-Q water, pH 7.0
Buffer H (see recipe in Reagents and Solutions)
10× galactosyltransferase labeling buffer (see recipe in Reagents and Solutions)
Galactosyltransferase, autogalactosylated (see Support Protocol 3)
Calf intestinal alkaline phosphatase
Unlabeled UDP-Gal
Stop solution: 10% (w/v) SDS/0.1 M EDTA
Speed-Vac evaporator
37°C incubator
100°C water bath
30 × 1–cm Sephadex G-50 column equilibrated in 50 mM ammonium formate/0.1% (w/v) SDS
Liquid scintillation counter
Additional reagents and equipment for acetone precipitation of protein (Lovrien and Matulis, 2001), PNGase F digestion of proteins (Powell, 2001), SDS-PAGE (Gallagher, 2001), and product analysis (see Basic Protocol 5)
Prepare the reaction
-
1
Denature the protein sample by adding DTT to 10 mM and SDS to 0.5% (w/v), then boiling the sample for 10 min.
-
2Decide how many reactions are going to be carried out and thus how much label will be needed (~1 to 2 μCi/reaction).A positive control (ovalbumin, 2 μg), a negative control (because galactosyltransferase can label itself), and a sample-minus-enzyme control will be needed.
-
3Remove solvent from label in a Speed-Vac evaporator or under a stream of nitrogen.Ethanol can inhibit the galactosyltransferase reaction, but if <4 μl is required, the label can be added directly to the reaction (final reaction volume, 550 μl).
-
4Resuspend an appropriate amount of label in 50 μl per reaction of 25 mM 5′-AMP.The AMP is included to inhibit possible phosphodiesterase reactions, which might compete for the label during the labeling experiment.
-
5Set up reactions as follows:
- Up to 50 μl protein sample (final concentration 0.5 to 5 mg/ml)
- 350 μl buffer H
- 55 μl 10× galactosyltransferase labeling buffer
- 50 μl UDP-[3H]Gal/5′-AMP mixture from step 4
- 30 to 50 μl autogalactosylated galactosyltransferase
- 1 to 4 U calf intestinal alkaline phosphatase
- Milli-Q water to a final volume of 550 μl.
- Reaction volumes can be scaled down to 55 μl.
-
6Incubate 2 hr at 37°C or overnight at 4°C.These are the typical conditions. Galactosyltransferase is active over a range of temperatures.
-
7Add unlabeled UDP-Gal to a final concentration of 0.5 to 1.0 mM and another 2 to 5 μl of galactosyltransferase. Incubate for an additional 2 hr at 37°C.This step is used when complete labeling of the GlcNAc is required, such as site mapping.
-
8
Add 50 μl stop solution to each sample and heat to 100°C for 5 min in a water bath.
Isolate the product
-
9Resolve the protein from unincorporated label using a Sephadex G-50 column equilibrated in 50 mM ammonium formate/0.1% w/v SDS. Collect 1-ml fractions.The column dimensions should be 1 × 30–cm, and the flow rate between 0.4 to 1 ml/min. Note, slower flow rates result in greater resolution. This step should be performed at room temperature.Size-exclusion chromatography using Sephadex G-50 (~30 cm) is traditionally used to desalt samples. However, TCA precipitation, spin filtration/buffer exchange, or other forms of size-exclusion chromatography (e.g., Cytiva Life Sciences PD-10 desalting column) can be used. The addition of carrier proteins, such as BSA (~67 kDa) and cytochrome c (~12.5 kDa), to samples and buffers will reduce the amount of protein lost due to nonspecific protein adsorption.
-
10Count a 50-μl aliquot of each fraction using a liquid scintillation counter.Approximately 2 × 106 dpm of [3H]Gal should be incorporated into 2 μg of ovalbumin.
-
11
Combine the void volume and lyophilize to dryness.
-
12
Resuspend the samples in 1 volume of Milli-Q water (200ml) and precipitate with 9 volumes of ice cold acetone (1800ml) (Lovrien and Matulis, 2001).
-
13Treat the samples with PNGase F (Powell, 2001), separate by SDS-PAGE, and detect by autoradiography (Gallagher, 2001). Alternatively, subject the samples to “product analysis” to confirm that the label was incorporated onto O-GlcNAc (see Basic Protocol 5).Numerous proteins, including ovalbumin, should be labeled by Gal-transferase and detected by autoradiography. O-GlcNAc-modified proteins should be reactive before and after PNGase F treatment.PNGase F treatment can be applied before or after Galactosyltransferase labeling.
SUPPORT PROTOCOL 3 AUTOGALACTOSYLATION OF GALACTOSYLTRANSFERASE
As galactosyltransferase contains N-linked glycosylation sites, it is necessary to block these before using this enzyme to probe other proteins for terminal GlcNAc.
Materials
10× galactosyltransferase labeling buffer (see recipe in Reagents and Solutions)
Galactosyltransferase (Millipore Sigma, G5507)
10,000 U/ml aprotinin
2-mercaptoethanol
UDP-Gal
Saturated ammonium sulfate: >17.4 g (NH4)2SO4 in 25 ml Milli-Q water
85% ammonium sulfate: 14 g (NH4)2SO4 in 25 ml Milli-Q water
Galactosyltransferase storage buffer prechilled (see recipe in Reagents and Solutions)
30- to 50-ml centrifuge tubes
Refrigerated centrifuge
Resuspend 25 U of galactosyltransferase in 1 ml of 1× galactosyltransferase labeling buffer.
- Transfer the sample into a 30- to 50-ml centrifuge tube.The centrifuge tubes selected should be able to withstand a centrifugal force of 15,000 × g.
- Remove a 5 μl aliquot for an activity assay.This is the “pre-gal” sample to be used in Support Protocol 4.
Add 10 μl of 10,000 U/ml aprotinin, 3.5 μl of 2-mercaptoethanol, and 1.5 to 3.0 mg of UDP-Gal.
Incubate the sample on ice for 30 to 60 min.
Add 5.66 ml of prechilled saturated ammonium sulfate in a dropwise manner over 10 minutes. Incubate on ice for 30 min.
Centrifuge 15 min at >10,000 × g, 4°C, and pour off the supernatant. Resuspend the pellet in 5 ml cold 85% ammonium sulfate and incubate on ice for 30 min.
Centrifuge 15 min at >10,000 × g, 4°C, and pour off the supernatant.
Resuspend the pellet in 1 ml of galactosyltransferase storage buffer and divide into 50-μl aliquots, saving 5 μl for an activity assay as the “auto-gal” sample. Assay that aliquot for activity (see Support Protocol 4).
Store the remaining aliquots up to 1 year at –20°C pending use in Alternate Protocols.
SUPPORT PROTOCOL 4 ASSAY OF GALACTOSYLTRANSFERASE ACTIVITY
As sample and activity may be lost during the autogalactosylation procedure, it is important to assess the activity of the enzyme.
Materials
1.0 mCi/ml UDP-[3H]Gal, (17.6 Ci/mmol; American Radiolabeled Chemicals) in 70% (v/v) ethanol
Nitrogen source
1× galactosyltransferase dilution buffer: galactosyltransferase storage buffer (see recipe in Reagents and Solutions) supplemented with 5 mg/ml BSA
10× galactosyltransferase labeling buffer (see recipe in Reagents and Solutions)
25 mM 5′-adenosine monophosphate (5′-AMP) in Milli-Q water, pH 7.0
“Pre-gal” sample aliquot (see Support Protocol 3, step 3) and “auto-gal” sample aliquot (see Support Protocol 3, step 9)
200 mM GlcNAc
Dowex AG1-X8 resin (PO4 form) slurry in 20% (v/v) ethanol
Speed-Vac evaporator
37°C incubator
Pasteur pipets
Glass wool
15-ml scintillation vials
Liquid scintillation counter
Dry 40 μl of 0.1 μCi/μl of UDP-[3H]Gal in a Speed-Vac evaporator or under a stream of nitrogen.
Resuspend in 90 μl of 25 mM 5′-AMP.
Make 1/1000, 1/10,000, and 1/100,000 serial dilutions of the “pre-gal” and “auto-gal” sample aliquots, in 1× galactosyltransferase dilution buffer. Using these dilutions, 200 mM GlcNAc, and 10× galactosyltransferase labeling buffer, prepare reaction mixtures as described in Table. 3.
Start the reaction by adding 10 μl of 0.05 μCi/μl UDP-[3H]Gal (see step 2) to each tube.
- Incubate the samples 30 min at 37°C.While the samples are incubating, prepare the columns.
- Pour 1 ml of Dowex AG1-X8 slurry (PO4 form) into 13 Pasteur pipets, each plugged with a small amount of glass wool.The glass wool prevents the resin from flowing out of the column. If too much glass wool is used, it will reduce the flow rate of the column. The glass wool plug should be 0.3- to 0.5-mm long and should not be over-compressed.
Wash with at least 3 ml of Milli-Q water. Do not let the columns run dry.
When almost all the Milli-Q water has eluted, place each column over a separate 15-ml scintillation vial.
Stop the reaction (still incubating from step 5) by adding 500 μl of Milli-Q water.
Load each sample onto the corresponding column and add a 500 μl water wash of the tube. Collect eluate as fraction A.
Elute with two 1 ml additions of Milli-Q water. Collect eluates as fractions B and C, respectively.
- Count 100 μl of the sample (Fractions A, B, and C) using a liquid scintillation counter. The activity can be expressed either as dpm 3H incorporation onto the GlcNAc residue, or as μmol 3H incorporation (1 μCi = 2.22 × 106 dpm).Notably, for UDP-Galactose with a specific activity of 17.6 Ci/mmol, 1000 DPM represents 25.8 fmol of Galactose added.Calculate the activity; one unit of activity (U) is defined as 1 μM of Gal transferred to GlcNAc per minute at 37°C.
Table 3.
Reaction Mixtures for Assay of Galactosyltransferase Activity
| Sample | Dilution | Vol. (μl) of diluted sample | Vol. (μl) of 200 mM GlcNAc | Vol. (μl) 10× Gal labeling buffer | Vol. (μl) Milli-Q water |
|---|---|---|---|---|---|
| Blank | 0 | 10 | 10 | 70 | |
| Pre-Gal | 1/1000 | 10 | 10 | 10 | 60 |
| 1/10,000 | 10 | 10 | 10 | 60 | |
| 1/100,000 | 10 | 10 | 10 | 60 | |
| Auto-Gal | 1/1000 | 10 | 10 | 10 | 60 |
| 1/10,000 | 10 | 10 | 10 | 60 | |
| 1/100,000 | 10 | 10 | 10 | 60 |
BASIC PROTOCOL 5 CHARACTERIZATION OF LABELED GLYCANS BY β-ELIMINATION AND CHROMATOGRAPHY
This protocol has three steps: (1) the release of carbohydrates as sugar alditols by reductive β-elimination; (2) desalting the sample, while confirming the size of the labeled sugar alditol(s); and (3) confirmation that the product is [3H] βGal1–4βGlcNAcitol (from galactosyltransferase labeling).
Materials
Radiolabeled glycoproteins (Alternate protocol 1)
β-elimination reagent: 1 M NaBH4/0.1 M NaOH (prepare fresh)
4 M acetic acid, pre-chilled on ice
1.5-ml screw-cap microcentrifuge tubes
37°C incubator
Additional reagents and equipment for acetone or methanol precipitation of proteins (Lovrien and Matulis, 2001), size-exclusion (gel-filtration) chromatography (Boysen and Hearn, 2001), and Dionex chromatography (Townsend et al., 1989; 1996; Townsend and Hardy, 1991) .
- Acetone- or methanol-precipitate the labeled proteins (Lovrien and Matulis, 2001) in 1.5-ml screw-cap microcentrifuge tubes.The β-elimination reaction is performed in screw-cap microcentrifuge tubes to prevent the lids from popping open during the lengthy incubation, which would result in evaporation of the sample. The tubes should be tightly sealed. Take care in opening the tubes at the end of the reaction, as gas generated during the reaction will escape.
- Resuspend the sample in 500 μl β-elimination reagent and incubate 18 hr at 37°C.After several hours, check that the pH is >13. Add more β-elimination reagent if needed.
Cool the sample on ice.
- Neutralize the reaction by adding 5 μl cold 4 M acetic acid in a stepwise manner. Check that the pH is between pH 6 and 7.Samples can be desalted either by chromatography on a Sephadex G-50 column (1 × 30 cm, equilibrated in 50 mM ammonium formate, 0.1% SDS) or by anion-exchange chromatography on a 1-ml Bio-Rad Dowex AG 50W-X2 200–400 mesh (H+ form) column equilibrated in water. Fractions containing [3H]GlcNAc or [3H]Gal are pooled and lyophilized. Residual NaBH4 is removed by washing the sample with methanol; NaBH4 is volatile in the presence of methanol and is removed in a Speed-Vac evaporator or under a stream on nitrogen (Fukuda, 1989).
- Resuspend the sugar alditols in Milli-Q water. Analyze by size-exclusion chromatography (Boysen and Hearn, 2001) or by Dionex chromatography (Townsend and Hardy, 1991; Townsend et al., 1996; 1989).To determine the size of the oligosaccharide, labeled glycans released by β-elimination are subjected to size-exclusion chromatography. Readers are referred to several standard methods using BioGel P4 (Kobata, 1994) or TSK Fractogel (Fukuda, 1989) chromatography. Alternatively, the Cytiva Life Sciences Superdex Peptide column, equilibrated in 30% (v/v) CH3CN, 0.1% (v/v) TFA, has been used to determine the sizes of oligosaccharides (R.N. Cole, pers. commun.). Galβ1–4GlcNAcitol should have a similar retention time to a disaccharide alditol standard.To determine the nature of the monosaccharide alditol or disaccharide alditol generated from either metabolic labeling or galactosyltransferase labeling, samples released by β-elimination can be analyzed by high-voltage paper electrophoresis or high-pH anion-exchange chromatography (HPAEC) with pulsed amperometric detection on a Dionex CarboPac PA100 column (Townsend et al., 1989; Hardy and Townsend, 1994).
