

Abstract
GNAO1 encephalopathy is a neurodevelopmental disorder with a spectrum of symptoms that include dystonic movements, seizures and developmental delay. While numerous GNAO1 mutations are associated with this disorder, the functional consequences of pathological variants are not completely understood. Here, we deployed the invertebrate C. elegans as a whole-animal behavioral model to study the functional effects of GNAO1 disorder-associated mutations. We tested several pathological GNAO1 mutations for effects on locomotor behaviors using a combination of CRISPR/Cas9 gene editing and transgenic overexpression in vivo. We report that all three mutations tested (G42R, G203R and R209C) result in strong loss of function defects when evaluated as homozygous CRISPR alleles. In addition, mutations produced dominant negative effects assessed using both heterozygous CRISPR alleles and transgenic overexpression. Experiments in mice confirmed dominant negative effects of GNAO1 G42R, which impaired numerous motor behaviors. Thus, GNAO1 pathological mutations result in conserved functional outcomes across animal models. Our study further establishes the molecular genetic basis of GNAO1 encephalopathy, and develops a CRISPR-based pipeline for functionally evaluating mutations associated with neurodevelopmental disorders.
Introduction
The human GNAO1 gene encodes Gαo, an α subunit of heterotrimeric G proteins that plays key roles in transducing G protein Coupled Receptor (GPCR) signals (1–3). Gαo is one of the most abundant membrane proteins in the brain. In the nervous system, Gαo plays important neuro-modulatory functions by coupling with various GPCRs, including dopamine, serotonin and opioid receptors (4–6). Mechanistically, the actions of Gαo in the nervous system are not completely understood, and a variety of signaling events and effectors that it influences have been described (7–10).
Numerous de novo GNAO1 mutations are associated with neurodevelopmental disorders, collectively referred to as GNAO1 encephalopathy (11–24). These encompass developmental and epileptic encephalopathy 17 (also called early infantile epileptic encephalopathy) [Online Mendelian Inheritance in Man (OMIM): 615473] (12,19), and neurodevelopmental disorder with involuntary movements [OMIM: 617493] (13–15). GNAO1 encephalopathy has a broad, emerging phenotypic spectrum. One core phenotype is impaired movement, which can include chorea, dystonia, and dyskinesia. Epilepsy and developmental delay are other common phenotypic characteristics.
Evaluation of GNAO-1 disorder-associated mutations in mice has recapitulated some of the phenotypes in GNAO1 encephalopathy including impaired movement and seizure susceptibility (25–27). Testing pathological GNAO1 mutations in other organisms will be valuable in assessing the conserved functional effects of these genetic perturbations, and their influence on movement. Gαo is highly conserved in invertebrates including the nematode C. elegans where its ortholog, G protein o-alpha subunit (GOA)-1, regulates locomotion (28,29). The extremely well-defined genetics of goa-1 in C. elegans makes this an ideal in vivo system for evaluating the functional impacts of pathological GNAO1 mutations.
To date, efforts to characterize GNAO1 pathological mutations at the molecular level have yielded conflicting results. An early study evaluated a pertussis toxin-insensitive version of Gαo using a heterologous cell-based assay (30). This placed pathological mutations in three categories: loss of function, gain of function, and normal function. Recent evaluation of unmodified Gαo indicated that GNAO1 mutations result in loss of function with several mutations reported to antagonize transduction of GPCR signals by acting as dominant negatives (27). Particularly notable are differing in vitro results with G203R, R209C and the less well characterized G42R mutation. These were described as gain or normal function initially (30), and were subsequently found to be loss of function and dominant negative (27). As a result of these differing conclusions, the functional effects and mechanisms of GNAO1 pathological mutations remain unresolved.
Intense interest has emerged in understanding Gαo function in the nervous system and developing intervention strategies for GNAO1 encephalopathy (31,32). Thus, there is a pressing need to use in vivo models to study the behavioral impacts of GNAO1 disorder-associated mutations. C. elegans provides an excellent opportunity to define the genetic mechanisms by which GNAO1 variants affect locomotor behavior, and could be key for resolving outstanding mechanistic issues pertaining to the molecular pathology of GNAO1 encephalopathy. Moreover, C. elegans has the potential to be developed as an in vivo platform capable of evaluating large numbers of GNAO1 variants, and could be used for genetic and small molecule screens targeting GNAO1.
In this study, we use C. elegans to evaluate the functional genetic effects of GNAO1 pathological mutations. The value of the C. elegans system is three-fold. It has a highly conserved Gαo ortholog, GOA-1 (28,33). C. elegans provided the first insight into Gαo function in the nervous system (34,35). Lastly, there are multiple, well-established behavioral paradigms to test Gαo function in worms (36–39). Thus, C. elegans is a simple, genetically well-defined system for evaluating the functional effects of GNAO1 mutations in vivo. We used CRISPR/Cas9 editing to model three controversial GNAO1 pathological mutations—G203R, R209C and G42R. Our results indicate that all three mutations result in loss of GOA-1/Gαo function. Further evaluation with monoallelic CRISPR mutations and transgenic overexpression experiments indicate that G42R and R209C mutations function as dominant negative alleles. Importantly, G42R dominant negative effects were confirmed upon its overexpression in the striatum and evaluation of motor behaviors in mice. Thus, in vivo functional genetics using multiple whole-animal models indicate that the pathological GNAO1 variants tested result in loss of function and act as dominant negative alleles. These findings showcase the value of the C. elegans genetic framework for investigating the molecular genetic effects of GNAO1 pathological mutations. Moreover, our results establish a cross-species functional genetic pipeline for evaluating mutations associated with neurodevelopmental disorders.
Results
Automated behavioral platform for evaluating Gαo function in C. elegans locomotion
We began by developing an automated platform for studying goa-1 function in vivo. Increasing or decreasing GOA-1 activity is well known to affect locomotor behavior producing hypoactive and hyperactive locomotion, respectively (Fig. 1A, B) (34,35,40). To validate our automated behavioral paradigms and ensure we can accurately resolve opposing locomotor phenotypes, we initially tested canonical goa-1 loss of function (LF) and gain of function (GF) mutants. The goa-1 LF allele (n363) was previously shown to be a null that lacks the goa-1 coding sequence (35,41). The goa-1 GF allele (n499) carries a classical GTPase inactivating mutation (R179C), which renders Gαo constitutively active (42–44). In automated locomotor assays on solid media, wild-type animals displayed an even pattern of sinusoidal movement (Fig. 1C). Consistent with prior observations, goa-1 GF mutants displayed flat waveform movement indicative of impaired locomotion (Fig. 1C). In contrast, we observed the opposite phenotype in goa-1 LF mutants—hyperactive movement consisting of exaggerated, compressed waveform (Fig. 1C). Quantitation confirmed significant, opposing locomotor deficits in goa-1 GF and LF mutants (Fig. 1D). Our second, automated behavioral paradigm evaluated swimming speed in liquid. Once again, goa-1 LF and GF mutants displayed opposing locomotor phenotypes in liquid (Fig. 1E, F). These results indicate that automated locomotor assays provide accurate, quantitative phenotypic readouts for genetic perturbations that clearly distinguish Gαo GF and LF activity.
Figure 1.
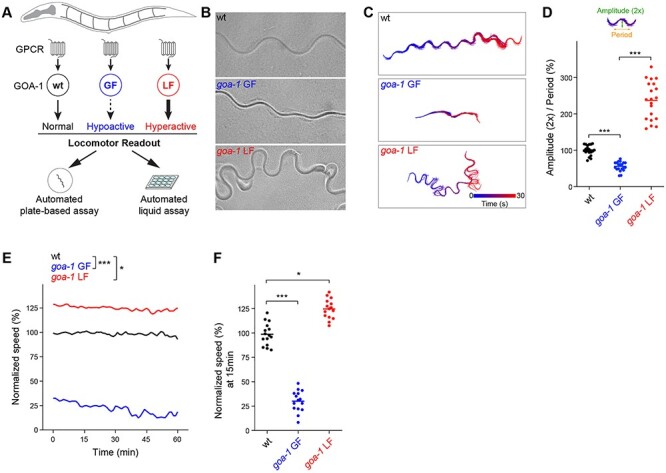
Automated behavioral tracking indicates that canonical goa-1 LF and GF mutants have opposing locomotor defects. (A) Schematic showing GOA-1/Gαo function in C. elegans locomotion and design of automated behavioral paradigms. (B) Representative images of C. elegans wave form traces in plate-based locomotion assays for indicated genotypes. (C) Representative traces of locomotor waveforms acquired using automated behavioral tracking for indicated genotypes. (D) Quantitation of locomotor waveforms for indicated genotypes (n = 20 animals per genotype). Shown are parameters (2× amplitude and period) used for quantitative analysis. (E) Time course of automated tracking in liquid locomotor assays for indicated genotypes (n = 15 wells; 60 ~ 75 total animals per genotype). (F) Quantitation of mean speed (line) and speed per well (circles) after 15 min of automated tracking for each genotype (n = 15 wells; 60–75 animals per genotype). In all cases, mean speed was calculated every minute for each well and normalized to wild type for baseline locomotion (5 min) and for body size. For panels D and F, comparisons represent one-way ANOVA followed by post hoc Bonferroni's test. For panel E, comparisons represent two-way ANOVA followed by post hoc Bonferroni's test. *P < 0.05, ***P< 0.001.
