

ABSTRACT
Macroautophagy/autophagy is a highly conserved process in eukaryotic cells. It plays a critical role in cellular homeostasis by delivering cytoplasmic cargos to lysosomes for selective degradation. OPTN (optineurin), a well-recognized autophagy receptor, has received considerable attention due to its multiple roles in the autophagic process. OPTN is associated with many human disorders that are closely related to autophagy, such as rheumatoid arthritis, osteoporosis, and nephropathy. Here, we review the function of OPTN as an autophagy receptor at different stages of autophagy, focusing on cargo recognition, autophagosome formation, autophagosome maturation, and lysosomal quality control. OPTN tends to be protective in most autophagy associated diseases, though the molecular mechanism of OPTN regulation in these diseases is not well understood. A comprehensive review of the function of OPTN in autophagy provides valuable insight into the pathogenesis of human diseases related to OPTN and facilitates the discovery of potential key regulators and novel therapeutic targets for disease intervention in patients with autophagic diseases.
Abbreviations: ATG: autophagy-related; APAP: acetaminophen; CALCOCO2/NDP52: calcium binding and coiled-coil domain 2; CC: coiled-coil; HACE1: HECT domain and ankyrin repeat containing E3 ubiquitin protein ligase 1; MYO6: myosin VI; IKBKG/NEMO: inhibitor of nuclear factor kappa B kinase regulatory subunit gamma; IKK: IκB kinase; LIR: LC3-interacting region; LZ: leucine zipper; MAP1LC3/LC3: microtubule associated protein 1 light chain 3; NFKB/NF-κB: nuclear factor kappa B subunit; OPTN: optineurin; PIK3C3: phosphatidylinositol 3-kinase catalytic subunit type 3; PINK1: PTEN induced kinase 1; PRKN: parkin RBR E3 ubiquitin protein ligase; RTECs: renal tubular epithelial cells; SQSTM1/p62: sequestosome 1; TBK1: TANK binding kinase 1; TOM1: target of myb1 membrane trafficking protein; UBD: ubiquitin-binding domain; ULK1: unc-51 like autophagy activating kinase 1; WIPI2: WD repeat domain, phosphoinositide interacting 2; ZF: zinc finger.
KEYWORDS: Autophagy, autophagosome formation, cargo recognition, diseases, lysosomal quality control, mitophagy, optineurin (OPTN)
Introduction
The lysosomal degradation pathway of autophagy is a highly conserved pathway in eukaryotic cells and plays an essential role in cellular, tissue, and organismal homeostasis by selectively removing dysfunctional organelles, intracellular bacteria, and aggregated proteins [1]. In general, cellular stress induces the formation of a cup-shaped structure called a phagophore that can mature into an autophagosome with the cooperation of autophagy-related (ATG) proteins and autophagy receptors. These autophagosomes and lysosomes fuse to form autolysosomes to ultimately degrade cargos [2]. The autophagic process is modulated sequentially and linked to numerous fundamental physiological functions [3].
OPTN (optineurin) is a conserved protein that can be found in humans, rhesus monkeys, rats, pigs, and many other species [4–6]. Generally speaking, OPTN plays roles in vesicular trafficking, NFKB/NF-κB (nuclear factor kappa B subunit) signaling and autophagy. In specific, OPTN has been identified as an autophagy receptor that connects the ubiquitinated autophagy substrates with MAP1LC3/LC3 (microtubule associated protein 1 light chain 3)-positive phagophore membranes [7]. Moreover, increasing evidence has shown that OPTN is also an autophagy inducer that initiates the autophagic process [8,9]. Studies have suggested that OPTN’s participation in autophagic initiation can start as early as the process of autophagosomal membrane formation [10,11]. These novel findings highlight the role of OPTN as a potential autophagy receptor with multiple functions during the autophagic process, rather than merely a common autophagy receptor that functions at a single stage of autophagy [12].
Mutations in the human OPTN gene have been found in many familial diseases, such as OPTNE478G in amyotrophic lateral sclerosis (ALS) and OPTNE50K in glaucoma [13–15]. OPTNE478G mutation occurs in its autophagy-associated ubiquitin-binding domain (UBD) [7,16], indicating the close connection between OPTN-induced autophagy and the disease of ALS. Moreover, low OPTN expression was detected in a subset of patients with Crohn disease [6]. These results indicate that OPTN may be involved in many biological processes and act as a key disease-driving gene. Given this new function of OPTN as an autophagy receptor, it may have implications for OPTN-associated diseases with novel autophagic mechanisms.
In this review, we first summarize the current knowledge of the protein structure and cellular function of OPTN, with an update of new OPTN interaction partners identified to act during autophagosome formation. Then, we give a comprehensive review of OPTN participation in the autophagic processes, including its role in cargo recognition, autophagosome formation, autophagosome maturation, lysosomal quality control, and autophagic degradation. We further discuss OPTN-induced autophagic mechanisms in various diseases, such as neurodegenerative diseases, inflammatory diseases, cancer, and nephropathy. Overall, an understanding of the autophagic functions of OPTN may have implications in the landscape and intervention of OPTN-associated human disorders.
The protein structure and cellular function of OPTN
The human OPTN protein is a 74-kDa scaffold protein comprised of 577 amino acids, and the mouse Optn gene codes for a 584–amino acid protein (67 kDa), which is 78% identical to human OPTN [4]. OPTN is a highly conserved protein that is expressed in many tissues including brain, liver, and heart and plays an important role in normal physiology and disease pathogenesis [4,17].
OPTN contains several structural domains, including two coiled-coil (CC) domains, a leucine zipper (LZ) domain, an LC3-interacting region (LIR), a ubiquitin-binding domain (UBD), and a zinc finger (ZF) domain. In OPTN, the LIR domain is an LC3-II binding site, and the UBD is the ubiquitinated cargo binding site. Both the LIR and UBD are critical regulatory elements in OPTN-associated autophagy. OPTN binds with LC3-II-conjugated-autophagic membranes via this LIR. Then, OPTN-bound-ubiquitinated cargos are enclosed by autophagic membranes and form autophagosomes for degradation in autolysosomes. Moreover, the LZ domain is also showing an autophagic function in interacting with ATG9A [10], which is the sole multi-spanning membrane protein among ATG proteins that is essential for autophagosome formation [18–20] (Figure 1).
Figure 1.
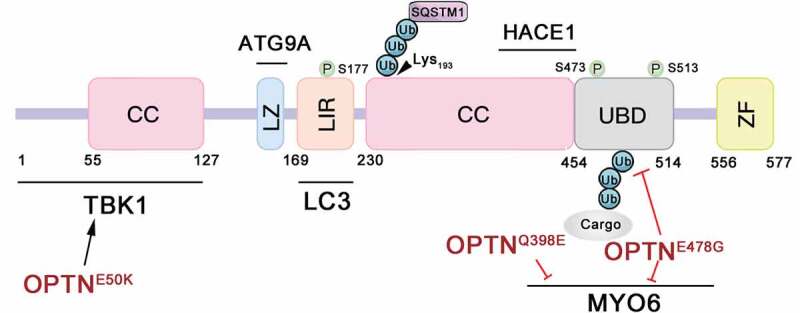
Schematic representation of OPTN functional domains and OPTN mutations. TBK1-binding domain (amino acids [aa] 1–127), RAB8-interacting region ([aa] 1–209), LC3-interacting region (LIR; [aa] 169–181), coiled-coil (CC) domains ([aa] 230–445), leucine zipper (LZ) domain and ATG9A-interacting region ([aa] 143–164), HACE1-interacting region ([aa] 411–456), MYO6-interacting region ([aa] 417–512), ubiquitin-binding domain (UBD; [aa] 454–514) and zinc finger (ZF; [aa] 556–577). OPTN can be phosphorylated on S177, S473, and S513 by TBK1 and ubiquitinated on Lys193 by HACE1.
Many biological functions of OPTN have been identified to date. First, OPTN has been found to colocalize with the Golgi apparatus and plays a vital role in the maintenance of Golgi integrity [14,21]. Second, OPTN shows strong homology with IKBKG/NEMO (inhibitor of nuclear factor kappa B kinase regulatory subunit gamma), a regulator of IKK/IκB kinase complexes [22]. To activate NFKB, IKBKG is recruited to the RIPs (poly-ubiquitin chains of receptor-interacting proteins) to form a functional complex with IKK [23,24]. As a homolog of IKBKG, OPTN competes for binding to the poly-ubiquitin chains of RIPs (poly-ubiquitin chains of receptor-interacting proteins), disrupts the formation of a functional IKK complex and negatively regulates NFKB signaling [22]. Third, an in vitro study identified the specific interaction between OPTN and LC3 using yeast two-hybrid and affinity-isolation assays [7]. This study provides a foundation for subsequent studies of OPTN in autophagy, which has received increasing attention in recent years.
OPTN can interact with a large number of proteins, and some of those interactions have been shown to be disrupted by mutations (Figure 1). For instance, OPTN can interact with ATG9A, LC3, MYO6 (myosin VI), TBK1 (TANK binding kinase 1) and the ubiquitin chains of cargos to execute its functions at multiple steps in autophagy [10,21,25,26]. Specifically, OPTN can be phosphorylated by TBK1 on serines 177 (S177), S473, and S513 [27,28]. OPTNE50K, a glaucoma-associated mutation, can abnormally enhance the interaction between OPTN and TBK1, leading to a blockage in autophagy [29]. The ALS-associated mutations OPTNQ398E and OPTNE478G disrupt the interaction between OPTN and MYO6, and OPTNE478G also loses the function of its UBD and thus fails to interact with the ubiquitinated cargos [7,16].
OPTN is a key regulator of autophagy
OPTN has been proposed to contribute to selective autophagy of depolarized mitochondria (mitophagy), protein aggregates (aggrephagy), and intracellular bacterial pathogens (xenophagy) through ubiquitin‐signaling. However, there have been no systematic and comprehensive reviews the authors are aware of highlighting the vital role of OPTN in autophagy fully. In this section we review the different stages of the autophagic process in which OPTN is actively involved, including cargo recognition, autophagosome formation, autophagosome maturation, lysosomal quality control, and autophagic degradation (Figure 2).
Figure 2.
