

Abstract
Bacterial dysbiosis is associated with Crohn’s disease (CD), a chronic intestinal inflammatory disorder thought to result from an abnormal immune response against intestinal bacteria in genetically susceptible individuals. However, it is unclear whether dysbiosis is a cause or consequence of intestinal inflammation and whether overall dysbiosis or specific bacteria trigger the disease. Here, we show that the combined deficiency of NOD2 and phagocyte NADPH oxidase, two CD susceptibility genes, triggers early-onset spontaneous TH1-type intestinal inflammation in mice with the pathological hallmarks of CD. Disease was induced by Mucispirillum schaedleri, a Gram-negative mucus-dwelling anaerobe. NOD2 and CYBB deficiencies led to marked accumulation of Mucispirillum, which was associated with impaired neutrophil recruitment and killing of the bacterium by luminal neutrophils. Maternal immunoglobulins against Mucispirillum protected mutant mice from disease during breastfeeding. Our results indicate that a specific intestinal microbe triggers CD-like disease in the presence of impaired clearance of the bacterium by innate immunity.
INTRODUCTION
Crohn’s disease (CD), one of the major forms of inflammatory bowel disease (IBD), is a complex disorder marked by chronic relapsing inflammation driven by CD4+ T helper 1 (TH1) cells (1, 2). Although CD can affect any part of the gastrointestinal tract, more than 60% of patients have colonic involvement, 20% have isolated colitis, and the remaining patients have both ileal and colonic disease (3). The prevalence of adult-onset IBD has plateaued in industrialized countries, but recent population-based studies from North America and Europe suggest a rapid increase in pediatric-onset IBD, particularly in children younger than 10 years of age (4–6). Lesions in very-early-onset IBD (VEOIBD), a subgroup of pediatric IBD that includes patients experiencing symptoms at an age less than 6 years, are often restricted to the colon (7). Thus, determining the factors that regulate host-microbial interactions and promote the development of colitis is of great clinical and scientific interest.
The pathogenesis of CD is complex and still poorly understood. CD is believed to be caused by a confluence of genetic and environmental factors that alter gut homeostasis, triggering disease in genetically susceptible individuals (8). Genetic studies have identified more than 200 susceptibility loci for IBD, roughly 40 of which are unique to CD (9), and many of them regulate host immune responses to bacteria (10). The most influential gene for CD susceptibility identified so far is nucleotide-binding oligomerization domain–containing protein 2 (NOD2) (11, 12). NOD2, a member of the intracellular NOD-like receptor family, is activated by muramyl dipeptide (MDP), a conserved motif in peptidoglycan from both Gram-negative and Gram-positive bacteria (13, 14). In response to MDP, NOD2 induces antimicrobial and pro-inflammatory responses that are impaired in the three common CD-associated NOD2 variants (R702W, G908R, and L1007insC) (14, 15). However, the great majority of individuals homozygous or compound heterozygous for NOD2 variants do not develop CD (16). Consistently, no spontaneous intestinal inflammation occurs in Nod2-deficient mice housed under specific pathogen–free (SPF) conditions (17, 18). Overall, these observations suggest that impaired NOD2 function leading to disease requires the presence of additional genetic hits and/or the presence of specific disease-causing microbes. CD is also associated with mutations in genes encoding components of the phagocyte NADPH [reduced form of nicotinamide adenine dinucleotide phosphate (NADP+)] oxidase complex that kill bacteria via oxidative burst (19, 20). Patients with NADPH gene mutations are more likely to have perianal disease and complications than children with CD without these mutations (21). In addition, up to 40% of patients with chronic granulomatous disease, a condition caused by homozygous mutations in CYBB/NOX2 or related NADPH subunits, develop CD-like disease in the colon (22). Thus, understanding how NOD2 and NADPH oxidase promote the development of CD is critical for understanding disease pathogenesis.
Patients with CD harbor a dysbiotic microbiota (23, 24). Consistent features of dysbiosis in CD are decreased community diversity and a shift in bacterial taxa, including a depletion of members of the phyla Firmicutes and Bacteroidetes and an expansion of Proteobacteria (25–28). A recent cohort study in pediatric treatment-naïve patients with CD reported that the extent of intestinal dysbiosis correlates with disease severity (29). However, it is unclear whether CD-associated dysbiosis plays a causal role or is secondary to inflammation. Despite these associations, searches for organisms causing CD have not led to the identification of a specific pathogen/pathobiont. One problem with identifying microbes that directly contribute to CD pathogenesis is that microbiota studies are typically performed in inflamed tissue, and thus, detection of rare causative microbes could be obscured by inflammation-driven dysbiosis. In this context, several studies have shown that defined communities of microbes or single organisms can trigger intestinal inflammation in genetically susceptible animal models (30–32). However, most or all of these findings rely on the identification of pathogenic bacteria in the inflamed intestine of susceptible mice or monoassociation of colitogenic microbes in germ-free animals. Thus, it is still unclear whether potentially pathogenic microbes can be identified before the development of intestinal inflammation. In addition, a microbial community or specific bacteria that can trigger disease with pathology comparable to that observed in humans in the presence of CD-associated mutations, such as NOD2, have yet to be identified. Here, we show that the combined deficiency of two CD-related genes (NOD2 and phagocyte NADPH oxidase/CYBB) triggers spontaneous colitis with the classical pathological hall-marks of CD. The development of CD-like disease is triggered by the presence of Mucispirillum schaedleri, a mucus-dwelling Gram-negative symbiotic bacterium. Mechanistically, the absence of NOD2 and CYBB led to a selective luminal and mucosal accumulation of Mucispirillum that was associated with impaired recruitment of neutrophils and killing of the bacterium by neutrophils.
RESULTS
Doubly deficient Nod2/Cybb mice develop spontaneous CD-like colitis when exposed to a specific microbiota
Defective NOD2 function is not sufficient to trigger CD (17, 18, 33). To assess whether impaired killing of bacteria can promote intestinal inflammation in the absence of Nod2, we crossed Nod2−/− mice with mutant mice deficient in CYBB (gp91phox) that clear bacteria poorly due to a profound defect in phagocyte oxidative burst generation (34). Because the microbiota has been linked to CD (23, 24), we fostered newborn wild-type (WT), Nod2−/−, Cybb−/−, and doubly deficient Nod2−/−Cybb−/− (DKO) mice to C57BL/6 mice purchased from either Taconic Biosciences (Tac) or the Jackson Laboratory (Jax), known to harbor different microbiota (Fig. 1A) (35). Unlike DKO mice fostered to Jax dams (Jax-DKO), both male and female DKO mice fostered to Tac dams (Tac-DKO) developed spontaneous colitis, shortly after weaning (~4 weeks of age), which was characterized by skip lesions with crypt architectural distortion, crypt abscesses, transmural inflammatory cell infiltrates, and narrow ulcers, which are pathological hallmarks of CD (Fig. 1, B to D). The CD-like inflammation was restricted to the ceca and colons and involved up to a third of the large intestine in Tac-DKO (Fig. 1C). CD-like inflammation in Tac-DKO mice was associated with marked increase in the fecal levels of lipocalin-2 (Lcn-2) (Fig. 1E), a marker of intestinal inflammation (36). Time course analysis of Lcn-2 levels revealed that Tac-DKO mice experienced a relapse of intestinal inflammation (12 weeks of age) when compared with Jax-DKO animals, albeit to a lesser extent than mice that were recently weaned (Fig. 1F). During the relapse, skip lesions with crypt architectural distortion and mucosal inflammatory cell infiltrates were found in the colons of Tac-DKO mice at older age (12 weeks of age) (fig. S1, A and B), indicating that the colonic lesions in these mice did not become confluent over time. Moreover, intestinal inflammation improves in Tac-DKO mice with aging (16 weeks of age and beyond) (Fig. 1F). No signs of intestinal inflammation were observed in either Nod2−/− or Cybb−/− mice fostered to the same Tac mothers (Fig. 1, C and E). About 30% of Tac-DKO mice did not display overt colitis immediately after weaning (Fig. 1E), but this subgroup of Tac-DKO mice developed intestinal inflammation within 2 to 3 weeks after weaning as assessed by fecal Lcn-2 (fig. S2, A and B). Collectively, these results indicate that the combined deficiency of Nod2 and NADPH oxidase activity triggers CD-like colitis in the presence of a specific microbiota.
Fig. 1. Doubly deficient Nod2/Cybb mice develop earlyonset spontaneous CD-like colitis when exposed to a specific microbiota.
