

Abstract
During animal embryogenesis, homeostasis and disease, tissues push and pull on their surroundings to move forward. Although the force-generating machinery is known, it is unknown how tissues exert physical stresses on their substrate to generate motion in vivo. Here, we identify the force transmission machinery, the substrate, and the stresses that a tissue, the zebrafish posterior lateral line primordium, generates during its migration. We find that the primordium couples actin flow through integrins to the basement membrane for forward movement. Talin/integrin-mediated coupling is required for efficient migration, and its loss is partly compensated for by increased actin flow. Using Embryogram, an approach to measure stresses in vivo, we show that the primordium’s rear exerts higher stresses than the front, suggesting that this tissue pushes itself forward with its back. This unexpected strategy likely also underlies the motion of other tissues in animals.
During development, homeostasis, and disease, cells and tissues move to form organs, seal wounds and hunt pathogens1. To move, cells generate force and interact with their surroundings to pull and push themselves forward. Force transmission from the actomyosin network to the surroundings has been molecularly characterized and precisely measured in cultured cells2,3. Cells use integrin-based adhesion complexes to couple the actomyosin network inside the cells to the substrates outside the cells and pull on their surroundings with forces around 3–30 pN across molecules4. Since many processes are altered when cells are removed from their physiological environment and placed in culture, it is largely unclear whether cells in living animals interact with their surrounding in the same manner and pull on their substrate with similar forces. To address these questions, we used the zebrafish posterior lateral line primordium as a model. The primordium is a tissue of about 140 cells that expresses the chemokine receptor Cxcr4b. It migrates directly under the skin from behind the ear to the tip of the tail and follows a gradient of the chemokine Cxcl12a along the body of the embryo5.
The primordium migrates on a basement membrane
To learn about the substrate that the primordium uses to push and pull itself forward, we inspected transverse sections of 30 hpf embryos at different locations along the primordium’s migratory route by transmission electron microscopy (TEM). Consistent with previous studies6,7, we found that the two-layered skin is separated from the underlying muscle by a 200 nm thick basement membrane (BM) in front of the migrating primordium (Extended Data Fig. 1a, e). At the position of the primordium, the migrating tissue separates the skin and the BM, such that the primordium’s basal side is juxtaposed to the BM while there is no BM detectable on the primordium’s apical side (Fig. 1a, Extended Data Fig. 1b-e). We confirmed these observations by inspecting the localization of the core BM component Laminin-γ1 tagged with superfolder GFP (LamC1-sfGFP) (Extended Data Fig. 1f, g). During its migration the primordium wedges itself between the skin and the muscle, and pushes the LamC1-sfGFP-labeled BM towards its basal side with little to no LamC1-sfGFP detectable between the primordium and the skin (Extended Data Fig. 1j). This separates the BM from the skin (Fig. 1b). Consistent with this, we find that the basal side of the skin labeled with fluorescently tagged E-Cadherin abuts the apical side of the primordium (Fig. 1c). Thus, the primordium migrates on top of a BM and underneath the skin (Fig, 1d).
Fig. 1. The primordium migrates on top of a basement membrane and directly under the skin.

a, TEM images of the skin (s), primordium (p, purple hue), the muscle (m), and BM (white arrows). n = 1 embryo. Scale bar = 1 μm. b, Optical sections through a primordium in a 31 hpf embryo expressing LamC1-sfGFP. Scale bar = 50 μm. c, Optical section through a primordium labeled with F-tractin-mCherry in a live 32 hpf embryo expressing Cdh1-sfGFP. n = 7 embryos. Scale bar = 25 μm. A: apical, B: basal. d, schematic illustration of the environment around the primordium.
The primordium requires the basement membrane for migration
If the skin and the BM serve as substrates for the primordium, they should also be required for the migration of the primordium. Indeed, surgical removal of the skin blocks primordium migration8. To assess the role of the skin in primordium migration in a less invasive manner, we depleted α-catenin tagged with Citrine (Ctnna1-Citrine) in the primordium using the degron system zGrad9 expressed from the cxcr4b promoter in the migrating primordium (Extended Data Fig. 2a-d). Ctnna1 links cadherin to the actin cytoskeleton10 and its depletion should abrogate cadherin-mediated cell-cell adhesion between the skin and the primordium. As previously reported11, depletion of Ctnna1-Citrine in the primordium affects sensory organ deposition and directionality of the cells in the primordium (Fig. 2a, b, Video 1). However, we find this does not affect the cells’ speed (Fig. 2c).
Fig. 2. Primordium migration requires an intact basement membrane.

a, Control and Ctnna1-depleted primordia (arrowheads) in 48 hpf embryos. Scale bar = 0.5 mm. Close-up of region indicated by a dashed square. b, Quantification of the migration distance for primordia shown in a. Individual data points, means and SD are indicated. **: p=0.0013 (two-tailed Mann-Whitney test). c, Speed of Ctnna1-depleted primordium cells. Solid line = median, dashed line = quartile. n = cell speeds from more than 7 primordia with each more than 100 cells). d, Left. TEM images of the ultrastructure of the BM between the skin (s) and the muscle (m) in control (n = 2) and lamC1−/− embryos (n = 1). White arrows indicate the BM. Scale bar = 2 μm. Right. Antibody staining against Collagen IV in control and lamC1−/− embryos. Scale bar = 50 μm. e, Quantification of Collagen IV filaments in control and lamC1−/− embryos. Individual data points, means and SD are indicated. **: p=0.0056 (two-tailed Mann-Whitney test). f, Strategy to express Cxcl12a in a few muscle cells in lamC1−/− embryos and siblings. g, Images of the migrating primordium in cxcl12a−/− and cxcl12a−/−; lamC1−/− 32 hpf embryos with clones in the trunk muscle that express mCherry (not shown) or Cxcl12a together with mCherry (not shown). Asterisks indicate the ear and arrowheads the primordium. Scale bar = 0.5 mm. h, Quantification of the distance migrated by the primordium in the indicated experimental conditions at 32 hpf. Individual data points, means and SD are indicated. ***: p=0.0002, ****: p<0.0001, n.s: p=0.0879 (two-tailed Mann-Whitney test). i, Principle of the Cxcl12a sensor. j, Images of the Cxcl12a sensor in primordia of cxcl12a−/− and cxcl12a−/−; lamC1−/− live embryos with clones in the muscle of the trunk that express mCherry or Cxcl12a. Scale bar = 20 μm. k, Quantification of the Cxcr4b-Kate-to-memGFP ratio in the primordia of embryos shown in j. Individual data points, means and SD are indicated. *: p=0.038, ***: p=0.0001, n.s: p=0.1349 (one way ANOVA followed by Holm-Sidak’s multiple comparison test). Note, controls are lamc1+/+ and lamc1−/+ embryos. For a, b, e, h, k, n = number of embryos.
To assess the role of the BM in primordium migration, we analyzed the migration of the primordium in lamC1 mutant embryos. In such embryos, the BM is disrupted or missing7, and inspection of the Collagen IV network and the BM confirms this observation (Fig. 2d, e). Since the lack of LamC1 also impairs the formation of the Cxcl12a-secreting stripe of cells that guides the primordium (Extended Data Fig. 2e), we assessed the ability of the primordium to migrate in lamC1 mutant embryos by generating an ectopic Cxcl12a source in the trunk muscles. The initial location of the primordium is not affected in lamC1 mutant embryos (Extended Data Fig. 2f, g). For this analysis, we also removed endogenous Cxcl12a to avoid competition between endogenous and ectopic chemokine sources (Fig. 2f). While local secretion of Cxcl12a from the muscle of cxcl12a mutant control embryos attracted the primordium, Cxcl12a failed to restore directed migration towards the chemokine sources in lamC1; cxcl12a double mutant embryos (Fig. 2g, h). In cxcl12a mutant control embryos with ectopic Cxcl12a in the muscle, the primordia did not travel the same distance as in wild-type embryos with or without ectopic clones (Extended Data Fig. 2h). The reason for this is that we cannot generate clones in the muscle all along the primordium’s migratory route. Importantly, ectopic Cxcl12a triggered the internalization of its receptor Cxcr4b in primordia of lamC1; cxcl12a double mutant embryos and cxcl12a mutant control embryos, indicating that the diffusion and presentation of Cxcl12a is not impaired in the absence of LamC1 and the BM (Fig. 2i-k, Extended Data Fig. 2i). Thus, an intact BM is required for directed primordium migration, indicating that the primordium relies mostly on the BM rather than the skin as a substrate for its migration—a notion that is corroborated by the increased levels of filamentous actin (F-actin) on the basal side of the cells (Fig. 1c).
The primordium cells form nascent adhesion-like clusters
To push, pull, and exert stresses on the BM, the primordium needs to adhere to the BM. Molecularly, cells can adhere to the BM through focal adhesions. Two core components of these large protein complexes are integrins and talins12. Integrins bind to specific BM components on the outside of the cell and—through talins and other adaptors—to the actin network inside the cell. To test whether the primordium uses focal adhesions to interact with the BM, we first identified the β-integrins and talins that the primordium expresses. Of the twelve β-integrins and three talins in zebrafish, integrin-β1b (itgb1b) and talin1 (tln1) were expressed throughout the primordium (Extended Data Fig. 3a, d). We therefore tagged itgb1b and tln1 with sfGFP and YPet at the endogenous locus and on a bacterial artificial chromosome (BAC) transgene, respectively (Extended Data Fig. 3b, e). Itgb1b-sfGFP and Tln1-YPet recapitulated the endogenous expression pattern (Extended Data Fig. 3c, f) and restored viability when placed in the respective mutant background (viable itgb1b-sfGFP/− and tln1:tln1-YPet; tln1−/− adults, n=20 and 5, respectively). While Itgb1b-sfGFP and Tln1-YPet were enriched at the myotendinous junctions of the muscle, Itgb1b-sfGFP localized fairly uniformly on the membranes of the primordium cells (Fig. 3a, Video 2), and Tln1-YPet was mostly cytoplasmic in the cells of the primordium (Fig. 3b, Video 2). Since Itgb1b-sfGFP and Tln1-YPet are also expressed by the surrounding skin and muscle, this expression could mask protein clustering on the membranes of the primordium cells. We therefore generated embryos in which only a few cells in the primordium expressed Itgb1b-sfGFP or Tln1-YPet together with membrane-tethered mCherry by blastomere transplantation (Fig. 3c). This analysis revealed that Itgb1b-sfGFP and Tln1-YPet formed short-lived clusters with a lifetime of less than 2 minutes on the basal sides of the cells in the primordium, often within the basal protrusions (Extended Data Fig. 4b, c). We also detected short-lived Itgb1b-sfGFP clusters on the apical side of the superficial cells (Extended Data Fig. 4a)—a layer of thin primordium cells that face the skin8,13. These clusters are probably induced by Fibronectin or proteoglycans—labeled by chondroitin sulfate—that are expressed around the primordium (Extended Data Figure 1k, l). Since talin links integrin to F-actin12, we asked whether clustered Itgb1b and Tln1 co-localized with F-actin. Chimeric analysis showed that this is the case; Itgb1b-sfGFP and Tln1-YPet clusters co-localized with F-tractin-labeled F-actin on the basal sides of primordium cells (Fig. 3d-f, Video 3). Control experiments showed that Itgb1b-sfGFP and, to a lesser degree, Tln1-YPet also co-localized with membrane-tethered mCherry, as expected for a transmembrane protein and a cytosolic protein, respectively (Extended Data Fig. 4d, e). Thus, the primordium cells form small, transient integrin/talin/F-actin clusters on their basal sides.
Fig. 3. β1-Integrin and Talin form small short-lived clusters at the basal sides of the primordium cells.

a, Expression of Itgb1b-sfGFP from the endogenous locus in a 33 hpf embryo. The image is a single slice from a z-stack. The primordium is outlined by a dotted, yellow line. Scale bar = 20 μm. b, Expression of Tln1-YPet from the tln1:tln1-YPet BAC transgene in a 33 hpf embryo. The image is a single slice from a z-stack. The primordium is outlined by a dotted, yellow line. Scale bar = 20 μm. c, Schematic of blastomere transplantation experiments. d, Intensity profiles of Itgb1b-sfGFP (left) and Tln1-YPet (right) with F-tractin-mCherry along the perimeter on the basal sides of the clones shown in (e) and (f) over time. e, Images of Itgb1b-sfGFP and F-tractin-mCherry localization at the basal side of a clone in the primordium over time. Images are single slices from a time-lapse movie (Video 3). Arrowheads indicate Itgb1b-sfGFP/F-tractin-mCherry clusters. Scale bar = 10 μm. f, Images of Tln1-YPet and F-tractin-mCherry localization at the basal side of a clone in the primordium over time. Images are single slices from a time-lapse movie (Video 3). Arrowheads indicate Tln1-YPet/F-tractin-mCherry clusters. Scale bar = 10 μm.
We corroborated the transient nature of the integrin clusters by measuring the mobility of integrin in the membrane through FRAP. Ligated integrin couples to the actin network and diffuses more slowly in the membrane than unligated integrin14,15. The mobility of integrin is therefore a measure of the degree of ligated integrins interacting with actin. Consistent with integrin function in muscle, the mobility of Itgb1b-sfGFP at the myotendinous junction increases when blocking ROCK-mediated actin network contractions (Extended Data Fig. 4f-h). In comparison to the myotendinous junction, the mobility of Itgb1b-sfGFP is higher in the cells of the primordium (Extended Data Fig. 4i-k), supporting the idea that integrin interacts with the actin network only transiently in this migrating tissue. In contrast to migrating cells in vitro16,17, these observations suggest that the primordium cells do not form long-lived focal adhesions but rather transient integrin clusters. When placed on a Laminin-coated surface, primordium cells formed large integrin and talin clusters along F-actin cables as observed in cultured cells16,17 (Extended Data Fig. 4l-n), indicating that primordium cells can form focal adhesions and stress fibers ex vivo but do not do so in vivo.
Integrin and talin are required for efficient migration
Next, we asked whether integrin and talin function are required for primordium migration. Since itgb1b and tln1—and possibly itgb1a, tln2a and tln2b—are expressed in the primordium, we generated mutants in these five genes (Extended Data Fig. 5a, Extended Data Fig. 6a). Phenotypic analysis showed that the primordium was less elongated and migrated more slowly in itgb1b mutant embryos than in wild-type controls (Fig. 4a-d, Extended Data Fig. 5b-f, Video 4). Since itgb1a−/+; itgb1b−/− and itgb1a−/−; itgb1b−/− embryos have severe morphogenesis defects (Extended Data Fig. 5b), we could not assess primordium migration in these genetic scenarios. Instead, we assessed whether Itgb1b is required within the tissue for migration by depleting Itgb1b-sfGFP in the primordium of itgb1a−/−; itgb1b-sfGFP/− embryos using zGrad (Fig. 4e, Extended Data Fig. 2a, Extended Data Fig. 5g, h). In such embryos, primordium migration was slowed to almost the same degree as in itgb1b mutant embryos (Fig. 4f, g), indicating that Itgb1b is required within the primordium for efficient migration with minor contribution from Itgb1a.
Fig. 4. β1-Integrin, Talin and their interaction are required for efficient primordium migration.

a, Maximum-projected z-stacks of migrating primordia in wild-type and itgb1b−/− embryos temporally color-coded as indicated. Scale bar = 100 μm. b, Cumulative primordium migration distance in control and itgb1b−/− embryos. Means and SDs are indicated. **: p=0.0053 (two-tailed t-test at the end point). c, Maximum-projected z-stacks of primordia in wild-type and the itgb1b−/− embryos. Scale bar = 25 μm. d, Primordium circularity in control and itgb1b−/− embryos. Means and SDs are indicated. *: p=0.035 (two-tailed t-test at the end point). e, Crossing scheme to generate embryos with primordium-specific depletion of Itgb1b-sfGFP. f, Primordium migration in 48 hpf embryos of indicated genotypes. Primordium-specific Itgb1 depletion refers to genotype shown in e. Arrows indicate primordia, arrowheads indicate the cxcr4b:zGrad transgene marker. Scale bar = 500 μm. g, Primordium migration distance of embryos shown in (f). Individual data points, means and SD are indicated. ****: p<0.0001 (one-way ANOVA followed by Tukey’s multiple comparison test). h, Crossing scheme to generate embryos with reduced Talin activity in the primordium. i, Maximum-projected z-stacks of migrating primordia in embryos of indicated genotypes color-coded for time as indicated. Scale bar = 100 μm. j, Cumulative primordium migration distance in cxcr4b:zGrad and (MZ)tln1−/−; (Z)tln2a+/− or +/+; (MZ)tln2b−/−; cxcr4b:zGrad embryos. Means and SD are indicated. ****: p<0.0001 (two-tailed t-test at the end point). k, Amino acid alignment of indicated Integrin-β1 cytoplasmic domains, the two NPxY motives are indicated in red. l, Itgb1b-sfGFP and Itgb1bΔNPxY-sfGFP localization in the primordium. Images are single transverse section from z-stacks. Scale bar = 25 μm. m, Quantification of the Itgb1b-sfGFP and Itgb1bΔNPxY-sfGFP distribution on the membrane of primordium cells along the apicobasal axis normalized to the fluorescence intensity of membrane-tethered mCherry. *: p=0.012 at 1.3 μm (two-tailed Welch’s t-test). Individual data points (pale dots), means (dots) and SDs are indicated. n, Primordium position in itgb1b:itgb1b-sfGFP/− and the itgb1b:itgb1bΔNPxY-sfGFP/− 48 hpf embryos. Arrows indicate primordia. Scale bar = 0.5 mm. o, Quantification of the primordium migration distance. Individual data points, means and SD are indicated. ****: p<0.0001 (two-tailed Mann-Whitney test). For b, d, g, j, m, o, n = number of embryos.
In talin single, double and triple mutant embryos, the primordium migrated normally and morphogenesis was mostly unaffected (Extended Data Fig. 6b, c). A possible explanation for the surprisingly mild defects in tln1−/−; tln2a−/−; tln2b−/− embryos could be the maternal contribution (M) of talin mRNA and talin protein to the embryo. To address this possibility, we generated zygotic (Z) Z tln1−/−; Z tln2a−/−; Z tln2b−/− embryos which also lacked the maternal contribution (M) of M tln1 and M tln2b (Extended Data Fig. 6d). However, such embryos had somitogenesis defects and disrupted cxcl12a expression along the migratory route of the primordium (Extended Data Fig. 6e-g), impeding the analysis of the role of talin in primordium migration. Next, we depleted Tln1-YPet in the primordium by expressing zGrad from the cxcr4b promoter in MZ tln1−/−; Z tln2a−/+ or Z tln2a+/+; MZ tln2b−/− embryos whose only source of Tln1-YPet was maternally deposited mRNA and protein (Fig. 4h). zGrad efficiently degraded Tln1-YPet (Extended Data Fig. 6h, i). This analysis showed that depleting most talin activity in the primordium slowed the migration of the primordium (Fig. 4i, j, Video 5) as reported for tln1 morphants13. We confirmed this observation by placing cells with strongly reduced talin activity in the migrating primordia by blastomere transplantation (Extended Data Fig. 6j). Compared to controls, such primordia also migrated slower (Extended Data Fig. 6k-m, Video 5).
Talin, and other cytosolic partners of integrin, bind to the NPxY motives in integrin’s cytoplasmic tail12. We therefore deleted the two NPxY motives in Itgb1b and tagged Itgb1bΔNPxY with sfGFP at its endogenous locus (Fig. 4k, Extended Data Fig. 7a). Compared to Itgb1b-sfGFP, Itgb1bΔNPxY-sfGFP localized less to the apical and basal sides of the cells in the primordium and its levels were reduced by 16% (Fig.4l, m). This was also observed for the myotendinous junctions (Extended Data Fig. 7b, c). Consistent with its more uniform distribution, Itgb1bΔNPxY-sfGFP was more mobile than Itgb1b-sfGFP in the membrane (Extended Data Fig. 7d-f). Similar to the global and tissue-specific loss of Itgb1b, Itgb1bΔNPxY-sfGFP also failed to support efficient primordium migration (Fig. 4n, o). This indicates that the integrin/talin complex is important for the primordium to move along its migratory route at normal speed.
β1-Integrin slows actin flow in the primordium
In migrating cells, the integrin-talin complex can couple F-actin flow inside the cell to the BM outside the cell and transduce force18. To test whether the cells in the primordium use such a clutch-like mechanism, we measured the speed of F-actin flow on the apical and basal sides of cells in the primordium of wild-type and itgb1b mutant embryos by labeling F-actin with F-tractin-mNeonGreen in a few cells of the primordium (Fig. 5a). This analysis showed that F-actin was concentrated in the front of wild-type cells; in the basal cells of the primordium at the basal sides of the cells (Fig. 5b), and in the apically located superficial cells of the primordium on the apical side of the cells (Extended Data Fig. 7g). The F-actin flow was halted or slowed towards the cells’ rear with a mean speed of 1.5 μm/min in the basal cells of the primordium (Fig. 5b, c, Video 6) while F-actin flowed faster in the superficial cells with a mean speed 7.4 μm/min (Fig. 5d, Video 6). Removal of Itgb1b function did not significantly affect the pattern or speed of actin flow in the superficial cells (Fig. 5d, Extended Data Fig. 7g , Video 6). In contrast, F-actin flow increased to 6.6 μm/min in itgb1b mutant basal cells of the primordium, and F-actin formed radial cables that were also observed in talin-depleted cells in culture19 (Fig. 5b, c, Video 6). In basal primordium cells, the actin polymerization rate—the sum of the actin flow rate and the rate of membrane protrusion—also increased from 2.8 μm/min in wild-type control cells to 7.6 μm/min in itgb1b mutant cells (Fig. 5c, Extended Data Fig. 7h, i). Thus, integrins couple force for primordium motility on the basal but not apical sides of cells in the primordium, and loss of force coupling through integrin results in an increase in the actin polymerization rate on the basal sides of the primordium cells.
Fig. 5. β1-Integrin couples cell-substrate adhesion to actin flow in the primordium.

