

Abstract
Although systemic antibiotics are critical in controlling infections and reducing morbidity and mortality, overuse of antibiotics is presumed to contribute to negative repercussions such as selection of antimicrobial-resistant organisms and collateral damage to commensal microbes. In a prospective, randomized study of four clinically relevant antibiotic regimens [doxycycline (20mg or 100mg), cephalexin, or trimethoprim/sulfamethoxazole], we investigated microbial alterations on skin after administration of systemic antibiotics to healthy human volunteers. Samples from different skin and oral sites as well as stool were collected before, during, and up to 1 year after antibiotic use, and shotgun metagenomic sequencing was performed. Taxonomic analysis showed that subjects receiving doxycycline 100mg and trimethoprim/sulfamethoxazole (TMP/SMX) exhibited greater changes to their skin microbial communities, as compared to those receiving other regimens, or untreated controls. Oral and stool microbiota also demonstrated fluctuations after antibiotics. Bacterial culturing in combination with whole-genome sequencing revealed specific emergence, expansion, and persistence of antibiotic-resistant staphylococci harboring tetK or tetL and dfrC or dfrG genes in all subjects who received doxycycline 100mg or TMP/SMX, respectively. Last, analysis of metagenomic data revealed an increase of genes involved in gene mobilization, indicating stress responses of microbes to antibiotics. Collectively, these findings demonstrate direct, long-lasting effects of antibiotics on skin microbial communities, highlighting the skin microbiome as a site for the development and persistence of antibiotic resistance and the risks of overprescribing.
One Sentence Summary:
Use of systemic antibiotics induces substantial changes in the microbiome and expansion of antimicrobial resistant bacteria.
Introduction
Antibiotics are essential in the treatment of infections with over 250 million outpatient oral antibiotics prescribed annually in the United States alone(1). Despite increased awareness of antibiotic stewardship, overprescribing continues to be a widespread issue(2, 3). The Centers for Disease Control and Prevention estimates more than 30% - and potentially up to 50% - of outpatient antibiotic prescriptions in the United States are unnecessary(1). Given the prevalence of overprescribing and the routine use of different classes of antibiotics for both infections and non-infectious chronic conditions, it is critical to understand the collateral damage of these prescribing habits.
Despite the skin’s constant exposure to the environment and other individuals, prior longitudinal studies have demonstrated that the skin microbiomes within healthy individuals are relatively stable from intervals of weeks to a few years(4, 5). Given intense interest in the biological relevance of the human microbiome, understanding how interventions may perturb the microbiome of the largest human organ over time is critically important. Although previous reports have suggested that systemic antibiotic usage can increase the prevalence of antibiotic-resistant microbes on skin(6–8), these studies focused on cultured isolates or targeted resistance genes. A limited number of skin microbiome studies have analyzed the effects of antibiotics(9, 10). Previous mouse studies have shown mixed results regarding the alteration of skin microbiome by systemic antibiotics (11, 12); however, skin microbiome alterations with clinically relevant antibiotic regimens routinely used for dermatologic conditions have not been deeply investigated. Two-week courses of cephalosporins (for example cephalexin) or trimethoprim/sulfamethoxazole are among first-line treatments for skin and soft-tissue infections. Oral doxycycline and other tetracyclines are commonly used to treat infection as well as chronic inflammatory conditions such as acne and rosacea. These chronic conditions are often treated with doxycycline 100mg twice daily for an average of 2–3 months – and sometimes years(13–15); low-dose doxycycline (20mg twice daily) has been suggested as a subantimicrobial alternative without notable antibacterial effects(16).
To determine if different classes of common antibiotic regimens result in short- and long-term alterations of the skin microbiome, we developed a pilot study to test the effects of distinct clinically relevant regimens of oral antibiotics on the human microbiome in healthy volunteers. By incorporating shotgun metagenomic microbiome analyses with bacterial cultivation and whole-genome sequencing, and skin biopsies to examine host changes, we sought to more deeply investigate how different commonly prescribed antibiotics might shape human microbial communities and development of antibiotic resistance.
Results
Changes in composition of microbial communities after antibiotic use
To investigate the effects of systemic antibiotics on an individual’s microbes and the antibiotic resistome, we recruited healthy volunteers (NCT01631617) to be randomized to receive one of four different clinically relevant antibiotic regimens [doxycycline 20mg (Doxy20) or 100mg (Doxy100) twice daily for 56 days, cephalexin 500mg three times daily for 14 days (Ceph), trimethoprim/sulfamethoxazole 160/800mg twice daily for 14 days (TMP/SMX)]. Because certain inflammatory diseases, infections, and exposures are associated with altered microbiomes(17–24), eligibility criteria were developed to reduce possible confounders, including chronic medical conditions, menopause, recent antibiotic use, and tobacco use (Fig. S1).
A total of 14 healthy volunteers completed one of four possible treatment regimens with a minimum of three subjects per regimen to detect whether an antibiotic regimen would result in differences in the skin microbiome. Given the relative intrapersonal stability of the skin microbiome in healthy individuals(4, 5) and early studies of stool and throats showing microbiome alterations are distinguishable in cohorts of three subjects treated with oral antibiotics(25, 26), our pilot study was designed to investigate whether the different antibiotic regimens commonly prescribed for dermatologic disorders could alter the skin microbiomes in healthy individuals. Swabs were collected longitudinally from 3 skin sites possessing distinct physiological and microbial characteristics(27) (Ac: antecubital crease, Ra: retroauricular crease, Vf: volar forearm), before, during, and after antibiotic use (≥6 time points per subject) (Fig. 1A, and data file S1).
Fig. 1. Changes and resilience of skin microbiome after systemic antibiotics.
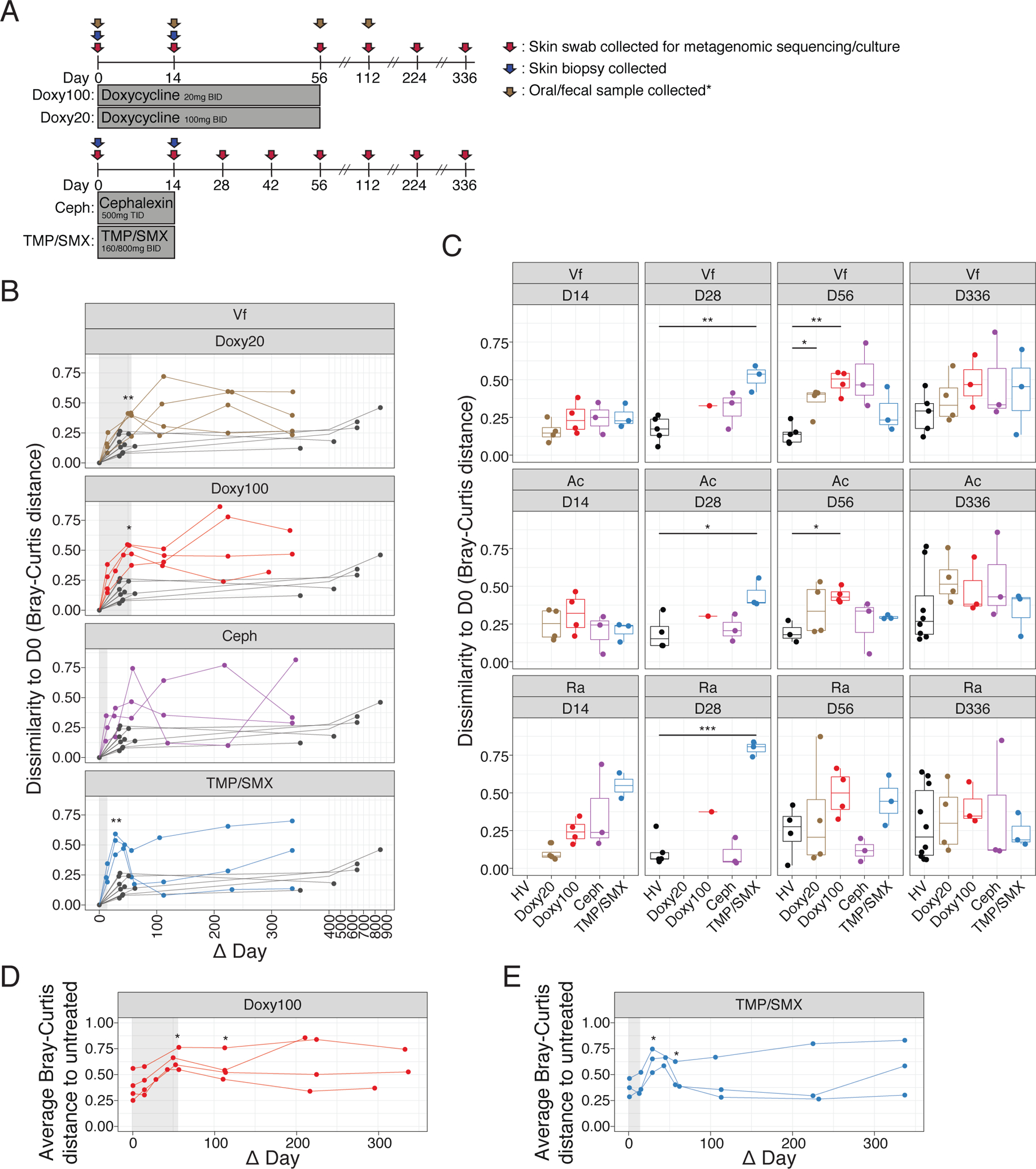
(A) Study design. Healthy subjects were randomized to 4 different antibiotic regimens. Skin swabs for metagenomic sequencing and in vitro culturing were collected at indicated time points (red arrows), skin biopsies for mass-spectrometry were collected at D0 and D14 (blue arrows) and oral/fecal samples were collected at D0, D14, D56 and D112 for doxycycline treated subjects (brown arrows). BID = twice daily; TID = three times daily. See Methods for details. *Fecal samples from Ceph and TMP/SMX were not sequenced. (B) Changes in microbiome similarity of Vf for each subject. Points indicate Bray-Curtis distance at each time point compared to D0 for each subject. Lines connect points from the same individual. Colored points/lines are data from subjects who received antibiotics, and gray points/lines indicate untreated healthy subjects (n=10, 5 samples excluded). Light gray shaded area marks the time period when subjects received antibiotics. (C) Bray-Curtis distances in (B) are summarized by time point and statistical significance determined using Wilcoxon rank-sum test (*P<0.05, **P<0.01, ***P<0.005); including Ac (n=10, 2 samples excluded) and Ra (n=10). (D) and (E) Average Bray-Curtis distance of Doxy100 subjects (D) and TMP/SMX subjects (E) to all of untreated healthy subjects (Vf). Statistical significance determined using Wilcoxon rank-sum test (*P<0.05, **P<0.01)
For the evaluable healthy volunteers, microbial compositions of each skin sample were profiled via shotgun metagenomic sequencing. In total, we obtained 4,896 million of non-human, quality-filtered paired-end total reads (median 11 million non-human reads per sample). As has been previously reported in healthy individuals (4, 28), skin microbial communities were predominated by bacteria (87.47% ± 1.62%), followed by fungi (11.52% ± 1.6%) and viruses (0.81% ± 0.31%) (Fig. S2). Deeper taxonomic analyses demonstrated Corynebacterium spp. (mainly C. tuberculostearium and C. simulans), Staphylococcus spp. (including S. epidermidis and S. hominis), Cutibacterium acnes, Cutibacterium and Staphylococcus phages, and Malassezia spp. (M. globosa and M. restricta) were the predominant microbes(28, 29) (Fig. S3, and data file S2).
