

ABSTRACT
Autophagic pathways cross with lipid homeostasis and thus provide energy and essential building blocks that are indispensable for liver functions. Energy deficiencies are compensated by breaking down lipid droplets (LDs), intracellular organelles that store neutral lipids, in part by a selective type of autophagy, referred to as lipophagy. The process of lipophagy does not appear to be properly regulated in fatty liver diseases (FLDs), an important risk factor for the development of hepatocellular carcinomas (HCC). Here we provide an overview on our current knowledge of the biogenesis and functions of LDs, and the mechanisms underlying their lysosomal turnover by autophagic processes. This review also focuses on nonalcoholic steatohepatitis (NASH), a specific type of FLD characterized by steatosis, chronic inflammation and cell death. Particular attention is paid to the role of macroautophagy and macrolipophagy in relation to the parenchymal and non-parenchymal cells of the liver in NASH, as this disease has been associated with inappropriate lipophagy in various cell types of the liver.
Abbreviations: ACAT: acetyl-CoA acetyltransferase; ACAC/ACC: acetyl-CoA carboxylase; AKT: AKT serine/threonine kinase; ATG: autophagy related; AUP1: AUP1 lipid droplet regulating VLDL assembly factor; BECN1/Vps30/Atg6: beclin 1; BSCL2/seipin: BSCL2 lipid droplet biogenesis associated, seipin; CMA: chaperone-mediated autophagy; CREB1/CREB: cAMP responsive element binding protein 1; CXCR3: C-X-C motif chemokine receptor 3; DAGs: diacylglycerols; DAMPs: danger/damage-associated molecular patterns; DEN: diethylnitrosamine; DGAT: diacylglycerol O-acyltransferase; DNL: de novo lipogenesis; EHBP1/NACSIN (EH domain binding protein 1); EHD2/PAST2: EH domain containing 2; CoA: coenzyme A; CCL/chemokines: chemokine ligands; CCl4: carbon tetrachloride; ER: endoplasmic reticulum; ESCRT: endosomal sorting complexes required for transport; FA: fatty acid; FFAs: free fatty acids; FFC: high saturated fats, fructose and cholesterol; FGF21: fibroblast growth factor 21; FITM/FIT: fat storage inducing transmembrane protein; FLD: fatty liver diseases; FOXO: forkhead box O; GABARAP: GABA type A receptor-associated protein; GPAT: glycerol-3-phosphate acyltransferase; HCC: hepatocellular carcinoma; HDAC6: histone deacetylase 6; HECT: homologous to E6-AP C-terminus; HFCD: high fat, choline deficient; HFD: high-fat diet; HSCs: hepatic stellate cells; HSPA8/HSC70: heat shock protein family A (Hsp70) member 8; ITCH/AIP4: itchy E3 ubiquitin protein ligase; KCs: Kupffer cells; LAMP2A: lysosomal associated membrane protein 2A; LDs: lipid droplets; LDL: low density lipoprotein; LEP/OB: leptin; LEPR/OBR: leptin receptor; LIPA/LAL: lipase A, lysosomal acid type; LIPE/HSL: lipase E, hormone sensitive type; LIR: LC3-interacting region; LPS: lipopolysaccharide; LSECs: liver sinusoidal endothelial cells; MAGs: monoacylglycerols; MAPK: mitogen-activated protein kinase; MAP3K5/ASK1: mitogen-activated protein kinase kinase kinase 5; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; MCD: methionine-choline deficient; MGLL/MGL: monoglyceride lipase; MLXIPL/ChREBP: MLX interacting protein like; MTORC1: mechanistic target of rapamycin kinase complex 1; NAFLD: nonalcoholic fatty liver disease; NAS: NAFLD activity score; NASH: nonalcoholic steatohepatitis; NPC: NPC intracellular cholesterol transporter; NR1H3/LXRα: nuclear receptor subfamily 1 group H member 3; NR1H4/FXR: nuclear receptor subfamily 1 group H member 4; PDGF: platelet derived growth factor; PIK3C3/VPS34: phosphatidylinositol 3-kinase catalytic subunit type 3; PLIN: perilipin; PNPLA: patatin like phospholipase domain containing; PNPLA2/ATGL: patatin like phospholipase domain containing 2; PNPLA3/adiponutrin: patatin like phospholipase domain containing 3; PPAR: peroxisome proliferator activated receptor; PPARA/PPARα: peroxisome proliferator activated receptor alpha; PPARD/PPARδ: peroxisome proliferator activated receptor delta; PPARG/PPARγ: peroxisome proliferator activated receptor gamma; PPARGC1A/PGC1α: PPARG coactivator 1 alpha; PRKAA/AMPK: protein kinase AMP-activated catalytic subunit; PtdIns3K: class III phosphatidylinositol 3-kinase; PtdIns3P: phosphatidylinositol-3-phosphate; PTEN: phosphatase and tensin homolog; ROS: reactive oxygen species; SE: sterol esters; SIRT1: sirtuin 1; SPART/SPG20: spartin; SQSTM1/p62: sequestosome 1; SREBF1/SREBP1c: sterol regulatory element binding transcription factor 1; TAGs: triacylglycerols; TFE3: transcription factor binding to IGHM enhancer 3; TFEB: transcription factor EB; TGFB1/TGFβ: transforming growth factor beta 1; Ub: ubiquitin; UBE2G2/UBC7: ubiquitin conjugating enzyme E2 G2; ULK1/Atg1: unc-51 like autophagy activating kinase 1; USF1: upstream transcription factor 1; VLDL: very-low density lipoprotein; VPS: vacuolar protein sorting; WIPI: WD-repeat domain, phosphoinositide interacting; WDR: WD repeat domain
KEYWORDS: Chaperone-mediated autophagy, fibrosis, hepatocellular carcinoma, macroautophagy, macrolipophagy, microautophagy, microlipophagy, nafld, nash, nonalcoholic fatty liver disease, nonalcoholic steatohepatitis
An overview of general autophagic mechanisms
The term autophagy, which is derived from the Greek words αυτος (“auto”) and φαγειν (“phagein”), meaning self-eating, includes a number of lysosomal degradative and recycling pathways that control both quality and quantity of proteins, lipids and organelles in eukaryotic cells. Upon starvation, autophagic pathways are triggered to compensate for the lack of nutrients and to balance energy deficiencies. In addition, numerous other types of cellular stress can induce autophagic pathways, including the accumulation of cytotoxic protein aggregates that are selectively degraded through autophagy. Likewise, cellular organelles, including LDs, can be degraded in lysosomes through autophagic processes to generate metabolites. Autophagy is therefore important in maintaining cellular homeostasis in both cells and tissues and plays a crucial role during development, adaptation to stress, and immune and metabolic responses. The three different autophagic pathways macroautophagy/autophagy, microautophagy and chaperone-mediated autophagy (CMA) are discussed in the following subchapters.
Macroautophagy
Macroautophagy is a highly conserved lysosomal degradation mechanism [1]. Unlike microautophagy or CMA, macroautophagy requires the formation of double-membrane vesicles called autophagosomes, which sequester intracellular components such as proteins, macromolecular complexes and organelles, but also invading pathogens [2]. Macroautophagy takes place in almost all eukaryotic cells at basal levels. However, it is most often a response to cellular stress induced by a wide variety of physiological stimuli, including nutritional deficiencies, growth factor withdrawal, oxidative and mechanical stress, as well as pathophysiological situations, including drug treatment and radiation therapy.
Macroautophagy begins with the formation of a precursor structure called the phagophore [3], which then elongates and closes to generate an autophagosome. The latter then matures and finally fuses with lysosomes to expose the engulfed cytosolic material to acidic lysosomal hydrolases for degradation. To different extents, various cellular organelles, including the endoplasmic reticulum (ER), Golgi complex, mitochondria, recycling endosomes, but also the plasma membrane, have been shown to supply the lipids required for the assembly of autophagosomes [4], a process executed by the autophagy related (ATG) proteins [5]. ATG genes were first discovered and characterized by Yoshinori Ohsumi, Daniel Klionsky and Michael Thumm [5–7]. During the process of autophagy, induction of autophagosome formation requires the activation of the ULK1/Atg1 (unc-51 like autophagy activating kinase 1) kinase complex [8], which integrates upstream molecular signals principally via two key kinases, the MTOR (mechanistic target of rapamycin kinase) complex 1 (MTORC1) and the PRKA/AMPK (protein kinase AMP-activated catalytic subunit) [9–11]. ULK1 kinase complex activation is essential to initiate the de novo formation of the phagophore, which takes place in specialized subdomains of the ER called omegasomes [12]. The latter are considered as the “cradle” for the forming nascent autophagosome [13]. Specifically, the ULK1 complex phosphorylates and activates the class III phosphatidylinositol 3-kinase (PtdIns3K) complex, composed of BECN1/Vps30/Atg6 (beclin 1), PIK3R4/VPS15 (phosphoinositide-3-kinase regulatory subunit 4), ATG14, PIK3C3/VPS34 (phosphatidylinositol 3-kinase catalytic subunit type 3), AMBRA1 (autophagy and beclin 1 regulator 1) and NRBF2 (nuclear receptor binding factor 2) [14], which generates phosphatidylinositol-3-phosphate (PtdIns3P). In turn, the generation of PtdIns3P mediates the association of specific PtdIns3P-binding proteins, such as ZFYVE1/DFCP1 (zinc finger FYVE-type containing 1) [12] and WIPI (WD repeat domain, phosphoinositide interacting) proteins [15]. The four WIPI proteins, WIPI1, WIPI2, WDR45B/WIPI3 (WD repeat domain 45B) and WDR45/WIPI4 (WD repeat domain 45) are the mammalian members of the β-propellers that bind PROPPIN (β-propellers that bind polyphosphoinositides) and function as conserved PtdIns3P effectors at the nascent autophagosome [16,17]. Thereby, WIPI2, assisted by WIPI1 [16], recruits ATG16L1 [18], which assemble with the ATG12–ATG5 conjugate into a multimeric complex [18] and initiate phagophore formation. In contrast, both WDR45B/WIPI3 and WDR45/WIPI4 control phagophore elongation, whereby WDR45 specifically associates with ATG2 proteins [16,19]. The ATG12–ATG5-ATG16L1 complex acts as an E3-like ligase for the conjugation of the members of the MAP1LC3/LC3 (microtubule associated protein 1 light chain 3) protein family to phosphatidylethanolamine [5]. The LC3 protein family comprises seven members, LC3A, LC3B, LC3B2 and LC3C, which are part of the MAP1LC3 subgroup, and GABARAP (GABA type A receptor-associated protein), GABARAPL1 (GABA type A receptor associated protein like 1) and GABARAPL2/GATE-16 (GABA type A receptor associated protein like 2), which belong to the GABARAP subgroup. LC3 proteins are important for autophagic cargo selection, phagophore elongation and closure. Complete autophagosomes finally contact lysosomes and fuse with them to form autolysosomes [20].
Macroautophagy can be either selective or nonselective. Nonselective macroautophagy involves random sequestration of cytoplasmic material by autophagosomes, whereas selective macroautophagy leads to the lysosomal degradation of specific cargoes and such pathways are named according to the type of cargo that is targeted, including aggregated proteins (aggrephagy), mitochondria (mitophagy), peroxisomes (pexophagy) and LDs (lipophagy) [21,22]. Selective types of macroautophagy rely on the so-called autophagy receptors that physically link the cargo to the phagophore [21,22] by Ub (ubiquitin) -dependent or Ub-independent cargo recognition [22]. The vast majority of macroautophagy receptors physically interact with the phagophore by selective binding to LC3 proteins present on the inner membrane of the phagophore. Thereby, short linear peptide sequences known as LC3-interacting regions (LIRs) in mammalian cells or Atg8-interacting motifs in yeast, mediate the effective tethering of the cargo to the forming vesicle, guaranteeing selective sequestration [21,22]. Ub-dependent macroautophagy receptors like SQSTM1/p62 (sequestosome 1), possess a Ub-binding domain that allows them to recognize and be recruited onto ubiquitinylated cargoes [21,22]. Ub-independent macroautophagy receptors, in contrast, are often membrane-associated and therefore present in the cargo that potentially can be targeted by selective autophagy [21,22]. While engagement of the Ub-dependent macroautophagy receptors is dictated by the ubiquitination of the cargo destined to degradation, those that are Ub-independent require additional regulation, which is often achieved through their phosphorylation [21–23].
Microautophagy
Microautophagy is a conserved process in which cytosolic components are directly engulfed and degraded by the vacuole in yeast or by endo-lysosomal compartments in mammals. Microautophagy cargoes include proteins and organelles, such as peroxisomes, mitochondria and portions of nucleus [24–26]. In mammalian cells two different mechanisms of lysosomal cargo sequestration have been reported, one involving protrusions of the lysosome limiting membrane and the other involving lysosomal invagination. However, given the fact that technical monitoring of microautophagy still presents a challenge, current investigations on microautophagy remain a difficult task [27]. Nevertheless, selective microautophagy-like uptake of protein cargo into late endosomes, referred to as endosomal microautophagy, has been described in mammalian cells [28,29], whereby cytosolic protein cargo are invaginated by late endosomes [27]. Such proteins harbor a KFERQ-like motif, which is recognized by the HSPA8/HSC70 [heat shock protein family A (Hsp70) member 8], to promote the translocation of the captured proteins into the lumen of a late endosome, or multivesicular body, in an endosomal sorting complex required for transport (ESCRT) III-dependent manner [30]. Subsequent fusion of the late endosomes/multivesicular body then delivers the translocated protein cargo into a lysosome for turnover [27].
Chaperone-mediated autophagy
CMA is a selective type of autophagy principally dedicated to the lysosomal degradation of cytosolic proteins [31]. The targeted proteins possess a KFERQ motif in their amino acid sequence [32], but unlike in endosomal microautophagy, are directly captured by lysosomes [27]. The KFERQ motif is recognized by the cytosolic chaperone HSPA8 and its co-chaperones, including STUB1/CHIP (STIP1 homology and U-box containing protein 1), DNAJB1/HSP40 [DnaJ heat shock protein family (Hsp40) member B1] and STIP1/HOP (stress induced phosphoprotein 1) [31], leading to the assembly of a cargo-chaperone complex [33]. This complex then binds the cytosolic tail of the LAMP2A (lysosomal associated membrane protein 2A), a protein only expressed in mammals [34], which mediates the unfolding and translocation of the targeted substrate through the lysosomal membrane [33]. During this transport, substrates are translocated one-by-one into the lysosomal lumen [35], via a process involving the multimerization of LAMP2A into a translocation-competent complex and the lysosomal resident pool of HSPA8 [36]. The substrates are then rapidly degraded by lysosomal proteases and LAMP2A disassembles to become available for a new cycle of substrate binding and translocation [35] .
Lipid droplets
LDs are conserved cellular organelles (Figure 1) that store neutral lipids [37] and fulfill critical functions in lipid and energy homeostasis [38]. In recent years, it has become apparent that LDs have a complex functional connection with autophagy [39], and that cellular energy levels can be elevated by employing free fatty acids (FFAs) gained from the selective breakdown of LDs through macroautophagy, referred to as macrolipophagy [40]. Macrolipophagy is a relatively new area in the field of autophagy, and will certainly provide us with exciting new information about its mechanism and regulation, but also its physiological relevance in the next few years. In fact, research on macrolipophagy has already begun to expand our understanding of the physiological relevance of autophagy. Macrolipophagy plays an important role in regulating lipid metabolism, and as part of lipolysis programs, can provide energy and the building blocks for membrane biosynthesis and lipid anabolism. In this latter context, macrolipophagy has an important role in the biosynthesis of the cholesterol-based lipidic hormone testosterone [41]. More recently, macrolipophagy-derived FFAs have been shown to undergo extracellular efflux via lysosomal exocytosis through a mechanism involving MCOLN1 (mucolipin TRP cation channel 1), a lysosomal Ca2+ channel that regulates lysosome-plasma membrane fusion [42]. These secreted lipids have been suggested to be important for cell-to-cell lipid exchange and signaling, but further studies are needed to better understand the significance of FFA efflux in tissue homeostasis and disease development [42]. Additionally, macrolipophagy is important for cell differentiation. For example, macrolipophagy-mediated lipolysis and subsequent generation of FFAs enables the shift from glycolysis to FFA β-oxidation, which generates the energy needed for neutrophil differentiation [43]. Finally, a study in the worm Caenorhabditis elegans showed that lipid signaling molecules, derived from lysosomal lipid hydrolysis, may modulate gene expression to extend lifespan [44].
Figure 1.
