

Abstract
D-type cyclins are central regulators of the cell division cycle and are among the most frequently deregulated therapeutic targets in human cancer1, but the mechanisms that regulate their turnover are still being debated2,3. Here, by combining biochemical and genetics studies in somatic cells, we identify CRL4AMBRA1 (also known as CRL4DCAF3) as the ubiquitin ligase that targets all three D-type cyclins for degradation. During development, loss of Ambra1 induces the accumulation of D-type cyclins and retinoblastoma (RB) hyperphosphorylation and hyperproliferation, and results in defects of the nervous system that are reduced by treating pregnant mice with the FDA-approved CDK4 and CDK6 (CDK4/6) inhibitor abemaciclib. Moreover, AMBRA1 acts as a tumour suppressor in mouse models and low AMBRA1 mRNA levels are predictive of poor survival in cancer patients. Cancer hotspot mutations in D-type cyclins abrogate their binding to AMBRA1 and induce their stabilization. Finally, a whole-genome, CRISPR–Cas9 screen identified AMBRA1 as a regulator of the response to CDK4/6 inhibition. Loss of AMBRA1 reduces sensitivity to CDK4/6 inhibitors by promoting the formation of complexes of D-type cyclins with CDK2. Collectively, our results reveal the molecular mechanism that controls the stability of D-type cyclins during cell-cycle progression, in development and in human cancer, and implicate AMBRA1 as a critical regulator of the RB pathway.
D-type cyclins (cyclin D1, cyclin D2 and cyclin D3) promote cell proliferation by activating CDK4 and CDK6, which, in turn, phosphorylate and inactivate members of the RB family of proteins1. To date, seven substrate receptors of CUL1–RING ubiquitin ligases (CRL1s) (the F-box proteins FBXO4, FBXO31, β-TrCP, FBXW8, SKP2, FBXL2 and FBXL8) and the anaphase-promoting complex/cyclosome (APC/C) have been proposed to target D-type cyclins for degradation3. However, Kanie et al.2 showed that cyclin D1 stability was unaffected in cells depleted of FBXO4, FBXO31, FBXW8 and SKP2 (alone or in combination) or in which CRL1 or APC/C activities were abrogated in the presence or absence of DNA damage. These results suggest the existence of one or more alternative bona fide ubiquitin ligase(s) targeting D-type cyclins.
Three screens for cyclin D1 regulators
Treatment of human cell lines with MLN4924, an inhibitor of all CRLs, induced accumulation and stabilization of D-type cyclins (Extended Data Fig. 1a, b). Silencing of either CUL4A and CUL4B or their associated subunit DDB1, but not other cullins, resulted in the accumulation of cyclin D1 and cyclin D3, and their phosphorylated forms (Fig. 1a and Extended Data Fig. 1c). Silencing of CUL1 or expression of a dominant-negative CUL1 mutant did not affect the levels of D-type cyclins in multiple cell lines (Extended Data Fig. 1d–h). To identify the substrate receptors of CRL4 complexes involved in the degradation of cyclin D1, we performed three orthogonal screens (Fig. 1b). First, we used a short interfering RNA (siRNA) approach in cells stably expressing fluorescently-tagged cyclin D1 (Extended Data Fig. 1i–k). We transfected oligonucleotides targeting all predicted human DDB1–CUL4-associated factors (DCAFs), the substrate receptors of CRL4 complexes4, and identified five hits (Fig. 1c). Using this approach, we identified five hits, four of which are shared between the two cell lines tested (AMBRA1, DDA1, ERCC8, and TOR1AIP2). Notably, silencing of ERCC8 revealed a peak in the accumulation of cyclin D1 at around 48–54 hours, which decreased at subsequent time points. Of the remaining three hits, silencing of AMBRA1 showed a consistent accumulation of cyclin D1 over time. Second, we used a proteomics approach, and identified six DCAFs that interacted with wild-type cyclin D1, but not with cyclin D1(T286A), a mutant insensitive to ubiquitin-mediated degradation3 (Fig. 1d, Extended Data Fig. 1l, Supplementary Table 1). Notably, AMBRA1 (also known as DCAF3) was the only interactor whose binding to cyclin D1 increased in the presence of MG132 or MLN4924, as expected for a ubiquitin ligase–substrate interaction. Third, we conducted a whole-genome CRISPR–Cas9 screen to identify global regulators of cyclin D1 and found two substrate receptors (Fig. 1e, Extended Data Fig. 1m). We identified AMBRA1 as the only hit common to all three screens (Fig. 1f).
Fig. 1 |. Three orthogonal screens identify CRL4AMBRA1 as a regulator of cyclin D1.
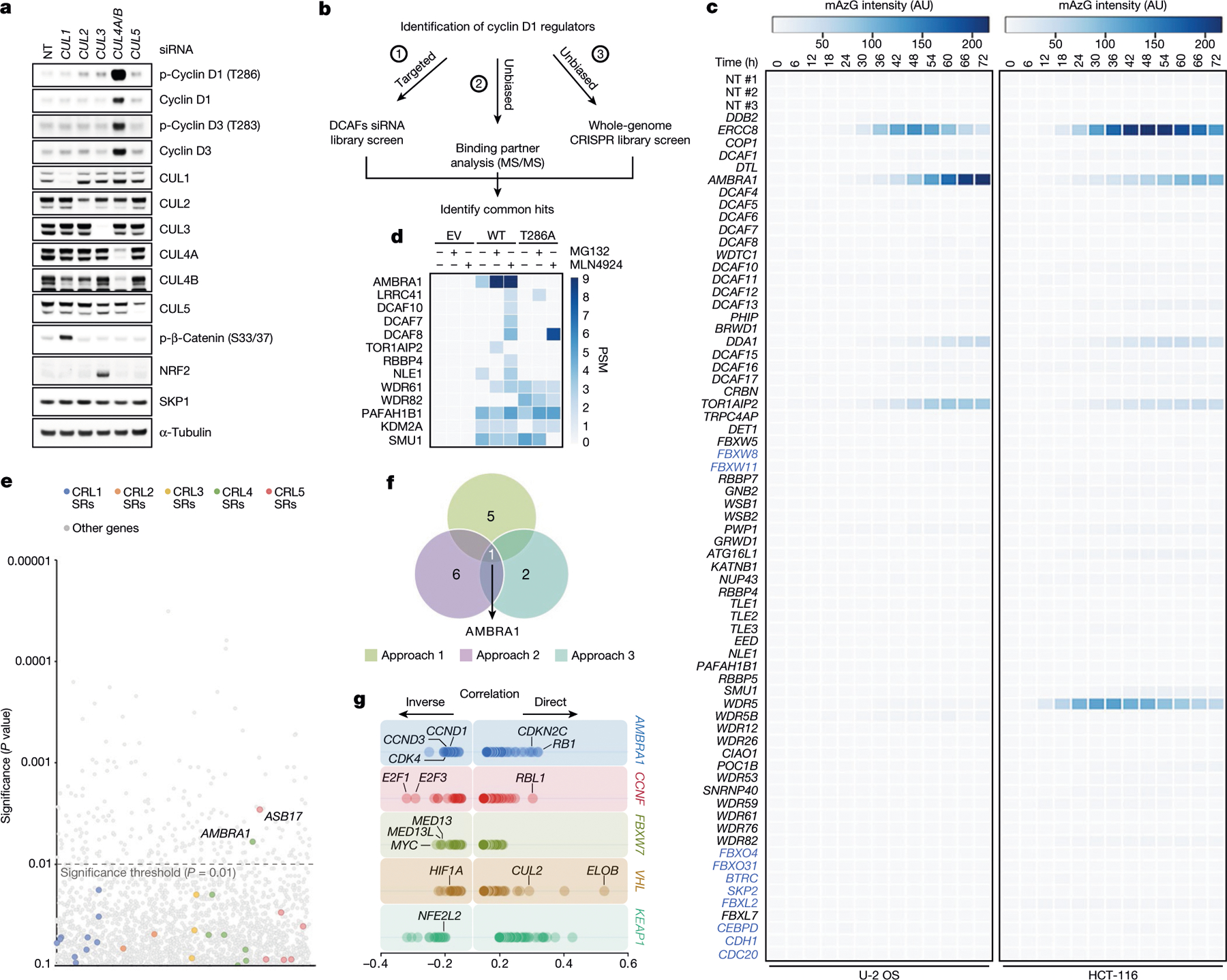
a, HCT-116 cells were transfected with a non-targeting (NT) siRNA or siRNAs against CUL1, CUL2, CUL3, CUL4A and CUL4B (CUL4A/B) or CUL5, and cell lysates were blotted with the indicated antibodies for proteins and phosphorylated (p-) proteins. This experiment was performed three times. b, Schematic representation of three orthogonal screens employed to identify regulators of cyclin D1. c, Heat map showing fluorescence intensity values in arbitrary units (AU), related to approach 1 in b. Genes encoding for the previously-reported regulators of D-type cyclins3 are reported in blue. This experiment was performed once in HCT-116 and once in U-2 OS cells. d, Heat map showing the number of peptide-spectrum matches (PSM) identified by LC-MS/MS matching substrate receptors from immunoprecipitates of empty vector (EV), wild-type (WT) cyclin D1, or cyclin D1(T286A), related to approach 2 in b. The full list of interactors is provided in Supplementary Table 1. e, Scatter plot of hits generated using approach 3 in b. This experiment was performed once, with three technical replicates. P values were calculated using the MAGeCK algorithm14. SR, substrate receptor. f, Venn diagram showing the number of hits obtained from the three screens in b. g, The top 100 CRISPR (Avana) Public 20Q1 pre-computed associations for AMBRA1 and selected substrate receptors that have a role in human cancer are depicted on the basis of their dependency profiles at genome scale (https://depmap.org/portal/; last accessed January 2020).
AMBRA1 targets D-type cyclins for degradation
A genetic interaction between AMBRA1 and the cyclin D–RB pathway was confirmed using depmap (https://depmap.org/) (Fig. 1g). Depletion of AMBRA1, but not of any of the other hits or of ubiquitin ligases reported to be involved in the degradation of D-type cyclins3, induced accumulation and stabilization of endogenous cyclin D1 and cyclin D3 (Fig. 2a, Extended Data Fig. 2a–k). Re-expression of AMBRA1 in AMBRA1−/− cells reduced the levels of D-type cyclins (Extended Data Fig. 2l). We tagged endogenous AMBRA1 with Flag-tagged minimal auxin-inducible degron (2×Flag–mAID)5 in HCT-116 human colon carcinoma cells. In all knock-in clones, auxin rapidly depleted AMBRA1 and induced cyclin D1 accumulation (Fig. 2b, Extended Data Fig. 3a–c). Auxin washout replenished AMBRA1, restoring cyclin D1 and cyclin D3 back to basal levels (Fig. 2c). The effects of AMBRA1 depletion were not synergistic with proteasome or CRL inhibition, and longer exposures of this or similar experiments revealed the presence of high-molecular-mass bands (Extended Data Fig. 3d–f). Immunopurification of cyclin D1 confirmed that loss of AMBRA1 prevents the poly-ubiquitylation of cyclin D1 (Fig. 2d). The C termini of D-type cyclins contain a highly conserved TP motif responsible for their ubiquitin-mediated degradation3 (Extended Data Fig. 4a, b). Mutations in this motif impaired the ability of D-type cyclins to interact with AMBRA1, consistent with their stabilization (Extended Data Fig. 4c–g). Cyclin D1 co-precipitated AMBRA1, but not FBXO4, FBXW8 or β-TrCP1, although these three substrate receptors co-precipitated with their respective substrates (Extended Data Fig. 4h–j). Similarly, Kanie et al. could not observe binding of cyclin D1 to FBXO4, FBXW8, β-TrCP1, FBXO31 or CDH1 in the absence or presence of DNA damage2. Conversely, we confirmed SKP2–cyclin D1 interaction, but this was independent of T286/P287 (Extended Data Fig. 4c, h), in agreement with the lack of cyclin D1 accumulation upon SKP2 silencing (Extended Data Fig. 2a, b). AMBRA1–cyclin D1 interaction was confirmed at the endogenous level, and removal of AMBRA1 did not induce accumulation of cyclin D1(T286A) (Extended Data Fig. 4k, l). The ratio of phosphorylated cyclin D1 to total cyclin D1 and the percentage accumulation of phosphorylated cyclin D1 compared with that of total cyclin D1 was higher in AMBRA1-depleted cells than in control cells (Extended Data Fig. 4m, n). Post-translational modification analysis confirmed that K269 is the major site of ubiquitylation in cyclin D16, and revealed the absence of this modification in cyclin D1(T286A) or when cells were treated with MLN4924 (Extended Data Fig. 4o). Incubation of a synthetic cyclin D1 phosphopeptide containing K269 and phosphorylated T286 with in vitro translated AMBRA1 resulted in the formation of higher-molecular-mass bands, which were ubiquitylated species, since they were not present when the phosphopeptide was incubated with ubiquitin(K0) (Extended Data Fig. 4p, q). AMBRA1 depletion abolished the S-phase degradation of cyclin D1, impaired degradation of D-type cyclins upon nutrient deprivation and genotoxic stress, promoted RB hyperphosphorylation, and induced an E2F-mediated increase in the populations of S and G2/M cells (Fig. 2e–g, Extended Data Fig. 5a–i). AMBRA1 and cyclin D1 were both detected in the nucleus, and AMBRA1 depletion accumulated nuclear cyclin D1 (Extended Data Fig. 5j). Leptomycin B treatment did not stabilize cyclin D1, and cyclin D1 fused to a nuclear-localization signal was destabilized (Extended Data Fig. 5k, l).
Fig. 2 |. AMBRA1 targets D-type cyclins for ubiquitin-mediated degradation, controlling cell cycle progression.
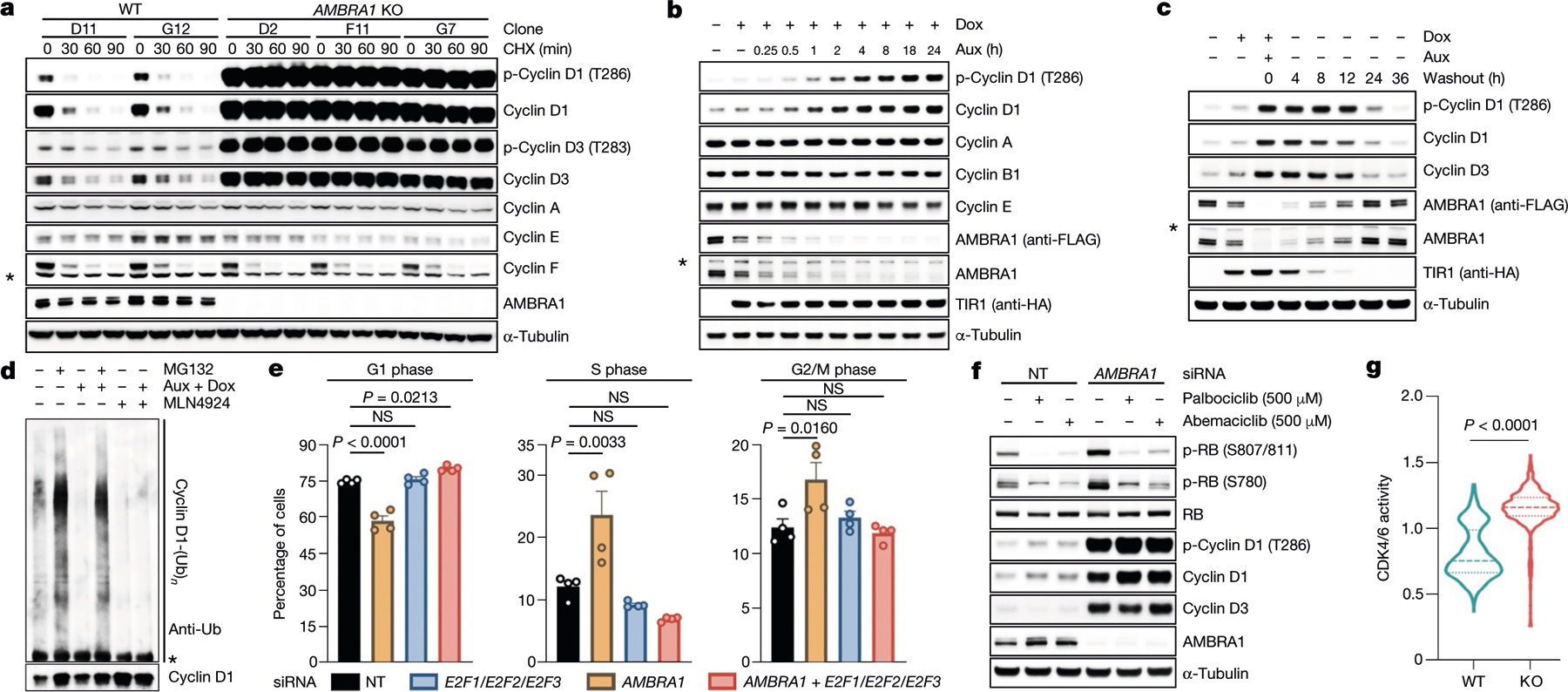
a, Three AMBRA1−/− (KO) and two AMBRA1+/+ HCT-116 clones were treated with cycloheximide (CHX) as indicated and lysates were blotted with the indicated antibodies. b, HCT-116 cells expressing 2×Flag–mAID–AMBRA1 (clone F1) were treated with DMSO or doxycycline (Dox) for 12 h before exposure to auxin (Aux). HA, haemagglutinin. c, HCT-116 cells expressing 2×Flag–mAID–AMBRA1 were treated as in b, washed, and cultured in growth medium as indicated. d, HCT-116 cells expressing 2×Flag–mAID–AMBRA1 were pre-treated with DMSO or doxycycline for 12 h, and exposed to combinations of auxin, doxycycline, MG132 or MLN4924 for 4 h. Endogenous cyclin D1 was immunoprecipitated in denaturing conditions and probed with ubiquitin (Ub) and cyclin D1 antibodies. e, HCT-116 cells were transfected with siRNAs as indicated (E2F1/E2F2/E2F3, E2F1, E2F2 and E2F3 siRNA; Extended Data Fig. 5h) before pulsing with EdU for 45 min. Cell cycle profiles were measured by flow cytometry. Data are mean ± s.e.m. (n = 4 biological replicates per group). Adjusted P values were calculated using a one-way ANOVA with Dunnett’s multiple comparisons test. NS, not significant. f, HCT-116 cells were transfected with a non-targeting (NT) siRNA or an siRNA against AMBRA1 before treatment with palbociclib or abemaciclib for 24 h. g, Ratio of cytoplasmic to nuclear CDK4/6 activity was measured in individual cells (Extended Data Fig. 5i). Violin plots show median (dashed line) and quartiles (dotted lines). P values were calculated using the non-parametric Mann–Whitney, unpaired t-test between AMBRA1 WT (n = 642 cells) and AMBRA1 KO (n = 738 cells). Unless otherwise noted, experiments were performed at least three independent times. Asterisks indicate non-specific bands.
AMBRA1 controls D-type cyclins in development
Cyclin D2 degron mutants have been identified in 15 individuals with megalencephaly–polymicrogyria–polydactyly–hydrocephalus (MPPH) syndrome, and have been shown to increase proliferation of neural progenitors (Extended Data Fig. 6a–c, Supplementary Table 2). Loss of Ambra1 leads to neural tube defects associated with increased cell proliferation and apoptosis, resulting in embryonic lethality7. Compared to wild-type mice, Ambra1gt/gt mice7 displayed accumulation of D-type cyclins, which correlated with an increase in RB phosphorylation and KI67 levels (Fig. 3a–c, Extended Data Fig. 6d, i–k). We administered abemaciclib to pregnant mice at embryonic day (E)8.5. Targeted liquid chromatography–mass spectrometry (LC-MS/MS) analysis detected the compound in the amniotic fluid and embryonic tissues (Extended Data Fig. 6e–h, Supplementary Figs. 3–5). Abemaciclib treatment decreased RB phosphorylation and rescued the hyperproliferative and neuronal phenotypes, including spina bifida and the overgrowth of telencephalic structures (Fig. 3b–d, Extended Data Fig. 6d, i).
Fig. 3 |. D-type cyclins accumulate in Ambra1gt/gt mice and their hyperproliferative and neurological phenotypes are attenuated by CDK4/6 inhibition.