BASIC PROTOCOL 6 DETECTION OF O-GLCNAC IN 96 WELL PLATES
The ability to detect O-GlcNAc in a 96 well plate format allows for the rapid optimization of treatments that alter protein O-GlcNAcylation levels in vivo. The method described below, a quantitative immunofluorescence assay, can be used directly in a 96 well plate reader or alternatively in cell imaging platforms. This protocol was optimized using three separate cell types (U2OS, mouse embryonic fibroblast, Cos-7); however, it is ideal to ensure that the O-GlcNAc levels of new cell types are within the linear range prior to assessing treatments that alter O-GlcNAc levels.
Materials
Wash buffer: 1x PBS
MES Buffer: 100mM MES, 1mM EDTA, 1mM MgCl2 pH6.9 (stored at 4ºC).
Fixation Buffer 1: 1:10 solution of MES buffer:methanol (stored at −20ºC).
This buffer rapidly dehydrates/precipitates protein while maintaining secondary structure. As methanol does not crosslink proteins there is less likelihood that protein epitopes are altered reducing antibody binding. Methanol also has the added effect of permeabilizing the cell membrane which leads to loss of some cytoplasmic content, particularly small molecules and lipophilic species.
Fixation Buffer 2: 4% v/v paraformaldehyde in 1X PBS (chill to 4°C).
Paraformaldehyde crosslinks proteins and nucleic acids with good preservation of the cellular spatial information and composition. Prolonged fixation increases the risk of epitope alteration and reduced antibody binding. Methanol-free paraformaldehyde is the recommended fixative for phalloidin stains.
Permeabilization solution: 0.5% (v/v) Triton-X-100 in 1X PBS.
Tween Wash Buffer (1X TBST): 1X TBS with 0.05% v/v Tween® 20.
Blocking buffer: 3% (w/v) BSA in TBST.
Competitive blocking buffer: 3% (w/v) BSA in TBST with 500mM GlcNAc.
Primary antibody dilutions:
CTD110.6 (Millipore SIGMA, MABS1254): 2μg/ml in 3% (w/v) BSA in 1X TBST.
RL2 (Millipore SIGMA, MABS157): 2μg/ml in 3% (w/v) BSA in 1X TBST.
Nuclear counter stain: SYTO 62 (5mM) Red Fluorescent Nucleic Acid Stain (Thermo Fisher Scientific, S11344), 1:2000 – 1:10000 (2.5μM – 0.5μM).
Optional: Actin counter stain – Alexa Fluor 647 Phalloidin (Thermo Fisher Scientific, A22287). Use only with paraformaldehyde fixed cells and use the manufacturer’s recommended working concentration. The working solution is made in the blocking buffer.
Fluorescent secondary antibody dilutions: Anti-mouse IgM Alexa Fluor 488 (Thermo Fisher Scientific, A-21042) and anti-rabbit IgG Alexa Fluor 488 (Thermo Fisher Scientific, A-32731/ Bethyl Laboratories A120–201D2) 2μg/mL – 4μg/mL – in 3% (v/w) BSA in 1X TBST. NOTE: Some of the secondary antibodies are at different concentrations (mg/mL). When choosing different secondary conjugates, it may be useful to try multiple dilutions or adjust the μg/mL concentration to match a previously validated dilution.
Aspirator, cell culture hood, incubator
Cells arrayed in a 96 well black wall clear bottom plate format (Corning, 3603).
Vortex with shaking attachment (or alternative)
Plate reader such as Spectramax plate reader (Molecular Devices)
Optional: Minimax 300 cell imager system
Aluminum Foil or plate covers
Ice
Prepare and plate the cells
-
1Split cells and resuspend to a concentration of 5×104 cells/ml.Note: Initially, it is critical to plate a dilution series of cells to ensure that signal will be within the linear range.
-
2Plate 100 μl in 96-well format (Black walled plate) including wells with media alone (blank).NOTE: It is critical to include blank wells as negative controls for nonspecific secondary antibody binding, and wells in which the signal can be competed away with free GlcNAc to provide a specificity control.
-
3
Allow cells to grow to 70–90% confluence.
-
4
Perform experimental manipulations as necessary, such as treatment with an inhibitor that would alter protein O-GlcNAcylation (See Basic Protocol 1).
Cell Fixation and Staining
-
5Wash away the media:
- Chill the wash buffer to 4ºC.
- Remove media from the microwell plate by aspiration.NOTE: Be sure to use a plastic 10μl pipette tip to cover the glass Pasteur pipette end. This helps prevent the loss of cells during aspiration.NOTE: Inverting the plate to remove the media and blotting the open face of the plate also reduces cell loss.
- Wash with 100 μl of wash buffer for 3 min and aspirate.NOTE: Add the solution carefully by pipetting down the side of the wells to avoid detaching the cells.
-
6Fix the cells:
- Immediately fix cells by addition of 100 μl of fixing solution (MES:Methanol or 4% (v/v) paraformaldehyde) and incubate the microwell plate on ice for 15 min.
- Remove the Fixation Solution by aspiration.
- Wash the cells with 100 μl wash buffer for 3 min (X 3).
- Proceed to the permeabilization step.NOTE: If the cells are not processed with primary antibodies on the same day, you can store the fixed cells in 100 μL of 1X PBS at 4°C (≤96 hours) until ready for immunostaining. When ready to process the cells aspirate the storage buffer and start from the permeabilization step.
-
7Permeabilize the cells:
- Add 100 μl of permeabilization buffer and incubate for 10 min at room temperature.
- Do not shake the plate
- Aspirate buffer.
- Add 100 μl of 1X TBST to the well, incubate for 3 min and aspirate (X 3).
-
8Block the cells:
- Carefully add 150 μl of Blocking Buffer down the side of the wells and incubate for 1.5 hr at room temperature with mild rocking.
-
9Wash the cells:
- Remove the blocking buffer by aspiration.
- Add 200 μl of 1X TBST to the well, incubate for 3 min and aspirate (X3).
-
10Counterstain the cells:
- Prepare the nuclear fluorescent stain as necessary. Some cells do not contain nuclei (red blood cells), whereas others can be multinuclear. In this case, actin is a more appropriate counterstain.
- Add 100 μl of the diluted counterstain to the cells and incubate for 30 min at room temperature.
- Aspirate the stain.
- Add 200 μl of 1X TBST to the well, incubate for 3 min, and aspirate (X3).Optional: The nuclear counterstain can be performed simultaneously with the blocking step by making the stain dilution in the blocking buffer.
-
11Add primary antibodies:
- Prepare O-GlcNAc antibody dilutions in blocking buffer and competitive blocking buffer as necessary.
- Add 100 μl of the desired primary antibody combination to the desired wells. The antibody solution should cover the bottom of each well.
- Incubate with primary antibody overnight at 4ºC with gentle shaking. Seal the plate around the edges with parafilm to reduce evaporation.NOTE: If you are using a primary antibody conjugated to a fluorophore, you should also cover the plate with foil to avoid photobleaching of the dye.
-
12Wash the cells:
- Add 200 μl of the 1X TBST washing solution by gently adding buffer down the side of the wells to avoid detaching the cells. Incubate for 3 min and aspirate (X3).
- Optional: Rock the plate gently during the wash steps.
-
13Add the secondary antibody:
- Dilute the fluorescent dye-labeled secondary antibodies in blocking buffer.NOTE: To lower nonspecific binding, add Tween® 20 to the diluted antibody to a final concentration of 0.2% v/v.
- Add 100 μl of the secondary antibody solution to each well and incubate for 60 min at room temperature.NOTE: Protect the plate from light during incubation.
-
14Wash the cells:
- Wash the plate with 1X TBST by gently adding 200 μL of buffer down the side of the wells to avoid detaching the cells.
- Incubate for 3 min at room temperature and aspirate the buffer (X 3).
- After the final wash turn the plate upside down and blot gently on paper towels to remove traces of wash buffer.
- Add 100 μL of 1X PBS per well for storage or imaging.NOTE: For best results, scan plate immediately; plates may also be stored at 4°C in commercial mounting media with antifade (protected from light).
- Seal the plate in parafilm to prevent evaporation and protect plates from light before imaging to ensure the highest sensitivity.
-
15Scan the plate:
- Before scanning, clean the bottom plate surface with a damp Kimwipe (water, not ethanol) to remove stains.
- Scan the plate using cell imaging functionality for fluorescence and transmitted light. Alternatively, scan the plate using the conventional fluorescence plate reader functionality.
- When storing plates after imaging, the plates should be sealed and remain protected from light at 4°C.
- The O-GlcNAc signal can be calculated by deducting signals from the equivalent competition well.NOTE: Well-to-well variability arising from variations in cell number can be addressed by rescaling the O-GlcNAc signal to the average counter stain intensity (DNA or actin) for that well.Note: When optimizing treatments, at signal to noise ratio of at least 3 is desirable.
BASIC PROTOCOL 7 ASSAY FOR OGT ACTIVITY
The detection and analysis of O-GlcNAc on proteins is only the first step in the analysis of O-GlcNAc and the protein(s) of interest. More important is determining the function of the modification. Protocols for the analysis of the enzymes that add and remove O-GlcNAc have been included in this unit, as they may aid in understanding the role of O-GlcNAc in a particular model. Recent examples where studies such as this have been critical include those which have shown the reciprocity between O-GlcNAc and O-phosphate on the C-terminal domain of RNA Pol II (Comer and Hart, 2001); studies showing elevated activity of enzymes in certain tissue/cell lines and tissue fractions (Whelan et al., 2008); and, finally, studies which have indicated that the enzymes responsible for the addition and removal of O-GlcNAc copurify with kinases and phosphatases (Wells et al., 2004).
O-GlcNAc transferase (OGT), or uridine diphospho-N-acetylglucosamine:polypeptide β-N-acetylglucosaminyltransferase, transfers GlcNAc to the hydroxyl groups of Ser and Thr residues of proteins and peptides using UDP-GlcNAc as a donor substrate (Haltiwanger et al., 1990; 1992; Kreppel et al., 1997). OGT activity is assayed by determining the rate at which [3H]GlcNAc is transferred to an acceptor peptide. A number of peptides have been identified as substrates for OGT in vitro, but a peptide (340PGGSTPVSSANMM352) from the α-subunit of casein kinase II (CKII) is an efficient in vitro substrate that is commonly used (Kreppel and Hart, 1999).
OGT activity can be assayed in crude preparations (Haltiwanger et al., 1990) or using recombinant protein (Kreppel and Hart, 1999). OGT activity is sensitive to salt inhibition and reducing agents, so it is important to desalt the preparation before assaying if high salt concentrations are present (see Support Protocol 5). Typically, 0.2 to 1 μg of purified recombinant protein is used per assay. To assay OGT activity in cell/tissue lysate, 10 to 50 μg of desalted lysate is required per assay.
Materials
Clear round-bottomed 96-well plate
0.1 mCi/ml UDP-[3H]GlcNAc (20 to 45 Ci/mmol; American Radiolabeled Chemicals) in 70% (v/v) ethanol
25 mM 5′-adenosine monophosphate (5′-AMP), in Milli-Q water, pH 7.0
Crude or purified OGT sample, desalted (see Support Protocol 5)
10× OGT assay buffer (see recipe in Reagents and Solutions)
CKII peptide substrate (+H2N-PGGSTPVSSANMM-COO–): dissolve in H2O to 10 mM and adjust to pH 7, if necessary
Note: OGT peptide substrates should be custom synthesized.
20U/μl Calf intestinal alkaline phosphatase (18009019; Thermo Fisher Scientific)
0.5mM Thiamet-G in 0.5M HEPES pH7.5
Quenching buffer: 50mM formic acid, 500mM NaCl
50 mM formic acid
Acetone
Methanol (HPLC-grade)
Scintillation fluid
Speed-Vac evaporator
Strata C18 96-well plate (25 mg/well; 8E-S001-CGB; Phenomenex)
Plates can be reused up to 10 times; however, plates should be precycled before use.
Scintillation counter
- Dry down an aliquot of UDP-[3H]GlcNAc in a Speed-Vac evaporator or under a stream of nitrogen just prior to use. Resuspend in an appropriate volume of 25 mM 5′-AMP, so that the concentration is 1 μCi/μl.5’-AMP is included in the assay to competitively inhibit any pyrophosphatase in the sample that will hydrolyze the UDP-GlcNAc.
- Prepare a master mix containing each of the following per reaction:5 μl of 10× OGT assay buffer5 μl of 10 mM CKII peptide substrate0.5 μl 1 μCi/μl UDP-[3 H]GlcNAc (0.5 μCi per reaction)1 μl of 0.5mM Thiamet-G0.25 μl of Calf intestinal alkaline phosphatase (5U per reaction)H20 to 20 μlOGT is feedback inhibited by a by-product of the reaction, UDP. Calf intestinal alkaline phosphatase degrades UDP and hence increases the efficiency of the reaction.