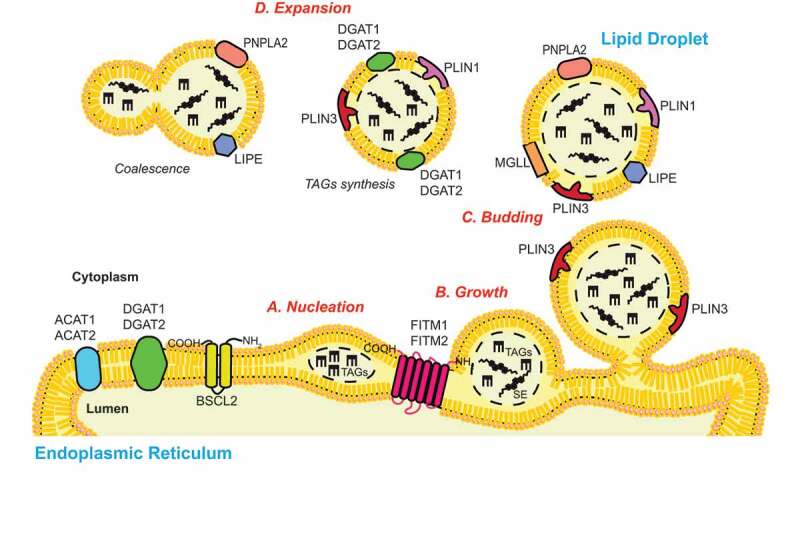
The biogenesis of LDs. The process of de novo LD biogenesis can be divided in three main discrete steps: (A) Nucleation, (B) growth and (C) budding. The nucleation step is characterized by the formation of an oil lens structure in between the two lipid bilayers of the ER limiting membrane, which is catalyzed by ER resident proteins such as FITM1/FITM2 and BSCL2/seipin. The growth of the nascent LDs starts with an accumulation of TAGs and SE, and involves a ripening phenomenon that regulates their size. The partial (in yeast) or complete (in mammalian cells) detachment of the LDs from the ER membrane defines the LDs budding. LDs can also expand (D) by increasing their size through the coalescence of LDs and/or the local synthesis of TAGs. Several enzymes involved in the biogenesis and function of LDs, which are discussed in the review, are indicated.
In the following, we discuss the current knowledge about the structure, biogenesis, function and catabolism of LDs, before extending to macrolipophagy and other autophagic pathways used to degrade LDs or LD-associated proteins.
The structure of lipid droplets
LDs are spherical organelles with a hydrophobic core composed by neutral lipids such as triacylglycerols (TAGs), diacylglycerols (DAGs) and sterol esters (SE). This core is coated with a monolayer of phospholipids, free cholesterol and lysophospholipids, into which specific proteins are inserted (Figure 1). The limiting membrane of LDs has the major role in maintaining the morphology and stability of LDs and also facilitates the interactions with other organelles [45]. Multiple proteins constantly interact with the surface of LDs, including members of the PLIN (perilipin) protein family, which are among the best characterized LD-associated proteins [46]. This protein family has five members: PLIN1 to PLIN5 [46]. While PLIN1 and PLIN4 are mainly expressed in white adipose tissue, PLIN2/adipophilin, and PLIN3/TIP47, are ubiquitously expressed. The major expression tissues of PLIN5, in contrast, are the cardiac and skeletal muscles, the brown adipose tissue and the liver [46–48]. PLIN proteins have an important role in regulating LD structure, function and catabolism through lipolysis and CMA [49]. In addition to the PLIN protein family, other proteins such as lipases are associated to LDs [45]. Among these, there is the PNPLA2/ATGL (patatin like phospholipase domain containing 2), the LIPE/HSL (lipase E, hormone sensitive type) and the MGLL/MGL (monoacylglycerol lipase), which together mediate lipolysis (see below). PNPLA2 is expressed in all tissues, with highest levels in adipose tissues. LIPE expression has also been found to be higher in adipose tissues, while MGLL is ubiquitously expressed [50]. Other lipases, including PNPLA3/adiponutrin (patatin like phospholipase domain containing 3) or PNPLA5 (patatin like phospholipase domain containing 5), which share significant sequence identity with PNPLA2, are also present on LDs and catalyze the hydrolysis of TAGs into DAGs [51]. ATG proteins have also been found on the surface of LDs. In particular, ATG2A, ATG14 and lipidated LC3 decorate LDs and are involved in the regulation of LD volume and/or distribution [52–54]. Interestingly, mutations in several LD-associated proteins that lead to a defect in expression and/or enzymatic activity, have been associated with human diseases [38].
The biogenesis of lipid droplets
LDs arise from the ER, where several neutral lipid biosynthetic enzymes are localized. Among these are the GPAT (glycerol-3-phosphate acyltransferase) enzymes, which catalyze the first step of the glycerol-3-phosphate pathway, the main cellular process that leads to the synthesis of TAGs in most cell types [55]. GPATs acylate glycerol-3-phosphate and acyl-coenzyme A (CoA) to generate lysophosphatidic acids, which are converted into phosphatidic acids by the 1-acyl glycerol-3-phosphate acyltransferase. Subsequently, LPIN (lipin) enzymes dephosphorylate the phosphatidic acids to generate DAGs, which are finally transformed into TAGs by diacylglycerol acyltransferases (DGATs) [56]. In the small intestine, liver and adipose tissue, TAGs can also be synthesized through the monoacylglycerol (MAG) pathway [57]. This pathway produces DAGs from MAGs and shares the final step with the glycerol-3-phosphate pathway. Other ER resident enzymes such as ACAT1 (acetyl-CoA acetyltransferase 1) and ACAT2, produce SE from sterols [38]. The process of de novo LD biosynthesis is divided in three major steps: (i) nucleation, (ii) growth and (iii) budding (Figure 1) [58]. Although the exact molecular mechanism of those different steps is still not yet fully understood, recent research has shed light on this important cellular process, and a consensus for the biogenesis of LDs is emerging. The nucleation step begins with the formation of an oil lens structure between the two leaflets of the ER membrane, which originates from the accumulation of the newly synthesized TAGs and SE [59,60]. The oil lens formation appears to be a lipid driven process, which does not rely on specific proteins as previously suggested [61]. It has been shown that the presence of non-bilayer lipids such as phosphatidic acids and DAGs in the ER bilayer increase the bilayer tension, impeding LD formation [61]. As a result, transformation of these lipids into TAGs has the opposite effect, i.e., inducing LD biogenesis. Nevertheless, two ER-resident protein families, the FITM/FIT (fat storage-inducing transmembrane protein) and the BSCL2/seipin (BSCL2 lipid droplet biogenesis associated, seipin) protein family, play an indirect role in the emergence of oil lens [62]. On the one hand, FITM proteins, in particular FITM2, are thought to be acyl-coenzyme A diphosphatases [63]. Their activity could promote the decrease of DAG level in the proximity of LDs’ budding site, at the cytosolic leaflet of the ER membrane, to help the unidirectional LD emergence [64]. On the other hand, BSCL2/seipins may regulate the local synthesis and distribution of phospholipids such as phosphatidic acid in the ER, and they interact and negatively control GPAT activity [65–68]. Normal levels and distribution of phosphatidic acid in the ER allow LDs to form and grow properly. Additional proteins that can modulate membrane rearrangements, including the ER membrane shaping proteins such as RTN (reticulon) and ATL/atlastin (atlastin GTPase) proteins or LD coat proteins such as PLIN3, play a role in biogenesis of LDs [38]. However, the exact mechanism by which those proteins promote the formation of LDs remains to be determined. The growth step is characterized by the phenomenon of ripening, a slow process that is promoted by the diffusion of molecules from the volume of smaller LDs toward larger LDs, and fusion [58]. This event is followed by the budding step, which is usually completed in mammalian cells, while in yeast, LDs remain associated with the ER (Figure 1) [59,69]. After budding LDs can still expand, either through LD-LD fusion, a process known as coalescence, transfer of TAGs from the ER to LDs via membrane contact sites, or local TAGs synthesis at the LD surface [70,71].
The functions of lipid droplets
An important function of LDs is to store non-esterified FFAs as inert TAGs, since an excess of FFAs in the cytoplasm would trigger the formation of harmful bioactive lipids, causing lipotoxicity [72]. Other main functions of LDs are to provide building blocks for the biosynthesis of cellular membranes and other lipid species, as well as for metabolic energy through FFA β-oxidation when nutrients are scarce [73]. LDs also play roles in the assembly of viruses such as hepatitis C virus, protein sequestration, and membrane trafficking and signaling [74].
One of the most interesting research areas on LDs is the characterization of membrane contact sites of LDs with other organelles, which are important for the fine-tuning of the regulation of LD functions. In addition to having membrane contact sites with ER, LDs physically communicate with peroxisomes to facilitate the transfer and the β-oxidation of very long chains and branched FFAs [75,76]. In addition, LDs often interact with mitochondria in a PLIN5-dependent manner in response to nutrient deficiencies in cultured cells, and to exercise in the skeletal muscles of animals [77,78]. These LD-mitochondria contact sites might help the transfer of FFAs for β-oxidation [73], although direct evidence for this event is lacking. Finally, LDs have also been functionally linked to macroautophagy. In this context, it has been shown that neutral lipid stores are mobilized from LDs to support the formation of the autophagosomes in both yeast and mammalian cells [51,79,80]. In summary, all these findings support our view on LDs, which are now considered to be highly dynamic organelles rather than inert fat bodies.
Catabolism of lipid droplets: neutral and acid lipolysis
The term lipolysis refers to the hydrolysis of the ester bonds in TAGs, to generate FFAs and glycerol. Cells are unable to uptake exogenous TAGs as they can only assimilate FFAs. Thus, lipolysis is essential to process dietary TAGs and lipoprotein-associated TAGs for the subsequent cellular uptake of FFAs from intestine and blood, respectively. In addition, lipolysis is required for the metabolism of TAGs endocytosed in association with lipoproteins from the blood, known as intracellular lipolysis. Intracellular lipolysis is also critical to catabolize TAGs stored in LDs in both adipose and non-adipose tissues. In contrast to adipose tissues, which secrete lipolysis products into the bloodstream, non-adipose tissues mobilize lipolysis products when cells require FFAs and glycerol to either generate energy or build new macromolecules [72,81].
Intracellular lipolysis can be divided into neutral and acid lipolysis, depending on the pH value and subcellular location where this process takes place. Neutral lipolysis occurs in the cytoplasm, at neutral pH, and it is mediated by a series of cytosolic lipases that directly act on the TAGs and cholesteryl esters stored in LDs. Neutral lipolysis of TAGs requires the consecutive action of the three neutral lipases (i) PNPLA2, which preferentially catalyzes the hydrolysis of TAGs into DAGs, (ii) LIPE, which mainly mediates the processing of DAGs into MAGs, as well as the hydrolysis of the ester bonds of other lipids such as cholesteryl esters, and (iii) MGLL, which catalyzes the hydrolysis of MAGs into glycerol [72]. In contrast, acid lipolysis takes place at acidic pH inside lysosomes, and is mediated by lipases such as the LIPA/LAL (lipase A, lysosomal acid), which are believed to hydrolyze TAGs and cholesteryl esters, while acid phospholipases and proteases break down LD-associated phospholipids and proteins, respectively. Acid lipolysis hydrolyzes lipids delivered into lysosomes by both receptor-mediated endocytosis of lipoproteins and lipophagy [72,82]. In this context, the finding that LDs could be delivered into lysosomes for turnover by macrolipophagy in 2009, was a ground-breaking finding in the field of both lipolysis and autophagy research [83], and added a new level of complexity to our view on the regulation of LD catabolism. In extension, the direct engulfment of LDs by endosomes or lysosomes/vacuoles, known as microlipophagy [84], as well as the degradation of LD-associated proteins by CMA, have been reported more recently [49].
Macrolipophagy
Similar to bulk nonselective autophagy, lipophagy can occur via both macro (Figure 2A) and microautophagic mechanisms (Figure 2B) depending on how LDs are transported into lysosomes/vacuoles [83,84]. Macrolipophagy involves the autophagosome-mediated sequestration of LDs and their subsequent delivery to lysosomes/vacuole for turnover [83], which were originally reported to occur in mouse hepatocytes that have been supplemented with oleate and then subjected to nutrient starvation [83]. Inhibition or loss of autophagy caused an increase in TAG and LD levels in vitro and in vivo, a decrease in TAG breakdown and a colocalization between ATG components and TAG or LD proteins [83]. Macrolipophagy has subsequently been described in many other mammalian cell types [41,85–93] and it therefore appears that this process contributes ubiquitously to the mobilization of lipids stored in LDs in higher eukaryotes [81].
Figure 2.
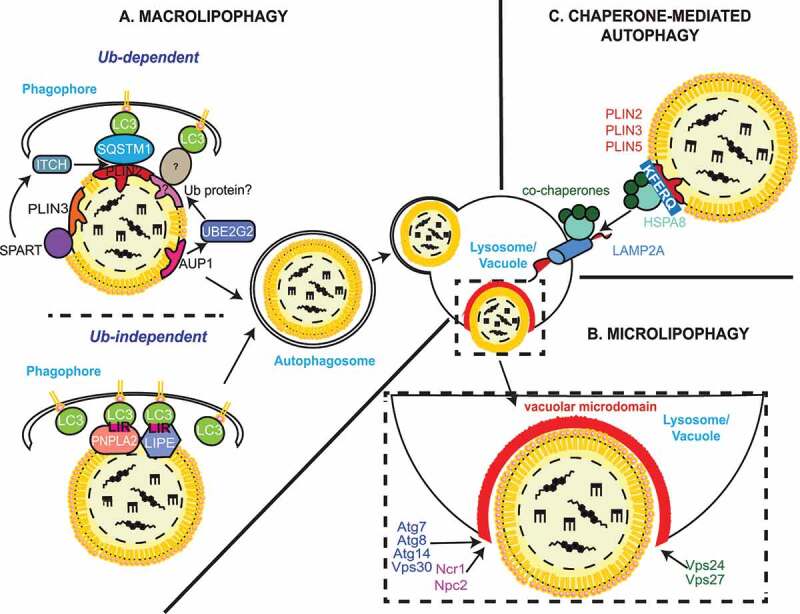
Autophagic processes of LDs. (A) Macrolipophagy involves the sequestration of LDs by autophagosomes and their subsequent delivery to lysosomes/vacuoles for turnover. LDs could be selectively recognized in both an Ub-dependent and -independent manner. On the one hand, during Ub-dependent macrolipophagy, the LDs surface protein AUP1 binds and recruits the Ub conjugating enzyme UBE2G2, which could represent the functional link to the subsequent Ub-dependent macroautophagic degradation of LDs. On the other hand, SPART binds the LDs-anchored protein PLIN3 and promotes the recruitment and activation of the Ub ligase ITCH/AIP4. ITCH polyubiquitinates PLIN2 (and potentially other LDs-associated proteins), leading to the association of the macroautophagy cargo receptor SQSTM1/p62, which triggers macrolipophagy. During Ub-independent microlipophagy, both PNPLA2/ATGL and LIPE/HSL, which are distributed on the phospholipid monolayer limiting LDs, interact with MAP1LC3A/LC3 via their LIR motifs, inducing the local, in situ, formation of a phagophore. It remains unknown whether the Ub-dependent and – independent macrolipophagy are mutually exclusive or act concomitantly/sequentially. RAB10, RAB7, EHBP1 and EHD2 have also been implicated in macrolipophagy. However, it is unclear whether they are playing a role in the Ub-dependent, the Ub–independent or both processes of macrolipophagy. (B) Microlipophagy has been better characterized in yeast S. cerevisiae. It involves the direct engulfment of LDs by vacuoles. Microlipophagy relies on liquid ordered and sterol enriched vacuolar microdomain formation. The sterol-transporting proteins Ncr1 and Npc2 are essential for the formation of these vacuolar microdomains and subsequent LD engulfment. The formation of these vacuolar microdomains also involve core ATG proteins such as Atg7, Atg8, Vps30/Atg6 and Atg14. Additionally, an ATG core machinery-independent microlipophagy has been described and requires ESCRT components such as Vps24 and Vps27 (C) During CMA of LDs-anchored proteins, the cytosolic chaperone HSPA8/HSC70, together with its co-chaperones, recognizes proteins possessing a KFERQ motif, including PLIN2, PLIN3 and PLIN5. This cargo-chaperone complex then binds LAMP2A present of the surface of lysosomes, which mediates the unfolding and translocation into the lumen of this organelle of the targeted cargo to be degraded. CMA-mediated degradation of PLIN proteins promotes the recruitment of PNPLA2/ATGL and ATG machinery components onto LDs, making CMA an upstream regulator of both neutral lipolysis and macrolipophagy.
As with macroautophagy, macrolipophagy requires ATG proteins, in particular those composing the highly conserved core machinery [83]. In mammals, sequestration of LDs appears to be through a piecemeal process, where phagophores sequester only a portion of the LDs [83]. Why phagophores may not engulf entire LDs is unclear, but it has been suggested that it is due to their enormous size [83]. In this context, it has been proposed that neutral lipolysis targets large LDs to (i) decrease their volume as well as to (ii) create small nascent LDs, generated by re-esterification of FFAs, which are then accessible for macrolipophagic sequestration [94]. Indeed, predominantly small LDs are targeted by the macrolipophagic machinery since lysosomal inhibition has been shown to lead to the accumulation of small LDs within autophagosomes [94]. Although the exact mechanism that allows lipophagic autophagosomes to preferentially sequester smaller LDs has yet to be deciphered, current evidence suggests that LDs could be selectively recognized in both an Ub-dependent and -independent manner.
Ub-dependent macrolipophagy
AUP1 (AUP1 lipid droplet regulating VLDL assembly factor) is a highly conserved and broadly expressed LD surface protein that is able to interact and thus recruit the UBE2G2/UBC7 (ubiquitin conjugating enzyme E2 G2) [95]. This could provide a possible molecular connection between LDs and ubiquitination as a putative lipophagy promoting signal, although a link with the subsequent Ub-dependent autophagic degradation of LDs remains uncertain (Figure 2A). Interestingly, Dengue virus induces deubiquitination of AUP1 and UBE2G2 [96], which is necessary to induce macrolipophagy and subsequent β-oxidation of the resulting FFAs to generate energy necessary for Dengue virus replication and virus production [96].
