

Abstract
Proline dehydrogenase (PRODH) catalyzes the first step of proline catabolism, the FAD-dependent oxidation of L-proline to Δ1-pyrroline-5-carboxylate. PRODH plays a central role in the metabolic rewiring of cancer cells, which has motivated the discovery of inhibitors. Here, we studied the inhibition of PRODH by 18 proline-like compounds to understand the structural and chemical features responsible for the affinity of the best-known inhibitor, S-(–)-tetrahydro-2-furoic acid (1). The compounds were screened, and then six were selected for more thorough kinetic analysis: cyclobutane-1,1-dicarboxylic acid (2), cyclobutanecarboxylic acid (3), cyclopropanecarboxylic acid (4), cyclopentanecarboxylic acid (16), 2-oxobutyric acid (17), and (2S)-oxetane-2-carboxylic acid (18). These compounds are competitive inhibitors with inhibition constants in the range of 1.4 – 6 mM, compared to 0.3 mM for 1. Crystal structures of PRODH complexed with 2, 3, 4, and 18 were determined. All four inhibitors bind in the proline substrate site, but the orientations of their rings differ from that of 1. The binding of 3 and 18 is accompanied by compression of the active site to enable nonpolar contacts with Leu513. Compound 2 is unique in that the additional carboxylate displaces a structurally conserved water molecule from the active site. Compound 18 also destabilizes the conserved water, but by an unexpected non-steric mechanism. The results are interpreted using a chemical double mutant thermodynamic cycle. This analysis revealed unanticipated synergism between ring size and hydrogen bonding to the conserved water. These structure-affinity relationships provide new information relevant to the development of new inhibitor design strategies targeting PRODH.
Introduction
Proline catabolism, the four-electron oxidation of proline to glutamate, occurs in the mitochondria and is catalyzed by two enzymes. Proline dehydrogenase (PRODH), the first enzyme, is a flavin-dependent enzyme that catalyzes the oxidation of proline to Δ1-pyrroline-5-carboxylate (P5C) (Fig. 1). P5C then undergoes nonenzymatic hydrolysis to form L-glutamate-γ-semialdehyde (GSAL), the substrate for the second enzyme in proline catabolism, GSAL dehydrogenase (GSALDH). GSALDH uses the cofactor NAD+ in the oxidation of GSAL to L-glutamate.
Fig. 1.
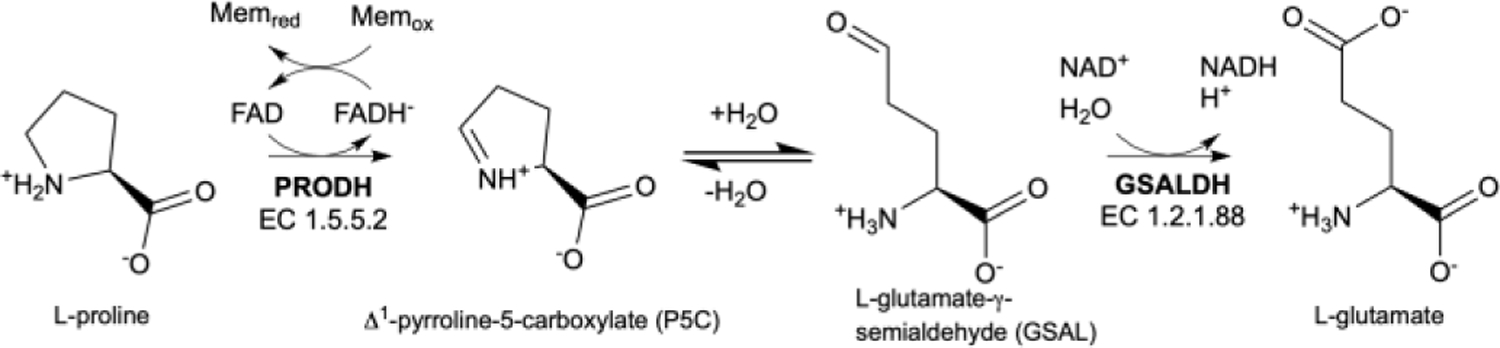
Enzymes and reactions of proline catabolism.
Proline metabolism is central to the metabolic rewiring of cancer cells. Proline catabolism, biosynthesis, and cycling have been implicated as metabolic pathways selectively altered in cancer cells providing ATP, macromolecules, and redox cofactors.1–3 PRODH acts as a tumor suppressor or an oncogene depending on the tumor type, and the environmental and metabolic context.4 Its oncogenic character has been revealed in non-small cell lung cancer, where increased PRODH expression promotes tumorigenesis by inducing epithelial to mesenchymal transition and several inflammatory genes.5 PRODH is also important in the rewiring of breast cancer cells leading to metastasis. Human metastatic tissue exhibits upregulated expression of PRODH compared with primary breast tumor tissue, and the inhibition of PRODH by S-(–)-tetrahydro-2-furoic acid (1 in Fig. 2) impairs metastasis formation in breast cancer mouse models.6 PRODH’s involvement in cancer metabolism is thought to manifest through the proline cycle, a substrate cycle composed of PRODH and the proline biosynthetic enzyme Δ1-pyrroline-5-carboxylate reductase 1 (PYCR1). PRODH forms half of the cycle by catalyzing the oxidation of proline to P5C, while PYCR1 catalyzes the reverse transformation, the NADPH-dependent reduction of P5C to L-proline. The proline cycle has been implicated in supporting ATP production, protein and nucleotide synthesis, anaplerosis, and redox homeostasis in cancer cells.2
Fig. 2.
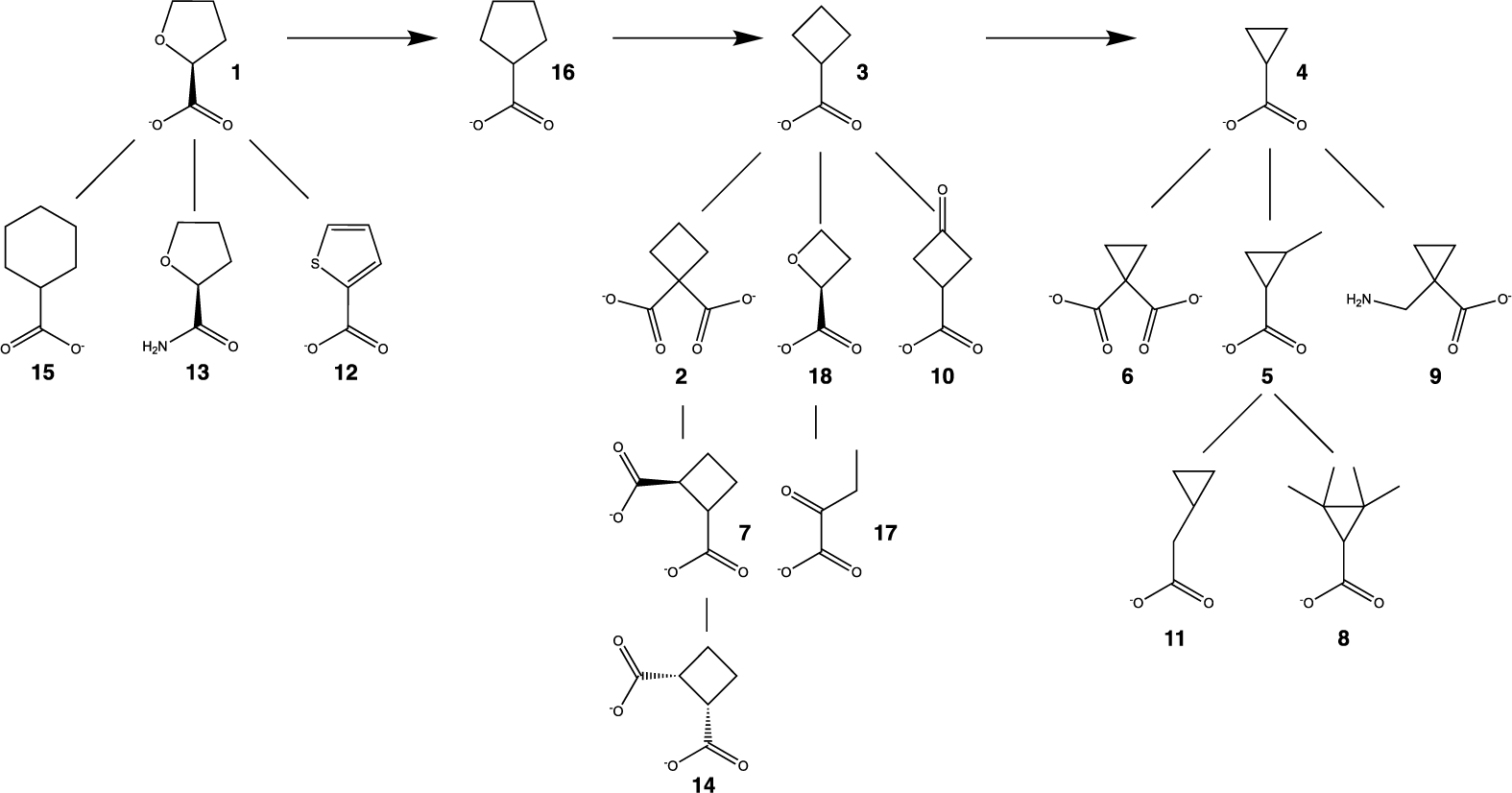
Chemical structures of the inhibitors used in this study.