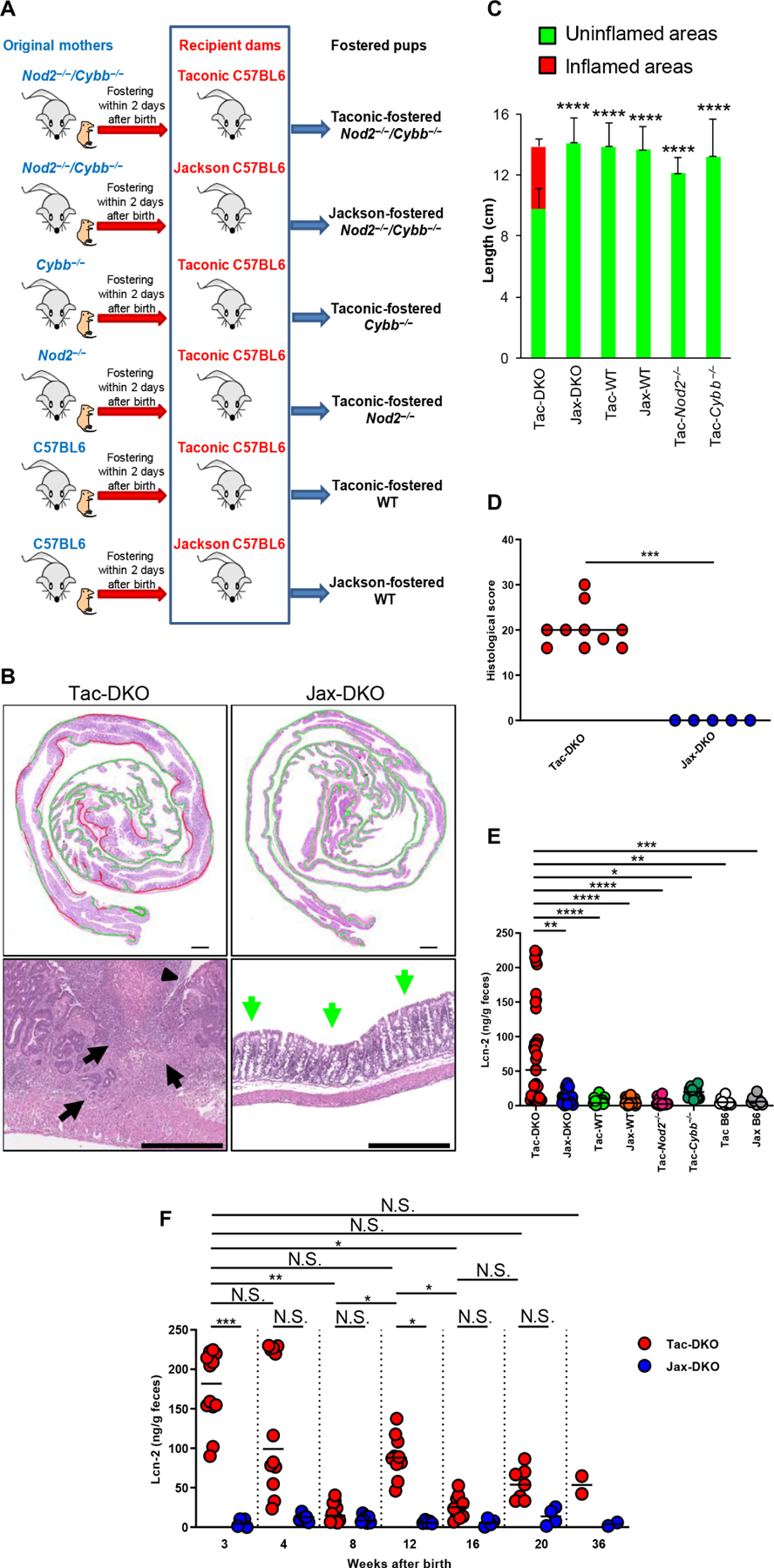
(A) Schematic representation of the experimental design for fostering experiments. WT or mutant pups were fostered to either Jackson (Jax) or Taconic (Tac) dams within 2 days after birth. Six groups of fostered mice were generated: Tac-DKO, Jax-DKO, Tac-WT, Jax-WT, Tac-Nod2−/−, and Tac-Cybb−/−. (B) Representative histology of H&E-stained colonic sections from ~4-week-old Jax-DKO and Tac-DKO mice. Top panels show colonic rolls with inflamed areas (red) and uninflamed areas (green). Scale bars, 1000 μm. Bottom panels show high-powered images. The arrowhead shows a narrow and deep ulcer. Black arrows indicate colonic transmural inflammation and regenerating crypt epithelia in Tac-DKO; green arrows show uninvolved colon in Jax-DKO. Scale bars, 500 μm. (C) Quantification of inflammatory involvement in colons of ~4-week-old fostered mice. Graphs indicate mean ± SD of at least five individual mice. One-way analysis of variance (ANOVA) followed by Tukey’s multiple comparisons test. **** P < 0.0001, data are from four independent experiments. (D) Histopathological scores of cecal and colonic tissues from Tac-DKO (n = 10) and Jax-DKO (n = 5) mice. Bars show median; data are from three independent experiments. Two-tailed Mann-Whitney U test. *** P = 0.0003. (E) Fecal Lcn-2 concentration in Tac-DKO (n = 38), Jax-DKO (n = 24), Tac-WT (n = 20), Jax-WT (n = 19), Tac-Nod2−/− (n = 16), Tac-Cybb−/− (n = 16), Tac C57BL/6 (Tac B6) (n = 10), and Jax C57BL/6 mice (Jax B6) (n = 10). Non-fostered Tac C57BL/6 (Tac B6) and Jax C57BL/6 (Jax B6) mice are included as controls. Bars show median; data are from three independent experiments. (F) Lcn-2 concentration was measured over time in fecal samples from Tac-DKO (n ≤ 12) and Jax-DKO (n ≤ 7) mice. Bars show median; data are pooled from three independent experiments. (E and F) Kruskal-Wallis test followed by Dunn’s post test. * P < 0.05; ** P < 0.01; *** P < 0.001; **** P < 0.0001; N.S., not significant.
CD-like inflammation in Nod2−/−Cybb−/− mice is TNFα dependent and characterized by TH1 immune responses
Because effector T cells, predominantly TH1 cells, play a central role in the pathogenesis of tissue damage in CD (1, 2), we next characterized the mucosal inflammatory infiltrates in this spontaneous model of CD-like colitis. Flow cytometry analysis revealed marked accumulation of CD4+ T cells and neutrophils in the inflamed intestine of Tac-DKO mice when compared with Jax-DKO, Tac-fostered WT (Tac-WT), Tac-fostered Nod2−/− (Tac-Nod2−/−), and Cybb−/− (Tac-Cybb−/−) mice (Fig. 2, A and B). No differences were observed between colitic Tac-DKO and the other mouse groups in the total number of CD8+ T cells, MHCIIhighGr1−CD11b+CD11c− (macrophages), MHCIIhighGr1−CD11b−CD11c+ (dendritic cells), CD11b+Ly6G−Ly6C+ (inflammatory monocytes), and CD19+ (B cells) in the intestinal tissue (fig. S3, A to E), except for B cells and CD8+ T cells that were more abundant in Tac-DKO mice when compared with Jax-DKO animals (fig. S3, D and E). A higher number of lamina propria mononuclear cells (LPMCs) was retrieved from the inflamed intestine of Tac-DKO mice, although the difference was statistically significant only when compared with Jax-fostered animals (fig. S3F). Analysis of LP celĺs revealed increased mRNA and protein levels of tumor necrosis factor–α (TNFα) and enhanced transcript levels of interferon-γ (IFN-γ) (Fig. 2, C to E). Consistently, evaluation of cytokine-producing LP T cells showed an increase in the percentage of IFN-γ–producing CD4+ T cells in colitic Tac-DKO mice when compared with other Tac- and Jax-fostered mice (Fig. 2F and fig. S4G). Additional analyses showed increased expression of interleukin-6 (IL-6) and IL-1β, but not IL-17A or IL-17F, in the inflamed intestine of Tac-DKO mice when compared with other fostered mice (fig. S4, A to E, and fig. S4H). Moreover, increased expression of Foxp3 was detected in the inflamed intestine of Tac-DKO mice when compared with Jax-DKO and Tac-WT animals (fig. S4F). Similar to patients with CD, in which anti-TNF agents are effective for the treatment of disease (37), administration of neutralizing TNFα antibody to colitic Tac-DKO mice inhibited intestinal inflammation (Fig. 2, G and H), which correlated with decreased levels of fecal Lcn-2 (Fig. 2I). Over-all, these results indicate that combined deficiency of Nod2 and Cybb triggers CD-like TH1-associated colitis when exposed to a specific microbiota.
Fig. 2. Doubly deficient Nod2/Cybb mice harboring Taconic microbiota develop TH1-type colitis.
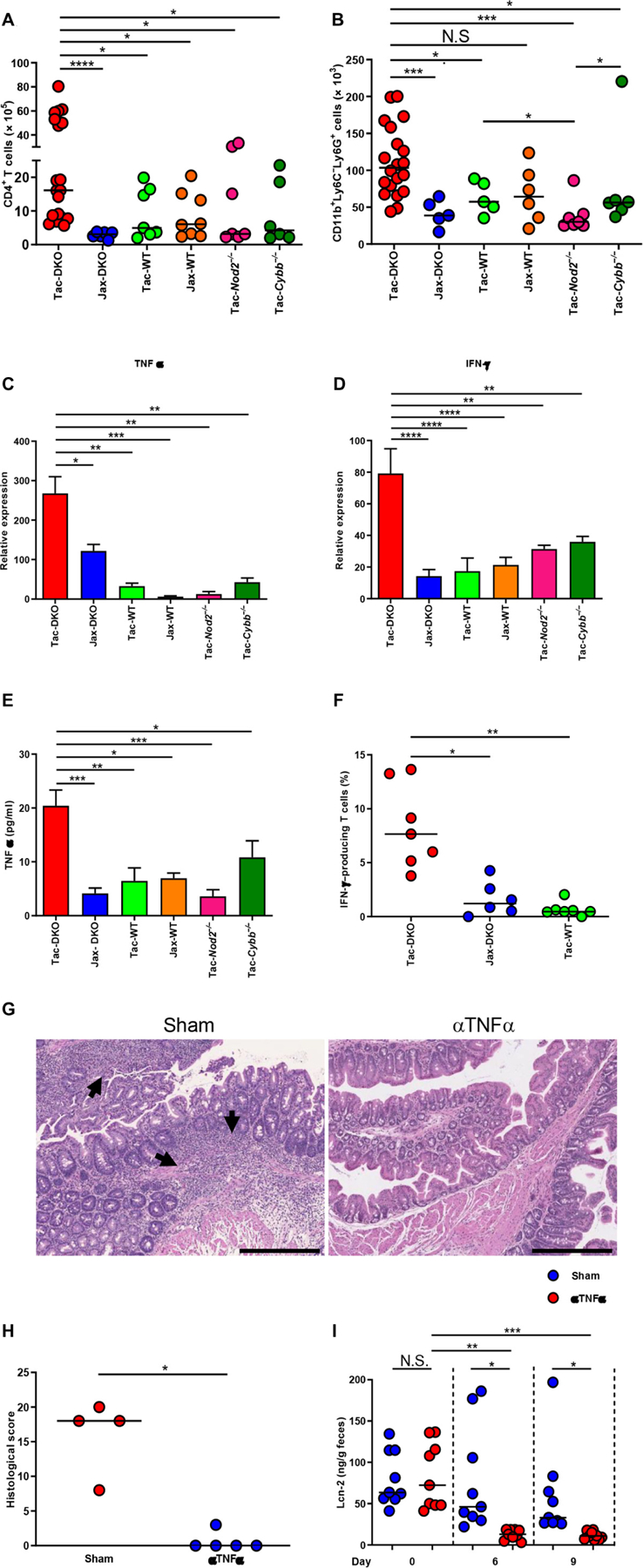
(A) Total number of LP CD4+ T cells from 4-to 6-week-old mice (Tac-DKO, n = 19; Jax-DKO, n = 6; Tac-WT, n = 7; Jax-WT, n = 8; Tac-Nod2−/−, n = 7; and Tac-Cybb−/−, n = 6). (B) Total number of LP neutrophils from 4- to 6-week-old mice (Tac-DKO, n = 20; Jax-DKO, n = 5; Tac-WT, n = 5; Jax-WT, n = 6; Tac-Nod2−/−, n = 7; and Tac-Cybb−/−, n = 6). (A and B) Bars show median; data are from five independent experiments. Kruskal-Wallis test followed by Dunn’s post test. *P < 0.05; ***P < 0.001; ****P < 0.0001. (C and D) LPMCs were isolated from 4- to 6-week-old fostered mice. Gene expression in LPMCs was normalized to glyceraldehyde-3-phosphate dehydrogenase (GAPDH) expression. Data are mean ± SEM of at least five individual mice; data are from four independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001 by one-way ANOVA followed by Tukey’s multiple comparisons test. (E) Total LPMCs from 4- to 6-week-old fostered mice were cultured for 12 hours. TNFα from culture supernatant was measured by ELISA (at least three individual mice each group). Data are mean ± SEM; data are from two independent experiments. *P < 0.05; **P < 0.01; ***P < 0.001 by one-way ANOVA followed by Tukey’s multiple comparisons test. (F)Percentages of IFN-γ–producing CD4+ T cells isolated from 4- to 6-week-old Tac-DKO (n = 7), Jax-DKO (n = 6), and Tac-WT (n = 7). Each dot represents an individual mouse. Bars show median; data are from three independent experiments. *P < 0.05; **P < 0.01 by Kruskal-Wallis test followed by Dunn’s multiple comparisons test. (G) Representative histology of H&E-stained colonic sections from Tac-DKO mice injected with anti-TNFα antibody (αTNFα) or control IgG (sham). Arrows show inflammatory cell infiltrate and epithelial crypt damage. Scale bars, 500 μm. (H) Histopathological scores of colonic tissue from anti–TNFα-treated Tac-DKO (n = 5) and from sham-treated Tac-DKO mice (n = 4) on day 9 after injection. Each dot represents an individual mouse. Bars show median; data are from two independent experiments; *P = 0.0159 by two-tailed Mann-Whitney U test. (I) Fecal Lcn-2 concentration in Tac-DKO mice treated with anti-TNFα antibody (n = 9) or control isotype–matched antibody (sham) (n = 9) before injection (day 0) and on day 6 and on day 9 after injection. Bars show median; data are from two independent experiments. Kruskal-Wallis test followed by Dunn’s post test. *P < 0.05; **P < 0.01; ***P < 0.001.