Another study has shown that the localization of the adaptor protein SPART/SPG20 (spartin) on LDs is stimulated in several cell lines fed with oleic acid, and that this association with LDs occurs through the binding of SPART C-terminal region with PLIN3 [97] (Figure 2A). Importantly, depletion of SPART causes an increase in both number and size of LDs in HEK 293 T cells fed with oleic acid [97]. SPART recruits homologous to E6-AP C terminus (HECT) Ub ligases though an interaction between its PPxY motif and the WW domain in the HECT ligases [97]. One of those ligases is WWP1 (WW domain containing E3 ubiquitin protein ligase 1), which interacts and monoubiquitinates SPART, triggering its degradation and preventing its accumulation in LDs [97]. The role of WWP1 in LD turnover has not been clearly defined yet, but it may be that by controlling SPART levels via ubiquitination, it permits to regulate macrolipophagy. Another HECT E3 Ub ligase is ITCH/AIP4 (itchy E3 ubiquitin protein ligase), which is both activated and recruited to LDs by SPART (Figure 2A). In this case, however, ITCH does not modify SPART but rather polyubiquitinates PLIN2 (Figure 2A), and potentially other LD-associated proteins [98]. PLIN2 negatively affects the turnover of the LD pool of TAGs [99]. Although these observations indicate that SPART- and ITCH-mediated ubiquitination of PLIN2 could initiate Ub-dependent macrolipophagy, more direct evidence supporting this notion need to be collected [97,98].
Immunofluorescence and co-immunoprecipitation experiments performed in a separate study revealed that rapamycin-induced autophagy in myocytes cultured in the presence of an equimolar mixture of oleate and palmitate, triggers the binding of SQSTM1 to PLIN2 on the surface of LDs [90] (Figure 2A). Under these conditions, an enhancement of the macroautophagic flux and a concomitant decrease in lipid content was observed, suggesting that SQSTM1 via PLIN2 may direct LD sequestration into autophagosomes [90]. In line with this finding, both SQSTM1- and LC3-positive autophagosomes have been found to colocalize with LDs in mouse AML12 hepatocytes that were acutely exposed to ethanol to trigger lipophagy [100], which is induced as a cytoprotective response against liver injury [101]. Moreover, SQSTM1 and LC3 signals overlapped with another LD protein PLIN1, and both SQSTM1 and LC3 association with LDs were significantly reduced upon PLIN1 depletion [100]. These observations suggest that ethanol-triggered macrolipophagy requires SQSTM1, and that PLIN1 could be another protein on the LD surface that is recognized by this selective macroautophagy receptor [100].
A recent study in Drosophila hepatocyte-like oenocytes and the human HepG2 hepatoma cell line further underlined the role of SQSTM1 in LD turnover by macrolipophagy upon nutrient starvation [102], and revealed that HDAC6 (histone deacetylase 6) is also involved in this process [102]. HDAC6 is required for both the transport of protein aggregates via the microtubule network to aggresomes (pericentriolar cytoplasmic inclusion bodies) [103], and the ATG machinery recruitment to aggresomes for their selective autophagic degradation [104,105]. In particular, HDAC6 depletion prevented LD breakdown, as did the overexpression of a HDAC6 mutant lacking its Ub binding domain but also SQSTM1 variants that cannot oligomerize and bind to Ub [102]. These observations suggest that either macrolipophagy employs the same machinery for the selective turnover of aggresomes by macroautophagy, or that SQSTM1-positive aggresome formation and recruitment of HDAC6 to these structures is essential for macrolipophagy. Additionally, knockdown of huntingtin, a scaffold protein involved in selective types of autophagy that physically interacts with SQSTM1 to facilitate its association with LC3 [106], results in significant LD accumulation in multiple cell types fed with oleic acid, further supporting the notion that huntingtin, SQSTM1 and ultimately Ub-dependent macrolipophagy are required for the lysosomal turnover of LDs [106]. Altogether, these data suggest that there could be a mechanism of Ub-dependent macrolipophagy that relies on SQSTM1, but more solid evidence is needed to support this concept.
Ub-independent macrolipophagy
Evidence suggests that LDs could be selectively targeted to macrolipophagy in a Ub-independent manner. Both PNPLA2 and LIPE, which are distributed on the phospholipid monolayer limiting LDs, contain several putative LIR motifs. Co-immunoprecipitation experiments have revealed that these proteins interact with LC3, and therefore could dock LDs onto the cytoplasmic surface of autophagosomes [89] (Figure 2A). Mutation of the LIR motif at amino acid positions 147–150 dissociates PNPLA2 from LDs and disrupts PNPLA2-mediated lipolysis [89], suggesting that the interaction with LC3 may be important for neutral lipolysis, but not necessarily showing that PNPLA2 mediates macrolipophagy [89]. Another possible scenario could be that autophagosomes recruit cytosolic lipases like PNPLA2 in a LIR motif-dependent manner, to consume large LDs until their size allows to be engulfed by an autophagosome. This assumption is supported by the recent study, which indicates that PNPLA2 targets large size LDs upstream of macrolipophagy [94]. Interestingly, it has been shown that PNPLA2 is necessary and sufficient for both autophagy and lipophagy induction in hepatocytes, thereby enabling control of hepatic LD catabolism and FFA β-oxidation [107]. In fact, while pharmacological inhibition and genetic knockdown of PNPLA2 reduces Atg gene expression, macroautophagic flux and lipophagy in hepatocytes, overexpressing PNPLA2 has the opposite effect and leads to an increase in TAG turnover [107]. The latter is mediated by an autophagy program because it is inhibited pharmacologically and genetically by chloroquine and ATG5 knockdown, respectively [107]. On the same line, blocking lysosomal lipid hydrolysis using the LIPA inhibitor LAListat, prevents TAG turnover and reduction in LD content caused by PNPLA2 overexpression. Interestingly, ablation of the SIRT1 (sirtuin 1) deacetylase, a well-established activator of autophagy through its deacetylation and activation of key ATG proteins [108], abrogated the increase in expression of Atg genes, macroautophagic flux, lipophagy and TAG turnover upon PNPLA2 overexpression in primary hepatocytes, suggesting that SIRT1 is required for PNPLA2-mediated macrolipophagy [107]. However, the mechanism by which PNPLA2 regulates SIRT1 is still under investigation [107]. Altogether, these data suggest that PNPLA2 could be an Ub-independent macroautophagy receptor that targets LDs for degradation, and therefore could also be a mechanism of Ub-independent macrolipophagy.
Small RAB GTPases in macrolipophagy induction
Another group of proteins that have also been linked to macrolipophagy are members of the RAB protein family. RAB proteins are small GTPases that act as switches and in their GTP-bound form, recruit effector molecules for the regulation of intracellular vesicle trafficking [109]. RAB7 plays a fundamental role in the control of late endocytic membrane trafficking [110] and autophagosomal maturation events [111,112], and moreover, decorates the surface of LDs and regulates macrolipophagy in mammalian cells [113,114]. The recruitment of RAB7 to both LDs and autophagosomal membranes is enhanced by the stimulation of lipolysis mediated by β-adrenergic receptor activation, while the depletion or inactivation of RAB7 under the same conditions inhibits macrolipophagy [114]. Similarly, it was found that RAB7 is essential for macrolipophagy in hepatocytes exposed to nutrient deficiency, since the knockdown of RAB7 or the overexpression of a dominant-negative RAB7 mutant leads to an accumulation of LDs [113]. Therefore, macrolipophagy is likely to require RAB7 under several conditions. In this context, it has recently been shown that RAB10 localizes to LDs and autophagosomal membranes in a RAB7-dependent manner under starvation conditions [115], and similar to RAB7, depletion of RAB10 results in LD accumulation in starved hepatocytes [115]. Furthermore, it was found that RAB10 is necessary for the recruitment of the complex formed by EHBP1/NACSIN (EH domain binding protein 1) and EHD2/PAST2 (EH domain containing 2) on LDs in order to initiate macrolipophagy. Thereby, EHBP1 serves as an adaptor protein for EHD2, a membrane-deforming dynamin-like ATPase, which has been suggested to promote the extension of the autophagosome membrane around LDs for engulfment [115].
Macrolipophagy regulation
The observation that both macroautophagy and lipolysis are regulated hormonally by insulin and glucagon, and that they are enhanced during starvation, led to the idea that macroautophagy may contribute to LD breakdown and to the first report on macrolipophagy [83]. Ever since, the regulation of macrolipophagy induction has been shown to be a complex cellular and physiological response triggered by starvation and FFA loading [83], as well as hormones such as thyroidine [116], adrenaline [114] and FGF21 (fibroblast growth factor 21) [117]. FGF21 is secreted by the liver and induces macrolipophagy in an autocrine/paracrine-dependent manner in hepatocytes [117,118]. Interestingly, exposure to cold induces autophagy in proopiomelanocortin neurons of the hypothalamus, which in turn activate macrolipophagy in the brown adipose tissue and liver through the sympathetic network [89].
The major cellular energy sensing pathways modulating autophagy, including the classic stress and nutrient-responsive pathways such us those downstream of MTORC1 and PRKA, also orchestrate macrolipophagy. MTORC1 inhibition drives macrolipophagy, reducing lipid accumulation in an autophagy-dependent manner in vivo [89], while PRKA can activate autophagy/macrolipophagy through various mechanisms, including stimulation of ULK1 [119], inhibition of MTORC1 [120] and activation of SIRT1 [121].
Macrolipophagy is also under major transcriptional control. The most notable transcription factors are the members of the microphthalmia family of basic helix-loop-helix – leucine-zipper transcription factors family of transcription factors, which includes TFEB (transcription factor EB) and TFE3 (transcription factor binding to IGHM enhancer 3). Both TFEB and TFE3 are master regulators of lysosomal function and macroautophagy [122,123], but are also crucial in lipid catabolism. TFEB directly upregulates the expression level of PPARGC1A/PGC1α (PPARG coactivator 1 alpha), a central player in lipid metabolism during liver starvation responses. PPARGC1A, but also TFEB, regulate lipid metabolism in the liver through the downstream nuclear receptor PPARA (peroxisome proliferator activated receptor, alpha) [124]. The effect of TFEB on lipid metabolism requires macrolipophagy since TFEB overexpression in mice fed with a high-fat diet (HFD) and in which autophagy is specifically suppressed in the liver by Atg7 deletion, does not rescue the observed increase in liver lipid content whereas it does in the wild type mice [124]. In contrast, TFE3 alleviates FFA exposure-mediated hepatocellular steatosis through macrolipophagy and PPARGC1A-mediated FFA β-oxidation, since inhibiting these pathways using small interfering RNA against Atg5 and PPARGC1A, respectively, dramatically reduces TFE3-mediated alleviation of hepatocellular steatosis [125]. Another transcription factor family involved in macrolipophagy regulation is the FOXO (forkhead box O) family. FOXO1 modulates lipid metabolism not only by inducing expression of PNPLA2 in adipocytes [126], but also by upregulating LIPA and inducing macrolipophagy upon nutrient depletion [127]. In addition, simultaneous depletion of FOX1, FOX3 and FOX4 in the liver leads to steatosis and elevated levels of TAGs in mice [128], a phenotype that can be reversed upon overexpression of Atg14 [129]. Finally, CREB1/CREB (cAMP responsive element binding protein 1) has been shown to also promote macrolipophagy upon nutrient deprival, while fed-state sensing NR1H4/FXR (nuclear receptor subfamily 1 group H member 4) inhibits this response [130]. Mechanistically, CREB1 upregulates key Atg genes, including Atg7, ULK1 and TFEB, by recruiting the coactivator CRTC2 (CREB regulated transcription coactivator 2). In presence of nutrients, NR1H4 trans-repressed these genes by disrupting the functional CREB1-CRTC2 complex [130]. A separate study showed that PPARA and NR1H4 compete for the same DNA binding sites in the ATG gene promoters, with opposite transcriptional outputs and starvation-induced PPARA activation suppresses NR1H4-mediated inhibition of macrolipophagy [131]. Taken together, these studies suggest that transcription factors and co-activators induced by fasting, such as TFEB, TFE3, PPARA, PPARGC1A, FOXO1, FOXO3, FOXO4 and CREB, promote macrolipophagy, whereas nutrient-induced transcription factors like NR1H4, suppress macrolipophagy.
Microlipophagy
A series of recent studies revealed that turnover of LDs can also be mediated by microlipophagy (Figure 2B). Indeed, in yeast lipophagy appears to actually not involve a macroautophagic process, at least in the tested conditions, but rather proceed through the direct engulfment of LDs by vacuoles [84,132,133]. The molecular requirements for yeast microlipophagy, however, are contradictory.
There are studies reporting that microlipophagy induced by nitrogen starvation, gradual glucose depletion or acute glucose deprivation, requires the core ATG machinery, even though this process presents microautophagic morphological features and proceeds in the absence of autophagosomes [24,84,133,134]. These works, however, obtained contrasting results regarding the implication of specific Atg proteins known to be exclusively involved in selective types of autophagy [84,133]. There have also been studies reporting that microlipophagy does not require core Atg proteins [132,135]. In particular, Atg7-independent microlipophagy triggered by acute lipid imbalance resulting from phosphatidylcholine biosynthesis defects and ER stress, involves Atg39, a reticulophagy receptor [136], and Vps24 (vacuolar protein sorting 24), an ESCRT component [132] (Figure 2B). The existence of a core ATG protein-independent and ESCRT-dependent microlipophagy was independently confirmed in a study showing that in the absence of another ESCRT component Vps27, microlipophagy is compromised during gradual nutrient depletion [135] (Figure 2B).
Yeast microlipophagy also relies on local changes in the vacuolar membrane. Gradual nutrient depletion-induced microlipophagy was first shown to be mediated by liquid-ordered raft-like sterol-enriched vacuolar microdomains, whose formation and integrity are critical for LDs translocation into the vacuole [133]. Subsequently, it was revealed that the yeast NPC (NPC intracellular cholesterol transporter) protein orthologs, Ncr1 and Npc2, are essential for both the formation of these raft-like vacuolar microdomains and LDs engulfment by vacuoles through microlipophagy, during both gradual nutrient depletion and nitrogen starvation [134] (Figure 2B). Mammalian NPC proteins, NPC1 and NPC2, bind the cholesterol derived from endocytosed lipoproteins and they transport this lipid from the lumen to the limiting membrane of lysosomes [137]. Thus, it is likely that NPC proteins promote microlipophagy by increasing the sterol in the limiting membrane of the vacuole [134]. The formation of these raft-like vacuolar microdomains upon nitrogen starvation depends on at least two core Atg components, Atg7 and Atg8 [134] (Figure 2B). This finding could imply an indirect involvement of macroautophagy in microlipophagy, as delivery of organelles and thus lipids by autophagosomes will supply the amounts of sterols necessary for the formation of the raft-like vacuolar microdomains. Intriguingly, the core ATG machinery component Atg14, translocates from ER exit sites onto sterol enriched raft-like vacuolar microdomains in response to acute glucose starvation that leads to PRKA activation, and there, together with Vps30/Atg6, facilitates microlipophagy initiation, including vacuole docking and internalization of LDs [24] (Figure 2B). Cells that cannot activate PRKA or lack either Atg14 or Vps30/Atg6, do not deliver LDs into the vacuole and fail to thrive under acute glucose starvation [24]. Interestingly, atg14Δ and vps30/atg6Δ knockout strains form little or no raft-like vacuolar domains, suggesting that these two proteins and possibly the PtdIns3K complex that are part of, play a key role in the remodeling of the vacuolar membrane under nutrient deprivation conditions [138] (Figure 2B). Ypt7, the yeast ortholog of RAB7, also associates with LDs and a recent study has revealed its involvement in the regulation of LD dynamics and membrane trafficking between the vacuole and LDs [139]. In the absence of Ypt7, FFA content and the number of LDs drastically increase when cells are deprived of nutrients [139]. It is well known that in the absence of Ypt7, fusion processes with the vacuole are impaired [140,141], including the one involving autophagosomes [142]. Thus, LD accumulation could be the consequence of an impairment of the vacuolar functions. However, it has also been observed by electron microscopy that LD morphology is significantly altered and the fusion between LDs appears to be blocked, revealing a key role for Ypt7 in LD dynamics as well [139].
According to the current evidence, different conditions lead to the induction of microlipophagy in yeast, and it appears that among few mechanisms of microlipophagy, some Atg machinery-dependent, exist. This is in line with the current view that there are at least three types of microautophagy, two taking place at the vacuoles/lysosomes and one at the endosomes [28]. Future studies are needed to unveil the precise mechanisms underlying microlipophagy and their regulation.
Chaperone-mediated autophagy and the degradation of lipid droplet surface proteins
Although CMA targets only proteins and not organelles such as LDs, several findings support a role for CMA in the catabolism of LDs by selectively breaking down proteins present on their surface [49,143]. In particular, PLIN2 and PLIN3, are the earliest LD-anchored proteins reported to be CMA substrates [49] (Figure 2C). Expression of CMA-resistant PLIN proteins, or CMA inhibition, e.g., in mouse liver, reduces the association of the neutral lipase PNPLA2 and core ATG proteins with LDs, which in turn causes an accumulation of LDs and a decrease in lipid β-oxidation [49]. Therefore, it is believed that CMA-mediated degradation of PLIN2 and PLIN3 stimulates macrolipophagy by promoting the recruitment of PNPLA2 and macrolipophagy machinery components on LDs. A follow-up study showed that PRKA phosphorylates PLIN2, an event that is required for CMA-mediated degradation of this protein [143]. More recently, it has been shown that PLIN5 is also a CMA substrate (Figure 2C) and that LD breakdown is impaired when CMA-mediated PLIN5 degradation is inhibited by LAMP2A depletion in mouse livers and HepG2 cells [144]. Overall, these studies present CMA-mediated degradation of PLIN proteins as an upstream regulatory event that is critical for both neutral lipolysis and induction of macrolipophagy, highlighting the important role of selective autophagy in the control of LD catabolism and the crosstalk between neutral lipolysis and lipophagy.