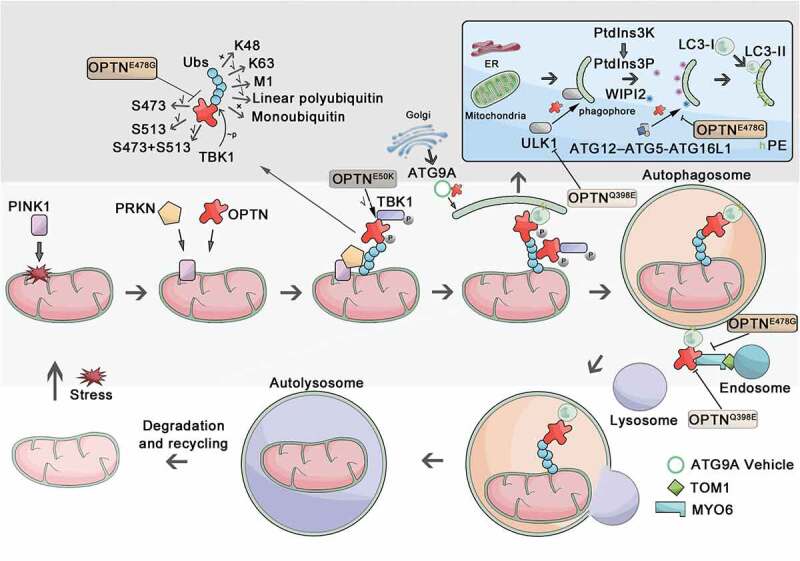
Schematic of OPTN’s roles in the autophagic process. In this figure, we take mitophagy as an example, and the cargo is mitochondrion. In response to stress, PINK1 is stabilized on the mitochondrial outer membrane (MOM), promoting the phosphorylation of PRKN on mitochondria. OPTN is recruited to the mitochondria immediately after PRKN recruitment and associates with ubiquitin chains (Ubs), which can be disrupted by mutation of OPTNE478G. The OPTN-TBK1 interaction can be abnormally enhanced by OPTNE50K, leading to the blockage of autophagy. Induction of autophagy results in the recruitment of ATG proteins to autophagy initiation sites, where OPTN can promote ULK1 translocation to the phagophore. This function can be disrupted by OPTNQ398E mutation. The ULK1 complex drives the downstream activation of PtdIns3K to trigger nucleation of the phagophore. Then, OPTN directs the lipidation of LC3 at the phagophore by recruiting ATG12–ATG5-ATG16L1, which can be disrupted by the OPTNE478G mutant. Also, OPTN can interact with ATG9A vesicles to seed local autophagosomal membrane formation. After this, the ubiquitinated-cargo-connected OPTN links cargo to autophagosomal membranes via binding to LC3. The autophagosomal membrane seals and gives rise to a double-layered vesicle called the autophagosome. OPTN directly interacts with MYO6, which can be disrupted by OPTNE478G and OPTNQ398E mutation. OPTN and MYO6 bind to TOM1, delivering endosomal membranes to autophagosomes, which is the ultimate maturation step for autophagosomes and is required for autophagosome-lysosome fusion. Finally, the autophagosome fuses with the lysosome and becomes the autolysosome for degrading and recycling cellular contents.
The role of OPTN in cargo recognition
It has become clear that specific cargos induce the initiation of autophagy in selective autophagy. Autophagy receptors recognize the ubiquitinated cargos and recruit scaffold proteins to these cargos [30–32]. As an autophagy receptor, OPTN acts to recognize ubiquitinated cargos, which forms the molecular basis for selective autophagy [33]. In selective autophagy, OPTN has to be recruited around these cargos and then recognizes such ubiquitinated cargo.
OPTN can be recruited to different kinds of cargos in different manners. In mitophagy, OPTN can be stabilized by PRKN (parkin RBR E3 ubiquitin protein ligase) on the surface of damaged mitochondria [16]. In the presence of PRKN in HeLa cells, OPTN was shown to be recruited to mitochondria right after PKRN translocation, and this recruitment was stabilized by the UBD binding with ubiquitinated mitochondria. In the absence of PRKN, OPTN puncta can only localize to damaged mitochondria transiently [16]. In addition to PRKN, PINK1 (PTEN induced kinase 1) can dynamically recruit OPTN to damaged mitochondria to activate mitophagy directly [34]. PINK1 works as a mitochondrial protein kinase that protects cells from stress-induced mitochondrial dysfunction [35]. While it has been shown that OPTN recruitment requires PINK1 kinase activity, but how PINK1 recruits OPTN remains unclear [34]. Together, these two studies highlight that PINK1 facilitates OPTN recruitment, and PRKN promotes OPTN stability in damaged mitochondria to activate mitophagy.
Like mitophagy, in xenophagy, intracellular bacterial pathogens are also targeted by cargo receptors via a ubiquitin-mediated pathway. At least four cargo receptors, namely CALCOCO2/NDP52 (calcium binding and coiled-coil domain 2), SQSTM1/p62 (sequestosome 1), NBR1 (NBR1 autophagy cargo receptor) and OPTN have been shown to bind ubiquitin chains on intracellular bacteria and direct ubiquitin-coated intracellular bacteria to autophagosomes [12,33,36,37]. Many E3 ubiquitin ligases, such as RNF166 (ring finger protein 166), LRSAM1 (leucine rich repeat and sterile alpha motif containing 1), PRKN and SMURF1 (SMAD specific E3 ubiquitin protein ligase 1), can localize to the surface of bacteria and ubiquitinate substrates [38–40]. However, how OPTN is recruited to ubiquitinated bacteria (cargos) to trigger autophagy is still an unclear process. Future studies of specific bacterial ubiquitinated substrates and their respective E3 enzymes may bring new insights into the molecular mechanism driving this process.
After being recruited, OPTN binds to ubiquitinated cargos. Interestingly, OPTN does not bind to linkage types of ubiquitin chains randomly. Rather, it has a binding preference for linear polyubiquitin chains and K63 chains rather than K48 chains or monoubiquitin modified substrates [29]. Crystal structure analysis revealed a complex of the OPTN UBD and linear diubiquitin, and the phosphorylation of OPTN plays a critical role in complex formation [41]. Furthermore, phosphorylation at S473 and dual phosphorylation of S473 and S513 in the UBD domain by TBK1 have been shown to enhance the binding of OPTN to multiple ubiquitin chains [27]. Moreover, using GST affinity-isolation assays, a study showed recently that phosphorylation of ubiquitin at S65 catalyzed by PINK1 prevents interaction with OPTN. However, OPTN UBD phosphorylation by TBK1 can partially rescue this loss of binding [27]. While many cargo-recognition studies have been done, how OPTN or other autophagy receptors selectively recognize the ubiquitin-modified cargos and how OPTN recognizes these cargos efficiently are still far from clear.
The role of OPTN in autophagosome formation
To form an autophagosome, OPTN binds to a ubiquitin-decorated cargo and links the ubiquitinated cargo to autophagosomal membranes via binding to LC3. Increasing evidence suggests that OPTN is actively participating in autophagosome formation steps, including the initiation of a phagophore, the biogenesis of a phagophore, and interaction with LC3 [42,43].
A variety of membrane sources and proteins are required for phagophore nucleation, and much evidence suggests that OPTN is involved in this process. At the early stage of PINK1-PRKN mitophagy, OPTN promotes the translocation of the ULK1 (unc-51 like autophagy activating kinase 1) complex to autophagy initiation sites. ULK1 is the first autophagy-specific complex recruited to autophagy initiation sites during autophagosome formation [34]. The ULK1 complex can drive the activation of the phosphatidylinositol 3-kinase (PtdIns3K) complex, which serves as the second kinase complex recruited to form an autophagosome [44–46]. The PIK3C3 (phosphatidylinositol 3-kinase catalytic subunit type 3) first generates a simple phosphoinositide, PtdIns3P, and then recruits the WIPI1 (WD repeat domain, phosphoinositide interacting 1)-WIPI2 (WD repeat domain, phosphoinositide interacting 2) effector to translocate to PtdIns(3,4,5)P3-enriched sites. Furthermore, WIPI1-WIPI2 binds to the ATG16L1 from the ATG12–ATG5-ATG16L1 complex, and then conjugates Atg8-family proteins to phagophore membranes [47,48]. In addition to ULK1 complex recruitment, OPTN can also form a complex with the WIPI2 and ATG12–ATG5 conjugate, facilitating the recruitment of the ATG12–ATG5-ATG16L1 complex to WIPI2-included phagophores [49]. The ATG12–ATG5-ATG16L1 complex is believed to act in the covalent modification of LC3 to the phosphatidylethanolamine (PE) in the phagophore membrane, suggesting that OPTN may also participate in the process of LC3 lipidation [50].
Interestingly, a recent study using a Fluoppi assay revealed that OPTN could interact with ATG9A vesicles to initiate local autophagosomal membrane formation, which is crucial for PRKN-mediated mitophagy [10]. ATG9, a sole transmembrane protein, provides a lipid/membrane source during the early steps of autophagosome formation [51]. However, it is enigmatic that ATG9-containing vesicles do not supply the bulk of the autophagosomal membrane in this process. Recently, Sawa-Makarska et al. recapitulated autophagosome formation’s initial steps using autophagy factors purified from yeast. This revealed that ATG9 vesicles serve as nucleators to establish membrane contact sites with a donor compartment such as the endoplasmic reticulum (ER) during the de novo formation of autophagosomes [11]. OPTN recruits ATG9 vesicles by interacting with ATG9A and cargos to initiate local autophagosomal membrane formation, indicating that OPTN is a very upstream regulator of autophagosomal membrane formation.
Generally speaking, OPTN is known to bind to Atg8-family proteins via its LIR domain, and this binding can be enhanced by the Ser177 phosphorylation of OPTN in the LIR domain by TBK1 [7]. The LIR motif within autophagy receptors is the main feature of selective autophagy models, in which autophagy receptors, such as SQSTM1, mediate the combination of Atg8-family protein associated membranes with the autophagic cargos. A previous study demonstrated that OPTN could recruit LC3 via its LIR domain and promote the lipidation of LC3 to the phagophore [50]. Unexpectedly, though OPTNF178A mutation led to loss of LC3 binding and impaired mitophagy, this mutant could still recruit all Atg8-family proteins, suggesting that the LIR domain of OPTN might be dispensable for LC3 recruitment in mitophagy [52]. This refreshing study showed that OPTN with a UBD mutation (OPTND474N) could still be recruited to depolarized mitochondria by Atg8-family protein-positive phagophores via the LIR domain, which could amplify the rate of autophagosome biogenesis after the initiation of autophagosome biogenesis [52].