Combined deficiency of Nod2 and Cybb led to a selective accumulation of the Mucispirillum pathobiont
The observation that a specific microbiota is required to elicit colitis in DKO mice suggested that particular bacterial species may trigger disease. To address this, we analyzed the microbiota composition in the feces of both Jax-and Tac-fostered mice. The α diversity did not differ between Tac- and Jax-fostered mice as measured by the Shannon diversity index (Fig. 3A). Furthermore, the α diversity was not correlated with fecal Lcn-2 levels in Tac-DKO mice (Fig. 3B). Analysis of β diversity using θyc distance that measures differences in taxonomic abundance profiles revealed a marked dissimilarity in the community structure of the gut microbiota between Jax and Tac mothers (Fig. 3C). After fostering to Tac mothers, Jax-DKO, Cybb−/−, Nod2−/−, and WT pups acquired a microbiota that resembled that of their foster Tac mothers independently of genotype (Fig. 3C). Because dysbiosis can be the result of intestinal inflammation, we analyzed the gut microbiota composition in Tac-DKO pups before the onset of colitis [Tac-DKO not inflamed (NI)] (fig. S5, A to E). Consistent with the known differences in the microbiota composition between Jax and Tac mice (35), linear discriminant analysis (LDA) effect size (LEfSe) methodology revealed the presence of multiple bacterial operational taxonomic units (OTUs) that were differentially abundant in the microbiota of Jax-DKO mice when compared with Tac-DKO mice before the onset of colitis (Tac-DKO NI) (fig. S6A). To narrow down the list of potential bacterial candidates and to identify colitogenic bacteria present in the Tac microbiota, we focused our analysis on Tac-fostered mice. Further LEfSe analysis revealed a remarkable increase (1349-fold) in one specific OTU in the microbiota of Tac-DKO NI mice but not in the microbial community of either Tac-Cybb−/− or Tac-Nod2−/−, when compared with Tac-WT animals (Fig. 3D and table S1). This specific OTU belongs to M. schaedleri, an obligate Gram-negative anaerobe that is a member of the Deferribacteraceae family and mucus inhabitant of the murine intestine (38). Mucispirillum was detected in Tac-fostered mice but not in Jax-fostered animals by quantitative real-time polymerase chain reaction (qPCR) analysis using bacterium-specific primers (Fig. 3E). Although Mucispirillum was detected in Tac-fostered single Nod2 and Cybb mutant mice, marked accumulation of Mucispirillum only occurred in colitic Tac-DKO mice and in the same animals before the onset of colitis (Fig. 3E). Consistent with the localization of this bacterium in the intestinal mucus layer (38), Mucispirillum was detected in colonic mucus scrapings harvested from Tac-fostered mice but not from Jax-fostered animals (fig. S6B). Accumulation of Mucispirillum in mucus scrapings occurred in Tac-DKO mice, regardless of the presence of intestinal inflammation, and in Tac-Nod2−/− animals, but less in Tac-Cybb−/− mice (fig. S6B). Furthermore, no differences in the abundance of another mucus-dwelling bacterium, namely, Akkermansia muciniphila, were found in colonic mucus among Jax- and Tac-fostered mice (fig. S6C), suggesting that the accumulation of Mucispirillum in intestinal mucus of Tac-fostered DKO mice is specific. Higher levels of Mucispirillum were observed in inflamed colonic areas versus uninflamed intestinal regions using laser microdissection in Tac-DKO mice (fig. S7, A and B), indicating that colonic inflammation is as sociated with accumulation of Mucispirillum within the intestinal wall in Nod2−/−Cybb−/− mice. In contrast, Mucispirillum was barely detected in spleens and livers of either Jax- or Tac-fostered mice (fig. S7, C and D), suggesting that this bacterium does not spread systemically in these mice. Consistent with previous studies (35), segmented filamentous bacterium (SFB) was detected in the ileum of Tac mice, and much less in the feces of WT and Cybb−/− Tac-fostered mice, but not in Jax-fostered animals (Fig. 3F). However, SFB was barely detected in the feces of Tac-DKO mice with or without colitis (Fig. 3F). Thus, although the overall gut microbial composition does not differ among all Tac-fostered mice, a unique bacterium Mucispirillum selectively accumulates in Nod2−/−/Cybb−/− mice that develop colitis. These results also indicate that the combined deficiency of Nod2 and Cybb is required for marked accumulation of Mucispirillum in the gut.
Fig. 3. Selective accumulation of the Mucispirillum pathobiont in doubly deficient Nod2/Cybb mice.
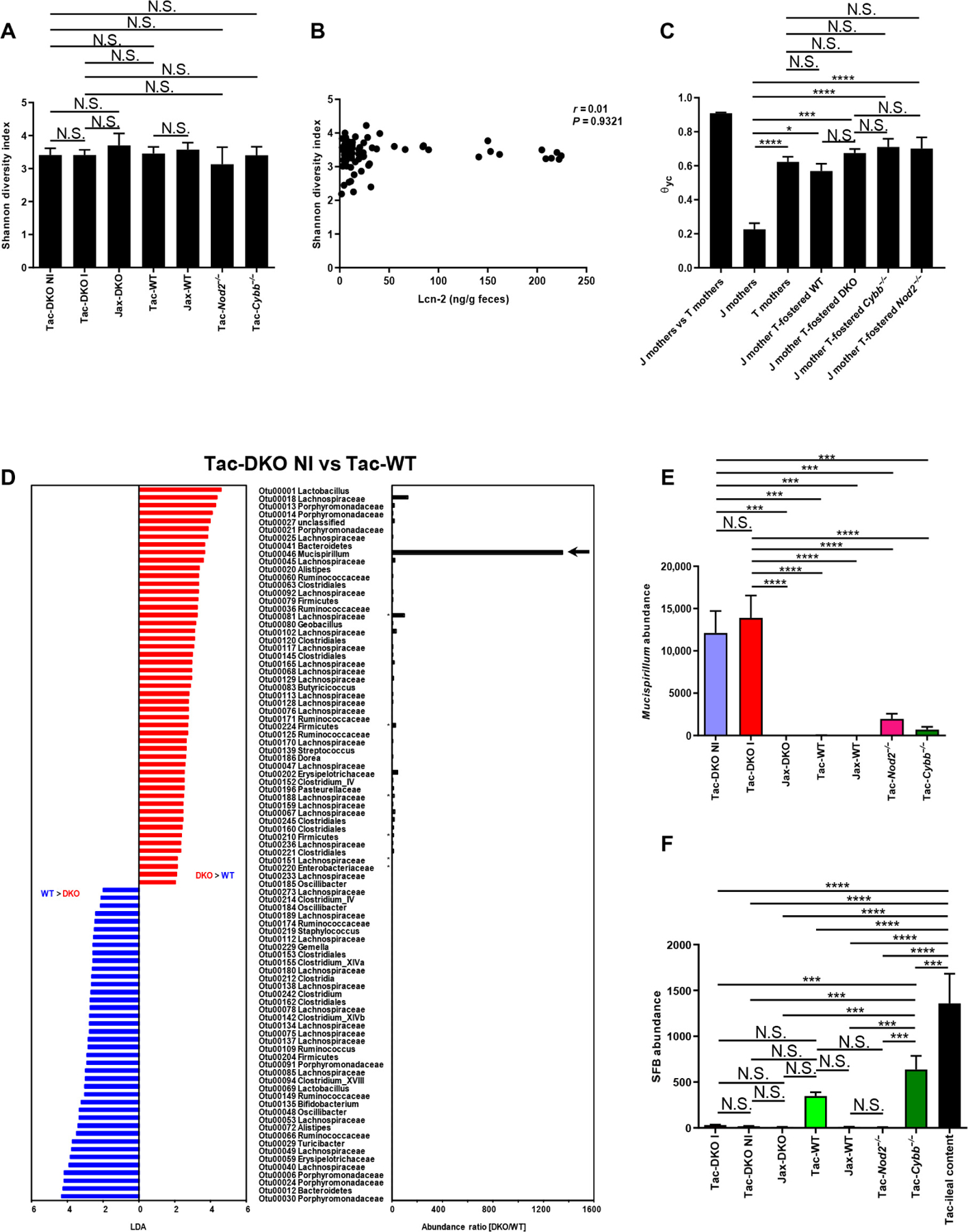
(A) Shannon’s diversity index of fecal samples from Tac- and Jax-fostered mice. Tac-DKO NI (n = 12), Tac-DKO I (n = 13), Jax-DKO (n = 10), Tac-WT (n = 7), Jax-WT (n = 7), Tac-Nod2−/− (n = 13), and Tac-Cybb−/− (n = 8). Results are mean ± SD. Data are pooled from two independent experiments. One-way ANOVA followed by Tukey’s multiple comparisons test. (B) Correlation between levels of fecal Lcn-2 and Shannon’s diversity index of fecal samples from Tac-DKO mice (n = 75). Data are pooled from five independent experiments. Spearman’s rank correlation. (C) Average θyc distance within or between groups of Jax mothers (J mothers, n = 4), Tac mothers (T mothers, n = 6), Jax-born DKO (J mother T-fostered DKO, n = 17), Cybb−/− (J mother T-fostered Cybb−/−, n = 8), Nod2−/− (J mother T-fostered Nod2−/−, n = 13), and WT mice (J mother T-fostered WT, n = 7) fostered to Tac mothers. Data are mean ± SEM; data are from two independent experiments. Kruskal-Wallis test followed by Dunn’s post test. *P < 0.05; ***P < 0.001; ****P < 0.0001. (D) LEfSe analysis shows bacterial OTUs that were differentially abundant between age-matched Tac-DKO mice before the onset of intestinal inflammation (Tac-DKO NI, n = 12) and Tac-WT animals (n = 7). The LDA score and the abundance ratio are shown. The arrow indicates the specific OTU that belongs to Mucispirillum. Asterisks indicate bacteria under detection (zero read) in 16S ribosomal RNA (rRNA) gene Illumina MiSeq analysis in denominator groups. Data are pooled from two independent experiments. (E) Presence of Mucispirillum in fecal DNA extracted from 3- to 4-week-old Tac-DKO NI (n = 12), Tac-DKO I (n = 13), Jax-DKO (n = 10), Tac-WT (n = 7), Jax-WT (n = 7), Tac-Nod2−/− (n = 13), and Tac-Cybb−/− (n = 8) and normalized to the universal 16S rRNA gene. Results are mean ± SEM; data are from two independent experiments. ***P < 0.001; ****P < 0.0001 by Kruskal-Wallis test followed by Dunn’s post test. (F) The presence of SFB in fecal DNA was quantified in fecal samples collected from 3- to 4-week-old Tac- and Jax-fostered mice. Results were normalized to the universal 16S rRNA gene. Small intestinal contents from Taconic C57BL/6 mice (Tac-ileal content) were used as positive control for the presence of SFB. Mean ± SEM of at least five mice. Data are pooled from two independent experiments. ***P < 0.001; ****P < 0.0001 by one-way ANOVA followed by Tukey’s multiple comparisons test.
