

Abstract
Genome-wide association studies (GWAS) have made clear that single-nucleotide variants (SNVs) that occur at multiple locations across the genome can be associated with a specific condition or trait, also known as a phenotype. Phenome-wide association studies (PheWAS) invert the idea of a GWAS by searching for phenotypes associated with specific SNVs across the range of thousands of human phenotypes, or the “phenome” (Figure). Analogous to GWAS, PheWAS have shown that specific genetic variations may be associated with multiple conditions and traits.
How It Works
The key requirement for PheWAS is a data set that includes a broad range of phenotypes ascertained in large numbers of patients. The technique was developed with electronic health records (EHRs) linked to DNA databases1–4 to find phenotypic associations with target SNVs. More recently, PheWAS has been used to analyze other types of data sets that include genetic information linked with extensive phenotype data. Example data sets include the UK Biobank, an epidemiologic cohort of 500 000 individuals with dense phenotypic data obtained by standardized questionnaires and other phenotyping methods (including incorporation of EHR data), and data held by the genetic testing company 23andMe, in which phenotype information has been solicited by questionnaires from more than 10 million participants.5
Initial validation of the PheWAS method was achieved through replication of known GWAS results by working backward from SNVs previously associated with a trait to determine whether those SNVs could be shown to be associated with the same phenotype among populations of individuals who had had phenotyping for many traits and conditions. For example, an early study conducted in the Electronic Medical Records and Genomics (eMERGE) network was a GWAS that included a population of 1317 individuals with hypothyroidism and 5053 controls without it. The study demonstrated significant associations between variants near FOXE1 and hypothyroidism (odds ratio [OR], 0.74 [95% CI, 0.67–0.82]; P = 3.96 × 10−9). A subsequent PheWAS of 13 617 individuals with greater than 200 000 patient-years of billing data identified statistically significant associations between the FOXE1 variants and hypothyroidism (n = 2108; OR, 0.76 [95% CI, 0.70–0.81];P = 2.7 × 10−13), as well as other thyroid-related phenotypes.3,4
As another example of validation of PheWAS, a study from the Emerge network2 examined the relationships among 3144 SNVs associated with human phenotypes in previous GWAS and 1358 phenotypes ascertained in EHRs that included 13 835 individuals and replicated 66% of previously reported GWAS results. This study also showed that the target SNVs could have associations with multiple phenotypes. For example, variants in IRF4, previously associated with hair and eye color, were significantly associated with actinic keratosis, nonmelanoma skin cancers, and susceptibility to sunburn. Discovery of previously unrecognized shared relationships may improve current understanding of shared mechanisms across diseases.2,6
Subsequent to the initial studies validating the approach, PheWAS had been used to validate the importance of genes studied in animal models for human disease: variants in the transcription factor ETV1, previously implicated in development of the cardiac conduction system in the mouse, were significantly associated with conduction system abnormalities in 26 256 adults of European ancestry and 3269 of African ancestry.7 Additionally, PheWAS has been applied to the drug development process. A study examined 25 SNVs in 19 genes encoding proteins that are known drug targets in up to 697 815 individuals with phenotyping for a large number of traits.5 The analysis replicated 75% of known GWAS associations with these 19 genes and identified new associations in 9. These new associations may identify potential adverse drug reactions or new clinical applications for existing drugs.
Although initial PheWAS explored associations among single SNVs and multiple phenotypes, any independent variable, including laboratory values, biomarkers, or even a disease or symptom of interest, can serve as the starting point for a PheWAS study. For example, although variability in gene expression is not conventionally captured in EHRs or other large data sets, it is possible to use combinations of SNVs to predict gene expression and to then use PheWAS to search for associations with predicted gene expression across human phenotypes. This approach, termed PrediXcan, has been used to identify statistically significant and previously unanticipated associations between predicted altered gene expression and human disease.8 Another example developed a multi-SNV predictor of thyroid-stimulating hormone (TSH) level from previous GWAS, and a PheWAS that included 37 154 individuals with dense genotyping in an EHR (and excluded patients with known thyroid disease) identified a significant association between low predicted TSH and hyperthyroidism and atrial fibrillation risk, supporting the idea that sub-clinical thyroid disease predisposes to the latter.9
Important Care Considerations
Researchers using PheWAS should be cautious about false-positive findings. Bonferroni correction for thresholds for statistical significance can be used to adjust for the number of phenotypes tested in a PheWAS. Novel associations should be validated by replication in independent sets or by entirely different human, animal, or cellular studies. As with GWAS, study results may be ancestry dependent (especially if target SNVs vary by ancestry) and may be confounded by unmeasured variables such as environmental influences on phenotypes.
