

Abstract
Metabolism and mechanics are intrinsically intertwined. External forces, sensed through the cytoskeleton or distortion of the cell and organelles, induce metabolic changes in the cell. The resulting changes in metabolism, in turn, feedback to regulate every level of cell biology including the mechanical properties of cells and tissues. Here, we examine the links between metabolism and mechanics, highlighting signaling pathways involved in the regulation and response to cellular mechanosensing. We consider how forces and metabolism regulate one another through nanoscale molecular sensors, micron-scale cytoskeletal networks, organelles, and dynamic biomolecular condensates. Understanding this crosstalk will create diagnostic and therapeutic opportunities for metabolic disorders such as cancer, cardiovascular pathologies and obesity.
In 1910, James B. Herrick reported mechanical deformations in the red blood cells (RBCs) of a patient with severe anaemia1, a condition we now know as sickle-cell anaemia (SCA). Forty years later, Linus Pauling elucidated the mechanism of SCA, revealing the molecular causes of a disease for the first time. In their paper entitled “Sickle Cell Anaemia, a Molecular Disease2“, Pauling and colleagues determined that it was the abnormalities in haemoglobin from SCA patients that led to disruption of cellular morphology and mechanics.3 In fact, a single point mutation causes sickle cell beta-haemoglobin (Hb) to polymerize in an oxygen-dependent manner.4 These polymers can exceed the diameter of red blood cells, distorting these cells to a characteristic sickle shape (figure 1).
Figure 1.
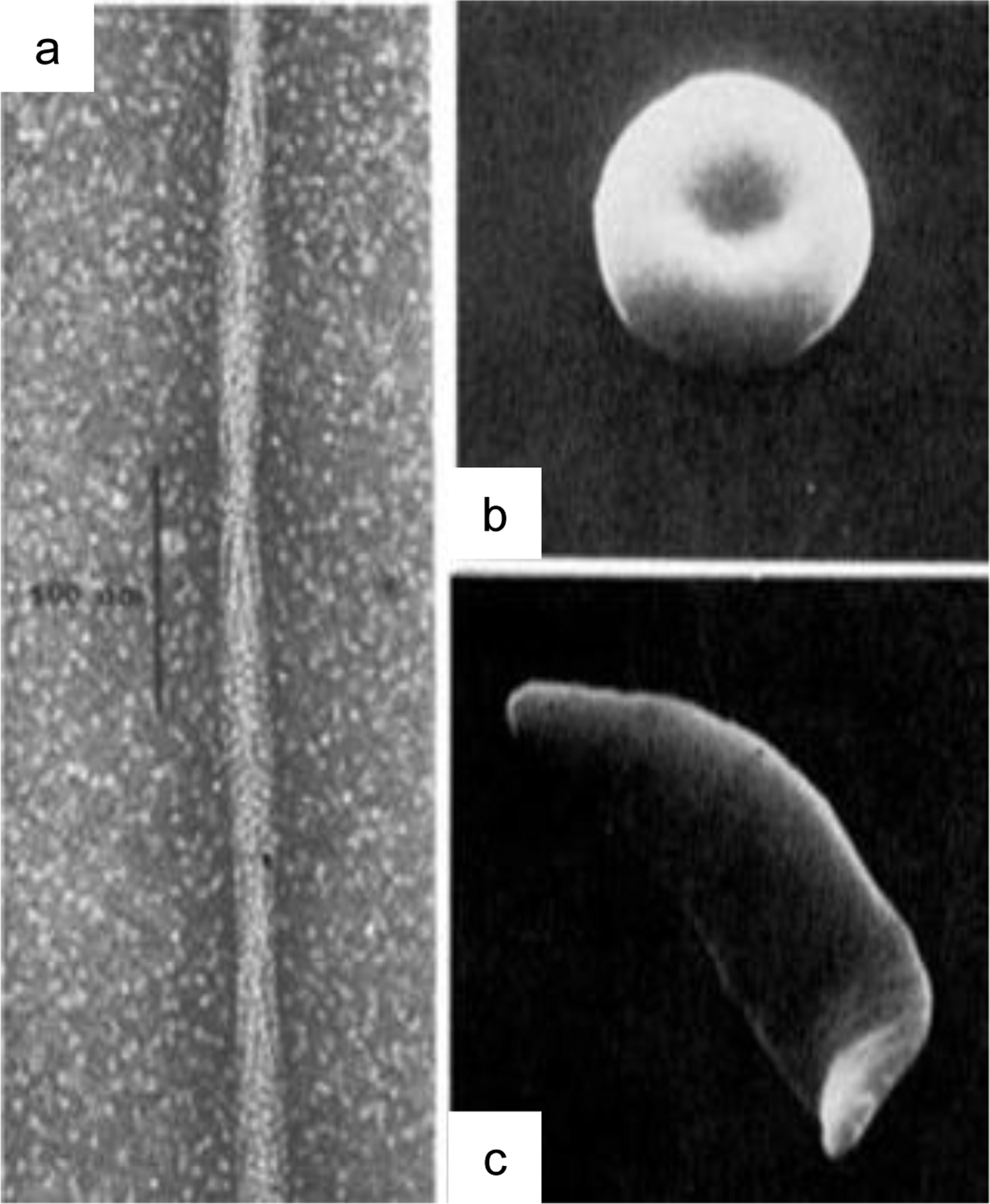
Polymerization of haemoglobin (Hb) drives sickle cell formation. (a) a single point mutation causes Hb to convert to a polymer in an oxygen and pH dependent manner. (b) normal shaped red blood cell. (c) Distortion of a red blood cell into a sickle shape. Reproduced with permission from Dykes, G., Crepeau, R. H. & Edelstein, S. J. Three-dimensional reconstruction of the fibres of sickle cell haemoglobin. Nature (1978) doi:10.1038/272506a04, and Eaton, W. A. & Hofrichter, J. Sickle Cell Hemoglobin Polymerization. Adv. Protein Chem. (1990) doi:10.1016/S0065-3233(08)60287-9.150
Amazingly, recent work has echoed these findings, showing that single mutations are sufficient to cause protein polymerization, especially if protein complexes are symmetric multimers, as is the case with many metabolic enzymes.5 Throughout the course of evolution, there have been many cases of enzymes transitioning to proteins with structural roles. For example, crystallins, the structural proteins in vertebrate eyes, derive from different enzymes in different species. Human alpha-crystallin is a small heat-shock protein, while argininosuccinate lyase has been co-opted as the delta 2 crystallin in ducks.6 Furthermore, many metabolic enzymes have been shown to polymerize in response to changing nutrient status7. Hexokinase shares a common fold with actin, and has been shown to polymerize in response to glucose and ATP.8
In light of these findings, an emerging hypothesis postulates that the modern cytoskeleton evolved from metabolic enzymes. Initially, polymerization may have served to regulate metabolic activity, but then the polymers themselves were co-opted to perform mechanical work. Thus, metabolism, polymerization, and mechanical processes could have been intrinsically connected throughout evolution. In what follows, we discuss the emerging links between cellular mechanical and metabolic processes. We organize our essay spatially, initially moving from the outside of the cell in towards the centre of the nucleus, and then in terms of scale, from the molecules to organelles to tissues. Finally, we discuss these issues in the context of disease.
Basic concepts in cellular mechanics and metabolism
The eukaryotic cytoskeleton is comprised of polymerizing proteins, including actin filaments (AF), microtubules (MT) and intermediate filaments (IF). The latter include nuclear lamins, which can dominate cellular stiffness and serve as a crucial focal point of mechanical sensing.9 To perform their functions, the cytoskeletal components integrate with multiple proteins (e.g. motor proteins) and cellular organelles. Of these, actin and tubulin are highly dynamic, and their assembly is regulated by binding to and hydrolysis of ATP and GTP respectively. Thus, actin and tubulin can be assembled (polymerization) and disassembled (depolymerization) to form different networks that organize cellular contents, connect the cell physically and biochemically to the external environment, generate coordinated forces that enable the cell to move, enable cells to adopt different shapes, and segregate chromosomes and organelles during cell division.10 The functions and properties of the cytoskeleton have been the focus of numerous excellent reviews11–13. The fundamental fact that actin and tubulin are enzymes (that hydrolyse phosphorylated nucleotides) represents an obvious coupling of mechanical and metabolic processes.
Cellular and tissue mechanics are determined by many factors beyond the cytoskeleton. At the most fundamental level, the high concentration of macromolecules found in cells creates unusual mechanical properties. Just like traffic suddenly grinding to a halt when a lane of the freeway is closed, dense colloids become non-Newtonian and highly viscous as they approach the “jamming transition”.14 Try making oobleck (2:1 corn-starch:water). The mechanical properties of this mix are liquid-like at rest, but become solid-like when sheered forcefully. Concentrated polymers can undergo gel transitions as they become entangled at high concentrations (this is why your car needs oil changes). Of course, the cell is a complex mixture of particles and polymers that all interact15. Crucially, this cellular milieu is also “active matter”; ATP consuming processes (e.g. molecular motor activity) constantly stir the cell.16 This crucial dependence upon energy-consuming processes to support the material properties of the cell highlights a fundamental biophysical connection between metabolism and mechanics. A dramatic illustration of this fact is that E. coli cells undergo a glass transition and become solid-like upon metabolic poisoning and associated macromolecular crowding.17 As discussed below, it is now thought that related phase transitions of proteins from particles to complexes have been co-opted as a central organizing principle of cell biology.
When we begin to consider the interrelationship between the interior and exterior of cells, all organisms rely on turgor pressure as a crucial determinant of mechanics. Thus, the flow of water into and out of cells, driven by active ion pumps and osmotic pressure, is key.18 Pumping ions is perhaps the one of the most energetically demanding task that cells undertake. For example, 20% of the ATP of a typical mammalian cell is used just to pump two ions: sodium and potassium.19 These monovalent ion fluxes, as well as proton gradients help control cell volume, and generate outward forces in the form of turgor pressure.20 These forces can range up to megapascals (> 100 psi); as a consequence, plants can exert sufficient force to grow through concrete.
In many organisms, the cell wall is crucial for mechanics. In this review, we focus on mammalian cells, which don’t have a cell wall, but nevertheless, the surface glycocalyx (a layer of glycoproteins and glycolipids that surrounds the cell) is a crucial determinant of the mechanical properties of cells21, and is highly responsive to metabolic cues. In addition to the glycocalyx, the plasma membrane (PM) is an important contributor to cell mechanics22 that is intrinsically linked to metabolism, as it provides a key platform for lipid biophysics to translate into metabolic cues.23 The PM is composed of a lipid bilayer that provides elastic rigidity to the cell; when a mechanical force is exerted on the membrane, either by stretching or bending, the lipid bilayer is slightly deformed, eliciting a mechanical restoring force that contributes to sculpting a stable membrane curvature.24 At the same time, the PM hosts mechanosensors (ion channels, junctional proteins and cell adhesion elements) that detect extracellular forces and initiate signaling cascades that culminate in rewiring of metabolism, as discussed below. Furthermore, the mechanical properties of metazoan tissues are determined to a large degree through the extracellular matrix (ECM). Importantly, the ECM has also been shown to regulate metabolic processes, such as glycolysis, as discussed below.25
Over the last two decades, research into metabolism has been reinvigorated, and the mechanisms by which metabolites regulate myriad cellular process have been slowly elucidated.26 For the purpose of exposition, we focus here on the mTORC1 and AMPK kinases, both essential energy sensors that have been frequently linked to mechanical processes. mTORC1 activation alters the phosphorylation state of many downstream targets to coordinate the transition from a resting state to growth by promoting anabolic processes, including protein27, lipid28 and nucleotide synthesis29, and by suppressing catabolic processes such as proteasome-dependent proteolysis and autophagy.30 Disturbances in any of these metabolic signaling pathways have been implicated in diseases such as metabolic syndrome,31 cancer32 and neurodevelopmental disorders.33
Whereas mTORC1 shifts the metabolic balance towards anabolism, AMP-activated protein kinase (AMPK) does the reverse.34 By phosphorylating downstream targets, AMPK inhibits energy-consuming, growth-promoting pathways and promotes catabolism of fatty acids and other bioenergetic fuels.35 Additionally, AMPK has been shown to regulate other cellular processes including autophagy, mitochondrial homeostasis and cell polarity.34,36
Mechanics and metabolism from the outside in
Junctional mechanics and regulation of metabolism
The outermost detection of cell-cell and cell-matrix forces is through cell surface protein assemblies such as cadherins at cell–cell junctions37, and integrins in large dynamic protein structures called focal adhesion complexes, which link the cell to the ECM. Upon activation, both integrins and cadherins form clusters and recruit a repertoire of proteins to initiate signaling cascades that culminate in the intracellular activation of the RhoA GTPase pathway (figure 2). RhoA modulates actin cytoskeleton remodelling, intracellular contractile forces and growth of the associated adhesion complex, allowing a cell that is under tension to respond to extracellular forces.38
Figure 2.
