

Abstract
One of the main challenges of Structure-based Virtual Screening (SBVS) is the incorporation of receptor’s flexibility, as its explicit representation in every docking run implies a high computational cost. Therefore, a common alternative to include the receptor’s flexibility is the approach known as ensemble docking. Ensemble docking consists of using a set of receptor conformations and performing the docking assays over each of them. However, there is still no agreement on how to combine the ensemble docking results to get the final ligand ranking. A common choice is to use consensus strategies to aggregate the ensemble docking scores, but these strategies exhibit slight improvement regarding the single-structure approach. Here, we claim that using machine learning methodologies over the ensemble docking results could improve the predictive power of SBVS. To test this hypothesis, four proteins were selected as study cases: CDK2, FXa, EGFR, and HSP90. Protein conformational ensembles were built from crystallographic structures, whereas the evaluated compound library comprised up to three benchmarking datasets (DUD, DEKOIS 2.0, CSAR-2012) and cocrystallized molecules. Ensemble docking results were processed through 30 repetitions of 4-fold cross-validation to train and validate two machine learning (ML) classifiers: Logistic regression and gradient boosting trees. Our results indicate that the ML classifiers significantly outperform traditional consensus strategies and even the best performance case achieved with single-structure docking. We provide statistical evidence that supports the effectiveness of ML to improve the ensemble docking performance.
Graphical Abstract
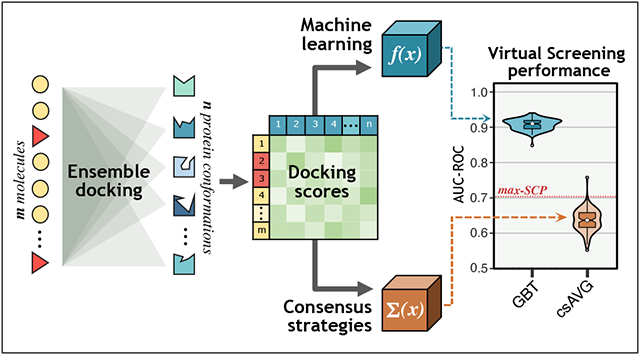
Introduction
Structure-based virtual screening (SBVS) is a key component in the early stage of drug discovery.1 SBVS comprises different in silico methodologies to filter a chemical compound library using the three-dimensional structure of the molecular target of interest (receptor). Commonly, the process consists of docking each compound (candidate ligand) to the receptor, predicting their binding mode and estimating their binding affinity through a scoring function.2 Subsequently, ligands are sorted by their binding score to the receptor, and the top-ranked ones are selected as the best candidates for further experimental analysis.2,3
To be computationally efficient, traditional docking tends to oversimplify the binding process, negatively affecting the predictive power of SBVS.4 One of the main drawbacks is that docking simulations usually rely on a single rigid conformation of the receptor, resulting in a poor representation of the receptor’s flexibility.4,5 Consequently, multiple approaches have been proposed to ameliorate this deficiency.5-9 Among these alternatives, ensemble docking is one of the most popular10,11 and has been successfully applied in various prospective studies,12-18 which support the motivation of using ensemble docking to improve SBVS.
Ensemble docking aims to represent the receptor’s binding site flexibility through a conformational ensemble.10 The assumption is that this structural diversity can lead to more accurate binding modes and explore broader chemical space.5 Thus, the most common implementation of ensemble docking consists of docking each ligand to multiple rigid conformations of the receptor.11,19 As a result, multiple docking scores are computed for each ligand, which finally must be aggregated to obtain a single compound score, or ranking. Nevertheless, it is still unclear what is the best methodology to perform this aggregation.20
Consensus strategies are a usual alternative to combine ensemble docking results.21 These strategies are data fusion rules22 similar to the consensus scoring schemes used to combine the scores from different docking tools.23,24 However, their effectiveness is target-dependent, varying among each docking study.25 Moreover, consensus strategies provide only modest improvement over the average performance of single-conformation docking19,26 and hardly exceed the best case’s performance of single-conformation docking.19,26 Previous studies also observed that adding more conformations to the ensemble tends to favor false-positive ligands, decreasing the SBVS predictive power.27-29
Here, we ask whether machine learning (ML) could be a better alternative than traditional consensus strategies to exploit ensemble docking results and improve the SBVS performance. ML has been widely and successfully employed in SBVS.30 Most of these models were developed using intermolecular features computed from experimental protein-ligand complexes31-34 and single-conformation docking results.34-37 Other studies have used ML to integrate the predictions of different scoring functions computed from a single binding mode.24,38,39 However, only a few studies,40,41 considered in the Results and Discussion section, have explored the direct application of ML classifiers over ensemble docking scores. Here, we extend these studies offering a statistical analysis comparing the ML models’ performance with some of the most used consensus strategies.
Therefore, to evaluate the suitability of combining ML and ensemble docking we evaluated four proteins as study cases: cyclin-dependent kinase 2 (CDK2), factor Xa (FXa), epidermal growth factor receptor (EGFR), and heat shock protein 90 (HSP90). These four proteins have been extensively used to test and validate SBVS methodologies,26,27,40,42-50 and have several crystallographic structures available.51 We selected CDK2 and FXa proteins because their binding sites are quite different; the CDK2 binding pocket is a deep hydrophobic cavity, while the FXa active site is a shallow and solvent-exposed hydrophobic groove.45 On the other hand, the EGFR and HSP90 proteins were suggested by one of our anonymous reviewers as a blind test.
We started by building the conformational ensembles of each of the four proteins. Then, ensemble docking simulations were carried out using ligands from three molecular libraries (DUD52,53, DEKOIS 2.054, and CSAR55) and cocrystallized compounds. Then, we used the raw docking scores to perform 30 repetitions of 4-fold cross-validation (30×4cv) to train and evaluate two ML algorithms: logistic regression (LR) and gradient boosting trees (GBT)56. The former is a basic linear classifier whereas the latter is a relatively new non-linear ensemble algorithm successfully employed in SBVS.24,57-60 We also considered three consensus strategies: the lowest score (csMIN), the average score (csAVG), and the geometric mean score (csGEO).21 csMIN and csAVG are among the commonly used consensus strategies,17,18,25-27,45,61 whereas csGEO was recently recommended as a better alternative to other strategies.21 Finally, we statistically compared the ML classifiers’ prediction power with that of the consensus strategies. We also explored different conformational selection criteria to evaluate how some associated properties and single-conformation predictions affected the ML performance.
Materials and methods
Figure 1 displays an overview of the methodology workflow used in the present study. Code implementation of this pipeline and the main output files are available on the Github repository provided in the Supporting Information section. Ensemble docking assays were performed using eight 16-core (Intel Xeon E5-2670v1) nodes of the Miztli supercomputer owned by the Universidad Autónoma de México. The rest of the analyses were done on a personal computer with 8 GB RAM and an Intel Core i7 CPU at 2.3 GHz.
Figure 1:

Overview of the methodology workflow (per target protein): data collection, ensemble docking, and 30×4cv to implement target-specific machine learning models over ensemble docking scores and compare their performance against traditional consensus strategies and single-conformation docking. *The CSAR-2012 dataset was available only for the CDK2 protein.
Protein conformational ensembles preparation
Crystal structures of each of the four protein targets were retrieved from the PDB51 using the ProDy library62. First, each target’s primary sequence was obtained from the Uniprot database (CDK2: P24941, FXa: P00742, EGFR: P00533, HSP90: P07900) and used to perform a blast-p against the PDB database. All PDB entries with a protein chain with a sequence identity equal to or greater than 95% were downloaded and processed to remove the solvent, small molecules, and other protein entities, to keep only the protein chain belonging to the target. When needed, the Modeller python module63 was employed to model missing loops without affecting the native geometry of the rest of the crystallographic structure. Subsequently, pdb4amber from the AmberTools1964 package was used to fill missing heavy atoms and standardize atom and residue names. Protonation states of ionizable residues were predicted at a pH 7.0 using the PDB2PQR software65.
Structural analysis of the protein ensembles
For each protein target, a conformational ensemble was constructed from its set of crystallographic structures incorporating the protein’s binding pocket, which was identified by the reference protein-ligand complex provided by the DUD dataset52. For CDK2 and FXa, all the available crystallographic structures were used to construct the conformational ensemble. Meanwhile, the EGFR and HSP90 conformational ensembles were built from 64 randomly selected conformations, obtained from all of their respective available crystallographic structures.
The python library pytraj66 was employed to compute a pairwise root-mean square deviation (RMSD) matrix, using the Cα atoms of each conformation’s binding pocket residues. Subsequently, the RMSD matrix was used to perform a classical multidimensional scaling (cMDS), a non-linear dimensionality reduction technique that displays the information of a distance matrix on a reduced Cartesian space67. The shape and volume of the binding pockets were measured using POVME 3.068. The pairwise similarity between pockets’ shapes was computed using the Tanimoto index. Hierarchical clustering and cMDS were performed using these results.
