

SUMMARY
Calcium-dependent inactivation (CDI) is a fundamental autoregulatory mechanism in CaV1 and CaV2 voltage-gated calcium channels. Although CDI initiates with the cytoplasmic calcium sensor, how this event causes CDI has been elusive. Here, we show that a conserved selectivity filter (SF) domain II (DII) aspartate is essential for CDI. Mutation of this residue essentially eliminates CDI and leaves key channel biophysical characteristics untouched. DII mutants regain CDI by placing an aspartate at the analogous SF site in DIII or DIV, but not DI, indicating that CaV SF asymmetry is key to CDI. Together, our data establish that the CaV SF is the CDI endpoint. Discovery of this SF CDI gate recasts the CaV inactivation paradigm, placing it squarely in the framework of voltage-gated ion channel (VGIC) superfamily members in which SF-based gating is important. This commonality suggests that SF inactivation is an ancient process arising from the shared VGIC pore architecture.
Graphical Abstract
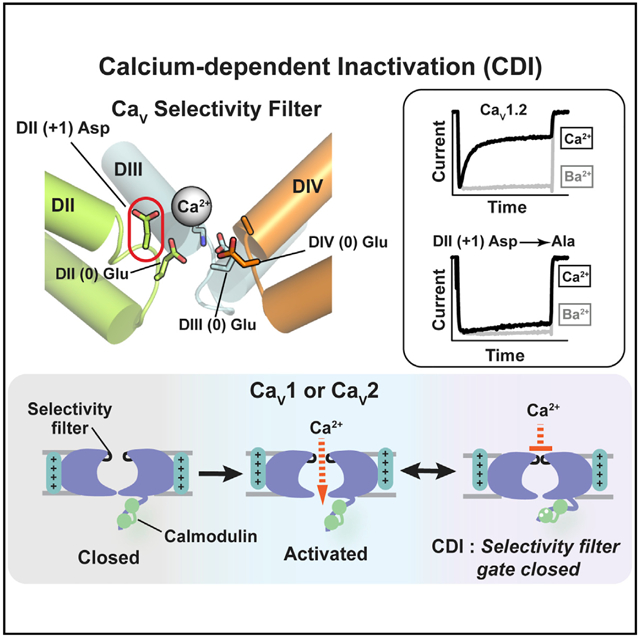
In Brief
Calcium-dependent inactivation (CDI) is essential for voltage-gated calcium channel (CaV) autoregulation. Abderemane-Ali et al. demonstrate that the CaV selectivity filter (SF) forms the CDI gate, suggesting an SF-based inactivation paradigm shared with other voltage-gated ion channel (VGIC) superfamily members.
INTRODUCTION
Voltage-gated calcium channels (CaVs) are multisubunit, macro-molecular complexes that control cellular calcium entry in response to membrane potential changes in the brain, nervous system, and heart (Catterall, 2011; Zamponi et al., 2015). Due to the central role of calcium in cellular signaling (Clapham, 2007) and the importance of CaVs as sources of Ca2+ influx that impact synaptic transmission, hormone release, vascular tone, muscle contraction, and gene expression (Nanou and Catterall, 2018; Simms and Zamponi, 2014; Zamponi et al., 2015), a multifaceted set of activity-dependent feedback regulation mechanisms shape CaV function. Chief among these is calcium-dependent inactivation (CDI), a process by which Ca2+ influx through CaV1 and CaV2 channels causes a cessation of ion conduction (Ben-Johny and Yue, 2014; Christel and Lee, 2011; Dunlap, 2007; Simms and Zamponi, 2014). Perturbations in CDI are involved in autism (Limpitikul et al., 2016), blindness (Singh et al., 2006), and cardiac arrhythmias (Alseikhan et al., 2002; Dick et al., 2016; Limpitikul et al., 2014, 2017; Mahajan et al., 2008; Morotti et al., 2012; Splawski et al., 2004, 2005), demonstrating that CDI is an important factor in diseases linked to CaV dysfunction.
The CaV pore-forming CaVα1 subunit is a 24 transmembrane segment protein comprising four repeats (DI–DIV) that are each made from six transmembrane helices that form the voltage sensor domain (SI–S4) and the pore domain (S5–S6) (Catterall et al., 2017; Wu et al., 2015, 2016). CaVα1 shares the overall architecture found throughout the voltage-gated ion channel (VGIC) superfamily but requires auxiliary subunits for proper function (Campiglio and Flucher, 2015; Dolphin, 2016). Complexes with two intracellular components that bind to the cytosolic I-II loop and C-terminal tail, the CaVβ subunit (Chen et al., 2004; Opatowsky et al., 2004; Van Petegem et al., 2004) and calmodulin (CaM) (Fallon et al., 2009; Kim et al., 2008, 2010; Mori et al., 2008; Van Petegem et al., 2005), respectively, are particularly important for activity-dependent feedback modulation by the two principal CaV inactivation mechanisms, voltage-dependent inactivation (VDI) (Cens et al., 2006; Stotz et al., 2004) and CDI (Ben-Johny and Yue, 2014; Cens et al., 2006; Christel and Lee, 2011; Halling et al., 2006).
The molecular origins of CDI have been extensively investigated, especially from the vantage point of the intracellular, CaM-based sensor apparatus and the role of the CaM-binding IQ domain (Ben-Johny and Yue, 2014; Fallon et al., 2005; Minor and Findeisen, 2010; Kim et al., 2008, 2010; Mori et al., 2008; Van Petegem et al., 2005). Although the involvement of this CaM-based sensor in CDI is clear (Ben-Johny and Yue, 2014; Minor and Findeisen, 2010; Kim et al., 2008, 2010; Lee et al., 1999; Mori et al., 2008; Peterson et al., 1999; Van Petegem et al., 2005; Zühlke et al., 1999), the fact that the N-terminal cytoplasmic domain (Ben Johny et al., 2013; Dick et al., 2008; Ivanina et al., 2000; Tadross et al., 2008) and the CaVβ/I-II loop complex (Almagor et al., 2012; Findeisen and Minor, 2009) also impact CDI has left open the question of how conformational changes in these cytoplasmic parts of the channel complex terminate Ca2+ influx (Babich et al., 2007; Barrett and Tsien, 2008; Cens et al., 2006; Findeisen and Minor, 2009; Kim et al., 2004; Tadross et al., 2010). The prevailing model suggests that the activation gate formed by the S6 helices of the channel pore acts as the CDI endpoint via an allosteric mechanism (Dick et al., 2016; Limpitikul et al., 2016; Tadross et al., 2010). Other studies have raised the idea of a close link between ion selectivity and CDI, based on the fact that CDI and selectivity properties are simultaneously affected by SF mutations or changes in extracellular Ca2+ concentrations (Babich et al., 2005; Zong et al., 1994) and on the observation that SF Gd3+ block appears mutually exclusive with inactivation (Babich et al., 2007). Further, the extent to which CDI and VDI share common pathways (Cens et al., 1999; Findeisen and Minor, 2009; Kim et al., 2004) or act by different mechanisms (Barrett and Tsien, 2008; Tadross et al., 2010) has been unclear.
We discovered that mutations in the conserved aspartate at the CaV1.2 domain II (DII) SF (+1) position, a site that is important for CaV SF Ca2+ binding and that forms an outer ion binding site in bacterial voltage-gated sodium channels (BacNaVs) and mammalian CaVs (Shaya et al., 2014), affect CDI. Mutation of this conserved aspartate can effectively eliminate CDI in CaV1.2, while sparing core biophysical properties including voltage-dependent activation, VDI, and ion selectivity. Analogous SF DII (+1) mutations in two other CaVs, a CaV1.3 variant bearing the most robust CDI among all CaVs (Huang et al., 2013; Singh et al., 2008; Xu and Lipscombe, 2001) and CaV2.1, a channel in which CDI relies on a different CaM lobe than in CaV1 s (DeMaria et al., 2001; Minor and Findeisen, 2010; Lee et al., 2003; Liang et al., 2003), demonstrate that the role of the DII (+1) aspartate in controlling CDI is both conserved and independent of the details of how the intracellular Ca2+ sensor acts. We also show that, in channels lacking CDI because the DII (+1) aspartate is mutated, one can restore CDI by placing an aspartate at the analogous SF (+1) sites of DIII or DIV but not DI. This observation is concordant with asymmetric functional roles for the four glutamates at the SF (0) position (Ellinor et al., 1995; Parent and Gopalakrishnan, 1995; Yang et al., 1993) and indicates that the change in the SF that drives CDI is asymmetric. Together, our data establish that the CaV SF is the endpoint gate for CDI and change the paradigm for understanding CaV inactivation mechanisms by placing it squarely within the framework of the growing list of VGIC superfamily members in which SF-based gating is central to function (Autzen et al., 2018; Bagriantsev et al., 2011; Cao et al., 2013; Cohen et al., 2008; Cuello et al., 2010, 2017; Liu et al., 1996; Lolicato et al., 2017; López-Barneo et al., 1993; Ogielska and Aldrich, 1999; Pavlov et al., 2005; Peters et al., 2013; Piechotta et al., 2011; Schewe et al., 2016; Steinberg et al., 2017). These findings support the idea that SF inactivation is an ancient process arising from the conserved VGIC pore architecture (Catterall et al., 2017) that not only affects slow inactivation, but also controls CDI in CaVs.
RESULTS
Single CaV1.2 Selectivity Filter Mutations Drastically Reduce CDI in Xenopus Oocytes
The SF sequences of each of the four CaV domains are highly similar to each other and to those found in BacNaVs (Payandeh and Minor, 2015; Ren et al., 2001; Shaya et al., 2014; Wu et al., 2015; Yue et al., 2002), which are thought to be a common ancestor of both voltage-gated sodium channels (NaVs) and CaVs (Koishi et al., 2004; Payandeh and Minor, 2015; Ren et al., 2001). All share the central glutamate residue (denoted position (0); Payandeh and Minor, 2015; Shaya et al., 2014) (Figure 1A) and a common architecture that supports the SF structure (Figure 1B) (Payandeh and Minor, 2015; Payandeh et al., 2011; Wu et al., 2015). The SF (0) position glutamates form a charged ring at the center of the CaV and BacNaV SFs (Payandeh and Minor, 2015; Payandeh et al., 2011; Wu et al., 2015, 2016) and play a key role in Ca2+ selectivity and ion permeation (Parent and Gopalakrishnan, 1995; Yang et al., 1993). In the case of CaVs, the four SF sequences are not identical. Further, even though the side-chain identity at the CaV SF position (0) is the same, each (0) position glutamate contributes differently to SF properties with the DIII (0) glutamate (CaV1.2 Glu 1115) being the most important contributor to ion selectivity (Ellinor et al., 1995; Kim et al., 1993; Mikala et al., 1993; Yang et al., 1993). A further asymmetry in the CaV SF occurs at DII position (+1).This site has a unique aspartate (CaV1.2 Asp 707) (Figure 1A) that is involved in the high-affinity binding of Ca2+ to the SF and whose neutralization by mutation has similar effects on Ca2+ binding as neutralization of the DIII position (0) glutamate (Shaya et al., 2014).
Figure 1. CaV1.2 Selectivity Filter Mutations in DII Affect CDI.
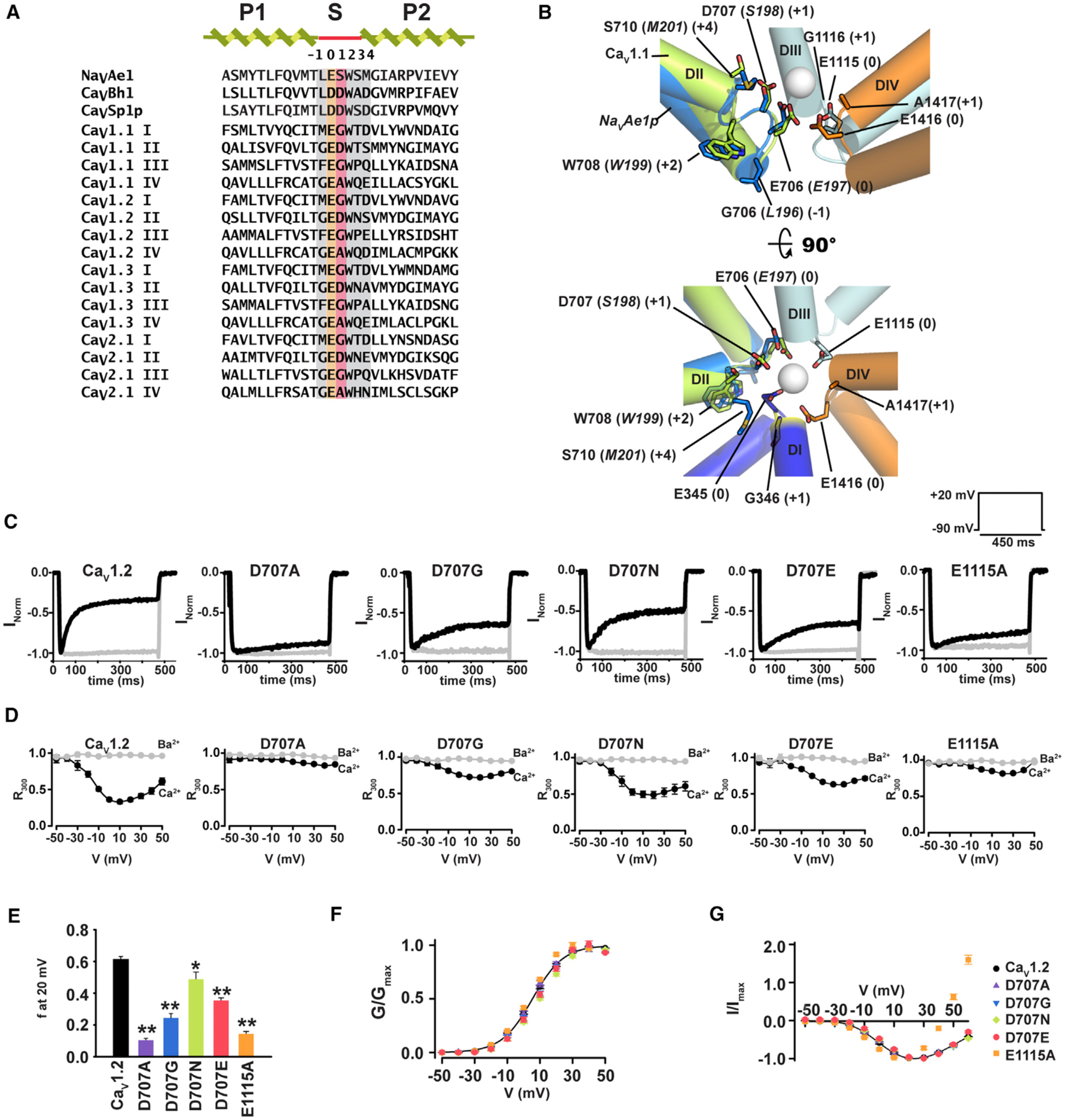
(A) Sequence alignment of pore helices and selectivity filters of NaVAe1 (Shaya et al., 2014), calcium-selective BacNaVs CaVBh1 (Yue et al., 2002), CaVSp1p (Shaya et al., 2011), and human CaV1 and CaV2 exemplars: CaV1.1 (UniProtKB: Q13698), CaV1.2 (UniProtKB: Q13936), CaV1.3 (UniProtKB: Q01668), and CaV2.1 (UniProtKB: O00555). SF sites (0) and (+1) are highlighted in orange and red, respectively. Other SF sites are highlighted in gray. Sequences are NaVAe1 185–211, CaVBh1 179–205, CaVSp1p 164–190, CaV1.1 DI 280–306, CaV1.1 DII 602–628, CaV1.1 DIII 1002–1028, CaV1.1 DIV 1311–1337, CaV1.2 DI 351–377, CaV1.2-DII 694–720; CaV1.2-DIII 1103–1129, CaV1.2-DIV 1404–1430, CaV1.3-DI: 343–369, CaV1.3-DII 704–730, CaV1.3 DIII 1100–1126, CaV1.3 DIV 1390–1416, CaV2.1-DI 306–332, CaV2.1 DII 656–682, CaV2.1 DIII 1448–1474, CaV2.1 DIV 1744–1770.