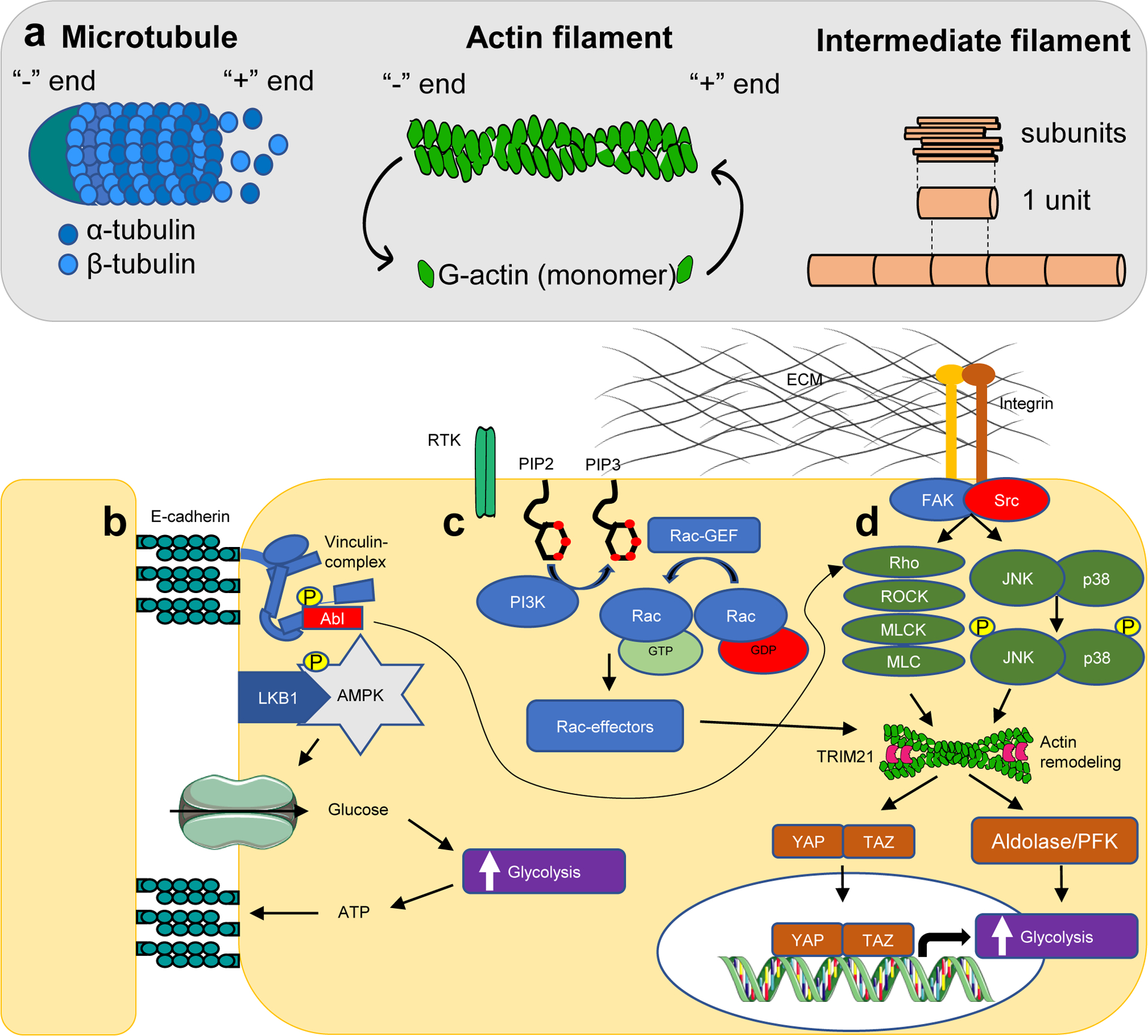
Intracellular mechanosensation pathways that accelerate glycolysis. (a) Cytoskeleton filaments and their dynamics. (b) Cell-cell adhesion forces are sensed through E-cadherin, which forms a complex with and activates liver kinase B1 (LKB1) as well as AMPK. AMPK and the membrane-cytoskeletal protein vinculin are phosphorylated by LKB1 and tyrosine-protein kinase Abl, respectively. Vinculin activity enhances actin remodelling through the Rho/ROCK pathway, whereas AMPK promotes glucose uptake and ATP production in order to maintain energy supply to adhesions. (c) Stimulation of receptor tyrosine kinases by growth factors activates PI3K, resulting in generation of PIP3, and recruitment and activation of GTP-Rac, which promotes actin fibre remodelling. (d) Cell-matrix forces are sensed through integrins in focal adhesion complexes, which promote actin remodelling through Rho/ROCK and JNK/p38 signaling. Actin fibre remodelling, integrates all three signaling pathways, and enhances transcriptional activity of YAP/TAZ as well as release of glycolytic enzymes in the cytosol. The cytoskeleton also sequesters TRIM21 from the cytosol, preventing proteasomal-mediated degradation of PFK. These processes culminate in acceleration of glucose metabolism.
FAK, focal adhesion kinase; MLC, myosin light chain; MLCK, myosin light-chain kinase; p38, P38 mitogen-activated protein kinase; PIP2, phosphatidylinositol 4,5-bisphosphate; PIP3, phosphatidylinositol (3,4,5)-trisphosphate; ROCK, Rho-associated kinase; RTK, receptor tyrosine kinase; Src, proto-oncogene tyrosine-protein kinase.
Recent studies have uncovered reciprocal regulation between integrins and critical controllers of cell metabolism, including mTORC1 and AMPK. In general, integrins control cell metabolism by ligand-induced modulation of the PI3K-Akt-mTORC1 signaling cascade, resulting in the upregulation of many metabolic pathways. Ligand-engaged integrins undergo internalization via the endo/exocytic pathway before being recycled back to the plasma membrane. For example, internalization and recycling of the α5β1 integrin, controlled by the Arf4 GTPase, was shown to be regulated by amino acid availability in an mTORC1-dependent manner.39 In turn, Arf4-mediated integrin internalization is required for mTORC1 lysosomal recruitment and activation, indicating the two-way regulatory interaction between mTORC1 and integrin trafficking. In addition, growth factor starvation in human mammary epithelial cells induces upregulation and endocytosis of β4-integrin along with its matrix substrate, laminin.40 Internalized laminin is degraded in lysosomes, resulting in increased intracellular levels of essential amino acids required for mTORC1 signaling and cell survival. In contrast, high nutrient conditions inhibit integrin activity through AMPK-dependent suppression of the integrin binding protein tensin.41 Loss of AMPK results in enhanced integrin activity, adhesion complex formation, and cell spreading, and enables cells to generate higher traction forces and increase fibronectin fibre formation.
Integrin signaling further interacts with metabolism by coupling mechanics to transcription. Forces are transduced to the nucleus through cytoskeletal connections to DNA via the LINC complex and nuclear lamina (see below)42 or via activation of mechanosensors in the cytosol.43 Either way, the transmission is facilitated by remodelling of the cytoskeleton, resulting in activation of transcription factors that regulate cell-type specific gene expression programs (figure 2).44
The cadherins are another crucial family of cell surface adhesion molecule that relay mechanical information and feed into metabolic decisions. Application of force directly to E-cadherin junctions between epithelial cells was shown to stimulate liver kinase B1 (LKB1) to activate AMPK.45 This in turn stimulates actomyosin contractility, glucose uptake and ATP production (see figure 2). The increase in ATP provides energy to reinforce the adhesion complex and actin cytoskeleton so that the cell can resist forces, which is required for efficient formation of an epithelial barrier.
Cytoskeletal regulation of metabolism
Many metabolic enzymes are known to interact with cytoskeletal proteins46 (e.g. F-actin, tubulin), but only recently have studies begun to unravel their role linking metabolic activity to cytoskeletal organization. The dynamic interactions of metabolic enzymes with microtubules and actin filaments supports the localization of these enzymes to areas of the cell with specific metabolic needs, allowing the cytoskeleton to respond to metabolic activity and ensure ATP production at cellular localities where demand is high. For instance, lung epithelial cells show locally increased actin cytoskeleton assembly and increased levels of phosphofructokinase (PFK), one of the key regulatory enzymes in glycolysis, on stiffer substrates47. PFK shows high-affinity for F-actin (linear polymers of globular actin subunits that occur as microfilaments in the cytoskeleton) and thus may provide a localized source of glycolytically derived energy. In addition, TRIM21, an E3 ubiquitin ligase, that regulates PFK degradation, is sequestered by F-actin rendering it inactive thus increasing PFK activity (figure 2). In this way, actin polymerization drives local ATP production to support cytoskeletal remodelling during mechanical stress.
In addition to its function as glycolytic enzyme, alpha-enolase constitutes a receptor for plasminogen on the cell surface, facilitating metastatic cancer invasion by promoting plasminogen activation into plasmin, a serine-protease involved in ECM degradation. In the presence of alpha-enolase, actin assembled into filaments localized closely to the cell surface. Silencing of alpha-enolase was shown to inhibit adhesion, invasion, and metastasis of pancreatic cancer cells, due to alterations in actin cytoskeleton organization, adhesion proteins, and integrin expression.48 Cancer cells having silenced alpha-enolase showed reduced actin filament organization, and actin relocalised adjacent to the nucleus, suggesting a profound and reorganization of the actin cytoskeleton by alpha-enolase.
Full activation of glycolysis by the phosphoinositide 3-kinase (PI3K)-pathway requires downstream activation of the serine/threonine kinase Akt as well as Rac-dependent actin remodelling (figure 2).49 Binding of insulin or growth factors to their receptors on the cell membrane activates PI3K resulting in generation of phosphatidylinositol-3,4,5-trisphosphate (PIP3) and recruitment and activation of GTP-Rac. GTP-Rac promotes actin fibre disassembly, causing the release of actin filament-associated metabolic enzyme aldolase, which catalyses the conversion of 6-carbon fructose molecules to the 3-carbon molecules glyceraldehyde and dihydroxyacetone, ultimately accelerating glycolysis (figure 2).49 Moreover, in response to stiff ECM, the small GTPase Rac controls the activity of YAP/TAZ50, crucial mechanosensors that are discussed below.