Nod2 and Cybb regulate neutrophil recruitment and killing to control Mucispirillum accumulation
Neutrophils can transmigrate into the intestinal lumen and engulf bacteria near the epithelium (39). To determine whether neutrophil accumulation in Tac-DKO mice (Fig. 2B) is a primary event or secondary to the development of inflammation, we analyzed the number of intestinal and luminal neutrophils in age-matched (4- to 5-week-old) Tac-DKO mice before the onset of inflammation (Tac-DKO NI) and colitic (Tac-DKO I) mice. Tac-DKO NI mice exhibited a reduced number of intestinal and luminal neutrophils when compared with Tac-WT animals (Fig. 4, A and B). To assess whether accumulation of Mucispirillum within neutrophils precedes the development of colitis, we isolated neutrophils from the intestinal luminal compartment and the LP of age-matched (~5-week-old) Jax- and Tac-fostered mice before the onset of colitis and analyzed the presence of the bacterium by qPCR. This analysis revealed that Mucispirillum was more abundant in both luminal and LP neutrophils from Tac-DKO NI mice when compared with other Tac-fostered mice and Jax-fostered ani mals (Fig. 4, C and D). Although no clear differences in the levels of Firmicutes and mucus-degrading A. muciniphila were observed in colonic neutrophils isolated from fostered mice (Fig. 4, E and F), Firmicutes exhibited a tendency to increase in LP neutrophils from Tac-DKO NI mice when compared with other Tac-fostered mice and Jax-fostered animals (Fig. 4E). Together, these data argue for an accumulation of Mucispirillum in intestinal neutrophils of Tac-DKO mice. Consistent with a role of Cybb in the phagosome oxidative burst (40), reactive oxygen species (ROS) were induced in WT and Nod2−/− neutrophils, but that activity was impaired in Cybb−/− and Cybb−/−Nod2−/− neutrophils in response to phorbol myristate acetate and to N-formyl-l-methionyl-l-leucyl-l-phenylalanine (fMLP) after priming with lipopolysaccharide (LPS) (Fig. 4G and fig. S8A). Consistent with previous studies (41), priming with LPS elicited very low ROS production in both WT and Nod2−/− neutrophils (fig. S8B); however, further stimulation with fMLP resulted in robust and comparable ROS production in LPS-primed WT and Nod2−/− neutrophils but not in Cybb−/− and Cybb−/−Nod2−/− neutrophils (fig. S8C). To assess the contribution of Nod2, we determined the number of intestinal neutrophils in all fostered mice in the LP and intestinal lumen. Consistent with previous studies that implicate Nod2 in the recruitment of immune cells (42, 43), our analysis revealed that Tac-DKO NI and Tac-Nod2−/− mice exhibited reduced numbers of intestinal neutrophils when compared with Tac-WT, Tac-Cybb−/−, and Jax-fostered WT animals (Jax-WT) (Fig. 4H). Similarly, Tac-Nod2−/− and Tac-DKO NI mice also exhibited a reduction in the number of neutrophils in the luminal compartment when compared with Tac-WT and Tac-Cybb−/− animals (Fig. 4I). No differences in the number of LP and luminal neutrophils were found between Tac-DKO and Jax-DKO mice (Fig. 4, H and I). In accordance with reduced neutrophil numbers, the expression of two neutrophil chemokines, Cxcl1 and Cxcl2, was decreased in colonic explants from Tac-DKO NI (Fig. 4, J and K). These results suggest that impairment of both neutrophil recruitment and NADPH oxidase activity is required for Mucispirillum accumulation and colitis in DKO mice. and Tac-Nod2−/− mice
Fig. 4. Nod2 and Cybb regulate neutrophil recruitment and killing to control Mucispirillum accumulation.
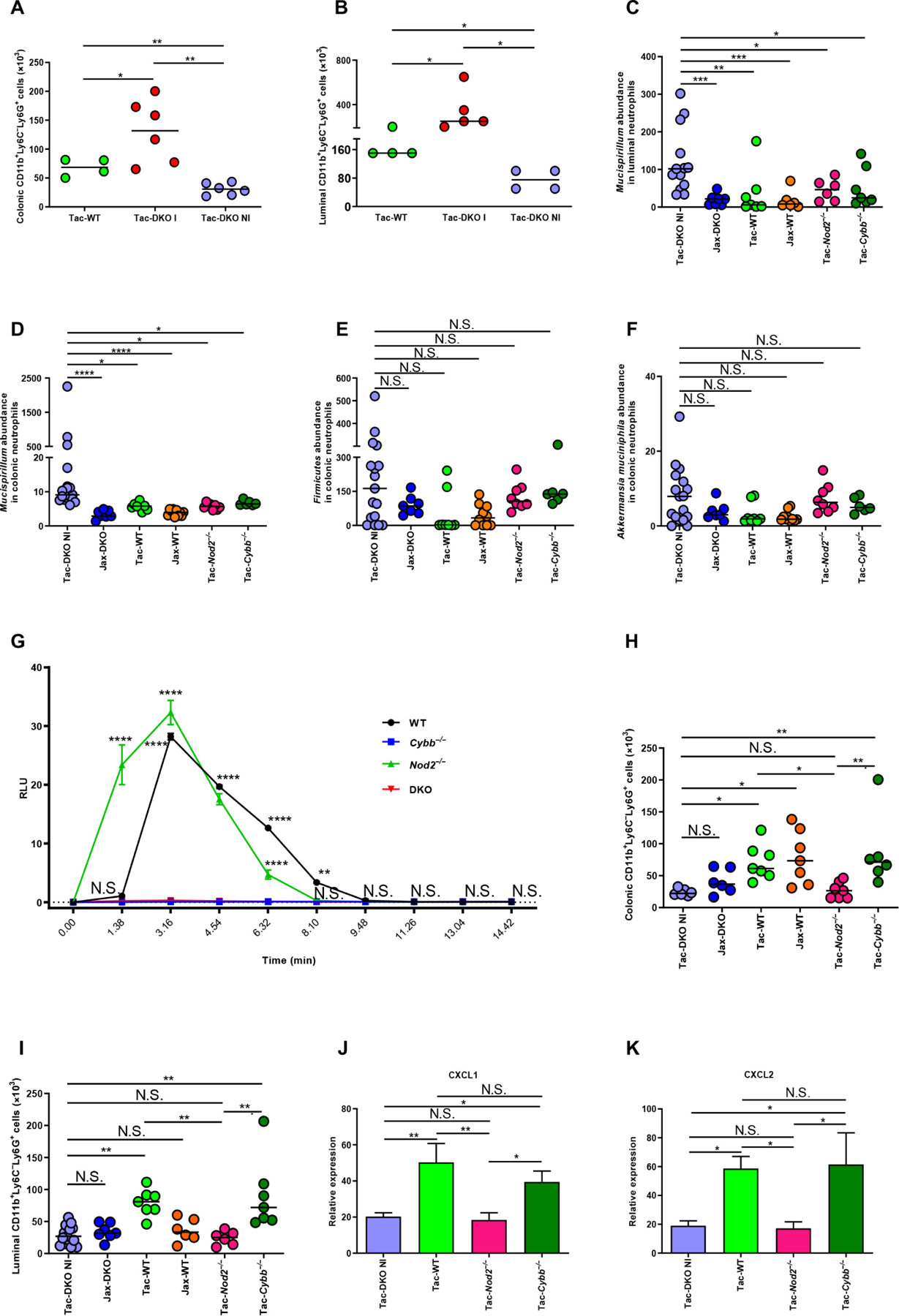
(A) Total number of colonic neutrophils isolated from ~4-week-old Tac-DKO NI (n = 6), Tac-DKO I (n = 6), and Tac-WT (n = 4). Bars show mean; data are from two independent experiments. *P < 0.05; **P < 0.01 by one-way ANOVA followed by Tukey’s multiple comparisons test. (B) Total number of luminal neutrophils isolated from ~4-week-old Tac-DKO NI (n = 4), Tac-DKO I (n = 5), and Tac-WT (n = 4). Bars show median; data are from two independent experiments. *P < 0.05 by Kruskal-Wallis test followed by Dunn’s post test. (C)Presence of Mucispirillum in luminal neutrophils isolated from ~5-week-old Tac-DKO NI (n = 13), Jax-DKO (n = 7), Tac-WT (n = 7), Jax-WT (n = 6), Tac-Nod2−/− (n = 6), and Tac-Cybb−/− (n = 7). Bars indicate median; data are from five independent experiments. (D) Presence of Mucispirillum in LP neutrophils (~5-week-old mice; Tac-DKO NI, n = 17; Jax-DKO, n = 7; Tac-WT, n = 8; Jax-WT, n = 10; Tac-Nod2−/−, n = 8; Tac-Cybb−/−, n = 6). Bars show median; data are from four independent experiments. (E and F) Presence of Firmicutes (E) and A. muciniphila (F) in LP neutrophils (~5-week-old mice; Tac-DKO NI, n = 17; Jax-DKO, n = 7; Tac-WT, n = 8; Jax-WT, n = 10; Tac-Nod2−/−, n = 8; and Tac-Cybb−/−, n = 6). Bars indicate median; data are from four independent experiments. (C to F) Results were normalized to GAPDH expression. Kruskal-Wallis test followed by Dunn’s post test. *P < 0.05; **P < 0.01; ***P < 0.001; ****P < 0.0001. (G) Representative chemiluminescence curves of ROS production frombone marrow–derived neutrophils isolated from Tac-WT (n = 3), Tac-Cybb−/− (n = 3), Tac-Nod2−/− (n = 3), and Tac-DKO (n = 3) mice activated by PMA. The relative light units (RLU) were monitored at 98-s intervals and shown up to 14.42 min. Results are mean ± SEM; data are from three independent experiments. **P < 0.01; ****P < 0.0001 by two-way ANOVA followed by Tukey’s multiple comparisons test. (H) Total number of LP neutrophils (~5-week-old mice; age-matched Tac-DKO NI, n = 6; Jax-DKO, n = 6; Tac-WT, n = 7; Jax-WT, n = 7; Tac-Nod2−/−, n = 7; and Tac-Cybb−/−, n = 6). Bars show median; data are from three independent experiments. (I) Absolute number of luminal neutrophils isolated from ~5-week-old, age-matched Tac-DKO NI (n = 15), Jax-DKO (n = 7), Tac-WT (n = 7), Jax-WT (n = 6), Tac-Nod2−/− (n = 6), and Tac-Cybb−/− (n = 7). Bars indicate median; data are from five independent experiments. (H and I) Kruskal-Wallis test followed by Dunn’s post test. *P < 0.05; **P < 0.01. (J and K) Gene expression in colonic tissues harvested from fostered mice. Results were normalized to GAPDH. Results are mean ± SEM of at least six individual mice; data are from two independent experiments. *P < 0.05; **P < 0.01 by one-way ANOVA followed by Tukey’s multiple comparisons test.