(B) Structural comparison of the NaVAe1 (PDB: 5HK7) (Arrigoni et al., 2016) SF (marine) with the CaV1.1 (PDB: 5GJV) (Wu et al., 2016) DII SF (green). Ca2+ from NaVAe1 (PDB: 4LTO) (Shaya et al., 2014) is shown as a sphere. DI (blue), DII (green), DIII (cyan), and DIV (orange) domains of CaV1.1 are shown. CaV1.1 and NaVAe1p residues are labeled in plain and italics, respectively. CaV numbering follows the CaV1.2 equivalent residues. SF positions are shown in parentheses. Side view (top) omits DI for clarity. DI (+1) position is highlighted in yellow.
(C) Exemplar normalized recordings at +20 mV in Ca2+ (black) or Ba2+ (gray) from Xenopus oocytes expressing CaV1.2 or indicated mutants.
(D) Fractional current remaining 300 ms post-depolarization (R300) as a function of the membrane potential for channels in (C).
(E) Average fraction CDI (f) at +20 mV, where f is the difference between Ca2+ and Ba2+ R300. *p < 0.015 and **p < 0.001 compared to CaV1.2. n values are in Table 1.
(F) Voltage-dependent activation curves for CaV1.2 (black circles), D707A (purple triangles), D707G (inverted blue triangles), D707N (green diamonds), D707E (red circles), and E1115A (orange squares).
(G) I–V relationships for the indicated channels. Symbols are the same as (F).
In the course of studying the functional properties of CaV1.2 SF mutants using two-electrode-voltage clamp of Xenopus oocytes (Figure S1A), we observed that replacement of the DII (+1) site, Asp 707, with alanine, glycine, asparagine, or glutamate decreased CDI. These effects manifested as varied degrees of reduction of the classic “U-shaped” response characteristic of CDI with D707A and D707N causing the greatest and mildest effects, respectively (Figures 1C and 1D).
To quantify changes in CDI, we measured the ratio between the current amplitude at the peak and 300 ms post-depolarization (R300) for both Ba2+ and Ca2+ as a function of voltage (Figure 1D) and compared the difference between Ba2+ and Ca2+ R300 values, f, for CaV1.2 and D707A/G/N/E mutants at the potential of maximal current, +20 mV (Peterson et al., 1999). This analysis revealed a rank order of D > N > E > G > A (f = 0.61 ±0.02, 0.49 ± 0.04, 0.35 ± 0.02, 0.24 ± 0.03, and 0.10 ± 0.01, respectively; Table 1), suggesting the importance of both the negative charge and the DII (+1) position side-chain geometry for CDI. The stronger CDI observed in D707N versus D707E indicates that other side-chain properties at the DII (+1) besides the negative charge are important for CDI. Experiments using a domain III position (0) mutant that reduces Ca2+ ion selectivity, E1115A (Ellinor et al., 1995; Mikala et al., 1993), also caused a large reduction in CDI (f = 0.14 ± 0.02) that was comparable to the Asp 707 mutant causing the most severe CDI reduction, D707A (Figures 1C–1E). None of the SF mutants that we tested affected voltage-dependent activation (Figure 1F). Consistent with its ability to reduce Ca2+ selectivity (Ellinor et al., 1995; Mikala et al., 1993), the E1115A mutant caused a shift in the reversal potential for Ca2+ relative to wild-type (Erev = 89.1 ± 4.0 mV and 49.8 ± 1.0 mV for CaV1.2 and E1115A, respectively) (Figure 1G). Such a change in Erev was notably absent from the Asp 707 mutants, all of which behaved similarly to wild-type (Erev = 92.8 ± 4.8, 94.8 ± 4.0, 87.8 ± 2.5, and 88.0 ± 8.1 mV for D707A, D707G, D707N, and D707E, respectively) (Figure 1G). E1115A reduced the ratio between Ba2+ and Ca2+ peak currents (IBa/ICa) to a value close to one, indicating that this mutation renders the SF unable to discriminate between Ba2+ and Ca2+, and consistent with a change in ion selectivity. By contrast, the ability to discriminate between Ba2+ and Ca2+ was preserved in all Asp 707 mutants, as IBa/ICa was either unaffected (D707A, D707G) or increased (D707N, D707E) (Figure S2A). Moreover, switching the CaVβ subunit to CaVβ3, a subunit that allows fast VDI (De Waard and Campbell, 1995; Stea et al., 1994) did not alter the relative impact of the Asp 707 mutations on CDI, a result demonstrating that SF mutation effects on CDI are independent of CaVβ (Figure S3). Overall, these observations indicate that Asp 707 mutations affect CDI but leave other biophysical properties unchanged.
Table 1.
Electrophysiological Parameters
| Channel | f or f′ | IBa/ICa | Erev | V1/2 | k | n | |
|---|---|---|---|---|---|---|---|
| Oocytes | CaV1.2 | 0.61 ± 0.02 | 1.72 ± 0.11 | 89.1 ± 4.0 | 5.4 ± 0.6 | 9.5 ± 0.3 | 8 |
| D707A | 0.10 ± 0.01 | 1.53 ± 0.05 | 92.8 ±4.8 | 5.2 ± 0.9 | 9.8 ± 0.5 | 13 | |
| D707G | 0.24 ± 0.03 | 1.94 ± 0.13 | 94.8 ± 4.0 | 7.2 ± 0.8 | 9.0 ± 0.6 | 5 | |
| D707N | 0.49 ± 0.04 | 2.63 ± 0.03 | 87.8 ± 2.5 | 10.0 ± 0.6 | 9.8 ± 0.5 | 5 | |
| D707E | 0.35 ± 0.02 | 3.58 ± 0.24 | 88.0 ± 8.1 | 8.5 ± 1.0 | 8.7 ± 0.5 | 6 | |
| E1115A | 0.14 ± 0.02 | 0.92 ± 0.01 | 49.8 ± 1.0 | 1.7 ± 0.3 | 8.6 ± 0.2 | 8 | |
| G364D-D707G | 0.30 ± 0.02 | 2.98 ± 0.26 | 88.7 ± 6.1 | 10.4 ± 1.2 | 9.3 ± 0.2 | 7 | |
| D707G-G1116D | 0.60 ± 0.02 | 2.28 ± 0.29 | 89.2 ± 4.3 | 4.8 ± 3.3 | 8.1 ± 0.6 | 6 | |
| D707A-A1417D | 0.45 ± 0.01 | 1.63 ± 0.14 | 83.9 ± 4.2 | 5.2 ± 1.1 | 8.9 ± 0.4 | 9 | |
| E1119A | 0.59 ± 0.02 | 1.73 ± 0.18 | 88.9 ± 3.6 | 6.3 ± 1.1 | 9.1 ± 0.2 | 6 | |
| D1420A | 0.60 ± 0.02 | 1.88 ± 0.23 | 79.3 ± 2.7 | 5.8 ± 1.9 | 10.0 ± 0.2 | 5 | |
| HEK293 | CaV 1.2 | 0.59 ± 0.02 | 1.57 ± 0.38 | 77.5 ± 3.0 | 7.1 ± 2.0 | 6.5 ± 0.6 | 8 |
| D707A | 0.17 ± 0.02 | 1.94 ± 0.21 | 80.5 ± 1.8 | 11.8 ± 0.5 | 8.7 ± 0.2 | 6 | |
| D707E | 0.40 ± 0.04 | 2.86 ± 0.52 | 73.3 ± 2.9 | 11.3 ± 2.1 | 6.8 ± 0.5 | 4 | |
| E1115A | 0.36 ± 0.04 | 1.00 ± 0.15 | 68.5 ± 1.5 | −0.5 ± 2.0 | 7.8 ± 0.5 | 6 | |
| CaV1.3 | 0.73 ± 0.01 | 1.58 ± 0.11 | 75.6 ± 3.0 | −8.5 ± 1.1 | 7.3 ± 0.5 | 6 | |
| D726A | 0.15 ± 0.05 | 1.85 ± 0.16 | 81.1 ± 1.3 | −8.6 ± 0.9 | 7.1 ± 0.4 | 8 | |
| E1121A | 0.63 ± 0.02 | 1.09 ± 0.08 | 61.9 ± 3.0 | −15.3 ± 1.7 | 7.8 ± 0.4 | 9 | |
| CaV2.1 | 0.26 ± 0.03 | 1.64 ± 0.39 | 46.9 ± 7.0 | −7.7 ± 0.8 | 3.2 ± 0.6 | 7 | |
| D667A | 0.04 ± 0.03 | 1.81 ± 0.12 | 56.4 ± 3.2 | −4.7 ± 1.4 | 3.8 ± 0.9 | 6 | |
| E1461A | 0.12 ± 0.02 | 1.07 ± 0.13 | 34.0 ± 1.2 | −8.8 ± 0.5 | 3.4 ± 0.2 | 5 |
f is defined as R300 for Ba2+ – R300 for Ca2+ where R300 = I300/I0 where I300 and I0 are current amplitudes at 300 ms post-depolarization and at the peak current, respectively. f′ is defined as R800 for Ba2+ – R800 for Ca2+ where R800 = I800/I0 where I800 and I0 are current amplitudes at 800 ms post-depolarization and at the peak current, respectively. f and f′ were determined at membrane potential of +20 and +10 mV, respectively. IBa/ICa is the Ba2+ and Ca2+ peak current amplitude ratio. Erev is the reversal potential. V1/2 is the midpoint of activation. k is the slope factor of the activation curve. Erev, V1/2, and k were determined using Ca2+ as the charge carrier. n is the number of experiments. Data were fit to I = Gmax * (Vm − Erev)/(1 + exp (V1/2 − Vm)/k), where I is the measured peak current at each test potential (Vm) and Gmax is the maximal macroscopic conductance. Data are expressed as mean values ± SEM.
SF Mutations Identify Elements that Selectively Affect CDI or VDI
CaVs have two principal inactivation mechanisms, CDI (Ben-Johny and Yue, 2014; Cens et al., 2006; Halling et al., 2006) and VDI (Cens et al., 2006; Stotz et al., 2004). Due to the strong slowing effects that CaVβ2a has on VDI (De Waard and Campbell, 1995; Olcese et al., 1994; Stea et al., 1994), VDI is largely absent in our initial experiments that uncovered the importance of the DII (+1) site in CDI (Figures 1C and 1D). Therefore, to examine the possible impact of changes at the DII (+1) SF position on VDI, we paired CaV1.2 channels bearing a set of DII (+1) mutations that had differing effects on CDI (D707A, D707E, and D707N) with the CaVβ isoform that imparts the fastest VDI, CaVβ3 (De Waard and Campbell, 1995; Stea et al., 1994), and measured the effects on VDI kinetics using Ba2+ as the charge carrier. These experiments showed that regardless of whether the DII (+1) SF mutant caused a strong (D707A), intermediate (D707E), or mild (D707N) effect on CDI, none caused changes in VDI (Figures 1C, 1D, S4A, and S4B).
Mutations at the SF (+4) site on the extracellular side of the pore above the SF (+1) and (0) positions (Figure 1B) affect inactivation in BacNaVs (Pavlov et al., 2005), bacterial channels sharing both structural and functional properties with CaVs and NaVs (Catterall et al., 2017; Payandeh and Minor, 2015). To ask whether mutations at the three homologous CaV1.2 SF (+4) positions bearing charged residues, DI Asp 367, DIII Glu 1119, and DIV Asp 1420 (Figure 1A), could affect CaV inactivation, we measured the effect of alanine mutations at each of these sites in CaV1.2 co-expressed with CaVβ3. Two of the three mutations,DIII (+4) E1119A and DIV (+4) D1420A, reduced both the rate and the extent of VDI. By contrast, DI (+4) D367A had no effect on VDI (Figures S4C and S4D). Interestingly, despite the clear effects of DIII (+4) E1119A and DIV (+4) D1420A on VDI, these mutations left CDI and other biophysical properties completely intact (Figure S5). These experiments demonstrate that removal of the conserved negative charges at two different levels of the SF architecture yields two distinct phenotypic outcomes. Removal of the negative charge at DII (+1) blunts CDI but spares VDI, whereas removal of the negative charge at DIII (+4) or DIV (+4) leaves CDI intact but reduces VDI. These results support the general idea that inactivation mechanisms are set by the shared nature of the pore domain architecture across diverse VGICs (Catterall et al., 2017). The ability of the DII (+1) mutants to blunt CDI but spare VDI and other biophysical properties differs from the consequences of cytoplasmic domain mutations that affect CDI but that also impact coupling to the CaVβ subunit and VDI (Findeisen and Minor, 2009), and S6 mutations that affect CDI but that also affect VDI and activation voltage dependency (Limpitikul et al., 2016; Tadross et al., 2010). Together, our observations suggest that the SF is an essential element in the CDI mechanism.
