

Abstract
We review the emerging method of super-resolved cryogenic correlative light and electron microscopy (srCryoCLEM). Super-resolution (SR) fluorescence microscopy and cryogenic electron tomography (CET) are both powerful techniques for observing subcellular organization, but each approach has unique limitations. The combination of the two brings the single-molecule sensitivity and specificity of SR to the detailed cellular context and molecular scale resolution of CET. The resulting correlative data is more informative than the sum of its parts. The correlative images can be used to pinpoint the positions of fluorescently labeled proteins in the high-resolution context of CET with nanometer-scale precision and/or to identify proteins in electron-dense structures. The execution of srCryoCLEM is challenging and the approach is best described as a method that is still in its infancy with numerous technical challenges. In this review, we describe state-of-the-art srCryoCLEM experiments, discuss the most pressing challenges, and give a brief outlook on future applications.
Keywords: cryogenic electron tomography, super-resolution microscopy, CLEM, electron microscopy, fluorescence microscopy, cryoEM
1. INTRODUCTION
The spatial arrangement of biomolecules plays a key role in reaction kinetics, structural integrity, cell signaling, and a range of other crucial cellular processes. Observation of this subcellular organization is critical for our understanding of cell and molecular biology. Richard Feynman once said, “It is very easy to answer many of these fundamental biological questions; you just look at the thing” (1, p. 24). The challenge is in how you look.
Cryogenic electron microscopy (cryoEM)—specifically a variant of cryoEM called cryogenic electron tomography (CET)—and fluorescence microscopy (FM) are powerful imaging methods for observing subcellular organization. In recent years, both methods have seen dramatic improvements in their obtainable resolution, which has profoundly impacted the imaging of biological systems. Historically, optical far-field imaging resolution has been restricted by the diffraction limit (DL). The DL as defined by Abbe (2) is
| 1. |
where dxy is the minimum resolvable lateral separation, λ is the excitation (bright-field) or emission (fluorescence) wavelength, n is the refractive index of the medium, α is half the maximum collection angle, and NA is the numerical aperture of the imaging system. For typical wavelengths in the visible range and high-NA objectives, this equates to ~200 nm. The DL means that images are fundamentally blurry below this spatial scale. The DL is larger in the axial dimension with a minimum resolvable separation of
| 2. |
which results in ~400 nm axial resolution. However, beginning in 1997 with the optical on-off switching of single molecules (3) and in 1999 with the first demonstration of stimulated emission depletion (STED) super-resolution (SR) microscopy (4), and later in 2006 with single-molecule-based SR methods (5–7), far-field fluorescence-based microscopy is now able to achieve resolution more than an order of magnitude beyond the DL in all three dimensions. SR microscopies with specific fluorescent labeling strategies now make it possible to investigate the organization of biomolecules in situ at the nanometer scale (8). These breakthroughs were recognized with the 2014 Nobel Prize in Chemistry.
In electron microscopy (EM), accelerated electrons are used in place of photons. These electrons are accelerated in the microscope using very large electric potentials in the range of hundreds of kilovolts and in rare cases as much as a megavolt. This extreme acceleration leads to de Broglie wavelengths for the electrons on the order of single picometers. Thus, the DL (see Equations 1 and 2), which still applies to EM, is not limiting the obtainable resolution. Instead, for decades EM was stuck in a regime of “blob-ology” with multi-nanometer-scale resolution due to a myriad of experimental challenges. Recently, cryoEM has undergone a resolution revolution with improvements in detector technology (9), electron microscope design (10), analysis methods, and sample preparation techniques (11). CryoEM now routinely achieves angstrom-scale structural resolution when images of many thousands of highly purified macromolecular complexes are averaged in an approach called single-particle cryoEM. These advances led to the 2017 Nobel Prize in Chemistry. CET, while different from the single-particle cryoEM approach, has also benefited from the same improvements.
Despite the improved resolution of both SR and CET in recent years, each suffers from unique limitations. Most notably, SR can pinpoint the positions of specifically labeled biomolecules but is only sensitive to fluorescently labeled molecular species. While the number of orthogonal labels used in any given experiment is ever increasing (12–14), in general SR lacks detailed cellular context. Conversely, CET provides detailed cellular context by being sensitive to the electron density of all biomolecules in the sample, but it lacks widely applicable, specific, and nonperturbative labeling methods. Without specific labeling strategies, CET does not produce the required contrast necessary to identify specific molecules that are smaller than a few hundred kilodaltons unless they are part of larger complexes or periodic arrays. This means that the majority of biomolecules of interest within cells are indiscernible by CET alone.
The combination of these strengths and weaknesses makes the two techniques an obvious synergistic match. Indeed, cryogenic correlative light and electron microscopy (cryoCLEM) has been performed for more than a decade (15), leveraging the specific labeling obtainable in FM and the detailed cellular context and resolution obtainable in cryoEM. However, the DL of traditional FM has hindered the utility of cryoCLEM due to the vastly different resolutions of the two imaging methods. The advent of SR provides a route to pinpointing the positions of distinct biomolecules within the context of cryoEM at the nanometer scale. Accurate registration of the SR and cryoEM data is essential to achieve this goal and demands minimal sample distortion during the time between the two imaging experiments. This requirement leads to SR approaches that can be performed under cryogenic conditions. Achieving SR of biological structures under cryogenic conditions in a manner compatible with cryoEM and with a quality that rivals room temperature SR is a current goal and a major focus of this review.
Many challenges remain before super-resolution cryoCLEM (srCryoCLEM) is fully realized, but due to the potential impact to our understanding of cell and molecular biology, recent progress in this area has been swift. There are currently a small but rapidly growing number of srCryoCLEM reports (16–20). To whet the appetite of the reader, several examples from recent work can be seen in Figure 1, spanning bacterial (Figure 1a,d) and eukaryotic cells (Figure 1b,c). The method of srCryoCLEM has been used to assign proteins to electron-dense structures (examples shown in Figure 1a,b,d) (16, 18–20) and to pinpoint the positions of specific proteins that are too small to be directly identified in CET (example shown in Figure 1c) (17, 19). Note the drastic difference in the ability of DL fluorescence and single-molecule fluorescence localizations to annotate CET data, as shown in the comparison in Figure 1d in an approach we have termed correlative imaging by annotation with single molecules (CIASM), pronounced “chasm” (19). These tantalizing images show that srCryoCLEM can provide both specificity for any biomolecule that can be fluorescently tagged as well as molecular-scale resolution and cellular context. This review describes the central ideas underlying how these images have been obtained in recent work and discusses current challenges facing this emerging approach. We begin with an overview of the CET and SR methods, discuss the numerous challenges one faces when combining these methods, and give a brief outlook regarding potential future developments and applications of srCryoCLEM.
Figure 1.

Examples from recent srCryoCLEM studies. In these overlays, the grayscale images are 2D slices from CET reconstructions, and the SR information, shown in a color overlay, identifies specifically labeled proteins. In panels a and b, there are additional colored features of the manual annotation of structures such as granules, protein fibrils, and membranes directly visible in the CET. (a) Two examples of srCryoCLEM imaging of the fusion construct VipA-PA-GFP (red/yellow) that binds the tubular structure (blue) in the bacterium Myxococcus xanthus. The insets below show the electron density associated with the blue tubular structures (16). Panel a adapted from Reference 16 with permission; copyright 2014 Springer Nature. (b, left) srCryoCLEM image of TOM20-Dronpa (color map) on top of CET data of a mitochondrion from HEK293 cells. (Right) TOM20-Dronpa localizations (green spheres) in 3D decorating the manually annotated mitochondrial membrane (purple) and cristae (blue) (17). Panel b adapted from Reference 17/CC-BY 4.0. (c, left) srCryoCLEM of the fusion construct rsEGFP2-MAP2 in U2OS cells. The color map approximates the density of rsEGFP2-MAP2 localizations. (Right) The same CET data displayed to the left absent fluorescence information showing the underlying microtubules (18). Panel c adapted from Reference 18/CC-BY 4.0. (d, left) DL overlay of a Caulobacter crescentus cell expressing the fusion construct PAmKate-McpA in which the color displays the pixelated fluorescence intensity. (Right) Single-molecule localizations of PAmKate-McpA (red circles) in which the precision of the localization is shown as the diameter of the circle (19). Panel d adapted from Reference 19/wCC-BY 4.0. Abbreviations: CET, cryogenic electron tomography; DL, diffraction limit; SR, super resolution; srCryoCLEM, super-resolution cryogenic correlative light and electron microscopy.
2. CRYOGENIC ELECTRON MICROSCOPY
There are many variants of EM used for imaging biological samples. These approaches vary in how the sample is prepared, how the electron beam interacts with the sample, and their suitability to study different types of samples. Reviewing all methods of EM that can be correlated with SR is beyond the scope of this review, and several useful summaries and protocols are already available (21–24). Instead, this review is restricted to cryoEM, defined as using transmission electron microscopy (TEM) to visualize samples that are both prepared and imaged under cryogenic conditions. CryoEM offers the highest preservation of structural detail and therefore the highest resolution for biological imaging. As defined, there are two main variants of cryoEM: (a) single-particle cryoEM (25–27), which analyzes thousands of copies of purified complexes such as proteins, nucleic acids, or viruses, and (b) CET (28, 29), which generates 3D tomographic reconstructions, typically of whole cells, thin sections of cells, or cellular fragments. Because all correlative imaging experiments to date correlate with CET and not single-particle cryoEM, we center our discussion around CET; however, there are potential applications of srCryoCLEM to single-particle cryoEM (30, 31).
2.1. Sample Preparation
In both single-particle and CET experiments, samples are prepared on TEM grids. These grids are typically composed of an underlying metallic mesh ~200 μm thick and 3 mm in diameter with a substrate adhered to the top of the metallic meshwork (see Figure 2a). There are numerous available substrates depending on the experiment, but most common is a thin layer (~10 nm) of amorphous carbon or formvar, a polyvinyl formal plastic. Because imaging through the substrate material limits the attainable contrast, the substrate covering is discontinuous, with micron-sized holes generated either randomly or in a gridlike pattern. There are numerous trade-offs to consider when choosing a TEM grid for a given experiment. For example, so-called lacey carbon grids provide larger holes in the substrate for more unobstructed viewing of the sample of interest but are less structurally stable than alternative gold substrates (32). In practice, the goals of a specific project dictate the choice of grid, and trial and error with different grid types is recommended. Of course, because fluorescence imaging is also performed on the sample, there must be minimal spurious background emission from the grid itself.
Figure 2.
