

Mammalian development, adult tissue homeostasis and the avoidance of severe diseases including cancer require a properly orchestrated cell cycle, as well as error-free genome maintenance. The key cell-fate decision to replicate the genome is controlled by two major signalling pathways that act in parallel–the MYC pathway and the cyclin D–cyclin-dependent kinase (CDK)–retinoblastoma protein (RB) pathway1,2. Both MYC and the cyclin D–CDK–RB axis are commonly deregulated in cancer, and this is associated with increased genomic instability. The autophagic tumour-suppressor protein AMBRA1 has been linked to the control of cell proliferation, but the underlying molecular mechanisms remain poorly understood. Here we show that AMBRA1 is an upstream master regulator of the transition from G1 to S phase and thereby prevents replication stress. Using a combination of cell and molecular approaches and in vivo models, we reveal that AMBRA1 regulates the abundance of D-type cyclins by mediating their degradation. Furthermore, by controlling the transition from G1 to S phase, AMBRA1 helps to maintain genomic integrity during DNA replication, which counteracts developmental abnormalities and tumour growth. Finally, we identify the CHK1 kinase as a potential therapeutic target in AMBRA1-deficient tumours. These results advance our understanding of the control of replication-phase entry and genomic integrity, and identify the AMBRA1–cyclin D pathway as a crucial cell-cycle-regulatory mechanism that is deeply interconnected with genomic stability in embryonic development and tumorigenesis.
AMBRA1 (activating molecule in beclin-1-regulated autophagy) is a scaffold factor that binds proteins involved in several cellular pathways3. We previously reported that AMBRA1 interacts with members of the cullin-RING family of E3 ubiquitin ligases to regulate the stability of key autophagy proteins4,5, and mediates PP2A-dependent dephosphorylation and degradation of the proto-oncogene c-MYC6. Furthermore, Ambra1 is essential for nervous system development in mice, and AMBRA1 deficiency causes embryonic lethality7. Inspired by the emerging links of AMBRA1 with cell proliferation, development and tumorigenesis3, here we set out to elucidate the mechanistic basis of the role(s) of AMBRA1 in cell-cycle regulation and the effects of AMBRA1 on neurogenesis, genomic integrity and cancer.
AMBRA1 controls proliferation through cyclin D
To complement and extend previous observations of enhanced proliferation upon downregulation of AMBRA16,7, we generated a nervous system conditional knockout mouse model (Ambra1 cKO) by selectively deleting Ambra1 in the columnar neuroepithelial cells at embryonic day (E)11 (Extended Data Fig. 1a). This delayed depletion of AMBRA1–as opposed to our previous Ambra1gt/gt-deficient mouse model characterized by systemic depletion of Ambra1–allowed us to examine later phases of neural development. Compared to wild-type mice, Ambra1 cKO mice showed an increase in the volume of the cortex and the lateral ventricles (Fig. 1a, Extended Data Fig. 1b, c), and this increase in volume was associated with an enhanced rate of proliferation in the whole E13.5 brain and in the olfactory bulbs of the E18.5 brain (Fig. 1b, Extended Data Fig. 1d, e). In addition, neural stem cells (NSCs) isolated from Ambra1 cKO mice showed increased levels of several cell-cycle regulatory proteins (Fig. 1c, Extended Data Fig. 1f, g), together with a higher clonogenic potential and replication rate (Fig. 1d, Extended Data Fig. 1h). Notably, levels of cyclins D1 and D2 and RB(pS807/811) (that is, RB phosphorylated at Ser807, Ser 811 or both sites) were highly increased in Ambra1 cKO cells both ex vivo and in vivo (Fig. 1c, e, Extended Data Fig. 1g, i–m), suggesting an AMBRA1-dependent modulation of cyclin D. Indeed, consistent with our previous results6, we found in neural cell lines ex vivo and in vitro that AMBRA1 directly binds to and regulates the stability of N-MYC via the phosphatase PP2A, thereby controlling the transcription of cyclins D1 and D2 (Extended Data Fig. 1n–r). Moreover, we noticed that both cyclin D1 and cyclin D2 are highly resilient to proteasomal degradation in Ambra1-deficient conditions (Fig. 1f, Extended Data Fig. 2a, b). In line with the fact that both MYC and D-type cyclins positively regulate the G1–S-phase transition8,9, Ambra1 cKO NSCs showed a shorter G1 phase, with faster entry into and a longer residence in S phase (Extended Data Fig. 2c). By reducing the activity of the cyclin D–CDK4 and CDK6 (CDK4/6) pathway through abemaciclib, we were able to restore proliferation to wild-type levels (Extended Data Fig. 2d), highlighting the importance of the accelerated G1–S transition in the phenotype that is driven by AMBRA1 depletion. In addition, we found that owing to the lack of Ambra1, deregulated cell-cycle progression is followed by increased cell death–a phenotype that was rescued by inhibition of cyclin D–CDK4/6 activity (Extended Data Fig. 2e, f). Of note, knockout of Ambra1 in neurodevelopment promotes the expansion of the staminal niche, and induces an unbalanced expression of SOX2+ multipotent NSCs at the expense of partially committed TBR2+ intermediate progenitors (Extended Data Fig. 2g). This phenotype was retained at a later embryonal stage, when knockout of Ambra1 led to a reduction in neuronal differentiation–confirming the observed delay in neurogenesis that is induced by overexpression of cyclin D110 (Extended Data Fig. 2h–j).
Fig. 1 |. AMBRA1 regulates cell proliferation by affecting the stability of D-type cyclins through interaction with DDB1 and CLR4.
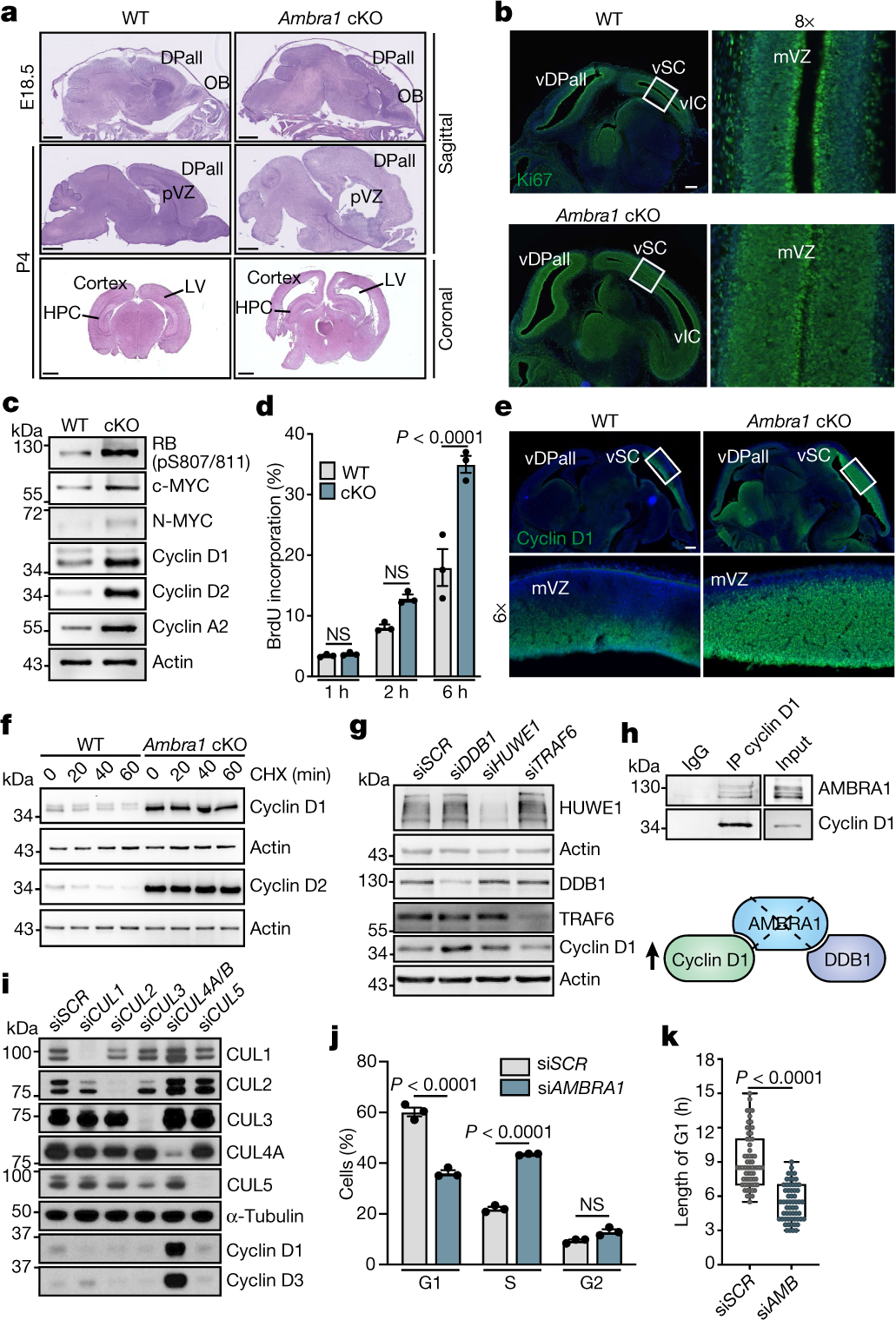
a, Haematoxylin and eosin (H&E)-stained wild-type (WT; Ambra1flox/flox) and Ambra1 cKO (Ambra1flox/flox:: Nestin-cre) brain sections. DPall, dorsal pallium; HPC, hippocampus; LV, lateral ventricle; OB, olfactory bulb; pVZ, pallium ventricular zone. P4, postnatal day 4. Scale bars, 400 μm (top); 1 mm (middle, bottom). b, Sagittal sections from wild-type and Ambra1 cKO E13.5 embryos, stained for Ki67 and Hoechst. Boxed regions are magnified to the right (8× magnification) (n = 5). mVZ, mesencephalic ventricular zone; vDPall, ventricular dorsal pallium; vIC, ventricular inferior colliculus; vSC, ventricular superior colliculus. Scale bar, 250 μm. c, Immunoblot of the indicated proteins from extracts of wild-type and Ambra1 cKO NSCs (n = 4). d, BrdU incorporation in wild-type and Ambra1 cKO NSCs; time points of BrdU administration are indicated (n = 3). e, Sagittal sections of wild-type and Ambra1 cKO E13.5 embryos, stained for cyclin D1 and Hoechst. Boxed regions are magnified below (6× magnification) (n = 5). Scale bar, 250 μm. f, Immunoblot of cyclin D1 and cyclin D2 after treating wild-type and Ambra1 cKO NSCs with cycloheximide (CHX) (n = 3). g, Immunoblot of cyclin D1 in U87-MG cells in which expression of the indicated E3 ubiquitin ligases was knocked down using small interfering RNA (siRNA) (n = 4). h, Top, co-immunoprecipitation of cyclin D1 and AMBRA1 in AMBRA1-overexpressing U87-MG cells (n = 3). IP, immunoprecipitation. Bottom, model of the regulation of cyclin D1 stability by AMBRA1. i, Immunoblot of cyclin D1 and cyclin D3 in U2OS cells in which expression of the indicated cullin proteins was knocked down using siRNA (n = 3). j, Percentage of cells in the indicated phase of the cell cycle (n = 3). Cells were immunostained for cyclin D1, and counterstained with EdU and Hoechst upon AMBRA1 mRNA interference. k, Box plots (centre line, median; box limits, 25th and 75th percentiles; whiskers, minimum and maximum) showing the length of the G1 phase in siAMBRA1 (n = 59 cells) and control siSCR (n = 63 cells) U2OS cells across three independent experiments. Unless otherwise stated, data are mean ± s.e.m.; n refers to biologically independent samples. Data were analysed using a two-sided one-way ANOVA followed by Sidak’s multiple comparisons test (d), one-way ANOVA followed by Sidak’s multiple comparisons test (j) or two-tailed unpaired t-test (k). NS, not significant. Quantifications of immunoblots are shown in Extended Data Figs. 1g, 2a, 3c.
Thus, our results indicate that AMBRA1 regulates the stability and abundance of D-type cyclins, with implications for G1–S control, cell proliferation, neurogenesis and cell death during neurodevelopment.
AMBRA1 regulates cyclin D through CRL4–DDB1
Next, we confirmed that the mechanism observed in mouse neurodevelopment is also applicable in the context of human neural cells (Extended Data Fig. 3a, b). In line with the fact that AMBRA1 regulates protein turnover by the proteasome through its direct interaction with several E3 ligases4,5,11, we found that DDB1 (part of the CRL4–DDB1 complex) is the specific E3 ligase that regulates AMBRA1-dependent cyclin D1 stability by interacting with AMBRA1 (Fig. 1g, h, Extended Data Fig. 3c–e). We also found that a phosphorylation-deficient mutant of cyclin D1 (T286A), which is resistant to proteasome degradation12, was unable to bind AMBRA1 (Extended Data Fig. 3f). Consistent with the Ambra1 cKO proliferative phenotype, knockdown of AMBRA1 in human U87-MG cells also led to an increase in cell division (Extended Data Fig. 3g).