- 3. Array mastermix in a round-bottomed 96 well plate and initiate reactions by adding 30 μl of desalted OGT.It is critical to include a negative control. A mimic of the CKII peptide where the Ser and Thr residues are replaced with Ala is appropriate, or, simply, a “no-peptide” control can be included.Recombinant His-OGT purified in-house diluted in desalting buffer is included as a positive control.The results generated are variable and the reactions should be set up in triplicate.
- Incubate for 30 min −1 hour at room temperature.An incubation time of 15 to 30 min at room temperature is usually sufficient.
Stop the reaction by adding 150 μl of quenching buffer, 50 mM formic acid, 500mM NaCl.
- Activate a C18 96-well plate (Phenomonex; 25mg resin/well) with 100% Acetone (1.5 ml) and then 100% methanol (1.5 ml). Equilibrate the plate in 50mM formic acid, 500mM NaCl (three times, 1.5ml each).It is also possible to use Sep-Pak C18 cartridges. The method is similar to that described below.
- Load the reaction (200 μl total) onto the 96-well plate. Wash the plate sequentially with 50mM formic acid containing 500mM NaCl, water, and then 50mM formic acid (two times with 1.5 ml each).The CKII peptide binds to the C18 resin. Unincorporated UDP-[3H]GlcNAc is eliminated by the wash.
Elute the peptide with 1.5 ml of 100% methanol.
- Add 0.75 mL of elution volume to 5 ml scintillation fluid and count 3H. Calculate OGT activity according to the following equations. The activity can be expressed either as dpm 3H incorporation onto the GlcNAc residue, or as μmol GlcNAc incorporated per minute per mg of protein.Notably, for UDP-GlcNAc with a specific activity of 60 Ci/mmol, 1000 DPM represents 7.54 fmol of GlcNAc added.
SUPPORT PROTOCOL 5 Desalting the O-GlcNAc Transferase
OGT activity is sensitive to salt inhibition (IC50 = 40 to 50 mM NaCl). It is important to desalt the enzyme preparation before setting up the assay if a high concentration of salt is present. Additionally, O-GlcNAcase activity is sensitive to detergent present in extraction buffers. The protocol described below is also used to desalt O-GlcNAcase prior to setting up assay reactions.
Materials
Zeba 96-well Spin Desalting Plates, 40K MWCO (87774; Thermo Fisher Scientific)
96-well wash plates
96-well collection plates
OGT desalting buffer (see recipe in Reagents and Solutions)
Sample volume range 20 – 100 μl
Ice
Place the desalting plate on top of the wash plate and centrifuge (1000xg, 2 min) to remove the storage solution.
Discard the flow-through and replace the desalting plate onto the wash plate.
- Add 250 μl of desalting buffer on top of the resin. Centrifuge and discard flow-through. Repeat this step 2 additional times.After each spin, make sure resin appears white and free of liquid. Poor sample recovery may result from incomplete centrifugation.
Blot the bottom of the desalting plate to remove excess liquid and place it on top of a new collection plate.
- Load the protein sample (20 – 100 μl) onto the resin. Centrifuge (1000xg, 3min) and retain the flow-through that contains the sample.Alternatively, Zeba Spin Desalting Columns, 7K MWCO (Thermo Fisher Scientific), PD-10 columns (Millipore SIGMA), or G50 Sephadex (Cytiva Life Sciences) can be used.
BASIC PROTOCOL 8 Assay for O-GlcNAcase Activity
O-GlcNAcase, also known as N-acetylglucosaminidase or hexosaminidase C (EC 3.2.1.52), is a cytosolic glycosidase specific for O-linked β-GlcNAc. The activity of O-GlcNAcase can be conveniently assayed in vitro with a fluorogenic substrate, 4-Methylumbelliferyl-GlcNAc (4MU-β-GlcNAc). The cleavage product, 4MU, is fluorescent with an emission at 460nm (Macauley et al., 2005).
Materials
Purified O-GlcNAcase (0.2 to 1 μg) or cell/tissue extract (10 to 50 μg, desalted).
10× O-GlcNAcase assay buffer (see recipe in Reagents and Solutions)
1M GalNAc
100mM 4MU-βGlcNAc in DMSO (Millipore SIGMA, M2133)
100mM 4MU-βGalNAc in DMSO (Millipore SIGMA, M9659)
4MU-βGalNAc is used to confirm that lysosomal hexosaminidases have been effectively inhibited.
Black flat-bottomed 96-well plate and sealing tape
OGT Desalting buffer (see recipe in Reagents and Solutions)
Free 4MU (4-methylumbelliferone) standards at 100 mM in DMSO
β-N-acetylhexosaminidasef at 5000 U/ml (New England Biolabs, P0721)
Quenching buffer: 200mM glycine pH 10.75
Centrifuge at 4°C
37°C incubator
Plate reader
- Prepare O-GlcNAcase.Native or recombinant O-GlcNAcase can be partially purified from animal tissues or cultured cells by several chromatographic steps (Dong and Hart, 1994; Gao et al., 2001). Alternatively, a crude enzyme preparation can be generated by passing cell extract over a 1 ml Con-A column. Most of the interfering acidic hexosaminidases are modified by N-linked sugars and bind to Con-A, while neutral O-GlcNAcase is in the flow-through (Izumi and Suzuki, 1983). Additionally, O-GlcNAcase activity is sensitive to detergent present in extraction buffers. Therefore, cell/tissue extracts or the enzyme preparation is desalted (see Support Protocol 5) prior to setting up the assay reaction. Free GalNAc is used in the assay to inhibit lysosomal hexosaminidases. The lysates are assayed against both 4MU-βGlcNAc and 4MU-βGalNAc.
Precool a 96-well plate or microcentrifuge tubes on ice.
- Set up reactions in triplicate in the precooled plate wells or tubes as follows:20 μl partially purified O-GlcNAcase enzyme or cell/tissue extract20 μl of desalting buffer is used as a negative control. 4MU-GlcNAc breaks down chemically. A blank reaction without enzyme should be included to determine the background.20 μl of β-N-acetylhexosaminidasef (diluted to 25U/mL in desalting buffer) is used as a positive control.
- Prepare a master mix containing each of the following per sample:5 μl 10× O-GlcNAcase assay buffer5 μl 1M GalNAc50 mM GalNAc (final) is included in the reaction to inhibit lysosomal hexosaminidases A and B which may be present in the enzyme preparation. O-GlcNAcase is not inhibited by 50 mM GalNAc.0.5 μl of 4MU-GlcNAcEach of the above reactions are repeated with 4MU-GalNAc as a substrate instead of 4MU-GlcNAc. 4MU-GalNAc acts as a negative control substrate that allows for an assessment of the activity of any contaminating lysosomal hexosaminidasesH2O 19.5 μl.
Add 30 μl of Mastermix to each sample
Prepare a 4MU standard curve in DMSO. Add 50 μL to each well and perform this in triplicate.
Quench one reaction per every triplicate reaction set up at time point zero with quenching buffer (200mM Glycine pH 10.75). This is included to determine the background cleavage of 4MU-GlcNAc that is not a result of O-GlcNAcase activity.
Mix well and cover.
- Incubate for 30–60 min at 37°C.Reactions should be optimized to keep the fluorescence intensity within the linear range of the plate reader. The authors find that 10 to 50 μg of cell/tissue extract used in a reaction of 50 μl, with a 1 hr incubation time, is appropriate.
At the end of the incubation, add 150 μL of Quenching buffer (200mM glycine pH 10.75) to the remaining reactions, including the 4MU standards.
Read the fluorescence intensity with an excitation wavelength at 368nm and an emission wavelength of 450 nm on a plate reader.
- Calculate O-GlcNAcase activity according to the following equation:moles of GlcNAc or GalNAc released = [(Average fluorescence intensity after 60min) – (fluorescence intensity at time point 0)]/slope of the 4MU standard curveOGA activity is normalized by subtracting the fluorescence signal resulting from lysosomal hexosaminidase contamination, which was assessed using 4MU-GalNAc.OGA activity is expressed in moles of GlcNAc released/mg of purified OGA or cell extract/min of total reaction time.
REAGENTS AND SOLUTIONS
Use deionized, distilled water in all recipes and protocol steps. For common stock solutions, see appendix 2e; for suppliers, see suppliers appendix.
Biosynthetic labeling medium
Glucose-free culture medium containing:
50 μCi/ml d-[6-3H]glucosamine (22 Ci/mmol; American Radiolabeled Chemicals; ART 0110A)
10% (v/v) FBS
Prepare fresh
Buffer H
50 mM HEPES, pH 6.8
50 mM NaCl
2% (v/v) Triton X-100
Store up to 1 month at room temperature
Citrate-phosphate buffer, pH 4.0, 2×
Dissolve 12.9 g citric acid monohydrate (mol. wt. 210) and 20.6 g disodium hydrogen phosphate heptahydrate (Na2HPO4∙6H2O) in 300 ml Milli-Q water. Bring volume to 500 ml. Divide into 10-ml aliquots and store up to 1 year at –20°C.
Galactosyltransferase labeling buffer, 10×
100 mM HEPES, pH 7.5
100 mM galactose
50 mM MnCl2
Store up to 1 month at 4°C
Galactosyltransferase storage buffer
2.5 mM HEPES, pH 7.4
2.5 mM MnCl2
50% (v/v) glycerol
Store up to 1 month at room temperature
Hexosaminidase reaction mixture, 2×
Per reaction:
25 μl 2× citrate-phosphate buffer (see recipe)
1 U N-acetyl-β-d-glucosaminidase (V-Labs)
0.01 U aprotinin
1 μg leupeptin
1 μg α2-macroglobulin
Prepare fresh
O-GlcNAcase assay buffer, 10×
500 mM sodium cacodylate, pH 6.4
3% (w/v) bovine serum albumin (BSA)
Prepare fresh
OGT assay buffer, 10×
500 mM sodium cacodylate, pH 6.0
10 mg/ml bovine serum albumin (BSA)
Prepare fresh
OGT desalting buffer
20 mM Tris∙Cl, pH 7.8 (appendix 2e)
1 mg/ml bovine serum albumin (BSA)
20% (v/v) glycerol
0.02% (w/v) NaN3
Store up to 1 week at 4°C
Protease inhibitors, 1000×
PIC 1, 1000×:
Dissolve the following in 10,000 U/ml aprotinin solution (Millipore SIGMA)
1 mg/ml leupeptin
2 mg/ml antipain
10 mg/ml benzamide
PIC 2, 1000×:
Prepare in DMSO
1 mg/ml chemostatin
2 mg/ml pepstatin
PMSF, 1000×:
0.1 M phenylmethylsulfonyl fluoride in 95% ethanol
Tris-buffered saline (TBS)
10 mM Tris∙Cl, pH 7.5 (appendix 2e)
150 mM NaCl
Store up to 1 month at room temperature
TBST
10 mM Tris∙HCl, pH 7.5 (appendix 2e)
150 mM NaCl
0.05% (v/v) Tween 20
Store up to 1 month at room temperature
WGA buffer
25 mM Tris∙HCl pH 7.8 (appendix 2e)
300 mM NaCl
5 mM CaCl2
1 mM MgCl2
Store up to 6 months at room temperature
WGA Gal elution buffer
Phosphate-buffered saline (PBS; appendix 2e) containing:
0.2% (v/v) NP-40
1 M d-(+)-galactose (Gal)
Store up to 1 week at 4°C
WGA GlcNAc elution buffer
Phosphate-buffered saline (PBS; appendix 2e) containing:
0.2% (v/v) NP-40
1 M N-acetylglucosamine (GlcNAc)
Store up to 1 week at 4°C
WGA storage buffer
25 mM Tris∙HCl, pH 7.8 (appendix 2e)
300 mM NaCl
5 mM CaCl2
1 mM MgCl2
0.05% (w/v) sodium azide
Store up to 1 year at 4°C
COMMENTARY
Background Information
(β)-d-1–4-galactosylaminyltransferase from bovine milk recognizes terminal N-acetylglucosamine (GlcNAc) residues and modifies them by the addition of a single Gal residue. Torres and Hart first used this enzyme in combination with UDP-[3H]Gal to demonstrate that bovine lymphocytes contain proteins modified by O-linked GlcNAc (Torres and Hart, 1984). Further refinements of this experiment led them to propose that the product, βGal1–4βGlcNAc, was the result of the galactosyltransferase recognizing and modifying a single GlcNAc residue O-linked to Ser/Thr residues of nuclear and cytoplasmic proteins(Holt and Hart, 1986). Since this report, many cytosolic and nuclear proteins from mammalian cells were shown to be modified by O-GlcNAc. This method has remained the “gold standard” technique for the detection of O-GlcNAc-modified proteins, as the label provides a “tag” for subsequent analyses, such as those described under Product characterization below (see Critical Parameters and Troubleshooting). This approach has been modified to include sugars with biorthogonal handles that can be further modified by probes allowing the detection and enrichment of glycans. This approach, colloquially referred to as Click-Chemistry, is marketed by Invitrogen and has been reviewed in depth(Kim, 2018). As described in Basic Protocol 1, methods such as WGA affinity and immunoblotting with GlcNAc-specific lectins and antibodies have become popular as simple techniques for the initial characterization of target proteins.
Critical Parameters and Troubleshooting
Extraction of proteins from cells
The O-GlcNAc modification can be removed from proteins either by cytosolic O-GlcNAcase or lysosomal hexosaminidases. The inclusion of inhibitors during the extraction and purification process will preserve the levels of O-GlcNAc on proteins. Commonly used inhibitors (Dong and Hart, 1994) include 1-amino-GlcNAc (1 mM), GlcNAc (100 mM), PUGNAc (5 μM), and Thiamet G (1 μM). Note that these may have to be removed, as they will act as inhibitors in other methods.