Maternal immunoglobulins confer protection against colitis in doubly deficient Nod2/Cybb mice
Doubly deficient Nod2/Cybb mice develop CD-like colitis shortly after weaning, when fostered to Tac mothers. Breastfeeding can protect against pediatric and adult-onset IBD (44, 45); however, the mechanisms involved remain unclear. Next, we assessed whether Tac-DKO mice are protected from disease development during breastfeeding. To test this hypothesis, we first assessed the presence of Mucispirillum-specific immunoglobulins (Igs) in the intestinal lumen of fostered mice. Mucispirillum-specific IgA was detected in the luminal contents of all Tac-fostered mice, but little or no IgA was observed in Jax-fostered animals (Fig. 5A). Furthermore, higher levels of Mucispirillum-specific IgA were found in Tac-DKO mice when compared with other Tac-fostered mice (Fig. 5A). Mucispirillum also elicited IgG responses in Tac-fostered animals (Fig. 5B), whereas total IgA and IgG were comparable in all animal groups (fig. S9, A and B). Although Mucispirillum was not observed in Tac-WT immediately after weaning (Fig. 3E), the bacterium was detected in older (5- to 6-week-old) Tac-WT mice (fig. S9C), thus explaining the presence of Mucispirillum-specific IgA and IgG in older Tac-WT mice (Fig. 5, A and B). Maternally derived microbiota-specific Igs are detectable in mice as early as 2 weeks after birth; their levels wane by 3 to 4 weeks of age and increase again in older mice due to their own antibody production (46). Consistently, time course analyses revealed that both Mucispirillum-specific IgA and IgG were low or undetectable at weaning; their production was increased by day 7 after weaning and returned to baseline by day 21, which inversely correlated with the levels of fecal Lcn-2 (Fig. 5, C to E). The low or absent Ig levels at weaning in fostered mice are presumably explained by the fact that maternally derived microbiota-specific Igs in the gut are acquired from breast milk after birth (47, 48), but such Igs are expected to be reduced when neonatal mice switched their diet to solid food by 3 weeks of age. Consistently, the presence of milk was detected in the stomach of 2-week-old but not 3-week-old pups (fig. S9, D and E). The high levels of fecal Lcn-2 at weaning time in Tac-DKO mice (Fig. 5E) are consistent with the recruitment of neutrophils to the intestine in response to bacterial accumulation and/or local invasion before the development of overt intestinal inflammation in Tac-DKO mice shortly after weaning (Fig. 1, B to D). Although the amounts of fecal Mucispirillum-specific Igs had declined by day 21 to 28 after weaning (Fig. 5, C and D), Igs against Mucispirillum were found in the serum of Tac-DKO mice (older than 7 weeks of age) but much less in age-matched Jax-DKO mice (fig. S9, F and G). The reason for the reduction of Mucispirillum-specific antibodies in the luminal content of Tac-DKO mice (Fig. 5, C and D) is unclear, but it could be explained by the binding of antibodies to Mucispirillum and the engulfment of Ig-coated bacteria by neutrophils. Consistent with this possibility, we found a reduced abundance of Mucispirillum in the feces of ~6-week-old Tac-DKO mice (fig. S9C) when compared with just weaned Tac-DKO mice (Fig. 3E).
Fig. 5. Maternal Igs protect doubly deficient Nod2/Cybb mice from the development of colitis.
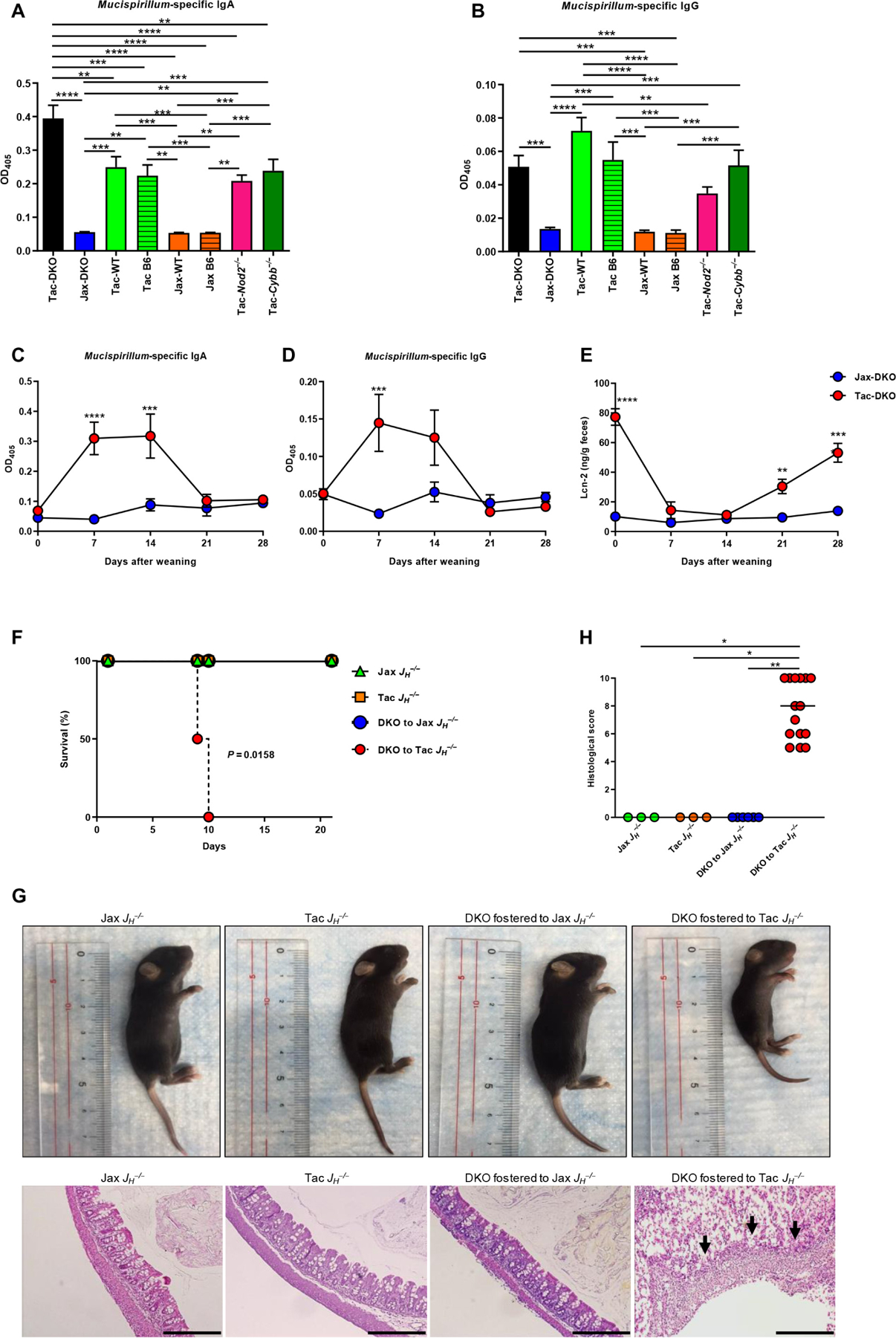
ELISA of Mucispirillum-specific IgA (A) and IgG (B) in the luminal content of fostered mice. Non-fostered Tac C57BL/6 (Tac B6) and Jax C57BL/6 (Jax B6) mice were included as controls. Data are mean ± SEM of at least six individual mice; data are from two independent experiments. **P < 0.01; ***P < 0.001;****P<0.0001byone-way ANOVA followed by Tukey’s multiple comparisons test. ELISA of Mucispirillum-specific IgA (C) and IgG (D) in the luminal content of Tac-DKO(n = 8) and Jax-DKO (n =6) mice. Results are mean ± SD; data are from two independent experiments. (E) Fecal Lcn-2 concentration in Tac-DKO (n = 8) and Jax-DKO (n = 6) mice. Data are mean ± SD; data are from two independent experiments. (C to E) Two-way ANOVA followed by Sidak’s multiple comparisons test. **P < 0.01; ***P < 0.001; ****P < 0.0001. (F) Mouse survival over time after fostering newborns DKO to either Jax JH−/− (DKO to Jax JH−/−, n = 7) or to Tac-exposed JH−/− mothers (DKO to Tac JH−/−, n = 15). Newborn Jax JH−/− (n = 7) and Tac JH−/− (n = 7) pups were included as controls. Data are from two independent experiments. Log-rank test. (G) Representative images (top) and H&E-stained colonic sections (bottom) from 9-day-old DKO fostered to Jax JH−/− or to Tac JH−/− mothers and Jax JH−/− and Tac JH−/− pups. Scale bars, 500 µm. (H) Histopathological scores of colonic tissues from (9-day-old) DKO fostered to Jax JH−/− (n = 6) or Tac JH−/− mothers (n = 15) and Jax JH −/− (n = 3) and Tac JH −/− mice (n = 3). Bars show median; data are from two independent experiments. *P < 0.05; **P < 0.01 by Kruskal-Wallis test followed by Dunn’s post test.