Dysregulation of autophagy and lipid droplet function in nonalcoholic steatohepatitis
The regulation of lipid metabolism by autophagy is critical for cellular and organismal homeostasis. Defects in autophagy and lipid metabolism are associated with a diverse set of human metabolic diseases, including liver diseases. In liver, the process of autophagy has been studied since the 1960’s and a large body of evidence identified hepatic macroautophagy as an important intrinsic mechanism relevant to human liver diseases [145,146], such as HCC and nonalcoholic fatty liver disease (NAFLD). In this context, impaired lipophagy has been discussed as a risk factor for NASH, a type of FLD considered an increasing threat to human health worldwide [147,148]. Impaired lipophagy has also been found to be associated with disease progression in NAFLD patients [149]. Hence, lipophagy regulators such as FGF21 and the members of the FOXO and TFEB protein families (see section 3.1.4) are considered as possible targets for future rational therapies that may combat NASH through the modulation of macroautophagy [150–154]. In the following, we discuss insights into the functional relationship between autophagy, macrolipophagy and NASH.
Nonalcoholic steatohepatitis
NASH is a metabolic disease of the liver characterized by hepatic lipid accumulation (steatosis) and inflammation [155]. NASH is a more progressive and severe stage of NAFLD and is often accompanied by fibrosis (Figure 3) [155]. NASH is associated with obesity, insulin resistance, hyperlipidemia and the metabolic syndrome [156] and a diagnosis of NASH is indicative for the need of a liver transplant later in life, due to the likelihood of liver failure or HCC development [157]. In general, steatosis is a major human health concern with approximately 25% of the world population suffering from NAFLD, and in the United States of America NAFLD and NASH affecting the 30% and 5% of the population, respectively [158].
Figure 3.
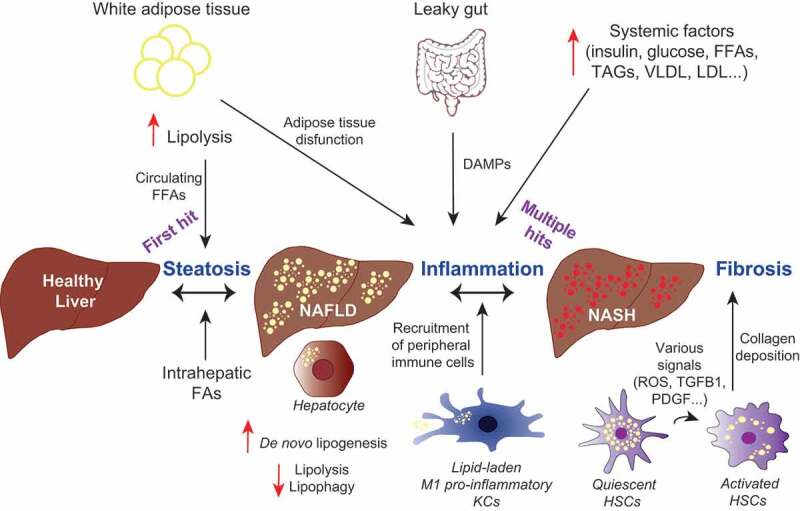
Dysregulated lipid metabolism plays a role in the development of NASH. Altered lipophagy, lipolysis and de novo lipogenesis in the liver contribute to NASH development through fat accumulation, and subsequent inflammation and fibrosis. A “first hit” of increased FFAs leads to accumulation of LDs in hepatocytes, a condition known as NAFLD. Additional “hits”, including inflammation and insulin resistance contribute to steatohepatitis (NASH), characterized by hepatocyte ballooning, lobular inflammation and fibrosis. Liver-resident macrophages (KCs) may acquire a pro-inflammatory phenotype due to the excess hepatic fat burden and mediate the inflammatory response through the recruitment of immune cells from the periphery. In response to hepatic injury, normally quiescent HSCs become activated by various signals. Enhanced lipophagy of vitamin A-storing LDs provides energy for HSCs activation, leading to hepatic fibrosis through HSCs-mediated collagen deposition.
The pathophysiology of NASH is complex and involves genetic, lifestyle and environmental factors [157]. The progression from NAFLD to NASH (Figure 3), and potentially liver failure or HCC [159], is non-linear, partially reversible, and may progress slowly or quickly over several decades [160] or it may not progress at all [156]. Accumulation of LDs within hepatocytes occurs as a result of a dysregulated lipid metabolism, which is closely associated with a metabolic syndrome whose features include obesity, insulin resistance, dyslipidemia and hypertension [156]. Histological features of NASH include steatosis, hepatocyte ballooning, lobular inflammation and sometimes fibrosis [157].
Two hypotheses have been made to explain the pathogenesis of NASH. The two-hit hypothesis considers lipid accumulation in hepatocytes to be regarded as the first hit, which precedes and sensitizes the liver to a second hit, characterized by mitochondrial dysfunction, oxidative stress, and release of inflammatory cytokines and adipokines [161]. The less simplistic multiple-hit hypothesis contemplates multiple parallel hits leading to NAFLD and NASH. Hepatic fat accumulation, insulin resistance, mitochondrial dysfunction, ER stress, gut microbiota, endotoxins, genetic and epigenetic factors as well as an altered metabolic crosstalk between different organs are taken into account [162].
NASH mouse models
Several mouse models of NASH have been developed and they can be categorized into diet-, genetic- and chemically-induced models (Table 1); each presenting its own advantages and disadvantages [163]. At present, there is no ideal mouse model to study the complexity of NASH observed in patients [163], as each currently available mouse model present differences in regard to histology, altered molecular pathways and genetics [164]. Often, the mouse model is chosen depending on the disease stage and disease aspect that will be experimentally addressed.
Table 1.
Mouse models of NASH
| Model | Mechanism | Pathophysiological characteristics | Suitability for NASH modeling | Autophagy/lipophagy assessments |
|---|---|---|---|---|
| Diet-induced models | ||||
| HFD | High fat intake (60% fat) leading to hepatic steatosis |
|
|
|
| MCD | High sucrose (40%), high fat (10%) and no methionine and choline |
|
|
|
| Choline-deficient diet | Similar to MCD, but without choline. Proteins are replaced by equal amounts of L-amino acids |
|
|
Not analyzed |
| Fructose diet | High-fructose added to a diet high in fat and cholesterol |
|
|
Not analyzed |
| FFC diet [260] | High saturated fat, high fructose and high cholesterol |
|
|
|
| Genetic models | ||||
| The lep−/-knockout mouse | Overeating animals because of a mutation in the Lep gene |
|
|
|
| The lpr−/-knockout mouse | Animal overeating because of a mutation in the Lpr gene |
|
|
|
| SREBF1/SREBP1c mouse | SREBF1/SREBP1c overexpression leads to increased DNL |
|
|
Not analyzed |
| The pten−/- null mouse | Pten is a tumor suppressor gene |
|
|
Not analyzed |
| Chemically-induced models | ||||
| CCl4 (together with HFD or a choline-deficient diet) | CCl4 causes acute hepatic injury and tissue remodeling through ROS production |
|
|
|
| Streptozocin (together with HFD or a choline-deficient diet) | Streptozotocin destroys pancreatic beta cells, leading to diabetes development |
|
|
|
| DEN [267] (together with HFD or a choline-deficient diet) |
Diethylnitrosamine (DEN) is a carcinogenic reagent that causes oxidative stress |
|
|
|
Dietary mouse models partially mimic human diets that lead to the development of NAFLD, and animals are typically subject to a methionine-choline deficient (MCD) diet, a choline-deficient, a high-fat or a fructose diet (Table 1). Mice subjected to these diets display similarities to the histological and pathologic features of human NAFLD and NASH to different degrees [165]. Genetic NAFLD and NASH mouse models are often used in combination with one of the abovementioned diets, as genetic models alone only result in the development of NAFLD, not NASH [166]. Chemical mouse models, in contrast, are more frequently used to study more severe NASH, fibrosis, cirrhosis and HCC. Mouse models are commonly utilized in the screening of potential NASH therapeutics.
Autophagy and lipophagy have been investigated in some of these mouse models, and results suggest that they play important roles in the development and progression of NAFLD and NASH [167–169]. The studies conducted so far mainly show a block in the macroautophagic flux in NASH triggered in mice through diet, genetics or chemicals [167–169]. Evidence indicates that nonselective macroautophagy, macrolipophagy and CMA are critically involved in the development of both NAFLD and NASH [49].
Lipid metabolism in NASH
The liver is a major site of lipid esterification and mitochondrial ß-oxidation [170]. FFAs from adipose tissue, diet and de novo lipogenesis are stored within LDs in hepatocytes. Dysregulated de novo lipogenesis (DNL), neutral lipolysis and macrolipophagy play a role in the development of NASH.
De novo lipogenesis in NASH
De novo lipogenesis contributes to only 5% of lipids in healthy individuals compared with 26% in NASH [171]. FFAs derived from DNL are either stored within LDs as TAG or secreted from the liver as very-low density lipoprotein (VLDL). Various studies in both mice and humans have demonstrated that increased hepatic DNL is associated with NAFLD, even when fasting [171,172]. DNL is regulated by two transcription factors: SREBF1/SREBP1c (sterol regulatory element binding transcription factor 1) and MLXIPL/ChREBP (MLX interacting protein like). These two transcription factors are activated by insulin and carbohydrates, respectively [173], leading to the expression of genes involved in FFA and TAG synthesis. Evidence indicates that altered expression of SREBF1 or MLXIPL contributes to NAFLD through increased DNL [174]. For example, a two-fold increase in SREBF1 expression has been found in NAFLD patient livers [172,175], while expression decreased with advanced NASH [176]. Expression of the lipogenic enzymes ACAC/ACC (acetyl-CoA carboxylase) 1 and FASN/FAS (fatty acid synthase), which are regulated by SREBF1, were also enhanced in NAFLD patients [172]. Severe NASH, i.e., fibrosis at stage 3–4, also termed burnt-out NASH, is associated with the loss of hepatic steatosis as well as other histological features of NASH [177]. Fat accumulation and SREBF1 expression have been negatively correlated with NASH progression and fibrosis stage [176], while the expression of DNL-associated genes regulated by SREBF1 such as FASN, ACAC1, ACAC2, DGAT1 and DGAT2 was reduced. The pathophysiological implications of SREBF1 reduction and reduced hepatic fat accumulation in severe NASH are currently unknown.
Furthermore, MLXIPL deficiency reduces DNL by decreasing the levels of key lipogenic enzymes, which results in a 65% reduction of the hepatic FFA biosynthetic rates [178]. Mice subjected to a high fructose diet were protected from hepatic steatosis when lacking MLXIPL, though they were more susceptible to liver damage due to increased cholesterol synthesis [179]. Conversely, MLXIPL overexpression in HFD fed mice caused increased DNL and hepatic TAG accumulation, while also displaying normal insulin levels and insulin signaling [180]. In liver biopsies from NASH patients, high MLXIPL expression correlated with steatosis and low MLXIPL expression with severe insulin resistance [180]. Altogether, these studies demonstrate that the expression of the lipogenic transcription factors SREBF1 and MLXIPL, which regulate DNL, is altered in NAFLD patients, but it is also stage dependent.
ACACs are regulated by SREBF1 and are the rate-limiting step in DNL [181]. Multiple studies have shown that ACAC inhibition reduces hepatic steatosis [159,182,183] and ACAC inhibitors are being investigated as a possible treatment for NASH [181]. Interestingly, ACAC inhibition reduced TGFB1/TGFβ (transforming growth factor beta 1)-stimulated hepatic stellate cells (HSC) activation and reduced fibrosis in a HFD diethylnitrosamine (DEN)-induced model, in rat choline-deficient diet and murine lep−/- NASH models [184] (Table 1). This study implicated a role of ACAC not only in hepatocyte TAG accumulation, but also in the activation of HSCs.
Lipolysis in NASH
Normally, circulating insulin prevents neutral lipolysis and promotes lipogenesis in adipose tissues after feeding [185]. Under starvation conditions, in contrast, catecholamine signaling promotes neutral lipolysis and FFAs are released for their redistribution to other organs to meet energy demands [185]. NASH is often accompanied by insulin resistance, which in turn increases neutral lipolysis in adipose tissues [186]. FFAs released in excess are transported to the liver where they contribute to lipid accumulation, leading to NAFLD [186]. Neutral lipolysis also occurs in hepatocytes. As mentioned, PNPLA2 is involved in the first step of neutral lipolysis, hence hydrolyzing TAGs into DAGs, and is highly expressed in adipose tissues and liver. A common single-nucleotide polymorphism in the PNPLA3 gene, a paralog of PNPLA2, increases the risk of developing NASH by three fold [187]. This isoleucine to methionine substitution at amino acid position 148 leads to a more pronounced sequestration of ABHD5/CGI-58 (abhydrolase domain containing 5) by PNPLA3, the shared co-factor of PNPLA2 and PNPLA3 [188], leading to a reduction of hepatic neutral lipolysis and an accumulation of fat in hepatocytes [189]. Studies in murine pnpla2−/- models support the notion of an association between reduced hepatic neutral lipolysis and hepatic steatosis. Mice lacking PNPLA2 have increased levels of TAGs in both adipose and non-adipose tissues [190]. In particular, the pnpla2−/- mice showed a 2.3-fold increase in hepatic TAG accumulation [190]. Another study showed elevated hepatic steatosis in pnpla2−/- mice with a 3-fold increase in hepatic TAG content [191]. Furthermore, adenoviral-mediated knockdown of hepatic PNPLA2 in mice led to steatosis regardless of whether the animals were fed with a HFD or standard chow [192]. Conversely, PNPLA2 overexpression in primary hepatocytes prevents lipid accumulation and augments TAG turnover [192].
Altogether, even if dysregulated neutral lipolysis appears to play a role in the development of NASH, data obtained from pnpla2−/- cells or mice should be interpreted with caution knowing that PNPLA2 could act as a macrolipophagy receptor.
Lipophagy in NASH
Hepatocyte macrolipophagy in NASH
ATG proteins have been found to be dysregulated in the livers of NASH mouse models. In genetic and dietary-induced NASH models, macroautophagy was found to be downregulated in the liver, particularly by Atg7 expression downregulation. Conversely, Atg7 overexpression in lep−/- mice (Table 1) prevented accumulation of lipids in hepatocytes through the stimulation of macrolipophagy [193]. In a HFD NASH mouse model, Atg5, Atg12 and Atg16L1 mRNA expression was found reduced [194]. Inhibition of autophagy by Atg5 knockdown or 3-methyladenine in cultured rat hepatocytes, in combination with either FFAs supplementation or MCD medium, increased LD accumulation [83]. Altogether, these studies suggest that ATG proteins can be downregulated in NASH and that induction of macrolipophagy in hepatocytes through ATG overexpression can reduce lipid accumulation.
In contrast, other studies have challenged this notion and implicate autophagy deficiency to be protective against HFD-induced obesity and insulin resistance. For example, hepatocyte-specific Atg7 deficiency attenuated steatosis in mice on a HFD [195]. Furthermore, hepatocyte-specific RB1CC1/FIP200 (RB1 inducible coiled-coil 1) deficiency resulted in a decrease in liver TAGs and a decrease in LDs in fasted condition compared to hepatocyte autophagy efficient mice. However, Sirius Red staining and mRNA expression levels showed increased fibrosis and inflammation [196]. Additional studies also show autophagy deficiency through hepatocyte-specific Atg7 or Atg5 deletion to reduce hepatic LD accumulation and protect against steatosis, but in the context of starvation rather than models of NASH [53,197–199]. To date, many studies have shown autophagy deficiency to contribute to hepatic steatosis and induction of macrolipophagy in hepatocytes to be beneficial in overcoming lipid accumulation. Intriguingly, few studies show autophagy deficiency to protect against HFD-induced steatosis. The discrepancies in these studies may be due to the different knockouts of Atg genes used. Further studies on how Atg genes participate in lipid metabolism might shed more light on the role of hepatocyte autophagy in the context of NAFLD/NASH.
A recent study highlighted the association of impaired autophagy with advanced disease in NAFLD patients. The number of autophagosomes and lipid-laden lysosomes (lipolysosomes) in liver increased with higher NAFLD activity score (NAS), while SQSTM1 correlated with NAS and fibrosis stage, indicating autophagy blockage [149]. Accumulation of SQSTM1 aggregates in hepatocytes of NAFLD patients has been confirmed in another study, where it also correlated with the number of autophagic vesicles, NAS and fibrosis [200]. The mechanism by which excess lipid accumulation may interfere with autophagy functions has been investigated in mouse studies. Electron microscopy examination of liver sections from mice on a HFD revealed a significant decrease in LD-containing macroautophagic intermediates, suggesting an impairment in lipophagy. Immunoblot analysis of isolated LDs showed that the association of LC3-II with LDs in HFD-mice was more prominent in the fed rather than the fasted state [83]. This apparent macrolipophagy defect caused by an excess of lipids may be due to the altered lipid composition of autophagosomes and/or lysosomes, which in turn affects their fusion [201]. This scenario indicates a harmful cycle between lipid accumulation and impaired autophagy.