We further expanded our findings to human non-neural RB-proficient cell types, namely U2OS and telomerase-immortalized BJ (BJ-hTERT) cells. Both inhibition of the cullin family as a whole by MLN4924 and depletion of individual cullin proteins confirmed that cullin 4A and 4B (two members of the CRL4–DDB1 complex) are responsible for the degradation of D-type cyclins (Fig. 1i, Extended Data Fig. 3h). Of note, AMBRA1-silenced cells accumulated in S phase (Fig. 1j) and showed increased levels of nuclear cyclin D1, along with markers of an accelerated G1–S transition (Extended Data Fig. 3i–k). Next, by using U2OS-FUCCI cells sorted into specific cell-cycle populations, we found that knockdown of AMBRA1 causes overexpression of cyclins D1 and A2, as well as hyperphosphorylation of RB at Ser807/Ser811 (Extended Data Fig. 3l), supporting a role for AMBRA1 in cell-cycle regulation. Consistently, AMBRA1-silenced fibroblasts and U2OS-FUCCI cells exhibited an accelerated entry into S phase and an overall shorter doubling time, compared to control cells (Fig. 1k, Extended Data Fig. 3m–r). Overall, our results thus indicate that AMBRA1 regulates the stability and abundance of D-type cyclins, and thereby affects their function, through the CRL4–DDB1 complex.
AMBRA1 loss leads to replication stress
The phenotype observed in neural cells prompted us to investigate the potential effects of AMBRA1 on genome stability. Indeed, downregulation of AMBRA1 caused an increase in endogenous DNA damage, mainly in S and G2 phases (Fig. 2a, Extended Data Fig. 4a, b). Of note, we could rule out that the DNA damage was related to the pro-autophagic role of AMBRA14,5,7,11,13 (Extended Data Fig. 4c–e). Impairment of autophagy has been shown to reduce the efficiency of homologous recombination14; consistently with our previous results, we showed that cells with knockdown of AMBRA1–in contrast to ATG7-knockdown cells–exhibit functional homologous recombination (Extended Data Fig. 4f). Furthermore, the formation of BRCA1 foci in AMBRA1-silenced cells after irradiation was enhanced (Extended Data Fig. 4g), probably reflecting the sum of endogenous and irradiation-induced lesions. Overall, these results indicate that AMBRA1 is essential to prevent spontaneous DNA damage.
Fig. 2 |. Depletion of AMBRA1 causes replication stress.
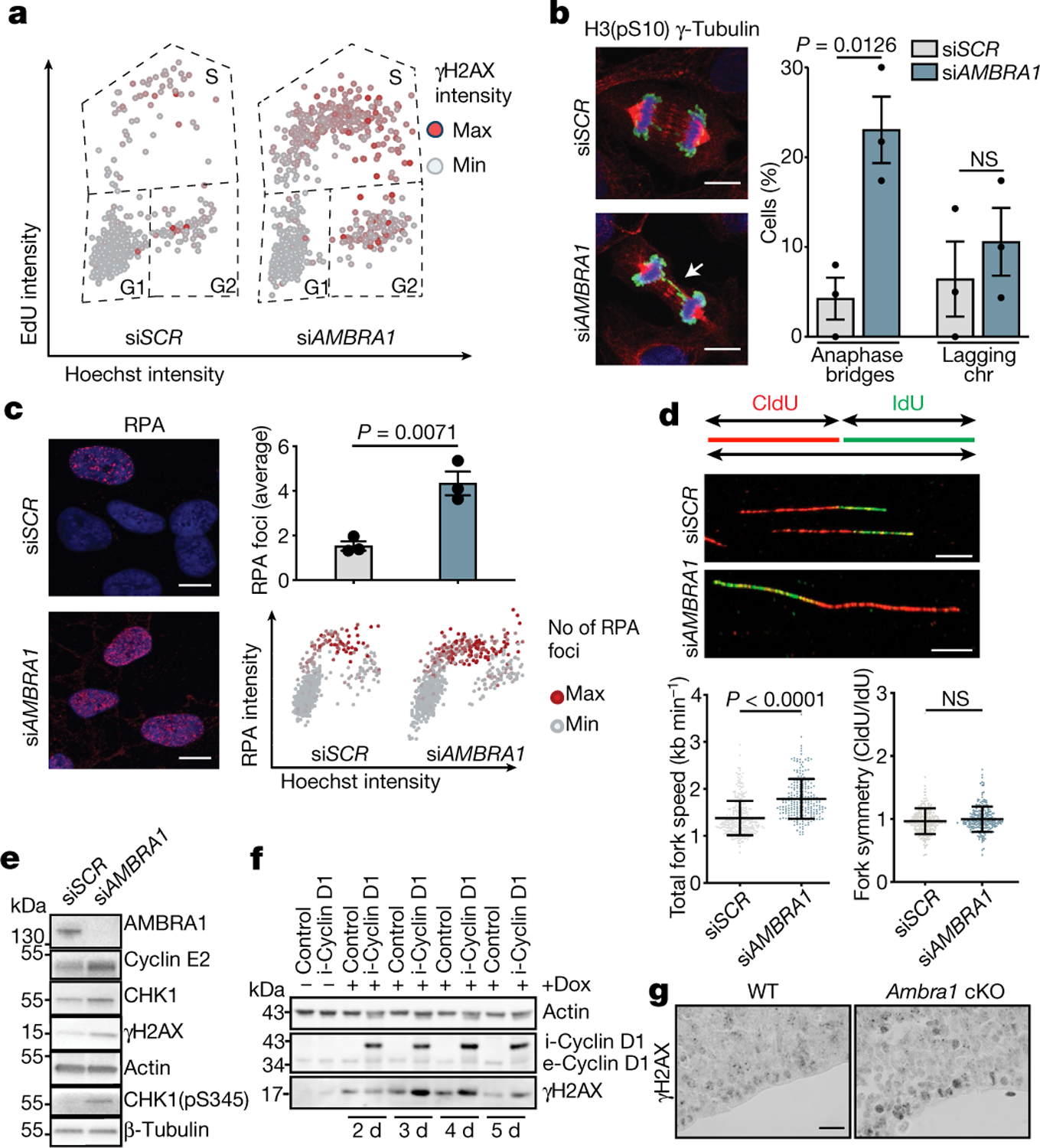
a, Scatter plots reporting single-cell γH2AX, EdU and Hoechst total nuclear intensities from AMBRA1-silenced (siAMBRA1) and control (siSCR) BJ-hTERT cells (siSCR, n = 716 cells; siAMBRA1, n = 715 cells; representative of three independent experiments). b, Left, AMBRA1-silenced and control U2OS cells stained for γ-tubulin (red), histone H3 phosphorylated at Ser10 (H3(pS10); green) and Hoechst (blue). Scale bars, 10 μm. Right, quantification of mitotic cells showing anaphase bridges and lagging chromosomes (chr) (n = 3). c, Left, AMBRA1-silenced and control BJ-hTERT cells immunostained for RPA (siSCR, n = 704 cells; siAMBRA1, n = 720 cells). Scale bars, 10 μm. Right, quantification of the average number of RPA foci and scatter plot of Hoechst intensity versus RPA intensity (n = 3). d, Top, DNA fibres from control and AMBRA1-silenced BJ-hTERT cells. Scale bars, 10 μm. Bottom, quantification of mean fork speed (kb min−1) and of fork symmetry analysis after incorporation and staining of 5-iodo-2’-deoxyuridine (IdU) and 5-chloro-2’-deoxyuridine (CldU). Scored forks: siSCR, n = 301; siAMBRA1, n = 233. Data are mean ± s.d. e, Immunoblot analysis of the indicated proteins in control or AMBRA1-silenced BJ-hTERT cells. f, Immunoblot analysis of cyclin D1 and γH2AX in control BJ-hTERT cells or cells in which cyclin D1 expression was induced by doxycyclin (dox) treatment for the indicated number of days. The prefixes i- and e- indicate the induced and the endogenous form of cyclin D1, respectively (n = 3). g, Sagittal sections of wild-type or Ambra1 cKO E13.5 embryos, stained for γH2AX antibody (n = 3). Scale bar, 40 μm. Quantification of immunohistochemistry is shown in Extended Data Fig. 5l. Actin or β-tubulin were used as loading controls. Unless otherwise stated, data are mean ± s.e.m.; n refers to biologically independent samples. Data were analysed using a two-tailed unpaired t-test (b, c) or two-tailed Mann–Whitney test (d).
Given that AMBRA1-silenced cells accumulate DNA damage in S and G2 phases, we then assessed potential mitotic defects. U2OS cells with knockdown of AMBRA1 showed prolonged mitosis and the formation of anaphase bridges, without displaying any differences in lagging chromosomes, followed by high rates of cell death (Fig. 2b, Extended Data Fig. 4h, i, Supplementary Videos 1, 2).
Notably, the occurrence of DNA damage in S phase, anaphase chromosome bridges and mitotic abnormalities, along with premature S-phase entry, are all associated with replication stress15–17. The replication stress phenotype is further evident in AMBRA1-silenced cells by the accumulation of RPA foci and the increased presence of 53BP1 bodies in G1 phase18 (Fig. 2c, Extended Data Fig. 4j). AMBRA1 depletion is associated with an increased replication fork speed without affecting fork symmetry (Fig. 2d), a phenotype that we previously showed is a mechanism that triggers replication stress and the DNA damage response19. Indeed, hydroxyurea-induced replication stress promotes robust γH2AX phosphorylation in AMBRA1-deficient cells (Extended Data Fig. 4k). Of note, downregulation of AMBRA1 induces increased levels of CHK1–a key kinase in the replication stress response–and hyperphosphorylation of CHK1 at Ser345, as well as increased levels of cyclin E2, a powerful oncogene20–22 (Fig. 2e, Extended Data Fig. 4l–n). Next, using a multi-method comparative approach, we found that knockdown of AMBRA1 correlates with a general upregulation of genes that are involved in DNA repair and transcriptional regulation of the G1–S transition (Extended Data Fig. 4o–s, Supplementary Tables 1, 2)–with E2F1 and MYC emerging among the top predicted activated transcriptional factors (Extended Data Fig. 5a). In cell-cycle-sorted AMBRA1-silenced FUCCI cells, we observed a cycle-phase-specific upregulation of genes related to the cell cycle and DNA repair (Extended Data Fig. 5b), consistently with the S-phase enrichment and replication stress phenotype that was seen upon downregulation of AMBRA1. The silencing of other autophagy genes does not induce upregulation of the levels of E2F1 protein, again indicating that the effect of AMBRA1 deficiency mainly reflects deregulated cell-cycle progression (Extended Data Fig. 5c).
Of note, inhibition of CDK4/6 activity rescues the DNA damage phenotype induced by AMBRA1 knockdown (Extended Data Fig. 5d), whereas overexpression of cyclin D1 induces increased proliferation and accumulation of cells in S phase (Extended Data Fig. 5e–h), together with a marked occurrence of DNA damage (Fig. 2f, Extended Data Fig. 5i). Notably, we found that overexpression of cyclin D1 causes an initial acceleration of the fork speed, followed by its marked decrease–mostly recapitulating the effect of AMBRA1 depletion (Extended Data Fig. 5j, k). Next, an analysis of DNA damage as measured by γH2AX levels in Ambra1 cKO embryos and NSCs confirmed that loss of Ambra1 also enhances endogenous replication stress during neurodevelopment (Fig. 2g, Extended Data Fig. 5l, m). Overall, these results reveal an unexpected role of AMBRA1 in preventing replication stress and the ensuing endogenous DNA damage and chromosomal instability. Such genome-destabilizing phenotypes, which are commonly observed in cancer, become unmasked when AMBRA1 is depleted, and in part reflect the loss of function of AMBRA1 in restraining the abundance of cyclin D.
AMBRA1 deficiency sensitizes to CHK1 inhibition
Given the relevance of our results to cancer, we next assessed the frequency of downregulation of AMBRA1 mRNA in a spectrum of human tumours. Notably, we identified the existence of ‘AMBRA1-low’ cancer subsets, which are characterized by an expression level of AMBRA1 that is much lower than the levels seen in corresponding healthy tissue (Extended Data Fig. 6a). In addition, in lung cancer datasets, AMBRA1 expression inversely correlates with the stemness signature (Extended Data Fig. 6b). Notably, AMBRA1 is frequently mutated in different types of cancer (Extended Data Fig. 6c, d), and its downregulation co-occurs with TP53 mutations (Extended Data Fig. 6e, f). AMBRA1-low expression is also associated with a worse prognosis in different cancer datasets (Extended Data Fig. 6g, h). On the basis of these findings, we investigated the effect of Ambra1 knockout in an inducible mouse model of lung cancer, conditionally driven by KrasG12D (Extended Data Fig. 7a–c). We observed a more aggressive growth phenotype with larger lesions in Ambra1flox/flox::KrasG12D/+ compared to Ambra1+/+::KrasG12D/+ lungs (Fig. 3a, Extended Data Fig. 7d). Furthermore, the Ki67-positive proliferative cell fraction is higher in Ambra1flox/flox::KrasG12D/+ mice and often found in tumour cell clusters, indicating the proliferative clones, compared with a more even pattern and lower frequency in Ambra1+/+::KrasG12D/+ mice (Fig. 3b, Extended Data Fig. 7e). Consistent with our results, Ambra1flox/flox:: KrasG12D/+ tumours exhibit a substantially higher increase in the levels of cyclin D1, MYC phosphorylated at Ser62 (MYC(pS62)) and markers associated with replication stress, when compared with Ambra1-proficient tumours (Fig. 3b, Extended Data Fig. 7e). Notably, human AMBRA1-low cancers also show higher expression of CHEK1 compared with corresponding healthy samples (Extended Data Fig. 8a, Supplementary Table 3).