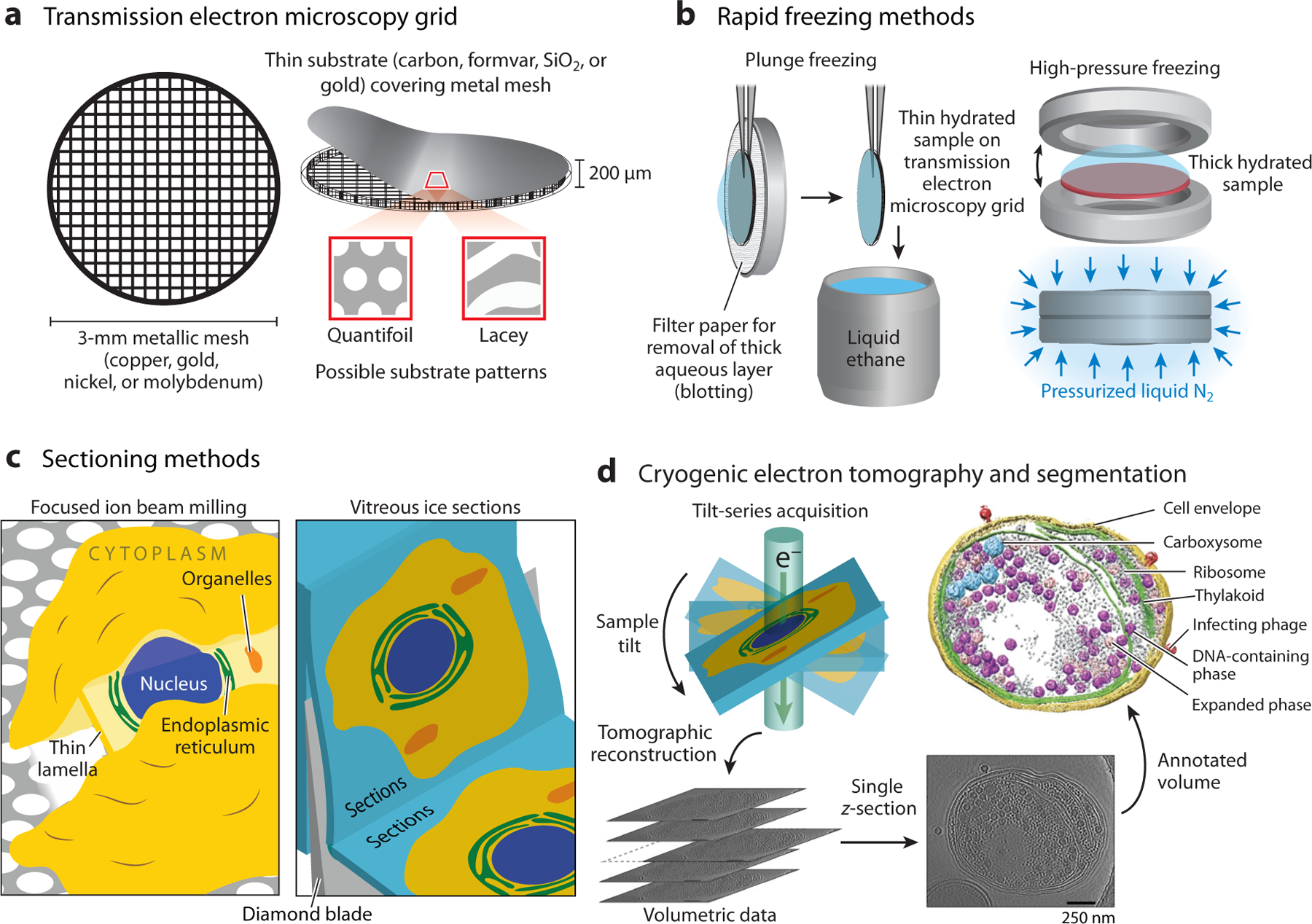
Cryogenic electron tomography (CET) sample preparation and data collection methods. (a) Transmission electron microscopy grids for cryogenic electron microscopy consist of a metallic mesh covered with a thin substrate layer. (b) Plunge freezing and high-pressure freezing are the most common methods used to rapidly freeze the sample. These methods preserve the sample in its native hydrated state encased in amorphous ice. (c) Thick samples cannot be directly analyzed due to the limited penetration of the electron beam. (Left) Focused ion beam milling uses a beam of gallium ions to ablate the sample to produce a thin lamella. (Right) Vitreous ice sectioning uses a diamond blade to cut serial thin sections. (d) CET data consists of multiple projections of the sample acquired by rotating the sample relative to the transmitted electron beam. These projections are then computationally reconstructed to produce a 3D volume that can be annotated to visualize features of interest. Data in panel d adapted with permission from Reference 151; copyright 2014 Springer Nature.
When preparing a biological sample, both single-particle cryoEM and CET rely on rapidly freezing the sample in its native hydrated state. The rapid freezing prevents the formation of crystalline ice that would scatter a transmitting electron beam; instead, rapid freezing produces an amorphous, or vitreous, ice that is largely transparent to the electron beam (33, 34). This rapid freezing can be accomplished through either plunge freezing (34, 35) or high-pressure freezing (HPF) (36, 37) (Figure 2b). In the plunge freezing approach, the sample can be adherent eukaryotic cells cultured on TEM grids or biological samples from solution such as prokaryotic cells, purified complexes, or cell fragments deposited on the TEM grid just prior to freezing. The sample is frozen by rapidly submerging the TEM grid in cryogenic liquid ethane or a liquid ethane/propane mixture; the high heat capacity, relatively high boiling point, and thermal conductivity of these liquids ensures extremely rapid cooling. This approach works well for samples less than ~10 μm in thickness. Thicker samples, such as tissues or multicellular organisms, do not cool fast enough to prevent the formation of crystalline ice. To address this, HPF slows the crystallization of ice by increasing the pressure to ~2,100 bar, allowing the vitrification of samples up to ~200 μm in thickness. Numerous sample geometries exist for HPF, including sapphire coverslips placed between two planchets (as shown in Figure 2b) as well as capillaries.
While plunge freezing and HPF can prepare samples that are <10 μm and ~10–200 μm thick, respectively, samples thicker than ~500 nm cannot be imaged directly with TEM due to the poor penetration of electrons. This constraint limits imaging to purified biomolecules, some prokaryotes, reconstituted systems, or the thin periphery of cells unless a sectioning method is used to thin the sample. There are two main methods currently employed for sectioning: (a) cryo-ultramicrotomy, in which sections <100 nm thick are cut from a vitreous sample using a diamond blade and collected on TEM grids for imaging, a process referred to as cryogenic electron microscopy of vitreous ice sections (CEMOVIS) (38), or (b) focused ion beam (FIB) milling, in which a beam of gallium ions ablates material to produce a thin lamella (~100–300 nm) from a thick vitreous sample directly on a TEM grid (see Figure 2c) (39–42). The FIB is combined with a cryogenic scanning electron microscope (cryoSEM) to track and guide the FIB milling process. Recently, FIB is increasingly favored over CEMOVIS, in part because it avoids structural distortions due to the diamond cutting (43) and, while still challenging, requires less demanding training. Intense engineering optimization of FIB milling to increase reliability has taken this method to a high art (44). Both approaches, however, require substantial amounts of specialized equipment and have low throughput.
2.2. Cryogenic Electron Tomography
After a cryogenic sample has been properly prepared on a TEM grid, the sample can be loaded into a cryogenic electron microscope. Low-magnification micrographs are taken to identify regions of interest for CET data collection. Once a suitable region has been identified, the collection of a CET data set begins with a series of images being taken for each field of view while the sample is held at different angles relative to the incident electron beam (28, 29, 45). These angles are in the range of ±60°; angles more extreme than this severely limit the transmission of electrons through the sample (see Figure 2d). This series of images, called a tilt-series, provides different projections of the sample typically separated by 1–5°. From these images a 3D volume can be reconstructed using mathematical methods such as weighted-back projection, Fourier inversion, or compressed sensing. The goals of CET are threefold: (a) determine the overall morphology of the biological specimen with nanometer resolution, (b) identify biomolecules of interest in situ, and (c) determine the structure of biomolecules of interest as they exist in situ at the highest possible resolution.
The main challenge facing cellular CET is this: How does one know precisely what biomolecules are being imaged? Specific and nonperturbative labeling compatible with CET remains challenging and is the focus of tremendous effort (46–48). It is precisely this labeling problem that srCryoCLEM hopes to address. A common approach is immunogold antibody labeling, but antibody binding requires possibly perturbative fixation and permeabilization steps. In CET alone, the identification of specific components comes down to what can be identified robustly from native contrast provided by the biomolecules themselves. For some large cellular structures (e.g., mitochondria, membranes, microtubules, and ribosomes), this contrast is sufficient to clearly identify and distinguish them in the tomographic reconstructions. Discernable features can be manually annotated in a process called segmentation (see Figure 2d). The annotation process can be incredibly time consuming to perform, easily taking weeks to annotate a single tomogram, but recent developments in the use of neural networks have somewhat mitigated this problem (49). One significant concern is the human bias introduced with this approach. The so-called ground truth for the composition of the tomogram is provided by the manual annotation, but of course there is variability from annotator to annotator (50). Despite these challenges, manual annotation can be a powerful method for visualizing approximately 10 subcellular structures simultaneously, but this is only a small subset of biomolecules present. A common method of identifying other, less distinguishable biomolecules is computationally searching a volume for high degrees of correlation to a model structure in a process known as template matching (45, 51–53). A threshold on the correlation can be used to identify the location and orientation of a biomolecule of interest, but great care must be taken to avoid template bias. This approach is improving rapidly and can now identify complexes as small as several hundred kilodaltons (54). Once many copies of a biomolecule of interest have been identified, either through manual segmentation or template matching, the extracted structures can be aligned and averaged to produce higher-resolution structures (55–58). While the current state-of-the-art abilities of CET are impressive, the majority of biomolecules are still not identifiable through either the manual segmentation or template matching methods.
3. SUPER-RESOLUTION METHODS COMPATIBLE WITH CRYOGENIC TEMPERATURES
The first single-molecule imaging studies in 1989 (59) and 1990 (60) utilized liquid helium temperatures, in which suitably chosen molecules could be selected for detailed study with a tunable laser (61, 62). Single-molecule imaging moved to room temperature in the mid-1990s (63, 64), greatly expanding the field from crystals and polymers to a range of biological and materials systems. The ability to use light to explore single emitting molecules led to many studies of local environments on the nanoscale without ensemble averaging (62, 65). These experiments were generally performed at the DL, but eventually it was recognized that different molecules being observed in the same DL volume could be distinguished, especially by switching molecules on and off (3, 66). These developments led to SR and the ability to observe structures beyond the DL with far-field optical methods.
As is true for EM, there are many varieties of SR. An examination of all possible SR methods is beyond the scope of this review but can be found elsewhere (67–72). Instead, we focus specifically on SR methods that have been demonstrated under cryogenic conditions. This includes image-scanning microscopy (ISM, also termed Airyscan by Carl Zeiss AG) (73, 74), structured illumination microscopy (SIM) (75), reversibly saturable optical fluorescence transition (RESOLFT) (76), photoactivated localization microscopy (PALM) (6), and stochastic optical reconstruction microscopy (STORM) (5) (see Figure 3). First, we briefly review the general principles behind each of these methods, followed in Section 4 by the challenges these approaches face under cryogenic conditions.
Figure 3.
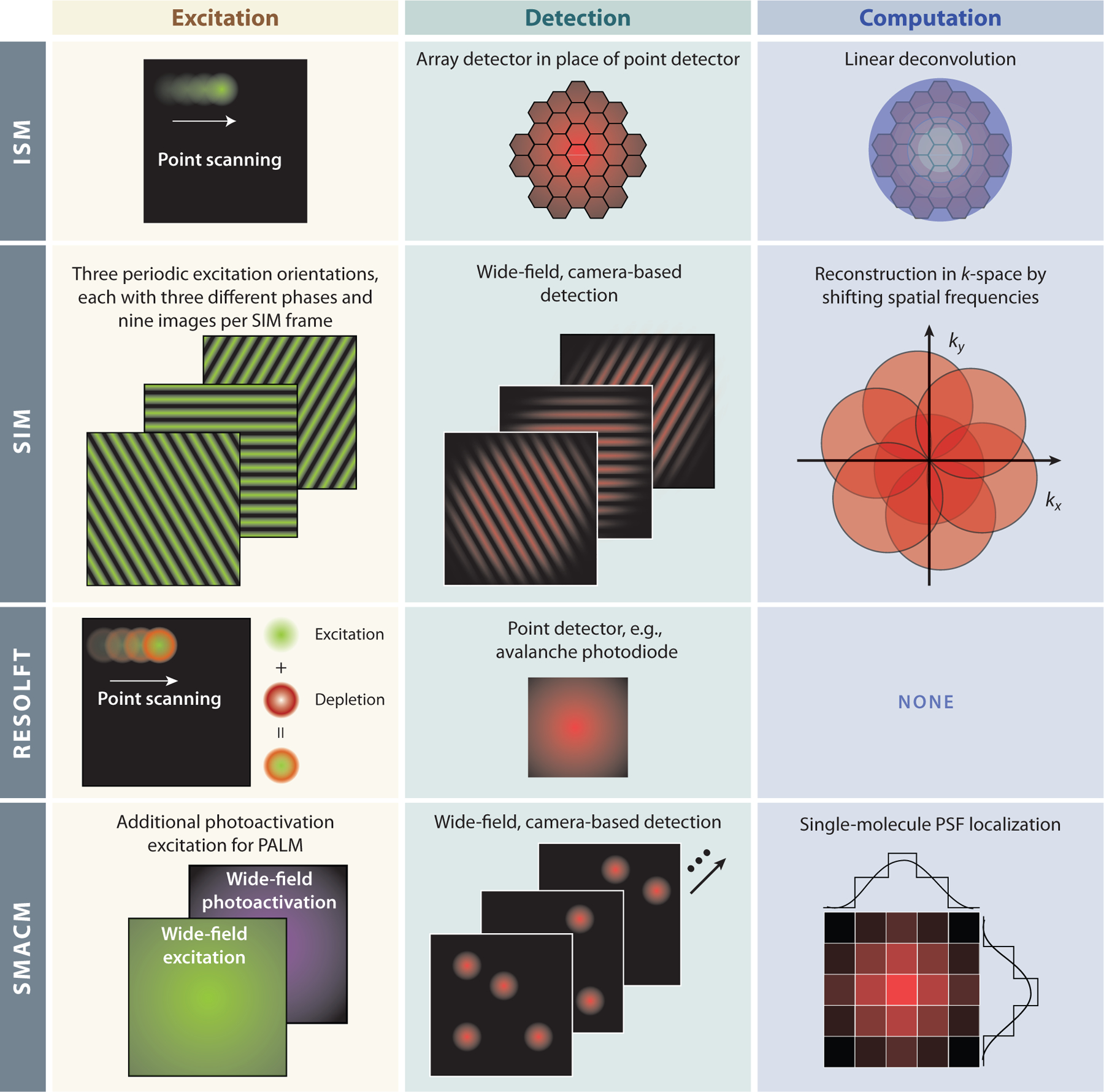
Different forms of super-resolution microscopy that have been demonstrated under cryogenic conditions. Abbreviations: ISM, image-scanning microscopy; PALM, photoactivated localization microscopy; PSF, point-spread function; RESOLFT, reversibly saturable optical fluorescence transition; SIM, structured illumination microscopy; SMACM, single-molecule active control microscopy.