The defects in autophagy caused by an excess nutrient supply may be an indirect result of altered kinase pathways, acting upstream of autophagy. For example, western diet-induced NAFLD in mice increased the expression of the mixed lineage kinase domain-like pseudokinase, a mediator of necroptotic cell death, and resulted in liver injury and inflammatory responses through inhibition of autophagy. Another kinase, the MAP3K5/ASK1 (mitogen-activated protein kinase kinase kinase 5) is an upstream regulator of the MAPK8/JNK (mitogen-activated protein kinase 8) and MAPK14/p38 (mitogen-activated protein kinase 8) MAPK signaling cascades [202] and it was discovered that in human livers MAP3K5 expression negatively correlates with hepatic lipid accumulation and NASH scores, and positively correlates with ATG5, ATG7 and ATG12 levels, suggesting a macroautophagy-linked protective role of hepatic MAP3K5 against NASH and fibrosis onset [203]. However, other studies have implicated MAP3K5 in NASH development, whereby MAP3K5 is overactive in NAFLD, leading to inflammation and fibrosis through increased MAPK8 and MAPK14 MAPK signaling [204]. Furthermore, MAP3K5 inhibition reduces hepatic lipid accumulation, inflammation and fibrosis, supporting a negative role of MAP3K5 in NASH [205,206]. Although MAP3K5 is implicated in NASH, it remains unclear whether it has a negative or positive effect on NASH progression.
Hepatic stellate cell macrolipophagy in NASH
HSCs are the major player in hepatic fibrosis [207,208]. In their quiescent state, HSCs are a major store of vitamin A, which is stored within their LDs [209]. Upon liver injury and in response to hepatocyte- and Kupffer cell (KC)-derived cytokines and CCL/chemokines (chemokine ligands) or other stimuli [208], HSCs undergo a transition to become myofibroblast-like [210]. Once activated, vitamin A stores are depleted through macrolipophagy [211]. Importantly, activated HSCs contribute to NASH progression though the deposition of type I and III collagen; leading to the formation of fibrotic tissues [209].
Evidence suggests a role for autophagy in the activation of quiescent HSCs in NASH. Increased macroautophagic flux has been demonstrated in activated HSCs, through the observation of increased LC3-II and decreased SQSTM1 protein levels [211,212]. Pharmacological inhibition of macroautophagy with chloroquine or 3-methyladenine, as well as small interfering RNA-mediated Atg5 or Atg7 knockdown, reduced the mRNA expression of fibrosis-associated genes in murine HSCs [212]. Moreover, mice with autophagy-deficient HSCs displayed attenuated liver fibrosis, as shown by a reduction of α-smooth muscle actin expression. The ß-oxidation of FFAs produced by macrolipophagy is required for HSC activation [212] as exogenous oleic acid allows macroautophagy-deficient HSCs to become activated, reacquiring the fibrotic phenotype [212]. Additional studies have shown that HSC activation is either increased or decreased through the induction or inhibition of macrolipophagy, respectively [213–217]. Recent studies unraveled a role of lipopolysaccharide (LPS)-associated activation of HSCs via macrolipophagy. Systemic LPS levels are elevated in human NAFLD because of the disrupted gut barrier, and is one of the factors that contributes to NASH progression through toll-like receptor 4-mediated activation of KCs and HSCs [218,219]. LPS was found to induce macrolipophagy in the murine LX-2 HSC cell line, while pharmacological inhibition of autophagy or Atg5 silencing reduced LDs decrease upon LPS-treatment [220]. Reactive oxygen species (ROS) levels are elevated in NASH patients [221] and have previously been linked to macroautophagy induction in response to chemotherapy [222]. ROS content of HSCs increased over a 7-day period and was abrogated upon treatment with ROS scavengers glutathione and N-acetyl-cysteine. Glutathione and N-acetyl-cysteine impaired autophagosome formation and macroautophagic flux, as well as binding between PtdIns3K complex and RAB25. Thus, the augmentation of ROS levels during HSC activation contributes to LD disappearance through increased PtdIns3K-RAB25 interaction and enhanced macrolipophagy, the effects of which can be avoided by ROS scavengers [223]. These studies implicate both ROS and LPS in the activation of HSCs through macrolipophagy.
All this compelling evidence shows that macrolipophagy leads to the activation of HSCs and subsequent fibrosis, rather than playing a protective role against NASH.
Hepatic macrophage macrolipophagy in NASH
Macroautophagy has a role in macrophage polarization toward their anti-inflammatory M2 type [224]. In mice fed with a HFD for 12 weeks, macroautophagic flux was decreased in peritoneal and bone-marrow derived macrophages (BMDMs) [224]. Mouse bone-marrow derived macrophages, but also Atg5-deficient KCs, showed polarization anomalies with an increase in the pro-inflammatory M1 and a decrease in the anti-inflammatory M2 population [225]. While HFD and LPS were not sufficient to cause inflammation in wild type mice, the specific ablation of macroautophagy in macrophages promoted a pro-inflammatory macrophage phenotype in the liver of animals, without altering macrophage number and steatosis [225]. Myeloid cell-specific atg5−/- deficient mice that were subjected to repeated injections of carbon tetrachloride (CCl4) to induce liver injury, had increased hepatic levels of IL1A/IL1-α (interleukin 1 alpha) and IL1B/IL1-β (interleukin 1 beta), enhanced infiltration of leucocytes in the liver and hepatocyte apoptosis, as well as exacerbation of liver fibrosis [224]. This result highlights the protective, anti-inflammatory role of macrophage basal autophagy in liver injury and fibrosis [224].
In NAFLD, KCs acquire a pro-inflammatory phenotype following their activation by danger/damage-associated molecular patterns (DAMPs) and other factors released by increased hepatic fat burden. Alternatively, KCs may become activated due to excess lipid accumulation and defective lipid processing [226]. In biopsies from NASH, but not NAFLD patients, hepatic macrophages form crown-like aggregates around LDs derived from dead hepatocytes, and these structures are similar to those found in adipose tissue in obese patients [227]. In an animal model of early NASH induced by a HFD for 16 weeks, KCs had a distinct phenotype, characterized by an accumulation of LDs, a dysregulation of lipid metabolism at the gene level [228] and an increase of cholesterol, ceramides and DAGs [228]. Importantly, enhanced lipid accumulation in KCs from HFD animals was accompanied by a pro-inflammatory phenotype, as they secreted more IFNA1/IFN-α (interferon alpha 1), TNF/TNF- α (tumor necrosis factor), IL10/CSIF (interleukin 10), CCL2/MCP1 (C-C motif chemokine ligand 2), and CCL5/RANTES (C-C motif chemokine ligand 5). Inhibition of lipogenesis in these KCs by 5-tetradecyloxy-2-furoic acid could reverse this pro-inflammatory phenotype. Taken together, accumulation of LDs in KCs influences their polarization toward a pro-inflammatory phenotype, which is a key event in the transition from NAFLD to NASH and fibrosis. Nevertheless, evidence suggests that fat-laden M1 macrophages may aid in the resolution of inflammation in advanced NASH, by expression of anti-inflammatory mediators such as ANNEXIN A1 [229]. Nonetheless, the relevance of using the M1/M2 macrophage classification is debatable; since M1 and M2 macrophages may represent the same subset of plastic macrophages during the course of disease, making it difficult to draw firm conclusions [226]. Thus, more studies are needed to elucidate the exact role of lipid dysregulation in the function of different hepatic macrophage subsets in the context of NASH assisting with the resolution of acute inflammation and the initiation of tissue healing through the stimulation of phagocytosis of apoptotic bodies [229]. Notably, apart from KCs being the main drivers of inflammation in NASH, they may also directly drive steatosis development, as KC depletion in rats has been shown to prevent HFD-induced steatosis [230]. In further support of this notion, in a HFD NAFLD model, LPS or FFA-driven activation of KCs mediated steatosis formation in hepatocytes via tumor necrosis factor signaling [231].
Liver sinusoidal endothelial cell macrolipophagy in NASH
Although much less studied compared to the other major liver cells types, liver sinusoidal endothelial cells (LSECs) appear to play a role in NASH pathogenesis. Changes in LSECs have been observed in the HFD and choline-deficient mouse models, where KCs and HSCs were activated, leading to a progression from simple steatosis to early NASH [232]. A recent study showed that macroautophagy is altered in LSECs from NASH patients [233]. In particular, the number of autophagosomes in LSECs from NASH patient liver biopsies was half of that in the samples from patients with no liver abnormalities or simple steatosis [233]. Primary LSECs specifically lacking Atg5 isolated from animals on a HFD, had major changes in inflammatory pathways at the gene expression level and a modest upregulation of angiogenic pathways [233]. The same animals exhibited increased fibrosis, mainly in the perisinusoidal area, higher markers of liver injury and liver inflammation, accompanied by an infiltration of inflammatory cells [233]. This data indicates that LSEC macroautophagy has a protective role in the context of NAFLD and NASH. In another study, LSECs from rats with CCl4-induced fibrosis displayed an augmented macroautophagic flux, which did not increase further in a more advanced fibrosis stage (6 weeks of CCl4 administration) [234]. This observation suggests a role for LSEC macroautophagy during early phases of liver injury [234]. Mildly fibrotic mice (1 week of CCl4 injections) with an LSEC-specific deletion of Atg7 exhibited endothelial dysfunction and an aggravation of fibrosis, due to activation of HSCs [234]. Overall, LSEC macroautophagy appears to elicit a protective effect in the context of NASH and liver injury, yet no studies have explored the role of LSEC macrolipophagy in this context.
Macrolipophagy in non-liver tissues over the course of NASH
Transition from simple steatosis to NASH can be triggered by intrahepatic and extrahepatic events (Figure 3). Extrahepatic events include adipose tissue dysfunction, intestinal dysbiosis and insulin resistance [155]. A recent study has highlighted the link between adipose tissue dysfunction and NASH pathogenesis [235]. Mice were subjected to a modified HFD and developed NAFLD or NASH of different severities, based on liver histology, alanine aminotransferase serum levels and hepatic TAG levels [235]. Gene expression profiling in liver by microarrays showed that among the mice who had the most severe histological features resembling NASH, there was a significant enhanced expression in genes involved in lipid metabolism, inflammation and fibrosis [235]. Additionally, these animals exhibited both high weight gain and high LEP/OB (leptin) levels in serum, and showed severe adipose tissue dysfunction and remodeling [235]. Adipose tissue dysfunction may thus promote progression of hepatic steatosis to NASH. Studies using adipose tissue from obese and non-obese patients showed that obesity is probably associated with an activation of autophagic flux in the visceral adipose tissue [236,237]. Direct evidence linking adipose tissue dysfunction, impaired lipid metabolism and NASH is missing, yet autophagy has a major role in adipocyte maturation via macrolipophagy and lipolysis-dependent mechanisms [238].
Increased lipolysis of the peripheral fat stored in adipose tissues reaches the liver as non-esterified FFAs, enhancing lipogenesis, reducing β-oxidation and downregulating the export of fat in the form of VLDL, thereby contributing to steatosis [239]. Increased plasma FFA levels result in an increase of intramyocellular lipids, which in turn leads to an impairment of insulin signaling, causing insulin resistance in the skeletal muscles of healthy subjects [240]. The size but not the number of individual subsarcolemmal LDs in human skeletal muscle fibers is associated with insulin resistance [241], but a direct association between LDs in skeletal muscle in the context of NASH is missing.
Outlook
LDs are dynamic cellular organelles that consist of a neutral lipid core which is surrounded by a phospholipid monolayer and LD-associated proteins, particularly of the PLIN protein family. LDs serve as platforms for energy storage and have multiple functions in lipid and energy homeostasis, membrane and hormone biosynthesis, cell signaling and differentiation. LDs are implicated in various metabolic diseases, such as obesity, atherosclerosis and FLD [242]. The latter is the world’s most common liver disease, characterized by excessive fat in the liver and either related to alcohol consumption or not. In this context, NASH is a subtype of FLD, which is also defined by the accumulation of liver fat and characterized by liver inflammation.
Intracellularly, fat is stored in LDs and there is a complex relationship between LDs and autophagy (Figure 1), as i) the autophagic machinery appears to be necessary for the formation of LDs, ii) LDs support the formation of autophagosomes, and especially iii) the autophagic machinery selectively breaks down LDs through (macro)lipophagy processes (Figure 2). In the context of FLD, an enhancement of lipophagic activity in the liver, possibly combined with drugs that act on the lipid biosynthetic pathways, could be viewed as a possible future therapeutic strategy to reduce the symptoms of FLD and stop its progression to NASH (Figure 3). However, as discussed in this review, autophagy modulation should be liver cell type-specific, since enhancement of autophagic activity may prove beneficial in hepatocytes, KCs and endothelial cells, while in HSCs it can lead to fibrosis [243].
In recent studies, strategies to increase autophagy, including exercise and dietary interventions [118,244,245], as well as the use of substances that modulate autophagy, including rapamycin [246], a novel TFEB inducer [154], herbal medicines [205,247] and marketed, FDA-approved drugs such as metformin [248], ezetimibe [151], irbesartan [249], pioglitazone [250] and exenatide [251], have shown that they counteract NASH in different animal models. Hence, stimulation of autophagy consistently resulted in improvements in NASH, demonstrated by decreased hepatic steatosis, inflammation, and fibrosis [118,151,154,205,244–251]. In this context, a new compound called MSL is of particular interest as it stimulates autophagy while acting on the TFEB pathway, and enhances the autophagic flux and the breakdown of LDs in the liver of lep−/- mice as well as HFD-fed mice [154].
Even though the use of compounds that improve NASH by induction of autophagy is an extremely attractive hypothesis, it needs further investigation as potential therapeutic benefits could actually be traced back to modulation of additional pathways since none of these strategies act solely on autophagy [252]. In addition, the targeted modulation of autophagy in certain cells or tissues is a formidable challenge, as our molecular understanding of how autophagy is differentially affected in certain cell types of an organ, such as the liver, is limited. For this understanding, the elucidation of the molecular players that discriminate between autophagy and closely related mechanisms such as LC3-associated endocytosis or LC3-associated phagocytosis, is essential for the discovery of autophagy-selective therapeutic windows [253]. The development of small molecules that specifically modulate selective forms of autophagy, such as lipophagy in the liver, will be essential to understand the molecular mechanisms of lipophagy and pave the way for effective therapies for NASH and other human liver diseases.
Acknowledgments
All the authors are part of DRIVE consortium, which is supported by a Marie Skłodowska-Curie ETN grant under the European Union’s Horizon 2020 Research and Innovation Programme (Grant Agreement No 765912). N.D. is also supported by the Agence Nationale pour la recherche (ANR) (R18176KK), CNRS and University de Paris. T.P.-C. is also funded by the Deutsche Forschungsgemeinschaft (DFG, German Research Foundation) under Germany’s Excellence Strategy Project-ID 390900677 - EXC 2180 (project 10076-1), DFG Project-ID 31490540 - TRR 209 (project B02), DFG Project-ID 323732846 - FOR 2625 (project 1), DFG Project-ID 259130777 - SFB 1177 (project E03). F.R. is also supported by ALW Open Programme (ALWOP.310), ENW KLEIN-1 (OCENW.KLEIN.118) and ZonMW TOP (91217002) grants.
All authors declare no conflict of interest.
Funding Statement
This work was supported by the H2020 Marie Skłodowska-Curie Actions [765912]; H2020 Marie Skłodowska-Curie Actions [765912]; H2020 Marie Skłodowska-Curie Actions [765912]; H2020 Marie Skłodowska-Curie Actions [765912]; DFG [EXC 2180 – 390900677]; CNRS; ANR [R18176KK]; University de Paris; ALW [ALWOP.310]; ENW [OCENW.KLEIN.118]; DFG [SFB/TR 209]; DFG [FOR2625]; DFG [SFB 1177]; ZonMw [91217002].
Disclosure statement
No potential conflict of interest was reported by the authors.