Although major emphasis has been placed on the interaction of actin with metabolic enzymes, a recent study suggests an important role for tubulin. Mechanical cues were shown to precisely coordinate glutamine metabolism with microtubule (MT) glutamylation, which modulates MT lattice stability to adjust the stiffness of the cytoskeleton and thereby adapt cell mechanics to the mechanical load of the environment.51 The metabolic enzyme tubulin glutamylase TTLL4 was identified as the major mediator of tubulin glutamylation in response to mechanical cues and directly interacts with microtubules by binding to the C-terminal tubulin tails.52 Depletion of TTLL4 enhanced MT dynamics, cell compliance and contractility, thereby impacting cell spreading, proliferation and migration, providing a clear link between metabolism and mechanics through MT dynamics.51
Overall, these studies demonstrate that the redistribution of metabolic enzymes in response to mechanical and/or metabolic signaling achieves rapid coordination of cytoskeletal dynamics and metabolic fluxes, while avoiding the time- and energy-consuming signaling cascades, transcriptional activation and biosynthesis of new enzyme molecules.
YAP/TAZ at the crossroad of mechanics and metabolism
Some of the best described integrators of mechanical and metabolic information are YAP and TAZ, which act as transcriptional co-activators for the TEA domain transcription factor (TEAD) family of transcription factors and play a key role in the transduction of mechanical signals from actin to the nucleus (figure 2).43,53 YAP/TAZ are effectors of the Hippo pathway, a signaling pathway that controls organ size in animals through the regulation of cell proliferation and apoptosis, and are regulated by several upstream kinases including MST1/2 and LATS1/2. MST1/2 are activated to phosphorylate and activate LATS1/2, which, in turn, phosphorylate YAP/TAZ promoting their degradation through the ubiquitin-proteasome system. When Hippo signaling is suppressed, YAP and TAZ are dephosphorylated leading to their stabilization and translocation to the nucleus, thus initiating transcriptional programs.
It is still unknown how mechanical signals are transmitted to the Hippo pathway. MST1/2 and LATS1/2 can bind to or colocalize with F-actin, suggesting that these kinases monitor cytoskeletal integrity.54 Mechanical signals may also directly affect LATS activity. In addition, Hippo-independent YAP/TAZ signaling occurs in many cellular contexts, such as cell migration and expansion55,56. Nuclear relay of mechanical signals through YAP/TAZ requires Rho GTPase activity and tension of the actomyosin cytoskeleton, but is independent of the Hippo/LATS cascade.57 In contact-inhibited cells (i.e. low mechanical stress), YAP/TAZ are inhibited by F-actin-capping and severing proteins (e.g. CapZ, Cofilin) resulting in diminished YAP/TAZ nuclear localization, transcriptional activity, and proliferation.58 This suggests that YAP/TAZ inhibition by cell mechanics entails a remodelling of the F-actin cytoskeleton distinct from LATS-induced YAP/TAZ inhibition, which requires a mechanically competent cytoskeleton. Direct physicochemical regulation of YAP/TAZ may occur as changes in molecular crowding favour liquid-liquid phase separation of YAP/TAZ59,60 (see below), and cellular mechanics (including cell spreading on stiff substrates61 or mechanical compression62) both drive changes in intracellular molecular crowding.
YAP and TAZ have been shown to couple mechanics to metabolic changes in multiple contexts. YAP/TAZ nuclear localization and activity is proportional to ECM stiffness, but is also regulated by multitude of other mechanical signals, such as cell-cell contact and cell geometry. In the context of pulmonary vascular stiffening, YAP/TAZ induces the metabolic enzymes glutaminase 1 (GLS1), pyruvate carboxylase (PC), and lactate dehydrogenase (LDHA) to promote glycolysis and glutaminolysis, thereby driving cell proliferation and providing energy during migration.63 Similarly, matrix stiffening activates YAP/TAZ to induce LDHA, GLUT1, and hexokinase II, which enhance aerobic glycolysis during the migration of hepatocellular carcinoma cells.64 Glycolysis can in turn sustain YAP/TAZ transcriptional activity by facilitating the interaction of YAP/TAZ with transcriptional co-activators, such as PFK1, which binds the YAP/TAZ TEADs and stabilizes their interaction with YAP/TAZ, thereby further reinforcing YAP/TAZ activation on a stiff ECM.65
YAP/TAZ mechanosignaling has also been found to regulate metabolism in both cancer cells and cancer-associated fibroblasts (CAFs) by increasing glutamine metabolism in response to a stiff ECM.66 It was shown that YAP/TAZ upregulates glutaminase to generate glutamate from glutamine, as well as the aspartate/glutamate SLC1A3 transporter, to exchange amino acids between cancer cells and CAFs. In this case, the cancer cells export glutamate in exchange for uptake of CAF-derived aspartate to fuel precursors for nucleotide synthesis, whereas CAFs import cancer cell-derived glutamate in order to sustain their ECM remodelling activity, exporting aspartate. This YAP/TAZ-dependent glutamate/aspartate crosstalk highlights an intercellular metabolic network in the tumour niche.
Recent evidence also indicates a role for YAP/TAZ in coupling of mechanical cues to autophagy; a process that can promote cell survival by generating important metabolites through recycling of macromolecules during conditions of metabolic stress.67 Cells plated at high densities are subject to contact-inhibition of proliferation, a fundamental property whereby cells cease proliferation and cell division upon reaching confluence.68 In these cells, YAP/TAZ fail to co-transcriptionally regulate the expression of actomyosin genes, resulting in the loss of F-actin stress fibres, which impairs autophagosome formation.69
Together, these findings unravel a complex interplay between YAP/TAZ and metabolism, in which YAP/TAZ actively coordinate intracellular metabolic programs by regulating expression of metabolic enzymes and nutrient transporters, whereas, in turn, YAP/TAZ activity is affected by metabolic cues, such as glucose, and metabolites with signaling activity. However, details remain to be elucidated and an integrated molecular understanding of YAP/TAZ, as central effectors of mechanotransduction being called into action via both Hippo pathway-dependent and -independent routes, is an important black box to elucidate in mechanobiology.
Organelle scale integration of mechanics and metabolism
Above we discuss molecular scale regulation, but cells also couple larger-scale, more global mechanical stresses to transcription. Changes in cell shape translate to altered membrane tension, which is coupled to ion fluxes through stretch-activated channels such as Piezo both on the plasma membrane and the ER.70,71 It was recently found that some force-activated transcription programs critically depend on nuclear shape.72 Force transmission leads to nuclear flattening, which stretches nuclear pores, reduces their mechanical resistance to molecular transport, and increases transcription factor nuclear import.55 Alterations to nuclear morphology also induce changes in the spatial organization of chromosomes, promoting their intermingling, and cells under tension reveal increased transcription of genes at the intermingling regions.73
The Golgi apparatus is also mechanosensitive and capable of transducing mechanical forces into biochemical signaling.74 Actomyosin relaxation, either by plating cells on soft substrates or by inhibiting the activity of myosins, inhibits lipin-1 activity, a phosphatidic acid phosphatase (PAP) in the Golgi that converts phosphatidic acid (PA) to diacylglycerol (DAG). Decreased DAG content in the Golgi membrane limits trafficking between the Golgi and endoplasmic reticulum, promoting the synthesis and accumulation of SREBPs in the Golgi. Protease-mediated cleavage of SREBP facilitates their translocation to the nucleus where they induce a transcription program leading to lipid synthesis.
Mitochondria, the key metabolic organelles in human cells, are structurally regulated by physical forces and the cytoskeleton.75,76 Very recently, it was found that cytoskeletal tension can induce a specific mitochondrial stress response, termed mitohormesis, which serves to activate oxidative stress resilience (OxSR) via solute carrier family 9 member A1 (SLC9A1) and (heat shock factor 1) HSF1.77 Mechanosensation by SLC9A1 and HSF1 was shown to stimulate mitochondrial reorganization (fragments with toroidal shapes) and metabolic reprogramming by increasing mitochondrial protein turnover and limiting respiration. These data indicate that cytoskeletal tension regulates mitochondrial function and suggests that mechanical forces influence tissue behaviour by modulating mitochondrial metabolism.
Linking metabolism and mechanics via phase separation
An important “organelle scale” coupling between mechanics and metabolism occurs through the biophysical process of phase separation, the process by which molecules with distinct chemical properties spontaneously demix. This physical organization of proteins and protein complexes into membrane-less compartments has received increasing attention over the last decade.78 Phase separation of cytoplasmic or nuclear macromolecular components results in the formation of condensates including gels, glasses (see E. coli above) and viscous lipid droplets. Here we focus on liquid-liquid phase separation, which is analogous to the separation of oil and water in a salad dressing.79 The interactions driving phase separation are often weak and distributed (e.g. electrostatic or dipole-dipole) and often mediated by intrinsically disordered proteins/regions (IDPs/IDRs) or other multivalent interactions. Importantly, when condensates assemble, they enrich certain molecules, while they exclude others.80,81 This allows condensates to dynamically organize and regulate myriad biochemical reactions.
Phase separation can dramatically affect molecular dynamics in cells, but can also perform mechanical work. Synthetic optogenetic platforms that use light to manipulate matter inside living cells, have recently been leveraged to elucidate how proteins assemble into different liquid and solid-like states.82,83 A CRISPR/Cas9-based optogenetic technology, CasDrop, revealed that growing nuclear condensates can mechanically pull genomic loci together, while excluding background chromatin. These condensates grow within softer genomic regions, due to lower mechanical energy cost of droplets deforming low-density genomic regions. Additionally, it has previously been shown that an elastic nuclear F-actin scaffold mechanically stabilizes ribonucleoprotein droplets (nucleoli and histone locus condensates) against gravitational forces in large cells.84 These studies begin to establish a link between intracellular mechanics and condensate formation.
In addition to the polymerization of metabolic enzymes discussed in the introduction, it has become apparent in the last decade that many metabolic enzymes and other regulators of metabolism form condensates in response to stresses (figure 3). One example is condensation of the translation initiation factor Ded1p, which promotes a switch from translation of housekeeping protein to translation of stress proteins.85 In S. cerevisiae and human hepatocarcinoma cells, hypoxia induces coalescence of glycolytic enzymes into ‘G bodies’.86 Cells containing G bodies showed increased glucose consumption, whereas cells unable to form G bodies exhibited growth defects and produce inviable daughter cells during cell division. These observations suggest that condensation of enzymes may help cells to survive and grow under low oxygen conditions.
Figure 3.