Fig. 3 |. AMBRA1 is a tumour suppressor and its loss is synthetic lethal with CHK1 inhibition.
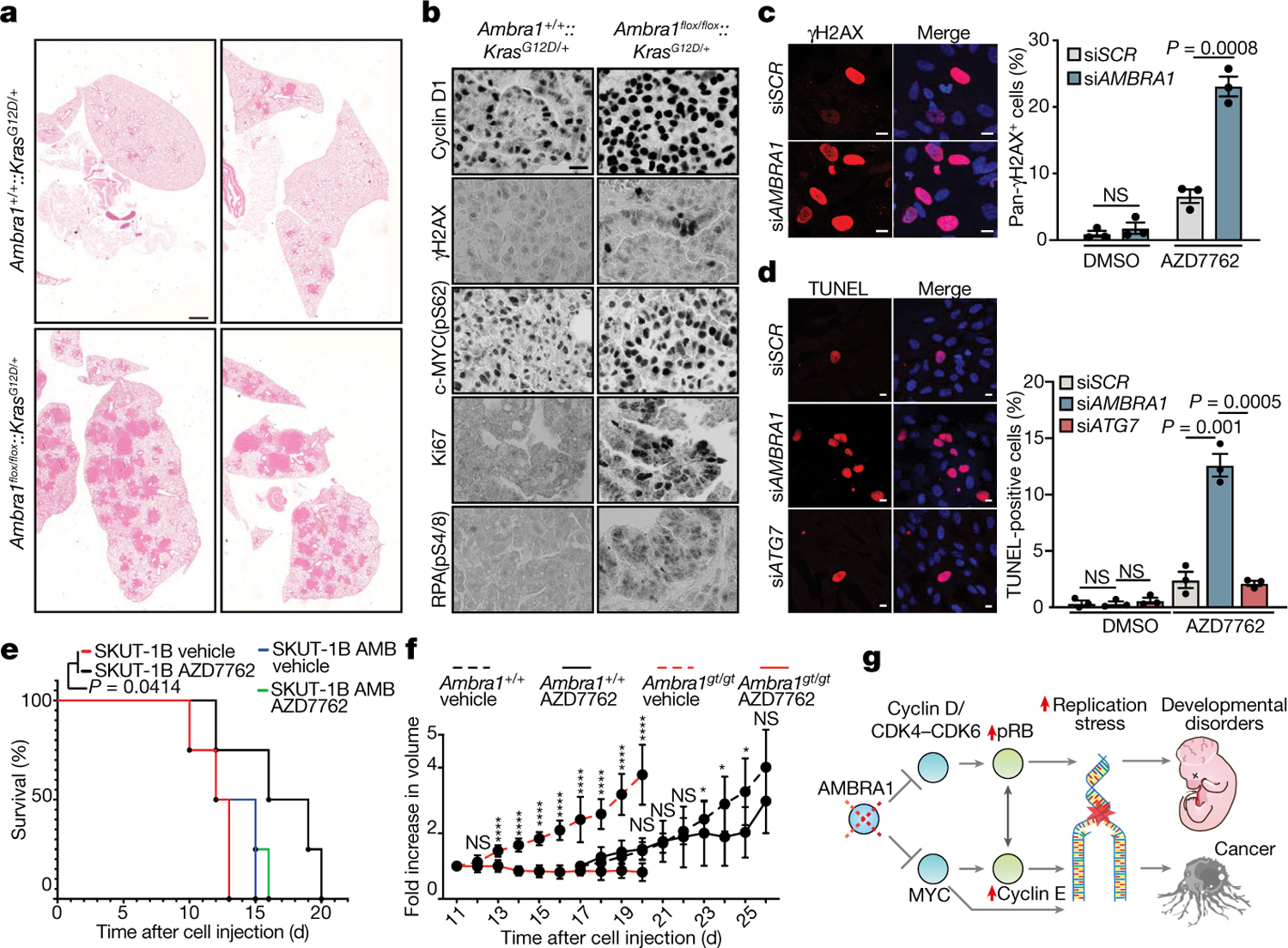
a, H&E-stained mouse lung sections showing neoplastic lesions in Ambra1-proficient (Ambra1+/+::KrasG12D/+) and Ambra1-deficient (Ambra1flox/flox::KrasG12D/+) lung tissue 20 weeks after adenoviral infection. Four different samples are shown and are representative of four mice. Scale bar, 1 mm. b, Immunohistochemistry analyses of Ki67, γH2AX, RPA(pS4/8), cyclin D1 and c-MYC(pS62) at 20 weeks after infection. Scale bar, 40 μm. c, Left, AMBRA1-silenced and control BJ-hTERT cells were treated with 100 nM AZD7762 for 24 h and immunostained for γH2AX and Hoechst. Scale bars, 5 μm. Right, quantification of pan-γH2AX-positive cells after inhibition of CHK1 by treatment with AZD7762 for 24 h (n = 3 independent experiments). d, Left, the indicated BJ-hTERT cells were treated with AZD7762 for 24 h and stained with TUNEL and Hoechst. Scale bars, 5 μm. Right, quantification of the average percentage of TUNEL-positive cells (n = 3 independent experiments). e, Survival curves of mice xenotransplanted with control sarcoma SKUT-1B cells or SKUT-1B cells reconstituted with AMBRA1 (SKUT-1B AMB). Mice were treated with vehicle or AZD7762 (n = 4 mice). f, Assessment of the volume of Ambra1+/+ and Ambra1gt/gt MEF xenografts in mice treated with AZD7762 or vehicle (Ambra1+/+ + vehicle, n = 8 for days 17–21, n = 7 from day 24; Ambra1+/+ + AZD7762, n = 8; Ambra1gt/gt + vehicle, n = 10; Ambra1gt/gt + AZD7762, n = 11 mice). Data are mean ± s.d. g, AMBRA1 regulates the G1–S-phase transition by mediating the degradation of cyclin D proteins and c-MYC. A defective AMBRA1–cyclin D axis causes a premature entry into S phase, leading to replication stress and genome instability. The increased DNA damage causes faster tumour growth and neurodevelopmental defects. pRB, phosphorylated RB. Quantification of immunohistochemistry is shown in Extended Data Fig. 7e. Unless otherwise stated, data are mean ± s.e.m. Data were analysed using a two-tailed unpaired t-test (c, d, f) or log-rank Mantel–Cox test (e). In f, day 23,*P = 0.0348; day 24, P = 0.0353; day 25, *P = 0.0228; ****P < 0.0001.
As treatment with hydroxyurea causes increased levels of γH2AX levels in AMBRA1-silenced cells, we wondered whether these cells could be more sensitive to anti-cancer drugs that target this key kinase in the response to replication stress. We found that inhibition of CHK1 exacerbates the endogenous DNA damage caused by AMBRA1 depletion, resulting in a robust increase of replication stress, DNA damage and cell death, mostly in S phase (Fig. 3c, d, Extended Data Fig. 8b–f). Furthermore, the vulnerability of AMBRA1-low cells to inhibition of the replication stress response is selective for CHK1 inhibition (Extended Data Fig. 8g), the effect of which specifically results in a strong decrease of replication fork speed and altered fork symmetry (Extended Data Fig. 8h). Such synthetic lethality can be observed with CHK1 inhibition in a set of non-small-cell lung cancer cell lines, further verifying the inverse correlation between AMBRA1 levels and sensitivity to CHK1 inhibition (Extended Data Fig. 8i, j). Moreover, in searching for a naturally AMBRA1-deficient cancer model, we found that SKUT-1B sarcoma cells–which show an aberrant overabundance of all three D-type cyclins23–lack AMBRA1 (Extended Data Fig. 8k). In this cell line, high levels of cyclin D are maintained despite cullin inhibition (Extended Data Fig. 8l), confirming the endogenous impairment of CRL4–DDB1-dependent degradation. Notably, in this system, reconstitution with wild-type AMBRA1, but not a mutant form of AMBRA1 that is unable to bind DDB1 (ΔWD40), is able per se to decrease the levels of cyclins D1 and D2 and rescue cell death induced by CHK1 inhibition (Extended Data Fig. 8m, n). To examine whether AMBRA1 deficiency sensitizes cells to CHK1 inhibition in vivo, SKUT-1B cells (AMBRA1-deficient or reconstituted with AMBRA1) were subcutaneously implanted in nude mice. Indeed, SKUT-1B AMBRA1-deficient grafts are highly sensitive to AZD7762, indicating that a stable downregulation of AMBRA1 can also lead to sensitivity to CHK1 inhibition in human cancer cells in vivo (Fig. 3e). Similarly, Ambra1gt/gt mouse embryonic fibroblasts (MEFs) transformed with RAS(V12) and E1A oncogenes6 are highly sensitive to CHK1 inhibition in vitro and in vivo (Fig. 3f, Extended Data Fig. 9a, b). Finally, and consistently with the model we propose (Fig. 3g), we found that overexpression of cyclin D1 is synthetically lethal with CHK1 inhibition (Extended Data Fig. 9c).
Altogether, these results indicate that replication stress caused by deregulation of AMBRA1-dependent pathways that control the G1–S transition, and the ensuing overabundance of D-type cyclins, lead to synthetic lethality with CHK1 inhibition, a vulnerability that could potentially be exploited in cancer treatments.
Discussion
The results we present herein advance our understanding of cell-cycle regulation. The key concept that emerges from our data is that the pleiotropic factor AMBRA1 has a previously unrecognized role as an upstream master gatekeeper that prevents the premature or excessive activation of the two parallel key pathways that drive the G1–S transition–namely the MYC pathway and the cyclin D–CDK4/6–RB–E2F axis, both of which upregulate the proto-oncogene cyclin E24 (Fig. 3g). The function of AMBRA1 in this context depends on its role as a DDB1–CUL4-associated factor (DCAF) that targets cyclin D for proteasomal degradation; indeed, the role of AMBRA1 as a DCAF parallels its role in binding and regulating the activity of the E3 ubiquitin ligases HUWE1 and TRAF65,11, with this defining AMBRA1 as a master regulator of fundamental proteostasis. Furthermore, we show that deregulation of the AMBRA1–cyclin D axis is an oncogenic event that evokes features of replication stress, leading to genomic instability and cancer.
Whereas the defective AMBRA1–cyclin D axis promotes faster tumour growth and may cause resistance to inhibitors of cyclin-D-associated CDK4/6 kinases, which are used to treat some cancers (see also two papers that are copublished with this one25,26), we found that AMBRA1-low cell lines and tumour models in vivo become more sensitive to inhibitors of CHK1. Such a synthetic lethality between AMBRA1 defects and CHK1 inhibition is likely to reflect the role of CHK1 signalling in allowing cells to tolerate replication stress, a scenario that is also consistent with multiple examples of tumours exhibiting high levels of replication stress and being sensitive to (pre)clinically tested CHK1 inhibitors27–31. Given that a major hurdle in cancer treatment is the need to pinpoint the subset(s) of patients with cancer who might most benefit from any emerging targeted treatment, we propose that defects in AMBRA1 might become a useful predictive biomarker in clinical oncology.
Our results also document the relevance of the G1–S regulatory function of AMBRA1 in neurodevelopment and embryogenesis, besides the already demonstrated autophagy-related role of AMBRA1 during development7. For this reason, our results may also shed new light on the involvement of defects in AMBRA1 in neurological disorders, such as autism32,33.
Overall, our findings thus highlight the importance of AMBRA1 as a hub that is capable of integrating diverse signalling pathways into complex cellular responses, with wide implications for a variety of severe diseases in humans.
Extended Data
Extended Data Fig. 1 |. AMBRA1 regulates cyclin D stability in NSCs.