Benchmarking libraries
The molecular library of each protein was constructed from different sources. The first source corresponds to the ligands crystallized at the protein’s binding pocket. Cocrystalized molecules were extracted from the retrieved protein conformations and were labeled as active compounds. The second source comprises two benchmarking sets: the charged-corrected version of the second release of the Directory of Useful Decoys (DUD) dataset52,53 and the DEKOIS 2.0 dataset54. Both datasets include active compounds experimentally proven to bind their respective target protein, as well as decoy molecules with similar physicochemical properties to their matching actives, but with a different 2D topology. Decoy molecules are assumed to be non-binders and will be referred to as inactive molecules in this work. The third source was the CSAR benchmark exercise 2012 dataset55, which incorporates experimentally confirmed active and inactive compounds targeting CDK2.
The benchmarking sets were downloaded from their respective web pages.69-71 Open Babel72 was employed for file conversion, protonation at pH 7.0, and 3D structure generation using the MMFF94 force field. Subsequently, Morgan fingerprints (with 2048 bits and radius 2) were computed for all molecules employing RDKit73. Tanimoto index was used to measure the fingerprint similarity between each pair of molecules. Those compounds sharing a similarity of 1.0 were considered duplicates, and only one of them was kept. Finally, a single dataset per protein was created by merging its corresponding cocrystallized and benchmarking sets, excluding duplicates.
Ensemble docking
Ensemble docking simulations were carried out using Vinardo, a modified Vina scoring function developed by Quiroga and Villarreal74 and implemented in the Smina software75. All protein structures and their corresponding molecules were prepared using AutoDockTools76 by merging non-polar hydrogens. The size and center of the docking grid box were specific for each protein target, but uniform across its conformations, which were previously aligned based on the Cα atoms of the residues comprising the binding pocket. The Smina’s exhaustiveness search criterion was set to 16, while the rest of the parameters were kept as default.
Feature Representation
For each protein, ensemble docking results were gathered into a matrix Xm×n, where n is the number of available conformations of that protein, and m is the number of compounds in its respective molecular library. Hence, each row represents a molecule (a sample), and each column represents a protein conformation (a feature). Each matrix element xi,j is the best docking score, as judged by Vinardo, between molecule i and protein conformation j. The X matrix (or a subset of it) served as input for the consensus scoring functions and the machine learning classifiers.
Ensemble Consensus Scoring
Three consensus strategies, namely csMIN, csAVG, and csGEO, were used to compare their performance against the ML classifiers. These strategies are aggregation functions that produce a single score per molecule from a row of X, i.e., an n-vector xi containing the ensemble docking scores of the molecule i across the n protein conformations. csMIN strategy returns the minimum value of xi, corresponding to the best raw docking score as judged by the Vinardo scoring function. csAVG and csGEO correspond to the arithmetic and the geometric means of the elements of xi, respectively. Each consensus strategy was applied over the scores of all molecules of a given dataset to get a single ranking list.
Machine Learning
Machine Learning Classifiers
Two supervised ML algorithms were implemented to develop target-specific binary classifiers to identify potential active molecules (binders) from inactive ones (non-binders or decoys) given a protein dataset. The chosen algorithms were Logistic Regression and Gradient Boosting Trees56. Logistic Regression (LR) is a basic linear classifier that applies the logistic function over a linear combination of weighted predictive features. Gradient Boosting is a tree-based ensemble algorithm where a set of decision trees are trained sequentially, fitting the new tree on the residual errors of the previous one. Additionally, a Dummy classifier (DClf) was implemented as a baseline to compare with LR and GBT. The DClf’s predictions are actually random guesses that respect the active/inactive distribution (stratified strategy)77. GBT was implemented by using the XGBoost library (v1.3.0)78, an optimized implementation of the Gradient Boosting algorithm, whereas the DClf and the LR classifier model were performed using the scikit-learn (v0.23.2) python library77.
Hyperparameter tuning
The ML classifiers were trained and tested using ensemble docking scores from the merged dataset, represented by X, along with the response variable y containing the active and inactive labels. The train-test split of the dataset was done by following a stratified hold-out strategy, where 75% of the samples were used for training and validation, while the remained 25% were held to test the model’s performance.
Hyperparameter tuning was done using the training dataset (75% of the data) with all features considered. For the LR classifier an exhaustive grid search was carried out, whereas a randomized grid search with 50 iterations was performed for the GBT classifier due to the large number of hyperparameters involved. At each grid point a 5-fold cross-validation was performed. The hyperparameters taken into account for each classifier are listed in Table S1. The evaluation metric employed was the area under the curve of the receiver operator characteristic (AUC-ROC). At the end of this stage, the best combination of hyperparameters was selected.
Machine learning model assessment with 30 repetitions of stratified 4-fold cross-validation
The performance of each ML classifier was evaluated using the whole dataset through 30 repetitions of stratified 4-fold cross-validation (30×4cv).79 At each cross-validation repetition, the data was randomly split into four folds. Then, during each fold, a new instance of the ML classifier was created using the hyperparameters selected by the hyperparameter tuning stage. Next, the ML classifier was trained with the 75% of the data (three folds), and evaluated over the remaining 25% (one fold). After the 30 repetitions, we registered the performance of the ML classifier over each of the 120 validation sets (data never saw by that particular ML model during its training). It is expected that the use of multiple cross-validation repetitions gives a more “robust” model assessment score than performing cross-validation just once.80,81
Additionally, the three consensus strategies were implemented over the same 120 validation sets evaluated by the ML classifiers. We also calculated the single-conformation performance (SCP) of the n protein conformations using the raw docking scores from the 120 validation sets. Finally, we computed the maximum and the average SCP from the 120×n SCP values obtained.
Y-randomization test
The robustness of the ML classifiers was evaluated through the y-randomization test. This procedure consists of randomly permuting the response variable y without altering the set of the independent variables (denoted by X). A new ML classifier is then trained and tested using this new dataset, and its performance is compared with that of the original model. Therefore, the new model works as a negative control that represents the action of chance when fitting the given data82.
In this work, the y-randomization test was applied to evaluate the ML algorithms and consensus strategies. Randomization was implemented over five percentages (c) of the target value: 0, 25, 50, 75, and 100. However, to keep class balance, these percentages were applied regarding the number of active labels. This means that, at a given c value and a dataset with a active molecules, only ⌊(c/100)a⌋ active labels are scrambled with ⌊(c/100)a⌋ inactive labels. Therefore, a 30×4cv procedure was performed at each percentage value, shuffling the equivalent number of target labels at each cross-validation repetition. For comparison, the consensus strategies were also applied over every validation set.
Feature Ranking and Feature Selection
Recursive feature elimination (RFE) was carried out using scikit-learn77 to rank the protein conformations by their contribution to the accurate prediction whether a molecule is a true binder. RFE is an algorithm that ranks the features by its importance, identifying a subset of those that maximize the model’s performance. To do so, RFE wraps a base ML classifier that computes how relevant each predictive variable is. Starting with all features in the training dataset, RFE performs a stepwise backward elimination of the less relevant features. Here, RFE was applied to remove one feature (conformation) per step until only one remained, allowing to obtain a feature ranking. The base classifier used was GBT, which applies the Information Gain metric to estimate feature importance.
Evaluation criteria
Two criteria were used to evaluate the SBVS methods’ performance: the AUC-ROC and the normalized enrichment factor (NEF). These measures were applied over the molecule ranking list predicted by each SBVS method. For the ML classifiers, these ranking lists were computed from the predicted probability of each molecule to be active.
The AUC-ROC is a robust criterion commonly used to evaluate SBVS results. It represents the probability that a randomly chosen active compound ranks higher than a randomly chosen inactive one83. An AUC-ROC of 1.0 indicates that all active molecules have been ranked at the top, meaning a perfect identification of the true binders, while an AUC-ROC of 0.5 represents a random selection.
Enrichment Factor (EF) addresses the early recognition problem by evaluating only the top χ fraction of the ranked compounds83. It is computed as follows:
| (1) |
where m is the total number of compounds in the dataset, a is the total number of true actives, and as (actives selected) indicates how many of the χm molecules are true actives. Therefore, the EF is an indicator of how enriched in actives is the top χ fraction, compared with a set of the same size with randomly selected elements83. Its value ranges from 0, when no actives are found in the top χm set of molecules, to 1/χ if χm ≥ a or m/a if χm < a. Because of this inconsistency in the EF upper bound value, we decided to use the normalized enrichment factor (NEF)84:
| (2) |
NEFχ value ranges from 0 to 1, simplifying its interpretation. Here, NEF was applied at χ = Ra, where Ra = a/m, i.e., the ratio of active molecules in the dataset.
Effect of the number of conformations on the performance of the SBVS methods
We evaluated the effect of the number of protein conformations over the ML classifiers’ performance. To do so, each ML classifier was trained and evaluated through a 30×4cv procedure using only a k = 2r number of features, for 1 ≤ r < log2n, i.e., with k duplicating its value at each new iteration, and n representing the protein ensemble size. Thus, at a given r value, the k selected conformations were drawn from the top of a given list of protein conformations ranked by a particular criterion. Four ranking lists were used for this analysis, along with a random selection used as reference:
BestSCP-rank: conformations ranked by its SCP using the raw docking scores, in descending order (conformations with higher AUC-ROC values appear at the beginning of the list).
WorstSCP-rank: conformations ranked by its SCP, in ascending order (conformations with lower AUC-ROC values appear at the beginning of the list).
LigMW-rank: conformations ranked by the molecular weight of its cocrystalized ligand, in descending order (apo conformations appear at the end of the list).
RFE-rank: conformations ranked as the best by the RFE method.
Random: The k conformations were randomly selected at each repetition.