a, Experimental design to assess actin flow. b, Images of F-tractin-mNeonGreen localization at the basal sides of wild-type and itgb1b−/− primordium cells (top). White arrows indicate the direction of migration. Scale bar = 2 μm. Images are single optical sections from Video 6. Kymographs of Video 6 along the dotted line indicated in top images (bottom). The dotted cyan and magenta lines indicate the rates of actin flow and protrusion, respectively. c, Quantification of the rates of actin flow (left) and actin polymerization (right) in the primordium basal cells. Individual data points (dots), means (horizontal lines) and SD (vertical lines) are indicated. ****: p<0.0001 (two-tailed Mann-Whitney test). d, Quantification of the rate of actin flow in superficial primordium cells. n.s.: p=0.3708 (two-tailed t-test). Individual data points (dots), means (horizontal lines) and SD (vertical lines) are indicated. For c, d, n = number of cells pooled from more than 5 primordia in each condition.
The primordium exerts highest stresses in its rear
If the primordium uses integrin and talin to pull on the BM and push itself forward, the primordium should exert stresses (force per area) on the BM and deform the BM. Such traction stresses have been measured for migrating cells in culture by imaging the displacement of fluorescent beads embedded in elastic surfaces or matrices and the bending of flexible cantilevers—collectively referred to as traction force microscopy20-28. To extend traction force microscopy to living embryos, we created optical landmarks on the BM and assessed how these landmarks are displaced as the primordium moves across them (Fig. 6a). Using a laser, we locally bleached LamC1-sfGFP in an approximately cylindrical volume in the BM (Extended Data Fig. 8a, b, Video 7). Little LamC1-sfGFP diffused back into the bleached cylinder (Extended Data Fig. 8c, d), and bleached cylinders remained clearly demarcated for two hours after photo-bleaching (Video 8) while untagged, extracellular mCherry filled bleached cylinders rapidly after bleaching (Extended Data Fig. 8c). This indicated that bleaching LamC1-sfGFP is a suitable approach to place local marks on the BM and monitor the BM’s deformation over time. We therefore bleached a hexagonal pattern of marks onto the LamC1-sfGFP-labeled BM in front of the migrating primordium and recorded the position of the marks as the primordium migrated across this pattern (Extended Data Fig. 8a, Video 8). To reconstruct the stresses from the displacement of the marks on the BM by the migrating primordium, we developed the analysis pipeline Embryogram (Extended Data Fig. 8e, f, https://zenodo.org/record/5762146#.Ya5X0y-B1QJ) inspired by the Cellogram algorithm 29. Embryogram identifies the bleached cylinders in the first frame of the time lapse. This assigns a point in space to each mark in the first frame. The points are connected using a variant of the iterative closest point algorithm to obtain a triangular mesh. This mesh is then deformed to follow the marks on the BM in the subsequent frames of the time lapse, leading to a time-sequence of triangular meshes that captures the deformation of the BM (Extended Data Fig. 8f). The time-varying mesh is used to compute displacements between the marks. To convert the displacements into stresses, we fill an axis-aligned box that contains the sample with a volumetric tetrahedral mesh. This mesh contains the surface mesh approximating the BM in the first frame of the time lapse. The stresses are then computed solving an elastic deformation of the volumetric mesh (for details see Supplementary Note 1). For this conversion, we determined the stiffness, or Young’s modulus, of the BM. We removed the skin above the BM and measured the BM stiffness by atomic force microscopy (AFM) (Extended Data Fig. 8g). In agreement with previous in vivo studies assessing stiffness at the micron-scale30,31, this analysis yielded a Young’s modulus for the BM of 566 ± 355 Pa (mean and SD) which was reduced to 321 ± 158 Pa (mean and SD) after collagenase treatment (Fig. 6b, c, Extended Data Fig. 8h-k), a value probably reflecting the stiffness of the underlying muscle (Extended Data Fig. 8i, m). Importantly, spontaneous twitches of skin cells contract the underlying BM. This causes the BM to buckle and wrinkle akin to the distortions of the substrate observed around cultured cells32 and in animals33. The wrinkles form and disappear in less than 2 min and tracking the optical marks indicates that the wrinkles do not cause lasting deformation of the BM (Extended Data Fig. 9a-e, Video 9). Similarly, repeated probing of the BM at the same location in deskinned embryos by AFM did not alter the stiffness measurements (Extended Data Fig. 8l). These observations suggest that the BM undergoes non-plastic deformations and can be approximated by a linear stress-strain relationship.
Fig. 6. Traction stress measurements indicate that the primordium exerts highest stresses in its rear.

a, Strategy to measure traction stresses in vivo. b, Representative force curve of BM stiffness measurement by AFM. Fit of the first 200 nm after the contact point at 0 μm to the Hertz model is indicated in cyan. Only the approach part of the force curve is shown. c, Quantification of the BM stiffness with and without Collagenase treatment. N = number of embryos, n = number of recorded approach curves. Individual data points, means, and SD are indicated ****: p<0.0001 (two-tailed Mann-Whitney test). d, Maximum-projected z-stack (Video 8). Scale bar = 25 μm. e, BM displacement by the primordium (grey) shown as a vector field along the X-, Y- and Z-axes (Video 10). The displacement vector magnitude is indicated as a color map. XY-view is shown from the basal side of the primordium. s: skin, m: muscle. Scale bar = 25 μm. f, BM displacement in the XY-plane by the primordium (grey) shown as a vector field (Video 10). Displacement field in the XY-plane is shown from the basal side of the primordium. Scale bar = 25 μm. Bottom panel is a magnification of the outlined region in top panel. Scale bars = 25 μm (top) and 5 μm (bottom), arrow = 50 μm (top) and 10 μm (bottom). g, Traction stress magnitudes on the BM indicated as color map (Video 10). Scale bar = 25 μm. h, Schematic of the approach to quantify the traction stresses and BM displacement vector direction. i, BM displacement vector angles with respect to the migration direction (0°). Mean, 25th–75th percentiles (box), and min/max (whiskers) are indicated N = number of embryos, n = number of bleached cylinders. ****: p<0.0001 and n.s.: p=0.6291. j, Pooled traction stresses exerted by the primordium on the BM. Magnitude in XY-plane (left) and along the Z-axis (right) with median (dashed line) and quartile (dotted line) are shown. *: p=0.0302, ****: p<0.0001, n.s.: p=0.2273 (two-tailed Mann-Whitney test). N = number of embryos, n = number of bleached cylinders analyzed. For c, i, j, n was used for statistical tests.
Using the Embryogram pipeline together with the stiffness measurements, we find that the primordium’s front cells pull slightly on the BM in fairly random directions. In contrast, the cells in the primordium’s middle and rear pull on the BM more strongly and more directional, displacing the BM sideways, backwards and downwards (Fig. 6d-f, Video 10). The traction stresses (the forces against the plane of the BM) reflect this displacement pattern. In the front, the mean traction stresses average 28 ± 22 Pa, and increase to 58 ± 49 Pa and 64 ± 51 Pa (mean and SD) in the middle and rear of the primordium, respectively, with higher traction stresses exerted preferentially along the sides of the primordium peaking at 600 Pa (Fig. 6g-j, Video 10). Similarly, the primordium generates high, mostly rearward-pointing stresses (stresses extracted in the direction of migration) along the sides and towards its rear (Extended Data Fig. 9h), where it also exerts the highest shear stresses on the BM (Extended Data Fig. 9i). Although the primordium moves at fairly constant speed34, it does not exert constant stresses on its substrate (Video 10), suggesting that its forward motion is the result of the average of the fluctuating stresses across the tissue. At the position where the primordium has passed, the BM returned to its original shape and the marks on the BM snapped back to their original position, indicating that the BM is not irreversibly deformed by the primordium (Extended Data Fig. 9f, g, Video 10). Also, we did not observe such BM displacements and stresses in controls in which we blocked primordium migration by ubiquitous over-expression of the primordium’s attractive guidance cue Cxcl12a (Fig. 6j, Extended Data Fig. 9j-o, Video 10). The observed stress distribution was also reflected in the wrinkling of the BM along the sides of the primordium with LamC1-sfGFP forming local clusters (Fig. 7a). These LamC1-sfGFP clusters were juxtaposed to F-actin clusters in the primordium (Fig. 7b) and specifically enriched around the primordium (Fig. 7c), suggesting that actin network contraction in the primordium locally pull on the BM and cause it to wrinkle. Together, these observations indicate that the front cells exert low and the rear cells high traction stresses. On a tissue level, the primordium moves in a continuous breaststroke-like manner with its front pushing the BM in fairly random directions, while its middle and rear strongly push the BM sideways and backwards. This stress pattern is consistent with theoretical predictions for adherent cell migration35.
Fig. 7. The basement membrane wrinkles around the primordium.

a, F-tractin-mCherry distribution in the primordium (top) and LamC1-sfGFP around the primordium (bottom) in a 32 hpf embryo. LamC1-GFP fluorescence intensity is pseudo-colored as a heat map. The image is a maximum-projected z-stack. Scale bar = 50 μm. b, Transverse section through the F-tractin-mCherry-expressing primordium and the underlying LamC1-sfGFP-labeled BM (left top). Corresponding image showing the LamC1-sfGFP fluorescence intensity as a heat map (left bottom). Arrows indicate apposed clusters of F-tractin-mCherry and LamC1-sfGFP. Images are single sections along the YZ-plane of a z-stack. Scale bar = 10 μm. Fluorescent intensity profiles of F-tractin-mCherry and LamC1-sfGFP of image shown in left along the Y-axis (right). Arrows indicate the position of the apposed clusters of F-tractin-mCherry and LamC1-sfGFP indicated by arrows in left. c, Quantification of the LamC1-sfGFP intensity within 3 μm-wide bands around the perimeter of the primordium (left) and at a distance of 6 μm from the primordium’s perimeter (right). ****: p<0.0001 (two-tailed paired t-test). n = number of embryos.
Actomyosin activity is highest in the primordium’s rear
The primordium generates higher stresses in the rear than in the front. This could be because there are more cells in the rear or because the cells in the rear pull stronger on the BM than in the front. Stronger pulling in the rear could be reflected in increased actomyosin activity, slower actin flow rates, increased integrin levels, more engaged integrin, stabler integrin clusters, and a greater stress dependence on integrins in the rear than in the front of the primordium. To test these possibilities, we first assessed the activity of actomyosin across the primordium. Since actomyosin activity correlates with the localization of the actin motor non-muscle myosin II into dots, we generated a myosin II reporter line for the primordium (Myl12.1-mScarlet). Consistent with previous studies in other contexts36, Myl12.1-mScarlet localized to dots in the cells of the primordium. These dots did not form a cable-like structure around the back of the primordium as in other contexts36 but were mostly localized to the basal sides of the cells and to the lateral sides in the middle to rear of the primordium (Fig. 8a), and enriched in the primordium’s rear (Fig. 8b). This suggests that the cells in the primordium’s rear and on its sides exert more force than the cells in the front. Actin flow across the primordium was the same (Fig. 8c, d). Itgb1b levels across the primordium were similar (Fig. 8e) and integrin engagement—judged by increased clustering of Itgb1b-sfGFP over Itgb1bΔNPxY-sfGFP compared to Itgb1b-tdTomato in embryos in which we modified the two endogenous itgb1b alleles—also showed no difference between the primordium’s front and back (Fig. 8f). Itgb1b-sfGFP clustering at the myotendinous junction was increased 4-fold compared to the talin-binding deficient control itgb1bΔNPxY-sfGFP (Extended Data Fig. 10a, b)—consistent with the idea that increased clustering of Itgb1b-sfGFP compared to Itgb1bΔNPxY-sfGFP reports integrin engagement. In contrast, the levels of phosphorylated Paxillin—a putative marker for integrin adhesion turnover37—but not total Paxillin were lower in the rear than the front of the primordium (Fig. 8j), suggesting that the primordium’s rear form longer-lived integrin-mediated adhesions. Consistent with this idea, the traction stresses in the plane of migration (XY-plane)—but not the traction stresses against the BM (Z-axis)—decreased more in the rear than in the front of primordia in embryos lacking Itgb1b compared to wild-type embryos but did not become equal in magnitude (Fig. 8g-i, Video 10). Thus, Itgb1b couples more force to the BM in the primordium’s rear than in its front, is dispensable for force transmission against BM, but does not account for all the difference in stresses between the front and the rear of the primordium. Together, these observations suggest that the cells in the primordium’s rear generate more force than the cell in its front, which—probably together with the higher cell number in the rear—accounts for the high stresses exerted by the rear of the primordium on the BM.
Fig. 8. The primordium generates larger forces in the rear.