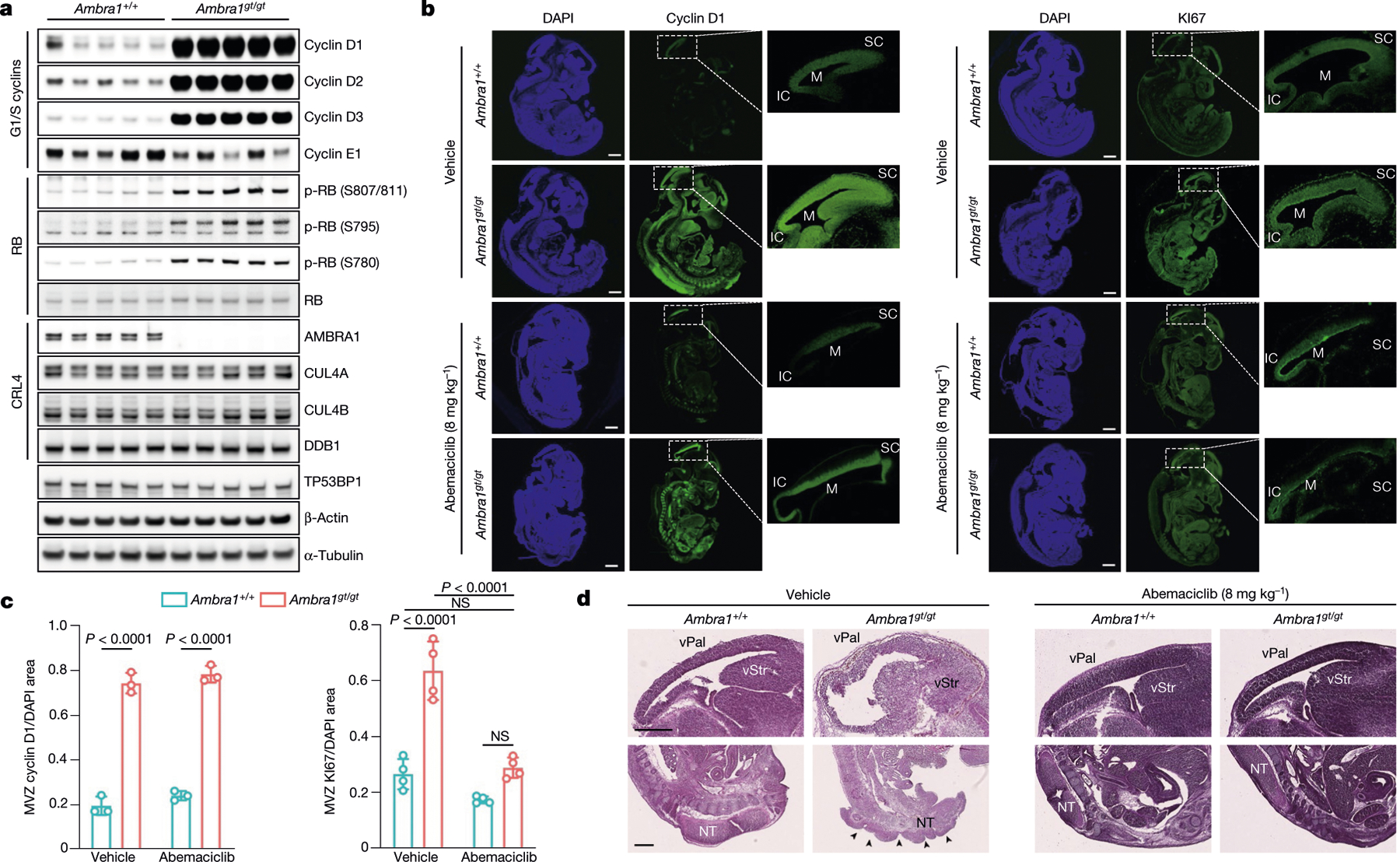
a, Immunoblotting of protein extracts of brain lysates from five E13.5 Ambra1+/+ and five Ambra1gt/gt mouse embryos. b, Representative immunofluorescent staining of E13.5 Ambra1+/+ and Ambra1gt/gt embryos collected from PBS-treated (top two rows) and abemaciclib-treated (bottom two rows) pregnant mice. M, mesencephalic ventricle; SC, superior colliculus; IC, inferior colliculus. Scale bars, 500 μm. c, Area of immunofluorescent staining for cyclin D1 and KI67 in the mesencephalic ventricular zone (MVZ) (data are mean ± s.d.; cyclin D1 n = 3; KI67 n = 4). P values by two-way ANOVA with Tukey’s multiple comparisons test. d, Representative sections stained with haematoxylin and eosin from PBS- (n = 3) or abemaciclib-treated (n = 7) E13.5 Ambra1+/+ and Ambra1gt/gt embryos. Top row, telencephalic structures: vStr (ventricular zone of striatum) and vPal (ventricular zone of pallium). Bottom row, caudal portion of the neural tube (NT). Arrows indicate the spina bifida phenotype. Scale bars, 500 μm. Unless otherwise noted, experiments were performed at least three independent times.
AMBRA1 is a tumour suppressor
Similar to other substrate receptors that act as tumour suppressors, AMBRA1 displayed a signature of cancer-associated mutations (Extended Data Fig. 7a), which suggests that AMBRA1 is a tumour suppressor gene8. Analysis of AMBRA1 mRNA and cyclin D1 protein levels revealed an inverse correlation in several tumour types (results validated by immunohistochemistry), and analysis of The Cancer Genome Atlas (TCGA) dataset (https://cancer.gov/tcga) revealed that low AMBRA1 expression is an unfavourable prognostic marker in several human cancers (Extended Data Fig. 7b–f). D-type cyclins are among the top therapeutic targets in human cancer9, and mutations in CCND1, CCND2 and CCND3 degrons have been identified in numerous malignancies (Extended Data Figs. 7g, 8, Supplementary Table 3). To evaluate whether AMBRA1 could be a tumour suppressor, we focused our studies on diffuse large B-cell lymphoma (DLBCL) because: (1) the large number of mutations in CCND3 degron in patients with DLBCL (Extended Data Fig. 8); (2) a forward genetic screen identified Ambra1 as the top putative tumour suppressor gene in DLBCL10; (3) when we examined the CRISPR gene-score distribution of six human DLBCL cell lines11, AMBRA1 was among the top tumour suppressor genes (Extended Data Fig. 9); (4) AMBRA1 mRNA levels are lower in DLBCL than in normal B-lymphocytes (https://www.oncomine.org/) (Extended Data Fig. 10a, b); (5) the AMBRA1 promoter is hypermethylated in DLBCL (Extended Data Fig. 10c); (6) low expression of AMBRA1 is an unfavourable prognostic marker in DLBCL (Extended Data Fig. 10d). TCGA dataset analysis revealed that mutations in the RB pathway (including in AMBRA1) account for 51% of cases (Extended Data Fig. 10e). Moreover, the transcriptional signatures of tumours bearing either deletion/missense mutations in AMBRA1 or amplifications/activating mutations in CCND3 cluster together, and this signature is enriched for genes belonging to E2F-mediated transcription and G1/S processes (Extended Data Fig. 10f,g). Notably, compared to parental cells, AMBRA1−/− lymphoma cells displayed a robust stabilization of cyclin D1 and cyclin D3 and generated tumours with increased volume and weight in NSG immunodeficient mice (Extended Data Fig. 10h–l).
AMBRA1 loss reduces sensitivity to CDK4/6 inhibitors
We performed a whole-genome, CRISPR–Cas9 screen aimed at identifying regulators of the response to CDK4/6 inhibitors in U-2 OS human osteosarcoma cells treated with palbociclib. AMBRA1 was identified as the top hit and the results were validated with palbociclib and abemaciclib in three cell lines (Extended Data Fig. 11a–c). Upon abemaciclib treatment, RB was more phosphorylated in AMBRA1-depleted cells than in control cells (Extended Data Fig. 11d, e, Fig. 2f). AMBRA1-knockout cells displayed increased interactions between D-type cyclins and CDK4, and also between D-type cyclins and CDK212 (Fig. 4a, Extended Data Fig. 11f–i). We also observed an increase in the formation of cyclin D1(T286A)–CDK2 complexes (Extended Data Fig. 11j). All three D-type cyclins formed an active complex with CDK2, which, in contrast to complexes with CDK4, were insensitive to palbociclib (Fig. 4b, Extended Data Fig. 12a–c). Silencing of CDK2 in AMBRA1-knockout cells resensitized them to both abemaciclib and palbociclib (Extended Data Fig. 12d, e). Next, we xenotransplanted parental and AMBRA1−/− DLBCL cells into NSG mice, and treated them with palbociclib at two different concentrations. While both doses efficiently reduced the growth of tumours from parental cells, the growth of AMBRA1−/− tumours was significantly reduced only when mice were treated with the higher concentration of palbociclib (Fig. 4c). Mice bearing AMBRA1−/− tumours had a shorter survival time than those bearing tumours from parental cells, and whereas both doses extended the survival of the mice bearing tumours from parental cells, the mice bearing AMBRA1−/− tumours were affected only by the higher concentration of palbociclib (Fig. 4d).
Fig. 4 |. AMBRA1 acts as a tumour suppressor and its absence reduces sensitivity to inhibitors of CDK4/6 by promoting the formation of D-type cyclins–CDK2 complexes.
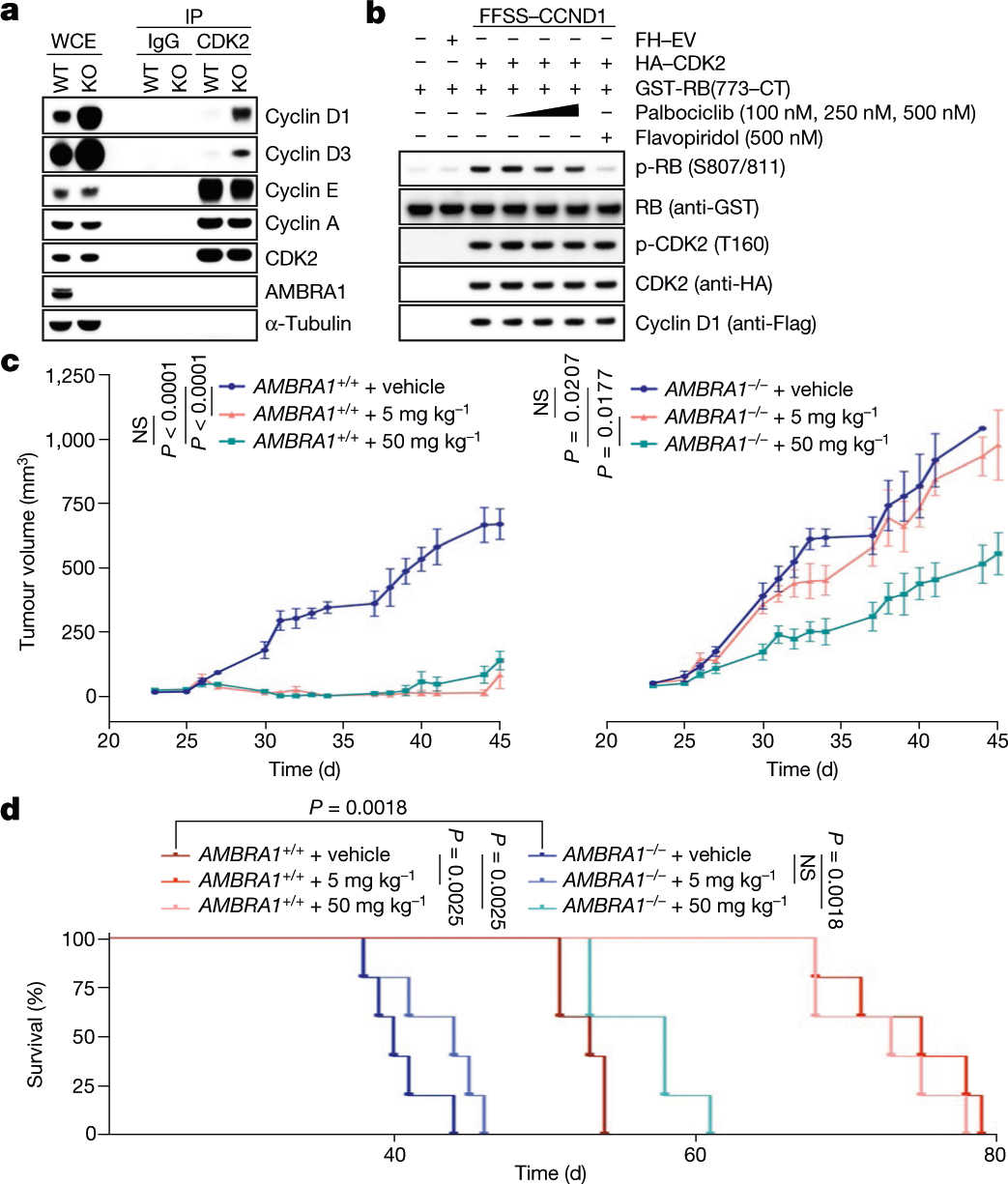
a, Endogenous CDK2 and IgG control immunoprecipitations (IPs) from parental and AMBRA1−/− HCT-116 cells. WCE, whole-cell extracts. b, HEK 293T cells were transfected as indicated. Immunopurified cyclin D1–CDK2 complexes were incubated with GST–RB(773-CT) and treated with palbociclib or flavopiridol. CT, C terminus. c, d, U-2932 cells expressing control short guide RNA (sgRNA) targeting luciferase or AMBRA1 were subcutaneously xenotransplanted in NSG mice and treated with PBS (vehicle) or palbociclib at the two indicated concentrations. c, Tumour volume. Data are mean ± s.e.m. (n = 5 per group at day 0). P values by unpaired, multiple-comparison t-test using the Holm–Sidak’s method, calculated at day 45 for the AMBRA1+/+ group and at day 40 for the AMBRA1−/− group (the latest time point where at least 3 mice per group were available). d, Survival curves. Experimental end point was declared when tumour volume reached 1,000 mm3 (n = 5 per group). P values by log-rank (Mantel–Cox) test. Unless otherwise noted, experiments were performed at least three independent times.
Discussion
Our study shows that CRL4AMBRA1 is the ubiquitin ligase that acts during normal cell cycle progression, as well as upon nutrient deprivation and genotoxic stress, to target all three D-type cyclins for ubiquitin-mediated degradation (Extended Data Fig. 12f). Moreover, our findings agree with previous studies that could not confirm a role for CRL1s and APC/C2,3 in the ubiquitin-mediated degradation of D-type cyclins. RB ensures proper neurogenesis and acts as a tumour suppressor1. Similarly, we found that, by regulating the degradation of D-type cyclins, AMBRA1 also warrants proper neurodevelopment and acts as a tumour suppressor in vivo. We also propose that decreased levels of AMBRA1 may be one of the mechanisms of desensitization to CDK4/6 inhibitors, not only by increasing the formation of D-type cyclins-CDK4/6 complexes, but also by increasing the formation of complexes with CDK2, which are insensitive to CDK4/6 inhibitors. Overall, these data highlight the need for the development of inhibitors that simultaneously target CDK2, CDK4 and CDK6, while avoiding the toxic inhibition of CDK113.
Online content
Any methods, additional references, Nature Research reporting summaries, source data, extended data, supplementary information, acknowledgements, peer review information; details of author contributions and competing interests; and statements of data and code availability are available at https://doi.org/10.1038/s41586-021-03445-y.
Methods
No statistical methods were used to predetermine sample size. The experiments were not randomized except that NSG mice were matched by age, and randomly assigned to specific treatment groups. No blinding was performed in experimental mouse interventions as knowledge of the treatment groups was required. Generally, the investigators were not blinded to allocation during experiments and outcome assessment, except when samples were assigned number labels, and randomized in order to remove downstream operator bias.
Cell culture
Cell lines were purchased from ATCC, except where indicated, and routinely monitored for Mycoplasma contamination using the Universal Mycoplasma Detection Kit (ATCC 30–1012K). HEK 293T (ATCC CRL-3216), hTERT RPE-1 (ATCC CRL-4000), T98G (ATCC CRL-1690) and NIH/3T3 (ATCC CRL-1658) cells were maintained in Dulbecco’s modified Eagle’s medium (DMEM) (Gibco). HCT-116 (ATCC CCL-247) and U-2 OS (ATCC HTB-96) cells were maintained in McCoy’s 5A medium (Gibco). PC-3 (ATCC CRL-1435), BJAB and U-2932 cells (both kindly provided to L.B. by A. Thomas-Tikhonenko, Y. Yang and L. Pasqualucci) were maintained in RPMI 1640 medium (Gibco), supplemented with 1 mM sodium pyruvate (Gibco). All media were supplemented with 10% fetal bovine serum (FBS) (Corning Life Sciences) and 1% penicillin/streptomycin/l-glutamine (Corning Life Sciences). HCT-116 2×Flag-mAID-AMBRA1 cells stably infected with pTRIPZ-HA-TIR1 were maintained in McCoy’s 5A medium supplemented with 10% Tet System Approved FBS (Takara, Clontech Laboratories) and 1% penicillin/streptomycin/l-glutamine (Corning Life Sciences). All cell lines were maintained at 37 °C and 5% CO2 in a humidified atmosphere. Where indicated, cells were treated with 0.1 mg ml−1 bafilomycin A1 (Santa Cruz Biotechnology), 20 μM chloroquine (Sigma-Aldrich), 100 μg ml−1 cycloheximide (Sigma-Aldrich), 0.4 μg ml−1 doxycycline (Sigma-Aldrich), 10 μM EdU (Thermo Fisher Scientific), 0.1 mM indole-3-acetic acid (auxin, Sigma-Aldrich), 20 nM leptomycin B (Cell Signaling), 10 μM MG132 (Peptides International) and 2.5 μM MLN4924 (Active Biochem). Where indicated, cells were nutrient deprived in Earle’s Balanced Salt Solution (EBSS) (Sigma-Aldrich) for 4 h before collection. Where indicated, cells were exposed to 5 mJ cm−2 UV irradiation for the indicated time points before collection.
Plasmids and antibodies
Homo sapiens cDNAs were amplified by PCR using Q5 High Fidelity DNA Polymerase (New England Biolabs) and sub-cloned into a variety of vector backbones, including modified pCDNA3.1 vectors containing N-terminal Flag, HA or Strep tags, pBABE-PURO retroviral vectors containing N-terminal Flag, HA, Strep, mAzG, eGFP or mPlum tags, pMSCV-PURO retroviral vectors containing N-terminal Flag or Strep tags, and pLVX-IRES-PURO or pLVX-IRES-mCherry lentiviral vectors containing N-terminal Flag or Strep-tags. FFSS indicates a tandem 2×Flag–2×Strep tag; SF indicates a Strep–Flag tag. lentiCas9-Blast was a gift from F. Zhang (Addgene plasmid # 52962)15. Site-directed mutagenesis was performed using KAPA HiFi DNA Polymerase (Kapa Biosystems). The following antibodies were used: β-actin (1:5,000, Sigma-Aldrich A5441), AMBRA1 (1:1,000, Proteintech Group 13762–1-AP), AMBRA1 (1:1,000, Santa Cruz Biotechnology sc-398204), AMBRA1 (1:1,000, Millipore ABC131), p-ATM (S1981) (1:1,000, Cell Signaling Technology 4526L), β-TrCP1 (1:1,000, Cell Signaling Technology 4394S), p-β-catenin (S33–37) (1:1,000, Cell Signaling Technology 9561S), CDC6 (1:1,000, Santa Cruz Biotechnology sc-9964), CDC20 (1:1,000, Cell Signaling Technology 4823S), CDK2 (1:1,000, Bethyl Laboratories A301–812A), p-CDK2 (T160) (1:1,000, Cell Signaling Technology 2561S), CDK4 (1:2,000, M.P. laboratory), CDT1 (1:1,000, Abcam ab70829), p-CHK1 (S317) (1:1,000, Cell Signaling Technology 2344S), p-CHK2 (T68) (1:1,000, Cell Signaling Technology 2661S), CUL1 (1:1,000, Life Technologies 71–8700), CUL2 (1:2,000, Bethyl Laboratories A302–475A), CUL3 (1:1,000, Bethyl Laboratories A301–109A), CUL4A (1:1,000, Bethyl Laboratories A300–739A), CUL4B (1:1,000, Bethyl Laboratories A303–863A), CUL5 (1:1,000, Bethyl Laboratories, A302–173A), cyclin A (1:5,000, M.P. laboratory), cyclin B1 (1:5,000, M.P. laboratory), cyclin D1 (1:1,000, Abcam ab16663), cyclin D1 (1:1,000, Santa Cruz Biotechnology sc-20044), cyclin D1 (1:1,000, Abcam ab134175), p-cyclin D1 (T286) (1:1,000, Cell Signaling Technology 3300S), cyclin D2 (1:1,000, Cell Signaling Technology 3741S), cyclin D3 (1:1,000, Thermo Fisher Scientific MA5–12717), p-cyclin D3 (T283) (1:1,000, Cell Signaling Technology 53966S), cyclin E (1:1,000, Santa Cruz Biotechnology sc-247), cyclin F (1:1,000, Santa Cruz Biotechnology sc-952), DCAF7 (1:1,000, Novus Biologicals NBP1–92589), DCAF8 (1:1,000, Bethyl Laboratories A301–556A), DDA1 (1:2,000, Proteintech Group 14995–1-AP), DDB1 (1:2,000, Bethyl Laboratories A300–462A), E2F1 (1:1,000, Cell Signaling Technology 3742S), E2F2 (1:1,000, Santa Cruz Biotechnology sc-632), E2F3 (1:1,000, GeneTex GTX11843), EMI1 (1:1,000, Proteintech Group 10872–1-AP), FBXO4 (1:1,000, Santa Cruz Biotechnology sc-376372), FBXO22 (1:1,000, GeneTex GTX117774), Flag (1:2,000, Sigma-Aldrich F1804), Flag (1:2,000, Sigma-Aldrich F7425), FZR1 (also known as CDH1, 1:1,000, Thermo Fisher Scientific 34–2000), GST (1:5,000, GE Healthcare 27457701), HA (1:2,000, Bethyl Laboratories A190–108A), LC-3 (1:5,000, Novus Biological NB100–2220), mAID (1:1,000, MBL International M214–3), NeuN (1:1,000, Cell Signaling Technology 24307S), NRF2 (1:1,000, Cell Signaling Technology 12721S), p21 (1:1,000, Cell Signaling Technology 2947S), p27 (1:1,000, BD Biosciences 610241), p53 (1:2,000, Santa Cruz Biotechnology sc-6243), p62 (1:5,000, MBL International PM045), RB (1:1,000, Cell Signaling Technology 9313S), RB (1:1,000, Cell Signaling Technology 9309S), p-RB (S780) (1:1,000, Cell Signaling Technology 8180S), p-RB (S780) (1:1,000, Cell Signaling Technology 9307S), p-RB (S795) (1:1,000, Cell Signaling Technology 9301S), p-RB (S807/811) (1:1,000, Cell Signaling Technology 8516S), p-RB (S807/811) (1:1,000, Cell Signaling Technology 9308S), RRM2 (1:1,000, Cell Signaling Technology 65939S), SKP1 (1:5,000, M.P. laboratory), SKP2 (1:1,000, Santa Cruz Biotechnology sc-1567), Tau (1:1,000, Invitrogen 13–6400), TP53BP1 (1:2,000, Abcam ab36823), α-tubulin (1:5,000, Sigma-Aldrich T6074), β-III tubulin (1:2,000, Invitrogen 480011), ubiquitin (1:1,000, Cell Signaling Technology 3933S).
Transient transfections and virus-mediated gene transfer
For gene silencing, ON-TARGETplus SMARTpool siRNA oligonucleotides (see Supplementary Table 4 for sequence details) were transfected using Lipofectamine RNAiMAX Reagent (Thermo Fisher Scientific), according to the manufacturer’s instructions. To achieve satisfactory RNAi-mediated depletion, siRNA oligonucleotides were transfected over two or three rounds. Plasmids were transiently transfected using Lipofectamine 3000 (Thermo Fisher Scientific), according to the manufacturer’s instructions, except for HEK 293T cells, which were transfected using polyethylenimine (PEI) (Polysciences). For retrovirus production, HEK 293T cells were transfected with pBABE or pMSCV vectors containing the genes of interest, in combination with pCMV-Gag-Pol and pCMV-VSV-G plasmids. For lentivirus production, HEK 293T cells were transfected with pLentiCRISPR, pLVX, or pTRIPZ vectors containing the genes of interest, in combination with pCMV delta R8.2 and pCMV-VSV-G. The virus-containing medium was collected 48 h after transfection, filtered using 0.22-μm sterile Millex-HV filter units (Millipore Sigma), and supplemented with 8 μg ml−1 polybrene (Sigma). Cells were infected by replacing their culture medium with the virus-containing medium for 8 h. Selection of transduced cells was carried out using either 1 μg ml−1 puromycin (Sigma), 10 μg ml−1 blasticidin S (Gibco) or FACS, depending on the vector selectable marker.