Single CaV1.2 SF Mutations Drastically Reduce CDI in Mammalian Cells
Our observations suggest two possible mechanisms by which the DII (+1) and DIII (0) SF mutants could diminish CDI. The first, consistent with the effects of the DIII (0) mutant E1115A, is that mutations at some SF sites cause ion selectivity changes that reduce Ca2+ ion influx and, thereby, reduce CDI. The second, suggested by the DII (+1) position Asp 707 mutations, is that key portions of the SF play an active role in CDI. Because the loss of the negatively charged residue at DII (+1) did not dramatically alter channel selectivity properties or activation voltage dependency, we pursued further characterization of this site to understand whether the CaV SF might have a previously unappreciated role in the CDI mechanism.
Alteration of the CaM binding IQ motif has different impacts on CaV1.2 CDI depending on whether oocytes or mammalian cells are used to measure function (Barrett and Tsien, 2008). Therefore, to test whether the consequences of SF mutations on CDI were similarly expression system dependent, we measured the properties of the two mutants that had the strongest effects on CDI reduction of CaV1.2 in oocytes, D707A and E1115A, as well as the mutant that maintained the SF DII (+1) negative charge, D707E, using whole-cell recordings from transfected HEK293 cells (Figures 2A and S1B). As in oocytes, D707A, D707E, and E1115A caused a substantial reduction in CDI (Figures 2A–2C). The magnitude of the CDI diminishment was less pronounced in the E1115A mutant compared to oocytes, whereas this effect was comparable between the two different expression systems for D707A and D707E (Table 1). Similar to oocytes, voltage-dependent activation and I-V curves of CaV1.2 and Asp 707 mutants were identical (Figures 2D and 2E). E1115A caused a small hyperpolarizing shift (−8 mV) in voltage-dependent activation, not seen in oocytes (Figure 2E; Table 1), and perturbed the reversal potential, although this change is smaller in HEK293 cells (−9 versus −39 mV for HEK293 cells and Xenopus oocytes, respectively) (Figures 1G and 2E; Table 1). In further support of differential effects of the mutants on ion selectivity, similar to oocytes, the ability to discriminate Ca2+ over Ba2+, reflected by IBa/ICa > 1, was conserved in the Asp 707 mutants but was eliminated in E1115A (Figure S2B; Table 1). Thus, in contrast to expression system-dependent differences in CDI at the level of the Ca2+ sensor (Barrett and Tsien, 2008), our results demonstrate that the SF mutation-induced CDI changes are expression system independent and provide additional support for the idea that the reduction in CDI caused by the DII (+1) mutants are not related to changes in the ion selectivity properties of the channel.
Figure 2. Domain II SF Mutations Reduce CDI in CaV1.2 Expressed in Mammalian Cells.
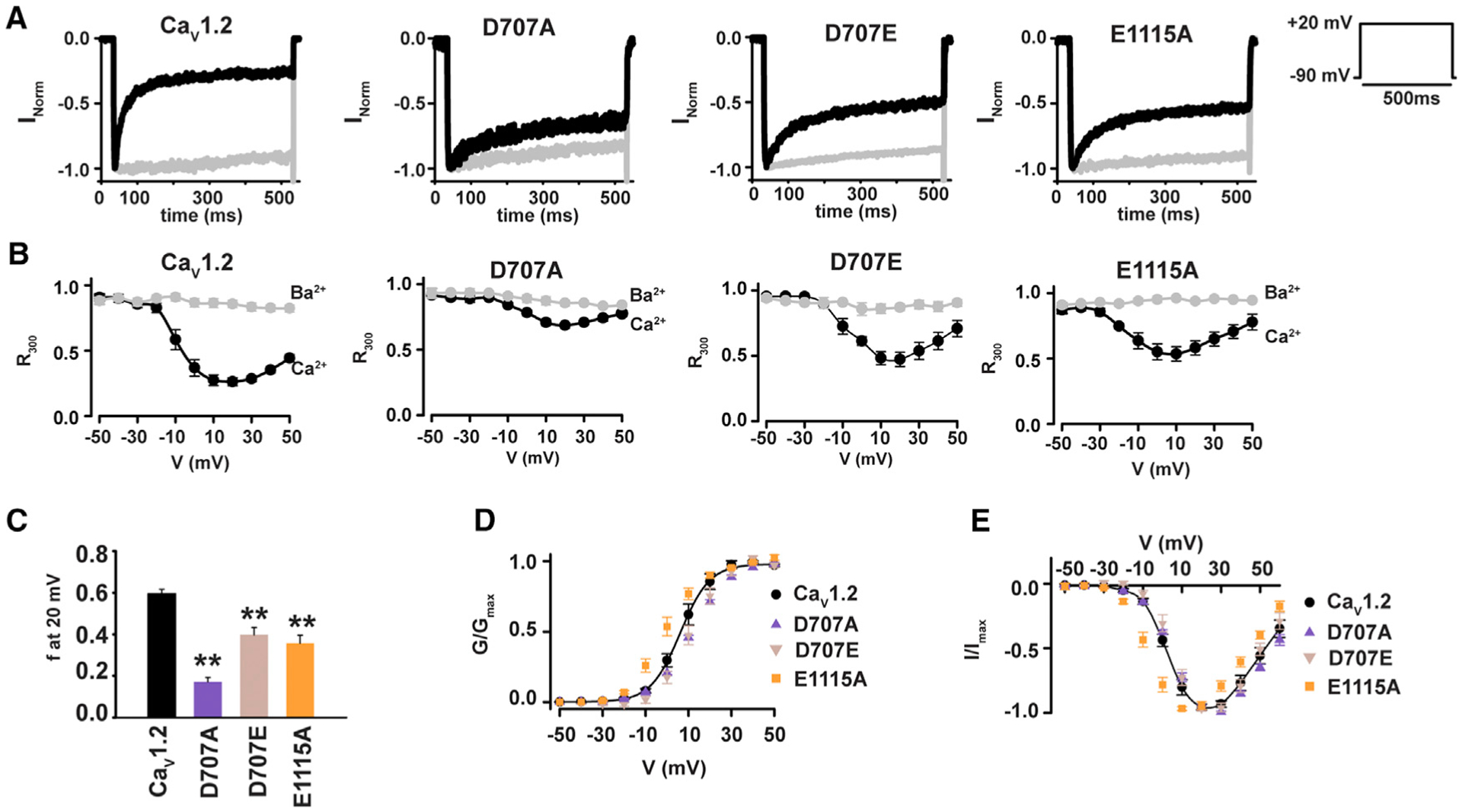
(A) Exemplar normalized recordings at +20 mV in Ca2+ (black) or Ba2+ (gray) from HEK293 cells expressing CaV1.2 or the indicated mutants.
(B) Fractional current remaining 300 ms post-depolarization (R300) as a function of the membrane potential for channels in (A).
(C) Average f values at 20 mV. **p < 0.001 compared to CaV1.2.
(D) Voltage-dependent activation curves for CaV1.2 (black circles), CaV1.2 D707A (purple triangles), CaV1.2 D707E (tan inverted triangles), and CaV1.2 E1115A (orange squares). Currents are normalized to the maximum peak current.
(E) I–V relationships for the indicated channels. Symbols are as in (D).
Lowered Ca2+ Influx in DII (+1) Mutants Does Not Cause Loss of CDI
Although the DII (+1) mutations did not cause ion selectivity changes, in principle, the effects on CDI could be caused by reduction of Ca2+ influx via a change in channel open probability or conductance. To investigate this possibility, we compared low-noise recordings from the DII (+1) mutants that exhibited the most severe, D707A, and the mildest, D707N, CDI loss with CaV1.2. Because of the challenges associated with single CaV channel recordings when Ca2+ is the charge carrier (Hess et al., 1984), we compared patches expressing CaV1.2, D707A, and D707N using Ba2+ as the charge carrier as in many prior studies (Adams et al., 2014; Bock et al., 2011; Dick et al., 2016; Doering et al., 2005; Hess et al., 1984; Tadross et al., 2008, 2013; Yang et al., 2015). These experiments showed single CaV1.2 channels having an ~1-pA current amplitude, consistent with prior reports (Dick et al., 2016). By contrast, in patches expressing either D707A or D707N, we could only observe measurable currents characteristic of multi-channel recordings with maximal current of ~2 pA (Figure 3A). These observations indicate that both the D707A and D707N decrease channel conductance relative to wild-type. To address whether there was a parallel reduction in Ca2+ conductance relative to the reduced level of CDI, we measured whole-cell Ca2+ currents of CaV1.2,D707A, and D707N (Figure 3B) and compared their current densities (Figure 3C). These data reveal similar current densities for D707A and D707N that are both reduced by ~80% relative to CaV1.2 (Figure 3C) and that are consistent with the reduced channel conductance assessed by the low-noise recordings (Figure 3A). The reduced single-channel conductance observed for D707A and D707N is in line with the ability of both mutants to cause similar reductions in Ca2+ binding to the channel pore (Shaya et al., 2014) and points to a key role of the DII (+1) site in ion conduction. Importantly, the data indicate that the shared decrease in ion conduction caused by the DII (+1) mutations cannot be the origin of the loss of CDI in D707A, as two channels having similar low conductances, D707A and D707N, have completely different CDI phenotypes (Figures 1C–1E).
Figure 3. CDI Loss in the SF (+1) Mutant D707A Is Unrelated to Insufficient Ca2+ Influx.
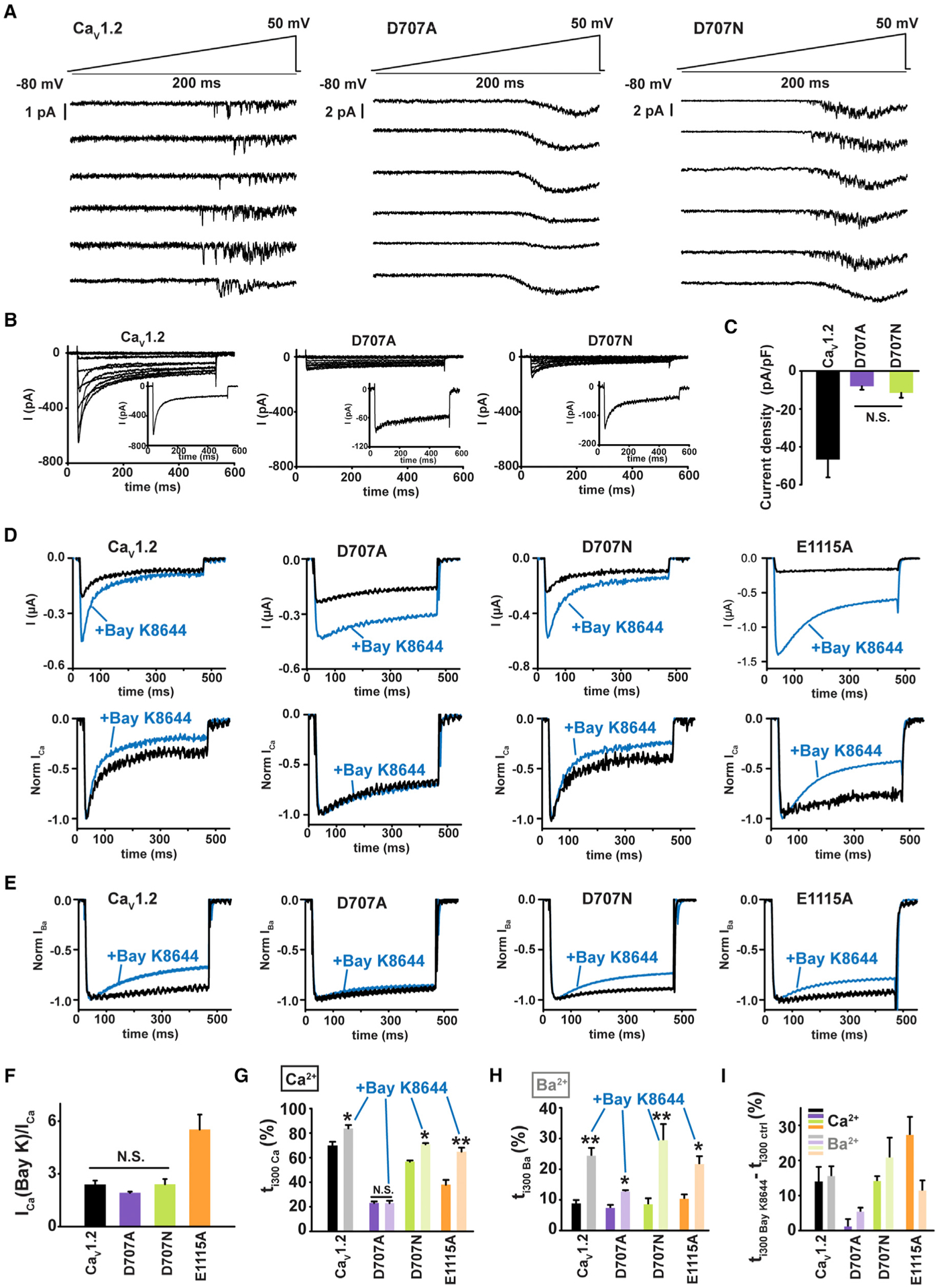
(A) Exemplar cell-attached low noise current recordings in response to a voltage ramp (top) for HEK293 cells expressing CaV1.2 or the indicated mutants. Each trace is from a different patch.
(B) Exemplar whole-cell current recordings from HEK293 cells expressing CaV1.2 or the indicated mutants. Currents were evoked with a multistep depolarization protocol from −50 to +50 mV using 10 mV increments and a −90 mV holding potential. Exemplar current traces at +20 mV are shown (inset).
(C) Current densities for CaV1.2 and the indicated mutants. N.S., not statistically different; n = 6–7.
(D) Exemplar Ca2+ currents from Xenopus oocytes expressing CaV1.2 or the indicated mutants, in response to a +20 mV depolarization in absence (black) or in presence of 5 μM Bay K8644 (blue). Raw traces (upper panel) and normalized traces (lower panel) illustrate Bay K8644 effects on the peak current and inactivation, respectively.
(E) Exemplar normalized Ba2+ currents from Xenopus oocytes expressing CaV1.2 or the indicated mutants, in response to a +20 mV depolarization in absence (black) or in presence of 5 μM Bay K8644 (blue).
(F) Ratio of Ca2+ current amplitudes in presence, ICa(Bay K), and in absence, ICa, of Bay K8644; n = 6–7.
(G and H) Percentage of inactivation 300 ms post-depolarization (ti300) in absence (dark bars) and presence of Bay K8644 (light bars) using (G) Ca2+ or (H) Ba2+ as charge carrier. *p < 0.01 and **p < 0.001 compared to the same construct without Bay K8644; n = 6–11.