a, Myl12.1-mScarlet distribution in the primordium. Middle, Myl12.1-mScarlet in the entire primordium. Bottom, Myl12.1-mScarlet on the primordium’s basal side pseudo-colored. Scale bar = 20 μm. b, Quantification of the basal Myl12.1-mScarlet intensity at indicated positions in the primordium. Data points, means, and SD are indicated. (one-way ANOVA followed by Tukey’s multiple comparison test). c, Images of F-tractin-mNeonGreen at the basal sides of primordium cells (top). Arrows, direction of migration. Scale bar = 2 μm. Kymographs along the dotted line indicated in top images (bottom). d, Actin flow rates in basal primordium cells. n = number of cells pooled from > 5 primordia. Data points, means, and SD are indicated. (two-tailed Mann-Whitney test). e, Distribution (top) and quantification (bottom) of Itgb1b-sfGFP on the primordium cell membranes. Itgb1b-sfGFP intensity is pseudo-colored. Scale bar = 20 μm. Mean and SD are indicated. f, Fluorescence intensity ratio images of Itgb1b-tdTomato to Itgb1b-sfGFP (top left) and Itgb1bΔNPxY-sfGFP (bottom left) on the membrane of primordium cells. Quantification of intensity ratios (right). Ratios are shown in pseudo-color. Mean and SD are indicated. g, BM displacement around the primordium (grey) in itgb1b−/− embryos shown as a vector field along the X-, Y- and Z-axes (Video 10). Vector magnitude is indicated as a color map. XY-view is from the basal side of the primordium. Scale bar = 25 μm. h, Traction stresses on the BM indicated as a color map (Video 10). Scale bar = 25 μm. i, Pooled traction stresses exerted by the primordium on the BM in itgb1b−/− embryos. Median (thick line) and quartile (thin line) are shown. Wild-type control is the same as Fig. 6j. (two-tailed Mann-Whitney test). N = number of embryos, n = number of bleached cylinders. n was used for statistics. j, Left, staining against Paxillin (top) and phosphorylated Paxillin (bottom) protein in primordia shown as a heat map. Right, quantification of the fluorescence intensity on the primordium membrane for Paxillin (top) and phosphorylated Paxillin (bottom). Mean and SD are indicated. Scale bar = 20 μm. k, Model for primordium motility. For b, e, f, j, n = number of embryos.
Another migrating tissue that is guided by Cxcl12 and propelled forward by its rear is the cranial neural crest (CNC) in Xenopus38. The CNC front cells are attracted by Cxcl12 and the rear cells form a supracellular cable around the back of the tissue. This cable contracts periodically and pushes rear cells forward while front cells become displaced to the side and then to the back (Extended Data Fig. 10e). In contrast, the primordium is attracted by a self-generated Cxcl12a gradient that extends almost across the entire tissue (Extended Data Fig. 10c, d)39,40, does not form a supracellular actomyosin cable across adherens junctions around its back (Fig. 8a, Extended Data Fig. 10h, i), does not periodically contract its rear (Extended Data Fig. 10f), and its cells do not intercalate but remain next to their neighbors during migration (Extended Data Fig. 10g, Video 1)11. Thus, although both tissues generate the highest forces in their rear, the underlying guidance and propulsion mechanisms are different and might represent two solutions to the same problem (Extended Data Fig. 10e)—how to propel a tissue through an animal.
Discussion
Together, this work elucidates how a tissue moves through a live animal. It provides three major insights. First, primordium cells link the force-generating actomyosin network to the BM through integrin clusters on their basal sides. These integrin clusters are less than 2 μm in size and form and disassemble in less than 2 minutes. This is in contrast to the larger and longer-lived focal adhesions that migrating cells in culture use to pull themselves forward16,17, and more reminiscent of nascent adhesions that form at the edge of protrusions and underneath spreading cells in culture37,41-43, suggesting that tissues in animals rely on transient rather than prolonged cell-substrate interactions for movement. Second, the primordium cells pull and deform the BM on their outside with maximal stresses around to 600 Pa. This is comparable to the average stress that migrating cells in culture exert on their substrates20-28, suggesting that stresses in this range are inherent to adherent migration in simplified and physiological scenarios. Consistent with this notion, retrograde actin flow is slowed or stalled in primordium cells, suggesting that most of the flow is converted into forward movement. Intriguingly, disrupting the coupling of actin flow across integrin results in increased flow. Such a compensatory response has also been observed in cultured dendritic cells and macrophages44. These cells also increase actin polymerization in response to decreased force coupling to the substrate to maintain forward movement—likely by coupling through other integrins and unspecific adhesion. Third, the primordium moves similar to a continuous breaststroke by pushing the BM downward, sideways and backwards. Counterintuitively, the primordium’s rear cells generate higher traction stresses than the front cells, probably because the rear cells need to overcome greater resistance in their environment than the front cells. One possible reason for the greater resistance is the front cells which the rear cells might need to push to move forward. Consistent with this idea, the front cells pull and push the BM in fairly random directions (Fig. 6i), suggesting that they contribute little to directed force generation in the tissue. Also, when severed from the rear cells, the front cells cease to move forward45. It thus seems that the rear pushes the front to propel the primordium forward, a scenario that is akin to a rear-engine-like design (Fig. 8k). Intriguingly, crawling Dictyostelium slugs, banana slugs, and garden snails also push stronger with their rears than their fronts on their surroundings46,47, indicating that this propulsion design is conserved across different length scales to drive tissue and animal movement.
Methods:
Data availability
Previously published genome assemblies as either GRCz10 or GRCz11 that were used here for the design of gRNA constructs are available for tln1, tln2a, tln2b, itgb1a and itgb1b under accession codes ENSDARG00000100729, ENSDARG00000017901, ENSDARG00000110973, ENSDARG00000071863 and ENSDARG00000104484, respectively. Source data have been provided in Source Data. All other data supporting the findings of this study are available from the corresponding authors on reasonable request.
Code availability
The code for Embryogram software was deposited with Zenodo (https://zenodo.org/record/5762146#.Ya5X0y-B1QJ)48. The codes for image analysis using ImageJ and R are provided as a zip file.
Zebrafish husbandry
This study was performed in accordance with the recommendations in the Guide for the Care and Use of Laboratory Animals of the National Institutes of Health. All of the animals were handled according to approved institutional animal care and use committee (IACUC) protocols (IA16-00788_AMEND202100320) of the NYU Grossman School of Medicine.
Zebrafish strains
Embryos were staged as previously described49. The cxcl12aat30516 allele was previously described50. The lamC1sa9866 allele was obtained from the Zebrafish International Resource Center (ZIRC, https://zebrafish.org) and contains a nonsense mutation resulting a premature stop codon51. Homozygous lamC1sa9866 mutants were identified by their shortened body axis7 or by PCR-based genotyping. Primers used for genotyping of lamC1sa9866 mutant fish are listed in Supplementary Table 1. The PCR product was digested with the restriction enzyme BsaI (New England Biolabs, R0535L) to yield a 168 bp fragment for the wild-type allele and a 120 bp and a 48 bp fragments for the mutant allele. The Tg(prim:mem-mCherry)52, Tg(cldnB:lyn2GFP)34, hsp70:cxcl12a53, TgBAC(cdh1:cdh1-TagRFP)9, TgBAC(cxcr4b:cxcr4b-Kate2-IRES-EGFP-CaaX)p740 and TgBAC(cxcr4b:zGrad)9, TgBAC(cdh1:cdh1-sfGFP exon16)11, TgBAC(cdh2:cdh2-mCherry exon16)11, Gt(ctnna1-citrine)Ct3a54,55, TgBAC(cxcr4b:h2a-mCherry)52, TgBAC(cxcr4b:cxcr4b-EGFP-IRES-Kate2-CaaX)p740, cdh1:cdh1-tdTomato56, itgb1b:itgb1b-tdTomato57 lines were previously described.
Generation of mutant alleles
To generate tln1, tln2a, tln2b, itgb1a and itgb1b mutants, we followed previously described CRISPR-Cas9-based gene editing protocols58. mRNA for Cas9 was synthesized by in vitro transcription with mMESSAGE mMACHINE T7 Transcription kit (Thermo Fisher Scientific, cat no. AM1344) using the linearized plasmid pST1374-NLS-flag-linker-Cas9 (Addgene 4475859) as a template. Three to four gRNAs were designed to target the coding sequence around the start codons in the cases where the genes were fully annotated in the Ensembl genome browser (GRCz10, www.ensembl.org). Otherwise, we designed gRNAs targeting the available coding sequence in the Ensembl genome browser (GRCz10). gRNA sequences were identified using the CRISPR guide design offered by Benchling (https://www.benchling.com/crispr/). The templates for gRNAs were synthesized by PCR. Briefly, a target sequence specific primer was designed which contained the T7 promoter sequence, the target sequence without the PAM site, and an overhang for primer annealing. A primer that coded for the chimeric gRNA backbone was designed. All primers were purchased from Integrated DNA Technologies (IDT, https://www.idtdna.com). The target sequence specific primer and the chimeric gRNA backbone primer were annealed, filled-in by Taq polymerase and amplified by PCR. The PCR products were column-purified using the QIAquick PCR Purification Kit (Qiagen, 28106) and subjected to in vitro transcription using the MEGAscript T7 Transcription kit (Thermo Fisher Scientific, AM1334) to obtain the gRNAs. Three to four gRNAs (final concentration of each gRNA: 200 ng/ul) were mixed with Cas9 mRNA (300 ng/ul) and 1 nl was injected into one-cell stage embryos. The injected embryos were raised to adulthood and out-crossed to wild-type adults. Embryos from these crosses were genotyped for potential mutations induced by gene editing to identify adults with germ line mutations. Embryos from adults carrying germ line mutations were raised and genotyped as adults by PCR and sequencing. Primers used are listed in Supplementary Table 1.
Generation and genotyping of the tln1d4 mutant
The gRNAs used to target tln1 are listed in Supplementary Table 1. The isolated mutant fish harbor a 4 bp deletion in tln1 exon 2 resulting in a frame shift that introduces a premature stop codon. tln1d4 mutant embryos recapitulate the previously described tln1 phenotype such as partially penetrant cardiac edema and embryonic lethality60. Note that tln1d4 mutant fish can be kept as homozygous adults in the presence of the BAC transgene TgBAC(tln1:tln1-YPet) which rescues the lack of tln1 activity. The tln1d4 allele was genotyped by amplifying the locus through PCR and digestion of the amplicon with the restriction enzyme Sau3AI (New England Biolabs, R0169L). The digest yields a 122 bp and a 53 bp fragment for the tln1 wild-type allele and in a 171 bp fragment for the tln1d4 allele. The primers used for genotyping by PCR are listed in the Supplementary Table 1.
Note that the copy number of the tln1 wild-type and the tln1d4 alleles with TgBAC(tln1:tln1-YPet) can be determined by the intensity of the bands of the digested amplicons on a 3% agarose gel with the above genotyping protocol.
Generation and genotyping of the tln2ai23 mutant:
The gRNAs used to target tln2a are listed in Supplementary Table 1. The isolated tln2a mutant line comprises a 23 bp insertion (1 bp deletion plus 24 bp insertion) in tln2a exon 3 resulting in a frame shift that causes a premature stop codon. We were not able to obtain tln2ai23 homozygous adult fish in Mendelian ratios, suggesting that tln2ai23 homozygous mutant fish die at some point between 5 dpf and 60 dpf. However, we occasionally recovered tln2ai23 homozygous adult fish. The tln2ai23 allele was genotyped by PCR followed by SrfI restriction digest (New England Biolabs, R0629L). The primers used for genotyping by PCR are listed in the Supplementary Table 1. Note that the forward primer introduces a SrfI target site in the mutant allele only. Thus, SrfI restriction digest of the PCR amplicons results in a 192 bp fragment for the tln2a wild-type allele and a 37 bp and a 155 bp fragments for tln2ai23 mutant allele.
Generation and genotyping of the tln2bd10 mutant:
The gRNAs used to target tln2b are listed in Supplementary Table 1. The isolated mutant harbors a 10 bp deletion resulting in a frame shift that causes a premature stop codon. The predicted Tln2bd10 mutant protein comprises only the first 658 amino acids of the total 2580 amino acids. Note that tln2bd10 mutants can be kept as homozygous adult fish. The tln2bd10 allele was genotyped by PCR. The primers used for genotyping by PCR are listed in the Supplementary Table 1.
Generation and genotyping of the itgb1ad34 mutant:
The gRNAs used to target itgb1a are listed in Supplementary Table 1. The isolated mutant contains a 34 bp deletion in itgb1a exon 3 resulting in a frame shift that causes a premature stop codon. itgb1ad34 mutants can be kept as homozygous adult fish. The itgb1ad34 allele was genotyped by PCR. The primers used for genotyping by PCR are listed in the Supplementary Table 1.
Generation and genotyping of the itgb1bi70 mutant:
The gRNAs used to target itgb1b are listed in Supplementary Table 1. The isolated mutant contains a 70 bp insertion resulting in a frame shift that introduces two successive premature stop codons. The predicted mutant protein comprises only the first 320 amino acids of the total 806 amino acids. itgb1bi70 homozygous mutant embryos display a shorter body axis (Extended Data Fig. 5b-d) and die as embryos consistent with previous reports61. The itgb1bi70 allele was genotyped by PCR. The primers used for genotyping by PCR are listed in the Supplementary Table 1.
Generation of transgenic lines
To generate BAC-mediated transgenes, we modified the BAC clone of interest with a galK-mediated BAC recombineering as previously described62,63. Briefly, we first modified the BAC clone of interest to include a transgensis marker and the tol2 sequences in the BAC backbone. Next, we modified the protein coding sequence of interest on the BAC to add a fluorescent protein or express coding sequence under the regulation of specific promoter. The final BAC was characterized by the EcoRI digestion and PCR followed by the sequencing around the modified sequences. The final BAC was purified with Nucleobond BAC 100 Kit (Takara Bio, 740579) and co-injected with 1 nl of 40 ng/ul tol2 mRNA into one-cell stage embryos. The stable transgenic line was established by out-crossing the adult fish injected with the BAC transgene and raising transgenic embryos.
TgBAC(lamC1:lamC1-sfGFP)
For the lamC1:lamC1-sfGFP transgene, we used the BAC clone CHORI-211-194I4 which was obtained from BACPAC Resources, Children’ Hospital Oakland Research Institute, CA, USA (bacpacorders@chori.org). This BAC spans 192,491 bp of genomic DNA and contains the lamC1 locus with about 50 kbp genomic sequence upstream of lamC1 exon 1 and about 35 kbp genomic sequence downstream of lamC1 exon 28. Transgenesis marker is cryaa:Cerulean. This transgene expresses full length Laminin-γ1 fused to sfGFP from the lamC1 promoter. The full name of this transgenic line is TgBAC(lamC1:lamC1-sfGFP)p1. Note that this transgene recapitulates the previously reported expression pattern of lamC1 mRNA expression64, partially rescues lamC1 homozygous mutant embryos (Extended Data Fig. 1h) and does not affect primordium migration (Extended Data Fig. 1i).
TgBAC(tln1:tln1-YPet)
For the tln1:tln1-YPet transgene, we used the BAC clone DKEY-42J10 which was obtained from ImaGenes GmbH, Germany (sales@imagenes-bio.de). The BAC clone spans 194,108 bp of genomic DNA. This includes 40 kb of genomic sequence upstream of the beginning of tln1 exon 1, tln1 exons 1 to 44, but lacks tln1 exons 45 to 56. To include the complete coding sequence of tln1 on the BAC we inserted the sequence of the missing tln1 exons (exons 45 to 56) directly downstream of exon 44. This design was guided by the annotated tln1 transcript tln1-202 (ENSDART00000166799.2, Ensembl). We inserted the coding sequence for YPet between the head and the rod domains of tln165, which is located in tln1 exon 13. As a transgenesis marker we inserted cryaa:dsRed in the BAC backbone. A cassette comprising tln1 exons 45-56 and the Kanamycin resistant gene KanR was inserted. The amino acid sequence around the YPet insertion is Gly-Ser-Val-X-Ala-Leu-Pro, where YPet and restriction enzyme sequences were inserted in frame at position X. The full name of this transgenic line is TgBAC(tln1:tln1-YPet)p2. Note that the expression pattern of this transgene recapitulates the in situ hybridization pattern against tln1 mRNA (Extended Data Fig. 3d, Extended Data Fig. 3f). This transgene rescued the lethality of tln1 homozygous mutant embryos.
TgBAC(cxcr4b:F-tractin-mCherry)
For the cxcr4b:F-tractin-mCherry transgene, we used the BAC clone DKEY-169F10 which was obtained from ImaGenes GmbH, Germany (sales@imagenes-bio.de). The BAC clone DKEY-169F10 contains a 60 kb genomic DNA fragment that spans the entire cxcr4b locus. As a transgenesis marker we used myl7:mScarlet. The sequence of F-tractin (rat inositol 1,4,5-triphosphate 3-kinase A (ITPKA) amino acids 10-52)66 together with a linker sequence (GLALPVAT) was used. The full name of this transgenic line is TgBAC(cxcr4b:F-tractin-mCherry)p3.
TgBAC(cxcr4b:F-tractin-mNeonGreen)
The BAC construct cxcr4b:F-tractin-mNeonGreen was generated as described above for the cxcr4b:F-tractin-mCherry line except that the BAC was modified to contain the mNeonGreen (mNG) instead of the mCherry coding sequence. The mNG coding sequence was PCR amplified from Addgene plasmid 9888667.
TgBAC(cxcr4b:myl12.1-mScarlet)
For the cxcr4b:myl12.1-mScarlet transgene, we used the DKEY-169F10 BAC clone described above. The transgenesis marker is myl7:mScarlet. The myl12.1 coding sequence was amplified from zebrafish cDNA by PCR. This transgene expresses the first five amino acids from cxcr4b exon 1 fused to Myl12.1-mScarlet from the cxcr4b promoter. The full name of this transgenic line is TgBAC(cxcr4b:myl12.1-mScarlet)p2.
TgBAC(cxcr4b:EGFP-CaaX)
For construction of cxcr4b:EGFP-CaaX BAC clone, we used the BAC clone DKEY-169F10 with the transgenesis marker myl7:mScarlet described above. This transgene expresses the first five amino acids from cxcr4b exon 1 fused to EGFP-CaaX from the cxcr4b promoter. The full name of this transgenic line is TgBAC(cxcr4b:EGFP-CaaX)p1.
Generation of knock-in strains
itgb1b:itgb1b-sfGFP
The itgb1b:itgb1b-sfGFP knock-in line was generated as described57. As a targeting cassette we used the described targeting plasmid for itgb1b:itgb1b-tdTomato57 but swapped the tdTomato coding sequence with sfGFP coding sequence. Note that the expression pattern of itgb1b:itgb1b-sfGFP recapitulates the pattern seen by in situ hybridization against itgb1b mRNA (Extended Data Fig. 3a, c). itgb1b:itgb1b-sfGFP homozygous and itgb1b:itgb1b-sfGFP/itgb1bi70 trans-heterozygous fish are adult viable.
itgb1b:itgb1bΔNPxY-sfGFP
The itgb1b:itgb1bΔNPxY-sfGFP knock-in line was generated as described above for the itgb1b:itgb1b-sfGFP line except that the sequence coding for the first to the second NPxY motif (NPIYKSAVTTVVNPKY, amino acid 777-792) was deleted in the plasmid containing the targeting cassette. We identified one itgb1b:itgb1bΔNPxY-sfGFP line and confirmed the correct knock-in event by sequencing PCR amplicons spanning the genomic insertion site.
Generation of plasmid constructs
Plasmids were generated by Gibson cloning68. To construct pDEST-tol2-acta1a-cxcl12a-t2a-mCherry and pDEST-tol2-acta1a-mCherry plasmids, the following plasmids were used as PCR templates: The plasmid backbone including the tol2 sites, the acta1a promoter and the cxcl12a coding sequence were amplified from pDestTol2pA269, pDEST-tol2-acta1a-GFP70 and pCS2-cxcl12a71, respectively. To construct pCS2(+)-YPet-ZF1, pCS2(+)-mNeonGreen-ZF1 and pCS2(+)-mCherry-ZF1, the following plasmids were used as templates: The plasmid backbone, the YPet, the mNeonGreen and mCherry sequences were amplified from pCS2(+)-sfGFP-ZF19 or pCS2(+), pUC19-tln1-5arm-YPet-3arm, the Addgene plasmid 9888667 and pDEST-tol2-hsp70l-secP-mCherry-SV40pA52, respectively. To construct pDEST-tol2-hsp70l-zGrad-t2a-mNeonGreen, the following plasmids were used as PCR templates: The plasmid backbone including tol2 sites, the hsp70l promoter and the zGrad coding sequence were amplified from pDEST-tol2-hsp70l-zGrad9 and the t2a-mNeonGreen sequence was amplified from the Addgene plasmid 9888667 with PCR primers that also included the t2a sequence. The pCS2(+)-lyn2mCherry plasmid was generated based on the pCS2(+)-lyn2EGFP plasmid (gift from Reinhard W. Köster)72.
In vitro mRNA transcription and mRNA injection
Linear templates for in vitro mRNA transcription were generated by restriction digest of plasmids or PCR on plasmids using primers containing a SP6 promoter sequence. mRNAs were transcribed using the mMESSAGE mMACHINE SP6 transcription Kit (Thermo Fisher Scientific, AM1340). Injection mixes contained 50 ng/μl mRNAs with 0.1% Phenol Red Solution (Lifetechnologies, 15100-043). 1 nl of the injection mix was injected in one-cell-stage embryos. tol2 mRNA was synthesized from a linearized pCS2FA-transposase plasmid69.
Embryo dissociation and primary zebrafish cell culture
Glass bottom dishes (MatTek, P35G-0-20-C) were coated with mouse Laminin protein (Corning, 354232). Briefly, 50 μg of mouse Laminin was mixed with Leibovitz L15 media without phenol red (Fisher, 21083027) and added to the glass bottom dishes. The dishes were incubated for 1 hour at room temperature and then washed with PBS (137 mM NaCl, 2.7 mM KCl, 10 mM Na2HPO4, 1.8 mM KH2PO4). 33 hpf embryos were manually dechorionated and the tails were dissected off in fish water (60 μg/mL of Instant Ocean Sea Salts (Instant Ocean, SS15-10)) supplemented with 0.4 mg/ml MS-222 anesthetic (Sigma, A5040-25g). The tails were harvested and transferred into a cell dissociation medium (0.05% Trypsin-EDTA (Invtrogen, 25200-056)). The tails were then incubated in the dissociation medium at 28°C for 20–30 min, pipetted up and down with a P1000 pipette tip every 5 min until the tails were dissociated into single cells. This cell suspension was filtered through a 70 μm nylon mesh (Fisher, 22-363-548). After a 3 to 4 washes with PBS, cells were resuspended in cell culture medium (Leibovitz L-15 medium without phenol red (Fisher, 21083027), 15% FBS (Fisher Scientific, cat no. A3160601), and 100 U/ml Penicillin-Streptomycin (Invitrogen, cat no. 15140-122)). The cell suspension was added to the Laminin-coated dishes and incubated at 28°C overnight to allow the cells to settle.
Immunofluorescence staining of cultured cells
Dissociated embryonic zebrafish cells seeded on Laminin-coated dishes were washed with PBS and fixed with 4% PFA in PBST at room temperature for 10 min. The cells were permeabilized with 0.5% Triton-X100 in PBS at room temperature for 10 min and blocked with 0.5% Bovine Serum Albumins (BSA, Millipore-Sigma, A4737-100G) in PBS at room temperature for 30 min, followed by incubation with the primary antibody overnight at room temperature. To detect Itgb1b-sfGFP and Tln1-YPet, we used the rabbit anti-GFP primary antibody (Torrey Pines Biolabs, NJ USA, cat no. TP401, lot no. 081211) at a dilution of 1:500 in 0.5% BSA/PBS. To detect F-tractin-mCherry, we used a sheep anti-mCherry primary antibody (custom made antibody by Covance52) at a dilution of 1:1000 in 0.5% BSA/PBS. The primary antibodies were detected with the following secondary antibodies at a 1:1000 dilution at room temperature for one hour: donkey anti-sheep-Cy3 (Jackson ImmunoResearch, PA USA, cat no. 713-166-147, lot no. 106361) and donkey anti-rabbit-Alexa488 (Jackson ImmunoResearch, PA USA, cat no. 711-546-152, lot no. 109010). The posterior lateral line cells were identified based on F-tractin-mCherry expression driven from the cxcr4b promoter. In the tails of 33 hpf embryos, the cxcr4b promoter drives high expression only in the posterior lateral line9,71. Imaging was performed using a spinning disk confocal Nikon W1 equipped with an Apo 60x NA 1.40 oil objective lens (Nikon, MRD01605). Images shown are maximum-projected z-stacks.
Antibody staining and quantification of Paxillin and p-Paxillin levels
To stain for F-actin, phospho-MLC, Fibronectin, Chondroitin sulfate, Paxillin and phospho-Paxillin (p-Y118) and the membrane of the primordium cells, we fixed 32 hpf TgBAC(cxcr4b:EGFP-CaaX) embryos in 4% PFA/PBST for 2 hrs (F-actin, phospho-MLC, Fibronectin, Chondroitin sulfate, phospho-Paxillin (p-Y118)) or overnight (Paxillin) at room temperature. Embryos were dehydrated in 100% methanol (Millipore-Sigma, cat no. 322415-100ML) overnight at −20°C, rehydrated using a series of 50% and 30% methanol in PBST and blocked in 1% BSA/PBST for 1 h at room temperature. Embryos fixed overnight, were permeabilized with Proteinase K (10 μg/mL in PBST) (Sigma Aldrich, cat no. P6556) for 5 min at room temperature and re-fixed with 4% PFA/PBST for 20 min at room temperature before blocking. The embryos were incubated in rabbit anti-Phospho-Myosin Light Chain 2 (Ser19) (1:100, Cell Signaling Technology, cat no. 3671, lot no. 6), rabbit anti-Fibronectin (1:100, Sigma, cat no. F3648, lot no. 0000090857), mouse anti-Chondroitin sulfate (1:100, Sigma, cat no. MA1-83055, lot no. WI3245117), mouse anti-Paxillin (1:100, BD Transductions, cat no. 610051, lot no. 1110894), or rabbit anti-Paxillin(pTyr118) (1:500, Novusbio, cat no. NBP2-24459, lot no. 031327 42D(P)) together with goat anti-GFP (1:500, Covance, custom made antibody40) overnight at 4°C. Embryos were washed four times with PBST and incubated with donkey anti-rabbit Cy3 (1:500, Jackson ImmunoResearch, cat no. 711-165-152, lot no. 102215) or goat anti-mouse Cy3 (1:500, Jackson ImmunoResearch, cat no. 115-165-003) together with donkey anti-goat Alexa488 (1:500, Jackson ImmunoResearch, cat no. 705-546-147, lot no. 110667) secondary antibodies in PBST. To stain for F-actin with phalloidin, embryos were fixed in 4% PFA overnight, permeabilized in 2% triton X100/PBS overnight at 4°C, blocked in 1% BSA/PBST for 1 h at room temperature. Subsequently, embryos were stained with AlexaFluore594 Phalloidin (1:100, Invitrogen, cat no. A12381, lot no. 2335606) in PBST overnight at 4°C. After four washes with PBST, embryos were imaged. Embryos were mounted in PBST. Images were taken on a Leica SP8 confocal microscope equipped with HyD detectors (Leica Microsystems) using a 40x (NA 1.1) objective with a sequential scan setting.
Quantification of Paxillin and p-Paxillin intensities were performed in a semi-automated manner with a custom-written macro in Fiji. Briefly, the primordium region was manually cropped. The macro generated a binary mask based on the EGFP-CaaX signal (auto-thresholding with the Iso-Data method). The mask was applied to the Paxillin or p-Paxillin channel and the signal on the primordium membrane was extracted. Next, three consecutive optical z-slices without apical constrictions were manually selected and average-projected. A rectangular ROI with 10 μm width and 100 μm length was manually defined from the tip of the primordium and the signal intensity profile was obtained along the front-to-rear axis of the primordium. We did not include the membrane at the leading edge of the primordium because the signal in this region could not be separated from the signal of the skin. Finally, the signal was normalized to the average intensity for each embryo and plotted.