Product characterization
Product characterization is a critical step to show that a protein is modified by O-GlcNAc and not other glycans. While many proteins modified by O-GlcNAc have been identified, there is evidence based on metabolic labeling (Medina et al., 1998) and lectin labeling studies (Hart et al., 1989) that suggests that O-GlcNAc is not the only intracellular carbohydrate post-translational modification. In addition, at least one peptide mimic of O-GlcNAc has been identified in cytokeratins (Shikhman et al., 1993; 1994). Moreover, many techniques used for breaking open cells also release proteins that are modified by complex N- and O-linked sugars, which may contain terminal GlcNAc. Many of the techniques described in this unit will recognize any terminal βGlcNAc residue, and it is important to perform the described controls, such as PNGase F digestion, to demonstrate specificity.
Product analysis is critical for metabolic labeling with glucosamine. While UDP-GlcNAc is the major product, glucosamine can enter other biosynthetic pathways, such as those used for amino acid synthesis. This issue was highlighted by studies of the SV40 large T antigen. Some researchers have found that the SV40 large T-antigen labels with a number of different tritiated carbohydrates. However, O-GlcNAc is the only carbohydrate post-translational modification of the SV40 large T antigen. The incorporation of glucosamine into amino acid biosynthetic pathways could be reduced by growing cells in the presence of excess nonessential amino acids (Medina et al., 1998).
Lastly, while galactosyltransferase is specific for terminal GlcNAc residues, researchers (Elling et al., 1999) have shown that galactosyltransferase will modify GlcNAc linked in either the α- or β-anomeric conformation. The authors of this protocol have shown that proteins modified by α-O-GlcNAc will be labeled using the procedure described (N. Zachara, unpub. observ.). While α-O-GlcNAc has not been identified in complex eukaryotes, it is a common modification of cell surface proteins of simple eukaryotes, such as trypanosomes and Dictyostelium. Product analysis, such as HPAEC of the sugar alditols, will resolve many of the issues discussed.
Understanding Results
Modulating the abundance of O-GlcNAc is a useful control for detection methodologies, as well as for testing the impact of O-GlcNAc on protein or pathway function. It is ideal to choose the shortest treatment with the lowest dose, as these treatments are less likely to have pleiotropic effects. To optimize treatments (Basic Protocol 1), inhibitor dose or application time can be varied. Figure 1 presents data in which inhibitor dose (Thiamet-G) has been modulated over 4h. In this example, there is a linear relationship between O-GlcNAc and inhibitor dose from (75nM). As previously published, elevating O-GlcNAc levels also augments the abundance of OGA (Kazemi et al., 2010; Park et al., 2017; Tan et al., 2020).
Figure 1. Determining inhibitor dose.
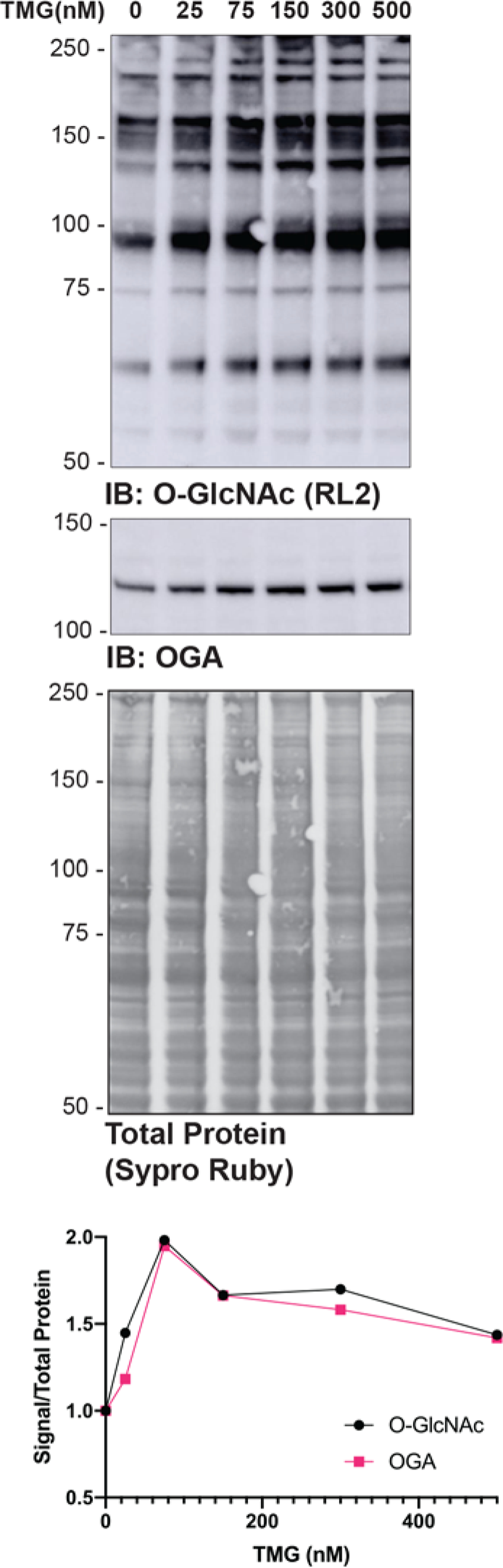
A) Mouse embryonic fibroblasts were treated with increasing doses of Thiamet-G (nM, 4h). Total cell lysates (10μg) were separated by SDS-PAGE, transferred to nitrocellulose, and total protein (Sypro™ Ruby Protein Blot stain), O-GlcNAc (RL2), and OGA (345; (Butkinaree et al., 2008)) were detected. Blots were developed using ECL and images captured with a GE Healthcare AI600 RGB. B) Quantification of O-GlcNAc (•, black) and OGA (■, pink) signals normalized to total protein.
When detecting O-GlcNAc by Immunoblot or lectin blot (Basic Protocol 2, Basic Protocol 3) it is critical to assess both linear range and specificity. Linear range will differ based on cell and tissue type, as well as the detection modality (ECL, fluorescence, reagents, film) and source of reagents (antibody concentration). Linear range is simply addressed by loading a dilution series of a protein of interest, detecting O-GlcNAc, and quantifying the O-GlcNAc and protein signals (Figure 2A). In this example, quantitation reveals that the signal is linear from 13–26 mg of protein. Specificity can be assessed by modulating O-GlcNAc levels (Basic Protocol 1, Support Protocol 1 and Support Protocol 2) and competing away primary antibody with free GlcNAc. This latter control is critical in immunoprecipitations and tissues, especially mouse tissues, in which immunoglobulin is present (Figure 2B). In the example shown (heart tissue) use of free GlcNAc or secondary alone, highlights two immune-reactive bands at ~70kDa that are mouse IgM heavy chain. A similar band at 55kDa is observed when probing with RL2, a mouse IgG.
Figure 2. Critical controls when detecting O-GlcNAc.
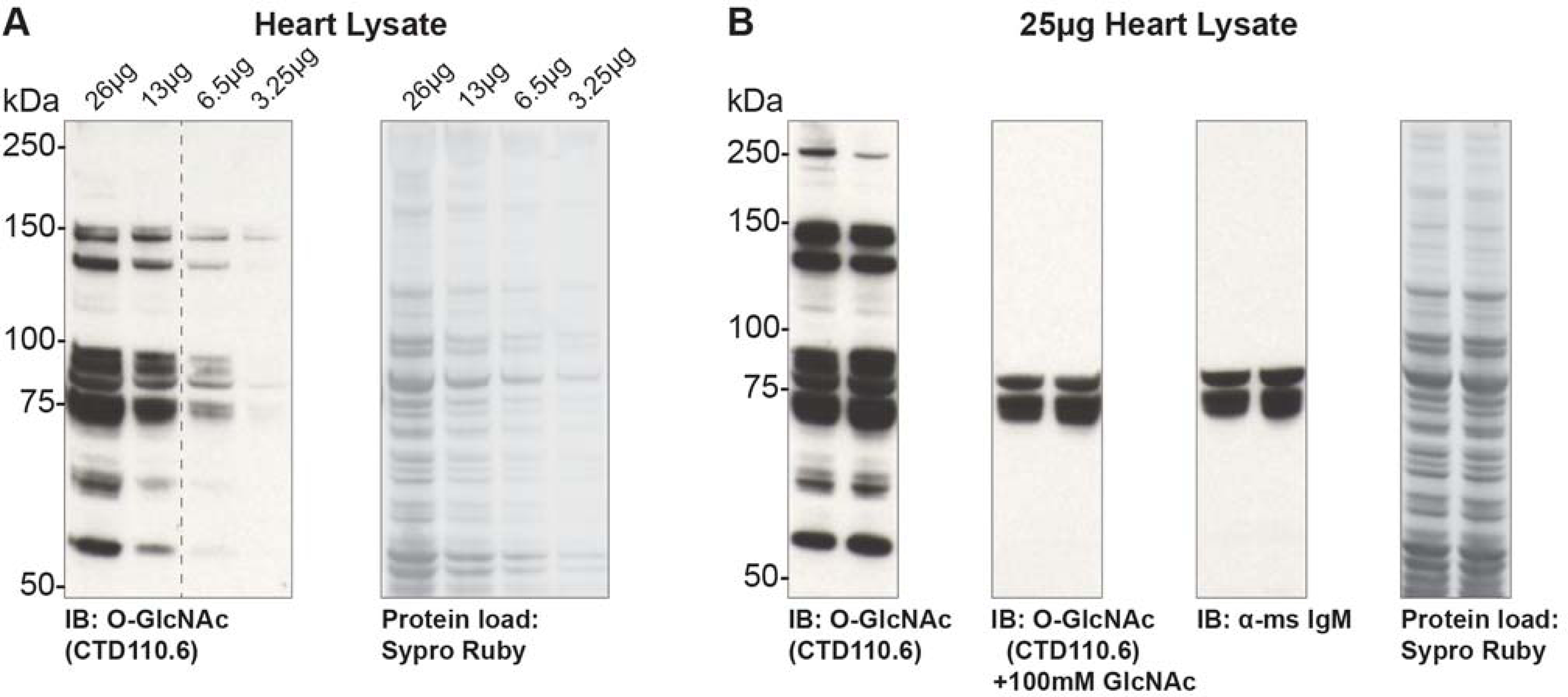
A) A dilution series of heart lysate (indicated) was separated by SDS-PAGE, transferred to nitrocellulose, and total protein (Sypro™ Ruby Protein Blot stain) and O-GlcNAc levels were detected. The dashed line represents the point at which the signal is no longer linear. B) 25μg of heart lysate was resolved by SDS-PAGE transferred to nitrocellulose, and total protein (Sypro™ Ruby Protein Blot stain), O-GlcNAc (CTD110.6), non-specific background (CTD110.6 + 100mM GlcNAc) and endogenous IgM-immunoglobulin (mouse IgM) were detected. Total protein was detected on a GE Healthcare AI600 RGB. Blots were develop using ECL and images captured on film.
There are numerous antibodies that can be used to detect the O-GlcNAc modification. Figure 3 presents representative data for 6 O-GlcNAc pan-specific antibodies. In addition to modulating O-GlcNAc levels (OGT WT and Null), the signal has been competed away with free GlcNAc. Confirmation of specific O-GlcNAc signals can be achieved by free GlcNAc competition and/or signal reduction with deletion of OGT. As previously reported, each of these antibodies displays display different preferences for substrates and this is represented by the different binding patterns (Snow et al., 1987; Teo et al., 2010; Lee et al., 2016; Reeves et al., 2014). Analogous data and controls should be obtained when using sWGA (Basic Protocol 3). Screening of antibodies and lectins in different models is warranted as some proteins, such as a-synuclein, are not well recognized by antibodies (Levine et al., 2019). For such proteins the use of Galactosyltransferase or metabolic labeling is necessary.
Figure 3 – Antibody detection of O-GlcNAc.

10μg of cell lysate from OGT wild-type (WT) and null cells was resolved by SDS-PAGE and transferred to nitrocellulose. Membranes were stained for total protein (Sypro™ Ruby Protein Blot stain) and A) CTD110.6 ± 100mM GlcNAc; B) RL2 ± 500mM GlcNAc; C) Multi-mAb – Cell Signaling Technologies (CST) ± 250mM GlcNAc; D) HGAC39 ± 250mM GlcNAc; E) HGAC49 ± 250mM GlcNAc; and F) HGAC85 ± 250mM GlcNAc. Total protein and immunoblots (developed with ECL) signals were captured with a GE Healthcare AI600 RGB.
A subset of pan-O-GlcNAc reactive antibodies cross-react with abbreviated N-linked glycans, as well as bisecting GlcNAc residues on N-linked glycans (Isono, 2011; Tashima and Stanley, 2014; Reeves et al., 2014). While the occurrence of the abbreviated N-linked glycan appears restricted to mammalian cells/tissues exposed to nutrient deprivation and protozoa, the ability to differentiate between an N- and O-linked GlcNAc residue is critical. Support protocol 1, represent such a method. In the example shown, cells have been exposed to nutrient deprivation (STV: 0mM Glucose, 1% FBS; 24h) which results in a robust increase in CTD110.6 signal that is sensitive to PNGase F (Figure 4A, lane 3). In control cells, the majority of the signal is sensitive to b-elimination suggesting that it arises from O-GlcNAc (Figure 4B). In contrast, a portion of the CTD110.6 signal in starved cells is resistant to b -elimination (Figure 4B). Sodium hydroxide will destroy protein epitopes. Thus, while a modest decrease in protein signals is expected (Compare actin in 4A and 4B), the signal should not be destroyed. Notably, a harsh b-elimination can impact N-linked glycans.