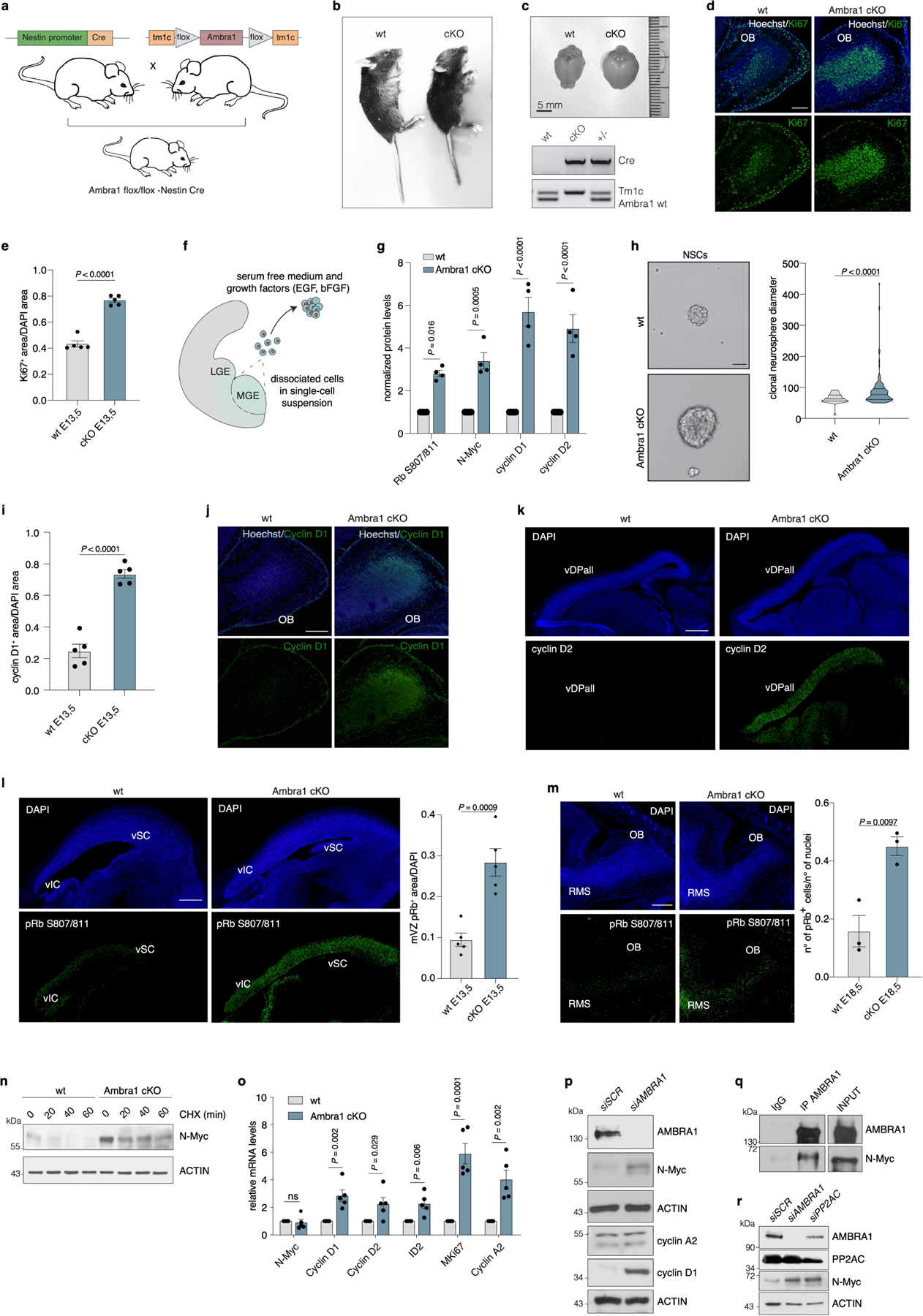
a, Schematic for production of the conditional knockout mouse model. b, c, Images of wild-type and Ambra1 cKO P21 mice (b) and brains (c). c, bottom, representative image of PCR amplification of Tm1c, Ambra1 and Cre. d, Wild-type and Ambra1 cKO olfactory bulbs in sagittal sections of E18.5 embryos, stained for Ki67 antibody and Hoechst (n = 3). e, Quantification of Ki67+ cell area in the whole brain of wild-type and Ambra1 cKO E13.5 embryos (sagittal sections shown in Fig. 1b) (n = 5). P value by two-tailed unpaired t-test. f, Representative scheme of NSCs extraction and cell culturing from mouse embryo medial ganglionic eminences (MGE). LGE, lateral ganglionic eminences. g, Densitometry quantification of normalized protein levels in wild-type and Ambra1 cKO NSCs shown in Fig. 1c (n = 4). P values by two-sided one-way ANOVA followed by Sidak’s multiple comparisons test. h, Left, representative images of NSCs extracted from mouse embryo medial ganglionic eminences. Right, violin plot of clonal neurosphere diameters in wild-type and Ambra1 cKO NSCs (n = 3; total of 128 neurospheres analysed for each condition). P value by two-tailed unpaired t-test. i, Whole-brain quantification of E13.5 wild-type and Ambra1 cKO cyclin D1 staining normalized over DAPI, represented in Fig. 1e. P value by two-tailed unpaired t-test (n = 5). j, Wild-type and Ambra1 cKO olfactory bulbs in sagittal sections of E18.5 embryos, stained for cyclin D1 antibody and Hoechst (n = 3). k, Sagittal sections of wild-type and Ambra1 cKO E13.5 embryos, stained for cyclin D2 (n = 3). l, Left, representative images of sagittal sections of the mesencephalic ventricular zone in wild-type and Ambra1 cKO E13.5 embryos, stained for RB(pS807/811) (n = 5). Right, quantification of RB(pS807/811-positive area in the mesencephalic ventricular zone of E13.5 wild-type and Ambra1 cKO embryos (n = 5). P value by two-tailed unpaired t-test. m, Left, representative images of RB(pS807/811) in sagittal sections of the olfactory bulb in wild-type and Ambra1 cKO E18.5 embryos. Right, quantification of the number of RB(pS807/811)-positive cells (n = 3). P value by two-tailed unpaired t-test. n, Immunoblot of N-MYC after cycloheximide treatment in wild-type and Ambra1 cKO NSCs (n = 3). o, Quantitative PCR with reverse transcription (qRT‒PCR) of NSCs; the investigated genes are at the bottom of the graph (n = 5). P values by two-tailed unpaired t-test. p, Immunoblot of control and AMBRA1silenced SH-SY5Y cells (n = 3). q, Immunoblot of AMBRA1 immunoprecipitation in SH-SY5Y cells. r, Immunoblot for AMBRA1, PP2AC and N-MYC in SH-SY5Y cells silenced for the indicated genes (n = 3). Unless otherwise stated, n refers to biologically independent samples. For immunoblots, actin was used as loading control. Data are mean ± s.e.m. Scale bars, 250 μm.
Extended Data Fig. 2 |. Ambra1 deficiency affects the cell cycle, cell death and neuronal differentiation.
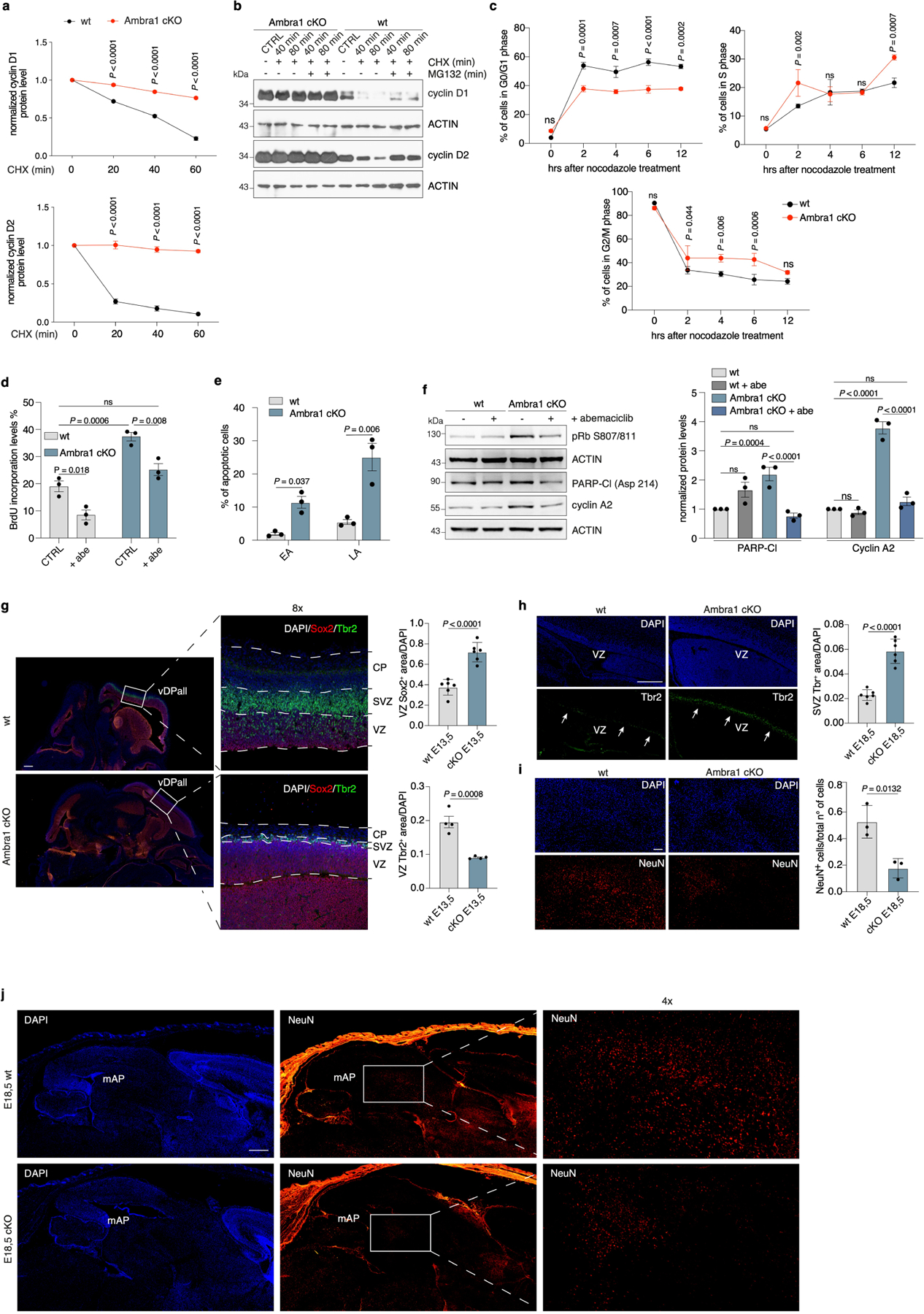
a, Densitometric quantification of cyclin D1 and D2 protein levels in the cycloheximide time course normalized over actin (n = 3). P values by two-sided one-way ANOVA followed by Sidak’s multiple comparisons test. b, Immunoblot of wild-type or Ambra1 cKO NSCs treated with cycloheximide and/or MG132 for the indicated times. (n = 3). c, Distribution of cell-cycle phases in NSCs after release from nocodazole treatment (n = 3). P values by two-sided one-way ANOVA followed by Sidak’s multiple comparisons test. d, Six-hour BrdU incorporation of passage-2 wild-type and Ambra1 cKO NSCs with or without abemaciclib treatment (n = 3). P values by two-sided one-way ANOVA followed by Tukey’s multiple comparisons test. e, Percentage of apoptotic cells in wild-type and Ambra1 cKO NSCs (n = 3). EA, early apoptotic; LA, late apoptotic. P values by two-sided one-way ANOVA followed by Sidak’s multiple comparisons test. f, Left, immunoblot of the indicated proteins in NSCs after abemaciclib treatment. Right, densitometry quantification of the indicated proteins (n = 3). P values by two-sided one-way ANOVA followed by Sidak’s multiple comparisons test. g, Left, representative images of sagittal sections from wild-type and Ambra1 cKO E13.5 embryos, stained for SOX2 and TBR2. Right, quantification of immunostained positive area (SOX, n = 6; TBR2, n = 4). P values by two-tailed unpaired t-test. h, Left, representative images of sagittal sections of wild-type and Ambra1 cKO E18.5 embryos, stained for TBR2. Right, quantification of immunostained positive area (n = 6). P value by two-tailed unpaired t-test. Arrows indicate TBR2+ cells in the subventricular zone. i, j, Representative images of sagittal sections of wild-type and Ambra1 cKO E18.5 embryos, stained for the neuronal marker NeuN. i, Left, higher magnification of the mesencephalic alar plate. Right, quantification of immunostained positive cells (n = 3). P value by two-tailed unpaired t-test. j, Lower magnification to better appreciate the uncropped quantified area (n = 3). Scale bar, 500 μm. Unless otherwise stated, n refers to biologically independent samples. For immunoblots, actin was used as loading control. Data are mean ± s.e.m. Unless otherwise noted, scale bars represent 250 μm.
Extended Data Fig. 3 |. The AMBRA1–cyclin D1 axis affects the cell cycle.