Quantification of filamentous Collagen-IV by immunofluorescence staining
To stain for Collagen-IV and the membrane of the primordium cells in lamC1 mutant and control embryos, we in-crossed cldnB:lyn2GFP; lamC1+/− fish and sorted for cldnB:lyn2GFP; lamC1−/− and cldnB:lyn2GFP; lamC1−/+ or cldnB:lyn2GFP; lamC1+/+ embryos at 30 hpf. One hour later, 31 hpf embryos were fixed with 4% PFA/PBST for 2 hrs at room temperature, then stored in 100% methanol (Millipore-Sigma, cat no. 322415-100ML) overnight at minus 20°C. Embryos were rehydrated using a series of 50% and 30% methanol in PBST and blocked in 1% BSA/PBST for 1 h at room temperature. The embryos were incubated in rabbit anti-collagen IV (1:200, ab6586, abcam, Cambridge UK) and goat anti-GFP (1:500, Covance, custom made antibody40) overnight at 4°C. Embryos were washed four times with PBST and incubated with donkey anti-rabbit Cy3 (1:500, Jackson ImmunoResearch, cat no. 711-165-152, lot no. 102215) and donkey anti-goat Alexa488 (1:500, Jackson ImmunoResearch, cat no. 705-546-147, lot no. 110667) secondary antibodies in PBST. Embryos were mounted in 0.5% low melt agarose/Ringer’s solution. Images were taken on a spinning disk confocal Nikon W1 microscope. The number of filament structures around the primordium was quantified using a custom-written macro in Fiji. Briefly, a 150 μm x 150 μm region of interest containing the entire primordium was manually defined. The red fluorescent channel of the z-stack representing the Collagen-IV signal was sum-projected and only the fluorescence values above 1.25 the mean fluorescence intensity of the image were kept. Filamentous structures were extracted using the Tubeness filter (https://www.longair.net/edinburgh/imagej/tubeness/) in Fiji with sigma set to 1.0. Then, the image was thresholded using Otsu’s method and the number of filaments was counted with the Analyze Particles command in Fiji (settings: limiting size = 50-Infinity, circularity = 0.0-0.3).
Whole mount in situ hybridization
The procedures for RNA probe synthesis and zebrafish embryo whole-mount in situ hybridization were performed as previously described73. The RNA probe against cxcl12a was previously described71. To synthesize the RNA probes against tln1 and tln2a, parts of the transcripts were PCR-amplified from cDNA synthesized from zebrafish embryos using the primers indicated in Supplementary Table 1 and cloned into the pCR2.1-TOPO vector (Thermo Fisher Scientific, cat no. 451641). The plasmids were linearized using BamHI-HF (New England Biolabs, cat no. R3136L), column-purified (Qiagen, QIAquick PCR Purification Kit, cat no. 28106), and in vitro transcribed using a DIG RNA Labeling Kit (Roche, cat no. 11277073910) together with a SP6/T7 Transcription Kit (Roche, cat no. 10999644001). To synthesize the RNA probes against tln2b, itgb1a, itgb1b, itgb1b.1, itgb1b.2, itgb2, itgb3a, itgb3b, itgb4, itgb5, itgb6 and itgb7 DNA templates were PCR-amplified from cDNA synthesized from maternal, 28 hpf or 33 hpf embryonic cDNA using the primers listed in Supplementary Table 1. The reverse primers harbor T7 promoter sequence at their a 5’ end so that the PCR products could directly be transcribed in vitro after column purification. The RNA probes were synthesized with the Roche DIG labeling mix (Roche, cat no. 11277073910) and detected using an anti-DIG antibody coupled to alkaline phosphatase (1:5000, Roche, cat no. 11093274910) and NBT/BCIP staining (Roche, cat no. 11681451001). Embryos were mounted on the 3% Methyl cellulose (Sigma Aldrich, cat no. M0512) and imaged on an Axioplan Microscope (Zeiss) equipped with an Axiocam (Zeiss) using a 10x (NA 0.5) objective lens.
Dissection of embryos and collagenase treatment
The head and the yolk of 28 hpf wild-type embryos were removed in fish water supplemented with 0.4 mg/ml MS-222 anesthetic using forceps. Dissected embryos were transferred into deskin media (Ca2+ free Ringer’s solution, 50 mM EDTA, 0.4 mg/ml MS-222 anesthetic). The skin of the embryos was peeled off under a Leica dissection scope (Leica, Wild M420 with light stand) using forceps. The deskinned embryos were then transferred into Leibovitz L-15 media (Fisher, cat no. 11415064) supplemented with 0.4 mg/ml MS-222 anesthetic. The collagenase (Collagenase, Purified, 4 ku, Worthington, cat no. LS005275) stock was prepared in Leibovitz L-15 media at a concentration of 1000 U/ml. The deskinned embryos were soaked in the collagenase solution (909 U/ml) supplemented with 0.4 mg/ml MS-222 anesthetic at the room temperature for 30 min. After the treatment, the collagenase was washed out by Leibovitz L-15 media supplemented with 0.4 mg/ml MS-222 anesthetic, then the embryos were quickly mounted for atomic force or confocal microscopy.
Atomic force microscopy measurements and data analysis
Deskinned embryos were glued to FluoroDish dishes (World Precision Instruments, FL USA, cat no. FD5040-100) using CELL-TAK (Corning, NY USA, cat no. 354240) and immersed in Leibovitz L-15 media supplemented with 0.4 mg/ml of the anesthetic MS-222 (Sigma Aldrich, cat no. A5040-25g). All Atomic force microscopy (AFM) measurements were carried out within 90 min after skin removal. The AFM measurements were performed on an Asylum Research MFP-3D-BIO Atomic Force Microscope using the Asylum Research software package Version IX (AR Software) as previously described74. The AR Software was used for cantilever calibration, force mapping, data export, and data visualization (Extended Data Fig. 8j-n). We used a spherical borosilicate glass bead probe with a 2.5 μm radius, a spring constant 0.07 N/m, a Young’s modulus of 68.0 GPa, and a Poison ratio of 0.19 (Novascan, PT-GS). In contrast to pyramidal probes, which probe structures at the nm length scale such as extracellular filaments, spherical probes work at the μm length scale and assess the global properties of the BM75. We used a trigger point of 1 nN force and an indentation velocity of 5.0 μm/s. Such a slow indentation velocity minimizes viscous effects. Two to three two-dimensional 8-by-8 (20 μm by 20 μm) square grids were probed per deskinned embryo. The probed area was located above the muscles that overlie the notochord in the center of the deskinned embryo’s trunk (Extended Data Fig. 8j).
The AFM measurements were analyzed using the Rasylum package (https://github.com/nstone8/Rasylum), which runs on the R programming language software environment (https://www.r-project.org/). We modified the Rasylum package in three ways. First, we included a batch-mode option to analyze a set of force curves automatically. Second, we modified the extractStiffness function in the Rasylum package to specify the length of the force curve that will be fit to the Hertz model. The original extractStiffness function fits the entire force curve from the contact point to the maximum deformation point to the Hertz model. Our modification allows the user to only fit a select part of the curve to the Hertz model (from the contact point to a user specified length of the deformation part of the force curve, in our case 200 nm and 500 nm). Third, we modified the extractStiffness function to report statistical parameters for the fit of the probe approach part of the force curve (beginning of force curve to contact point) to a linear model and the indentation part of the force curve to the Hertz model (contact point to end of force curve/point of maximum deformation or the end point defined by the user). These statistical parameters were used as quality criteria to include or reject force curves. For the calculation of Young’s modulus, the sample Poisson ratio was assumed to be 0.45. The reduced Young’s modulus E* was obtained by fitting the first 200 nm of the approach curve past the contact point to the Hertz model
where F is the loading force, E* is the reduced Young’s modulus, r is the radius of the spherical probe used, and δ is the indentation. The sample Young’s modulus Es was calculated using
where νi is the indenter’s Poisson ratio, νs is the sample Poisson ratio, and Ei is the indenter’s Young’s modulus.
Due to debris after skin removal and the curvature of the BM above the muscle, some force curves were of low quality. To automatically select high quality force curves, we applied three criteria. The first criterion was the slope of the fit of the baseline of the approach curve (start of approach curve to contact point) to a linear equation. The second criterion was the residuals between the measured and fitted curve of the baseline of the approach curve to a linear equation. The third criterion was the P-value of the fit of the approach curve to the Hertz model from the contact point to 200 nm or 500 nm into the sample. Only force curves with a baseline slope between minus 10 to plus 10 pN/μm, baseline sum-of-squared-residuals (SSR) smaller than 0.1 nN2, and P-values smaller than 1.0e-14 were included to determine the overall stiffness of the BM. These criteria select for force curves that have a flat baseline approach curve and a clearly defined contact point. Extended Data Fig. 8n shows representative force curves that meet and do not meet these criteria.
Electron microscopy
lamC1−/− embryos were generated by in-crossing lamC1−/+ fish. 30 hpf embryos were fixed in EM fixative containing 2% paraformaldehyde, 2% glutaraldehyde in 0.1 M sodium cacodylate buffer at room temperature for 2 hrs and then overnight at 4°C. Fixed embryos were rinsed with 0.1 M sodium cacodylate buffer and post-fixed with 1% OsO4 in 0.1 M cacodylate buffer, followed by block-staining with 1% uranyl acetate aqueous solution overnight at 4°C. The samples were rinsed with water, dehydrated in a graded series of ethanol, infiltrated with propylene oxide/Epon mixtures and finally embedded in EMbed812 (Electron Microscopy Sciences, PA USA). 70 nm sections were cut and mounted on 200 copper mesh grids and stained with uranyl acetate and lead citrate. Imaging was performed on a Talos120C transmission electron microscope (Thermo Fisher Scientific, Hillsboro, OR) with Gatan (4k x 4k) OneView Camera (Gatan, Inc., Pleasanton, CA). The primordium cells were pseudo-colored using Adobe Illustrator 2020 (Adobe).
Ectopic expression of Cxcl12a from the trunk muscle cells
1 nl of 10 ng/μl pDEST-tol2-acta1a-cxcl12a-t2a-mCherry plasmid DNA or pDEST-tol2-acta1a-mCherry plasmid DNA was injected together with 40 ng/μl tol2 mRNA into one-cell stage embryos obtained from the following crosses: lamC1+/−; cxcl12a−/−; cldnB:lyn2GFP in-cross, lamC1+/+; cxcl12a−/−; cldnB:lyn2GFP in-cross, wild type in-cross and lamC1+/−; cxcl12a−/−; cxcr4b:cxcr4b-Kate2-IRES-GFP-CaaX in-cross. lamC1−/− mutant embryos were identified by morphology.
Live imaging with a Leica SP8 confocal system and image analysis
The live imaging experiments detailed below were performed on a Leica SP8 confocal microscope system equipped with a 40x (NA 1.1), a 20x (NA 0.7) and a 20x (NA 0.5) objectives. Samples were kept at 28°C with a heated stage (Warner Instruments, Quick Exchange Heated Base, cat no. QE-1) for long time time-lapse imaging. The power of the laser lines was calibrated using a power meter (X-Cite Power Meter Model, Lumen Dynamics, cat no. XR2100). For quantifying the fluorescence intensity, image acquisition was carried out using the photon-counting mode. Embryos were mounted in 0.5% low melt agarose (National Diagnostics, cat no. EC-205)/Ringer’s solution supplemented with 0.4 mg/ml MS-222 anesthetic on a cover-slip or a plastic dish.
itgb1b mutant and control embryos were generated by crossing itgb1b:itgb1b-sfGFP/−; prim:mem-mCherry fish to itgb1b:itgb1b-sfGFP/− fish. Such embryos were imaged for 10 hours with the following settings: z-step size 5.0 μm, time interval 30 min, duration 9.5 h. Mutants were identified by the absence of Itgb1b-sfGFP expression. Note that itgb1b mutants produced by this cross showed a slightly stronger overall morphological defect than mutants generated by in-crossing itgb1b−/+ fish, indicating that maternal Itgb1b rescues the lack of zygotic Itgb1b slightly better than maternal Itgb1b-sfGFP mRNA and protein.
To image the localization of Itgb1bΔNPxY-sfGFP in the primordium cells, 34 hpfitgb1b:itgb1bΔNPxY-sfGFP; prim:mem-mCherry and itgb1b:itgb1b-sfGFP; prim:mem-mCherry control embryos were mounted. The z-step size was 0.42 μm. The power of the 488 nm and 594 nm laser lines was calibrated to 95 μW and 29 μW, respectively. The GFP-to-mCherry fluorescence intensity ratio in the basal to apical axis was obtained by a custom-written macro in Fiji. Briefly, the macro generated a cell-membrane mask based on the mCherry channel using the Default thresholding method in Fiji. This mask was applied to the GFP and the mCherry channels and the signal intensities were obtained for each z-slices. To analyze the intensity profile of Itgb1b-sfGFP in the primordium cells along the front-to-back axis, we semi-automatically quantified the signal intensity of Itgb1b-sfGFP in the membrane of the primordium cells using custom-written macros in Fiji. Briefly, we first manually aligned the primordium along the x,y,z axes such that the three central slices in the z-stack (= 1.2 μm) neither contained the apical constrictions nor the lateral line nerve but contained the tip and the rear of the primordium. Next, the macro generated a binary mask for the cell membrane using membrane-mCherry fluorescence and the “default” thresholding method in Fiji. The mask was applied to the GFP channel, the three central slices were average-projected, the masked GFP channel was resliced from the primordium’s tip, the z-profile was obtained and plotted.
To image the localization of F-tractin-mCherry in the primordium cells with skin cells labeled with Cdh1-sfGFP, 33 hpf TgBAC(cxcr4b:F-tractin-mCherry); TgBAC(cdh1:cdh1-sfGFP) embryos were mounted. The z-step size was 0.42 μm. The power of the 488 nm and 594 nm laser lines were calibrated to 68 μW and 14 μW, respectively. The two channels were sequentially scanned.
To analyze the ratio of Itgb1b-tdTomato/ Itgb1b-sfGFP and Itgb1b-tdTomato/ Itgb1bΔNPxY-sfGFP fluorescence intensities, itgb1b:itgb1b-sfGFP; itgb1b:itgb1b-tdTomato and itgb1b:itgb1bΔNPxY-sfGFP; itgb1b:itgb1b-tdTomato embryos were mounted at 32 hpf. The z-step size was 0.76 μm and the pinhole set to 2.0 airy units. The power of the 488 nm and 546 nm laser lines was calibrated to 38 μW and 30 μW, respectively. Two channels were sequentially scanned. The ratio analysis was carried out in a semi-automated manner with a custom-written macro in Fiji. Briefly, the red channel was divided by the green channel to generate the ratio image. The membrane mask was generated as follows. The red channel and the green channels were added to generate a single channel image, then this single channel image was thresholded with the “moments” method in Fiji and was binarized to generate a mask image. The mask was then applied to the ratio image. The middle slice without the apical constrictions or lateral line nerve was manually defined. A line with a width of 30 pixels (= 8.52 μm) was drawn from the tip of the primordium to the rear and the intensity profile along this line was obtained. To analyze the ratio in the MTJs and lateral sides of the muscle cells, we used the ratio image described above. The single slice at the bottom of the primordium in the z-stack was manually chosen. The mean ratio at MTJs was quantified by drawing a line with a width of 10-pixel (= 2.84 μm). The mean ratio at the lateral sides of the muscle cells was quantified by manually encircling the muscle cells between two MTJs.
To image the localization of Cdh2-mCherry in the primordium cells, 32 hpf TgBAC(cxcr4b:EGFP-CaaX); TgBAC(cdh2:cdh2-mCherry) embryos were mounted. The z-step size was set to 1.0 μm with a pinhole of 2.0 airy units. The power of the 488 nm and 561 nm laser lines was calibrated to 27 μW and 1.52 mW, respectively. The two channels were sequentially scanned. The localization of Cdh2-mCherry in the primordium cells was obtained by extracting the fluorescence signal of Cdh2-mCherry only on the membrane of the primordium cells using a binary mask generated based on the EGFP-CaaX fluorescence signal and using the “default” thresholding method in Fiji.
To assess the chemokine Cxcl12a signaling gradient across the primordium, TgBAC(cxcr4b:cxcr4b-EGFP-IRES-Kate2-CaaX)p7 embryos were mounted. The z-step size was set to 0.7 μm with a pinhole of 2.0 airy units. The power of the 488 nm and 594 nm laser lines was calibrated to 30 μW and 64 μW, respectively. The ratio of mem-Kate2 to Cxcr4b-EGFP was analyzed with a previously published custom-written ImageJ macro76.
To observe LamC1-sfGFP localization in the apical side of the primordium, TgBAC(lamC1:lamC1-sfGFP);cdh1:cdh1-tdTomato/+ embryos at 32 hpf were mounted. The z-step size was set to 0.42 μm with a pinhole of 1.0 airy units. The power of the 488 nm and 546 nm laser lines was calibrated to 136 μW and 108 μW, respectively. The two channels were sequentially scanned.
To obtain embryos for long-term live imaging (7 h) of primordium migration in tln1−/−; tln2a+/+ or tln2a+/−; tln2b−/−; TgBAC(cxcr4b:zGrad)/+; prim:mem-mChery embryos and control prim:mem-mCherry; TgBAC(cxcr4b:zGrad)/+ embryos, we crossed tln1−/−; tln2a+/−; tln2b+/−; TgBAC(tln1:tln1-YPet)/+; TgBAC(cxcr4b:zGrad)/+; prim:mem-mCherry male fish to tln1−/−; tln2a+/−; tln2b−/−; TgBAC(tln1:tln1-YPet)/+; prim:mem-mCherry female fish. The control embryos were obtained by crossing prim:mem-mCherry fish to TgBAC(cxcr4b:zGrad)/+ fish. tln1−/− mutants were identified by the absence of Tln1-YPet expression at 28 hpf. Mutant embryos and control embryos were mounted in the same dish. The following settings were used: z-step size 6.0 μm, time interval 20 min, duration 8.0 h. Imaged embryos were digested and genotyped for TgBAC(cxcr4b:zGrad) transgene, tln2a and tln2b.
To quantify zGrad-mediated Tln1-YPet, Itgb1b-sfGFP and Ctnna1-Citrine degradation with zGrad expressed from the cxcr4b promoter, TgBAC(tln1:tln1-YPet)/+; prim:mem-mCherry embryos and TgBAC(tln1:tln1-YPet)/+; prim:mem-mCherry; TgBAC(cxcr4b:zGrad)/+ embryos, itgb1b:itgb1b-sfGFP/+; prim:mem-mCherry embryos and itgb1b:itgb1b-sfGFP/+; prim:mem-mCherry; TgBAC(cxcr4b:zGrad)/+ embryos, ctnna1-citrine/+; prim:mem-mCherry embryos and ctnna1-citrine/+; prim:mem-mCherry; TgBAC(cxcr4b:zGrad)/+ embryos were sorted at 29 hpf. Five to six embryos of each genotype were mounted at 33 to 34 hpf. The laser power was calibrated to 45 μW (for Itgb1b-sfGFP) and 90 μW (Tln1-YPet and Ctnna1-Citrine) for the 488 nm laser line and 10 μW for the 594 nm laser line. The z-step size was set to 0.76 μm. To quantify the signal intensity of Tln1-YPet, Itgb1b-sfGFP and Ctnna1-Citrine in the primordium, we used a custom-written ImageJ macro. Briefly, a binary mask for the primordium was generated using membrane-mCherry fluorescent signal and the “default” thresholding method followed by the “filling holes” function. The binary mask was applied to the z-stack images and images were sum-projected. The fluorescence signal was obtained only in the masked area. To remove the contribution of autofluorescence from the embryo, we imaged three prim:mem-mCherry embryos with the same setting above and quantified the autofluorescence signal intensity with the same macro. The mean autofluorescence signal intensity from three embryos was subtracted from the experimental data.
To observe the localization of Myl12.1-mScarlet in the primordium cells, TgBAC(cxcr4b:myl12.1-mScarlet); TgBAC(cxcr4b:EGFP-CaaX) embryos were mounted at 32 hpf. The z-step size was 0.42 μm with pinhole 1.0 airy units. The power of the 488 nm and 546 nm laser lines were calibrated to 105 μW and 112 μW, respectively. The two channels were sequentially scanned. Basally localized Myl12.1-mScarlet was analyzed with a custom-written macro in Fiji. Briefly, we first aligned the primordium with the x, y, z axes. The macro includes only the bottom half of the z-stack such that the stack did not contain the apical constrictions. Then, the stack was maximum-projected. The primordium area was masked by a binary mask generated as follows: The EGFP-CaaX channel was maximum-projected, thresholded by the “Huang” method and the holes were filled. Then, 1.7 μm was eroded to eliminate the Myl12.1-mScarlet signal localized at the periphery of the primordium. The value (mean+stdev) of Myl12.1-mScarlet was subtracted from the Myl12.1-mScarlet intensity to highlight only accumulated Myosin II punctae. Total intensity of Myl12.1-mScarlet punctae in the front, middle and rear region of the primordium was manually obtained. The total intensity of Myl12.1-mScarlet punctae was normalized by the area of the front, middle and rear of the primordium. Finally, these values were divided by the mean for normalization and plotted.
To quantify the migration speed of Ctnna1-depleted primordium cells, five ctnna1:ctnna1-citrine/ctnna1:ctnna1-citrine; TgBAC(cxcr4b:zGrad)/+; TgBAC(cxcr4b:H2A-mCherry) embryos and five ctnna1:ctnna1-citrine/ctnna1:ctnna1-citrine; TgBAC(cxcr4b:H2A-mCherry) control embryos were mounted. Images were acquired every 2 min for a duration of 30 to 200 min. The pinhole was set to 1 airy unit.
To track the cell nuclei in the primordium, we imported the time-lapse videos into the software Imaris Version 8.0 (Bitplane, Oxford Instruments). As in the original recordings, the dimensions were set to x = 0.378 μm, y = 0.378 μm, z = 1.00 μm and t = 120 s. To track the cell nuclei the spot tool in the Imaris software was used with a specified cell diameter of 3.78 μm and default background subtraction activated. The histogram of the spot quality was adjusted such that about 70 to 80% of the cells were selected. The cell nuclei were tracked using the autoregressive motion method with a maximum distance that an object can move between two consecutive time points set to 20 μm for the red channel (H2A-mCherry). The maximum gap size—maximum number of consecutive time points that are allowed to be missing in order to join track fragments—was set to two. After running the tracking algorithm the filter for track length was set to select 75 to 85% of the longest tracks and the tracks were manually curated to remove tracked nuclei that do not belong to the primordium or represent dying cells and to delete or correct track segments. For dividing cells, only one daughter cell was tracked. To correct for stage-drift a muscle cell nucleus was tracked and chosen as a reference point. The x, y, z, t values and the track IDs were exported and the position of the reference cells was subtracted from all tracked cells at each time point. With the exported data, the speed of each cell in the indicated genotype was pooled.
To observe receptor internalization induced by ectopic Cxcl12a secreted from the muscle cells, 25–26 hpf embryos were imaged. The power of the 488 nm and 561 nm laser lines was calibrated to 152 μW and 110 μW, respectively. The z-step size was set to 2.0 μm. We manually cropped the z-stack such that it only contained the primordium. We then generated a binary mask based on the GFP channel and applied it to the RFP and GFP channels. The GFP and RFP channels were then sum projected, the mean intensity of each channel was measured and the RFP mean intensity was divided by the GFP mean intensity using a custom-written macro in Fiji.
The tagged protein expression patterns of TgBAC(lamC1:lamC1-sfGFP), TgBAC(tln1:tln1-YPet), itgb1b:itgb1b-sfGFP and itgb1b:itgb1bΔNPxY-sfGFP were imaged in 28 hpf embryos. The z-step size was set to 1.5 μm. The images were stitched together using the auto tiling feature in the LAS X Life Science Microscope Software (Leica) and sum-projected.
Live imaging with a Nikon W1 spinning disk confocal system and image analysis
The live imaging experiments detailed below were performed using a Nikon W1 spinning disk confocal microscope that was equipped with an Apo LWD 40X NA 1.15 objective lens (Nikon, cat no. MRD77410) and a SR HP Plan Apo 100X NA 1.45 objective lens (Nikon, MRD01905). The 100X objective was used only for the actin flow experiments. All other experiments were performed using the 40X objective. The samples for live time-lapse imaging experiments were kept at 30°C using a Tokai Hit incubation system STXG-TIZWX-SET. The embryos were mounted in 0.5% low melt agarose (National Diagnostics, cat no. EC-205)/Ringer’s solution supplemented with 0.4 mg/ml MS-222 anesthetic on a glass-bottom dish.
All fluorescent recovery after photobleaching (FRAP) experiments were performed using 32 to 34 hpf embryos. For the analysis of Itgb1b-sfGFP at the myotendinous junction, embryos were treated with 50 μM Rockout (Rho Kinase Inhibitor III, MilliporeSigma, cat no. 555553-10MG) in 1% DMSO or control-treated with 1% DMSO for 3 hrs. The circular ROI with a 3 μm radius was selected and bleached with a 405 nm laser. Images with a fixed z-position were taken every 0.5 sec for 30 sec or 1 min. Two regions of interest per embryo were recorded. For simultaneous FRAP of LamC1-sfGFP and sec-mCherry, 24 hpf lamC1:lamC1-sfGFP; hsp70l:sec-mCherry embryos were heat shocked at 39.5°C for 1 h to induce sec-mCherry expression from the heat shock promoter. To bleach LamC1-sfGFP and sec-mCherry, a circular ROI with a 6 μm radius was selected and bleached with a 405 nm laser. Images with a fixed z-position were taken every 100 ms for 30 sec. For long-term LamC1-sfGFP FRAP, three to six circular ROI with a 6 μm radius per embryo were selected and bleached with a 405 nm laser. Z-stack images were taken every 10 min for 50 min. All FRAP analyses were performed using the mean intensity values of the regions of interest extracted by the time-measurement setting in NIS-Elements (Nikon) except for the long-term LamC1-sfGFP FRAP. For the long-term LamC1-sfGFP FRAP, we first sum-projected the z-stacks and corrected for photo-bleaching using the mean fluorescent intensity of the image for each time point. The circular regions of interest with a 6 μm radius were manually selected and tracked over time. The mean intensity in the regions of interest was then measured and plotted. For all other FRAP analyses, we first normalized the fluorescent intensity of the bleached region at each time point Ix to the difference between the fluorescent intensity before bleaching (Imax) and the minimal fluorescent intensity after bleaching (Imin) using the following relation
where I is the normalized fluorescence intensity. We then corrected for overall photo-bleaching by dividing the normalized fluorescent intensity I by the overall rate of photo-bleaching. To calculate the overall rate of photo-bleaching we randomly picked a 30 μm × 30 μm region outside of the bleached region and extracted the mean intensity over time from four movies for each experimental setting and averaged them. The overall photo-bleaching rate was calculated by dividing the average intensity of each time point by the average intensity of the first time point.
To locally photo-bleach LamC1-sfGFP, the ROIs for bleaching were defined by a custom-written macro in Fiji and saved as a binary tiff image with 1024 x 1024 pixel resolution. The tiff image was imported into the Nikon NIS-Elements AR software as an ROI for the photo-bleaching experiments. The Fiji macro creates a hexagonal pattern with 20 x 20 dots (= 120 μm x 120 μm), each of which is separated from its neighboring dots by 6 μm. A single dot was 2-pixel wide (= 0.65 μm). Bleaching was performed with a 473 nm laser in a single plane.