Using day 0 (D0, prior to antibiotic use) as the baseline for each subject, the relative changes in the composition of microbial species after antibiotic use in each individual were quantified by Bray-Curtis distance (30), which calculates dissimilarity between two ecological communities and considers both membership and relative abundance of each member of the community. Analysis of skin microbiome data from untreated healthy subjects followed over a similar duration [short-interval (1–2 months, n=10) and long-interval (>1 year, n≥5, excluding samples with low sequencing quality)] were included to demonstrate homeostatic stability of unperturbed skin microbiomes(4). Microbiomes of individuals on Doxy100 (at D56, P<0.05, Wilcoxson rank-sum test) and TMP/SMX (at D28 and D42, P<0.05, Wilcoxson rank-sum test) were significantly dissimilar from baseline after oral antibiotics compared to untreated individuals (Fig. 1, B and C, and Fig. S4), with the exception of the Ra site after Doxy100 treatment. The results indicate that the composition and relative abundances of the individual taxa were altered with oral antibiotics to a greater extent than typically observed in untreated healthy individuals. The skin microbiomes of study subjects in the 3–7 weeks prior to oral antibiotic ingestion were similar to D0 and shifted only after exposure to systemic antibiotics (Fig. S4, C and D), further showing that the perturbations of the skin microbiome were due to systemic antibiotics and not from stochastic changes in the skin microbiomes. Of note, qPCR showed the relative quantity of bacterial genomic DNA was not significantly (P >= 0.05, Wilcoxson rank-sum test) reduced by antibiotics, suggesting the alteration was not due to the reduced biomass (Fig. S4, E).
To further examine antibiotic-induced microbial alterations, microbiomes of antibiotic recipients and untreated subjects were evaluated by principal coordinate analysis (Fig. S5A). Although alterations of skin microbiomes during and after antibiotic use were evident, especially for Doxy100 and TMP/SMX, inter-personal variation also contributed to the differences among samples (Fig. S5B). To delineate the changes of the microbiomes as compared to an unperturbed status, we calculated the average Bray-Curtis distance between the data from all antibiotic study subjects to all of the untreated subject samples (Fig. 1, D and E, and Fig. S5, C to E). Microbiomes of Doxy100 subjects significantly shifted away from the untreated samples at days 56 and 112 (P<0.05), and at days 28, 42, and 56 (P<0.05) for TMP/SMX. Although the skin microbiomes in subjects who received TMP/SMX (14-day regimen) had greater Bray-Curtis dissimilarities compared to their own baseline microbiomes and the untreated cohort before day 42, the skin microbiomes exhibited significant resilience – defined as a return to the baseline state – after day 42. In contrast, Doxy100 subjects exhibited profound and prolonged skin microbiome changes persisting beyond 200 days – potentially due to a longer treatment duration of 56 days (Fig. 1, B to E). Altogether, the results demonstrated substantial alteration and resilience of skin microbiomes after systemic antibiotics, with varying magnitudes dependent on subjects and regimens. Mass spectrometry analysis from skin biopsies revealed that all antibiotics were readily detectable in the outermost areas – dermis and epidermis – of the skin in all tested subjects (data file S3), thereby explaining or directly influencing the lasting impact and effects of antibiotics on the skin microbiome.
To understand the effects of systemic antibiotics on other distinct microbial niches, we investigated changes in oral and gut microbiomes, as it is known that each body site hosts unique microbial communities (31, 32). Although oral microbial communities became relatively dissimilar during and after doxycycline use in comparison to baseline states, (Fig. S6A), a lack of comparable controls limited robust comparisons. Changes in the fecal microbiome (Fig. S6B) in doxycycline recipients were also observed but the degree of alteration varied among subjects. It has been shown that the effects of antimicrobials on the gut microbiome are heavily context dependent(33–37) with respect to antimicrobial type, administration route, regimen duration, host health status, and so on., Antimicrobial regimens in this cohort were selected based on their typical usage in dermatological practice, which would be anticipated to have antimicrobial activity in the skin. Unlike the reduced microbial diversities observed in the gut after gastrointestinal disease-directed antibiotics(33, 34), skin microbial community diversities increased after Doxy100 and TMP/SMX regimens (Fig. S7A). In subjects receiving Doxy100, increased gut microbial diversity was due to an influx of new species (Fig. S7, B to D).
Emergence and selective expansion of doxycycline-resistant staphylococci on skin
We next focused on investigating antibiotic resistance by culturing and sequencing bacterial isolates from pre- and post-antibiotic skin swabs (Fig. 2A). Addition of doxycycline 2μg/ml to culture media selected the growth of culturable bacteria collected from the pre-antibiotic skin of Doxy100 and Doxy20 subjects (Fig. 2B and C, and Fig. S8, A to D). Recoverable bacteria from on- and post-antibiotic samples of all Doxy100 and two of the four Doxy20 subjects (Subjects 1 and 3) grew in the presence of doxycycline (Fig. 2B and C), indicating emergence of doxycycline-resistant species on the skin of these subjects. Resistant species from those individuals were predominantly staphylococci, including S. epidermidis, followed by S. hominis (Fig. 2B and C). Similarly, TMP-resistant staphylococci grew from all TMP/SMX individuals (Subjects 12, 13, and 14) only after antibiotic use (Fig. 2D, and Fig. S8E).
Fig. 2. Expansion and emergence of antibiotic-resistant S. epidermidis within skin-associated microbial communities.
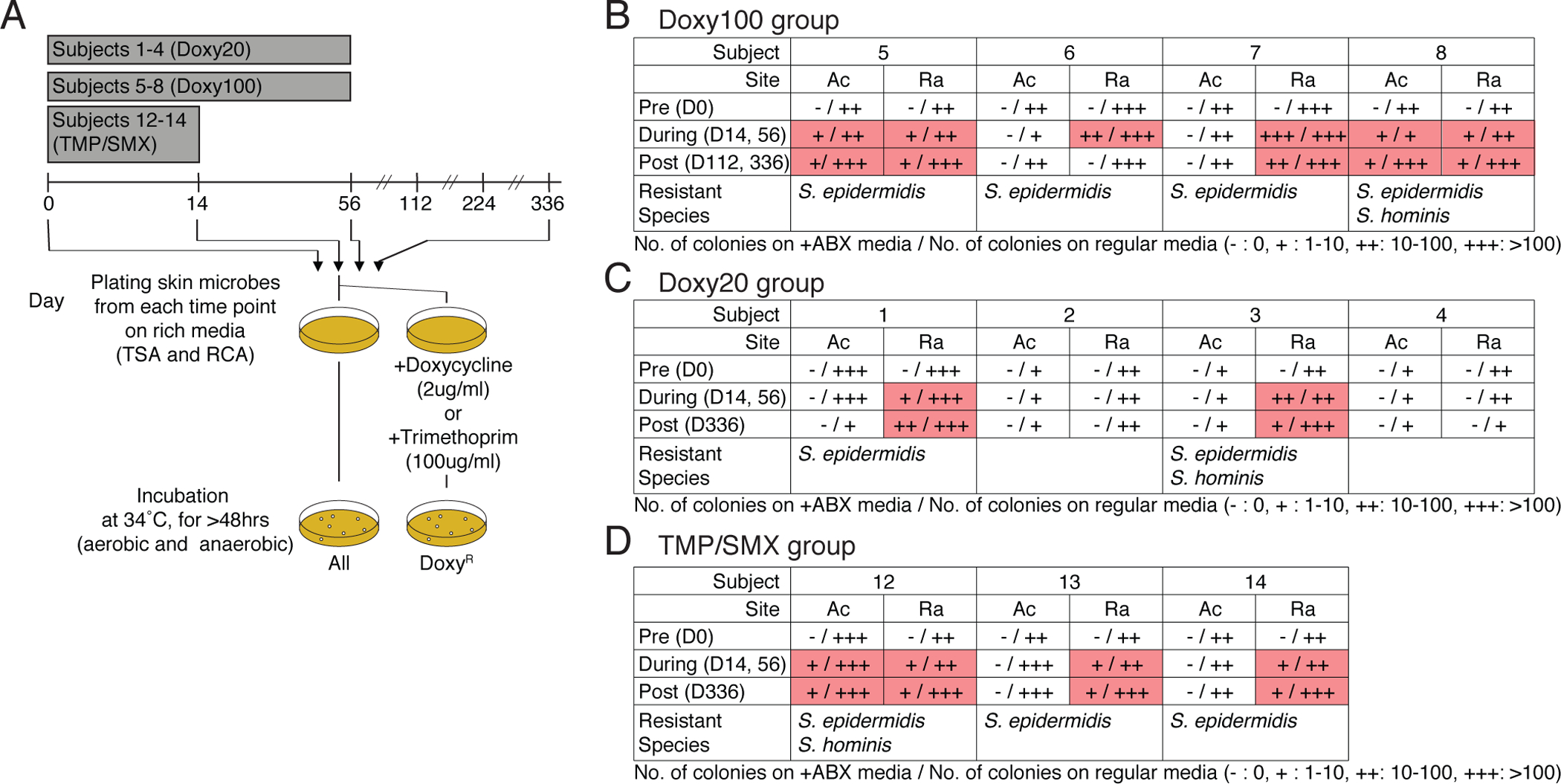
(A) Experimental design for in vitro culturing of doxycycline-resistant (DoxyR) or TMP-resistant (TMPR)skin bacteria. Skin swabs from D0, D14, D56, D112, and D336 were plated on TSB and RCA agar plate (with or without Doxy 2ug/ml or 100ug/ml), and incubated under aerobic and anaerobic conditions, respectively. After 72 hours (up to 120 hours to allow for minor and slow growing microbes to make visible colony), colonies were counted, grown independently, DNA extracted, and sequenced. (B) to (D) Summary tables for in vitro culture results of (B) Doxy100 subjects, (C) Doxy20 subjects and (D) TMP/SMX subjects. (+: 1–10 colonies, ++: 10–100 colonies, +++: >100 colonies, pink boxes highlight presence of DoxyR colonies). Cells with read shading indicate where resistant colonies were found. Species of isolated resistant bactera are listed at the bottom of each table.
To explore strain diversity and the dynamics of acquisition of doxycycline-resistant S. epidermidis, we sequenced S. epidermidis isolates from pre- and post-antibiotic timepoints from all 8 individuals in the Doxy100 and Doxy20 groups, regardless of antibiotic resistance (>19 isolates per subject). Then sequences were compared and a phylogeny was constructed for all the isolates of each individual (Fig 3A and B, and Fig. S9A to F). As previously proposed from metagenomic analyses (4, 28), S. epidermidis colonization of an individual was multiphyletic, simultaneously harboring strains spanning multiple sequence types (ST, defined by multi-locus sequence typing (MLST)). In addition, the presence of genes that confer doxycycline resistance were analyzed, and the minimum inhibitory concentration (MIC) to doxycycline of each isolate was measured in vitro. All doxycycline-resistant S. epidermidis isolates (MIC >= 2μg/ml) harbored either plasmid-associated tetK or tetL genes, encoding efflux pumps known to confer resistance to tetracyclines (38). Notably, doxycycline-resistant S. epidermidis strains were only isolated after subjects received doxycycline and continued to be isolated at D336 (Fig. 3A and B, and Fig. S9A and F), indicating that the antibiotic served as the selective pressure for doxycycline-resistant S. epidermidis. In addition, doxycycline-resistant isolates from Doxy100 subjects had higher MIC values to doxycycline than those from Doxy20 subjects (Fig. 3A and B, and Fig. S9A and D).
Fig. 3. Phylogenetic analyses and dynamics of S. epidermidis skin isolates in Doxy100 subjects.