(I) Difference in ti300 induced by Bay K8644 in Ca2+(dark bars) and Ba2+ (light bars).
To investigate whether reduced Ca2+ influx was the cause of CDI loss in D707A, we used the CaV agonist Bay K8644 that increases single-channel open times (Hess et al., 1984) to enhance Ca2+ influx through the channel. Bay K8644 application to Xenopus oocytes expressing CaV1.2, D707A, D707N, or the mutant E1115A, in which changes in ion selectivity reduce Ca2+ influx (Ellinor et al., 1995; Mikala et al., 1993), showed clear augmentation in Ca2+ current amplitude for all cases, consistent with an increase in channel openings and higher Ca2+ influx through individual channels (Hess et al., 1984) (Figures 3D and 3F). This Bay K8644-enhanced Ca2+ current was accompanied by a more complete inactivation for CaV1.2, D707N, and E1115A as measured by the percentage of inactivation 300 ms post-activation (ti300) (Figures 3D and 3G), consistent with prior studies of Bay K8644 on CaV1.2 in Xenopus oocytes (Noceti et al., 1998). However, for CaV1.2 and D707N, the equivalent level of increased inactivation was also observed when Ba2+ was used as charge carrier, indicating that this effect is a consequence of Bay K8644 itself rather than a response to the increased Ca2+ influx due to an undersaturated CaM Ca2+ sensor (Figures 3D, 3H, and 3I). By contrast, for the mutant having compromised selectivity, E1115A, CDI was enhanced beyond the level expected from the Bay K8644 effect in Ba2+ (Figure 3I). Notably, Bay K8644-induced inactivation in Ca2+ is absent in the D707A mutant (Figures 3D, 3E, and 3G–3I). Hence, even though the magnitude of the Bay K8644-induced Ca2+ current increase was similar for CaV1.2, D707A, and D707N (Figure 3F), the enhanced Ca2+-influx was not able to induce CDI for D707A (Figures 3E and 3G). Taken together, these observations establish that CDI loss in the D707A mutant is not caused by a reduced Ca2+ influx and strongly suggest that SF DII (+1) position plays an active role in CDI.
A Single CaV1.3 SF DII (+1) Mutation Drastically Reduces CDI
The aspartate at the SF DII (+1) site is conserved among all CaVs (Figure 1A) (Payandeh and Minor, 2015; Shaya et al., 2014). CDI occurs in high voltage-activated CaVs (CaV1s and CaV2s) (Liang et al., 2003), and, among the L-type (CaV1) channels, the CaV1.342a variant exhibits the most pronounced CDI (Huang et al., 2013; Singh et al., 2008; Xu and Lipscombe, 2001). Therefore, to examine the general importance of the DII (+1) aspartate for CDI, we asked whether removal of this aspartate would affect a channel in which CDI is particularly strong. We compared CaV1.342a CDI with a DII (+1) mutant and a DIII (0) mutant (D762A and E1121A, respectively) that correspond to the mutants that were most effective at reducing CaV1.2 CDI. Measurement of the functional properties of these channels in HEK293 cells showed that, as with the analogous CaV1.2 mutants, the DII (+1) mutation D726A drastically reduced CDI, whereas the DIII (0) mutation E1121A caused only a slight reduction in CDI (f = 0.73 ± 0.01, 0.15 ± 0.05, and 0.63 ± 0.02 for CaV1.3, D726A, and E1121A, respectively; Figures 4A–4C; Table 1). Similar to our observations for CaV1.2, the DII (+1) mutant D726A had no effect on channel selectivity or the activation voltage dependency (Figures 2D and 2E; Table 1), whereas the DIII (0) mutant, E1121A, caused a slight leftward shift of both the reversal potential and the activation curve, comparable to the effects of E1115A on CaV1.2 (Figures 2D, 2E, 4D, and 4E; Table 1). In addition, unlike the DII (+1) D726A change, the DIII (0) E1121A substitution caused a significant reduction in the peak current IBa/ICa ratio to ~1, indicating a loss in the ability to discriminate between Ba2+ and Ca2+ (Figure S2C; Table 1). Hence, as with CaV1.2, the effects of neutralizing the DIII (0)glutamate with respect to the observed CDI reduction are consistent with a change in ion selectivity. By contrast, neutralization of the DII (+1) aspartate spared all of the tested CaV1.3 biophysical parameters, except CDI. These results demonstrate the general importance of the DII (+1) aspartate in CaV1 channels and lend further support to the hypothesis that changes in the SF have a direct role in CDI.
Figure 4. Domain II SF Mutations Reduce CDI in CaV1.3 and CaV2.1.
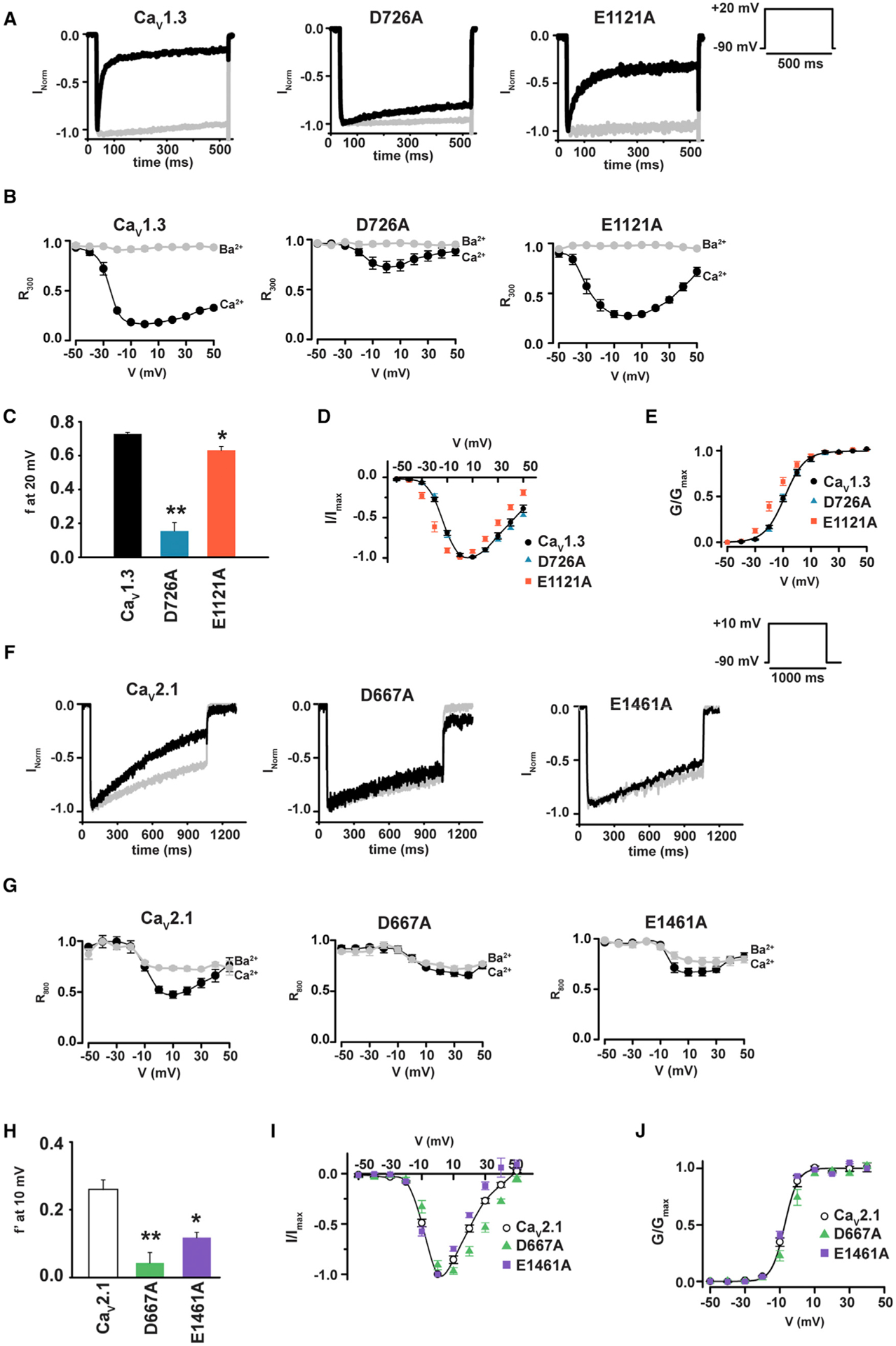
(A) Exemplar normalized recordings at +20 mV in Ca2+ (black) or Ba2+ (gray) from HEK293 cells expressing CaV1.3 or the indicated mutants.
(B) Fractional current remaining 300 ms post-depolarization (R300) as a function of the membrane potential for (A).
(C) Average f values at +20 mV. *p < 0.01 and **p < 0.001 compared to CaV1.3.
(D) I–V relationships for CaV1.3 (black circles), CaV1.3 D707A (blue triangles), and CaV1.3 E1121A (red orange squares).
(E) Voltage-dependent activation curves for the indicated channels. Symbols are as in (D).
(F) Exemplar normalized recordings at +10 mV in Ca2+ (black) or Ba2+ (gray) from HEK293 cells expressing CaV2.1 or the indicated mutants.
(G) Fractional current remaining 800 ms post-depolarization (R800) as a function of the membrane potential for (F).
(H) Average f′ values at +10 mV, where is f′ the difference between Ca2+ and Ba2+ R800 values. *p < 0.01 and **p < 0.001 compared to CaV2.1.
(I) I–V relationships for CaV2.1 (open circles), CaV2.1 D667A (green triangles), and CaV2.1 E1461A (purple squares).
(J) Voltage-dependent activation curves for the indicated channels. Symbols are as in (I).
SF DII (+1) Aspartate Is Generally Important for CDI
CaV2 channels also have CDI (DeMaria et al., 2001; Lee et al., 2003; Liang et al., 2003), and, similar to CaV1s, this process relies on CaM:IQ domain interactions (Ben-Johny and Yue, 2014; Dunlap, 2007; Minor and Findeisen, 2010). However, the roles of the CaM lobes are inverted between the two subfamilies, with the C-lobe controlling CDI in CaV1 (Peterson et al., 1999) but the N-lobe controlling CDI in CaV2s (DeMaria et al., 2001; Lee et al., 2003; Liang et al., 2003). To ask whether the conserved DII (+1) aspartate is important for CaV CDI regardless of which CaM lobe governs CDI, we measured the effects of mutants equivalent to CaV1.2 DII (+1) D707A and DIII (0) E1115A in a CaV2 family representative, CaV2.1 (D667A and E1461A, respectively). To quantify changes in CaV2.1 CDI, we measured the ratio between the current amplitude at peak and 800 ms post-depolarization (R800) for both Ba2+ and Ca2+ as a function of voltage (DeMaria et al., 2001) (Figures 4F and 4G) and compared the difference between Ba2+ and Ca2+ R800, f′, for CaV2.1 and mutants at the potential of maximal current, +10 mV (DeMaria et al., 2001). As in the case of CaV1.2 and CaV1.3, DIII (0) neutralization caused a CDI reduction (f′ = 0.26 ± 0.03 and 0.12 ± 0.02 for CaV2.1 and E1461A, respectively; Figure 4H) and did not affect activation voltage dependency. However, this site (0) mutation did induce a −13 mV reversal potential shift and reduced peak current IBa/ICa ratio to ~1 (Figures 4H–4J and S2D; Table 1), consistent with an ion selectivity change. By contrast, neutralization of the CaV2.1 DII (+1) residue essentially eliminated CDI (f′ = 0.04 ± 0.03; Figure 4H) and did not affect the activation voltage dependency or the peak current IBa/ICa ratio (Figures 4J and S2D; Table 1), although it did cause a +10 mV reversal potential shift (Figure 4I). The clear loss of CaV2.1 CDI caused by the D667A mutation demonstrates the universal role of the CaV SF in CDI and establishes that this role is independent of the details of how the CDI is initiated by CaM. Together with the CaV1 results, these data support the idea that the SF is involved in the final step of the CaV CDI process.
CDI Requires a Negative Charge at SF (+1) Position on DII, DIII, or DIV
The four CaV SF domains contribute an identical set of negatively charged glutamate side chains at the (0) position but a set of non-equivalent residues at the (+1) site (G, D, G, and A for DI–DIV, respectively) (Figures 1A and 1B). Given the crucial role we found for the DII (+1) aspartate in CDI (Figures 5A and 5B), and the fact that it is the sole negatively charged amino acid at this CaV SF level, we asked whether the DII (+1) position had a special role in CDI or whether CDI would be preserved if the aspartate were moved to other (+1) positions around the SF. Hence, we created a set of mutants that placed an aspartate at the DI, DIII, or DIV (+1) sites. To preserve the SF amino acid composition, each of these mutants exchanged the amino acid from the host site into the DII (+1) site to create the following swap mutants: DIAsp (G364D/D707G), DIIIAsp (D707G/G1116D), and DIVAsp (D707A/A1417D) (Figure 5C). We recorded whole-cell currents from Xenopus oocytes injected with these constructs using Ca2+ and Ba2+ as charge carriers and compared these mutants with the corresponding single-point changes at DII (+1) (Figures 5C and 5D). Exchange of the SF (+1) aspartate between the DII and DI positions, DIAsp, yielded channels that had severely diminished CDI that was equivalent to the D707G mutant (Figures 5C–5E), indicating that the DI (+1) site aspartate was unable to restore CDI in the face of the loss of the aspartate at the DII (+1) position. By contrast, exchange of the SF (+1) aspartate from the DII to DIII positions, DIIIAsp, or from the DII to DIV positions, DIVAsp, resulted in complete or near complete CDI restoration, respectively (Figures 5C–5E). These results demonstrate the ability of a SF (+1) aspartate to preserve CDI when located on DIII or DIV but not DI. To test whether reduced Ca2+ influx was the cause of the compromised CDI in DIAsp, we examined the consequences of Bay K8644-enhanced Ca2+ influx on the SF (+1) swap mutants. Bay K8644 application increased Ca2+ currents by ~2-fold in all three (+1) swap mutants, similar to results on other SF mutants (Figures 3C, 3F, and 3G), but failed to enhance CDI in the channel having reduced CDI, DIAsp (Figures S6A, S6C, and S6D). Studies using Ba2+ showed that, as with our other SF (+1) Bay K8644 experiments (Figure 3), inactivation is increased in a Ca2+-independent manner (Figures S6B, S6E, and S6F). These results indicate that reduced CDI of DIAsp is not caused by insufficient Ca2+ influx but is linked to a change in the ability of the SF to reach an inactivated state. The preservation of CDI in the DIIIAsp and DIVAsp swaps demonstrates that the single changes at DII (+1), D707G and D707A, do not preclude CDI provided that either the DIII or DIV domain provides the SF position (+1) negative charge. The fact that CDI occurs only when the (+1) negative charge resides on DII, DIII, or DIV, but not DI, supports the idea of asymmetric functional roles for the four elements of the SF (Ellinor et al., 1995; Parent and Gopalakrishnan, 1995; Yang et al., 1993) and indicates that the mere presence of a negative charge at the SF (+1) level is insufficient to support CDI.