The consensus strategies were also applied over the same validation sets used for the ML models. The AUC-ROC and NEFχ criteria were used to measure the performance of each SBVS method
Statistical Analysis
Statistical analyses were performed to detect significant differences between the performance of the compared SBVS methods. The AUC-ROC and the NEF values from the 30 ×4cv results were evaluated for each protein using the Friedman test, a non-parametric alternative to the one-way ANOVA with repeated measures. The Friedman test ranks the SBVS methods by their average performance among the same validation sets (within-subjects effect) and evaluates the null-hypothesis that states that all SBVS methods have similar ranks, implying an equal performance.85 Subsequently, pairwise comparisons between methods performances were carried out using the Nemenyi post-hoc test86 and Critical Difference (CD) plots,87 which were used to visualize significant differences between SBVS methods. We decided to perform non-parametric tests because the normality and sphericity assumptions were not met. Results from the Shapiro-Wilk normality test88 and the Mauchly’s test of sphericity89 are shown in Table S2 and Table S3, respectively. Additionally, the Kruskal-Wallis test90 for independent samples and the Dunn’s post hoc test91 were implemented to compare different randomization percentages during the y-randomization assay of each SBVS method. All statistical analyses were carried out in R using the tidyverse92 and rstatix93 packages.
Results and discussion
Datasets
Protein conformational ensembles
The final CDK2 and FXa conformational ensembles comprised 402 and 136 PDB structures, respectively. On the other hand, EGFR and HSP90 conformational ensembles were limited to 64 conformations, which were selected randomly from among all their available crystallographic structures. The selection and the preparation process of these ensembles, and the list of PDB entries, are documented as Jupyter notebooks available in the Supporting Information.
Molecular libraries and ensemble docking simulation
The final molecular library of each protein target was obtained by merging its respective cocrystallized molecules with compounds from different benchmarking sets for molecular docking; the four targets’ libraries included molecules from the DUD and the DEKOIS 2.0 datasets, and the CDK2’s also included compounds from the CSAR dataset. The CDK2 merged library comprised 3,466 molecules, of which 415 were actives, giving an active/total ratio (Ra) of 0.12. Similarly, the FXa merged library had 6,233 molecules (300 actives, Ra = 0.05), the EGFR merged library had 15,510 molecules (585 actives, Ra = 0.04), and the HSP90 merged library had 2,302 molecules (256 actives, Ra = 0.11). Figure S1 presents a breakdown of each merged library, indicating the initial number of molecules inside each original dataset and the number of duplicates between them. The merged library and the conformational ensemble of each protein were used to perform the ensemble docking simulations. Considering the four proteins, a total of 3,390,844 docking runs were carried out.
Machine learning over ensemble docking scores improves SBVS performance
To test whether the ensemble docking performance could be improved by using ML, we carried out a 30×4cv analysis using the raw docking scores of ensemble docking simulations. For this purpose, two ML classifiers were evaluated: LR and GBT. LR and GBT were selected as two representative shallow learning algorithms; LR is a basic linear ML classifier while GBT is a more complex, ensemble-based, ML algorithm that has previously applied to combine docking scores from multiple scoring functions.24 Selecting only two ML classifiers and three consensus strategies allowed us to simplify the analysis when comparing their performance against traditional consensus strategies and single-conformation docking. Similarly, we selected only three consensus strategies: csMIN, csAVG, and csGEO. We take this decision because our main objective was to assess the applicability of ML over ensemble docking results, setting the stage for future research that could incorporate more sophisticated ML algorithms and input features. Figure 1 shows an outline of the whole process.
ML models outperform traditional consensus strategies
The two ML methods had better performance than the consensus strategies. This difference is not only statistically significant but large enough to be important in a drug discovery campaign. This is evident from Figure 2a that displays the results of the 30×4cv analysis in terms of the AUC-ROC. Violin boxplots show the distribution of the AUC-ROC values reached by each SBVS method over the 120 validation sets. Additionally, the Dclf results are shown as a reference, having an expected behavior with AUC-ROC values close to 0.5. Most of the pair-wise differences between the SBVS methods were statistically significant, as indicated by the Friedman test results (Table 1) and the Nemenyi post-hoc test (Figure S3). The CD plots of Figure S2a allow us to visualize these results, showing each SBVS method’s average rank and highlighting those that were not significantly different, at α = 0.01. Such was the case of the csAVG and csGEO strategies, which did not show significant differences in the case of CDK2 and FXa; this result differs from that reported by Bajusz et al.21, who observed better performance of csGEO over csAVG. On the contrary, the median AUC-ROC values of the ML models were significantly higher than any of the consensus strategies.
Figure 2:

Results from the 30×4cv analysis. (a) AUC-ROC values. (b) NEF values. Violin boxplots showing the distribution of the performance values of each SBVS method across the 30 repetitions. The white points within the boxes indicate the value of the median, and the notches represent the 95% confidence interval around it. Outliers are shown as black points. The max SCP (single-conformation performance) and the avg SCP dashed lines indicate the maximum and the average performance, respectively, achieved by a single conformation using the raw docking scores from the 120 × n validation sets generated during the 30×4cv analysis. The translucent gray area surrounding the avg SCP value represents one standard deviation from the average SCP. The dotted black lines indicate the expected performance of a random classifier.
Table 1:
Statistical significance of performance differences among methods during the 30×4cv analysis, evaluated with the Friedman test
| Protein | Evaluation criterion | n | p-value | |
|---|---|---|---|---|
| CDK2 | AUC-ROC | 120 | 586.98 | 1.3e–124 |
| NEF0.12 | 120 | 576.43 | 2.5e–122 | |
| FXa | AUC-ROC | 120 | 584.77 | 3.9e–124 |
| NEF0.05 | 120 | 542.02 | 6.7e–115 | |
| EGFR | AUC-ROC | 120 | 600.0 | 2.0e–127 |
| NEF0.04 | 120 | 577.94 | 1.2e–122 | |
| HSP90 | AUC-ROC | 120 | 572.82 | 1.5e–121 |
| NEF0.11 | 120 | 522.47 | 1.1e–110 |
n is number of validation sets (samples) and is the Friedman’s Chi-square with 5 degrees of freedom.
We also evaluated the 30×4cv results using the NEF metric to address the SBVS methods’ early enrichment. The NEF results showed a similar behavior as the AUC-ROC (Figure 2b). The median Dclf’s performance Ra was equal to the respective Ra value matching the NEF value we would expect by chance at the given dataset. The Friedman test suggested significant differences between the NEF medians of the SBVS methods for both proteins (Table 1). These differences are exposed by the Nemenyi post-hoc test (Figure S4) and the CD plots (Figure S2b), indicating that the ML classifiers had significantly higher NEF median values than the consensus strategies.
ML models outperform the best case of single-conformation docking
ML models also outperform the best performance obtained with single-conformation docking. SCP values were obtained by measuring the AUC-ROC and the NEF values from the n protein conformations across the 120 validation sets of the 30×4cv analysis. This led to 120 × n SCP values for each evaluation criterion. The maximum SCP and the average SCP values are shown as dotted lines in Figures 2a and 2b. The consensus strategies, except by the csMIN over the CDK2 and the EGFR datasets, had higher median performances than the respective average SCP. This result is consistent with earlier reports where similar consensus strategies19,25,26,28,43,44,94,95 and other ensemble aggregation procedures29,96 showed an improvement with respect to the average SCP. Particularly, Korb et al.26 proved that the csAVG strategy tends to have better performance than the average case with single-conformation docking. Rueda et al.28 observed the same behavior when using the csMIN strategy over the ensemble docking results of a reduced number of conformations.
However, some studies19,23,25,26,28,29,94,97 also demonstrated that these ensemble-based procedures generally perform worse than the maximum SCP. Similarly, in our study, none of the consensus strategies surpassed the maximum SCP. By contrast, the two ML classifiers achieved better AUC-ROC and NEF values than the maximum SCP of the corresponding dataset. The only exception was the LR classifier on the FXa dataset, which median NEF value was equal to its respective max SCP. Overall, this is a noticeable result considering that the max SCP is the best observed performance achievable with a single conformation. Hence, surpassing the max SCP may indicate that the ML models are able to combine the results of multiple conformations (features) improving the individual predictions.
30x4cv with default ML hyperparameters and ligand efficiency scores
We repeated the 30×4cv employing the LR and GBT algorithms with their default hyperparameters (Table S1). Figure S5 depicts the results of this analysis. As expected, the ML models with the default hyperparameters show a decrease in their performance regarding the ML models with optimized hyperparameters (Figure 2). Nevertheless, the performances of the ML algorithms are still significantly better than the consensus strategies and achieve higher values than the SCP (Figure S6).
We also performed the 30×4cv using the ligand efficiency (LE) scores, which were computed by dividing the raw docking scores between the number of heavy atoms of each molecule. LE is a normalization strategy – applied both over experimental and computational binding affinity measures98 – intending to lessen the bias of some scoring functions towards large compounds. This bias is often denoted by the correlation between docking scores with ligand size.99,100
The 30×4cv results with LE scores are shown in Figure S7. Overall, these results are similar to those of the 30 × 4cv using raw docking scores (Figure 2), since ML classifiers also significantly outperformed the consensus strategies when LE scores were used (see CD plots in Figure S8). However, compared with the 30×4cv results using raw docking scores (Figure 2), the application of LE scores had a greater impact on the consensus strategies performance than on the ML models performance. Particularly, the GBT AUC-ROC and NEF values were practically the same when using raw docking scores (Figure 2) or the LE scores (Figure S7). This implies that the behavior of the GBT classifier was less affected by any possible bias related to the scoring function and the ligand size.