References
- [1].Antonioli M, Di Rienzo M, Piacentini M, et al. Emerging mechanisms in initiating and terminating autophagy. Trends Biochem Sci. 2017;42(1):28–41. . [DOI] [PubMed] [Google Scholar]
- [2].Yu L, Chen Y, Tooze SA.. Autophagy pathway: Cellular and molecular mechanisms. Autophagy. 2018;14(2):207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Mizushima N, Yoshimori T, Ohsumi Y.. The role of Atg proteins in autophagosome formation. Annu Rev Cell Dev Biol. 2011;27(1):107–132. [DOI] [PubMed] [Google Scholar]
- [4].Klionsky DJ, Schulman BA. Dynamic regulation of macroautophagy by distinctive ubiquitin-like proteins. Nat Struct Mol Biol. 2014;21(4):336–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Tsukada M, Ohsumi Y. Isolation and characterization of autophagy-defective mutants of Saccharomyces cerevisiae. FEBS Lett. 1993;333(1–2):169–174. [DOI] [PubMed] [Google Scholar]
- [6].Thumm M, Egner R, Koch B, et al. Isolation of autophagocytosis mutants of Saccharomyces cerevisiae. FEBS Lett. 1994;349(2):275–280. . [DOI] [PubMed] [Google Scholar]
- [7].Harding TM, Morano KA, Scott SV, et al. Isolation and characterization of yeast mutants in the cytoplasm to vacuole protein targeting pathway. J Cell Biol. 1995;131(3):591–602. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Wong PM, Puente C, Ganley IG, et al. The ULK1 complex: sensing nutrient signals for autophagy activation. Autophagy. 2013;9(2):124–137. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Hosokawa N, Hara T, Kaizuka T, et al. Nutrient-dependent mTORC1 association with the ULK1-Atg13-FIP200 complex required for autophagy. Mol Biol Cell. 2009;20(7):1981–1991. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Nazio F, Cecconi F. mTOR, AMBRA1, and autophagy: an intricate relationship. Cell Cycle. 2013;12(16):2524–2525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Jung CH, Jun CB, Ro SH, et al. ULK-Atg13-FIP200 complexes mediate mTOR signaling to the autophagy machinery. Mol Biol Cell. 2009;20(7):1992–2003. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Axe EL, Walker SA, Manifava M, et al. Autophagosome formation from membrane compartments enriched in phosphatidylinositol 3-phosphate and dynamically connected to the endoplasmic reticulum. J Cell Biol. 2008;182(4):685–701. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Roberts R, Ktistakis NT. Omegasomes: PI3P platforms that manufacture autophagosomes. Essays Biochem. 2013;55:17–27. [DOI] [PubMed] [Google Scholar]
- [14].He C, Levine B. The Beclin 1 interactome. Curr Opin Cell Biol. 2010;22(2):140–149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Mercer TJ, Gubas A, Tooze SA. A molecular perspective of mammalian autophagosome biogenesis. J Biol Chem. 2018;293(15):5386–5395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Bakula D, Muller AJ, Zuleger T, et al. WIPI3 and WIPI4 beta-propellers are scaffolds for LKB1-AMPK-TSC signalling circuits in the control of autophagy. Nat Commun. 2017;8(1):15637. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Proikas-Cezanne T, Takacs Z, Donnes P, et al. WIPI proteins: essential PtdIns3P effectors at the nascent autophagosome. J Cell Sci. 2015;128(2):207–217. . [DOI] [PubMed] [Google Scholar]
- [18].Dooley HC, Razi M, Polson HE, et al. WIPI2 links LC3 conjugation with PI3P, autophagosome formation, and pathogen clearance by recruiting Atg12-5-16L1. Mol Cell. 2014;55(2):238–252. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Behrends C, Sowa ME, Gygi SP, et al. Network organization of the human autophagy system. Nature. 2010;466(7302):68–76. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Galluzzi L, Baehrecke EH, Ballabio A, et al. Molecular definitions of autophagy and related processes. Embo J. 2017;36(13):1811–1836. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Gatica D, Lahiri V, Klionsky DJ. Cargo recognition and degradation by selective autophagy. Nat Cell Biol. 2018;20(3):233–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Kirkin V, Rogov VV. A diversity of selective autophagy receptors determines the specificity of the autophagy pathway. Mol Cell. 2019;76(2):268–285. [DOI] [PubMed] [Google Scholar]
- [23].Farre JC, Subramani S. Mechanistic insights into selective autophagy pathways: lessons from yeast. Nat Rev Mol Cell Biol. 2016;17(9):537–552. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Seo AY, Lau PW, Feliciano D, et al. AMPK and vacuole-associated Atg14p orchestrate mu-lipophagy for energy production and long-term survival under glucose starvation. Elife. 2017;6:e21690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [25].Roberts P, Moshitch-Moshkovitz S, Kvam E, et al. Piecemeal microautophagy of nucleus in Saccharomyces cerevisiae. Mol Biol Cell. 2003;14(1):129–141. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Lemasters JJ. Variants of mitochondrial autophagy: types 1 and 2 mitophagy and micromitophagy (Type 3). Redox Biol. 2014;2:749–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Tekirdag K, Cuervo AM. Chaperone-mediated autophagy and endosomal microautophagy: joint by a chaperone. J Biol Chem. 2018;293(15):5414–5424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Oku M, Sakai Y. Three distinct types of microautophagy based on membrane dynamics and molecular machineries. Bioessays. 2018;40(6):e1800008. [DOI] [PubMed] [Google Scholar]
- [29].Sahu R, Kaushik S, Clement CC, et al. Microautophagy of cytosolic proteins by late endosomes. Dev Cell. 2011;20(1):131–139. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Mejlvang J, Olsvik H, Svenning S, et al. Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J Cell Biol. 2018;217(10):3640–3655. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Kaushik S, Cuervo AM. Chaperone-mediated autophagy: a unique way to enter the lysosome world. Trends Cell Biol. 2012;22(8):407–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Dice JF. Peptide sequences that target cytosolic proteins for lysosomal proteolysis. Trends Biochem Sci. 1990;15(8):305–309. [DOI] [PubMed] [Google Scholar]
- [33].Kaushik S, Cuervo AM. The coming of age of chaperone-mediated autophagy. Nat Rev Mol Cell Biol. 2018;19(6):365–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Cuervo AM, Dice JF. A receptor for the selective uptake and degradation of proteins by lysosomes. Science. 1996;273(5274):501–503. [DOI] [PubMed] [Google Scholar]
- [35].Bandyopadhyay U, Kaushik S, Varticovski L, et al. The chaperone-mediated autophagy receptor organizes in dynamic protein complexes at the lysosomal membrane. Mol Cell Biol. 2008;28(18):5747–5763. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Cuervo AM, Dice JF, Knecht E. A population of rat liver lysosomes responsible for the selective uptake and degradation of cytosolic proteins. J Biol Chem. 1997;272(9):5606–5615. [DOI] [PubMed] [Google Scholar]
- [37].Martin S, Parton RG. Lipid droplets: a unified view of a dynamic organelle. Nat Rev Mol Cell Biol. 2006;7(5):373–378. [DOI] [PubMed] [Google Scholar]
- [38].Olzmann JA, Carvalho P. Dynamics and functions of lipid droplets. Nat Rev Mol Cell Biol. 2019;20(3):137–155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Ward C, Martinez-Lopez N, Otten EG, et al. Autophagy, lipophagy and lysosomal lipid storage disorders. Biochim Biophys Acta. 2016;1861:269–284. [DOI] [PubMed] [Google Scholar]
- [40].Weidberg H, Shvets E, Elazar Z. Lipophagy: selective catabolism designed for lipids. Dev Cell. 2009;16(5):628–630. [DOI] [PubMed] [Google Scholar]
- [41].Ma Y, Zhou Y, Zhu YC, et al. Lipophagy contributes to testosterone biosynthesis in male rat Leydig cells. Endocrinology. 2018;159(2):1119–1129. . [DOI] [PubMed] [Google Scholar]
- [42].Cui W, Sathyanarayan A, Lopresti M, et al. Lipophagy-derived fatty acids undergo extracellular efflux via lysosomal exocytosis. Autophagy. 2020;19:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [43].Riffelmacher T, Clarke A, Richter FC, et al. Autophagy-dependent generation of free fatty acids is critical for normal neutrophil differentiation. Immunity 2017;47(3):466–480 e5. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Folick A, Oakley HD, Yu Y, et al. Aging. lysosomal signaling molecules regulate longevity in Caenorhabditis elegans. Science. 2015;347:(6217):83–86. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Natarajan SK, Rasineni K, Ganesan M, et al. Structure, function and metabolism of hepatic and adipose tissue lipid droplets: implications in alcoholic liver disease. Curr Mol Pharmacol. 2017;10(3):237–248. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Itabe H, Yamaguchi T, Nimura S, et al. Perilipins: a diversity of intracellular lipid droplet proteins. Lipids Health Dis. 2017;16(1):83. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [47].Trevino MB, Mazur-Hart D, Machida Y, et al. Liver perilipin 5 expression worsens hepatosteatosis but not insulin resistance in high fat-fed mice. Mol Endocrinol. 2015;29(10):1414–1425. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [48].Zhang E, Cui W, Lopresti M, et al. Hepatic PLIN5 signals via SIRT1 to promote autophagy and prevent inflammation during fasting. J Lipid Res. 2020;61(3):338–350. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Kaushik S, Cuervo AM. Degradation of lipid droplet-associated proteins by chaperone-mediated autophagy facilitates lipolysis. Nat Cell Biol. 2015;17(6):759–770. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Bolsoni-Lopes A, Alonso-Vale MI. Lipolysis and lipases in white adipose tissue - An update. Arch Endocrinol Metab. 2015;59(4):335–342. [DOI] [PubMed] [Google Scholar]
- [51].Dupont N, Chauhan S, Arko-Mensah J, et al. Neutral lipid stores and lipase PNPLA5 contribute to autophagosome biogenesis. Curr Biol. 2014;24(6):609–620. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [52].Velikkakath AK, Nishimura T, Oita E, et al. Mammalian Atg2 proteins are essential for autophagosome formation and important for regulation of size and distribution of lipid droplets. Mol Biol Cell. 2012;23(5):896–909. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Shibata M, Yoshimura K, Furuya N, et al. The MAP1-LC3 conjugation system is involved in lipid droplet formation. Biochem Biophys Res Commun. 2009;382(2):419–423. . [DOI] [PubMed] [Google Scholar]
- [54].Pfisterer SG, Bakula D, Frickey T, et al. Lipid droplet and early autophagosomal membrane targeting of Atg2A and Atg14L in human tumor cells. J Lipid Res. 2014;55(7):1267–1278. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Takeuchi K, Reue K. Biochemistry, physiology, and genetics of GPAT, AGPAT, and lipin enzymes in triglyceride synthesis. Am J Physiol Endocrinol Metab. 2009;296(6):E1195–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [56].Shi Y, Cheng D. Beyond triglyceride synthesis: the dynamic functional roles of MGAT and DGAT enzymes in energy metabolism. Am J Physiol Endocrinol Metab. 2009;297(1):E10–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Yu J, Loh K, Song ZY, et al. Update on glycerol-3-phosphate acyltransferases: the roles in the development of insulin resistance. Nutr Diabetes. 2018;8(1):34. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Thiam AR, Foret L. The physics of lipid droplet nucleation, growth and budding. Biochim Biophys Acta. 2016;1861(8):715–722. [DOI] [PubMed] [Google Scholar]
- [59].Jacquier N, Choudhary V, Mari M, et al. Lipid droplets are functionally connected to the endoplasmic reticulum in Saccharomyces cerevisiae [Research Support, Non-U.S. Gov’t]. J Cell Sci. 2011;124(14):2424–2437. . [DOI] [PubMed] [Google Scholar]
- [60].Kassan A, Herms A, Fernandez-Vidal A, et al. Acyl-CoA synthetase 3 promotes lipid droplet biogenesis in ER microdomains. J Cell Biol. 2013;203(6):985–1001. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Ben M’barek K, Ajjaji D, Chorlay A, et al. ER membrane phospholipids and surface tension control cellular lipid droplet formation. Dev Cell. 2017;41(6):591–604 e7. . [DOI] [PubMed] [Google Scholar]
- [62].Gao M, Huang X, Song BL, et al. The biogenesis of lipid droplets: Lipids take center stage. Prog Lipid Res. 2019;75:100989. [DOI] [PubMed] [Google Scholar]
- [63].Becuwe M, Bond LM, Pinto AFM, et al. FIT2 is an acyl-coenzyme A diphosphatase crucial for endoplasmic reticulum homeostasis. J Cell Biol. 2020Oct5 219;(10)e202006111. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Choudhary V, Golani G, Joshi AS, et al. Architecture of lipid droplets in endoplasmic reticulum is determined by phospholipid intrinsic curvature. Curr Biol. 2018;28(6):915–926 e9. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [65].Yan R, Qian H, Lukmantara I, et al. Human SEIPIN binds anionic phospholipids. Dev Cell. 2018;47(2):248–256 e4. . [DOI] [PubMed] [Google Scholar]
- [66].Fei W, Shui G, Gaeta B, et al. Fld1p, a functional homologue of human seipin, regulates the size of lipid droplets in yeast. J Cell Biol. 2008;180(3):473–482. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [67].Szymanski KM, Binns D, Bartz R, et al. The lipodystrophy protein seipin is found at endoplasmic reticulum lipid droplet junctions and is important for droplet morphology. Proc Natl Acad Sci U S A. 2007;104(52):20890–20895. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [68].Wang H, Becuwe M, Housden BE, et al. Seipin is required for converting nascent to mature lipid droplets. Elife 2016;5:e16582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [69].Wilfling F, Wang H, Haas JT, et al. Triacylglycerol synthesis enzymes mediate lipid droplet growth by relocalizing from the ER to lipid droplets. Dev Cell. 2013;24(4):384–399. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].Choudhary V, Ojha N, Golden A, et al. A conserved family of proteins facilitates nascent lipid droplet budding from the ER. J Cell Biol. 2015;211(2):261–271. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [71].Walther TC, Chung J, Farese RV Jr.. Lipid droplet biogenesis. Annu Rev Cell Dev Biol. 2017;33(1):491–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [72].Zechner R, Zimmermann R, Eichmann TO, et al. FAT SIGNALS–lipases and lipolysis in lipid metabolism and signaling. Cell Metab. 2012;15(3):279–291. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Rambold AS, Cohen S, Lippincott-Schwartz J. Fatty acid trafficking in starved cells: regulation by lipid droplet lipolysis, autophagy, and mitochondrial fusion dynamics. Dev Cell. 2015;32(6):678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Zehmer JK, Huang Y, Peng G, et al. A role for lipid droplets in inter-membrane lipid traffic. Proteomics. 2009;9(4):914–921. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Dirkx R, Vanhorebeek I, Martens K, et al. Absence of peroxisomes in mouse hepatocytes causes mitochondrial and ER abnormalities. Hepatology. 2005;41(4):868–878. . [DOI] [PubMed] [Google Scholar]
- [76].Binns D, Januszewski T, Chen Y, et al. An intimate collaboration between peroxisomes and lipid bodies. J Cell Biol. 2006;173(5):719–731. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Zhang H, Wang Y, Li J, et al. Proteome of skeletal muscle lipid droplet reveals association with mitochondria and apolipoprotein a-I. J Proteome Res. 2011;10(10):4757–4768. . [DOI] [PubMed] [Google Scholar]
- [78].Wang H, Sreenivasan U, Gong DW, et al. Cardiomyocyte-specific perilipin 5 overexpression leads to myocardial steatosis and modest cardiac dysfunction. J Lipid Res. 2013;54(4):953–965. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [79].Shpilka T, Welter E, Borovsky N, et al. Lipid droplets and their component triglycerides and steryl esters regulate autophagosome biogenesis. Embo J. 2015;34(16):2117–2131. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [80].Ogasawara Y, Cheng J, Tatematsu T, et al. Long-term autophagy is sustained by activation of CCTbeta3 on lipid droplets. Nat Commun. 2020;11(1):4480. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [81].Wang CW. Lipid droplets, lipophagy, and beyond. Biochim Biophys Acta. 2016;1861(8):793–805. [DOI] [PubMed] [Google Scholar]
- [82].Zechner R, Madeo F, Kratky D. Cytosolic lipolysis and lipophagy: two sides of the same coin. Nat Rev Mol Cell Biol. 2017;18(11):671–684. [DOI] [PubMed] [Google Scholar]
- [83].Singh R, Kaushik S, Wang Y, et al. Autophagy regulates lipid metabolism. Nature. 2009;458(7242):1131–1135. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Van Zutphen T, Todde V, De Boer R, et al. Lipid droplet autophagy in the yeast Saccharomyces cerevisiae. Mol Biol Cell. 2014;25(2):290–301. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Ouimet M, Franklin V, Mak E, et al. Autophagy regulates cholesterol efflux from macrophage foam cells via lysosomal acid lipase. Cell Metab. 2011;13(6):655–667. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Kaushik S, Arias E, Kwon H, et al. Loss of autophagy in hypothalamic POMC neurons impairs lipolysis. EMBO Rep. 2012;13(3):258–265. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Khaldoun SA, Emond-Boisjoly MA, Chateau D, et al. Autophagosomes contribute to intracellular lipid distribution in enterocytes. Mol Biol Cell. 2014;25(1):118–132. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [88].Cairo M, Villarroya J, Cereijo R, et al. Thermogenic activation represses autophagy in brown adipose tissue. Int J Obes (Lond). 2016;40(10):1591–1599. . [DOI] [PubMed] [Google Scholar]