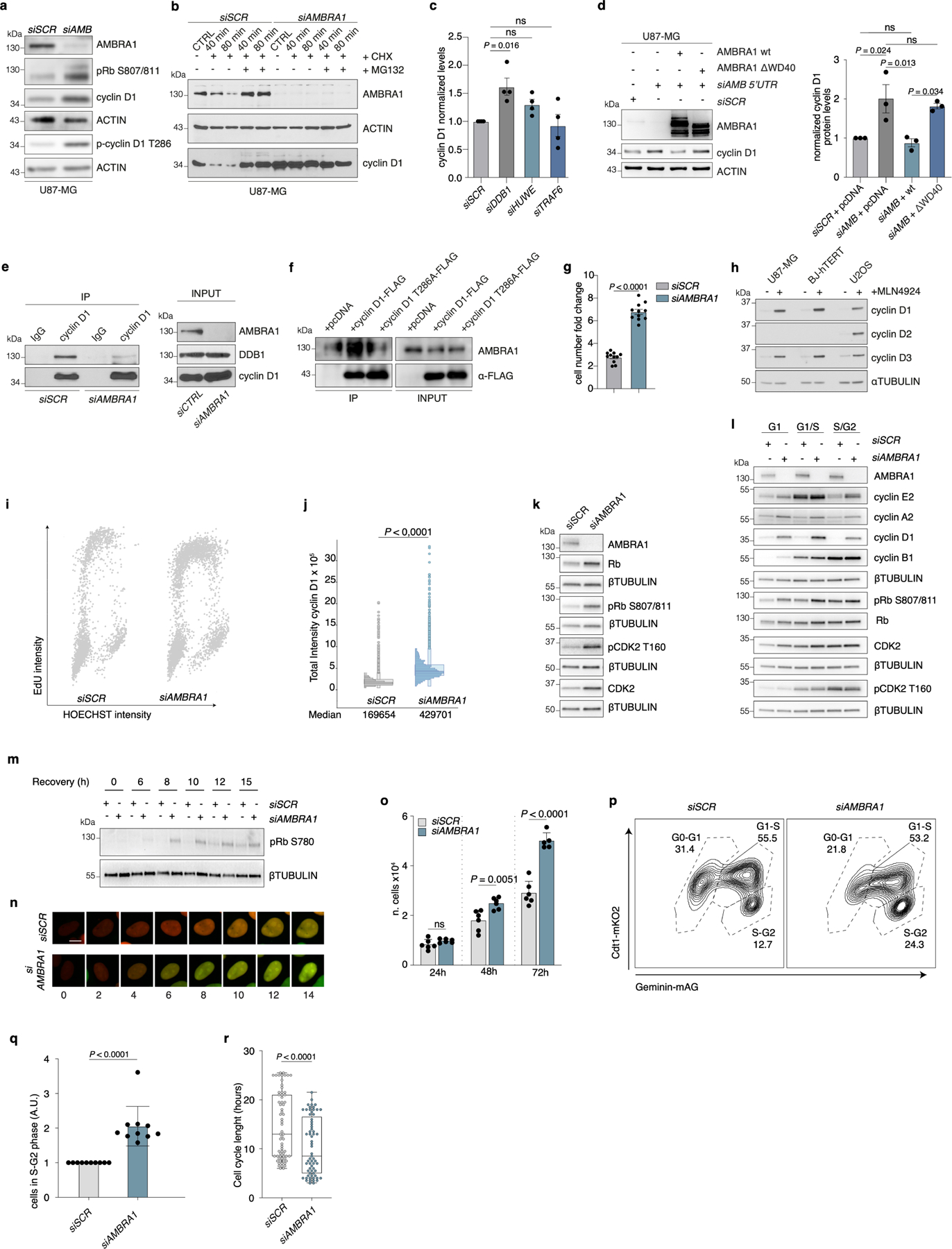
a, Immunoblot of control or AMBRA1-silenced U87-MG cells for the indicated proteins (n = 3). b, Immunoblot of cyclin D1 in control or AMBRA1-silenced U87-MG cells treated with cycloheximide and/or MG132 for the indicated times (n = 3). c, Analysis of densitometry for the cyclin D immunoblot in U87-MG cells, silenced for the indicated genes, shown in Fig. 1g (n = 4). P values by one-way ANOVA followed by Dunnett’s multiple comparisons test. d, Left, immunoblot of cyclin D1 in U87-MG cells silenced for AMBRA1 expression and overexpressing empty vector (pcDNA), wild-type AMBRA1 or AMBRA1(ΔWD40). Right, analysis from densitometry (n = 3). P values by one-way ANOVA followed by Tukey’s multiple comparisons test. e, Immunoblot analysis of cyclin D1 immunoprecipitation from protein extracts of control and AMBRA1-silenced U87-MG cells (n = 3). f, Co-immunoprecipitation of AMBRA1 in U87-MG cells transiently overexpressing empty vector, cyclin D1–Flag or cyclin D1(T286A)–Flag. Cells were treated with MG132 for 3 h before lysis (n = 3). g, Fold change in the number of cells in control or AMBRA1-silenced U87-MG cells (n = 11). P value by two-tailed unpaired t-test. h, Immunoblot of the indicated proteins of U87MG, BJ-hTERT and U2OS cells that were untreated or treated with MLN4924 for 4 h (n = 3). i, j, Cells immunostained with cyclin D1, EdU antibody and counterstained with Hoechst. i, Scatter plots reporting single-cell total nuclear intensities of EdU versus Hoechst (cells examined over three independent experiments: siSCR, n = 3,279; siAMBRA1, n = 3,608 cells). j, Box plots (centre line, median; box limits, 25th and 75th percentile) indicating total cyclin D1 nuclear intensities (siSCR, n = 3,279; siAMBRA1, n = 3,608 cells. median siSCR = 169,654; siAMBRA1 = 429,623). k, l, Immunoblot of cell-cycle markers in control and AMBRA1-silenced BJ-hTERT cells (k) and cell-cycle-sorted U2OS-FUCCI cells (l) (n = 3). m, Immunoblot of the indicated proteins in AMBRA1-silenced BJ-hTERT cells synchronized by 24-h serum starvation. Cells were collected after the indicated starvation recovery time points (n = 3). n, Representative images of live-cell imaging of control and AMBRA1-silenced U2OS-FUCCI cells from 0 to 14 h with a 2-h interval between different images. The length of the G1 phase is shown in Fig. 1k (n = 3). Scale bar, 5 μm. o, Cell proliferation in control or AMBRA1-silenced BJ-hTERT cells (24 h and 48 h n = 6; 72 h siSCR n = 6, siAMBRA1 n = 5). p, q, Control or AMBRA1-silenced U2OS-FUCCI cells. p, Representative contour plot. q, Fold increase of cells present in S–G2 phase in AMBRA1-downregulated cells with respect to control cells (n = 10). r, Box plots (centre line, median; box limits, 25th and 75th percentile; whiskers, minimum and maximum) showing the cell-cycle length of siSCR (n = 65; median = 13) or siAMBRA1 (n = 65; median = 8.5) U2OS-FUCCI cells examined over three independent experiments. Unless otherwise stated, n refers to biologically independent samples; data are mean ± s.e.m. Data were analysed using a two-tailed unpaired t-test (g, o, q) or two-tailed Mann–Whitney test (j, r). For immunoblots, actin or β-tubulin were used as loading control.
Extended Data Fig. 4 |. AMBRA1 deficiency causes replication stress.
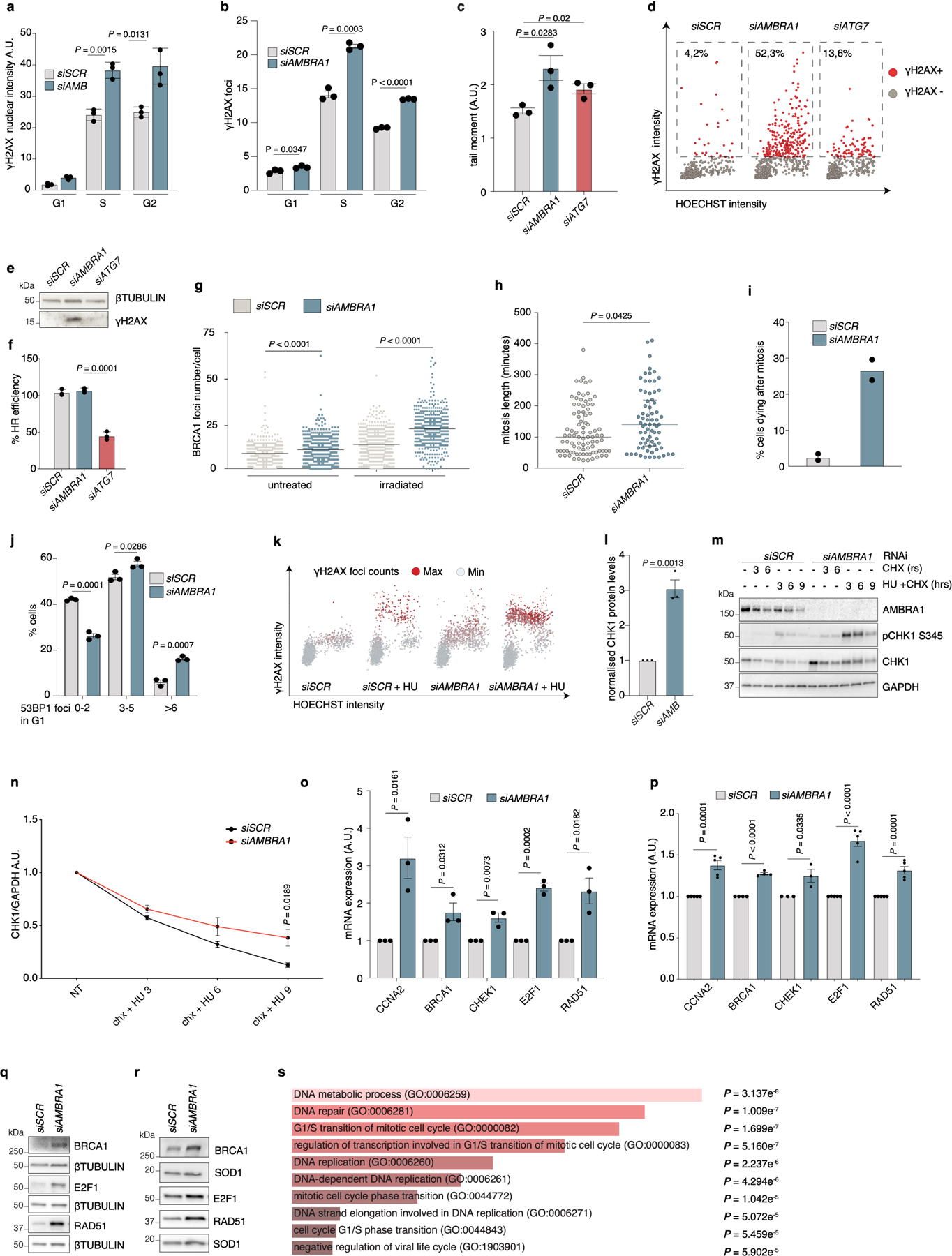
a, Total γH2AX nuclear intensity in the different cell-cycle phases of BJ-hTERT cells (n = 3). Data are mean ± s.d. b, Average number of γH2AX foci in control or AMBRA1-silenced U2OS cells (n = 3). c, Alkaline comet assay of control, AMBRA1- and ATG7-silenced U2OS cells (n = 3). d, Scatter plots showing γH2AX versus Hoechst total nuclear intensities from immunostainings of control, AMBRA1- and ATG7-silenced BJ-hTERT cells. The proportion of γH2AX-positive cells (red, arbitrary cut-off) is indicated (siSCR, n = 721; siAMBRA1, n = 725; siATG7, n = 733 cells examined over 3 independent experiments). e, Immunoblot of γH2AX in control, AMBRA1- and ATG7-silenced BJ-hTERT cells (n = 3). f, Homologous recombination (HR) efficiency in control, AMBRA1- and ATG7-silenced U2OS cells (n = 3). Data are mean ± s.d. g, Number of BRCA1 foci per nucleus in control and AMBRA1-silenced U2OS cells either untreated or treated with 3-Gy irradiation, stained against BRCA1 (n = 500 cells examined over 3 independent experiments, centre indicates the mean). h, Time in mitosis in control (n = 91 cells examined over 3 independent experiments) or AMBRA1-silenced (n = 72 cells examined over 3 independent experiments) cells. Bars represent median and interquartile range. i, Dying cells upon mitotic exit as evaluated by time-lapse imaging (n = 2 independent experiments; more than 60 cells per condition). j, Distribution of 53BP1 nuclear foci in G1 U2OS cells (n = 3). k, Total γH2AX versus Hoechst intensity in control and AMBRA1-silenced BJ-hTERT cells that were untreated or treated with 2 mM hydroxyurea (HU) for 2 h (siSCR, n = 2,481; siAMBRA1, n = 2,237; siSCR + HU, n = 2,484; siAMBRA1 + HU, n = 2,281 cells; scatter plots are representative of n = 3 independent experiments). l, Quantification of normalized protein levels of CHK1 represented in Fig. 2e (n = 3). m, n, BJ-hTERT cells as in Extended Data Fig. 4k treated with cycloheximide or with cycloheximide and 2 mM hydroxyurea. m, Immunoblot analysis of the indicated proteins in total cell lysates. n, Quantification of normalized CHK1 protein expression levels (n = 4). o, p, qRT–PCR analyses of the indicated genes in control or AMBRA1-silenced BJ-hTERT (o) and U2OS (p) cells, respectively (CCNA2, E2F1 and RAD51 n = 5; BRCA1 n = 4; CHEK1 n = 3). q, r, Immunoblot analysis of the indicated proteins in control or AMBRA1-silenced U2OS (q) and BJ-hTERT (r) cells (n = 3 in both conditions). s, Gene ontology (GO) biological processes (2018) from enrichment analysis of DEA (Differential Expression Analysis) genes from RNA sequencing (RNA-seq) experiments. DEA originating from three RNA-seq independent experiments was used as input for the web-based software EnrichR34,35. P values computed using Fisher’s exact test; clearer bars show a smaller P value. Unless otherwise stated, n refers to biologically independent samples; data are mean ± s.e.m. Data were analysed using a two-tailed unpaired t-test (a, b, c, f, j, l, n, o, p) or two-tailed Mann–Whitney test (g, h). For immunoblots, β-tubulin, SOD1 or GADPH were used as loading control.
Extended Data Fig. 5 |. AMBRA1 deficiency causes replication stress.