The involvement of the proline cycle in cancer cell metabolism has motivated the development of inhibitors targeting PRODH and PYCR1. The proline analog 1 is the best characterized reversible inhibitor of PRODH. Compound 1 (Ki ~ 0.2–1.0 mM)7,8 has proven to be useful for investigating the proline cycle in cancer cells and mouse models of cancer.6,9 Other small carboxylic acids, such as L-lactic acid (Ki ~ 1 mM) and acetic acid (Ki ~ 30 mM), are also inhibitors of PRODH.10,11 Irreversible inactivators of PRODH, which covalently modify the N5 of the FAD of PRODH, have also been investigated. These include N-propargylglycine,9,12–14 thiazolidine-2-carboxylate,14,15 and 1,3-dithiolane-2-carboxylate.16 The latter compound is unique in that the inactivation mechanism is photoinduced by blue light. Inhibitors of PYCR1 have been less studied and include the proline analog N-formyl-L-proline17 and pargyline derivatives.18,19
Motivated by the observation that proline analogs inhibit PRODH, we screened 18 cycloalkyl carboxylic acids and related compounds as probes to explore the active site (Fig. 2). The inhibition constants for six of the compounds were determined and found to be in the range of 1 – 6 mM. The crystal structures of four of the compounds in complex with PRODH were determined to understand the basis for inhibition. Our results provide insight into structure-affinity relationships of proline analogs targeting PRODH, including the importance of the carboxylate group, the tolerance of the active site for inhibitor ring size, and synergism between inhibitor ring size and hydrogen bonding to a conserved active site water molecule.
Experimental
Materials
The following compounds were bought from Sigma: (S)-(−)-tetrahydro-2-furoic acid (1) product number 527890, cyclobutane-1,1-dicarboxylic acid (2) product number C95803, cyclobutanecarboxylic acid (3) product number C95609, cyclopropanecarboxylic acid (4) product number C116602, 2-methylcyclopropanecarboxylic acid (5) product number 209759, cyclopropane-1,1-dicarboxylic acid (6) product number 343412, transcyclobutane-1,2-dicarboxylic acid (7) product number 28684, 2,2,3,3-tetramethylcyclopropanecarboxylic acid (8) product number 301566, 1-(aminomethyl)cyclopropanecarboxylic acid (9) product number CDS015451, 3-oxocyclobutanecarboxylic acid (10) product number CDS004694, cyclopropylacetic acid (11) product number CDS000888, 2-thiophenecarboxylic acid (12) product number T32603, (S)-(−)-tetrahydrofuran-2-carboxamide (13) product number 573310, cis-cyclobutane-1,2-dicarboxylic acid (14) product number 28682, tetrahydropyran-2-carboxylic acid (15) product number CDS015473, cyclopentanecarboxylic acid (16) product number C112003, 2-oxobutyric acid (17) product number 68217, (2S)-oxetane-2-carboxylic acid (18) product number SY3H6E416CAC.
Protein Expression and Purification
The PRODH domain of the bifunctional PRODH-GSALDH enzyme proline utilization A (PutA) from Escherichia coli was used for crystallization and kinetic assays. A PRODH domain construct containing residues 86–630 of E. coli PutA (PutA86–630) with a C-terminal His tag was expressed and purified using a protocol developed from previous studies of E. coli PutA PRODH domain constructs.11,20–22 PutA86–630 was overexpressed in E. coli strain BL21(DE3) pLysS. Cells were grown in TB media for 3 hours at 37 °C and induced with 0.5 mM isopropyl β-D-1-thiogalactopyranoside for 3 hours at 37 °C. Harvested cells were resuspended in a buffer containing 20 mM Tris pH 7.9, 5 mM imidazole, 500 mM NaCl, and 10% v/v glycerol (buffer A) and then lysed via sonication in the presence of 1 mM FAD and EDTA-free protease inhibitor tablets (Thermo Fisher). Centrifugation at 16000 rpm for 1 hour at 4 °C was performed to remove insoluble material. The lysate was then purified by gravity-flow chromatography on a column containing Ni2+-NTA resin (Qiagen) pre-equilibrated with buffer A. The column was washed with buffer A and eluted with buffer A supplemented with 500 mM imidazole. Elution fractions containing PutA86–630 were identified on SDS-PAGE, pooled, spiked with 1 mM FAD, and dialyzed into 70 mM Tris pH 8.1, 2 mM EDTA, and 10% v/v glycerol. Anion exchange with a 5 mL HiTrap Q column was performed the next day using a gradient of KCl (0–0.5 M over 20 column volumes). PutA86–630 was collected in the flow-through, while contaminating proteins were retained by the resin. PutA86–630 was dialyzed into 70 mM Tris pH 7.5, 5 mM EDTA, and 10% v/v glycerol. The protein concentration was estimated with a Bradford assay and confirmed with absorbance at 280 nm. The purified enzyme was distributed in 50 μL aliquots in 1.5 mL microcentrifuge tubes and stored at −80 °C.
Enzyme Activity Assays
Kinetic measurements were performed in a 96-well plate in a BioTek Epoch 2 microplate spectrophotometer at room temperature in a buffer containing 20 mM MOPS pH 7.5 and 10 mM MgCl2. The assay monitors the production of P5C as an adduct formed with ortho-aminobenzaldehyde (o-AB), which is detected by absorbance at 443 nm (ε443 = 2.59 mM−1cm−1).23 Menadione was used as the electron acceptor to reoxidize the reduced FAD of PRODH, enabling catalytic cycling. An initial assessment of enzyme inhibition by a panel of compounds was performed at one substrate concentration using 200 mM L-proline, 4 mM o-AB, 0.15 mM menadione, 63 nM PutA86–630, and 5 mM of the potential inhibitor. For compounds 1 - 4 and 16-18, kinetic measurements were performed at varied L-proline (0–500 mM) and inhibitor (0–10 mM) concentrations. L-proline and inhibitors were spotted on the plate and a master mix including enzyme, menadione, o-AB, and buffer was added to the plate by multichannel pipette to initiate the reaction. The initial rate was determined from linear regression of the first 10 minutes of the progress curve using Origin v9.7.0.188 software. The initial rate data as functions of both substrate and inhibitor concentrations were fit globally to a competitive inhibition model using Origin. Kinetic constants from fitting are listed in Table 1.
Table 1.
Inhibition and kinetic constants
| Compound | Ki (mM) | Km (mM) | kcat (s−1) | kcat/Km (M−1s−1) |
|---|---|---|---|---|
| 1 | 0.28 ± 0.02 | 86.6 ± 7.84 | 1.16 ± 0.03 | 13.0 ± 1.3 |
| 2 | 1.5 ± 0.2 | 123.8 ± 20.3 | 0.90 ± 0.05 | 7.2 ± 1.6 |
| 3 | 1.9 ± 0.2 | 121.0 ± 11.6 | 1.47 ± 0.05 | 12.2 ± 1.6 |
| 4 | 3.1 ± 0.3 | 87.5 ± 8.3 | 1.20 ± 0.03 | 13.7 ± 1.7 |
| 16 | 6.3 ± 0.7 | 118.6 ± 10.4 | 1.35 ± 0.04 | 11.4 ± 1.3 |
| 17 | 1.8 ± 0.1 | 84.5 ± 6.1 | 1.14 ± 0.02 | 13.5 ± 0.1 |
| 18 | 1.4 ± 0.1 | 136.2 ± 10.7 | 1.42 ± 0.04 | 10.4 ± 0.9 |
Crystallization and Cryoprotection
Crystals were grown at 20 °C in a hanging drop setup with 20 mg/mL protein. A drop ratio of 2 μL:2 μL for protein: reservoir solution was used. The reservoir solution contained 50–175 mM sodium citrate pH 6.2 and 20–26% PEG 3350. Crystals were harvested, soaked in reservoir solution supplemented with 50 mM 2 for a few minutes, cryoprotected with reservoir solution supplemented with 20% PEG 200, and flash-cooled in liquid nitrogen. Crystals for the complexes with 3, 4, and 18 were grown using a reservoir solution containing 100 mM sodium citrate pH 5.6, 5.75, 5.9 or 6.0 and 18–28% PEG 3000. Crystals were harvested, soaked with reservoir solution supplemented with 100 mM 3, 50 mM 4, or 50 mM 18 for a few minutes, and then cryoprotected with reservoir solution supplemented with 20% PEG 200 and flash-cooled in liquid nitrogen.