PheWAS relies on high-throughput phenotype definitions that are prone to misclassification among cases and controls, which increases the chance of type 2 error. Many PheWAS studies rely on billing codes, which introduce potential errors and variability across sites. The potential for false-negative results makes it difficult to interpret a “null PheWAS” (ie, one that does not yield any significant associations after correction). Current implementations of PheWAS have not yet taken advantage of the full richness of data sets available in EHRs or in epidemiologic cohorts. Thus, for example, laboratory data have generally not been incorporated into PheWAS phenotypes.
Potential Value
PheWAS has been developed during the last decade as a research tool. One application is to validate GWAS results (as described earlier). Another is to better understand genetic contributions to human disease, and to begin to identify shared mechanisms across diseases. The approach provides validation for important biologic findings and may have an important role in drug development and repurposing. In addition, PheWAS can be applied to all variants available in a data set, producing a PheWAS × GWAS catalog of associations for exploratory analyses. PheWeb10 is a tool to visualize these large data sets, and presents PheWAS × GWAS catalogs for UK Biobank, the Michigan Genomics Initiative, and the Finnish resource Finnish Metabolic Sequencing (FinMetSeq).
Evidence Base
PheWAS are not yet used in clinical care. The evidence base for research use of PheWAS is literature derived and, in parallel to GWAS, it is clear that large numbers of study participants enable the identification of statistically robust, and thus likely more biologically compelling, associations.
Conclusions
PheWAS is a powerful technique that could help facilitate new insights about how genetic variation relates to phenotypic variation across human populations. As increasing numbers of researchers make use of PheWAS to drive scientific discovery, the hope is that these discoveries will translate to better understanding of genetic contributions to various diseases and long-term improvement in clinical care.
Figure. Genome-Wide Association Study (GWAS) Compared With Phenome-Wide Association Study (PheWAS).
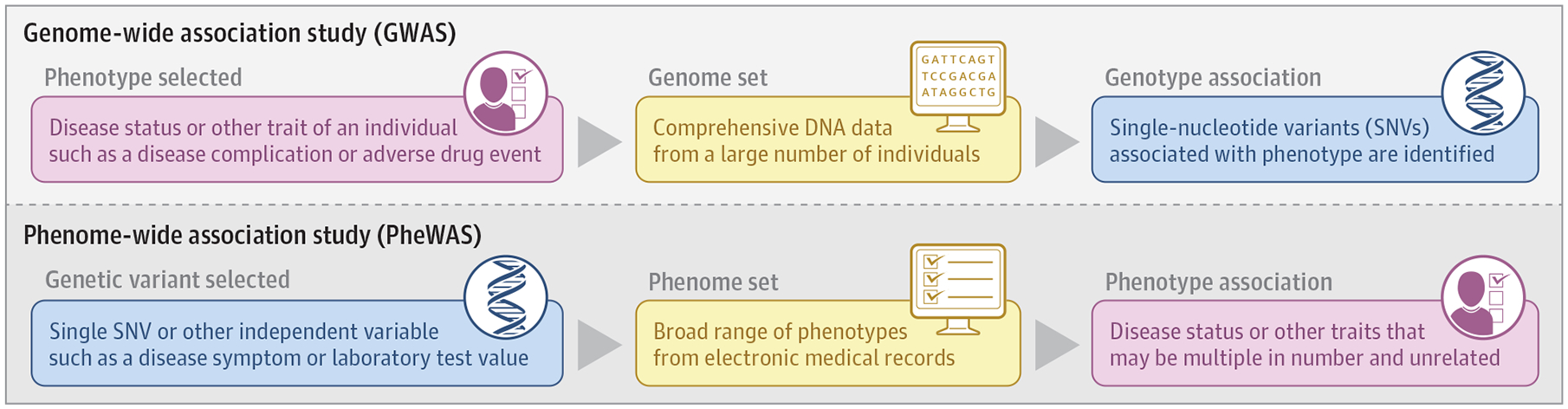
A GWAS (top) contrasted with a PheWAS (bottom). A GWAS starts with families or populations in which individuals have been assigned affected or unaffected status for a disease or other trait, such as a complication of a disease or an adverse outcome during drug treatment, and searches for associated genetic variants. A PheWAS starts with a genetic variant and searches across a set of curated human phenotypes (the “phenome”) to identify associated phenotypes. The “input function” for the PheWAS can be a single genetic variant or sets of variants or other traits.