Quantitative PCR with reverse transcription
RNA extraction, reverse transcription and quantitative PCR reactions were performed as previously described16. In brief, total RNA was extracted using RNeasy Mini Kit (QIAGEN). cDNA was generated using RNA to cDNA EcoDry Premix (Double Primed) (Takara/Clontech Laboratories) according to manufacturer’s instruction. The quantitative PCR reaction was carried out using PowerUp SYBR Green (Thermo Fisher Scientific) with the Thermo Fisher QuantStudio 3 Real-Time PCR system in a 96-well format. ROX was used as a reference dye for fluorescent signal normalization and well-to-well optical variations correction. Bar graphs represent the relative ratios of target genes to housekeeping gene values. See Supplementary Table 4 for primer sequence details.
RNAi library screen
A RNAi ON-TARGETplus SMARTpool Cherry-pick Library was purchased from Dharmacon/Horizon Discovery, including oligonucleotides targeting all DCAF genes17 (see Supplementary Table 4 for details). The library was resuspended in 1× siRNA Buffer (Dharmacon, Horizon Discovery) to a final 2 μM stock concentration. HCT-116 and U-2 OS cells, previously transduced with pBabe-mAzG-CCND1, were reverse transfected in a 96-well plate with siRNA:DharmaFECT 2 (Dharmacon, Horizon Discovery) complex using 20 nM siRNA and 0.1 μl of DharmaFECT 2 per well, according to the manufacturer’s instructions. siRNA:DharmaFECT 2 was complexed in Opti-MEM (Thermo Fisher Scientific). Cells were seeded in FluoroBrite DMEM (Thermo Fisher Scientific) supplemented with 10% FBS (Corning Life Sciences) and 1% penicillin/streptomycin/l-glutamine (Corning Life Sciences). Images were acquired every 6 h for a total of 72 h using a Cytation 5 Imaging Reader (BioTek), equipped with a humidified BioSpa 8 Automated Incubator (BioTek) set at 37 °C and 5% CO2. Phase contrast imaging was used to generate a cell mask. Total fluorescence intensity was calculated at each time point within the cell mask, and normalized to the first time point (time = 0). Images were analysed with Gen5 (version 3.0).
Immunoprecipitation and immunoblotting
HEK 293T cells were transiently transfected using PEI (Polysciences); HCT-116 and U-2 OS where transiently transfected with Lipofectamine 3000 (Thermo Fisher Scientific). Where indicated, 48 h after transfection, cells were treated with MG132 or MLN4924 for 4 h before collection. Cells were collected and lysed in lysis buffer (50 mM Tris-Cl pH 7.5, 150 mM NaCl, 10% glycerol, 1 mM EDTA, 1 mM EGTA, 5 mM MgCl2 and 0.2% NP-40), supplemented with protease inhibitors (Complete ULTRA, Roche), phosphatase inhibitors (PhosSTOP, Roche), and 1 mM 1,4-dithiothreitol (DTT) (Sigma-Aldrich). The insoluble fraction was removed by centrifugation (20,000g) for 15 min at 4 °C. Protein concentrations in cell lysates were normalized using the Pierce BCA Protein Assay Kit (Thermo Fisher Scientific), according to the manufacturer’s instructions. Immunoprecipitations of Flag-tagged proteins (including endogenous 2×Flag–mAID–AMBRA1) were carried out using Flag–M2 agarose beads (Sigma-Aldrich) for 2 h at 4 °C. The beads were then washed 4 times in lysis buffer and elution of the precipitated complexes was carried out using 3×Flag peptide (Sigma-Aldrich). For denaturing immunoprecipitation of endogenous cyclin D1, HCT-116 cells were collected and lysed with lysis buffer supplemented with 1% SDS. The lysates were boiled for 10 min. The lysates were then diluted with lysis buffer to 0.1% SDS. Cyclin D1 was immunoprecipitated for 2 h at 4 °C using an anti-cyclin D1 antibody (1:50, Abcam Cat. No. ab16663) mixed with Dynabeads Protein G (Thermo Fisher Scientific). Mouse IgG isotype (Thermo Fischer Scientific) was used a negative control. Endogenous immunoprecipitations of cyclin D1, cyclin D3, CDK2 and CDK4 were carried out for 2 h at 4 °C using anti-cyclin D1 (1:100, Abcam ab134175), anti-cyclin D3 (1:50, Santa Cruz Biotechnology sc-6283), anti-CDK2 (1:100, Bethyl Laboratories A301–812A) and anti-CDK4 (1:100, M.P. Laboratory) antibodies, respectively. For downstream immunoblotting, immunoprecipitates were eluted in NuPAGE LDS sample buffer (Thermo Fisher Scientific) supplemented with β-mercaptoethanol (Sigma-Aldrich) and incubated at 95 °C for 5 min. Whole-cell lysates and immunoprecipitates were separated by SDS–PAGE and transferred to 0.45-μm Immobilon-P PVDF membranes (Millipore Sigma) for western blotting. Transfer efficiency and equal protein amounts or loading were verified by Ponceau S (Sigma-Aldrich) staining. For the immunoblotting of multiple protein targets, membranes were cut into strips corresponding to the target’s molecular mass using a Feather Disposable Scalpel (Thermo Fisher Scientific), and containing at least one molecular weight marker. Membranes were then blocked in 5% milk/TBST for 1 h at room temperature and incubated with the indicated primary antibodies at 4 °C overnight. For enhanced chemiluminescence, the detection of proteins was accomplished using the appropriate secondary antibodies conjugated to horseradish peroxidase (anti-mouse and anti-rabbit, GE Healthcare Life Sciences; anti-goat, Santa Cruz Biotechnology) at a 1:5,000 dilution in 5% milk/TBST for 1 h at room temperature. Immunoblots were developed using SuperSignal enhanced chemiluminescence (Thermo Fisher Scientific) using the ImageQuant LAS 4000 (GE Healthcare Life Sciences) image analyser. For fluorescent western blotting, proteins were detected with the appropriate fluorescently labelled, secondary antibodies (IRDye 680RD and IRDye 800CW, LI-COR) at a 1:10,000 dilution in 5% milk/TBST for 1 h at room temperature. Fluorescent western blot signals were detected using the ChemiDoc MP Imaging System (Bio-Rad). Western blot images were acquired with a linear or sigmoid gradation conversion curve. When necessary, membranes were stripped with stripping buffer (Restore PLUS Western Blot Stripping Buffer, Thermo Scientific) for 15 min at room temperature, and reblotted with a different primary antibody. Stripping efficiency was confirmed by redeveloping the membranes after incubation with the stripping buffer (see Supplementary Fig. 6 for example). Images were assembled and rescaled using Adobe Photoshop (version 21.1.0).
Mass spectrometry for proteomics
The immunoprecipitated samples were filtered with Ultrafree centrifugal filters (Millipore Sigma) and concentrated using Amicon Ultra 3,000 NMWL centrifugal filters (Millipore Sigma), according to the manufacturer’s instructions. The samples were then resuspended in NuPAGE LDS Sample Buffer (Novex). The proteins were reduced with 2 μl of 0.2 M DTT for 1 h at 57 °C at pH 7.5. Next, the proteins were alkylated with 2μl of 0.5 M iodoacetamide (Sigma) for 45 min at room temperature in the dark. The samples were loaded on a NuPAGE 4–12% Bis-Tris Gel 1.0 mm (Life Technologies) and run for 20 min at 200 V. The gel was stained with GelCode Blue Stain Reagent (Thermo Fisher Scientific). The gel lanes were excised, cut into 1 mm3 pieces and destained for 15 min in a 1:1 (v/v) solution of methanol and 100 mM ammonium bicarbonate. The buffer was exchanged and the samples were destained for another 15 min. This was repeated for another 3 cycles. The gel plugs were dehydrated by washing with acetonitrile, and further dried by placing them in a SpeedVac for 20 min. 500 ng of sequencing grade modified trypsin (Promega) was added directly to the dried gel pieces followed by enough 100mM ammonium bicarbonate to cover the gel pieces. The gel plugs were allowed to shake at room temperature and digestion proceeded overnight. The digestion was halted by adding a slurry of R2 50 μm Poros beads (Applied Biosystems) in 5% formic acid and 0.2% trifluoroacetic acid (TFA) to each sample at a volume equal to that of the ammonium bicarbonate added for digestion. The samples were allowed to shake at 4 °C for 120 min. The beads were loaded onto C18 ziptips (Millipore Sigma), equilibrated with 0.1% TFA, using a microcentrifuge for 30 s at 6,000 rpm. The beads were washed with 0.5% acetic acid. Peptides were eluted with 40% acetonitrile in 0.5% acetic acid followed by 80% acetonitrile in 0.5% acetic acid. The organic solvent was removed using a SpeedVac concentrator and the sample reconstituted in 0.5% acetic acid. Each sample was loaded onto a trap column (Acclaim PepMap 100 pre-column, 75 μm × 2 cm, C18, 3 μm, 100 Å, Thermo Scientific) connected to an analytical column (EASY-Spray column, 50 m × 75 μm internal diameter, PepMap RSLC C18, 2 μm, 100 Å, Thermo Scientific) using the autosampler of an Easy nLC 1000 (Thermo Fisher Scientific) with solvent A consisting of 2% acetonitrile in 0.5% acetic acid and solvent B consisting of 80% acetonitrile in 0.5% acetic acid. The peptide mixture was gradient eluted into the Orbitrap Lumos mass spectrometer (Thermo Fisher Scientific) using the following gradient: a 2–31% solvent B in 60 min, 31–40% solvent B in 10 min, followed by 40–100% solvent B in 10 min. The samples were acquired on the Orbitrap Lumos using the following parameters: full MS spectra resolution of 120,000 (at m/z 200), an AGC target of 4 × 105, maximum ion time of 50 ms, scan range from 400 to 1,500 m/z. Following each full MS scan, low resolution MS/MS spectra were acquired using the TopSpeed algorithm for a 3 s duty cycle. The MS/MS spectra were collected in the ion trap in rapid scan mode, with an AGC target of 1 × 104, maximum ion time of 30 ms, one microscan, 2 m/z isolation window, normalized collision energy of 32 and a dynamic exclusion of 30 s. To identify binding partners, all acquired MS2 spectra were searched against a UniProt human database using Sequest HT within Proteome Discoverer 1.4 (Thermo Fisher Scientific). The search parameters were as follows: precursor mass tolerance ± 10 ppm, fragment mass tolerance ± 0.02 Da, digestion parameters trypsin allowing two missed cleavages, fixed modification of carbamidomethyl on cysteine, variable modification of oxidation on methionine, and variable modification of deamidation on glutamine and asparagine and a 1% peptide and protein false discovery rate searched against a decoy database. The results were filtered to only include proteins identified by at least two unique peptides. To quantify the post-translational modification sites, the MS/MS spectra were searched against a given protein sequence of interest using Byonic18 (Protein Metrics). The mass tolerance was set to 10ppm for MS1 and 0.4 Da for MS2 searches. A false discovery rate of 1% was applied on the protein level. The Byonic search included fixed modification of carboxymethyl on cysteine and variable modifications of oxidation on methionine, ubiquitination at lysine and phosphorylation at serine, threonine and tyrosine residues. The spectra of the peptides of interest were manually verified to localize the phosphorylation and ubiquitination sites. For relative quantification of the modified sites the semi-automatic program Byologic (Protein Metrics) was used to obtain the area under the curve (AUC) for the extracted ion chromatogram of each peptide isoform. The AUC was manually verified and adjusted if appropriate for each isoform. The percent modification was obtained by dividing the isoform AUC by the total AUC for the peptide.
Whole-genome CRISPR–Cas9 screens
HCT-116 (previously transduced with pBabe-mAzG-CCND1) and U-2 OS cells were transduced with lentiCas9-Blast, a gift from F. Zhang (Addgene plasmid #52962)15, and selected with 10 μg ml−1 blasticidin S (Gibco). The Human GeCKOv2 CRISPR-knockout pooled library was a gift from F. Zhang (Addgene #1000000049). The library was amplified according to the amplification protocol provided by the F. Zhang laboratory (https://media.addgene.org/cms/filer_public/b5/fd/b5fde702-d02c-4873–806f-24ac28b2a15a/geckov20_library_amplification_protocol_1.pdf) and deep sequenced using Illumina MiSeq (80 cycles forward read + 8 bp for the index read) to verify representation and pool complexity. To produce lentivirus, HEK 293T cells were seeded in twenty, 15-cm dishes and co-transfected with the lentiGuide-Puro plasmid library, pCMV delta R8.2 and pCMV-VSV-G. After 48 h, the supernatant was filtered using 0.22-μm sterile Millex-HV filter units (Millipore Sigma), concentrated by ultracentrifugation at 24,000 rpm for 2 h at 4 °C, and resuspended in DMEM supplemented with 10% FBS. Aliquots were stored at −80 °C. To achieve a multiplicity of infection of 0.3–0.5, 3 × 106 HCT-116 and U-2 OS cells were tested by spinfecting in a 12-well plate with different volumes of virus supplemented with 8 μg ml−1 polybrene (Sigma). The next day, each well was split into two replicate wells. To calculate percent transduction, one duplicate well was treated with 1 μg ml−1 puromycin (Sigma) for 3 days. Per cent transduction was calculated as cell count from the puromycin-treated cells divided by the cell count from untreated cells multiplied by 100. The volume of virus which gave a multiplicity of infection of 0.4 was chosen for large-scale screening. For the identification of genes involved in the regulation of cyclin D1, 1 × 108 HCT-116 (previously transduced with pBabe-mAzG-CCND1 and lentiCas9-Blast) were spinfected as described above, seeding 3 × 106 cells per well in 12-well plates. The day after spinfection, wells were pooled in larger 15-cm dishes and selected for 3 days with 1 μg ml−1 puromycin (Sigma). To select for guide RNAs (gRNAs) that induced an increase in the levels of fluorescently-tagged cyclin D1, cells were split into two groups. One group was used as reference, while the second group was sorted by FACS to enrich for cells that expressed high levels of mAzG as compared to parental cells not infected with the CRISPR–Cas9 library. Cells were cultured for seven additional days and genomic DNA was extracted with a Blood & Cell Culture Midi kit (QIAGEN). For the identification of genes whose loss confers resistance to palbociclib, 1 × 108 U-2 OS cells (previously transduced with lentiCas9-Blast) were spinfected as described above, seeding 3 × 106 cells per well in 12-well plates. The day after spinfection, wells were pooled in larger 15-cm dishes and selected for 3 days with 1 μg ml−1 puromycin (Sigma). Cells were then split into two groups. One group was treated with 500 nM palbociclib (Sigma-Aldrich) for 60 days, while the other group was treated with vehicle (DMSO). Genomic DNA was extracted with a Blood & Cell Culture Midi kit (QIAGEN). For each sample we performed 15 separate PCR reactions using NEBNext Q5 Hot Start HiFi PCR Master Mix (New England Biolabs) (see Supplementary Table 4 for primer details). PCR products were loaded onto an agarose gel, purified, mixed and quantified. Pooled libraries were sequenced on a NextSeq 500 (Illumina), either a mid-output or a high-output cartridge, as single read 81, and index read of 8, PhiX spike-in, 20%. Illumina primers were used as sequencing primers. Upon demultiplexing, FASTQ files were processed and trimmed using Cutadapt (https://doi.org/10.14806/ej.17.1.200) to retrieve the unique sgRNA sequence. sgRNA reads were aligned to the library using the MAGeCK algorithm14.
CRISPR–Cas9 genome editing
CRISPR–Cas9 genome editing techniques were carried out as previously described19–21. In brief, to generate AMBRA1-knockout cells, optimal gRNA target sequences closest to the genomic target site were designed using the Benchling CRISPR Genome Engineering tool (https://www.benchling.com). To generate 2×Flag-mAID-AMBRA1 knock-in HCT-116 cells, an optimal gRNA target sequence closest to the genomic target site and a ~3 kb homologous recombination (HR) donor template were designed using the Benchling CRISPR Genome Engineering tool. The HR donor template was designed to introduce a 2×Flag-mAID tag in frame with the N terminus of AMBRA1, in the following order: methionine-FLAG-linker-FLAG-linker-mAID-linker-AMBRA1, and was purchased as a synthetic gene from Genewiz. For transient transfection, AMBRA1 gRNA #3 (see Supplementary Table 4 for sequence details) were cloned into pSpCas9(BB)-2A-GFP (PX458), a gift from F. Zhang (Addgene plasmid #48138)22. Lentiviruses encoding gRNAs targeting human AMBRA1 were subcloned into a Lenti-Guide-mCherry vector (see Supplementary Table 4 for sequence details). For AMBRA1 knockout in hTERT RPE-1, HCT-116, U-2 OS and T98G, cells were seeded into 10-cm dishes at approximately 70% confluency, and transfected with 5 μg of gRNA-containing PX458 plasmid, using Lipofectamine 3000 (Life Technologies). For 2×Flag-mAID-AMBRA1 knockin, HCT-116 cells were seeded into 10-cm dishes at approximately 70% confluency, and transfected with 2.5 μg of gRNA-containing PX458 plasmid and 2.5 μg of HR donor template, using lipofectamine 3000 (Life Technologies). Two days after transfection, GFP-positive cells were sorted using the Beckman Coulter MoFlo XDP cell sorter (100-μm nozzle), and 15,000 cells were plated on a 15-cm dish. 8–10 days later, single cell clones were picked, dissociated in trypsin-EDTA (Sigma-Aldrich) for 5 min, and plated into individual wells of a 96-well plate for genotyping. Genomic DNA was collected using QuickExtract (Epicentre). Genotyping PCRs were performed with MyTaq HS Red Mix (Bioline), using primers surrounding the genomic target sites (see Supplementary Table 4 for primer details). The resulting PCR products were then purified and sequenced to determine the presence of an insertion or deletion event. To further validate the mutational status of candidate clones, the PCR products were subjected to TOPO-TA Cloning (Invitrogen), and sequenced in order to distinguish the amplified products of distinct alleles. Fifty bacterial colonies for each TOPO-TA cloning reaction were sequenced and aligned to the corresponding wild-type template in Benchling to confirm that all alleles were correctly targeted. Clones positive to insertion or deletion events were then validated by western blot. For infection of BJAB and U-2932 cells, lentiviruses expressing either control sgRNA targeting luciferase or sgRNA targeting AMBRA1 were produced in HEK 293T. After infection, mCherry-positive BJAB and U-2932 cells were selected by FACS and validated by western blot.
Immunofluorescence and live-cell microscopy
For immunofluorescence microscopy, cells were cultured on coverslips, washed in PBS, and fixed in 4% paraformaldehyde (PFA) in PBS for 10 min at room temperature. Cells were permeabilized with PBS/0.1% Triton X-100 for 10 min and blocked for 1 h in blocking buffer (PBS/0.05% Triton X-100 containing 5% FBS and 3% BSA) before incubation with the indicated primary antibodies in blocking buffer. Alexa-555 and Alexa-488-conjugated secondary antibodies were from Thermo Fischer Scientific and used at 1:2000 dilution. Slides were mounted in ProLong Diamond Antifade Mountant with DAPI (Thermo Fisher Scientific). Imaging was performed using a DeltaVision Elite inverted microscope system (Applied Precision), using a 60× objective (numerical aperture 1.42) from Olympus or a Zeiss LSM 800 microscope using a Plan-Apochromat 63×/1.40 oil objective. For live-cell microscopy, cells were seeded on Nunc LabTek II Chambered Coverglass 24 h before imaging. On the day of imaging, cells were incubated in FluoroBrite DMEM (Thermo Fisher Scientific) supplemented with 25 mM HEPES, 10% FBS (Corning Life Sciences) and 1% penicillin/streptomycin/l-glutamine (Corning Life Sciences). Imaging was performed using a DeltaVision Elite inverted microscope system (Applied Precision) with softWoRx (version 7.0.0), using a 60× objective (numerical aperture 1.42) from Olympus, or a Zeiss Axio Observer with Zeiss Zen (version 2.1), using an A-Plan 20× objective (numerical aperture 0.35) equipped with an Axio 506 monocamera. To analyse changes in mean nuclear intensity, each nucleus was manually annotated by its starting frame (first frame in which a nucleus is visible), location at starting frame, and frame of initial PCNA foci formation. Tracking was then performed automatically by an in-house Python script. This script segments all objects in an image via thresholding and finds the closest object with a similar size and shape to the object identified in the image from the previous frame. The script continues until it can no longer identify any objects meeting the size–shape criteria within the distance cut-off. The tracks were outputted as movies for manual examination of their quality. Any nuclei where tracking failed or that were tracked for less than 10 h were excluded. The mean intensity for each nucleus at each time point was recorded during tracking. The tracks were aligned by setting the time of PCNA foci formation to 0 h for all tracks. The confidence intervals were calculated using the lineplot method of the Python package Seaborn, which uses the bootstrap method23 to find the confidence interval. To measure cytoplasmic to nuclear intensity as a readout of CDK4/6 activity, ImageJ (version 1.52p) was used, as previously described24.
Flow cytometry
For analysis of DNA replication, cells were pulsed with EdU (10 μM final concentration, Thermo Fisher Scientific) for 45 min, washed in PBS and fixed in 4% PFA. Detection of EdU was accomplished using the Click-iT Plus EdU Alexa Fluor 488 Flow Cytometry Assay Kit (Thermo Fisher Scientific) according to manufacturer’s instructions. For analysis by flow cytometry, cells were counterstained with propidium iodide (10 μg ml−1 final concentration, Sigma-Aldrich) diluted in RNase (20 μg ml−1 final concentration, Qiagen) containing PBS. Flow cytometry data were collected using the SP6800 Spectral Analyzer (Sony Biotechnology) using the SP6800 Spectral Analyzer software (version 1.6.3.7151). Flow cytometry data were analysed using the FlowJo software (version 10.7.1). A representative FACS gating strategy is provided in Supplementary Fig. 2.