X-ray Crystal Structure Determination
X-ray diffraction data were collected at Advanced Photon Source beamline 24-ID-E using an Eiger-16 M detector and 24-ID-C using an Eiger2 X 16 M detector. The data were processed with XDS24 and AIMLESS25. The space group is I222, and the asymmetric unit contains one PutA86–630 chain. We note this is the same crystal form used previously for structural studies of the E. coli PutA PRODH domain.13,20,26,27 Data processing statistics are summarized in Table 2.
Table 2.
X-ray diffraction data collection and refinement statistics
| Compound 2 | Compound 3 | Compound 4 | Compound 18 | |
|---|---|---|---|---|
|
| ||||
| Space group | I222 | I222 | I222 | I222 |
| Unit cell parameters (Å) | a = 72.80 | a = 73.08 | a = 72.99 | a = 72.85 |
| b = 141.82 | b = 141.74 | b = 141.57 | b = 140.98 | |
| c = 146.64 | c = 146.30 | c = 144.97 | c = 145.96 | |
| Wavelength (Å) | 0.97918 | 0.97918 | 0.97918 | 0.97918 |
| Resolution (Å) | 101.26 – 2.31 | 101.94 – 1.71 | 73.15 – 1.72 | 64.72 – 2.25 |
| (2.31 – 2.19) | (1.71 – 1.68) | (1.72 – 1.69) | (2.32 – 2.25) | |
| Observationsa | 183109 (26444) | 631755 (23468) | 639699 (29360) | 191098 (18009) |
| Unique reflectionsa | 38519 (5542) | 85566 (3890) | 85319 (4112) | 35985 (3268) |
| Rmerge(I)a | 0.110 (2.344) | 0.076 (2.843) | 0.119 (2.989) | 0.114 (1.315) |
| Rmeas(I)a | 0.139 (2.764) | 0.082 (3.105) | 0.127 (3.218) | 0.126 (1.451) |
| Rpim(I)a | 0.063 (1.247) | 0.030 (1.224) | 0.045 (1.170) | 0.053 (0.600) |
| Mean I/σa | 10.3 (1.0) | 15.0 (0.6) | 15.2 (0.6) | 6.3 (1.0) |
| CC1/2a | 0.997 (0.234) | 0.998 (0.333) | 0.999 (0.200) | 0.993 (0.595) |
| Completeness (%)a | 99.4 (99.7) | 99.4 (88.8) | 99.7 (94.6) | 99.8 (99.9) |
| Multiplicitya | 4.8 (4.8) | 7.4 (6.0) | 7.5 (7.1) | 5.3 (5.5) |
| No. of protein residues | 499 | 499 | 499 | 503 |
| No. of atoms | ||||
| Protein | 3708 | 3740 | 3732 | 3760 |
| FAD | 53 | 53 | 53 | 53 |
| Inhibitor | 10 | 7 | 6 | 7 |
| Water | 57 | 219 | 247 | 58 |
| R cryst a | 0.223 (0.386) | 0.207 (0.645) | 0.213 (0.658) | 0.208 (0.339) |
| R free a,b | 0.257 (0.404) | 0.234 (0.574) | 0.240 (0.653) | 0.243 (0.351) |
| rmsd bonds (Å) | 0.007 | 0.007 | 0.006 | 0.007 |
| rmsd angles (°) | 0.874 | 0.869 | 0.859 | 0.858 |
| Ramachandran plotc | ||||
| Favored (%) | 96.75 | 98.78 | 97.57 | 97.59 |
| Outliers (%) | 0.00 | 0.00 | 0.00 | 0.00 |
| Clashscore (PR)c | 3.92 (99) | 1.86 (99) | 1.60 (99) | 3.31 (99) |
| Molprobity score (PR)c | 1.76 (94) | 1.22 (98) | 1.11 (99) | 1.63 (97) |
| Average B (Å2) | ||||
| Protein | 69.1 | 43.7 | 40.3 | 70.2 |
| FAD | 48.2 | 26.8 | 23.5 | 48.1 |
| Inhibitor | 53.3 | 30.1 | 31.4 | 51.0 |
| Conserved water | N/A | 33.9 | 30.7 | N/A |
| All water | 50.7 | 40.3 | 37.9 | 52.1 |
| Coord. error (Å)d | 0.38 | 0.32 | 0.34 | 0.36 |
| PDB code | 7MWT | 7MWU | 7MWV | 7SQN |
Values for the outer resolution shell of data are given in parenthesis.
5% test set.
From MolProbity. The percentile ranks (PR) for Clashscore and MolProbity score are given in parentheses.
Maximum likelihood-based coordinate error estimate from PHENIX.
A 1.85 Å resolution structure of a PutA86–630 variant complexed with 1 (PDB ID 3E2R)26 was used as a starting model for crystallographic refinement of the enzyme complexed with 2 in PHENIX.28,29 Refinements of the complexes with 3 and 4 were started from the final model of the complex with 2. Refinement of the complex with 18 was started from the final model of the complex with 3. The B-factor model consisted of one TLS group per protein chain and isotropic B-factors for all non-hydrogen atoms. Interactive model building was performed with COOT.30 SMILES strings for inhibitors were used as the input to ELBOW31 to generate the coordinates and restraint files used during refinement. The structures were validated using MolProbity and the wwPDB validation service.32,33 Modeling of ligands was validated with polder omit maps.34 Refinement statistics are summarized in Table 2.
Results
Compound Screening
For this study, we used the PRODH domain of the bifunctional PRODH-GSALDH enzyme proline utilization A (PutA) from E. coli, which we used previously to investigate the reversible inhibition of PRODH by 1, L-lactic acid, and acetic acid.11 The PutA PRODH domain construct used here (residues 86–630, PutA86–630) is a good model system for inhibitor testing because of its crystallizability (routinely better than 2.0 Å resolution)26,27 and the high sequence conservation of PRODH active sites across bacteria and eukaryotes.2 We note that human PRODH is an inner mitochondrial membrane protein and is challenging to isolate for structural and kinetic study.
The inhibition of PutA86–630 by several cycloalkyl carboxylic acids and related compounds was investigated to provide insight into the nature of chemical structures that bind the PRODH active site (Fig. 2). The goal of obtaining a deeper understanding of inhibitory effects of 1 drove the design of the panel of compounds. Starting with 1 and making iterative alterations to the chemical structures, we formulated a list of low molecular weight compounds to explore the active site of PRODH (Fig. 2).
An initial assessment was performed with the inhibitor at a concentration of 5 mM and the substrate L-proline at 200 mM (~2Km). Under these conditions, the enzyme activity in the presence of 1 is 12% of the no-inhibitor control (Fig. 3). Although none of the compounds achieved a level of inhibition similar to 1, the results provide structure-activity relationships that help explain the activity of 1. For example, removal of the O atom in the ring of 1, as in 16, or increasing the ring size to six, as in 15, severely decreases inhibition. Substituting an amide for the carboxylate (13) also decreases inhibition. These results show that interactions with both the tetrahydrofuran and carboxylate group are essential to the activity of 1.
Fig. 3.
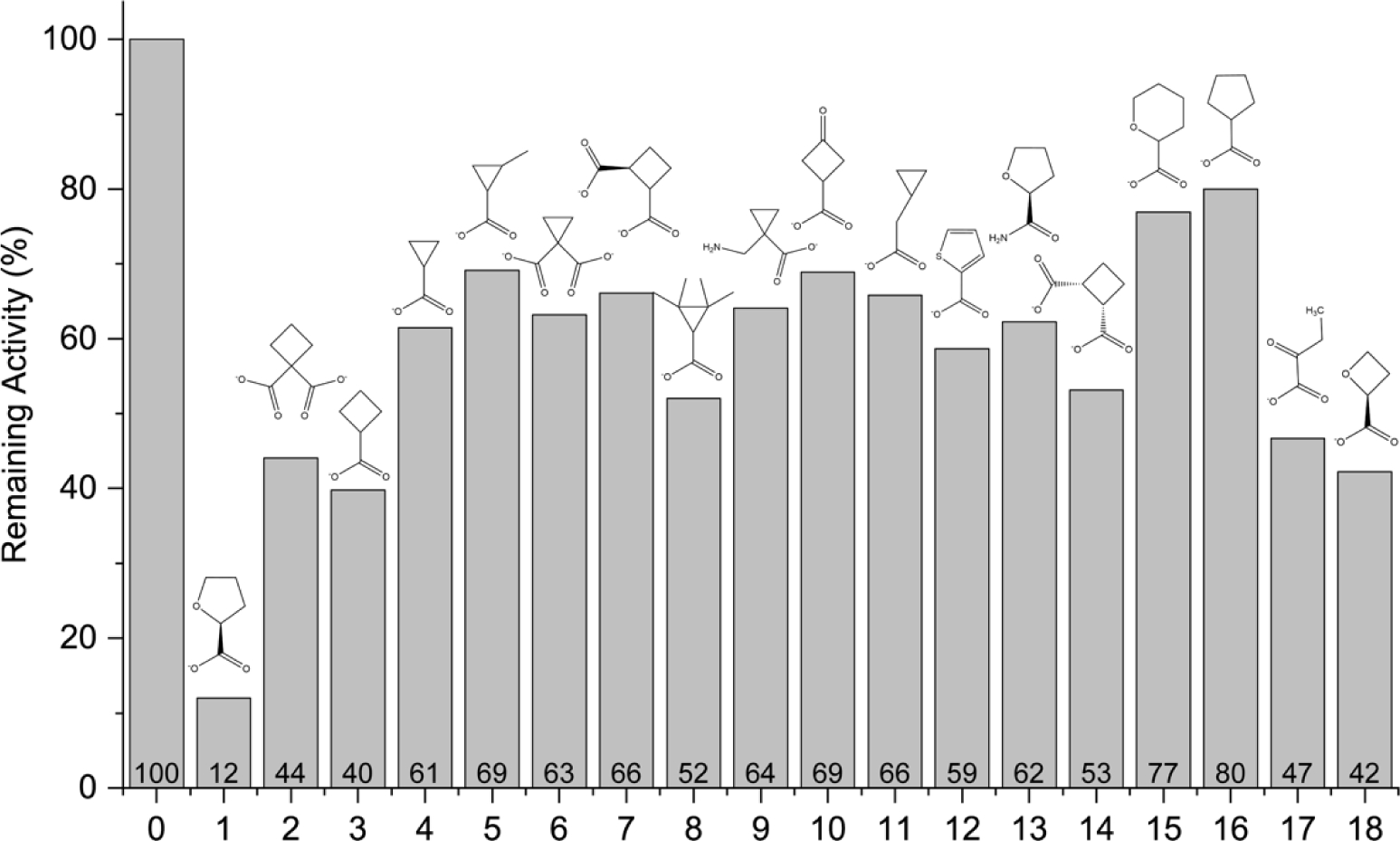
Initial screening of compounds for inhibition of PRODH activity. The bars represent the enzyme activity of PutA86–630 measured at a single L-proline substrate concentration (200 mM) and the compound present at 5 mM. The data are normalized to the activity in the absence of any inhibitor (compound 0). Compound 1 was included as a positive control. The percent activity relative to the no-inhibitor control is labeled above the x-axis.