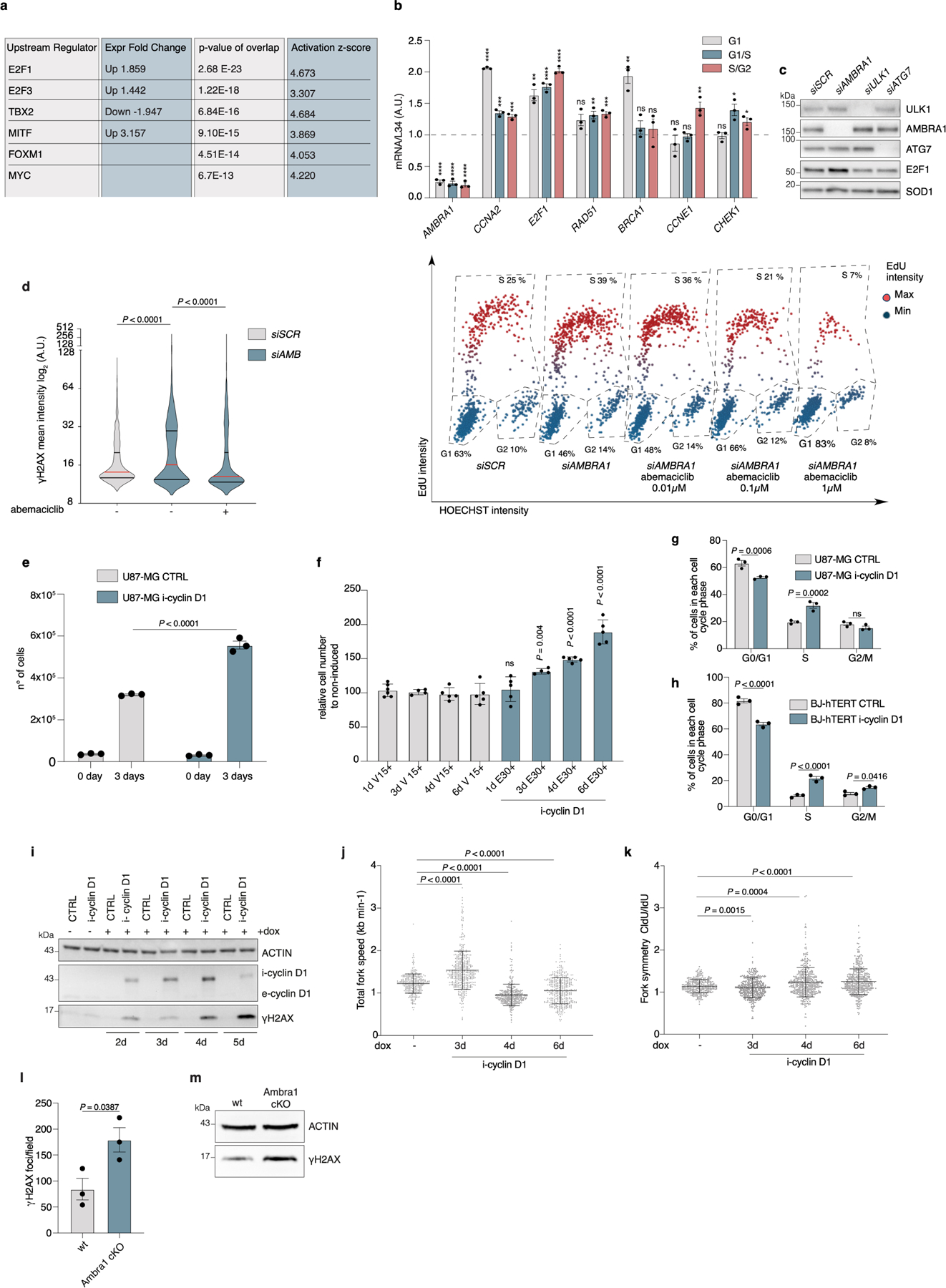
a, Analysis of DEA genes (from n = 3 independent RNA-seq experiments) predicting the transcription factor activated after depletion of AMBRA1. b, qRT–PCR analyses of the indicated genes in control or AMBRA1-silenced U2OS-FUCCI cells sorted for the different cell-cycle phases (n = 3) c, Immunoblot for the indicated proteins in U2OS cells interfered for the indicated autophagy regulators (n = 3). d, Left, violin plot of γH2AX nuclear mean intensity in control and AMBRA1-silenced BJ-hTERT cells that were untreated or treated with 0.1 μM abemaciclib for 48 h. Right, representative scatter plot of single-cell γH2AX nuclear mean intensity versus Hoechst, and cell cycle phase gating strategies from control and AMBRA1silenced BJ-hTERT cells treated with abemaciclib (n = 643 cells). e, Cell count of control U87-MG cells or U87-MG cells with inducible cyclin D1 expression, three days after stimulation with dox (n = 3). f, Cell count of control BJ-hTERT cells or BJ-hTERT cells with inducible cyclin D1 expression at the indicated time points after stimulation with dox, normalized over non-induced cells (1-d V15+, n = 6; 3-d V15+, 3-d E30+, n = 4; 1-d E30+, 4-d V15+, 4-d E30+, 6-d V15+ and 6-d E30+, n = 5). V15+: dox-treated control cells; E30+: dox-treated cyclin D1-inducible cells. g, h, Percentage of cells in each cell-cycle phase in U87-MG (g) and BJ-hTERT (h) cells, control or with inducible cyclin D1 expression, untreated or 48 h after doxycycline stimulation (n = 3). i, Immunoblot for the indicated proteins in control U87-MG cells or U87-MG cells with inducible cyclin D1 expression at the indicated time points with or without dox stimulation (n = 3). j, k, Mean fork speed (j) (kb min−1) and fork symmetry analysis (k) of DNA fibres from control BJ-hTERT cells and BJ-hTERT cells with inducible cyclin D1 expression treated as in Fig. 2d (scored forks: − dox, n = 312; 3-d dox, n = 449; 4-d dox, n = 429; 6-d dox, n = 426). Data are mean ± s.d. l, Quantification of immunohistochemistry staining in Fig. 2g (n = 3 mice). m, Immunoblot for the indicated proteins in wild-type or Ambra1 cKO NSCs (n = 3). Unless otherwise stated, n refers to biologically independent samples; data are mean ± s.e.m. Data were analysed using a two-tailed unpaired t-test (b, l), two-tailed Mann–Whitney test (d, j, k), two-way ANOVA followed by Sidak’s multiple comparisons test (e, g, h) or one-way ANOVA followed by Sidak’s multiple comparisons test (f). Exact P values are provided in the ‘Statistical analysis and data reproducibility’ section of the Supplementary Methods.
Extended Data Fig. 6 |. Bioinformatics analysis of AMBRA1 in cancer.
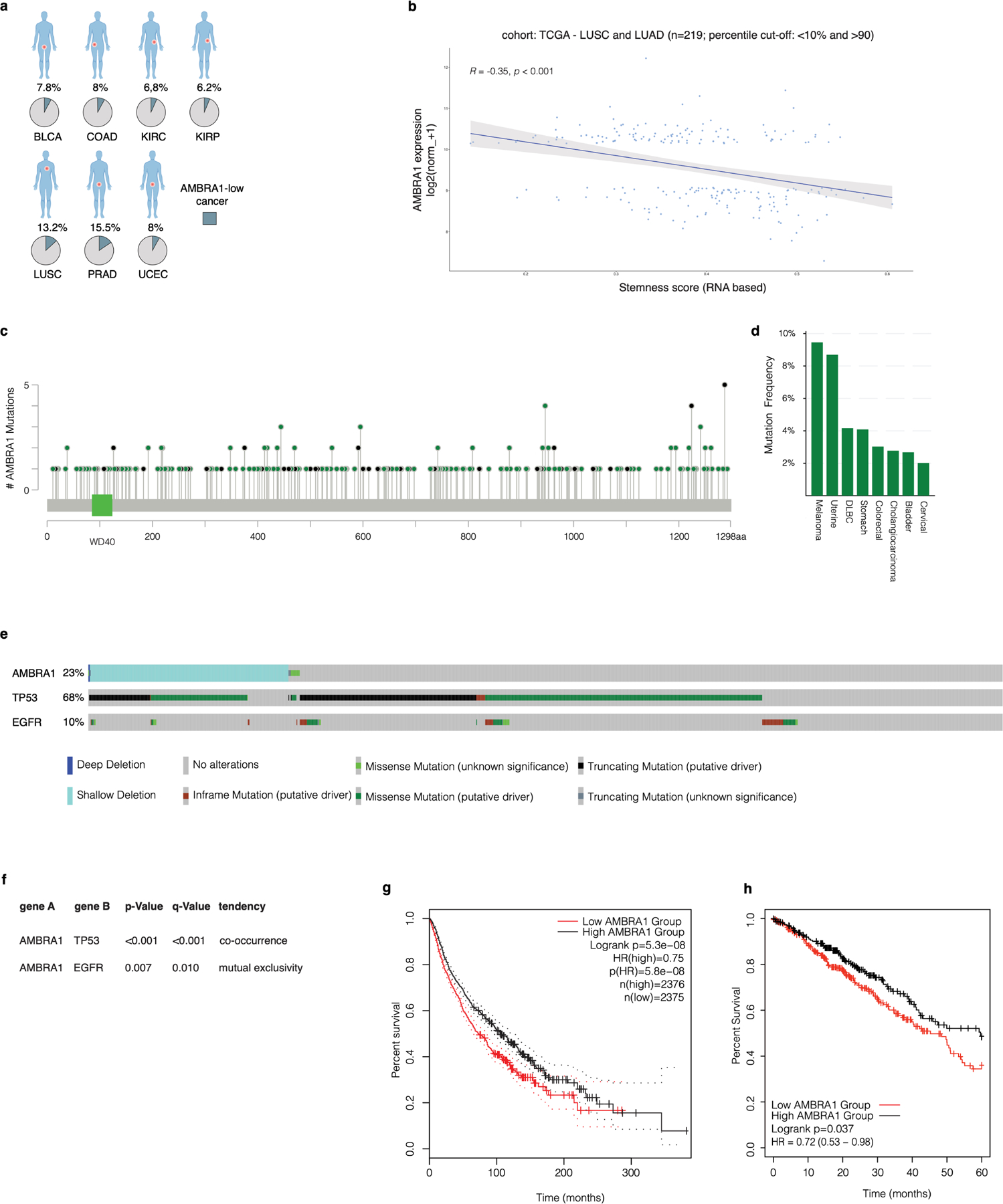
a, Bioinformatics analysis of expression data from the TCGA database. Pie charts show the percentage of AMBRA1-low cancers (light blue) with respect to the total (grey) in the indicated datasets. BLCA, bladder urothelial carcinoma; COAD, colon adenocarcinoma; KIRC, kidney renal clear cell carcinoma; KIRP, kidney renal papillary cell carcinoma; LUSC, lung squamous cell carcinoma; PRAD, prostate adenocarcinoma; UCEC, uterine corpus endometrial carcinoma. b, Xena correlation analysis of AMBRA1 mRNA expression and stemness score. The shaded area in the plot indicates the confidence interval (95%). c, Lolliplots showing the distribution of AMBRA1 mutations annotated in TCGA Pan-Cancer Atlas Studies datasets. d, Frequency of AMBRA1 mutations (expressed as a percentage) in TCGA Pan-Cancer Atlas Studies datasets. The cut-off was selected at 2%. e, Oncoprint of AMBRA1 alterations (homodeletions, shallow deletions, mutations), and TP53 and EGFR mutations from TCGA Pan-Lung Cancer datasets. f, Mutual exclusivity and co-occurrence analysis of the indicated genes from TCGA Pan-Lung Cancer datasets. P values derived from one-sided Fisher’s exact test. g, Kaplan–Meier analysis of patients in the Pan-Cancer Atlas Studies database was generated based on the expression level of AMBRA1 (low, below 20%; high, above 80%). Plot was downloaded from the online database GEPIA36 (http://gepia2.cancer-pku.cn/#analysis). P values derived from one-sided log-rank Mantel–Cox test). h, Kaplan–Meier analysis of overall survival based on RNA-seq analysis of AMBRA1 mRNA levels using the KM-plotter37 lung adenocarcinoma database.
Extended Data Fig. 7 |. AMBRA1 controls tumour growth in a mouse model of lung cancer.