Funding/Support:
Supported in part by grants from the National Institutes of Health (NIH): P50 GM115305, U01 HG08672, and U01HG011181.
Conflict of Interest Disclosures:
Ms Bastarache and Drs Denny and Roden reported receiving licensing fees for phenome-wide association studies from Nashville Biosciences, an electronic health record data analysis company wholly owned by Vanderbilt University Medical Center. This fee applies only to Nashville Biosciences’ use of phenome-wide association studies to analyze the Vanderbilt biobank BioVU. Dr Roden also reported serving on the Nashville Biosciences scientific advisory board but does not receive any compensation in this role.
Role of the Funder/Sponsor:
Granting agencies had no role in the preparation, review, or approval of the manuscript; and decision to submit the manuscript for publication.
Footnotes
Additional Information: The phenome-wide association study method is in the public domain.
REFERENCES
- 1.Denny JC, Ritchie MD, Basford MA, et al. PheWAS: demonstrating the feasibility of a phenome-wide scan to discover gene-disease associations. Bioinformatics. 2010;26(9):1205–1210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Denny JC, Bastarache L, Ritchie MD, et al. Systematic comparison of phenome-wide association study of electronic medical record data and genome-wide association study data. Nat Biotechnol. 2013;31(12):1102–1110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Bush WS, Oetjens MT, Crawford DC. Unravelling the human genome-phenome relationship using phenome-wide association studies. Nat Rev Genet. 2016;17(3):129–145. doi: 10.1038/nrg.2015.36 [DOI] [PubMed] [Google Scholar]
- 4.Denny JC, Bastarache L, Roden DM. Phenome-wide association studies as a tool to advance precision medicine. Annu Rev Genomics Hum Genet. 2016;17:353–373. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Diogo D, Tian C, Franklin CS, et al. Phenome-wide association studies across large population cohorts support drug target validation. Nat Commun. 2018;9(1):4285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Verma A, Bang L, Miller JE, et al. ; DiscovEHR Collaboration. Human-disease phenotype map derived from PheWAS across 38,682 individuals. Am J Hum Genet. 2019;104(1):55–64. doi: 10.1016/j.ajhg.2018.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shekhar A, Lin X, Liu FY, et al. Transcription factor ETV1 is essential for rapid conduction in the heart. J Clin Invest. 2016;126(12):4444–4459. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Unlu G, Gamazon ER, Qi X, et al. GRIK5 genetically regulated expression associated with eye and vascular phenomes: discovery through iteration among biobanks, electronic health records, and zebrafish. Am J Hum Genet. 2019;104(3):503–519. doi: 10.1016/j.ajhg.2019.01.017 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Salem JE, Shoemaker MB, Bastarache L, et al. Association of thyroid function genetic predictors with atrial fibrillation: a phenome-wide association study and inverse-variance weighted average meta-analysis. JAMA Cardiol. 2019;4(2):136–143. doi: 10.1001/jamacardio.2018.4615 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Gagliano Taliun SA, VandeHaar P, Boughton AP, et al. Exploring and visualizing large-scale genetic associations by using PheWeb. Nat Genet. 2020;52(6):550–552. doi: 10.1038/s41588-020-0622-5 [DOI] [PMC free article] [PubMed] [Google Scholar]