The compounds tested included carboxylates of cyclopropane and cyclobutane to explore the potential of inhibitors with rings smaller than the substrate L-proline. Generally, the cyclobutyl compounds exhibited better inhibition than the cyclopropyl derivatives. In particular, 2 as well as 3 and 18, reduced enzyme activity by over 2-fold relative to the no-inhibitor control (Fig. 3). In contrast, none of the cyclopropyl compounds achieved better than 50% inhibition. Including a second carboxylate group (2, 6, 7, 14) did not improve inhibition compared to the mono-carboxylate parents (3, 4).
Cyclobutane carboxylate (3) inhibited PRODH almost twice as much as cyclopentane carboxylate (16), which led us to hypothesize that 18 had potential to be a better inhibitor than 1. Surprisingly, 18 was no more effective than 3 in the single-point assay (Fig. 3). Thus, tetrahydrofuran is preferred over oxetane.
Compound 17 was included to test an acyclic analog of 18 and for its structural similarity to L-lactate, a known inhibitor of PRODH. 17 was less effective than 18, which perhaps reflects a larger conformational entropy penalty for binding 17.
We tested 10 as an example of a compound with the same atom inventory as 1 but with a different three-dimensional structure (i.e., functional group isomer). 10 showed very little inhibition, suggesting the oxo group does not engage hydrogen bonding partners as effectively as the heteroatom O of 1.
Estimation of Inhibition Constants
Six compounds were selected for additional kinetic analysis to estimate the inhibition constants for comparison to our reference compound 1. Compounds 2, 3, 17, and 18 were chosen because they caused significant inhibition in the single-point assay (>50% inhibition, Fig. 3). 4 and 16 were included for comparison to 3 to investigate the influence of cycloalkyl ring size on ligand recognition. For each compound, the initial rate as functions of L-proline and inhibitor concentration were fit globally to the competitive model (Fig. 4). Table 1 lists the Ki values from global fitting.
Fig. 4.
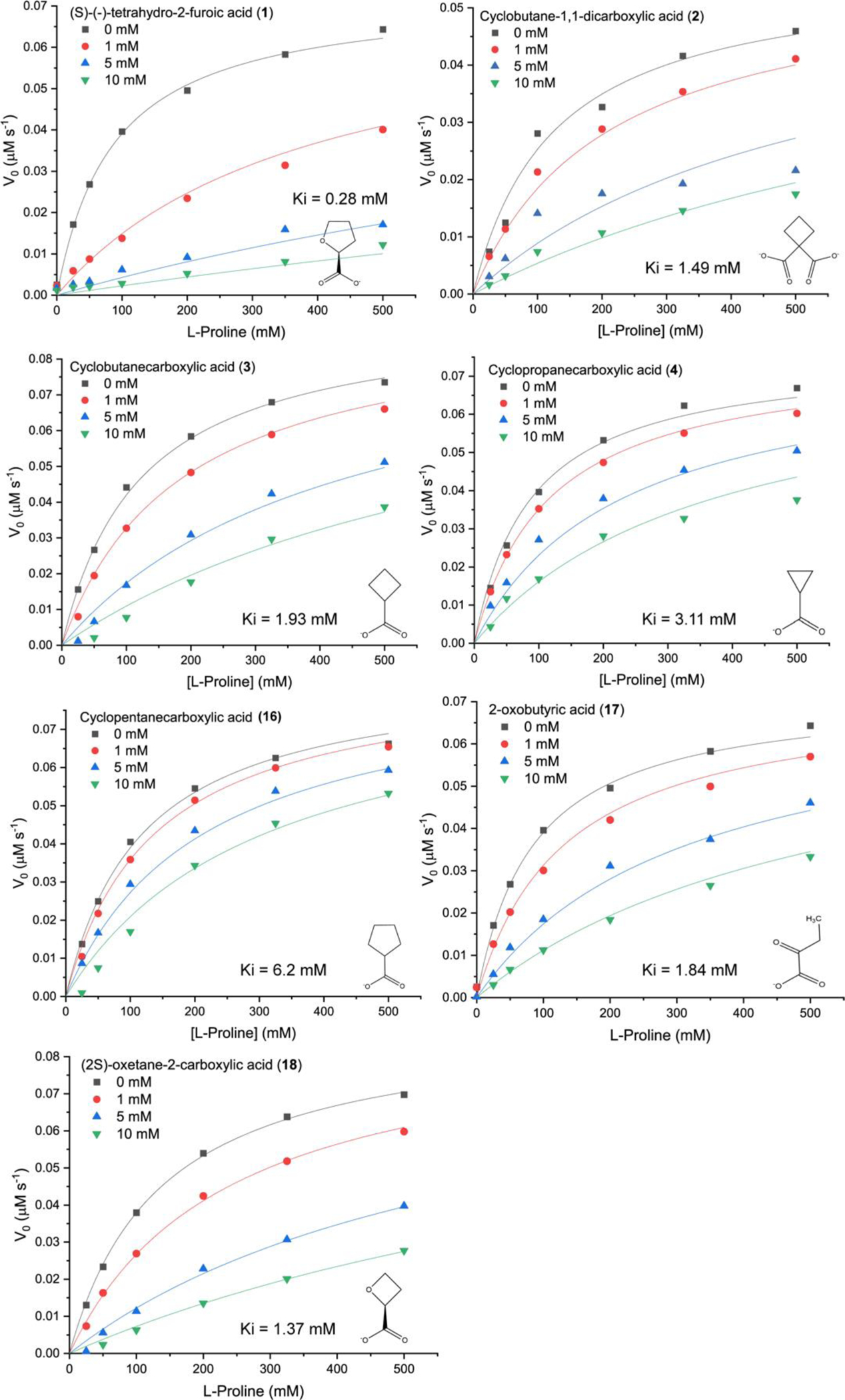
Inhibition of the PRODH activity of PutA86–630 by 1, 2, 3, 4, 16, 17, and 18. The assays were performed at room temperature with 0–500 mM L-proline, 4 mM o-AB, 0.15 mM menadione, and 63 nM PutA86–630 in a buffer containing 20 mM MOPS pH 7.5 and 10 mM MgCl2. The data for each inhibitor were analyzed by global fitting to the competitive inhibition model using Origin software. Kinetic constants are listed in Table 1.
Compound 1 has a Ki of 0.3 mM, consistent with previous studies.7,8 In the comparison of cycloalkyl ring size, cyclobutane carboxylate (3) is preferred with a Ki of 1.9 mM, followed by cyclopropane carboxylate (4, Ki = 3.1 mM), and cyclopentane carboxylate (16, Ki of 6.2 mM). Incorporation of an O atom into the ring of the preferred cycloalkane carboxylate (i.e., 18) or the addition of a second carboxylate (2) lowered Ki by about 20% from 1.9 mM to ~1.5 mM. The acyclic compound 17 inhibits comparably to 3. These results are generally consistent with the single-point survey data (Fig. 3).