- [89].Martinez-Lopez N, Garcia-Macia M, Sahu S, et al. Autophagy in the CNS and periphery coordinate lipophagy and lipolysis in the brown adipose tissue and liver. Cell Metab. 2016;23(1):113–127. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Lam T, Harmancey R, Vasquez H, et al. Reversal of intramyocellular lipid accumulation by lipophagy and a p62-mediated pathway. Cell Death Discov. 2016;2(1):16061. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Minami S, Yamamoto T, Takabatake Y, et al. Lipophagy maintains energy homeostasis in the kidney proximal tubule during prolonged starvation. Autophagy. 2017;13(10):1629–1647. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [92].Miceli C, Roccio F, Penalva-Mousset L, et al. The primary cilium and lipophagy translate mechanical forces to direct metabolic adaptation of kidney epithelial cells. Nat Cell Biol. 2020;22(9):1091–1102. . [DOI] [PubMed] [Google Scholar]
- [93].Kaini RR, Sillerud LO, Zhaorigetu S, et al. Autophagy regulates lipolysis and cell survival through lipid droplet degradation in androgen-sensitive prostate cancer cells. Prostate. 2012;72(13):1412–1422. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Schott MB, Weller SG, Schulze RJ, et al. Lipid droplet size directs lipolysis and lipophagy catabolism in hepatocytes. J Cell Biol. 2019;218(10):3320–3335. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [95].Spandl J, Lohmann D, Kuerschner L, et al. Ancient ubiquitous protein 1 (AUP1) localizes to lipid droplets and binds the E2 ubiquitin conjugase G2 (Ube2g2) via its G2 binding region. J Biol Chem. 2011;286(7):5599–5606. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [96].Zhang J, Lan Y, Li MY, et al. Flaviviruses exploit the lipid droplet protein AUP1 to trigger lipophagy and drive virus production. Cell Host Microbe. 2018;23(6):819–831 e5. . [DOI] [PubMed] [Google Scholar]
- [97].Eastman SW, Yassaee M, Bieniasz PD. A role for ubiquitin ligases and Spartin/SPG20 in lipid droplet turnover. J Cell Biol. 2009;184(6):881–894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Hooper C, Puttamadappa SS, Loring Z, et al. Spartin activates atrophin-1-interacting protein 4 (AIP4) E3 ubiquitin ligase and promotes ubiquitination of adipophilin on lipid droplets. BMC Biol. 2010;8(1):72. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [99].Tsai TH, Chen E, Li L, et al. The constitutive lipid droplet protein PLIN2 regulates autophagy in liver. Autophagy. 2017;13(7):1130–1144. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Wang L, Zhou J, Yan S, et al. Ethanol-triggered lipophagy requires SQSTM1 in AML12 hepatic cells. Sci Rep. 2017;7(1):12307. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [101].Ding WX, Li M, Chen X, et al. Autophagy reduces acute ethanol-induced hepatotoxicity and steatosis in mice. Gastroenterology. 2010;139(5):1740–1752. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [102].Yan Y, Wang H, Wei C, et al. HDAC6 regulates lipid droplet turnover in response to nutrient deprivation via p62-mediated selective autophagy. J Genet Genomics. 2019;46(4):221–229. . [DOI] [PubMed] [Google Scholar]
- [103].Johnston JA, Ward CL, Kopito RR. Aggresomes: a cellular response to misfolded proteins. J Cell Biol. 1998;143(7):1883–1898. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [104].Iwata A, Riley BE, Johnston JA, et al. HDAC6 and microtubules are required for autophagic degradation of aggregated huntingtin. J Biol Chem. 2005;280(48):40282–40292. . [DOI] [PubMed] [Google Scholar]
- [105].Boyault C, Zhang Y, Fritah S, et al. HDAC6 controls major cell response pathways to cytotoxic accumulation of protein aggregates. Genes Dev. 2007;21(17):2172–2181. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Rui YN, Xu Z, Patel B, et al. Huntingtin functions as a scaffold for selective macroautophagy. Nat Cell Biol. 2015;17(3):262–275. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Sathyanarayan A, Mashek MT, Mashek DG. ATGL promotes autophagy/lipophagy via SIRT1 to control hepatic lipid droplet catabolism. Cell Rep. 2017;19(1):1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [108].Lee IH, Cao L, Mostoslavsky R, et al. A role for the NAD-dependent deacetylase Sirt1 in the regulation of autophagy. Proc Natl Acad Sci U S A. 2008;105(9):3374–3379. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Stenmark H. Rab GTPases as coordinators of vesicle traffic. Nat Rev Mol Cell Biol. 2009;10(8):513–525. [DOI] [PubMed] [Google Scholar]
- [110].Vitelli R, Santillo M, Lattero D, et al. Role of the small GTPase Rab7 in the late endocytic pathway. J Biol Chem. 1997;272(7):4391–4397. . [DOI] [PubMed] [Google Scholar]
- [111].Gutierrez MG, Munafo DB, Beron W, et al. Rab7 is required for the normal progression of the autophagic pathway in mammalian cells. J Cell Sci. 2004;117(13):2687–2697. . [DOI] [PubMed] [Google Scholar]
- [112].Jager S, Bucci C, Tanida I, et al. Role for Rab7 in maturation of late autophagic vacuoles. J Cell Sci. 2004;117(20):4837–4848. . [DOI] [PubMed] [Google Scholar]
- [113].Schroeder B, Schulze RJ, Weller SG, et al. The small GTPase Rab7 as a central regulator of hepatocellular lipophagy. Hepatology. 2015;61(6):1896–1907. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [114].Lizaso A, Tan KT, Lee YH. beta-adrenergic receptor-stimulated lipolysis requires the RAB7-mediated autolysosomal lipid degradation. Autophagy. 2013;9(8):1228–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Li Z, Schulze RJ, Weller SG, et al. A novel Rab10-EHBP1-EHD2 complex essential for the autophagic engulfment of lipid droplets. Sci Adv. 2016;2(12):e1601470. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [116].Sinha RA, You SH, Zhou J, et al. Thyroid hormone stimulates hepatic lipid catabolism via activation of autophagy. J Clin Invest. 2012;122(7):2428–2438. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [117].Byun S, Seok S, Kim YC, et al. Fasting-induced FGF21 signaling activates hepatic autophagy and lipid degradation via JMJD3 histone demethylase. Nat Commun. 2020;11(1):807. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Gao Y, Zhang W, Zeng LQ, et al. Exercise and dietary intervention ameliorate high-fat diet-induced NAFLD and liver aging by inducing lipophagy. Redox Biol. 2020;36:101635. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Kim J, Kundu M, Viollet B, et al. AMPK and mTOR regulate autophagy through direct phosphorylation of Ulk1. Nat Cell Biol. 2011;13(2):132–141. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [120].Kim J, Kim YC, Fang C, et al. Differential regulation of distinct Vps34 complexes by AMPK in nutrient stress and autophagy. Cell. 2013;152(1–2):290–303. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Canto C, Gerhart-Hines Z, Feige JN, et al. AMPK regulates energy expenditure by modulating NAD+ metabolism and SIRT1 activity. Nature. 2009;458(7241):1056–1060. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [122].Raben N, Puertollano R. TFEB and TFE3: linking lysosomes to cellular adaptation to stress. Annu Rev Cell Dev Biol. 2016;32(1):255–278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [123].Napolitano G, Ballabio A. TFEB at a glance. J Cell Sci. 2016;129(13):2475–2481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Settembre C, De Cegli R, Mansueto G, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop. Nat Cell Biol. 2013;15(6):647–658. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Xiong J, Wang K, He J, et al. TFE3 alleviates hepatic steatosis through autophagy-induced lipophagy and PGC1alpha-mediated fatty acid beta-oxidation. Int J Mol Sci. 2016;17(3):387. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Chakrabarti P, Kandror KV. FoxO1 controls insulin-dependent adipose triglyceride lipase (ATGL) expression and lipolysis in adipocytes. J Biol Chem. 2009;284(20):13296–13300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Lettieri Barbato D, Tatulli G, Aquilano K, et al. FoxO1 controls lysosomal acid lipase in adipocytes: implication of lipophagy during nutrient restriction and metformin treatment. Cell Death Dis. 2013;4(10):e861. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [128].Tao R, Wei D, Gao H, et al. Hepatic FoxOs regulate lipid metabolism via modulation of expression of the nicotinamide phosphoribosyltransferase gene. J Biol Chem. 2011;286(16):14681–14690. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Xiong X, Tao R, DePinho RA, et al. The autophagy-related gene 14 (Atg14) is regulated by forkhead box O transcription factors and circadian rhythms and plays a critical role in hepatic autophagy and lipid metabolism. J Biol Chem. 2012;287(46):39107–39114. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [130].Seok S, Fu T, Choi SE, et al. Transcriptional regulation of autophagy by an FXR-CREB axis. Nature. 2014;516(7529):108–111. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Lee JM, Wagner M, Xiao R, et al. Nutrient-sensing nuclear receptors coordinate autophagy. Nature. 2014;516(7529):112–115. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Vevea JD, Garcia EJ, Chan RB, et al. Role for lipid droplet biogenesis and microlipophagy in adaptation to lipid imbalance in yeast. Dev Cell. 2015;35(5):584–599. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [133].Wang CW, Miao YH, Chang YS. A sterol-enriched vacuolar microdomain mediates stationary phase lipophagy in budding yeast. J Cell Biol. 2014;206(3):357–366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Tsuji T, Fujimoto M, Tatematsu T, et al. Niemann-Pick type C proteins promote microautophagy by expanding raft-like membrane domains in the yeast vacuole. Elife. 2017;6:e25960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Oku M, Maeda Y, Kagohashi Y, et al. Evidence for ESCRT- and clathrin-dependent microautophagy. J Cell Biol. 2017;216(10):3263–3274. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Mochida K, Oikawa Y, Kimura Y, et al. Receptor-mediated selective autophagy degrades the endoplasmic reticulum and the nucleus. Nature 2015;522(7556):359–362. . [DOI] [PubMed] [Google Scholar]
- [137].Wang ML, Motamed M, Infante RE, et al. Identification of surface residues on Niemann-Pick C2 essential for hydrophobic handoff of cholesterol to NPC1 in lysosomes. Cell Metab. 2010;12(2):166–173. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [138].Kihara A, Noda T, Ishihara N, et al. Two Distinct Vps34 Phosphatidylinositol 3–Kinase Complexes Function in Autophagy and Carboxypeptidase Y Sorting inSaccharomyces cerevisiae. J Cell Biol. 2001;152(3):519–530. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [139].Bouchez I, Pouteaux M, Canonge M, et al. Regulation of lipid droplet dynamics in Saccharomyces cerevisiae depends on the Rab7-like Ypt7p, HOPS complex and V1-ATPase. Biol Open. 2015;4(7):764–775. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [140].Haas A, Scheglmann D, Lazar T, et al. The GTPase Ypt7p of Saccharomyces cerevisiae is required on both partner vacuoles for the homotypic fusion step of vacuole inheritance. Embo J. 1995;14(21):5258–5270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [141].Wichmann H, Hengst L, Gallwitz D. Endocytosis in yeast: evidence for the involvement of a small GTP-binding protein (Ypt7p). Cell. 1992;71:1131–1142. [DOI] [PubMed] [Google Scholar]
- [142].Kim J, Dalton VM, Eggerton KP, et al. Apg7p/Cvt2p is required for the cytoplasm-to-vacuole targeting, macroautophagy, and peroxisome degradation pathways. Mol Biol Cell. 1999;10(5):1337–1351. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [143].Kaushik S, Cuervo AM. AMPK-dependent phosphorylation of lipid droplet protein PLIN2 triggers its degradation by CMA. Autophagy. 2016;12(2):432–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [144].Ma SY, Sun KS, Zhang M, et al. Disruption of Plin5 degradation by CMA causes lipid homeostasis imbalance in NAFLD. Liver Int. 2020;40(10):2427–2438. . [DOI] [PubMed] [Google Scholar]
- [145].Madrigal-Matute J, Cuervo AM. Regulation of liver metabolism by autophagy. Gastroenterology. 2016;150(2):328–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [146].Ueno T, Komatsu M. Autophagy in the liver: functions in health and disease. Nat Rev Gastroenterol Hepatol. 2017;14:170–184. [DOI] [PubMed] [Google Scholar]
- [147].Musso G, Cassader M, Gambino R. Non-alcoholic steatohepatitis: emerging molecular targets and therapeutic strategies. Nat Rev Drug Discov. 2016;15:249–274. [DOI] [PubMed] [Google Scholar]
- [148].Kounakis K, Chaniotakis M, Markaki M, et al. Emerging roles of lipophagy in health and disease. Front Cell Dev Biol. 2019;7:1–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [149].Carotti S, Aquilano K, Zalfa F, et al. Lipophagy impairment Is associated with disease progression in NAFLD. Front Physiol. 2020;11:850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [150].Zhu S, Wu Y, Ye X, et al. FGF21 ameliorates nonalcoholic fatty liver disease by inducing autophagy. Mol Cell Biochem. 2016;420(1–2):107–119. . [DOI] [PubMed] [Google Scholar]
- [151].Kim SH, Kim G, Han DH, et al. Ezetimibe ameliorates steatohepatitis via AMP activated protein kinase-TFEB-mediated activation of autophagy and NLRP3 inflammasome inhibition. Autophagy. 2017;13(10):1767–1781. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [152].Pan X, Zhang Y, Kim HG, et al. FOXO transcription factors protect against the diet-induced fatty liver disease. Sci Rep. 2017;7(1):1–12. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [153].Zhang H, Yan S, Khambu B, et al. Dynamic MTORC1-TFEB feedback signaling regulates hepatic autophagy, steatosis and liver injury in long-term nutrient oversupply. Autophagy. 2018;14(10):1779–1795. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [154].Lim H, Lim YM, Kim KH, et al. A novel autophagy enhancer as a therapeutic agent against metabolic syndrome and diabetes. Nat Comm. 2018;9(1):1438. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [155].Schuster S, Cabrera D, Arrese M, et al. Triggering and resolution of inflammation in NASH. Nat Rev Gastroenterol Hepatol. 2018;15:349–364. [DOI] [PubMed] [Google Scholar]
- [156].Fotbolcu H, Zorlu E. Nonalcoholic fatty liver disease as a multi-systemic disease. World J Gastroenterol. 2016;22(16):4079–4090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [157].Noureddin M, Sanyal AJ. Pathogenesis of NASH: the impact of multiple pathways. Curr Hepatol Rep. 2018;17(4):350–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [158].Cotter TG, Rinella M. Nonalcoholic fatty liver disease 2020: the state of the disease. Gastroenterology. 2020;158(7):1851–1864. [DOI] [PubMed] [Google Scholar]
- [159].Goedeke L, Bates J, Vatner DF, et al. Acetyl-CoA carboxylase inhibition reverses NAFLD and hepatic insulin resistance but promotes hypertriglyceridemia in rodents. Hepatology. 2018;68(6):2197–2211. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [160].Calzadilla Bertot L, Adams LA. The natural course of non-alcoholic fatty liver disease. Int J Mol Sci. 2016;17(5):774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [161].Day CP, James OF. Steatohepatitis: a tale of two “hits”? Gastroenterology. 1998;114(842–5). 10.1016/S0016-5085(98)70599-2 [DOI] [PubMed] [Google Scholar]
- [162].Buzzetti E, Pinzani M, Tsochatzis EA. The multiple-hit pathogenesis of non-alcoholic fatty liver disease (NAFLD). Metabolism. 2016;65(8):1038–1048. [DOI] [PubMed] [Google Scholar]
- [163].Hansen HH, Feigh M, Veidal SS, et al. Mouse models of nonalcoholic steatohepatitis in preclinical drug development. Drug Discov Today. 2017;22(11):1707–1718. . [DOI] [PubMed] [Google Scholar]
- [164].Van Herck MA, Vonghia L, Francque SM. Animal models of nonalcoholic fatty liver disease-A starter’s guide. Nutrients. 2017;9(10):1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [165].Farrell G, Schattenberg JM, Leclercq I, et al. Mouse models of nonalcoholic steatohepatitis: toward optimization of their relevance to human nonalcoholic steatohepatitis. Hepatology. 2019;69(5):2241–2257. . [DOI] [PubMed] [Google Scholar]
- [166].Santhekadur PK, Kumar DP, Sanyal AJ. Preclinical models of non-alcoholic fatty liver disease. J Hepatol. 2018;68(2):230–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [167].Guo J, Fang W, Chen X, et al. Upstream stimulating factor 1 suppresses autophagy and hepatic lipid droplet catabolism by activating mTOR. FEBS Lett. 2018;592(16):2725–2738. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [168].Machado MV, Michelotti GA, Xie G, et al. Mouse models of diet-induced nonalcoholic steatohepatitis reproduce the heterogeneity of the human disease. PLoS One. 2015;10(5):e0127991. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [169].Song YM, Lee YH, Kim JW, et al. Metformin alleviates hepatosteatosis by restoring SIRT1-mediated autophagy induction via an AMP-activated protein kinase-independent pathway. Autophagy. 2015;11(1):46–59. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [170].Gluchowski NL, Gabriel KR, Chitraju C, et al. Hepatocyte deletion of triglyceride-synthesis enzyme Acyl CoA: diacylglycerol acyltransferase 2 reduces steatosis without increasing inflammation or fibrosis in mice. Hepatology. 2019;70(6):1972–1985. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [171].Donnelly KL, Smith CI, Schwarzenberg SJ, et al. Sources of fatty acids stored in liver and secreted via lipoproteins in patients with nonalcoholic fatty liver disease. J Clin Invest. 2005;115(5):1343–1351. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [172].Kohjima M, Enjoji M, Higuchi N, et al. Re-evaluation of fatty acid metabolism-related gene expression in nonalcoholic fatty liver disease. Int J Mol Med. 2007;20(3):351–358. [PubMed] [Google Scholar]
- [173].Steneberg P, Sykaras AG, Backlund F, et al. Hyperinsulinemia enhances hepatic expression of the fatty acid transporter Cd36 and provokes hepatosteatosis and hepatic insulin resistance. J Biol Chem. 2015;290(31):19034–19043. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [174].Ipsen DH, Lykkesfeldt J, Tveden-Nyborg P. Molecular mechanisms of hepatic lipid accumulation in non-alcoholic fatty liver disease. Cell Mol Life Sci. 2018;75(18):3313–3327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [175].Pettinelli P, Videla LA. Up-regulation of PPAR-gamma mRNA expression in the liver of obese patients: an additional reinforcing lipogenic mechanism to SREBP-1c induction. J Clin Endocrinol Metab. 2011;96(5):1424–1430. [DOI] [PubMed] [Google Scholar]