Biallelic CRISPR/Cas9 editing of GNAO1 pathological mutations in C. elegans results in loss of GOA-1 function
After establishing locomotor behavioral paradigms with canonical goa-1 LF and GF alleles, we functionally evaluated goa-1 harboring mutations orthologous to human GNAO1 pathological mutations (Fig. 2A). Sequence analysis identified very high evolutionary conservation (>80% identical amino acid sequence) between C. elegans GOA-1 and human Gαo/GNAO1 (Supplementary Material, Fig. S1) (28,33). Indeed, several GNAO1 mutations that we examined (11), map to conserved, identical residues in C. elegans GOA-1 (Supplementary Material, Fig. S1). To test the functional effects of pathological variants in vivo, we used CRISPR/Cas9 editing to introduce GNAO1 disorder-associated mutations into both alleles of endogenous goa-1 (Fig. 2A; Supplementary Material, Fig. S2A, C, E). We focused on three representative GNAO1-related mutations: G42R (18), G203R (12,14,19) and R209C (15,16,19) that were previously found to have differing outcomes using in vitro assays (Fig. 2B) (27,30). Intended CRISPR edits were confirmed by DNA sequencing (Supplementary Material, Fig. S2B, D, F).
Figure 2.
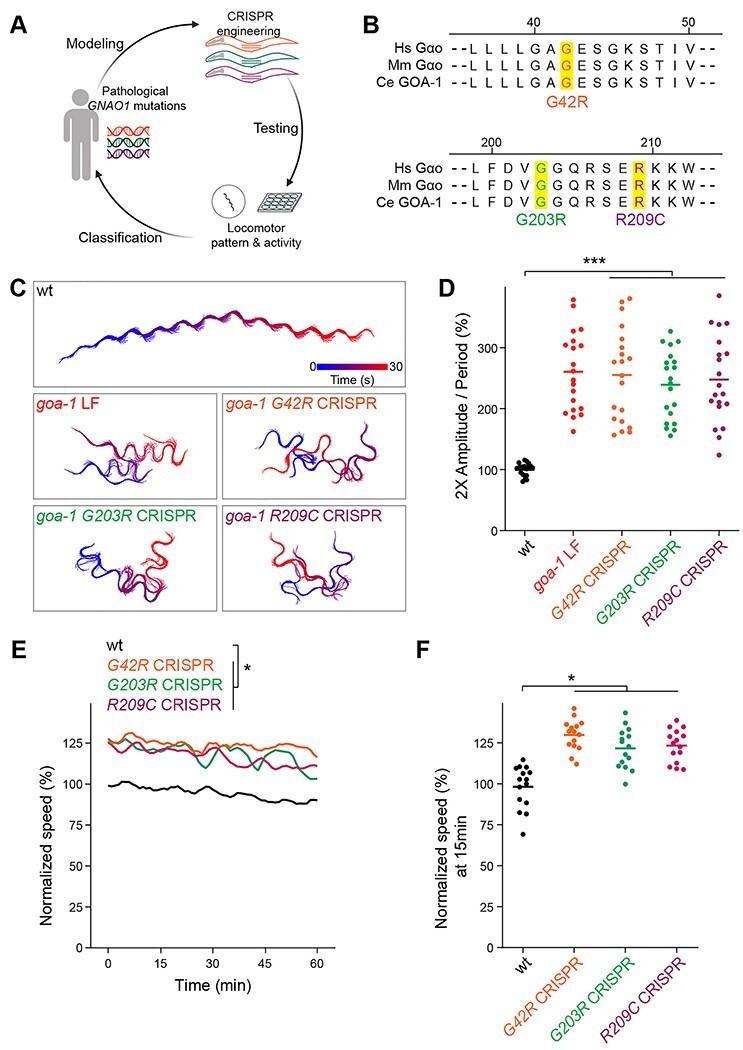
CRISPR editing GNAO1 pathological mutations into conserved GOA-1 residues results in loss of function. (A) Experimental pipeline for functional evaluation of GNAO1 pathological variants using CRISPR editing of goa-1/Gαo in C. elegans. (B) Evolutionary conservation of key regions and residues edited by CRISPR in Gαo from humans (Hs) mice (Mm) and C. elegans (Ce). (C) Representative automated behavioral traces in plate-based locomotor assays for indicated genotypes. (D) Quantitation of plate-based locomotor assays for indicated genotypes (n = 20 animals per genotype). (E) Time course of automated tracking in liquid locomotor assays for indicated genotypes (n = 15 wells; 60–75 total animals per genotype). (F) Quantitation of mean speed (line) and speed per well (circles) after 15 min of automated tracking for each genotype (n = 15 wells; 60–75 animals per genotype). In all cases, mean speed was calculated every minute for each well and normalized to wild type for baseline locomotion (5 min) and for body size. For panels D and F, comparisons represent one-way ANOVA followed by post hoc Bonferroni's test. For panel E, comparisons represent two-way ANOVA followed by post hoc Bonferroni's test. *P < 0.05, ***P < 0.001.
Automated tracking of locomotor patterns on solid media revealed that all three homozygous CRISPR mutants displayed hyperactive locomotor waveforms that were indistinguishable from goa-1 LF mutants (Fig. 2C). Quantitation confirmed significant hyperactivity in CRISPR animals carrying GNAO1-related pathological mutations compared to wild-type animals (Fig. 2D). G42R, G203R, and R209C homozygous CRISPR mutants also showed significant levels of continuous hyperactivity in automated locomotor assays performed in liquid (Fig. 2E, F). This once again phenocopied the behavior of goa-1 LF mutants (Fig. 1E, F). Thus, pathological GNAO1-related mutations result in locomotor behavior consistent with goa-1 LF with biallelic, homozygous gene editing.
GNAO1 pathological mutations phenocopy Gαo loss of function during pharmacological manipulation of the motor circuit
Mechanistically, GOA-1 transduces GPCR signals to negatively regulate presynaptic acetylcholine (Ach) release from motor neurons to control C. elegans locomotion (37,38,45). The C. elegans motor circuit can be pharmacologically manipulated with aldicarb, an acetylcholinesterase (AchE) inhibitor. Aldicarb causes accumulation of Ach, hypercontraction of muscles and paralysis (Fig. 3A). Because GOA-1 negatively regulates presynaptic Ach release, goa-1 LF mutants have augmented Ach release that leads to aldicarb hypersensitivity (Fig. 3A).
Figure 4.