Common Features of the Poses of Inhibitors
Crystal structures of PutA86–630 inhibited by 2, 3, 4, and 18 were determined (Table 2). All the inhibitors occupy the known proline substrate site, between the FAD isoalloxazine and the α8 helix, as shown for 2 (Fig. 5). Helix α8 is notable for containing the conserved sequence motif YXXRRXXET/N, which provides residues that interact with L-proline (Fig. 5B). Electron density was strong for all the inhibitors (Fig. 6), and all were modelled at an occupancy of 1.0 with reasonable B-factors, i.e., similar to that of the FAD (Table 2).
Fig. 5.
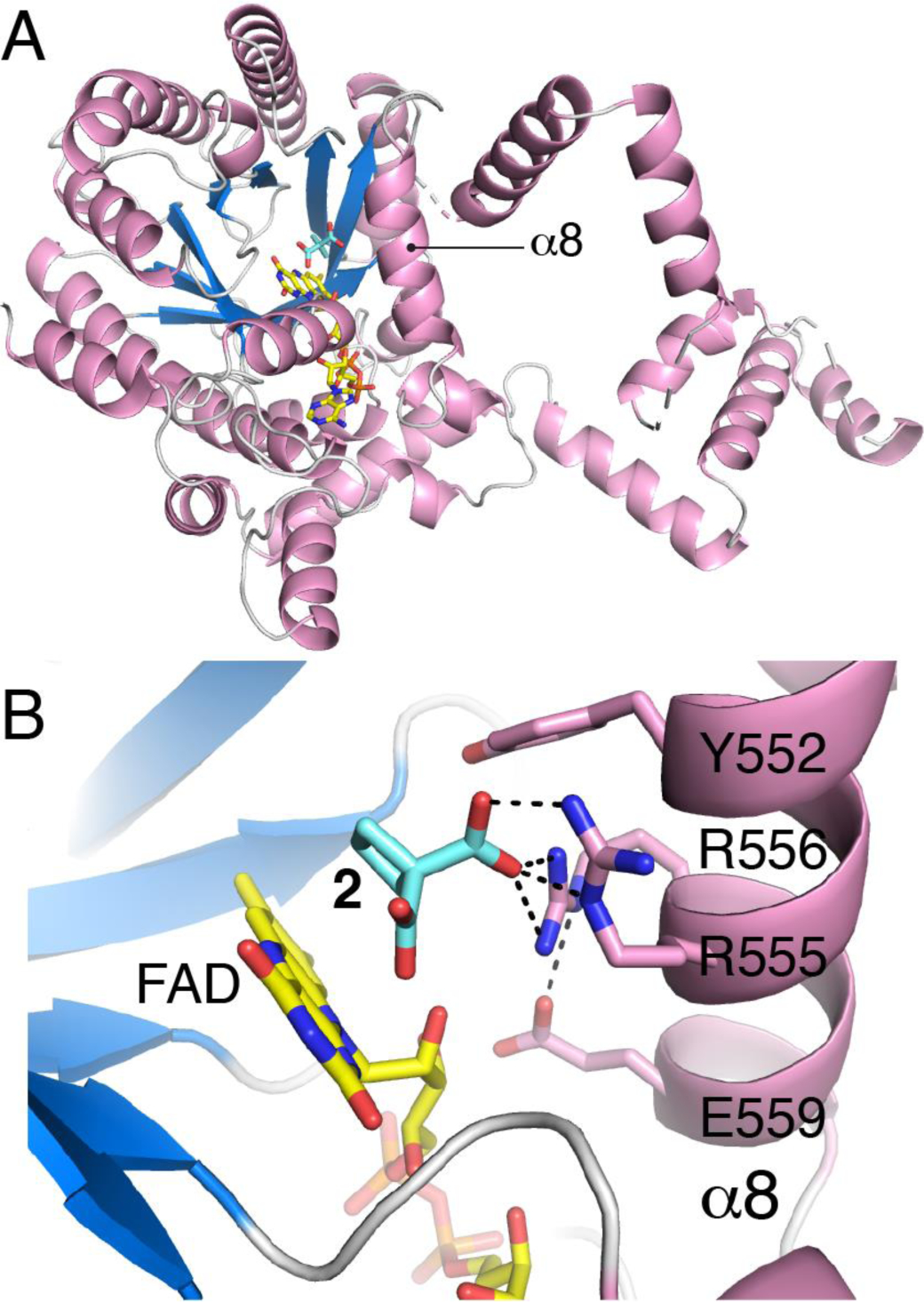
Structure of PutA86–630 complexed with 2. (A) Cartoon drawing showing the location of the active site within the protein fold. (B) Close-up view of the active site showing 2 and side chains of the YXXRRXXET/N motif of α8.
Fig. 6.
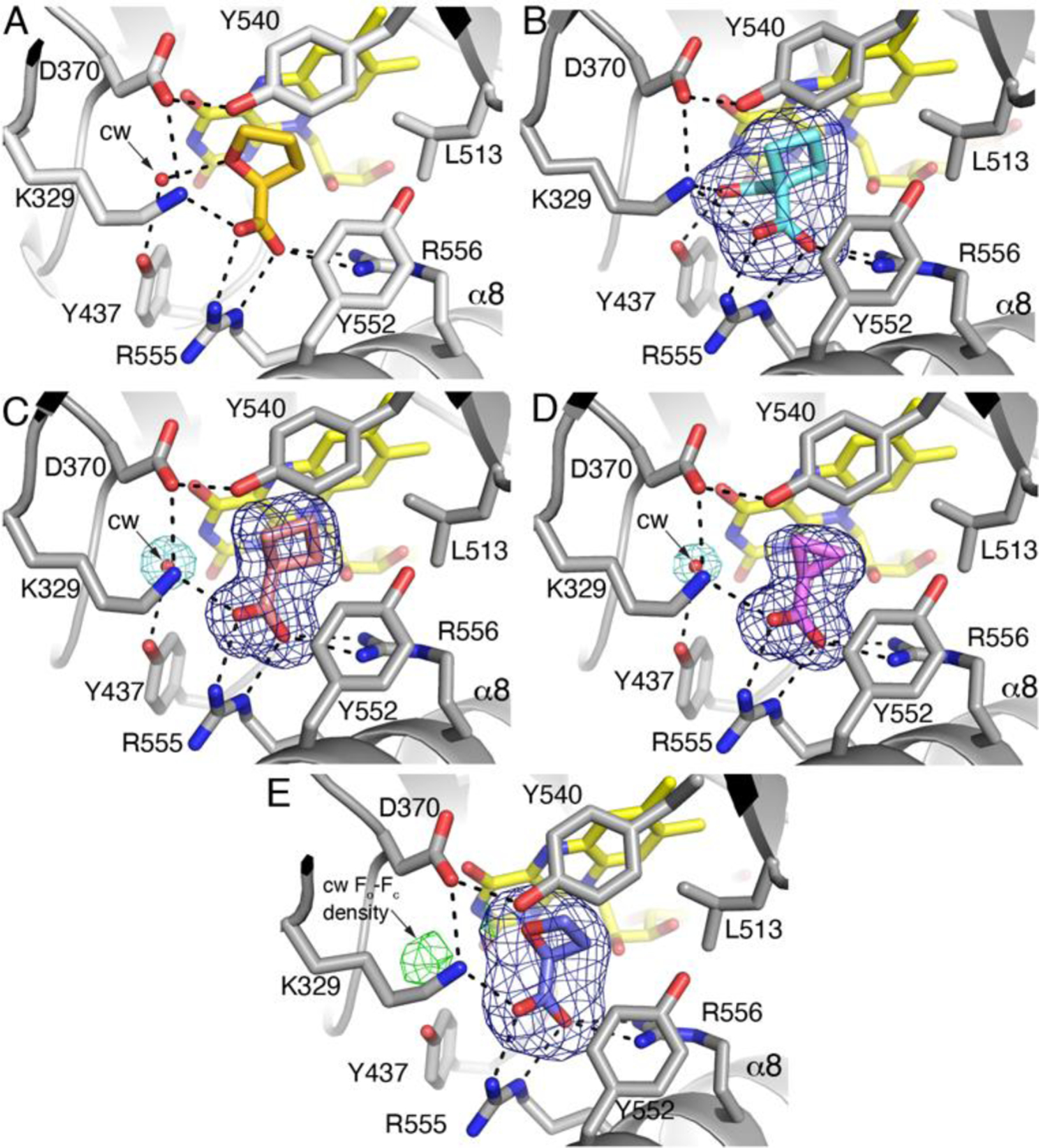
Interactions and electron density for inhibitors bound to PutA86–630. (A) 1, from PDB ID 1TIW11. (B) Compound 2. The blue mesh represents a polder omit map (4σ). (C) Compound 3. The blue mesh represents a polder omit map (4σ). The aquamarine mesh represents the final refined 2Fo-Fc map (1σ) and indicates the conserved water molecule (cw). (D) Compound 4. The blue mesh represents a polder omit map (4σ). The aquamarine mesh represents the final refined 2Fo-Fc map (1σ) and indicates the conserved water molecule (cw). (E) Compound 18. The blue mesh represents a polder omit map (5σ). The green mesh represents Fo-Fc density (2.5σ) at the location expected for the conserved water molecule (cw).