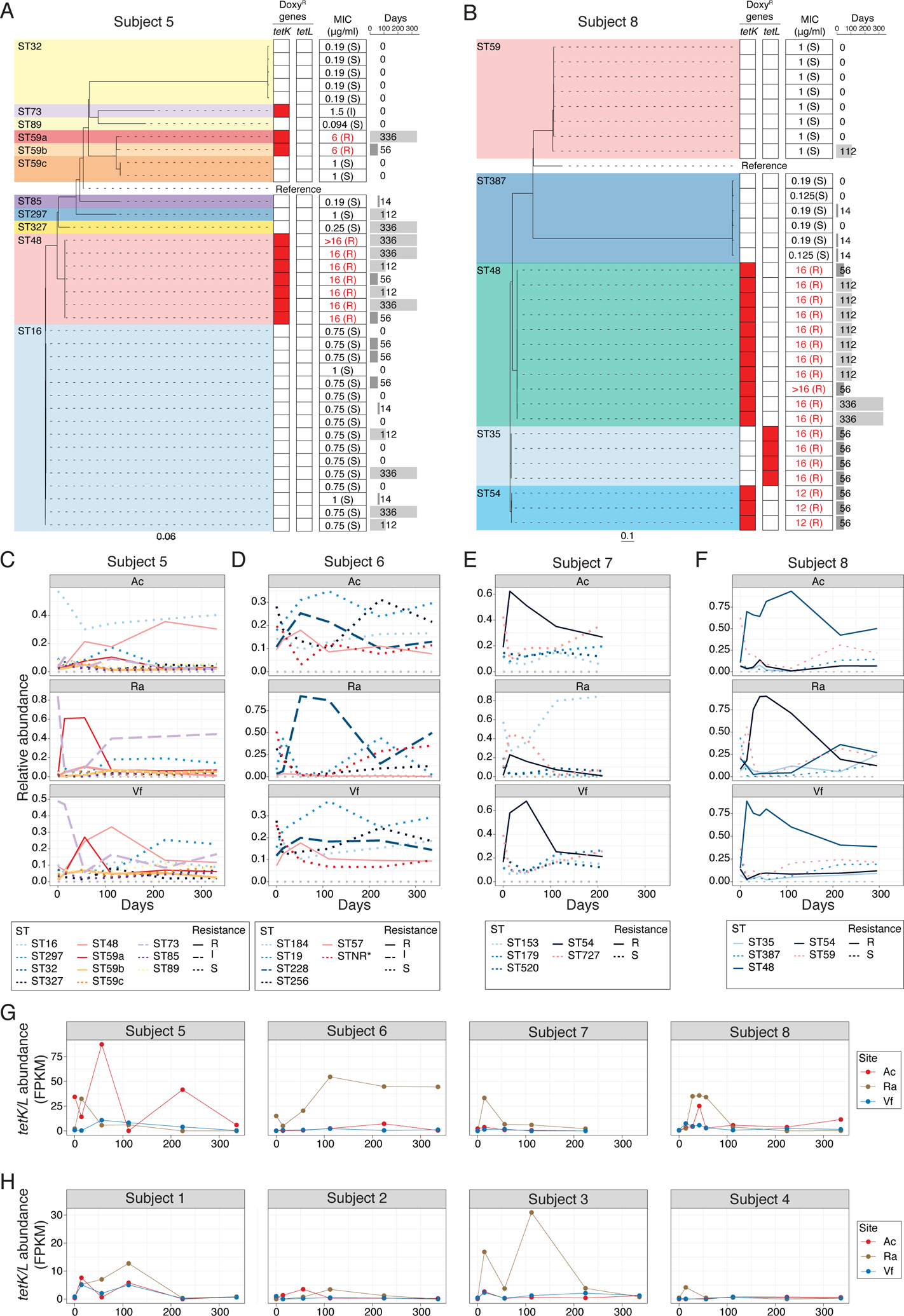
(A) and (B) Phylogeny of S. epidermidis isolates from Subject 5 (A), and 8 (B). Dashed lines connect with doxycycline-resistant genes profiles (heatmap), with red denoting presence of specific resistant genes. Minimum inhibitory concentration (MIC) for doxycyline is summarized on the middle column. Time point (days) of isolation for each isolates are indicated on the right panel. STNR*: allele combination not reported in MLST allele database (MLST.net). (C) to (F) Changes of relative abundances of S. epidermidis STs from metagenomic data of Doxy100 subjects. Subject 5 (C), 6 (D), 7 (E) and 8 (F). Relative abundances of each ST were estimated by BIB (see Methods for detail). (G) and (H) Changes of relative abundances of tetK/L from metagenomic data of Doxy100 (G) and Doxy20 (H) groups. FPKM (fragments per kilobase per million reads) was calculated based on the number of reads mapped at high confidence to either tetK or tetL to the total of non-human reads (see Methods).
To systematically investigate the dynamics of doxycycline-resistant S. epidermidis strains during and after antibiotics, we estimated relative changes of each ST in each subject using a tool deploying a probabilistic model to infer a ratio of different genomes from metagenomic data(39). As expected from the culture results, expansion and persistence of doxycycline resistant STs were observed in all Doxy100 subjects (Fig. 3C to F) and 2 subjects in Doxy20 groups (Fig. S9G to J). In subjects 5 and 8, the predominant doxycycline-resistant strains were different on each body site, consistent with differences in microbial communities observed in anatomically distinct skin sites (5, 28) and strongly suggesting that antibiotic-driven selection is also influenced by the skin site characteristics. This site-dependent observation also was reflected in the abundances of tetK and tetL genes measured in each subject and site (Fig. 3, G and H). Collectively, these results clearly demonstrated the selection, expansion, and persistence of antibiotic-resistant strains on skin both during and after systemic antibiotic use. There were no detectable tetK and tetL genes in the metagenomic data of other body sites from the same subjects, such as oral, gut, and vaginal tract, indicating site- and niche-specific selection of resistance genes and bacteria (Fig S10).
Emergence and selective expansion of trimethoprim-resistant staphylococci on skin
Analysis of genomes of S. epidermidis isolates from Subject 12, 13 and 14, who developed TMP-resistant staphylococci, revealed a core chromosomal point mutation (F99Y) in the dihydrofolate reductase (dfrA) gene or an additional TMP-resistant dfrG allele which emerged and expanded after TMP/SMX usage (Fig. 4, A to C). Similar to the expansion of doxycycline-resistant strains in Doxy100, expansion of TMP-resistant S. epidermidis was observed when relative changes of STs were inferred from metagenomics (Fig. 4, D to F). Together, whole-genome sequencing of isolates and targeted metagenomic analyses demonstrated selective expansion of TMP-resistant bacteria on skin, similar to doxycycline.
Fig. 4. Dynamics of S. epidermidis strains in TMP/SMX group.
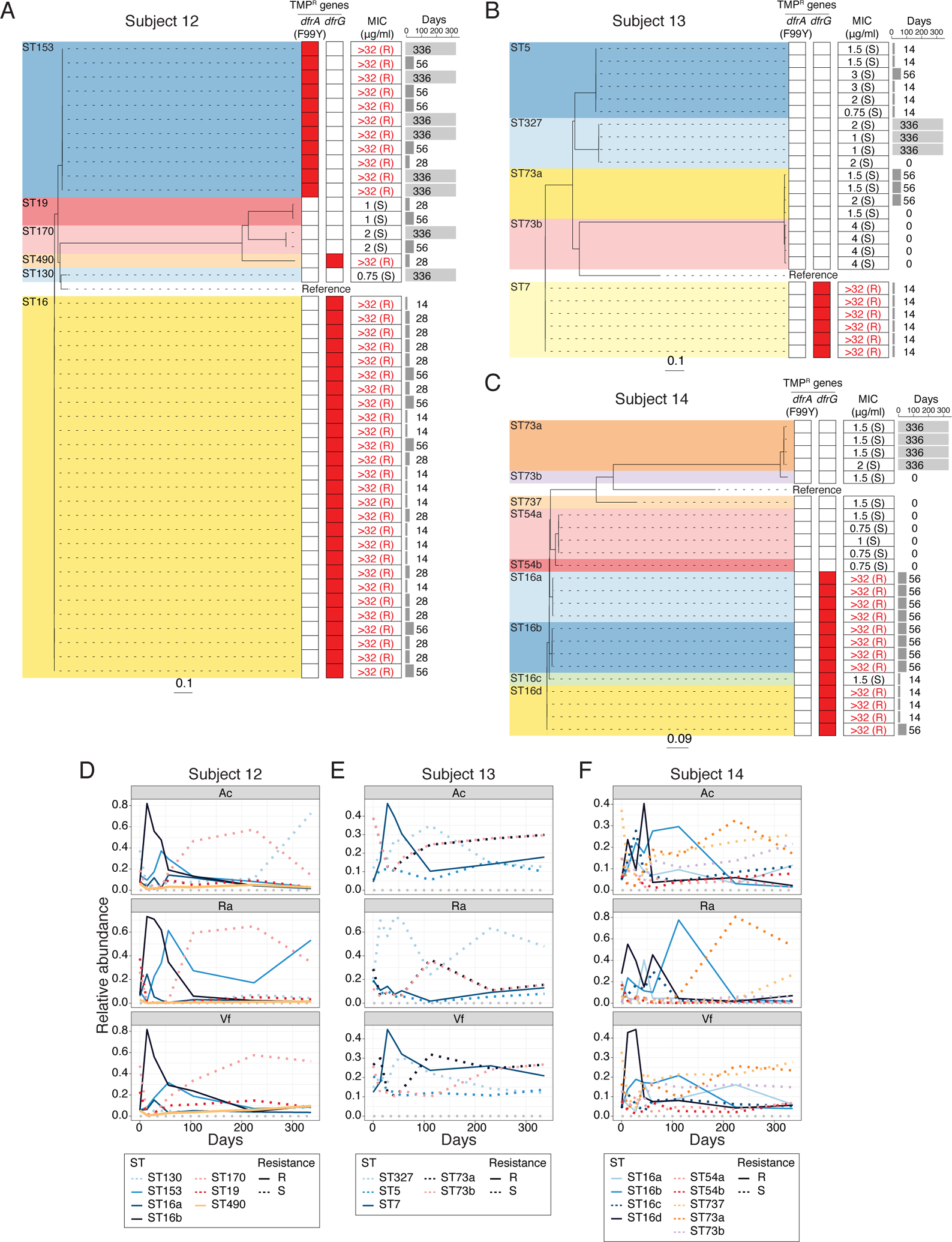
(A) to (C) Phylogeny of S. epidermidis isolates from Subject 12 (A), 13 (B) and 14 (C). Dashed lines connect with TMP-resistant genes profiles (heatmap), with red denoting presence of specific resistant genes. Minimum inhibitory concentration (MIC) for TMP is summarized on the middle column. Time point (days) of isolation for each isolates are indicated on the right pane l. (D) to (F) Changes of relative abundances of S. epidermidis STs from metagenomic data of TMP/SMX subjects. Subject 12 (D), 13 (E) and 14 (F). Relative abundances of each ST were estimated by BIB (see Methods for detail).
Increased gene mobilization observed after antibiotic use
Previous studies have described that antibiotic usage is associated with changes in the functional capacity of the microbiome(40). To increase the resolution of this analysis, we performed de novo assembly of the unmapped shotgun metagenomic reads and then classified functional metagenomic gene clusters (MGCs) from this expanded reference (Fig. S11, A and B). 31 MGCs (14 increased/17 decreased) from Doxy100 subject samples were significantly changed (adj. P<0.05) at D56, as compared to D0 and D14 (Fig. 5A). Among 14 increased MGCs, 5 clusters were annotated as transposable elements or contained putative functions for DNA integration (GO:0015074) (Fig. 5B, and data file S4). This observation is consistent with previous findings that gene mobilization in bacteria is associated with stress, including antimicrobials(41, 42). Similarly, samples from TMP/SMX subjects revealed 134 and 237 MGCs increased and decreased, respectively, at D42 compared to D0 and D14 (Fig. 5C); more than 50% of the increased MGCs contained GO terms for DNA integration (GO:0015074) (Fig 5D, and data file S5).