In summary, during the autophagosome formation, OPTN recruits the ULK1 complex to initiate phagophore biogenesis and then directs LC3 lipidation by recruiting the ATG12–ATG5-ATG16L1 complex to phagophores [34,49]. Subsequently, OPTN recruits ATG9 vesicles to assist local autophagosomal membrane formation and promotes autophagosomal membrane recruitment to cargos by interacting with LC3 [10,53]. Autophagosome is generally formed by the interaction between autophagy receptors and LC3, which is a common feature of autophagy receptors such as OPTN, SQSTM1, and CALCOCO2 [2]. However, the recruitment of the ULK1 complex, the ATG12–ATG5-ATG16L1 complex and ATG9 vesicles is a unique function of OPTN, suggesting that it acts as a multifunctional autophagy receptor in autophagosome formation.
The role of OPTN in autophagosome maturation
OPTN plays a vital role in the maturation of autophagosomes through its interactions with MYO6 and LC3 family members. The fusion of autophagosome with lysosome is a crucial step in autophagy [54]. MYO6 is the only known unconventional myosin motor that moves in the opposite direction along actin filaments [55]. Recent studies have also clarified the importance of MYO6 in autophagy and mitochondrial homeostasis. Defects in MYO6 cause impeded autophagy and the accumulation of mitophagosomes [56]. Moreover, MYO6 can form actin cages to ensure the sequestration of damaged mitochondria [57]. Additionally, OPTN directly interacts with MYO6, binding with TOM1 (target of myb1 membrane trafficking protein) to deliver endosomal membranes to autophagosomes, which is required for autophagosome-lysosome fusion [58]. MYO6 or TOM1 dysfunction can reduce the autophagosomal delivery of endocytic cargo and block maturation from autophagosomes to autolysosomes [25,59,60]. Consistently, a similar molecular process can also be found during the maturation of S. typhimurium-containing autophagosomes [61]. Via a non-conventional LIR domain, CALCOCO2/NDP52 and OPTN can promote autophagosome maturation by interacting with several Atg8 orthologs, such as LC3A/B and GABARAPL2 (GABA type A receptor associated protein like 2), but not via an interaction with LC3C [61]. Another autophagy receptor, TAX1BP1 (Tax1 binding protein 1), also mediates the maturation of autophagosome by binding to MYO6, suggesting that OPTN is not the only autophagy receptor involved in the maturation of autophagosome [62]. However, whether OPTN is highly conserved in the maturation of autophagosomes is worth further attention.
The role of OPTN in lysosomal quality control
The lysosome is the central organelle for cellular degradation and recycling in eukaryotic cells. Autophagosomes fuse with lysosomes containing degrading enzymes in autophagy, leading to autophagosome content digestion. Though the lysosome’s functions have often been overlooked, they play an essential role in ensuring autophagy proceeds without a hitch. A recent study employed correlative light-electron microscopy (CLEM) in alpha synuclein-stimulated microglial and found that instead of SQSTM1 and CALCOCO2, OPTN was recruited to ubiquitinated lysosomes in cases of lysosomal quality impairment. In this study, OPTN restored the lysosomal quality control by lysophagy, a kind of selective autophagy mediating the degradation of lysosomes [63]. Autophagy induction is frequently associated with the upregulation of hydrolase synthesis and lysosome biogenesis [64]. When lysosomes are unstable or their functions are impaired, autophagy inhibitors tend to lessen the stress of autophagy on dysfunctional lysosomes by reducing autophagic cargo delivery to lysosomes rather than reducing an excessively aggressive autophagic process [64]. Generally, when lysosomal function is impaired, cells cannot tolerate robust autophagy induction, and autophagic flux is consequently inhibited [64]. Collectively, OPTN contributes to successful lysosomal quality control, which guarantees autophagic flux.
The role of OPTN as a substrate of autophagy
It has been established that OPTN can be degraded by the autophagic process and the ubiquitin-proteasome system. HACE1 has been identified as the E3 ubiquitin ligase responsible for the ubiquitylation of OPTN by yeast two-hybrid [28]. Moreover, endogenous OPTN can be stabilized by the lysosomal inhibitor bafilomycin A1, but not the proteasome inhibitor bortezomib [28]. Although another study demonstrated that the ubiquitin-proteasome system contributes to the major degradation of exogenous OPTN, OPTN can also be stabilized by bafilomycin A1 [65]. Lysosomes participate in the autophagosome-lysosome fusion in autophagy as well as the endosomal system. The results of OPTN accumulation by bafilomycin A1 illustrates autophagy might be a system for OPTN degradation. Further investigations should be performed to define the degradation mechanisms regulating OPTN.
OPTN mediates autophagic dysfunction in diseases
Autophagy dysfunction is associated with many pathological conditions, such as cancer, neurodegenerative diseases, and microbial infection, making autophagy a promising target for disease intervention. Particularly, OPTN-mediated autophagy-dysfunction is closely related to a variety of diseases (Table 1). Understanding the autophagic mechanisms of OPTN dysfunction in diseases may ultimately pave the road for novel disease intervention methods.
Table 1.
The different types of OPTN related diseases and autophagy related mechanisms
| Type of disease | Diseases | Mechanism | Reference |
|---|---|---|---|
| Neurodegenerative diseases | Glaucoma | OPTNE50K disrupts the interaction with RAB8 and induces oxidative stress; OPTNE50K enhances the interaction with TBK1 but reduces autolysosomes; OPTNE50K disrupts the oligomeric state of OPTN | [29,82,133,134] |
| ALS | OPTNE478G disrupts the ubiquitin-binding function of OPTN and affects PRKN-mediated mitophagy | [16] | |
| PD | Increased OPTN expression and colocalized puncta in PD-relevant brain regions with specific impairments of autophagy | [135] | |
| HD | Impaired autophagic clearance of aggregates colocalized with OPTN | [96,136–138] | |
| NIID | Unclear, abnormal accumulation of OPTN and its binding partner MYO6 in intranuclear inclusions of NIID | [87,136] | |
| Inflammatory diseases | CD | Clearance of intracellular bacteria by OPTN mediated xenophagy | [95,96] |
| PDB | There are several PDB genes (including OPTN) with functions related to the autophagic system | [106] | |
| RA | OPTN is upregulated while the related autophagy mechanism is unclear | [110] | |
| OP | optn deficiency caused inhibition of selectively degradation of FABP3 by autophagy | [120] | |
| ALF | OPTN is upregulated accompanied with autophagy activation after APAP treatment | [122] | |
| Cancer | Lung cancer | HACE1-OPTN axis mediated autophagy | [28] |
| Pancreatic cancer | OPTN involved chaperone-mediated autophagy (CMA) | [124] | |
| Nephropathy | DN | OPTN mediated mitophagy | [128,129] |
| AKI | OPTN mediated mitophagy | [130] |
AKI: acute kidney injury; ALF: acute liver failure; ALS: amyotrophic lateral sclerosis; CD: Crohn disease; DN: diabetic nephropathy; HD: Huntington disease; NIID, neuronal intranuclear inclusion disease; OP: osteoporosis; PD: Parkinson disease; PDB: Paget disease of bone; RA: rheumatoid arthritis.
OPTN-mediated autophagy alleviates neurodegenerative diseases
The most prevalent pathological feature of neurodegenerative diseases is the aggregation of proteins and dysfunctional mitochondria, which are both primarily degraded by autophagy. Many autophagy-associated genetic mutants have been found in neurodegenerative diseases, such as the OPTNE50K mutant in glaucoma and the OPTNE478G mutant in ALS. Abnormal protein aggregates, along with compromised autophagy, are found to co-occur in these neurodegenerative diseases. However, the mechanism of OPTN-mutant-related pathology in neurodegenerative diseases is still unclear. Herein, we provide a summary of OPTN-mediated autophagic mechanisms in these diseases.
Glaucoma
Glaucoma is the second leading cause of blindness in the world [14]. The name of “optineurin” was designated when the disease was first identified to correspond to one of the genes encoding the glaucoma form of the “optic neuropathy inducing” protein [14]. Mutations in the coding region of OPTN are associated with 16.7% of families with hereditary primary open-angle glaucoma [14]. The OPTNE50K mutant has been frequently and widely studied in glaucoma. Transgenic mice with the OPTNE50K mutation present a glaucoma phenotype, including a loss of retinal ganglion cells (RGCs) and the thinning of various cell layers of the retina and gliosis [66], which further confirms the association between OPTNE50K and glaucoma. Moreover, OPTN expression is significantly increased in human anterior segment organ cultures treated with glaucoma-related stimuli such as elevated-intraocular pressure (IOP), TNF (tumor necrosis factor) and dexamethasone [67], also indicating OPTN’s involvement in the occurrence of glaucoma.
Many studies have tried to elucidate the mechanism of OPTNE50K induced glaucoma. It has been reported that the E50K mutation can abnormally enhance OPTN’s interaction with TBK1 while disrupting its interaction with RAB8, leading to compromised autophagy and increased oxidative stress [68–71]. In different studies, there have been some disputes of OPTNE50K-mediated autophagy in glaucoma. Shim et al. presented the idea that OPTNE50K can induce mitophagy in RGCs. Increased mitophagosome number, upregulated LC3-II expression, and LC3 puncta were observed in the glial lamina axons of OPTNE50K−tg mice [72,73]. In this case, the accumulation of mitophagosomes and LC3 may have been caused by either autophagic activation or blockage of downstream steps in autophagy [74]. Therefore, further investigations are needed to determine if OPTNE50K can induce mitophagy. In another study, Madhavi et al. used an mCherry-GFP-LC3B plasmid to monitor autophagy. Once the mCherry-GFP-LC3B plasmid entered into autolysosomes, only red fluorescence was detected while the GFP fluorescence was quenched in the acidic autolysosomes. In amino acid starvation conditions, OPTNE50K retinal cells showed fewer autolysosomes indicated by reduced red fluorescence, suggesting the inhibition of autophagic flux by OPTNE50K [75]. In addition, OPTNE50K colocalized more often with autophagosomes than wild-type OPTN. The autophagy activator rapamycin treatment significantly inhibited OPTNE50K induced retinal cell death, indicating that OPTNE50K disrupted autophagy in retinal cells [75].