To determine the role of maternal Mucispirillum-specific Igs in protection against disease during nursing, we performed fostering experiments using JH−/− mothers, which lack mature B cells (49), and focused our analyses on earlier time points (i.e., from birth until 21 days of age) as compared with the fostering experiments using WT dams in Figs. 1 to 4. After colonization of Jax JH−/− mice with Mucispirillum by cohousing with Tac mice (fig. S10, A and B), newborn DKO mice were fostered to either Jax- or Tac-cohoused JH−/− mothers. DKO newborns fostered to JH−/− Tac mice, which lacked Igs (fig. S10, E and F), succumbed to disease within 10 days after fostering, whereas all DKO fostered to Jax JH−/− mothers survived (Fig. 5F). DKO mice fostered to Tac JH−/− mothers exhibited decreased size, marked destruction of the intestinal epithelium, and inflammatory cell infiltrates in the colon (Fig. 5, G and H). No macroscopic or histological alterations were observed in newborn DKO mice fostered to Jax JH−/− mothers or non-fostered Tac JH−/− or Jax JH−/− pups (Fig. 5, G and H). To further identify the class of Igs that are important in disease protection, we fostered DKO newborns to IgA-deficient mothers or dams deficient in the neonatal Fc receptor (FcRn) that mediates placental and perinatal IgG transport, as well as protection of IgG from catabolism (fig. S10, A and C to F) (50). Unlike DKO newborns fostered to Tac-cohoused JH −/− mice, newborn DKO mice fostered to Tac-cohoused IgA−/− or FcRn−/− mothers survived and did not display any histological intestinal alterations (fig. S11, A to F) at weaning time (21-day-old pups). Collectively, these results suggest that maternal IgA and IgG against Mucispirillum act redundantly to confer protection against colitis in doubly deficient Nod2/Cybb mice.
Mucispirillum induces intestinal inflammation in doubly deficient Nod2/Cybb mice
We next determined the ability of Mucispirillum to induce intestinal inflammation in DKO mice. To this end, adult Jax DKO, Cybb−/−, Nod2−/−, and WT mice were pretreated with vancomycin and then orally gavaged with Mucispirillum. Oral challenge with Mucispirillum resulted in increased levels of fecal Lcn-2 (Fig. 6A) and colon shortening in DKO mice (Fig.6B), but not in Cybb−/−, Nod2−/−, and WT mice, despite comparable colonization in the four groups of animals (fig. S12A). Histological evaluation of Mucispirillum-infected mice confirmed the development of inflammation in the large intestine of DKO mice (Fig. 6, C and D). Oral challenge with a Gram-negative, obligate anaerobe, Bacteroides uniformis, did not induce intestinal inflammation in Jax-DKO (fig. S12, B to E), despite successful colonization (fig. S12F). To further assess the role of B cells in the development of colitis, we pretreated DKO mice with vancomycin and then injected them intraperitoneally with anti-CD20 antibody to deplete B cells, followed by intragastric administration of Mucispirillum. Because anti-CD20 antibodies do not target antibody-secreting long-lived plasma cells, only Jax-DKO mice, devoid of Mucispirillum, were depleted of B cells and then orally gavaged with the bacterium. Administration of anti-CD20 antibody resulted in selective depletion of peripheral blood B cells (fig. S13A). Unlike control IgG-injected DKO mice in which Mucispirillum-specific IgA and IgG were detected in the luminal content 7 days after bacterial challenge, CD20-depleted DKO mice did not mount robust anti-Mucispirillum IgA and IgG responses (fig. S13B). Oral challenge with Mucispirillum resulted in increased levels of fecal Lcn-2 on day 5 after the initial bacterial gavage in both control IgG-treated and CD20-depleted DKO mice (Fig. 6E). However, on day 10 after infection, control IgG-treated mice recovered from the intestinal inflammation, whereas the CD20-depleted group exhibited further increase in fecal levels of Lcn-2 (Fig. 6E), which was associated with colon shortening (Fig. 6F) and higher levels of Mucispirillum colonization (fig. S13C). These results indicate that Mucispirillum triggers colitis selectively in DKO mice and suggest that Igs against the bacterium protect neonates against the development of intestinal inflammation.
Fig. 6. Mucispirillum triggers intestinal inflammation in doubly deficient Nod2/Cybb mice.
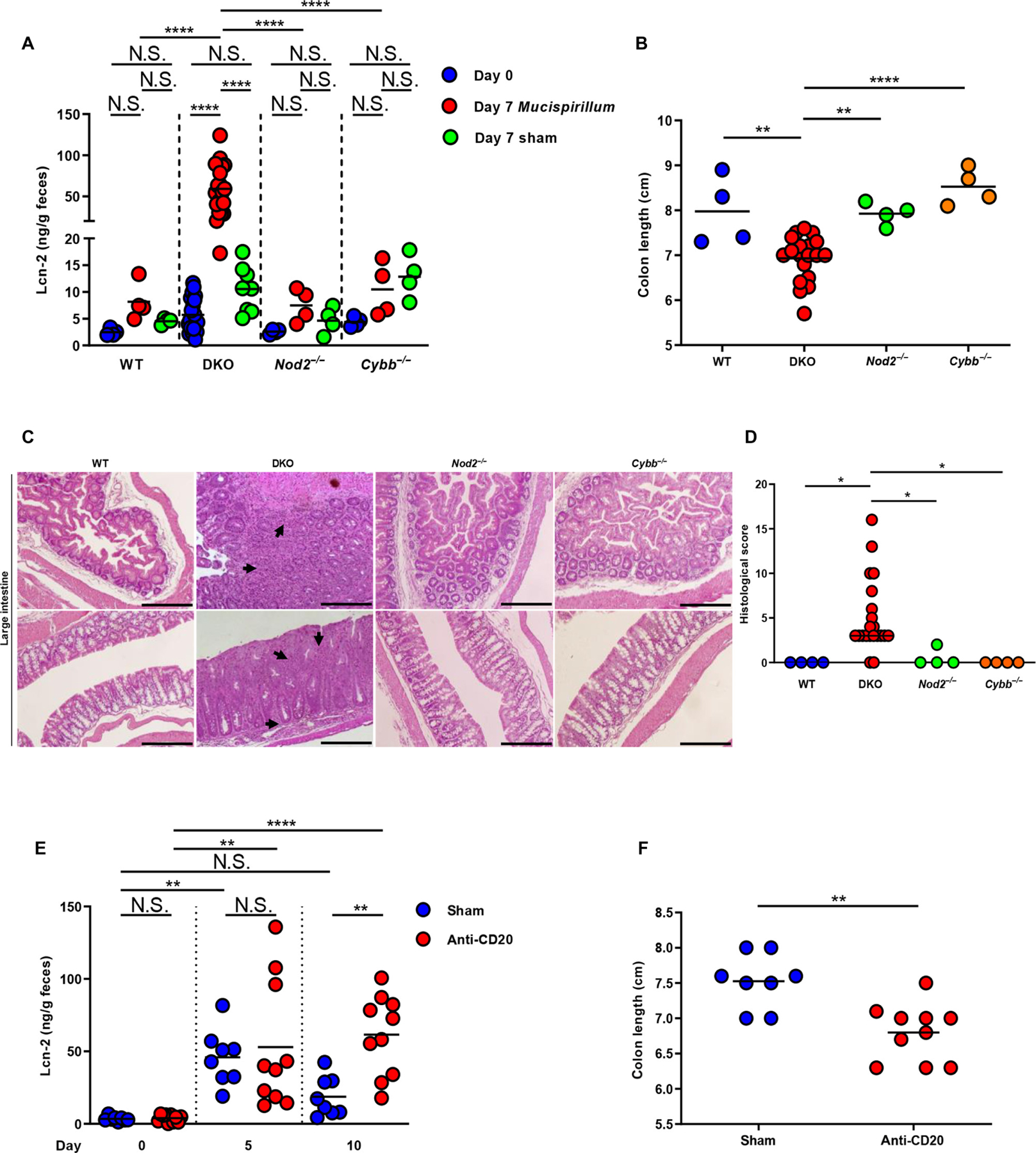
SPF Jax WT (n = 4), DKO (n = 21), Nod2−/− (n = 4), and Cybb−/− (n = 4) mice were orally gavaged with either Mucispirillum or culture broth (sham) every other day up to 7 days. Lcn-2 concentration (A) was measured in fecal samples before (day 0) and 7 days after the first oral gavage. Colon length (B) was determined on day 7 after infection with Mucispirillum. (A and B) Bars show mean; data are from four independent experiments. **P < 0.01; ****P < 0.0001 by one-way ANOVA followed by Tukey’s multiple comparisons test. (C) Representative histology of H&E-stained sections from large intestines of Mucispirillum-infected WT, DKO, Nod2−/−, and Cybb−/− mice (on day 7 after infection). Arrows show inflammatory cell infiltrate. Scale bars, 500 μm. (D) Histopathological scores of cecal and colonic tissues from Mucispirillum-infected WT (n = 4), DKO (n = 21), Nod2−/− (n = 4), and Cybb−/− (n = 4) mice (on day 7 after infection). Bars show median; data are from four independent experiments. *P < 0.05 by Kruskal-Wallis test followed by Dunn’s post test. (E and F) SPF Jax-DKO mice were treated with an anti-CD20 monoclonal antibody (anti-CD20) (n = 10) or control antibody (sham) (n = 8) and then orally gavaged with Mucispirillum every other day up to 10 days. Fecal Lcn-2 concentration (E) was measured before (day 0), on day 5, and on day 10 after oral infection with Mucispirillum. Colon length (F) was measured in anti–CD20-treated DKO (n = 10) and in sham-treated DKO mice (n = 8) on day 10 after infection. Bars show mean; data are from two independent experiments. One-way ANOVA followed by Tukey’s multiple comparisons test. **P < 0.01; ****P < 0.0001 in (E); **P = 0.013 by two-tailed unpaired t test in (F).
DISCUSSION
A major question in the IBD field is whether general dysbiosis or accumulation of specific pathobionts can trigger intestinal inflammation in individuals harboring IBD susceptibility genes. We show here that mice deficient in Nod2 and Cybb, two genes involved in CD susceptibility (11, 12, 19), develop spontaneous colitis with the pathology hallmarks of CD. Exposure to a specific member of the gut microbiota, Mucispirillum, triggered CD-like disease. The colitis observed in Tac-DKO mice bears a notable resemblance to VEOIBD, a form of CD that mostly affects the colon of young children and has been associated with NADPH oxidase and NOD2 mutations (19, 51).