Figure 4. Differentiation of N-linked and O-linked glycans.
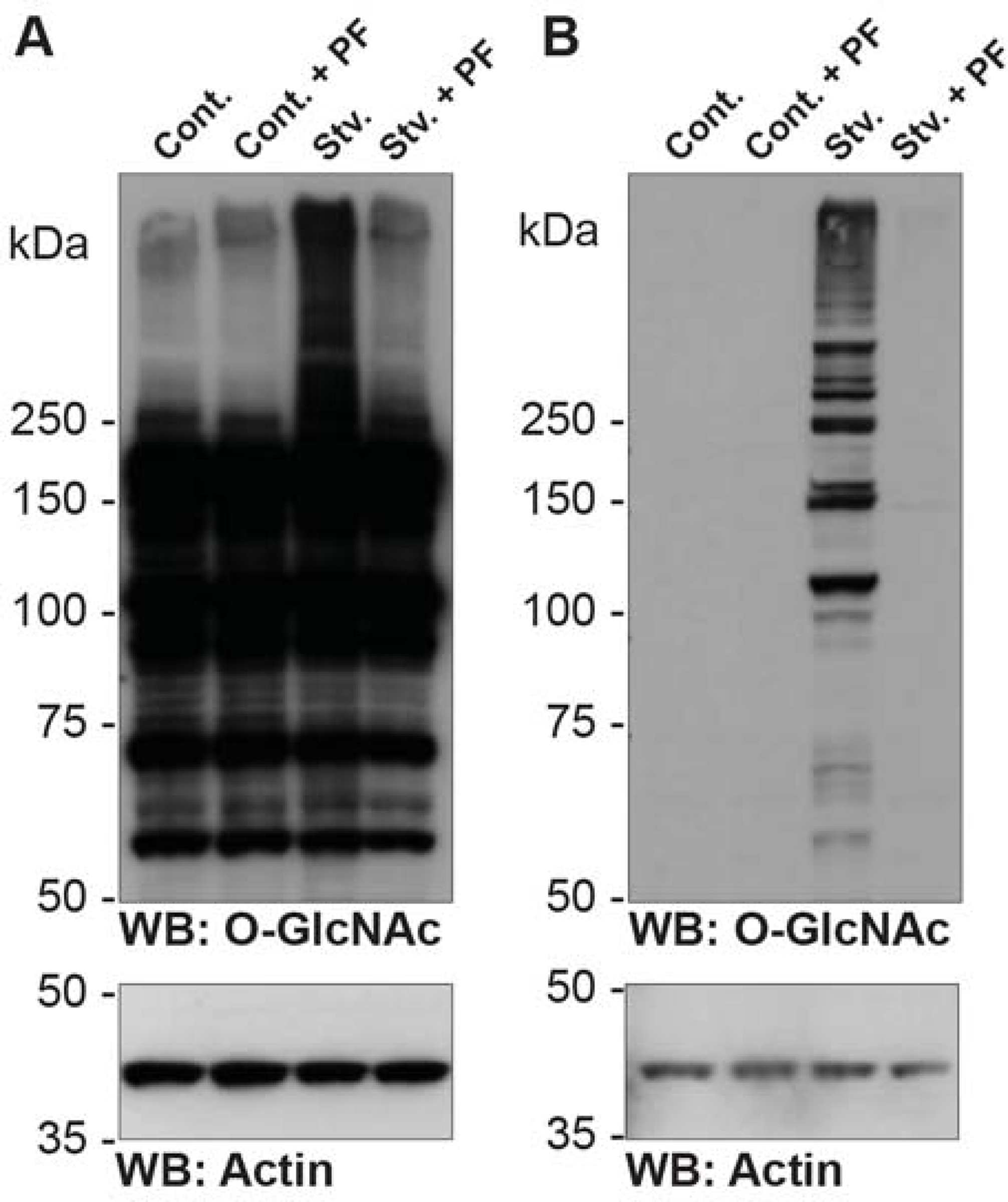
Lysates from cells exposed to control conditions or nutrient deprivation (STV: 0mM glucose, 1% dialyzed FBS) were treated with ±PNGase F (PF), prior to separation by SDS-PAGE and transferred to PVDF. Membranes were incubated in A) water or B) 55mM NaOH overnight at 40°C. After blocking, membranes were probed for O-GlcNAc (CTD110.6) or Actin. Blots were developed using ECL and images captured on film. These data are adapted from Reeves et al., 2014 (Reeves et al., 2014).
Galactosyltransferase labeling is a powerful approach for product characterization, identification of glycosylation sites, and of O-GlcNAc modified-proteins. Proteins labeled with 3H Gal should first be separated from the majority of unincorporated label using size exclusion chromatography (Figure 5A). A negative control reaction should be used to identify the retention time of 3H-UDP-Gal. Fractions with signal higher than the blank should be combined and the protein precipitated (TCA, Acetone, Methanol). In the example shown, better resolution from the unincorporated label could be achieved by increasing the length of the column. Proteins are subsequently separated by SDS-PAGE and the 3H-Gal detected by autoradiography. Ovalbumin is a useful control, as it is both labeled by Galactosyltransferase and carries one glycan that is sensitive to PNGase F. Labeled proteins can be further assessed using a range of approaches such as product analysis (Basic Protocol 6), protein purification, and manual Edman degradation.
Figure 5. Galactosyltransferase Labeling.
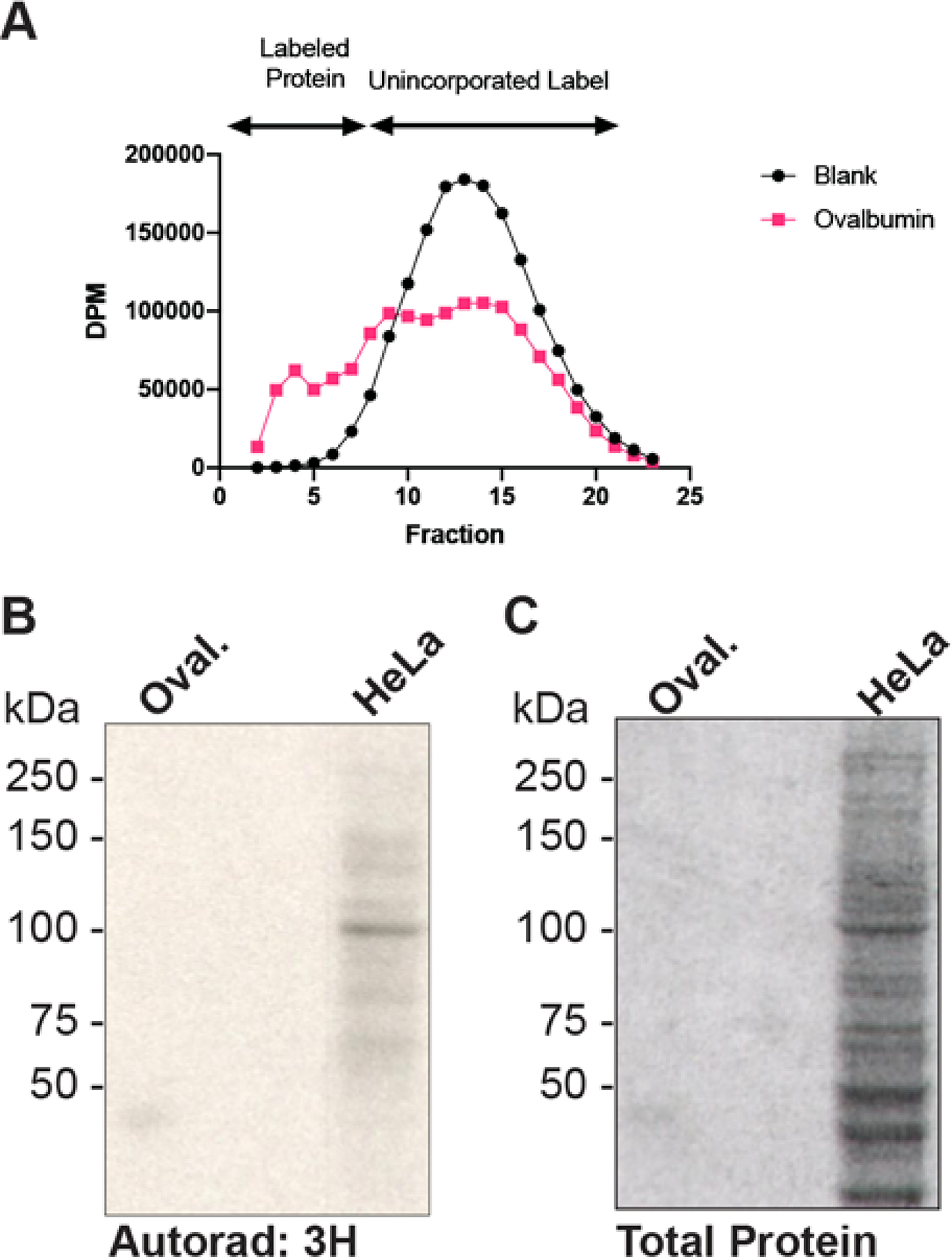
A) Ovalbumin (•, pink) or a blank (■, black) were labeled with Gal-T and 3H-UDP-Gal. Reactions were desalted using a G50 column. Fractions were counted and those with significant signal above the blank were combined and acetone precipitated. B) Ovalbumin (51,722 DPM) and HeLa cell lysates (18,295 DPM) labeled with Gal-T were separated by SDS-PAGE and stained with Coomassie blue (total protein). The resulting gel was dried and exposed to autoradiography film (1 week) with the aid of an intensifying screen.
O-GlcNAc-modified proteins can be enriched using antibody or lectin affinity chromatography (Basic Protocol 4) (Lee et al., 2016). It should be noted that as these experiments are performed under native conditions, unmodified proteins that interact with O-GlcNAcylated proteins will also be enriched. Capturing O-GlcNAcylated proteins from nuclear and cytosolic lysates reduces the possibility of enriching proteins modified by N-linked glycans. When combining this approach with TNT®, fractions eluting from affinity columns should be subsequently separated by SDS-PAGE and detected by autoradiography to confirm that radioactive signal arises from a protein with the appropriate molecular weight.
Basic Protocol 6 reports conditions for detecting O-GlcNAc in 96 well plates that should be readily adapted for immunofluorescence. In Figure 6A, OGT wild-type and null cells have been stained with CTD110.6. As expected, lower raw fluorescent signals are observed for O-GlcNAc in OGT null cells. As OGT null cells grow more slowly that OGT wildtype cells, the cell number is also reduced (Figure 6A, middle panel) (Kazemi et al., 2010). To generate the final data (Figure 6A, lower panel), raw CTD110.6 signals were corrected for total cell number using the nuclear stain. In Figure 6B, U2OS cells treated ±Thiamet-G have been stained using CTD110.6 resulting in a stronger signal. Similar to immunoblotting, incubation of the primary antibody with free GlcNAc provides an excellent specificity control. In Figure 6, incubation with free GlcNAc significantly reduces the signal. To generate final data, the raw fluorescent signal for CTD110.6 +100mM GlcNAc is subtracted from the raw fluorescent signal for CTD110.6. Finally, as in Figure 6A, the specific CTD110.6 signal is corrected for cell number or total protein using a nuclear stain. Alternatively, the CTD110.6 signal could be normalized to total protein using a stain for a housekeeping protein such as actin.
Figure 6. In Cell Western Blotting.