- [176].Nagaya T, Tanaka N, Suzuki T, et al. Down-regulation of SREBP-1c is associated with the development of burned-out NASH. J Hepatol. 2010;53(4):724–731. . [DOI] [PubMed] [Google Scholar]
- [177].Yoshioka Y, Hashimoto E, Yatsuji S, et al. Nonalcoholic steatohepatitis: cirrhosis, hepatocellular carcinoma, and burnt-out NASH. J Gastroenterol. 2004;39(12):1215–1218. . [DOI] [PubMed] [Google Scholar]
- [178].Iizuka K, Bruick RK, Liang G, et al. Deficiency of carbohydrate response element-binding protein (ChREBP) reduces lipogenesis as well as glycolysis. Proc Natl Acad Sci U S A. 2004;101(19):7281–7286. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [179].Zhang D, Tong X, VanDommelen K, et al. Lipogenic transcription factor ChREBP mediates fructose-induced metabolic adaptations to prevent hepatotoxicity. J Clin Invest. 2017;127(7):2855–2867. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [180].Benhamed F, Denechaud PD, Lemoine M, et al. The lipogenic transcription factor ChREBP dissociates hepatic steatosis from insulin resistance in mice and humans. J Clin Invest. 2012;122(6):2176–2194. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [181].Alkhouri N, Lawitz E, Noureddin M, et al. GS-0976 (Firsocostat): an investigational liver-directed acetyl-CoA carboxylase (ACC) inhibitor for the treatment of non-alcoholic steatohepatitis (NASH). Expert Opin Investig Drugs. 2020;29(2):135–141. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [182].Gapp B, Jourdain M, Bringer P, et al. Farnesoid X Receptor Agonism, Acetyl-Coenzyme A Carboxylase Inhibition, and Back Translation of Clinically Observed Endpoints of De NovoLipogenesis in a Murine NASH Model. Hepatol Commun. 2020;4(1):109–125. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [183].Kim CW, Addy C, Kusunoki J, et al. Acetyl CoA carboxylase inhibition reduces hepatic steatosis but elevates plasma triglycerides in mice and humans: a bedside to bench investigation. Cell Metab. 2017;26(3):576. . [DOI] [PubMed] [Google Scholar]
- [184].Bates J, Vijayakumar A, Ghoshal S, et al. Acetyl-CoA carboxylase inhibition disrupts metabolic reprogramming during hepatic stellate cell activation. J Hepatol. 2020;73(4):896–905. . [DOI] [PubMed] [Google Scholar]
- [185].Petersen MC, Shulman GI. Mechanisms of insulin action and insulin resistance. Physiol Rev. 2018;98:2133–2223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [186].Tilg H, Moschen AR, Roden M. NAFLD and diabetes mellitus. Nat Rev Gastroenterol Hepatol. 2017;14(1):32–42. [DOI] [PubMed] [Google Scholar]
- [187].Salameh H, Hanayneh MA, Masadeh M, et al. PNPLA3 as a genetic determinant of risk for and severity of non-alcoholic fatty liver disease spectrum. J Clin Transl Hepatol. 2016;4(3):175–191. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [188].Wang Y, Kory N, BasuRay S, et al. PNPLA3, CGI-58, and inhibition of hepatic triglyceride hydrolysis in mice. Hepatology. 2019;69(6):2427–2441. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [189].Li JZ, Huang Y, Karaman R, et al. Chronic overexpression of PNPLA3I148M in mouse liver causes hepatic steatosis. J Clin Invest. 2012;122(11):4130–4144. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [190].Haemmerle G, Lass A, Zimmermann R, et al. Defective lipolysis and altered energy metabolism in mice lacking adipose triglyceride lipase. Science. 2006;312(5774):734–737. . [DOI] [PubMed] [Google Scholar]
- [191].Wu JW, Wang SP, Alvarez F, et al. Deficiency of liver adipose triglyceride lipase in mice causes progressive hepatic steatosis. Hepatology. 2011;54(1):122–132. . [DOI] [PubMed] [Google Scholar]
- [192].Ong KT, Mashek MT, Bu SY, et al. Adipose triglyceride lipase is a major hepatic lipase that regulates triacylglycerol turnover and fatty acid signaling and partitioning. Hepatology. 2011;53(1):116–126. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [193].Yang L, Li P, Fu S, et al. Defective hepatic autophagy in obesity promotes ER stress and causes insulin resistance. Cell Metab. 2010;11(6):467–478. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [194].Okada H, Takabatake R, Honda M, et al. Peretinoin, an acyclic retinoid, suppresses steatohepatitis and tumorigenesis by activating autophagy in mice fed an atherogenic high-fat diet. Oncotarget. 2017;8(25):39978–39993. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [195].Kim KH, Jeong YT, Oh H, et al. Autophagy deficiency leads to protection from obesity and insulin resistance by inducing Fgf21 as a mitokine. Nat Med. 2013;19(1):83–92. . [DOI] [PubMed] [Google Scholar]
- [196].Ma D, Molusky MM, Song J, et al. Autophagy deficiency by hepatic FIP200 deletion uncouples steatosis from liver injury in NAFLD. Mol Endocrinol. 2013;27(10):1643–1654. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [197].Kwanten WJ, Vandewynckel YP, Martinet W, et al. Hepatocellular autophagy modulates the unfolded protein response and fasting-induced steatosis in mice. Am J Physiol Gastrointest Liver Physiol. 2016;311(4):G599–G609. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [198].Li Y, Chao X, Yang L, et al. Impaired fasting-induced adaptive lipid droplet biogenesis in liver-specific Atg5-deficient mouse liver is mediated by persistent nuclear factor-like 2 activation. Am J Pathol. 2018;188(8):1833–1846. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [199].Takahashi SS, Sou YS, Saito T, et al. Loss of autophagy impairs physiological steatosis by accumulation of NCoR1. Life Sci Alliance. 2020;3(1):e201900513. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [200].Fukushima H, Yamashina S, Arakawa A, et al. Formation of p62-positive inclusion body is associated with macrophage polarization in non-alcoholic fatty liver disease. Hepatol Res. 2018;48(9):757–767. . [DOI] [PubMed] [Google Scholar]
- [201].Koga H, Kaushik S, Cuervo AM. Altered lipid content inhibits autophagic vesicular fusion. Faseb J. 2010;24(8):3052–3065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [202].Hattori K, Naguro I, Runchel C, et al. The roles of ASK family proteins in stress responses and diseases. Cell Commun Signal. 2009;7(1):9. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [203].Challa TD, Wueest S, Lucchini FC, et al. Liver ASK1 protects from non-alcoholic fatty liver disease and fibrosis. EMBO Mol Med. 2019;11(10):e10124. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [204].Wang PX, Ji YX, Zhang XJ, et al. Corrigendum: targeting CASP8 and FADD-like apoptosis regulator ameliorates nonalcoholic steatohepatitis in mice and nonhuman primates. Nat Med. 2017;23(10):1241. . [DOI] [PubMed] [Google Scholar]
- [205].Ye P, Xiang M, Liao H, et al. Dual-specificity phosphatase 9 protects against nonalcoholic fatty liver disease in mice through ASK1 suppression. Hepatology. 2019;69(1):76–93. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [206].Wang Y, Wen H, Fu J, et al. Hepatocyte TNF receptor-associated factor 6 aggravates hepatic inflammation and fibrosis by promoting lysine 6-linked polyubiquitination of apoptosis signal-regulating kinase 1. Hepatology. 2020;71(1):93–111. . [DOI] [PubMed] [Google Scholar]
- [207].Shiratori Y, Ichida T, Geerts A, et al. Modulation of collagen synthesis by fat-storing cells, isolated from CCl4- or vitamin A-treated rats. Dig Dis Sci. 1987;32(11):1281–1289. . [DOI] [PubMed] [Google Scholar]
- [208].Tsuchida T, Friedman SL. Mechanisms of hepatic stellate cell activation. Nat Rev Gastroenterol Hepatol. 2017;14(7):397–411. [DOI] [PubMed] [Google Scholar]
- [209].Wake K. “Sternzellen” in the liver: perisinusoidal cells with special reference to storage of vitamin A. Am J Anat. 1971;132(4):429–462. [DOI] [PubMed] [Google Scholar]
- [210].Hellerbrand C, Stefanovic B, Giordano F, et al. The role of TGFbeta1 in initiating hepatic stellate cell activation in vivo. J Hepatol. 1999;30(1):77–87. . [DOI] [PubMed] [Google Scholar]
- [211].Thoen LF, Guimaraes EL, Dolle L, et al. A role for autophagy during hepatic stellate cell activation. J Hepatol. 2011;55(6):1353–1360. . [DOI] [PubMed] [Google Scholar]
- [212].Hernández-Gea V, Ghiassi-Nejad Z, Rozenfeld R, et al. Autophagy releases lipid that promotes fibrogenesis by activated hepatic stellate cells in mice and in human tissues. Gastroenterology. 2012;142(4):938–946. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [213].Meng D, Li Z, Wang G, et al. Carvedilol attenuates liver fibrosis by suppressing autophagy and promoting apoptosis in hepatic stellate cells. Biomed Pharmacother. 2018;108:1617–1627. [DOI] [PubMed] [Google Scholar]
- [214].Bai F, Huang Q, Nie J, et al. Trolline ameliorates liver fibrosis by inhibiting the NF-κB pathway, promoting HSC apoptosis and suppressing autophagy. Cell Physiol Biochem. 2017;44(2):436–446. . [DOI] [PubMed] [Google Scholar]
- [215].Liu N, Feng J, Lu X, et al. Isorhamnetin inhibits liver fibrosis by reducing autophagy and inhibiting extracellular matrix formation via the TGF-. Mediators Inflamm. 2019;2019:6175091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [216].Zhang XL, Chen ZN, Huang QF, et al. Methyl helicterate inhibits hepatic stellate cell activation through modulation of apoptosis and autophagy. Cell Physiol Biochem. 2018;51(2):897–908. . [DOI] [PubMed] [Google Scholar]
- [217].Qiu S, Xu H, Lin Z, et al. The blockade of lipophagy pathway is necessary for docosahexaenoic acid to regulate lipid droplet turnover in hepatic stellate cells. Biomed Pharmacother. 2019;109:1841–1850. [DOI] [PubMed] [Google Scholar]
- [218].Paik YH, Schwabe RF, Bataller R, et al. Toll-like receptor 4 mediates inflammatory signaling by bacterial lipopolysaccharide in human hepatic stellate cells. Hepatology. 2003;37(5):1043–1055. . [DOI] [PubMed] [Google Scholar]
- [219].Takahashi M, Ogasawara K, Takeda K, et al. LPS induces NK1.1+ alpha beta T cells with potent cytotoxicity in the liver of mice via production of IL-12 from Kupffer cells. J Immunol. 1996;156(7):2436–2442. [PubMed] [Google Scholar]
- [220].Chen M, Liu J, Yang W, et al. Lipopolysaccharide mediates hepatic stellate cell activation by regulating autophagy and retinoic acid signaling. Autophagy. 2017;13(11):1813–1827. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [221].Koliaki C, Szendroedi J, Kaul K, et al. Adaptation of hepatic mitochondrial function in humans with non-alcoholic fatty liver is lost in steatohepatitis. Cell Metab. 2015;21(5):739–746. . [DOI] [PubMed] [Google Scholar]
- [222].Pallichankandy S, Rahman A, Thayyullathil F, et al. ROS-dependent activation of autophagy is a critical mechanism for the induction of anti-glioma effect of sanguinarine. Free Radic Biol Med. 2015;89:708–720. [DOI] [PubMed] [Google Scholar]
- [223].Zhang Z, Zhao S, Yao Z, et al. Autophagy regulates turnover of lipid droplets via ROS-dependent Rab25 activation in hepatic stellate cell. Redox Biol. 2017;11:322–334. [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- [224].Lodder J, Denaes T, Chobert MN, et al. Macrophage autophagy protects against liver fibrosis in mice. Autophagy. 2015;11(8):1280–1292. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [225].Liu K, Zhao E, Ilyas G, et al. Impaired macrophage autophagy increases the immune response in obese mice by promoting proinflammatory macrophage polarization. Autophagy. 2015;11(2):271–284. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [226].Remmerie A, Scott CL. Macrophages and lipid metabolism. Cell Immunol. 2018;330:27–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [227].Ioannou GN, Haigh WG, Thorning D, et al. Hepatic cholesterol crystals and crown-like structures distinguish NASH from simple steatosis. J Lipid Res. 2013;1061:19–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [228].Leroux A, Ferrere G, Godie V, et al. Toxic lipids stored by Kupffer cells correlates with their pro-inflammatory phenotype at an early stage of steatohepatitis. J Hepatol. 2012;57(1):141–149. . [DOI] [PubMed] [Google Scholar]
- [229].Jindal A, Bruzzì S, Sutti S, et al. Fat-laden macrophages modulate lobular inflammation in nonalcoholic steatohepatitis (NASH). Exp Mol Pathol. 2015;99(1):155–162. . [DOI] [PubMed] [Google Scholar]
- [230].Huang W, Metlakunta A, Dedousis N, et al. Depletion of liver Kupffer cells prevents the development of diet-induced hepatic steatosis and insulin resistance. Diabetes. 2010;59(2):347–357. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [231].Diehl KL, Vorac J, Hofmann K, et al. Kupffer cells sense free fatty acids and regulate hepatic lipid metabolism in high-fat diet and inflammation. Cells. 2020;9(10):2258. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [232].Miyao M, Kotani H, Ishida T, et al. Pivotal role of liver sinusoidal endothelial cells in NAFLD/NASH progression. Lab Invest. 2015;95(10):1130–1144. . [DOI] [PubMed] [Google Scholar]
- [233].Hammoutene A, Biquard L, Lasselin J, et al. A defect in endothelial autophagy occurs in patients with non-alcoholic steatohepatitis and promotes inflammation and fibrosis. J Hepatol. 2020;72(3):528–538. . [DOI] [PubMed] [Google Scholar]
- [234].Ruart M, Chavarria L, Campreciós G, et al. Impaired endothelial autophagy promotes liver fibrosis by aggravating the oxidative stress response during acute liver injury. J Hepatol. 2019;70(3):458–469. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [235].Duval C, Thissen U, Keshtkar S, et al. Adipose tissue dysfunction signals progression of hepatic steatosis towards nonalcoholic steatohepatitis in C57Bl/6 mice. Diabetes. 2010;59(12):3181–3191. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [236].Kovsan J, Bluher M, Tarnovscki T, et al. Altered autophagy in human adipose tissues in obesity. J Clin Endocrinol Metab. 2011;96(2):E268–77. . [DOI] [PubMed] [Google Scholar]
- [237].Kosacka J, Kern M, Kloting N, et al. Autophagy in adipose tissue of patients with obesity and type 2 diabetes. Mol Cell Endocrinol. 2015;409:21–32. [DOI] [PubMed] [Google Scholar]
- [238].Zhang X, Wu D, Wang C, et al. Sustained activation of autophagy suppresses adipocyte maturation via a lipolysis-dependent mechanism. Autophagy. 2019;16(9):1668–1682. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [239].Tacke F. Targeting hepatic macrophages to treat liver diseases. J Hepatol. 2017;66(6):1300–1312. [DOI] [PubMed] [Google Scholar]
- [240].Mendez-Sanchez N, Cruz-Ramon VC, Ramirez-Perez OL, et al. New aspects of lipotoxicity in nonalcoholic steatohepatitis. Int J Mol Sci. 2018;19(7):2034. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [241].Nielsen J, Christensen AE, Nellemann B, et al. Lipid droplet size and location in human skeletal muscle fibers are associated with insulin sensitivity. Am J Physiol Endocrinol Metab. 2017;313(6):E721–E730. . [DOI] [PubMed] [Google Scholar]
- [242].Onal G, Kutlu O, Gozuacik D, et al. Lipid droplets in health and disease. Lipids Health Dis. 2017;16(1):128. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [243].Allaire M, Rautou PE, Codogno P, et al. Autophagy in liver diseases: time for translation? J Hepatol. 2019;70(5):985–998. . [DOI] [PubMed] [Google Scholar]
- [244].Kim KE, Jung Y, Min S, et al. Caloric restriction of db/db mice reverts hepatic steatosis and body weight with divergent hepatic metabolism. Sci Rep. 2016;6(1):30111. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [245].Pi H, Liu M, Xi Y, et al. Long-term exercise prevents hepatic steatosis: a novel role of FABP1 in regulation of autophagy-lysosomal machinery. Faseb J. 2019;33(11):11870–11883. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [246].Chen R, Wang Q, Song S, et al. Protective role of autophagy in methionine-choline deficient diet-induced advanced nonalcoholic steatohepatitis in mice. Eur J Pharmacol. 2016;770:126–133. [DOI] [PubMed] [Google Scholar]
- [247].Kim JH, Sim HA, Jung DY, et al. Poria cocus wolf extract ameliorates hepatic steatosis through regulation of lipid metabolism, inhibition of ER stress, and activation of autophagy via AMPK activation. Int J Mol Sci. 2019;20(19):4801. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [248].Li YL, Li XQ, Wang YD, et al. Metformin alleviates inflammatory response in non-alcoholic steatohepatitis by restraining signal transducer and activator of transcription 3-mediated autophagy inhibition in vitro and in vivo. Biochem Biophys Res Commun. 2019;513(1):64–72. . [DOI] [PubMed] [Google Scholar]
- [249].Zhong J, Gong W, Lu L, et al. Irbesartan ameliorates hyperlipidemia and liver steatosis in type 2 diabetic db/db mice via stimulating PPAR-gamma, AMPK/Akt/mTOR signaling and autophagy. Int Immunopharmacol. 2017;42:176–184. [DOI] [PubMed] [Google Scholar]
- [250].Hsiao PJ, Chiou HC, Jiang HJ, et al. Pioglitazone enhances cytosolic lipolysis, beta-oxidation and autophagy to ameliorate hepatic steatosis. Sci Rep. 2017;7(1):9030. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [251].Shao N, Yu XY, Ma XF, et al. Exenatide delays the progression of nonalcoholic fatty liver disease in C57BL/6 mice, which may involve inhibition of the NLRP3 inflammasome through the mitophagy pathway. Gastroenterol Res Pract. 2018;2018:1864307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [252].Zhang Y, Sowers JR, Ren J. Targeting autophagy in obesity: from pathophysiology to management. Nat Rev Endocrinol. 2018;14:356–376. [DOI] [PubMed] [Google Scholar]
- [253].Wan J, Weiss E, Ben Mkaddem S, et al. LC3-associated phagocytosis protects against inflammation and liver fibrosis via immunoreceptor inhibitory signaling. Sci Transl Med. 2020;12(539):eaaw8523. . [DOI] [PubMed] [Google Scholar]