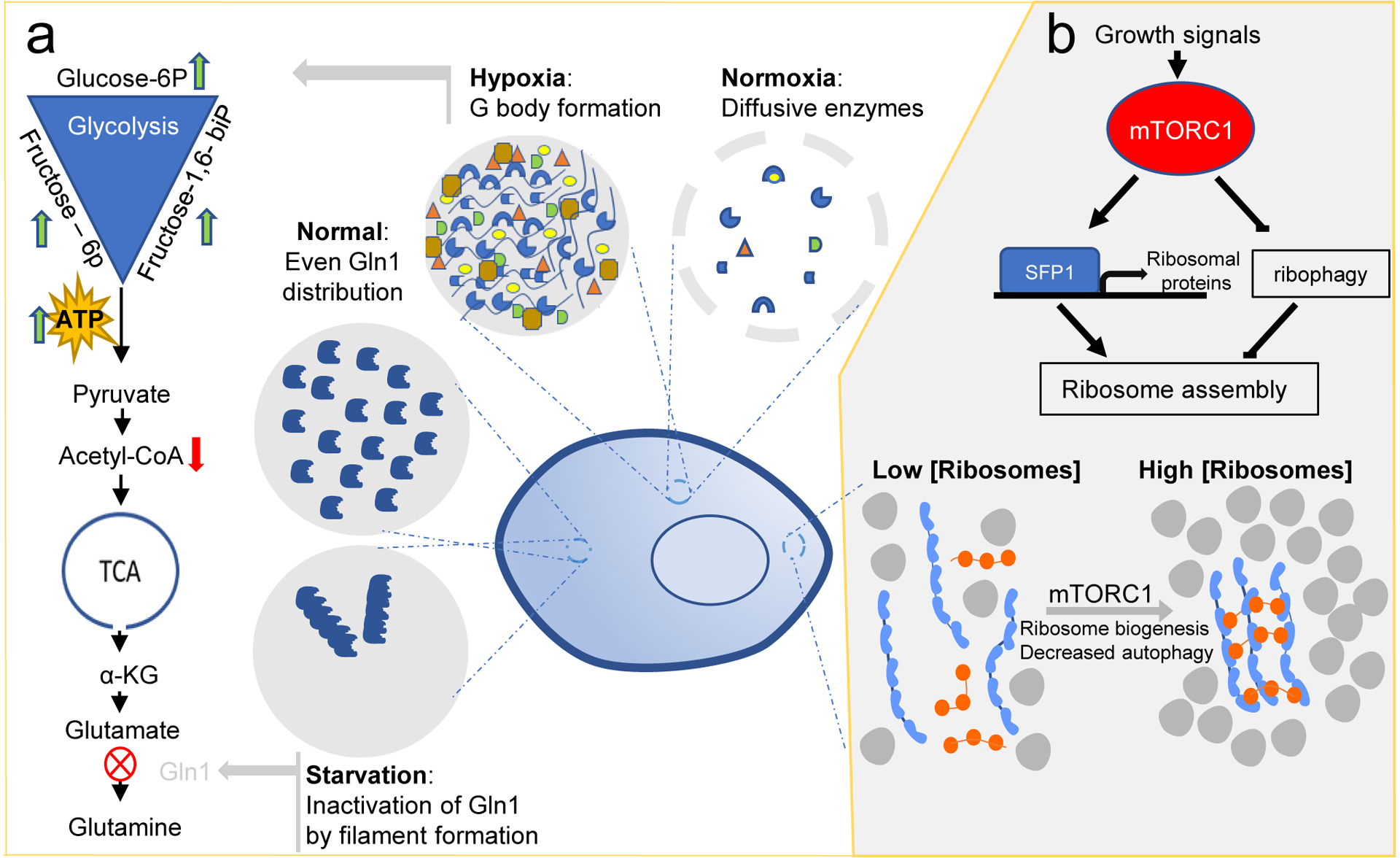
Reciprocal regulation of phase separation and cellular metabolism. (a) Hypoxia induces condensation of glycolytic enzymes into ‘G bodies’. G bodies promote cycling through the entire glycolytic pathway, reflected by increased levels of glucose-6-phosphate, fructose-6-phosphate and fructose-1,6-bisphosphate, and decreased levels of acetyl-CoA. Under normoxia, glycolytic enzymes remain diffusive in the cytoplasm. Starvation induces filament formation of glutamine synthetase (Gln1) and promotes its inactivation and storage in the cytoplasm. In the presence of nutrients, Gln1 is evenly distributed through the cytoplasm. (b) In response to growth signals, mTORC1 (blue) tunes macromolecular crowding by modulating ribosome concentration (grey) in the cytoplasm. Higher crowding favors phase separation and molecular assembly. In the absence of growth signals, ribosome concentration is decreased by ribophagy. glucose-6P, glucose-6-phosphate; fructose-6P, fructose-6-phosphate; fructose-1,6-biP, fructose-1,6-bisphosphate; acetyl-CoA, acetyl coenzyme-A; α-KG, α-ketoglutarate; SFP1, transcription factor SFP1.
Glucose withdrawal in budding yeast triggers assembly of TORC1 into large assemblies.87 Assembly inactivates TORC1, suggesting a mechanism to preserve energy during glucose deprivation. Conversely, TORC1 activity has been shown to generally regulate condensate assembly in cells. Tracking of genetically encoded multimeric nanoparticles (GEMs) provided a rapid assay to quantify macromolecular crowding in cells. GEMs are homomultimeric scaffolds fused to a fluorescent protein that self-assemble into bright, stable particles of defined size and shape. Using GEMs together with genetic and pharmacological perturbations led to the discovery that mTORC1 controls macromolecular crowding inside cells by tuning ribosome concentration.88 Macromolecular crowding favours phase separation, therefore mTORC1 activity couples metabolic sensing to condensation in cells. Extrapolating from these studies, mTORC1-dependent changes in molecular crowding may also impact the dynamics of many processes, including cytoskeletal elements, through the physicochemical mechanisms of depletion attraction and viscosity effects. Indeed, it has been shown that microtubule dynamics strongly depend upon crowding both in vitro89, and in cells.90
Stress-induced phase transitions can propagate to the whole-cell scale in yeast. Similar to the glass transition in E. coli, the widespread assembly of higher order structures in starved yeast cells has additional remarkable consequences for cell mechanics: the cytoplasm of starved cells transitions from liquid to solid.91,92 This solid-like state makes yeast cells so rigid that they can keep their shape in the absence of a cell wall. These material changes may protect cells from mechanical stress, or may be a mechanism for energy conservation. The assembly of higher-order structures through condensation is an emerging explanation for how cells rapidly adapt metabolism as well as the mechanical properties to sudden changes in the environment.
Genome folding and metabolic reprogramming
Finally, mechanics and metabolism are integrated at the level of genome folding and chromatin state. At the micron scale, genome organization is orchestrated by multiple mechanisms, including association with the nuclear lamina, which connects to the cytoskeleton and thus the extracellular environment via the LINC complexes93, and perhaps by phase separation of heterochromatin.94,95
The global mechanical properties of the nucleus have been shown to be strongly dependent on chromatin state. During the earliest stages of stem-cell differentiation, nuclei exhibit highly unusual auxetic mechanics where compression in one axis drives compression in an orthogonal axis rather than spreading, and compression-induced stiffening. This auxetic behaviour is driven by histone acetylation, as inhibition of histone deacetylases (HDACs) causes stem cells to adopt the auxetic phenotype.96 On the other hand, increased methylation and heterochromatin formation leads to softening of the nucleus. In epithelial cells, oscillatory stretch leads to opening of Piezo-1 channels in the ER, which activates histone methylases, thus changing the mechanical properties of the nucleus. This softening of the nucleus is necessary to prevent DNA damage during oscillatory stress (e.g. in beating cardiomyocytes).70 The metabolic state of the cell, in turn, can also directly impact chromatin state and mechanical properties of the nucleus by supplying co-factors for histone-modifying enzymes, such as histone acetyl transferase (HAT) and HDAC enzymes, which depend on acetyl-Co-enzyme A and NAD+ cofactors respectively.97
The genome is a dramatic example of the importance of “active matter” introduced above. The metabolic activity of ATP and metabolite production impacts the mechanics of chromatin through an intricate system of molecular motors and mechanical constraints. Transcription is one of the most significant contributors to the active dynamics of chromatin. RNA-polymerase holoenzymes are some of the most powerful and energy-demanding molecular motors, generating larger forces than kinesin- or myosin-type motors.98 Polymerase activity repositions nucleosomes and generates torsional stress behind the transcription bubble.99 These mechanical effects drive genomic spatial organization and dynamics, which in turn can regulate metabolic transcription programs. For example, CCCTC-binding factor (CTCF) is a key architectural protein that forms complexes at metabolic promoters to create DNA loops, organizing protein interactions and regulating epigenetic marks.100 Metabolic factors, in turn, can regulate CTCF mediated DNA looping.101,102
Together, these findings indicate that mechanics and metabolism are integrated at the genome level, whereby genomic spatial organization and dynamics regulate metabolic transcription programs, and metabolic states, in turn, influence genomic organization in normal and pathogenic gene programming states.
Mechanics and metabolism in disease
Cancer
The field of cancer metabolism is enormous and an intense area of ongoing research103,104. Here, we highlight a few key areas of dysregulation at the mechano-metabolic interface, again stepping from the outside toward the centre of the cell.
Tissue-scale mechanical changes and metabolic reprogramming
During cancer progression, the ECM becomes deregulated and disorganized105 leading to tissue stiffening. Changes in osmolytes and vasculature can increase interstitial fluid pressure, while excess growth within a confined space cause the build-up of mechanical solid compressive stress.106 In addition to hyperproliferation of tumour cells, tumour infiltrating cells (e.g. fibroblasts and immune cells) further remodel the tumour microenvironment leading to dramatic mechanical changes. Cancer cells must adapt both mechanically and metabolically to survive, proliferate and disseminate in this environment. Metabolic reprogramming is part of this adaptation and also feeds back to modify the ECM and the mechanical environment.
In fibrotic pancreatic cancers, the stiff tumour ECM has been shown to stimulate YAP-mediated upregulation of cytoplasmic creatine kinase B (CKB), as well as directing arginine-derived carbon into creatine biosynthesis and away from the urea cycle.107 These processes promote creatine-phosphagen ATP-recycling, in which CKB regulates the phosphorylation of creatine to phosphocreatine, which in turn transfers phosphate to ADP to regenerate ATP, thus providing a non-mitochondrial energy source for cancer cells. In non-small-cell lung carcinoma (NSCLC), ECM stiffening induces proline metabolism and consequently cell proliferation.108 Kindlin-2, a focal adhesion component that is essential for cell–ECM signaling, was found to localize to mitochondria and stimulate proline synthesis by upregulating pyrroline-5-carboxylate reductase 1 (PYCR1). As proline is the main building block for collagen, the kindlin-2-PYCR1 axis could play a crucial role in collagen synthesis and ECM remodelling, and consequently lung tumour progression.108 Uptake of pyruvate in breast cancer drives the TCA cycle to enhance the production of α-ketoglutarate.109 This metabolite enhances the activity of the enzyme collagen prolyl-4-hydroxylase, which promotes ECM remodelling. Thus, tissue stiffening can reprogram metabolism, which in turn can further perturb tissue mechanics in a vicious cycle, a mechanochemical axis that remains a crucial underexplored aspect of oncogenesis.
Mechanical effects on migration and metastasis
The most dangerous feature of cancer is the progression to metastasis, leading to cancer dissemination beyond the primary site. Metastasis requires both reprograming of cells to attain a migratory phenotype, typically through the epithelial to mesenchymal transition (EMT), and metabolic and mechanical adaptation of cells to facilitate migration within the dense, stiff tumour microenvironment.
Multiple studies have shown that mechanical perturbations can drive EMT. In pancreatic cancer, ECM matrix rigidity promotes elements of EMT, including downregulation of E-cadherin expression and increased nuclear localization of YAP and TAZ, driving cells towards a mesenchymal phenotype.110 Independent of YAP/TAZ, integrin-dependent mechanosensation events in breast cancer lead to release of the mechanosensor TWIST1 from its cytoplasmic anchor G3BP2, to enter the nucleus and drive transcriptional events of EMT.111 These examples suggest that multiple independent mechanosensation pathways can drive EMT in response to the altered mechanical properties of the tumour microenvironment.