3.1. Fluorescent Labeling
To resolve the positions of specific biomolecules of interest with any SR method, the molecules must first either be natively fluorescent (which is rarely the case) or be labeled with a fluorescent marker, which is why the development of SR has gone hand in hand with the development of biocompatible fluorescent labels and labeling strategies. There are now many different methods for labeling biomolecules of interest. The most common methods include (a) genetically encoded protein fusions to fluorescent proteins (77–79); (b) genetically encoded self-labeling protein tags, such as HaloTag® and SNAP-tag®, with exogenous dyes (80–83); (c) and immunofluorescence labeling using nanobodies (84–86) or primary and secondary antibodies. Any SR method requires dense labeling to achieve high resolution. The labeling density imposes a limit on the obtainable resolution independent of how precisely an individual emitter’s position can be determined. This interplay between the resolving power of the optical system and the number of locations is captured by error metrics such as the Fourier ring/shell correlation (87). For srCryoCLEM studies, the fluorescent labeling strategy needs to be nonperturbative to the ultrastructure of the biological sample. If chemical fixation and permeabilization of the sample are required for fluorescent labeling, as is typical of immunolabeling, the purpose of performing srCryoCLEM is largely defeated. Additionally, the fluorescent label itself needs to not perturb the function and organization of the labeled molecular species. Usually the addition of a fairly small molecule (1–3 nm), such as a fluorescent protein fusion, does not perturb the underlying biology, but this should be checked with controls whenever possible.
3.2. Image-Scanning Microscopy
ISM is a confocal laser-scanning microscopy approach that exceeds the DL by a factor of 1.7 in each dimension but is still fundamentally limited in obtainable resolution (70, 74). It exceeds the DL by replacing the small confocal pinhole and point detector found in a confocal microscopy detection path with a relatively large 1.25–airy unit (AU) pinhole and an array of point detectors. This provides efficient light collection, and each point detector acts as an effective 0.2-AU pinhole. This provides a resolution 1.4× beyond the DL; however, by further deconvolving the signal on the detector, the resolution is improved to 1.7× beyond the DL. This method of SR is compatible with low excitation intensities (~1–10 W/cm2) and fluorescent labels for standard FM (i.e., no additional switching, blinking, or other properties are required). Additionally, this approach has been heavily commercialized by Zeiss (Airyscan) for applications at cryogenic temperatures (20, 88).
3.3. Structured Illumination Microscopy
Like ISM, SIM exceeds the DL but remains theoretically bounded unless more complicated nonlinear sample responses are exploited (70, 89, 90). As originally demonstrated, SIM can exceed the lateral DL by a factor of two (75), but an extension of this method also applies to the axial dimension (91). The approach works by interfering multiple beams to generate a diffraction-limited low intensity (~1–10 W/cm2) periodic excitation pattern at the sample. The effect of this illumination is best thought of in reciprocal space, or k-space, which describes the spatial frequencies observable by the optical system. The periodic excitation effectively shifts high-spatial frequency components that are beyond the DL to lower spatial frequencies within the DL, much like Moiré fringes. Three images with differing phases of the periodic excitation are required to recover higher spatial frequencies along one axis, which corresponds to the orientation of the periodic excitation. To generate a reconstruction with nearly isotropic resolution, the periodic excitation is rotated. Typically, three different directions for the periodic excitation are chosen, each separated by 60°, and in each orientation three images of differing phase are required for a total of nine images per SIM frame. These nine images are then used for the computational reconstruction of high-resolution images by shifting the high spatial frequencies to their appropriate locations in k-space. The computational reconstruction is nontrivial and, if care is not taken, can easily result in artifacts in the reconstructed images (92).
3.4. Reversible Saturable Optical Fluorescence Transitions
RESOLFT is an umbrella term for multiple SR methods largely developed in the lab of Stefan Hell (93). RESOLFT techniques include stimulated emission depletion (STED), ground-state depletion (GSD), and RESOLFT with switchable fluorescent labels. All RESOLFT experiments use a so-called depletion beam with a specific spatial pattern that depletes a population of molecules in a given state by saturating a transition from that state to a new state and in doing so inhibits fluorescence emission. Each of these RESOLFT variations differs only in the states and transitions being exploited. STED depletes the excited state by saturating a stimulated emission transition from the excited to the ground state before fluorescence occurs, GSD depletes the ground state by transitioning to a long-lived dark state (typically the triplet state), and RESOLFT with switchable fluorescent labels depletes one of two spectrally distinct ground states accessible by photoswitching. In addition to the depletion beam, all RESOLFT experiments use an excitation beam to induce fluorescence from the undepleted labels. Importantly, the depletion and excitation beams spatially overlap at the sample. The most common spatial pattern for the depletion beam is a toroidal (donut) shape generated by shaping the wave front to produce destructive interference at its focus. By increasing the intensity of this toroidal depletion beam, all molecules except those at the very center of the torus are driven to a nonfluorescent state. This process effectively shrinks the volume of molecules that are allowed to fluoresce and therefore improves the resolution (94, 95). Unlike ISM and SIM, the resolution of a RESOLFT experiment is, in theory, limitless. However, in practice, applications of the technique achieve resolution on the order of tens of nanometers. The obtainable (2D) resolution in a RESOLFT experiment can be described mathematically as a modification of Abbe’s diffraction limit
| 3. |
where Is is the saturation intensity that reduces the probability of emission by a factor of two and I is the depletion beam intensity used in the experiment, typically many times the saturation intensity (93, 95, 96). The saturation intensity,
| 4. |
is experimentally determined but can be written in terms of the absorption cross section of the depletion transition, σ, and the lifetime of the state, τ, which is either the state being depleted, in the case of STED, or the state being populated, in the case of GSD and RESOLFT with switchable fluorescent labels. One of the major concerns for STED under cryogenic conditions is the depletion intensities required for high resolution. Following Equation 4, one can see that the shorter the lifetime, τ, the greater the saturation intensity. Singlet-excited states typically have lifetimes on the order of nanoseconds. Because STED relies on interactions with the short-lived singlet-excited state, the intensities used in STED can be in excess of 100 MW/cm2, which can lead to heating of the cryogenic sample. GSD can operate at intensities that are thousands of times lower due to the longer lifetimes of the triplet and other dark states, and RESOLFT with switchable fluorescent labels can use depletion beams that are 109 times lower. However, among all of the RESOLFT approaches, only STED has been demonstrated at cryogenic temperatures (96).
3.5. Photoactivated Localization Microscopy and Stochastic Optical Reconstruction Microscopy
Both PALM and STORM are single-molecule-based approaches to SR that fall under the category of what we call single-molecule active control microscopy [SMACM (97)]. These methods are sometimes referred to as single-molecule localization microscopy (SMLM), but the latter term also includes single-molecule tracking measurements that are seldom performed under cryogenic conditions for obvious reasons. These SMACM approaches have two key concepts: (a) localization of individual emitters by the regression (fitting) of a model function to the microscope’s measured point-spread function (PSF), that is, a DL image of a single molecule’s emission, and (b) active control over the emissive state to maintain the requisite sparsity to identify individual emitters for localization and to change the subset of emitters that are emissive. In SMACM approaches, videos of a fluorescently labeled sample are acquired in which these two key concepts allow for the precise localization of different subsets of emitters in each frame. These localizations are then used to generate a pointillist SR reconstructed image. The experimenter must select a control mechanism to maintain the required emitter sparsity in any frame and to stochastically switch the set of emitters that are in the emissive state from frame to frame. These active control mechanisms might include photoactivation of a new sparse set of emitters, as in the case of PALM (6, 7), or the fluorescent labels may blink randomly due to recovery from some long-lived dark state, as in the case of STORM (5).
The precision with which an individual emitter can be localized is in leading order inversely proportional to the square root of the number of photons detected from that individual emitter before photobleaching (an irreversible photochemical reaction leading to a permanent dark state). This is simply an extension of standard error statistics (central limit theorem) in which each photon serves as an independent estimate for the true emitter’s location. One expression for the expected precision obtained using the standard PSF modeled as a Gaussian function and fit using a least-squares regression was described analytically by Thompson et al. (98) [with a later correction by Mortensen et al. (99)] as
| 5. |
where σ is the standard deviation of the localization and is a measure of the localization precision, σPSF is the standard deviation of the PSF modeled as a Gaussian, a2 is the area of a pixel, and Nbg and Nsig are the number of background photons/pixel and total signal photons, respectively. Typically, the ratio of Nbg to Nsig is low enough that the second term can be neglected, recovering the expected scaling. Normally, this localization procedure is strictly in the lateral dimensions; however, modifications to the microscope can alter the shape of the PSF to encode axial localization information, which provides 3D localization information (71, 100). Excitation intensities are typically high (~1–10 kW/cm2) to drive more molecules into dark states in STORM-like blinking methods or to collect as many photons from as many individual emitters in the shortest amount of time possible. Many software platforms exist to process movies of single emitters into SMACM reconstructions (101, 102).
4. CHALLENGES IN PERFORMING SUPER-RESOLUTION CRYOGENIC CORRELATIVE LIGHT AND ELECTRON MICROSCOPY
The goal of any srCryoCLEM experiment is to overlay SR and cryoEM information. Accurate registration of these two data sets demands that minimal sample distortions occur between the two imaging methods, thus requiring that the sample be cryogenically prepared prior to the SR imaging. The main challenge of an srCryoCLEM experiment is therefore the performance of SR under cryogenic conditions. The typical workflow for several recent srCryoCLEM experiments is shown in Figure 4. The experiments begin by rapidly freezing a fluorescently labeled specimen using either plunge freezing or HPF. The transition temperature between vitreous ice, which is largely transparent in EM, and crystalline ice, which scatters an electron beam, is ~135 K. An irreversible transition to crystalline ice occurs rapidly above this temperature, requiring that the sample be maintained below ~135 K during the acquisition of SR, optional sectioning steps, and EM. Note that the SR images must be acquired prior to EM due to the destructive nature of high-energy electron-scattering events that lead to significant sample and fluorophore bleaching.
Figure 4.

Experimental workflow for the conduction of a super-resolution cryogenic correlative light and electron microscopy (srCryoCLEM) experiment. Black arrows show possible paths through the workflow, and colored arrows show workflows that have been used by specific researchers.