Figure 5. CaV1.2 CDI Requires a Negative Charge at the DII, DIII, or DIV SF (+1) Position.
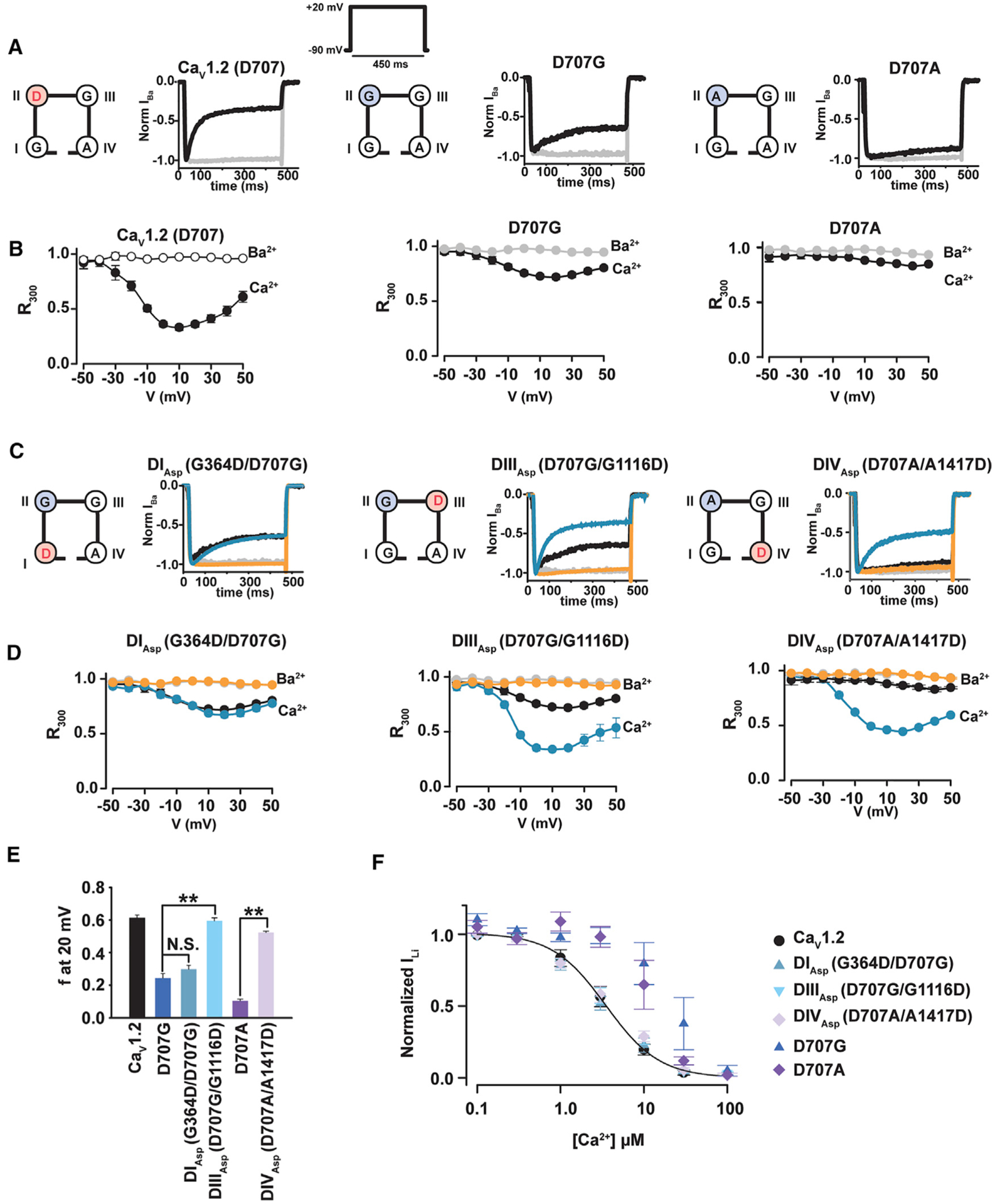
(A) Exemplar normalized recordings at +20 mV in Ca2+ (black) or Ba2+ (gray) from Xenopus oocytes expressing the CaV1.2 or the indicated mutants.
(B) Fractional current remaining 300 ms post-depolarization (R300) as a function of the membrane potential for channels in (A). Data in (A) and (B) are identical to Figures 1A and 1B.
(C) Exemplar normalized recordings at +20 mV in Ca2+ (blue) or Ba2+ (orange) from Xenopus oocytes expressing the indicated CaV1.2 mutants moving the negative charge at the (+1) position of the selectivity filter from domain II to domains I, III, or IV. Ca2+ (black) and Ba2+ (gray) currents from CaV1.2 are shown for comparison.
(D) Fractional current remaining 300 ms post-depolarization (R300) as a function of the membrane potential for channels in (C).
(E) Average f values at 20 mV. **p < 0.001 compared to the corresponding single mutant.
(F) Dose-response curves for Ca2+ block of Li+ currents for CaV1.2, DIAsp G364D/D707G (blue triangles), DIIIAsp D707G/G1116D (inverted blue triangles), and DIVAsp D707A/A1417D (lavender diamonds). Each data point at each Ca2+ concentration is normalized to the current at 3 nM Ca2+ and averaged for n = 7–9 oocytes.
To probe whether the swap mutations affected the ability of the channel to bind Ca2+, we measured Ca2+ block of Li+ currents (Shaya et al., 2014; Yang et al., 1993). Interestingly, all three aspartate-swap mutants behaved identically to CaV1.2, indicating that the affinity for Ca2+ was preserved regardless of the domain that housed the (+1) aspartate (Figure 5F). This result contrasts the reduction in Ca2+ binding caused by the single-point mutants at the DII (+1) position (Figure 5F) (Shaya et al., 2014). The observation that Ca2+ binding is preserved when the aspartate is placed at any of the four possible site (+1) positions contrasts with the domain selective results for CDI that these mutants cause. This finding indicates that the involvement of the SF in CDI is not strictly related to its ability to bind Ca2+.These results together with the differential contributions of the individual (+1) sites to CDI support the idea that the SF has a direct role in CDI.
DISCUSSION
CDI is an essential feature of high voltage-activated CaVs (CaV1s and CaV2s) that serves as an activity-dependent autoregulatory mechanism for limiting Ca2+ influx (Christel and Lee, 2011; Dunlap, 2007; Liang et al., 2003; Simms and Zamponi, 2014). This process contributes to autism (Limpitikul et al., 2016), vision (Singh et al., 2006), and cardiac action potential duration (Alseikhan et al., 2002; Dick et al., 2016; Mahajan et al., 2008; Morotti et al., 2012; Splawski et al., 2004, 2005) and is impacted by alternative splicing (Bartels et al., 2018; Shen et al., 2006; Tan et al., 2011), RNA editing (Huang et al., 2012), as well as mutations in CaM associated with long QT syndrome (Limpitikul et al., 2014, 2017). Because CDI is central to both the biophysical and physiological functions of CaVs, its molecular origins have been extensively investigated, especially from the vantage point of the intracellular, CaM-based sensor and the role of the IQ domain (Ben-Johny and Yue, 2014; Minor and Findeisen, 2010; Kim et al., 2008, 2010; Mori et al., 2008; Van Petegem et al., 2005). Although the involvement of this CaM-based sensor in CDI is firmly established, how conformational changes of this cytoplasmic element result in the cessation of ion flow through the channel and the exact CDI endpoint have remained unresolved (Babich et al., 2007; Barrett and Tsien, 2008; Cens et al., 2006; Findeisen and Minor, 2009; Kim et al., 2004; Tadross et al., 2010). Our observations provide the first evidence that the CaV SF is a central element of the CDI mechanism and constitutes the CDI gate.
Three CaV channel intracellular elements contribute to CDI: the N-terminal cytoplasmic domain (Ben Johny et al., 2013; Dick et al., 2008; Ivanina et al., 2000; Tadross et al., 2008), the CaVβ/I-II loop complex (Almagor et al., 2012; Findeisen and Minor, 2009), and the C-terminal tail:CaM complex (Ben-Johny and Yue, 2014; Minor and Findeisen, 2010; Kim et al., 2008, 2010; Lee et al., 1999; Mori et al., 2008; Peterson et al., 1999; Van Petegem et al., 2005; Zühlke et al., 1999), with the C-terminal tail:CaM complex serving as the Ca2+ sensor that initiates CDI. Given this functionally interconnected network of domains that influence CDI, there has been an ongoing search to identify the molecular endpoints of CaV CDI (Barrett and Tsien, 2008; Benmocha Guggenheimer et al., 2016; Tadross et al., 2010). There has been much focus on the S6 pore lining helices as a candidate for the CDI endpoint (Benmocha Guggenheimer et al., 2016; Raybaud et al., 2006; Tadross et al., 2010; Tadross and Yue, 2010), as these transmembrane helices form the channel intracellular gate (Wu et al., 2015, 2016) and are directly linked to two of the three domains that affect CDI. Given the involvement of many channel parts in CDI, an allosteric framework has been used to try to understand the actions of disease mutants on CDI (Dick et al., 2016; Limpitikul et al., 2016; Tadross et al., 2010). An alternative proposal is that the SF may participate in CDI through an ion-blocking model in which Ca2+affinity is increased in the inactivated state (Babich et al., 2007). Our observations that SF DII (+1) site mutations such as D707A eliminate CDI but spare biophysical parameters related to the activation process are inconsistent with the proposal that the activation gate formed by S6 is the CDI endpoint (Tadross et al., 2010). Nevertheless, S6 is likely to be critical for coupling to the various intracellular domains that contribute to CDI and may mediate direct coupling between the activation gate and the SF-based CDI gate, similar to its role in other VGIC superfamily members (Ader et al., 2009; Cuello et al., 2017; Imai et al., 2010; Panyi and Deutsch, 2006; Peters et al., 2013). Further, in contrast to the proposal that CDI occurs through an increased affinity of the SF for Ca2+ (Babich et al., 2007), we find that mutations that remove the aspartate at the SF DII (+1) site can have identical Ca2+ affinities but exhibit very different degrees of CDI (Figures 1 and 5) (Shaya et al., 2014). Together, our findings establish that the CaV SF serves as the CDI gate and recast the paradigm for understanding the CaV CDI mechanism with a focus on the SF.
The pore domain of all VGIC superfamily members is made from four subunits arranged around the central ion-conducting pore (Yu et al., 2005) that share a common architectural fold comprising two transmembrane helices, the SF, and a short pore helix that supports the SF architecture (Catterall et al., 2017; Payandeh and Minor, 2015). Within the context of this shared architecture, studies of diverse VGIC superfamily members have begun to uncover a central role for SF in inactivation mechanisms, being best characterized in diverse potassium channel types including: KcsA (Cuello et al., 2010, 2017), Kvs (Liu et al., 1996; López-Barneo et al., 1993; Ogielska and Aldrich, 1999; Peters et al., 2013), and K2Ps (Bagriantsev et al., 2011; Cohen et al., 2008; Lolicato et al., 2017; Piechotta et al., 2011; Schewe et al., 2016). Additionally, structural observations of asymmetric BacNaV SF conformations (Catterall et al., 2017; Payandeh et al., 2012) and functional studies of SF (+4) position mutants (Pavlov et al., 2005) have suggested a role for the SF in BacNaV VDI that may be shared with eukaryotic NaVs and CaVs (Catterall et al., 2017). Studies of members of the TRP channel branch, TRPV1 (Cao et al., 2013; Steinberg et al., 2017) and TRPM4 (Autzen et al., 2018), also support the idea that the SF constitutes a gate. Hence, the discovery that CaV CDI also relies on a SF-based mechanism sets CaVs squarely within the growing list of VGICs in which SF-based gating is central to function. The apparent widespread role of the SF in the inactivation processes of VGIC superfamily members suggests that these SF-based inactivation mechanisms capitalize on a fundamental pore domain (PD) property that predates the evolutionary divergence in ion selectivity and gating cue responses among the VGIC superfamily branches. This prevalence of SF-based inactivation mechanisms seems likely to have its origins in the shared ancient structure that forms the PD.
Elements from the cytoplasmic side of the channel drive conformational changes that stop ion permeation for both CDI and VDI. Whether CaV CDI and VDI share a common mechanism has been unresolved (Barrett and Tsien, 2008; Cens et al., 2006; Findeisen and Minor, 2009; Kim et al., 2004; Tadross et al., 2010). Our studies support the idea that both processes involve the SF. Notably, we find that there are selective effects on CDI or VDI depending on which parts of the SF structure are altered (Figures 1 and S4), supporting the idea that CDI and VDI have different endpoints even though they are affected by common channel elements (Barrett and Tsien, 2008; Findeisen and Minor, 2009; Tadross et al., 2010). The VDI effects match those reported for BacNaVs (Pavlov et al., 2005) and support the idea that there is a common SF-based mechanism underlying VDI in BacNaVs, CaVs, and NaVs (Catterall et al., 2017). Considering our new findings of the importance of the SF in CDI together with prior cytoplasmic domain studies (Almagor et al., 2012; Ben Johny et al., 2013; Dick et al., 2008; Findeisen and Minor, 2009; Ivanina et al., 2000; Kim et al., 2008, 2010; Lee et al., 1999; Mori et al., 2008; Peterson et al., 1999; Tadross et al., 2008; Van Petegem et al., 2005; Zühlke et al., 1999), we propose the following model in which SF conformational changes constitute the end stage of a CDI process that is initiated by CaM on the cytoplasmic side of the channel (Figure 6). Upon activation by voltage, Ca2+ influx through the channel is sensed by CaM and initiates a set of conformational rearrangements that end with a conformational change in the SF that stops ion flow. Although the exact conformational changes underling CaV SF inactivation and complete accounting of the residues involved remain to be elaborated and ultimately will require structural studies of the channel trapped in various states, our data indicate that this process exploits the previously identified functional asymmetry in the CaV SF (Ellinor et al., 1995; Parent and Gopalakrishnan, 1995; Yang et al., 1993). In this regard, it is interesting to note that the SF DII (+1) aspartate seems capable of making interactions that involve other SF domain residues (Cheng et al., 2010) and that we can restore CDI to DII (+1) SF mutants by supplying an aspartate at the equivalent site in two of the three other domains (Figure 5).