Robustness of the ML classifiers
We implemented the y-randomization test to assess the robustness of the ML classifiers. This test is commonly used to validate QSAR and QSPR models where the number of features is high, increasing the risk of overfitting and chance correlation.82,101 The procedure consists of breaking any possible relationship between predictive and response variables to train and evaluate new ML models (y-randomized models). Then, the y-randomized models’ performance is compared with that of the original model.82 If they are not significantly different, we should conclude that the features and the labels are independent, meaning that the original performance was obtained by chance correlation.101
Herein, y-randomization was done at different percentages of the response variable following by a 30×4cv procedure at each. Subsequently, we evaluated the ML models and consensus strategies performance using the validation sets. Figure 3 shows the test results of the four proteins regarding the AUC-ROC metric. Each panel displays how the performance of each SBVS method changes as a response to the percentage of shuffled labels. When the shuffling percentage is equal to zero, i.e., no permutation at all, the SBVS methods have the same performance shown in Figure 2. However, their performance decrease as the shuffling percentage increases, leading to values close to those expected by random chance when the shuffle is 100%. A similar behavior is observed when using the NEF metric (Figure S9). For each SBVS method, the statistical analyses confirm that the observed differences among the five randomization percentages are significant (see Table S4 and S5), indicating that the effect of chance correlation can be neglected. This is a favorable result, as it suggests that the ML models are learning to differentiate between active and decoy molecules through underlying patterns of the ensemble docking scores.
Figure 3:

Results of the y-randomization test showing the SBVS methods’ average AUC-ROC values at different percentages of active/inactive shuffled labels. Error bars indicate standard deviations. The csAVG and csGEO strategies had practically the same average and standard deviation values. Results of the NEF metric are shown in Figure S9.
Comparison with related studies
Previous studies have also tested the application of ML models over ensemble docking scores. Tian et al.40 employed a Naïve Bayes classifier (NBC) using three proteins as study cases: ALK, VEGFR2X, and CDK2. The NBC showed a high prediction power, with a 0.843 AUC-ROC value for the CDK2 dataset using a protein ensemble of eight conformations. However, this result cannot be directly compared with ours because they employed a different docking tool (Glide) and a distinct molecular library, including actives and decoys from the BindingDB and the ChemBridge databases, respectively. However, they did not compute the single-conformation AUC-ROC using the raw docking scores, making it difficult to compare how much the NBC improved the performance with respect to the SCP.
More recently, Wong95 compared the performance of an NBC against the SCP, and two consensus strategies; csAVG and csMIN. The author employed Autodock Vina to dock the EGFR’s DUD-E dataset to 34 EGFR protein conformations. The ensemble scores were then used to train and validate the NBC through a single repetition of 3-fold cross-validation. The AUC-ROC values of the csAVG and csMIN strategies were 0.605 and 0.630, respectively, while the best single-conformation AUC-ROC was 0.684. The NBC had an AUC-ROC of 0.685, slightly higher than the consensus strategies and similar to the SCP. Later, Chandak et al.41 explored additional ML models, like LR and Random Forest. These ML models, applied over the same EGFR’s dataset, achieved AUC-ROC values around 0.86, outperforming the previous results. Moreover, Chandak et al.41 extended this methodology to other 20 protein targets using 11 conformations per each of them. Unfortunately, the comparison with the SCP and with the consensus strategies was omitted for these protein targets.
Thus, our results confirm and extend those of Tian et al.40 and Wong et al.41,95 about the applicability of ML to improve the ensemble docking performance. Furthermore, we provide statistical evidence indicating that the ML classifiers significantly outperform commonly used consensus strategies and even the best case with a single-conformation docking. Lastly, the y-randomization test result suggests a true relationship between the ensemble docking scores and the discrimination of active from inactive inhibitors that traditional strategies are unable to recognize.
The effect of conformational selection over the ML models’ performance
Two of the main challenges of Ensemble Docking are how to select the best suitable protein structures and how many of them should be used.10,44,102 Different selection methodologies have been proposed to get the optimal set of conformations that maximizes the ensemble docking results, including geometrical clustering,45,94,103,104 machine learning,105 and knowledge-based methods.26,44 Nevertheless, there is no consensus about which structural or energetic properties might help identify the best suitable conformations for ensemble docking.10,19,26 Herein, we evaluated the SBVS methods through a series of 30×4cv analyses combining different ensemble sizes with five selection criteria. The aim was to measure how much the ML classifier performance was affected by these two variables, i.e., ensemble size and selection criteria.
ML models’ performance increases with the number of conformations
Contrary to the consensus strategies, the ML models’ performance improved as more conformations were added to the protein ensemble. Moreover, the RFE-rank was the best sequence for choosing conformations, surpassing the max SCP after using only eight conformations. As an example, Figure 4 allows us to compare the GBT classifier results with those of the csMIN; the two SBVS methods with the highest performance disparity. As can be observed, the csMIN’s performance does not improve as more conformations are added to the ensemble (left panels of Figures 4a, 4b, 4c, and 4d). The only exception is the WorstSCP-rank selection, as its average performance rises with the ensemble size. This is expected because the WorstSCP-rank prioritizes conformations with the worst AUC-ROC, meaning that the features used at lower k values are closer to be random guesses. On the contrary, with the rest of the selection criteria, the csMIN’s performance keeps the same, or even gets worse; a trend that is also observed in the other two consensus strategies (Figure S10). However, the effect is more evident with the csMIN strategy (especially for CDK2 and EGFR) as it is more susceptible to false positives as the number of conformations increases.27 This could partially explain why csMIN had worse results than the average SCP in the case of the CDK2 protein, using the 402 conformations (Figure 2).
Figure 4:

Comparison between the average performance (AUC-ROC) of the csMIN (squares) and the GBT ML classifier (diamonds) using only k protein conformations. Five different selection criteria are compared. Error bars indicate standard deviations. As a reference, the max SCP dashed line indicates the maximum performance achieved by a single conformation using the raw docking scores from the 120 × n validation sets generated during the 30×4cv analysis, where all n conformations were considered. (a) CDK2 protein results. (b) FXa protein results (k = 128 values are omitted for clarity). (c) EGFR protein results. (d) HSP90 protein results. An extended version of these results can be consulted in Figure S10 and S11.
Unlike the consensus strategies, the GBT’s AUC-ROC value increases with the number of conformations no matter the selection criterion used (right panels of Figures 4a, 4b, 4c, and 4d). A similar trend is observed for the LR classifier (Figure S10), although GBT has a steeper increment from few conformations. Wong et al.41,95 noticed the same behavior with other ML classifiers, using up to 34 conformations of the EGFR protein. Moreover, Tian et al.40 reported that their best NBC was the one that involved more structures. This a remarkable result considering that most of the ensemble docking procedures have concluded that adding more conformations to an ensemble reduces the docking efficiency.27-29,44
Which protein conformations enhance the ML models performance?
Here, we compared five selection criteria to take k protein conformations as input features: Random, BestSCP-rank, WorstSCP-rank, LigMW-rank, and RFE-rank. The Random selection serves as a baseline and represents the case when is impossible to know in advance which conformations could give the best enrichment individually. On the contrary, the BestSCP-rank and the WorstSCP-rank pick the k conformations with the best and the worst SCP, respectively. LigMW-rank prioritizes the protein conformations bound to ligands with the largest molecular weight. Lastly, the RFE-rank is the list of conformations sorted by their importance for the GBT classifier, as judged by the RFE procedure.
When comparing the SBVS methods in terms of the Random selection (GBT panels of Figure 4), we can see that, as k increases, the GBT classifier provides a noticeable improvement in all protein datasets regarding the csMIN strategy. Furthermore, on average, the GBT classifier surpassed the max SCP (in terms of AUC) after using 16 randomly chosen conformations (blue line of GBT panels, Figure 4). Instead, with the Random selection, the LR classifier requires more conformations to exceed the max SCP; 32 conformations for EGFR and HSP90, and 64 for CDK2 and EGFR (Figure S10). Interestingly, when the LigMW-rank is combined with the GBT classifier it has a similar (CDK2 and HSP90) or even better (FXa and EGFR) average performance than the Random selection, mainly when few conformations were used (yellow line of GBT panels, Figure 4). This is consistent with the results of Rueda et al.28 and Korb et al.26, who observed favorable enrichment when using ensembles with conformations having the largest ligands.
Meanwhile, the best suitable rank for the csMIN strategy is the BestSCP-rank (red line of csMIN panels, Figure 4), which achieved better results than the Random selection at any k value. The same effect appears with the csAVG and csGEO strategies, as much as for the AUC-ROC (Figure S10) and the NEF metrics (Figure S11). This is in line with previous reports showing that the best ensembles are usually those comprised of conformations that individually give the best performance.27,29,44 However, the ML classifiers do not give the best results when trained with conformations from the BestSCP-rank, particularly when k is above 4 conformations. At higher k values, the GBT classifier got lower AUC-ROC values with the BestSCP-rank (red line of GBT panels, Figure 4) than with the Random selection. This effect is more prominent with the LR classifier. As shown in Figures S10 and S11, with the BestSCP-rank, the LR performance remains the same at low k values and improves only after being trained with more than 64 conformations. The above means that LR only improves after conformations with lower SCP values are added to the ensemble.