The conduction of an srCryoCLEM experiment has numerous challenges. Here we focus on those challenges that are unique to srCryoCLEM. We find the main challenges to be (a) sample geometry for maintaining cryogenic conditions during fluorescent imaging, (b) devitrification due to optical excitation, (c) control over the emissive state of the fluorophores, and (d) accurate registration of the SR and cryoEM data sets in three dimensions. We summarize these challenges as well as various solutions to these problems and their advantages and shortcomings.
4.1. Sample Geometry
srCryoCLEM experiments require a cryogenic stage for FM, and many designs have been explored (15, 103, 104). Cryostages can be divided into two broad categories: (a) open cryostages, which are open to the atmosphere and cool the sample with cold nitrogen vapor and a strong thermal connection to a liquid nitrogen reservoir, and (b) closed cryostages, which maintain the sample under vacuum and cool the sample with a strong thermal connection to a liquid nitrogen or liquid helium reservoir. The choice of cryostage design also has a major impact on the choice of imaging objective. While cryogenic SR typically uses lower-NA air-spaced objectives, higher-NA immersion objectives are possible and are also discussed.
4.1.1. Open cryostage solutions.
One of the most common cryostages on the market today is the CMS196 from Linkam Scientific Instruments Ltd. This stage is designed for FM imaging at 77 K specifically for correlative studies and can hold up to three TEM grids at a time on a built-in x-y translation stage. Many of the srCryoCLEM experiments to date have used this stage (18–20). Leica also sells a cryostage with a thoughtful grid transfer system as part of its cryogenic fluorescence imaging platform, which has been used in numerous cryoCLEM experiments (105). Both systems work on upright microscopes, and in both designs, a microscope objective is lowered through an opening at the top of the stage and into the cryogenic environment. This opening in the lid allows for unobstructed viewing of the sample and enables the use of higher-NA objectives, typically 0.7–0.9 NA, but it also makes these cryostages more susceptible to ice contamination from moisture in the ambient air condensing on the TEM grid. To limit this contamination, both cryostages have a close fit between their objectives and the lid of the cryostage, and the stage environment is maintained at positive pressure from the boil-off of the liquid nitrogen reservoir. In both stage designs, the sample is held on top of a copper block that is cooled both by thermal contact to liquid nitrogen and by cold nitrogen vapor. The advantages of the open cryostage design include commercial options, lack of vacuum equipment, and straightforward sample loading and unloading. These advantages are tempered by the increased risk of ice contamination and potential drift (106) due to thermal gradients and the flow of liquid nitrogen.
4.1.2. Closed cryostage solutions.
To our knowledge there are no current commercial closed cryostage solutions for srCryoCLEM. There are numerous examples of bespoke closed cryostages that operate on samples under vacuum (107, 108), including those used in the first observations of single molecules in superfluid He (109), but these do not easily facilitate the loading and unloading of a vitrified sample. Modification of commercial cryostages to facilitate vitrified samples has been successful. For example, Hoffman et al. (110) acquired superb cryogenic SR data using a modified ST-500 cryostat from the Janis Research Company. Many details regarding the modifications made to the cryostat can be found in their supporting information (110). Another approach presented by Li et al. (111) used a vacuum interlock system similar to those found on electron microscopes and a commercial cryoEM sample holder. This approach has the advantage that the same sample holder can be used directly in the EM without any additional transfer steps. The advantages of custom-made closed cryostages are the greatly reduced risk of ice contamination, the room temperature objectives, and, in the work of Hoffman et al. (110), the ability to operate at variable temperatures down to 4 K using liquid helium. These advantages are tempered by the lack of commercial options as well as the general requirement of longer working distance and lower-NA objectives to image through the vacuum windows. However, with very careful engineering, working distances of just 1.3 mm and NA 0.85 have been achieved (110).
4.1.3. High-numerical-aperture objectives.
In our discussion of closed versus open cryostages, we have assumed that the objective is largely held at room temperature, with only the tip being in the cryogenic environment for the open cryostage design. In addition to the low-temperature, single-molecule spectroscopy studies in the early 1990s (61), there are several examples of modern attempts to incorporate the objective completely within the cryogenic environment, including a liquid propane immersion objective (112), solid immersion lenses (113), and an all-reflective hemispherical objective lens (114). Bringing the objective lens into the cryogenic environment permits the use of higher NA and reduces the risk of ice contamination by eliminating an opening directly above the sample, as is the case in the open cryostage design. However, Nahmani et al. (115) also demonstrated the use of an immersion objective with the objective maintained at room temperature outside the cryogenic environment. Increased NA improves light collection by increasing the angle of the cone of light collected by the objective and improves the obtainable resolution according to Equations 1 and 2. While this improved NA will benefit all forms of SR shown in Figure 3, it will have the most profound impact on ISM and SIM, both of which are fundamentally limited by the DL. For example, following Equations 1 and 2, the maximum achievable lateral and axial resolution using ISM for emission at 600 nm and a 0.8-NA, air-spaced objective is just ~220 nm and ~550 nm, respectively, but with a 1.3-NA propane immersion objective is ~140 nm laterally and ~275 nm axially.
4.2. Devitrification Restrictions
Local devitrification due to the optical excitation intensity used to generate high emission rates or light-induced blinking in FM poses a significant constraint on the performance of srCryoCLEM on TEM grids. Samples prepared and imaged on sapphire coverslips do not have the same difficulties, likely due to improved thermal conductivity and reduced absorption by the substrate (110), but these coverslips are incompatible with direct follow-up via CET. The exact optical excitation intensity that leads to devitrification on TEM grids is not well-defined and is likely a function of sample thickness, grid substrate, and excitation wavelength. Some studies have determined that devitrification occurs with intensities of 300–600 W/cm2 when the sample is illuminated for just 5 min (16, 18), or 100 W/cm2 when illuminated for hours (19). Still other studies state that using a formvar substrate rather than the more popular carbon substrate allows for 1,750 W/cm2 of continuous illumination (17). However, formvar can suffer from charging effects during EM, resulting in electron beam–induced motion of the sample, and commercial formvar grids exhibit far more autofluorescence.
In any case, all of these excitation intensities are substantially lower than the intensities used in typical room temperature blinking-based STORM experiments, which are often in excess of 10 kW/cm2, or the 10 MW/cm2 peak intensities needed for even low-power STED depletion beams (70, 116, 117). This restriction on illumination intensity makes the ISM and SIM approaches highly favorable due to their low excitation intensity requirements. This restriction can make cryogenic SMACM-based SR a very slow process in comparison to room temperature experiments, because the photon detection rate and rate of optically driven transitions scale linearly with intensity (19, 106). This restriction is even more damaging for STED-based SR, in which the obtainable resolution is directly dependent on the intensity of the depletion beam (see Equation 3). While cryogenic STED has achieved a 4× resolution improvement using ~30 MW/cm2 to image fluorescent polystyrene beads on glass coverslips (96), it is yet to be demonstrated that STED can be conducted on TEM grids in a manner compatible with cryoEM. Other RESOLFT-based approaches operate with much lower intensity and could be more viable for srCryoCLEM but have not yet been explored.
4.3. Photophysics Under Cryogenic Conditions
Four key effects of cryogenic conditions impact the implementation of cryogenic SR. First, the diffusion of molecular oxygen is limited in frozen samples. Molecular oxygen plays a crucial role in both the photobleaching of fluorescent labels and the quenching of fluorescent labels in the triplet state. Limiting oxygen diffusion reduces the quantum yield of photobleaching and increases the effective triplet-state lifetime. Second, fluorescent labels are rigidly held in vitreous ice, eliminating the usual room temperature assumption that the emission is isotropic due to free rotation of the label. Instead, under cryogenic conditions, molecules exhibit a rigid dipole emission pattern that can shift the apparent location of detected fluorescence relative to isotropic emission. Third, the reduced thermal energy and lack of diffusion under cryogenic conditions limits the applicability of many photoswitching and blinking mechanisms employed at room temperature. Fourth, transition line widths narrow by many orders of magnitude at extreme cryogenic temperatures, and this effect may be used as an active control mechanism unique to experiments at a few K (118, 119). We discuss each of these effects as well as their impact on cryogenic SR in more detail in this section.
4.3.1. Reduced photobleaching quantum yields and increased triplet-state lifetimes.
Photobleaching is often an undesirable process for fluorescence imaging, because it limits the number of photons collected from an emitter and reduces the resolution obtainable in an SR experiment or possibly distorts the resulting images. However, photobleaching is also a necessary part of some SR methods, most notably PALM. In a PALM experiment, as discussed in Section 3.5, emitters are initially in a dark state and then are stochastically photoactivated into an emissive state. These labels must photobleach before new emitters are photoactivated to prevent overlapping emission from multiple emitters, which would impair the ability to separately localize single molecules. Ironically, one of the challenges emerging under cryogenic conditions is that emitters persist in the emissive state for too long (19). Unlike at room temperature, photobleaching cannot be relied upon to turn off an emitter for PALM-based approaches. Because PALM is a popular method for srCryoCLEM (16–19), we need to briefly consider how the process of photobleaching changes under cryogenic conditions.
Photobleaching is often mediated through the generation of reactive oxygen species (ROS) (120, 121), which form in two main ways. First, molecular oxygen is a triplet in its ground state, allowing it to interact with fluorescent molecules in a photoexcited triplet state. An interaction between these two states has a high likelihood of triplet–triplet annihilation, yielding the molecular oxygen in an excited singlet state and the fluorophore back in its ground state (120). This highly reactive singlet oxygen species can then react with the fluorophore through many reaction pathways, such as bond breaking or the interruption of conjugation leading to photobleaching. Second, fluorophores in the excited triplet state can donate an electron to molecular oxygen and produce a superoxide radical that, like singlet oxygen, is highly reactive and can lead to photobleaching.
Under cryogenic conditions, there is less diffusion of molecular oxygen, and the photobleaching reactions performed by ROS have been shown to be temperature dependent (122). The reduced quantum yield for photobleaching in the absence of oxygen can be quite dramatic. For example, photobleaching becomes nearly inconsequential for the organic dye terrylene in a crystalline host absent molecular oxygen. It has been demonstrated at 5 K that single dye molecules remain emissive for many minutes under illumination intensities in excess of 100 kW/cm2, and even at room temperature, protected fluorophores can emit for tens of hours under 2 kW/cm2 (123, 124). While these are rather extreme situations, many organic dyes under cryogenic conditions have reported quantum yields for photobleaching that are less than 1% of their room temperature values (122, 125, 126). Similar results have been obtained for some fluorescent proteins (106, 112, 127); in order for SMACM-based SR to fully exploit this reduction in photobleaching, great care must be taken to merge photons from individual emitters that may emit across many frames with long but not permanent dark periods under cryogenic conditions (19, 110).
While molecular oxygen and ROS play a crucial role in photobleaching, the generation of ROS also provides a quenching pathway for fluorophores in a photoexcited triplet state (120). In the absence of this quenching pathway, the lifetime of the triplet state can increase by several orders of magnitude, from microseconds to milliseconds (128). Importantly, the quantum yield for entering the triplet state following excitation is largely temperature independent and is typically on the order of ~0.1% for fluorescent labels (128). The low excitation intensities that can be used before devitrification lead to emitters spending the majority of their time in their ground state even with this increase in triplet-state lifetime. Therefore, this increase in triplet lifetime is likely inconsequential for most SR approaches until higher excitation intensities can be used. However, the increased triplet-state lifetime does appear to have a large impact on the acquisition of STED images at cryogenic temperatures (96). Supposing the problems of devitrification are solved, the high intensities needed for STED and long triplet lifetimes quickly lead to a buildup of molecules in the triplet state. Although this buildup of triplets is precisely what is desired in a GSD approach, this approach to cryogenic SR is unexplored at the moment.