In vitro ubiquitylation
C-terminal 3×Flag-tagged AMBRA1 was in vitro translated (IVT) using the TNT T7 Quick Coupled Transcription/Translation System (Promega) according to the manufacturer’s recommendations. In vitro ubiquitylation reaction of synthetic phosphorylated (T286) cyclin D1 peptide (Yenzym) by 3×Flag-tagged AMBRA1 was carried out in buffer containing 50 mM Tris HCL pH 7.5, 5 mM MgCl2, 1 mM DTT, 1 mM ATP, 0.1 μM E1 (Boston Biochem), 0.25 μM UBC H5C (Boston Biochem), 2.5 μM MG132, 0.2 nM okadaic acid, 0.2 μM NaV, and either 5 μM His-ubiquitin K0 (Boston Biochem) or 10 μM Myc-ubiquitin (Boston Biochem) and 3 μM ubiquitin aldehyde (Boston Biochem). Reactions were incubated at 30 °C for the indicated times, and terminated by adding NuPAGE LDS sample buffer (Thermo Fisher Scientific) followed by incubation for 5 min at 95 °C. Samples were resolved by SDS–PAGE and immunoblotted as indicated.
In vitro kinase assay
HEK 293T cells were transiently (co)transfected for 48 h with vectors expressing FFSS-tagged cyclin D1, FFSS-tagged cyclin D2, FFSS-tagged cyclin D3, HA-tagged CDK2, HA-tagged CDK4, or FH-tagged empty vector, in the combinations indicated. Complexes were purified by double immunoprecipitation in the following order: anti-Flag immunoprecipitation, elution with 3×Flag peptide, then anti-HA immunoprecipitation. Complexes bound to the beads were pre-washed in NEBuffer for Protein Kinases (PK, New England Biolabs B6022) and incubated with 0.5 μg human recombinant Rb (773-end) with an N-terminal GST tag, expressed in an Escherichia coli expression system (Sigma-Aldrich SRP0256). Reactions were carried out in NEBuffer for PK containing 0.1 mM NaV, 0.2 nM okadaic acid, 10 μM ATP in the presence or absence of either palbociclib isethionate (Sigma-Aldrich) or flavopiridol (Selleck Chemicals) at the concentrations indicated for 30 min at 30 °C, before stopping the reaction with Laemmli buffer. Samples were resolved by SDS–PAGE and immunoblotted as indicated.
Mouse and animal procedures
All animal work was performed following the ethical guidelines and protocols approved by the Institutional Animal Care and Use Committee of the University of Pennsylvania (IACUC Protocol 805157), and in accordance with the ethical international, EU and national formal approval obtained from the Danish Health Authorities (Dyreforsøgstilsynet, 2018–15-0201–01391) at the Danish Cancer Society Research Center. NSG mice were purchased from the Jackson Laboratory (Cat. No 005557). Ambra1gt/+ mice were previously described7. To obtain Ambra1gt/gt embryos, three- to six-month-old Ambra1gt/+ mice were intercrossed, and vaginal plug inspection was carried out to determine the time of mating. For the assessment of morphological phenotypes and for immunofluorescence staining, abemaciclib mesylate (8 mg kg−1, Selleck Chemicals) or PBS (vehicle) were administered from E8.5 to E13.5 by daily oral gavage using a disposable oral gavage needle. To measure abemaciclib by LC-MS/MS, abemaciclib mesylate was administered by daily oral gavage at two concentrations (8 mg kg−1, 16 mg kg−1) from E8.5 to E13.5, and at one concentration (100 mg kg−1) from E12.5 to E13.5. Mice were scarified and the conceptuses from the gravid uteri were exposed to aspirate a sample of amniotic fluid for targeted LC-MS/MS analysis, using a 28G insulin syringe. The amniotic fluid samples were centrifuged at 2,700g for 10 min at 4 °C to remove cellular debris, and then stored at −80 °C for subsequent analysis. Maternal peripheral blood was collected by retro-orbital aspiration using heparinized capillary tubes (Thermo Fisher Scientific), and centrifuged at 2,700g for 10 min at 4 °C to separate the plasma fraction. Plasma was collected and stored at −80 °C for subsequent analysis. Maternal brains and embryonic tissue samples (mainly from the limbs) were collected, washed in PBS, and stored at −80 °C for subsequent analysis. For immunoblotting of brain lysates, E13.5 Ambra1+/+ and Ambra1gt/gt mouse brain were collected and homogenized in lysis buffer (50 mM Tris-Cl pH 7.5, 320 mM sucrose, 10% glycerol, 1% NP-40) supplemented with protease inhibitors (Complete ULTRA, Roche) and phosphatase inhibitors (PhosSTOP, Roche).
Mass spectrometry for metabolomics
Amniotic fluid.
For amniotic fluid samples, the abemaciclib extraction was carried out on each sample in a ratio of 50 μl sample per 1 ml of 50% methanol, 0.1% Trifluoroacetic acid (TFA). The sample in extraction buffer mixture was subjected to a homogenization step with zirconium disruption beads (0.5 mm, RPI) and homogenized for 10 min at 4 °C in a BeadBlaster with a 30 s on, 30 s off pattern. The homogenized sample was centrifuged at 21,000g for 3 min and a fixed volume of the supernatant (450 μl, all samples) was transferred to a 1.5-ml microfuge tube for speed vacuum concentration, no heating. The dried sample was then resuspended in 100% methanol (50 μl) for LC-MS analysis.
Plasma samples.
For plasma samples, a solid phase extraction (SPE) procedure was used to extract abemaciclib and remove partial haemolysis products. The SPE cartridge (Hypersep SPE 500 mg/2.8 ml C18) was conditioned with 1 ml of 100% methanol, 0.1% TFA, and then equilibrated with 1 ml of HPLC grade water with 0.1% TFA. Then, 50 μl of plasma was homogenized with 950 μl of HPLC water with 0.1% TFA using the same method described above and a fixed volume of the homogenized mixture (900 μl) was loaded onto the SPE cartridge. Elution was carried out with two fractions (each 1 ml) of 50% methanol with 0.1% TFA. The two fractions for each sample were pooled and dried to completion by speed vacuum, then resuspended in 100% methanol and analysed by LC-MS.
Brain tissue.
For the tissue samples, the abemaciclib extraction was carried out on each sample in a ratio of 50 mg per ml of HPLC grade water with 0.1% TFA, as with the plasma samples. After homogenization (same as for amniotic fluid and plasma), 300 μl of the sample mixture was then mixed with 700 μl of HPLC grade water with 0.1% TFA before loading. The rest of the SPE procedure was identical to the plasma SPE procedure described above.
Standard curve quantification.
For both the amniotic fluid and plasma samples, an abemaciclib standard curve ranging from 10 nM to 1,000 nM was prepared in blank amniotic fluid (undosed controls) or blank plasma. These standard curve samples were extracted and analysed side-by-side with the experimental samples to account for matrix specific effects in the quantification. For tissue samples, no blank matrix control was suitable, so an external standard sample of abemaciclib in 100% methanol (1,000 nM) was used to confirm target detection. For the LC-MS analysis, three independent data acquisition sequences were carried out; plasma samples, amniotic fluid samples, and brain tissue samples. For each sequence, the order of acquisition (samples and standard curve points) was randomized to mitigate sequence bias, and each sample was preceded by a wash and blank injection to assess potential carryover. The sample data were interpolated against the respective standard curve in GraphPad Prism (v7.03).
Liquid chromatography.
The LC column was an ACQUITY UPLC BEH Phenyl (1.0 × 50 mm, 1.7 μm) coupled to a Dionex Ultimate 3000 system and the column oven temperature was set to 25 °C for the gradient elution. A flow rate of 200 μl min−1 was used with the following buffers: (A) HPLC-grade water with 0.1% formic acid, and (B) acetonitrile with 0.1% formic acid. The gradient profile was as follows; 0–35% B (0–10 min), 35–75% B (10–15 min), 75–99% B (15–16.5 min), 99–0% B (16.5–16.75 min), 0–0% B (16.75–20 min). Injection volume was set to 1 μl for all analyses (20 min total run time per injection).
Mass spectrometry.
MS analyses were carried out by coupling the LC system to a Thermo Q Exactive HF mass spectrometer operating in heated electrospray ionization mode. Method duration was 20 min with a polarity switching data-dependent Top 3 method for both positive and negative modes. A dedicated tandem MS (MS2) scan was programmed to target abemaciclib ([M+2H]2+, 254.1432 m/z, isolation window of ± 0.2 m/z). Spray voltage for both positive and negative modes was 3.5 kV and capillary temperature was set to 320 °C with a sheath gas rate of 35, aux gas of 5. The full MS scan for both polarities used 120,000 resolution with an AGC target of 3 × 106 and a maximum IT of 100 ms, and the scan range was from 95–1,000 m/z. Tandem MS spectra for both positive and negative mode used a resolution of 15,000, AGC target of 1 × 105, maximum IT of 50 ms, isolation window of 0.4 m/z, isolation offset of 0.1 m/z, and 3-way multiplexed normalized collision energies (nCE) of 10, 35, 80. The minimum AGC target was 5 × 104 with an intensity threshold of 1 × 106. All data were acquired in profile mode.
Mass spectrometry imaging.
Fresh frozen embryos were sectioned on a single slide at 10-μm thickness and thaw-mounted on conductive indium tin oxide IntelliSlides from Bruker Scientific. Slides were stored at −80 °C before analysis and dried in a vacuum desiccator before applying 2,5-dihydroxybenzoic acid MALDI matrix. Uniform layers of matrix were deposited in 12 spray passes over the tissue, at 15 mg ml−1 in 70% acetonitrile + 0.1% TFA using the TM Sprayer from HTX Technologies. Data acquisition was performed with the dual source timsTOF FleX mass spectrometer from Bruker Scientific equipped with the 10kHz Smartbeam 3D laser and the dual trapped ion mobility separation cell. Detection settings were determined for abemaciclib using authentic standard (10 μM) spotted onto InteliSlides and the MALDI MS/MS pattern was confirmed against the LC-MS/MS data: MALDI ionization mode [M+H]+ at 507.2791 m/z ( ± 15 ppm) with a mobility (1/K0) of 1.177 ( ± 1%). Images of abemaciclib were acquired using FlexImaging software at 75-μm spatial resolution with M5 small laser profile and ScanBeam on, and the acquisition region was manually selected around the tissues using the registered optical image with a 1-mm border beyond the tissue. The ions generated in the MALDI source were separated in the trapped ion mobility cell using 90-ms ramp time and 100% duty cycle. Data analysis and visualization was performed in SCiLS Lab software, FlexImaging and TIMS Data Viewer from Bruker Scientific. Tissue distribution of the drug and any isobaric molecules was visualized separately based on their specific mobility values (1/K0) extracted from TIMS Data Viewer. Data quality for each region was assessed using shared lipid features.
Immunofluorescence and haematoxylin and eosin staining
For immunofluorescent staining, embryos were fixed overnight in 4% PFA at 4 °C. Embryos were then washed three times in cold PBS, and dehydrated for cryopreservation in 30% sucrose in PBS at 4 °C, until sinking of the embryos. Upon full dehydration, embryos were moved to a 1:1 mixture of OCT compound (Thermo Fisher Scientific) and 30% sucrose/PBS, and gently rocked for 2 h. Embryos were then embedded in cryomolds that were pre-chilled in an isopentane (Sigma-Aldrich) bath on dry ice. Sections were obtained using a cryostat, re-hydrated in PBS, and blocked in 1× blocking buffer (Abcam) with 0.3% Triton X-100 for 1 h at room temperature in a humidified chamber. Primary antibodies were diluted in blocking buffer and incubated overnight at 4 °C. The following antibodies were used: cyclin D1 (1:200, Abcam ab16663), cyclin D2 (1:50, Santa Cruz Biotechnology sc-56305), KI67 (1:200, Abcam ab15580), and pRb (S807/811) (1:200, Cell Signaling Technology 8516S). Secondary antibodies were anti-rabbit (Alexa Fluor 488, Thermo Fischer Scientific A-11008) and anti-mouse (Alexa Fluor 555, Thermo Fischer Scientific A-21422), and used at 1:300 in 1× blocking buffer (Abcam) for 2 h at room temperature in a humidified chamber. Sections were then washed three times in PBS, incubated with DAPI in PBS for 10 min, and mounted with Fluoromount Aqueous Mounting Medium (Sigma-Aldrich). The signal-positive area for cyclin D1 and KI67 intensity levels from the mesencephalic ventricular zone (MVZ) was quantified using ImageJ (version 1.52p) and normalized over the signal-positive area for DAPI intensity levels, setting the same intensity threshold among different conditions. For H&E staining, embryos were fixed overnight in 4% PFA at 4 °C. Embryos were then washed three times in cold PBS, and dehydrated in 30% sucrose in PBS at 4 °C, until sinking of the embryos. Upon full dehydration, embryos were moved to a 1:1 mixture of OCT compound (Thermo Fisher Scientific) and 30% sucrose/PBS, and gently rocked for 2 h. Embryos were then embedded in cryomolds that were pre-chilled in an isopentane (Sigma-Aldrich) bath on dry ice. Sections were obtained using a cryostat and rehydrated in PBS, followed by a wash in ddH2O. Sections were then immersed in Gill’s Haematoxylin No. 2 Solution (Bio-Optica, 05–06014/L) for 30 s. Sections were washed in ddH2O, followed by 0.5% alcoholic eosin (Diapath, C0353). Sections were dehydrated with one, 10-s wash in 90% ethanol, and three, 10-s washes with 100% ethanol. Lastly, sections were immersed in Diasolv (Diapath, H0315) three times.
Immunohistochemistry of human tissues
Chromogenic immunohistochemistry for human AMBRA1 was performed on plasma-thrombin cell blocks prepared from wild-type and knockout AMBRA1 HCT-116 cell lines as previously described25. Cell blocks were fixed in 10% Neutral Buffered formalin for 48 h, processed and paraffin embedded. Four-micrometre sections were collected from both cell blocks on to the same Plus microscope slides (Fisher Scientific Cat. No. 22–042-924) and stored at room temperature before use. Anonymized ovarian tumour tissue microarray (TMA) construction and sectioning was performed according to best practices26. Unconjugated mouse anti-human AMBRA1 clone G-6 (Santa Cruz Biotechnology sc-398204, lot no. G1217, RRID: AB_2861324), and unconjugated rabbit polyclonal anti-human AMBRA1 (Proteintech Group 13762–1, lot no. 00005112 and 00017852, RRID: AB_10642007) were tested27. Unconjugated rabbit anti-human cyclin D1 clone SP4 (Ventana Medical Systems 790–4508, lot no. B08078, RRID: AB_2335988) was used. Chromogenic immunohistochemistry was performed on a Ventana Medical Systems Discovery Ultra platform using Ventana’s reagents and detection kits unless otherwise noted. Cyclin D1 (IVD) was used according to the manufacturer’s instructions28,29. AMBRA1 antibodies were optimized by first determining antigen retrieval requirements followed by serial dilution of primary antibody to establish immunohistochemistry assay conditions. In brief, slides were deparaffinized online and endogenous peroxidase activity was blocked with 3% hydrogen peroxide for 4 min. Antigen retrieval for both AMBRA1 antibodies was performed using Cell Conditioner 1 (Tris-EDTA pH8.5): 24 min at 95 °C for Santa Cruz Biotechnology and 48 min at 95 °C for Proteintech Group antibodies. Both antibodies were diluted 1:100 in Ventana antibody diluent (Ventana Medical Systems 760–018) and incubated for 12 and 6 h at room temperature, respectively. Primary antibody was detected using either goat anti-rabbit or goat anti-mouse horseradish peroxidase-conjugated multimer incubated for 8 min. The complex was visualized with 3,3 diaminobenzidene for 8 min and enhanced with copper sulfate for 4 min. All slides were washed in distilled water, counterstained with haematoxylin, dehydrated and mounted with permanent medium. Proteintech Group AMBRA1 conditions were modified (increased primary time to 10 h and goat anti-mouse horseradish peroxidase conjugated multimer incubation to 12 min) and applied to the ovarian TMA. Analysis and scoring of the TMA was performed by either automated image analysis software (ImmunoRatio, version 1.0c)30 for ImageJ (version 1.52o), or by visually determining the intensity of nuclear immunoreactivity (0, no expression; 1, faint expression and 2, mild–moderate expression) for AMBRA1. Images were obtained using an Olympus BX43 microscope fitted with an Olympus DP27 camera using Olympus CellSens Entry (version 1.18).
Xenotransplantation experiments
Either female or male, eight-week-old NSG mice received subcutaneous flank injections of 1 × 107 BJAB or U-2932 cells that expressed control sgRNA targeting luciferase or sgRNA targeting AMBRA1 (sgRNA#5) in 100 μl sterile PBS. Where indicated, mice were administered palbociclib isethionate (5 mg kg−1 or 50 mg kg−1 as indicated in Fig. 4; Selleckchem S1579) or PBS (as vehicle) via intraperitoneal injection for five consecutive days per week, until experimental end point. Treatment was started when tumours grew to 100 mm3. Experimental end point was considered when tumour volume reached 1,000 mm3 (U-2932) or exceeded 2,000 mm3 (BJAB), in accordance with the University of Pennsylvania IACUC guidelines. The institutional committee maximal tumour limits (4.2 cm3) were not exceeded. Tumour burden was monitored daily by palpation and visual inspection, and measured by caliper at the days indicated in the Source Data. Tumour volume was calculated as following: V = (L × W × W)/2, where V is tumour volume, L is tumour length and W is tumour width. Tumour weight was measured on the excised tumours at the experimental end point using an analytical scale.
Survival data analysis
To determine the association of AMBRA1 expression with survival in human cancer, we performed survival analysis using Transcriptome Profiling data (HTSeq–FPKM) from all available cohorts in the TCGA database whose sample number is greater than or equal to 80 (n = 10,547). The expression and clinical data were directly downloaded using the TCGAbiolinks package (v2.12.6) in R (v3.6.0). The RNA expression data of AMBRA1 was divided into high and low expression groups on the basis of either 20% or 50% quantiles. Kaplan–Meier survival curves analysis with log-rank testing were conducted and visualized using the survival package (v3.1) in R. In order to corroborate the survival data analysis from TCGA, we calculated a meta P value by combining the log-rank testing P values from each individual cohort analysed above, using the Fisher’s method. The association of AMBRA1 expression with survival in the diffuse large B-cell lymphoma (DLBCL) cohort was generated by combining the data from GEO accession GSE2350131 (n = 69) and GEO accession GSE1084632 (n = 414) downloaded from Oncomine (https://www.oncomine.org). To concatenate these two studies, we centre scaled the expression levels of AMBRA1 using the Affymetrix probe ‘52731_at’ for each study in R (scale function with the parameter centre = TRUE) and divided into high and low AMBRA1 expression on the basis of 10% quantiles, individually. Survival analysis was then performed as in the above method.
Differential expression analysis
Differential expression analysis from TCGA (DLBC cohort, n = 48) was conducted between two groups of samples as previously described33, and the list of the 300 most differentially expressed genes (two-sided Mann–Whitney U test, top and bottom 150 genes of the test statistics) was used to construct a heat map with the whole data set. Samples were clustered using the hierarchical clustering algorithm based on the complete linkage method and implemented on the Euclidean distance. Heat map and hierarchical clustering were generated and performed using the pheatmap package (v1.0.12) in R. Gene enrichment analysis of the differentially expressed genes was performed using Enrichr (https://amp.pharm.mssm.edu/Enrichr/) with BioPlant (2019), Reactome (2016) and NCI-Nature (2016) databases. Differential expression analyses of DLBCL patients from Compagno et al.34 (Affymetrix probe ‘222189_at,’ n = 64) and Brune et al.35 (Affymetrix probe ‘222189_at,’ n = 36) were generated in Oncomine (https://www.oncomine.org).
Correlation, mutation and methylation analyses
To determine the correlation between AMBRA1 mRNA expression levels as measured by RNA-Seq (FPKM-UQ) and cyclin D1 protein expression levels as measured by reverse phase protein array (RPPA), we performed the Pearson correlation analysis across individual TCGA cohorts and plotted the figures using ggpubr package (v0.4.0) in R. OncoPrint map of the TCGA genetic alterations (DLBC cohort) was downloaded from cBioPortal (https://www.cbioportal.org/) 36,37. CCND1, CCND2 and CCND3 mutation profiles were gathered and combined from cBioPortal (curated set of non-redundant studies), COSMIC (https://cancer.sanger.ac.uk/cosmic) 38 and supplementary table 8 from Schmitz et al.39. Redundant samples among the three data sets were removed by using their unique sample ID. Lolliplots showing the amino acid position and its mutation numbers were generated using the ggplot2 package (v3.2.1) in R. AMBRA1, FBXO11, CCNF and KEAP1 mutation profiles were downloaded from cBioPortal (curated set of non-redundant studies). CRISPR (Avana) Public 20Q1 pre-computed associations were downloaded from the Depmap portal (https://depmap.org/portal/). As a positive control for this method, we evaluated the dependencies of the genes encoding other established CRL substrate receptors that have a role in human cancer (that is, CCNF, FBXW7, VHL and KEAP1)40–43 and found that they inversely correlate with those of their substrates. Methylation data was downloaded from the TCGA (DLBC cohort) using SMART App (http://www.bioinfo-zs.com/smartapp) 44. CpG-aggregated methylation values (β-values, median aggregation method) were plotted in GraphPad Prism 8 (v8.3.1).
Statistical analyses
Data were analysed using GraphPad Prism 9.0.0 software, unless noted otherwise. Datasets were analysed using two-tailed statistical tests described in the figure legends.
Reporting summary
Further information on research design is available in the Nature Research Reporting Summary linked to this paper.
Data availability
All accession codes, unique identifiers, and web links for publicly available datasets are available within the article and in the Reporting Summary. Those who carried out the original analysis and collection of the data generated by the DECIPHER community bear no responsibility for the further analysis or interpretation of the data. Mass spectrometry data for cyclin D1 immunopurifications are provided in Supplementary Table 1. The full scanned images obtained by electrophoretic separation are provided in Supplementary Fig. 1. All original data are available from the corresponding authors upon request. Source data are provided with this paper.
Code availability
Custom scripts for image analysis are provided at GitHub (https://github.com/FenyoLab/CRL4AMBRA1_img_analysis). Custom scripts for cancer-related analyses are provided at GitHub (https://github.com/Lesdormis/AMBRA1_analysis).
Extended Data
Extended Data Fig. 1 |. CRL1 does not contribute to the degradation of D-type cyclins.