The binding poses of the inhibitors are very similar (Fig. 6). In each structure, the carboxylate group of the inhibitor ions pairs with Arg555 and Arg556 of α8, as well as Lys329. Additional residues surround the aliphatic part of the inhibitor, including Leu513, Tyr552, and Tyr540. This mode of binding is very similar to that of 1 (Fig. 6A, PDB ID 1TIW11).
The Conserved Active Site Water Molecule
A conserved active site water molecule is present in the complexes of 3 and 4 (Fig. 6C, 6D). This water molecule is present in ligand-free PRODH structures, implying it is a conserved structural feature of the active site (e.g., PDB IDs 6X9A35 and 4NM936). The conserved water molecule plays a key role in the recognition of 1 by bridging the O heteroatom of the inhibitor and Tyr437 via a hydrogen bond (Fig. 6A). Obviously, 3 and 4 do not have a hydrogen bonding group to engage the conserved water molecule. Nevertheless, the electron density for the conserved water is strong in these structures (Fig. 6C, 6D) and the refined B-factor is below the average for water molecules in the respective structure (Table 2).
The conserved water molecule is absent in the complex with 2. The extra carboxylate of 2 occupies the conserved water site and forms a hydrogen bond with Tyr437 (Fig. 7A). Apparently, the binding of 2 is accompanied by the dissociation of the conserved water molecule. The increase in entropy associated with release of the water molecule into the bulk solvent presumably contributes to the affinity of 2.
Fig. 7.
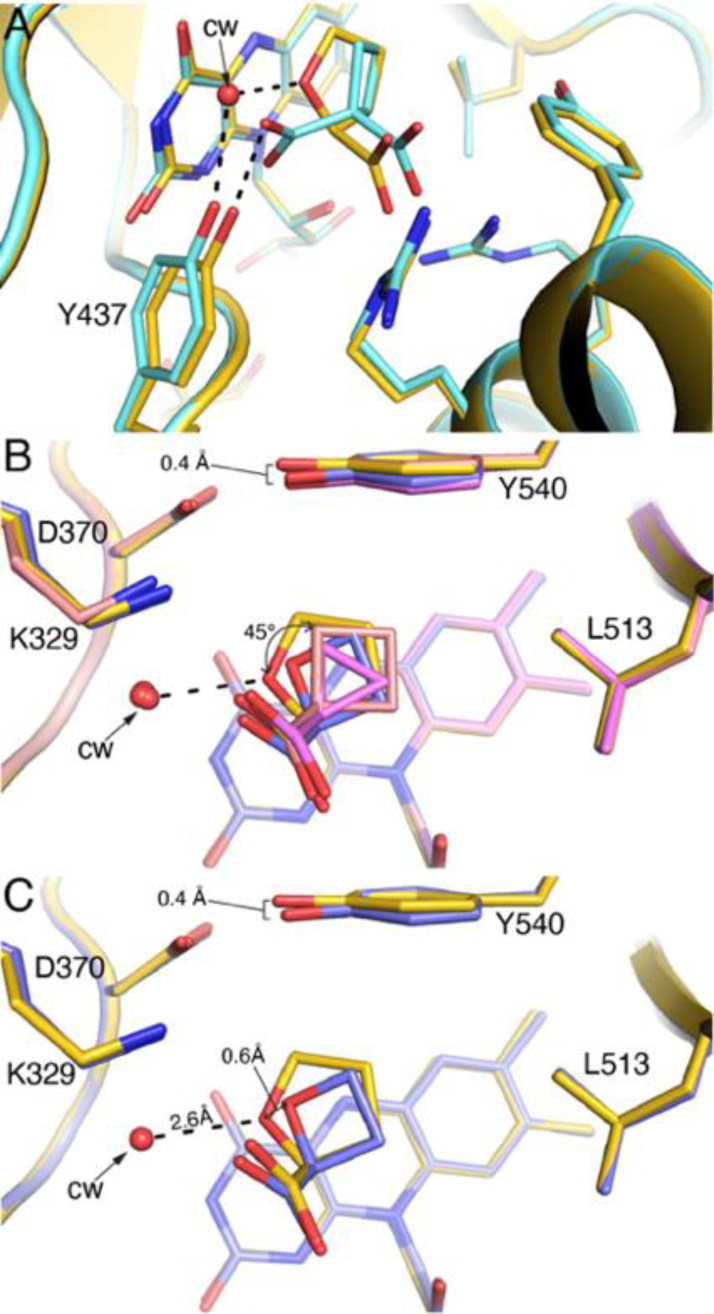
Superposition of the PutA86–630-inhibitor complexes. (A) Comparison of the active site complexed with 1 (gold, PDB ID 1TIW11) and 2 (light blue). (B) Comparison of the active site complexed with 1 (gold, PDB ID 1TIW11), 3 (salmon), and 4 (pink), and 18 (slate). The curved arrow denotes the difference in orientation of the rings of 3/4 relative to compound 1. (C) Comparison of the active site complexed with 1 (gold, PDB ID 1TIW11) and 18 (slate).
Paradoxically, the conserved water molecule is also absent in the complex with 18, despite this inhibitor containing a hydrogen bonding heteroatom like 1. The electron density at the location of the conserved water was very weak. Fo-Fc density was observed at 2.5σ (Fig. 6E), but corresponding 2Fo-Fc electron density was lacking at any contour level. Refinement of a water molecule at this location resulted in a B-factor of 56 Å2, which is higher than the mean water B-factor of 52 Å2. More significantly, the refined 2Fo-Fc map showed no feature for the water molecule when contoured at 0.5σ, and the Fo-Fc map had a negative feature at −2.5σ encasing the water molecule. Based on these observations and calculations, we did not include the conserved water molecule in the structure and concluded that either it is absent or very weakly bound.
Influence of Ring Size on Inhibitor Pose
The active site accommodates inhibitors with ring sizes of 3 – 5 with minimal change in the protein structure. One perceptible difference is that Tyr540 tilts 0.4 Å toward the ligand binding site in the complexes with 4 and with 18 (Fig. 7B). This inward deflection allows Tyr540 to pack against the rings of these compounds, diminishing open space around the inhibitor.
The poses of compounds 3 and 4 differ somewhat from the reference compound, 1. The rings of 3 and 4 rotate by ~45° away from the conserved water molecule, toward Leu513 (Fig. 7B). The apparent rotation enables the nonpolar rings of these compounds to form favorable interactions with Leu513 while avoiding an unfavorable interaction with the conserved water molecule.
The pose of compound 18 is somewhat surprising in that its O heteroatom does not align with that of 1. Instead, the C3 atoms of the two inhibitors are superimposed and the O heteroatom of 18 is shifted by 0.6 Å from that of 1 (Fig. 7C). Like compounds 3 and 4, the pose of 18 appears to favor nonpolar interactions with Leu513 while avoiding the conserved water site. As mentioned in the previous section, the conserved water molecule is either absent or very weakly bound in the complex with 18. If the conserved water molecule is refined at the expected location, the predicted hydrogen bonding distance to 18 is 3.5 Å, which is not optimal for a hydrogen bond and much longer than the hydrogen bond distance of 2.6 Å for compound 1 (Fig. 7C). It appears that the binding of 18 destabilizes the conserved water molecule.
Discussion
The growing evidence for the proline cycle enzymes as cancer therapy targets motivates the discovery of chemical probes of PRODH and PYCR1.2,3,37 The exploration of how their active sites recognize chemical matter may inform inhibitor design. Here, we performed a focused screen of compounds related to 1, the best characterized reversible inhibitor of PRODH. The panel of compounds enabled us to dissect the contributions of the structural and chemical features of 1 to binding affinity, including the carboxylate group, the heteroatom O, and ring size.
The carboxylate of 1 is clearly important for affinity. Substituting the carboxylate of 1 with an amide (13) markedly reduced affinity and confirms the importance of the ionic interactions with the conserved arginine residues of helix α8. Indeed, all known reversible inhibitors and covalent inactivators of PRODH contain a carboxylate group. This result suggests that the investigation of compounds with carboxylate mimics, such as phosphonates and sulfonates, may be productive.
Our data provide insight into the contributions of the O heteroatom and ring size of 1 for affinity, which is represented by a chemical double mutant thermodynamic cycle (Fig. 8). This type of cycle is analogous to the double mutant thermodynamic cycles used to analyze site-directed mutagenesis of proteins.38,39 In this case, the two “mutagenesis” steps are removing the O heteroatom from 1 to generate 16, and reducing the ring size of 1 by one carbon to compound 18. The double mutation thus converts 1 to 3. The ΔΔG values for each leg of the cycle are obtained from the inhibition Ki values.
Fig. 8.