4.3.2. Rigid dipole orientation.
The emission pattern from a fluorescent label in the far field is an expanding toroidal pattern centered along the emission dipole (129). This anisotropic emission pattern is known to cause localization errors for rigid emitters in SMACM experiments (130, 131), but these errors can be corrected and the emission pattern can be exploited to measure the orientation of the fluorescent label and the structure of the sample (132, 133). Typical room temperature SMACM experiments with floppy labels (134) assume that the detected emitters are free to rotate and that the average emission collected is isotropic. Under cryogenic conditions, the molecules are no longer free to rotate. There are some efforts to exploit this rigidity using polarized excitation to achieve SR (135). This rigidity does not impact the acquisition of ISM, SIM, or RESOLFT images because these methods still average over many emitters. The rigidity of dipoles does pose a concern for SMACM-based SR because the resultant localization errors can be in excess of 100 nm, especially for emitters slightly out of focus (131). Thankfully, the extent of these localization errors increases dramatically with the collection of supercritical light, light collected beyond an NA of 1.0, as is the case for oil immersion objectives at room temperature with an NA of 1.4. Weisenburger et al. (108) have shown that if the emission goes from high- to low-index media, as is typical for open cryostage and most closed cryostage designs, and light is collected with a 0.9-NA objective, the localization error is just 0.075 nm, even for emitters 100 nm out of focus. The effect of NA on the localization error suggests that while high-NA immersion objectives can be useful for all SR approaches, their implementation in SMACM adds complexity but also the prospect of additional information (132, 133). Finally, if this orientation-induced lateral shift becomes a concern, a purely optical method has been proposed (136) and demonstrated (137) that can use a commercially available optical element, a vortex half-wave plate, to remove the shift for all dipole orientations.
4.3.3. Control mechanisms under cryogenic conditions.
Perhaps the greatest challenge facing the conduction of resolution-unlimited cryogenic SR is the ability to control the photophysical state of the emitter under cryogenic conditions. The resolution-limited methods of ISM and SIM demand very little by way of control over the emitter, making them promising as routine methods for srCryoCLEM. RESOLFT- and SMACM-based approaches have much more restrictive demands. Fundamentally, both RESOLFT and SMACM rely on manipulating the state of the emitter in either a directed manner (RESOLFT) or stochastically (SMACM). Several methods that have been explored under cryogenic conditions are photoswitching including photoactivation, blinking by accessing long-lived reversible dark states, and narrow excitation line widths. Figure 5b,c illustrates aspects of these control mechanisms under cryogenic conditions. We discuss each approach briefly, as well as the challenges and progress that has been made.
Figure 5.
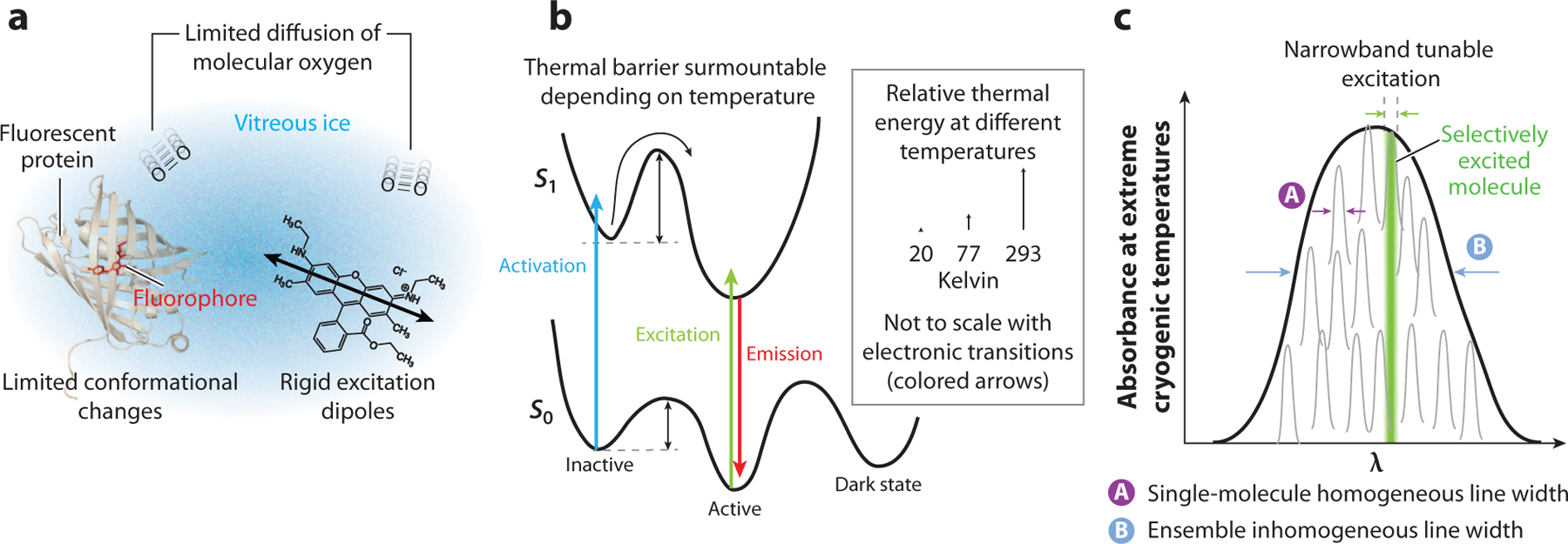
Changes in the photophysical and photochemical landscape under cryogenic temperatures. (a) Fluorescent labels such as (left) a fluorescent protein or (right) a small molecule dye held in a solid host such as vitreous ice have limited conformational space to explore, and excitation/emission dipoles are held rigidly (double black arrow), leading to polarized excitation and emission. The solid host also limits the diffusion of molecular oxygen, which plays a key role in both photobleaching and triplet-state quenching. (b) Cryogenic temperatures limit thermal energy, reducing the ability to overcome thermal barriers (black arrows) in photoswitching reaction pathways on the excited- and ground-state manifolds. Optical electronic transitions are shown by colored arrows: blue for activation, green for fluorescence excitation, and red for fluorescence emission. (c) Extreme cryogenic temperatures, <4 K, can lead to narrow single-molecule absorption line widths. The local environment for each absorber is different and can shift these absorption bands, leading to each absorber (black profiles) having its own unique resonance wavelength that can be specifically excited with a narrowband tunable laser (green) to produce fluorescence. Sparse excitation using the narrow homogeneous line widths has been demonstrated as a viable active control mechanism for super resolution using organic dyes in crystalline hosts but has not been explored as a control mechanism for biological environments.
4.3.3.1. Photoswitching.
The majority of SMACM-based srCryoCLEM experiments to date have used multicolor excitation to drive photoswitching either between a dark and a bright state or between two spectrally distinct bright states (16–19). RESOLFT using photoswitchable labels is unexplored for srCryoCLEM at the moment but relies on similar control mechanisms (76, 138). The challenge in photoswitching labels under cryogenic temperatures is that many photoswitching mechanisms rely on large conformational changes or have switching pathways that are restricted in the solid phase when thermal energy is limited (139, 140). Complicating matters is the fact that photoswitchable labels may have numerous accessible long-lived states under cryogenic conditions, and ensemble measurements of photoswitching capture the behavior of a heterogeneous population of fluorescent labels. This complication makes empirical statements of whether a label switches or not based on a bulk study insufficient and leads to conflicting results in the literature. For example, both PAmCherry and Dronpa fluorescent proteins are reported as switchable and not switchable under cryogenic conditions (16–18, 106). Instead of classifying emitters as switching or not switching, the quantum yield of photoswitching needs to be determined, or, at least, the relative efficiency of switching compared to room temperature needs to be quantified (106, 141). Labels with efficient switching both into and out of an emissive state at cryogenic temperatures, possibly with different colors, are needed to improve cryogenic SR. Identifying the metastable states available to fluorescent labels under cryogenic temperatures can be challenging, and the use of time-resolved spectroscopies such as transient absorption might be helpful for identifying accessible states in the future.
4.3.3.2. Blinking by accessing long-lived reversible dark states.
Transitions to long-lived dark states can also be used for the requisite control needed for SMACM (specifically STORM) and RESOLFT (specifically GSD-based) approaches. It is worth considering what is required of the dark state. In a STORM approach, with 1,000 emitters in the DL volume, the vast majority, ~99.9%, of emitters in the field of view must be in the dark state at any given time to isolate the fluorescence contribution from an individual emitter. Further, emissive molecules must remain in the bright state until a sufficient number of photons have been collected for localization. Until the devitrification challenges discussed in Section 4.2 are solved, the limited excitation intensities that can be used restrict the rate at which emitters can be excited and therefore the rate at which emitted photons can be detected to several kilohertz, assuming typical absorption cross sections on the order of 10−16 cm2. Thus, without any additional experimental complexity, the dark states must have lifetimes on the order of seconds. This long of a dark-state lifetime excludes the use of the triplet state as the dark state for most dyes, even with extended triplet lifetimes in the absence of diffuse molecular oxygen under cryogenic conditions. Instead, some other long-lived state, possibly conformational or charge-separated states, must be accessed, and there must be reversible return from this state to the emissive state. Dark states meeting these requirements do appear to exist. For example, in the cryogenic SR work of Hoffman et al. (110), fluorescent labels in cells on sapphire coverslips were shelved into long-lived dark states. For unknown reasons, this shelving worked best under kilowatt per centimeter squared illumination intensities after extended initial illumination, and the nature of the dark states was not explored in depth.
The requirements for a dark state in a GSD experiment are slightly different from those for a STORM (blinking) experiment. Exceeding the DL by a factor of 10 in a RESOLFT experiment demands that the ratio of I to Is be on the order of 100 following Equation 3. Again, the devitrification concerns detailed previously restrict the usable values of I to hundreds of watts per square centimeter; thus, Is is restricted to watts per square centimeter. Saturation intensities of this order have already been demonstrated at room temperature using reversibly switchable proteins (138) and even lower than this at cryogenic temperatures (142). Following Equation 4, the use of dark states with lifetimes on the order of a millisecond or shorter (similar to triplet states under cryogenic conditions) would work only if there was a large absorption cross section to enter the dark state. Considering spin effects on optical transitions, it is unlikely that a triplet could be optically populated in one step, making the direct use of triplet states for cryogenic GSD challenging. More theoretical and experimental work needs to be done to explore the potential of GSD for future srCryoCLEM studies.
4.3.3.3. Narrow excitation line widths.
A potential mechanism to control the emissive state of fluorescent labels for SMACM-based SR is unique to extreme cryogenic conditions. This mechanism is the selective excitation of fluorescent labels based on the narrow bandwidth of purely electronic transitions at 4 K and below and is the same effect that was exploited for the first optical detection of single molecules (109). The absorption line width of a single fluorescent molecule can be many orders of magnitude narrower at liquid helium temperatures than at room temperature, on the order of 1 kHz–100 MHz (143). This homogeneous line width can be orders of magnitude narrower than the inhomogeneous line width, which is the width of the ensemble of transition energies resulting from many different molecules, each with their own local environment that shifts the transition energy. By tuning a narrow excitation laser across the frequencies spanned by the inhomogeneous line width, molecules are selectively excited when in resonance with the excitation source. The inhomogeneous to homogeneous ratio could provide the necessary control and sparsity needed for SMACM-based imaging. This control mechanism was demonstrated in initial experiments on organic dyes in crystalline hosts by van Oijen et al. (118, 144) and elaborated on by Naumov et al. (119) in work achieving ~3 × 105 localizations with an inhomogeneous to homogeneous ratio of 106. The behavior of fluorescent labels in amorphous hosts, as is the case in srCryoCLEM of biological samples, can be more complex, but there may be enough spectral selectivity to enable SMACM (145). Green fluorescent protein, for instance, has been shown to have homogeneous line widths less than 30 GHz and inhomogeneous line widths hundreds of times that when embedded in glycerol glasses at 1.6 K (146). This spectral selection method of active control at extreme cryogenic temperatures is currently underexplored for biological systems but is a promising route for future srCryoCLEM studies.
4.4. Registration Methods
Regardless of the quality of the SR and CET data, the power of srCryoCLEM can only be realized if the two data sets can be accurately registered. In an srCryoCLEM experiment, there are two essential registration processes. The first is a crude registration, often done by eye at the electron microscope, that occurs after the acquisition of the SR data but before the acquisition of high-resolution CET data. This crude registration is to ensure that the same region imaged in SR is also imaged in CET. TEM finder grids have an alphanumeric grid system built into the underlying metallic mesh that can be useful for this step, but the pattern of the deposited biological sample, such as cells, can also be used to guide the process. When an additional sectioning step such as FIB milling is required, more sophisticated registration approaches are often employed. These approaches have relied on gold or polystyrene beads that can be seen in FM as well as by the cryoSEM that monitors the FIB milling process (20, 147). These methods of registration work well for guiding the sectioning process laterally, but accurate axial guidance is the subject of ongoing work.