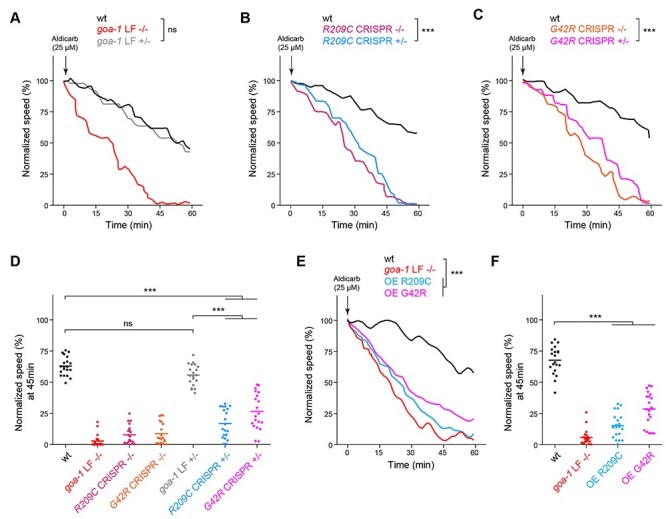
Multiple genetic approaches demonstrate GNAO1 pathological mutations functions as dominant negative alleles in C. elegans. (A) Automated aldicarb assay showing homozygous animals carrying a goa-1 LF null allele are hypersensitive to aldicarb. In contrast, heterozygous goa-1 LF+/− animals show normal aldicarb responses compared to wild type. (B) Automated aldicarb assays show both heterozygous and homozygous goa-1 R209C CRISPR mutants are hypersensitive to aldicarb. (C) Automated aldicarb assays show both heterozygous and homozygous goa-1 G42R CRISPR mutants are hypersensitive to aldicarb. (D) Quantitation of mean speed (line) and speed per well (circles) after 45 min of tracking in automated aldicarb assays for indicated genotypes. (E) Automated aldicarb assay showing transgenic overexpression of GOA-1 R209C and G42R induce aldicarb hypersensitivity. (F) Quantitation of mean speed (line) and speed per well (circles) after 45 min of tracking in automated aldicarb assays for indicated genotypes. In all cases, mean speed was calculated every minute for each well and normalized to baseline locomotion (10 min prior to addition of aldicarb) for each genotype. For E and F, data shown is from 5 transgenic lines for each genotype. Data for individual transgenic lines is shown in Supplementary Material, Fig. S3. For panels A-C and E, comparisons represent two-way ANOVA followed by post hoc Bonferroni's test. For panels D and F, comparisons represent one-way ANOVA followed by post hoc Bonferroni's test. For all experiments, n = 20 wells; 80–100 animals per genotype. ***P < 0.001.
Figure 3.
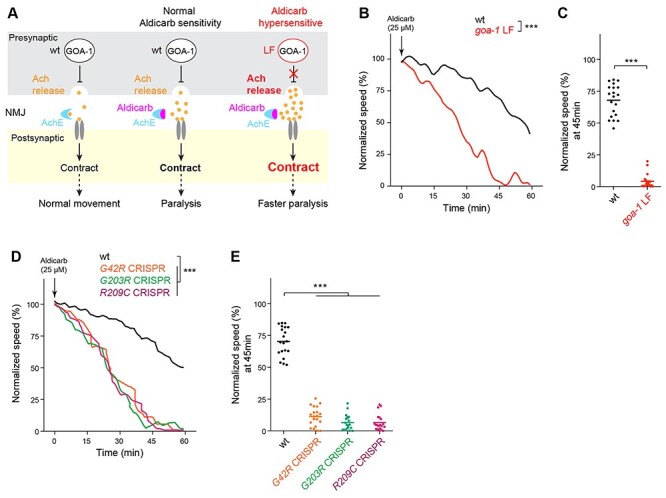
GNAO1 pathological mutations cause loss of function effects during pharmacological manipulation of the C. elegans motor circuit. (A) Diagram illustrating pharmacological manipulation of the C. elegans motor circuit using the acetylcholinesterase inhibitor aldicarb (left, middle). goa-1 LF increases excitatory Ach release in the C. elegans motor circuit resulting in hypersensitivity to aldicarb (right). (B) Automated aldicarb assay shows canonical goa-1 LF mutants are hypersensitive to aldicarb. Arrow indicates drug application. (C) Quantitation of mean speed (line) and speed per well (circles) after 45 min of tracking in automated aldicarb assays for indicated genotype. (D) Automated aldicarb assays show three CRISPR edited mutants that are homozygous for GNAO1 pathological mutations (G42R, G203R, R209C) display aldicarb hypersensitivity. (E) Quantitation of mean speed (line) and speed per well (circles) after 45 min of tracking in automated aldicarb assays for indicated genotypes. In all cases, mean speed was calculated every minute for each well and normalized to baseline locomotion (10 min prior to addition of aldicarb) for each genotype. For panels B and D, comparisons represent two-way ANOVA followed by post hoc Bonferroni's test. For panel C, comparison represents two-tailed unpaired Student’s t-test. For panel E, comparisons represent one-way ANOVA followed by post hoc Bonferroni's test. For all experiments, n = 20 wells; 80 ~ 100 animals per genotype. ***P < 0.001.
To measure the impact of GNAO1 variants, we employed our previously developed automated liquid assay for aldicarb (46). This automated approach has higher throughput, greater quantitative accuracy, and is unbiased. These are substantial improvements over manual, plate-based approaches traditionally used. As expected, exposure to aldicarb suppressed locomotor activity of wild-type animals (Fig. 3B, C). goa-1 LF mutants showed substantial hypersensitivity as evidenced by faster paralysis (Fig. 3B, C). Importantly, all three CRISPR edited mutants carrying GNAO1-related mutations exhibited dramatic aldicarb hypersensitivity when tested as homozygous alleles (Fig. 3D, E). Thus, results from pharmacological manipulation of the motor circuit further demonstrate that three GNAO1 disorder-associated mutations (G42R, G203R, and R209C) result in strong loss of goa-1 function.
Monoallelic GNAO1-related mutations produce dominant negative effects in C. elegans
In a clinical setting, GNAO1 mutations are heterozygous. As a result, mutant versions of Gαo are expressed simultaneously with wild-type protein. To better model genetic changes that occur in GNAO1 disorders, we CRISPR edited monoallelic GNAO1 pathological mutations into goa-1 to test if this is sufficient to cause phenotypes.
To begin, it was essential to evaluate heterozygous animals carrying a canonical goa-1 LF null allele (goa-1 LF +/−). goa-1 LF +/− animals respond normally to aldicarb similar to wild-type animals (Fig. 4A, D). This differs from goa-1 LF homozygous mutants (goa-1 LF −/−) which are aldicarb hypersensitive (Fig. 4A, D). Thus, goa-1 does not display haploinsufficiency in our system. In contrast, heterozygous R209C CRISPR +/− mutants showed hypersensitive aldicarb responses that were significant compared to wild-type animals (Fig. 4B, D), and resembled homozygous R209C CRISPR −/− mutants (Fig. 4B, D). Heterozygous G42R CRISPR +/− mutants also displayed significant aldicarb hypersensitivity compared to wild type (Fig. 4C, D), and were similar to homozygous G42R CRISPR −/− mutants (Fig. 4C, D). Importantly, both heterozygous G42R +/− and R209C +/− animals showed significant aldicarb hypersensitivity compared to goa-1 LF +/− heterozygotes, which carry a single copy of a goa-1 LF null allele (Fig. 4D). Taken in the context of our previous observations, which indicated that G42R and R209C mutations do not affect Gαo protein expression (27), these results suggest that G42R and R209C mutations act as dominant negative alleles that antagonize the function of wild-type GOA-1/Gαo.
Figure 5.
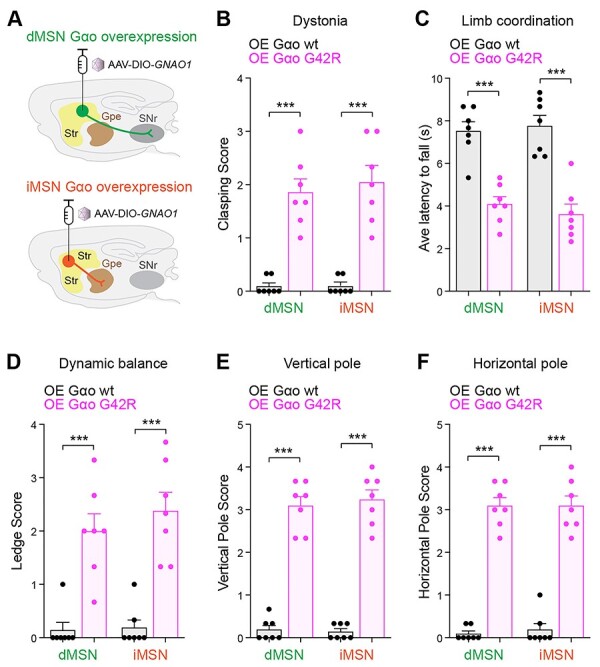
Overexpression of GNAO1 G42R in two populations of striatal neurons impairs locomotor behaviors in mice. (A) Schematic showing adeno-associated viral (AAV) particle delivery and overexpression of wt or G42R Gαo in two striatal neuron populations, dMSN and iMSN. (B) Quantitation of hindlimb clasping which indicates increased dystonia. (C) Quantitation of limb coordination based on latency to fall in backward walking test. (D, E & F) Quantitation of three motor coordination and balance tests (D) ledge test, (E) vertical pole test and (F) horizontal pole test. Comparisons represent two-tailed unpaired Student’s t-test. n = 7 animals per genotype. Error bars are SEM. ***P < 0.001.