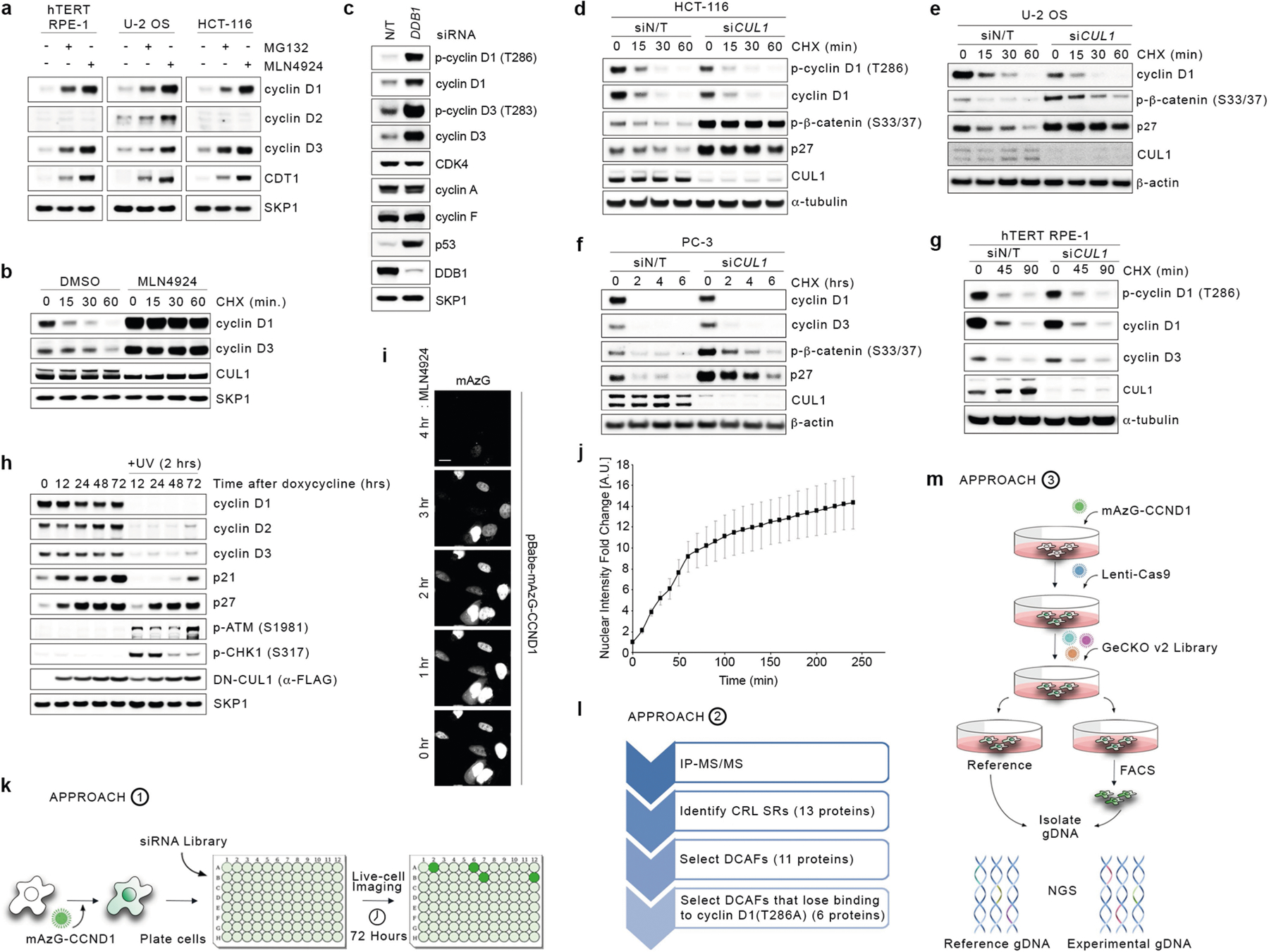
a, hTERT RPE-1, U-2 OS and HCT-116 cells were treated with MG132 or MLN4924 for 4 h before collection. Cell extracts were immunoblotted for the indicated proteins. b, HCT-116 cells were treated for 3 h with vehicle (DMSO) or MLN4924 followed by cycloheximide for the indicated times before collection. Cell extracts were immunoblotted for the indicated proteins. c, HCT-116 cells were transfected with a non-targeting (N/T) siRNA or an siRNA against DDB1 for three rounds before lysis and immunoblotting for the indicated proteins. d, HCT-116 cells were transfected with a non-targeting siRNA or an siRNA against CUL1 for three rounds followed by treatment with cycloheximide for the indicated times before collection. Cell extracts were immunoblotted as indicated. e, U-2 OS cells were transfected with a non-targeting siRNA or an siRNA against CUL1 for three rounds followed by treatment with cycloheximide for the indicated times before collection. Cell extracts were immunoblotted as indicated. f, PC-3 cells were transfected with a non-targeting (N/T) siRNA or an siRNA against CUL1 for three rounds followed by treatment with cycloheximide for the indicated times before collection. Cell extracts were immunoblotted as indicated. g, hTERT RPE-1 cells were transfected with a non-targeting siRNA or an siRNA against CUL1 for three rounds followed by treatment with cycloheximide for the indicated times before collection. Cell extracts were immunoblotted as indicated. h, U-2 OS cells were infected with a doxycycline-inducible lentivirus expressing a dominant-negative mutant of CUL1 (DN-CUL1). Cells were treated with doxycycline for the indicated times. Where indicated, cells were exposed to UV (5 mJ cm−2) irradiation for 2 h before collection. Cell extracts were immunoblotted as indicated. i, Representatives images of U-2 OS cells infected with a retrovirus expressing mAzG-CCND1 and treated with MLN4924 for the indicated times. Cells were monitored via live-cell imaging. Scale bar, 20 μm. j, Quantification of relative mAzG-CCND1 nuclear intensity fold change in arbitrary units (A.U.) from single cells tracked over time as in i. Data are mean ± s.d., n = 34 cells. k, Schematic representation of approach 1, aimed at measuring the accumulation of cyclin D1 upon silencing of all human DCAFs and the previously-reported regulators of D-type cyclins45–52 in HCT-116 and U-2 OS cells. l, Schematic representation of approach 2, aimed at identifying the binding partners of FFSS-tagged wild-type cyclin D1 and FFSS-tagged cyclin D1(T286A) compared with empty vector. HCT-116 cells were treated with DMSO, MG132 or MLN4924 for 4 h before lysis and immunoprecipitation with an anti-Flag resin, followed by elution with a 3×Flag peptide. Eluted samples were then analysed by LC-MS/MS for the identification of binding partners. m, Schematic representation of approach 3, aimed at identifying global regulators of cyclin D1 in HCT-116 cells using the whole-genome, CRISPR–Cas9 human GECKOv2 pooled library. Unless otherwise noted, experiments were performed at least three independent times.
Extended Data Fig. 2 |. Validation of candidate regulators of D-type cyclins.
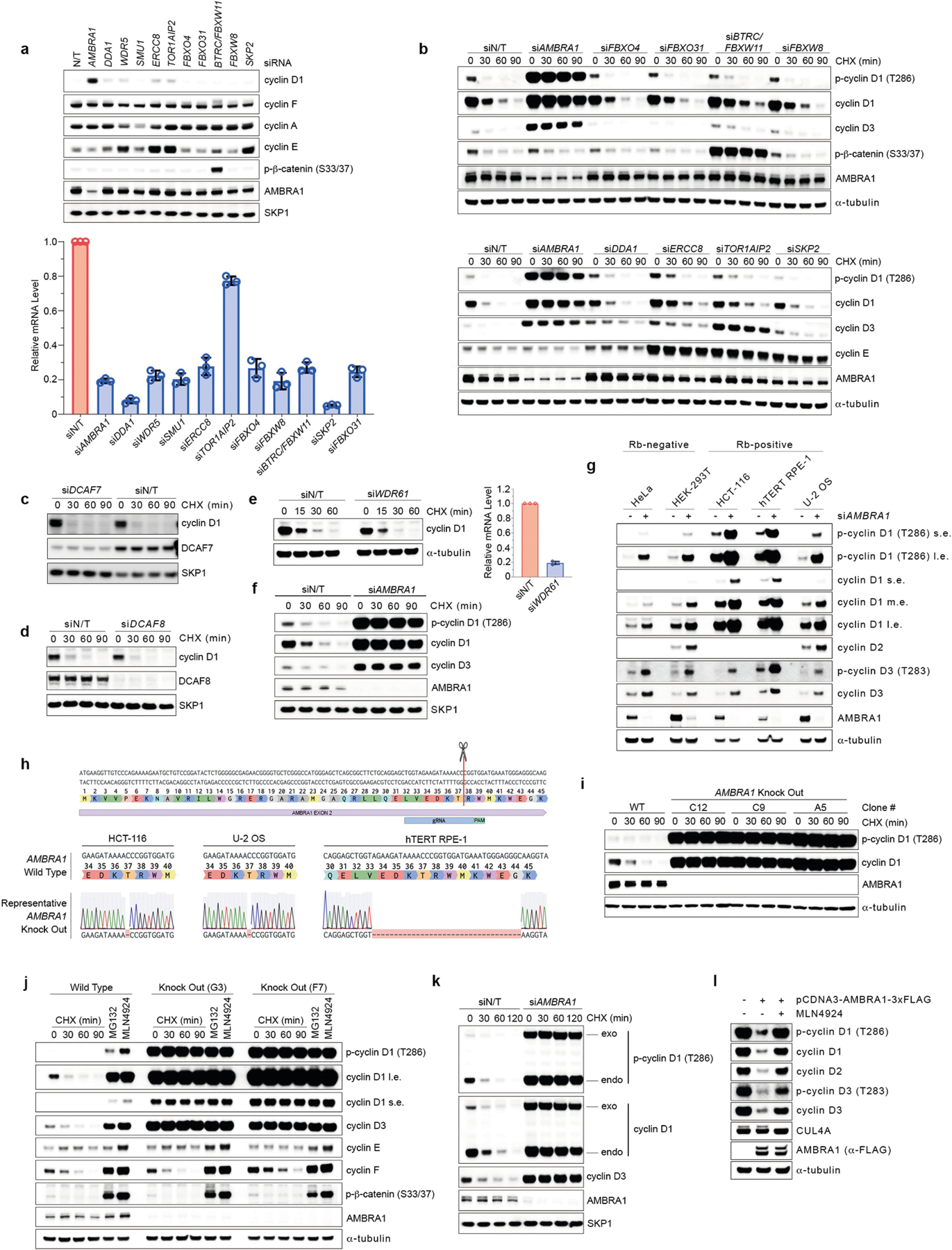
a, HCT-116 cells were transfected with a non-targeting siRNA or siRNAs against the indicated targets for two rounds before collection. Cell extracts were immunoblotted as indicated (upper panel). RNA extracts were also collected to measure the relative mRNA levels for each indicated target by quantitative PCR with reverse transcription (RT–qPCR). Each blue column represents the mRNA levels of the target indicated by the respective siRNA and normalized to its corresponding mRNA levels in cells transfected with a non-targeting siRNA (bottom). Data are mean mRNA levels ± s.d., n = 3 independent experiments. b, HCT-116 cells were transfected with a non-targeting siRNA or siRNAs against the indicated targets for two rounds followed by treatment with cycloheximide for the indicated times before collection. Cell extracts were immunoblotted as indicated. c, HCT-116 cells were transfected with a non-targeting siRNA or an siRNA against DCAF7 followed by treatment with cycloheximide for the indicated times before collection. Cell extracts were immunoblotted as indicated. d, U-2 OS cells were transfected with a non-targeting siRNA or an siRNA against DCAF8 followed by treatment with cycloheximide for the indicated times before collection. Cell extracts were immunoblotted as indicated. e, U-2 OS cells were transfected with a non-targeting siRNA or an siRNA against WDR61 followed by treatment with cycloheximide for the indicated times before collection. Cell extracts were immunoblotted as indicated (left). RNA extracts were also collected to measure the relative mRNA levels of WDR61 by RT–qPCR (right). Data are mean mRNA levels ± s.d., n = 3 independent experiments. f, hTERT RPE-1 cells were transfected with a non-targeting siRNA or an siRNA against AMBRA1 followed by treatment with cycloheximide for the indicated times before collection. Cell extracts were immunoblotted as indicated. g, HeLa, HEK 293T, HCT-116, hTERT RPE-1 and U-2 OS cells were transfected with a non-targeting siRNA or an siRNA against AMBRA1 before collection. Cell extracts were immunoblotted for the indicated proteins (l.e., long exposure; m.e., medium exposure; s.e., short exposure). h, Schematic representation of the AMBRA1 genomic locus and gRNA target location. Exon 2 refers to the human AMBRA1 gene in XM_005253009 (GRCh38. p13, NCBI gene: 55626). Representative AMBRA1 wild-type genomic DNA template and knockout mutant sequences identified by TOPO-TA cloning of PCR products from HCT-116, U-2 OS and hTERT-RPE1 cells are depicted. i, Three AMBRA1−/− and one AMBRA1+/+ hTERT RPE-1 clones were treated with cycloheximide for the indicated times, after which cell extracts were immunoblotted for the indicated proteins. j, Two AMBRA1−/− and one AMBRA1+/+ T98G clones were treated with either cycloheximide for the indicated times, MG132 or MLN4924 (both for 4 h) before collection. Cell extracts were then immunoblotted for the indicated proteins. k, HCT-116 cells were infected with a retrovirus expressing mAzG-CCND1 and transfected with a non-targeting siRNA or an siRNA against AMBRA1 for two rounds. Cells were then treated with cycloheximide for the indicated times, after which cell extracts were immunoblotted for the indicated proteins (endo, endogenous; exo, exogenous). l, AMBRA1−/− U-2 OS cells were transiently transfected with a vector expressing a C-terminal 3×Flag-tagged AMBRA1 or an empty vector for 96 h. Where indicated, cells were treated with MLN4924 for 4 h before lysis. Cell extracts were immunoblotted as indicated. Unless otherwise noted, experiments were performed at least three independent times.
Extended Data Fig. 3 |. Depletion of AMBRA1 using the auxin-inducible degron system.
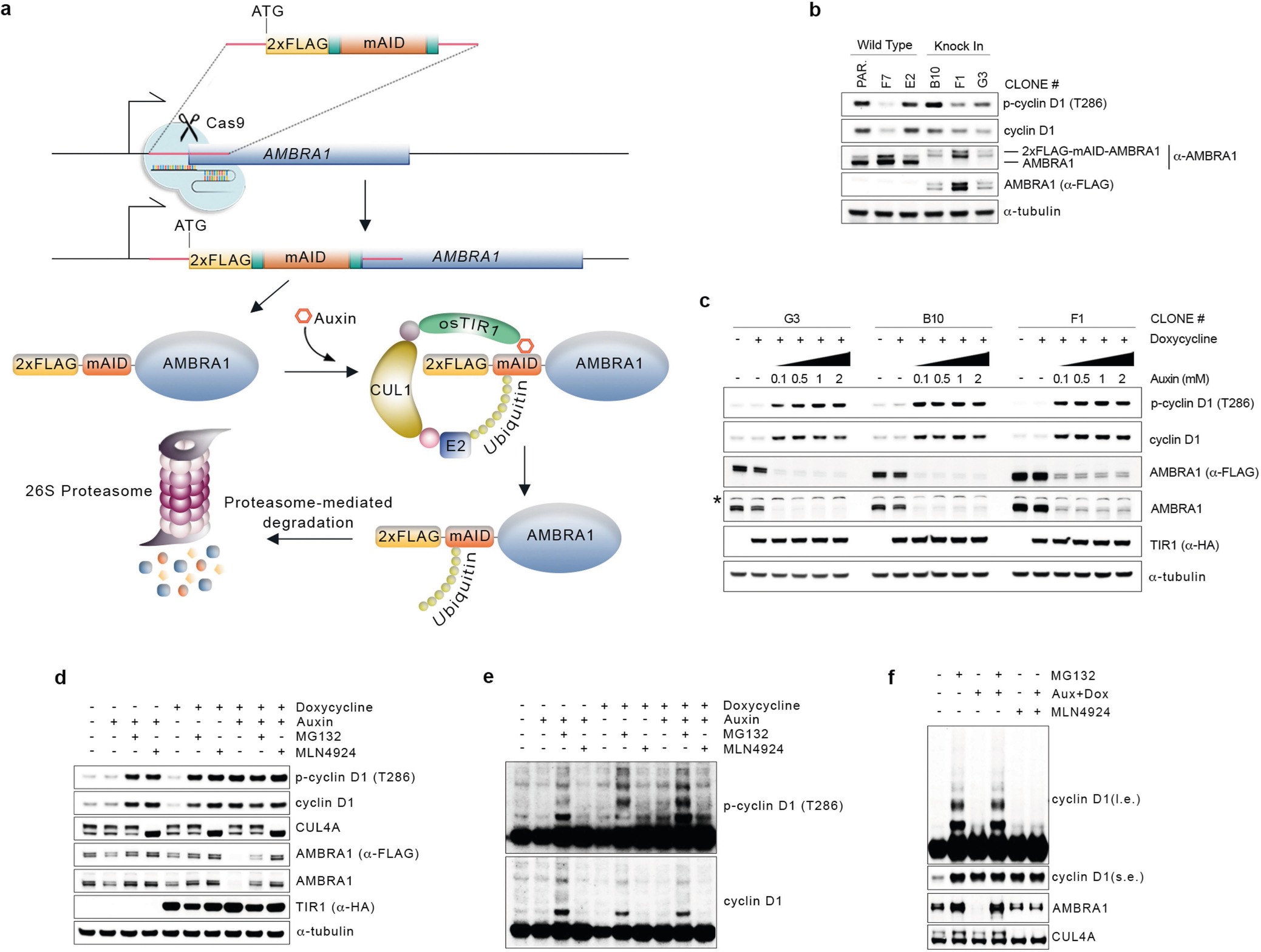
a, Schematic representation of the CRISPR–Cas9-based strategy to insert an N-terminal, 2×Flag-tagged minimal AID (mAID) in the endogenous locus of AMBRA1 (5′-end of exon 2) in HCT-116 cells. Cells were infected with a doxycycline-inducible lentivirus expressing Oryza sativa TIR1 (osTIR1). b, Three knock-in clones were obtained as in a and compared to wild-type clones for proper endogenous tagging of AMBRA1 by immunoblotting. Note the higher molecular weight of 2×Flag-mAID-tagged AMBRA1. c, Three knock-in clones were treated with doxycycline with or without increasing doses of auxin for 12 h. Cell extracts were immunoblotted as indicated. The asterisk indicates a non-specific band. d, One clone of 2×Flag-mAID-AMBRA1 HCT-116 cells (clone #F1) was treated with either DMSO or doxycycline for 12 h, and/or auxin, MG132 and MLN4924 (alone or in combination) for 4 h as indicated. Cell extracts were immunoblotted as indicated. e, Longer exposure from the cyclin D1 and phospho-cyclin D1 (T286) immunoblots described in d. f, Whole-cell extracts from 2×Flag-mAID-AMBRA1 HCT-116 cells pre-treated with either DMSO or doxycycline for 12 h, and exposed to combinations of auxin, MG132 and/or MLN4924 as indicated for 4 h, related to Fig. 2d. Cell extracts were immunoblotted as indicated. Unless otherwise noted, experiments were performed at least three independent times.
Extended Data Fig. 4 |. Mutations in the TP motif of D-type cyclins protect them from AMBRA1-mediated degradation.

a, Protein sequence alignment of the extreme C termini of D-type cyclins from different source organisms. Amino acids highlighted in red correspond to the highly conserved TP motif. Asterisks indicate positions that are fully conserved. Colons indicate conservation between groups of strongly similar properties. The dot indicates conservation between groups of weakly similar properties. The alignment was performed in UniProt (https://www.uniprot.org/) using the Clustal Omega algorithm. UniProt identifiers are shown on the left. Source organisms: HUMAN, H. sapiens; BOVIN, Bos taurus; CANLF, Canis familiaris; MOUSE, Mus musculus; RAT, Rattus norvegicus; DANRE, Danio rerio; BRAFL, Branchiostoma floridae; STRPU, Strongylocentrotus purpuratus; CRAGI, Crassostrea gigas; LOTGI, Lottia gigantean; OCTBM, Octopus bimaculoides; STRMM, Strigamia maritima; BOMMO, Bombyx mori; ATTCE, Atta cephalotes; APIME, Apis mellifera; CULQU, Culex quinquefasciatus; HYDVU, Hydra vulgaris; NEMVE, Nematostella vectensis; TRIAD Trichoplax adhaerens. b, Multiple sequence logo of the alignment performed in a generated using WebLogo 3 (http://weblogo.threeplusone.com)53. c, HCT-116 cells were infected with lentiviruses expressing either FFSS-tagged empty vector (EV) or FFSS-tagged wild-type or mutant cyclin D1. Cells were treated with MLN4924 before lysis and affinity purification (AP) with an anti-STREP resin. Proteins were immunoblotted as indicated. d, HEK 293T cells were transiently transfected with plasmids expressing either SF-tagged empty vector or SF-tagged D-type cyclins wild-type or mutants. Cells were treated with MLN4924 before lysis and immunoprecipitation with an anti-Flag resin. Proteins were immunoblotted as indicated. e, HCT-116 cells were infected with retroviruses expressing FFSS-tagged wild-type or mutant cyclin D1. Cells were treated with cycloheximide for the indicated times before collection. Cell extracts were immunoblotted as indicated. f, HCT-116 cells were infected with retroviruses expressing FFSS-tagged wild-type or mutant cyclin D2. Cells were treated with cycloheximide for the indicated times before collection. Cell extracts were immunoblotted as indicated. g, HCT-116 cells were infected with retroviruses expressing FFSS-tagged wild-type or mutant cyclin D3. Cells were treated with cycloheximide for the indicated times before collection. Cell extracts were immunoblotted as indicated. h, HCT-116 cells were transfected with vectors expressing SF-tagged CDC25A, SF-tagged p27, SF-tagged cyclin D1, SF-tagged cyclin D1(T286A) or empty vector. Cells were treated with MLN4924 for 4 h before collection. Lysates were used for an immunoprecipitation with an anti-Flag resin. Proteins were immunoblotted as indicated. i, HCT-116 cells were transfected with vectors expressing HA-tagged FBXO4, SF-tagged TRF1, SF-tagged cyclin D1, SF-tagged cyclin D1(T286A) or empty vector. Cells were treated with MLN4924 for 4 h before collection. Lysates were used for an affinity purification with an anti-Strep resin. Proteins were immunoblotted as indicated. The asterisk indicates a non-specific band. j, HCT-116 cells were transfected with vectors expressing HA-tagged FBXW8, SF-tagged IRS1, SF-tagged cyclin D1, SF-tagged cyclin D1(T286A) or empty vector. Cells were treated with MLN4924 for 4 h before collection. Lysates were used for an immunoprecipitation with an anti-Flag resin. Proteins were immunoblotted as indicated. k, Parental and 2×Flag–mAID–AMBRA1 HCT-116 cells were lysed and their extracts were used for an immunoprecipitation with an anti-Flag resin. Proteins were immunoblotted as indicated. l, One clone of 2×Flag–mAID–AMBRA1 HCT-116 cells (clone F1) was infected with retroviruses expressing GFP-CCND1 or GFP-CCND1(T286A). Cells were treated with doxycycline for 12 h, before exposure to auxin for 4 h before collection as indicated. Control cells were treated with DMSO. Cell extracts were immunoblotted as indicated. m, Parental and AMBRA1−/− HCT-116 and U-2 OS cells were lysed and their extracts were blotted as indicated. The accumulation of cyclin D1 and phosphorylated cyclin D1 (T286) is reported as their corresponding intensity ratios between AMBRA1−/− and parental cells. Data are mean ± s.e.m. (n = 3 biological replicates per group). n, Three different AMBRA1−/− HCT-116 clones (clones D2, F11 and G7), 2×Flag-mAID-AMBRA1 HCT-116 cells (clone F1) and HCT-116 cells transfected with a non-targeting siRNA or an siRNA against AMBRA1 were lysed and their extracts were immunoblotted as indicated (either by employing fluorescently labelled secondary antibodies or by enhanced chemiluminescence). Untransfected CCND1−/− HCT-116 cells or CCND1−/− HCT-116 cells transfected with vectors expressing untagged cyclin D1(WT) or untagged cyclin D1(T286A) were used as control. Where indicated (black triangles), samples were serially diluted before electrophoresis and immunoblotting for easier visual comparison of cyclin D1 and phosphorylated cyclin D1 (T286) levels between groups. The analysis of normalized phosphorylated cyclin D1 (T286) over total cyclin D1 fluorescent intensity levels are provided as bar graphs. The ratios were calculated from lane 2 and lane 4 from each biological group in the fluorescent immunoblot. o, HCT-116 cells were infected with lentiviruses expressing FFSS-tagged cyclin D1 or FFSS-tagged cyclin D1(T286A). Cells were treated with DMSO (N/T) or MLN4924 for 4 h before lysis and immunoprecipitation with an anti-Flag resin, followed by elution with a 3×Flag peptide. Eluted samples were then analysed by LC-MS/MS for the identification of post-translational modifications. The table reports all identified sites in wild-type cyclin D1 and cyclin D1(T286A) that were modified with a di-Gly/114.0429 signature. p, Synthetic, unphosphorylated (amino acids PKAAEEEEEEEEEVDLACTPTDVRDVDI) and phosphorylated (T286) (amino acids PKAAEEEEEEEEEVDLAC-pT-PTDVRDVDI) peptides (2.5 μg each) corresponding to the extreme C terminus of cyclin D1 were spotted on a nitrocellulose membrane before immunoblotting with an anti-phosphorylated cyclin D1 (T286) antibody. q, Synthetic, phosphorylated (T286) peptide corresponding to the sequence of cyclin D1 indicated in p was incubated with in vitro transcribed and translated AMBRA1 from rabbit reticulocytes extracts for the indicated times in the presence of wild-type ubiquitin (Ub) or a mutant of ubiquitin [Ub(K0)] in which all lysine residues have been mutated to arginine. Reactions were stopped with Laemmli buffer. Proteins were immunoblotted as indicated. Unless otherwise noted, experiments were performed at least three independent times.