Fig. 5. Increased gene mobilization of skin microbiomes with antibiotic use.
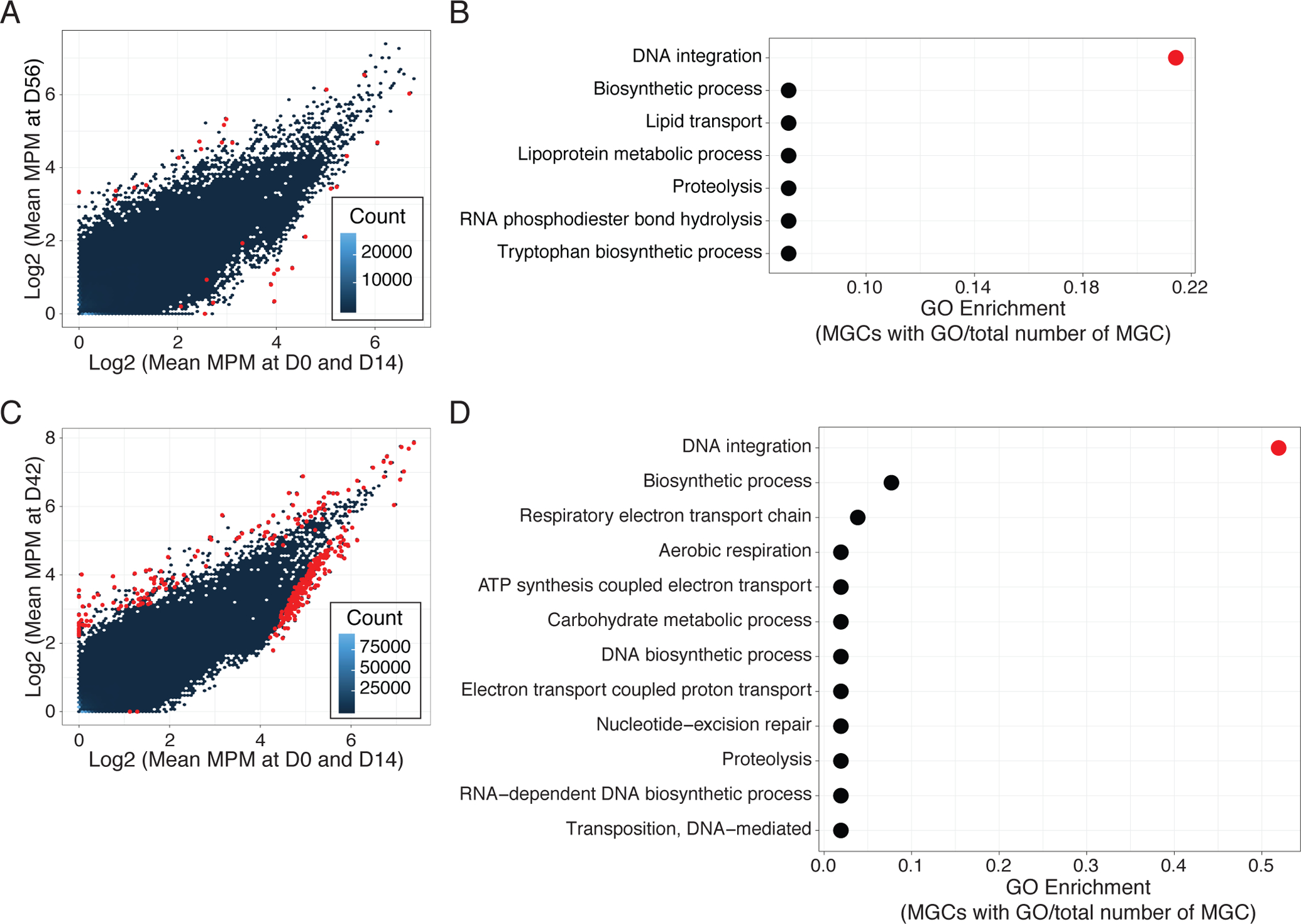
(A) and (C) Comparison of MGC relative abundances between D0 and D56 in Doxy100 subjects (A) and D0 and D42 in TMP/SMX subjects (C). The x-axis indicates log-transformed mean MGC abundances (MGC abundance per million reads (MPM), see Methods for detail) at D0 and D14 and the y-axis indicates log-transformed mean MPM at D56 (A) and D42 (C). Hexbin (blue to dark blue colors) indicate the number of MGCs in each hexagon and red points indicate significantly increased or decreased MGCs (adjusted P<0.05). (B) and (D) Analysis for Gene Ontology (GO, biological process) enrichment of increased MGCs in Doxy100 subjects (B) and in TMP/SMX subjects (D). GO enrichment was calculated by dividing of MGC with indicated GO term to the all differentially abundant MGC (x-axis). Red color indicates P<0.05 (Fisher’s exact test, see Methods).
Discussion
Systemic antibiotics have undoubtedly shaped modern medicine by reducing mortality from invasive infection, yet the repercussions of current widespread antibiotic overprescribing should be closely examined. In this pilot study, we randomized healthy human subjects to several clinically relevant antibiotic regimens and showed that human skin microbial communities can be substantially altered. The degree and duration of the change, and emergence of antimicrobial resistant bacteria differed based on type and dose of antibiotics, with inter-personal variability.
Perturbations of the human microbiome by antibiotics have been most often studied in the gut (33, 40, 43–45). These gut microbiome studies consistently demonstrated decreased community diversity, outgrowth of minor taxa, and changes of the resistome in the gut after oral antibiotics, albeit depending on the agent and duration. Only a few sequencing-based studies have investigated the changes of skin microbial populations after usage of systemic antimicrobials(11, 34, 46), reporting a negligible effect of antibiotics on the skin microbiome. Conversely, culture-based studies showed long-term alterations of the commensal skin microbiota by antibiotics, with decreasing numbers of commensal Staphylococcus and Cutibacterium bacteria and increasing numbers of antibiotic-resistant microbes(6, 7, 47, 48). Consistent with culture-based evidence, our current work demonstrates that antibiotics can disrupt the homeostasis of skin microbial communities and elicit critical changes in the resistome, with some changes persisting almost one year after discontinuation. The antibiotics used in this study are routinely used to treat not only various systemic and cutaneous infections but also common inflammatory disorders; therefore, our results raise serious concerns about collateral damage to the skin microbiome with antibiotic usage.
A prior longitudinal study in healthy volunteers demonstrated that although the intrapersonal skin microbiomes were relatively stable over short and long time intervals, the stability was dependent on the specific anatomical skin sites and the skin microbial communities were individual-specific(4). Consistent with the previously reported individuality of the human skin microbiome, we also uncovered noticeable heterogeneity in the changes of skin microbiome across different individuals and sites. One could speculate that multiple interconnected factors may contribute to the variations, including dynamic changes of antibiotic concentrations on skin during the course of treatment; physiological, chemical, and physical characteristics of different skin sites; individual genetic and environmental exposure variations; inherent differences in skin microbial communities before antibiotic usage in individual subjects; and individual-specific differences in antibiotic metabolism and bioavailability. Despite the myriad potential contributors to the microbiome changes in each subject, we observed that, in general, doxycycline-resistant S. epidermidis isolated from individuals who received doxycycline 20mg had lower doxycycline MICs than isolates from Doxy100 subjects, indicating effective concentrations of antibiotics on skin is a critical factor. With regards to anatomical site variation, Ra skin is more sebaceous, or oily, than Ac and Vf skin and houses higher relative abundances of lipophilic bacteria or strains that harbor more genes related to utilizing fatty acids as an energy source or that are tolerant to high fatty acid concentrations. The lipophilic nature of doxycycline also likely contributes to different local concentrations of antibiotics at anatomical sites with varied abundances of skin lipids. In addition, it is important to note that some skin microbiome changes might not be reflected in the results due to inherent limitations in metagenomic sampling and analysis pipelines. A further limitation is the small cohort of subjects who fulfilled the eligibility criteria and were able to comply with study requirements. A future study with a larger cohort would be important to more deeply explore the mechanisms of interindividual variation and antimicrobial resistance.
As proposed in previous reports(49), antibiotic use introduces selective pressure for the emergence and expansion of antimicrobial resistant bacteria on skin. A unique aspect of our study is providing not only evidence of shifts in microbial communities with shotgun metagenomic sequencing but also genetic insights into antimicrobial resistance at an high resolution through combined whole-genome isolate sequencing and phenotypic profiling. We demonstrated that doxycycline and trimethoprim use promoted the selection and expansion of staphylococcal species harboring resistance. One important question beyond the scope of resolution of this study is whether the resistant microbes were present before or acquired after antibiotic usage. Culturing from the skin surface did not reveal selectively resistant strains prior to antibiotic receipt. However, this does not preclude a resistant strain deeper within the skin adnexal structures or at abundances below detection. Although a resistant strain was predicted to exist before the treatments, multiple resistance determinants and strains were identified in each subject, suggesting a complex combination of different acquisition routes: expansion of a low abundant pre-colonizer, de novo colonization and selection post-treatment, or intra-/inter-species resistance transmission. A previous report showed antibiotic stress can trigger bacterial competence and eventually promote horizontal gene transfer (50), suggesting resistance transmission is likely occurring in this context. In this regard, it is interesting that doxycycline-sensitive ST73s (from subjects 1, 3, 4, and 5), even with intact tetK gene, were isolated from pretreated skin, strongly suggesting that these can potentially serve as donors for resistance gene transfer. As mentioned above, although various possible mechanisms for the emergence of antibiotic-resistant exist, the observation that all Doxy100 and TMP/SMX individuals acquired resistant microbes relatively quickly suggests that the antibiotic-resistant – against doxycycline and trimethoprim in this study – microorganisms and mechanisms are widely spread in the community, and capable of colonizing and expanding on susceptible individuals. Individuals on antibiotics also may be a source of resistant microbial spread to a wider population as skin is constantly shedding into the environment and skin microbes are a dominant component of indoor and man-made structures (51–53).
We also showed that the antibiotics were detectable in the outermost compartment of skin. Excreted antibiotics potentially remain on the skin for an extended period of time due to the presence of sebum on skin and the lipophilicity of some antibiotics (for example doxycycline and trimethoprim), the relatively gradual turnover of keratinocytes, and lack of active metabolism of antibiotics on skin. Doxycycline has been shown to have higher bioavailability in skin (54, 55), and we showed concentrations of doxycycline in the epidermal layer of skin of Doxy100 subjects were comparable to – or even slightly higher than – the in vitro MIC of staphylococci. Additionally, in a discrepancy between strain abundance approximation and tetK/L gene abundance, ST48 on the Ac of subjects 5 and 8 remained relatively high until the last day of the study (~300 days), but tetK/L expression dropped to near 0 at that time point. This may be due to the technical or sensitivity differences between the two measurements. However, genetic adaptation of strains after the cessation of antibiotics potentially contributed, although we did not observe decreased MIC or significant genetic changes from ST48 strains isolated from later time points as compared to earlier time points. Therefore, selection, expansion, and persistence of different resistant strains may be a function of multiple parameters, including concentrations of antibiotics at a given time and location and the fitness costs of a specific resistance mechanism for each organism.
