

Abstract
Increasing antimicrobial resistance compels the search for next-generation inhibitors with differing or multiple molecular targets. In this regard, energy conservation in Mycobacterium tuberculosis has been clinically validated as a promising new drug target for combatting drug-resistant strains of M. tuberculosis. Here, we show that HM2-16F, a 6-substituted derivative of the FDA-approved drug amiloride, is an anti-tubercular inhibitor with bactericidal properties comparable to the FDA-approved drug bedaquiline (BDQ; Sirturo®) and inhibits the growth of bedaquiline-resistant mutants. We show that HM2-16F weakly inhibits the F1Fo-ATP synthase, depletes ATP, and affects the entry of acetyl-CoA into the Krebs cycle. HM2-16F synergizes with the cytochrome bcc-aa3 oxidase inhibitor Q203 (Telacebec) and co-administration with Q203 sterilizes in vitro cultures in 14 days. Synergy with Q203 occurs via direct inhibition of the cytochrome bd oxidase by HM2-16F. This study shows that amiloride derivatives represent a promising discovery platform for targeting energy generation in drug-resistant tuberculosis.
Subject terms: Pathogens, Drug development
Derivatives of the FDA-approved drug, amiloride, can eliminate drug-resistant Mycobacterium tuberculosis in vitro by interfering with bacterial energy conservation.
Introduction
Tuberculosis (TB) is a leading cause of mortality globally, with over one million deaths annually1. The emergence of multidrug-resistant (MDR), extensively drug-resistant (XDR) and totally drug-resistant (TDR) strains of Mycobacterium tuberculosis are resulting in extremely limited treatment options2. Current drugs for treating drug-resistant TB disease are slow-acting and treating drug-sensitive strains requires the use of up to four drugs for at least six months3. At present, MDR-TB is treated with a combination of eight to ten drugs lasting 18-24 months4. Bedaquiline (BDQ; Sirturo®) was approved by the US FDA in 2012 for the treatment of adults with pulmonary MDR-TB5,6. BDQ targets the energy-generating machinery (F1Fo-ATP synthase) of M. tuberculosis7,8 marking energy generation a compelling target space for antimicrobial drug development9.
BDQ is generally bactericidal and can kill highly drug-resistant mycobacterial species and dormant bacilli10,11. It acts quickly compared to most TB drugs, but still requires many weeks of therapy and BDQ-resistance has been reported, including in treatment-naïve populations12,13. BDQ binds to the c-subunit rotor in the membrane-embedded part of the F1Fo-ATP synthase8, inhibiting the enzyme by tightly binding to the a–c subunit interface14 and decreasing intracellular ATP levels15,16. BDQ also dissipates the ΔpH component of the proton-motive force in mycobacteria17,18. This depends on the target-based accumulation of BDQ and leads to an uncoupled microenvironment around the F1Fo-ATP synthase18. Targeting multiple mechanisms within energy generation thus appears to be key for developing efficacious anti-tubercular compounds19.
Targeting respiratory complexes earlier in the process of oxidative phosphorylation has begun to provide an understanding on how multi-targeting respiratory therapies can be designed. Telacebec® (Q203;20), has been developed as an inhibitor of M. tuberculosis cytochrome bcc:aa3 terminal oxidase, one of two terminal oxidases that catalyze the terminal reduction of oxygen during cellular respiration20. Although bacteriostatic on its own, Q203 is rapidly and potently bactericidal when a secondary terminal oxidase, cytochrome bd, is deleted21. We recently demonstrated that the cytochrome bd inhibitor ND-011992 strongly synergizes with Q203 and the combination can kill antibiotic-tolerant hypoxic M. tuberculosis22. However, the less-than-optimal pharmacokinetic properties of ND-011992 make it unsuitable for development and alternative chemical scaffolds targeting cytochrome bd are required. Notably, Q203 itself shows no synergy with BDQ23. It is unknown whether compounds can be developed to exploit the best properties of both BDQ and terminal oxidase inhibition.
Several studies have identified new scaffolds for developing next-generation F1Fo-ATP synthase inhibitors, such as squaramides24 and quinoline arylsulfonamides25. However, these compounds have limited pre-existing data regarding their clinical safety and efficacy. In contrast, few studies have identified inhibitors of cytochrome bd. We hypothesized that bioisosterism between BDQ and amiloride could be exploited to identify amiloride-based inhibitors of the mycobacterial F1Fo-ATP synthase and thus mycobacterial growth. Amilorides as a drug class were originally identified as potassium-sparing diuretics in 196726. The parent drug, amiloride, is still in use today as tablets (MidamorTM). In this study, we investigated the ability of a previously synthesized 6-substituted amiloride derivative, HM2-16F27, to function as an antimycobacterial F1Fo-ATP synthase inhibitor. HM2-16F has previously been optimized to eliminate the diuretic activity of amiloride while having promising pharmacological properties for the treatment of cancers driven by the urokinase-type plasminogen activator27. Here, we found that HM2-16F shows comparable killing kinetics to BDQ and displays a different resistance profile at the level of the F1Fo-ATP synthase c-ring, although its direct inhibition of the F1Fo-ATP synthase was found to be comparatively poor. We show that HM2-16F possesses activity as a cytochrome bd inhibitor and potently synergizes with Q203, comparable to ND-01199222. We propose that amilorides represent a promising scaffold for antitubercular drug development, particularly in combination with Q203. HM2-16F may be a promising starting point for developing compounds that target both the F1Fo-ATP synthase and terminal oxidases of M. tuberculosis.
Results
Amiloride derivatives are selectively active against the growth and survival of mycobacterial species
We hypothesized that bioisosterism between BDQ and amiloride could be exploited to identify amiloride-based inhibitors of the mycobacterial F1Fo-ATP synthase and thus mycobacterial growth. Amiloride (Fig. 1a) and the more hydrophobic 5-substituted derivatives 5-(N-ethyl-N-isopropyl)-amiloride (EIPA) and 5-(N,N-hexamethylene)amiloride (HMA, Fig. 1b) were tested for their ability to inhibit the growth of M. tuberculosis and a selection of bacterial pathogens (Table 1). Amiloride and EIPA were able to inhibit the growth of M. tuberculosis at 64–256 μM while a further drop in the minimal inhibitory concentration (MIC) was noted for HMA (32 μM, Table 1). Bedaquiline and isoniazid were included as positive controls in these experiments and exhibited the expected MIC values for these inhibitors (Table 1). Both amiloride and HMA were able to inhibit ATP synthesis in inverted membrane vesicles (IMVs) of M. smegmatis at concentrations comparable to their MIC (Fig. 1c and Table 1). In support of this, HMA was able to outcompete the canonical c-ring covalent inhibitor DCCD for inhibition of the M. phlei F1Fo-ATP synthase c-subunit (Fig. 1d). The compounds were poorly active against the growth of other bacterial pathogens, suggesting some selectivity towards mycobacteria (Table 1).
Fig. 1. Inhibition of the mycobacterial F1Fo-ATP synthase by amiloride and hexamethylene amiloride.

a Structure of amiloride and b 5-(N,N-hexamethylene)amiloride (HMA). c ATP synthesis activity of NADH-energized M. smegmatis inverted membrane vesicles (IMV), treated with either 64 μM of amiloride, 64 μM of HMA, or 2 μM of BDQ as indicated. Error bars represent standard deviation from triplicate biologically independent measurements. d Time course of DCCD labelling in purified M. phlei c-rings in the presence of the indicated amounts of HMA. Data are representative of duplicate measurements.
Table 1.