Figure 6. CaV CDI Model.
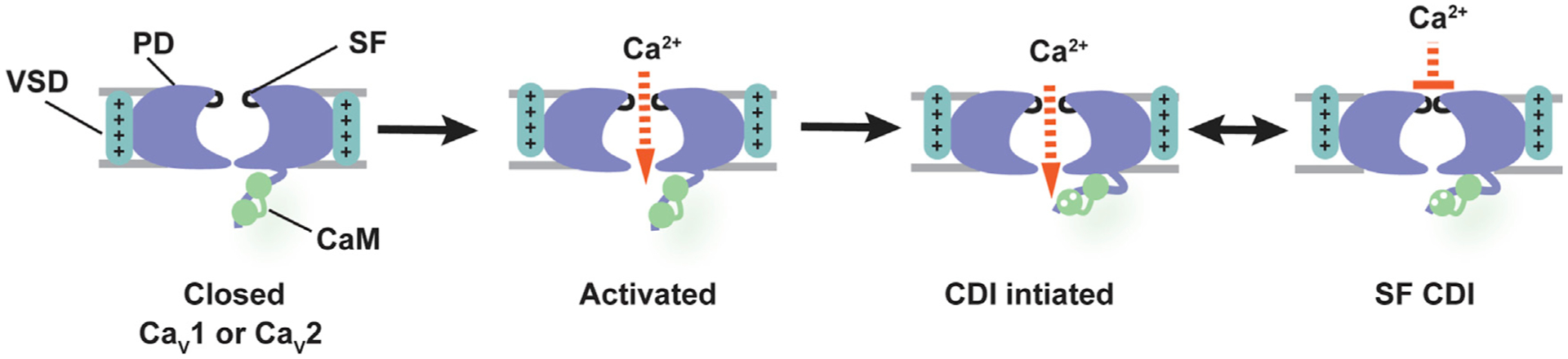
Closed channels (left) are activated by depolarization that includes voltage sensor domain (VSD) activation and pore opening. Flow of Ca2+ through the channel leads to Ca2+ (white circles) binding to CaM (green) that initiates CDI. CDI results in a selectivity filter (SF) conformational change that obstructs ion flow. Pore domain, PD (violet), voltage sensor domain, VSD (cyan), and SF (black) are labeled. Gray bars indicate membrane. Other intracellular elements that affect CDI such as the N-terminal cytoplasmic domain and CaVβ/I-II loop complex are not shown.
Our studies provide clear evidence that the CaV SF plays a central role in the CDI process by forming the CDI gate. The evidence for this CaV SF gate, together with the presence of an inner gate formed by the S6 helices (Benmocha Guggenheimer et al., 2016; Raybaud et al., 2006; Tadross et al., 2010; Tadross and Yue, 2010; Wu et al., 2015, 2016), establishes that CaVs use two gates to control their activity, similar to other VGIC superfamily members (Autzen et al., 2018; Cao et al., 2013; Cuello et al., 2017; Steinberg et al., 2017). This finding creates a new framework for addressing how the two CaV gates interact, whether they are coupled in a manner similar to structurally related potassium channels (Cuello et al., 2017), whether there are commonalities in SF-based inactivation mechanisms between channels that have wide SFs such as CaVs versus those that intimately contact the permeant ions such as potassium channels (Cuello et al., 2017), and how the intracellular components that contribute to CDI (Almagor et al., 2012; Ben Johny et al., 2013; Ben-Johny and Yue, 2014; Dick et al., 2008; Findeisen and Minor, 2009; Minor and Findeisen, 2010; Ivanina et al., 2000; Kim et al., 2008, 2010; Lee et al., 1999; Mori et al., 2008; Peterson et al., 1999; Tadross et al., 2008; Van Petegem et al., 2005; Zühlke et al., 1999) affect structural transitions in the SF gate. Such issues may be important for understanding how CaV disease mutations that impact CDI act (Dick et al., 2016; Limpitikul et al., 2014, 2016, 2017; Singh et al., 2006; Splawski et al., 2004, 2005) and how such functional defects may be overcome by pharmacological intervention.
STAR★METHODS
CONTACT FOR REAGENT AND RESOURCE SHARING
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Daniel L. Minor, Jr. (daniel.minor@ucsf.edu).
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Oocytes collection
Oocytes were harvested from female Xenopus laevis frogs purchased from Nasco and housed in the UCSF Laboratory Animal Resource Center (LARC) facilities. The use of these Xenopus oocytes was approved by IACUC (protocol approval # AN178461–01) and experiments were performed in accordance with University of California guidelines and regulations.
Cell Culture
Human embryonic kidney cells (HEK293) were purchased from ATCC (CRL-1573) and were grown at 37°C under 5% CO2, in a Dulbecco’s modified Eagle’s medium (DMEM) supplemented with 10% fetal bovine serum (FBS), 10% L-glutamine, and antibiotics (100 IU mL−1 penicillin and 100 mg mL–1 streptomycin) (University of California, San Francisco Cell Culture Facility). The sex of cell line is not determined.
METHOD DETAILS
Molecular Biology
Human CaV1.2 (α1C77, GenBank: Z34815), human CaV2.1 (α1A, GenBank: NM_001127221.1), rat Cav1.3 (α1D, GenBank: AF370009.1), rat CaVβ2a (GenBank: NM_053851), rabbit CaVβ3 (GenBank: NM_001101715.2), and rabbit CaVα2δ-1 (GenBank: NM_001082276.1) were used for both patch clamp and two-electrode voltage clamp experiments. CaV1.2 mutations were introduced by two separate PCR reactions. First the region of interest was PCR amplified using pcDNA3.1 Cav1.2 as template. The PCR product was then subcloned into pcDNA3.1 by restriction-ligation. The new plasmid containing the region of interest was then used as template to introduce the desired mutation using the QuikChange Site-Directed Mutagenesis Kit (Stratagene). The region of interest containing the desired mutation was then subcloned back into the pcDNA3.1 Cav1.2 to form the mutant full-length channel using the following restriction sites: NheI-HpaI, HpaI-PpuMI, KpnI-AgeI, and AgeI-FseI, for DI, DII, DIII, and DIV mutants, respectively. CaV2.1 and CaV1.3 mutants were made using the QuikChange Site-Directed Mutagenesis Kit (Agilent). All mutants were validated by complete sequencing of the genes encoding for the proteins of interest.
Two-electrode voltage clamp electrophysiology
Linearized cDNA was translated into capped mRNA using the T7 mMessenger kit (Ambion). We injected 50 nL of CaV1.2α1, CaVβ2a or CaVβ3 and CaVα2δ-1 mRNA at a 1:1:1 molar ratio into Xenopus oocytes. Two-electrode voltage clamp experiments were performed 2–3 days post-injection.
Oocytes were injected with 50 nL of 100 mM BAPTA four minutes before recording, to minimize calcium-activated chloride currents as previously described (Findeisen and Minor, 2009). For recording of Ca2+ or Ba2+ currents, bath solutions contained 40 mM CaCl2 or 40 mM BaCl2, respectively, 50 mM NMDG-Cl, 1 mM KOH, 10 mM HEPES, adjusted to pH 7.4 with HNO3. Measurements of Ca2+ block of Li+ currents followed previously described protocols (Shaya et al., 2014). The bath solution contained 100 mM LiOH, Ca(NO3)2 at test concentrations between 3 nM and 100 μM, and 10 mM HEPES, adjusted to pH 7.4 with HNO3. Ca2+ concentrations were verified using a Ca2+ electrode. A solution having a nominal 3 nM free Ca2+ concentration was used as control condition and contained 170 μM Ca(NO3)2 and 15 mM ethylene glycol-bis(2-aminoethylether)-N, N,N′,N′-tetraacetic acid (EGTA). Electrodes were filled with 3 M KCl and had resistances of 0.3–1.0 MΩ. Recordings were conducted at room temperature from a holding potential of 90 mV. Leak currents were subtracted using a P/4 protocol.
Whole-cell Patch-clamp electrophysiology
HEK293 cells were transfected (in 35-mmdiameter wells) with Lipofectamine 2000 (Invitrogen, Carlsbad, CA, USA) and plated onto coverslips coated with Matrigel (BD Biosciences, San Diego, CA, USA).
Cells were transfected using a total of 4.4 μg DNA having a ratio by weight of 2:1.6:0.4:0.4 of CaVα1:CaVβ2a:CaVα2δ-1:sv40 T-antigen plasmids. The SV40 T-antigen plasmid was used to increase channel expression. Transfected cells were identified visually using an enhanced green fluorescent protein (EGFP) expression in the second cassette of the plasmid expressing the β2a subunit. Whole cell patch clamp (Hamill et al., 1981) was used to record Ca2+ and Ba2+ currents at room temperature (23 ± 2°C) 48–72 h post-transfection. Data acquisition was performed using pCLAMP 9 (Molecular Devices, Sunnyvale, CA, USA) and an Axopatch 200B amplifier (Molecular Devices). Pipettes were pulled from borosilicate glass capillaries (TW150F-3; World Precision instruments, Sarasota, FL, USA) and polished (MF-900 microforge; Narishige, Tokyo, Japan) to obtain 2–3 MΩ resistances. Sixty to eighty percent of the voltage error due to the series resistance was compensated, and leak currents were subtracted using a P/4 protocol. For CaV1.2 and CaV1.3 experiments, the pipette solution contained 120 mM NMDG-Cl, 1 mM MgCl2, 5 mM EGTA, 4 mM Mg-ATP, 42 mM HEPES (pH 7.3 adjusted with Methane sulfonic acid). Bath solution contained 40 mM CaCl2 or 40 mM BaCl2, 1 mM MgCl2, 105 mM Tris (pH 7.3 adjusted with Methane sulfonic acid). For CaV2.1 experiments, the pipette solution contained 120 mM NMDG-Cl, 1 mM MgCl2, 0.5 mM EGTA, 2 mM Mg-ATP, 60 mM HEPES (pH 7.3 adjusted with Methane sulfonic acid). Bath solution contained 10 mM CaCl2 or 10 mM BaCl2, 1 mM MgCl2, 150 mM Tris (pH 7.3 adjusted with Methane sulfonic acid).
Single-channel recordings
HEK cells were maintained and transfected as described above. Cell-attached configuration of the patch clamp technique (Hamill et al., 1981) was used to record Ba2+ currents from single or multiple CaV1.2 channels at room temperature (23 ± 2°C) 48–72 h post-transfection. Data acquisition was performed using pCLAMP 9 (Molecular Devices, Sunnyvale, CA, USA) and an Axopatch 200B amplifier (Molecular Devices). Pipettes were pulled from quartz glass capillaries (QF100-70-7.5; Sutter Instrument, Novato, CA, USA) using a Laser-Based Micropipette puller (P-2000, Sutter Instrument, Novato, CA, USA), and filled with 140 mM TEA-Cl, 40 mM BaCl2, 10 mM HEPES (pH 7.4 adjusted with TEA-OH). To zero membrane potential, the bath solution contained 132 mM K glutamate, 5 mM KCl, 5 mM NaCl, 3 mM MgCl2, 2 mM EGTA, 10 mM glucose, 20 mM HEPES (pH 7.4 adjusted with KOH) (Dick et al., 2016). Recordings were low pass filtered with a cutoff frequency of 2 kHz and digitized at 50 ms. Patches were stimulated by a voltage ramp from −80 mV to 50 mV over the duration of 200 ms. The leak for each trace was subtracted using a linear fit added to an exponential fit.
Data Analysis
All results are from at least two independent oocyte batches or at least two independent transfections. Data were analyzed with Clampfit 10.6 (Axon Instruments). Activation curves were obtained by fitting the data with the following Boltzmann equation: I/Imax = 1/(1+exp((V0.5−Vm)/k)), where V0.5 is the half-activation potential, Vm is the membrane potential, and k is the slope factor. Dose–response curves were calculated as follows: Ix/I3nMCa2+ = 1/(1 + x/IC50), where Ix is the current amplitude at the Ca2+ concentration x, I3nMCa2+ is the current amplitude at 3 nM and IC50 is the half-maximal inhibitory concentration. VDI time constant (τιναχτ) was determined at a test potential of +20 mV using the formula I = A exp (−t/τ) + C, where I is the recorded current, A is the peak current, C is the residual current at steady state, and t is the time. Current density was determined as the ratio between current amplitude (pA) and the membrane capacitance (pF).
QUANTIFICATION AND STATISTICAL ANALYSIS
All the details of data analysis and statistical analysis can be found in the Method Details and figure/table legends. All data values are presented as mean ± SEM ‘n’ represents the number of cells. Statistical significance of the observed effects was assessed by Student’s t test, using SigmaStat 3.1 software. p < 0.01 was considered significant, unless otherwise stated.