Short-term live imaging (1 h) of the migration of the primordium in itgb1b−/−; cldnB:lyn2GFP embryos and control embryos was performed using 32 hpf embryos. itgb1b−/− embryos and control (sibling) embryos were sorted according to their body length before mounting. Images with a 40 μm z-stack with a step size 0.8 μm were taken every 5 min for 1 hour with a multi-position setting. Imaged embryos were digested and genotyped for itgb1b as described above.
To image the deformation of the BM by skin basal cells, 32 hpf TgBAC(lamC1:lamC1-sfGFP) embryos injected with lyn2-mCherry mRNA at the 1-cell stage were mounted at 32 hpf. First, the LamC1-sfGFP-labeled BM was photo-bleached to generate a hexagonal dot pattern. Second, the location of the bleach pattern was imaged with the following parameters: z-step size 0.4 μm, time interval 1 min, duration 10 min.
To image the deformation of the BM by the primordium, 32 hpf TgBAC(lamC1:lamC1-sfGFP); prim:mem-mCherry embryos, TgBAC(lamC1:lamC1-sfGFP); prim:mem-mCherry; hsp70l:cxcl12a control embryos , and TgBAC(lamC1:lamC1-sfGFP); prim:mem-mCherry; itgb1b−/− were mounted. First, the LamC1-sfGFP-labeled BM was photo-bleached to generate a hexagonal dot pattern. Second, the location of the bleach pattern was imaged with the following parameters: z-step size 0.4 μm, time interval 5 min, duration 120 min. The multi-position setting was used to image multiple embryos at the same time and in the same dish. The TgBAC(lamC1:lamC1-sfGFP); prim:mem-mCherry; hsp70l:cxcl12a control embryos were heat shocked at 39.5°C for 1 hour at 28 hpf to induce Cxcl12a expression and block primordium migration before imaging. The genotype of itgb1b−/− embryos was confirmed by PCR after live imaging.
To image the accumulation of LamC1-sfGFP under the primordium, 32 hpfTgBAC(lamC1:lamC1-sfGFP); TgBAC(cxcr4b:F-tractin-mCherry) embryos were mounted. The green and the red channels were imaged sequentially to prevent fluorescence bleed-through. To quantify the LamC1-sfGFP intensity around the edge of the primordium and beyond the primordium, we used a semi-automated custom-written macro in Fiji. z-stacks were maximum-projected. The outer circumference of the primordium was manually traced using the primordium-specific mCherry signal as a guide. The encircled area was filled and thresholded. The thresholded image was duplicated. The first duplicate was 5 times eroded from its outline inward (=1.625 μm). The second duplicate was dilated 5 times from its outline outward. Then the two images were subtracted from each other. This generated a 10 pixel-wide (3.25 μm) annulus-like area that we used as a binary mask. Next, the bright LamC1-sfGFP signal at the myotendinous junctions was thresholded using the LamC1-sfGFP signal and converted to a binary mask. This mask was dilated two times to remove the myotendinous junction signal from the analysis. These two masks were applied to the LamC1-sfGFP channel of the original image to extract the signal intensity of the LamC1-sfGFP around the primordium. To obtain the LamC1-sfGFP signal beyond the edge of the primordium (control measurement), the thresholded image of the primordium’s outer circumference was duplicated two times. The first and second duplicate was dilated 30 and 20 times, respectively, and subtracted from each other to generate a 10 pixel-wide (3.25 μm) annulus-like area 6.5 μm away from the primordium’s outer circumference as a binary mask. We plotted the mean intensity of the LamC1-sfGFP signal in each region from individual embryos.
Analysis of actin flow
To image actin flow in single primordium superficial and basal cells, we co-injected BAC DNA coding for cxcr4b:F-tractin-mNG with 1 nl of 40 ng/ul tol2 mRNA into one-cell stage wild-type and itgb1b−/− embryos also transgenic for prim:mem-mCherry. The itgb1b−/− embryos were generated by crossing itgb1b:itgb1b-sfGFP/−; prim:mem-mCherry to itgb1b:itgb1b-sfGFP/− and sorting for embryos lacking Itgb1b-sfGFP expression at 24 hpf. The embryos were mounted at 32 to 34 hpf. Clones in the primordium were identified based on mCherry and mNeonGreen expression and imaged on their basal side collecting single planes every 2 sec for 3 min.
Actin flow analysis was performed by generating kymographs using Fiji. Briefly, a one pixel-wide 20 μm region of interest line was drawn manually from the cell’s center outward across the protrusion on the basal side. The kymograph was generated using the KymoResliceWide plugin provided by Eugene Katrukha and Laurie Young (https://imagej.net/KymoResliceWide). In singly labeled cells, the front of the actin flow and the front of the protrusion were identified visually, manually traced, and the actin flow rate and protrusion rate were extracted from the slopes of the trace lines. The actin polymerization rate was calculated by subtracting the actin flow rate from the protrusion rate. To image and analyze actin flow in single primordium basal cells across the front to rear axis of the primordium, we prepared injected embryos as described above. To identify the location of F-tractin-mNG expressing cells, we first imaged a z-stack of the whole primordium. Then, switched to the 100x objective to image the actin flow. The location of F-tractin-mNG expressing cells was later identified based on the whole primordium z-stack image. To image F-actin retrograde flow in the tip cells, we used TgBAC(cxcr4b:F-tractin-mCherry); TgBAC(cxcr4b:EGFP-CaaX) embryos. The imaging conditions and analysis were identical.
Blastomere transplantation and imaging of primordia with clones expressing Itgb1b-sfGFP and Tln1-YPet
We transplanted 20 to 50 cells from donor embryos at the 1000 to 8000-cell stage into host embryos of the same stage. All host embryos were wild type. Donor embryos were transgenic for TgBAC(tln1:tln1-Ypet) or itgb1b:itgb1b-sfGFP and Tg(prim:mem-mCherry) or TgBAC(cxcr4b:F-tractin-mCherry). At 28hpf, we isolated embryos that contained donor cells in the primordium based on the expression of the Tg(prim:mem-mCherry) or TgBAC(cxcr4b:F-tractin-mCherry) transgenes. To observe clones in the superficial cells of the primordium, we used itgb1b:itgb1b-sfGFP; TgBAC(cxcr4b:F-tractin-mCherry) as donor embryos. Chimeric embryos were mounted at 32–34 hpf. Images were collected every 30 sec for 10 min as z-stacks with a z-step size of 1.0 μm. The green and red fluorescent channels were sequentially scanned to prevent fluorescent bleed-through.
Blastomere transplantation and imaging of primordia with clones depleted in Talin activity
We transplanted 20 to 50 cells from donor embryos at the 1000 to 8000-cell stage into host embryos of the same stage. All host embryos carried Tg(cldnB:lyn2GFP). Control donor embryos were transgenic for Tg(prim:mem-mCherry). Talin-depleted donor embryos were obtained from an in-cross of tln1−/−; tln2a+/−; tln2b−/−; TgBAC(tln1:tln1-YPet)/+; Tg(prim:mem-mCherry) fish. Wild-type and Talin-depleted donor embryos were injected with 50 ng/μl zGrad mRNA at the one-cell stage. 32-34 hpf embryos with chimeric primordia were mounted. Images were collected every 5 min for 2 hours as Z-stacks with a Z-step size of 1.0 μm. The green and red fluorescent channels were sequentially scanned to prevent fluorescent bleed-through. Cumulative migration distance was quantified by manually tracking the tip of the primordia using the Manual Tracking plugin in Fiji. Kymographs were drawn using the KymoResliceWide plugin in Fiji.
Analysis of rear contraction and migration
To analyze rear contractions in the primordium, TgBAC(cxcr4b:EGFP-CaaX) embryos were mounted at 33 hpf. Images were collected every 30 sec for 30 min as z-stacks with a z-step size of 1.0 μm. As a proxy for the rear cells, we tracked the apical constriction site located in the rear of the primordium (> 65 μm from the tip) using the sum-projected EGFP-CaaX signal and the TrackMate plugin in Fiji. The contraction of the rear cells was analyzed as follows: First, we duplicated the middle nine slices (= 8.0 μm) of the z-stack and sum-projected them. Next, we subtracted the background from the projected image using 5 pixels in Fiji to enhance the membrane signal. Third, we applied the “Huang” thresholding method to this image and binarized it. Finally, we manually identified two vertices, which were located three cells apart from each other, located at the periphery of the rear region of the primordium, and manually tracked their locations over time.
Live imaging with a Leica 165M FC Fluorescent Stereo Microscope
The experiments detailed below were performed using a Leica 165M FC Fluorescent Stereo Microscope equipped with a Leica DFC345 FX camera.
To assess the migration distance of the primordium in embryos in different genetic scenarios (cldnB:lyn2GFP; lamC1−/− and cldnB:lyn2GFP embryos with clones in the muscle expressing mCherry from the acta1a promoter, TgBAC(lamC1:lamC1-sfGFP)), 26 hpf (initial distance), 32 hpf (assessment of migration) and 48 hpf embryos, respectively, were imaged. The TgBAC(lamC1:lamC1-sfGFP) copy number was determined based on the intensity of the sfGFP fluorescence. Distance measurements were performed using Fiji.
To measure the migration distance of the primordium in itgb1b−/− embryos, 54 hpf embryos were generated by crossing itgb1b:itgb1b-sfGFP/−; prim:mem-mCherry fish with itgb1b:itgb1b-sfGFP/− fish. itgb1b−/− embryos were identified by the absence of Itgb1b-sfGFP expression. The primordium migration distance was measured using Fiji. Note that the primordium reaches the tip of the tail by 42 hpf in wild-type embryos49.
To analyze the distance that the primordium migrated in various mutant backgrounds at 48 hpf, the following crosses were set up to obtain embryos of the indicated genotypes. After imaging, the embryos were digested and genotyped if required.
(Z) tln1−/− embryos: In-cross of tln1−/−; TgBAC(tln1:tln1-YPet)/+; prim:mem-mCherry fish, embryos were sorted against Tln1-YPet and for mem-mCherry.
(Z) tln2a−/− embryos: In-cross of tln2a+/−; cldnB:lyn2GFP fish, after the experiment embryos were genotyped for tln2a.
(MZ) tln2b−/− embryos: In-cross of tln2b−/−; cldnB:lyn2GFP fish.
(Z) tln1−/−; (Z) tln2a−/− embryos: In-cross of tln1−/−; TgBAC(tln1:tln1-YPet)/+; tln2a+/−; prim:mem-mCherry, embryos were sorted against Tln1-YPet and for mem-mCherry. After the experiment embryos were genotyped for tln2a.
(Z) tln1−/−; (MZ) tln2b−/− embryos: In-cross of tln1−/−; TgBAC(tln1:tln1-YPet)/+; tln2b−/−; prim:mem-mCherry fish, embryos were sorted against Tln1-YPet and for mem-mCherry.
(Z) tln2a−/−; (MZ) tln2b−/− embryos: In-cross of tln2a+/−; tln2b−/−; cldnB:lyn2GFP fish, after the experiment embryos were genotyped for tln2a.
(Z) tln1−/−; (Z) tln2a−/−; (MZ) tln2b−/− embryos: In-cross of tln1−/−; TgBAC(tln1:tln1-YPet)/+; tln2a+/−; tln2b−/−; prim:mem-mCherry fish, embryos were sorted against Tln1-YPet and for mem-mCherry. After the experiment, embryos were genotyped for tln2a.
(MZ) itgb1a−/− embryos: In-cross of itgb1a−/−; cldnB:lyn2GFP fish.
(Z) itgb1b−/− embryos: In-cross of itgb1b−/+; cldnB:lyn2GFP fish. After the experiment the embryos were genotyped for itgb1b.
(MZ) itgb1a−/−; (Z) itgb1b−/+ embryos: Cross of itgb1a−/− female fish to itgb1a−/+; itgb1b+/−; cldnB:lyn2GFP male fish. After the experiment the embryos were genotyped for itgb1a and itgb1b.
(Z) itgb1a−/+; (Z) itgb1b−/− and (Z) itgb1a−/−; (Z) itgb1b−/− embryos: Cross of itgb1a−/+; itgb1b:itgb1b-sfGFP/−; prim:mem-mCherry fish to itgb1a−/+; itgb1b:itgb1b-sfGFP/− fish, embryos were sorted against Itgb1b-sfGFP and for mem-mCherry. After the experiment the embryos were genotyped for itgb1a.
ctnna1-citrine/ctnna1-citrine and ctnna1-citrine/ctnna1-citrine; TgBAC(cxcr4b:zGrad)/+ embryos: In-cross of ctnna1-citrine/+; TgBAC(cxcr4b:zGrad)/+; prim:mem-mCherry fish, embryos were sorted against homozygous Ctnna1-Citrine by fluorescent intensity and for mem-mCherry. Embryos with zGrad expression were identified by mScarlet expression in the heart.
To assess the degradation of sfGFP by zGrad, we injected an mRNA injection mix containing 50 ng/μl of sfGFP-ZF1 mRNA, 50 ng/μl of mCherry-ZF1 mRNA with or without 50 ng/μl of zGrad mRNA into one-cell stage embryos. To assess the degradation of YPet by zGrad, we injected an mRNA injection mix containing 50 ng/μl of YPet-ZF1 mRNA, 50 ng/μl of mCherry-ZF1 mRNA with or without 50 ng/μl of zGrad mRNA into one-cell stage embryos. As a control, we injected an mRNA injection mix containing 50 ng/μl of mNG-ZF1 mRNA, 50 ng/μl of mCherry-ZF1 mRNA with or without 50 ng/μl of zGrad mRNA into one-cell stage embryos. Injected embryos at 8hpf were dechorionated and imaged under the microscope. Preparation of mRNAs is described previously9.
Live imaging with an Axioplan Microscope
The experiments detailed below were performed using an Axioplan Microscope (Zeiss) equipped with an Axiocam (Zeiss) and a 5x (NA 0.25) objective.
Documentation of the phenotype of lamC1−/−; TgBAC(lamC1:lamC1-sfGFP) embryos. The TgBAC(lamC1:lamC1-sfGFP) was identified based on sfGFP fluorescence. The lamC1 mutant embryos were identified and scored based on the morphological defects at 48 hpf.
Documentation of the phenotype of itgb1b−/− embryos. itgb1b−/− embryos were generated by crossing itgb1b:itgb1b-sfGFP/−; prim:mem-mCherry fish to itgb1b:itgb1b-sfGFP/− fish. itgb1b−/− embryos were identified by the absence of Itgb1b-sfGFP fluorescence.
Primordium migration distance quantification
To quantify the cumulative migration distance of the primordium, time-lapse image sequences were maximum- or sum-projected and the tip of the primordium was tracked using the Manual Tracking plugin provided by Fabrice Cordelieres in Fiji (https://imagej.nih.gov/ij/plugins/manual-tracking.html).
Circularity analysis of the primordium
To quantify the circularity of the primordium, we limited the length of the primordium to the first 100 μm from the tip of the primordium. z-stacks were sum-projected. Using Fiji, the primordium region was manually cropped based on the intensity of Lyn2-GFP fluorescence. Then, a median filter with a radius of six pixel was applied and the background was subtracted using a rolling ball radius of 100 pixels. Images were rendered binary using the Huang thresholding algorithm to obtain a clear outline of the primordium. Finally, we quantified the circularity of the primordium for each time point using the ‘Analyze Particles’ macro in Fiji. The circularity C is defined as
where A is the primordium’s area and P is the perimeter.
Analysis of Itgb1b-sfGFP and Tln1-YPet localization in primordium cells
We analyzed the spatial distribution of Itgb1b-sfGFP and Tln1-YPet with respect to F-tractin-mCherry and membrane-tethered mCherry at the basal side of small clones of labeled primordium cells in two ways. First, we chose a single slice at the basal side of the cells in a clone. The clone contour was manually selected based on the F-tractin-mCherry or membrane-mCherry fluorescence using a 5 pixel-wide (= 1.625 μm) segmented line to obtain the fluorescent intensity profile at each time point for the green and red channels in Fiji. Second, we assessed the co-localization of the fuorescently tagged proteins using the coloc2 plugin in Fiji. Before applying the coloc2 plugin, we processed the images as follows: Due to photo-bleaching only the first ten time points were included for analysis. Images were rotated such that the direction of migration aligned with the x-axis. Then the images were re-sliced to obtain transverse sections in the xz-plane. This resulted in images with the basal sides of the cells pointing upward (z-axis of imaging) and the direction of migration pointing to the right. Next, a median filter with 1 pixel width was applied and the images were rotated again such that the basal sides of the cells aligned horizontally. An ROI with a width of 3.25 μm from the basal membrane inward was manually defined as the basal region of cells and used for the coloc2 analysis in Fiji with a custom-written macro. The macro compiles the individual ROIs of the basal region from each time point, creates a mask based on the sum intensities of the green and red channels using the Default method in Fiji. The individual ROIs were also compiled into a single image for each channel as a montage as shown in (Extended Data Fig. 4d). The macro then calculates the degree of co-localization, the Li’s ICQ value, using the coloc2 function in Fiji. Li’s ICQ is calculated as follows77. For each pixel in the ROI, the product of the difference of intensity and the mean intensity for each channel is calculated ((Ch1-mean(Ch1))*(Ch2-mean(Ch2)). Then, the number of pixels with a positive product are normalized to the total pixel number and 0.5 is subtracted. Therefore, Li’s ICQ ranges from −0.5 (signals perfectly segregated) to 0.5 (signals perfectly overlap). We performed this analysis for three consecutive xz-slices per clone, pooled the data for display in the panel.
Generation of embryos lacking most Talin activity
Control wild-type embryos and embryos from an in-cross of tln1−/−; tln2a+/−; tln2b−/−; TgBAC(tln1:tln1-YPet)/+; Tg(prim:mem-mCherry) fish were injected with 50 ng/μl zGrad mRNA at the one-cell stage. At 28 hpf, embryos were fixed with 4% PFA in PBST. After imaging, individual embryos were genotyped for tln2a.
Analysis of skin-generated basement membrane wrinkles and traction
A 30 μm x 30 μm ROI centered on a transient increase in LamC1-sfGFP fluorescence intensity at the third time point in a 4-D stack spanning five time points with an 1 min interval was manually defined. The third point was set to 0 min. To visualize the LamC1-sfGFP increase as a graph, the stack was max-projected along the z-axis. The fluorescent intensity profile was extracted along a 10 μm line ROI across the LamC1-sfGFP increase at the 0 min time point. The same fluorescent intensity profiles along the same line ROI were obtained for the −1 min and +1 min time points. The fluorescence intensity was normalized to the mean of the −1 min time point. Traction was analyzed using Embryogram, and calculated and visualized with ParaView (ParaView-5.8.1, Kitware). For the traction stress calculation a Young’s modulus of 566.7 Pa was used for the BM based on the AFM measurements (Fig. 6c) and a Poisson ratio of 0.45. Traction was obtained using the −2 min time point as a reference for the undeformed BM. To quantify the temporal change of traction stresses around the local increases in LamC1-sfGFP, we averaged the traction stresses measured at the three marks closest to the LamC1-sfGFP increase. Particle Image Velocimetry (PIV) analysis of the basal skin cells was performed for detecting the displacement of the membrane from −1 min to 0 min using the PIVlab plugin (version 2.38 by William Thielicke) in MATLAB (Version 9.9, MathWorks). Every second vector was visualized on the cell membrane image at the −1 min time point. The displacement of the BM was obtained by Embryogram. The 3D quiver plots of the displacement of the BM from −2 min (reference) to 0 min with the scale factor 3 were drawn using ParaView (ParaView-5.8.1, Kitware).
Analysis of primordium-generated traction stresses and angles of BM deformation
Four-dimensional confocal z-stacks were denoised by applying a median filer (width 2 pixels) and analyzed in Embryogram. In Embryogram, an area containing a well-defined bleach pattern that was in front of the primordium at the first time point of the time lapse was manually selected. Bleached cylinders with radii between 2–8 pixels (=0.65–2.6 μm) were identified and tracked over time in the time lapse and manually curated in Embryogram. To subtract rigid motions, we manually selected cylinders to calculate the global displacement. If the bleach pattern spanned the horizontal myoseptum and extended past the primordium on either side, we used the two rows of bleached cylinders furthest from the horizontal myoseptum on both sides of the bleach pattern. If the bleach pattern did not span the horizontal myoseptum, we used the two rows of bleached cylinders furthest from the horizontal myoseptum on one side of the embryo only. To perform the Finite Element Analysis, we constructed volumetric meshes on both sides of the BM to represent the skin and the muscle above and below the primordium, used up-sampling of 2, discretization order of 2, a Young’s modulus of 566.7 Pa and a Poisson ratio of 0.45. We excluded the cylinders on the edge of the bleach pattern from the analysis because these cylinders were often tracked incorrectly.
To analyze the angles between the displacement vectors and the direction of migration, and the traction stresses under the primordium, we exported data files containing 1) the X, Y and Z coordinates, 2) the displacement vector, and 3) the traction stress vector for each extrapolated vertex from ParaView (ParaView-5.8.1, Kitware). Then, we semi-manually selected vertices in front, middle, and rear part of the primordia defined as the 0–15 μm, 15–65 μm and 65 < from the tip of the primordium, respectively. For this analysis, we used the 40, 60 and 80 min time points of the time lapse videos. We also included cylinders with a distance of less than 10 μm to the primordium in the analysis. For controls, we used cylinders that are at a distance less than 20 μm from the horizontal myoseptum on the dorsal and ventral sides along the migratory route of the primordium. This corresponds to the maximal width of the primordium. In Fiji, we maximum-projected the primordium channel, manually traced the primordium edge, converted it to a binary mask and dilated the mask 10 μm. Then, we applied this binary mask to the maximum-projected BM channel at the first time point. The approximate coordinates of the cylinders within the mask were manually recorded in Fiji. Using a custom-written R script, we compared these approximate cylinder-coordinates to the actual cylinder-coordinates obtained from the tracking in Embryogram by minimizing the distance between these two, and extracted information of actual cylinders only in the front, middle and rear of the primordium. BM displacement angles were analyzed based on the displacement of the bleached cylinder in the XY-plane in relation to the direction of primordium migration or the horizontal myoseptum in the case of the controls. The direction of primordium migration/horizontal myoseptum was obtained manually in Fiji. Half-circle polar diagrams were drawn using MATLAB. The cosines of these angles are shown in Fig. 6i. The magnitude of traction stress in the X- and Y-directions and in the Z-direction were extracted from cylinder located as described above.
3-dimensional and 2-dimensional displacement vectors, the magnitude of the traction stresses, the components of the stress tensors, and the magnitude of the stresses in the direction of primordium migration/horizontal myoseptum were visualized using ParaView (ParaView-5.8.1, Kitware). To calculate the stresses in the direction of primordium migration, we first obtained the vector for the direction of primordium migration in 3D using Fiji. We next calculated the cross-products (cross-products 1) between this directional vector and the normal vectors to the surface of the BM. To obtain the vectors normal to the primordium direction, we calculated the cross-products (cross-products 2) between cross-products 1 and the normal vectors to the surface of the BM. Finally, we generated stress vectors by multiplying cross-products 2 with the stress tensor obtained by the Embryogram simulation. This calculation was done in Paraview. The confocal stack of the primordium channel was binarized using the Li’s thresholding method in Fiji and superimposed. The data were visualized in ParaView.
Nuclear tracking of the primordium cells
To analyze the direction of primordium cells, we used previously published cell tracking data11. The imaging condition and tracking method using Imaris Version 8.0 (Bitplane, Oxford Instruments) were same as above. Tracking data for three primordia were exported and analyzed using the R environment. Briefly, the cosine between the directions of two given cells at a given time-frame was obtained. We performed this analysis for all the combinations of two cells for all the time-frames and pooled data. The trajectories of the primordium cells were also drawn using R.
Statistical tests
All the statistical tests were performed using Prism 9 (GraphPad software). We first tested the normality of distribution in each group with a Kolmogorov-Smirnov (KS) test. When p>0.05 in the KS test was obtained for a multiple comparison, we performed an ordinary one-way ANOVA test followed by a multiple comparison test specified in the figure legend, for a comparison of two groups, we performed an F-test to compare variances. When p>0.05 in the F-test was obtained in the two-group comparison, we performed a two-tailed t-test. Otherwise, we performed a two-tailed t-test with Welch’s correction (Welch’s t-test). When we obtained p<0.05 in the initial KS test for either the multiple comparison or two-group comparison, we subsequently performed a two-tailed Mann-Whitney test between two groups. The qualitative TEM experiments (Fig. 1a, 2d and Extended Data Fig. 1b) one embryo was analyzed. For Fig. 2g and h, Fig. 4a, b, c and d, and Fig, 8a, all embryos originate from a single experiment. All other experiments have been performed two or more times.
Extended Data
Extended Data Fig. 1. Ultrastructure of the basement membrane along the migratory route of the primordium and the characterization of the TgBAC(lamC1:lamC1-sfGFP) line.
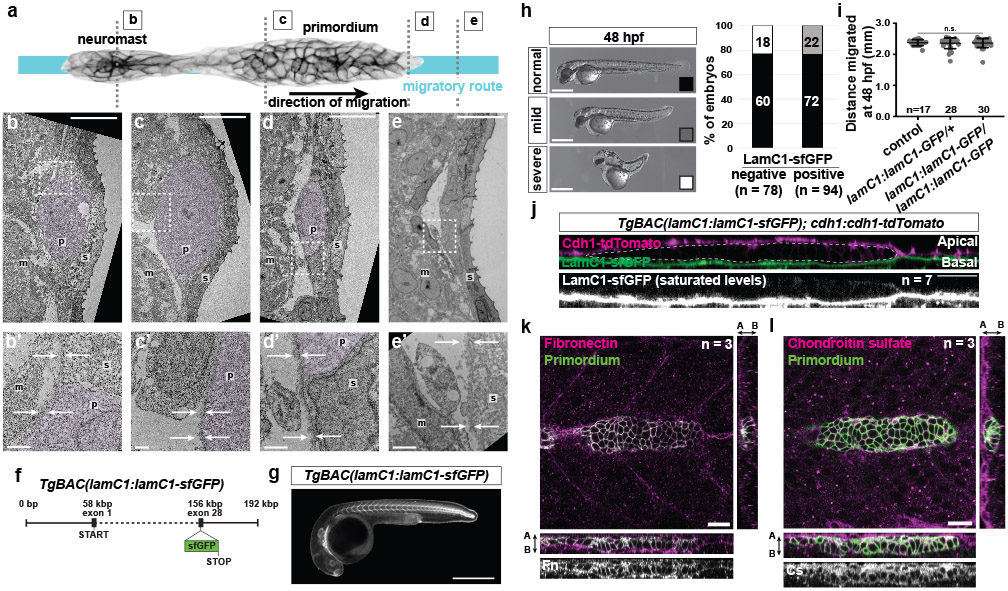
a, Overview of the primordium. Dotted lines indicate the location of the cross-sections shown in (b-e). b-e, TEM images of cross-sections at the level of the most recently deposited neuromast (b), at the level of the primordium’s rear (c), at the level of the primordium’s front (d), and in front of the primordium (e). Scale bars = 10 μm. b’-e’, Magnification of area outlined by a dotted line in (b-e). Scale bars = 1 μm. The skin (s), primordium (p, purple hue), the muscle (m), and the BM (arrows) are indicated. n = 1 embryo. f, Schematic of the TgBAC(lamC1:lamC1-sfGFP) transgene. g, Image of the expression of LamC1-sfGFP from TgBAC(lamC1:lamC1-sfGFP) transgene in a 28 hpf embryo. The image is a sum-projected z-stack. Scale bar = 0.5 mm. h, The TgBAC(lamC1:lamC1-sfGFP) transgene partly rescues the lamC1 mutant phenotype. Crosses from lamC1−/+; lamC1:lamC1-sfGFP to lamC1−/+ fish resulted in embryos with three different phenotypes shown on the left. Quantification of the phenotypic categories from these crosses for non-transgenic embryos and embryos expressing LamC1-sfGFP are shown on the right. Note that the mild phenotype correlates with the presence of LamC1-sfGFP and the severe phenotype represents the lamC1 mutant phenotype. Scale bars = 0.5 mm. i, Quantification of the primordium migration in the presence of different copy numbers of the TgBAC(lamC1:lamC1-sfGFP) transgene. Data points, means, and SD are indicated. n.s.: p=0.6514 (non-transgenic vs. lamC1:lamC1-sfGFP/+), p=0.7842 (non-transgenic vs. lamC1:lamC1-sfGFP/lamC1:lamC1-sfGFP) (two-tailed Mann-Whitney test). j, Cross-section along apical-basal axis of a primordium (dotted line in top panel) of embryos expressing LamC1-sfGFP (BM) and the Cdh1-tdTomato (skin). The LamC1-sfGFP signal is enhanced to saturated levels (bottom panel). Scale bars = 25 μm. k, Images of slices from a z-stack of 32 hpf TgBAC(cxcr4b:EGFP-CaaX) embryos stained for Fibronectin and GFP. Orthogonal views are shown. l, Images of slices from a z-stack of 32 hpf TgBAC(cxcr4b:EGFP-CaaX) embryos stained for Chondroitin sulfate and GFP. Orthogonal views are shown. For h, i, n= number of embryos.
Extended Data Fig. 2. Depletion of Ctnna1-Citrine by zGrad and characterization of the lamC1 mutants.