Minimum inhibitory concentrations (MIC, μM) of amiloride and derivatives against M. tuberculosis and selected bacterial pathogens.
| Compound | Mycobacterium tuberculosis H37Rv | Escherichia coli | Staphylococcus aureus | Enterococcus faecalis |
|---|---|---|---|---|
| Amiloride | 256 | >512 | >512 | >512 |
| HMA | 32 | 128 | 128 | 512 |
| EIPA | 64 | >512 | >512 | 512 |
| HM2-16F | 4 | 256 | >256 | 256 |
| Bedaquiline | 0.2 | ND | ND | ND |
| Isoniazid | 0.2 | ND | ND | ND |
All MIC values are reported in micromolar final concentration. ND = not determined as inactive against these bacteria. All strains are listed in Supplementary Table 1.
Amiloride is an approved potassium-sparing diuretic with a low maximum daily dose (~20 mg/day)26; an activity that would be incompatible with long-term treatment of tuberculosis patients. We previously reported on a 6-substituted derivative (HM2-16 F; Fig. 2a) that shows no inhibition of the human epithelial sodium channel (ENaC) in vitro and minimal diuretic or antikaliuretic properties in rats27. HM2-16F was found to be more potent growth inhibitor against M. tuberculosis, showing a 64- and 8-fold reduction in MIC compared to amiloride and HMA, respectively (Table 1). HM2-16F was also highly selective for M. tuberculosis, minimally inhibiting the growth of Escherichia coli, Staphylococcus aureus or Enterococcus faecalis (Table 1). HM2-16F was bactericidal towards M. tuberculosis at 5X MIC (20 μM), achieving 1.5 log10 CFU mL-1 killing of cells over 15 days (Fig. 2b). The efficacy and kinetics of killing were comparable to BDQ at 10X MIC (2 μM, Fig. 2b), and did not show generation of persisters/resisters in comparison to treatment with isoniazid at 10X MIC (2 μM, Fig. 2b). HM2-16F was also able to prevent the survival of M. bovis in THP-1 macrophages in a manner comparable to BDQ (Fig. 2c). Under hypoxia (non-replicating cultures), both HM2-16F and BDQ had no bactericidal activity over the time course of 30 days (Supplementary Fig. 1).
Fig. 2. HM2-16F is bactericidal towards mycobacteria in vitro and in macrophages.
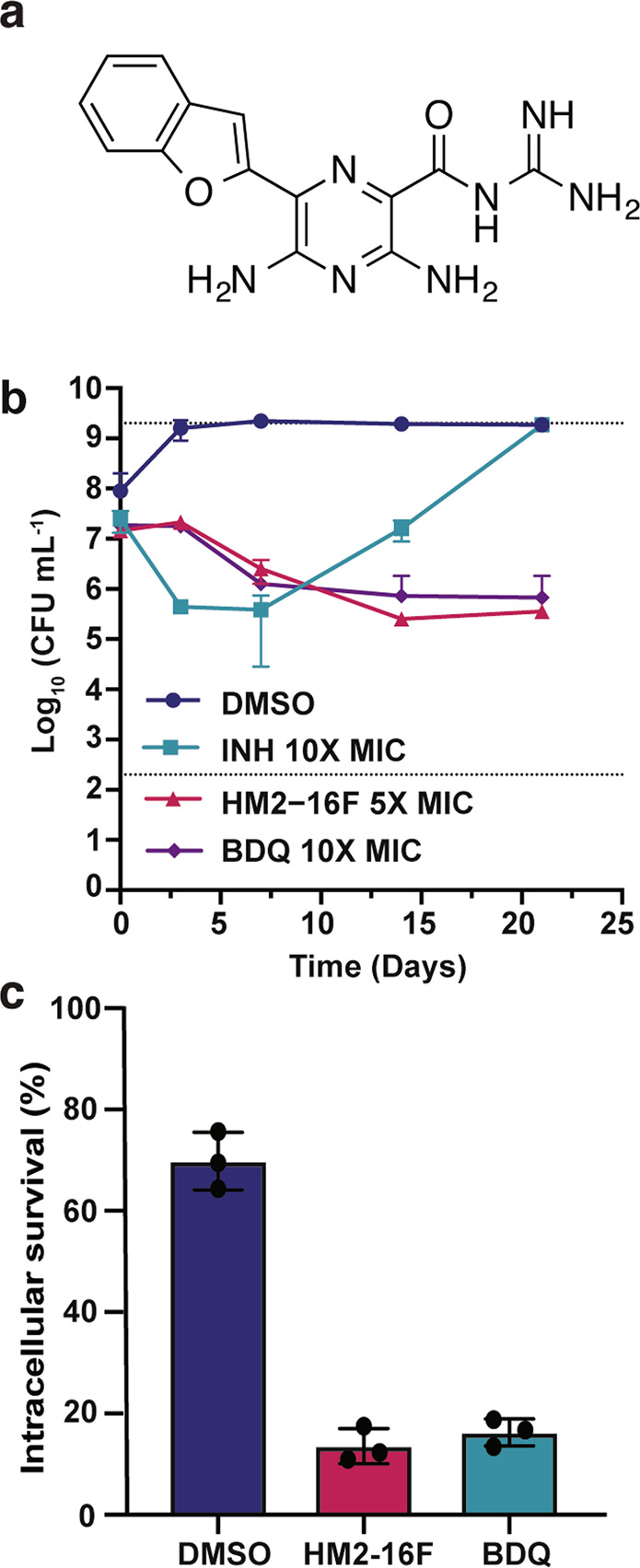
a Structure of HM2-16F. b Survival of M. tuberculosis after treatment with: isoniazid (INH 2 μM, 10× MIC), HM2-16F (20 μM, 5× MIC) and bedaquiline (BDQ 2 μM, 10X MIC). c Survival of M. bovis BCG in human THP-1 macrophages after treatment with 5 μM HM2-16F and BDQ. Error bars indicate standard deviation from three biological independent experiments.
Interaction of HM2-16F with the F1Fo-ATP synthase
To investigate whether HM2-16F targets the mycobacterial ATP synthase at the whole-cell level, we used CRISPR interference (CRISPRi) to generate a transcriptional knockdown of the a-subunit of the F1Fo-ATP synthase (atpB gene) in M. tuberculosis mc26230. This atpB knockdown was previously shown to reduce transcription by ~80 fold and increase sensitivity to BDQ, demonstrating its utility for validating inhibitors of the ATP synthase28. Addition of increasing concentrations of ATc, which induces CRISPRi activation and atpB transcriptional repression28, reduced the MIC of HM2-16F by ~7 fold (MIC = 34 and 4 μM with 0 and 100 ng/ml ATc respectively), while a non-targeting single-guide RNA (sgRNA) did not affect the MIC (Figs. 3a, b and Supplementary Figs. 2 and 3). Viability assays showed a similar trend for killing of cells in an equivalent experiment (Fig. 3c). This suggests that the transcriptional repression of the mycobacterial F1Fo-ATP synthase reduced the concentration of HM2-16F required to inhibit M. tuberculosis. HM2-16F (10 μM) was only able to weakly outcompete the c-ring inhibitor DCCD for binding at the M. phlei c-ring (Fig. 3d), compared to the potent inhibition displayed by BDQ (10 μM, 50X MIC), suggesting that HM2-16F results in only modest binding/inhibition of the F1Fo-ATP synthase.
Fig. 3. Interactions of HM2-16F with the mycobacterial F1Fo-ATP synthase.