On the other hand, the WorstSCP-rank always gave worse results than the Random selection in both the csMIN and the GTB methods (green line of Figure 4). The same occurs with the LR classifier and the other two consensus strategies (Figure S10 and S11). However, in all protein datasets, the GBT classifier was able to take advantage even form the WorstSCP-rank, reaching the max SCP AUC-ROC after using the top 32 WorstSCP-rank conformations (green line of GBT panels of Figure 4). The above means that even when using the worst 32 conformations, the GBT model can combine their individual predictions and beat the SCP of the best conformation. In contrast, the LR classifier does not show this behavior, which could suggest that the improvement with the WorstSCP-rank is only achievable by a non-linear model such as GBT (see Figure S12 for a more detailed visualization).
Finally, the GBT model performance had the best results when the RFE selection method was used (black line of GBT panels, Figure 4), outperforming the respective max SCP AUC-ROC values with only eight conformations. The results with the RFE-rank were equivalent with the NEF metric (Figure S10). However, for the FXa protein, the average NEF value of the GBT model surpass the max SCP value after 32 conformations (Figure S11).
ML classifiers benefit from structurally diverse conformations with different levels of SCP
Figure 5 shows the distribution of the top eight conformations selected by the RFE procedure for each protein. The left panels display the cMDS projection of the protein conformations obtained from the similarity between their respective active sites’ shape. Therefore, the closer two given conformations are, the more similar their cavities are. Likewise, the swarm plots (Figure 5, right panels) show the conformations’ distribution according to their AUC-ROC value obtained by single-conformation docking assays. The purpose of these plots was to offer a straightforward visualization to determine if the RFE selected conformations shared conformational properties or had similar individual performances. Figure S13 shows two additional cMDS projections per protein. These cMDS were computed from the pairwise RMSD matrix considering Cα atoms from two different subsequences: protein secondary structure residues and protein pocket’s residues. Finally, we carried out y-randomization tests using only the 8 RFE selected conformations. Figure S14 shows these results, confirming the ML models’ robustness even with a reduced number of conformations.
Figure 5:

Top 8 (red orange to black points) protein conformations selected by the RFE procedure using GBT as a base estimator. (a) CDK2 protein: 402 conformations. (b) FXa protein: 136 confs. (c) EGFR protein: 64 confs. (d) HSP90 protein: 64 confs. Left panels: Classical Multidimensional Scaling (cMDS) over the protein pocket’s shape. Each point represents a protein conformation. The point’s size is proportional to the protein pocket’s volume, computed by POVME3. Additional cMDS projections using Cα RMSD values are shown in Figure S13. Right panels: Swarm plots showing the SCP (AUC-ROC) value obtained by each protein conformation according to its performance using the raw docking scores from the whole dataset. The top eight RFE-rank conformations are colored from red to black according to their rank position.
Despite the information provided by cMDS analyses, we were unable to identify structural patterns among the top conformations selected by the RFE procedure. Instead, it seems that the GBT classifier tends to prefer structurally diverse ensembles; the top eight RFE-rank conformations shown in Figure 5 and Figure S13 are spread over the subspace of the cMDS projections. Moreover, in all four proteins, the top RFE-rank conformations also show different levels of prediction power. The swarm plots in Figure 5 show the individual performance of the top eight RFE-rank conformations. As can be seen, these conformations no necessarily have the highest SCP. Instead, in some cases they have the lowest SCP. Consequently, the ML models seem to benefit from conformations capable of discriminating among different sets of active and decoy molecules.
ML classifiers provide a better aggregation of single-conformation predictions
In some cases, the ML classifiers correctly identify active molecules that are poorly ranked by the consensus strategies. This is shown in Tables S6 to S13 and Figures S17 to S22 of the Supporting Information. These results were obtained by integrating the predictions of the SBVS methods after the 30×4cv analysis. Taking the CDK2 protein as an example, Tables S6 and S7 show the top nine molecules identified by the GBT classifier and by the csMIN strategy, respectively. Interestingly, some of the top nine molecules of GBT – all of them actives – were poorly ranked by the consensus strategies (Table S6). This means that, while these active molecules were challenging for the traditional strategies and for the single-conformation docking, the GBT was able to correctly identify them as true positives. Similar results are also observed for the other three protein datasets (Tables S8 to S13). Additionally, Figure S23 shows the pair-wise Kendall’s tau correlations among the molecular rankings obtained by two the ML classifiers and the three consensus strategies. Low correlation values among the five ensemble methods can be observed in all protein datasets. This indicates that, regarding the consensus strategies, the ML classifiers perform a different and better combination of the single-conformation predictions.
It is worth remembering that the ML classifiers are not directly learning from binding patterns or conformational properties but from the individual docking scores obtained by each protein conformation. From this perspective, the combination of the scoring function and a given conformation works as an individual predictor, while the ML classifier (GBT or LR) operates like a meta-learner algorithm that combines all the individual predictions, forming a stacking ensemble.106 Stacked generalization is an ensemble learning technique where the individual predictions of first-level ensemble members (base learners) are combined using a second-level ML algorithm – known as blender or meta-learner.106,107 Similarly, the consensus strategies can be saw as variants of a more basic ensemble learning method known as voting; the csAVG strategy is an example of unweighted soft voting.108 Nevertheless, these voting schemes are only recommended if the base learners perform comparably well.109 However, that is not the case for the protein conformations because they could have a wide performance range (EGFR), or an average SCP below 0.6 of AUC-ROC (for CDK2, FXa, and HSP90). See avg SCP in Figure 2 and SCP in the swarm plots of Figure 5. The above could explain the poor performance of the consensus strategies – even with the top conformations of the BestSCP-rank. Instead, the GBT classifier beat the max SCP even when using the worst 32 conformations, which individually were slightly better than random guesses (Figure S12).
Conclusions
In this work, we proposed that the application of ML techniques can improve the ensemble docking performance. Specifically, we asked if ML algorithms could be used to effectively aggregate the ensemble docking scores and lead to better predictions than those obtained with traditional consensus strategies. To test this idea, we selected two ML classifiers, GBT and LR, to evaluate four protein datasets as study cases: CDK2, FXa, EGFR, and HSP90. We performed ensemble docking simulations using the protein’s respective conformational ensembles and molecular libraries. Subsequently, we carried out a series of 30×4cv analyses to (i) compare the ML classifiers’ prediction power with traditional procedures, like consensus strategies and single-conformation docking; (ii) evaluate the robustness of the ML models; and (iii) determine the effect of the number and the selection of protein conformations on the ML models’ performance.
Our results showed that the two ML classifiers significantly outperform the traditional consensus strategies and surpass the best observed performance achieved by a single conformation (max SCP). In general, GBT gave significantly better results than LR, giving favorable performances even with a reduced number of conformations. Moreover, contrary to the traditional consensus strategies, the ML classifiers’ performance increased with the number of structures included in the ensemble.
Although our primary goal was not to develop ML models for production use, the trained ML models employed here are available in the GitHub repository (Supporting Information). However, the major weakness of these ML models is that they are target-specific, and their application is limited to their respective proteins. The extensiveness of the followed methodology is also restricted to those targets with available crystallographic structures and large sets of known actives. Moreover, when SBVS benchmarking sets are used for training ML models, it is not always clear if they have learned from receptor-ligand interactions or, instead, from ligand biases inherent to the benchmarking sets.100,110-112 Although we employed docking scores as the input features – assuming them as protein-ligand features –, overestimation due to the dataset bias is still possible.100 Consequently, we would like to emphasize that any performance improvement shown by our ML models only makes sense regarding the baselines used for comparison: the consensus strategies and the SCP values computed from the same dataset.
The main contribution of the present study is supporting the use of ensemble docking along with ML techniques. Here, we have used the ensemble docking scores, computed by the Smina-Vinardo scoring function, as the input features of the ML algorithms. Nevertheless, future research could continue exploring other sources of protein conformations and other scoring functions – such as ML-based ones30,113 – to work as the base learners of the stacking ensemble. Moreover, more explicit protein-ligand features33,114-117 could serve to make the most from the multiple binding modes obtained from the protein’s conformational ensemble. Hence, we believe that no matter how simple or complex an ML may be, the incorporation of multiple receptor conformations could enhance them by capturing different binding patterns from all the potential inhibitors evaluated during the screening process.5,11,118 Altogether, we hope this work will serve as a basis to develop more complex and general ML models that can effectively exploit the receptor’s flexibility for SBVS.
Supplementary Material
Acknowledgement
The authors thank LANCAD-UNAM-DGTIC-286 and PAPIIT-DGAPA-UNAM-IG200320 grants. CAB and JRL acknowledge the support of CONACyT under grant A1-S-20638. JRL was supported by the Programa de Doctorado en Nanociencias at CICESE-UNAM and by CONACyT. Authors also thank to the anonymous reviewers for their comments and thoughtful suggestions, which substantially helped to improve the manuscript. M.K.G. acknowledges funding from National Institute of General Medical Sciences (GM061300 and GM100946). These findings are solely of the authors and do not necessarily represent the views of the NIH
Footnotes
Disclosure
M.K.G. has an equity interest in and is a cofounder and scientific advisor of VeraChem LLC.