Following this crude registration and the acquisition of high-resolution CET data, a fine registration is performed that overlays the SR and CET data. This registration uses fiducials that are visible in both imaging modalities, such as fluorescent polystyrene beads (16–18, 148), as well as the patterned substrate of some TEM grids (19, 31). The latter has the advantages of being present at high density throughout the sample and not producing additional background fluorescence. To date, the fine registration process has been strictly lateral for srCryoCLEM studies. This registration process is often broken into two steps. First, the SR is aligned to a low-magnification cryoEM micrograph, because the field of view for high-magnification CET is too small to contain an adequate number of fiducials visible in FM. Second, the low-magnification cryoEM micrograph is registered to the high-magnification region of interest using gold beads as fiducials that can be seen in sufficient numbers in both the low- and high-magnification micrographs. Gold beads in the range of 15–50 nm in diameter are usually sufficient to be seen clearly in the low-magnification electron micrograph. The computation of the image transformations based on the recovered fiducial positions can be accomplished using custom software. Several functions exist in MATLAB from MathWorks for the registration of two images based on the position of fiducials in each image (19). There are also some published software tools available (149), and even Adobe Photoshop has been used (148). While the minimum number of control-point pairs required to compute a similarity transformation that performs only rotation and scaling is three pairs, we find that this number is insufficient for accurate lateral registration (19). Ideally, 10 or more control-point pairs localized to the regions being correlated in the srCryoCLEM image are used for accurate registration. Again, this registration using fiducials has been performed only in the lateral dimensions. In work in which axial information has been displayed, this was accomplished by aligning the average recovered axial position of a feature in SR with the average axial position of a structure from CET (17, 19). Axial registration can also be accomplished using control-point pairs if a sufficient number of such pairs are determined accurately in all three dimensions (150). While straightforward theoretically, in practice it is challenging to obtain a sufficient number of precise pairs, and as such this is an area of ongoing work.
5. OUTLOOK
In the previous sections, a variety of challenges have been described, and a number of potential solutions have been presented. Without a doubt, the greatest challenge facing the field of srCryoCLEM is improved control (with limited optical intensities) over the emissive state of fluorophores necessary for SMACM-based SR to recover a high density of localizations. Multicolor photoswitching mechanisms need to be explored, possibly with the assistance of cryogenic transient absorption spectroscopy to determine the different transitions and dark states available to fluorophores under cryogenic temperatures. The current challenge to achieving this control makes ISM and SIM promising techniques for routine srCryoCLEM in the short term. The use of immersion objectives would greatly improve the resolution of these two approaches. Devitrification poses another significant challenge to resolution-unlimited SR approaches. Both RESOLFT and SMACM approaches would benefit from higher allowed excitation intensities. This could be enabled by a systematic study of different grid substrates to identify those that absorb less and dissipate more heat. Cryogenic immersion objectives may also reduce devitrification by increasing heat capacity and thermal conduction.
At this time, there is much to be done, and as always, the creativity of the many researchers in the field as they attack these problems will very likely lead to solutions and progress. The tantalizing images shown in Figure 1 can easily be expected to improve, and greatly increased insight can be anticipated.
ACKNOWLEDGMENTS
The authors thank Jesús Galaz-Montoya for a critical reading of the manuscript. This article was supported in part by the National Institute of General Medical Sciences (grant R35-GM118067).
Footnotes
DISCLOSURE STATEMENT
The authors are not aware of any affiliations, memberships, funding, or financial holdings that might be perceived as affecting the objectivity of this review.
LITERATURE CITED
- 1.Feynman R 1960. There’s plenty of room at the bottom. Caltech Eng. Sci 23:22–36 [Google Scholar]
- 2.Abbe E 1873. Beiträge zur Theorie des Mikroskops und der mikroskopischen Detektion [Contributions to the theory of the microscope and microscopic detection]. Arch. Mikroskop. Anat 9:413–68 [Google Scholar]
- 3.Dickson RM, Cubitt AB, Tsien RY, Moerner WE. 1997. On/off blinking and switching behavior of single molecules of green fluorescent protein. Nature 388:355–58 [DOI] [PubMed] [Google Scholar]
- 4.Klar TA, Hell SW. 1999. Subdiffraction resolution in far-field fluorescence microscopy. Opt. Lett 24:954–56 [DOI] [PubMed] [Google Scholar]
- 5.Rust MJ, Bates M, Zhuang X. 2006. Sub-diffraction-limit imaging by stochastic optical reconstruction microscopy (STORM). Nat. Methods 3:793–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Betzig E, Patterson GH, Sougrat R, Lindwasser OW, Olenych S, et al. 2006. Imaging intracellular fluorescent proteins at nanometer resolution. Science 313:1642–45 [DOI] [PubMed] [Google Scholar]
- 7.Hess ST, Girirajan TPK, Mason MD. 2006. Ultra-high resolution imaging by fluorescence photoactivation localization microscopy. Biophys. J 91:4258–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Moerner WE. 2007. New directions in single-molecule imaging and analysis. PNAS 104:12596–602 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Li X, Mooney P, Zheng S, Booth CR, Braunfeld MB, et al. 2013. Electron counting and beam-induced motion correction enable near-atomic-resolution single-particle cryo-EM. Nat. Methods 10:584–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Danev R, Buijsse B, Khoshouei M, Plitzko JM, Baumeister W. 2014. Volta potential phase plate for in-focus phase contrast transmission electron microscopy. PNAS 111:15635–40 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Earl LA, Falconieri V, Milne JL, Subramaniam S. 2017. Cryo-EM: beyond the microscope. Curr. Opin. Struct. Biol 46:71–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lehmann M, Lichtner G, Klenz H, Schmoranzer J. 2016. Novel organic dyes for multicolor localization-based super-resolution microscopy. J. Biophoton 9:161–70 [DOI] [PubMed] [Google Scholar]
- 13.Schueder F, Lara-Gutiérrez J, Beliveau BJ, Saka SK, Sasaki HM, et al. 2017. Multiplexed 3D super-resolution imaging of whole cells using spinning disk confocal microscopy and DNA-PAINT. Nat. Commun 8:2090. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zheng Q, Lavis LD. 2017. Development of photostable fluorophores for molecular imaging. Curr. Opin. Chem. Biol 39:32–38 [DOI] [PubMed] [Google Scholar]
- 15.Schwartz CL, Sarbash VI, Ataullakhanov FI, McIntosh JR, Nicastro D. 2007. Cryo-fluorescence microscopy facilitates correlations between light and cryo-electron microscopy and reduces the rate of photobleaching. J. Microsc 227:98–109 [DOI] [PubMed] [Google Scholar]
- 16.Chang YW, Chen S, Tocheva EI, Treuner-Lange A, Lobach S, et al. 2014. Correlated cryogenic photoactivated localization microscopy and cryo-electron tomography. Nat. Methods 11:737–39 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Liu B, Xue Y, Zhao W, Chen Y, Fan C, et al. 2015. Three-dimensional super-resolution protein localization correlated with vitrified cellular context. Sci. Rep 5:13017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Tuijtel MW, Koster AJ, Jakobs S, Faas FGA, Sharp TH. 2019. Correlative cryo super-resolution light and electron microscopy on mammalian cells using fluorescent proteins. Sci. Rep 9:1369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Dahlberg PD, Saurabh S, Sartor AM, Wang J, Mitchell PG, et al. 2020. Cryogenic single-molecule fluorescence annotations for electron tomography reveal in situ organization of key proteins in Caulobacter. PNAS 117:13937–44 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Wu G, Mitchell PG, Galaz-Montoya JG, Hecksel CW, Sontag EM, et al. 2020. Multi-scale 3D cryo-correlative microscopy for vitrified cells. bioRxiv 107771. 10.1101/2020.05.21.107771 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Sochacki KA, Shtengel G, van Engelenburg SB, Hess HF, Taraska JW. 2014. Correlative super-resolution fluorescence and metal-replica transmission electron microscopy. Nat. Methods 11:305–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Hauser M, Wojcik M, Kim D, Mahmoudi M, Li W, Xu K. 2017. Correlative super-resolution microscopy: new dimensions and new opportunities. Chem. Rev 117:7428–56 [DOI] [PubMed] [Google Scholar]
- 23.Mateos JM, Barmettler G, Doehner J, Ojeda Naharros I, Guhl B, et al. 2017. Correlative super-resolution and electron microscopy to resolve protein localization in zebrafish retina. J. Vis. Exp 129:e56113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Kopek BG, Paez-Segala M, Shtengel G, Sochacki KA, Sun MG, et al. 2017. Diverse protocols for correlative super-resolution fluorescence imaging and electron microscopy of chemically fixed samples. Nat. Protoc 12:916–46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Elmlund D, Elmlund H. 2015. Cryogenic electron microscopy and single-particle analysis. Annu. Rev. Biochem 84:499–517 [DOI] [PubMed] [Google Scholar]
- 26.Cheng Y, Grigorieff N, Penczek PA, Walz T. 2015. A primer to single-particle cryo-electron microscopy. Cell 161:438–49 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ognjenović J, Grisshammer R, Subramaniam S. 2019. Frontiers in cryo electron microscopy of complex macromolecular assemblies. Annu. Rev. Biomed. Eng 21:395–415 [DOI] [PubMed] [Google Scholar]
- 28.Baumeister W, Grimm R, Walz J. 1999. Electron tomography of molecules and cells. Trends Cell Biol. 9:81–85 [DOI] [PubMed] [Google Scholar]
- 29.Asano S, Engel BD, Baumeister W. 2016. In situ cryo-electron tomography: a post-reductionist approach to structural biology. J. Mol. Biol 428:332–43 [DOI] [PubMed] [Google Scholar]
- 30.Wolff G, Hagen C, Grünewald K, Kaufmann R. 2016. Towards correlative super-resolution fluorescence and electron cryo-microscopy. Biol. Cell 108:245–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Dahlberg PD, Perez D, Su Z, Chiu W, Moerner WE. 2020. Cryogenic correlative single-particle photoluminescence spectroscopy and electron tomography for investigation of nanomaterials. Angew. Chem. Int. Ed 59:15642–48 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Russo CJ, Passmore LA. 2014. Electron microscopy: ultrastable gold substrates for electron cryomicroscopy. Science 346:1377–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dubochet J, McDowall AW. 1981. Vitrification of pure water for electron microscopy. J. Microsc 124:3–4 [Google Scholar]
- 34.Dubochet J, Adrian M, Chang J, Homo J, Lepault J, et al. 1988. Cryo-electron microscopy of vitrified specimens. Q. Rev. Biophys 21:129–228 [DOI] [PubMed] [Google Scholar]
- 35.Dobro MJ, Melanson LA, Jensen GJ, McDowall AW. 2010. Plunge freezing for electron cryomicroscopy. In Methods in Enzymology, Vol. 481, ed. Jensen GJ, pp. 63–82. Amsterdam: Elsevier; [DOI] [PubMed] [Google Scholar]