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Bacterial and Virus Strains | ||
| Dh5α competent E. coli | MCLAB | Cat#DA-196 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| ethylene glycol-bis(β-aminoethyl ether)-N,N,N’,N’-tetraacetic acid (EGTA) | Sigma-Aldrich | Cat#E4378 |
| 1,2-Bis(2-aminophenoxy)ethane-N,N,N’,N’-tetraacetic acid tetrakis (BAPTA) | Santa Cruz Biotechnology | Cat#sc-202076 |
| Dulbecco’s modified Eagle’s medium (DMEM) | GIBCO | Cat#11965-092 |
| fetal bovine serum (FBS) | GIBCO | Cat#16140-071 |
| L-glutamine | GIBCO | Cat#25030-081 |
| penicillin/streptomycin | UCSF Cell Culture Facility | Cat#CCFGK004-153K01 |
| Lipofectamine 2000 | Invitrogen | Cat#11668-019 |
| Matrigel | BD Biosciences | Cat#354234 |
| Critical Commercial Assays | ||
| T7 mMessenger kit | Thermo Fisher Scientific | Cat# AM1344 |
| QuikChange Site-Directed Mutagenesis Kit | Agilent | Cat#200515 |
| Experimental Models: Cell Lines | ||
| HEK293 | ATCC | Cat#CRL-1573 |
| Xenopus oocytes | This study | N/A |
| Experimental Models: Organisms/Strains | ||
| Xenopus Laevis | Nasco | Cat# LM00531 |
| Recombinant DNA | ||
| human CaV1.2/pcDNA3.1 | Findeisen and Minor, 2009 | N/A |
| human CaV1.2 D707A/pcDNA3.1 | Shaya et al., 2014 | N/A |
| human CaV1.2 D707N/pcDNA3.1 | Shaya et al., 2014 | N/A |
| human CaV1.2 D707G/pcDNA3.1 | Shaya et al., 2014 | N/A |
| human CaV1.2 D707E/pcDNA3.1 | This study | N/A |
| human CaV1.2 E1115A/pcDNA3.1 | This study | N/A |
| human CaV1.2 D367A/pcDNA3.1 | This study | N/A |
| human CaV1.2 E1119A/pcDNA3.1 | This study | N/A |
| human CaV1.2 D1420A/pcDNA3.1 | This study | N/A |
| human CaV1.2 G364D/D707G/pcDNA3.1 | This study | N/A |
| human CaV1.2 D707G/G1116D/pcDNA3.1 | This study | N/A |
| human CaV1.2 D707A/A1417D/pcDNA3.1 | This study | N/A |
| rat CaV1.342a/pcDNA6/V5-His ABC | Xu and Lipscombe, 2001 | Addgene#26577 |
| rat CaV1.3 D726A/pcDNA6/V5-His ABC | This study | N/A |
| rat CaV1.3 E1121A/pcDNA6/V5-His ABC | This study | N/A |
| human CaV2.1/pcDNA3.1 | Kim et al., 2008 | N/A |
| human CaV2.1 D667A/pcDNA3.1 | This study | N/A |
| human CaV2.1 E1461A/pcDNA3.1 | This study | N/A |
| rat CaVβ2a/pTracer-CMV2-GFP | Findeisen and Minor, 2009 | N/A |
| rabbit CaVβ3/pSport | This study | N/A |
| rabbit CaVα2δ-1/pcDNA3.1 | Findeisen and Minor, 2009 | N/A |
| sv40 T-antigen/Bluescribe | addgene | Cat#21826 |
| Software and Algorithms | ||
| pCLAMP 9 | Molecular Devices | N/A |
| Clampfit 10 | Molecular Devices | N/A |
| SigmaStat 3.1 | Systat | N/A |
Highlights.
CaV selectivity filter forms the calcium-dependent inactivation (CDI) endpoint
Conserved CaV domain II selectivity filter (+1) aspartate plays an active role in CDI
CaV selectivity filter asymmetry is important for CDI
CaVs gating relies on an SF-based gating framework shared among the VGIC superfamily
ACKNOWLEDGMENTS
We thank M. Grabe and L. Jan for comments on the manuscript. This work was supported by grant NIH-NHLBI R01-HL080050 to D.L.M. and a Marcel Bleustein-Blanchet Foundation fellowship to F.A.-A.
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental Information includes six figures and can be found with this article online at https://doi.org/10.1016/j.neuron.2019.01.011.
DECLARATION OF INTERESTS
The authors declare no competing interests.
REFERENCES
- Adams PJ, Ben-Johny M, Dick IE, Inoue T, and Yue DT (2014). Apocalmodulin itself promotes ion channel opening and Ca(2+) regulation. Cell 159, 608–622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ader C, Schneider R, Hornig S, Velisetty P, Vardanyan V, Giller K, Ohmert I, Becker S, Pongs O, and Baldus M (2009). Coupling of activation and inactivation gate in a K+-channel: Potassium and ligand sensitivity. EMBO J. 28, 2825–2834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Almagor L, Chomsky-Hecht O, Ben-Mocha A, Hendin-Barak D, Dascal N, and Hirsch JA (2012). The role of a voltage-dependent Ca2+ channel intracellular linker: A structure-function analysis. J. Neurosci 32, 7602–7613. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alseikhan BA, DeMaria CD, Colecraft HM, and Yue DT (2002). Engineered calmodulins reveal the unexpected eminence of Ca2+ channel inactivation in controlling heart excitation. Proc. Natl. Acad. Sci. USA 99, 17185–17190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arrigoni C, Rohaim A, Shaya D, Findeisen F, Stein RA, Nurva SR, Mishra S, Mchaourab HS, and Minor DL Jr. (2016). Unfolding of a temperature-sensitive domain controls voltage-gated channel activation. Cell 164, 922–936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autzen HE, Myasnikov AG, Campbell MG, Asarnow D, Julius D, and Cheng Y (2018). Structure of the human TRPM4 ion channel in a lipid nano-disc. Science 359, 228–232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babich O, Isaev D, and Shirokov R (2005). Role of extracellular Ca2+ in gating of CaV1.2 channels. J. Physiol 565, 709–715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Babich O, Matveev V, Harris AL, and Shirokov R (2007). Ca2+-dependent inactivation of CaV1.2 channels prevents Gd3+ block: does Ca2+ block the pore of inactivated channels? J. Gen. Physiol 129, 477–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bagriantsev SN, Peyronnet R, Clark KA, Honoré E, and Minor DL Jr. (2011). Multiple modalities converge on a common gate to control K2P channel function. EMBO J. 30, 3594–3606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barrett CF, and Tsien RW (2008). The Timothy syndrome mutation differentially affects voltage-and calcium-dependent inactivation of CaV1.2 L-type calcium channels. Proc. Natl. Acad. Sci. USA 105, 2157–2162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bartels P, Yu D, Huang H, Hu Z, Herzig S, and Soong TW (2018). Alternative splicing at N terminus and domain I modulates CaV1.2 inactivation and surface expression. Biophys. J 114, 2095–2106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben-Johny M, and Yue DT (2014). Calmodulin regulation (calmodulation) of voltage-gated calcium channels. J. Gen. Physiol 143, 679–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ben Johny M, Yang PS, Bazzazi H, and Yue DT (2013). Dynamic switching of calmodulin interactions underlies Ca2+ regulation of CaV1.3 channels. Nat. Commun 4, 1717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Benmocha Guggenheimer A, Almagor L, Tsemakhovich V, Tripathy DR, Hirsch JA, and Dascal N (2016). Interactions between N and C termini of α1C subunit regulate inactivation of CaV1.2 L-type Ca(2+) channel. Channels (Austin) 10, 55–68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bock G, Gebhart M, Scharinger A, Jangsangthong W, Busquet P, Poggiani C, Sartori S, Mangoni ME, Sinnegger-Brauns MJ, Herzig S, et al. (2011). Functional properties of a newly identified C-terminal splice variant of Cav1.3 L-type Ca2+ channels. J. Biol. Chem 286, 42736–42748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campiglio M, and Flucher BE (2015). The role of auxiliary subunits for the functional diversity of voltage-gated calcium channels. J. Cell. Physiol 230, 2019–2031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cao E, Liao M, Cheng Y, and Julius D (2013). TRPV1 structures in distinct conformations reveal activation mechanisms. Nature 504, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA (2011). Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol 3, a003947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Catterall WA, Wisedchaisri G, and Zheng N (2017). The chemical basis for electrical signaling. Nat. Chem. Biol 13, 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cens T, Restituito S, Galas S, and Charnet P (1999). Voltage and calcium use the same molecular determinants to inactivate calcium channels. J. Biol. Chem 274, 5483–5490. [DOI] [PubMed] [Google Scholar]
- Cens T, Rousset M, Leyris JP, Fesquet P, and Charnet P (2006). Voltage- and calcium-dependent inactivation in high voltage-gated Ca(2+) channels. Prog. Biophys. Mol. Biol 90, 104–117. [DOI] [PubMed] [Google Scholar]
- Chen YH, Li MH, Zhang Y, He LL, Yamada Y, Fitzmaurice A, Shen Y, Zhang H, Tong L, and Yang J (2004). Structural basis of the alpha1-beta subunit interaction of voltage-gated Ca2+ channels. Nature 429, 675–680. [DOI] [PubMed] [Google Scholar]
- Cheng RC, Tikhonov DB, and Zhorov BS (2010). Structural modeling of calcium binding in the selectivity filter of the L-type calcium channel. Eur. Biophys. J 39, 839–853. [DOI] [PubMed] [Google Scholar]
- Christel C, and Lee A (2011). Ca(2+)-dependent modulation of voltage-gated Ca(2+) channels. Biochim. Biophys. Acta 1820, 1243–1252. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clapham DE (2007). Calcium signaling. Cell 131, 1047–1058. [DOI] [PubMed] [Google Scholar]
- Cohen A, Ben-Abu Y, Hen S, and Zilberberg N (2008). A novel mechanism for human K2P2.1 channel gating. Facilitation of C-type gating by protonation of extracellular histidine residues. J. Biol. Chem 283, 19448–19455. [DOI] [PubMed] [Google Scholar]
- Cuello LG, Jogini V, Cortes DM, and Perozo E (2010). Structural mechanism of C-type inactivation in K(+) channels. Nature 466, 203–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cuello LG, Cortes DM, and Perozo E (2017). The gating cycle of a K+ channel at atomic resolution. eLife 6. Published online November 22, 2017. 10.7554/eLife.28032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Waard M, and Campbell KP (1995). Subunit regulation of the neuronal alpha 1A Ca2+ channel expressed in Xenopus oocytes. J. Physiol 485, 619–634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeMaria CD, Soong TW, Alseikhan BA, Alvania RS, and Yue DT (2001). Calmodulin bifurcates the local Ca2+ signal that modulates P/Q-type Ca2+ channels. Nature 411, 484–489. [DOI] [PubMed] [Google Scholar]
- Dick IE, Tadross MR, Liang H, Tay LH, Yang W, and Yue DT (2008). A modular switch for spatial Ca2+ selectivity in the calmodulin regulation of CaV channels. Nature 451, 830–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dick IE, Joshi-Mukherjee R, Yang W, and Yue DT (2016). Arrhythmogenesis in Timothy syndrome is associated with defects in Ca(2+)-dependent inactivation. Nat. Commun 7, 10370. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doering CJ, Hamid J, Simms B, McRory JE, and Zamponi GW (2005). Cav1.4 encodes a calcium channel with low open probability and unitary conductance. Biophys. J 89, 3042–3048. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dolphin AC (2016). Voltage-gated calcium channels and their auxiliary subunits: physiology and pathophysiology and pharmacology. J. Physiol 594, 5369–5390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dunlap K (2007). Calcium channels are models of self-control. J. Gen. Physiol 129, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ellinor PT, Yang J, Sather WA, Zhang JF, and Tsien RW (1995). Ca2+ channel selectivity at a single locus for high-affinity Ca2+ interactions. Neuron 15, 1121–1132. [DOI] [PubMed] [Google Scholar]
- Fallon JL, Halling DB, Hamilton SL, and Quiocho FA (2005). Structure of calmodulin bound to the hydrophobic IQ domain of the cardiac Ca(v)1.2 calcium channel. Structure 13, 1881–1886. [DOI] [PubMed] [Google Scholar]
- Fallon JL, Baker MR, Xiong L, Loy RE, Yang G, Dirksen RT, Hamilton SL, and Quiocho FA (2009). Crystal structure of dimeric cardiac L-type calcium channel regulatory domains bridged by Ca2+* calmodulins. Proc. Natl. Acad. Sci. USA 106, 5135–5140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Findeisen F, and Minor DL Jr. (2009). Disruption of the IS6-AID linker affects voltage-gated calcium channel inactivation and facilitation. J. Gen. Physiol 133, 327–343. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halling DB, Aracena-Parks P, and Hamilton SL (2006). Regulation of voltage-gated Ca2+ channels by calmodulin. Sci. STKE 2006, er1. [DOI] [PubMed] [Google Scholar]
- Hamill OP, Marty A, Neher E, Sakmann B, and Sigworth FJ (1981). Improved patch-clamp techniques for high-resolution current recording from cells and cell-free membrane patches. Pflugers Arch. 391, 85–100. [DOI] [PubMed] [Google Scholar]
- Hess P, Lansman JB, and Tsien RW (1984). Different modes of Ca channel gating behaviour favoured by dihydropyridine Ca agonists and antagonists. Nature 311, 538–544. [DOI] [PubMed] [Google Scholar]
- Huang H, Tan BZ, Shen Y, Tao J, Jiang F, Sung YY, Ng CK, Raida M, Köhr G, Higuchi M, et al. (2012). RNA editing of the IQ domain in Ca(v)1.3 channels modulates their Ca2+-dependent inactivation. Neuron 73, 304–316. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang H, Yu D, and Soong TW (2013). C-terminal alternative splicing of CaV1.3 channels distinctively modulates their dihydropyridine sensitivity. Mol. Pharmacol 84, 643–653. [DOI] [PubMed] [Google Scholar]
- Imai S, Osawa M, Takeuchi K, and Shimada I (2010). Structural basis underlying the dual gate properties of KcsA. Proc. Natl. Acad. Sci. USA 107, 6216–6221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ivanina T, Blumenstein Y, Shistik E, Barzilai R, and Dascal N (2000). Modulation of L-type Ca2+ channels by gbeta gamma and calmodulin via interactions with N and C termini of alpha 1C. J. Biol. Chem 275, 39846–39854. [DOI] [PubMed] [Google Scholar]
- Kim MS, Morii T, Sun LX, Imoto K, and Mori Y (1993). Structural determinants of ion selectivity in brain calcium channel. FEBS Lett. 318, 145–148. [DOI] [PubMed] [Google Scholar]
- Kim J, Ghosh S, Nunziato DA, and Pitt GS (2004). Identification of the components controlling inactivation of voltage-gated Ca2+ channels. Neuron 41, 745–754. [DOI] [PubMed] [Google Scholar]
- Kim EY, Rumpf CH, Fujiwara Y, Cooley ES, Van Petegem F, and Minor DL Jr. (2008). Structures of CaV2 Ca2+/CaM-IQ domain complexes reveal binding modes that underlie calcium-dependent inactivation and facilitation. Structure 16, 1455–1467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Findeisen F, and Minor DL Jr. (2010). Calmodulin interactions with CaV1 and CaV2 voltage-gated calcium channel IQ domains. In Handbook of Metalloproteins, Messerschmidt A, ed. (John Wiley; ). [Google Scholar]
- Koishi R, Xu H, Ren D, Navarro B, Spiller BW, Shi Q, and Clapham DE (2004). A superfamily of voltage-gated sodium channels in bacteria. J. Biol. Chem 279, 9532–9538. [DOI] [PubMed] [Google Scholar]