Overexpressing GNAO1-related mutations causes dominant negative effects in C. elegans
To further test dominant negative activity of GNAO1-related mutations, we performed transgenic overexpression studies. To do so, we used a pan-neuronal promoter to transgenically overexpress GOA-1 G42R or R209C in wild-type animals. Indeed, we observed aldicarb hypersensitivity with overexpression of GOA-1 R209C or G42R (Fig. 4E, F;Supplementary Material, Fig. S3). This is similar to what occurs in goa-1 LF−/− animals (Fig. 4E, F). Thus, transgenic overexpression experiments in C. elegans provide further evidence that GNOA1 pathological mutations act mechanistically as dominant negative alleles.
Overexpression of pathological GNAO1 G42R mutation impairs motor behaviors in mice
Layers of independent genetic studies in C. elegans demonstrated that G42R is a dominant negative allele. To test whether this is also the case in the mammalian brain, we performed viral expression studies coupled with behavioral evaluation in mice. In the mammalian striatum, Gαo functions in D1 and D2 dopamine receptor-expressing medium spiny neurons (MSN) to control movement (27,47,48). Therefore, we tested how overexpressing a representative Gαo with dominant negative effects, G42R, affects a range of motor behaviors. Gαo constructs were overexpressed with circuit specificity in either direct MSNs (dMSNs) or indirect MSNs (iMSNs) using adeno-associated viral (AAV) particles, which were stereotaxically injected into the dorsal striatum of animals containing two wild-type copies of Gαo (Fig. 5A).
Figure 6.
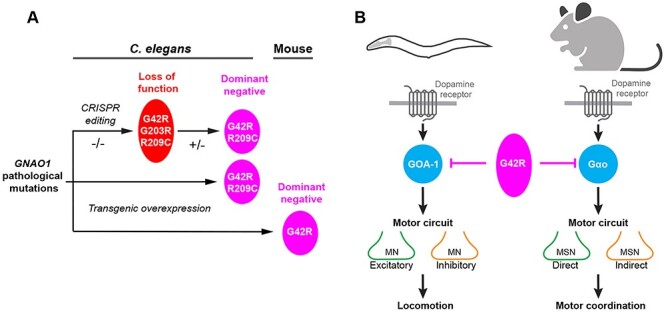
Summary of functional genetic pipeline and outcomes with GNAO1 pathological mutations using C. elegans and mice. (A) Schematic summarizing cross-species genetic pipeline using CRISPR and transgenic approaches to evaluate GNAO1 pathological mutations in vivo. G42R, G203R and R209C disorder-associated mutations were found to impair Gαo/GOA-1 function. G42R and R209C mutations result in dominant negative effects across multiple functional genetic assays in C. elegans and mice. (B) Summary showing dominant negative effects of GNAO1 G42R pathological variant across C. elegans and mice, and similarities between Gαo function in locomotor behaviors of both C. elegans and mice.
We surveyed a wide array of motor behaviors in these animals beginning with spontaneous hind limb clasping, which evaluates dystonic movement. G42R Gαo overexpression in both dMSNs and iMSNs increased hindlimb clasping, while overexpressing wild-type Gαo had no effect (Fig. 5B). Next, we turned to a backward walking task to evaluate limb coordination. This was impaired by overexpressing the G42R Gαo variant, but not wild-type Gαo, in either dMSNs or iMSNs (Fig. 5C). To solidify these findings, we evaluated balance and motor coordination using three additional behavioral tasks: the ledge test (Fig. 5D; Supplementary Material, Movie S1-S2), the vertical pole test (Fig. 5E) and the horizontal pole test (Fig. 5F; Supplementary Material, Movie S3-S4). G42R Gαo overexpressed in dMSNs or iMSNs significantly impaired performance in all three assays compared to overexpression of wild-type Gαo (Fig. 5D-F). Thus, overexpressing G42R Gαo in either major population of dopamine-modulated striatal neurons led to profound deficits in movement control. Taken as a whole, our results with both heterozygous CRISPR alleles and transgenic overexpression demonstrate that GNOA1 pathological mutations have dominant negative effects across multiple animal models.
Discussion
GNAO1 encephalopathy is characterized by de novo heterozygous mutations in the GNAO1 gene. This emerging neurodevelopmental disorder displays a broad spectrum of symptoms including impaired motor coordination, developmental delay and epilepsy. To date, a fundamental question regarding GNAO1 encephalopathy remains unanswered: What are the functional effects of GNAO1 pathological mutations? Here, we use a range of locomotor behavioral readouts across model systems to demonstrate several outcomes (Fig. 6A, B). First, all three GNAO1 pathological mutations we tested (G42R, G203R and R209C) result in loss of function when evaluated as homozygous alleles. Second, G42R and R209C disorder-associated mutations function as dominant negative alleles. This was demonstrated in C. elegans using the two principal approaches for dominant negative genetic classification, evaluation of heterozygous alleles and overexpression in wild-type animals. Finally, overexpression of the G42R Gαo construct in striatal MSN populations resulted in impaired motor coordination. This complements our prior finding that overexpression of the G203R and R209C Gαo constructs result in similar outcomes in rodents (27). Thus, collective results from multiple, whole-animal behavioral models indicate that the G42R and R209C pathological mutations function as dominant negative alleles in vivo.
Functional evaluation of GNAO1 pathological mutations is essential, but grew more pressing when recent studies drew different conclusions about how these mutations affect Gαo activity (27,30). For example, G42R and R209C/H were found to have opposite effects (loss versus gain of function) in cell-based signaling assays. This has led to confusion about the functional genetic nature of GNAO1 pathological mutations. Our study aimed to provide clarity on this important topic by bringing another in vivo behavioral genetic model, the invertebrate C. elegans, to bear on this question for the first time.
As an initial foray into understanding the functional effects of GNAO1 pathological variants in the C. elegans system, we focused on three representative mutations. 1) G203R, the first mutation identified in GNAO1 encephalopathy (12,19). 2) R209C, which affects a residue subject to four different disorder-associated mutations (R209C, R209H, R209G, R209L) (11). 3) The rare variant G42R (18). We pursued G42R because independent cell-based studies arrived at opposing conclusions about its effect on Gαo activity (27,30), and this pathological variant has not been tested in vivo. We evaluated these three mutations in a range of locomotor behaviors, as well as testing how they affect pharmacological manipulation of the C. elegans motor circuit. We found that G203R, R209C and G42R disorder-associated mutations all result in loss of function as homozygous alleles. A result that is consistent with our previous study that evaluated these three GNAO1 mutations in cell-based signaling assays with Gαo (27). Further findings here using well-established, behavioral genetic assays for Gαo function in C. elegans indicate that G42R and R209C function as dominant negative alleles. Importantly, our results demonstrate that G42R dominant negative activity also occurs in a wide range of motor assays in mice. Thus, dominant negative activity is a conserved genetic mechanism of action for the G42R mutation.
Our previous cell-based work provided insight into how different GNAO1 pathological variants can affect GPCR signaling (27). The R209C and G42R pathological mutations that we show here have dominant negative effects in vivo influence Gαo via distinct effects on GPCR signaling based on studies in transfected cells. They both inhibit dissociation of Gβγ upon activation. In addition, the G42R mutation also disrupts heterotrimer formation and impairs its association with activated GPCR (27). Thus, G protein activation/deactivation cycles are disrupted in both cases. R209C was further found to have dominant negative effects in both cell based and rodent behavioral assays (27). Our findings here using the C. elegans model provide further evidence that the R209C pathological variant utilizes a dominant negative mechanism (Fig. 6). The G42R pathological mutation was not tested in vivo using rodents in our previous study. Here, we show for the first time in both C. elegans and rodents that this mutation also relies upon a dominant negative mechanism (Fig. 6). Our findings for G42R are consistent with prior predictions that this mutation might act as a dominant negative based on results from filamentous fungi (49). The exact signaling mechanism by which G42R elicits dominant negative activity remains to be determined. Collectively, our findings and these prior observations indicate that GNAO1 pathological variants can differentially affect GPCR signaling to cause dominant negative genetic outcomes.