Extended Data Fig. 5 |. Loss of AMBRA1 regulates D-type cyclins during normal cell cycle, upon nutrient deprivation and after genotoxic stress.
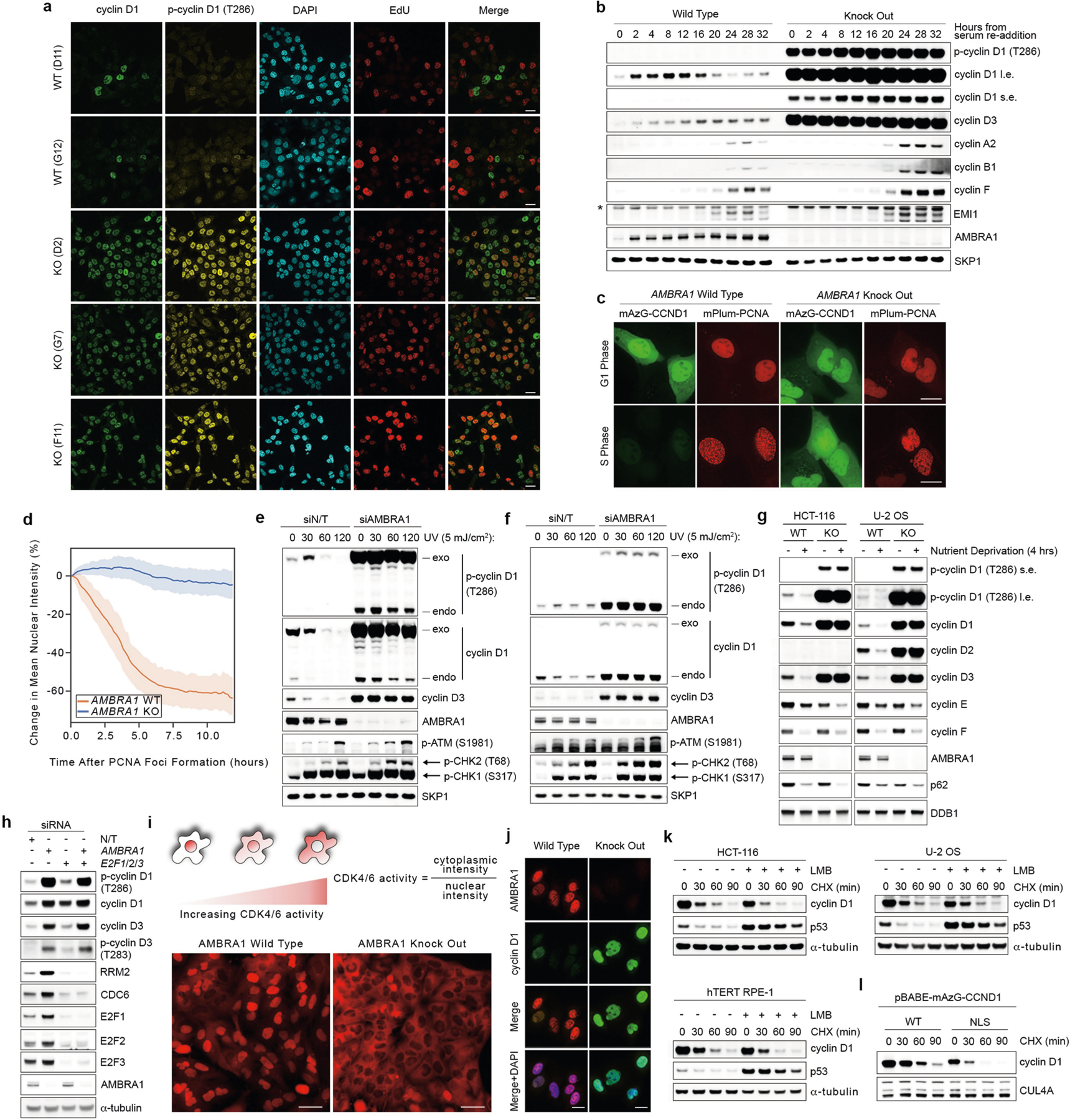
a, Three AMBRA1−/− and two AMBRA1+/+ clones were pulsed with EdU for 45 min before fixation with 4% paraformaldehyde, and immunofluorescent staining for the indicated proteins. Detection of EdU was accomplished using the Click-iT Plus EdU Alexa Fluor 647 Imaging Kit (Thermo Fisher Scientific). DAPI was used to detect DNA. Scale bars, 20 μm. b, AMBRA1+/+ and AMBRA1−/− T98G cells were serum-deprived for 72 h. Cells were then released with the re-addition of 10% FBS and collected at the indicated times. Cell extracts were immunoblotted as indicated. c, Representative images of AMBRA1+/+ and AMBRA1−/− U-2 OS cells stably infected with retroviruses expressing mAzG-CCND1 and mPlum-PCNA and monitored via live-cell imaging. Note that mPlum-PCNA forms nuclear foci during S phase. Scale bar, 20 μm. d, Quantification of mAzG-CCND1 changes in mean nuclear intensity in single cells tracked over time related to c. Plots represent percentage change from t = 0 (PCNA foci formation). Lines show mean and the shaded regions represent the 95% confidence interval. AMBRA1 WT, n = 12 cells; AMBRA1 KO, n = 34 cells. e, U-2 OS cells were infected with a retrovirus expressing mAzG-cyclin D1, and transfected with a non-targeting siRNA or an siRNA against AMBRA1 for two rounds. Cells were then exposed to UV (5 mJ cm−2) irradiation for the indicated times before collection. Cell extracts were immunoblotted as indicated. f, HCT-116 cells were infected with a retrovirus expressing mAzG-cyclin D1, and transfected with a non-targeting siRNA or an siRNA against AMBRA1 for two rounds. Cells were then exposed to UV (5 mJ cm−2) irradiation for the indicated times before collection. Cell extracts were immunoblotted as indicated. g, AMBRA1+/+ and AMBRA1−/− HCT-116 and U-2 OS cells were maintained in normal medium or nutrient-deprived in EBSS for 4 h before collection. Cell extracts were immunoblotted as indicated. h, HCT-116 cells were transfected with a non-targeting siRNA or combinations of siRNAs against AMBRA1, E2F1, E2F2 and E2F3 as indicated for three rounds before lysis and immunoblotting for the indicated proteins. i, T98G cells were transfected with an sgRNA control or an sgRNA targeting AMBRA1. AMBRA1−/− clones positive for gene editing were pooled together. Parental and AMBRA1−/− pooled T98G cells were then infected with a CDK4/6 activity reporter54 and seeded in glass-bottom plates. Three days after seeding, cells were stained with Hoechst 33342 before imaging. Scale bars, 50 μm. j, AMBRA1+/+ and AMBRA1−/− U-2 OS cells were fixed in 4% paraformaldehyde and immunostained as indicated. DAPI was used to detect DNA. Scale bars, 20 μm. k, HCT-116, U-2 OS and hTERT RPE-1 cells were treated with leptomycin B (LMB) for 4 h before exposure to cycloheximide for the indicated times. Cell extracts were immunoblotted as indicated. l, U-2 OS cells were infected with retroviruses expressing either mAZG-tagged wild-type cyclin D1 or cyclin D1 fused to a nuclear localization signal (NLS). Cells were then treated with cycloheximide for the indicated times before collection. Cell extracts were immunoblotted as indicated. Unless otherwise noted, experiments were performed at least three independent times.
Extended Data Fig. 6 |. Report of MPPH-derived mutations in CCND2 and evidence of the ability of abemaciclib to cross the murine placental barrier.
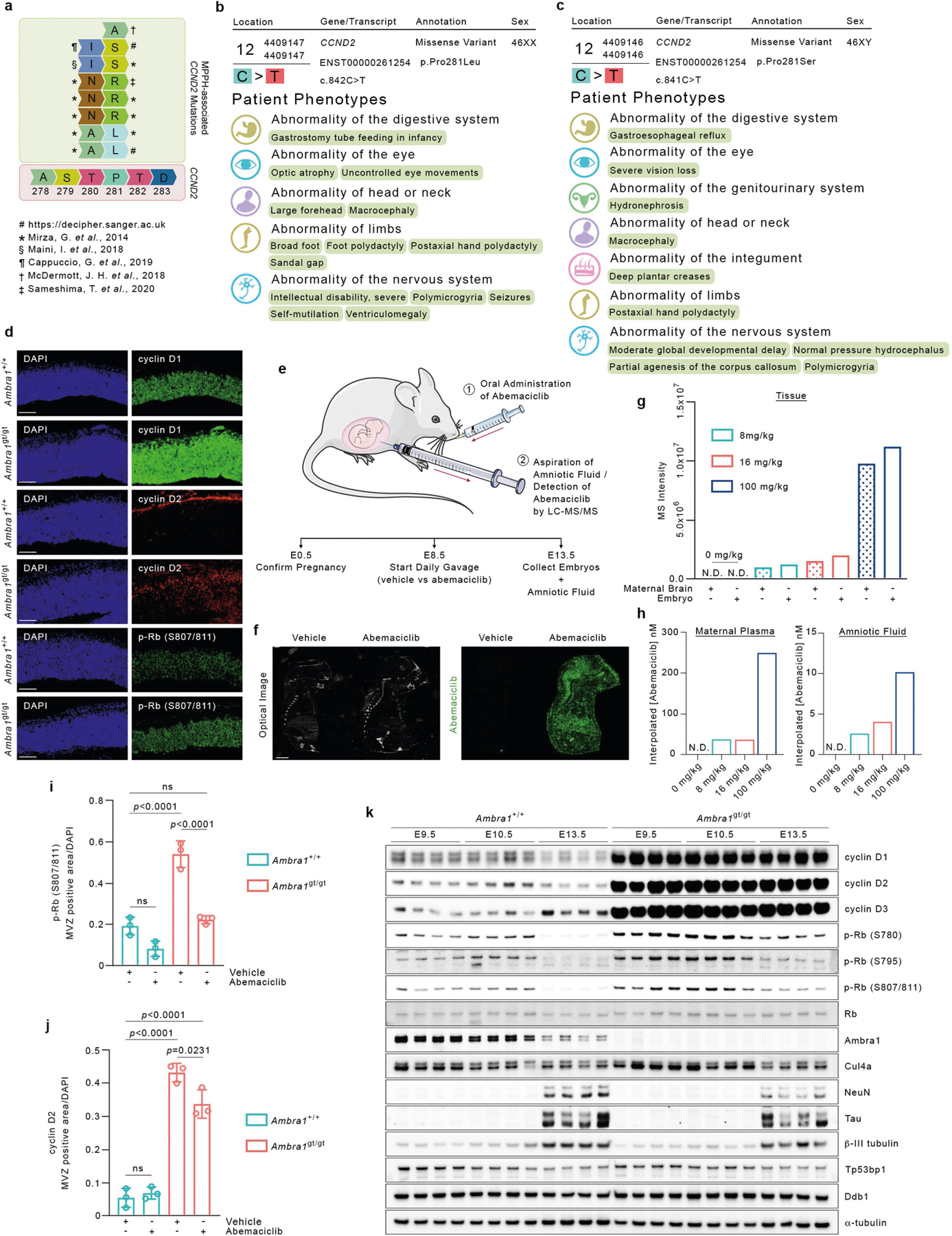
a, Graphical representation of MPPH-associated CCND2 mutations affecting T280 and P281 in cyclin D255–60. b, Genotype and clinical phenotypes of patient 295112 (https://decipher.sanger.ac.uk) 59. c, Genotype and clinical phenotypes of patient 305416 (https://decipher.sanger.ac.uk) 59. d, Representative immunofluorescent sections (20× magnification) of the mesencephalic ventricular zone of E13.5 Ambra1+/+ and Ambra1gt/gt embryos stained for cyclin D1, cyclin D2 or p-RB (S807/811). Scale bars, 100 μm. e, Experimental timeline of the daily, oral administration of abemaciclib to pregnant Ambra1gt/+ mice (from E8.5 to E13.5) (1), and collection of amniotic fluid via a 28G insulin syringe from the amniotic sacs of E13.5 embryos for subsequent LC-MS/MS analysis (2). f, Mass spectrometry imaging of abemaciclib in mouse embryos. Left, optical registration image of the slide with each section before MALDI imaging. Right, extracted ion mobility image for abemaciclib (green) as detected based on authentic standards [M+H]+ at 507.2791 m/z (±15 ppm) with a mobility (1/K0) of 1.177 (±1%), maternal dose of 100 mg kg−1. Scale bar, 2 mm. This experiment was performed once. g, h, MS intensity of abemaciclib in tissue samples (g), interpolated abemaciclib concentration in maternal plasma (h, left) and interpolated abemaciclib concentration in amniotic fluid (h, right). Maternal brain, embryonic tissue, amniotic fluid and maternal plasma samples were obtained from mice administrated with daily dosages of abemaciclib mesylate at 8 mg kg−1 or 16 mg kg−1 from E8.5 to E13.5, and at 100 mg kg−1 from E12.5 to E13.5. The abemaciclib MS intensity shown for tissue samples was corrected based on a 3× signal-to-noise cut-off with a floor of 10,000 using the blank controls. The abemaciclib concentration shown in both the amniotic fluid and plasma sample was interpolated based on the standard curve prepared in blank amniotic fluid and blank plasma, respectively. N.D., not detected. This experiment was performed once. i, j, E13.5 Ambra1+/+ and Ambra1gt/gt embryos were collected from pregnant mice treated with vehicle (PBS) or 8 mg kg−1 abemaciclib. Immunofluorescence from sections was quantified to show area of p-RB (S807/811) (i) and cyclin D2 (j) staining in the mesencephalic ventricular zone (MVZ). Data are mean ± s.d. p-Rb (S807/811), n = 3; cyclin D2, n = 3. Adjusted P values were calculated using a two-way ANOVA with Tukey’s multiple comparisons test. k, Protein extracts derived from E9.5, E10.5 and E13.5 Ambra1+/+ and Ambra1gt/gt embryo heads (four independent embryos per group) were immunoblotted for the indicated proteins. Unless otherwise noted, experiments were performed at least three independent times.
Extended Data Fig. 7 |. AMBRA1 and D-type cyclins in human cancer.
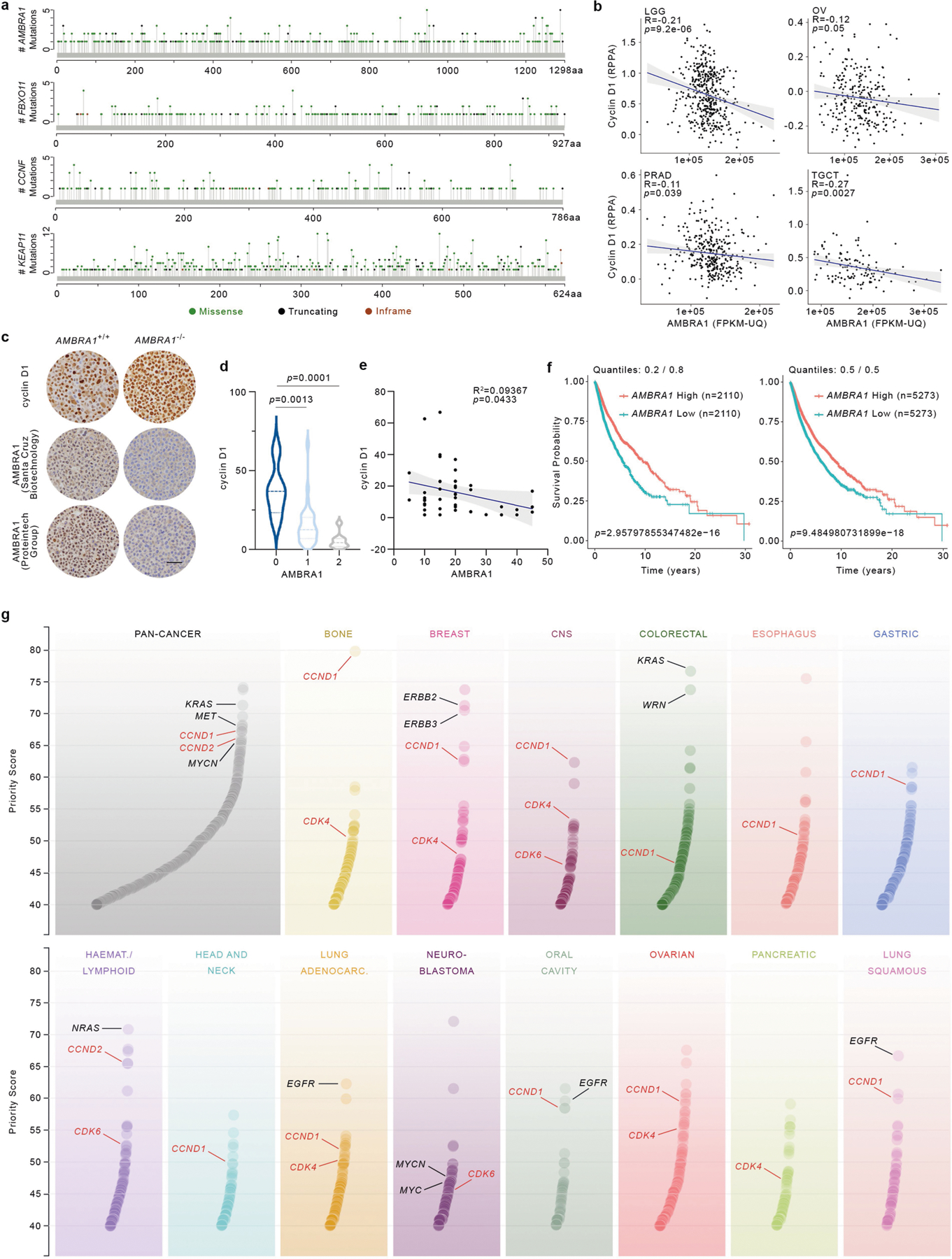
a, Lolliplots showing amino acid positions and numbers of mutations in AMBRA1, FBXO11, CCNF and KEAP1 in pan-cancer samples generated from cBioPortal (https://www.cbioportal.org/) and using the curated set of non-redundant studies36,37 (last accessed: December 2019). b, Scatter plot of cyclin D1 protein levels (RPPA) and mRNA expression levels of AMBRA1 (FPKM-UQ) in different TCGA cohorts. LGG, n = 435; OV, n = 257; PRAD, n = 344; TGCT, n = 124. Pearson correlation analysis was performed to calculate P and R values. The shaded area represents the 95% confidence interval of the regression line. c, Representative immunohistochemistry image fields of AMBRA1−/− and AMBRA1+/+ HCT-116 plasma-thrombin cell blocks stained with either a rabbit polyclonal anti-human AMBRA1 antibody (Proteintech Group 13762–1, lot nos. 00005112 and 00017852, RRID: AB_10642007), a mouse anti-human AMBRA1 clone G-6 antibody (Santa Cruz Biotechnology sc-398204, lot no. G1217, RRID: AB_2861324), or a rabbit anti-human cyclin D1 clone SP4 antibody (Ventana Medical Systems 790–4508, lot no. B08078, RRID: AB_2335988). Scale bar, 50 μm. This experiment was performed once. d, A TMA containing normal and tumour ovarian human specimens was immunostained with antibodies to AMBRA1 and cyclin D1. Violin plots show automated quantification of cyclin D1 intensity levels and pathological scoring of nuclear AMBRA1 intensity levels (0, no expression; 1, faint expression; 2, mild or moderate expression) (n = 44 biologically independent samples). Adjusted P values were calculated using a one-way ANOVA with Dunnett’s multiple comparisons test. e, The graph shows automated quantification of cyclin D1 and AMBRA1 intensity levels from the TMA described in d. n = 44 biologically independent samples. Pearson correlation analysis was performed to calculate P and R2 values. The shaded area represents the 95% confidence interval of the regression line. f, Kaplan–Meier plots showing survival probability of human cancer patients partitioned in low AMBRA1 mRNA levels (n = 2,110 for quantiles 0.2/0.8, and n = 5,273 for quantiles 0.5/0.5) and high AMBRA1 mRNA levels (n = 2,110 for quantiles 0.2/0.8, and n = 5,273 for quantiles 0.5/0.5; TCGA (https://cancer.gov/tcga)). P values were calculated using the log-rank test. Meta P values: 0.0389 (for quantiles 0.2/0.8), 0.0475 (for quantiles 0.5/0.5). g, Priority targets from pan-cancer and cancer-type analyses, as previously described9. CCND1, CCND2, CDK4 and CDK6 are highlighted in red. Select oncogenes are in black.