a, Principle of zGrad-mediated protein degradation. b, Left: 8 hpf embryos injected with sfGFP-ZF1 mRNA and mCherry-ZF1 mRNA with or without co-injected zGrad mRNA. Middle: 8 hpf embryos injected with YPet-ZF1 mRNA and mCherry-ZF1 mRNA with or without co-injected zGrad mRNA. Right: 8 hpf embryos injected with mNeonGreen-ZF1 mRNA and mCherry-ZF1 mRNA with or without co-injected zGrad mRNA. n ≥ 20 embryos. Scale bar: 1 mm. c, Single confocal slices of primordia in prim:mem-mCherry; ctnna1:ctnna1-citrine control (left) and prim:mem-mCherry; ctnna1:ctnna1-citrine; cxcr4b:zGrad 32 hpf embryos (right). Lower panels show the Ctnna1-Citrine fluorescence as a heat map. Scale bar = 20 μm. d, Quantification of the Ctnna1-Citrine fluorescence intensity in control and zGrad-expressing embryos at 32 hpf. Data points, means, and SD are indicated. ****: p<0.0001 (two-tailed Welch’s t-test). e, Expression of cxcl12a in control (wild-type or lamc1−/+) and lamC1 mutant 30 hpf embryos. Bracket indicates the location of interrupted cxcl12a expression domain. Scale bar = 0.5 mm. f, mCherry-expressing clones in muscle of 26 hpf control (wild-type or lamC1−/+) and lamC1 mutant embryos also transgenic for cldnB:lyn2GFP. Arrowheads indicate the position of primordium. Scale bar = 0.5 mm. g, Quantification of the distance from the ear to the first somite in the indicated genotypes at 26 hpf. Data points, means, and SD are indicated. n.s.: p=0.5516 (two-tailed Mann-Whitney test). h, Images of the primordium in wild-type and cxcl12a−/− 32 hpf embryos with clones in the trunk muscle that express Cxcl12a together with mCherry (not shown) (Left). Asterisks indicate the ear and arrowheads the primordium. Scale bar = 0.5 mm. Quantification of the distance migrated by the primordium in the indicated experimental conditions at 32 hpf (Right). Data points, means, and SD are indicated. ****: p<0.0001 (One-way ANOVA followed by Tukey’s multiple comparison test). i, Cross-sectional images of the Cxcl12a sensor in primordia of cxcl12a−/− and cxcl12a−/−; lamC1−/− embryos with clones in the muscle of the trunk that express mCherry or Cxcl12a. Quantification shown in Fig. 2k. Scale bar = 20 μm. For d, e, g, h, n = number of embryos.
Extended Data Fig. 3. β-integrin and talin expression analysis.