Among the different regimens tested, both subtherapeutic (Doxy20) and therapeutic (Doxy100) doses of doxycycline altered the skin microbiome. Higher concentrations of doxycycline were measured in the skin of the Doxy100 subjects as compared to Doxy20. Moreover, doxycycline-resistant S. epidermidis isolated from individuals who received doxycycline 20mg had lower MIC to doxycycline than isolates from Doxy100 subjects. Studies have focused on the effects of subtherapeutic administration of antibiotics in animal feed on the development of antibiotic-resistant bacteria in gut microbiota. Although a recent study of subtherapeutic tetracycline doses did not demonstrate increased abundances of tetM resistance genes in animal stools compared to the therapeutic group(56), prior work has shown increases in the abundance of antimicrobial resistance microbes in stool from cattle fed subtherapeutic antibiotics(57). Results of the current study strongly suggest that subtherapeutic doses may provide weaker selection pressure for the emergence and expansion of resistance on skin. Given the shedding of human keratinocytes into the environment, additional studies of how antibiotic regimens may induce antimicrobial resistance in humans and spread the resistance into the environment are important.
Analysis of metagenomic sequencing data from various body sites revealed that tetK- and tetL-harboring microbes were expanded only on skin, but not in gut or oral sites. This could be partly due to the differences in epithelial site characteristics because gut and oral microbiota likely experience more direct exposure to doxycycline or trimethoprim, with potentially higher antibiotic concentrations and for a relatively shorter duration of time as a result of gut transit times and oral salivary production, leading to a lower likelihood of exposure to persistent subtherapeutic antibiotic concentrations. The other potential reason is that the dominant microbes in intestinal tracts, such as species belonging to Bacteroides, Proteobacteria, and anaerobic Firmicutes, preferentially harbor alternate resistance determinants. Supporting this hypothesis, studies have shown that the types of tetracycline resistant genes more prevalent among intestinal microbes are tetQ, tetW, tetO, tetX, and tet32, but not tetK or tetL(58, 59). We also observed expansion of tet32 in the gut of Doxy100 subjects. Furthermore, although there was systemic absorption of these antibiotics, the fact that the antibiotic types and regimens used in this study are effective against skin infections and conditions correspond with the more appreciable alterations of the skin microbiome than other sites.
Using de novo assembly and functional annotation from metagenomic data, we observed enrichment of the DNA mobilization related genes in the skin microbiome after antibiotic use. Gene mobilization and phage activation have been shown as stress and SOS responses of bacteria, including antibiotic-induced stress. The subsequent increased gene mobilization has been shown to contribute to horizontal gene transfer(60), including virulence and antibiotic resistance determinants(42, 60, 61). Therefore, stress responses – gene mobilization – of skin microbes during antibiotics treatment may contribute to the expansion of resistance.
Our study provides a systematic framework for understanding the alterations in skin microbiome based on specific perturbations. With the integrative approach of metagenomic sequencing and whole-genome sequencing of the cultured bacteria, this study highlights expansion of antimicrobial resistant S. epidermidis, one of the most predominant species in skin microbial communities and important nosocomial pathogens. Our findings demonstrate that the skin, continually shedding microbiota into the environment(51–53), is an important niche for selection and persistence of antimicrobial resistant organisms, and an understudied reservoir for spread of antimicrobial resistance(62, 63).
Materials and Methods
Study design
This study was designed as a randomized single-center, longitudinal, interventional pilot study to compare the effects of three different antibiotic classes with four standard oral regimens [doxycycline 20 mg twice daily for 56 days, doxycycline 100 mg twice daily for 56 days, cephalexin 500mg three times daily for 14 days, and trimethoprim/sulfamethoxazole (TMP/SMX; 160/800 mg twice daily for 14 days)] on the human microbiome for up to one year. Eligibility criteria included healthy adults aged between 18 and 50 years old with ability to comply with antibiotic administration, microbiome sampling procedures, and longitudinal follow-up after ingestion of antibiotics for up to 1 year. Exclusion criteria included systemic antibiotics in the 12 months preceding baseline sampling, history of atopic dermatitis or asthma, known allergy to drugs related to interventional agents, family history of toxic epidermal necrolysis, known immunodeficiencies or chronic past or present medical/dermatological conditions, pregnant or lactating females, females with symptoms and/or hormone levels consistent with perimenopause or menopause, smokers or subjects using smokeless tobacco products, and inability to comply with seven-day skin preparatory regimen prior to each skin sampling (avoidance of topical antimicrobials, swimming/hot tubs, and topical emollients and cosmetics for seven days; avoidance of showering and/or bathing for 24 hours). The eligibility criteria were developed to reduce potential confounders because several diseases (for example acne and eczema), infections, physiological factors such as menopause, and exposures such as. tobacco use are associated with altered microbiomes (17–24). The study complied with institutional guidelines including all relevant ethical regulations and included a safety monitoring plan and safety reporting requirements during subject enrollment. The study was approved by the Institutional Review Board of the National Cancer Institute (https://clinicaltrials.gov/ct2/show/NCT01631617) in June 2012. Safety reporting of systemic, including cutaneous, toxicity criteria utilized the revised NCI Common Terminology Criteria for Adverse Events (CTCAE) version 4.0.
After enrollment, the study was designed for subjects to undergo investigator-blinded randomization to receive one of the four antibiotic regimens. Prior to starting antibiotics, baseline visits included microbiome sample collection (skin, oral, vaginal, and stool samples), blood draws, and skin biopsy. Subjects were administered open-label antibiotic regimens. In addition to review of systems to evaluate for adverse events, general skin examination, and microbiome sample collection (skin, oral, vaginal, and stool samples) at subsequent clinic visits, pill counts were completed during the antibiotic administration period. Skin biopsies were collected again at day 14. Blood draws for antibiotic serum levels were performed at day 14 (and additionally at day 56 for doxycycline-randomized subjects) with witnessed ingestion of the antibiotics 60 minutes (TMP/SMX) or 90 minutes (doxycycline, cephalexin) prior to blood draws and collected in red top plain Vacutainer tubes (Becton, Dickinson and Company).
Microbiome alterations after antibiotics and Shannon diversity index differences between baseline and day 14 were selected as the primary outcome measures with exploratory outcomes including over-/under-representation of certain microbial taxa as well as within-subject analyses. Based on existing knowledge of intrapersonal skin microbiome relatively stability within healthy individuals(4, 5) and prior studies of stool and throats showing microbiome alterations in cohorts of three treated with oral antibiotics particularly notable in contrast to untreated controls(25, 26), a minimum of 3 evaluable sets of data for each antibiotic regimen for this pilot study was determined to be sufficient to investigate if the antibiotic regimens would alter the skin microbiome. This pilot cohort of healthy volunteers receiving one of the four antibiotic regimens was anticipated to have a 50% drop-out rate with an intention of 3 evaluable datasets for each regimen, thus the recruitment goal was a total of 24 healthy subjects.
Between November 2012 and December 2018, healthy adults were recruited to participate. A total of 564 potential subjects were screened (Fig. S1): 320 were determined to be ineligible (unable to comply with protocol requirements, chronic past or present medical conditions, outside eligible age range, use of systemic antibiotics in the prior 12 months, tobacco use, allergies to antibiotics used in the study, or lactation) and 244 declined to participate (not interested after hearing about the study, not able to attend study visits, or not willing to participate in study instructions, sample collection, or antibiotic ingestion).
Of the 22 subjects enrolled, written informed consent was obtained. Three subsequently declined to participate; therefore, 19 were randomized to four different clinically-relevant antibiotic regimens [doxycycline 20mg (Doxy20) or 100mg (Doxy100) twice daily for 56 days, cephalexin 500mg three times daily for 14 days (Ceph), trimethoprim/sulfamethoxazole 160/800mg twice daily for 14 days (TMP/SMX)]. Two healthy volunteers declined to begin their allocated antibiotic regimen, two healthy volunteers terminated their antibiotic regimen early due to side effects (gastrointestinal side effects in 1 subject on doxycycline 20 mg twice daily and in 1 subject on doxycycline 100mg twice daily), and one healthy volunteer was removed from the study due to multiple missed doses noted during the first pill count.
Evaluable subjects were 14 men and women (mean age 33.1 ± 7.3 years); one of the 14 evaluable subjects developed a non-life-threatening cutaneous hypersensitivity reaction to TMP/SMX after completion of the regimen. Some subjects declined skin biopsies, and several subjects were unable to comply with stool collection procedures. Subject and sample metadata are presented in data file S1.
Sample collection, DNA preparation and metagenomic sequencing.
Prior to, during, and after antibiotic administration, subjects underwent sample collection. Sterile swabs (Epicentre) were premoistened with 100μl of yeast cell lysis buffer (Lucigen) and rubbed vigorously on pre-specified skin sites for 20 seconds. For oral (Buccal cavity) samples, dry sterile swabs (Epicentre) were used to rub pre-specified oral sites for 20 seconds and immediately placed in 100μl of yeast cell lysis buffer (Lucigen). All collected swabs were immediately stored at −80°C. At the same time, skin swabs for culture were also collected from bilateral sites and stored in 250 μl of Fastidious Broth (Remel) supplemented with 20% glycerol, at −80°C. Negative control swabs were collected for each patient visit. Stool samples were self-collected by subjects, placed in sterile containers (Sarstedt, NC0705093), and immediately stored in −20°C until transferred to laboratory −80°C freezers for long-term storage.
For shotgun metagenomics sequencing, samples from three skin sites representing diverse physiological characteristics were obtained: moist (antecubital crease, Ac); dry (volar forearm, Vf); and sebaceous (retroauricular crease, Ra). Procedures for DNA extraction, library generation, and sequencing were done as previously described for the skin and oral samples(28). Briefly, samples were incubated in yeast cell lysis buffer (MasterPure Yeast DNA Purification Kit, Lucigen) and Ready-Lyse (Lucigen) for 30 min at 37°C, then mechanically disrupted using 5 mm stainless steel beads (Qiagen) in a Tissuelyser (Qiagen) for 2 min, 30Hz. Samples were incubated for 60 min at 65°C, placed on ice for 5 min, and debris spun down after treatment with MPC protein precipitation reagent. Supernatants were combined with 350μL of 100% ethanol and column purified using the Invitrogen PureLink Genomic DNA. Last, samples were eluted in 30μl of PCR-water (Qiagen). Control swabs also underwent the same DNA extraction processes and sequencing along with experimental samples, and no apparent contamination from either reagents or experimental procedures was observed. For stool samples, frozen fecal matter (1–2g) was pulverized using cooled BioPulverizer (BioSpec products). Then, DNeasy powersoil kit was used to extract DNA from 250ug of pulverized stool with manufactures instruction. Nextera XT (Illumina) library kits were used to generate Illumina libraries per manufacturer’s instructions. Libraries were sequenced on an Illumina HiSeq and NovaSeq 6000 at the NIH Intramural Sequencing Center to a target of 30 to 100 million clusters of 2 × 125bp reads. In total, we obtained 14.7 billion total reads and 4.9 billion of non-human, quality filtered paired-end total reads (median 11 million reads per sample).
Untreated subject data from a previous publication
Data from our previous publication (4) were used as ‘untreated controls’. Initial enrollment criteria for the prior and current studies were largely identical, except concerns regarding allergies to antibiotics and ability to comply to antibiotic ingestion and clinic visit frequency. In the prior study, samples were collected longitudinally such that the span between time 1 and time 2 was 10–30 months, while 5–10 weeks separated time 2 and time 3. Samples for both studies were collected at the same facility and processed with same methods; including DNA and sequencing library preparation and sequencing data processing (see above for details). Among the 12 subjects in the prior publication(4), data from 2 subjects were excluded because the same two subjects also participated in the current study (Subject 12 and 13 are HV08 and HV10 in the previous study, respectively). Also, due to the low sequencing quality (total non-human reads < 50,000), 5 samples from Vf and 2 samples from Ac were excluded.