After EMT, the process of migration is energetically costly, especially in the perturbed tumour microenvironment.112 The cytoskeletal dynamics that generate and relay the forces that propel cell migration have been estimated to consume up to 50% of the cell’s ATP budget.113 Some of the metabolic reprogramming described above may help supply the requisite ATP. There is consensus that motile aggressive mesenchymal cells exhibit increased aerobic glycolysis114, and that the Warburg Effect, which is a hallmark of many cancer cells115, may induce migratory or invasive behavior.116 For instance, mesenchymal prostate cancer cells exhibit higher glycolytic activity relative to their epithelial precedents. This higher glycolysis is associated with increased rates of cytoskeletal remodeling, greater cell traction forces and higher migration speeds.117
Together, these studies indicate that metastatic cancer cells reprogram various metabolic pathways in order to migrate to and invade secondary tissues. However, even in non-cancerous migratory processes such as cellular unjamming and EMT in the neural crest, the migrating cell layers exhibit increased glycolytic flux in much of the same manner as migrating mesenchymal cells.118
Cellular mechanics
Altered mechanics are a hallmark of metastatic cells: more than two-thirds of metastatic cells exhibit lower stiffness119. The origins of these mechanical changes are the subject of ongoing investigation, and are likely to be a consequence of a combination of altered cytoskeletal activity, glycocalyx composition and organelle properties, including chromatin state (see below), all of which are strongly impacted by metabolic state.120,121 A crucial, often underappreciated, determinant of the mechanical properties of cells and tissues is the cell surface glycocalyx. This incredibly dense network of glycoproteins and sugar polymers that is secreted into the ECM and presented at the cell surface dramatically alters the physical properties of tumours and cancer cells. For example, cell surface mucins such as Muc1, Muc4 or Muc16 are highly overexpressed in most breast, pancreatic and ovarian cancers, especially in metastatic cells.122 These large, highly glycosylated proteins can extend 200 nm from the cell surface and generate large entropic pressures that drive increased membrane curvature and tubulation (figure 4). Mucin transcription is stimulated by mechanical perturbation and their glycosylation state is highly sensitive to the metabolic state of the cell. Due to their crucial role in cell surface interaction with the environment, mucins are regulated by multiple metabolites123 and are therefore a crucial mechanism linking cell mechanics to metabolism.
Figure 4.
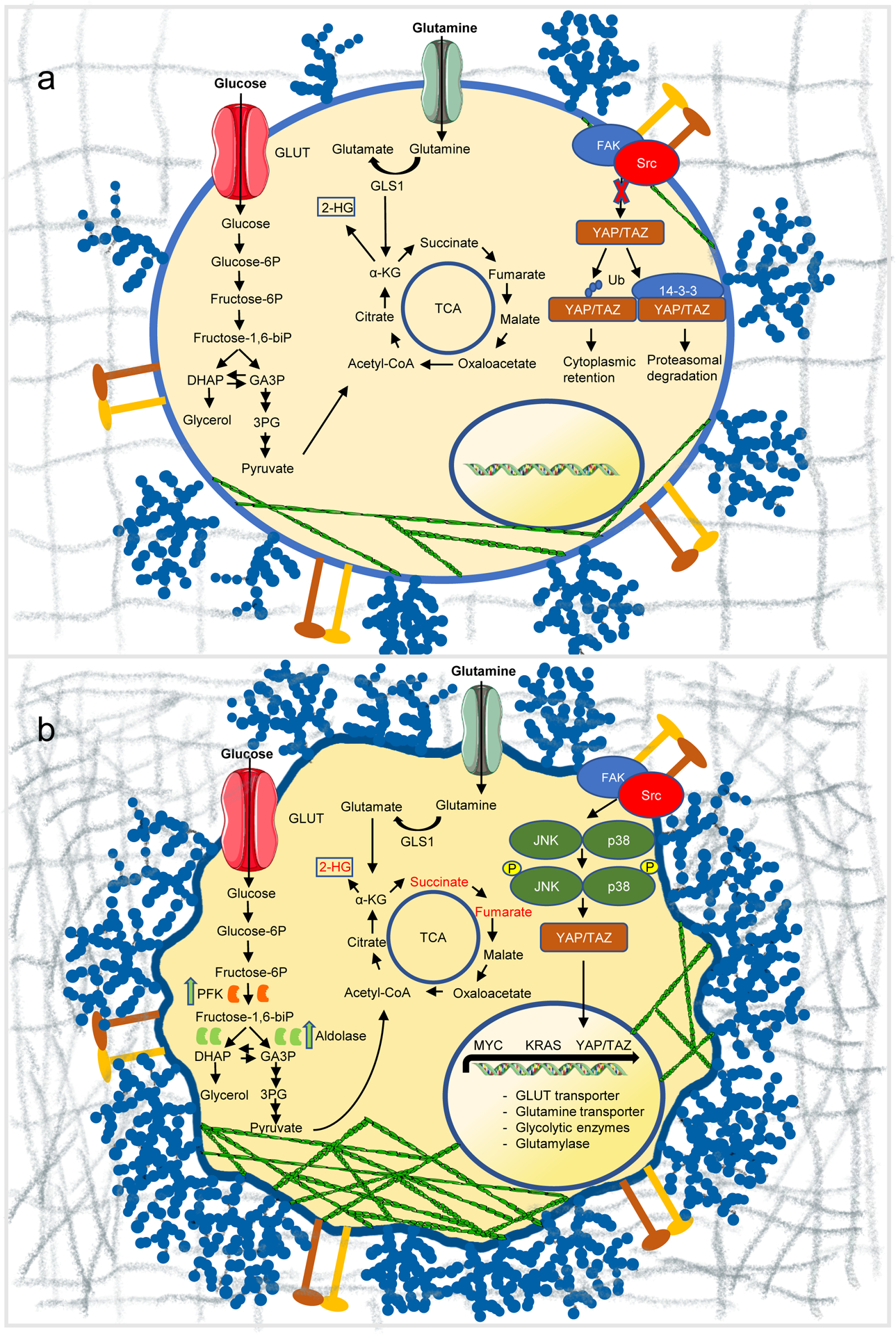
Reciprocal regulation of mechanics and metabolism in cancer. (a) Under normal conditions, the compliant extracellular matrix suppresses JNK/p38 signaling, and promotes either retainment of inactive YAP/TAZ in the cytosol or its degradation by proteasomes. Cells show reduced actin cytoskeleton assembly and express normal levels of surface glycocalyx. (b) In cancer, extracellular matrix stiffening accelerates glycolysis through JNK/p38 and YAP/TAZ-mediated metabolic reprogramming, and induces cytoskeleton remodelling, resulting in the release of glycolytic enzymes in the cytosol, thereby promoting glucose metabolism. Transcriptional programs are perturbed, promoting metabolic reprogramming, including glucose and glutamine metabolism. Glutamine metabolic and glycolytic end-products feed into the TCA cycle, increasing the abundance of oncometabolites fumarate, succinate, and 2-HG (red). The surface glycocalyx is frequently overexpressed generating large entropic pressures that drive increased membrane curvature and tubulation. GLUT, glucose transporter; GLS1, glutaminase 1; Ub, ubiquitin.
Cytoskeleton
Hyperactivation of PI3K as a consequence of constitutively active receptor tyrosine kinases (RTKs)124 results in increased cytoskeleton remodelling, which in turn regulates metabolic pathways. Filamentous actin usually sequesters aldolase. Therefore, increased actin dynamics can cause excess free, cytoplasmic aldolase that drives glycolysis, promoting cancer cell proliferation (figure 4).49 Cells growing on relaxed matrices usually target PFK for proteasome-mediated degradation leading to downregulation of glycolysis, but this mechanism is overridden in cancer cells transformed with oncogenic KRAS, and MYC or silenced TP53.47 Thus, perturbed crosstalk between the matrix, cytoskeleton and key metabolic enzymes may explain how cancer cells maintain high glycolytic rates despite continuous mechanical alterations to the tumour tissue.
Nuclear mechanics
The nucleus is the stiffest organelle in the cell, partly due to the mechanically robust lamina. The stiffness of nuclei has been shown to decrease as cancer cells progress to more metastatic states.121,125 These mechanical changes facilitate migration in constricted 3D environments but also lead to frequent nuclear rupture121,126 and DNA damage that drives genome instability.120 Apart from their crucial mechanical roles, lamins also directly organize the genome and coordinate transcriptional regulation of many genes, including metabolic enzymes. For example, 12-lipoxygenase expression is increased in cancer cells, resulting in conversion of polyunsaturated fatty acid to pro-carcinogenic metabolites.127 Mutations in lamins also cause dysregulation of mTORC1, autophagy, and adipogenesis.128 mTORC1 and its downstream targets are over-activated in cells derived from patients suffering from metabolic laminopathies, resulting in an abnormal autophagy, and downregulation of genes that regulate adipogenesis. In this way, lamins couple nuclear mechanical properties and metabolic states and act as mechanosensors.
Clinical prospects
Mechano-metabolic coupling in cancer cells is emerging as an important concept in drug development. Although several different approaches have been explored, only a small number of effective mechanical and metabolic inhibitors have been developed so far for use in cancer therapy.129,130 Moreover, altered cell and ECM mechanical properties are thought to contribute to drug resistance.131 More holistic studies of the interplay between mechanical and metabolic features of the tumour microenvironment are likely to suggest therapeutic strategies and perhaps explain the variable success of current treatments. For example, surgical intervention has dramatic mechanical impacts – how might the changes in tissue mechanics, inflammation, cellular metabolism and scarring impact the likely effects of subsequent chemotherapy?
Cardiovascular disease
The interplay between cell mechanics and metabolism has been extensively studied in cardiovascular pathologies. Atherosclerotic plaque formation frequently initiates at branches and bends in arteries that are exposed to irregular, disturbed patterns of blood flow.132 Irregular shear stress has been shown to upregulate Rac1 activity, which mediates both cytoskeletal alignment as well as the upregulation of intercellular cell adhesion molecule 1 (ICAM-1), the latter through activation of nuclear factor-kappa-B (NF-κB). NF-κB activation requires metabolic generation of reactive oxygen species (ROS).133
Non-uniform shear stress also stimulates integrin-mediated upregulation of RhoA and subsequent YAP activation. Similar to ROS, YAP enhances JNK signaling and upregulates pro-inflammatory genes such as NF-κB.134 NF-κB is a central inflammatory mediator that plays a major role in the progression of atherosclerosis by inducing upregulation of pro-inflammatory cytokines, chemotactic factors and focal enrichment of adhesion molecules, thereby promoting monocyte recruitment into the arterial wall of an atherosusceptible site.135
In ischemia, shear stress causes endothelial plasma membrane depolarization via ATP-sensitive K+ channel closure, inducing PI3K/Akt-mediated signaling transduction cascade resulting in NADPH oxidase activation and ROS production.136 Recently, application of mechanical fluctuations to vascular smooth muscle cells was shown to regulate mitochondrial bioenergetics by tuning the structure of mitochondrial network, ensuring ATP production via the electron transport chain and minimizing ROS production.137 Pathological high or low mechanical strain reduced ATP production and increased ROS production, promoting oxidative stress and metabolic disturbances. These data indicate the importance of optimal mechanical homeostasis in the prevention of metabolic disturbance and cardiovascular disease.
Connective tissue diseases
Fibrosis in human tissues is observed in response to hypoxia as well as mechanical shear, indicating the importance of both metabolism and mechanosensing in these pathologies. Impaired YAP/YAZ mechanotransduction is implicated in fibrotic diseases, including viral hepatitis induced liver fibrosis or Lupus-induced lung and kidney fibrosis. YAP/TAZ promote TGFβ signaling, thereby inducing kidney and liver tissue fibrosis, which then further activates YAP/TAZ in a viscous cycle. Liver damage caused by viral hepatitis results in increases in collagen deposition and smooth muscle actin expression, which are associated with the generation of myofibroblasts, key players in fibrotic disease. Associated with these mechanical alterations, dysregulated glycolysis has been reported in experimental models of lung, kidney and liver fibrosis.138 Upregulation of glutaminolysis and enhanced fatty acid oxidation are also drivers of fibroblast activation. For example, glutaminolysis is increased in TGFβ-stimulated fibroblasts. This leads to enhanced conversion of glutamine to glutamate which promotes the stabilization of collagen via mTORC1 signaling; inhibition of glutamine conversion ameliorates TGFβ-induced lung fibrosis. Verteporfin, a small-molecule that inhibits YAP/TAZ and TGF-β/Smad crosstalk, illustrates promising mechano-interference efficacy in preclinical kidney and hepatic fibrosis models139,140Also, Rho/ROCK is an upstream regulator of YAP/TAZ and the therapeutic effects of ROCK inhibitors in preclinical fibrosis models may reflect, in part, reduced YAP/TAZ activation141,142 The processes that link metabolism and mechanics thus appear as promising targets for future drugs to manage fibrosis.