a Knockdown of the ATP synthase operon (sgRNA targeting atpB – pCi74) was induced with the indicated amounts of ATc and the growth relative to the vehicle control was determined. Error bars represent standard deviation from three biological independent experiments. b Growth after 10 days and IC50 of HM2-16F as a function of F1Fo-ATP synthase knockdown (sgRNA targeting atpB – pCi74). Error bars represent 95% confidence interval. c Viability of M. tuberculosis after 10 days was determined in an analogous experiment to panel a at 0, 10 and 100 ng mL−1. d Time course of DCCD labelling in purified M. phlei c-rings in the presence of the indicated amounts of compounds. Data are representative of duplicate measurements. e HM2-16F (purple carbons) docked into the BDQ binding site of mycobacterial F1Fo-ATP synthase c-ring (PDB ID: 4V1F). The high-resolution c-ring-BDQ structure is shown in grey as a reference. The protein is shown as an electrostatic potential surface (red – electronegative, white – neutral, blue – electropositive; generated in PyMOL). Amino acid positions refer to the M. phlei protein. The top three ranked poses are shown in Supplementary Figure 5.
The structure of the M. phlei c-ring bound to BDQ revealed that BDQ mimics the a-subunit arginine, which temporarily interacts with the c-ring Glu65 carboxylate side chain during the ion translocation cycle8. Although BDQ and HM2-16F both have arginine mimetic groups, HM2-16F is otherwise structurally dissimilar. We, therefore, sought to understand the mechanism of HM2-16F and its dissimilarity to BDQ. Firstly, we attempted to isolate HM2-16F-resistant mutants in M. tuberculosis mc26230. While mutants were able to be isolated at a frequency of 1.45 × 10−6, these did not show mutations in the F1Fo-ATP synthase. Instead, we identified 4 separate mutations in the transcription factor Rv3066 (R38S, G134fs, G157R and E173*), which regulates the Mmr efflux pump, with these mutants showing ~4-fold increases in MBC to HM2-16F (Supplementary Figure 4). Notably, Mmr is not reported to contribute to efflux-mediated BDQ resistance, which is typically ascribed to the MmpL5 pump (regulated by Rv0678)29. Furthermore, HM2-16F cross-resistance was not observed in a BDQ-resistant mutant carrying a mutation in the ATP synthase c-ring subunit (AtpE(A63P); Fig. 4 and Table 2) or in the efflux pump regulator Rv0678 (Rv0678(G65fs)), which is cross-resistant to clofazimine29. Taken together, these results suggests that HM2-16F has only a weak interaction with the F1Fo-ATP synthase that may not necessarily drive its primary mode of action. To understand the basis of this difference, we performed molecular docking simulations with HM2-16F against the recently solved M. phlei F1Fo-ATP synthase8. The acidic glutamate residue (E65; M. phlei numbering) was used as the centre of the binding site as well as a hydrogen-bonding constraint on the assumption that the acylguanidine of HM2-16F mimics the binding of the basic dimethylamine in BDQ. The docked structure (Fig. 3e and Supplementary Fig. 5) suggests that HM2-16F binds to the F1Fo-ATP synthase in a very different way to BDQ (e.g., additional interactions with F58; M. phlei numbering) that may reduce its affinity for this binding site. The putative binding pose suggests that mutations affecting BDQ binding might be tolerated by HM2-16F, consistent with our mutational data (Fig. 4).
Fig. 4. Bedaquiline-resistant mutants of M. tuberculosis are not cross-resistant to HM2-16F.

The M. tuberculosis strains mc26206 (WT), AtpE(A63P), and Rv0678(G65fs) were grown in the presence of increasing concentrations of: a BDQ, b HM2-16F or c clofazimine and growth measured after 10 days. Error bars represent standard deviation from four biological independent experiments.
Table 2.
Minimum inhibitory concentrations (MIC, μM) of selected inhibitors against wild-type and mutant M. tuberculosis mc26206 strains.
| Compound | WT | AtpE(A63P) | Rv0678(G65fs) |
|---|---|---|---|
| BDQ | 0.3 | 8 | 4 |
| HM2-16F | 9 | 5 | 12 |
| Clofazimine | 0.9 | 0.5 | 3 |
All MIC values are reported in micromolar final concentration. All strains are listed in Supplementary Table 1. WT = wild-type M. tuberculosis mc26206.
Metabolite profiles of M. tuberculosis treated with HM2-16F
To identify alternative targets of HM2-16F, we challenged M. tuberculosis H37Rv with varying concentrations of HM2-16F (1–10× MIC) and identified changes in intracellular metabolites by LC/MS-MS (Supplementary Fig. 6). We considered two different scenarios for biological significance: (1) sub-saturated changes in metabolites, where changes are in a dose-dependent manner30 and (2) saturated changes, where the three measurements showed a consistent fold-change in metabolites regardless of HM2-16F concentration.
HM2-16F caused a saturating or sub-saturating change in 37 metabolites (Fig. 5a, b and Supplementary Data 1). Of the sub-saturated effects, 11 metabolites decreased in a dose-dependent manner, while 5 were increased (Fig. 5a, c and Supplementary Data 1). For saturated effects, HM2-16F caused a consistent decrease in 16 metabolites, while 5 metabolites increased (Fig. 5b, c and Supplementary Data 1). HM2-16F affected the total nucleotide pool in M. tuberculosis: ATP, ADP, AMP, and adenine concentrations were all decreased (Fig. 5c, d), while CTP and uridine showed a saturated increase (Fig. 5c). Guanine concentrations increased in a dose-dependent manner, while TMP decreased (Fig. 5c). These changes suggest an inability to regenerate ATP and impaired biosynthesis of nucleoside pools.
Fig. 5. Metabolomic profiling of HM2-16F-treated cultures of M. tuberculosis indicates HM2-16F inhibits respiration and induces cellular reductive stress.

a Global profile of metabolites with dose-dependent changes, as assessed by log-linear regressions. b Global profile of metabolites with dose-independent changes, as assessed by two sample t-tests with Benjamini & Hochberg False Discovery Rate (FDR) adjustment. c Fold changes of selected metabolites, classified according to whether the metabolite changed in a dose-dependent (increasing/decreasing) or dose-independent manner. d Changes in adenine nucleotide/nucleoside pools. e Relative changes in the indicated metabolites. Raw data are available in Supplementary Data 1.
Additionally, HM2-16F caused a reduction of carbon entry into the TCA cycle. Acetyl-CoA was consistently increased at each concentration of HM2-16F (Fig. 5c, e), phosphoenolpyruvate (PEP) was initially increased, but the response declined with increasing concentrations of HM2-16F (Fig. 5c, e). Collectively, these data indicate that flux through the oxidative decarboxylation steps of the TCA cycle was reduced. In support of this, γ-aminobutyric acid (GABA) increased in a dose-dependent manner (Fig. 5c, e), suggesting that there was lower flux in the GABA shunt, a major pathway for bypassing steps in the TCA cycle in M. tuberculosis31. Together with the depletion of ATP:ADP, this suggests that F1Fo-ATP synthase inhibition, mediated by HM2-16F, has resulted in either consequent or concomitant inhibition of the electron transport chain and cellular reductive stress that feedback onto energy cofactors, reducing equivalents and alternative central metabolic pathways.
Cytochrome bd inhibition enables HM2-16F to sterilize M. tuberculosis cultures in combination with Q203
As the metabolomic profiling suggested induction of reductive stress, we next examined the potential of HM2-16F to synergize with inhibitors of other protein complexes in the M. tuberculosis respiratory pathway. Clear evidence of HM2-16F synergy was observed with the quinol:cytochrome c oxidoreductase inhibitor Q203 (Fig. 6). Treatment of M. tuberculosis mc26230 with 2.5-fold MIC of HM2-16F effectively sterilized cultures in the presence of 10-fold MIC of Q203 (Fig. 6a, limit of detection of 100 CFU mL−1). The combination is more potent than Q203 alone (Fig. 6a). In contrast, BDQ is not synergistic with Q20323, suggesting that HM2-16F interacts with other cellular targets that synergize with inhibition of the cytochrome bcc-aa3 oxidase.