It is worth mentioning that OPTNE50K mutant retinal cells show abnormal insolubility, and present small dot-like deposits in the outer plexiform and inner nuclear layers. Studies have suggested that the insoluble aggregation in OPTNE50K mutant retinal cells may be related to an enhanced TBK1 interaction with OPTNE50K. As shown in this study, abnormal insolubility could be resolved by BX795, a specific TBK1 inhibitor [29,68]. Though the mechanism of OPTNE50K mediated autophagy in glaucoma is elusive, we think that an enhanced interaction with TBK1 may reduce the binding affinity of OPTN to its other interaction partners. In addition, as a delayed glaucoma diagnosis may lead to more severe symptoms without effective treatment [76], it is worth testing if TBK1 inhibitors could act as a potential medication for OPTNE50K-related glaucoma.
Amyotrophic lateral sclerosis
ALS is a progressive neurodegenerative disease characterized by the degeneration of motor neurons in the primary motor cortex, brain stem, and spinal cord [77]. Many genes have been reported to be associated with familial ALS/fALS, including SOD1, TBK1, SQSTM1, and OPTN [78–80]. Several types of OPTN mutations occur in ALS patients, including a homozygous deletion of exon 5, a homozygous Q398X nonsense mutation, a heterozygous E478G missense mutation, and a novel Q454E missense mutation. Both E478G and Q454E are located in the UBD of OPTN [13,81]. OPTNE478G and OPTNQ454E mutations result in impaired mitophagy after carbonyl cyanide 3-chlorophenylhydrazone/CCCP treatment in HeLa cells [82]. Moreover, optn−/- mice exhibit accumulated mitochondria in the hypoglossal nerve axons, which further implicates the involvement of OPTN-mediated autophagy in ALS development [83].
The OPTNE478G mutation is located in the UBD region, which abolishes OPTN’s binding to ATG12–ATG5 conjugates, ubiquitin, and MYO6 and leads to severe disruption in several stages of autophagy [7,49]. OPTNE478G mutation impairs the ability of OPTN to form autophagosomes probably because it disrupts complex formation between OPTN and ATG12–ATG5 conjugates, which further undermines the recruitment of the ATG12–ATG5-ATG16L1 complex to WIPI2-positive structures and leads to defective autophagy [49]. In addition, OPTNE478G mutation disrupts ubiquitin binding and thus impairs autophagosomal membrane recruitment [7]. In HeLa cells, OPTNE478G consistently reduces the capacity of LC3 recruitment to mitochondria and slows autophagosome formation [16]. It is worth noting that OPTNE478G can also cause a defect in linear chain ubiquitin-binding capacity, resulting in the dysregulation of IKK-mediated NFKB activation and the consequent aggravation of the pathogenesis of ALS [84]. In addition, the interaction between OPTN and MYO6 is an essential process for facilitating autophagosome maturation in autophagy [25]. A potential mechanism could be that OPTNE478G mutants disrupt the association of OPTN with MYO6 [16]. Therefore, OPTNE478G mutation causes less colocalization with MYO6, which inhibits autophagosome-lysosome fusion and results in autophagy impairment. In summary, although how OPTNE478G causes defective autophagy is becoming clearer, the connection between ALS pathogenesis and defective autophagy caused by OPTNE478G is still not resolved.
Other neurodegenerative diseases
In addition to the ubiquitin-dependent mechanism, OPTN can also recognize and actively participate in the degradation of HTT exon 1 Q103 aggregation in Huntington disease (HD) and SOD1G93C aggregation in ALS through its C-terminal CC domain in a ubiquitin-independent manner, suggesting a diverse role of OPTN in autophagic clearance in different neurodegenerative diseases [85]. Moreover, OPTN is frequently observed in the protein inclusions of neurons and glia in Parkinson disease (PD) and neuronal intranuclear inclusion disease (NIID) [86–88]. We theorize that this OPTN positive protein inclusion accumulation may be mainly caused by defective autophagy, leading to neurodegeneration. Moreover, several investigations have shown that OPTN deficiency leads to progressive demyelination in the CNS (central nervous system), indicating a connection between OPTN and demyelination diseases [83,89]. Nevertheless, it is not clear whether OPTN-associated autophagy is involved in demyelination, and further studies are required in this field to resolve this important question.
Interestingly, SQSTM1 also acts as an autophagy receptor to remove the abnormal protein aggregates and destroy organelles to protect form neuron death in several neurodegenerative diseases such as PD, AD, HD, and ALS [90]. Except for working as an autophagy receptor like SQSTM1, OPTN with specific mutations can also cause defective autophagosome nucleation and autophagosome maturation in ALS [25,49], indicating the multiple functions of OPTN as an autophagy receptor during disease pathology.
OPTN activates autophagy to reduce inflammation
A growing body of evidence suggests that autophagy plays a protective role in inflammatory diseases. In addition, several inflammatory diseases have been reported to be closely related to OPTN. Given that OPTN has a direct role in inflammation, its autophagic functions in inflammation are often underestimated. Here, we review current research on the association of OPTN with inflammatory diseases, aiming to disclose the autophagic mechanism of OPTN in these diseases.
Crohn disease
Crohn disease (CD) is characterized by chronic inflammation in the gut, with aberrant immune responses to gut microbiota during disease pathogenesis [91]. Patients with CD may experience an abnormal intestinal bacteria overgrowth, and bacterial infection control is critical in the management of CD. On the molecular level, xenophagy is a type of selective autophagy that specifically targets intracellular pathogens [92]. Many genetic studies have demonstrated that mutation of autophagy-associated genes can increase susceptibility to CD, which reveals the critical role of autophagy in intestinal homeostasis [93]. Susceptibility loci for this disease include a coding variant in ATG16L1 (ATG16L1T300A) and polymorphisms in IRGM (immunity related GTPase M), both of which are autophagy-related genes, implicating a role for xenophagy in intestinal homeostasis and disease [94]. Interestingly, in macrophages, OPTN expression was shown to decrease in a subpopulation of CD patients revealed by a microarray study. Additionally, macrophages from both optn-deficient mice and OPTNlow patients showed lower levels of TNF and IL6 (interleukin 6) after HkEc (heat-killed Escherichia coli) stimulation than control cells [6]. Moreover, optn-deficient mice were shown to be more susceptible to Citrobacter colitis, E. coli peritonitis, and Salmonella infection [95]. Also, optn-knockdown zebrafish infected with Salmonella experience higher mortality risk [96]. In intestinal epithelial cells, impaired autophagy always results in more susceptibility to intestinal inflammation [97]. To restrict bacterial proliferation, OPTN, which mediates xenophagy, competes with IKBKG, and triggers local NFKB activation, allowing for ubiquitin-coated bacteria binding [98]. However, a deficiency in OPTN would not affect NFKB activation after treatment with TNF, lipopolysaccharides or polyinosinic-polycytidylic acid/poly(I:C) [99]. Together, these data indicate that OPTN-mediated xenophagy is essential for restricting bacterial proliferation in the pathogenesis of CD.
Paget disease of bone
Paget disease of bone (PDB) is a common bone disorder characterized by disorganized bone remodeling with unknown pathogenesis [100]. The most likely cause of PDB is chronic paramyxovirus infection in genetically susceptible people [101]. Genome-wide association studies (GWAS) reveal a locus situated on chromosome 10p13, the locus of OPTN, which showed significant association with familial PDB. Therefore, the risk haplotype can be associated with rare allele(s) within OPTN that markedly increases susceptibility to PDB [102,103]. Consistently, it has been reported that 100% of aged optn−/- mice present the clinical features of human PDB patients, including polyostotic osteolytic lesions, mixed-phase lesions, and elevated serum ALP (alkaline phosphatase) levels [104]. Mechanistically, optn-deficiency enhances osteoclast differentiation, which significantly decreases the production of type I interferon (IFN) and type I IFN signaling through IFNAR/IFNα/βR (interferon alpha and beta receptor), and meanwhile increases NFKB activity through a CYLD-dependent pathway [104,105]. Although current mechanistic studies mainly focused on the OPTN modulation of NFKB and IFN signaling [105], OPTN mediated autophagy may also be involved in the progression of this disease. In addition to OPTN, PDB is also related to several other genes in the autophagic system, including SQSTM1, VCP (valosin containing protein), and ATG16L1, which suggests a strong connection between autophagy and PDB [106–108]. For instance, a SQSTM1 mutant in PDB has been recently reported to induce autophagy blockage [109], suggesting autophagy’s potential role in PDB progression. Future studies focusing on OPTN or SQSTM1 in PDB will provide further insight into the relationship between autophagy and PDB.
Rheumatoid Arthritis
OPTN has also been associated with rheumatoid arthritis (RA), a chronic erosive polyarthritis characterized by systemic inflammatory autoimmunity. OPTN was determined to functionally downregulate TNFSF11 (TNF superfamily member 11), a protein primarily responsible for the development of bone erosions in RA patients [110]. OPTN knockdown in rheumatoid arthritis synovial fibroblasts results in increased osteoclast formation when co-cultured with monocytes from RA patients [110]. Additionally, cytokines derived from immune responses, such as TNF and IFNG/IFN-γ, have been shown to increase OPTN expression in a mimic of inflammatory conditions to develop RA joint [111,112]. The finding above provides further evidence that OPTN may play a protective role in RA pathogenesis. Given that autophagy degradation of polyubiquitinated proteins is one of the fundamental mechanisms for maintaining rheumatoid arthritis synovial fibroblast homeostasis [113], autophagy tends to negatively regulate the innate immune signaling pathway by degrading the cytoplasmic signaling complexes in RA. The current opinion is that OPTN inhibits NFKB-induced TNFSF11 (TNF superfamily member 11) expression [22,114]; however, whether OPTN-mediated autophagy affects RA progression should also be examined.