Abnormal interactions between microbes and the immune system have been identified as the core defect, leading to chronic inflammation in CD (52). Genetic studies have revealed that a large subset of CD susceptibility genes, including NOD2, regulate bacterial recognition/clearance, suggesting that impaired innate immune function by mutation can lead to disease (10). Disease in DKO mice required the presence of Mucispirillum in the microbiota and was not triggered in susceptible Nod2−/−Cybb−/− mice with Jax microbiota that lacks this bacterium. Thus, genetic defects that impair microbial clearance can result in highly specific accumulation of rare bacterial symbionts with pathogenic traits such as Mucispirillum that can trigger colitis. The development of disease in our model requires marked accumulation of this bacterium that was observed in DKO mice, but much less in single mutants, arguing that a threshold level of Mucispirillum accumulation is important for the development of CD-like colitis. In this context, previous studies showed a combinatory effect of Nod2 and neutrophil dysfunction in worsening intestinal inflammation in mice (53). However, unlike our spontaneous colitis model, the study by Han and colleagues (53) required chemically induced epithelial injury to elicit intestinal inflammation.
The cecal and colonic involvement of inflammation in this mouse model of CD is consistent with the fact that this bacterium is a strict anaerobe that resides in the cecum and colon (38). Given the large numbers of microorganisms that colonized the human gut, it is possible that certain disease-causing bacterial species accumulate and trigger disease in the presence of CD-associated NOD2 or CYBB mutations alone. Consistent with this notion, up to 40% of patients with chronic granulomatous disease, which is caused by homozygous mutations in CYBB or related NAPDH subunits, develop CD-like disease (22). Because Mucispirillum is a murine species, it is unlikely that this bacterium triggers CD in humans. However, bacteria related to Mucispirillum, such as Proteobacteria (54), have been found to be increased in the microbiota of adult and juvenile patients with CD (27–29). Our results indicate that assessment of the microbiota before the onset of intestinal inflammation is critical to reveal disease-causing symbionts. Mucispirillum resides in the intestinal mucus layer of rodents and harbors some virulence traits, including a type VI secretion system and putative effector proteins (54). Consistent with these virulent features, we found marked accumulation of Mucispirillum within the inflamed colonic tissue of Tac-DKO mice. Thus, localization near the epithelium and/or the presence of virulence genes may play a role in the ability of the bacterium to locally invade the intestinal tissue and trigger colitis in the absence of Nod2 and Cybb. No signs of systemic spread of Mucispirillum were found in Tac-DKO mice. Similar to human IBD (37), CD-like colitis induced in our model was treated effectively with anti-TNF therapy, suggesting that the DKO model of CD could be used to assess the ability of potential drugs and biologicals to inhibit inflammation in preclinical studies.
In WT mice, the accumulation and intestinal invasion of Mucispirillum are neutralized by Nod2 and phagosome NAPDH activity. The protective mechanisms appear to involve the recruitment of neutrophils to the LP, a step regulated by Nod2 at the steady state, and transcytosis of neutrophils into the intestinal lumen where the bacterium is engulfed and killed via a Cybb-dependent oxidative burst. Single deficiency of Nod2 and Cybb leads to Mucispirillum accumulation to a lesser extent than that observed in DKO mice and was not sufficient to trigger colitis in the presence of Tac microbiota. These results suggest that additional Nod2-dependent mechanisms for bacterial clearance operate in Cybb−/− mice. Although previous studies suggested that Nod2 can regulate ROS production in intestinal epithelial cells and bone marrow–derived macrophages (55, 56), our findings do not support an important role for Nod2 in regulating ROS generation in neutrophils. Consistent with our data, Deshmukh and colleagues (57) showed that NOD2 does not regulate ROS generation in neutrophils stimulated with Staphylococcus aureus, suggesting that NOD2 regulates ROS generation in a cell type–specific manner and/or only under certain experimental conditions.
Impaired recruitment of neutrophils to intestinal inflammatory sites has been observed in patients with CD (58, 59). However, further work is needed to understand the role of neutrophils in colitis and the mechanism by which NOD2 regulates the recruitment of neutrophils in the intestine. The accumulation of Mucispirillum detected in the colonic mucus layer of Tac-Nod2−/− mice may be the consequence of impaired luminal transcytosis of neutrophils, but other mechanisms including impaired secretion of mucins may play a role (60). Our data suggest that the accumulation of Mucispirillum within the intestinal tissue, rather than in the mucus layer, plays a pivotal role in triggering inflammatory disease. Impaired clearance of Mucispirillum in the intestinal tissue appears to drive colitogenic TH1 CD4+ T cells in Nod2−/−Cybb−/− mice, which is also observed in patients with CD (1, 2). Consistent with recent studies showing that extrathymically generated regulatory T cells play a role in the establishment of border-dwelling bacteria such as Mucispirillum during community assembly (61), our results show enhanced expression of Foxp3 in the intestine of Mucipirillum-enriched Tac-DKO mice. However, further work is needed to understand the role, if any, of regulatory T cells in colitis triggered by Mucispirillum in Tac-DKO mice.
The role of Nod2 in the regulation of the composition of the microbiota is controversial. Some studies showed an altered composition of the gut microbiota with a net increase in the abundance of Bacteroidetes and Firmicutes in Nod2−/− mice (62, 63). However, other studies using littermate mice did not identify substantial differences in the composition of the gut microbial community in Nod2−/− mice (64, 65). A previous study showed that Nod2 deficiency gives rise to subtle intestinal abnormalities that correlate with the expansion of the commensal Bacteroides vulgatus (66). Unlike Mucispirillum in DKO mice, B. vulgatus did not elicit spontaneous inflammation but exacerbated intestinal inflammation after epithelial damage induced by piroxicam treatment in Nod2−/− mice. Thus, B. vulgatus promotes inflammation in Nod2−/− mice but does not trigger spontaneous CD-like inflammation in Nod2 mutant mice.
We found that Mucispirillum elicits IgA and IgG immune responses in mice harboring the bacterium. These studies are consistent with a previous report that showed that Mucispirillum evades T cell–independent IgA production but elicits T cell–dependent IgA responses (67). The induction of IgA and IgG production by Mucispirillum likely reflects the localization of the bacterium near the epithelium and its ability to elicit adaptive immune responses. Our findings suggest a protective role of maternal Igs in breast milk against the development of CD-like colitis. Previous evidence suggested that breastfeeding during infancy protects from pediatric and adult-onset IBD (44). However, further work is needed to establish the mechanism by which breastfeeding protects against the development of IBD. Nevertheless, the observation that maternal Igs confer protection against colitis suggests that vaccines may be potential therapeutic approaches for patients with CD once colitogenic bacterial species are identified in humans.
MATERIALS AND METHODS
Study design
The aim of this study was to understand the link between CD-like colitis and the microbiota using a mouse model that is based on genes involved in CD susceptibility. The experimental design involved in vivo and in vitro experiments, including fostering experiments, bacterial in vivo challenge, administration of neutralizing antibodies, histological analysis, laser microdissection, enzyme-linked immunosorbent assay (ELISA), chemiluminescence assay, flow cytometry, 16S rRNA gene sequencing, and qPCR analysis. The sample size (at least three to five mice per group) for the in vivo experiments was determined to be the optimal size for statistical analysis while allowing for independent repeats. Animals were randomly assigned to experimental groups during administration of anti-TNFα antibody, B cell depletion, and Mucispirillum and B. uniformis infections. The investigators were not blinded to allocation during experiments and analyses unless otherwise indicated. Experimental replication is indicated in the figure legends.
Experimental model and mice
Mice
C57BL/6, Nod2−/−, Cybb−/−, and FcRn−/− mice were originally purchased from the Jackson Laboratory and bred in-house at the University of Michigan. C57BL/6 mice were also purchased from Taconic Biosciences. All mice were maintained in SPF conditions. JH−/− mice were provided by M. Cascalho (University of Michigan), and IgA−/− mice were provided by B. Arulanandam (UT Health San Antonio). Doubly deficient Nod2/Cybb mice were generated by breeding single Nod2−/− and Cybb−/− mice for six generations. All mice were on the C57BL/6 background. All animal studies were performed according to approved protocols by the University of Michigan Committee on the Use and Care of Animals.
Cohousing and fostering experiments
For cohousing experiments, Jax JH−/−, IgA−/−, and FcRn−/− breeding pairs were converted in trio breeding cages by adding a Nod2/Cybb−/− female harboring Taconic microbiota. Mice were cohoused for more than 4 weeks before performing fostering experiments. For fostering experiments, the day on which pups were born was considered day 0 and pups were fostered between day 0 and day 2. Foster mothers were selected on the basis of the presence of a healthy and well-fed litter that was within 1 or 2 days of age of the fostered pups. Foster mothers were removed from the cage and placed in a holding pen, whereas the original litter was euthanized. The fostered pups (whole litters, both males and females) were gently covered with the nesting material and bedding of the foster mother’s cage. Mothers were then introduced in their original cage with the fostered pups. To avoid potential cage effects, we performed several experiments using the same Jax or Tac C57BL/6 mothers for fostering multiple
Intestinal histopathology and disease evaluation
Fostered mice (both males and females) were euthanized at ~4 weeks of age, and the terminal ileum, cecum, and colon were flushed with phosphate-buffered saline (PBS) (Gibco), fixed in 10% formalin (Fisher Scientific), and then processed for hematoxylin and eosin (H&E) staining. Histologic evaluation was performed in a blinded fashion, using a scoring system described previously with some modifications (68). Briefly, a three- to four-point scale was used to denote the severity of inflammation (0, none; 1, mild; 2, moderate; and 3, severe), the level of involvement (0, none; 1, mucosa; 2, mucosa and submucosa; and 3, transmural), and the extent of epithelial/crypt damage (0, none; 1, basal 1/3; 2, basal 2/3; 3, crypt loss; 4, crypt and surface epithelial destruction). Each variable was evaluated in the involved areas and then multiplied by a factor reflecting the percentage of the cecum/colon involved (0 to 10%, 11 to 40%, 41 to 70%, and 71 to 100%), and lastly summed to obtain the overall score. Measurement of the extension of colonic inflammatory involvement was performed using Aperio ImageScope version 12.1.0.5029 (Aperio Technologies Inc.).
Laser capture microdissection
The Leica LMD6 Laser Microdissection (Leica Microsystems) was used to capture uninflamed and inflamed colonic areas from the intestines of 4-week-old Tac-DKO mice. Formalin-fixed, paraffin-embedded colonic sections (8 μm thick) were deposited on polyethylene terephthalate (PET) membrane FrameSlides (Leica Microsystems) and then deparaffinized in xylene followed by rehydration by 100–95-70% ethanol. DNA was extracted from the laser-captured areas using the Arcturus PicoPure DNA Extraction Kit (Applied Biosystems) with the addition of glass beads to disrupt the bacterial cells.