Conclusion and future perspectives
Interest in the reciprocal regulation of cellular mechanics and metabolism has grown extensively over the last decade. We now know that metabolism is interwoven with cellular mechanics at multiple scales. Nanoscale molecular sensors at the cell surface relay mechanical information through the cytoskeleton and transcriptional programs, and feed into metabolic pathways to increase energy production, allowing cells to resist and generate mechanical forces. At the micron-scale, cytoskeletal networks facilitate local enrichment of metabolic enzymes to provide high rates of metabolic activity where demand is high, while deformation of organelles, such as the nucleus, Golgi apparatus and mitochondria, induces transcriptional changes that rewire metabolism. The resulting alterations in metabolites, in turn, regulate every level of cell biology including the mechanical properties of cells and tissues.
Much remains to be discovered. One frontier is the single-cell analysis of mechano-metabolic coupling. Many current approaches homogenize complex tissues, with a consequent loss of information about the unique behaviour of individual cells, including cellular metabolism, growth, and proliferation.143,144. Cutting edge technological advances are beginning to address these issues. For example, combining single-cell metabolic modalities with single-cell force spectroscopy (SCFS) techniques enables rapid and automatic linking of metabolic profiles to multiple mechanical parameters (figure 5). Single-cell mass spectrometry-based approaches also show great promise: recently, researchers were able to measure metabolic changes in response to a drug in a single cell.145 SCFS techniques such as atomic force microscopy and optical tweezers have proven crucial for providing physical insight into the mechanical properties of cells146–148 and offer a unique opportunity to unravel inter- and intracellular heterogeneity.149 Understanding the crosstalk between mechanics and metabolism at the singe-cell level will likely create diagnostic and therapeutic opportunities for metabolic disorders such as cancer, cardiovascular pathologies and obesity.
Figure 5.
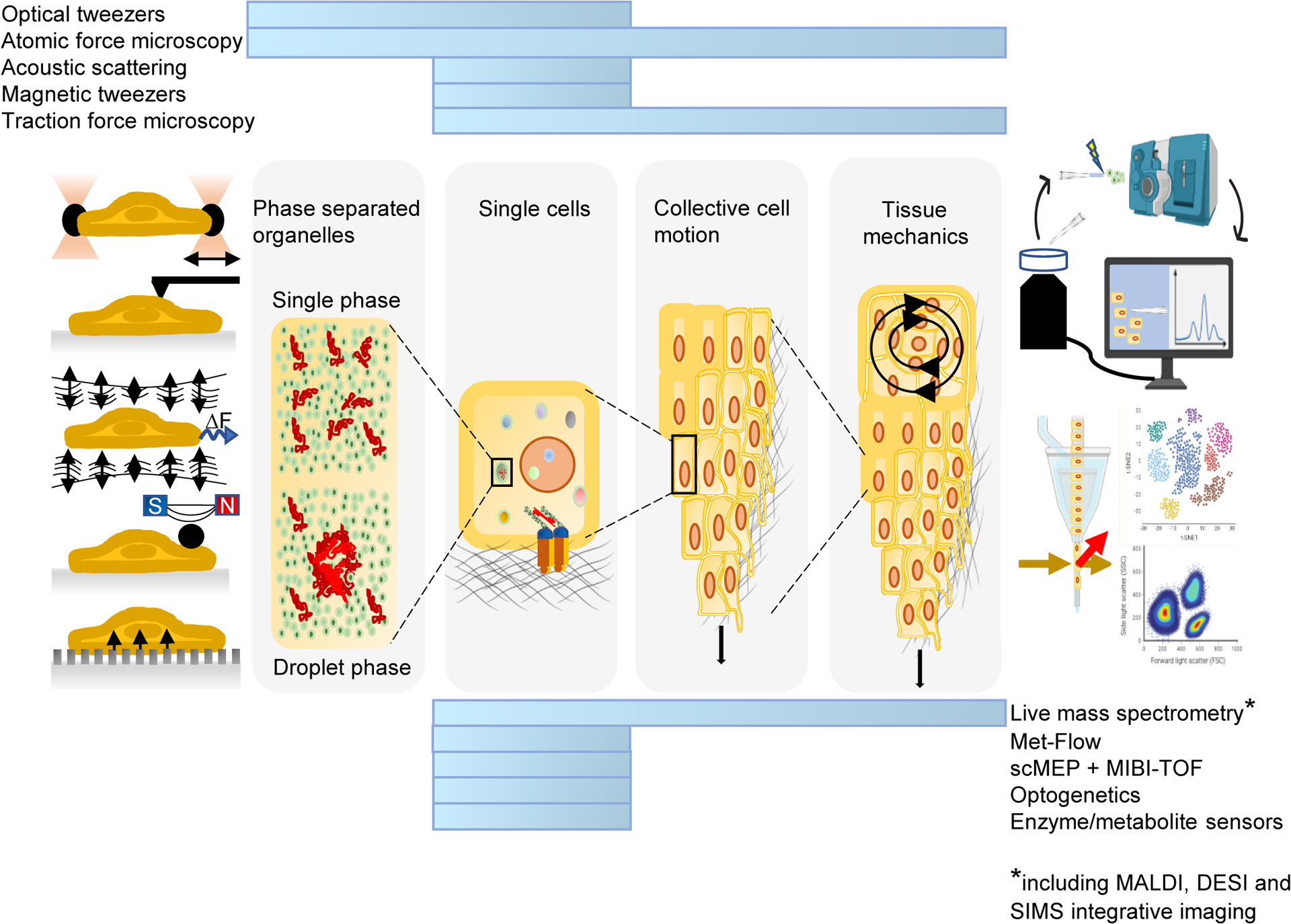
Integration of metabolic technologies with force spectroscopy modalities correlate metabolic profiles to mechanics at different scales (blue bars) and in variable conditions. Future advances may improve the application of these techniques to single cells.
Met-Flow, a high-parameter flow cytometry method utilizing antibodies against metabolic proteins; scMEP, single-cell metabolic regulome profiling; MIBI-TOF, multiplexed ion beam imaging by time of flight; MALDI, matrix-assisted laser desorption/ionization; DESI, desorption electrospray ionization; SIMS, secondary ion mass spectrometry. Mass spectrometer and flow cytometry illustrations were generated using BioRender.
Conceptually, an intriguing emerging field is that of phase separation. Although several groups have shown that membrane-less condensates perform mechanical work, and that metabolic enzymes and other regulators of metabolism can undergo condensate formation, very little else is known. This field is likely to be a productive area of research in the near future. Furthermore, the study of the material properties of cells as soft-condensed matter, far from equilibrium will yield deeper insights. We have made amazing progress since the first description of sickle-cell anaemia as a molecular disorder at the intersection of metabolism and mechanics. The coming years are sure to yield many more exciting discoveries at the intersection of metabolism and mechanosensing.
Footnotes
Competing interests
The authors declare no competing financial interests.
References
- 1.Herrick JB Peculiar elongated and sickle-shaped red blood corpuscles in a case of severe anemia. Arch. Intern. Med VI, 517–521 (1910). [PMC free article] [PubMed] [Google Scholar]
- 2.Pauling L, Itano HA, Singer SJ & Wells IC Sickle cell anemia, a molecular disease. Science (80-.) 110, 543–548 (1949). [DOI] [PubMed] [Google Scholar]
- 3.Li X, Dao M, Lykotrafitis G & Karniadakis GE Biomechanics and biorheology of red blood cells in sickle cell anemia. J. Biomech 50, 34–41 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Dykes G, Crepeau RH & Edelstein SJ Three-dimensional reconstruction of the fibres of sickle cell haemoglobin. Nature 272, 506–510 (1978). [DOI] [PubMed] [Google Scholar]
- 5.Garcia-Seisdedos H, Empereur-Mot C, Elad N & Levy ED Proteins evolve on the edge of supramolecular self-assembly. Nature 548, 244–247 (2017). [DOI] [PubMed] [Google Scholar]
- 6.Sampaleanu LM, Vallée F, Slingsby C & Howell PL Structural studies of duck δ1 and δ2 crystallin suggest conformational changes occur during catalysis. Biochemistry 40, 2732–2742 (2001). [DOI] [PubMed] [Google Scholar]
- 7.Barry RM & Gitai Z Self-assembling enzymes and the origins of the cytoskeleton. Current Opinion in Microbiology 14, 704–711 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stoddard PR et al. Polymerization in the actin ATPase clan regulates hexokinase activity in yeast. Science (80-.) 367, 1039–1042 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kirby TJ & Lammerding J Emerging views of the nucleus as a cellular mechanosensor. Nature Cell Biology 20, 373–381 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Mashaghi A & Dekker C Systems and synthetic biology approaches to cell division. Systems and Synthetic Biology 8, 173–178 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Janmey PA & Lindberg U Cytoskeletal regulation: Rich in lipids. Nature Reviews Molecular Cell Biology 5, 658–666 (2004). [DOI] [PubMed] [Google Scholar]
- 12.Fletcher DA & Mullins RD Cell mechanics and the cytoskeleton. Nature 463, 485–492 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Pollard TD Genomics, the cytoskeleton and motility. Nature 409, 842–843 (2001). [DOI] [PubMed] [Google Scholar]
- 14.Trappe V, Prasad V, Cipelletti L, Segre PN & Weitz DA Jamming phase diagram for attractive particles. Nature 411, 772–775 (2001). [DOI] [PubMed] [Google Scholar]
- 15.Kasza KE et al. The cell as a material. Current Opinion in Cell Biology 19, 101–107 (2007). [DOI] [PubMed] [Google Scholar]
- 16.Bursac P et al. Cytoskeletal remodelling and slow dynamics in the living cell. Nat. Mater 4, 557–561 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Parry BR et al. The bacterial cytoplasm has glass-like properties and is fluidized by metabolic activity. Cell 156, 183–194 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Freeman SA et al. Lipid-gated monovalent ion fluxes regulate endocytic traffic and support immune surveillance. Science (80-.) 367, 301–305 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.PAUL J Carbohydrate and Energy Metabolism. in Cells and Tissues in Culture 239–276 (1965). doi: 10.1016/b978-1-4831-9797-5.50014-7. [DOI] [Google Scholar]
- 20.Rojas ER & Huang KC Regulation of microbial growth by turgor pressure. Current Opinion in Microbiology 42, 62–70 (2018). [DOI] [PubMed] [Google Scholar]
- 21.Shurer CR et al. Physical Principles of Membrane Shape Regulation by the Glycocalyx. Cell 177, 1757–1770.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Mashaghi A et al. Label-free characterization of biomembranes: From structure to dynamics. Chemical Society Reviews 43, 887–900 (2014). [DOI] [PubMed] [Google Scholar]
- 23.Van Meer G, Voelker DR & Feigenson GW Membrane lipids: Where they are and how they behave. Nature Reviews Molecular Cell Biology 9, 112–124 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Groves JT Membrane Mechanics in Living Cells. Developmental Cell 48, 15–16 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Sullivan WJ et al. Extracellular Matrix Remodeling Regulates Glucose Metabolism through TXNIP Destabilization. Cell 175, 117–132.e21 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rinschen MM, Ivanisevic J, Giera M & Siuzdak G Identification of bioactive metabolites using activity metabolomics. Nature Reviews Molecular Cell Biology 20, 353–367 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Valvezan AJ & Manning BD Molecular logic of mTORC1 signalling as a metabolic rheostat. Nature Metabolism 1, 321–333 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Peterson TR et al. MTOR complex 1 regulates lipin 1 localization to control the srebp pathway. Cell 146, 408–420 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Robitaille AM et al. Quantitative phosphoproteomics reveal mTORC1 activates de novo pyrimidine synthesis. Science (80-.) 339, 1320–1323 (2013). [DOI] [PubMed] [Google Scholar]
- 30.Liu GY & Sabatini DM mTOR at the nexus of nutrition, growth, ageing and disease. Nature Reviews Molecular Cell Biology 21, 183–203 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Catania C, Binder E & Cota D MTORC1 signaling in energy balance and metabolic disease. International Journal of Obesity 35, 751–761 (2011). [DOI] [PubMed] [Google Scholar]
- 32.Dancey J MTOR signaling and drug development in cancer. Nature Reviews Clinical Oncology 7, 209–219 (2010). [DOI] [PubMed] [Google Scholar]
- 33.Costa-Mattioli M & Monteggia LM mTOR complexes in neurodevelopmental and neuropsychiatric disorders. Nature Neuroscience 16, 1537–1543 (2013). [DOI] [PubMed] [Google Scholar]
- 34.Herzig S & Shaw RJ AMPK: Guardian of metabolism and mitochondrial homeostasis. Nature Reviews Molecular Cell Biology 19, 121–135 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Mihaylova MM & Shaw RJ The AMPK signalling pathway coordinates cell growth, autophagy and metabolism. Nature Cell Biology 13, 1016–1023 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Zhang CS & Lin SC AMPK promotes autophagy by facilitating mitochondrial fission. Cell Metabolism 23, 399–401 (2016). [DOI] [PubMed] [Google Scholar]
- 37.Rübsam M et al. E-cadherin integrates mechanotransduction and EGFR signaling to control junctional tissue polarization and tight junction positioning. Nat. Commun 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hall A Rho GTpases and the actin cytoskeleton. Science 279, 509–514 (1998). [DOI] [PubMed] [Google Scholar]
- 39.Rainero E et al. Ligand-Occupied Integrin Internalization Links Nutrient Signaling to Invasive Migration. Cell Rep. 10, 398–413 (2015). [DOI] [PubMed] [Google Scholar]