Osteoporosis
Osteoporosis is an inflammatory disease characterized by osteoporotic bone loss during aging [115]. In the bones of aged mice, osteopenia is associated with lower autophagic activity, which can be alleviated by autophagy activation [116–118]. Recent studies have shown that OPTN plays dual roles in osteogenesis. OPTN inhibits inflammation-related STAT1 (signal transducer and activator of transcription 1) signaling to promote osteoblast differentiation [119]. OPTN deficient mice display impaired autophagic activity, decreased bone mass, and reduced bone rigidity [120]. Mechanistically, optn deficiency in mesenchymal stem cells impairs autophagy and inhibits selective recognition and degradation of adipogenesis and osteogenesis-related FABP3 (fatty acid binding protein 3), leading to decreased osteogenesis, increased adipogenesis, and a phenotype with elevated senescence [120]. Though autophagy is known to prevent cellular senescence during aging, the molecular mechanism remains elusive, especially the effect of OPTN-regulated autophagy on senescence phenotypes.
Other inflammatory diseases
Autophagy is closely related to hepatic inflammatory diseases, including drug-induced liver injury, virus-induced liver injury, nonalcoholic fatty liver disease, and liver fibrosis. In most liver diseases, autophagy fights to keep cells alive in the presence of “life-threatening” stimuli [121]. Specifically, increased OPTN expression in mice livers injured by acetaminophen (APAP) overdose were found to be accompanied by increased autophagic flux, indicating that OPTN mediated autophagy may also play a role in the progression of acute liver injury [122]. Another study shows that OPTN downregulates TCR (T Cell Receptor)-induced NFKB activation and TNF production to regulate T cell activation during autoimmune diseases [123], revealing a new biological function of OPTN in inflammatory diseases.
Taken together, it is clear that OPTN functions in these inflammatory diseases. Mechanistically, on the one hand, OPTN directly modulates inflammatory signaling pathways independent of autophagy [105]. On the other hand, OPTN-mediated autophagy can remove intracellular bacteria to regulate inflammation indirectly [98]. However, it is still confusing whether OPTN regulates other inflammatory processes such as inflammatory cytokine secretion in an autophagy-dependent manner. It is worth noting that, similar to OPTN, SQSTM1 also plays a role in PDB, RA, and APAP-induced acute liver injury, indicating a general role of autophagy receptors in inflammatory diseases.
OPTN-mediated autophagy in cancer progression
OPTN is regarded as a versatile factor in the progression of cancer in general. HACE1 can ubiquitinate OPTN on Lys193 and then form an autophagic complex with SQSTM1. This is a critical step in HACE1-activated autophagy to accelerate the total cellular autophagic flux and facilitate tumor suppression, suggesting that OPTN is a tumor suppressor in lung cancer [28]. The Cancer Genome Atlas (TCGA) database analysis showed high expression of the OPTN gene across several tumor types, especially in pancreatic and renal cancers [124]. Furthermore, OPTN knockdown had a limited effect on the proliferation of different human pancreatic cancer cell lines, but it could significantly increase cell migration and reduce colony formation. Although the authors of this study discussed the role of OPTN knockdown induced autophagy inhibition in increased cell migration and apoptosis, the precise mechanism is still obfuscated [124].
It has been reported that sustained SQSTM1 expression resulting from autophagy defects promotes tumor formation [125]. SQSTM1 overexpression in clear cell renal cell carcinoma/ccRCC lines was reported to promote resistance to oxidative stress and increased tumor formation [126]. These data suggest an oncogenic role of SQSTM1 during tumorigenesis. Unlike SQSTM1, OPTN suppresses tumor growth by activating autophagy in lung cancer [28], while optn knockdown induces apoptosis in pancreatic cancer cells [124], suggesting dual roles of OPTN in different tumors. In summary, in contrast to SQSTM1, OPTN-mediated autophagy has different effects on tumorigenesis.
OPTN enhances mitophagy to protect nephropathy
Diabetic nephropathy (DN) is one of the most severe and frequent chronic complications of type 1 diabetes [127]. Recent studies have revealed that OPTN presents anti-senescent and protective effects in diabetic nephropathy through enhancing mitophagy [128]. Renal OPTN expression has been shown negatively correlate with tubulointerstitial injury scores in clinical specimens [128]. Moreover, OPTN expression is negatively correlated with serum creatinine levels and positively correlated with eGFR (estimated glomerular filtration rates), suggesting a close relationship between OPTN and DN progression. Interestingly, the expression of OPTN in human renal tubular epithelial cells (RTECs) is always negatively correlated with the senescence marker CDKN2A/p16 (cyclin dependent kinase inhibitor 2A). OPTN in mouse RTECs enhances mitophagosome formation, leading to the downregulation of cellular senescence markers, such as CDKN2A, CDKN1A/p21 (cyclin dependent kinase inhibitor 1A), SA-GLB1/β-gal (senescence-associated galactosidase beta 1), and senescence-associated heterochromatin foci [128,129]. Though OPTN-induced mitophagy can negatively regulate RTEC’s senescence, it is not clear if cellular senescence or OPTN-mediated mitophagy is a critical factor of DN. Therefore, more definitive studies are needed. Similarly, OPTN-mediated mitophagy may also be protective in AKI (acute kidney injury) induced by sepsis, one of the major types of AKI with extremely high morbidity and mortality rates [130]. Although the protective role of OPTN-mediated mitophagy in kidney injury has been well established, further study is needed to decipher how OPTN enhances mitophagy to protect the kidney from injury.
Perspectives
Except for an autophagy receptor, OPTN regulates autophagosome nucleation, autophagosome maturation, lysosomal quality control, and autophagic degradation, making OPTN a multifunctional receptor in the autophagic process. OPTNE478G mutant-related autophagic flux disruption cannot be restored by activating other autophagy receptors, indicating the essential role of OPTN in the autophagic process [34]. Moreover, OPTN is linked to various diseases through its autophagic roles. The pathogenesis of disorders related to OPTN mutation or dysfunction, unfortunately, is not yet fully understood. Although the autophagic mechanisms mediated by OPTN are very well clarified in ALS, Crohn disease, and lung cancer, the studies in other diseases are relatively superficial and only show correlations between OPTN and these diseases.
Interestingly, several different mutations of OPTN with autophagic function loss are associated with different diseases, such as OPTNE478G in ALS and OPTNE50K in glaucoma [13,66]. We think this is because of the heterogenicity of OPTN’s function in different kinds of cells or tissues. Therefore, the choice of specific target cells is quite important in any etiological and mechanistic studies of different diseases.
Collectively, OPTN usually plays a protective role in diseases via OPTN-mediated autophagy, such as in lung cancer, neurodegenerative diseases, osteoporosis, and kidney injury. Coincidently, these diseases all appear with low autophagic activity, which might be associated with either mutation or low expression of OPTN [131,132]. Unexpectedly, our unpublished data have shown that OPTN may act as a negative regulator in experimental autoimmune encephalomyelitis pathology via an autophagy-independent mechanism. Thus, OPTN may play distinctive roles in different diseases, depending on its primary molecular function in each respective condition.
In summary, the multiple roles of OPTN in the autophagy process are of great significance, and new tools are required to monitor autophagy flux to better clarify the precise function of OPTN. Given the close relationship of OPTN and autophagy-related diseases, OPTN can be a key regulator and a potential novel target for disease intervention, and will require further studies in the future.
Funding Statement
This work was supported by the National Natural Science Foundation of China [82073857]; Natural Science Foundation of Zhejiang Province [LR21H310001].
Disclosure statement
The authors declare no competing interests.