- 36.Sartori N, Richter K, Dubochet J. 1993. Vitrification depth can be increased more than 10-fold by high-pressure freezing. J. Microsc 172:55–61 [Google Scholar]
- 37.Studer D, Graber W, Al-Amoudi A, Eggli P. 2001. A new approach for cryofixation by high-pressure freezing. J. Microsc 203:285–94 [DOI] [PubMed] [Google Scholar]
- 38.Al-Amoudi A, Chang J, Leforestier A, McDowall A, Salamin LM, et al. 2004. Cryo-electron microscopy of vitreous sections. EMBO J. 23:3583–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Rigort A, Bäuerlein FJB, Villa E, Eibauer M, Laugks T, et al. 2012. Focused ion beam micromachining of eukaryotic cells for cryoelectron tomography. PNAS 109:4449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Villa E, Schaffer M, Plitzko JM, Baumeister W. 2013. Opening windows into the cell: focused-ion-beam milling for cryo-electron tomography. Curr. Opin. Struct. Biol 23:771–77 [DOI] [PubMed] [Google Scholar]
- 41.Marko M, Hsieh C, Schalek R, Frank J, Mannella C. 2007. Focused-ion-beam thinning of frozen-hydrated biological specimens for cryo-electron microscopy. Nat. Methods 4:215–17 [DOI] [PubMed] [Google Scholar]
- 42.Wagner FR, Watanabe R, Schampers R, Singh D, Persoon H, et al. 2020. Preparing samples from whole cells using focused-ion-beam milling for cryo-electron tomography. Nat. Protoc 15:2041–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Bouchet-Marquis C, Hoenger A. 2011. Cryo-electron tomography on vitrified sections: a critical analysis of benefits and limitations for structural cell biology. Micron 42:152–62 [DOI] [PubMed] [Google Scholar]
- 44.Xu CS, Hayworth KJ, Lu Z, Grob P, Hassan AM, et al. 2017. Enhanced FIB-SEM systems for large-volume 3D imaging. eLife 6:e25916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Plitzko J, Baumeister WP. 2019. Cryo-electron tomography. In Springer Handbook of Microscopy: Electron and Ion Microscopy, ed. Hawkes PW, Spence JCH, pp. 189–228. Cham, Switz.: Springer [Google Scholar]
- 46.Mercogliano CP, DeRosier DJ. 2006. Gold nanocluster formation using metallothionein: mass spectrometry and electron microscopy. J. Mol. Biol 355:211–23 [DOI] [PubMed] [Google Scholar]
- 47.Martell JD, Deerinck TJ, Lam SS, Ellisman MH, Ting AY. 2017. Electron microscopy using the genetically encoded APEX2 tag in cultured mammalian cells. Nat. Protoc 12:1792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Oda T, Kikkawa M. 2013. Novel structural labeling method using cryo-electron tomography and biotin–streptavidin system. J. Struct. Biol 183:305–11 [DOI] [PubMed] [Google Scholar]
- 49.Chen M, Dai W, Sun SY, Jonasch D, He CY, et al. 2017. Convolutional neural networks for automated annotation of cellular cryo-electron tomograms. Nat. Methods 14:983–85 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Hecksel CW, Darrow MC, Dai W, Galaz-Montoya JG, Chin JA, et al. 2016. Quantifying variability of manual annotation in cryo-electron tomograms. Microsc. Microanal 22:487–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Wietrzynski W, Schaffer M, Tegunov D, Albert S, Kanazawa A, et al. 2020. Charting the native architecture of Chlamydomonas thylakoid membranes with single-molecule precision. eLife 9:e53740. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Frangakis AS, Bohm J, Forster F, Nickell S, Nicastro D, et al. 2002. Identification of macromolecular complexes in cryoelectron tomograms of phantom cells. PNAS 99:14153–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Hrabe T, Chen Y, Pfeffer S, Cuellar LK, Mangold A, Förster F. 2012. PyTom: a python-based toolbox for localization of macromolecules in cryo-electron tomograms and subtomogram analysis. J. Struct. Biol 178:177–88 [DOI] [PubMed] [Google Scholar]
- 54.Albert S, Wietrzynski W, Lee CW, Schaffer M, Beck F, et al. 2020. Direct visualization of degradation microcompartments at the ER membrane. PNAS 117:1069–80 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Himes BA, Zhang P. 2018. emClarity: software for high-resolution cryo-electron tomography and subtomogram averaging. Nat. Methods 15:955–61 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Jin J, Galaz-Montoya J, Sherman MB, Sun SY, Goldsmith CS, et al. 2018. Neutralizing antibodies inhibit chikungunya virus budding at the plasma membrane. Cell Host Microbe 24:417–28.e5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Bharat TA, Scheres SH. 2016. Resolving macromolecular structures from electron cryo-tomography data using subtomogram averaging in RELION. Nat. Protoc 11:2054–65 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Galaz-Montoya JG, Flanagan J, Schmid MF, Ludtke SJ. 2015. Single particle tomography in EMAN2. J. Struct. Biol 190:279–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kador L, Horne DE, Moerner WE. 1990. Optical detection and probing of single dopant molecules of pentacene in a p-terphenyl host crystal by means of absorption spectroscopy. J. Phys. Chem 94:1237–48 [Google Scholar]
- 60.Orrit M, Bernard J. 1990. Single pentacene molecules detected by fluorescence excitation in a p-terphenyl crystal. Phys. Rev. Lett 65:2716–19 [DOI] [PubMed] [Google Scholar]
- 61.Moerner WE, Basché T. 1993. Optical spectroscopy of single impurity molecules in solids. Angew. Chem. Int. Ed 32:457–76 [Google Scholar]
- 62.Basché T, Moerner WE, Orrit M, Wild UP, eds. 1997. Single-Molecule Optical Detection, Imaging, and Spectroscopy. Basel, Switz.: Verlag-Chemie [Google Scholar]
- 63.Betzig E, Chichester RJ. 1993. Single molecules observed by near-field scanning optical microscopy. Science 262:1422–25 [DOI] [PubMed] [Google Scholar]
- 64.Shera EB, Seitzinger NK, Davis LM, Keller RA, Soper SA. 1990. Detection of single fluorescent molecules. Chem. Phys. Lett 174:553–57 [Google Scholar]
- 65.Rigler R, Orrit M, Basché T, eds. 2001. Single Molecule Spectroscopy: Nobel Conference Lectures. Springer Ser. Chem. Phys Berlin: Springer-Verlag [Google Scholar]
- 66.Moerner WE, Plakhotnik T, Irngartinger T, Croci M, Palm V, Wild UP. 1994. Optical probing of single molecules of terrylene in a Shpol’kii matrix: a two-state single-molecule switch. J. Phys. Chem 98:7382–89 [Google Scholar]
- 67.Hell SW. 2009. Microscopy and its focal switch. Nat. Methods 6:24–32 [DOI] [PubMed] [Google Scholar]
- 68.Sahl SJ, Moerner WE. 2013. Super-resolution fluorescence imaging with single molecules. Curr. Opin. Struct. Biol 23:778–87 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Schermelleh L, Heintzmann R, Leonhardt H. 2010. A guide to super-resolution fluorescence microscopy. J. Cell Biol 190:165–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Schermelleh L, Ferrand A, Huser T, Eggeling C, Sauer M, et al. 2019. Super-resolution microscopy demystified. Nat. Cell Biol 21:72–84 [DOI] [PubMed] [Google Scholar]
- 71.von Diezmann A, Shechtman Y, Moerner WE. 2017. Three-dimensional localization of single molecules for super-resolution imaging and single-particle tracking. Chem. Rev 117:7244–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Huang B, Bates M, Zhuang X. 2009. Super resolution fluorescence microscopy. Annu. Rev. Biochem 78:993–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Müller CB, Loman A, Pacheco V, Koberling F, Willbold D, et al. 2008. Precise measurement of diffusion by multi-color dual-focus fluorescence correlation spectroscopy. Eur. Phys. Lett 83:46001 [Google Scholar]
- 74.Mueller CB, Enderlein J. 2010. Image scanning microscopy. Phys. Rev. Lett 104:198101. [DOI] [PubMed] [Google Scholar]
- 75.Gustafsson MGL. 2000. Surpassing the lateral resolution limit by a factor of two using structured illumination microscopy. J. Microsc 198:82–87 [DOI] [PubMed] [Google Scholar]
- 76.Hell SW. 2005. Fluorescence nanoscopy: breaking the diffraction barrier by the RESOLFT concept. NanoBiotechnology 1:296 [Google Scholar]
- 77.Shaner NC, Steinbach PA, Tsien RY. 2005. A guide to choosing fluorescent proteins. Nat. Methods 2:905–9 [DOI] [PubMed] [Google Scholar]
- 78.Fernández-Suárez M, Ting AY. 2008. Fluorescent probes for super-resolution imaging in living cells. Nat. Rev. Mol. Cell Biol 9:929–43 [DOI] [PubMed] [Google Scholar]
- 79.Endesfelder U, Malkusch S, Flottmann B, Mondry J, Liguzinski P, et al. 2011. Chemically induced photoswitching of fluorescent probes—a general concept for super-resolution microscopy. Molecules 16:3106–18 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.O’Hare HM, Johnsson K, Gautier A. 2007. Chemical probes shed light on protein function. Curr. Opin. Struct. Biol 17:488–94 [DOI] [PubMed] [Google Scholar]
- 81.van de Linde S, Heilemann M, Sauer M. 2012. Live-cell super-resolution imaging with synthetic fluorophores. Annu. Rev. Phys. Chem 63:519–40 [DOI] [PubMed] [Google Scholar]
- 82.Saurabh S, Perez AM, Comerci CJ, Shapiro L, Moerner WE. 2016. Super-resolution imaging of live bacteria cells using a genetically directed, highly photostable fluoromodule. J. Am. Chem. Soc 138:10398–401 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Grimm JB, English BP, Choi H, Muthusamy AK, Mehl BP, et al. 2016. Bright photoactivatable fluorophores for single-molecule imaging. Nat. Methods 13:985–88 [DOI] [PubMed] [Google Scholar]
- 84.Ries J, Kaplan C, Platonova E, Eghlidi H, Ewers H. 2012. A simple, versatile method for GFP-based super-resolution microscopy via nanobodies. Nat. Methods 9:582–84 [DOI] [PubMed] [Google Scholar]
- 85.Muyldermans S 2013. Nanobodies: natural single-domain antibodies. Annu. Rev. Biochem 82:775–97 [DOI] [PubMed] [Google Scholar]
- 86.Mikhaylova M, Cloin BMC, Finan K, van den Berg R, Teeuw J, et al. 2015. Resolving bundled microtubules using anti-tubulin nanobodies. Nat. Commun 6:7933. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Banterle N, Bui KH, Lemke EA, Beck M. 2013. Fourier ring correlation as a resolution criterion for super-resolution microscopy. J. Struct. Biol 183:363–67 [DOI] [PubMed] [Google Scholar]
- 88.Huff J 2015. The Airyscan detector from ZEISS: confocal imaging with improved signal-to-noise ratio and super-resolution. Nat. Methods 12:i–ii [Google Scholar]
- 89.Wu Y, Shroff H. 2018. Faster, sharper, and deeper: structured illumination microscopy for biological imaging. Nat. Methods 15:1011–19 [DOI] [PubMed] [Google Scholar]
- 90.Heintzmann R, Huser T. 2017. Super-resolution structured illumination microscopy. Chem. Rev 117:13890–908 [DOI] [PubMed] [Google Scholar]
- 91.Gustafsson MGL, Shao L, Carlton PM, Wang CJR, Golubovskaya IN, et al. 2008. Three-dimensional resolution doubling in wide-field fluorescence microscopy by structured illumination. Biophys. J 94:4957–70 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Lal A, Shan C, Xi P. 2016. Structured illumination microscopy image reconstruction algorithm. IEEE J. Sel. Top. Quantum Electron 22:50–63 [Google Scholar]
- 93.Hell SW. 2007. Far-field optical nanoscopy. Science 316:1153–58 [DOI] [PubMed] [Google Scholar]
- 94.Westphal V, Hell SW. 2005. Nanoscale resolution in the focal plane of an optical microscope. Phys. Rev. Lett 94:143903. [DOI] [PubMed] [Google Scholar]
- 95.Harke B, Keller J, Ullal CK, Westphal V, Schönle A, Hell SW. 2008. Resolution scaling in STED microscopy. Opt. Express 16:4154–62 [DOI] [PubMed] [Google Scholar]
- 96.Giske A 2007. CryoSTED microscopy - a new spectroscopic approach for improving the resolution of STED microscopy using low temperature. PhD Thesis, Heidelberg Univ., Heidelberg, Ger. [Google Scholar]