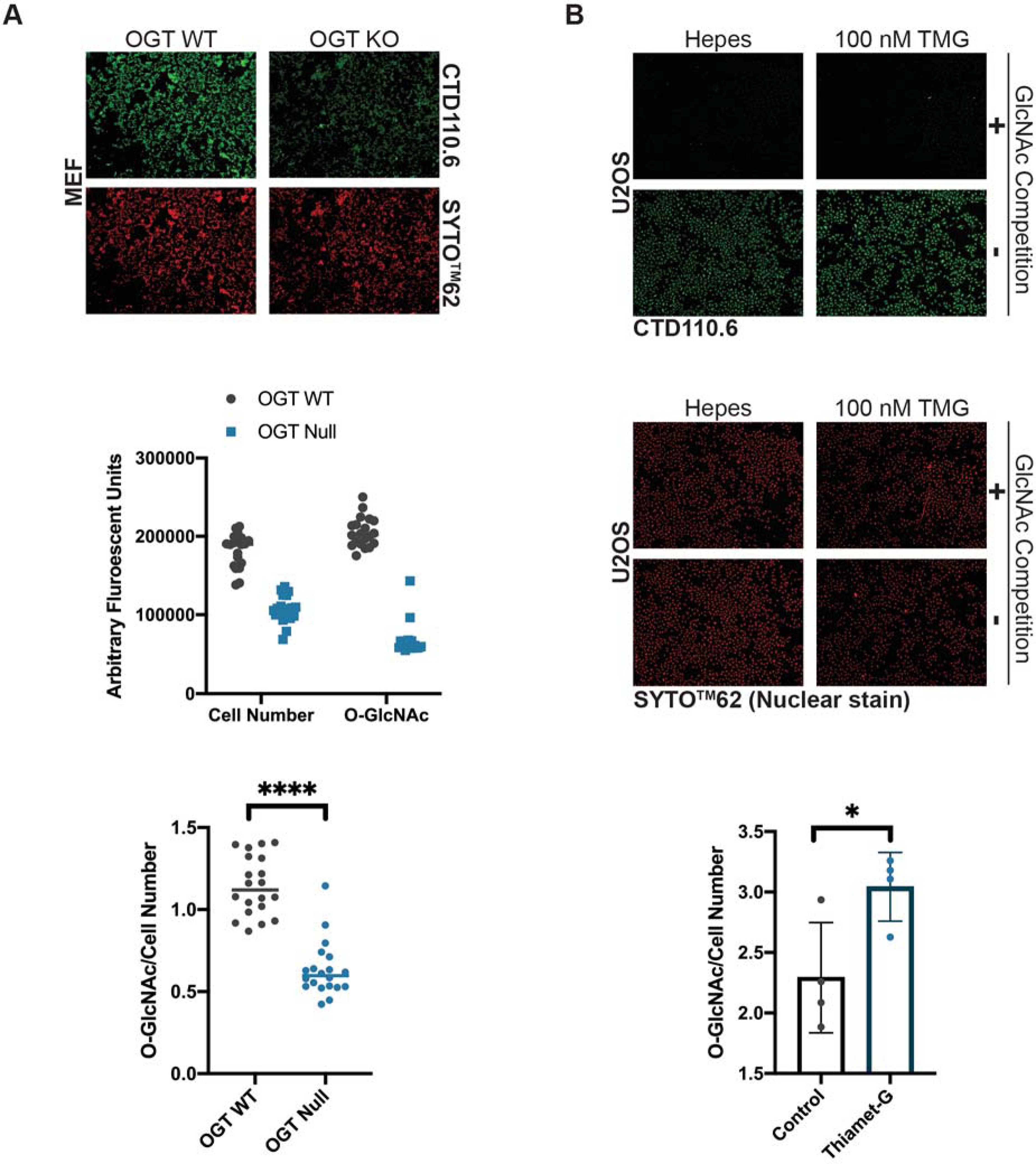
A) O-GlcNAc or cell number were detected in OGT WT (•, black) or null (•, blue) cells with CTD110.6 and SYTO™62. Raw fluorescent numbers for cell number and O-GlcNAc are plotted in the middle panel. O-GlcNAc levels corrected for cell number are plotted in the lower panel. P-values are the result of a student’s T-test of 20 technical replicates. **** p<0.001. B) O-GlcNAc or cell number were detected in U2OS cells treated with Vehicle (•, black) or TMG (100nM, 4h; •, blue) with CTD110.6 (upper panel) and SYTO™62 (middle panel). The O-GlcNAc signal corrected for cell number is plotted in the lower panel. Error bars represent one standard deviation. P-values are the result of a student’s T-test of 4 technical replicates. * p<0.05. Fluorescent intensity was assessed using a Molecular Devices SpectraMax i3x and fluorescent cell images (4x) with a Molecular Devices SpectraMax MiniMax 300™ Imaging Cytometer.
Figure 7 presents the raw data, an example calculation, and processed data for both OGT and OGA assays (Figure 7 A-D). To calculate OGT activity:
Triplicates are used to address variability and are averaged;
To assess autoglycosylation a “no peptide control is used. OGT Activity (DPM) = (Signal: CK2-peptide) – (Signal: No-Peptide-control);
- To calculate DPM of GlcNAc added per minute per mg:(DPM/Assay time in minutes) x [1000/protein per well in mg].
Based on the specific activity of the 3H-UDP-GlcNAc – DPM are converted to fmoles.
Figure 7 – Representative data from OGT and OGA assays.
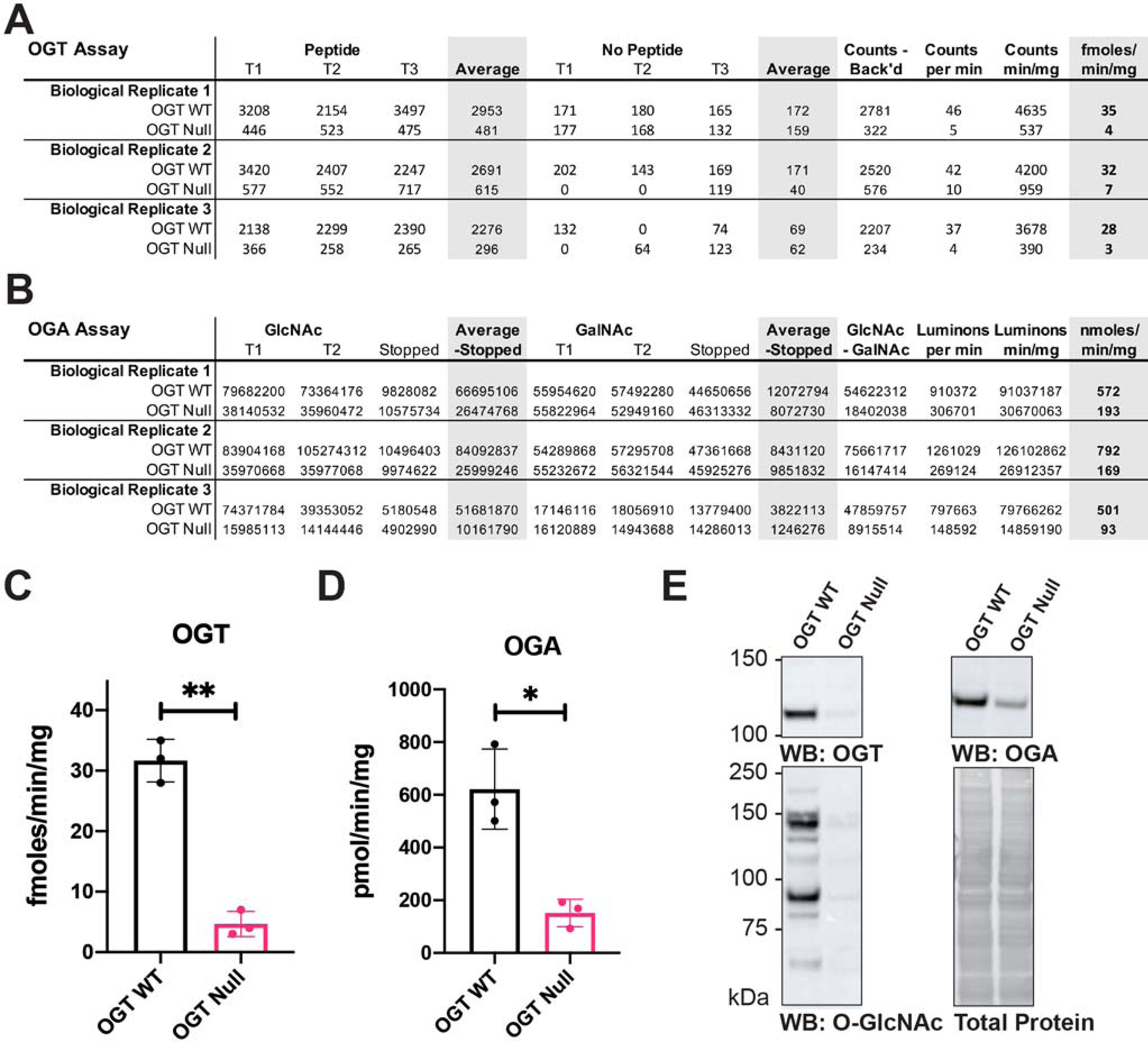
A) OGT activity in 10μg of desalted lysate from OGT Wild-type (WT) and null cells was assayed in triplicate in the presence and absence of CK2 substrate peptide. The incorporation of 3H-GlcNAc was assessed using liquid scintillation counting (DPM). Activity was subsequently calculated using 10μg of protein, an assay length of 60 min, and 7.54fmol of GlcNAc per 1000 DPM. B) OGA activity in 10μg of desalted lysate from OGT Wild-type (WT) and null cells was assayed in triplicate using the pseudosubstrates 4MU-β-GlcNAc and 4MU-β-GalNAc. One replicate was stopped using glycine at time=0 (Stopped). 4MU released was assessed in a fluorescent plate reader and quantified using a 4MU standard curve. Activity was subsequently calculated using 10μg of protein, an assay length of 60 min, and 159222 luminons per nmol (experimentally derived standard curve). C) Plotted OGT activity assays. D) Plotted OGA activity assays; E. Lysates from OGT WT and Null cells (10μg) were separated by SDS-PAGE and electroblotted to nitrocellulose. The following were detected using a GE Healthcare AI600 RGB: Total Protein (Sypro™ Ruby Protein Blot stain), O-GlcNAc (CTD110.6), OGT (Sigma, DM17), and OGA (345; (Butkinaree et al., 2008)). Error bars represent one Standard Deviation. P-values are the result of a student’s T-test (2 tailed). * p<0.05, **p<0.01.
Notably, a low signal to noise suggests that the OGT assay buffer has expired or that the assay length or protein amount should be increased. Initial experiments should assess linear range, by altering protein amounts present in the assay. In this example, and as we expect, OGT activity is depressed in the OGT null.
With regard to OGA assays:
The reaction stopped at T=0min is used to assess the spontaneous breakdown of 4MU-GlcNAc/GalNAc and is background subtracted from the average of the duplicates;
4MU-GalNAc is used to assess activity from lysosomal hexosaminidases. As such, to assess OGA activity, the averaged 4MU-GalNAc signal is background subtracted from the 4MU-GlcNAc signal.
Finally, activity is expressed in nmoles/min/mg by dividing the luminons by the assay length, adjusting for protein (here, multiplying by 100 as 10μg of lysate was used), and then converted to nmoles using an experimentally derived standard curve.
In this example, and as we expect, OGA activity is depressed in the OGT null. The importance of using the 4MU-GalNAc pseudo-substrate is highlighted in this example. First, readers will note that deletion of OGT which impacts OGA abundance only significantly alters total luminons in the 4MU-GlcNAc samples. Secondly, readers will note that in spite of the GalNAc included in the assay, there is a significant difference between the 4MU-GalNAc replicates and the time=0 sample. Like OGT assays, readers should ensure that they are within the linear range.
Time Considerations
Detection of O-GlcNAc proteins using antibodies (see Basic Protocol 2) and lectins (see Basic Protocol 3) will take ~2 to 3 days after the extraction of the proteins from cells. Samples and controls must be treated with PNGase F and/or hexosaminidase (1 hr to overnight), before SDS-PAGE and blotting. In either case, overnight incubation at 4°C provides the best signal-to-noise ratio.
WGA affinity chromatography of low-copy-number proteins will take 2 to 3 days. The TNT® protein expression and WGA affinity chromatography can be completed in 1 day; subsequent analysis of the product by SDS-PAGE will take 1 to 2 days depending on the label used and the amount of label incorporated into proteins eluting from the WGA-agarose.
Autogalactosylation of the galactosyltransferase and subsequent analysis of the activity will take 1 to 2 days. As the enzyme is stable for 6 to 12 months at –20°C, autogalactosylation does not need to be repeated for each analysis. Galactosyltransferase labeling of the proteins can take several hours to overnight, though optimization of the conditions may take a few days. The subsequent analysis, as well as desalting (dependent on the technique used), PNGase F digestion (1 hr to overnight), precipitation of protein (3 hr to overnight), SDS-PAGE, and autoradiography (1 to 10 days), can take up to 2 weeks. The length of time allotted for product analysis is dependent on the methods chosen but will almost certainly require 7 to 10 days. Further analysis, including digestion of labeled proteins and subsequent purification of peptides, will take at least 3 days.
Similar to indirect immunofluorescence methods, the O-GlcNAc In-cell western using antibodies will take 2–3 days to complete following user-defined experiments. The O-GlcNAc primary antibodies require overnight incubation for efficient binding but secondary antibody staining and analysis can be performed in a day.
OGT assays (Basic Protocol 7) are somewhat complicated and typically take 2 days to complete. Notably, after the OGT assay is stopped, it can be stored at –20°C for several weeks before desalting. O-GlcNAcase assays are straightforward and typically take 1 day.
Acknowledgments
Original research in the author’s laboratories were supported by the National Institutes of Health (K12HL141952, U01 CA230978, and RO1HL139640 to N.E.Z). G.W.Hart receives a percentage of royalties received by the university on sales of the CTD 110.6 antibody. The terms of these arrangement are in accordance with the conflict of interest policy at The Johns Hopkins University.
Abbreviations:
- 4MU
4 methylumberlliferyl
- BSA
Bovine serum albumin
- DON
diazo-5-oxonorleucine
- ECL
enhanced chemiluminescence
- Gal
Galactose
- GFAT
glutamine fructose-6-phosphate amidotransferase
- GlcNAc
N-acetylglucosamine
- HRP
Horse radish peroxidase
- HS-TBST
high salt TBST
- OGA
O-GlcNAcase
- O-GlcNAc
O-linked N-acetylglucosamine
- OGT
O-GlcNAc transferase
- PBS
Phosphate buffered saline
- PVDF
polyvinylidene difluoride
- sWGA
succinylated wheat germ agglutinin
- RCA1
Ricinus communis agglutinin 1
- RRL
Rabbit reticulocyte lysate
- STZ
Streptozotocin,
- TBS
Tris-buffered saline
- TCA
Trichloroacetic acid
- TNT
in vitro transcription translation
- WGA
wheat germ agglutinin.
Footnotes
The terms of these arrangement are in accordance with the conflict of interest policy at The Johns Hopkins University.