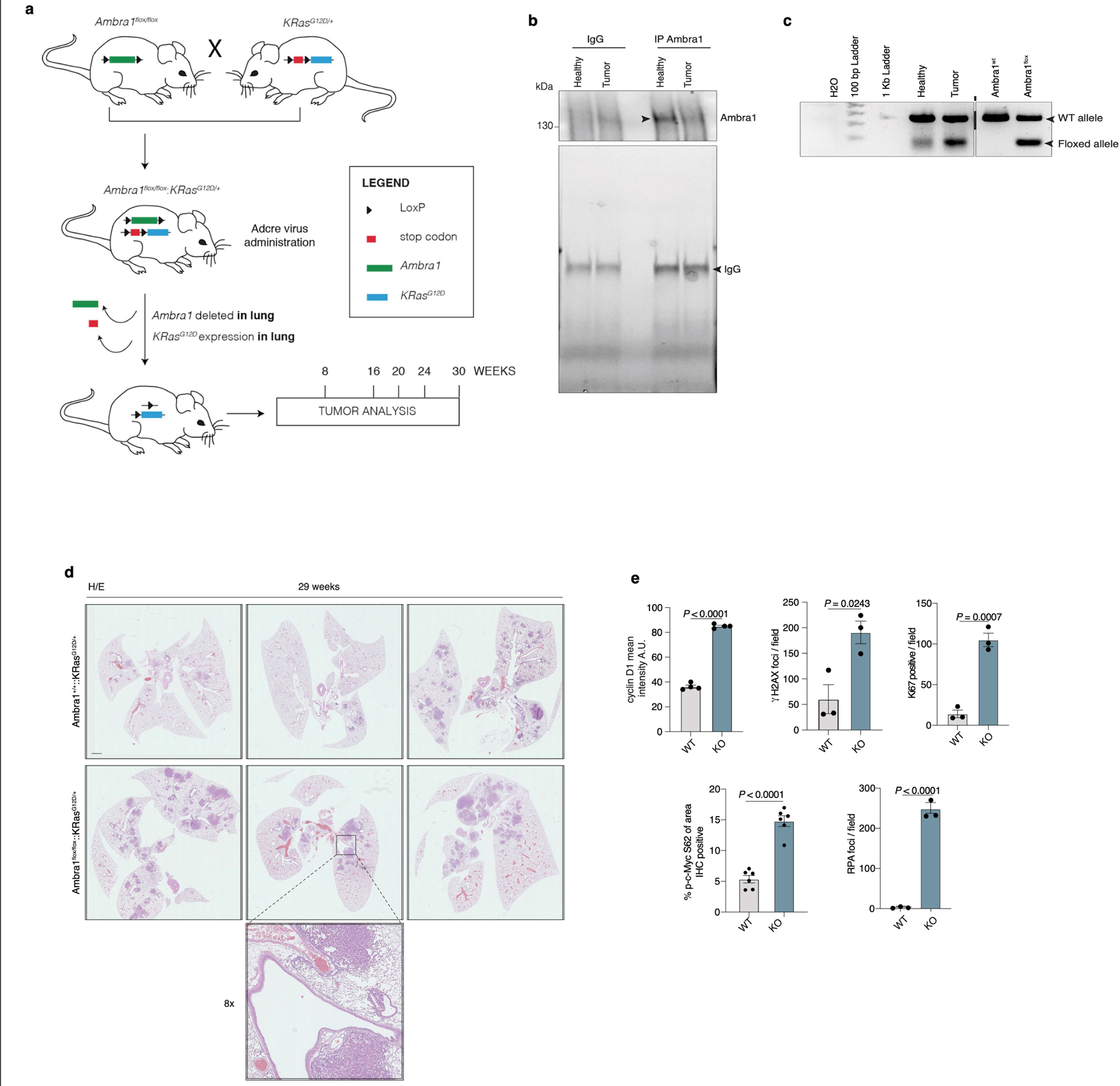
a, Schematic representations of the mouse model and initial testing of the system. The KrasG12D transgenic mouse is mated with the conditional Ambra1flox/flox mouse to produce the Ambra1+/+::KrasG12D/+ and the Ambra1flox/flox::KrasG12D/+ genotypes. Lung-specific expression of oncogenic KrasG12D and deletion of Ambra1 is induced by intranasal inoculation with defective adenoviral particles carrying the Cre recombinase. b, Immunoblot analysis of AMBRA1 immunoprecipitation from tissue lung samples from Ambra1flox/flox::KrasG12D/+ mice 16 weeks after administration of AdenoCre (n = 3). c, The expression of the Ambra1 floxed allele after Cre administration was verified by RT–PCR performed in lung tissue samples as in c (n = 3). Primers were designed to distinguish wild-type and floxed alleles. d, Representative examples of H&E images of fixed lungs. Bottom, Magnification of the bronchus, highlighting the tumour initiation site. Scale bar, 1 mm. e, Quantification of immunohistochemistry staining in Fig. 3b (Ki67, n = 3; γH2AX, n = 3; RPA(pS4/8), n = 3; cyclin D1, n = 4; c-Myc(pS62), n = 3 in two independent tumours for each condition). Unless otherwise stated, n refers to biologically independent samples; data are mean ± s.e.m. P values for γH2AX and cyclin D1 by two-tailed Welch t-test; P values for Ki67, c-MYC(pS62), RPA(pS4/8) by two-tailed unpaired t-test.
Extended Data Fig. 8 |. AMBRA1 deficiency is synthetic lethal with CHK1 inhibitors.
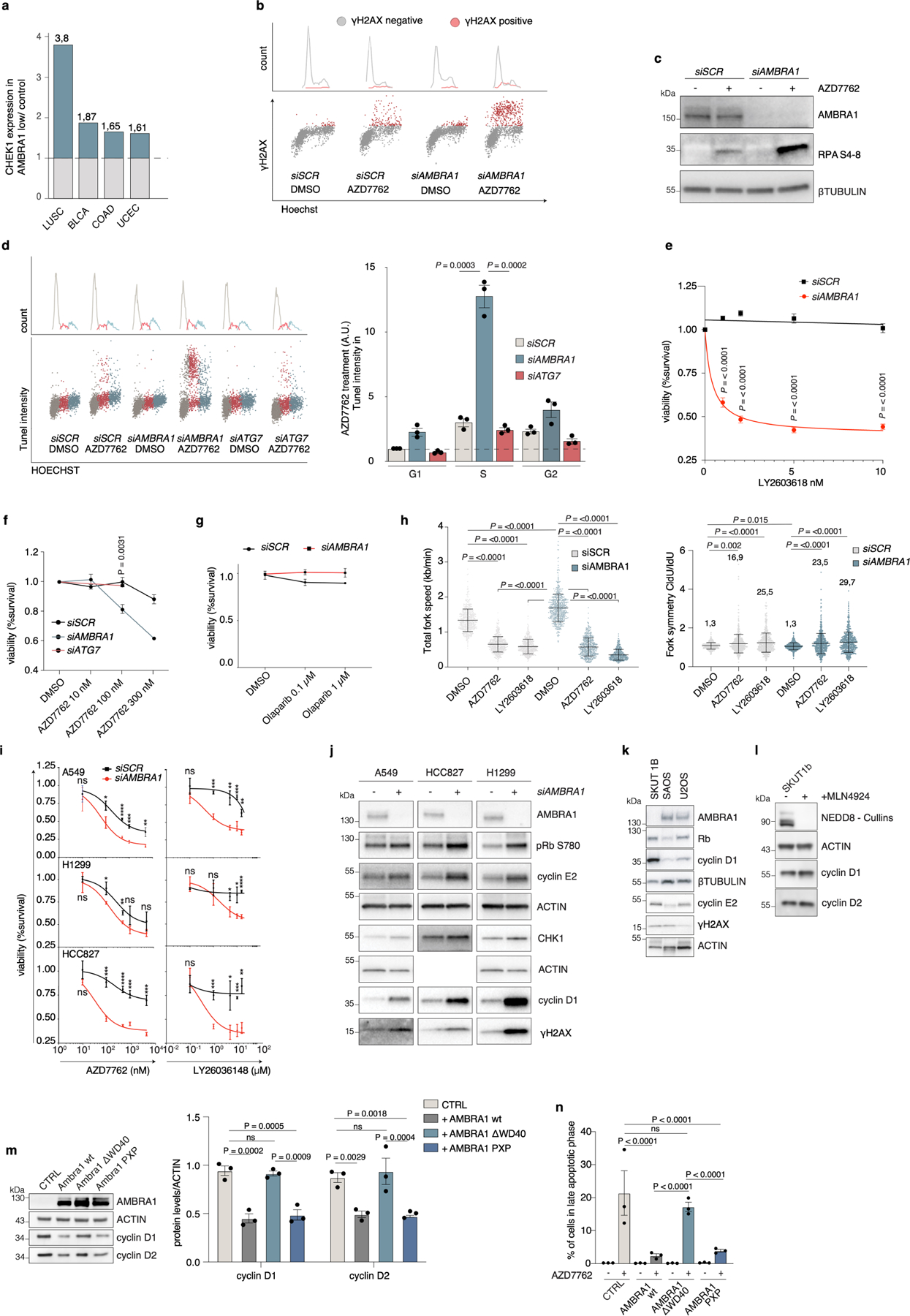
a, Ratio between CHEK1 expression in the AMBRA1-low subpopulation of cancers with respect to normal tissue. b, Gating strategy for Fig. 3c. Bottom, scatter plots of total nuclear DNA intensity versus γH2AX intensity (siSCR DMSO, n = 1,850; siSCR AZD, n = 1,716; siAMBRA1 DMSO, n = 1,866; siAMBRA1 AZD, n = 1,731 cells; representative of three independent experiments). The γH2AX-positive cells (arbitrary cut-off) are indicated in red. Top, Hoechst nuclear intensity versus counts. γH2AX-positive cells are indicated by the red line. c, Immunoblot of AMBRA1, RPA(pS4/8) and β-tubulin in control or AMBRA1-silenced BJ-hTERT cells that were untreated or treated with AZD7762. d, Left, gating strategy for the quantification on the right. Top, Hoechst nuclear intensity versus counts. Bottom, scatter plots reporting single-cell total nuclear DNA intensity versus TUNEL intensity. Right, TUNEL-positive cells in the different cell phases calculated based on Hoechst intensity (n = 3). P values by two-tailed unpaired t-test. e, Viability analysis of control and AMBRA1-silenced BJ-hTERT cells treated with the indicated concentrations of LY2603618 for 24 h (n = 3). P values by two-stage step-up (Benjamini, Krieger and Yekutieli). f, Cell viability in control, AMBRA1- and ATG7-silenced BJ-hTERT cells that were untreated or treated with AZD7762 for 24 h (n = 4 for Control and treatments with 100 nM AZD7762). P value by two-tailed unpaired t-test. g, Cell viability in control and AMBRA1-silenced BJ-hTERT cells that were untreated or treated with olaparib for 24 h (n = 3). Analysis by two-tailed unpaired t-test. h, Fork symmetry analysis from BJ-hTERT cells treated for 24 h with 100 nM AZD7762 or 5 μM LY2603618 (scored forks: siSCR DMSO, n = 533; siSCR AZD, n = 560; siSCR LY, n = 548; siAMBRA1 DMSO, n = 601; siAMBRA1 AZD, n = 543; siAMBRA1 LY, n = 548). P values by two-tailed Mann–Whitney test. Data are mean ± s.d. i, Cell viability analysis of control and AMBRA1-silenced A549, H11299 and HCC827 lung cancer cell lines treated with the indicated concentrations of AZD7762 and LY2603618 (n = 3) for 24 h. P values by two-stage step-up (Benjamini, Krieger and Yekutieli). Data are mean ± s.d. j, Immunoblot of the indicated proteins in control or AMBRA1-silenced A549, HCC827 and H1299 cells (n = 3). k, Immunoblot of sarcoma cell lines (n = 3). l, Immunoblot of SKUT-1B cells treated with the inhibitor MLN4924 for 4 h (n = 3). m, Left, immunoblot of SKUT-1B cells reconstituted with wild-type AMBRA1 or mutant AMBRA1(ΔWD40) or AMBRA1(PXP). Right, densitometry quantification of the indicated normalized protein levels (n = 3). P values by two-sided one-way ANOVA followed by Sidak’s multiple comparisons test. n, Late apoptosis analysis in SKUT-1B cells reconstituted with wild-type AMBRA1, AMBRA1(ΔWD40) or AMBRA1(PXP) and treated with 200 nM AZD7762 for 24 h (n = 3). P values by two-sided one-way ANOVA followed by Tukey’s multiple comparisons test. Unless otherwise stated, n refers to biologically independent samples; data are mean ± s.e.m. *P < 0.05, **P < 0.01, ***P < 0.001, ****P < 0.0001. Exact P values are provided in the ‘Statistical analysis and data reproducibility’ section of the Supplementary Methods.
Extended Data Fig. 9 |. AMBRA1 deficiency is synthetic lethal with CHK1 inhibitors in vivo.
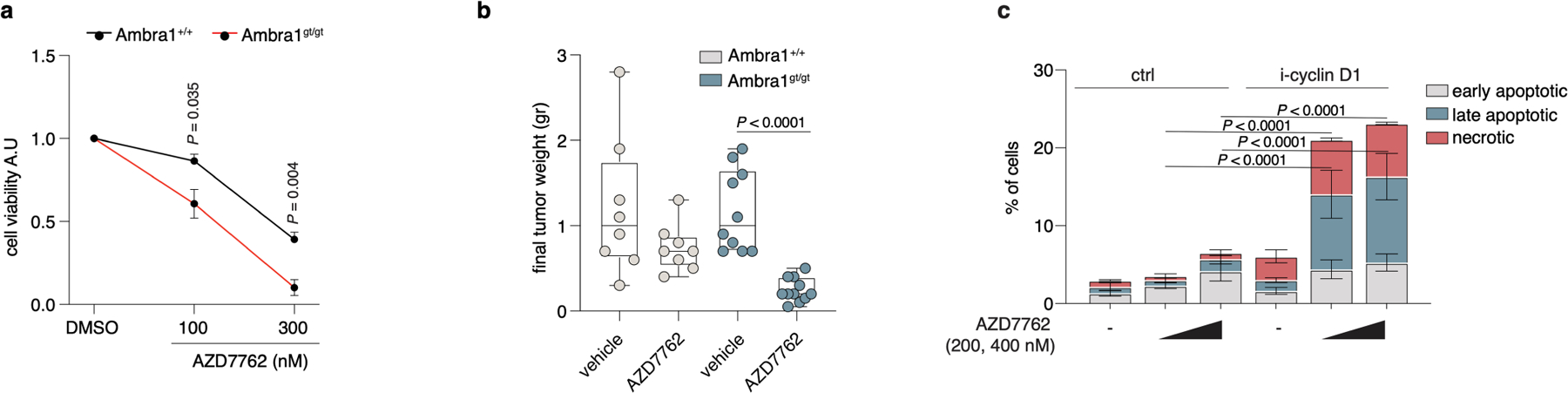
a, Cell viability of Ambra1+/+ and Ambra1gt/gt MEFs treated with AZD7762 or vehicle for 24 h (n = 4 independent experiments). P values by two-tailed unpaired t-test. b, Box plots (centre line, median; box limits, 25th and 75th percentile; whiskers, minimum and maximum) indicating weight of Ambra1+/+ and Ambra1gt/gt MEF xenografts referred to in Fig. 3f (Ambra1+/+ + vehicle, n = 8; Ambra1+/+ + AZD7762, n = 8; Ambra1gt/gt + vehicle, n = 10; Ambra1gt/gt + AZD7762, n = 11 mice). P values by two-tailed unpaired t-test. c, Cell death percentage in control U87-MG cells or overexpressing cyclin D1, either untreated or treated with AZD7762 for 24 h; mean ± s.e.m. (n = 3 independent experiments). P values by two-sided one-way ANOVA followed by Tukey’s multiple comparisons test. Unless otherwise stated, data are mean ± s.d.