- 97.Moerner WE. 2012. Microscopy beyond the diffraction limit using actively controlled single molecules. J. Microsc 246:213–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Thompson RE, Larson DR, Webb WW. 2002. Precise nanometer localization analysis for individual fluorescent probes. Biophys. J 82:2775–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mortensen KI, Churchman LS, Spudich JA, Flyvbjerg H. 2010. Optimized localization analysis for single-molecule tracking and super-resolution microscopy. Nat. Methods 7:377–81 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Huang B, Wang W, Bates M, Zhuang X. 2008. Three-dimensional super-resolution imaging by stochastic optical reconstruction microscopy. Science 319:810–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Sage D, Pham T-A, Babcock H, Lukes T, Pengo T, et al. 2019. Super-resolution fight club: assessment of 2D and 3D single-molecule localization microscopy software. Nat. Methods 16:387–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ovesny M, Krizek P, Borkovec J, Svindrych Z, Hagen GM. 2014. ThunderSTORM: a comprehensive ImageJ plug-in for PALM and STORM data analysis and super-resolution imaging. Bioinformatics 30:2389–90 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Sartori A, Gatz R, Beck F, Rigort A, Baumeister W, Plitzko JM. 2007. Correlative microscopy: bridging the gap between fluorescence light microscopy and cryo-electron tomography. J. Struct. Biol 160:135–45 [DOI] [PubMed] [Google Scholar]
- 104.van Driel LF, Valentijn JA, Valentijn KM, Koning RI, Koster AJ. 2009. Tools for correlative cryo-fluorescence microscopy and cryo-electron tomography applied to whole mitochondria in human endothelial cells. Eur. J. Cell Biol 88:669–84 [DOI] [PubMed] [Google Scholar]
- 105.Schorb M, Gaechter L, Avinoam O, Sieckmann F, Clarke M, et al. 2017. New hardware and workflows for semi-automated correlative cryo-fluorescence and cryo-electron microscopy/tomography. J. Struct. Biol 197:83–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Dahlberg PD, Sartor AM, Wang J, Saurabh S, Shapiro L, Moerner WE. 2018. Identification of PAmKate as a red photoactivatable fluorescent protein for cryogenic super-resolution imaging. J. Am. Chem. Soc 140:12310–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Weisenburger S, Jing B, Hänni D, Reymond L, Schuler B, et al. 2014. Cryogenic colocalization microscopy for nanometer-distance measurements. Chem. Phys. Chem 15:763–70 [DOI] [PubMed] [Google Scholar]
- 108.Weisenburger S, Boening D, Schomburg B, Giller K, Becker S, et al. 2017. Cryogenic optical localization provides 3D protein structure data with Angstrom resolution. Nat. Methods 14:141–44 [DOI] [PubMed] [Google Scholar]
- 109.Moerner WE, Kador L. 1989. Optical detection and spectroscopy of single molecules in a solid. Phys. Rev. Lett 62:2535–38 [DOI] [PubMed] [Google Scholar]
- 110.Hoffman DP, Shtengel G, Xu CS, Campbell KR, Freeman M, et al. 2020. Correlative three-dimensional super-resolution and block-face electron microscopy of whole vitreously frozen cells. Science 367:eaaz5357. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Li S, Ji G, Shi Y, Klausen LH, Niu T, et al. 2018. High-vacuum optical platform for cryo-CLEM (HOPE): a new solution for non-integrated multiscale correlative light and electron microscopy. J. Struct. Biol 201:63–75 [DOI] [PubMed] [Google Scholar]
- 112.Le Gros M, McDermott G, Uchida M, Knoechel C, Larabell C. 2009. High-aperture cryogenic light microscopy. J. Microsc 235:1–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Wang L, Bateman B, Zanetti-Domingues LC, Moores AN, Astbury S, et al. 2019. Solid immersion microscopy images cells under cryogenic conditions with 12 nm resolution. Commun. Biol 2:74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Inagawa H, Toratani Y, Motohashi K, Nakamura I, Matsushita M, Fujiyoshi S. 2015. Reflecting microscope system with a 0.99 numerical aperture designed for three-dimensional fluorescence imaging of individual molecules at cryogenic temperatures. Sci. Rep 5:12833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Nahmani M, Lanahan C, DeRosier D, Turrigiano GG. 2017. High-numerical-aperture cryogenic light microscopy for increased precision of superresolution reconstructions. PNAS 114:3832. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Gustavsson A-K, Petrov PN, Lee MY, Shechtman Y, Moerner WE. 2018. 3D single-molecule super-resolution microscopy with a tilted light sheet. Nat. Commun 9:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Vicidomini G, Bianchini P, Diaspro A. 2018. STED super-resolved microscopy. Nat. Methods 15:173–82 [DOI] [PubMed] [Google Scholar]
- 118.van Oijen AM, Köhler J, Schmidt J, Müller M, Brakenhoff GJ. 1998. 3-Dimensional super-resolution by spectrally selective imaging. Chem. Phys. Lett 292:183–87 [Google Scholar]
- 119.Naumov AV, Gorshelev AA, Vainer YG, Kador L, Köhler J. 2009. Far-field nanodiagnostics of solids with visible light by spectrally selective imaging. Angew. Chem. Int. Ed 48:9747–50 [DOI] [PubMed] [Google Scholar]
- 120.Kozankiewicz B, Orrit M. 2014. Single-molecule photophysics, from cryogenic to ambient conditions. Chem. Soc. Rev 43:1029–43 [DOI] [PubMed] [Google Scholar]
- 121.Zheng Q, Jockusch S, Zhou Z, Blanchard SC. 2014. The contribution of reactive oxygen species to the photobleaching of organic fluorophores. Photochem. Photobiol 90:448–54 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Zondervan R, Kulzer F, Kol’chenk MA, Orrit M. 2004. Photobleaching of rhodamine 6G in poly(vinyl alcohol) at the ensemble and single-molecule levels. J. Phys. Chem. A 108:1657–65 [Google Scholar]
- 123.Banasiewicz M, Wiącek D, Kozankiewicz B. 2006. Structural dynamics of 2,3-dimethylnaphthalene crystals revealed by fluorescence of single terrylene molecules. Chem. Phys. Lett 425:94–98 [Google Scholar]
- 124.Werley CA, Moerner WE. 2006. Single-molecule nanoprobes explore defects in spin-grown crystals. J. Phys. Chem. B 110:18939–44 [DOI] [PubMed] [Google Scholar]
- 125.Hulleman CN, Li W, Gregor I, Rieger B, Enderlein J. 2018. Photon yield enhancement of red fluorophores at cryogenic temperatures. Chem. Phys. Chem 19:1774–80 [DOI] [PubMed] [Google Scholar]
- 126.Weisenburger S, Jing B, Renn A, Sandoghdar V. 2013. Cryogenic localization of single molecules with angstrom precision. In Proceedings SPIE, Nanoimaging and Nanospectroscopy, Vol. 8815. San Diego, CA: Int. Soc. Opt. Photonics [Google Scholar]
- 127.Kaufmann R, Schellenberger P, Seiradake E, Dobbie IM, Jones EY, et al. 2014. Super-resolution microscopy using standard fluorescent proteins in intact cells under cryo-conditions. Nano Lett. 14:4171–75 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Ha T, Tinnefeld P. 2012. Photophysics of fluorescent probes for single-molecule biophysics and super-resolution imaging. Annu. Rev. Phys. Chem 63:595–617 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Dickson RM, Norris DJ, Moerner WE. 1998. Simultaneous imaging of individual molecules aligned both parallel and perpendicular to the optic axis. Phys. Rev. Lett 81:5322–25 [Google Scholar]
- 130.Enderlein J, Toprak E, Selvin PR. 2006. Polarization effect on position accuracy of fluorophore localization. Opt. Express 14:8111–20 [DOI] [PubMed] [Google Scholar]
- 131.Engelhardt J, Keller J, Hoyer P, Reuss M, Staudt T, Hell SW. 2011. Molecular orientation affects localization accuracy in superresolution far-field fluorescence microscopy. Nano Lett. 11:209–13 [DOI] [PubMed] [Google Scholar]
- 132.Backlund MP, Lew MD, Backer AS, Sahl SJ, Grover G, et al. 2012. Simultaneous, accurate measurement of the 3D position and orientation of single molecules. PNAS 109:19087–92 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Backer AS, Lee MY, Moerner WE. 2016. Enhanced DNA imaging using super-resolution microscopy and simultaneous single-molecule orientation measurements. Optica 3:659–66 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Lew MD, Backlund MP, Moerner WE. 2013. Rotational mobility of single molecules affects localization accuracy in super-resolution fluorescence microscopy. Nano Lett. 13:3967–72 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Hulleman CN, Huisman M, Moerland RJ, Grünwald D, Stallinga S, Rieger B. 2018. Fluorescence polarization control for on-off switching of single molecules at cryogenic temperatures. Small Methods 2:1700323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Lew MD, Moerner WE. 2014. Azimuthal polarization filtering for accurate, precise, and robust single-molecule localization microscopy. Nano Lett. 14:6407–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Backlund MP, Arabi A, Petrov PN, Arabi E, Saurabh S, et al. 2016. Removing orientation-induced localization biases in single-molecule microscopy using a broadband metasurface mask. Nat. Photon 10:459–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Schwentker MA, Bock H, Hofmann M, Jakobs S, Bewersdorf J, et al. 2007. Wide-field subdiffraction RESOLFT microscopy using fluorescent protein photoswitching. Microsc. Res. Tech 70:269–80 [DOI] [PubMed] [Google Scholar]
- 139.Subach FV, Malashkevich VN, Zencheck WD, Xiao H, Filonov GS, et al. 2009. Photoactivation mechanism of PAmCherry based on crystal structures of the protein in the dark and fluorescent states. PNAS 106:21097–102 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Regis Faro A, Carpentier P, Jonasson G, Pompidor G, Arcizet D, et al. 2011. Low-temperature chromophore isomerization reveals the photoswitching mechanism of the fluorescent protein Padron. J. Am. Chem. Soc 133:16362–65 [DOI] [PubMed] [Google Scholar]
- 141.Regis Faro A, Adam V, Carpentier P, Darnault C, Bourgeois D, de Rosny E. 2010. Low-temperature switching by photoinduced protonation in photochromic fluorescent proteins. Photochem. Photobiol. Sci 9:254–62 [DOI] [PubMed] [Google Scholar]
- 142.Ambrose WP, Basché T, Moerner WE. 1991. Detection and spectroscopy of single pentacene molecules in a p-terphenyl crystal by means of fluorescence excitation. J. Chem. Phys 95:7150–63 [Google Scholar]
- 143.Moerner WE, ed. 1988. Persistent Spectral Hole-Burning: Science and Applications. Top. Curr. Phys Vol. 44. Berlin: Springer [Google Scholar]
- 144.van Oijen AM, Köhler J, Schmidt J, Müller M, Brakenhoff GJ. 1999. Far-field fluorescence microscopy beyond the diffraction limit. J. Opt. Soc. Am. A 16:909–15 [Google Scholar]
- 145.Völker S 1987. Optical linewidths and dephasing of organic amorphous and semi-crystalline solids studied by hole burning. J. Lumin 36:251–62 [Google Scholar]
- 146.Creemers TMH, Lock AJ, Subramaniam V, Jovin TM, Voelker S. 2000. Photophysics and optical switching in green fluorescent protein mutants. PNAS 97:2974–78 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Arnold J, Mahamid J, Lucic V, de Marco A, Fernandez J, et al. 2016. Site-specific cryo-focused ion beam sample preparation guided by 3D correlative microscopy. Biophys. J 110:860–69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Tuijtel MW, Koster AJ, Faas FG, Sharp TH. 2019. Correlated cryo super-resolution light and cryo-electron microscopy on mammalian cells expressing the fluorescent protein rsEGFP2. Small Methods 3:1900425 [Google Scholar]
- 149.Paul-Gilloteaux P, Heiligenstein X, Belle M, Domart M, Larijani B, et al. 2017. eC-CLEM: flexible multidimensional registration software for correlative microscopies. Nat. Methods 14:102–3 [DOI] [PubMed] [Google Scholar]
- 150.Gahlmann A, Ptacin JL, Grover G, Quirin S, von Diezmann ARS, et al. 2013. Quantitative multicolor subdiffraction imaging of bacterial protein ultrastructures in 3D. Nano Lett. 13:987–93 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Dai W, Fu C, Khant HA, Ludtke SJ, Schmid MF, Chiu W. 2014. Zernike phase-contrast electron cryotomography applied to marine cyanobacteria infected with cyanophages. Nat. Protoc 9:2630–42 [DOI] [PMC free article] [PubMed] [Google Scholar]