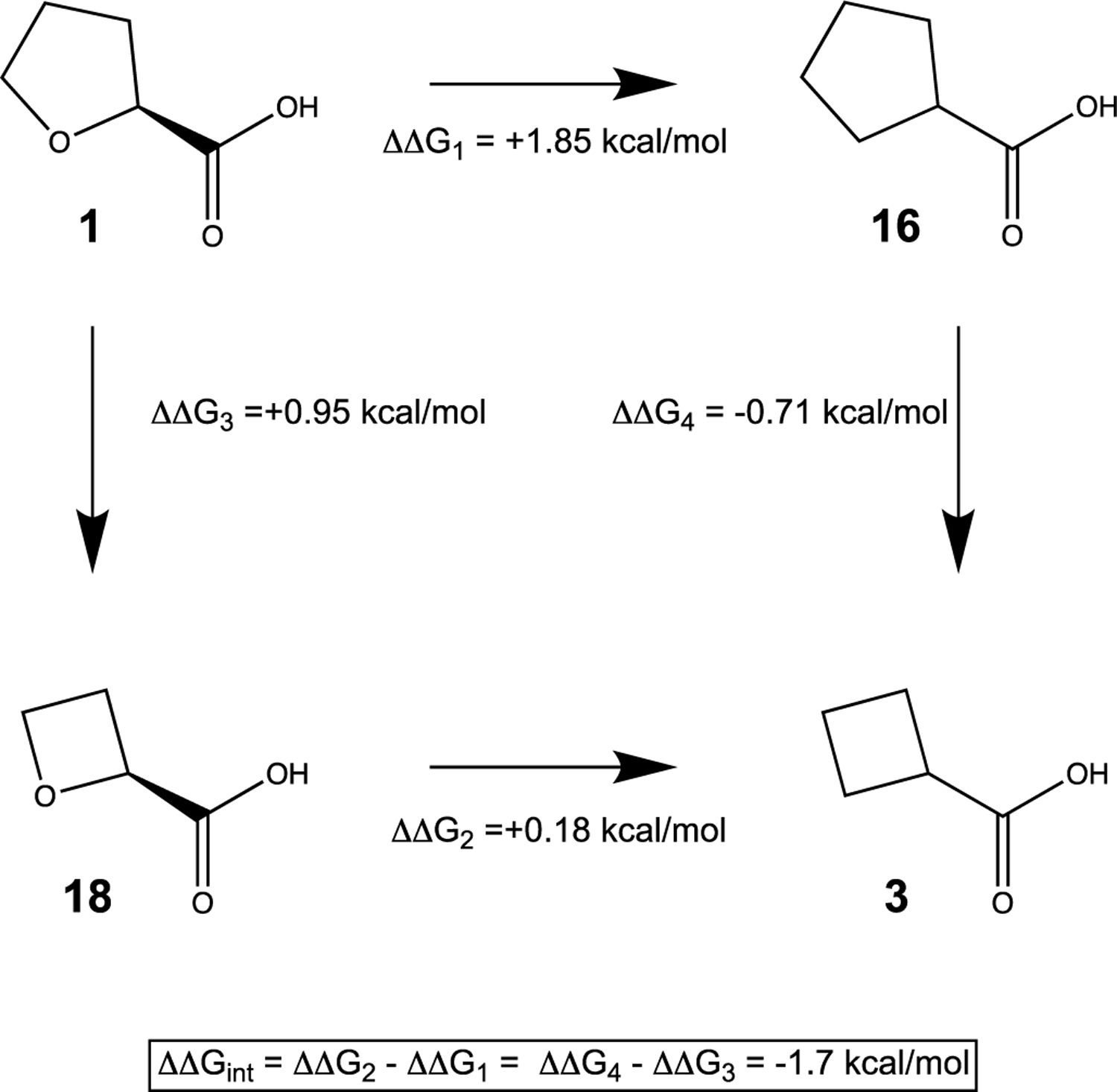
Chemical double mutant cycle expressing the relationship between hydrogen bonding capacity and ring size of 1. The ΔΔG values for the chemical mutations were calculated from the Ki values of the inhibitors.
Starting from 1, removing the O heteroatom to generate 16 resulted in a 20-fold decrease in Ki, implying that the hydrogen bond with the conserved water molecule contributes almost 2 kcal/mol to the binding energy (Fig. 8). This result provides another example of the importance of water molecules at the protein-inhibitor interface.40 In contrast, removing the O heteroatom from the 4-membered ring of 18 to generate 3 resulted in only a modest loss of binding energy of about 0.2 kcal/mol. Decreasing the ring size of 1 by one C atom to 18 increased Ki 5-fold, which amounts to a loss of 0.95 kcal/mol of binding energy. In contrast, decreasing the cyclopentane of 16 to cyclobutane (3) improved affinity by 0.71 kcal/mol. These results suggests that the strength of the hydrogen bond between the O heteroatom and the conserved water molecule depends on ring size, i.e., that these two elements of molecular recognition interact thermodynamically. This interaction may be obtained from the thermodynamic cycle as −1.7 kcal/mol (Fig. 8), which indicates that the hydrogen bond is stronger in the context of the 5-membered ring than in the 4-membered ring. This is consistent with the structure showing that 18 is not positioned optimally to hydrogen bond to a water molecule in the conserved site (Fig. 7C), and as a result, the conserved water molecule is either absent or very weakly bound in the complex with 18.
Five appears to be the optimal ring size for the O-heterocyclic inhibitors. Both expanding the ring size to six (15) or decreasing it to four (18) decreased affinity. In contrast, we found that four is the optimal ring size for the cycloalkane carboxylate inhibitors. For example, decreasing the ring size from five in cyclopentane carboxylate (16) to four in cyclobutane carboxylate (3) improved affinity by 0.7 kcal/mol; however, decreasing the ring size further to cyclopropane carboxylate (4) decreased affinity by 0.3 kcal/mol. These results are consistent with ring size and hydrogen bonding to the conserved water contributing synergistically to binding affinity. These structure-affinity relationships may be useful for future inhibitor design targeting PRODH.
Acknowledgements
This work was supported by NIGMS, National Institutes of Health, Grant R01GM132640. A.N.B. is the recipient of a Wayne L. Ryan Fellowship through The Ryan Foundation. We thank Jonathan Schuermann for help with X-ray diffraction data collection. This work is based upon research conducted at the Northeastern Collaborative Access Team beamlines, which are funded by the National Institute of General Medical Sciences from the National Institutes of Health (P30 GM124165). The Eiger 16M detector on 24-ID-E is funded by a NIH-ORIP HEI grant (S10OD021527). This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02-06CH11357.
Abbreviations
- GSAL
L-glutamate-γ-semialdehyde
- GSALDH
L-glutamate-γ-semialdehyde dehydrogenase
- o-AB
ortho-aminobenzaldehyde
- P5C
Δ1-pyrroline-5-carboxylate
- PRODH
proline dehydrogenase
- PutA
proline utilization A
- PutA86–630
protein containing residues 86–630 of Escherichia coli proline utilization A
- PYCR
Δ1-pyrroline-5-carboxylate reductase
Footnotes
Values for the outer resolution shell of data are given in parenthesis.
5% test set.
From MolProbity. The percentile ranks (PR) for Clashscore and MolProbity score are given in parentheses.
Maximum likelihood-based coordinate error estimate from PHENIX.
Conflicts of interest
There are no conflicts to declare.
Accession Codes
The coordinates and structure factor amplitudes of all the structures have been deposited to the PDB with the accession codes 7MWT, 7MWU, 7MWV and 7SQN.