Extended Data Fig. 8 |. C-terminal cancer mutations affecting cyclin D1, cyclin D2 and cyclin D3.
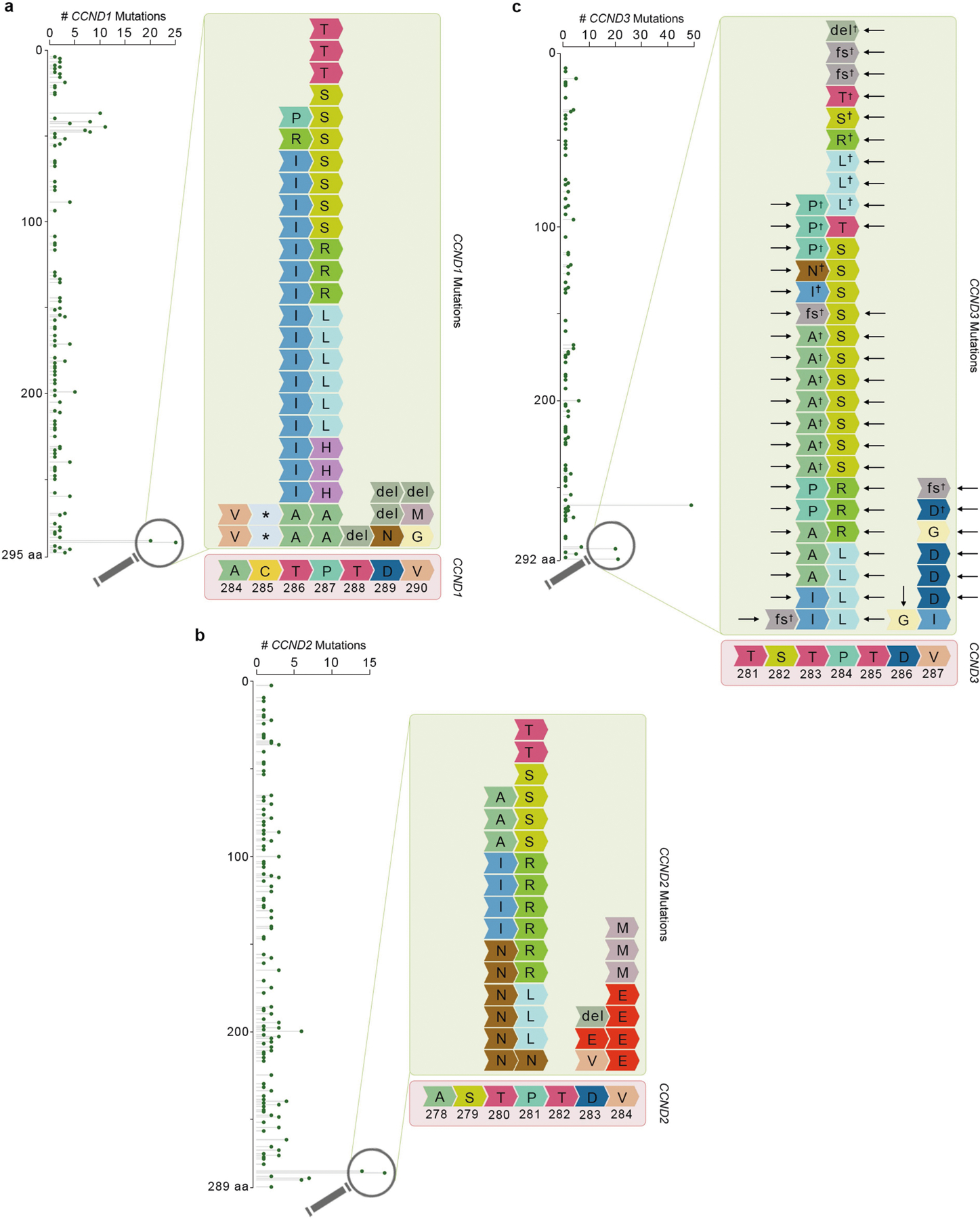
a–c, Lolliplots showing the amino acid positions and their mutation numbers in the degron motif of CCND1 (a), CCND2 (b) and CCND3 (c) in pan-cancer, gathered and combined from cBioPortal (curated set of non-redundant studies)36,37, COSMIC (https://cancer.sanger.ac.uk/cosmic) 38, and Schmitz et al.39 (indicated by the dagger). Black arrows in c indicate mutations identified in haematopoietic and lymphoid malignancies. del, deletion; fs, frameshift; *, stop codon. Last accessed October 2019.
Extended Data Fig. 9 |. AMBRA1 CRISPR gene score distribution in DLBCL.
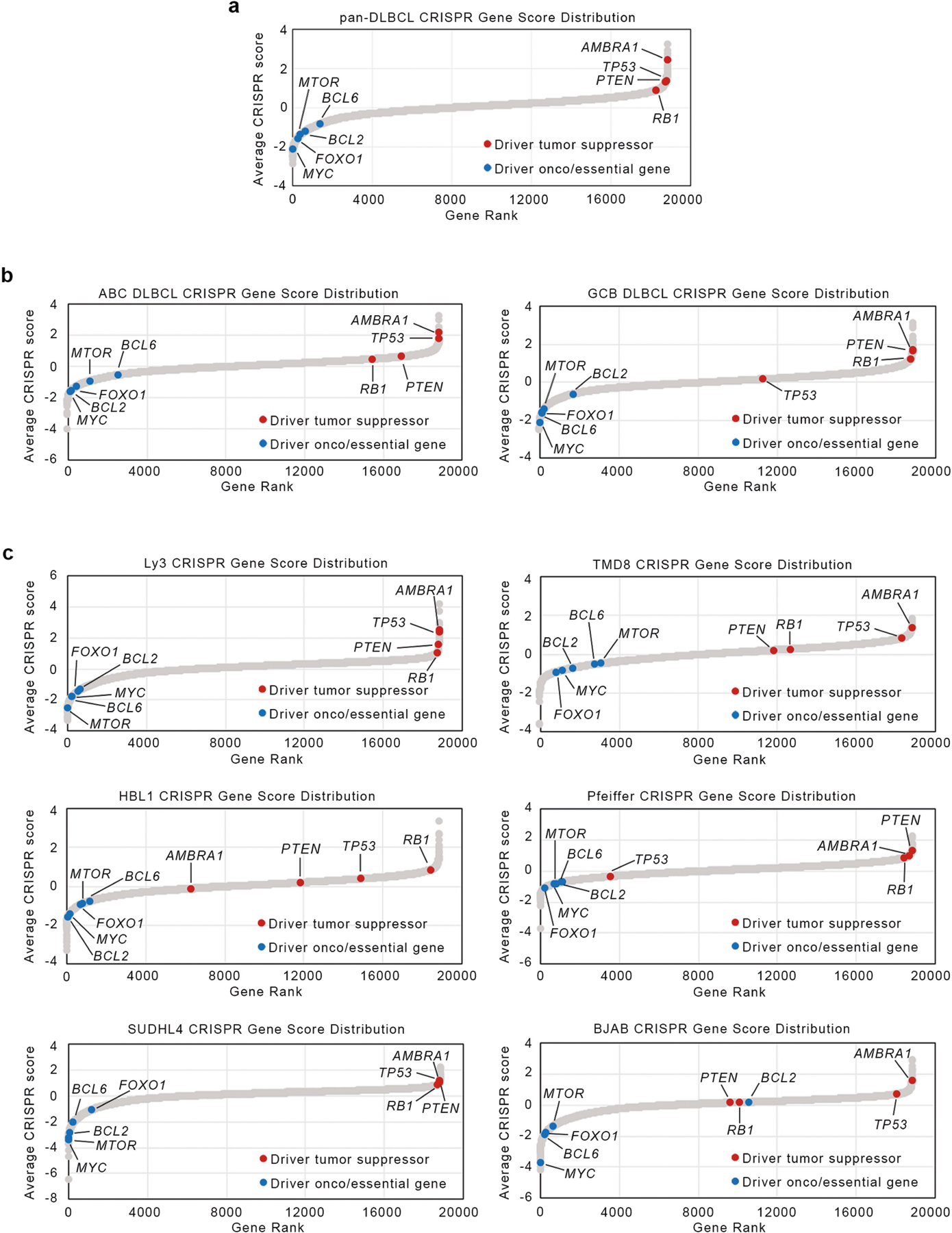
a–c, Average pan- (a), subtype-specific (b) and cell-type specific (c) DLBCL CRISPR gene score distribution, as reported11. ABC, activated B cell-like; GCB, germinal centre B cell-like. Putative oncogenes are depicted in blue; putative tumour suppressors are depicted in red.
Extended Data Fig. 10 |. AMBRA1 is a tumour suppressor in DLBCL.
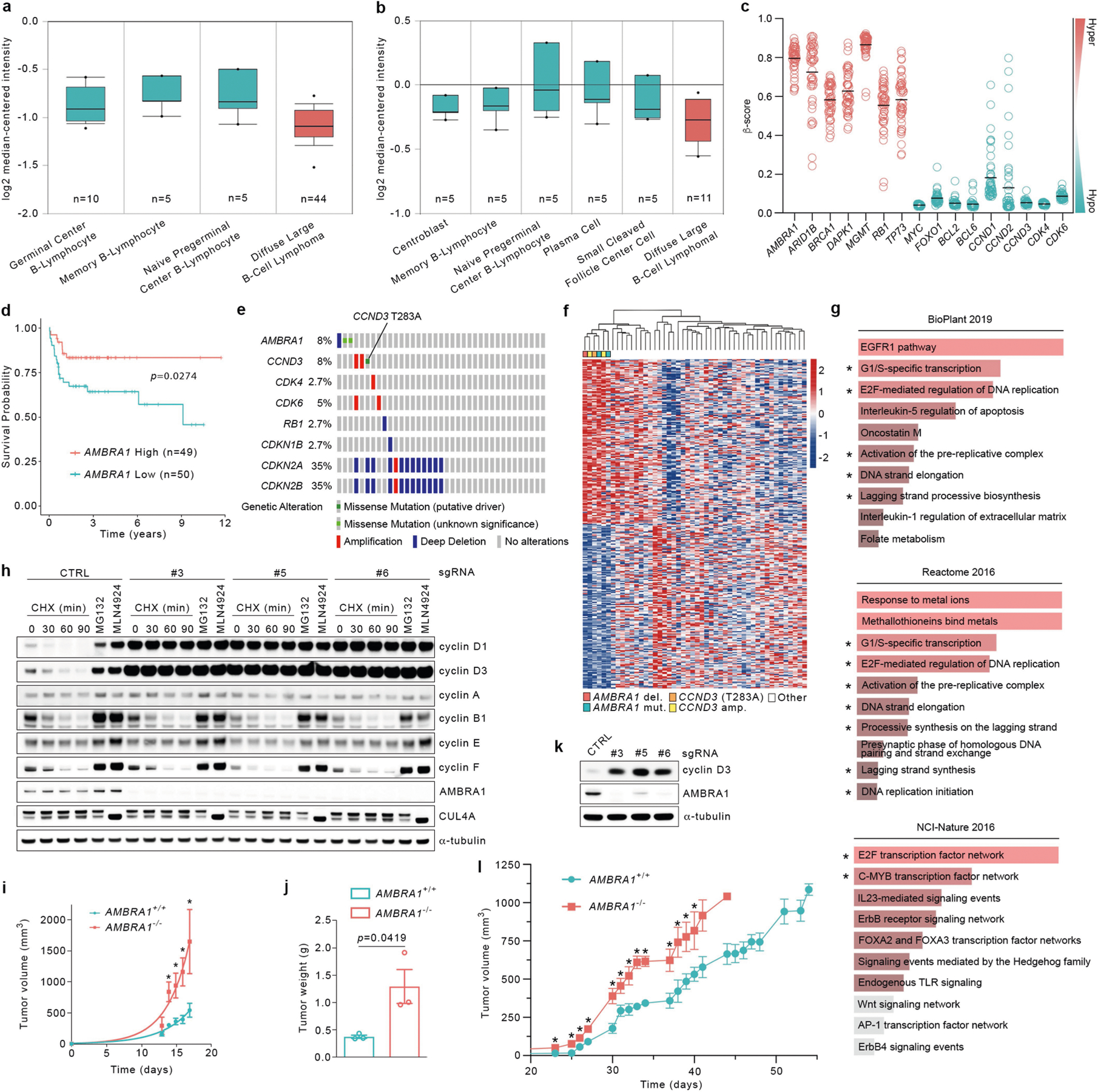
a, Differential expression of AMBRA1 in DLBCL compared with normal B-lymphocytes from Compagno et al.34. P = 9.91 × 10−6. b, Differential expression of AMBRA1 in DLBCL compared with normal B-lymphocytes from Brune et al.35. P = 0.003. a, b, Dots indicate maxima and minima; whiskers indicate 90th and 10th percentiles; box edges indicate 75th and 25th percentiles; and centre lines indicate median; P values were calculated using a Student’s t-test, as described (https://www.oncomine.org). c, TCGA-derived, CpG-aggregated methylation values (β-values) of AMBRA1 and other established hypermethylated genes in cancer (depicted in red) compared with hypomethylated genes (depicted in green) in DLBCL. Solid lines denote the mean of β-values for each group. d, Kaplan–Meier plot showing survival probability of patients with DLBCL partitioned in low AMBRA1 mRNA levels (n = 50) and high AMBRA1 mRNA levels (n = 49), combining GEO number GSE2350131 and GEO number GSE1084632, using probe 52731_at (https://www.oncomine.org) (0.10/0.90 quantiles). P values by log-rank test. e, OncoPrint map of the genetic alterations in AMBRA1 and selected RB-pathway genes; TCGA DLBC cohort. f, Heat map showing hierarchical clustering of transcriptional signatures of tumours bearing alterations in either AMBRA1 or CCND3, derived from TCGA DLBC cohort. del, deletion; mut, mutation; amp, amplification; other, tumours bearing unaltered AMBRA1 and CCND3. AMBRA1 (RefSeq XM_005253009) mutations: R439K and D1287G. CCND3 (RefSeq NM_001760) mutation: T283A. g, Gene enrichment analysis of the top 300 most differentially expressed genes in patients harbouring alterations in AMBRA1 and CCND3 from TCGA (DLBC cohort) was performed using Enrichr (https://amp.pharm.mssm.edu/Enrichr/) with BioPlant (2019), Reactome (2016) and NCI-Nature (2016) databases. Asterisks indicate pathways regulated by D-type cyclins. h, BJAB cells were infected with lentiviruses expressing either one of three independent sgRNAs targeting AMBRA1 or one sgRNA targeting luciferase as control (CTRL). Upon FACS sorting, cell populations were treated with cycloheximide for the indicated times, MG132 or MLN4924 (for 4 h) before collection. Cell extracts were immunoblotted for the indicated proteins. i, BJAB cells expressing either control sgRNA targeting luciferase or sgRNA targeting AMBRA1 were xenotransplanted in NSG mice via subcutaneous flank injections. Tumour volume was calculated by caliper measurement. Data are mean tumour volume ± s.d. (n = 3 per group); nonlinear (exponential growth) fitted curves. P values by unpaired, multiple-comparison t-test using the Holm–Sidak method. Day 0: NS; day 13: P = 0.1885]; day 14 P = 0.0045; day 15: P = 0.0083; day 16: P = 0.0048; day 17: P = 0.0223. *P < 0.05. j, The weight of tumours described in i was measured on excised tumours at the experimental end point. Data are mean tumour weight ± s.e.m. (n = 3 per group). P values by unpaired t-test. k, U-2932 cells were infected with lentiviruses expressing either one of three independent sgRNAs targeting AMBRA1 or one sgRNA targeting luciferase as control (CTRL). Upon FACS sorting, cell populations were collected, and cell extracts were immunoblotted as indicated. l, U-2932 expressing either control sgRNA targeting luciferase or sgRNA targeting AMBRA1 were xenotransplanted in NSG mice via subcutaneous flank injections. Tumour volume was calculated by caliper measurement. Data are mean tumour volume ± s.e.m. (n = 5 per group at day 0). P values by unpaired, multiple-comparison t-test using the Holm–Sidak method, until day 40 (the latest time point where at least 3 mice per group were available). Day 23: P = 0.0008; day 25: P = 0.0191; day 26: P = 0.0133; day 27: P = 0.0041; day 30: P = 0.0079; day 31: P = 0.0292; day 32: P = 0.0148; day 33: P = 0.0002; day 34: P = 0.0001; day 37: P = 0.0165; day 38: P = 0.0306; day 39: P = 0.0240; day 40: P = 0.0401. *P < 0.05. These data are from the same dataset shown in Fig. 4c. Unless otherwise noted, experiments were performed at least three independent times.
Extended Data Fig. 11 |. Loss of AMBRA1 promotes the formation of CDK2–D-type cyclins complexes.
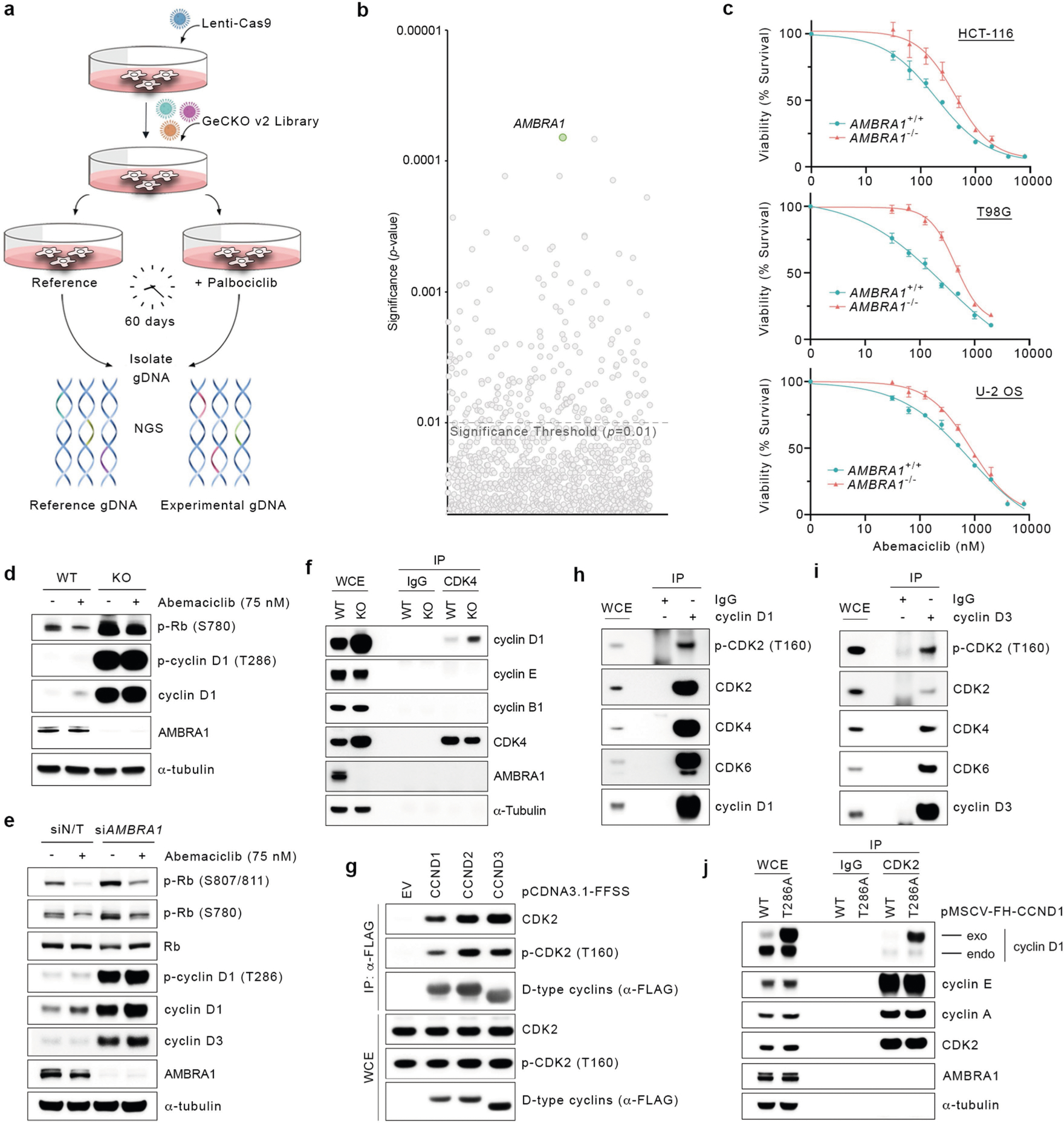
a, Schematic representation of the experiment used to identify global regulators of the response to CDK4/6 inhibitors in U-2 OS cells using a whole-genome, CRISPR–Cas9 human GECKOv2 pooled library. b, Scatter plot of the candidate hits generated from a. This experiment was performed once, with four technical replicates. P values were calculated using the MAGeCK algorithm14. c, Parental and AMBRA1−/− HCT-116, T98G or U-2 OS cells were treated with various concentrations of abemaciclib. Survival curves were generated using the AlamarBlue assay. Data are mean survival percentage ± s.d. (n = 3 biological replicates per group). The signal from each condition was normalized to an untreated control to adjust for any relative growth difference. d, AMBRA1+/+ and AMBRA1−/− T98G cells were treated with abemaciclib (75 nM) or DMSO for 24 h before collection and immunoblotting for the indicated proteins. e, HCT-116 cells were transfected with a non-targeting siRNA or an siRNA against AMBRA1 for three rounds before treatment with abemaciclib (75 nM) or DMSO for 24 h. Cells were collected and their extracts were immunoblotted for the indicated proteins. f, Parental and AMBRA1−/− HCT-116 cells were collected, and their extracts were used for an immunoprecipitation with an anti-CDK4 antibody or control IgG isotype. Proteins were immunoblotted as indicated. g, HEK 293T were transfected with plasmids expressing either FFSS-tagged cyclin D1, cyclin D2 and cyclin D3 or empty vector before lysis and immunoprecipitation with an anti-Flag resin. Proteins were immunoblotted as indicated. h, AMBRA1−/− HCT-116 cells were collected, and their extracts were used for an immunoprecipitation with an anti-cyclin D1 antibody or control IgG isotype. Proteins were immunoblotted as indicated. i, AMBRA1−/− HCT-116 cells were collected, and their extracts were used for an immunoprecipitation with an anti-cyclin D3 antibody or control IgG isotype. Proteins were immunoblotted as indicated. j, HCT-116 cells were infected with retroviruses expressing either FH-tagged cyclin D1(WT) or cyclin D1(T286A), and their extracts were used for immunoprecipitation with anti-CDK2 antibody or control IgG isotype. Proteins were immunoblotted as indicated. Unless otherwise noted, experiments were performed at least three independent times.