References
- [1].Zaffagnini G, Martens S.. Mechanisms of Selective Autophagy. J Mol Biol. 2016. May 8;428(9Pt A):1714–1724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [2].Johansen T, Lamark T.. Selective autophagy mediated by autophagic adapter proteins. Autophagy. 2011. Mar;7(3):279–296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [3].Tesseraud S, Avril P, Bonnet M, et al. Autophagy in farm animals: current knowledge and future challenges. Autophagy. 2020. Jul;30:1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Rezaie T, Sarfarazi M. Molecular cloning, genomic structure, and protein characterization of mouse optineurin. Genomics. 2005. Jan;85(1):131–138. [DOI] [PubMed] [Google Scholar]
- [5].Xia B, Yu J, He T, et al. Lactobacillus johnsonii L531 ameliorates enteritis via elimination of damaged mitochondria and suppression of SQSTM1-dependent mitophagy in a Salmonella infantis model of piglet diarrhea. Faseb J. 2020. Feb;34(2):2821–2839. . [DOI] [PubMed] [Google Scholar]
- [6].Smith AM, Sewell GW, Levine AP, et al. Disruption of macrophage pro-inflammatory cytokine release in Crohn’s disease is associated with reduced optineurin expression in a subset of patients. Immunology. 2015. Jan;144(1):45–55. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [7].Wild P, Farhan H, McEwan DG, et al. Phosphorylation of the autophagy receptor optineurin restricts Salmonella growth. Science. 2011. Jul 8;333(6039):228–233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [8].Ying H, Yue BY. Optineurin: the autophagy connection. Exp Eye Res. 2016. Mar;144:73–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Shen XA, Ying HY, Qiu Y, et al. Processing of Optineurin in Neuronal Cells. J Biol Chem. 2011. Feb 4;286(5):3618–3629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [10].Yamano K, Kikuchi R, Kojima W, et al. Critical role of mitochondrial ubiquitination and the OPTN-ATG9A axis in mitophagy. J Cell Biol. 2020. Sep 7;219(9). doi: 10.1083/jcb.201912144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Sawa-Makarska J, Baumann V, Coudevylle N, et al. Reconstitution of autophagosome nucleation defines Atg9 vesicles as seeds for membrane formation. Science. 2020. Sep 4;369(6508):1206-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [12].Stolz A, Ernst A, Dikic I. Cargo recognition and trafficking in selective autophagy. Nat Cell Biol. 2014. Jun;16(6):495–501. [DOI] [PubMed] [Google Scholar]
- [13].Maruyama H, Morino H, Ito H, et al. Mutations of optineurin in amyotrophic lateral sclerosis. Nature. 2010. May 13;465(7295):223–226. [DOI] [PubMed] [Google Scholar]
- [14].Rezaie T, Child A, Hitchings R, et al. Adult-onset primary open-angle glaucoma caused by mutations in optineurin. Science. 2002. Feb 8;295(5557):1077–1079. [DOI] [PubMed] [Google Scholar]
- [15].Aung T, Rezaie T, Okada K, et al. Clinical features and course of patients with glaucoma with the E50K mutation in the optineurin gene. Invest Ophthalmol Vis Sci. 2005. Aug;46(8):2816–2822. [DOI] [PubMed] [Google Scholar]
- [16].Wong YC, Holzbaur EL.. Optineurin is an autophagy receptor for damaged mitochondria in parkin-mediated mitophagy that is disrupted by an ALS-linked mutation. Proc Natl Acad Sci U S A. 2014. Oct 21;111(42):E4439–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Rezaie T, Waitzman DM, Seeman JL, et al. Molecular cloning and expression profiling of optineurin in the rhesus monkey. Invest Ophthalmol Vis Sci. 2005. Jul;46(7):2404–2410. . [DOI] [PubMed] [Google Scholar]
- [18].Lang T, Reiche S, Straub M, et al. Autophagy and the cvt pathway both depend on AUT9. J Bacteriol. 2000. Apr;182(8):2125–2133. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Noda T, Kim J, Huang WP, et al. Apg9p/Cvt7p is an integral membrane protein required for transport vesicle formation in the Cvt and autophagy pathways. J Cell Biol. 2000. Feb 7;148(3):465–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Young ARJ, Chan EYW, Hu XW, et al. Starvation and ULK1-dependent cycling of mammalian Atg9 between the TGN and endosomes. J Cell Sci. 2006. Sep 15;119(18):3888–3900. [DOI] [PubMed] [Google Scholar]
- [21].Park BC, Shen X, Samaraweera M, et al. Studies of optineurin, a glaucoma gene: golgi fragmentation and cell death from overexpression of wild-type and mutant optineurin in two ocular cell types. Am J Pathol. 2006. Dec;169(6):1976–1989. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Zhu G, Wu CJ, Zhao Y, et al. Optineurin negatively regulates TNFalpha- induced NF-kappaB activation by competing with NEMO for ubiquitinated RIP. Curr Biol. 2007. Aug 21;17(16):1438–1443. [DOI] [PubMed] [Google Scholar]
- [23].Yamaoka S, Courtois G, Bessia C, et al. Complementation cloning of NEMO, a component of the IkappaB kinase complex essential for NF-kappaB activation. Cell. 1998. Jun 26;93(7):1231–1240. [DOI] [PubMed] [Google Scholar]
- [24].Clark K, Nanda S, Cohen P. Molecular control of the NEMO family of ubiquitin-binding proteins. Nat Rev Mol Cell Biol. 2013. Oct;14(10):673–685. [DOI] [PubMed] [Google Scholar]
- [25].Tumbarello DA, Waxse BJ, Arden SD, et al. Autophagy receptors link myosin VI to autophagosomes to mediate Tom1-dependent autophagosome maturation and fusion with the lysosome. Nat Cell Biol. 2012. Oct;14(10):1024–1035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Hattula K, Peranen J. FIP-2, a coiled-coil protein, links Huntingtin to Rab8 and modulates cellular morphogenesis. Curr Biol. 2000. Dec 14;10(24):1603–1606. [DOI] [PubMed] [Google Scholar]
- [27].Heo JM, Ordureau A, Paulo JA, et al. The PINK1-PARKIN Mitochondrial Ubiquitylation Pathway Drives a Program of OPTN/NDP52 Recruitment and TBK1 Activation to Promote Mitophagy. Mol Cell. 2015. Oct 1;60(1):7–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Liu Z, Chen P, Gao H, et al. Ubiquitylation of autophagy receptor Optineurin by HACE1 activates selective autophagy for tumor suppression. Cancer Cell. 2014. Jul 14;26(1):106–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [29].Li F, Xie X, Wang Y, et al. Structural insights into the interaction and disease mechanism of neurodegenerative disease-associated optineurin and TBK1 proteins. Nat Commun. 2016. Sep 13;7(1):12708. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Smith MD, Wilkinson S. CCPG1, an unconventional cargo receptor for ER-phagy, maintains pancreatic acinar cell health. Mol Cell Oncol. 2018;5(5):5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [31].Turco E, Witt M, Abert C, et al. FIP200 Claw Domain Binding to p62 Promotes Autophagosome Formation at Ubiquitin Condensates. Mol Cell. 2019. Apr 18;74(2):330-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [32].Vargas JNS, Wang CX, Bunker E, et al. Spatiotemporal Control of ULK1 Activation by NDP52 and TBK1 during Selective Autophagy. Mol Cell. 2019. Apr 18;74(2):347-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Rogov V, Dotsch V, Johansen T, et al. Interactions between autophagy receptors and ubiquitin-like proteins form the molecular basis for selective autophagy. Mol Cell. 2014. Jan 23;53(2):167–178. [DOI] [PubMed] [Google Scholar]
- [34].Lazarou M, Sliter DA, Kane LA, et al. The ubiquitin kinase PINK1 recruits autophagy receptors to induce mitophagy. Nature. 2015. Aug 20;524(7565):309–314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Valente EM, Abou-Sleiman PM, Caputo V, et al. Hereditary early-onset Parkinson’s disease caused by mutations in PINK1. Science. 2004. May 21;304(5674):1158–1160. [DOI] [PubMed] [Google Scholar]
- [36].Deretic V. Autophagy as an innate immunity paradigm: expanding the scope and repertoire of pattern recognition receptors. Curr Opin Immunol. 2012. Feb;24(1):21–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Mejlvang J, Olsvik H, Svenning S, et al. Starvation induces rapid degradation of selective autophagy receptors by endosomal microautophagy. J Cell Biol. 2018. Oct 1;217(10):3640–3655. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Heath RJ, Goel G, Baxt LA, et al. RNF166 Determines Recruitment of Adaptor Proteins during Antibacterial Autophagy. Cell Rep. 2016. Nov 22;17(9):2183–2194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Franco LH, Nair VR, Scharn CR, et al. The Ubiquitin Ligase Smurf1 Functions in Selective Autophagy of Mycobacterium tuberculosis and Anti-tuberculous Host Defense. Cell Host Microbe. 2017. Jan 11;21(1):59–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Huett A, Heath RJ, Begun J, et al. The LRR and RING domain protein LRSAM1 is an E3 ligase crucial for ubiquitin-dependent autophagy of intracellular Salmonella Typhimurium. Cell Host Microbe. 2012. Dec 13;12(6):778–790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [41].Li FX, Xu DC, Wang YL, et al. Structural insights into the ubiquitin recognition by OPTN (optineurin) and its regulation by TBK1-mediated phosphorylation. Autophagy. 2018;14(1):66–79. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [42].Song GJ, Jeon H, Seo M, et al. Interaction between optineurin and Rab1a regulates autophagosome formation in neuroblastoma cells. J Neurosci Res. 2018. Mar;96(3):407–415. . [DOI] [PubMed] [Google Scholar]
- [43].Evans CS, Holzbaur ELF. Degradation of engulfed mitochondria is rate-limiting in Optineurin-mediated mitophagy in neurons. Elife. 2020. Jan;14;9(18):1713–1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [44].Zhou X, Babu JR, Da Silva S, et al. Unc-51-like kinase 1/2-mediated endocytic processes regulate filopodia extension and branching of sensory axons. Proc Natl Acad Sci U S A. 2007. Apr 3;104(14):5842–5847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [45].Ganley IG, Lam Du H, Wang J, et al. ULK1.ATG13.FIP200 complex mediates mTOR signaling and is essential for autophagy. J Biol Chem. 2009. May 1;284(18):12297–12305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [46].Mercer CA, Kaliappan A, Dennis PB. A novel, human Atg13 binding protein, Atg101, interacts with ULK1 and is essential for macroautophagy. Autophagy. 2009. Jul;5(5):649–662. [DOI] [PubMed] [Google Scholar]
- [47].Dikic I, Elazar Z. Mechanism and medical implications of mammalian autophagy. Nat Rev Mol Cell Bio. 2018. Jun;19(6):349–364. [DOI] [PubMed] [Google Scholar]