Fig. 6. HM2-16F synergizes with Q203 by inhibiting cytochrome bd oxidase.

a Cell killing (viability) of M. tuberculosis with increasing amounts of both Q203 and HM2-16F after 14 days challenge with both compounds. Error bars indicate standard deviation from three independent experiments. b Inhibition of the M. tuberculosis cytochrome bd oxidase in IMVs from M. smegmatis mc2155 ΔcydAB with either pYUB28b (empty vector) or pLHcyd- MtbCydABDC+. IMVs were pre-inhibited with 500 nM TB47 for 1 min as indicated. Oxygen consumption was initiated with 5 mM malate (final concentration) as the sole electron donor. The indicated treatment: 0.2× MIC aurachin D (AuD) (top panel 6b), 10× MIC HM2-16F (middle panel b) and 500× MIC BDQ (bottom panel b) was added once malate energized oxygen consumption reached steady state (~2 min after malate addition). Error bars represent standard deviation from triplicate measurements. Asterisks indicate significance compared to the corresponding malate energized OCR *** = p < 0.001, ** = p < 0.01, ns = p > 0.05 (One-way ANOVA, Tukey multiple comparisons, 95% CI). c Titration of the indicated compounds in malate-energized IMVs of M. smegmatis mc2155 ΔcydAB pLHcyd- MtbCydABDC+ pre-inhibited with 500 nM TB47 (final concentration). Error bars indicate standard error from four technical replicates.
Recently, we demonstrated that the cytochrome bd inhibitor ND-011992 potently synergizes with Q203 to induce bactericidal killing22. We, therefore, hypothesized that HM2-16F might also inhibit cytochrome bd oxidase. We assessed the ability of HM2-16F to inhibit M. tuberculosis cytochrome bd (CydABDC+) heterologously expressed in a markerless M. smegmatis cytochrome bd mutant (Δcyd pLHcyd-MtbCydABDC+) (Supplementary Table 1)32. IMVs prepared from M. smegmatis ∆cydAB harbouring an empty vector control (pYUB28b) exhibited high rates of oxygen consumption when energized with malate (Fig. 6b). When M. smegmatis ∆cydAB was complemented with the cydABDC operon from M. tuberculosis (MtbCydABDC+) the OCR increased significantly suggesting the cytochrome bd oxidase from M. tuberculosis was functional in M. smegmatis (Fig. 6b). When HM2-16F (10× MIC) was added to malate-energized IMVs of M. smegmatis ∆cydAB-pYUB28b no significant effect was observed on the OCR (Fig. 6b, top panel). However, addition of HM2-16F to M. smegmatis ∆cydAB-MtbCydABDC+ caused significant inhibition of the OCR suggesting this inhibition was dependent on the presence of cytochrome bd. When IMVs were preincubated with the potent cytochrome bcc-aa3 oxidase inhibitor TB4730, the OCR in the M. smegmatis ∆cydAB-pYUB28b mutant was completely inhibited demonstrating that oxygen consumption in this genetic background was entirely mediated by cytochrome bcc-aa3 oxidase. Oxygen consumption was restored in M. smegmatis ∆cydAB-MtbCydABDC+ that was TB47-insensitive as this was mediated by cytochrome bd, and HM2-16F addition inhibited this OCR (Fig. 6b, top panel). The combination of TB47 with HM2-16F was effective in shutting down the OCR of M. smegmatis ∆cydAB-MtbCydABDC+. The same experiments were performed with the specific ATP synthase inhibitor bedaquiline (Fig. 6b, middle panel) and the known cytochrome bd oxidase inhibitor, aurachin D32 (Fig. 6b, bottom panel). In contrast to HM2-16F that exhibited a remarkable specificity for cytochrome bd, bedaquiline and aurachin D showed inhibition of OCR in the M. smegmatis ∆cydAB-pYUB28b genetic background. This inhibition of OCR was observed throughout the other treatments suggesting both inhibitors impacted negatively on the OCR in a cytochrome bd-independent manner (Fig. 6b, middle and bottom panel).
HM2-16F was found to inhibit cytochrome bd activity with an IC50 of 21.2 μM (Supplementary Table 2; Fig. 6c), close to its MIC and comparable to the concentrations used in synergy experiments (Fig. 6A). BDQ was also able to inhibit cytochrome bd (Fig. 6c and Supplementary Table 2), however, the concentrations required were orders of magnitude higher than its MIC and not as specific towards cytochrome bd as HM2-16F (Fig. 6b). The effects of BDQ are likely not relevant to its bacteriostatic or bactericidal action. These findings support that HM2-16F synergizes with Q203 through direct cytochrome bd inhibition.
Discussion
The F1Fo-ATP synthase of M. tuberculosis is a validated drug target, as demonstrated by the clinical approval of BDQ and of its utility in treating drug-resistant tuberculosis infection. Nevertheless, concerns about BDQ’s safety6 and the isolation of resistant mutants12,13 highlight the need for next-generation inhibitors. Amilorides are a class originally identified in 1967 as potassium-sparing diuretics that act on ENaCs in the kidney26. Numerous studies since then have shown that amilorides have a multitude of other activities resulting from their arginine-mimetic acylguanidine moiety27,33–36. We postulated that the acylguanidine of amilorides could make them F1Fo-ATP synthase inhibitors since it is known that the dimethylamino group of BDQ functions as an arginine mimetic in its interaction with F1Fo-ATP synthase c-ring7. In this study, we found that the 6-substituted amiloride derivative HM2-16F functions weakly as an M. tuberculosis F1Fo-ATP synthase inhibitor, but more potently functions as an inhibitor of the M. tuberculosis cytochrome bd oxidase. With no diuretic activity and better M. tuberculosis inhibitory activity than amiloride, we propose that 6-substituted amiloride derivatives like HM2-16F present a promising scaffold for developing alternative drugs to BDQ with different resistance profiles.
Arginine mimetics are present in a broad range of compounds. Not all arginine mimics could be F1Fo-ATP synthase inhibitors as this would be inconsistent with the favourable selectivity indices that have been obtained for BDQ7, which itself has a lower structural resemblance to the arginine side chain than amilorides or other aryl/alkylguanidines. Instead, and in line with the BDQ-bound structure of the M. phlei c-ring8, we propose that steric hindrance at the membrane-embedded a–c subunit interface selects for only certain guanidine-mimetic chemotypes. Along these lines, amiloride derivatives targeting the M. tuberculosis F1Fo-ATP synthase may possess a similar range of selectivity. In support of this, AtpEA63P mutants were not cross-resistant to HM2-16F and this likely indicates that HM2-16F does not utilize the Asp32 water-mediated bonding network to efficiently bind to the c-ring, unlike BDQ8.
The effect of HM2-16F on total cellular metabolites showed many parallels to previous metabolomic and transcriptomic profiling of BDQ37. For example, GABA was found to increase in abundance following BDQ treatment on a timescale comparable to our metabolomic profiling37. Additionally, one of the M. smegmatis pyruvate dehydrogenases (MSMEG_4712) is upregulated 12-fold in response to BDQ17, consistent with our observation of increased abundance of acetyl-CoA following HM2-16F treatment. Finally, proteomic analysis of BDQ-treated M. tuberculosis found a time-dependent increase in abundance of PEP carboxykinase (Rv021115), consistent with the increased abundance of PEP seen with HM2-16F. Taken together, these data suggest that the effects of HM2-16F and BDQ are comparable at a metabolic level. In conjunction with our CRISPRi interference data of the atp operon knockdown, this may suggest that HM2-16F has an ability to indirectly affect the F1Fo-ATP synthase or directly targets a different molecular site of the F1Fo-ATP synthase. More studies are required to understand the direct or indirect interaction of HM2-16F with the mycobacterial F1Fo-ATP synthase.