Taxonomic profiling of metagenomic data.
Taxonomic classifications were performed as previously described(28). Quality processed reads that did not match to the hg19 human reference genome were mapped against a database of 2,349 bacterial, 389 fungal, 4,695 viral, and 67 archaeal reference genomes using Bowtie 2 (version 2.3.2) --very-sensitive parameter(64). Read hit counts were not normalized by genome size. Reads were scaled to 100 to calculate relative abundance of each species.
Pipelines for functional analysis of metagenomic data.
First, non-human metagenomic reads from Doxy_std and TMP/SMX groups were mapped to bacterial, viral, fungal, or archaeal reference genomes with Bowtie 2(64). Regions where reads were mapped were extracted from the reference (‘Mapped regions’, Fig. S12B). Reads that were not mapped to bacterial, viral, fungal, or archaeal reference genomes (‘Unmapped reads’) were extracted, and then de novo assembled using SPAdes(65) genome assembler (version 3.9), with --meta parameter, with k-mer lengths of 21, 33, 55, 77. After assembly, contigs shorter than 500 nucleotides (nt) were discarded. Open reading frames (ORFs) were predicted using Prodigal(66), from both extracted mapped regions and unmapped contigs. ORFs that share more than 90% identity were merged into one representative ORF using CD-HIT(67) to remove redundant sequences, and nucleotide sequences for representative ORFs were used as a database for later analysis. Then, metagenomic reads were mapped back to All_ORF_DB, using usearch(68) and the relative abundance of each ORF in each sample was calculated with following formula;
(RAi: Relative abundance of ORFi per million reads, Xi: number of reads that mapped to ORFi, li: length of ORFi, in kilobase) Of note, this formula is the same formula for transcript per million reads (TPM) in RNA-seq analysis, but referred differently as ‘relative ORF abundance per million reads (or OPM)’ here. To cluster ORFs with similar function, homologous ORFs from All_ORF_DB were grouped with CD-HIT, with --e 0.4 parameter, and each ORF cluster referred as ‘metagenomic gene cluster (MGC)’. For Doxy_std, from 51 metagenomic data, 457,304 MGCs were identified, and 563,153 MGCs from 69 metagenomic data of TMP/SMX. OPM values for the ORFs belong to the same MGC were summed and represented as an abundance of each MGC (MGC abundance per million reads, MPM). To find differentially abundant MGCs, Limma package was used with MPM table as an input (MGCs with adjusted P<0.05 were selected for further analysis)(69). A schematic diagram for the pipeline is shown in Fig. S12.
For gene ontology (GO) enrichment analysis, a representative amino acid sequence from each differentially abundant MGC was extracted and GO terms for the sequences were obtained via OmicsBox (BioBam) Gene Ontology Annotation workflow. To find significantly enriched GO terms, Fischer’s exact test was performed with 100 randomly selected MGCs as a reference with 100 bootstraps, after which the median P value was used.
Measuring antibiotics concentration from blood.
Cephalexin, sulfamethoxazole, and doxycycline serum concentration testing was performed by Mayo Clinic Laboratories. Certain samples had doxycycline serum concentrations measured with a validated LC-MS/MS assay with a calibration range of 2 – 2000 ng/mL that utilized a simple acetonitrile protein-precipitation. Tetracycline was used as an internal standard and this assay was performed and assessed per FDA Guidance.
Laser Capture Microdissection of skin biopsy
Frozen skin biopsy samples were embedded in 10% gelatin (Sigma) and cryosectioned (CM1810, Leica) at 5 μm thickness for histology and 25 μm thickness for laser capture microdissection (LMD7, Leica). Hematoxylin and eosin (H&E) staining was carried out according to the manufacturer’s protocol (Thermo) and the correct orientation was assessed prior to Liquid Chromatography Mass Spectrometry (LCM). The stratum corneum and epidermis, superficial dermis, and deep dermis were dissected from multiple serial sections until 3 million μm2 tissue was obtained from each region of interest, per biopsy sample. Sections were taken either side of sections dedicated to LCM to ensure histological structures of interest did not change throughout the serial sections. Pre- and post-LCM scans were taken from each section dissected using the 5x magnification on the LMD7 scope (Fig. S12). H&E-stained sections were scanned using a Pannoramic Desk digital slide scanner and images were generated in CaseViewer (3DHistech).
Antibiotics quantitation by high pressure liquid chromatography coupled to tandem mass spectrometry (HPLC-MS/MS)
Doxycycline, cephalexin, trimethoprim, sulfamethoxazole, and verapamil were purchased from Sigma Aldrich. Doxycycline-d3 internal standard (IS) was purchased from Toronto Research Chemicals. Drug free CD1-Mouse skin was used as a surrogate to human skin to build standard curves. Neat 1 mg/mL DMSO stocks for trimethoprim, sulfamethoxazole, doxycyline, and cephalexin were serial diluted in 50/50 acetonitrile (CAN)/Milli-Q water to create neat standards. Control tissue homogenate was created by adding 25.6 parts PBS buffer: 1 part tissue (26.7x dilution) and shaking the samples using a Fisher Bead Mill for 1 minute at 6000 rpm with zirconia beads. Standard (STD), quality control (QC), and control samples for trimethoprim, sulfamethoxazole, and cephalexin were extracted by adding 2 μL of blank homogenate, 10 μL of neat standard, and 50 μL of extract solvent containing 50/50 ACN/MeOH and 10 ng/mL Verapamil IS. STD, QC, and control samples for doxycycline were extracted by adding 2 uL of matrix matched homogenate, 10 μL of Neat standard, 10 μL of 5 ng/mL doxycyline-d3 IS, and 50 μL of extract solvent containing 33% trichloroacetic acid. Laser micro-dissected study samples were extracted similarly to standards using 2 μL of PBS in place of tissue homogenate. Extracts were bath sonicated 10 minutes and centrifuged at 4000 rpm for 5 minutes. 50μL of supernatant was transferred to a 96-well plate for HPLC-MS/MS analysis.
LC-MS/MS analysis was performed on a Sciex Applied Biosystems Qtrap 6500+ triple-quadrupole mass spectrometer coupled to a Shimadzu Nexera X2 UHPLC system to quantify each drug in plasma. Chromatography was performed on an Agilent Zorbax SB-C8 column (2.1×30 mm; particle size, 3.5 μm) using a reverse phase gradient elution with aqueous. Trimethoprim, sulfamethoxazole, and cephalexin used Milli-Q deionized water with 0.1% formic acid for the aqueous mobile phase and 0.1% formic acid in ACN for the organic mobile phase. Doxycyline used Milli-Q deionized water with 0.1% formic acid and 0.1% heptaflurobutyric acid for the aqueous mobile phase and 0.1% formic acid and 0.1% heptafluorobutyric in ACN for the organic mobile phase. Multiple-reaction monitoring (MRM) of precursor/fragment transitions in electrospray positive-ionization mode was used to quantify the analytes. MRM transitions of 291.00/230.20, 254.10/156.00, 347.988/158.100, 455.2/165.2, 445.20/428.20, and 449.00/432.00 were used for trimethoprim, sulfamethoxazole, cephalexin, verapamil, doxycyline, and doxycyline-d3 respectively. Sample analysis was accepted if the concentrations of the quality control samples were within 20% of the nominal concentration. Data processing was performed using Analyst software (version 1.6.2; Applied Biosystems Sciex). A more detailed method description is available in the previously published protocol(70).
In vitro culturing of skin bacteria for antibiotics-resistance analysis.
A schematic diagram for culture strategy is shown in Fig. 2A. First, 25μl of skin swab-contained fastidious broth were plated on tryptic soy agar (TSA) and reinforced clostridial agar (RCA), for aerobic and anaerobic culture, respectively, and incubated at 34°C for >72 hours (up to 120 hours to obtain minor and slow-growing microbes). Antibiotic-resistant microbes were cultured from antibiotic-containing (doxycycline 2μg/ml or trimethoprim 100μg/ml) plates and grown in either of the condition described above in the presence of antibiotics. Of note, whole-genome shotgun sequencing of the culture plate from the baseline of Subject 5 (Fig. S13, A and B) showed that multiple bacterial species including Staphylococcus, Cutibacterium and Corynebacterium spp. were cultivatable, demonstrating that subsequent growth of only Staphylococcus spp. as doxycycline-resistant was due to the development of resistance as opposed to potential bias of culturing methods in species recovery. Minimum inhibitory concentrations of selected isolates MIC test strip (doxycycline and trimethoprim, Liofilchem).
Whole-genome shotgun sequencing from cultured bacteria.
All cultured microbes were collected in toto from culture plates (see above) with 1ml PBS and a cell spreader (plate swipe). Bacterial cells were pelleted with centrifugation at 5,000rpm, 10min. DNA was extracted with Maxwell tissue DNA kit following the manufacturer’s instructions. DNA libraries were prepared using Nextera XT Library Kit and sequenced on Illumina MiSeq (2 × 250 bp). Raw sequence data were demultiplexed into sample-specific fastq files using bcl2fastq conversion software from Illumina. Adaptor-trimmed, high-quality reads were then used to calculate relative abundances of microbes, using MetaPhlAn2(71).
Whole-genome sequencing of individual isolates.
Individual isolates were picked from culture plates (see above) and streaked on a new plate, to ensure single clone. After 16 hours, a single colony from each streaked plate was picked, and DNA was extracted with Maxwell tissue DNA kit following manufacturer’s instructions. DNA libraries were prepared using Nextera XT Library Kit and sequenced on Illumina MiSeq (2 × 250 bp). Raw sequence data were demultiplexed into sample-specific fastq files using bcl2fastq conversion software from Illumina. Multilocus sequence typing (MLST) was performed using SRST2(72) with MLST database from (https://pubmlst.org/sepidermidis/, https://pubmlst.org/shominis/).
For phylogenetic analysis of S. epidermidis, reads from each isolate were mapped, and variants called using Snippy core (https://github.com/tseemann/snippy), with S. epidermidis ATCC12228 (accession no: GCA_000007645.1) as a reference. Predicted recombination was identified using Gubbins(73). Core genome SNPs, following removal of recombination, were then used to construct a phylogeny with FastTree(74). Resistance gene content in isolate sequencing data was detected by mapping to a reference database of known AMR determinants, as derived from previous reports(75, 76), using SRST2(72).
Calculating the relative abundances of STs from metagenomic data.
Assembled genomes of isolates (using SPADES(65)) for each subject were clustered with dRep (77) with 99.99% average nucleotide identity to cluster sequences and then select a representative ‘reference’. Then, metagenomic data were then aligned to the reference of each individual and abundance of each strain cluster was inferred using BIB(39).
Abundance calculation of tetK and tetL genes from metagenomic data.
Human-filtered metagenomic reads were aligned to tetK (WP_053028219.1), tetL (WP_014638220.1), dfrC (WP_002473616.1 or WP_000175735.1), and dfrG (AQR07665) genes using usearch(68). The number of reads that were confidently mapped (>90% of query coverage and identity, and >200 of bit score) to the genes were then used to calculate fragments per kilobase per million reads (FPKM);
(Xi: number of reads that mapped to genei, total reads: number of non-human, and quality filtered total reads, li: length of genei (tetK: 1,206bp, tetL: 1,287bp, dfrC: 477bp and 486bp, dfrG: 498bp)).
Statistical analysis.