a, Expression analysis of β-integrins in the migrating primordium by in situ hybridization on 32 hpf embryos. Note that itgb8 could not be amplified from embryonic cDNA. Arrows indicate expression in the primordium. Scale bar = 0.5 mm. b, Schematics of the itgb1b locus, the itgb1b targeting cassette, and the modified itgb1b locus. c, Itgb1b-sfGFP expression in a 28 hpf itgb1b:itgb1b-sfGFP embryo. The image is a sum-projected z-stack. Scale bar = 0.5 mm. d, in situ hybridization against the three zebrafish talin genes on 32 hpf embryos. Arrow indicates enriched talin expression in the primordium. Scale bar = 0.5 mm. e, Schematic of the TgBAC(tln1:tln1-YPet) transgene and its protein product. f, Tln1-YPet expression in a 28 hpf tln1:tln1-YPet embryo. The image is a sum-projected z-stack. Scale bar = 0.5 mm.
Extended Data Fig. 4. Integrin-β1b and Talin1 dynamics in cells of the primordium.

a, Localization of Itgb1b-sfGFP and F-tractin-mCherry at the apical side of superficial cells in the primordium. The images are single optical slices. Arrowheads indicate Itgb1b-sfGFP clustering. Scale bar = 10 μm. b, Localization of Itgb1b-sfGFP (top) and Tln1-YPet (bottom) with membrane-mCherry at the basal sides of cells in clones in the primordium imaged over time taken from Video 3. The images are single optical slices. Arrowheads indicate Itgb1b-sfGFP and Tln1-YPet clustering. Scale bar = 10 μm. c, Intensity profiles of Itgb1b-sfGFP (left) and Tln1-YPet (right) together with membrane-tethered mCherry along the contours of clones at indicated times taken from Video 3. Arrows indicate Itgb1b-sfGFP and Tln1-YPet clusters that do not coincide with membrane-tethered mCherry clustering. Representative profile of 5 or more imaged cells. d, Montage of 10 consecutive images of the basal sides of the clones. The images are single transverse sections from a time lapse video. Scale bar = 10 μm. e, Quantification of co-localization of Itgb1b-sfGFP and Tln1-YPet with F-tractin-mCherry and membrane tethered mCherry. Li’s ICQ co-localization indices of 0.5 and −0.5 indicate perfectly co-localized and perfectly anti-co-localized signals, respectively. n = number of cells. Data points, means, and SD are indicated. Three data points were analyzed from the same embryo. **: p=0.0015 (two-tailed t-test). f, Images from time-lapse video after photo-bleaching of Itgb1b-sfGFP at the myotendinous junction of embryos treated with DMSO or 50 μM Rockout. GFP intensities are pseudo-colored as a heat map. Scale bars = 10 μm. g, Graph of Itgb1b-sfGFP fluorescence intensity over time before and after photo-bleaching in embryos treated with DMSO or 50 μM Rockout. The fluorescence intensities are normalized to the minimal intensities after photo-bleaching. Dots indicate mean intensities and error bars are SD. n = number of experiments, N = number of embryos. h, Plot of the percent recovery of Itgb1b-sfGFP fluorescence intensity at 28 sec after photo-bleaching in embryos treated with DMSO or 50 μM Rockout. Data points, means, and SD are indicated. ****: p<0.0001 (two-tailed Welch’s t-test). n = number of experiments (used for statistical test), N = number of embryos. i, Images from time-lapse video after photo-bleaching of Itgb1b-sfGFP in the primordium (left) and at the myotendinous junction (right). Fluorescence intensities are pseudo-colored as a heat map. Scale bars = 10 μm. j, Graph of Itgb1b-sfGFP fluorescence intensity over time before and after photo-bleaching in the primordium and at the myotendinous junction. The fluorescence intensities are normalized to the minimal intensities after photo-bleaching. Dots indicate mean intensities and error bars are SD. n = number of experiments, N = number of embryos. k, Plot of the percent recovery of Itgb1b-sfGFP fluorescence intensity at 27 sec after photo-bleaching in the primordium and at the myotendinous junction. n = number of experiments (used for statistical test), N = number of embryos. Data points, means, and SD are indicated. ****: p<0.0001 (two-tailed Welch’s t-test). l, Experimental design to culture primordium cells. m, Antibody staining against Itgb1b-GFP and F-tractin-mCherry on cultured primordium cells. Arrowheads indicate actin stress fibers (m’) with Itgb1b-GFP clusters (arrows in m”) in the cell center and in protrusions (m”’). Scale bars = 20 μm (m-m”) and 1 μm (m”’). n, Antibody staining against Tln1-YPet and F-tractin-mCherry on cultured primordium cells. Arrowheads indicate actin stress fibers (n’) with Tln1-YPet clusters (arrows in n”) in the cell center and in protrusions (n”’). Scale bars = 20 μm (n-n”) and 1 μm (n”’). Images are max-projected z-stacks. Close-ups (right panels) are magnifications of the regions indicated by dotted squares in the middle panels.
Extended Data Fig. 5. β1-integrin mutational analysis.

a, Schematic of the itgb1a and itgb1b alleles. d and i denote deletion and insertion, respectively. b, Primordium migration and morphology defects in embryos with different levels of integrin-β1 activity at 48 hpf. M and Z denote maternal and zygotic mutants, respectively. Arrows indicate the position of the primordium. Scale bar = 0.5 mm. c, Quantification of the primordium migration distance (left), the body length (middle), and primordium migration distance normalized to body length (right) in 48 hpf itgb1 mutant embryos. n = number of embryos. Data points, means, and SD are indicated. ****: p<0.0001 (one-way ANOVA followed by Tukey’s multiple comparison test, left plot), **: p=0.0026 (two-tailed Welch’s t-test, middle plot), and ***: p=0.0002 (two-tailed t-test, right plot). d, Overall morphology of control (wild-type or itgb1b−/+) and itgb1b mutant embryos at 24 hpf. Scale bar = 0.5 mm. e, Primordium migration in control (wild-type or itgb1b−/+) and itgb1b mutant embryos at 54 hpf. Scale bar = 0.5 mm. The arrows indicate the position of the primordium and the arrowheads indicate the position of the tip of the tail. f, Quantification of primordium migration in control (wild-type or itgb1b−/+) and itgb1b mutant embryos at 54 hpf. Data points, means, and SD are indicated. n = number of embryos. ****: p<0.0001 (two-tailed Welch’s t-test). g, prim:mem-mCherry; itgb1b:itgb1b-sfGFP control (left) and prim:mem-mCherry; itgb1b:itgb1b-sfGFP; cxcr4b:zGrad 33 hpf embryos (right). Lower panels show the Itgb1b-sfGFP fluorescence as a heat map. Scale bar = 25 μm. Images are single confocal slices from a z-stack. h, Quantification of the Itgb1b-sfGFP fluorescence intensity in control and zGrad-expressing embryos at 33 hpf. Data points, means, and SD are indicated. ****: p<0.0001 (two-tailed Welch’s t-test). n = number of embryos.
Extended Data Fig. 6. Generation and characterization of talin mutant and analysis of primordium migration in embryos or primordia with depleted Talin activity.

a, Schematic of the tln1, tln2a and tln2b mutant alleles. The sequence around the deletions (d) and insertions (i) are shown. The start codons are indicated for tln1d4 and tln2ai23, and the premature stop codon for tln2ai23. b, Primordium migration distance in 48 hpf embryos with different levels of talin activity. Scale bar = 0.5 mm. c, Quantification of the primordium migration distance (left), the body length (middle), and primordium migration distance normalized to body length (right) in tln mutants at 48 hpf. Data points, means, and SD are indicated. ****: p<0.0001 (one-way ANOVA with Tukey’s multiple comparisons test, left plot), ***: p=0.0002 (two-tailed Welch’s t-test, middle plot). n = number of embryos. d, Crosses to generate embryos with depleted Talin activity. e, in situ hybridization against cxcl12a mRNA on 28 hpf wild-type (top) and Talin-depleted (bottom) embryos injected with zGrad mRNA. Scale bar = 0.5 mm. f, Quantification of the percentage of control and Talin-depleted embryos with perturbed cxcl12a expression along the horizontal myoseptum in 28 hpf embryos. n = number of embryos. g, Quantification of the body length in control and Talin-depleted 28 hpf embryos. n = number of embryos. Data points, means, and SD are indicated. **: p=0.0063, ****: p<0.0001, n.s.:p=0.1610 (two-tailed Mann-Whitney test). h, prim:mem-mCherry; tln1:tln1-YPet control (left) and prim:mem-mCherry; tln1:tln1-YPet; cxcr4b:zGrad 33 hpf embryos (right). Lower panels show the Tln1-YPet fluorescence as a heat map. Scale bar = 25 μm. Images are single confocal slices from a z-stack. i, Quantification of the Tln1-YPet fluorescence intensity in control and zGrad-expressing embryos at 33 hpf. Data points, means, and SD are indicated. ****: p<0.0001 (two-tailed t-test). n = number of embryos. j, Experimental strategy to generate embryos with Talin-depleted clones in the primordium. k, Migration of wild-type primordia with clones of control cells (top) and Talin-depleted cells (bottom). Images are maximum-projected z-stacks from Video 5. The dotted lines indicate the location of primordium tip. Scale bar = 20 μm. l, Kymographs of migrating chimeric primordia shown in (k) and Video 5. m, Quantification of the cumulative migration distance of primordia with clones of control cells and Talin-depleted cells. Dots are means, error bars are SD. *: p=0.03131, p=0.03046 and p=0.04856 (45, 50 and 55 min in the graph) (two-tailed t-test). n = number of embryos.
Extended Data Fig. 7. Generation and characterization of itgb1b:Itgb1bΔNPxY-sfGFP mutant knock-in line and F-actin retrograde flow analysis.
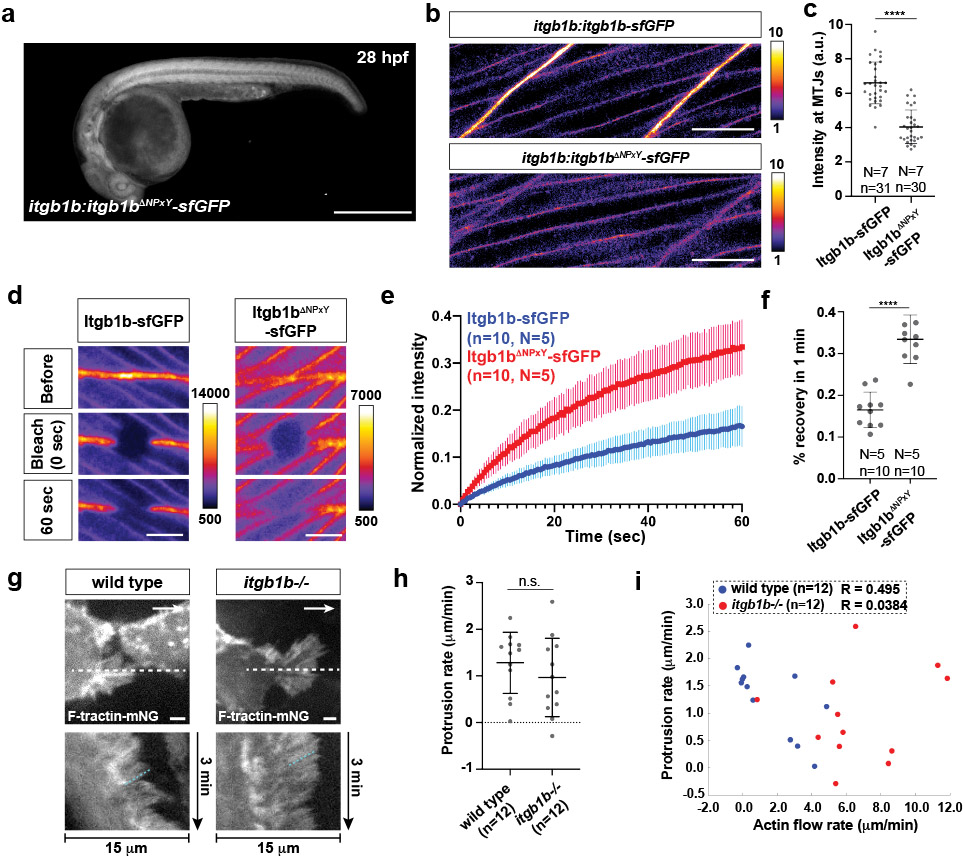
a, Itgb1bΔNPxY-sfGFP expression in a 28 hpf itgb1bΔNPxY-sfGFP embryo. Image is sum-projected z-stacks. Scale bars = 0.5 mm. b, Distribution of Itgb1b-sfGFP and Itgb1bΔNPxY-sfGFP in muscle of 33 hpf embryos. Images are single z-slices through muscle at the myotendinous junction imaged and scaled identically. The GFP intensity is pseudo-colored as a heat map. Scale bars = 20 μm. c, Quantification of the Itgb1b-sfGFP and Itgb1bΔNPxY-sfGFP fluorescence intensities at the myotendinous junction (MTJ). Data points, means, and SD are indicated. ****: p<0.0001 (two-tailed Mann-Whitney test). n = number of experiments (used for statistical analysis), N = number of embryos. d, Images from time-lapse video after photo-bleaching of Itgb1b-sfGFP and Itgb1bΔNPxY-sfGFP at the myotendinous junction. GFP intensities are pseudo-colored as heat maps. Scale bars = 10 μm. e, Graph of Itgb1b-sfGFP and Itgb1bΔNPxY-sfGFP fluorescence intensities over time before and after photo-bleaching. The GFP fluorescence intensities are normalized to the minimal intensities after photo-bleaching. Dots indicate mean intensities and error bars are SD. n = number of experiments, N = number of embryos. f, Plot of the percent recovery of Itgb1b-sfGFP and Itgb1bΔNPxY-sfGFP fluorescence intensities at 1 min after photo-bleaching shown in e. n = number of experiments (used for statistical analysis), N = number of embryos. Data points, means, and SD are indicated. ****: p<0.0001 (two-tailed t-test). g, Images of F-tractin-mNeonGreen localization at the apical sides of wild-type and itgb1b−/− primordium superficial cells (top). White arrows indicate the direction of migration. Scale bar = 2 μm. Images are single optical sections from Video 6. Kymographs of Video 6 along the dotted line indicated in top images (bottom). The dotted cyan line indicates the rate of actin flow. h, Protrusion rates in wild-type and itgb1b mutant primordium basal cells. Data points, means, and SD are indicated. n.s.: p=0.3167 (two-tailed t-test). n = number of cells. i, Plot of the protrusion rate versus the actin flow rate in individual primordium basal cells. n = number of cells.
Extended Data Fig. 8. LamC1-sfGFP mobility, Embryogram workflow, and basement membrane stiffness measurements.
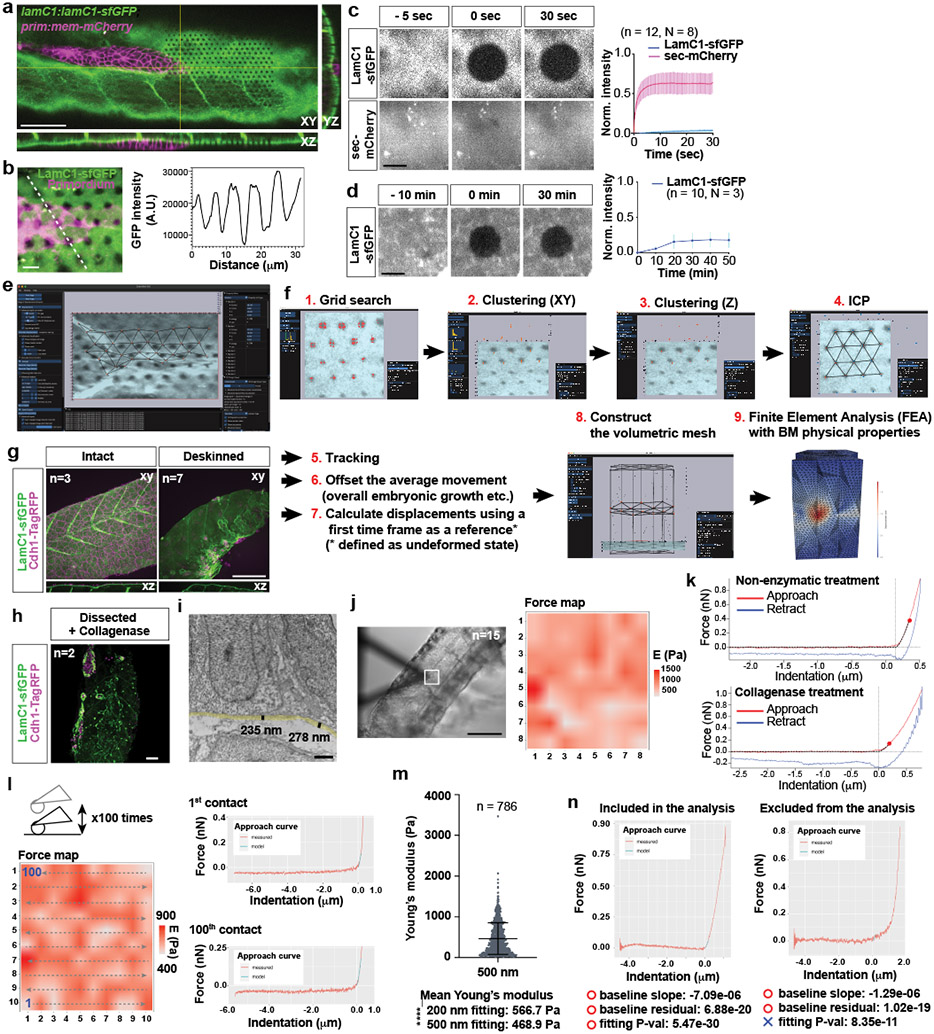
a, Optical sections along the indicated planes of a z-stack of the primordium and the BM labeled with LamC1-sfGFP from Video 7. LamC1-sfGFP was bleached in front of the primordium in a hexagonal pattern. Scale bar = 50 μm. b, Hexagonal bleach pattern on LamC1-sfGFP-labeled BM underneath the migrating primordium (left). Dotted line indicates location of intensity profile shown on right. Scale bar = 5 μm. The image is a maximum-projected z-stack. c, FRAP analysis of LamC1-sfGFP and extracellular mCherry in heat-shocked hsp70l:sec-mCherry; lamC1:lamC1-sfGFP embryos. Images from the time course are shown on the left and quantification of fluorescence recovery is shown on the right. Scale bar = 10 μm, error bars = SD, n = measurements from N embryos, dots = means, n was used for statistical analysis. d, Extended FRAP analysis of LamC1-sfGFP over 50 min in lamC1:lamC1-sfGFP embryos. Images from time course are shown on the left and quantification of fluorescence recovery is shown on the right. Scale bar = 10 μm, error bars = SD, dots = means, n = measurements from N embryos. n was used for statistical analysis. e, Image of Embryogram application user interface. f, In Embryogram, candidate locations for the bleached markers are identified by a grid search (1), clustered in the XY-plane (2), and then along the Z-axis (3). We match these candidates with a regular hexagonal grid using the iterative closest point algorithm (4). Markers are tracked in subsequent frames using optical flow and numerical optimization. The user can manually offset rigid body motions caused by the movement of the microscope, sample movement or sample growth (6). The displacement of each dot is calculated using the mesh for the first time frame as the relaxed reference (7). To perform finite element analysis (FEA), the user constructs a volumetric tetrahedral mesh above, below or both (8) and inputs the Young’s modulus and the Poisson ratio of the material. The results of the FEA can be exported and visualized in other software packages such as ParaView (9). For detail see Supplementary Note 1. g, 28 hpf embryos with labeled skin and BM before and after surgical skin removal. Images are maximum-projected z-stacks. Scale bar = 100 μm. h, Deskinned and collagenase-treated lamC1:lamC1-sfGFP; cdh1:cdh1-TagRFP embryo. Image is a maximum-projected z-stack. Scale bar = 50 μm. i, TEM-image of the BM underneath the primordium. The semi-transparent yellow line traces the BM and the black lines indicate the thickness of the BM. Scale bar = 1 μm. j, Bright-field image of a deskinned embryo tail with the cantilever during an AFM measurement (left). A grid of 8×8 squares (20 μm by × 20 μm) on the BM was probed for its stiffness (square in left image) and the resultant stiffness map is shown on the right. Scale bar = 1 mm. k, Representative force curves showing the approach (red) and retraction (blue) curves for a deskinned embryo (top) and a collagenase-treated deskinned embryo (bottom). Cross-hairs indicate contact point position and force. Red dots on the approach curves indicate the first 200 nm from the contact point. The fit to the baseline and the Hertz model is indicated by a dotted black line. l, Analysis of the effect of repeated probing of the same area by AFM. The left image is a montage of the stiffness values obtained for the same location after measurements 1 to 100. The order of the measurements is indicated by the arrows. The force curves for the first and 100th measurements are shown on the right. The fit to the Hertz model is indicated in cyan. m, Quantification of the stiffness of the BM of deskinned embryos when fitting the first 500 nm after the contract point to the Hertz model. Data points, mean and SD are shown. Values for the fit of the first 200 nm to the Hertz model are shown for comparison. ****: p<0.0001 (two-tailed Mann-Whitney test). n, Representative force curves that meet (left) and do not meet (right) the indicated quality criteria.
Extended Data Fig. 9. Distribution of stresses under the skin, under the primordium, and in the absence of the primordium.