- 40.Muranen T et al. Starved epithelial cells uptake extracellular matrix for survival. Nat. Commun 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Georgiadou M et al. AMPK negatively regulates tensin-dependent integrin activity. J. Cell Biol 216, 1107–1121 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Simon DN & Wilson KL The nucleoskeleton as a genome-associated dynamic ‘network of networks’. Nature Reviews Molecular Cell Biology 12, 695–708 (2011). [DOI] [PubMed] [Google Scholar]
- 43.Panciera T, Azzolin L, Cordenonsi M & Piccolo S Mechanobiology of YAP and TAZ in physiology and disease. Nature Reviews Molecular Cell Biology 18, 758–770 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Uhler C & Shivashankar GV Regulation of genome organization and gene expression by nuclear mechanotransduction. Nature Reviews Molecular Cell Biology 18, 717–727 (2017). [DOI] [PubMed] [Google Scholar]
- 45.Bays JL, Campbell HK, Heidema C, Sebbagh M & Demali KA Linking E-cadherin mechanotransduction to cell metabolism through force-mediated activation of AMPK. Nat. Cell Biol 19, 724–731 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates that LKB1-mediated activation of AMPK is a key player in a junctional contractility pathway that increases glucose uptake and ATP synthesis to resist physiological forces.
- 46.Knull HR & Walsh JL Association of Glycolytic Enzymes with the Cytoskeleton. in Current Topics in Cellular Regulation vol. 33 15–30 (1992). [DOI] [PubMed] [Google Scholar]
- 47.Park JS et al. Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 578, 621–626 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper describes a mechanism by which stiffness in the ECM promotes the reorganization of actin cytoskeleton filaments to enhance glycolysis.
- 48.Principe M et al. Alpha-enolase (ENO1) controls alpha v/beta 3 integrin expression and regulates pancreatic cancer adhesion, invasion, and metastasis. J. Hematol. Oncol 10, 1–13 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Hu H et al. Phosphoinositide 3-Kinase Regulates Glycolysis through Mobilization of Aldolase from the Actin Cytoskeleton. Cell 164, 433–446 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper reveals a mechanism by which signal transduction via PI3K allows for the physical dissociation of the glycolytic enzyme aldolase from F-actin into the cytoplasm where it accelerates glucose metabolism.
- 50.Panciera T et al. Reprogramming normal cells into tumour precursors requires ECM stiffness and oncogene-mediated changes of cell mechanical properties. Nat. Mater (2020) doi: 10.1038/s41563-020-0615-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Torrino S et al. Biophysical forces rewire cell metabolism to guide microtubule-dependent cell mechanics. bioRxiv (2020) doi: 10.1101/2020.03.10.985036. [DOI] [Google Scholar]
- 52.Natarajan K, Gadadhar S, Souphron J, Magiera MM & Janke C Molecular interactions between tubulin tails and glutamylases reveal determinants of glutamylation patterns. EMBO Rep. 18, 1013–1026 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Totaro A, Panciera T & Piccolo S YAP/TAZ upstream signals and downstream responses. Nature Cell Biology 20, 888–899 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Visser-Grieve S et al. LATS1 tumor suppressor is a novel actin-binding protein and negative regulator of actin polymerization. Cell Research 21, 1513–1516 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Elosegui-Artola A et al. Force Triggers YAP Nuclear Entry by Regulating Transport across Nuclear Pores. Cell 171, 1397–1410.e14 (2017). [DOI] [PubMed] [Google Scholar]
- 56.Sorrentino G et al. Glucocorticoid receptor signalling activates YAP in breast cancer. Nat. Commun 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Dupont S et al. Role of YAP/TAZ in mechanotransduction. Nature 474, 179–184 (2011). [DOI] [PubMed] [Google Scholar]
- 58.Aragona M et al. A mechanical checkpoint controls multicellular growth through YAP/TAZ regulation by actin-processing factors. Cell 154, 1047–1059 (2013). [DOI] [PubMed] [Google Scholar]; This paper identified F-actin-capping/severing proteins as essential gatekeepers that limit YAP/TAZ activity in cells experiencing low mechanical stresses.
- 59.Cai D et al. Phase separation of YAP reorganizes genome topology for long-term YAP target gene expression. Nat. Cell Biol 21, 1578–1589 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lu Y et al. Phase separation of TAZ compartmentalizes the transcription machinery to promote gene expression. Nat. Cell Biol 22, 453–464 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Guo M et al. Cell volume change through water efflux impacts cell stiffness and stem cell fate. Proc. Natl. Acad. Sci. U. S. A 114, E8618–E8627 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Li Y et al. Volumetric Compression Induces Intracellular Crowding to Control Intestinal Organoid Growth via Wnt/β-Catenin Signaling. Cell Stem Cell 28, 63–78.e7 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Bertero T et al. Vascular stiffness mechanoactivates YAP/TAZ-dependent glutaminolysis to drive pulmonary hypertension. J. Clin. Invest 126, 3313–3335 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Liu QP, Luo Q, Deng B, Ju Y & Bin Song G. Stiffer matrix accelerates migration of hepatocellular carcinoma cells through enhanced aerobic glycolysis via the MAPK-YAP signaling. Cancers (Basel). 12, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Enzo E et al. Aerobic glycolysis tunes YAP / TAZ transcriptional activity. EMBO J. 34, 1349–1370 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Bertero T et al. Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab. 29, 124–140.e10 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper identified a metabolic crosstalk between cancer associated fibroblasts (CAF) and cancer cells in which CAF-derived aspartate sustains cancer cell proliferation, while cancer cell-derived glutamate balances the redox state of CAFs to promote ECM remodeling.
- 67.Rabinowitz JD & White E Autophagy and metabolism. Science 330, 1344–1348 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Eagle H & Levine EM Growth regulatory effects of cellular interaction. Nature 213, 1102–1106 (1967). [DOI] [PubMed] [Google Scholar]
- 69.Pavel M et al. Contact inhibition controls cell survival and proliferation via YAP/TAZ- autophagy axis. Nat. Commun 9, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ellefsen KL et al. Myosin-II mediated traction forces evoke localized Piezo1-dependent Ca2+ flickers. Commun. Biol 2, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang T, Chi S, Jiang F, Zhao Q & Xiao B A protein interaction mechanism for suppressing the mechanosensitive Piezo channels. Nat. Commun 8, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Wang Y, Nagarajan M, Uhler C & Shivashankar GV Orientation and repositioning of chromosomes correlate with cell geometry-dependent gene expression. Mol. Biol. Cell 28, 1997–2009 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Belyaeva A, Venkatachalapathy S, Nagarajan M, Shivashankar GV & Uhler C Network analysis identifies chromosome intermingling regions as regulatory hotspots for transcription. Proc. Natl. Acad. Sci. U. S. A 114, 13714–13719 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Romani P et al. Extracellular matrix mechanical cues regulate lipid metabolism through Lipin-1 and SREBP. Nat. Cell Biol 21, 338–347 (2019). [DOI] [PubMed] [Google Scholar]
- 75.Moore AS, Wong YC, Simpson CL & Holzbaur ELF Dynamic actin cycling through mitochondrial subpopulations locally regulates the fission-fusion balance within mitochondrial networks. Nat. Commun 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Helle SCJ et al. Mechanical force induces mitochondrial fission. Elife 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Tharp K et al. Adhesion-mediated mechanosignaling forces mitohormesis. bioRxiv (2020) doi: 10.1101/2020.03.06.979583. [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper demonstrates that cells sense the physical properties of the ECM and activate a mitochondrial stress response which adaptively tunes mitochondrial function via SLC9A1-dependent ion exchange and HSF1-dependent transcription.
- 78.Hyman AA, Weber CA & Jülicher F Liquid-liquid phase separation in biology. Annu. Rev. Cell Dev. Biol 30, 39–58 (2014). [DOI] [PubMed] [Google Scholar]
- 79.Dolgin E What lava lamps and vinaigrette can teach us about cell biology. Nature 555, 300–302 (2018). [DOI] [PubMed] [Google Scholar]
- 80.Weber SC & Brangwynne CP Getting RNA and protein in phase. Cell 149, 1188–1191 (2012). [DOI] [PubMed] [Google Scholar]
- 81.Hyman AA & Simons K Beyond oil and water - Phase transitions in cells. Science 337, 1047–1049 (2012). [DOI] [PubMed] [Google Scholar]
- 82.Shin Y et al. Spatiotemporal Control of Intracellular Phase Transitions Using Light-Activated optoDroplets. Cell 168, 159–171.e14 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bracha D et al. Mapping Local and Global Liquid Phase Behavior in Living Cells Using Photo-Oligomerizable Seeds. Cell 175, 1467–1480.e13 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Feric M & Brangwynne CP A nuclear F-actin scaffold stabilizes ribonucleoprotein droplets against gravity in large cells. Nat. Cell Biol 15, 1253–1259 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Iserman C et al. Condensation of Ded1p Promotes a Translational Switch from Housekeeping to Stress Protein Production. Cell 181, 818–831.e19 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Jin M et al. Glycolytic Enzymes Coalesce in G Bodies under Hypoxic Stress. Cell Rep. 20, 895–908 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Prouteau M et al. TORC1 organized in inhibited domains (TOROIDs) regulate TORC1 activity. Nature 550, 265–269 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Delarue M et al. mTORC1 Controls Phase Separation and the Biophysical Properties of the Cytoplasm by Tuning Crowding. Cell 174, 338–349.e20 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]; This work discovered that mTORC1 controls macromolecular crowding inside cells by tuning ribosome concentration.