References
- 1.Phang JM, Proline Metabolism in Cell Regulation and Cancer Biology: Recent Advances and Hypotheses, Antioxid Redox Signal, 2019, 30, 635–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Tanner JJ, Fendt SM and Becker DF, The Proline Cycle As a Potential Cancer Therapy Target, Biochemistry, 2018, 57, 3433–3444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bogner AN, Stiers KM and Tanner JJ, Structure, biochemistry, and gene expression patterns of the proline biosynthetic enzyme pyrroline-5-carboxylate reductase (PYCR), an emerging cancer therapy target, Amino Acids, 2021, 53, 1817–1834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Burke L, Guterman I, Palacios Gallego R, Britton RG, Burschowsky D, Tufarelli C and Rufini A, The Janus-like role of proline metabolism in cancer, Cell Death Discov, 2020, 6, 104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Liu Y, Mao C, Wang M, Liu N, Ouyang L, Liu S, Tang H, Cao Y, Liu S, Wang X, Xiao D, Chen C, Shi Y, Yan Q and Tao Y, Cancer progression is mediated by proline catabolism in non-small cell lung cancer, Oncogene, 2020, 39, 2358–2376. [DOI] [PubMed] [Google Scholar]
- 6.Elia I, Broekaert D, Christen S, Boon R, Radaelli E, Orth MF, Verfaillie C, Grünewald TGP and Fendt S-M, Proline metabolism supports metastasis formation and could be inhibited to selectively target metastasizing cancer cells, Nature Communications, 2017, 8, 15267. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Zhu W, Gincherman Y, Docherty P, Spilling CD and Becker DF, Effects of proline analog binding on the spectroscopic and redox properties of PutA, Arch. Biochem. Biophys, 2002, 408, 131–136. [DOI] [PubMed] [Google Scholar]
- 8.White TA, Krishnan N, Becker DF and Tanner JJ, Structure and kinetics of monofunctional proline dehydrogenase from Thermus thermophilus, J. Biol. Chem, 2007, 282, 14316–14327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Scott GK, Yau C, Becker BC, Khateeb S, Mahoney S, Jensen MB, Hann B, Cowen BJ, Pegan SD and Benz CC, Targeting Mitochondrial Proline Dehydrogenase with a Suicide Inhibitor to Exploit Synthetic Lethal Interactions with p53 Upregulation and Glutaminase Inhibition, Mol Cancer Ther, 2019, 18, 1374–1385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Kowaloff EM, Phang JM, Granger AS and Downing SJ, Regulation of proline oxidase activity by lactate, Proc. Natl. Acad. Sci. U S A, 1977, 74, 5368–5371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Zhang M, White TA, Schuermann JP, Baban BA, Becker DF and Tanner JJ, Structures of the Escherichia coli PutA proline dehydrogenase domain in complex with competitive inhibitors, Biochemistry, 2004, 43, 12539–12548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.White TA, Johnson WH Jr., Whitman CP and Tanner JJ, Structural basis for the inactivation of Thermus thermophilus proline dehydrogenase by N-propargylglycine, Biochemistry, 2008, 47, 5573–5580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Srivastava D, Zhu W, Johnson WH Jr., Whitman CP, Becker DF and Tanner JJ, The structure of the proline utilization a proline dehydrogenase domain inactivated by N-propargylglycine provides insight into conformational changes induced by substrate binding and flavin reduction, Biochemistry, 2010, 49, 560–569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Scott GK, Mahoney S, Scott M, Loureiro A, Lopez-Ramirez A, Tanner JJ, Ellerby LM and Benz CC, N-Propargylglycine: a unique suicide inhibitor of proline dehydrogenase with anticancer activity and brain-enhancing mitohormesis properties, Amino Acids, 2021, 53, 1927–1939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Campbell AC, Becker DF, Gates KS and Tanner JJ, Covalent Modification of the Flavin in Proline Dehydrogenase by Thiazolidine-2-Carboxylate, ACS Chem Biol, 2020, 15, 936–944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Campbell AC, Prater AR, Bogner AN, Quinn TP, Gates KS, Becker DF and Tanner JJ, Photoinduced Covalent Irreversible Inactivation of Proline Dehydrogenase by S-Heterocycles, ACS Chemical Biology, 2021, 16, 2268–2279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Christensen EM, Bogner AN, Vandekeere A, Tam GS, Patel SM, Becker DF, Fendt SM and Tanner JJ, In crystallo screening for proline analog inhibitors of the proline cycle enzyme PYCR1, J Biol Chem, 2020, 295, 18316–18327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Milne K, Sun J, Zaal EA, Mowat J, Celie PHN, Fish A, Berkers CR, Forlani G, Loayza-Puch F, Jamieson C and Agami R, A fragment-like approach to PYCR1 inhibition, Bioorg Med Chem Lett, 2019, 29, 2626–2631. [DOI] [PubMed] [Google Scholar]
- 19.Forlani G, Sabbioni G, Ragno D, Petrollino D and Borgatti M, Phenyl-substituted aminomethylene-bisphosphonates inhibit human P5C reductase and show antiproliferative activity against proline-hyperproducing tumour cells, J Enzyme Inhib Med Chem, 2021, 36, 1248–1257. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Nadaraia S, Lee YH, Becker DF and Tanner JJ, Crystallization and preliminary crystallographic analysis of the proline dehydrogenase domain of the multifunctional PutA flavoprotein from Escherichia coli, Acta Crystallogr, 2001, D57, 1925–1927. [DOI] [PubMed] [Google Scholar]
- 21.Zhang W, Zhang M, Zhu W, Zhou Y, Wanduragala S, Rewinkel D, Tanner JJ and Becker DF, Redox-induced changes in flavin structure and roles of flavin N(5) and the ribityl 2’-OH group in regulating PutA-membrane binding, Biochemistry, 2007, 46, 483–491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Moxley MA, Tanner JJ and Becker DF, Steady-state kinetic mechanism of the proline:ubiquinone oxidoreductase activity of proline utilization A (PutA) from Escherichia coli, Arch. Biochem. Biophys, 2011, 516, 113–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Tanner JJ, Antioxidants and Redox Signaling, 2019, 30, 650–673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kabsch W, XDS, Acta Crystallogr. D Biol. Crystallogr, 2010, 66, 125–132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Evans PR and Murshudov GN, How good are my data and what is the resolution?, Acta Crystallogr. D Biol. Crystallogr, 2013, 69, 1204–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Ostrander EL, Larson JD, Schuermann JP and Tanner JJ, A conserved active site tyrosine residue of proline dehydrogenase helps enforce the preference for proline over hydroxyproline as the substrate, Biochemistry, 2009, 48, 951–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhu W, Haile AM, Singh RK, Larson JD, Smithen D, Chan JY, Tanner JJ and Becker DF, Involvement of the beta3-alpha3 loop of the proline dehydrogenase domain in allosteric regulation of membrane association of proline utilization A, Biochemistry, 2013, 52, 4482–4491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Adams PD, v Afonine P, Bunkoczi G, Chen VB, Davis IW, Echols N, Headd JJ, Hung LW, Kapral GJ, Grosse-Kunstleve RW, McCoy AJ, Moriarty NW, Oeffner R, Read RJ, Richardson DC, Richardson JS, Terwilliger TC and Zwart PH, PHENIX: a comprehensive Python-based system for macromolecular structure solution, Acta Crystallogr. D Biol. Crystallogr, 2010, 66, 213–221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.v Afonine P, Grosse-Kunstleve RW, Echols N, Headd JJ, Moriarty NW, Mustyakimov M, Terwilliger TC, Urzhumtsev A, Zwart PH and Adams PD, Towards automated crystallographic structure refinement with phenix.refine, Acta Crystallogr. D Biol. Crystallogr, 2012, 68, 352–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Emsley P, Lohkamp B, Scott WG and Cowtan K, Features and development of Coot, Acta Cryst. D Biol. Crystallogr, 2010, 66, 486–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Moriarty NW, Grosse-Kunstleve RW and Adams PD, electronic Ligand Builder and Optimization Workbench (eLBOW): a tool for ligand coordinate and restraint generation, Acta Crystallogr. D Biol. Crystallogr, 2009, 65, 1074–1080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Chen VB, Arendall WB 3rd, Headd JJ, Keedy DA, Immormino RM, Kapral GJ, Murray LW, Richardson JS and Richardson DC, MolProbity: all-atom structure validation for macromolecular crystallography, Acta Crystallogr. D Biol. Crystallogr, 2010, D66, 12–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gore S, Sanz Garcia E, Hendrickx PMS, Gutmanas A, Westbrook JD, Yang H, Feng Z, Baskaran K, Berrisford JM, Hudson BP, Ikegawa Y, Kobayashi N, Lawson CL, Mading S, Mak L, Mukhopadhyay A, Oldfield TJ, Patwardhan A, Peisach E, Sahni G, Sekharan MR, Sen S, Shao C, Smart OS, Ulrich EL, Yamashita R, Quesada M, Young JY, Nakamura H, Markley JL, Berman HM, Burley SK, Velankar S and Kleywegt GJ, Validation of Structures in the Protein Data Bank, Structure, 2017, 25, 1916–1927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Liebschner D, v Afonine P, Moriarty NW, Poon BK, v Sobolev O, Terwilliger TC and Adams PD, Polder maps: improving OMIT maps by excluding bulk solvent, Acta Crystallogr D Struct Biol, 2017, 73, 148–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Campbell AC, Bogner AN, Mao Y, Becker DF and Tanner JJ, Structural analysis of prolines and hydroxyprolines binding to the l-glutamate-gamma-semialdehyde dehydrogenase active site of bifunctional proline utilization A, Arch Biochem Biophys, 2021, 698, 108727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Singh H, Arentson BW, Becker DF and Tanner JJ, Structures of the PutA peripheral membrane flavoenzyme reveal a dynamic substrate-channeling tunnel and the quinone-binding site, Proceedings of the National Academy of Sciences of the United States of America, 2014, 111, 3389–3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Geng P, Qin W and Xu G, Proline metabolism in cancer, Amino Acids 2021, 2021, 1–9. [DOI] [PubMed] [Google Scholar]
- 38.Cockroft SL and Hunter CA, Chemical double-mutant cycles: Dissecting non-covalent interactions, Chemical Society Reviews, 2007, 36, 172–188. [DOI] [PubMed] [Google Scholar]
- 39.Liu Y and Liu SY, Exploring the strength of a hydrogen bond as a function of steric environment using 1,2-azaborine ligands and engineered T4 lysozyme receptors, Organic & biomolecular chemistry, 2019, 17, 7002–7006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Samways ML, Taylor RD, Bruce Macdonald HE and Essex JW, Water molecules at protein-drug interfaces: computational prediction and analysis methods, Chemical Society reviews, 2021, 50, 9104–9120. [DOI] [PubMed] [Google Scholar]