a, Images of LamC1-sfGFP (left) and basal skin cell membranes (middle) from Video 9. The LamC1-sfGFP intensity is pseudo-colored as a heat map. The area outlined by a dotted line was analyzed using Embryogram to calculate the traction stresses (right) pseudo-colored as temperature map (right). The arrowhead indicates a spot of transient accumulation of LamC1-sfGFP. Images are maximum-projected z-stacks. Scale bar = 5 μm. b, Quiver plots of the BM displacement at 0 min in the XY- and XZ-planes. The XZ-plane quiver plot shows a subset of the vector field outlined by the orange rectangle. The magnitude of the vectors was increased by a factor of 3 for visualization purposes. Scale bar = 5 μm. c, Image of cell membrane at −1 min with arrows indicating the direction of movement from time point −1 min to 0 min as determined by PIV. Vector magnitudes are magnified three-times. Scale bar = 5 μm. d, Quantification of traction stresses. Traction stresses at the three vertices closest to a given wrinkle were averaged. Individual data points are shown. Individiual data points are indicated. **: p=0.0027 (−1 min vs. 0 min), p=0.0043 (0 min vs. 2 min) and n.s.: p=0.1479 (0 min vs. 1 min), p=0.7341 (−1 min vs. 2 min) (two-tailed paired t-test). n = number of measured cells, N = number of embryos, n was used for statistical analysis. e, Quantification of LamC1-sfGFP accumulation during BM wrinkling. Intensity profiles were obtained from a line plot across the BM wrinkle at 0 min indicated by the arrowhead in a, and from line plots at the same location of the images at the time points −1 min and 1 min. Intensities were normalized to the mean intensities at time point −1 min. Mean and SD are shown. n = number of measurements, N = number of embryos. f, Deformation of the BM before, during, and after primordium (magenta) migration. Images are from Video 8. The white arrow indicates the direction of migration. Scale bar = 5 μm. g, Quantification of the displacement of bleached marks (yellow circles 1–4 in f) relative to control bleached marks (cyan circles in f). h, Quiver plot of the stresses in the direction of primordium migration. The magnitude of the stress vectors is color-coded. i, Distribution of the tensile and shear stresses around the migrating primordium outlined by a dotted line. The value of each unique component of the stress tensor is colored as a temperature map. The X and Y direction are indicated. The Z direction is orthogonal to the image plane. j, Experimental design of the stress analysis with blocked primordium migration. k, Images of a heat-shocked control embryo at 0 min and 80 min of Video 8. The dotted line indicates the region used for the analysis. Images are maximum-projected z-stacks. Scale bar = 50 μm. l, Quiver plot of the displacement vectors shown along the Z, Y and X axes. The magnitude of the displacement vectors is color coded. Scale bar = 10 μm. m, Quiver plot of the displacement vectors projected in the XY-plane. The magnitude of the vectors was increased twofold. Scale bar = 10 μm. n, Distribution of the traction stress magnitudes color-coded using a temperature map. Scale bar = 10 μm. o, Quiver plot of the stresses in the direction of horizontal myoseptum. The magnitude of the stress vectors is color-coded. Scale bar = 10 μm. (l–o) Data correspond to the at the 80 min time point of Video 10.
Extended Data Fig. 10. The primordium is a continuously migrating tissue.
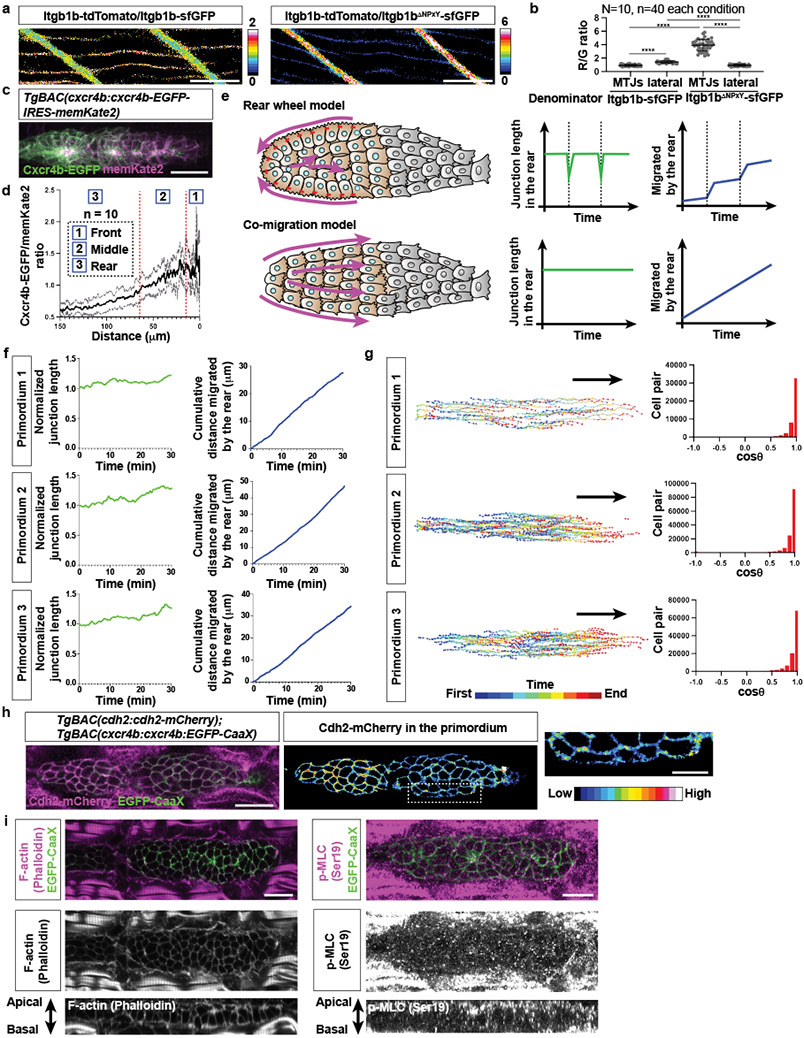
a, Itgb1b-tdTomato-to-Itgb1b-sfGFP (left) and Itgb1b-tdTomato-to-Itgb1bΔNPxY-sfGFP (right) ratio images in trunk muscle cells. Images are single optical slices from z-stacks. Ratios are color-coded as indicated. Scale bar = 25 μm. b, Quantification of ratios Itgb1b-tdTomato to Itgb1b-sfGFP and Itgb1b-tdTomato to Itgb1bΔNPxY-sfGFP at the myotendinous junction and lateral sides of muscle cells. Data points, means, and SD are indicated. ****: p<0.0001 (two-tailed Mann-Whitney test). n = number of measurements at indicated locations, N = number of embryos, n was used for statistical analysis. c, Image of Cxcr4b-EGFP and membrane-tethered Kate2 expressed from the Cxcl12a sensor in the primordium. Image is a maximum-projection of a z-stack. Scale bar = 25 μm. d, Quantification of the Cxcr4b-EGFP/Kate2 ratio across the primordium. Mean (black line) and SD (gray lines) are shown. n = number of embryos. e, Illustrations and predictions for two models of tissue migration. f, Quantification of junction length (left) and the cumulative migration distance over time for three primordia. g, Trajectories of individual primordium cells (left) and frequency plots for angles between any two given cell velocity vectors (right). h, Localization of Cadherin-2-mCherry and membrane-tethered EGFP in the primordium (left). Cdh2-mCherry fluorescence intensity pseudo-colored as a temperature map (middle) and on the primordium’s rear at higher magnification (right). Images are single confocal slice from the z-stack. Scale bars = 25 μm (left) and 10 μm (right). i, Images of slices from a z-stack of 32 hpf TgBAC(cxcr4b:EGFP-CaaX) embryos stained for F-actin (left) or phospho-MLC (right) and GFP. Orthogonal views are shown.
Supplementary Material
Video 1. Migration of wild-type and Ctnna1-depleted primordia. Time lapse of the nuclei of migrating primordia in ctnna1:ctnna1-citrine/ctnna1:ctnna1-citrine; cxcr4b:h2a-mCherry (top) and ctnna1:ctnna1-citrine/ctnna1:ctnna1-citrine; cxcr4b:h2a-mCherry; cxcr4b:zGrad embryos (bottom). Time stamp in minutes. Each time frame is a sum-projected z-stack. The video starts at 36 hpf. Scale bar = 50 μm.
Video 2. z-stacks of Itgb1b-sfGFP and Tln1-YPet protein localization. z-stack of the primordium from lateral (apical side of primordium) to medial (basal side of primordium) in wild-type embryos transgenic for itgb1b:itgb1b-sfGFP; prim:mem-mCherry (top) and TgBAC(tln1:tln1-YPet); prim:mem-mCherry (bottom). Scale bar = 50 μm, Z-slice = 1.01 μm.
Video 3. Time lapses of the dynamics of Itgb1b-sfGFP and Tln1-YPet proteins with the cell membrane or F-actin in a clone of cells in the primordium. Time lapse of cell clones expressing Itgb1b-sfGFP and membrane-tethered mCherry (top left), Tln1-YPet and membrane-tethered mCherry (bottom left), Itgb1b-sfGFP and F-tractin-mCherry (top right) and Tln1-YPet and F-tractin-mCherry (bottom right) in unlabeled primordia of 32 hpf chimeric embryos. The Itgb1b-sfGFP and Tln1-YPet fluorescence intensities with the membrane-tethered mCherry or F-tractin-mCherry fluorescence intensity (left), the Itgb1b-sfGFP and Tln1-YPet fluorescence intensities only (middle), and the membrane-tethered mCherry fluorescence intensity or F-actin-mCherry only (right) are shown. Scale bar = 5 μm. Time stamp in sec. Each time frame is a single slice at the basal side of the cells from a z-stack.
Video 4. Time lapses of migrating primordia in wild-type and itgb1b mutant embryos. (Top) Time lapse of migrating primordia in wild-type (top) and itgb1b mutant embryos (lower top) labeled with the prim:mem-mCherry transgene. Scale bars = 50 μm. Each time frame is a sum-projected z-stack. The video starts at 34 hpf. (Bottom) High temporally resolved time lapse of primordium migration in wild-type (upper bottom) and itgb1b mutant embryos (bottom) labeled with the cldnB:lyn2GFP transgene. Scale bars = 30 μm. Time stamp in min. Each time frame is a maximum-projected z-stack. The video starts at 32 hpf.
Video 5. Time lapses of migrating primordia with reduced Talin activity. (Top) Time lapse of migrating primordia in cxcr4b:zGrad control (top) and (MZ)tln1−/−; (Z)tln2a+/−; (MZ)tln2b−/−; cxcr4b:zGrad embryos (lower top). The primordia are labeled with the prim:mem-mCherry transgene. Scale bar = 100 μm, time stamp in min. Each time frame is a sum-projected z-stack. The time lapses start at 32 hpf.
(Bottom) Time lapse of wild-type primordia labeled with the cldnB:lyn2GFP transgene (green) containing wild-type (upper bottom) and Talin-depleted cells (bottom) labeled with the prim:mem-mCherry transgene (magenta) at the tip of the primordium. Scale bar = 20 μm, time stamp in min. Each time frame is a maximum-projected z-stack. The videos start at 36 hpf.
Video 6. Time lapses of F-actin dynamics in single wild-type and itgb1b mutant primordium basal cells and superficial cells. Time lapse of F-actin dynamics in single basal cells (top) and superficial cells (bottom) expressing the F-actin marker F-tractin-mNeonGreen from the injected cxcr4b:F-tractin-mNeonGreen BAC construct in wild-type primordia (left) and itgb1b mutant primordia (right). Scale bar = 2 μm, time stamp in sec. Each time frame is a single slice at the cell’s basal side from an z-stack. Images were thresholded using the Huang algorithm in Fiji to extract pixels with high signal. The videos start at 33-36 hpf.
Video 7. Z-stack of LamC1-sfGFP-labeled basement membrane with bleach marks. z-stack of a 32 hpf lamC1:lamC1-sfGFP; prim:mem-mCherry embryo in which LamC1-sfGFP was bleached in a hexagonal pattern to optically mark the BM in front of the migrating primordium. Optical section thickness is 0.4 μm. Scale bar = 50 μm.
Video 8. Time lapses of primordia with the basement membrane marked by a bleached pattern in wild-type, Cxcl12a over-expressing, itgb1b mutant embryos. Time Lapse of lamC1:lamC1-sfGFP; prim:mem-mCherry (top), lamC1:lamC1-sfGFP; prim:mem-mCherry; hsp70:cxcl12a (middle) and itgb1b−/−; lamC1:lamC1-sfGFP; prim:mem-mCherry (bottom) embryos with bleach marks in front of the primordium at the first time point. Montage of orthogonal views of the z-stack are shown along the cross-hair for each time point (XY-, XZ-, YZ-planes). Note that the view of the XY-plane is a maximum-projection of the z-stack while the views of the XZ- and YZ-planes are single slices through the z-stack. Ubiquitous Cxcl12a expression was induced by a heat shock to block primordium migration (middle). Scale bar = 25 μm, time stamp in min.
Video 9. Time lapse of basement membrane wrinkling and traction stresses. Montage of time lapses showing the membrane of the skin cells labeled by membrane-mCherry expressed from injected lyn2mCherry mRNA and the LamC1-sfGFP-labeled basement membrane bleach pattern (TgBAC(lamC1:lamC1-sfGFP), left), the LamC1-sfGFP signal intensities as a fire map (middle left), membrane-mCherry signal intensities as a gray scale (middle right), and the magnitude of the traction stresses as a temperature map (right). Note that the first time frame of the time lapse serves as the reference for the undeformed BM. Therefore, no stresses can be calculated for the −2 min time frame. Images are maximum-projected z-stack. Scale bar = 5 μm, time stamp in min.
Video 10. Basement membrane deformation and stresses in control, wild-type and itgb1b mutant embryos. Montage of time lapses showing the displacement vector field (left), the displacement vector field projected onto the XY-plane (middle left), the traction stress magnitudes (middle right), and the stress vectors calculated in the direction of migration (right) together with the migrating primordium (top), in the control condition with blocked primordium migration (middle) and in itgb1b mutant embryos (bottom). For the displacement vector field the orthogonal views of the XZ- (left, bottom) and YZ-planes are also shown (left, left). Note that the magnitude of the displacement vectors are increased twofold for visibility, the magnitude of the traction stresses are indicated as a temperature map, the magnitude of the stress vectors calculated in the direction of migration are indicated as a color code, and the XY-planes are shown from the basal side of the primordium. Video 8 shows the original time lapses that were used for this analysis. Scale bar = 25 μm.
ImageJ-based and R-based codes used for image analysis.
Acknowledgments:
We thank R. Lehmann, L. Christiaen, D. Rifkin, M. Schober, J. Torres-Vázquez, W. Qian, P. Vagni, S. Lau and T. Colak-Champollion for critical comments, T. Gerson, T. Colak-Champollion and A. Feitzinger for reagents, T. Gerson, J. Proietti and S. Pirani for excellent fish care, N. Paknejad for advice on AFM, M. Cammer and Y. Deng for advice on microscopy, A. Liang, C. Petzold and K. Dancel-Manning for consultation and assistance with TEM work, and A. Ferrari and N. Chala for AFM consultation. The use of the NYULH DART Microscopy Laboratory (P30CA016087) and the Memorial Sloan Kettering Molecular Cytology Core Facility (P30 CA008748) is gratefully acknowledged. For providing the zebrafish knockout allele lamC1sa9866, we thank the Zebrafish International Resource Center (ZIRC). For providing the cdh1:cdh1-tdTomato line, we thank M. Cronan and D. Tobin. This work was supported by NIH grant NS102322 (H.K.), by an NYSTEM fellowship C322560GG (N.Y.), by an American Heart Association fellowship 20PRE35180164 (N.Y.), in part through the NYU IT High Performance Computing resources, services, and staff expertise, the NSF CAREER award 1652515 (D.N.), the NSF grants IIS-1320635 (D.N.), OAC-1835712 (D.N.), OIA-1937043 (D.N.), CHS-1908767 (D.N.), CHS-1901091 (D.N.), a gift from Adobe Research (D.N.), a gift from nTopology (D.N.), and a gift from Advanced Micro Devices, Inc (D.N.).
Footnotes
Competing interests:
No competing interests declared.
References:
- 1.Friedl P & Gilmour D Collective cell migration in morphogenesis, regeneration and cancer. Nat Rev Mol Cell Biol 10, 445–457, doi: 10.1038/nrm2720 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Alert R & Trepat X Physical Models of Collective Cell Migration. Annu Rev Conden Ma P 11, 77–101, doi: 10.1146/annurev-conmatphys-031218-013516 (2020). [DOI] [Google Scholar]
- 3.Yamada KM & Sixt M Mechanisms of 3D cell migration. Nat Rev Mol Cell Biol 20, 738–752, doi: 10.1038/s41580-019-0172-9 (2019). [DOI] [PubMed] [Google Scholar]
- 4.Ringer P, Colo G, Fassler R & Grashoff C Sensing the mechano-chemical properties of the extracellular matrix. Matrix Biol 64, 6–16, doi: 10.1016/j.matbio.2017.03.004 (2017). [DOI] [PubMed] [Google Scholar]
- 5.Dalle Nogare D & Chitnis AB A framework for understanding morphogenesis and migration of the zebrafish posterior Lateral Line primordium. Mech Dev 148, 69–78, doi: 10.1016/j.mod.2017.04.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Metcalfe WK Sensory neuron growth cones comigrate with posterior lateral line primordial cells in zebrafish. J Comp Neurol 238, 218–224, doi: 10.1002/cne.902380208 (1985). [DOI] [PubMed] [Google Scholar]
- 7.Parsons MJ et al. Zebrafish mutants identify an essential role for laminins in notochord formation. Development 129, 3137–3146 (2002). [DOI] [PubMed] [Google Scholar]
- 8.Dalle Nogare DE, Natesh N, Vishwasrao HD, Shroff H & Chitnis AB Zebrafish Posterior Lateral Line primordium migration requires interactions between a superficial sheath of motile cells and the skin. Elife 9, doi: 10.7554/eLife.58251 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Yamaguchi N, Colak-Champollion T & Knaut H zGrad is a nanobody-based degron system that inactivates proteins in zebrafish. Elife 8, doi: 10.7554/eLife.43125 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Leckband DE & de Rooij J Cadherin adhesion and mechanotransduction. Annu Rev Cell Dev Biol 30, 291–315, doi: 10.1146/annurev-cellbio-100913-013212 (2014). [DOI] [PubMed] [Google Scholar]
- 11.Colak-Champollion T et al. Cadherin-Mediated Cell Coupling Coordinates Chemokine Sensing across Collectively Migrating Cells. Curr Biol 29, 2570–2579 e2577, doi: 10.1016/j.cub.2019.06.061 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sun Z, Costell M & Fassler R Integrin activation by talin, kindlin and mechanical forces. Nat Cell Biol 21, 25–31, doi: 10.1038/s41556-018-0234-9 (2019). [DOI] [PubMed] [Google Scholar]
- 13.Olson HM & Nechiporuk AV Lamellipodia-like protrusions and focal adhesions contribute to collective cell migration in zebrafish. Dev Biol 469, 125–134, doi: 10.1016/j.ydbio.2020.10.007 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Shibata AC et al. Archipelago architecture of the focal adhesion: membrane molecules freely enter and exit from the focal adhesion zone. Cytoskeleton (Hoboken) 69, 380–392, doi: 10.1002/cm.21032 (2012). [DOI] [PubMed] [Google Scholar]
- 15.Rossier O et al. Integrins beta1 and beta3 exhibit distinct dynamic nanoscale organizations inside focal adhesions. Nat Cell Biol 14, 1057–1067, doi: 10.1038/ncb2588 (2012). [DOI] [PubMed] [Google Scholar]
- 16.Parsons JT, Horwitz AR & Schwartz MA Cell adhesion: integrating cytoskeletal dynamics and cellular tension. Nat Rev Mol Cell Biol 11, 633–643, doi: 10.1038/nrm2957 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.De Pascalis C & Etienne-Manneville S Single and collective cell migration: the mechanics of adhesions. Mol Biol Cell 28, 1833–1846, doi: 10.1091/mbc.E17-03-0134 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Case LB & Waterman CM Integration of actin dynamics and cell adhesion by a three-dimensional, mechanosensitive molecular clutch. Nat Cell Biol 17, 955–963, doi: 10.1038/ncb3191 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Zhang X et al. Talin depletion reveals independence of initial cell spreading from integrin activation and traction. Nat Cell Biol 10, 1062–1068, doi: 10.1038/ncb1765 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Balaban NQ et al. Force and focal adhesion assembly: a close relationship studied using elastic micropatterned substrates. Nat Cell Biol 3, 466–472, doi: 10.1038/35074532 (2001). [DOI] [PubMed] [Google Scholar]
- 21.Tan JL et al. Cells lying on a bed of microneedles: an approach to isolate mechanical force. Proc Natl Acad Sci U S A 100, 1484–1489, doi: 10.1073/pnas.0235407100 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Butler JP, Tolic-Norrelykke IM, Fabry B & Fredberg JJ Traction fields, moments, and strain energy that cells exert on their surroundings. Am J Physiol Cell Physiol 282, C595–605, doi: 10.1152/ajpcell.00270.2001 (2002). [DOI] [PubMed] [Google Scholar]
- 23.Legant WR et al. Multidimensional traction force microscopy reveals out-of-plane rotational moments about focal adhesions. Proc Natl Acad Sci U S A 110, 881–886, doi: 10.1073/pnas.1207997110 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Maskarinec SA, Franck C, Tirrell DA & Ravichandran G Quantifying cellular traction forces in three dimensions. Proc Natl Acad Sci U S A 106, 22108–22113, doi: 10.1073/pnas.0904565106 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Trichet L et al. Evidence of a large-scale mechanosensing mechanism for cellular adaptation to substrate stiffness. Proc Natl Acad Sci U S A 109, 6933–6938, doi: 10.1073/pnas.1117810109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Legant WR et al. Measurement of mechanical tractions exerted by cells in three-dimensional matrices. Nat Methods 7, 969–971, doi: 10.1038/nmeth.1531 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Serra-Picamal X et al. Mechanical waves during tissue expansion. Nature Physics 8, 628–U666, doi: 10.1038/nphys2355 (2012). [DOI] [Google Scholar]
- 28.Brugues A et al. Forces driving epithelial wound healing. Nat Phys 10, 684–691, doi: 10.1038/Nphys3040 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lendenmann T et al. Cellogram: On-the-Fly Traction Force Microscopy. Nano Lett 19, 6742–6750, doi: 10.1021/acs.nanolett.9b01505 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Pearson JR et al. ECM-Regulator timp Is Required for Stem Cell Niche Organization and Cyst Production in the Drosophila Ovary. PLoS Genet 12, e1005763, doi: 10.1371/journal.pgen.1005763 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SN et al. ECM stiffness regulates glial migration in Drosophila and mammalian glioma models. Development 141, 3233–3242, doi: 10.1242/dev.106039 (2014). [DOI] [PubMed] [Google Scholar]
- 32.Harris AK, Wild P & Stopak D Silicone rubber substrata: a new wrinkle in the study of cell locomotion. Science 208, 177–179, doi: 10.1126/science.6987736 (1980). [DOI] [PubMed] [Google Scholar]
- 33.Hagedorn EJ et al. The netrin receptor DCC focuses invadopodia-driven basement membrane transmigration in vivo. J Cell Biol 201, 903–913, doi: 10.1083/jcb.201301091 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Haas P & Gilmour D Chemokine signaling mediates self-organizing tissue migration in the zebrafish lateral line. Dev Cell 10, 673–680, doi: 10.1016/j.devcel.2006.02.019 (2006). [DOI] [PubMed] [Google Scholar]
- 35.Bergert M et al. Force transmission during adhesion-independent migration. Nat Cell Biol 17, 524–529, doi: 10.1038/ncb3134 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Clarke DN & Martin AC Actin-based force generation and cell adhesion in tissue morphogenesis. Curr Biol 31, R667–R680, doi: 10.1016/j.cub.2021.03.031 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Zaidel-Bar R, Milo R, Kam Z & Geiger B A paxillin tyrosine phosphorylation switch regulates the assembly and form of cell-matrix adhesions. J Cell Sci 120, 137–148, doi: 10.1242/jcs.03314 (2007). [DOI] [PubMed] [Google Scholar]
- 38.Shellard A, Szabo A, Trepat X & Mayor R Supracellular contraction at the rear of neural crest cell groups drives collective chemotaxis. Science 362, 339–343, doi: 10.1126/science.aau3301 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Dona E et al. Directional tissue migration through a self-generated chemokine gradient. Nature 503, 285–289, doi: 10.1038/nature12635 (2013). [DOI] [PubMed] [Google Scholar]
- 40.Venkiteswaran G et al. Generation and dynamics of an endogenous, self-generated signaling gradient across a migrating tissue. Cell 155, 674–687, doi: 10.1016/j.cell.2013.09.046 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Choi CK et al. Actin and alpha-actinin orchestrate the assembly and maturation of nascent adhesions in a myosin II motor-independent manner. Nat Cell Biol 10, 1039–1050, doi: 10.1038/ncb1763 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Changede R, Xu X, Margadant F & Sheetz MP Nascent Integrin Adhesions Form on All Matrix Rigidities after Integrin Activation. Dev Cell 35, 614–621, doi: 10.1016/j.devcel.2015.11.001 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Nayal A et al. Paxillin phosphorylation at Ser273 localizes a GIT1-PIX-PAK complex and regulates adhesion and protrusion dynamics. J Cell Biol 173, 587–589, doi: 10.1083/jcb.200509075 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Renkawitz J et al. Adaptive force transmission in amoeboid cell migration. Nat Cell Biol 11, 1438–1443, doi: 10.1038/ncb1992 (2009). [DOI] [PubMed] [Google Scholar]
- 45.Dalle Nogare D et al. Leading and trailing cells cooperate in collective migration of the zebrafish posterior lateral line primordium. Development 141, 3188–3196, doi: 10.1242/dev.106690 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Lai JH, del Alamo JC, Rodriguez-Rodriguez J & Lasheras JC The mechanics of the adhesive locomotion of terrestrial gastropods. J Exp Biol 213, 3920–3933, doi: 10.1242/jeb.046706 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Rieu JP, Barentin C, Maeda Y & Sawada Y Direct mechanical force measurements during the migration of Dictyostelium slugs using flexible substrata. Biophys J 89, 3563–3576, doi: 10.1529/biophysj.104.056333 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Yamaguchi N et al. A rear-engine drives adherent tissue migration in vivo. Zenodo, doi: 10.5281/zenodo.5762146 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Kimmel CB, Ballard WW, Kimmel SR, Ullmann B & Schilling TF Stages of embryonic development of the zebrafish. Dev Dyn 203, 253–310, doi: 10.1002/aja.1002030302 (1995). [DOI] [PubMed] [Google Scholar]
- 50.Valentin G, Haas P & Gilmour D The chemokine SDF1a coordinates tissue migration through the spatially restricted activation of Cxcr7 and Cxcr4b. Curr Biol 17, 1026–1031, doi: 10.1016/j.cub.2007.05.020 (2007). [DOI] [PubMed] [Google Scholar]
- 51.Kettleborough RN et al. A systematic genome-wide analysis of zebrafish protein-coding gene function. Nature 496, 494–497, doi: 10.1038/nature11992 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Wang J et al. Anosmin1 Shuttles Fgf to Facilitate Its Diffusion, Increase Its Local Concentration, and Induce Sensory Organs. Dev Cell 46, 751–766 e712, doi: 10.1016/j.devcel.2018.07.015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Knaut H, Blader P, Strahle U & Schier AF Assembly of trigeminal sensory ganglia by chemokine signaling. Neuron 47, 653–666, doi: 10.1016/j.neuron.2005.07.014 (2005). [DOI] [PubMed] [Google Scholar]
- 54.Trinh le A et al. A versatile gene trap to visualize and interrogate the function of the vertebrate proteome. Genes Dev 25, 2306–2320, doi: 10.1101/gad.174037.111 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Zigman M, Trinh le A, Fraser SE & Moens CB Zebrafish neural tube morphogenesis requires Scribble-dependent oriented cell divisions. Curr Biol 21, 79–86, doi: 10.1016/j.cub.2010.12.005 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Cronan MR & Tobin DM Endogenous Tagging at the cdh1 Locus for Live Visualization of E-Cadherin Dynamics. Zebrafish 16, 324–325, doi: 10.1089/zeb.2019.1746 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Levic DS, Yamaguchi N, Wang S, Knaut H & Bagnat M Knock-in tagging in zebrafish facilitated by insertion into non-coding regions. Development 148, doi: 10.1242/dev.199994 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Gagnon JA et al. Efficient mutagenesis by Cas9 protein-mediated oligonucleotide insertion and large-scale assessment of single-guide RNAs. PLoS One 9, e98186, doi: 10.1371/journal.pone.0098186 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Shen B et al. Generation of gene-modified mice via Cas9/RNA-mediated gene targeting. Cell Res 23, 720–723, doi: 10.1038/cr.2013.46 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Wu Q et al. Talin1 is required for cardiac Z-disk stabilization and endothelial integrity in zebrafish. FASEB J 29, 4989–5005, doi: 10.1096/fj.15-273409 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Iida A, Wang Z, Hirata H & Sehara-Fujisawa A Integrin beta1 activity is required for cardiovascular formation in zebrafish. Genes Cells 23, 938–951, doi: 10.1111/gtc.12641 (2018). [DOI] [PubMed] [Google Scholar]
- 62.Fuentes F, Reynolds E, Lewellis SW, Venkiteswaran G & Knaut H A Plasmid Set for Efficient Bacterial Artificial Chromosome (BAC) Transgenesis in Zebrafish. G3 (Bethesda) 6, 829–834, doi: 10.1534/g3.115.026344 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Warming S, Costantino N, Court DL, Jenkins NA & Copeland NG Simple and highly efficient BAC recombineering using galK selection. Nucleic Acids Res 33, e36, doi: 10.1093/nar/gni035 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Sztal T, Berger S, Currie PD & Hall TE Characterization of the laminin gene family and evolution in zebrafish. Dev Dyn 240, 422–431, doi: 10.1002/dvdy.22537 (2011). [DOI] [PubMed] [Google Scholar]
- 65.Austen K et al. Extracellular rigidity sensing by talin isoform-specific mechanical linkages. Nat Cell Biol 17, 1597–1606, doi: 10.1038/ncb3268 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Johnson HW & Schell MJ Neuronal IP3 3-kinase is an F-actin-bundling protein: role in dendritic targeting and regulation of spine morphology. Mol Biol Cell 20, 5166–5180, doi: 10.1091/mbc.E09-01-0083 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Mastop M et al. Characterization of a spectrally diverse set of fluorescent proteins as FRET acceptors for mTurquoise2. Sci Rep 7, 11999, doi: 10.1038/s41598-017-12212-x (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Gibson DG et al. Enzymatic assembly of DNA molecules up to several hundred kilobases. Nat Methods 6, 343–345, doi: 10.1038/nmeth.1318 (2009). [DOI] [PubMed] [Google Scholar]
- 69.Kwan KM et al. The Tol2kit: a multisite gateway-based construction kit for Tol2 transposon transgenesis constructs. Dev Dyn 236, 3088–3099, doi: 10.1002/dvdy.21343 (2007). [DOI] [PubMed] [Google Scholar]
- 70.Higashijima S, Okamoto H, Ueno N, Hotta Y & Eguchi G High-frequency generation of transgenic zebrafish which reliably express GFP in whole muscles or the whole body by using promoters of zebrafish origin. Dev Biol 192, 289–299, doi: 10.1006/dbio.1997.8779 (1997). [DOI] [PubMed] [Google Scholar]
- 71.Knaut H, Werz C, Geisler R, Nusslein-Volhard C & Tubingen Screen C A zebrafish homologue of the chemokine receptor Cxcr4 is a germ-cell guidance receptor. Nature 421, 279–282, doi: 10.1038/nature01338 (2003). [DOI] [PubMed] [Google Scholar]
- 72.Koster RW & Fraser SE Tracing transgene expression in living zebrafish embryos. Dev Biol 233, 329–346, doi: 10.1006/dbio.2001.0242 (2001). [DOI] [PubMed] [Google Scholar]
- 73.Thisse C & Thisse B High-resolution in situ hybridization to whole-mount zebrafish embryos. Nat Protoc 3, 59–69, doi: 10.1038/nprot.2007.514 (2008). [DOI] [PubMed] [Google Scholar]
- 74.Zoellner H, Paknejad N, Manova K & Moore MA A novel cell-stiffness-fingerprinting analysis by scanning atomic force microscopy: comparison of fibroblasts and diverse cancer cell lines. Histochem Cell Biol 144, 533–542, doi: 10.1007/s00418-015-1363-x (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Loparic M et al. Micro- and nanomechanical analysis of articular cartilage by indentation-type atomic force microscopy: validation with a gel-microfiber composite. Biophys J 98, 2731–2740, doi: 10.1016/j.bpj.2010.02.013 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lau S et al. A negative-feedback loop maintains optimal chemokine concentrations for directional cell migration. Nat Cell Biol 22, 266–273, doi: 10.1038/s41556-020-0465-4 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Li Q et al. A syntaxin 1, Galpha(o), and N-type calcium channel complex at a presynaptic nerve terminal: analysis by quantitative immunocolocalization. J Neurosci 24, 4070–4081, doi: 10.1523/JNEUROSCI.0346-04.2004 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Video 1. Migration of wild-type and Ctnna1-depleted primordia. Time lapse of the nuclei of migrating primordia in ctnna1:ctnna1-citrine/ctnna1:ctnna1-citrine; cxcr4b:h2a-mCherry (top) and ctnna1:ctnna1-citrine/ctnna1:ctnna1-citrine; cxcr4b:h2a-mCherry; cxcr4b:zGrad embryos (bottom). Time stamp in minutes. Each time frame is a sum-projected z-stack. The video starts at 36 hpf. Scale bar = 50 μm.
Video 2. z-stacks of Itgb1b-sfGFP and Tln1-YPet protein localization. z-stack of the primordium from lateral (apical side of primordium) to medial (basal side of primordium) in wild-type embryos transgenic for itgb1b:itgb1b-sfGFP; prim:mem-mCherry (top) and TgBAC(tln1:tln1-YPet); prim:mem-mCherry (bottom). Scale bar = 50 μm, Z-slice = 1.01 μm.
Video 3. Time lapses of the dynamics of Itgb1b-sfGFP and Tln1-YPet proteins with the cell membrane or F-actin in a clone of cells in the primordium. Time lapse of cell clones expressing Itgb1b-sfGFP and membrane-tethered mCherry (top left), Tln1-YPet and membrane-tethered mCherry (bottom left), Itgb1b-sfGFP and F-tractin-mCherry (top right) and Tln1-YPet and F-tractin-mCherry (bottom right) in unlabeled primordia of 32 hpf chimeric embryos. The Itgb1b-sfGFP and Tln1-YPet fluorescence intensities with the membrane-tethered mCherry or F-tractin-mCherry fluorescence intensity (left), the Itgb1b-sfGFP and Tln1-YPet fluorescence intensities only (middle), and the membrane-tethered mCherry fluorescence intensity or F-actin-mCherry only (right) are shown. Scale bar = 5 μm. Time stamp in sec. Each time frame is a single slice at the basal side of the cells from a z-stack.
Video 4. Time lapses of migrating primordia in wild-type and itgb1b mutant embryos. (Top) Time lapse of migrating primordia in wild-type (top) and itgb1b mutant embryos (lower top) labeled with the prim:mem-mCherry transgene. Scale bars = 50 μm. Each time frame is a sum-projected z-stack. The video starts at 34 hpf. (Bottom) High temporally resolved time lapse of primordium migration in wild-type (upper bottom) and itgb1b mutant embryos (bottom) labeled with the cldnB:lyn2GFP transgene. Scale bars = 30 μm. Time stamp in min. Each time frame is a maximum-projected z-stack. The video starts at 32 hpf.
Video 5. Time lapses of migrating primordia with reduced Talin activity. (Top) Time lapse of migrating primordia in cxcr4b:zGrad control (top) and (MZ)tln1−/−; (Z)tln2a+/−; (MZ)tln2b−/−; cxcr4b:zGrad embryos (lower top). The primordia are labeled with the prim:mem-mCherry transgene. Scale bar = 100 μm, time stamp in min. Each time frame is a sum-projected z-stack. The time lapses start at 32 hpf.
(Bottom) Time lapse of wild-type primordia labeled with the cldnB:lyn2GFP transgene (green) containing wild-type (upper bottom) and Talin-depleted cells (bottom) labeled with the prim:mem-mCherry transgene (magenta) at the tip of the primordium. Scale bar = 20 μm, time stamp in min. Each time frame is a maximum-projected z-stack. The videos start at 36 hpf.
Video 6. Time lapses of F-actin dynamics in single wild-type and itgb1b mutant primordium basal cells and superficial cells. Time lapse of F-actin dynamics in single basal cells (top) and superficial cells (bottom) expressing the F-actin marker F-tractin-mNeonGreen from the injected cxcr4b:F-tractin-mNeonGreen BAC construct in wild-type primordia (left) and itgb1b mutant primordia (right). Scale bar = 2 μm, time stamp in sec. Each time frame is a single slice at the cell’s basal side from an z-stack. Images were thresholded using the Huang algorithm in Fiji to extract pixels with high signal. The videos start at 33-36 hpf.
Video 7. Z-stack of LamC1-sfGFP-labeled basement membrane with bleach marks. z-stack of a 32 hpf lamC1:lamC1-sfGFP; prim:mem-mCherry embryo in which LamC1-sfGFP was bleached in a hexagonal pattern to optically mark the BM in front of the migrating primordium. Optical section thickness is 0.4 μm. Scale bar = 50 μm.
Video 8. Time lapses of primordia with the basement membrane marked by a bleached pattern in wild-type, Cxcl12a over-expressing, itgb1b mutant embryos. Time Lapse of lamC1:lamC1-sfGFP; prim:mem-mCherry (top), lamC1:lamC1-sfGFP; prim:mem-mCherry; hsp70:cxcl12a (middle) and itgb1b−/−; lamC1:lamC1-sfGFP; prim:mem-mCherry (bottom) embryos with bleach marks in front of the primordium at the first time point. Montage of orthogonal views of the z-stack are shown along the cross-hair for each time point (XY-, XZ-, YZ-planes). Note that the view of the XY-plane is a maximum-projection of the z-stack while the views of the XZ- and YZ-planes are single slices through the z-stack. Ubiquitous Cxcl12a expression was induced by a heat shock to block primordium migration (middle). Scale bar = 25 μm, time stamp in min.
Video 9. Time lapse of basement membrane wrinkling and traction stresses. Montage of time lapses showing the membrane of the skin cells labeled by membrane-mCherry expressed from injected lyn2mCherry mRNA and the LamC1-sfGFP-labeled basement membrane bleach pattern (TgBAC(lamC1:lamC1-sfGFP), left), the LamC1-sfGFP signal intensities as a fire map (middle left), membrane-mCherry signal intensities as a gray scale (middle right), and the magnitude of the traction stresses as a temperature map (right). Note that the first time frame of the time lapse serves as the reference for the undeformed BM. Therefore, no stresses can be calculated for the −2 min time frame. Images are maximum-projected z-stack. Scale bar = 5 μm, time stamp in min.
Video 10. Basement membrane deformation and stresses in control, wild-type and itgb1b mutant embryos. Montage of time lapses showing the displacement vector field (left), the displacement vector field projected onto the XY-plane (middle left), the traction stress magnitudes (middle right), and the stress vectors calculated in the direction of migration (right) together with the migrating primordium (top), in the control condition with blocked primordium migration (middle) and in itgb1b mutant embryos (bottom). For the displacement vector field the orthogonal views of the XZ- (left, bottom) and YZ-planes are also shown (left, left). Note that the magnitude of the displacement vectors are increased twofold for visibility, the magnitude of the traction stresses are indicated as a temperature map, the magnitude of the stress vectors calculated in the direction of migration are indicated as a color code, and the XY-planes are shown from the basal side of the primordium. Video 8 shows the original time lapses that were used for this analysis. Scale bar = 25 μm.
ImageJ-based and R-based codes used for image analysis.
Data Availability Statement
Previously published genome assemblies as either GRCz10 or GRCz11 that were used here for the design of gRNA constructs are available for tln1, tln2a, tln2b, itgb1a and itgb1b under accession codes ENSDARG00000100729, ENSDARG00000017901, ENSDARG00000110973, ENSDARG00000071863 and ENSDARG00000104484, respectively. Source data have been provided in Source Data. All other data supporting the findings of this study are available from the corresponding authors on reasonable request.