Literature Cited
- Amano J, and Kobata A. 1989. Quantitative conversion of mucin-type sugar chains to radioactive oligosaccharides. Methods in enzymology 179:261–270. [DOI] [PubMed] [Google Scholar]
- Banerjee S, Robbins PW, and Samuelson J. 2009. Molecular characterization of nucleocytosolic O-GlcNAc transferases of Giardia lamblia and Cryptosporidium parvum. Glycobiology 19:331–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boysen RI, and Hearn MT 2001. HPLC of peptides and proteins. Current protocols in protein science / editorial board, John E. Coligan ... [et al.] Chapter 8:Unit8.7–8.7.40. [DOI] [PubMed]
- Brew K, Vanaman TC, and Hill RL 1968. The role of alpha-lactalbumin and the A protein in lactose synthetase: a unique mechanism for the control of a biological reaction. Proceedings of the National Academy of Sciences of the United States of America 59:491–497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brister MA, Pandey AK, Bielska AA, and Zondlo NJ 2014. OGlcNAcylation and Phosphorylation Have Opposing Structural Effects in tau: Phosphothreonine Induces Particular Conformational Order. Journal of the American Chemical Society 136:3803–3816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Butkinaree C, Cheung WD, Park S, Park K, Barber M, and Hart GW 2008. Characterization of beta-N-acetylglucosaminidase cleavage by caspase-3 during apoptosis. The Journal of biological chemistry 283:23557–23566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Caldwell SA, Jackson SR, Shahriari KS, Lynch TP, Sethi G, Walker S, Vosseller K, and Reginato MJ 2010. Nutrient sensor O-GlcNAc transferase regulates breast cancer tumorigenesis through targeting of the oncogenic transcription factor FoxM1. Oncogene 29:2831–2842. [DOI] [PubMed] [Google Scholar]
- Cameron A, Giacomozzi B, Joyce J, Gray A, Graham D, Ousson S, Neny M, Beher D, Carlson G, O’Moore J, et al. 2013. Generation and characterization of a rabbit monoclonal antibody site-specific for tau O-GlcNAcylated at serine 400. FEBS letters 587:3722–3728. [DOI] [PubMed] [Google Scholar]
- Chalkley RJ, Thalhammer A, Schoepfer R, and Burlingame AL 2009. Identification of protein O-GlcNAcylation sites using electron transfer dissociation mass spectrometry on native peptides. Proceedings of the National Academy of Sciences 106:8894–8899. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou TY, Hart GW, and Dang CV 1995. c-Myc is glycosylated at threonine 58, a known phosphorylation site and a mutational hot spot in lymphomas. The Journal of biological chemistry 270:18961–18965. [DOI] [PubMed] [Google Scholar]
- Comer FI, and Hart GW 2001. Reciprocity between O-GlcNAc and O-phosphate on the carboxyl terminal domain of RNA polymerase II. Biochemistry 40:7845–7852. [DOI] [PubMed] [Google Scholar]
- Comer FI, Vosseller K, Wells L, Accavitti MA, and Hart GW 2001. Characterization of a Mouse Monoclonal Antibody Specific for O-Linked N-Acetylglucosamine. Analytical Biochemistry 293:169–177. [DOI] [PubMed] [Google Scholar]
- Dennis RJ, Taylor EJ, Macauley MS, Stubbs KA, Turkenburg JP, Hart SJ, Black GN, Vocadlo DJ, and Davies GJ 2006. Structure and mechanism of a bacterial beta-glucosaminidase having O-GlcNAcase activity. Nature Structural & Molecular Biology 13:365–371. [DOI] [PubMed] [Google Scholar]
- Dong DL, and Hart GW 1994. Purification and characterization of an O-GlcNAc selective N-acetyl-beta-D-glucosaminidase from rat spleen cytosol. The Journal of biological chemistry 269:19321–19330. [PubMed] [Google Scholar]
- Duk M, Ugorski M, and Lisowska E. 1997. beta-Elimination of O-glycans from glycoproteins transferred to immobilon P membranes: method and some applications. Analytical Biochemistry 253:98–102. [DOI] [PubMed] [Google Scholar]
- Elbaum MB, and Zondlo NJ 2014. OGlcNAcylation and Phosphorylation Have Similar Structural Effects in α-Helices: Post-Translational Modifications as Inducible Start and Stop Signals in α-Helices, with Greater Structural Effects on Threonine Modification. Biochemistry 53:2242–2260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Elling L, Zervosen A, Gallego RG, Nieder V, Malissard M, Berger EG, Vliegenthart JF, and Kamerling JP 1999. UDP-N-Acetyl-alpha-D-glucosamine as acceptor substrate of beta-1,4-galactosyltransferase. Enzymatic synthesis of UDP-N-acetyllactosamine. Glycoconjugate journal 16:327–336. [DOI] [PubMed] [Google Scholar]
- Freeze HH 2001. Lectin affinity chromatography. Current protocols in protein science / editorial board, John E. Coligan ... [et al.] Chapter 9:Unit 9.1–9.1.9. [DOI] [PubMed]
- Fukuda M. 1989. Characterization of O-linked saccharides from cell surface glycoproteins. Methods in enzymology 179:17–29. [DOI] [PubMed] [Google Scholar]
- Gallagher S. 2001. Immunoblot detection. Current protocols in protein science / editorial board, John E. Coligan ... [et al.] Chapter 10:Unit 10.10–10.10.12. [DOI] [PubMed]
- Gao Y, Parker GJ, and Hart GW 2000. Streptozotocin-induced beta-cell death is independent of its inhibition of O-GlcNAcase in pancreatic Min6 cells. Archives of biochemistry and biophysics 383:296–302. [DOI] [PubMed] [Google Scholar]
- Gao Y, Wells L, Comer FI, Parker GJ, and Hart GW 2001. Dynamic O-glycosylation of nuclear and cytosolic proteins: cloning and characterization of a neutral, cytosolic beta-N-acetylglucosaminidase from human brain. The Journal of biological chemistry 276:9838–9845. [DOI] [PubMed] [Google Scholar]
- Gloster TM, Zandberg WF, Heinonen JE, Shen DL, Deng L, and Vocadlo DJ 2011. hijacking a biosynthetic pathway yields a glycosyltransferase inhibitor within cells. Nature Chemical Biology 7:174–181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Greis KD, and Hart GW 1998. Analytical methods for the study of O-GlcNAc glycoproteins and glycopeptides. Methods in molecular biology (Clifton, N.J.) 76:19–33. [DOI] [PubMed] [Google Scholar]
- Greis KD, Hayes BK, Comer FI, Kirk M, Barnes S, Lowary TL, and Hart GW 1996. Selective detection and site-analysis of O-GlcNAc-modified glycopeptides by beta-elimination and tandem electrospray mass spectrometry. Analytical Biochemistry 234:38–49. [DOI] [PubMed] [Google Scholar]
- Gross BJ, Kraybill BC, and Walker S. 2005. Discovery of O-GlcNAc transferase inhibitors. Journal of the American Chemical Society 127:14588–14589. [DOI] [PubMed] [Google Scholar]
- Halim A, Larsen ISB, Neubert P, Joshi HJ, Petersen BL, Vakhrushev SY, Strahl S, and Clausen H. 2015. Discovery of a nucleocytoplasmic O-mannose glycoproteome in yeast. Proceedings of the National Academy of Sciences:201511743. [DOI] [PMC free article] [PubMed]
- Haltiwanger RS 1998. Modulation of O-Linked N-Acetylglucosamine Levels on Nuclear and Cytoplasmic Proteins in Vivo Using the Peptide O-GlcNAc-beta -N-acetylglucosaminidase Inhibitor O-(2-Acetamido-2-deoxy-Dglucopyranosylidene)amino-N-phenylcarbamate. Journal of Biological Chemistry 273:3611–3617. [DOI] [PubMed] [Google Scholar]
- Haltiwanger RS, Blomberg MA, and Hart GW 1992. Glycosylation of nuclear and cytoplasmic proteins. Purification and characterization of a uridine diphospho-N-acetylglucosamine:polypeptide beta-N-acetylglucosaminyltransferase. The Journal of biological chemistry 267:9005–9013. [PubMed] [Google Scholar]
- Haltiwanger RS, Holt GD, and Hart GW 1990. Enzymatic addition of O-GlcNAc to nuclear and cytoplasmic proteins. Identification of a uridine diphospho-N-acetylglucosamine:peptide beta-N-acetylglucosaminyltransferase. The Journal of biological chemistry 265:2563–2568. [PubMed] [Google Scholar]
- Han C, Gu Y, Shan H, Mi W, Sun J, Shi M, Zhang X, Lu X, Han F, Gong Q, et al. 2017. O-GlcNAcylation of SIRT1 enhances its deacetylase activity and promotes cytoprotection under stress. Nature Communications 8:1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Han I, Oh ES, and Kudlow JE 2000. Responsiveness of the state of O-linked N-acetylglucosamine modification of nuclear pore protein p62 to the extracellular glucose concentration. Biochemical Journal 350 Pt 1:109–114. [PMC free article] [PubMed] [Google Scholar]
- Hardy MR, and Townsend RR 1994. High-pH anion-exchange chromatography of glycoprotein-derived carbohydrates. Methods in enzymology 230:208–225. [DOI] [PubMed] [Google Scholar]
- Hart GW, Haltiwanger RS, Holt GD, and Kelly WG 1989. Glycosylation in the nucleus and cytoplasm. Annual review of biochemistry 58:841–874. [DOI] [PubMed] [Google Scholar]
- Hart GW, Slawson C, Ramirez-Correa G, and Lagerlof O. 2011. Cross talk between O-GlcNAcylation and phosphorylation: roles in signaling, transcription, and chronic disease. Annual review of biochemistry 80:825–858. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hayes BK, Greis KD, and Hart GW 1995. Specific isolation of O-linked N-acetylglucosamine glycopeptides from complex mixtures. Analytical Biochemistry 228:115–122. [DOI] [PubMed] [Google Scholar]
- Heart E, Choi WS, and Sung CK 2000. Glucosamine-induced insulin resistance in 3T3-L1 adipocytes. American journal of physiology. Endocrinology and metabolism 278:E103–12. [DOI] [PubMed] [Google Scholar]
- Holt GD, and Hart GW 1986. The subcellular distribution of terminal N-acetylglucosamine moieties. Localization of a novel protein-saccharide linkage, O-linked GlcNAc. The Journal of biological chemistry 261:8049–8057. [PubMed] [Google Scholar]
- Holt GD, Snow CM, Senior A, Haltiwanger RS, Gerace L, and Hart GW 1987. Nuclear pore complex glycoproteins contain cytoplasmically disposed O-linked N-acetylglucosamine. The Journal of Cell Biology 104:1157–1164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Immunoprecipitation. 2001. Immunoprecipitation. Current protocols in protein science / editorial board, John E. Coligan ... [et al.] Chapter 9:Unit 9.8. [DOI] [PubMed]
- Isono T. 2011. O-GlcNAc-Specific Antibody CTD110.6 Cross-Reacts with N-GlcNAc2-Modified Proteins Induced under Glucose Deprivation. PLoS ONE 6:e18959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Izumi T, and Suzuki K. 1983. Neutral beta-N-acetylhexosaminidases of rat brain. Purification and enzymatic and immunological characterization. The Journal of biological chemistry 258:6991–6999. [PubMed] [Google Scholar]
- Jiang J, Lazarus MB, Pasquina L, Sliz P, and Walker S. 2011. A neutral diphosphate mimic crosslinks the active site of human O-GlcNAc transferase. Nature Publishing Group 8:72–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamemura K, Hayes BK, Comer FI, and Hart GW 2002. Dynamic interplay between O-glycosylation and O-phosphorylation of nucleocytoplasmic proteins: alternative glycosylation/phosphorylation of THR-58, a known mutational hot spot of c-Myc in lymphomas, is regulated by mitogens. The Journal of biological chemistry 277:19229–19235. [DOI] [PubMed] [Google Scholar]
- Kazemi Z, Chang H, Haserodt S, McKen C, and Zachara NE 2010. O-Linked -N-acetylglucosamine (O-GlcNAc) Regulates Stress-induced Heat Shock Protein Expression in a GSK-3 -dependent Manner. Journal of Biological Chemistry 285:39096–39107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khidekel N, Ficarro SB, Clark PM, Bryan MC, Swaney DL, Rexach JE, Sun YE, Coon JJ, Peters EC, and Hsieh-Wilson LC 2007. Probing the dynamics of O-GlcNAc glycosylation in the brain using quantitative proteomics. Nature Chemical Biology 3:339–348. [DOI] [PubMed] [Google Scholar]
- Kim EJ 2018. Chemical Reporters and Their Bioorthogonal Reactions for Labeling Protein O-GlcNAcylation. Molecules (Basel, Switzerland) 23:2411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EJ, Kang DO, Love DC, and Hanover JA 2006. Enzymatic characterization of O-GlcNAcase isoforms using a fluorogenic GlcNAc substrate. Carbohydrate research 341:971–982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kobata A. 1994. Size fractionation of oligosaccharides. Methods in enzymology 230:200–208. [DOI] [PubMed] [Google Scholar]
- Kreppel LK, and Hart GW 1999. Regulation of a cytosolic and nuclear O-GlcNAc transferase. Role of the tetratricopeptide repeats. The Journal of biological chemistry 274:32015–32022. [DOI] [PubMed] [Google Scholar]
- Kreppel LK, Blomberg MA, and Hart GW 1997. Dynamic glycosylation of nuclear and cytosolic proteins. Cloning and characterization of a unique O-GlcNAc transferase with multiple tetratricopeptide repeats. The Journal of biological chemistry 272:9308–9315. [DOI] [PubMed] [Google Scholar]