Extended Data Fig. 12 |. Loss of AMBRA1 desensitizes cells to CDK4/6 inhibitors.
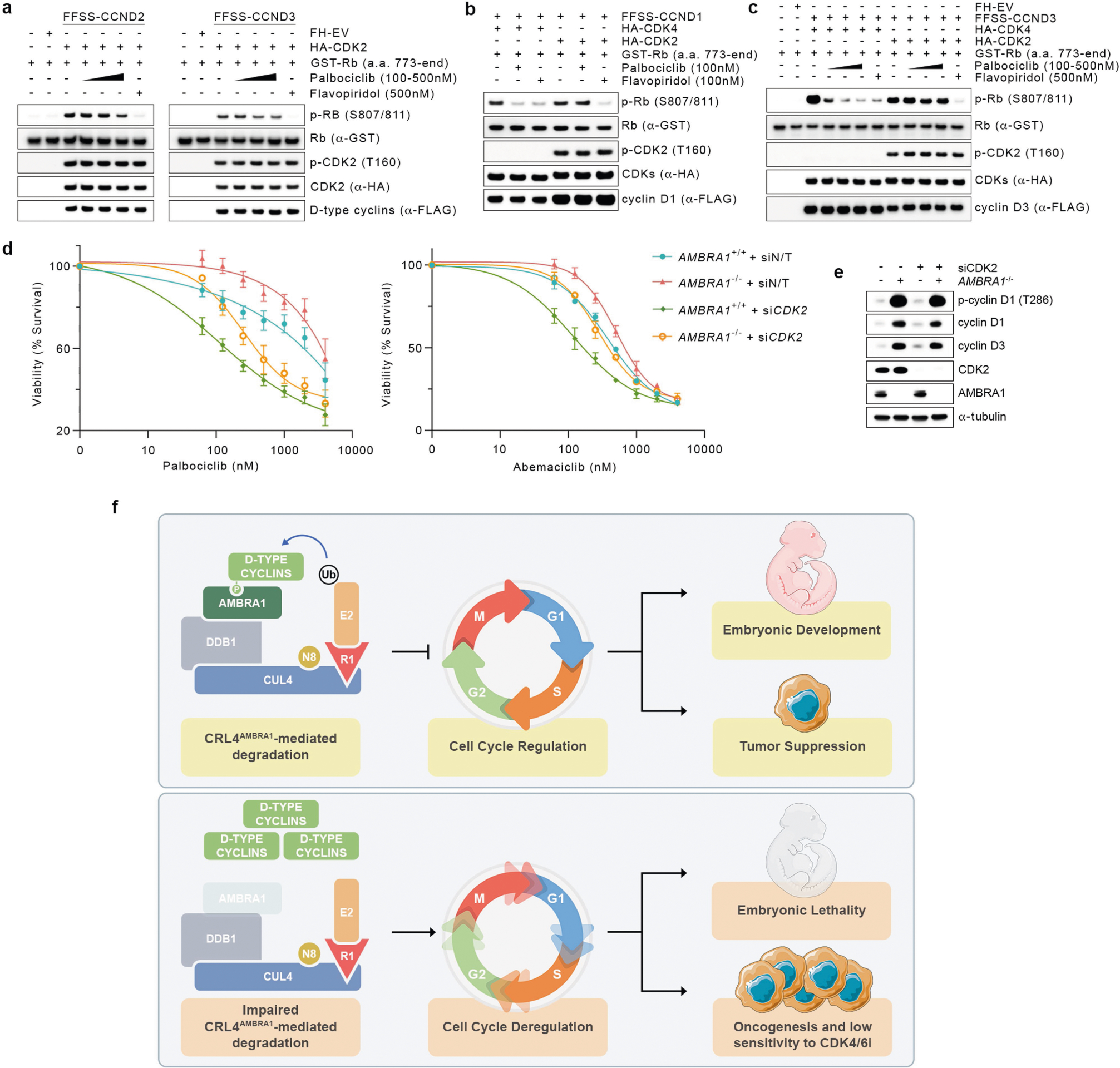
a, HEK 293T cells were (co)transfected for 48 h with FFSS-tagged cyclin D2 or FFSS-tagged cyclin D3, HA-tagged CDK2, or FH-tagged empty vector, in the combinations indicated. After a double-immunoprecipitation, cyclin D–CDK2 complexes were incubated with GST–RB(773-CT) for 30 min with or without 100 nM, 250 nM or 500nM palbociclib, or 500 nM flavopiridol, before stopping the reaction with Laemmli buffer. Proteins were immunoblotted as indicated. b, HEK 293T cells were (co)transfected for 48 h with FFSS-tagged cyclin D1, HA-tagged CDK2, HA-tagged CDK4 or FH-tagged empty vector in the combinations indicated. After a double immunoprecipitation, the purified cyclin D1–CDK4 and cyclin D1–CDK2 complexes were incubated with GST–RB(773-CT) for 30 min with or without 100 nM palbociclib or 100 nM flavopiridol, before stopping the reaction with Laemmli buffer. Proteins were immunoblotted as indicated. c, HEK 293T cells were (co)transfected for 48 h with FFSS-tagged cyclin D3, HA-tagged CDK2, HA-tagged CDK4, or FH-tagged empty vector in the combinations indicated. After a double immunoprecipitation (anti-Flag immunoprecipitation, elution with 3×Flag peptide, then anti-HA immunoprecipitation), cyclin D3–CDK4 and cyclin D3–CDK2 complexes bound to the beads were incubated with GST–RB(773-CT) for 30 min with or without 100 nM, 250 nM or 500nM palbociclib, or 500 nM flavopiridol, before stopping the reaction with Laemmli buffer. Proteins were immunoblotted as indicated. d, Parental and AMBRA1−/− HCT-116 cells were transfected with a non-targeting siRNA or an siRNA against CDK2 for three rounds before treatment with various concentrations of palbociclib (left) or abemaciclib (right). Survival curves were generated using the AlamarBlue assay. Data are mean survival percentage ± s.d. (n = 3 biological replicates per group). The signal from each condition was normalized to an untreated control to adjust for any relative growth difference. e, Before treatment, cell extracts from d were immunoblotted as indicated. f, Working model: by modulating the levels of D-type cyclins, CRL4AMBRA1 controls the execution of cell-cycle progression, thus ensuring proper embryonic development and tumour suppression (top). Impairment of CRL4AMBRA1-mediated degradation of D-type cyclins (for example, alterations or downregulation of AMBRA1 or mutations in the TP motif of D-type cyclins) deregulates cell-cycle progression, and results in embryonic lethality, increased tumorigenic potential, and reduced sensitivity to CDK4/6 inhibitors (bottom). Unless otherwise noted, experiments were performed at least three independent times.
Supplementary Material
Acknowledgements
We thank G. V. Pires, L. Moro, V. Donato, A. Zeke, A. Peschiaroli, M. Swartling, K. Avrampou, J. Wang, E. Hernandez and A. Tsirigos for their contribution to this work, and R. Abraham, S. Dann, K. Nakayama and W. Wei for critically reading the paper. This study makes use of data generated by the DECIPHER community. A full list of centres that contributed to the generation of the data is available from https://decipher.sanger.ac.uk/about/stats and via email from decipher@sanger.ac.uk. Funding for the DECIPHER project was provided by Wellcome. We thank E. Rosser and M. Lees from the Great Ormond Street Hospital for Children NHS Foundation Trust in London, UK, for providing the information on two patients with mutations in CCND2, deposited at DECIPHER (https://decipher.sanger.ac.uk). The Proteomics Laboratory, the Genome Technology Center (GTC), the Experimental Pathology Research Laboratory, the Cytometry and Cell Sorting Laboratory, and the Metabolomics Laboratory at NYU Langone Health are partially supported by the Cancer Center Support Grant P30CA016087 at the Laura and Isaac Perlmutter Cancer Center. The NYULH Center for Biospecimen Research and Development, Histology and Immunohistochemistry Laboratory (RRID:SCR_018304) is supported in part by the Laura and Isaac Perlmutter Cancer Center Support Grant, NIH/NCI P30CA016087, and the National Institutes of Health S10 Grants, NIH/ORIP S10OD01058 and S10OD018338. The authors thank C. I. Silvescu for data acquisition and processing of the mass spectrometry images. D.S. thanks F. Simoneschi, P. Simoneschi, D. Guidi and E. Guidi for continuous support. M.P. thanks T. M. Thor and T. B. Balduur for continuous support. This work was funded by grants from the National Institutes of Health (R01-CA76584 and R35-GM136250) to M.P. and (R01-CA207513-01) to L.B. The F.C. laboratory is supported by grants from the Danish Cancer Society (KBVU R72-A4408, R146-A9364, R231-A14034 to F.C. and R146-A9471 to V.C.), the Novo Nordisk Foundation (NNF13OC0007559, NNF16OC0022544), the Lundbeck Foundation (R233-2016-3360 to F.C. and R209-2015-3505 to V.C.), the LEO Foundation (LF17024 to F.C.), Associazione Italiana per la Ricerca sul Cancro (AIRC project IG 2019 no. 23543 to F.C.), the Italian Ministry of Research (MIUR, project PRIN 2017 Radius), and the Italian Ministry of Health (Ricerca Corrente to F.N.). V.C. is supported by Fondazione Umberto Veronesi. E.M. is an adjunct professor at UniCamillus—Saint Camillus International University of Health Sciences in Rome, Italy. F.B. is supported by a Young Investigator Grant from the Alex’s Lemonade Stand Foundation. M.P. is an investigator with the Howard Hughes Medical Institute.
Footnotes
Competing interests M.P. is a consultant for and has financial interests in Coho Therapeutics, CullGen, Kymera Therapeutics and SEED Therapeutics. M.P. is a cofounder of Coho Therapeutics, and is a member of the scientific advisory board of CullGen, Kymera Therapeutics and SEED Therapeutics, and is a consultant for Santi Therapeutics. The other authors declare no competing interests.
Additional information
Supplementary information The online version contains supplementary material available at https://doi.org/10.1038/s41586-021-03445-y.
Peer review information Nature thanks Piotr Sicinski and the other, anonymous, reviewer(s) for their contribution to the peer review of this work. Peer reviewer reports are available.
Reprints and permissions information is available at http://www.nature.com/reprints.
References
- 1.Malumbres M & Barbacid M Cell cycle, CDKs and cancer: a changing paradigm. Nat. Rev. Cancer 9, 153–166 (2009). [DOI] [PubMed] [Google Scholar]
- 2.Kanie T et al. Genetic reevaluation of the role of F-box proteins in cyclin D1 degradation. Mol. Cell. Biol. 32, 590–605 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Qie S & Diehl JA Cyclin D degradation by E3 ligases in cancer progression and treatment. Semin. Cancer Biol. 67, 159–170 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Reichermeier KM et al. PIKES analysis reveals response to degraders and key regulatory mechanisms of the CRL4 network. Mol. Cell 77, 1092–1106.e9 (2020). [DOI] [PubMed] [Google Scholar]
- 5.Natsume T, Kiyomitsu T, Saga Y & Kanemaki MT Rapid protein depletion in human cells by auxin-inducible degron tagging with short homology donors. Cell Rep. 15, 210–218 (2016). [DOI] [PubMed] [Google Scholar]
- 6.Barbash O, Egan E, Pontano LL, Kosak J & Diehl JA Lysine 269 is essential for cyclin D1 ubiquitylation by the SCFFbx4/αB-crystallin ligase and subsequent proteasome-dependent degradation. Oncogene 28, 4317–4325 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Fimia GM et al. Ambra1 regulates autophagy and development of the nervous system. Nature 447, 1121–1125 (2007). [DOI] [PubMed] [Google Scholar]
- 8.Davoli T et al. Cumulative haploinsufficiency and triplosensitivity drive aneuploidy patterns and shape the cancer genome. Cell 155, 948–962 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Behan FM et al. Prioritization of cancer therapeutic targets using CRISPR–Cas9 screens. Nature 568, 511–516 (2019). [DOI] [PubMed] [Google Scholar]
- 10.Rahrmann EP et al. Sleeping Beauty screen identifies RREB1 and other genetic drivers in human B-cell lymphoma. Mol. Cancer Res. 17, 567–582 (2019). [DOI] [PubMed] [Google Scholar]
- 11.Reddy A et al. Genetic and functional drivers of diffuse large B cell lymphoma. Cell 171, 481–494 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chytil A et al. Construction of a cyclin D1–Cdk2 fusion protein to model the biological functions of cyclin D1–Cdk2 complexes. J. Biol. Chem. 279, 47688–47698 (2004). [DOI] [PubMed] [Google Scholar]
- 13.Asghar U, Witkiewicz AK, Turner NC & Knudsen ES The history and future of targeting cyclin-dependent kinases in cancer therapy. Nat. Rev. Drug Discov. 14, 130–146 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li W et al. MAGeCK enables robust identification of essential genes from genome-scale CRISPR/Cas9 knockout screens. Genome Biol. 15, 554 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
References
- 15.Sanjana NE, Shalem O & Zhang F Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Reynders M et al. PHOTACs enable optical control of protein degradation. Sci. Adv. 6, eaay5064 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Lee J & Zhou P DCAFs, the missing link of the CUL4–DDB1 ubiquitin ligase. Mol. Cell 26, 775–780 (2007). [DOI] [PubMed] [Google Scholar]
- 18.Bern M, Kil YJ & Becker C Byonic: advanced peptide and protein identification software. Curr. Protoc. Bioinformatics Chapter 13, Unit13.20 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Donato V et al. The TDH–GCN5L1–Fbxo15–KBP axis limits mitochondrial biogenesis in mouse embryonic stem cells. Nat. Cell Biol. 19, 341–351 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Jeong YT et al. The ULK1–FBXW5–SEC23B nexus controls autophagy. eLife 7, e42253 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Kuchay S et al. PTEN counteracts FBXL2 to promote IP3R3- and Ca2+-mediated apoptosis limiting tumour growth. Nature 546, 554–558 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ran FA et al. Genome engineering using the CRISPR–Cas9 system. Nat. Protoc. 8, 2281–2308 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Dragicevic P in Modern Statistical Methods for HCI (eds Robertson J & Kaptein M) 291–330 (Springer, 2016). [Google Scholar]
- 24.Róna G et al. Dynamics of re-constitution of the human nuclear proteome after cell division is regulated by NLS-adjacent phosphorylation. Cell Cycle 13, 3551–3564 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cecchetelli A, Kwan R, Krauter S & Chiriboga L Minimum concentration of cells required for the preparation of compact plasma-thrombin cell blocks. J. Histotechnol. 35, 68–70 (2012). [Google Scholar]
- 26.Glinsmann-Gibson B et al. Recommendations for tissue microarray construction and quality assurance. Appl. Immunohistochem. Mol. Morphol. (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Miki Y et al. AMBRA1, a novel α-synuclein-binding protein, is implicated in the pathogenesis of multiple system atrophy. Brain Pathol. 28, 28–42 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Lewitowicz P et al. Tumor digital masking allows precise patient triaging: a study based on Ki-67 scoring in gastrointestinal stromal tumors. Scanning 2018, 7807416 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Lorenzo-Martín LF et al. VAV2 signaling promotes regenerative proliferation in both cutaneous and head and neck squamous cell carcinoma. Nat. Commun. 11, 4788 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Tuominen VJ, Ruotoistenmäki S, Viitanen A, Jumppanen M & Isola J ImmunoRatio: a publicly available web application for quantitative image analysis of estrogen receptor (ER), progesterone receptor (PR), and Ki-67. Breast Cancer Res. 12, R56 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Shaknovich R et al. DNA methylation signatures define molecular subtypes of diffuse large B-cell lymphoma. Blood 116, e81–e89 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Lenz G et al. Molecular subtypes of diffuse large B-cell lymphoma arise by distinct genetic pathways. Proc. Natl Acad. Sci. USA 105, 13520–13525 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.D’Angelo F et al. The molecular landscape of glioma in patients with Neurofibromatosis 1. Nat. Med. 25, 176–187 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Compagno M et al. Mutations of multiple genes cause deregulation of NF-κB in diffuse large B-cell lymphoma. Nature 459, 717–721 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Brune V et al. Origin and pathogenesis of nodular lymphocyte-predominant Hodgkin lymphoma as revealed by global gene expression analysis. J. Exp. Med. 205, 2251–2268 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Cerami E et al. The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov. 2, 401–404 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Gao J et al. Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal. 6, pl1 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tate JG et al. COSMIC: the Catalogue Of Somatic Mutations In Cancer. Nucleic Acids Res. 47 (D1), D941–D947 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schmitz R et al. Burkitt lymphoma pathogenesis and therapeutic targets from structural and functional genomics. Nature 490, 116–120 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Clijsters L et al. Cyclin F controls cell-cycle transcriptional outputs by directing the degradation of the three activator E2Fs Mol. Cell 74, 1264–1277 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Yada M et al. Phosphorylation-dependent degradation of c-Myc is mediated by the F-box protein Fbw7. EMBO J. 23, 2116–2125 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Maxwell PH et al. The tumour suppressor protein VHL targets hypoxia-inducible factors for oxygen-dependent proteolysis. Nature 399, 271–275 (1999). [DOI] [PubMed] [Google Scholar]
- 43.Kubbutat MH, Jones SN & Vousden KH Regulation of p53 stability by Mdm2. Nature 387, 299–303 (1997). [DOI] [PubMed] [Google Scholar]
- 44.Li Y, Ge D & Lu C The SMART app: an interactive web application for comprehensive DNA methylation analysis and visualization. Epigenetics Chromatin 12, 71 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Lin DI et al. Phosphorylation-dependent ubiquitination of cyclin D1 by the SCFFBX4-αB crystallin complex. Mol. Cell 24, 355–366 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Okabe H et al. A critical role for FBXW8 and MAPK in cyclin D1 degradation and cancer cell proliferation. PLoS ONE 1, e128 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Santra MK, Wajapeyee N & Green MR F-box protein FBXO31 mediates cyclin D1 degradation to induce G1 arrest after DNA damage. Nature 459, 722–725 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Wei S et al. A novel mechanism by which thiazolidinediones facilitate the proteasomal degradation of cyclin D1 in cancer cells. J. Biol. Chem. 283, 26759–26770 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Yu ZK, Gervais JL & Zhang H Human CUL-1 associates with the SKP1/SKP2 complex and regulates p21(CIP1/WAF1) and cyclin D proteins. Proc. Natl Acad. Sci. USA 95, 11324–11329 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Chen BB, Glasser JR, Coon TA & Mallampalli RK FBXL2 is a ubiquitin E3 ligase subunit that triggers mitotic arrest. Cell Cycle 10, 3487–3494 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Chen BB et al. F-box protein FBXL2 targets cyclin D2 for ubiquitination and degradation to inhibit leukemic cell proliferation. Blood 119, 3132–3141 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pawar SA et al. C/EBPdelta targets cyclin D1 for proteasome-mediated degradation via induction of CDC27/APC3 expression. Proc. Natl Acad. Sci. USA 107, 9210–9215 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Crooks GE, Hon G, Chandonia JM & Brenner SE WebLogo: a sequence logo generator. Genome Res. 14, 1188–1190 (2004). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Yang HW et al. Stress-mediated exit to quiescence restricted by increasing persistence in CDK4/6 activation. eLife 9, e44571 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cappuccio G, Ugga L, Parrini E, D’Amico A & Brunetti-Pierri N Severe presentation and complex brain malformations in an individual carrying a CCND2 variant. Mol. Genet. Genomic Med. 7, e708 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maini I et al. A novel CCND2 mutation in a previously reported case of megalencephaly and perisylvian polymicrogyria with postaxial polydactyly and hydrocephalus. Neuropediatrics 49, 222–224 (2018). [DOI] [PubMed] [Google Scholar]
- 57.McDermott JH et al. Hypoglycaemia represents a clinically significant manifestation of PIK3CA- and CCND2-associated segmental overgrowth. Clin. Genet. 93, 687–692 (2018). [DOI] [PubMed] [Google Scholar]
- 58.Mirzaa G et al. De novo CCND2 mutations leading to stabilization of cyclin D2 cause megalencephaly-polymicrogyria-polydactyly-hydrocephalus syndrome. Nat. Genet. 46, 510–515 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Firth HV et al. DECIPHER: Database of Chromosomal Imbalance and Phenotype in Humans Using Ensembl Resources. Am. J. Hum. Genet. 84, 524–533 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Sameshima T, Morisada N, Egawa T, Kugo M & Iijima K MPPH syndrome with aortic coarctation and macrosomia due to CCND2 mutations. Pediatr. Int. 62, 115–117 (2020). [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All accession codes, unique identifiers, and web links for publicly available datasets are available within the article and in the Reporting Summary. Those who carried out the original analysis and collection of the data generated by the DECIPHER community bear no responsibility for the further analysis or interpretation of the data. Mass spectrometry data for cyclin D1 immunopurifications are provided in Supplementary Table 1. The full scanned images obtained by electrophoretic separation are provided in Supplementary Fig. 1. All original data are available from the corresponding authors upon request. Source data are provided with this paper.