Our results with C. elegans show that all three disorder-associated mutations we tested, via CRISPR editing of the native goa-1/Gαo locus, result in abnormal locomotor behavior. Thus, prior rodent studies and our findings here now point to emerging, common principles for how GNAO1 pathological variants affect Gαo function in vivo. This is quite reasonable given several similarities between locomotor behaviors in C. elegans and rodent models. The C. elegans motor circuit (consisting of both excitatory cholinergic and inhibitory GABAergic motor neurons) and GABAergic striatal neurons (consisting of dMSNs and iMSNs) that regulate motor coordination in mice are both sensitive to dopaminergic modulation (Fig. 6B) (40,50–52). In both C. elegans and mice, loss of Gαo function leads to hyperactive locomotion (34,35,53). Finally, Gαo signaling inhibits neuronal activity in C. elegans and mammals (2,38,41,54,55). It is likely that these conserved features of C. elegans and rodent locomotor programs have worked to our advantage in profiling GNAO1 variants.
The present study has not explored how GNAO1 pathological mutations affect Gαo function in different types of neurons in C. elegans. Our findings with pharmacological manipulation of the worm motor circuit using the acetylcholinesterase inhibitor aldicarb likely reflects functional effects of Gαo in cholinergic motor neurons. This is supported by single-cell transcriptional profiling of the C. elegans nervous system that showed enriched expression of Gαo/GOA-1 in cholinergic neurons (56), and prior studies with Gαo and aldicarb (37). However, it is notable that Gαo is also enriched in GABAergic and dopaminergic neurons of C. elegans (56). Cell-specific CRISPR editing could enable functional evaluation of GNAO1 variants specifically in cholinergic, GABAergic or dopaminergic neurons. While this would be technically challenging, it may be an important next step for research on GNAO1 pathological mutations using C. elegans.
Our study here focused on how GNAO1 pathological mutations affect locomotor behaviors in C. elegans and rodents. This is because one principal phenotype in GNAO1 encephalopathy is impaired movement. However, it important to note that seizures also significantly contribute to symptoms of GNAO1 encephalopathy. To date, much less is known about how pathological GNAO1 variants increase risk of seizures. Interestingly, C. elegans has emerged as a valuable model to study seizures (57–61). Our findings here show that GNAO1 pathological mutations can be evaluated using C. elegans with outcomes that are relevant to mammals. This encourages further studies in C. elegans aimed at evaluating how GNAO1 variants affect seizures.
Overall, our findings demonstrate that C. elegans is a valuable in vivo system for evaluating the functional genetic effects of GNAO1 pathological mutations. This contributes to growing evidence that C. elegans has utility for studying the molecular genetic basis of neurodevelopmental disorders (62–68). Indeed, C. elegans could be an ideal tool for functionally evaluating the large numbers of GNAO1 pathological variants identified to date. A list of mutations that seems likely to grow with time, as will the challenge of functional classification.
Materials and Methods
C. elegans strains and genetics
C. elegans strains were maintained using standard protocols and were generated using the N2 isolate. The following transgene and mutant alleles were used: muIs32 [Pmec-7GFP], goa-1 (n499, gain-of-function), goa-1 (n363, loss-of-function/null), goa-1 G42R CRISPR (bgg44), goa-1 G203R CRISPR (bgg45) and goa-1 R209C CRISPR (bgg46). See Supplementary Material, Tables S1-S3 for specific details about alleles, transgenic strains, CRISPR/Cas9 reagents and transgene microinjection conditions.
CRISPR/Cas9 gene editing
CRISPR/Cas9 gene editing with ribonucleoprotein complexes and homology-directed repair was used to engineer goa-1 with human GNAO1 pathological mutations (69). In brief, 42 nt-length crRNA, 74 nt-length tracrRNA, and repair template (ssDNA containing ~ 35 nt homology arms) were synthesized (Dharmacon and IDT). Recombinant 6 × His-Cas9 protein was purified from E. coli BL21. Assembled Cas9-crRNA-tracrRNA complexes with repair templates (Supplementary Material, Table S2; Supplementary Material, Fig. S2) were pre-incubated for 15 min at 37°C and microinjected into C. elegans. dpy-10 co-CRISPR was used to facilitate the isolation of gene-edited animals. All CRISPR gene edits were confirmed by PCR genotyping (with restriction enzyme digest) and DNA sequencing (Supplementary Material, Fig. S2). To mitigate possible off-target effects of CRISPR editing, all CRISPR edited animals were outcrossed four times to wild-type animals.
Molecular biology
GOA-1 expressing plasmids were generated as follows. N2 cDNA was obtained by RT-PCR (SuperScriptTM IV First-Strand Synthesis System, Invitrogen) and then wild-type goa-1 cDNA was amplified using High-Fidelity DNA Polymerase (iProof, Bio-Rad). goa-1 cDNA was cloned into pCR8 vector and underwent point-mutagenesis to create G42R and R209C mutations. pCR8-based goa-1 (wt, G42R and R209C) entry vectors were recombined with destination vector (pBG-GY152) containing the pan-neuronal expression promoter Prgef-1. All plasmids were confirmed by DNA sequencing.
Tests for dominant negative effects in C. elegans
We evaluated dominant-negative effects using goa-1+/− (LF, G42R or R209C) hermaphrodites. Heterozygous animals were generated by crossing males containing a transgenic selection marker, muIs32 (Pmec-7GFP), with homozygous goa-1 alleles. Heterozygotes F1 animals were isolated using the transgenic GFP reporter and evaluated for locomotor behavior and pharmacological manipulation of the motor circuit using aldicarb.
To evaluate dominant-negative effects by transgenic overexpression, GOA-1 G42R or R209C were overexpressed using transgenic extrachromosomal arrays. GOA-1 transgenes were expressed using the pan-neuronal rgef-1 promoter. For each genotype, 5 independent transgenic lines were isolated and tested in aldicarb assays.
C. elegans automated behavioral assays
C. elegans were synchronized (egg laying for 4 h) and grown at 20°C to adulthood. All experiments were performed at room temperature. Multi-Worm Tracker (MWT) was used to analyze animal behaviors (46).
For the plate-based locomotion assay, 5 adults were placed on a single NGM plate without food and tracks were monitored for 5 min. Waveform traces for single animals were generated using custom-written scripts. The parameters of amplitude and period were measured by ImageJ from images. The measure of 2× amplitude/period was used to quantify waveform traces. For each genotype, data was collected from 20 animals obtained from 4 independent experiments.
For the liquid locomotor swimming assay, adult animals were placed in 15 μL assay buffer (M9 + 0.01% Tween-20) on the lid of a 96 well plate. Each assay well contained 4–5 animals. Tracking was initiated after 5 min of baseline recording, paused to add 15 μL assay buffer, and tracking was resumed continuously for 60 min. Mean speed was calculated every minute for each well using custom-written scripts and normalized to baseline based on wild-type animals and also normalized for body size. For each genotype, data was collected from 15 wells obtained from 3 independent experiments.
For the aldicarb assay, adults were placed in 20 μL assay buffer in the lid of a 96 well plate. Each well contained 4–5 animals. Tracking was initiated after 10 min of baseline recording, paused to add 10 μL of aldicarb (75 μM; Aldicarb PESTANAL®, Sigma) and resumed for continuous tracking for 60 min. Mean speed was calculated every minute for each well using custom-written scripts and normalized to baseline locomotion and body size for each genotype. For each genotype, data was collected from 20 wells obtained from 4 independent experiments.
Mouse strains
All experimental procedures and work utilizing mice were approved by The Scripps Research Institute’s IACUC committee in compliance with guidelines set by the NIH. The mice were maintained under standard housing conditions in a pathogen-free facility under a 12/12 light/dark cycle where all mice had continuous access to food and water. Drd1aCre (Drd1-Cre; EY262; stock# 017264-UCD) and Drd2Cre (Drd2-Cre; ER43; Stock #: 017268-UCD) mouse lines were obtained from the Mutant Mouse Resource & Research Centers (MMRRC). Behavioral studies utilized both male and female mice. All experiments were performed on mice between 3–5 months old.
Mouse behavioral studies
Hindlimb clasping
As previously described (70), mice (males and females, approximately 3–5 months old) were held by base of tail, lifted in the air, and observed for 30 s. Animals were scored as follows: no clasping (0), clasping of 1 hindlimb part of the time (1), clasping of 1 hindlimb the entire time (2), clasping of both hindlimbs part of the time (3) and clasping of both hindlimbs the entire time (4). Animals were tested and scored once a day for 3 days.