All statistical analyses were performed using R software. For all boxplots, center lines represent the median, lower and upper box limits represent the first and third quartiles, respectively (interquartile range), whiskers represent the maximal values up to 1.5 times interquartile range, and all values beyond this range are defined as outliers. The nonparametric Wilcoxon rank-sum test with Bonferroni multiple comparison correction was used to determine statistically significant (P<0.05) differences unless otherwise indicated. In addition, when samples from the same individual were compared (for example, comparing D0 and D56 of Doxy100 group), the ‘paired=T’ parameter was used. Similarity between microbial communities was assessed using the Bray-Curtis index or the Yue–Clayton theta index(30).
Supplementary Material
Acknowledgments:
The authors wish to acknowledge the contribution of all study participants. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health. This study utilized the computational resources of the NIH HPC Biowulf Cluster (http://hpc.nih.gov). We thank members of the NCI CCR Laboratory of Integrative Cancer Immunology, Microbiome and Genetics Core for assistance with whole-genome sequencing, Loreto Abusleme and Teresa Wild for assistance with oral samples, members in Department of Laboratory Medicine (DLM) of NIH for performing antibiotic susceptibility profiling of isolates. We also thank Valentina Pillai, Amynah Pradhan, Bryan Higgins, Sheila Phang, Sharon Osgood, Monica Taylor, Brian Wilson, Iryna Chub, Weng-Ian Ng, and other members of the Kong and Segre labs for their underlying efforts.
Funding:
This work is supported by the NIH Intramural Research Programs of the National Institute of Arthritis and Musculoskeletal and Skin Diseases, National Cancer Institute, National Institute of Dental and Craniofacial Research, and National Human Genome Research Institute. Additional sequencing funding support was provided by the National Institute of Allergy and Infectious Diseases CARB-X (Combating Antibiotic Resistant Bacteria) Program (JAS), and shared instrumentation grant S10-OD023524 (VD).
Footnotes
Competing interests: The authors declare that they have no competing interests.
Data and materials availability:
All data associated with this study are present in the paper or supplementary materials. All raw sequencing data have been uploaded to SRA with the BioProject accession number PRJNA604820. The custom code is available at DOI: 10.5281/zenodo.5733506.
References and Notes:
- 1.Centers for Disease Control and Prevention. Outpatient antibiotic prescriptions — United States. (2017).
- 2.Morgan DJ, Dhruva SS, Coon ER, Wright SM, Korenstein D, 2019 Update on Medical Overuse: A Review. JAMA Intern Med, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Pouwels KB, Dolk FCK, Smith DRM, Robotham JV, Smieszek T, Actual versus ‘ideal’ antibiotic prescribing for common conditions in English primary care. J Antimicrob Chemother 73, 19–26 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Oh J, Byrd AL, Park M, N. C. S. Program, Kong HH, Segre JA, Temporal Stability of the Human Skin Microbiome. Cell 165, 854–866 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA, Topographical and temporal diversity of the human skin microbiome. Science 324, 1190–1192 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Brown JM, Poston SM, Resistance of propionibacteria to antibiotics used in the treatment of acne. J Med Microbiol 16, 271–280 (1983). [DOI] [PubMed] [Google Scholar]
- 7.Kurokawa I, Nishijima S, Asada Y, The antibiotic susceptibility of Propionibacterium acnes: a 15-year bacteriological study and retrospective evaluation. J Dermatol 15, 149–154 (1988). [DOI] [PubMed] [Google Scholar]
- 8.Nakase K, Yoshida A, Saita H, Hayashi N, Nishijima S, Nakaminami H, Noguchi N, Relationship between quinolone use and resistance of Staphylococcus epidermidis in patients with acne vulgaris. The Journal of dermatology 46, 782–786 (2019). [DOI] [PubMed] [Google Scholar]
- 9.SanMiguel AJ, Meisel JS, Horwinski J, Zheng Q, Grice EA, Topical Antimicrobial Treatments Can Elicit Shifts to Resident Skin Bacterial Communities and Reduce Colonization by Staphylococcus aureus Competitors. Antimicrob Agents Chemother 61, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chien AL, Tsai J, Leung S, Mongodin EF, Nelson AM, Kang S, Garza LA, Association of Systemic Antibiotic Treatment of Acne With Skin Microbiota Characteristics. JAMA Dermatol 155, 425–434 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Naik S, Bouladoux N, Wilhelm C, Molloy MJ, Salcedo R, Kastenmuller W, Deming C, Quinones M, Koo L, Conlan S, Spencer S, Hall JA, Dzutsev A, Kong H, Campbell DJ, Trinchieri G, Segre JA, Belkaid Y, Compartmentalized control of skin immunity by resident commensals. Science 337, 1115–1119 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kobayashi T, Glatz M, Horiuchi K, Kawasaki H, Akiyama H, Kaplan DH, Kong HH, Amagai M, Nagao K, Dysbiosis and Staphyloccus aureus Colonization Drives Inflammation in Atopic Dermatitis. Immunity 42, 756–766 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zouboulis CC, Piquero-Martin J, Update and future of systemic acne treatment. Dermatology 206, 37–53 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Bienenfeld A, Nagler AR, Orlow SJ, Oral Antibacterial Therapy for Acne Vulgaris: An Evidence-Based Review. Am J Clin Dermatol 18, 469–490 (2017). [DOI] [PubMed] [Google Scholar]
- 15.Odsbu I, Selmer R, Stalsby Lundborg C, Blix HS, Increased prescribing of systemic tetracyclines and isotretinoin for treatment of acne. J Antimicrob Chemother 72, 1510–1515 (2017). [DOI] [PubMed] [Google Scholar]
- 16.Walker C, Preshaw PM, Novak J, Hefti AF, Bradshaw M, Powala C, Long-term treatment with sub-antimicrobial dose doxycycline has no antibacterial effect on intestinal flora. J Clin Periodontol 32, 1163–1169 (2005). [DOI] [PubMed] [Google Scholar]
- 17.Fitz-Gibbon S, Tomida S, Chiu BH, Nguyen L, Du C, Liu M, Elashoff D, Erfe MC, Loncaric A, Kim J, Modlin RL, Miller JF, Sodergren E, Craft N, Weinstock GM, Li H, Propionibacterium acnes strain populations in the human skin microbiome associated with acne. J Invest Dermatol 133, 2152–2160 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Kong HH, Oh J, Deming C, Conlan S, Grice EA, Beatson MA, Nomicos E, Polley EC, Komarow HD, Murray PR, Turner ML, Segre JA, Temporal shifts in the skin microbiome associated with disease flares and treatment in children with atopic dermatitis. Genome Res 22, 850–859 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Tirosh O, Conlan S, Deming C, Lee-Lin S-Q, Huang X, Barnabas BB, Bouffard GG, Brooks SY, Marfani H, Dekhtyar L, Guan X, Han J, Ho S.-l., Legaspi R, Maduro QL, Masiello CA, McDowell JC, Montemayor C, Mullikin JC, Park M, Riebow NL, Schandler K, Scharer C, Schmidt B, Sison C, Stantripop S, Thomas JW, Thomas PJ, Vemulapalli M, Young AC, Su HC, Freeman AF, Segre JA, Kong HH, N. C. S. Program, Expanded skin virome in DOCK8-deficient patients. Nature Medicine, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Chng KR, Tay ASL, Li C, Ng AHQ, Wang J, Suri BK, Matta SA, McGovern N, Janela B, Wong XFCC, Sio YY, Au BV, Wilm A, Florez De Sessions P, Lim TC, Tang MBY, Ginhoux F, Connolly JE, Lane EB, Chew FT, Common JEA, Nagarajan N, Whole metagenome profiling reveals skin microbiome-dependent susceptibility to atopic dermatitis flare. Nat Microbiol, (2016). [DOI] [PubMed] [Google Scholar]
- 21.Kong HH, Oh J, Conlan S, Deming C, Grice EA, Turner ML, Segre JA, Temporal shifts in the skin microbiome associated with atopic dermatitis disease flares and treatment. Journal of Investigative Dermatology 132, S113–S113 (2012). [Google Scholar]
- 22.Gliniewicz K, Schneider GM, Ridenhour BJ, Williams CJ, Song Y, Farage MA, Miller K, Forney LJ, Comparison of the Vaginal Microbiomes of Premenopausal and Postmenopausal Women. Front Microbiol 10, 193 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Vieira AT, Castelo PM, Ribeiro DA, Ferreira CM, Influence of Oral and Gut Microbiota in the Health of Menopausal Women. Front Microbiol 8, 1884 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Forslund K, Hildebrand F, Nielsen T, Falony G, Le Chatelier E, Sunagawa S, Prifti E, Vieira-Silva S, Gudmundsdottir V, Krogh Pedersen H, Arumugam M, Kristiansen K, Voigt AY, Vestergaard H, Hercog R, Igor Costea P, Kultima JR, Li J, Jorgensen T, Levenez F, Dore J, H. I. T. c. Meta, Nielsen HB, Brunak S, Raes J, Hansen T, Wang J, Ehrlich SD, Bork P, Pedersen O, Disentangling type 2 diabetes and metformin treatment signatures in the human gut microbiota. Nature 528, 262–266 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dethlefsen L, Huse S, Sogin ML, Relman DA, The pervasive effects of an antibiotic on the human gut microbiota, as revealed by deep 16S rRNA sequencing. PLoS Biol 6, e280 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Jakobsson HE, Jernberg C, Andersson AF, Sjolund-Karlsson M, Jansson JK, Engstrand L, Short-term antibiotic treatment has differing long-term impacts on the human throat and gut microbiome. PLoS One 5, e9836 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Grice EA, Kong HH, Conlan S, Deming CB, Davis J, Young AC, N. C. S. Program, Bouffard GG, Blakesley RW, Murray PR, Green ED, Turner ML, Segre JA, Topographical and temporal diversity of the human skin microbiome. Science 324, 1190–1192 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Oh J, Byrd AL, Deming C, Conlan S, N. C. S. Program, Kong HH, Segre JA, Biogeography and individuality shape function in the human skin metagenome. Nature 514, 59–64 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Byrd AL, Belkaid Y, Segre JA, The human skin microbiome. Nat Rev Microbiol 16, 143–155 (2018). [DOI] [PubMed] [Google Scholar]
- 30.Yue JC, Clayton MK, A similarity measure based on species proportions. Communications in Statistics-Theory and Methods 34, 2123–2131 (2005). [Google Scholar]