- 89.Wieczorek M, Chaaban S & Brouhard GJ Macromolecular crowding pushes catalyzed microtubule growth to near the theoretical limit. Cell. Mol. Bioeng 6, 383–392 (2013). [Google Scholar]
- 90.Molines AT et al. Physical properties of the cytoplasm modulate the rates of microtubule growth and shrinkage. bioRxiv (2020) doi: 10.1101/2020.10.27.352716. [DOI] [Google Scholar]
- 91.Munder MC et al. A pH-driven transition of the cytoplasm from a fluid- to a solid-like state promotes entry into dormancy. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Joyner RP et al. A glucose-starvation response regulates the diffusion of macromolecules. Elife 5, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Maurer M & Lammerding J The Driving Force: Nuclear Mechanotransduction in Cellular Function, Fate, and Disease. Annu. Rev. Biomed. Eng 21, 443–468 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Gibson BA et al. Organization of Chromatin by Intrinsic and Regulated Phase Separation. Cell 179, 470–484.e21 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Falk M et al. Heterochromatin drives compartmentalization of inverted and conventional nuclei. Nature 570, 395–399 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Pagliara S et al. Auxetic nuclei in embryonic stem cells exiting pluripotency. Nat. Mater 13, 638–644 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This study discovered an unexpected biophysical phenotype of the nuclei of mouse embryonic stem cells that are transitioning towards differentiation: contrary to the behaviour of most known materials, the nucleus of the cell expands when stretched.
- 97.Reid MA, Dai Z & Locasale JW The impact of cellular metabolism on chromatin dynamics and epigenetics. Nature Cell Biology 19, 1298–1306 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Marko JF & Poirier MG Micromechanics of chromatin and chromosomes. in Biochemistry and Cell Biology vol. 81 209–220 (2003). [DOI] [PubMed] [Google Scholar]
- 99.Kouzine F, Liu J, Sanford S, Chung HJ & Levens D The dynamic response of upstream DNA to transcription-generated torsional stress. Nat. Struct. Mol. Biol 11, 1092–1100 (2004). [DOI] [PubMed] [Google Scholar]
- 100.Peñ A-Hernández R et al. Genome-wide targeting of the epigenetic regulatory protein ctcf to gene promoters by the transcription factor TFII-I. Proc. Natl. Acad. Sci. U. S. A 112, E677–E686 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Tyrakis PA et al. S-2-hydroxyglutarate regulates CD8+ T-lymphocyte fate. Nature 540, 236–241 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Chisolm DA et al. CCCTC-Binding Factor Translates Interleukin 2- and α-Ketoglutarate-Sensitive Metabolic Changes in T Cells into Context-Dependent Gene Programs. Immunity 47, 251–267.e7 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Biancur DE & Kimmelman AC The plasticity of pancreatic cancer metabolism in tumor progression and therapeutic resistance. Biochimica et Biophysica Acta - Reviews on Cancer 1870, 67–75 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Faubert B, Solmonson A & DeBerardinis RJ Metabolic reprogramming and cancer progression. Science 368, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Lu P, Weaver VM & Werb Z The extracellular matrix: A dynamic niche in cancer progression. Journal of Cell Biology 196, 395–406 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Wirtz D, Konstantopoulos K & Searson PC The physics of cancer: The role of physical interactions and mechanical forces in metastasis. Nature Reviews Cancer 11, 512–522 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Papalazarou V et al. The creatine–phosphagen system is mechanoresponsive in pancreatic adenocarcinoma and fuels invasion and metastasis. Nat. Metab 2, 62–80 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Guo L et al. Kindlin-2 links mechano-environment to proline synthesis and tumor growth. Nat. Commun 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Elia I et al. Breast cancer cells rely on environmental pyruvate to shape the metastatic niche. Nature 568, 117–121 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Rice AJ et al. Matrix stiffness induces epithelial-mesenchymal transition and promotes chemoresistance in pancreatic cancer cells. Oncogenesis 6, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wei SC et al. Matrix stiffness drives epithelial-mesenchymal transition and tumour metastasis through a TWIST1-G3BP2 mechanotransduction pathway. Nat. Cell Biol 17, 678–688 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Carey SP et al. Comparative mechanisms of cancer cell migration through 3D matrix and physiological microtracks. Am. J. Physiol. - Cell Physiol 308, C436–C447 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Bernstein BW & Bamburg JR Actin-ATP hydrolysis is a major energy drain for neurons. J. Neurosci 23, 1–6 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Gatenby RA & Gillies RJ Why do cancers have high aerobic glycolysis? Nature Reviews Cancer 4, 891–899 (2004). [DOI] [PubMed] [Google Scholar]
- 115.Hanahan D & Weinberg RA Hallmarks of cancer: The next generation. Cell 144, 646–674 (2011). [DOI] [PubMed] [Google Scholar]
- 116.Lu J The Warburg metabolism fuels tumor metastasis. Cancer and Metastasis Reviews 38, 157–164 (2019). [DOI] [PubMed] [Google Scholar]
- 117.Shiraishi T et al. Glycolysis is the primary bioenergetic pathway for cell motility and cytoskeletal remodeling in human prostate and breast cancer cells. Oncotarget 6, 130–143 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Bhattacharya D, Azambuja AP & Simoes-Costa M Metabolic Reprogramming Promotes Neural Crest Migration via Yap/Tead Signaling. Dev. Cell 53, 199–211.e6 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Cross SE, Jin YS, Rao J & Gimzewski JK Nanomechanical analysis of cells from cancer patients. Nat. Nanotechnol (2007) doi: 10.1038/nnano.2007.388. [DOI] [PubMed] [Google Scholar]
- 120.Irianto J et al. DNA Damage Follows Repair Factor Depletion and Portends Genome Variation in Cancer Cells after Pore Migration. Curr. Biol 27, 210–223 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Denais CM et al. Nuclear envelope rupture and repair during cancer cell migration. Science (80-.) 352, 353–358 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Paszek MJ et al. The cancer glycocalyx mechanically primes integrin-mediated growth and survival. Nature 511, 319–325 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]; This paper highlights the, often underappreciated, role of surface covering sugar-conjugated proteins (glycocalyx) in regulating cell survival during tumour spread.
- 123.Pothuraju R et al. Mechanistic and functional shades of mucins and associated glycans in colon cancer. Cancers 12, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Engelman JA Targeting PI3K signalling in cancer: Opportunities, challenges and limitations. Nature Reviews Cancer 9, 550–562 (2009). [DOI] [PubMed] [Google Scholar]
- 125.Xia Y, Pfeifer CR & Discher DE Nuclear mechanics during and after constricted migration. Acta Mechanica Sinica/Lixue Xuebao 35, 299–308 (2019). [Google Scholar]
- 126.Raab M et al. ESCRT III repairs nuclear envelope ruptures during cell migration to limit DNA damage and cell death. Science (80-.) 352, 359–362 (2016). [DOI] [PubMed] [Google Scholar]
- 127.Shureiqi I & Lippman SM Lipoxygenase modulation to reverse carcinogenesis. Cancer Research 61, 6307–6312 (2001). [PubMed] [Google Scholar]
- 128.Charar C & Gruenbaum Y Lamins and metabolism. Clinical Science 131, 105–111 (2017). [DOI] [PubMed] [Google Scholar]
- 129.Galluzzi L, Kepp O, Heiden MGV & Kroemer G Metabolic targets for cancer therapy. Nature Reviews Drug Discovery 12, 829–846 (2013). [DOI] [PubMed] [Google Scholar]
- 130.Surcel A et al. Pharmacological activation of myosin ii paralogs to correct cell mechanics defects. Proc. Natl. Acad. Sci. U. S. A 112, 1428–1433 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hamidi H & Ivaska J Every step of the way: integrins in cancer progression and metastasis. Nat. Rev. Cancer 1–16 (2018) doi: 10.1038/s41568-018-0038-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Kwak BR et al. Biomechanical factors in atherosclerosis: Mechanisms and clinical implications. European Heart Journal 35, 3013–3020 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Tzima E et al. Activation of Rac1 by shear stress in endothelial cells mediates both cytoskeletal reorganization and effects on gene expression. EMBO J. 21, 6791–6800 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Huang Y et al. Integrin-YAP/TAZ-JNK cascade mediates atheroprotective effect of unidirectional shear flow. Nature 540, 579–582 (2016). [DOI] [PubMed] [Google Scholar]
- 135.Liu T, Zhang L, Joo D & Sun SC NF-κB signaling in inflammation. Signal Transduction and Targeted Therapy 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Chatterjee S et al. Membrane depolarization is the trigger for PI3K/Akt activation and leads to the generation of ROS. Am. J. Physiol. - Hear. Circ. Physiol 302, (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Bartolák-Suki E & Suki B Tuning mitochondrial structure and function to criticality by fluctuation-driven mechanotransduction. Sci. Rep 10, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Henderson NC, Rieder F & Wynn TA Fibrosis: from mechanisms to medicines. Nature 587, 555–566 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Szeto SG et al. YAP/TAZ are mechanoregulators of TGF-b-smad signaling and renal fibrogenesis. J. Am. Soc. Nephrol 27, 3117–3128 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Martin K et al. PAK proteins and YAP-1 signalling downstream of integrin beta-1 in myofibroblasts promote liver fibrosis. Nat. Commun 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Knipe RS, Tager AM & Liao JK The rho kinases: Critical mediators of multiple profibrotic processes and rational targets for new therapies for pulmonary fibrosis. Pharmacol. Rev 67, 103–117 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Zhou Y et al. Inhibition of mechanosensitive signaling in myofibroblasts ameliorates experimental pulmonary fibrosis. J. Clin. Invest 123, 1096–1108 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Altschuler SJ & Wu LF Cellular Heterogeneity: Do Differences Make a Difference? Cell 141, 559–563 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Evers TMJ et al. Deciphering Metabolic Heterogeneity by Single-Cell Analysis. Anal. Chem 91, 13314–13323 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Ali A, Abouleila Y & Germond A An integrated raman spectroscopy and mass spectrometry platform to study single-cell drug uptake, metabolism, and effects. J. Vis. Exp 2020, (2020). [DOI] [PubMed] [Google Scholar]
- 146.Iskratsch T, Wolfenson H & Sheetz MP Appreciating force and shape-the rise of mechanotransduction in cell biology. Nature Reviews Molecular Cell Biology 15, 825–833 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Polacheck WJ & Chen CS Measuring cell-generated forces: A guide to the available tools. Nature Methods 13, 415–423 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Sheikh-Hasani V et al. Atorvastatin treatment softens human red blood cells: an optical tweezers study. Biomed. Opt. Express 9, 1256 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Efremov YM, Cartagena-Rivera AX, Athamneh AIM, Suter DM & Raman A Mapping heterogeneity of cellular mechanics by multi-harmonic atomic force microscopy. Nat. Protoc 13, 2200–2216 (2018). [DOI] [PubMed] [Google Scholar]
- 150.Eaton WA & Hofrichter J Sickle Cell Hemoglobin Polymerization. Adv. Protein Chem 40, 63–279 (1990). [DOI] [PubMed] [Google Scholar]