The potent synergy seen with HM2-16F and Q203 serves as a major point of difference to BDQ. BDQ can function as an ionophore17,18,38 and is not synergistic with Q203 in vitro23. However, HM2-16F displayed strong inhibition of the cytochrome bd oxidase that allowed it to affect synergistic killing with TB47, a validated inhibitor of the cytochrome bcc-aa3 oxidase30. Overall, we propose a model (Fig. 7) where HM2-16F inhibits cytochrome bd and, to a lesser or indirect extent, the F1Fo-ATP synthase. Disruption of quinol regeneration can affect the primary dehydrogenases of the respiratory chain, leading to dysregulated regeneration of reducing equivalents that can lead to a systemic failure of cellular redox reactions. The addition of TB47 (or Q203 – Telacebec) removes the only alternative pathway that mycobacteria can use to escape this inhibition of oxygen consumption leading to cell death (Fig. 7). HM2-16F could create alternative treatment options for patients unable to take BDQ, such as those taking other drugs that prolong the QT interval39. Overall, we propose that amilorides are a promising class for developing an alternative to BDQ where toxicity or resistance profiles might preclude BDQ’s usage. In this regard, the amiloride scaffold appeals as an ideal starting point for selective optimization of side activity programs aimed at producing new respiratory inhibitors and antimycobacterial drugs.
Fig. 7. Summary of the action of HM2-16F at the level of the respiratory chain.

HM2-16F inhibits cytochrome bd and, to a lesser or indirect extent, the F1Fo-ATP synthase. Disruption of quinol regeneration can affect the primary dehydrogenases of the respiratory chain leading to dysregulated regeneration of reducing equivalents that can lead to a systemic failure of cellular redox reactions. The addition of Q203 (or TB47), direct inhibitors of the cytochrome bcc-aa3 oxidase branch, removes the only alternative pathway that mycobacteria can use to escape this inhibition of oxygen consumption leading to cell death.
Methods
Bacterial strains, media, and growth conditions
Bacterial strains used in this study are listed in Supplementary Table 1. For M. smegmatis mc2155 growth, cells were grown in modified Hartman’s de Bont medium40, containing 0.2% glycerol as the sole carbon and energy source. Tween80 was omitted for determination of the MIC. For M. tuberculosis strains mc2623041 and mc2620642 growth (Supplementary Table 1), cells were grown in Middlebrook 7H9 broth supplemented with OADC (0.005% oleic acid, 0.5% bovine serum albumin, 0.2% dextrose, 0.085% catalase), 0.05% tyloxapol (Sigma) and 25 μg/mL pantothenic acid. The growth of mc26206 was also supplemented with leucine (50 µg/mL). Unless otherwise specified, the following growth procedures were followed: M. smegmatis was grown in 125 mL conical flasks, containing 25 mL media, and M. tuberculosis was grown in 30 mL inkwells, containing 10 mL media. M. smegmatis and M. tuberculosis were grown with agitation at 200 rpm and 140 rpm respectively. All cultures were maintained at 37 °C. To obtain hypoxic cultures of M. tuberculosis, 30 mL of complete 7H9 media was inoculated at an optical density (OD600) of 0.05 in 100 mL serum vials (Wheaton) that were stopped and capped40. Cultures were monitored until methylene blue (1.5 μg/mL) control indicator decolorized to signify cells has reached hypoxia (~7–10 Days). Once cultures became hypoxic, cells were challenged with BDQ and HM2-16F at 10× MIC and 5× MIC respectively with the inclusion of a DMSO only control, all experiment were done in biological triplicate. Colony counts (cfu/ml) to determine cell viability were taken on days 0, 3, 7, 14, 21, and 28 after compound challenge and plated on complete 7H11 agar.
Inhibition and killing assays
MIC values were performed by serial dilution in 96-well plates in growth medium. Unless otherwise specified, MIC is defined as the lowest concentration to inhibit all observable growth. Log phase cultures were used to inoculate each dilution to a final OD600 of 0.005. Cultures were incubated at 37 °C, 200 rpm. Unless otherwise specified, the MIC was determined from the visual presence or absence of growth after 2 days (M. smegmatis), 7 days (M. tuberculosis H37Rv), 10 days (M. tuberculosis mc26206) or 1 day (all other strains) (Supplementary Table 1). Controls containing no compound or compound dissolution solvent (DMSO) alone were included in all experiments.
Infection of differentiated human THP-1 macrophages and drug susceptibility assays ex vivo
The human monocytic cell line THP-1 was cultured in standard RPMI 1640 macrophage medium supplemented with 10% inactivated fetal bovine serum and 1 mM sodium pyruvate at 37 °C with 5% CO243. THP-1 monocytes (5 × 105 cells/well) were differentiated overnight using 20 ng/mL phorbol myristate acetate (PMA) and seeded in a 96 well-plate. The next day, differentiated macrophages were infected with a mid-logarithmic phase culture of Mycobacterium bovis BCG (Pasteur 1173P2)44 (OD 0.4–0.8) at a multiplicity of infection of 10:1 (10 bacteria/1 cell). Infection was allowed to proceed for 5 h. Cells were then washed 4 times with pre-warmed complete RPMI to remove extracellular bacilli. RPMI media containing compounds at varying concentrations was added to the infected cells and incubated for 1–3 days at 37 °C with 5% CO2. At various times, the infected cells were lysed in distilled water containing 1% Tween80 for 5 min at room temperature to determine the number of CFU/mL on Middlebrook 7H11 + OADC agar. All cells used as inocula were washed in saline (0.85% NaCl). To rule out adverse effects on THP-1 cell physiology leading to indirect effects on M. bovis BCG replication in these cells, Alamar Blue assays45 were performed to determine if HM2-16F was toxic to THP-1 cells. THP-1 cells were grown in 96-well plates and treated with compounds at a range of concentrations (0.05–500 μM) for 48 h. The percentage of cell viability was determined by subtracting values for non-treated infected cells from the non-infected cells.
Isolation of bedaquiline and HM2-16F resistant mutants
M. tuberculosis mc26230 resistant mutants against HM2-16F and mc26206 resistant mutants against BDQ were isolated as follows46. Cultures were grown for approximately 2 weeks, in Middlebrook 7H9 + OADC medium, to an OD600 of approx. 0.4–1 (i.e., ~1 × 108 CFU/mL). Cells (100 µL) at approximate densities of 1 × 108, 1 × 107, 1 × 106 and 1 × 105 CFU/mL were plated on 7H11 + OADC agar containing either 20 µM HM2-16F (5x the solid media MIC) or 1, 3, or 11 µM BDQ (3, 9, and 30× MIC). Plates were incubated at 37 °C until colonies appeared, approximately 4 weeks.
Whole Genome sequencing and single nucleotide polymorphism identification
Genomic DNA was extracted from M. tuberculosis using the MoBio UltraClean Microbial DNA isolation Kit. Ten mL of late log phase culture was harvested, resuspended in 300 µL MoBio bead binding buffer, and heat-inactivated for 15 min at 95 °C. Manufacturer’s guidelines were followed to extract genomic DNA from the heat-inactivated culture. Whole-genome sequencing (Illumina 150 bp paired-end reads) was performed at Otago Genomics and reads were mapped to the wild type H37RV genome using Geneious.