Backwards walking
Mice (males and females, approximately 3–5 months old) were placed into RotaRod apparatus (IITC Life Science Inc., Woodland Hills, CA USA) and made to walk backwards. RotaRod was fitted to ensure that mice could not turn and walk forward. Animals had to walk backwards from 1 s (beginning at 8.1 RPM) to 10 s (ending at 9.15 RPM). Each mouse was tested once a day for 3 days. Latency to fall was recorded.
Ledge test
As previously described (70), mice (males and females, approximately 3–5 months old) were individually placed onto lip of house cage (Allentown Inc., Allentown, NJ USA) and observed for balancing and movement. Animals were scored as followed: balancing and walking well (0), good balance but teetering walk (1), teetering in balance and walk (2), teetering in balance but unable to walk (3) and falling off (4). Animals were tested and scored once a day for 3 days.
Vertical pole
As previously described (71), mice (males and females, approximately 3–5 months old) were placed nose facing up on a wooden pole (1 cm diameter) at 50 cm in height from bottom of mouse cage (Allentown Inc., Allentown NJ USA). In order to successfully complete this task with a score of 0, subjects had to turn around (nose facing down) and proceed down the pole. Subjects that turned around and climbed down the pole with some difficulty (scored 1), climbed down the pole without turning around (2), slid down the pole (3) and fell off the pole (4). Due to the nature of this study, there was no cut off time. Animals were tested and scored three times on the same day.
Horizontal pole
As previously described (72), mice (males and females, approximately 3–5 months old) were placed 50 cm away from home cage (facing towards home cage) on a 1 cm diameter wooden pole. Mice were scored as followed: normal gait and balance to home cage (0), normal gait but unbalanced to home cage (1), both poor gait and balance to home cage (2), unable to complete task due to lack of movement (3) and falling off pole (4). Cut-off time for sessions was 120 s. Animals were tested and scored three times on the same day.
Adeno-associated viruses (AAV) and stereotaxic injections
Mice were anesthetized with isoflurane and their head fixed on a Kopf stereotaxic apparatus. Animals were kept warm (~37°C) for the whole duration of the surgery via a heating pad connected to a DC temperature controller provided with a feedback system (FHC Inc.). Eye lubricant was applied to prevent corneal drying during surgery. Adeno-associated virus (AAV) encoding the fluorescent protein EYFP (AAV5-EF1a-DIO-EYFP) was obtained from the Vector Core at the University of North Carolina at Chapel Hill (UNC Vector Core, USA). AAV encoding GNAO1 variants (AAV9-Syn-DIO-Gao-IRES-mCherry) were obtained from VectorBuilder (Chicago, IL). Viral injections were targeted to the dorsal striatum (AP +0.7, ML ±1.5 relative to bregma, DV -1.7 relative to dura) of Drd1aCre (to target dMSN) or Drd2Cre (to target iMSN). Injection volume (300 nl) and flow rate (50 nl/min) were controlled with an injection pump (Cemyx Nanojet, USA). The needle was left in place for 5 min after the injection and then slowly withdrawn. Mice were allowed to recover for at least 15 days before behavioral experiments.
Supplementary Material
Acknowledgements
The authors would like to thank Ms Natalia Martemyanova for producing and maintaining mice examined in this study.Conflict of Interest statement. K.A.M. serves on the scientific advisory boards of the Bow Foundation and the Child's Cure Research Foundation.
Contributor Information
Dandan Wang, Center for Integrative Brain Research, Seattle Children’s Research Institute, Seattle, WA 98101, USA.
Maria Dao, Department of Neuroscience, The Scripps Research Institute, Jupiter, FL 33458, USA.
Brian S Muntean, Department of Neuroscience, The Scripps Research Institute, Jupiter, FL 33458, USA.
Andrew C Giles, Department of Neuroscience, The Scripps Research Institute, Jupiter, FL 33458, USA.
Kirill A Martemyanov, Department of Neuroscience, The Scripps Research Institute, Jupiter, FL 33458, USA.
Brock Grill, Center for Integrative Brain Research, Seattle Children’s Research Institute, Seattle, WA 98101, USA; Department of Pediatrics, University of Washington School of Medicine, Seattle, WA, USA; Department of Pharmacology, University of Washington School of Medicine, Seattle, WA, USA.
Funding
The National Institutes of Health (grants R01 DA048036 to B.G. and K.A.M.), R01 DA036596 (to K.A.M.); Bow Foundation grant (B.S.M.).
References
- 1. Hepler, J.R. and Gilman, A.G. (1992) G proteins. Trends Biochem. Sci., 17, 383–387. [DOI] [PubMed] [Google Scholar]
- 2. Jiang, M. and Bajpayee, N.S. (2009) Molecular mechanisms of go signaling. Neurosignals, 17, 23–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Sternweis, P.C. and Robishaw, J.D. (1984) Isolation of two proteins with high affinity for guanine nucleotides from membranes of bovine brain. J. Biol. Chem., 259, 13806–13813. [PubMed] [Google Scholar]
- 4. de Oliveira, P.G., Ramos, M.L.S., Amaro, A.J., Dias, R.A. and Vieira, S.I. (2019) Gi/o-protein coupled receptors in the aging brain. Front. Aging Neurosci., 11, 89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Masuho, I., Ostrovskaya, O., Kramer, G.M., Jones, C.D., Xie, K. and Martemyanov, K.A. (2015) Distinct profiles of functional discrimination among G proteins determine the actions of G protein-coupled receptors. Sci. Signal., 8, ra123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Hescheler, J., Rosenthal, W., Trautwein, W. and Schultz, G. (1987) The GTP-binding protein, Go, regulates neuronal calcium channels. Nature, 325, 445–447. [DOI] [PubMed] [Google Scholar]
- 7. VanDongen, A.M., Codina, J., Olate, J., Mattera, R., Joho, R., Birnbaumer, L. and Brown, A.M. (1988) Newly identified brain potassium channels gated by the guanine nucleotide binding protein Go. Science, 242, 1433–1437. [DOI] [PubMed] [Google Scholar]
- 8. Ewald, D.A., Pang, I.H., Sternweis, P.C. and Miller, R.J. (1989) Differential G protein-mediated coupling of neurotransmitter receptors to Ca2+ channels in rat dorsal root ganglion neurons in vitro. Neuron, 2, 1185–1193. [DOI] [PubMed] [Google Scholar]
- 9. Purvanov, V., Koval, A. and Katanaev, V.L. (2010) A direct and functional interaction between Go and Rab5 during G protein-coupled receptor signaling. Sci. Signal., 3, ra65. [DOI] [PubMed] [Google Scholar]
- 10. Solis, G.P., Bilousov, O., Koval, A., Luchtenborg, A.M., Lin, C. and Katanaev, V.L. (2017) Golgi-resident Galphao promotes protrusive membrane dynamics. Cell, 170, 939, e924–955. [DOI] [PubMed] [Google Scholar]
- 11. Schirinzi, T., Garone, G., Travaglini, L., Vasco, G., Galosi, S., Rios, L., Castiglioni, C., Barassi, C., Battaglia, D., Gambardella, M.L.et al. (2019) Phenomenology and clinical course of movement disorder in GNAO1 variants: results from an analytical review. Parkinsonism Relat. Disord., 61, 19–25. [DOI] [PubMed] [Google Scholar]
- 12. Nakamura, K., Kodera, H., Akita, T., Shiina, M., Kato, M., Hoshino, H., Terashima, H., Osaka, H., Nakamura, S., Tohyama, J.et al. (2013) De novo mutations in GNAO1, encoding a Galphao subunit of heterotrimeric G proteins, cause epileptic encephalopathy. Am. J. Hum. Genet., 93, 496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Ananth, A.L., Robichaux-Viehoever, A., Kim, Y.M., Hanson-Kahn, A., Cox, R., Enns, G.M., Strober, J., Willing, M., Schlaggar, B.L., Wu, Y.W.et al. (2016) Clinical course of six children with GNAO1 mutations causing a severe and distinctive movement disorder. Pediatr. Neurol., 59, 81–84. [DOI] [PubMed] [Google Scholar]