- [48].Yu L, Chen Y, Tooze SA. Autophagy pathway: cellular and molecular mechanisms. Autophagy. 2018;14(2):207–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [49].Bansal M, Moharir SC, Sailasree SP, et al. Optineurin promotes autophagosome formation by recruiting the autophagy-related Atg12-5-16L1 complex to phagophores containing the Wipi2 protein. J Biol Chem. 2018. Jan 5;293(1):132–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [50].Dooley HC, Razi M, Polson HEJ, et al. WIPI2 Links LC3 Conjugation with PI3P, Autophagosome Formation, and Pathogen Clearance by Recruiting Atg12-5-16L1. Mol Cell. 2014. Jul 17;55(2):238–252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [51].Reggiori F, Tucker KA, Stromhaug PE, et al. The Atg1-Atg13 complex regulates Atg9 and Atg23 retrieval transport from the pre-autophagosomal structure. Dev Cell. 2004. Jan;6(1):79–90. . [DOI] [PubMed] [Google Scholar]
- [52].Padman BS, Nguyen TN, Uoselis L, et al. LC3/GABARAPs drive ubiquitin-independent recruitment of Optineurin and NDP52 to amplify mitophagy. Nat Commun. 2019. Jan 24;10(1):408. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [53].Richter B, Sliter DA, Herhaus L, et al. Phosphorylation of OPTN by TBK1 enhances its binding to Ub chains and promotes selective autophagy of damaged mitochondria. Proc Natl Acad Sci U S A. 2016. Apr 12;113(15):4039–4044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [54].Tian X, Teng J, Chen J. New insights regarding SNARE proteins in autophagosome-lysosome fusion. Autophagy. 2020. Sep;24:1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [55].Buss F, Spudich G, Kendrick-Jones K-J-J. MYOSIN VI: cellular Functions and Motor Properties. Annu Rev Cell Dev Biol. 2004;20(1):649–676. [DOI] [PubMed] [Google Scholar]
- [56].Schubert T, Gleiser C, Heiduschka P, et al. Deletion of myosin VI causes slow retinal optic neuropathy and age-related macular degeneration (AMD)-relevant retinal phenotype. Cell Mol Life Sci. 2015. Nov;72(20):3953–3969. . [DOI] [PMC free article] [PubMed] [Google Scholar]
- [57].Kruppa AJ, Kishi-Itakura C, Masters TA, et al. Myosin VI-Dependent Actin Cages Encapsulate Parkin-Positive Damaged Mitochondria. Dev Cell. 2018. Feb 26;44(4):484-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [58].Hu S, Guo Y, Wang Y, et al. Structure of Myosin VI/Tom1 complex reveals a cargo recognition mode of Myosin VI for tethering. Nat Commun. 2019. Aug 1;10(1):3459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [59].Sundaramoorthy V, Walker AK, Tan V, et al. Defects in optineurin- and myosin VI-mediated cellular trafficking in amyotrophic lateral sclerosis. Hum Mol Genet. 2015. Jul 1;24(13):3830–3846. [DOI] [PubMed] [Google Scholar]
- [60].O’Loughlin T, Masters TA, The BF. MYO6 interactome reveals adaptor complexes coordinating early endosome and cytoskeletal dynamics. Embo Rep. 2018. Apri;19(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [61].Verlhac P, Gregoire IP, Azocar O, et al. Autophagy receptor NDP52 regulates pathogen-containing autophagosome maturation. Cell Host Microbe. 2015. Apr 8;17(4):515–525. [DOI] [PubMed] [Google Scholar]
- [62].Hu SC, Wang YL, Gong YK, et al. Mechanistic Insights into Recognitions of Ubiquitin and Myosin VI by Autophagy Receptor TAX1BP1. J Mol Biol. 2018. Sep 14;430(18):3283–3296. [DOI] [PubMed] [Google Scholar]
- [63].Bussi C, Peralta Ramos JM, Arroyo DS, et al. Alpha-synuclein fibrils recruit TBK1 and OPTN to lysosomal damage sites and induce autophagy in microglial cells. J Cell Sci. 2018. Nov 30;131(23):jcs226241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [64].Ghavami S, Shojaei S, Yeganeh B, et al. Autophagy and apoptosis dysfunction in neurodegenerative disorders. Prog Neurobiol. 2014;112:24–49. [DOI] [PubMed] [Google Scholar]
- [65].Mao J, Xia Q, Liu C, et al. A critical role of Hrd1 in the regulation of optineurin degradation and aggresome formation. Hum Mol Genet. 2017. May 15;26(10):1877–1889. [DOI] [PubMed] [Google Scholar]
- [66].Meng Q, Xiao Z, Yuan H, et al. Transgenic mice with overexpression of mutated human optineurin(E50K) in the retina. Mol Biol Rep. 2012. Feb;39(2):1119–1124. [DOI] [PubMed] [Google Scholar]
- [67].Vittitow JL, Borras T. Expression of optineurin, a glaucoma-linked gene, is influenced by elevated intraocular pressure. Biochem Biophys Res Commun. 2002. Oct 18;298(1):67–74. [DOI] [PubMed] [Google Scholar]
- [68].Minegishi Y, Iejima D, Kobayashi H, et al. Enhanced optineurin E50K-TBK1 interaction evokes protein insolubility and initiates familial primary open-angle glaucoma. Hum Mol Genet. 2013. Sep 1;22(17):3559–3567. [DOI] [PubMed] [Google Scholar]
- [69].Chi ZL, Akahori M, Obazawa M, et al. Overexpression of optineurin E50K disrupts Rab8 interaction and leads to a progressive retinal degeneration in mice. Hum Mol Genet. 2010. Jul 1;19(13):2606–2615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [70].De Marco N, Buono M, Troise F, et al. Optineurin increases cell survival and translocates to the nucleus in a Rab8-dependent manner upon an apoptotic stimulus. J Biol Chem. 2006. Jun 9;281(23):16147–16156. [DOI] [PubMed] [Google Scholar]
- [71].Chalasani ML, Radha V, Gupta V, et al. A glaucoma-associated mutant of optineurin selectively induces death of retinal ganglion cells which is inhibited by antioxidants. Invest Ophthalmol Vis Sci. 2007. Apr;48(4):1607–1614. [DOI] [PubMed] [Google Scholar]
- [72].Shim MS, Kim KY, Noh M, et al. Optineurin E50K triggers BDNF deficiency-mediated mitochondrial dysfunction in retinal photoreceptor cell line. Biochem Biophys Res Commun. 2018. Sep 18;503(4):2690–2697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [73].Shim MS, Takihara Y, Kim KY, et al. Mitochondrial pathogenic mechanism and degradation in optineurin E50K mutation-mediated retinal ganglion cell degeneration. Sci Rep. 2016. Sep 22;6(1):33830. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [74].Chu Y, Kang Y, Yan C, et al. LUBAC and OTULIN regulate autophagy initiation and maturation by mediating the linear ubiquitination and the stabilization of ATG13. Autophagy. 2020. Jun;26:1–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [75].Chalasani ML, Kumari A, Radha V, et al. E50K-OPTN-induced retinal cell death involves the Rab GTPase-activating protein, TBC1D17 mediated block in autophagy. PloS One. 2014;9(4):e95758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [76].Weinreb RN, Aung T, Medeiros FA. Weinreb RN, Aung T, Medeiros FA. The Pathophysiology and . Treatment of Glaucoma A Review. Jama-J Am Med Assoc.2014. May 14;311(18):1901–1911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [77].Al-Chalabi A, Hardiman O. The epidemiology of ALS: a conspiracy of genes, environment and time. Nat Rev Neurol. 2013. Nov;9(11):617–628. [DOI] [PubMed] [Google Scholar]
- [78].Rosen DR. Mutations in Cu/Zn superoxide dismutase gene are associated with familial amyotrophic lateral sclerosis. Nature. 1993. Jul 22;364(6435):362. [DOI] [PubMed] [Google Scholar]
- [79].Fecto F, et al. Mutations in Familial and Sporadic Amyotrophic Lateral Sclerosis. Arch Neurol. 2011. Nov;68(11):1440–1446. [DOI] [PubMed] [Google Scholar]
- [80].Freischmidt A, Wieland T, Richter B, et al. Haploinsufficiency of TBK1 causes familial ALS and fronto-temporal dementia. Nat Neurosci. 2015. May;18(5):631–636. [DOI] [PubMed] [Google Scholar]
- [81].Van Blitterswijk M, Van Vught PW, Van Es MA, et al. Novel optineurin mutations in sporadic amyotrophic lateral sclerosis patients. Neurobiol Aging. 2012. May;33(5):1016 e1–7. [DOI] [PubMed] [Google Scholar]
- [82].Chernyshova K, Inoue K, Yamashita SI, et al. Glaucoma-Associated Mutations in the Optineurin Gene Have Limited Impact on Parkin-Dependent Mitophagy. Invest Ophthalmol Vis Sci. 2019. Aug 1;60(10):3625–3635. [DOI] [PubMed] [Google Scholar]
- [83].McCall AL, Dhindsa JS, Pucci LA, et al. Respiratory pathology in the Optn(-/-) mouse model of Amyotrophic Lateral Sclerosis. Respir Physiol Neurobiol. 2020. Aug;14(282):103525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [84].Nakazawa S, Oikawa D, Ishii R, et al. Linear ubiquitination is involved in the pathogenesis of optineurin-associated amyotrophic lateral sclerosis. Nat Commun. 2016 Aug;7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [85].Korac J, Schaeffer V, Kovacevic I, et al. Ubiquitin-independent function of optineurin in autophagic clearance of protein aggregates. J Cell Sci. 2013. Jan 15;126(2):580–592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [86].Wise JP Jr., Cannon J. From the Cover: alterations in Optineurin Expression and Localization in Pre-clinical Parkinson’s Disease Models. Toxicol Sci. 2016. Oct;153(2):372–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [87].Nakamura M, Murray ME, Lin WL, et al. Optineurin immunoreactivity in neuronal and glial intranuclear inclusions in adult-onset neuronal intranuclear inclusion disease. Am J Neurodegener Dis. 2014;3(2):93–102. [PMC free article] [PubMed] [Google Scholar]
- [88].Cupidi C, Dijkstra AA, Melhem S, et al. Refining the Spectrum of Neuronal Intranuclear Inclusion Disease: a Case Report. J Neuropathol Exp Neurol. 2019. Jul 1;78(7):665–670. [DOI] [PubMed] [Google Scholar]
- [89].Ito Y, Ofengeim D, Najafov A, et al. RIPK1 mediates axonal degeneration by promoting inflammation and necroptosis in ALS. Science. 2016. Aug 5;353(6299):603–608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [90].Ma S, Attarwala IY, Xie XQ. SQSTM1/p62: a Potential Target for Neurodegenerative Disease. ACS Chem Neurosci. 2019. May 15;10(5):2094–2114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [91].Veauthier B, Hornecker JR. Crohn’s Disease: diagnosis and Management. Am Fam Physician. 2018. Dec 1;98(11):661–669. [PubMed] [Google Scholar]