Antimicrobial susceptibility of atpB transcriptional knockdown strains
Sensitivity of M. tuberculosis (mc26230) strains expressing an sgRNA targeting atpB28 or non-targeting control47 to HM2-16F were determined. Briefly, cultures were inoculated into 96-well plates containing supplemented Middlebrook 7H9 broth (OADC + PAN + KAN), at a starting OD600 of 0.005, in a total starting volume of 100 µL. Anhydrotetracycline (ATc) at 0, 10, or 100 ng/mL was added to alternating rows, and HM2-16F was dispensed from a 9-point, three-fold dilution gradient to each well, with a maximum of 2% DMSO present. Ninety-six-well plates were incubated without shaking at 37 °C for 10 days. On day 10, OD600 values were measured in a Varioskan LUX microplate reader, and minimum inhibitory concentration (MIC) values were determined. To determine the effects on bacterial viability, culture was removed from desired wells on day 10 and diluted along a four-point, ten-fold dilution curve. Five µL was spotted onto 7H11 media, incubated at 37 °C and colony-forming units were determined after four weeks.
MALDI-MS-based competition studies of ATP synthase inhibitors with N,N’-dicyclohexylcarbodiimide (DCCD)
Purified M. phlei c-ring in 0.6% lauryldimethylamine oxide (LDAO) (Sigma)8 was used to assay the competition of dicyclohexylcarbodiimide (DCCD) with BDQ or HM2-16F. The concentrated sample (6.5 mg/mL) was diluted to 0.1 mg/mL using 20 mM cacodylate/trimethylamine/NH3 (pH 7.5). Compounds, solubilized in DMSO, were added to the indicated final concentrations. Samples were incubated for 1 h at room temperature before adding 25 mM DCCD. Aliquots were removed at several time points (0–45 min), directly mixed in a 1:1 ratio with 2′,4′,-dihydroxyacetophenone matrix, and applied onto a ground steel MALDI target in duplicates. MALDI mass spectra were acquired in the mass range of 5–20 kDa on a Bruker Autoflex III Smartbeam MALDI-TOF mass spectrometer using optimized ionization, ion optics, and detector settings. Spectra were recalibrated using the near-neighbour method with a calibrant mixture (Bruker Protein Calibration standard 1, Bruker Daltonics, Bremen). All spectra were evaluated and recalibrated using the software Bruker FlexAnalysis 3.3 (Build 75). After background substraction (TopHat), smoothing (Savitzky-Golay, width 1 m/z, 3 cycles) and peak picking (Centroid, s/n 5) the intensities of the c-monomer and the DCCD-labelled species were used for calculating the efficiency of DCCD binding. The “labelling efficiency” was calculated as the intensity ratio of the DCCD-bound species to the sum of the labelled and unlabelled species. All values were calculated from 3 to 4 experimental replications and technical MALDI measurement duplicates.
Molecular docking
The 3D structure of HM2-16F was created and minimized using Avogadro (v1.2.0)48. Docking was carried out using GOLD v5.7.2. Compounds were docked into the BDQ binding site of the ATP synthase c-ring structure (PDB ID: 4V1F,8) with Glu65 used as the centre of the binding site as well as a protein hydrogen bonding constraint. Nine poses were produced and 3 fell within 1.5 Å RMSD. Poses that failed to interact with E65 were excluded.
Liquid chromatography-mass spectrometry (LC-MS) metabolomics
Filter-cultured M. tuberculosis strain H37Rv was first grown for 5 days in 7H10 agar media to expand biomass, and then moved to fresh 7H10 medium containing compounds or a vehicle control (DMSO) for a 24 h exposure30. M. tuberculosis metabolism was quenched by plunging M. tuberculosis–laden filters into extraction buffer (acetonitrile: methanol: H2O = 40:40:20), which was precooled to −40 °C on dry ice49. M. tuberculosis metabolites were then extracted by mechanical lysis with zirconia beads in Precellys tissue homogenizer under continuous cooling at or below 2 °C. Extracted M. tuberculosis metabolites were analyzed by high performance liquid chromatography-coupled mass spectrometry using an Agilent 1290 HPLC and Accurate Mass 6220 TOF or 6520 qTOF mass spectrometer50. M. tuberculosis metabolites were identified based on curated accurate mass-retention time identifiers, and quantified using Agilent Quantitative Analysis software and Agilent Profinder software with a mass tolerance of < 0.005 Da.
Oxygen consumption in inverted membranes vesicles (IMVs)
IMVs of the M. smegmatis mc2155 Δcyd pLHcyd (overproducing cytochrome bd oxidase from M. tuberculosis), or the corresponding empty vector control (Δcyd pYUB28b) (Supplementary Table 1) were prepared as follows. A single colony of the production strain (Δcyd pLHcyd or Δcyd pYUB28b) was used to inoculate LBT media containing 50 µg/ml hygromycin B. This culture was grown for 72 h at 37 °C and 200 rpm to achieve maximum cytochrome bd oxidase production and cells were harvested by centrifugation at 4000 × g for 20 min at 4 °C. Cells were resuspended in buffer containing 50 mM Tris-HCl (pH 7.4), 5 mM magnesium chloride, 0.05% Tween 80 and 0.1 mg/ml Pefabloc SC (5 ml buffer/1 g cells). Cells were disrupted using a high-pressure homogenizer (Avestin Emulsiflex C3) via several passages (4–6) at 22,000 psi. The lysate was centrifuged at 10,000 × g for 20 min to remove cell debris and unlysed cells. The supernatant of the low-speed centrifugation step was subsequently centrifuged at 225,000 × g for 90 min. Pelleted IMVs were resuspended in buffer (50 mM Tris, 100 mM KCl, 5 mM MgCl2, pH 7.5) and 5 mM malate was used to initiate oxygen consumption that was measured using an Oroboros O2k fluorespirometer. For titrations, 500 nM TB47 (an inhibitor of cytochrome bcc:aa3 oxidase complex30) was added prior to the experiment. IMVs were used at a protein concentration 12.5 μg mL−1. Stepwise titrations were performed using the Oroboros O2k TIP2k automatic injection micropump, with rates measured at 120 s intervals between each injection. Data are normalized to protein concentrations that were estimated by BCA assay (Thermo), using a BSA standard.
ATP synthesis in inverted membrane vesicles
ATP synthesis in IMVs was carried out at 37 °C in 1 ml of 50 mM Tris-HCl (pH 8.0) buffer containing 5 mM MgCl2 and 100 mM KCl with constant stirring. Approximately 0.1 mg mL−1 of IMVs were incubated with stirring at 37 °C for 2 min, followed by incubation in the presence of 1 mM NADH for 2 min. When performing inhibition experiments, test inhibitor was added 5 min prior to the addition of NADH. ATP synthesis was initiated with the concurrent addition of 0.75 mM ADP and 2.5 mM potassium phosphate (pH 8.0). At various time intervals, 100 µl aliquots were removed and transferred to 400 µl of stop solution (1% trichloroacetic acid, 2 mM EDTA plus 200 µM CCCP). Each sample was diluted 500-fold in water prior to the measurement of ATP. The amount of ATP was determined by the luciferin-luciferase assay as follows. Each sample was diluted into 400 µl of Tris acetate buffer (50 mM Tris acetate, pH 7.8, 2 mM EDTA, 50 mM MgCl2) in a luminometer tube. Luciferin-luciferase reagent (50 µL, Sigma) was added to the tube, and the fluorescence monitored with a chemiluminometer (FB 12 luminometer; Berthold)51. The amount of ATP synthesized was calculated from a standard curve performed on the day of each set of ATP measurements. For each individual experimental set, the presence of background ATP was measured using non-energized vesicles (no NADHJ) and subtracted from total ATP measured.
Statistics and reproducibility
All statistical analysis was performed in R version 4.0.2. IC50 values and 95% confidence intervals were determined by fitting 4-parameter logistic regressions using the package nplr. Pairwise comparison and multiple comparison corrections were performed using estimated marginal means in the package emmeans. For metabolomics analysis, linear regressions were modelled to the log2–log10 transformed data (i.e., log2(Fold Change) vs log10([Drug])) for each metabolite. The p-value derived from the F-statistic was used to evaluate if the two variables were linearly related. Otherwise, t-test comparisons with Benjamini & Hochberg False Discovery Rate adjustments were performed.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this article.