- Lee A, Miller D, Henry R, Paruchuri VDP, O’Meally RN, Boronina T, Cole RN, and Zachara NE 2016. Combined Antibody/Lectin Enrichment Identifies Extensive Changes in the O-GlcNAc Sub-proteome upon Oxidative Stress. Journal of Proteome Research 15:4318–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Levine PM, Galesic A, Balana AT, Mahul-Mellier A-L, Navarro MX, De Leon CA, Lashuel HA, and Pratt MR 2019. α-Synuclein O-GlcNAcylation alters aggregation and toxicity, revealing certain residues as potential inhibitors of Parkinson’s disease. Proceedings of the National Academy of Sciences 116:1511–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu T-W, Zandberg WF, Gloster TM, Deng L, Murray KD, Shan X, and Vocadlo DJ 2018. Metabolic Inhibitors of O-GlcNAc Transferase That Act In Vivo Implicate Decreased O-GlcNAc Levels in Leptin-Mediated Nutrient Sensing. Angewandte Chemie International Edition 57:7644–7648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lovrien RE, and Matulis D. 2001. Selective precipitation of proteins. Current protocols in protein science / editorial board, John E. Coligan ... [et al.] Chapter 4:Unit 4.5–4.5.36. [DOI] [PubMed]
- Ma Z, and Vosseller K. 2014. Cancer metabolism and elevated O-GlcNAc in oncogenic signaling. The Journal of biological chemistry 289:34457–34465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macauley MS, Whitworth GE, Debowski AW, Chin D, and Vocadlo DJ 2005. O-GlcNAcase uses substrate-assisted catalysis: kinetic analysis and development of highly selective mechanism-inspired inhibitors. The Journal of biological chemistry 280:25313–25322. [DOI] [PubMed] [Google Scholar]
- Martin SES, Tan Z-W, Itkonen HM, Duveau DY, Paulo JA, Janetzko J, Boutz PL, Törk L, Moss FA, Thomas CJ, et al. 2018. Structure-Based Evolution of Low Nanomolar O-GlcNAc Transferase Inhibitors. Journal of the American Chemical Society 140:13542–13545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matsuoka Y, Matsuoka Y, Shibata S, Yasuhara N, and Yoneda Y. 2002. Identification of Ewing’s sarcoma gene product as a glycoprotein using a monoclonal antibody that recognizes an immunodeterminant containing O-linked N-acetylglucosamine moiety. Hybridoma and hybridomics 21:233–236. [DOI] [PubMed] [Google Scholar]
- Medina L, Grove K, and Haltiwanger RS 1998. SV40 large T antigen is modified with O-linked N-acetylglucosamine but not with other forms of glycosylation. Glycobiology 8:383–391. [DOI] [PubMed] [Google Scholar]
- Monsigny M, Sene C, Obrenovitch A, Roche AC, Delmotte F, and Boschetti E. 1979. Properties of succinylated wheat-germ agglutinin. European Journal of Biochemistry 98:39–45. [DOI] [PubMed] [Google Scholar]
- Muha V, Williamson R, Hills R, McNeilly AD, McWilliams TG, Alonso J, Schimpl M, Leney AC, Heck AJR, Sutherland C, et al. 2019. Loss of CRMP2 O-GlcNAcylation leads to reduced novel object recognition performance in mice. Open Biology 9:190192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nandi A, Sprung R, Barma DK, Zhao Y, Kim SC, Falck JR, and Zhao Y. 2006. Global identification of O-GlcNAc-modified proteins. Analytical Chemistry 78:452–458. [DOI] [PubMed] [Google Scholar]
- Ngoh GA, Facundo HT, Hamid T, Dillmann W, Zachara NE, and Jones SP 2008. Unique Hexosaminidase Reduces Metabolic Survival Signal and Sensitizes Cardiac Myocytes to Hypoxia/Reoxygenation Injury. Circulation research 104:41–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ortiz-Meoz RF, Jiang J, Lazarus MB, Orman M, Janetzko J, Fan C, Duveau DY, Tan Z-W, Thomas CJ, and Walker S. 2015. A Small Molecule That Inhibits OGT Activity in Cells. ACS Chemical Biology 10:1392–1397. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park S-K, Zhou X, Pendleton KE, Hunter OV, Kohler JJ, O’Donnell KA, and Conrad NK 2017. A Conserved Splicing Silencer Dynamically Regulates O-GlcNAc Transferase Intron Retention and O-GlcNAc Homeostasis. Cell reports 20:1088–1099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Powell LD 2001. Inhibition of N-linked glycosylation. Current protocols in protein science / editorial board, John E. Coligan ... [et al.] Chapter 12:Unit 12.3–12.3.9. [DOI] [PubMed]
- Rao X, Duan X, Mao W, Li X, Li Z, Li Q, Zheng Z, Xu H, Chen M, Wang PG, et al. 2015. O-GlcNAcylation of G6PD promotes the pentose phosphate pathway and tumor growth. Nature Communications 6:8468–8477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reeves RA, Lee A, Henry R, and Zachara NE 2014. Characterization of the specificity of O-GlcNAc reactive antibodies under conditions of starvation and stress. Analytical Biochemistry 457:8–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roos MD, Xie W, Su K, Clark JA, Yang X, Chin E, Paterson AJ, and Kudlow JE 1998. Streptozotocin, an analog of N-acetylglucosamine, blocks the removal of O-GlcNAc from intracellular proteins. Proceedings of the Association of American Physicians 110:422–432. [PubMed] [Google Scholar]
- Roquemore EP, Chou TY, and Hart GW 1994. Detection of O-linked N-acetylglucosamine (O-GlcNAc) on cytoplasmic and nuclear proteins. Methods in enzymology 230:443–460. [DOI] [PubMed] [Google Scholar]
- Schindler M, Hogan M, Miller R, and DeGaetano D. 1987. A nuclear specific glycoprotein representative of a unique pattern of glycosylation. The Journal of biological chemistry 262:1254–1260. [PubMed] [Google Scholar]
- Schnedl WJ, Ferber S, Johnson JH, and Newgard CB 1994. STZ transport and cytotoxicity. Specific enhancement in GLUT2-expressing cells. Diabetes 43:1326–1333. [DOI] [PubMed] [Google Scholar]
- Shan H, Sun J, Shi M, Liu X, Shi Z, Yu W, and Gu Y. 2018. Generation and characterization of a site-specific antibody for SIRT1 O-GlcNAcylated at serine 549. Glycobiology 28:482–487. [DOI] [PubMed] [Google Scholar]
- Shanmugasundaram B, Debowski AW, Dennis RJ, Davies GJ, Vocadlo DJ, and Vasella A. 2006. Inhibition of O-GlcNAcase by a gluco-configured nagstatin and a PUGNAc-imidazole hybrid inhibitor. Chemical communications (Cambridge, England) 0:4372–4374. [DOI] [PubMed] [Google Scholar]
- Shikhman AR, Greenspan NS, and Cunningham MW 1993. A subset of mouse monoclonal antibodies cross-reactive with cytoskeletal proteins and group A streptococcal M proteins recognizes N-acetyl-beta-D-glucosamine. Journal of immunology (Baltimore, Md. : 1950) 151:3902–3913. [PubMed] [Google Scholar]
- Shikhman AR, Greenspan NS, and Cunningham MW 1994. Cytokeratin peptide SFGSGFGGGY mimics N-acetyl-beta-D-glucosamine in reaction with antibodies and lectins, and induces in vivo anti-carbohydrate antibody response. Journal of immunology (Baltimore, Md. : 1950) 153:5593–5606. [PubMed] [Google Scholar]
- Slawson C, Zachara NE, Vosseller K, Cheung WD, Lane MD, and Hart GW 2005. Perturbations in O-linked β- N-Acetylglucosamine Protein Modification Cause Severe Defects in Mitotic Progression and Cytokinesis. The Journal of biological chemistry 280:32944–32956. [DOI] [PubMed] [Google Scholar]
- Snow CM, Senior A, and Gerace L. 1987. Monoclonal antibodies identify a group of nuclear pore complex glycoproteins. The Journal of Cell Biology 104:1143–1156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan Z-W, Fei G, Paulo JA, Bellaousov S, Martin SES, Duveau DY, Thomas CJ, Gygi SP, Boutz PL, and Walker S. 2020. O-GlcNAc regulates gene expression by controlling detained intron splicing. Nucleic Acids Research 48:5656–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tashima Y, and Stanley P. 2014. Antibodies That Detect O-Linked -D-N-Acetylglucosamine on the Extracellular Domain of Cell Surface Glycoproteins. Journal of Biological Chemistry 289:11132–11142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teo CF, Ingale S, Wolfert MA, Elsayed GA, Nöt LG, Chatham JC, Wells L, and Boons G-J 2010. glycopeptide-specific monoclonal antibodies suggest new roles for O-glcnac. Nature Methods 6:338–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Toleman CA, Schumacher MA, Yu S-H, Zeng W, Cox NJ, Smith TJ, Soderblom EJ, Wands AM, Kohler JJ, and Boyce M. 2018. Structural basis of O-GlcNAc recognition by mammalian 14–3-3 proteins. Proceedings of the National Academy of Sciences 115:5956–5961. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Torres CR, and Hart GW 1984. Topography and polypeptide distribution of terminal N-acetylglucosamine residues on the surfaces of intact lymphocytes. Evidence for O-linked GlcNAc. The Journal of biological chemistry 259:3308–3317. [PubMed] [Google Scholar]
- Townsend RR, and Hardy MR 1991. Analysis of glycoprotein oligosaccharides using high-pH anion exchange chromatography. Glycobiology 1:139–147. [DOI] [PubMed] [Google Scholar]
- Townsend RR, Basa LJ, and Spellman MW 1996. Identification and characterization of glycopeptides in tryptic maps by high-pH anion-exchange chromatography. Methods in enzymology 271:135–147. [DOI] [PubMed] [Google Scholar]
- Townsend RR, Hardy MR, Cumming DA, Carver JP, and Bendiak B. 1989. Separation of branched sialylated oligosaccharides using high-pH anion-exchange chromatography with pulsed amperometric detection. Analytical Biochemistry 182:1–8. [DOI] [PubMed] [Google Scholar]
- Trinidad JC, Barkan DT, Gulledge BF, Thalhammer A, Sali A, Schoepfer R, and Burlingame AL 2012. Global identification and characterization of both O-GlcNAcylation and phosphorylation at the murine synapse. Molecular & Cellular Proteomics 11:215–229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turner JR, Tartakoff AM, and Greenspan NS 1990. Cytologic assessment of nuclear and cytoplasmic O-linked N-acetylglucosamine distribution by using anti-streptococcal monoclonal antibodies. Proceedings of the National Academy of Sciences of the United States of America 87:5608–5612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vosseller K, Trinidad JC, Chalkley RJ, Specht CG, Thalhammer A, Lynn AJ, Snedecor JO, Guan S, Medzihradszky KF, Maltby DA, et al. 2006. O-linked N-acetylglucosamine proteomics of postsynaptic density preparations using lectin weak affinity chromatography and mass spectrometry. Molecular & cellular proteomics : MCP 5:923–934. [DOI] [PubMed] [Google Scholar]
- Wells L, Kreppel LK, Comer FI, Wadzinski BE, and Hart GW 2004. O-GlcNAc transferase is in a functional complex with protein phosphatase 1 catalytic subunits. The Journal of biological chemistry 279:38466–38470. [DOI] [PubMed] [Google Scholar]
- Whelan SA, Lane MD, and Hart GW 2008. Regulation of the O-linked beta-N-acetylglucosamine transferase by insulin signaling. The Journal of biological chemistry 283:21411–21417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu S-L, Chalkley RJ, Wang Z-Y, and Burlingame AL 2012. Identification of O-linked β-D-N-acetylglucosamine-modified proteins from Arabidopsis. Methods in molecular biology (Clifton, N.J.) 876:33–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi W, Clark PM, Mason DE, Keenan MC, Hill C, Goddard WA, Peters EC, Driggers EM, and Hsieh-Wilson LC 2012. Phosphofructokinase 1 Glycosylation Regulates Cell Growth and Metabolism. Science 337:975–980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuzwa SA, Macauley MS, Heinonen JE, Shan X, Dennis RJ, He Y, Whitworth GE, Stubbs KA, McEachern EJ, Davies GJ, et al. 2008. A potent mechanism-inspired O-GlcNAcase inhibitor that blocks phosphorylation of tau in vivo. Nature Chemical Biology 4:483–490. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Shan X, Macauley MS, Clark T, Skorobogatko Y, Vosseller K, and Vocadlo DJ 2012. Increasing O-GlcNAc slows neurodegeneration and stabilizes tau against aggregation. Nature Chemical Biology 8:393–399. [DOI] [PubMed] [Google Scholar]
- Yuzwa SA, Yadav AK, Skorobogatko Y, Clark T, Vosseller K, and Vocadlo DJ 2011. Mapping O-GlcNAc modification sites on tau and generation of a site-specific O-GlcNAc tau antibody. Amino acids 40:857–868. [DOI] [PubMed] [Google Scholar]
- Zentella R, Sui N, Barnhill B, Hsieh W-P, Hu J, Shabanowitz J, Boyce M, Olszewski NE, Zhou P, Hunt DF, et al. 2017. The Arabidopsis O-fucosyltransferase SPINDLY activates nuclear growth repressor DELLA. Nature Publishing Group 13:479–485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Y, Shan X, Yuzwa SA, and Vocadlo DJ 2014. The emerging link between O-GlcNAc and Alzheimer disease. The Journal of biological chemistry 289:34472–34481. [DOI] [PMC free article] [PubMed] [Google Scholar]