- 14. Kulkarni, N., Tang, S., Bhardwaj, R., Bernes, S. and Grebe, T.A. (2016) Progressive movement disorder in brothers carrying a GNAO1 mutation responsive to deep brain stimulation. J. Child Neurol., 31, 211–214. [DOI] [PubMed] [Google Scholar]
- 15. Danti, F.R., Galosi, S., Romani, M., Montomoli, M., Carss, K.J., Raymond, F.L., Parrini, E., Bianchini, C., McShane, T., Dale, R.C.et al. (2017) GNAO1 encephalopathy: broadening the phenotype and evaluating treatment and outcome. Neurol. Genet., 3, e143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kelly, M., Park, M., Mihalek, I., Rochtus, A., Gramm, M., Perez-Palma, E., Axeen, E.T., Hung, C.Y., Olson, H., Swanson, L.et al. (2019) Spectrum of neurodevelopmental disease associated with the GNAO1 guanosine triphosphate-binding region. Epilepsia, 60, 406–418. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kim, S.Y., Shim, Y., Ko, Y.J., Park, S., Jang, S.S., Lim, B.C., Kim, K.J. and Chae, J.H. (2020) Spectrum of movement disorders in GNAO1 encephalopathy: in-depth phenotyping and case-by-case analysis. Orphanet J. Rare Dis., 15, 343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Zhu, X., Petrovski, S., Xie, P., Ruzzo, E.K., Lu, Y.F., McSweeney, K.M., Ben-Zeev, B., Nissenkorn, A., Anikster, Y., Oz-Levi, D.et al. (2015) Whole-exome sequencing in undiagnosed genetic diseases: interpreting 119 trios. Genet. Med., 17, 774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Saitsu, H., Fukai, R., Ben-Zeev, B., Sakai, Y., Mimaki, M., Okamoto, N., Suzuki, Y., Monden, Y., Saito, H., Tziperman, B.et al. (2016) Phenotypic spectrum of GNAO1 variants: epileptic encephalopathy to involuntary movements with severe developmental delay. Eur. J. Hum. Genet., 24, 129–134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Euro, E.-R.E.S.C. and Epilepsy Phenome/Genome, P. and Epi, K.C. (2014) De novo mutations in synaptic transmission genes including DNM1 cause epileptic encephalopathies. Am. J. Hum. Genet., 95, 360–370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Talvik, I., Moller, R.S., Vaher, M., Vaher, U., Larsen, L.H., Dahl, H.A., Ilves, P. and Talvik, T. (2015) Clinical phenotype of De novo GNAO1 mutation: case report and review of literature. Child Neurol. Open, 2, 2329048X15583717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Menke, L.A., Engelen, M., Alders, M., Odekerken, V.J., Baas, F. and Cobben, J.M. (2016) Recurrent GNAO1 mutations associated with developmental delay and a movement disorder. J. Child Neurol., 31, 1598–1601. [DOI] [PubMed] [Google Scholar]
- 23. Marce-Grau, A., Dalton, J., Lopez-Pison, J., Garcia-Jimenez, M.C., Monge-Galindo, L., Cuenca-Leon, E., Giraldo, J. and Macaya, A. (2016) GNAO1 encephalopathy: further delineation of a severe neurodevelopmental syndrome affecting females. Orphanet J. Rare Dis., 11, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Muir, A.M., Myers, C.T., Nguyen, N.T., Saykally, J., Craiu, D., De Jonghe, P., Helbig, I., Hoffman-Zacharska, D., Guerrini, R., Lehesjoki, A.E.et al. (2019) Genetic heterogeneity in infantile spasms. Epilepsy Res., 156, 106181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Feng, H., Larrivee, C.L., Demireva, E.Y., Xie, H., Leipprandt, J.R. and Neubig, R.R. (2019) Mouse models of GNAO1-associated movement disorder: allele- and sex-specific differences in phenotypes. PLoS One, 14, e0211066. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 26. Larrivee, C.L., Feng, H., Quinn, J.A., Shaw, V.S., Leipprandt, J.R., Demireva, E.Y., Xie, H. and Neubig, R.R. (2020) Mice with GNAO1 R209H movement disorder variant display Hyperlocomotion alleviated by risperidone. J. Pharmacol. Exp. Ther., 373, 24–33. [DOI] [PubMed] [Google Scholar]
- 27. Muntean, B.S., Masuho, I., Dao, M., Sutton, L.P., Zucca, S., Iwamoto, H., Patil, D.N., Wang, D., Birnbaumer, L., Blakely, R.D.et al. (2021) Galphao is a major determinant of cAMP signaling in the pathophysiology of movement disorders. Cell Rep., 34, 108718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Koelle, M.R. (2018) Neurotransmitter signaling through heterotrimeric G proteins: insights from studies in C. elegans. WormBook, 2018, 1–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Bastiani, C. and Mendel, J. (2006) Heterotrimeric G proteins in C. elegans. WormBook, 2006, 1–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Feng, H., Sjogren, B., Karaj, B., Shaw, V., Gezer, A. and Neubig, R.R. (2017) Movement disorder in GNAO1 encephalopathy associated with gain-of-function mutations. Neurology, 89, 762–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Feng, H., Khalil, S., Neubig, R.R. and Sidiropoulos, C. (2018) A mechanistic review on GNAO1-associated movement disorder. Neurobiol. Dis., 116, 131–141. [DOI] [PubMed] [Google Scholar]
- 32. Carecchio, M. and Mencacci, N.E. (2017) Emerging monogenic complex hyperkinetic disorders. Curr. Neurol. Neurosci. Rep., 17, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Lochrie, M.A., Mendel, J.E., Sternberg, P.W. and Simon, M.I. (1991) Homologous and unique G protein alpha subunits in the nematode Caenorhabditis elegans. Cell. Regul., 2, 135–154. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Mendel, J.E., Korswagen, H.C., Liu, K.S., Hajdu-Cronin, Y.M., Simon, M.I., Plasterk, R.H. and Sternberg, P.W. (1995) Participation of the protein Go in multiple aspects of behavior in C. elegans. Science, 267, 1652–1655. [DOI] [PubMed] [Google Scholar]
- 35. Segalat, L., Elkes, D.A. and Kaplan, J.M. (1995) Modulation of serotonin-controlled behaviors by Go in Caenorhabditis elegans. Science, 267, 1648–1651. [DOI] [PubMed] [Google Scholar]
- 36. Koelle, M.R. and Horvitz, H.R. (1996) EGL-10 regulates G protein signaling in the C. elegans nervous system and shares a conserved domain with many mammalian proteins. Cell, 84, 115–125. [DOI] [PubMed] [Google Scholar]
- 37. Nurrish, S., Segalat, L. and Kaplan, J.M. (1999) Serotonin inhibition of synaptic transmission: Galpha(0) decreases the abundance of UNC-13 at release sites. Neuron, 24, 231–242. [DOI] [PubMed] [Google Scholar]
- 38. Miller, K.G., Emerson, M.D. and Rand, J.B. (1999) Goalpha and diacylglycerol kinase negatively regulate the Gqalpha pathway in C. elegans. Neuron, 24, 323–333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Robatzek, M., Niacaris, T., Steger, K., Avery, L. and Thomas, J.H. (2001) eat-11 encodes GPB-2, a Gbeta(5) ortholog that interacts with G(o)alpha and G(q)alpha to regulate C. elegans behavior. Curr. Biol., 11, 288–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Chase, D.L., Pepper, J.S. and Koelle, M.R. (2004) Mechanism of extrasynaptic dopamine signaling in Caenorhabditis elegans. Nat. Neurosci., 7, 1096–1103. [DOI] [PubMed] [Google Scholar]
- 41. Maher, K.N., Swaminathan, A., Patel, P. and Chase, D.L. (2013) A novel strategy for cell-autonomous gene knockdown in Caenorhabditis elegans defines a cell-specific function for the G-protein subunit GOA-1. Genetics, 194, 363–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Topalidou, I., Chen, P.A., Cooper, K., Watanabe, S., Jorgensen, E.M. and Ailion, M. (2017) The NCA-1 and NCA-2 ion channels function downstream of Gq and Rho to regulate locomotion in Caenorhabditis elegans. Genetics, 206, 265–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Wong, Y.H., Federman, A., Pace, A.M., Zachary, I., Evans, T., Pouyssegur, J. and Bourne, H.R. (1991) Mutant alpha subunits of Gi2 inhibit cyclic AMP accumulation. Nature, 351, 63–65. [DOI] [PubMed] [Google Scholar]
- 44. Takida, S., Fischer, C.C. and Wedegaertner, P.B. (2005) Palmitoylation and plasma membrane targeting of RGS7 are promoted by alpha o. Mol. Pharmacol., 67, 132–139. [DOI] [PubMed] [Google Scholar]