Supplementary information
Description of additional Supplementary Files (new)
Acknowledgements
C.-Y.C., G.C., M.M., and M.J.K. were funded by the Health Research Council of New Zealand that primarily supported this work. K.H. was funded by the Royal Society of New Zealand Marsden Fund. T.M. was supported by the Wellcome Trust [WT110068/Z/15/Z]. The authors have no conflict of interest to declare.
Author contributions
C.-Y.C. performed MIC testing against standard strains (Table 1) and viability experiments (Figs. 2b, 5a). N.W. performed MIC testing against resistant strains (Table 2 and Fig. 4). N.W. and M.M. performed knockdown experiments (Fig. 3a). K.R. performed metabolomic experiments, with analysis performed by KH (Fig. 5). A.M. and C.-Y.C. performed macrophage experiments (Fig. 2c), with assistance from E.H. and M.B. L.P., J.L., and T.M. performed DCCD labelling (Figs. 1d, 3b). K.H. and Z.S.T. performed ATP synthesis assays (Fig. 1c). J.T. and T.M. performed docking analysis (Fig. 3c). K.H. and L.K. performed cytochrome bd oxidase assays (Fig. 6b, c), using sampled prepared by H.O.-R. and K.K. L.H. cloned the Cyd expression construct. B.J.B., R.B., and M.J.K. synthesized HM2-16F and advised on experiments. G.M.C. and M.J.K. conceived this project and obtained the primary funding from the HRC for this work. C.-Y.C., C.A., L.K., K.H., E.H., M.B., B.J.B., M.J.K., and G.C. contributed to manuscript direction. K.H. drafted the manuscript, analyzed data and formulated conclusions with co-authors. All authors contributed to manuscript revisions.
Peer review
Peer review information
Communications Biology thanks Yusuke Minato, Martin (I) Voskuil, and Lev Ostrer for their contribution to the peer review of this work. Primary Handling Editor: George Inglis. Peer reviewer reports are available.
Data availability
Raw metabolomic data are presented in Supplementary Data 1. Source data underlying main figures are presented in Supplementary Data 2. Sequencing data are available from NCBI BioProject under the accession numbers PRJNA802611 and PRJNA802880. All other data are available from the corresponding author upon reasonable request.
Competing interests
The authors declare no competing interests.
Footnotes
Publisher’s note Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
These authors contributed equally: Kiel Hards, Chen-Yi Cheung.
Contributor Information
Michael J. Kelso, Email: mkelso@uow.edu.au
Gregory M. Cook, Email: Greg.cook@otago.ac.nz
Supplementary information
The online version contains supplementary material available at 10.1038/s42003-022-03110-8.
References
- 1.World Health Organization. Global Tuberculosis Report 2019 (Geneva: World Health Organization, 2019).
- 2.Dheda K, et al. Global control of tuberculosis: from extensively drug-resistant to untreatable tuberculosis. Lancet Respir. Med. 2014;2:321–338. doi: 10.1016/S2213-2600(14)70031-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.World Health Organization. Guidelines for the Treatment of Tuberculosis: Fourth Edition (World Health Organization, 2010).
- 4.World Health Organization. Guidelines for the Programmatic Management of Drug-Resistant Tuberculosis: Emergency Update 2008 (Geneva: World Health Organization, 2008).
- 5.Jones D. Tuberculosis success. Nat. Rev. Drug Discov. 2013;12:175–176. doi: 10.1038/nrd3957. [DOI] [PubMed] [Google Scholar]
- 6.Cohen J. Approval of novel TB drug celebrated with restraint. Science. 2013;339:130–130. doi: 10.1126/science.339.6116.130. [DOI] [PubMed] [Google Scholar]
- 7.Andries K, et al. A diarylquinoline drug active on the ATP synthase of Mycobacterium tuberculosis. Science. 2005;307:223–227. doi: 10.1126/science.1106753. [DOI] [PubMed] [Google Scholar]
- 8.Preiss L, et al. Structure of the mycobacterial ATP synthase Fo rotor ring in complex with the anti-TB drug bedaquiline. Sci. Adv. 2015;1:1–8. doi: 10.1126/sciadv.1500106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hards K, Cook GM. Targeting bacterial energetics to produce new antimicrobials. Drug Resist. Updat. 2018;36:1–12. doi: 10.1016/j.drup.2017.11.001. [DOI] [PubMed] [Google Scholar]
- 10.Diacon AH, et al. Randomized pilot trial of eight weeks of bedaquiline (TMC207) treatment for multidrug-resistant tuberculosis: long-term outcome, tolerability, and effect on emergence of drug resistance. Antimicrob. Agents Chemother. 2012;56:3271–3276. doi: 10.1128/AAC.06126-11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Diacon AH, et al. The diarylquinoline TMC207 for multidrug-resistant tuberculosis. N. Eng. J. Med. 2009;360:2397–2405. doi: 10.1056/NEJMoa0808427. [DOI] [PubMed] [Google Scholar]
- 12.Andries K, et al. Acquired resistance of Mycobacterium tuberculosis to bedaquiline. PloS One. 2014;9:e102135. doi: 10.1371/journal.pone.0102135. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Villellas C, et al. Unexpected high prevalence of resistance-associated Rv0678 variants in MDR-TB patients without documented prior use of clofazimine or bedaquiline. J. Antimicrob. Chemother. 2017;72:684–690. doi: 10.1093/jac/dkw502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Guo H, et al. Structure of mycobacterial ATP synthase bound to the tuberculosis drug bedaquiline. Nature. 2021;589:143–147. doi: 10.1038/s41586-020-3004-3. [DOI] [PubMed] [Google Scholar]
- 15.Koul A, et al. Delayed bactericidal response of Mycobacterium tuberculosis to bedaquiline involves remodelling of bacterial metabolism. Nat. Commun. 2014;5:3369. doi: 10.1038/ncomms4369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Koul A, et al. Diarylquinolines are bactericidal for dormant mycobacteria as a result of disturbed ATP homeostasis. J. Biol. Chem. 2008;283:25273–25280. doi: 10.1074/jbc.M803899200. [DOI] [PubMed] [Google Scholar]
- 17.Hards K, et al. Bactericidal mode of action of bedaquiline. J. Antimicrob. Chemother. 2015;70:1–10. doi: 10.1093/jac/dkv054. [DOI] [PubMed] [Google Scholar]
- 18.Hards K, et al. Ionophoric effects of the antitubercular drug bedaquiline. Proc. Natl Acad. Sci. USA. 2018;115:7326–7331. doi: 10.1073/pnas.1803723115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Li K, et al. Multitarget drug discovery for tuberculosis and other infectious diseases. J. Med. Chem. 2014;57:3126–3139. doi: 10.1021/jm500131s. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Pethe K, et al. Discovery of Q203, a potent clinical candidate for the treatment of tuberculosis. Nat. Med. 2013;19:1157–1160. doi: 10.1038/nm.3262. [DOI] [PubMed] [Google Scholar]
- 21.Kalia NP, et al. Exploiting the synthetic lethality between terminal respiratory oxidases to kill Mycobacterium tuberculosis and clear host infection. Proc. Natl Acad. Sci. USA. 2017;114:7426–7431. doi: 10.1073/pnas.1706139114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lee, B. S. et al. Dual inhibition of the terminal oxidases eradicates antibiotic-tolerant Mycobacterium tuberculosis. EMBO Mol. Med.13, e13207 (2020). [DOI] [PMC free article] [PubMed]