Supplementary Material
Acknowledgements
E. Maiani is an Adjunct Professor at UniCamillus–Saint Camillus International University of Health Sciences. The F.C. laboratory is supported by grants from the Danish Cancer Society (KBVU R72-A4408, R146-A9364, R231-A14034 to F.C.; R146-A9471 to V. Cianfanelli; R146-A9414 to G.F.; R204-A12424 to D.D.Z.), the Novo Nordisk Foundation (NNF13OC0007559, NNF16OC0022544), the Lundbeck Foundation (R233–2016-3360 to F.C.; R209–2015-3505 to V. Cianfanelli), the LEO Foundation (LF17024 to F.C. and E. Papaleo; LF-OC-19–000004 to D.D.Z.), the Associazione Italiana per la Ricerca sul Cancro (AIRC project IG 2019 #23543 to F.C.; #22811 to L.L.; 5×1000 #9962 and AIRC IG 2018 #21724 to F.L.), the Italian Ministry of Research (MIUR, project PRIN 2017 FS5SHL Radius) and the Italian Ministry of Health (Ricerca Corrente to F.L. and F.N.). This work was also supported by the European Union’s Horizon 2020 research and innovation program (Marie Sklodowska-Curie grant agreement 642295 (MEL-PLEX)). D.D.Z. is supported by the Melanoma Research Alliance (MRA 620385). The F.C., J. Bartek and E. Papaleo laboratories in Copenhagen are part of the Center of Excellence for Autophagy, Recycling and Disease (CARD), funded by the Danmarks Grundforskningsfond (DNRF125). L.L. is supported by FPRC 5×1000 Ministero della Salute 2015. V. Cianfanelli, C.M. and M.B. are supported by the Fondazione Umberto Veronesi. M.P. is funded by grants from the National Institute of Health (R01-CA76584 and R35-GM136250) and is an investigator with the Howard Hughes Medical Institute; the work of E. Papaleo is supported by the Carlsberg Foundation Distinguished Fellowship (CF18–0314); work in the G.V. group is supported by the PI15/00339 grant, integrated into the State Plan for R&D + I20132016 and funded by the Instituto de Salud Carlos III (ISCIII) and the European Regional Development Fund (ERDF), by the Marie Skłodowska-Curie Innovative Training Network (ITN) action TRAIN (GA 721532) funded by the European Commission (H2020) and by grants from Voices Against Brain Cancer and ‘Fundació La Marató de TV3’ (20134031). J. Bartek, J. Bartkova and A.M.-M. are supported by grants from the Danish Cancer Society (R204-A12617-B153), the Novo Nordisk Foundation (16854 and 0060590), the Danish Council for Independent Research (DFF-7016–00313), the Lundbeck Foundation (R266–2017-4289), the Swedish Research Council (VR-MH 2014–46602-117891–30) and the Swedish Cancerfonden (170176). We thank P. Bonaldo and P. Braghetta for the generation of the Ambra1flox/flox mouse model, V. Turcanova for help with cloning and mutagenesis, Plaisant S.r.l. (Castel Romano) and the Danish Cancer Society animal facilities for help with in vivo experiments, V. Tocco and the FACS facility members for technical help in the flow cytometry analysis and C. Rodolfo for his help and support. G.M. is grateful to A. M. Gatta and V. Milletti for their support.
Footnotes
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-021-03422-5.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Code availability
All of the computer scripts and source codes used to generate and analyse the results from The Cancer Genome Atlas (TCGA) analyses presented in Extended Data Figs. 6a, 8a are available at https://github.com/ELELAB/AMBRA_low.
Competing interests M.P. is a consultant for and has financial interests in Coho Therapeutics, CullGen, Kymera Therapeutics and SEED Therapeutics. M.P. is a cofounder of Coho Therapeutics, is on the Scientific Advisory Board of CullGen and Kymera Therapeutics, and is a consultant for Santi Therapeutics. The other authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-021-03422-5.
Peer review information Nature thanks Piotr Sicinski and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Reprints and permissions information is available at http://www.nature.com/reprints.
Data availability
Data from the Kaplan–Meier analysis in Extended Data Fig. 6g, h referenced during the study are available in a public repository from the websites (http://kmplot.com/ and http://gepia2.cancer-pku.cn/#analysis). AMBRA1 expression data and the stemness score (RNA-based) were downloaded from the Xena platform (http://xena.ucsc.edu/). The graph and map of AMBRA1 mutations in TCGA Pan-Cancer Atlas studies were downloaded from cBioPortal (https://www.cbioportal.org/). The original uncropped immunoblot data that support the findings of this study are available in Supplementary Fig. 1. A representative gating strategy for fluorescence-activated cell sorting (FACS) analysis is included in Supplementary Fig. 2. Source data are provided with this paper.
References
- 1.Sherr CJ & Sicinski P in D-type Cyclins and Cancer (eds Hinds PW & Brown NE) 1–26 (2018).
- 2.Otto T & Sicinski P Cell cycle proteins as promising targets in cancer therapy. Nat. Rev. Cancer 17, 93–115 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Cianfanelli V et al. Ambra1 at a glance. J. Cell Sci 128, 2003–2008 (2015). [DOI] [PubMed] [Google Scholar]
- 4.Antonioli M et al. AMBRA1 interplay with cullin E3 ubiquitin ligases regulates autophagy dynamics. Dev. Cell 31, 734–746 (2014). [DOI] [PubMed] [Google Scholar]
- 5.Nazio F et al. mTOR inhibits autophagy by controlling ULK1 ubiquitylation, self-association and function through AMBRA1 and TRAF6. Nat. Cell Biol 15, 406–416 (2013). [DOI] [PubMed] [Google Scholar]
- 6.Cianfanelli V et al. AMBRA1 links autophagy to cell proliferation and tumorigenesis by promoting c-Myc dephosphorylation and degradation. Nat. Cell Biol 17, 20–30 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fimia GM et al. Ambra1 regulates autophagy and development of the nervous system. Nature 447, 1121–1125 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Baldin V, Lukas J, Marcote MJ, Pagano M & Draetta G Cyclin D1 is a nuclear protein required for cell cycle progression in G1. Genes Dev 7, 812–821 (1993). [DOI] [PubMed] [Google Scholar]
- 9.Mateyak MK, Obaya AJ & Sedivy JM c-Myc regulates cyclin D-Cdk4 and -Cdk6 activity but affects cell cycle progression at multiple independent points. Mol. Cell. Biol 19, 4672–4683 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Lange C, Huttner WB & Calegari F Cdk4/cyclinD1 overexpression in neural stem cells shortens G1, delays neurogenesis, and promotes the generation and expansion of basal progenitors. Cell Stem Cell 5, 320–331 (2009). [DOI] [PubMed] [Google Scholar]
- 11.Di Rita A et al. HUWE1 E3 ligase promotes PINK1/PARKIN-independent mitophagy by regulating AMBRA1 activation via IKKα. Nat. Commun 9, 3755 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Diehl JA, Zindy F & Sherr CJ Inhibition of cyclin D1 phosphorylation on threonine-286 prevents its rapid degradation via the ubiquitin-proteasome pathway. Genes Dev 11, 957–972 (1997). [DOI] [PubMed] [Google Scholar]
- 13.Di Bartolomeo S et al. The dynamic interaction of AMBRA1 with the dynein motor complex regulates mammalian autophagy. J. Cell Biol 191, 155–168 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liu EY et al. Loss of autophagy causes a synthetic lethal deficiency in DNA repair. Proc. Natl Acad. Sci. USA 112, 773–778 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Burrell RA et al. Replication stress links structural and numerical cancer chromosomal instability. Nature 494, 492–496 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fragkos M & Naim V Rescue from replication stress during mitosis. Cell Cycle 16, 613–633 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Wilhelm T et al. Spontaneous slow replication fork progression elicits mitosis alterations in homologous recombination-deficient mammalian cells. Proc. Natl Acad. Sci. USA 111, 763–768 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Lukas C et al. 53BP1 nuclear bodies form around DNA lesions generated by mitotic transmission of chromosomes under replication stress. Nat. Cell Biol 13, 243–253 (2011). [DOI] [PubMed] [Google Scholar]
- 19.Maya-Mendoza A et al. High speed of fork progression induces DNA replication stress and genomic instability. Nature 559, 279–284 (2018). [DOI] [PubMed] [Google Scholar]
- 20.Petermann E, Woodcock M & Helleday T Chk1 promotes replication fork progression by controlling replication initiation. Proc. Natl Acad. Sci. USA 107, 16090–16095 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Feijoo C et al. Activation of mammalian Chk1 during DNA replication arrest: a role for Chk1 in the intra-S phase checkpoint monitoring replication origin firing. J. Cell Biol 154, 913–923 (2001). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Bartkova J et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 444, 633–637 (2006). [DOI] [PubMed] [Google Scholar]
- 23.Lukas J, Bartkova J, Rohde M, Strauss M & Bartek J Cyclin D1 is dispensable for G1 control in retinoblastoma gene-deficient cells independently of cdk4 activity. Mol. Cell. Biol 15, 2600–2611 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bartek J & Lukas J Pathways governing G1/S transition and their response to DNA damage. FEBS Lett 490, 117–122 (2001). [DOI] [PubMed] [Google Scholar]
- 25.Chaikovsky AC et al. The AMBRA1 E3 ligase adaptor regulates the stability of cyclin D. Nature 10.1038/s41586-021-03474-7 (2021). [DOI] [PMC free article] [PubMed]
- 26.Simoneschi D et al. CRL4AMBRA1 is a master regulator of D-type cyclins. Nature 10.1038/s41586-021-03445-y (2021). [DOI] [PMC free article] [PubMed]
- 27.Murga M et al. Exploiting oncogene-induced replicative stress for the selective killing of Myc-driven tumors. Nat. Struct. Mol. Biol 18, 1331–1335 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Syljuåsen RG et al. Inhibition of human Chk1 causes increased initiation of DNA replication, phosphorylation of ATR targets, and DNA breakage. Mol. Cell. Biol 25, 3553–3562 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Sen T et al. CHK1 inhibition in small-cell lung cancer produces single-agent activity in biomarker-defined disease subsets and combination activity with cisplatin or olaparib. Cancer Res 77, 3870–3884 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bartek J, Mistrik M & Bartkova J Thresholds of replication stress signaling in cancer development and treatment. Nat. Struct. Mol. Biol 19, 5–7 (2012). [DOI] [PubMed] [Google Scholar]
- 31.Toledo LI, Murga M & Fernandez-Capetillo O Targeting ATR and Chk1 kinases for cancer treatment: a new model for new (and old) drugs. Mol. Oncol 5, 368–373 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Dere E et al. Heterozygous Ambra1 deficiency in mice: a genetic trait with autism-like behavior restricted to the female gender. Front. Behav. Neurosci 8, 181 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.La Barbera L, Vedele F, Nobili A, D’Amelio M & Krashia P Neurodevelopmental disorders: functional role of ambra1 in autism and schizophrenia. Mol. Neurobiol 56, 6716–6724 (2019). [DOI] [PubMed] [Google Scholar]
- 34.Kuleshov MV et al. Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–W97 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen EY et al. Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tang Z et al. GEPIA: a web server for cancer and normal gene expression profiling and interactive analyses. Nucleic Acids Res 45, W98–W102 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Nagy A, Gyöngyi M & Győrffy B Pancancer survival analysis of cancer hallmark genes. Sci. Rep 11, 6047 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data from the Kaplan–Meier analysis in Extended Data Fig. 6g, h referenced during the study are available in a public repository from the websites (http://kmplot.com/ and http://gepia2.cancer-pku.cn/#analysis). AMBRA1 expression data and the stemness score (RNA-based) were downloaded from the Xena platform (http://xena.ucsc.edu/). The graph and map of AMBRA1 mutations in TCGA Pan-Cancer Atlas studies were downloaded from cBioPortal (https://www.cbioportal.org/). The original uncropped immunoblot data that support the findings of this study are available in Supplementary Fig. 1. A representative gating strategy for fluorescence-activated cell sorting (FACS) analysis is included in Supplementary Fig. 2. Source data are provided with this paper.