- 45. Zhen, M. and Samuel, A.D. (2015) C. elegans locomotion: small circuits, complex functions. Curr. Opin. Neurobiol., 33, 117–126. [DOI] [PubMed] [Google Scholar]
- 46. Giles, A.C., Desbois, M., Opperman, K.J., Tavora, R., Maroni, M.J. and Grill, B. (2019) A complex containing the O-GlcNAc transferase OGT-1 and the ubiquitin ligase EEL-1 regulates GABA neuron function. J. Biol. Chem., 294, 6843–6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Jiang, M., Spicher, K., Boulay, G., Wang, Y. and Birnbaumer, L. (2001) Most central nervous system D2 dopamine receptors are coupled to their effectors by Go. Proc. Natl. Acad. Sci. U. S. A., 98, 3577–3582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Tecuapetla, F., Jin, X., Lima, S.Q. and Costa, R.M. (2016) Complementary contributions of striatal projection pathways to action initiation and execution. Cell, 166, 703–715. [DOI] [PubMed] [Google Scholar]
- 49. Bosch, D.E., Willard, F.S., Ramanujam, R., Kimple, A.J., Willard, M.D., Naqvi, N.I. and Siderovski, D.P. (2012) A P-loop mutation in Galpha subunits prevents transition to the active state: implications for G-protein signaling in fungal pathogenesis. PLoS Pathog., 8, e1002553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Allen, A.T., Maher, K.N., Wani, K.A., Betts, K.E. and Chase, D.L. (2011) Coexpressed D1- and D2-like dopamine receptors antagonistically modulate acetylcholine release in Caenorhabditis elegans. Genetics, 188, 579–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Xu, M., Moratalla, R., Gold, L.H., Hiroi, N., Koob, G.F., Graybiel, A.M. and Tonegawa, S. (1994) Dopamine D1 receptor mutant mice are deficient in striatal expression of dynorphin and in dopamine-mediated behavioral responses. Cell, 79, 729–742. [DOI] [PubMed] [Google Scholar]
- 52. Baik, J.H., Picetti, R., Saiardi, A., Thiriet, G., Dierich, A., Depaulis, A., Le Meur, M. and Borrelli, E. (1995) Parkinsonian-like locomotor impairment in mice lacking dopamine D2 receptors. Nature, 377, 424–428. [DOI] [PubMed] [Google Scholar]
- 53. Jiang, M., Gold, M.S., Boulay, G., Spicher, K., Peyton, M., Brabet, P., Srinivasan, Y., Rudolph, U., Ellison, G. and Birnbaumer, L. (1998) Multiple neurological abnormalities in mice deficient in the G protein Go. Proc. Natl. Acad. Sci. U. S. A., 95, 3269–3274. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kleuss, C., Hescheler, J., Ewel, C., Rosenthal, W., Schultz, G. and Wittig, B. (1991) Assignment of G-protein subtypes to specific receptors inducing inhibition of calcium currents. Nature, 353, 43–48. [DOI] [PubMed] [Google Scholar]
- 55. Goldenstein, B.L., Nelson, B.W., Xu, K., Luger, E.J., Pribula, J.A., Wald, J.M., O'Shea, L.A., Weinshenker, D., Charbeneau, R.A., Huang, X.et al. (2009) Regulator of G protein signaling protein suppression of Galphao protein-mediated alpha2A adrenergic receptor inhibition of mouse hippocampal CA3 epileptiform activity. Mol. Pharmacol., 75, 1222–1230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Taylor, S.R., Santpere, G., Weinreb, A., Barrett, A., Reilly, M.B., Xu, C., Varol, E., Oikonomou, P., Glenwinkel, L., McWhirter, R.et al. (2021) Molecular topography of an entire nervous system. Cell, 84, 4329–4347.e23. doi: 10.1016/j.cell.2021.06.023. Epub 2021 Jul 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Williams, S.N., Locke, C.J., Braden, A.L., Caldwell, K.A. and Caldwell, G.A. (2004) Epileptic-like convulsions associated with LIS-1 in the cytoskeletal control of neurotransmitter signaling in Caenorhabditis elegans. Hum. Mol. Genet., 13, 2043–2059. [DOI] [PubMed] [Google Scholar]
- 58. Zhu, B., Mak, J.C.H., Morris, A.P., Marson, A.G., Barclay, J.W., Sills, G.J. and Morgan, A. (2020) Functional analysis of epilepsy-associated variants in STXBP1/Munc18-1 using humanized Caenorhabditis elegans. Epilepsia, 61, 810–821. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Jones, A., Barker-Haliski, M., Ilie, A.S., Herd, M.B., Baxendale, S., Holdsworth, C.J., Ashton, J.P., Placzek, M., Jayasekera, B.A.P., Cowie, C.J.A.et al. (2020) A multiorganism pipeline for antiseizure drug discovery: identification of chlorothymol as a novel gamma-aminobutyric acidergic anticonvulsant. Epilepsia, 61, 2106–2118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60. Risley, M.G., Kelly, S.P., Jia, K., Grill, B. and Dawson-Scully, K. (2016) Modulating behavior in C. elegans using electroshock and antiepileptic drugs. PLoS One, 11, e0163786. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Wong, S.Q., Jones, A., Dodd, S., Grimes, D., Barclay, J.W., Marson, A.G., Cunliffe, V.T., Burgoyne, R.D., Sills, G.J. and Morgan, A. (2018) A Caenorhabditis elegans assay of seizure-like activity optimised for identifying antiepileptic drugs and their mechanisms of action. J. Neurosci. Methods, 309, 132–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Bessa, C., Maciel, P. and Rodrigues, A.J. (2013) Using C. elegans to decipher the cellular and molecular mechanisms underlying neurodevelopmental disorders. Mol. Neurobiol., 48, 465–489. [DOI] [PubMed] [Google Scholar]
- 63. Dexter, P.M., Caldwell, K.A. and Caldwell, G.A. (2012) A predictable worm: application of Caenorhabditis elegans for mechanistic investigation of movement disorders. Neurotherapeutics, 9, 393–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Kepler, L.D., McDiarmid, T.A. and Rankin, C.H. (2020) Habituation in high-throughput genetic model organisms as a tool to investigate the mechanisms of neurodevelopmental disorders. Neurobiol. Learn. Mem., 171, 107208. [DOI] [PubMed] [Google Scholar]
- 65. Locke, C.J., Williams, S.N., Schwarz, E.M., Caldwell, G.A. and Caldwell, K.A. (2006) Genetic interactions among cortical malformation genes that influence susceptibility to convulsions in C. elegans. Brain Res., 1120, 23–34. [DOI] [PubMed] [Google Scholar]
- 66. Tong, X.J., Hu, Z., Liu, Y., Anderson, D. and Kaplan, J.M. (2015) A network of autism linked genes stabilizes two pools of synaptic GABA(a) receptors. Elife, 4, e09648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Opperman, K.J., Mulcahy, B., Giles, A.C., Risley, M.G., Birnbaum, R.L., Tulgren, E.D., Dawson-Scully, K., Zhen, M. and Grill, B. (2017) The HECT family ubiquitin ligase EEL-1 regulates neuronal function and development. Cell Rep., 19, 822–835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Wong, W.R., Brugman, K.I., Maher, S., Oh, J.Y., Howe, K., Kato, M. and Sternberg, P.W. (2019) Autism-associated missense genetic variants impact locomotion and neurodevelopment in Caenorhabditis elegans. Hum. Mol. Genet., 28, 2271–2281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Paix, A., Folkmann, A., Rasoloson, D. and Seydoux, G. (2015) High efficiency, homology-directed Genome editing in Caenorhabditis elegans using CRISPR-Cas9 ribonucleoprotein complexes. Genetics, 201, 47–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Guyenet, S.J., Furrer, S.A., Damian, V.M., Baughan, T.D., La Spada, A.R. and Garden, G.A. (2010) A simple composite phenotype scoring system for evaluating mouse models of cerebellar ataxia. J. Vis. Exp., 39, 1787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Matsuura, K., Kabuto, H., Makino, H. and Ogawa, N. (1997) Pole test is a useful method for evaluating the mouse movement disorder caused by striatal dopamine depletion. J. Neurosci. Methods, 73, 45–48. [DOI] [PubMed] [Google Scholar]
- 72. Farr, T.D., Liu, L., Colwell, K.L., Whishaw, I.Q. and Metz, G.A. (2006) Bilateral alteration in stepping pattern after unilateral motor cortex injury: a new test strategy for analysis of skilled limb movements in neurological mouse models. J. Neurosci. Methods, 153, 104–113. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.