- 23.Lamprecht DA, et al. Turning the respiratory flexibility of Mycobacterium tuberculosis against itself. Nat. Commun. 2016;7:12393. doi: 10.1038/ncomms12393. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Tantry SJ, et al. Discovery of Imidazo[1,2-a]pyridine ethers and squaramides as selective and potent inhibitors of mycobacterial adenosine triphosphate (ATP) synthesis. J. Med. Chem. 2017;60:1379–1399. doi: 10.1021/acs.jmedchem.6b01358. [DOI] [PubMed] [Google Scholar]
- 25.Singh S, et al. Novel, potent, orally bioavailable and selective mycobacterial ATP synthase inhibitors that demonstrated activity against both replicating and non-replicating M. tuberculosis. Bioorg. Med. Chem. 2015;23:742–752. doi: 10.1016/j.bmc.2014.12.060. [DOI] [PubMed] [Google Scholar]
- 26.Baer JE, Jones CB, Spitzer SA, Russo HF. The potassium-sparing and natriuretic activity of n-amidino-3,5-diamino-6-chloropyrazinecar-boxamide hydrochloride dihydrate (amiloride hydrochloride) J. Pharm. Exp. Ther. 1967;157:472 LP–472485. [PubMed] [Google Scholar]
- 27.Buckley BJ, et al. 6-Substituted amiloride derivatives as inhibitors of the urokinase-type plasminogen activator for use in metastatic disease. Bioorg. Med. Chem. Lett. 2019;29:126753. doi: 10.1016/j.bmcl.2019.126753. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.McNeil MB, Ryburn HWK, Harold LK, Tirados JF, Cook GM. Transcriptional inhibition of the F1Fo-type ATP synthase has bactericidal consequences on the viability of mycobacteria. Antimicrob. Agents Chemother. 2020;64:e00492–20. doi: 10.1128/AAC.00492-20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hartkoorn RC, Uplekar S, Cole ST. Cross-resistance between Clofazimine and Bedaquiline through Upregulation of MmpL5 in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2014;58:2979–2981. doi: 10.1128/AAC.00037-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Lu X, et al. Pyrazolo[1,5-a]pyridine inhibitor of the respiratory cytochrome bcc complex for the treatment of drug-resistant tuberculosis. ACS Infect. Dis. 2018;5:239–249. doi: 10.1021/acsinfecdis.8b00225. [DOI] [PubMed] [Google Scholar]
- 31.Rhee KY, et al. Central carbon metabolism in Mycobacterium tuberculosis: An unexpected frontier. Trends Microbiol. 2011;19:307–314. doi: 10.1016/j.tim.2011.03.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Safarian S, et al. The cryo-EM structure of the bd oxidase from Mycobacterium tuberculosis reveals a unique structural framework and enables rational drug design to combat tuberculosis. Nat. Commun. 2021;12:5236. doi: 10.1038/s41467-021-25537-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Baconguis I, Bohlen CJ, Goehring A, Julius D, Gouaux E. X-ray structure of acid-sensing ion channel 1-snake toxin complex reveals open state of a Na(+)-selective channel. Cell. 2014;156:717–729. doi: 10.1016/j.cell.2014.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Buckley BJ, et al. 6-Substituted hexamethylene amiloride (HMA) derivatives as potent and selective inhibitors of the human urokinase plasminogen activator for use in cancer. J. Med. Chem. 2018;61:8299–8320. doi: 10.1021/acs.jmedchem.8b00838. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Counillon L, Scholz W, Lang HJ, Pouysségur J. Pharmacological characterization of stably transfected Na+/H+ antiporter isoforms using amiloride analogs and a new inhibitor exhibiting anti-ischemic properties. Mol. Pharm. 1993;44:1041–1045. [PubMed] [Google Scholar]
- 36.Kleyman TR, Cragoe EJ. Amiloride and its analogs as tools in the study of ion transport. J. Membr. Biol. 1988;105:1–21. doi: 10.1007/BF01871102. [DOI] [PubMed] [Google Scholar]
- 37.Wang Z, et al. Mode-of-action profiling reveals glutamine synthetase as a collateral metabolic vulnerability of M. tuberculosis to bedaquiline. Proc. Natl Acad. Sci. USA. 2019;116:19646–19651. doi: 10.1073/pnas.1907946116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Feng X, et al. Antiinfectives targeting enzymes and the proton motive force. Proc. Natl Acad. Sci. USA. 2015;112:E7073–E7082. doi: 10.1073/pnas.1521988112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Janssen Therapeutics, Division of Janssen Products, LP. (2020).
- 40.Berney M, Weimar MR, Heikal A, Cook GM. Regulation of proline metabolism in mycobacteria and its role in carbon metabolism under hypoxia. Mol. Microbiol. 2012;84:664–681. doi: 10.1111/j.1365-2958.2012.08053.x. [DOI] [PubMed] [Google Scholar]
- 41.Sambandamurthy VK, et al. A pantothenate auxotroph of Mycobacterium tuberculosis is highly attenuated and protects mice against tuberculosis. Nat. Med. 2002;8:1171–1174. doi: 10.1038/nm765. [DOI] [PubMed] [Google Scholar]
- 42.Tufariello JM, et al. Enhanced specialized transduction using recombineering in Mycobacterium tuberculosis. mBio. 2014;5:e01179–14. doi: 10.1128/mBio.01179-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Vogt G, Nathan C. In vitro differentiation of human macrophages with enhanced antimycobacterial activity. J. Clin. Invest. 2011;121:3889–3901. doi: 10.1172/JCI57235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Brosch R, et al. Genome plasticity of BCG and impact on vaccine efficacy. Proc. Natl Acad. Sci. USA. 2007;104:5596–5601. doi: 10.1073/pnas.0700869104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Yajko DM, et al. Colorimetric method for determining MICs of antimicrobial agents for Mycobacterium tuberculosis. J. Clin. Microbiol. 1995;33:2324–2327. doi: 10.1128/jcm.33.9.2324-2327.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.McNeil MB, Dennison DD, Shelton CD, Parish T. In vitro isolation and characterization of oxazolidinone-resistant Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2017;61:e01296–17. doi: 10.1128/AAC.01296-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.McNeil MB, Cook GM. Utilization of CRISPR interference to validate MmpL3 as a drug target in Mycobacterium tuberculosis. Antimicrob. Agents Chemother. 2019;63:e00629–19. doi: 10.1128/AAC.00629-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Hanwell MD, et al. Avogadro: An advanced semantic chemical editor, visualization, and analysis platform. J. Cheminform. 2012;4:17. doi: 10.1186/1758-2946-4-17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Nandakumar M, Prosser GA, De Carvalho LPS, Rhee K. Metabolomics of Mycobacterium tuberculosis. Methods Mol. Biol. 2015;1285:105–115. doi: 10.1007/978-1-4939-2450-9_6. [DOI] [PubMed] [Google Scholar]
- 50.Eoh H, Rhee KY. Multifunctional essentiality of succinate metabolism in adaptation to hypoxia in Mycobacterium tuberculosis. Proc. Natl Acad. Sci. USA. 2013;110:6554–6559. doi: 10.1073/pnas.1219375110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.McMillan DGG, Keis S, Dimroth P, Cook GM. A specific adaptation in the subunit of thermoalkaliphilic F1Fo-ATP synthase enables ATP synthesis at high pH but not at neutral pH values. J. Biol. Chem. 2007;282:17395–17404. doi: 10.1074/jbc.M611709200. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Description of additional Supplementary Files (new)
Data Availability Statement
Raw metabolomic data are presented in Supplementary Data 1. Source data underlying main figures are presented in Supplementary Data 2. Sequencing data are available from NCBI BioProject under the accession numbers PRJNA802611 and PRJNA802880. All other data are available from the corresponding author upon reasonable request.