Supporting Information Available
Details about the statistical analyses, the ML models’ hyperparameters, the molecular libraries, and the VS methods performance.
Data and Software Availability
The datasets used here are available in the following GitHub repository: jRicciL/ML-ensemble-docking. This repository also contains Jupyter notebooks and additional python and R scripts to reproduce the results and figures. The third-party python libraries used for the analyses are also listed.
References
- (1).Cerqueira NM; Gesto D; Oliveira EF; Santos-Martins D; Brás NF; Sousa SF; Fernandes PA; Ramos MJ Receptor-Based Virtual Screening Protocol for Drug Discovery. Arch. Biochem. Biophys 2015, 582, 56–67. [DOI] [PubMed] [Google Scholar]
- (2).Kitchen DB; Decornez H; Furr JR; Bajorath J Docking and Scoring in Virtual Screening for Drug Discovery: Methods and Applications. Nat. Rev. Drug Discovery 2004, 3, 935–949. [DOI] [PubMed] [Google Scholar]
- (3).Pinzi L; Rastelli G Molecular Docking: Shifting Paradigms in Drug Discovery. Int. J. Mol. Sci 2019, 20, 4331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Scior T; Bender A; Tresadern G; Medina-Franco JL; Martínez-Mayorga K; Langer T; Cuanalo-Contreras K; Agrafiotis DK Recognizing Pitfalls in Virtual Screening: A Critical Review. J. Chem. Inf. Model 2012, 52, 867–881. [DOI] [PubMed] [Google Scholar]
- (5).Lexa KW; Carlson HA Protein Flexibility in Docking and Surface Mapping. Q. Rev. Biophys 2012, 45, 301–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Totrov M; Abagyan R Flexible Ligand Docking to Multiple Receptor Conformations: a Practical Alternative. Curr. Opin. Struct. Biol 2008, 18, 178–184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).B-Rao C; Subramanian J; Sharma SD Managing Protein Flexibility in Docking and Its Applications. Drug Discovery Today 2009, 14, 394–400. [DOI] [PubMed] [Google Scholar]
- (8).Durrant JD; McCammon JA Computer-Aided Drug-Discovery Techniques That Account for Receptor Flexibility. Curr. Opin. Pharmacol 2010, 10, 770–774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).Feixas F; Lindert S; Sinko W; McCammon JA Exploring the Role of Receptor Flexibility in Structure-Based Drug Discovery. Biophys. Chem 2014, 186, 31–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Amaro RE; Baudry J; Chodera J; Demir O; McCammon JA; Miao Y; Smith JC Ensemble Docking in Drug Discovery. Biophys. J 2018, 114, 2271–2278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Sørensen J; Demir Ö; Swift RV; Feher VA; Amaro RE In Molecular Docking to Flexible Targets; Kukol A, Ed.; Methods in Molecular Biology; Springer New York: New York, NY, 2015; Vol. 1215; pp 445–469. [DOI] [PubMed] [Google Scholar]
- (12).Schames JR; Henchman RH; Siegel JS; Sotriffer CA; Ni H; McCammon JA Discovery of a Novel Binding Trench in HIV Integrase. J. Med. Chem 2004, 47, 1879–1881. [DOI] [PubMed] [Google Scholar]
- (13).Cheng LS; Amaro RE; Xu D; Li WW; Arzberger PW; McCammon JA Ensemble-Based Virtual Screening Reveals Potential Novel Antiviral Compounds for Avian Influenza Neuraminidase. J. Med. Chem 2008, 51, 3878–3894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Amaro RE; Schnaufer A; Interthal H; Hol W; Stuart KD; McCammon JA Discovery of Drug-Like Inhibitors of an Essential RNA-Editing Ligase in Trypanosoma Brucei. Proc. Natl. Acad. Sci 2008, 105, 17278–17283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Durrant JD; Hall L; Swift RV; Landon M; Schnaufer A; Amaro RE Novel Naphthalene-Based Inhibitors of Trypanosoma Brucei RNA Editing Ligase 1. PLoS Neglected Trop. Dis 2010, 4, e803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (16).Chan AH; Wereszczynski J; Amer BR; Yi SW; Jung ME; McCammon JA; Clubb RT Discovery of S Taphylococcus Aureus Sortase A Inhibitors Using Virtual Screening and the Relaxed Complex Scheme. Chem. Biol. Drug Des 2013, 82, 418–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Miao Y; Goldfeld DA; Moo EV; Sexton PM; Christopoulos A; McCammon JA; Valant C Accelerated Structure-Based Design of Chemically Diverse Allosteric Modulators of a Muscarinic G Protein-Coupled Receptor. Proc. Natl. Acad. Sci 2016, 113, E5675–E5684. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (18).Ochoa R; Watowich SJ; Flórez A; Mesa CV; Robledo SM; Muskus C Drug Search for Leishmaniasis: a Virtual Screening Approach by Grid Computing. J. Comput.-Aided Mol. Des 2016, 30, 541–552. [DOI] [PubMed] [Google Scholar]
- (19).Craig IR; Essex JW; Spiegel K Ensemble Docking into Multiple Crystallographically Derived Protein Structures: An Evaluation Based on the Statistical Analysis of Enrichments. J. Chem. Inf. Model 2010, 50, 511–524. [DOI] [PubMed] [Google Scholar]
- (20).Cavasotto CN; Abagyan RA Protein Flexibility in Ligand Docking and Virtual Screening to Protein Kinases. J. Mol. Biol 2004, 337, 209–225. [DOI] [PubMed] [Google Scholar]
- (21).Bajusz D; Rácz A; Héberger K Comparison of Data Fusion Methods as Consensus Scores for Ensemble Docking. Molecules 2019, 24, 2690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Willett P Combination of Similarity Rankings Using Data Fusion. J. Chem. Inf. Model 2013, 53, 1–10. [DOI] [PubMed] [Google Scholar]
- (23).Palacio-Rodríguez K; Lans I; Cavasotto CN; Cossio P Exponential Consensus Ranking Improves the Outcome in Docking and Receptor Ensemble Docking. Sci. Rep 2019, 9, 5142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Ericksen SS; Wu H; Zhang H; Michael LA; Newton MA; Hoffmann FM; Wildman SA Machine Learning Consensus Scoring Improves Performance Across Targets in Structure-Based Virtual Screening. J. Chem. Inf. Model 2017, 57, 1579–1590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Bottegoni G; Rocchia W; Rueda M; Abagyan R; Cavalli A Systematic Exploitation of Multiple Receptor Conformations for Virtual Ligand Screening. PLoS One 2011, 6, e18845. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Korb O; Olsson TSG; Bowden SJ; Hall RJ; Verdonk ML; Liebeschuetz JW; Cole JC Potential and Limitations of Ensemble Docking. J. Chem. Inf. Model 2012, 52, 1262–1274. [DOI] [PubMed] [Google Scholar]
- (27).Barril X; Morley SD Unveiling the Full Potential of Flexible Receptor Docking Using Multiple Crystallographic Structures. J. Med. Chem 2005, 48, 4432–4443. [DOI] [PubMed] [Google Scholar]
- (28).Rueda M; Bottegoni G; Abagyan R Recipes for the Selection of Experimental Protein Conformations for Virtual Screening. J. Chem. Inf. Model 2010, 50, 186–193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Xu M; Lill MA Utilizing Experimental Data for Reducing Ensemble Size in Flexible-Protein Docking. J. Chem. Inf. Model 2012, 52, 187–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (30).Li H; Sze KH; Lu G; Ballester PJ Machine-Learning Scoring Functions for Structure-Based Virtual Screening. Wiley Interdiscip. Rev.: Comput. Mol. Sci 2021, 11, 1–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Ballester PJ; Mitchell JBO A Machine Learning Approach to Predicting Protein-Ligand Binding Affinity with Applications to Molecular Docking. Bioinformatics 2010, 26, 1169–1175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Durrant JD; McCammon JA NNScore 2.0: A Neural-Network Receptor-Ligand Scoring Function. J. Chem. Inf. Model 2011, 51, 2897–2903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Li H; Leung KS; Wong MH; Ballester PJ Improving Autodock Vina Using Random Forest: The Growing Accuracy of Binding Affinity Prediction by the Effective Exploitation of Larger Data Sets. Mol. Inf 2015, 34. [DOI] [PubMed] [Google Scholar]
- (34).Wallach I; Dzamba M; Heifets A AtomNet: A Deep Convolutional Neural Network for Bioactivity Prediction in Structure-Based Drug Discovery. 2015, 1–11. [Google Scholar]
- (35).Pereira JC; Caffarena ER; dos Santos CN Boosting Docking-Based Virtual Screening with Deep Learning. J. Chem. Inf. Model 2016, 56, 2495–2506. [DOI] [PubMed] [Google Scholar]