- [92].Lopes F, Keita AV, Saxena A, et al. ER-stress mobilization of death-associated protein kinase-1-dependent xenophagy counteracts mitochondria stress-induced epithelial barrier dysfunction. J Biol Chem. 2018. Mar 2;293(9):3073–3087. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [93].Lassen KG, Xavier RJ. Mechanisms and function of autophagy in intestinal disease. Autophagy. 2018;14(2):216–220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [94].Murthy A, Li Y, Peng I, et al. A Crohn’s disease variant in Atg16l1 enhances its degradation by caspase 3. Nature. 2014. Feb 27;506(7489):456-+. [DOI] [PubMed] [Google Scholar]
- [95].Slowicka K, Vereecke L, Mc Guire C, et al. Optineurin deficiency in mice is associated with increased sensitivity to Salmonella but does not affect proinflammatory NF-kappaB signaling. Eur J Immunol. 2016. Apr;46(4):971–980. [DOI] [PubMed] [Google Scholar]
- [96].Chew TS, O’Shea NR, Sewell GW, et al. Optineurin deficiency in mice contributes to impaired cytokine secretion and neutrophil recruitment in bacteria-driven colitis. Dis Model Mech. 2015. Aug 1;8(8):817–829. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [97].Tschurtschenthaler M, Adolph TE. The Selective Autophagy Receptor Optineurin in Crohn’s Disease. Front Immunol. 2018;9:766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [98].Van Wijk SJL, Fricke F, Herhaus L, et al. Linear ubiquitination of cytosolic Salmonella Typhimurium activates NF-kappaB and restricts bacterial proliferation. Nat Microbiol. 2017. May;8(2):17066. [DOI] [PubMed] [Google Scholar]
- [99].Gleason CE, Ordureau A, Gourlay R, et al. Polyubiquitin binding to optineurin is required for optimal activation of TANK-binding kinase 1 and production of interferon beta. J Biol Chem. 2011. Oct 14;286(41):35663–35674. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [100].Appelman-Dijkstra NM, Papapoulos SE. Paget’s disease of bone. Best Pract Res Clin Endocrinol Metab. 2018. Oct;32(5):657–668. [DOI] [PubMed] [Google Scholar]
- [101].Cundy T. Paget’s disease of bone. Metabolism. 2018. Mar;80:5–14. [DOI] [PubMed] [Google Scholar]
- [102].Albagha OM, Visconti MR, Alonso N, et al. Genome-wide association study identifies variants at CSF1, OPTN and TNFRSF11A as genetic risk factors for Paget’s disease of bone. Nat Genet. 2010. Jun;42(6):520–524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [103].Erdmann J, Kessler T, Munoz Venegas L, et al. A decade of genome-wide association studies for coronary artery disease: the challenges ahead. Cardiovasc Res. 2018. Jul 15;114(9):1241–1257. [DOI] [PubMed] [Google Scholar]
- [104].Wong SW, Huang BW, Hu XX, et al. Global deletion of optineurin results in altered type I IFN signaling and abnormal bone remodeling in a model of Paget’s disease. Cell death differ. 2020. Jul16;27(1):71–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [105].Obaid R, Wani SE, Azfer A, et al. Optineurin Negatively Regulates Osteoclast Differentiation by Modulating NF-kappaB and Interferon Signaling: implications for Paget’s Disease. Cell Rep. 2015. Nov 10;13(6):1096–1102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [106].Usategui-Martin R, Garcia-Aparicio J, Corral-Gudino L, et al. Polymorphisms in Autophagy Genes Are Associated with Paget Disease of Bone. PloS One. 2015. Jun 1;10(6). DOI: 10.1371/journal.pone.0128984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [107].Hocking LJ, Lucas GJA, Daroszewska A, et al. Domain-specific mutations in sequestosome 1 (SQSTM1) cause familial and sporadic Paget’s disease. Hum Mol Genet. 2002. Oct 15;11(22):2735–2739. [DOI] [PubMed] [Google Scholar]
- [108].Badadani M, Nalbandian A, Watts GD, et al. VCP Associated Inclusion Body Myopathy and Paget Disease of Bone Knock-In Mouse Model Exhibits Tissue Pathology Typical of Human Disease. PloS One. 2010. Oct 5;5(10):e13183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [109].Usategui-Martin R, Gestoso-Uzal N, Calero-Paniagua I, et al. A mutation in p62 protein (p. R321C), associated to Paget’s disease of bone, causes a blockade of autophagy and an activation of NF-kB pathway. Bone. 2020. Apr;133:115265. [DOI] [PubMed] [Google Scholar]
- [110].Lee WS, Kato M, Sugawara E, et al. Protective Role of Optineurin Against Joint Destruction in Rheumatoid Arthritis Synovial Fibroblasts. Arthritis Rheumatol. 2020. Sep;72(9):1493–1504. [DOI] [PubMed] [Google Scholar]
- [111].Sudhakar C, Vaibhava V, Swarup G. IRF-1-binding site in the first intron mediates interferon-gamma-induced optineurin promoter activation. Biochem Biophys Res Commun. 2013. Jul 19;437(1):179–184. [DOI] [PubMed] [Google Scholar]
- [112].Senoo K, Yamashiro K, Yamamoto T, et al. Expression of optineurin isolated from rat-injured dental pulp and the effects on inflammatory signals in normal rat kidney cells (vol 106, pg 135, 2017). Odontology. 2018. Apr;106(2):223. [DOI] [PubMed] [Google Scholar]
- [113].Kato M, Ospelt C, Gay RE, et al. Dual role of autophagy in stress-induced cell death in rheumatoid arthritis synovial fibroblasts. Arthritis Rheumatol. 2014. Jan;66(1):40–48. [DOI] [PubMed] [Google Scholar]
- [114].Okada Y, Wu D, Trynka G, et al. Genetics of rheumatoid arthritis contributes to biology and drug discovery. Nature. 2014. Feb 20;506(7488):376-+. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [115].Montalcini T, Romeo S, Ferro Y, et al. Osteoporosis in chronic inflammatory disease: the role of malnutrition. Endocrine. 2013. Feb;43(1):59–64. [DOI] [PubMed] [Google Scholar]
- [116].Chen K, Yang YH, Jiang SD, et al. Decreased activity of osteocyte autophagy with aging may contribute to the bone loss in senile population. Histochem Cell Biol. 2014. Sep;142(3):285–295. [DOI] [PubMed] [Google Scholar]
- [117].Ma Y, Qi M, An Y, et al. Autophagy controls mesenchymal stem cell properties and senescence during bone aging. Aging cell. 2018 Feb;17(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [118].Onal M, Piemontese M, Xiong J, et al. Suppression of autophagy in osteocytes mimics skeletal aging. J Biol Chem. 2013. Jun 14;288(24):17432–17440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [119].Mizuno N, Iwata T, Ohsawa R, et al. Optineurin regulates osteoblastogenesis through STAT1. Biochem Biophys Res Commun. 2020. May 14;525(4):889–894. [DOI] [PubMed] [Google Scholar]
- [120].Liu ZZ, Hong CG, Hu WB, et al. Autophagy receptor OPTN (optineurin) regulates mesenchymal stem cell fate and bone-fat balance during aging by clearing FABP3. Autophagy. 2020;4:1–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [121].Rautou PE, Mansouri A, Lebrec D, et al. Autophagy in liver diseases. J Hepatol. 2010. Dec;53(6):1123–1134. [DOI] [PubMed] [Google Scholar]
- [122].Li F, Qiu Y, Xia F, et al. Dual detoxification and inflammatory regulation by ceria nanozymes for drug-induced liver injury therapy. Nano Today. 2020. Dec;35:100925. [Google Scholar]
- [123].Montecalvo A, Watkins SC, Orange J, et al. Inducible turnover of optineurin regulates T cell activation. Mol Immunol. 2017. May;85:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [124].Ali DM, Ansari SS, Zepp M, et al. Optineurin downregulation induces endoplasmic reticulum stress, chaperone-mediated autophagy, and apoptosis in pancreatic cancer cells. Cell Death Discov. 2019;5(1):128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [125].Mathew R, Karp CM, Beaudoin B, et al. Autophagy suppresses tumorigenesis through elimination of p62. Cell. 2009. Jun 12;137(6):1062–1075. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [126].Li L, Shen C, Nakamura E, et al. SQSTM1 is a pathogenic target of 5q copy number gains in kidney cancer. Cancer Cell. 2013. Dec 9;24(6):738–750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [127].Papadopoulou-Marketou N, Chrousos GP, Kanaka-Gantenbein C. Diabetic nephropathy in type 1 diabetes: a review of early natural history, pathogenesis, and diagnosis. Diabetes-Metab Res. 2017. Feb;33(2):e2841. [DOI] [PubMed] [Google Scholar]
- [128].Chen K, Dai H, Yuan J, et al. Optineurin-mediated mitophagy protects renal tubular epithelial cells against accelerated senescence in diabetic nephropathy. Cell Death Dis. 2018. Jan 24;9(2):105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [129].Chen K, Feng L, Hu W, et al. Optineurin inhibits NLRP3 inflammasome activation by enhancing mitophagy of renal tubular cells in diabetic nephropathy. Faseb J. 2019. Mar;33(3):4571–4585. [DOI] [PubMed] [Google Scholar]
- [130].Wang Y, Zhu J, Liu Z, et al. The PINK1/PARK2/optineurin pathway of mitophagy is activated for protection in septic acute kidney injury. Redox Biol. 2020. Oct 23;38:101767. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [131].Levine B, Kroemer G. Biological Functions of Autophagy Genes: a Disease Perspective. Cell. 2019. Jan 10;176(1–2):11–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [132].Heemels MT. Neurodegenerative diseases. Nature. 2016. Nov 10;539(7628):179. [DOI] [PubMed] [Google Scholar]
- [133].Gao J, Ohtsubo M, Hotta Y, et al. Oligomerization of optineurin and its oxidative stress- or E50K mutation-driven covalent cross-linking: possible relationship with glaucoma pathology. PloS One. 2014;9(7):e101206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [134].Nagabhushana A, Chalasani ML, Jain N, et al. Regulation of endocytic trafficking of transferrin receptor by optineurin and its impairment by a glaucoma-associated mutant. Bmc Cell Biol. 2010. Jan 19;11(1):4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [135].Wise JP, Price CG, Amaro JA, et al. Autophagy Disruptions Associated With Altered Optineurin Expression in Extranigral Regions in a Rotenone Model of Parkinson’s Disease. Front Neurosci-Switz. 2018. May 16;12:289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [136].Mori F, Tanji K, Toyoshima Y, et al. Optineurin immunoreactivity in neuronal nuclear inclusions of polyglutamine diseases (Huntington’s, DRPLA, SCA2, SCA3) and intranuclear inclusion body disease. Acta Neuropathol. 2012. May;123(5):747–749. [DOI] [PubMed] [Google Scholar]
- [137].Schwab C, Yu S, McGeer EG, et al. Optineurin in Huntington’s disease intranuclear inclusions. Neurosci Lett. 2012. Jan 6;506(1):149–154. [DOI] [PubMed] [Google Scholar]
- [138].Anborgh PH, Godin C, Pampillo M, et al. Inhibition of metabotropic glutamate receptor signaling by the huntingtin-binding protein optineurin. J Biol Chem. 2005. Oct 14;280(41):34840–34848. [DOI] [PubMed] [Google Scholar]