- 31.C. Human Microbiome Project, Structure, function and diversity of the healthy human microbiome. Nature 486, 207–214 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Costello EK, Lauber CL, Hamady M, Fierer N, Gordon JI, Knight R, Bacterial community variation in human body habitats across space and time. Science 326, 1694–1697 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dethlefsen L, Relman DA, Incomplete recovery and individualized responses of the human distal gut microbiota to repeated antibiotic perturbation. Proc Natl Acad Sci U S A 108 Suppl 1, 4554–4561 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Abeles SR, Jones MB, Santiago-Rodriguez TM, Ly M, Klitgord N, Yooseph S, Nelson KE, Pride DT, Microbial diversity in individuals and their household contacts following typical antibiotic courses. Microbiome 4, 39 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Panda S, El khader I, Casellas F, Lopez Vivancos J, Garcia Cors M, Santiago A, Cuenca S, Guarner F, Manichanh C, Short-term effect of antibiotics on human gut microbiota. PLoS One 9, e95476 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Willmann M, Vehreschild M, Biehl LM, Vogel W, Dorfel D, Hamprecht A, Seifert H, Autenrieth IB, Peter S, Distinct impact of antibiotics on the gut microbiome and resistome: a longitudinal multicenter cohort study. BMC Biol 17, 76 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Vrieze A, Out C, Fuentes S, Jonker L, Reuling I, Kootte RS, van Nood E, Holleman F, Knaapen M, Romijn JA, Soeters MR, Blaak EE, Dallinga-Thie GM, Reijnders D, Ackermans MT, Serlie MJ, Knop FK, Holst JJ, van der Ley C, Kema IP, Zoetendal EG, de Vos WM, Hoekstra JB, Stroes ES, Groen AK, Nieuwdorp M, Impact of oral vancomycin on gut microbiota, bile acid metabolism, and insulin sensitivity. J Hepatol 60, 824–831 (2014). [DOI] [PubMed] [Google Scholar]
- 38.Grossman TH, Tetracycline Antibiotics and Resistance. Cold Spring Harb Perspect Med 6, a025387 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Sankar A, Malone B, Bayliss SC, Pascoe B, Meric G, Hitchings MD, Sheppard SK, Feil EJ, Corander J, Honkela A, Bayesian identification of bacterial strains from sequencing data. Microb Genom 2, e000075 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Langdon A, Crook N, Dantas G, The effects of antibiotics on the microbiome throughout development and alternative approaches for therapeutic modulation. Genome Med 8, 39 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Laureti L, Matic I, Gutierrez A, Bacterial Responses and Genome Instability Induced by Subinhibitory Concentrations of Antibiotics. Antibiotics (Basel) 2, 100–114 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Baharoglu Z, Mazel D, SOS, the formidable strategy of bacteria against aggressions. FEMS Microbiol Rev 38, 1126–1145 (2014). [DOI] [PubMed] [Google Scholar]
- 43.Palleja A, Mikkelsen KH, Forslund SK, Kashani A, Allin KH, Nielsen T, Hansen TH, Liang S, Feng Q, Zhang C, Pyl PT, Coelho LP, Yang H, Wang J, Typas A, Nielsen MF, Nielsen HB, Bork P, Wang J, Vilsboll T, Hansen T, Knop FK, Arumugam M, Pedersen O, Recovery of gut microbiota of healthy adults following antibiotic exposure. Nat Microbiol 3, 1255–1265 (2018). [DOI] [PubMed] [Google Scholar]
- 44.Jernberg C, Lofmark S, Edlund C, Jansson JK, Long-term ecological impacts of antibiotic administration on the human intestinal microbiota. ISME J 1, 56–66 (2007). [DOI] [PubMed] [Google Scholar]
- 45.Willing BP, Russell SL, Finlay BB, Shifting the balance: antibiotic effects on host-microbiota mutualism. Nat Rev Microbiol 9, 233–243 (2011). [DOI] [PubMed] [Google Scholar]
- 46.Zhang M, Jiang Z, Li D, Jiang D, Wu Y, Ren H, Peng H, Lai Y, Oral antibiotic treatment induces skin microbiota dysbiosis and influences wound healing. Microb Ecol 69, 415–421 (2015). [DOI] [PubMed] [Google Scholar]
- 47.Marples RR, Kligman AM, Ecological effects of oral antibiotics on the microflora of human skin. Arch Dermatol 103, 148–153 (1971). [PubMed] [Google Scholar]
- 48.Eady EA, Cove JH, Holland KT, Cunliffe WJ, Superior antibacterial action and reduced incidence of bacterial resistance in minocycline compared to tetracycline-treated acne patients. Br J Dermatol 122, 233–244 (1990). [DOI] [PubMed] [Google Scholar]
- 49.Williamson DA, Monecke S, Heffernan H, Ritchie SR, Roberts SA, Upton A, Thomas MG, Fraser JD, High usage of topical fusidic acid and rapid clonal expansion of fusidic acid-resistant Staphylococcus aureus: a cautionary tale. Clin Infect Dis 59, 1451–1454 (2014). [DOI] [PubMed] [Google Scholar]
- 50.Slager J, Kjos M, Attaiech L, Veening JW, Antibiotic-induced replication stress triggers bacterial competence by increasing gene dosage near the origin. Cell 157, 395–406 (2014). [DOI] [PubMed] [Google Scholar]
- 51.Lax S, Smith DP, Hampton-Marcell J, Owens SM, Handley KM, Scott NM, Gibbons SM, Larsen P, Shogan BD, Weiss S, Metcalf JL, Ursell LK, Vazquez-Baeza Y, Van Treuren W, Hasan NA, Gibson MK, Colwell R, Dantas G, Knight R, Gilbert JA, Longitudinal analysis of microbial interaction between humans and the indoor environment. Science 345, 1048–1052 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Hsu T, Joice R, Vallarino J, Abu-Ali G, Hartmann EM, Shafquat A, DuLong C, Baranowski C, Gevers D, Green JL, Morgan XC, Spengler JD, Huttenhower C, Urban Transit System Microbial Communities Differ by Surface Type and Interaction with Humans and the Environment. mSystems 1, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Weiss H, Hertzberg VS, Dupont C, Espinoza JL, Levy S, Nelson K, Norris S, T. FlyHealthy Research, The Airplane Cabin Microbiome. Microb Ecol 77, 87–95 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pal A, Matzneller P, Gautam A, Osterreicher Z, Wulkersdorfer B, Reiter B, Stimpfl T, Zeitlinger M, Target site pharmacokinetics of doxycycline for rosacea in healthy volunteers is independent of the food effect. Br J Clin Pharmacol 84, 2625–2633 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Cunha BA, Domenico P, Cunha CB, Pharmacodynamics of doxycycline. Clin Microbiol Infect 6, 270–273 (2000). [DOI] [PubMed] [Google Scholar]
- 56.Keijser BJF, Agamennone V, van den Broek TJ, Caspers M, van de Braak A, Bomers R, Havekes M, Schoen E, van Baak M, Mioch D, Bomers L, Montijn RC, Dose-dependent impact of oxytetracycline on the veal calf microbiome and resistome. BMC Genomics 20, 65 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Alexander TW, Yanke LJ, Topp E, Olson ME, Read RR, Morck DW, McAllister TA, Effect of subtherapeutic administration of antibiotics on the prevalence of antibiotic-resistant Escherichia coli bacteria in feedlot cattle. Appl Environ Microbiol 74, 4405–4416 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Hu Y, Yang X, Qin J, Lu N, Cheng G, Wu N, Pan Y, Li J, Zhu L, Wang X, Meng Z, Zhao F, Liu D, Ma J, Qin N, Xiang C, Xiao Y, Li L, Yang H, Wang J, Yang R, Gao GF, Wang J, Zhu B, Metagenome-wide analysis of antibiotic resistance genes in a large cohort of human gut microbiota. Nat Commun 4, 2151 (2013). [DOI] [PubMed] [Google Scholar]
- 59.van Schaik W, The human gut resistome. Philos Trans R Soc Lond B Biol Sci 370, 20140087 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Lindsay JA, Staphylococcus aureus genomics and the impact of horizontal gene transfer. Int J Med Microbiol 304, 103–109 (2014). [DOI] [PubMed] [Google Scholar]
- 61.Goerke C, Koller J, Wolz C, Ciprofloxacin and trimethoprim cause phage induction and virulence modulation in Staphylococcus aureus. Antimicrob Agents Chemother 50, 171–177 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Miller YW, Eady EA, Lacey RW, Cove JH, Joanes DN, Cunliffe WJ, Sequential antibiotic therapy for acne promotes the carriage of resistant staphylococci on the skin of contacts. J Antimicrob Chemother 38, 829–837 (1996). [DOI] [PubMed] [Google Scholar]
- 63.Chng KR, Li C, Bertrand D, Ng AHQ, Kwah JS, Low HM, Tong C, Natrajan M, Zhang MH, Xu L, Ko KKK, Ho EXP, Av-Shalom TV, Teo JWP, Khor CC, Meta SUBC, Chen SL, Mason CE, Ng OT, Marimuthu K, Ang B, Nagarajan N, Cartography of opportunistic pathogens and antibiotic resistance genes in a tertiary hospital environment. Nat Med, (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Langmead B, Salzberg SL, Fast gapped-read alignment with Bowtie 2. Nat Methods 9, 357–359 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bankevich A, Nurk S, Antipov D, Gurevich AA, Dvorkin M, Kulikov AS, Lesin VM, Nikolenko SI, Pham S, Prjibelski AD, Pyshkin AV, Sirotkin AV, Vyahhi N, Tesler G, Alekseyev MA, Pevzner PA, SPAdes: a new genome assembly algorithm and its applications to single-cell sequencing. J Comput Biol 19, 455–477 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Hyatt D, Chen GL, Locascio PF, Land ML, Larimer FW, Hauser LJ, Prodigal: prokaryotic gene recognition and translation initiation site identification. BMC Bioinformatics 11, 119 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Li W, Godzik A, Cd-hit: a fast program for clustering and comparing large sets of protein or nucleotide sequences. Bioinformatics 22, 1658–1659 (2006). [DOI] [PubMed] [Google Scholar]
- 68.Edgar RC, Search and clustering orders of magnitude faster than BLAST. Bioinformatics 26, 2460–2461 (2010). [DOI] [PubMed] [Google Scholar]
- 69.Ritchie ME, Phipson B, Wu D, Hu Y, Law CW, Shi W, Smyth GK, limma powers differential expression analyses for RNA-sequencing and microarray studies. Nucleic Acids Res 43, e47 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Zimmerman M, Blanc L, Chen PY, Dartois V, Prideaux B, Spatial Quantification of Drugs in Pulmonary Tuberculosis Lesions by Laser Capture Microdissection Liquid Chromatography Mass Spectrometry (LCM-LC/MS). J Vis Exp, (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Truong DT, Franzosa EA, Tickle TL, Scholz M, Weingart G, Pasolli E, Tett A, Huttenhower C, Segata N, MetaPhlAn2 for enhanced metagenomic taxonomic profiling. Nat Methods 12, 902–903 (2015). [DOI] [PubMed] [Google Scholar]
- 72.Inouye M, Dashnow H, Raven LA, Schultz MB, Pope BJ, Tomita T, Zobel J, Holt KE, SRST2: Rapid genomic surveillance for public health and hospital microbiology labs. Genome Med 6, 90 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Croucher NJ, Page AJ, Connor TR, Delaney AJ, Keane JA, Bentley SD, Parkhill J, Harris SR, Rapid phylogenetic analysis of large samples of recombinant bacterial whole genome sequences using Gubbins. Nucleic Acids Res 43, e15 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Price MN, Dehal PS, Arkin AP, FastTree: computing large minimum evolution trees with profiles instead of a distance matrix. Mol Biol Evol 26, 1641–1650 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Aanensen DM, Feil EJ, Holden MT, Dordel J, Yeats CA, Fedosejev A, Goater R, Castillo-Ramirez S, Corander J, Colijn C, Chlebowicz MA, Schouls L, Heck M, Pluister G, Ruimy R, Kahlmeter G, Ahman J, Matuschek E, Friedrich AW, Parkhill J, Bentley SD, Spratt BG, Grundmann H, S. R. L. W. G. European, Whole-Genome Sequencing for Routine Pathogen Surveillance in Public Health: a Population Snapshot of Invasive Staphylococcus aureus in Europe. mBio 7, (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Lee JYH, Monk IR, Goncalves da Silva A, Seemann T, Chua KYL, Kearns A, Hill R, Woodford N, Bartels MD, Strommenger B, Laurent F, Dodemont M, Deplano A, Patel R, Larsen AR, Korman TM, Stinear TP, Howden BP, Global spread of three multidrug-resistant lineages of Staphylococcus epidermidis. Nat Microbiol 3, 1175–1185 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are present in the paper or supplementary materials. All raw sequencing data have been uploaded to SRA with the BioProject accession number PRJNA604820. The custom code is available at DOI: 10.5281/zenodo.5733506.