- Lee A, Wong ST, Gallagher D, Li B, Storm DR, Scheuer T, and Catterall WA (1999). Ca2+/calmodulin binds to and modulates P/Q-type calcium channels. Nature 399, 155–159. [DOI] [PubMed] [Google Scholar]
- Lee A, Zhou H, Scheuer T, and Catterall WA (2003). Molecular determinants of Ca2+/calmodulin-dependent regulation of Cav2.1 channels. Proc. Natl. Acad. Sci. USA 100, 16059–16064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang H, DeMaria CD, Erickson MG, Mori MX, Alseikhan BA, and Yue DT (2003). Unified mechanisms of Ca2+ regulation across the Ca2+ channel family. Neuron 39, 951–960. [DOI] [PubMed] [Google Scholar]
- Limpitikul WB, Dick IE, Joshi-Mukherjee R, Overgaard MT, George AL Jr., and Yue DT (2014). Calmodulin mutations associated with long QT syndrome prevent inactivation of cardiac L-type Ca(2+) currents and promote proarrhythmic behavior in ventricular myocytes. J. Mol. Cell. Cardiol 74, 115–124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limpitikul WB, Dick IE, Ben-Johny M, and Yue DT (2016). An autism-associated mutation in CaV1.3 channels has opposing effects on voltage-and Ca(2+)-dependent regulation. Sci. Rep 6, 27235. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Limpitikul WB, Dick IE, Tester DJ, Boczek NJ, Limphong P, Yang W, Choi MH, Babich J, DiSilvestre D, Kanter RJ, et al. (2017). A precision medicine approach to the rescue of function on malignant calmodulinopathic long-QT syndrome. Circ. Res 120, 39–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Jurman ME, and Yellen G (1996). Dynamic rearrangement of the outer mouth of a K+ channel during gating. Neuron 16, 859–867. [DOI] [PubMed] [Google Scholar]
- Lolicato M, Arrigoni C, Mori T, Sekioka Y, Bryant C, Clark KA, and Minor DL Jr. (2017). K2P2.1 (TREK-1)-activator complexes reveal a cryptic selectivity filter binding site. Nature 547, 364–368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- López-Barneo J, Hoshi T, Heinemann SH, and Aldrich RW (1993). Effects of external cations and mutations in the pore region on C-type inactivation of Shaker potassium channels. Receptors Channels 1, 61–71. [PubMed] [Google Scholar]
- Mahajan A, Sato D, Shiferaw Y, Baher A, Xie LH, Peralta R, Olcese R, Garfinkel A, Qu Z, and Weiss JN (2008). Modifying L-type calcium current kinetics: consequences for cardiac excitation and arrhythmia dynamics. Biophys. J 94, 411–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mikala G, Bahinski A, Yatani A, Tang S, and Schwartz A (1993). Differential contribution by conserved glutamate residues to an ion-selectivity site in the L-type Ca2+ channel pore. FEBS Lett. 335, 265–269. [DOI] [PubMed] [Google Scholar]
- Minor DL Jr., and Findeisen F (2010). Progress in the structural understanding of voltage-gated calcium channel (CaV) function and modulation. Channels (Austin) 4, 459–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mori MX, Vander Kooi CW, Leahy DJ, and Yue DT (2008). Crystal structure of the CaV2 IQ domain in complex with Ca2+/calmodulin: high-resolution mechanistic implications for channel regulation by Ca2+. Structure 16, 607–620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morotti S, Grandi E, Summa A, Ginsburg KS, and Bers DM (2012). Theoretical study of L-type Ca(2+) current inactivation kinetics during action potential repolarization and early afterdepolarizations. J. Physiol 590, 4465–4481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nanou E, and Catterall WA (2018). Calcium channels, synaptic plasticity, and neuropsychiatric disease. Neuron 98, 466–481. [DOI] [PubMed] [Google Scholar]
- Noceti F, Olcese R, Qin N, Zhou J, and Stefani E (1998). Effect of bay K 8644 (-) and the beta2a subunit on Ca2+-dependent inactivation in alpha1C Ca2+ channels. J. Gen. Physiol 111, 463–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ogielska EM, and Aldrich RW (1999). Functional consequences of a decreased potassium affinity in a potassium channel pore. Ion interactions and C-type inactivation. J. Gen. Physiol 113, 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olcese R, Qin N, Schneider T, Neely A, Wei X, Stefani E, and Birnbaumer L (1994). The amino terminus of a calcium channel beta subunit sets rates of channel inactivation independently of the subunit’s effect on activation. Neuron 13, 1433–1438. [DOI] [PubMed] [Google Scholar]
- Opatowsky Y, Chen CC, Campbell KP, and Hirsch JA (2004). Structural analysis of the voltage-dependent calcium channel beta subunit functional core and its complex with the alpha 1 interaction domain. Neuron 42, 387–399. [DOI] [PubMed] [Google Scholar]
- Panyi G, and Deutsch C (2006). Cross talk between activation and slow inactivation gates of Shaker potassium channels. J. Gen. Physiol 128, 547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent L, and Gopalakrishnan M (1995). Glutamate substitution in repeat IV alters divalent and monovalent cation permeation in the heart Ca2+ channel. Biophys. J 69, 1801–1813. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlov E, Bladen C, Winkfein R, Diao C, Dhaliwal P, and French RJ (2005). The pore, not cytoplasmic domains, underlies inactivation in a prokaryotic sodium channel. Biophys. J 89, 232–242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J, and Minor DL Jr. (2015). Bacterial voltage-gated sodium channels (BacNa(V)s) from the soil, sea, and salt lakes enlighten molecular mechanisms of electrical signaling and pharmacology in the brain and heart. J. Mol. Biol 427, 3–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J, Scheuer T, Zheng N, and Catterall WA (2011). The crystal structure of a voltage-gated sodium channel. Nature 475, 353–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Payandeh J, Gamal El-Din TM, Scheuer T, Zheng N, and Catterall WA (2012). Crystal structure of a voltage-gated sodium channel in two potentially inactivated states. Nature 486, 135–139. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peters CJ, Fedida D, and Accili EA (2013). Allosteric coupling of the inner activation gate to the outer pore of a potassium channel. Sci. Rep 3, 3025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peterson BZ, DeMaria CD, Adelman JP, and Yue DT (1999). Calmodulin is the Ca2+ sensor for Ca2+ -dependent inactivation of L-type calcium channels. Neuron 22, 549–558. [DOI] [PubMed] [Google Scholar]
- Piechotta PL, Rapedius M, Stansfeld PJ, Bollepalli MK, Ehrlich G, Andres-Enguix I, Fritzenschaft H, Decher N, Sansom MS, Tucker SJ, and Baukrowitz T (2011). The pore structure and gating mechanism of K2P channels. EMBO J. 30, 3607–3619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raybaud A, Dodier Y, Bissonnette P, Simoes M, Bichet DG, Sauvé R, and Parent L (2006). The role of the GX9GX3G motif in the gating of high voltage-activated Ca2+ channels. J. Biol. Chem 281, 39424–39436. [DOI] [PubMed] [Google Scholar]
- Ren D, Navarro B, Xu H, Yue L, Shi Q, and Clapham DE (2001). A prokaryotic voltage-gated sodium channel. Science 294, 2372–2375. [DOI] [PubMed] [Google Scholar]
- Schewe M, Nematian-Ardestani E, Sun H, Musinszki M, Cordeiro S, Bucci G, de Groot BL, Tucker SJ, Rapedius M, and Baukrowitz T (2016). A non-canonical voltage-sensing mechanism controls gating in K2P K(+) channels. Cell 164, 937–949. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaya D, Kreir M, Robbins RA, Wong S, Hammon J, Brüggemann A, and Minor DL Jr. (2011). Voltage-gated sodium channel (NaV) protein dissection creates a set of functional pore-only proteins. Proc. Natl. Acad. Sci. USA 108, 12313–12318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaya D, Findeisen F, Abderemane-Ali F, Arrigoni C, Wong S, Nurva SR, Loussouarn G, and Minor DL Jr. (2014). Structure of a prokaryotic sodium channel pore reveals essential gating elements and an outer ion binding site common to eukaryotic channels. J. Mol. Biol 426, 467–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shen Y, Yu D, Hiel H, Liao P, Yue DT, Fuchs PA, and Soong TW (2006). Alternative splicing of the Ca(v)1.3 channel IQ domain, a molecular switch for Ca2+-dependent inactivation within auditory hair cells. J. Neurosci 26, 10690–10699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simms BA, and Zamponi GW (2014). Neuronal voltage-gated calcium channels: structure, function, and dysfunction. Neuron 82, 24–45. [DOI] [PubMed] [Google Scholar]
- Singh A, Hamedinger D, Hoda JC, Gebhart M, Koschak A, Romanin C, and Striessnig J (2006). C-terminal modulator controls Ca2+-dependent gating of Ca(v)1.4 L-type Ca2+ channels. Nat. Neurosci 9, 1108–1116. [DOI] [PubMed] [Google Scholar]
- Singh A, Gebhart M, Fritsch R, Sinnegger-Brauns MJ, Poggiani C, Hoda JC, Engel J, Romanin C, Striessnig J, and Koschak A (2008). Modulation of voltage- and Ca2+-dependent gating of CaV1.3 L-type calcium channels by alternative splicing of a C-terminal regulatory domain. J. Biol. Chem 283, 20733–20744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Sharpe LM, Decher N, Kumar P, Bloise R, Napolitano C, Schwartz PJ, Joseph RM, Condouris K, et al. (2004). Ca(V)1.2 calcium channel dysfunction causes a multisystem disorder including arrhythmia and autism. Cell 119, 19–31. [DOI] [PubMed] [Google Scholar]
- Splawski I, Timothy KW, Decher N, Kumar P, Sachse FB, Beggs AH, Sanguinetti MC, and Keating MT (2005). Severe arrhythmia disorder caused by cardiac L-type calcium channel mutations. Proc. Natl. Acad. Sci. USA 102, 8089–8096, discussion 8086–8088. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stea A, Tomlinson WJ, Soong TW, Bourinet E, Dubel SJ, Vincent SR, and Snutch TP (1994). Localization and functional properties of a rat brain alpha 1A calcium channel reflect similarities to neuronal Q- and P-type channels. Proc. Natl. Acad. Sci. USA 91, 10576–10580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steinberg X, Kasimova MA, Cabezas-Bratesco D, Galpin JD, Ladronde-Guevara E, Villa F, Carnevale V, Islas L, Ahern CA, and Brauchi SE (2017). Conformational dynamics in TRPV1 channels reported by an encoded coumarin amino acid. eLife 6. Published online December 5, 2017. 10.7554/eLife.28626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stotz SC, Jarvis SE, and Zamponi GW (2004). Functional roles of cytoplasmic loops and pore lining transmembrane helices in the voltage-dependent inactivation of HVA calcium channels. J. Physiol 554, 263–273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadross MR, and Yue DT (2010). Systematic mapping of the state dependence of voltage- and Ca2+-dependent inactivation using simple open-channel measurements. J. Gen. Physiol 135, 217–227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadross MR, Dick IE, and Yue DT (2008). Mechanism of local and global Ca2+ sensing by calmodulin in complex with a Ca2+ channel. Cell 133, 1228–1240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadross MR, Ben Johny M, and Yue DT (2010). Molecular endpoints of Ca2+/calmodulin- and voltage-dependent inactivation of Ca(v)1.3 channels. J. Gen. Physiol 135, 197–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tadross MR, Tsien RW, and Yue DT (2013). Ca2+ channel nanodomains boost local Ca2+ amplitude. Proc. Natl. Acad. Sci. USA 110, 15794–15799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan BZ, Jiang F, Tan MY, Yu D, Huang H, Shen Y, and Soong TW (2011). Functional characterization of alternative splicing in the C terminus of L-type CaV1.3 channels. J. Biol. Chem 286, 42725–42735. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F, Clark KA, Chatelain FC, and Minor DL Jr. (2004). Structure of a complex between a voltage-gated calcium channel beta-subunit and an alpha-subunit domain. Nature 429, 671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Van Petegem F, Chatelain FC, and Minor DL Jr. (2005). Insights into voltage-gated calcium channel regulation from the structure of the CaV1.2 IQ domain-Ca2+/calmodulin complex. Nat. Struct. Mol. Biol 12, 1108–1115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu J, Yan Z, Li Z, Yan C, Lu S, Dong M, and Yan N (2015). Structure of the voltage-gated calcium channel Cav1.1 complex. Science 350, aad2395. [DOI] [PubMed] [Google Scholar]
- Wu J, Yan Z, Li Z, Qian X, Lu S, Dong M, Zhou Q, and Yan N (2016). Structure of the voltage-gated calcium channel Ca(v)1.1 at 3.6 Å resolution. Nature 537, 191–196. [DOI] [PubMed] [Google Scholar]
- Xu W, and Lipscombe D (2001). Neuronal Ca(V)1.3alpha(1) L-type channels activate at relatively hyperpolarized membrane potentials and are incompletely inhibited by dihydropyridines. J. Neurosci 21, 5944–5951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang J, Ellinor PT, Sather WA, Zhang JF, and Tsien RW (1993). Molecular determinants of Ca2+ selectivity and ion permeation in L-type Ca2+ channels. Nature 366, 158–161. [DOI] [PubMed] [Google Scholar]
- Yang G, Shi Y, Yu J, Li Y, Yu L, Welling A, Hofmann F, Striessnig J, Juntti-Berggren L, Berggren PO, and Yang SN (2015). CaV1.2 and CaV1.3 channel hyperactivation in mouse islet β cells exposed to type 1 diabetic serum. Cell. Mol. Life Sci 72, 1197–1207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu FH, Yarov-Yarovoy V, Gutman GA, and Catterall WA (2005). Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol. Rev 57, 387–395. [DOI] [PubMed] [Google Scholar]
- Yue L, Navarro B, Ren D, Ramos A, and Clapham DE (2002). The cation selectivity filter of the bacterial sodium channel, NaChBac. J. Gen. Physiol 120, 845–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zamponi GW, Striessnig J, Koschak A, and Dolphin AC (2015). The physiology, pathology, and pharmacology of voltage-gated calcium channels and their future therapeutic potential. Pharmacol. Rev 67, 821–870. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zong S, Zhou J, and Tanabe T (1994). Molecular determinants of calcium-dependent inactivation in cardiac L-type calcium channels. Biochem. Biophys. Res. Commun 201, 1117–1123. [DOI] [PubMed] [Google Scholar]
- Zühlke RD, Pitt GS, Deisseroth K, Tsien RW, and Reuter H (1999). Calmodulin supports both inactivation and facilitation of L-type calcium channels. Nature 399, 159–162. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.