- (36).Wójcikowski M; Ballester PJ; Siedlecki P Performance of machine-learning scoring functions in structure-based virtual screening. Sci. Rep 2017, 7, 46710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (37).Ragoza M; Hochuli J; Idrobo E; Sunseri J; Koes DR Protein-Ligand Scoring with Convolutional Neural Networks. J. Chem. Inf. Model 2017, 57, 942–957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Klon AE; Glick M; Davies JW Combination of a Naive Bayes Classifier with Consensus Scoring Improves Enrichment of High-Throughput Docking Results. J. Med. Chem 2004, 47, 4356–4359. [DOI] [PubMed] [Google Scholar]
- (39).Tian S; Sun H; Li Y; Pan P; Li D; Hou T Development and Evaluation of an Integrated Virtual Screening Strategy by Combining Molecular Docking and Pharmacophore Searching Based on Multiple Protein Structures. J. Chem. Inf. Model 2013, 53, 2743–2756. [DOI] [PubMed] [Google Scholar]
- (40).Tian S; Sun H; Pan P; Li D; Zhen X; Li Y; Hou T Assessing an Ensemble Docking-Based Virtual Screening Strategy for Kinase Targets by Considering Protein Flexibility. J. Chem. Inf. Model 2014, 54, 2664–2679. [DOI] [PubMed] [Google Scholar]
- (41).Chandak T; Mayginnes JP; Mayes H; Wong CF Using Machine Learning to Improve Ensemble Docking for Drug Discovery. Proteins: Struct., Funct., Bioinf 2020, 88, 1263–1270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (42).Thomas MP; McInnes C; Fischer PM Protein Structures in Virtual Screening: A Case Study with CDK2. J. Med. Chem 2006, 49, 92–104. [DOI] [PubMed] [Google Scholar]
- (43).Osguthorpe DJ; Sherman W; Hagler AT Generation of Receptor Structural Ensembles for Virtual Screening Using Binding Site Shape Analysis and Clustering. Chem. Biol. Drug Des 2012, 80, 182–193. [DOI] [PubMed] [Google Scholar]
- (44).Swift RV; Jusoh SA; Offutt TL; Li ES; Amaro RE Knowledge-Based Methods to Train and Optimize Virtual Screening Ensembles. J. Chem. Inf. Model 2016, 56, 830–842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (45).Campbell AJ; Lamb ML; Joseph-McCarthy D Ensemble-Based Docking Using Biased Molecular Dynamics. J. Chem. Inf. Model 2014, 54, 2127–2138. [DOI] [PubMed] [Google Scholar]
- (46).Moroy G; Sperandio O; Rielland S; Khemka S; Druart K; Goyal D; Perahia D; Miteva MA Sampling of Conformational Ensemble for Virtual Screening Using Molecular Dynamics Simulations and Normal Mode Analysis. Future Med. Chem 2015, 7, 2317–2331. [DOI] [PubMed] [Google Scholar]
- (47).Greenidge PA; Kramer C; Mozziconacci J-C; Sherman W Improving Docking Results via Reranking of Ensembles of Ligand Poses in Multiple X-Ray Protein Conformations with MM-GBSA. J. Chem. Inf. Model 2014, 54, 2697–2717. [DOI] [PubMed] [Google Scholar]
- (48).Evangelista W; Weir RL; Ellingson SR; Harris JB; Kapoor K; Smith JC; Baudry J Ensemble-Based Docking: From Hit Discovery to Metabolism and Toxicity Predictions. Bioorg. Med. Chem 2016, 24, 4928–4935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (49).Li L; Khanna M; Jo I; Wang F; Ashpole NM; Hudmon A; Meroueh SO Target-Specific Support Vector Machine Scoring in Structure-Based Virtual Screening: Computational Validation, In Vitro Testing in Kinases, and Effects on Lung Cancer Cell Proliferation. J. Chem. Inf. Model 2011, 51, 755–759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (50).Shen C; Hu Y; Wang Z; Zhang X; Zhong H; Wang G; Yao X; Xu L; Cao D; Hou T Can Machine Learning Consistently Improve the Scoring Power of Classical Scoring Functions? Insights into the Role of Machine Learning in Scoring Functions. Briefings Bioinf. 2021, 22, 497–514. [DOI] [PubMed] [Google Scholar]
- (51).Berman HM; Westbrook J; Feng Z; Gilliland G; Bhat TN; Weissig H; Shindyalov IN; Bourne PE The Protein Data Bank. Nucleic Acids Res. 2000, 28, 235–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (52).Huang N; Shoichet BK; Irwin JJ Benchmarking Sets for Molecular Docking. J. Med. Chem 2006, 49, 6789–6801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (53).Armstrong MS; Morris GM; Finn PW; Sharma R; Moretti L; Cooper RI; Richards WG ElectroShape: Fast Molecular Similarity Calculations Incorporating Shape, Chirality and Electrostatics. J. Comput.-Aided Mol. Des 2010, 24, 789–801. [DOI] [PubMed] [Google Scholar]
- (54).Bauer MR; Ibrahim TM; Vogel SM; Boeckler FM Evaluation and Optimization of Virtual Screening Workflows with DEKOIS 2.0 - A Public Library of Challenging Docking Benchmark Sets. J. Chem. Inf. Model 2013, 53, 1447–1462. [DOI] [PubMed] [Google Scholar]
- (55).Dunbar JB; Smith RD; Damm-Ganamet KL; Ahmed A; Esposito EX; Delproposto J; Chinnaswamy K; Kang Y-N; Kubish G; Gestwicki JE et al. CSAR Data Set Release 2012: Ligands, Affinities, Complexes, and Docking Decoys. J. Chem. Inf. Model 2013, 53, 1842–1852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (56).Friedman JH Stochastic Gradient Boosting. Computational Statistics & Data Analysis 2002, 38, 367–378. [Google Scholar]
- (57).Wang B; Zhao Z; Nguyen DD; Wei G-W Feature Functional Theory–Binding Predictor (FFT–BP) for the Blind Prediction of Binding Free Energies. Theor. Chem. Acc 2017, 136, 55. [Google Scholar]
- (58).Cang Z; Mu L; Wei G-W Representability of Algebraic Topology for Biomolecules in Machine Learning Based Scoring and Virtual Screening. PLoS Comput. Biol 2018, 14, e1005929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Ashtawy HM; Mahapatra NR Task-Specific Scoring Functions for Predicting Ligand Binding Poses and Affinity and for Screening Enrichment. J. Chem. Inf. Model 2018, 58, 119–133. [DOI] [PubMed] [Google Scholar]
- (60).Shen C; Ding J; Wang Z; Cao D; Ding X; Hou T From Machine Learning to Deep Learning: Advances in Scoring Functions for Protein–Ligand Docking. WIREs Computational Molecular Science 2020, 10. [Google Scholar]
- (61).Amaro RE; Baron R; McCammon JA An Improved Relaxed Complex Scheme for Receptor Flexibility in Computer-Aided Drug Design. J. Comput.-Aided Mol. Des 2008, 22, 693–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (62).Bakan A; Meireles LM; Bahar I ProDy: Protein Dynamics Inferred from Theory and Experiments. Bioinformatics 2011, 27, 1575–1577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Sali a. MODELLER: A Program for Protein Structure Modeling Release 9.12, R9480. Rockefeller University; 2013, 779–815. [Google Scholar]
- (64).Case D; Ben-Shalom I; Brozell S; Cerutti D; Cheatham T; Cruzeiro V; Darden T; Duke R; Ghoreishi D; Gilson M et al. University of California, San Francisco; 2018. [Google Scholar]
- (65).Dolinsky TJ; Czodrowski P; Li H; Nielsen JE; Jensen JH; Klebe G; Baker NA PDB2PQR: Expanding and Upgrading Automated Preparation of Biomolecular Structures for Molecular Simulations. Nucleic Acids Res. 2007, 35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Roe DR; Cheatham TE PTRAJ and CPPTRAJ: Software for Processing and Analysis of Molecular Dynamics Trajectory Data. J. Chem. Theory Comput 2013, 9, 3084–3095. [DOI] [PubMed] [Google Scholar]
- (67).Torgerson WS Multidimensional Scaling: I. Theory and Method. Psychometrika 1952, 17, 401–419. [Google Scholar]
- (68).Wagner JR; Sørensen J; Hensley N; Wong C; Zhu C; Perison T; Amaro RE POVME 3.0: Software for Mapping Binding Pocket Flexibility. J. Chem. Theory Comput 2017, 13, 4584–4592. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (69).DUD Dataset: Partial Charges for DUD Molecules Recalculated by Inhibox. 2010; http://dud.docking.org/inhibox.html, (accessed on Jun 20, 2021). [Google Scholar]
- (70).DEKOIS 2.0 Dataset. 2015; http://www.dekois.com/, (accessed on Jun 20, 2021).
- (71).CSAR-2012 Dataset. 2013; http://www.csardock.org/, (accessed on Jun 20, 2021).
- (72).O’Boyle NM; Banck M; James CA; Morley C; Vandermeersch T; Hutchison GR Open Babel: An Open Chemical Toolbox. J. Cheminf 2011, 3, 33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (73).RDKit: Open-Source Cheminformatics. 2020; http://www.rdkit.org, (accessed on May 12, 2020).
- (74).Quiroga R; Villarreal MA Vinardo: A Scoring Function Based on Autodock Vina Improves Scoring, Docking, and Virtual Screening. PLoS One 2016, 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (75).Koes DR; Baumgartner MP; Camacho CJ Lessons Learned in Empirical Scoring with Smina from the CSAR 2011 Benchmarking Exercise. J. Chem. Inf. Model 2013, 53, 1893–1904. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (76).Morris GM; Huey R; Lindstrom W; Sanner MF; Belew RK; Goodsell DS; Olson AJ AutoDock4 and AutoDockTools4: Automated Docking with Selective Receptor Flexibility. J. Comput. Chem 2009, 30, 2785–2791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Pedregosa F; Varoquaux G; Gramfort A; Michel V; Thirion B; Grisel O; Blondel M; Prettenhofer P; Weiss R; Dubourg V et al. Scikit-Learn: Machine Learning in Python. Journal of Machine Learning Research 2011, [Google Scholar]
- (78).Chen T; Guestrin C XGBoost: A Scalable Tree Boosting System. 2016, 785–794. [Google Scholar]
- (79).Refaeilzadeh P; Tang L; Liu H Encyclopedia of Database Systems; Springer US: Boston, MA, 2009; pp 532–538. [Google Scholar]
- (80).Kim J-H Estimating Classification Error Rate: Repeated Cross-Validation, Repeated Hold-Out and Bootstrap. Computational Statistics & Data Analysis 2009, 53, 3735–3745. [Google Scholar]
- (81).Raschka S Model Evaluation, Model Selection, and Algorithm Selection in Machine Learning. 2018, 1–49. [Google Scholar]
- (82).Rücker C; Rücker G; Meringer M Y-Randomization and Its Variants in QSPR/QSAR. J. Chem. Inf. Model 2007, 47, 2345–2357. [DOI] [PubMed] [Google Scholar]
- (83).Truchon JF; Bayly CI Evaluating Virtual Screening Methods: Good and Bad Metrics for the “Early Recognition” Problem. J. Chem. Inf. Model 2007, 47, 488–508. [DOI] [PubMed] [Google Scholar]
- (84).Liu S; Alnammi M; Ericksen SS; Voter AF; Ananiev GE; Keck JL; Hoffmann FM; Wildman SA; Gitter A Practical Model Selection for Prospective Virtual Screening. J. Chem. Inf. Model 2019, 59, 282–293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (85).Friedman M A Comparison of Alternative Tests of Significance for the Problem of M Rankings. The Annals of Mathematical Statistics 1940, 11, 86–92. [Google Scholar]
- (86).Nemenyi PB Distribution-Free Multiple Comparisons. Ph.D. thesis, Princeton University, 1963. [Google Scholar]
- (87).Demšar J Statistical Comparisons of Classifiers over Multiple Data Sets. Journal of Machine Learning Research 2006, 7, 1–30. [Google Scholar]
- (88).Shapiro SS; Wilk MB An Analysis of Variance Test for Normality (Complete Samples). Biometrika 1965, 52, 591–611. [Google Scholar]
- (89).Mauchly J Significance Test for Sphericity of a Normal N-Variate Distribution. The Annals of Mathematical Statistics. The Annals of Mathematical Statistics 1940, 11, 204–209. [Google Scholar]
- (90).Kruskal WH; Wallins WW Use of Ranks in One-Criterion Variance Analysis. J. Am. Stat. Assoc 1952, 47, 583–621. [Google Scholar]
- (91).Dunn O Multiple Comparisons Using Rank Sums. Technometrics 1964, 6, 241–256. [Google Scholar]
- (92).Wickham H; Averick M; Bryan J; Chang W; McGowan LD; François R; Grolemund G; Hayes A; Henry L; Hester J et al. Welcome to the {Tidyverse}. Journal of Open Source Software 2019, 4, 1686. [Google Scholar]
- (93).Kassambara A Rstatix: Pipe-Friendly Framework for Basic Statistical Tests. 2020; https://cran.r-project.org/package=rstatix. [Google Scholar]
- (94).Strecker C; Meyer B Plasticity of the Binding Site of Renin: Optimized Selection of Protein Structures for Ensemble Docking. J. Chem. Inf. Model 2018, 58, 1121–1131. [DOI] [PubMed] [Google Scholar]
- (95).Wong CF Improving Ensemble Docking for Drug Discovery by Machine Learning. J. Theor. Comput. Chem 2019, 18, 19–22. [Google Scholar]
- (96).Huang S-Y; Zou X Ensemble Docking of Multiple Protein Structures: Considering Protein Structural Variations in Molecular Docking. Proteins: Struct., Funct., Bioinf 2006, 66, 399–421. [DOI] [PubMed] [Google Scholar]
- (97).Ben Nasr N; Guillemain H; Lagarde N; Zagury JF; Montes M Multiple Structures for Virtual Ligand Screening: Defining Binding Site Properties-Based Criteria to Optimize the Selection of the Query. J. Chem. Inf. Model 2013, 53, 293–311. [DOI] [PubMed] [Google Scholar]
- (98).García-Sosa AT; Hetényi C; Maran U Drug efficiency indices for improvement of molecular docking scoring functions. J. Comput. Chem 2010, 31, 174–184. [DOI] [PubMed] [Google Scholar]
- (99).Cosconati S; Forli S; Perryman AL; Harris R; Goodsell DS; Olson AJ Virtual Screening with AutoDock: Theory and Practice. Expert Opin. Drug Discovery 2010, 5, 597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Ballester PJ Selecting Machine-Learning Scoring Functions for Structure-Based Virtual Screening. Drug Discovery Today: Technol. 2019, 32–33, 81–87. [DOI] [PubMed] [Google Scholar]
- (101).Ojala M; Garriga GC Permutation Tests for Studying Classifier Performance. 2009 Ninth IEEE International Conference on Data Mining. 2009; pp 908–913. [Google Scholar]
- (102).Teodoro ML Conformational Flexibility Models for the Receptor in Structure Based Drug Design. Curr. Pharm. Des 2003, 9, 1419–1431. [DOI] [PubMed] [Google Scholar]
- (103).Osguthorpe DJ; Sherman W; Hagler AT Exploring Protein Flexibility: Incorporating Structural Ensembles from Crystal Structures and Simulation into Virtual Screening Protocols. J. Phys. Chem. B 2012, 116, 6952–6959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (104).Basciu A; Malloci G; Pietrucci F; Bonvin AMJJ; Vargiu AV Holo-Like and Druggable Protein Conformations from Enhanced Sampling of Binding Pocket Volume and Shape. J. Chem. Inf. Model 2019, 59, 1515–1528. [DOI] [PubMed] [Google Scholar]
- (105).Akbar R; Jusoh SA; Amaro RE; Helms V ENRI: A Tool for Selecting Structure-Based Virtual Screening Target Conformations. Chem. Biol. Drug Des 2017, 89, 762–771. [DOI] [PubMed] [Google Scholar]
- (106).Wolpert DH Stacked Generalization. Neural Networks 1992, 5, 241–259. [Google Scholar]
- (107).Ting KM; Witten IH Issues in Stacked Generalization. Journal of Artificial Intelligence Research 1999, 10, 271–289. [Google Scholar]
- (108).Dietterich TG Machine-Learning Research. AI Magazine 1997, 18, 97–97. [Google Scholar]
- (109).Witten IH; Frank E; Hall MA; Pal CJ Data Mining: Practical Machine Learning Tools and Techniques; Elsevier, 2011; pp 1–621. [Google Scholar]
- (110).Réau M; Langenfeld F; Zagury JF; Lagarde N; Montes M Decoys Selection in Benchmarking Datasets: Overview and Perspectives. Front. Pharmacol. 2018, 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (111).Chen L; Cruz A; Ramsey S; Dickson CJ; Duca JS; Hornak V; Koes DR; Kurtzman T Hidden Bias in the DUD-E Dataset Leads to Misleading Performance of Deep Learning in Structure-Based Virtual Screening. PLoS One 2019, 14, 1–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (112).Sieg J; Flachsenberg F; Rarey M In Need of Bias Control: Evaluating Chemical Data for Machine Learning in Structure-Based Virtual Screening. J. Chem. Inf. Model 2019, 59, 947–961. [DOI] [PubMed] [Google Scholar]
- (113).Ain QU; Aleksandrova A; Roessler FD; Ballester PJ Machine-Learning Scoring Functions to Improve Structure-Based Binding Affinity Prediction and Virtual Screening. Wiley Interdiscip. Rev.: Comput. Mol. Sci 2015, 5, 405–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Durrant JD; McCammon JA BINANA: A Novel Algorithm for Ligand-Binding Characterization. J. Mol. Graphics Modell 2011, 29, 888. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Arciniega M; Lange OF Improvement of Virtual Screening Results by Docking Data Feature Analysis. J. Chem. Inf. Model 2014, 54, 1401–1411. [DOI] [PubMed] [Google Scholar]
- (116).Wójcikowski M; Zielenkiewicz P; Siedlecki P Open Drug Discovery Toolkit (ODDT): a New Open-Source Player in the Drug Discovery Field. J. Cheminf 2015, 7, 26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Ye W-L; Shen C; Xiong G-L; Ding J-J; Lu A-P; Hou T-J; Cao D-S Improving Docking-Based Virtual Screening Ability by Integrating Multiple Energy Auxiliary Terms from Molecular Docking Scoring. J. Chem. Inf. Model 2020, 60, 4216–4230. [DOI] [PubMed] [Google Scholar]
- (118).De Vivo M; Masetti M; Bottegoni G; Cavalli A Role of Molecular Dynamics and Related Methods in Drug Discovery. J. Med. Chem 2016, 59, 4035–4061. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.