Enzyme-linked immunosorbent assay
For measurement of Mucispirillum-specific IgA and IgG by ELISA, Mucispirillum cultures were washed, heat-killed at 85°C for 1 hour, and resuspended in 10 ml of carbonate/bicarbonate (pH 9.6) buffer, and 100 ml was added to each well of a 96-well ELISA plate for overnight coating at 4°C. Wells were blocked with 1% (w/v) bovine serum albumin (Fisher Scientific) in PBS for 2 hours at room temperature. Fecal samples were resuspended with sterile PBS (100 mg/ml) and filtered through a 40-μm cell strainer to remove debris, and 200 ml was added to each well. To detect serum levels of Mucispirillum-specific Igs, we diluted mouse sera at 1:200 and incubated them over-night at 4°C. The presence of Mucispirillum-specific IgA and IgG was detected by alkaline phosphatase–conjugated polyclonal goat anti-mouse IgG or IgA antibodies (Southern Biotechnology Associates). Plates were developed using p-nitrophenyl phosphate substrate (Southern Biotechnology Associates), and OD405 (optical density at 405 nm) values were determined. To measure total IgA and IgG, we coated ELISA plates with unlabeled goat anti-mouse IgA or IgG (Southern Biotechnology Associates). For measurement of fecal Lcn-2, feces were resuspended with sterile PBS (100 mg/ml), vortexed for 5 min, and then centrifuged for 10 min at 12,000 rpm at 4°C. Lcn-2 levels were determined in fecal supernatants using a Duoset murine Lcn-2 ELISA kit (R&D Systems). ELISAs for cytokines were performed by the University of Michigan ELISA Core.
Supplementary Material
Fig. S2. Kinetics of intestinal inflammation in doubly deficient Nod2/Cybb mice.
Fig. S3. Cellular infiltrates in the large intestine of Tac- and Jax-fostered mice.
Fig. S1. Skip lesions in older Tac-fostered Nod2−/−Cybb−/− mice.
Fig. S5. Characterization of Tac-fostered doubly deficient Nod2/Cybb mice before the development of colitis.
Fig. S6. Analysis of the gut microbiota in Tac- and Jax-fostered doubly deficient Nod2/Cybb mice and quantification of mucus-dwelling bacteria in fostered mice.
Fig. S4. CD-like inflammation in Nod2−/−Cybb−/− mice is characterized by TH1 immune responses.
Fig. S8. ROS production after microbial stimulation.
Fig. S7. Increased abundance of Mucispirillum in inflamed colonic areas in Nod2−/−Cybb−/− mice.
Fig. S9. Immunoglobulin production in fostered mice.
Fig. S11. Mucispirillum-specific IgA and IgG cooperate in conferring protection against colitis in doubly deficient Nod2/Cybb mice.
Fig. S10. Mucispirillum-specific colonization and Ig production in JH−/−, IgA−/− and FcRn−/− mice
Fig. S12. Specificity of Mucispirillum-induced intestinal inflammation in doubly deficient Nod2/Cybb mice.
Fig. S13. Efficiency of B cell depletion in doubly deficient Nod2/Cybb mice.
Table S1. The abundances and LEfSe data of individual OTUs in Jax- and Tac-fostered mice. File S1. Data in tabular format.
Acknowledgments:
We thank L. Haynes and C. Reynolds for animal husbandry, M. Cascalho and B. Arulanandam for mutant mice, J. O’Rourke for bacterial strain, T. Moore for anti-TNFα antibody, J. Whitfield for ELISAs, Genentech for anti-CD20 antibody (provided under MTA), G. Chen for critical reading of the manuscript, P. Kuffa and N. A. Pudlo for technical assistance, and the Microbial Systems Molecular Biology Laboratory, the Center for Live Cell Imaging and Pathology Slide Scanning Service, and Flow Cytometry Core Laboratory at the University of Michigan Medical School for support.
Funding:
This work was supported by NIH grants R01DK61707 and R01DK091191 (to G.N.), Senior Research and Career Development Awards from the Crohn’s and Colitis Foundation of America (to N.I. and R.C., respectively), and EMBO Long-Term Fellowship and Scholarship for training abroad from the Italian Group for the Study of Inflammatory Bowel Diseases (to R.C.).
Footnotes
Competing interests: The authors declare that they have no competing interests.
SUPPLEMENTARY MATERIALS
immunology.sciencemag.org/cgi/content/full/4/34/eaaw4341/DC1
Data and materials availability:
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The microbiota FASTQ files have been deposited in the NCBI database under the accession numbers PRJNA489470 and PRJNA488108.
REFERENCES AND NOTES
- 1.Baumgart DC, Sandborn WJ, Crohn’s disease. Lancet 380, 1590–1605 (2012). [DOI] [PubMed] [Google Scholar]
- 2.Bouma G, Strober W, The immunological and genetic basis of inflammatory bowel disease. Nat. Rev. Immunol 3, 521–533 (2003). [DOI] [PubMed] [Google Scholar]
- 3.Hedrick TL, Friel CM, Colonic crohn disease. Clin. Colon Rectal Surg 26, 84–89 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Benchimol EI, Guttmann A, Griffiths AM, Rabeneck L, Mack DR, Brill H, Howard J, Guan J, To T, Increasing incidence of paediatric inflammatory bowel disease in Ontario, Canada: Evidence from health administrative data. Gut 58, 1490–1497 (2009). [DOI] [PubMed] [Google Scholar]
- 5.Herrinton LJ, Liu L, Lewis JD, Griffin PM, Allison J, Incidence and prevalence of inflammatory bowel disease in a Northern California managed care organization, 1996–2002. Am. J. Gastroenterol 103, 1998–2006 (2008). [DOI] [PubMed] [Google Scholar]
- 6.Burisch J, Jess T, Martinato M, Lakatos PL; ECCO -EpiCom, The burden of inflammatory bowel disease in Europe. J. Crohns Colitis 7, 322–337 (2013). [DOI] [PubMed] [Google Scholar]
- 7.Moran CJ, Very early onset inflammatory bowel disease. Semin. Pediatr. Surg 26, 356–359 (2017). [DOI] [PubMed] [Google Scholar]
- 8.Xavier RJ, Podolsky DK, Unravelling the pathogenesis of inflammatory bowel disease. Nature 448, 427–434 (2007). [DOI] [PubMed] [Google Scholar]
- 9.Liu JZ, van Sommeren S, Huang H, Ng SC, Alberts R, Takahashi A, Ripke S, Lee JC, Jostins L, Shah T, Abedian S, Cheon JH, Cho J, Daryani NE, Franke L, Fuyuno Y, Hart A, Juyal RC, Juyal G, Kim WH, Morris AP, Poustchi H, Newman WG, Midha V, Orchard TR, Vahedi H, Sood A, Sung JJY, Malekzadeh R, Westra H-J, Yamazaki K, Yang S-K; International Multiple Sclerosis Genetics Consortium; International IBD Genetics Consortium, Barrett JC, Franke A, Alizadeh BZ, Parkes M, B K T, Daly MJ, Kubo M, Anderson CA, Weersma RK, Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet 47, 979–986 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.McGovern DP, Kugathasan S, Cho JH, Genetics of inflammatory bowel diseases. Gastroenterology 149, 1163–1176.e2 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ogura Y, Bonen DK, Inohara N, Nicolae DL, Chen FF, Ramos R, Britton H, Moran T, Karaliuskas R, Duerr RH, Achkar J-P, Brant SR, Bayless TM, Kirschner BS, Hanauer SB, Nuñez G, Cho JH, A frameshift mutation in NOD2 associated with susceptibility to Crohn’s disease. Nature 411, 603–606 (2001). [DOI] [PubMed] [Google Scholar]
- 12.Hugot J-P, Chamaillard M, Zouali H, Lesage S, Cézard J-P, Belaiche J, Almer S, Tysk C, O’Morain CA, Gassull M, Binder V, Finkel Y, Cortot A, Modigliani R, Laurent-Puig P, Gower-Rousseau C, Macry J, Colombel J-F, Sahbatou M, Thomas G, Association of NOD2 leucine-rich repeat variants with susceptibility to Crohn’s disease. Nature 411, 599–603 (2001). [DOI] [PubMed] [Google Scholar]
- 13.Girardin SE, Boneca IG, Viala J, Chamaillard M, Labigne A, Thomas G, Philpott DJ, Sansonetti PJ, Nod2 is a general sensor of peptidoglycan through muramyl dipeptide (MDP) detection. J. Biol. Chem 278, 8869–8872 (2003). [DOI] [PubMed] [Google Scholar]
- 14.Inohara N, Ogura Y, Fontalba A, Gutierrez O, Pons F, Crespo J, Fukase K, Inamura S, Kusumoto S, Hashimoto M, Foster SJ, Moran AP, Fernandez-Luna JL, Nuñez G, Host recognition of bacterial muramyl dipeptide mediated through NOD2. Implications for Crohn’s disease. J. Biol. Chem 278, 5509–5512 (2003). [DOI] [PubMed] [Google Scholar]
- 15.van Heel DA, Ghosh S, Butler M, Hunt KA, Lundberg AM, Ahmad T, McGovern DP, Onnie C, Negoro K, Goldthorpe S, Foxwell BM, Mathew CG, Forbes A, Jewell DP, Playford RJ, Muramyl dipeptide and toll-like receptor sensitivity in NOD2-associated Crohn’s disease. Lancet 365, 1794–1796 (2005). [DOI] [PubMed] [Google Scholar]
- 16.Brant SR, Wang M-H, Rawsthorne P, Sargent M, Datta LW, Nouvet F, Shugart YY, Bernstein CN, A population-based case-control study of CARD15 and other risk factors in Crohn’s disease and ulcerative colitis. Am. J. Gastroenterol 102, 313–323 (2007). [DOI] [PubMed] [Google Scholar]
- 17.Kobayashi KS, Chamaillard M, Ogura Y, Henegariu O, Inohara N, Nuñez G, Flavell RA, Nod2-dependent regulation of innate and adaptive immunity in the intestinal tract. Science 307, 731–734 (2005). [DOI] [PubMed] [Google Scholar]
- 18.Kim Y-G, Shaw MH, Warner N, Park J-H, Chen F, Ogura Y, Nuñez G, Cutting edge: Crohn’s disease-associated Nod2 mutation limits production of proinflammatory cytokines to protect the host from Enterococcus faecalis-induced lethality. J. Immunol 187, 2849–2852 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Muise AM, Xu W, Guo C-H, Walters TD, Wolters VM, Fattouh R, Lam GY, Hu P, Murchie R, Sherlock M, Gana JC; NEOPICS, Russell RK, Glogauer M, Duerr RH, Cho JH, Lees CW, Satsangi J, Wilson DC, Paterson AD, Griffiths AM, Silverberg MS, Brumell JH, NADPH oxidase complex and IBD candidate gene studies: Identification of a rare variant in NCF2 that results in reduced binding to RAC2. Gut 61, 1028–1035 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Roberts RL, Hollis-Moffatt JE, Gearry RB, Kennedy MA, Barclay ML, Merriman TR, Confirmation of association of IRGM and NCF4 with ileal Crohn’s disease in a population-based cohort. Genes Immun 9, 561–565 (2008). [DOI] [PubMed] [Google Scholar]
- 21.Denson LA, Jurickova I, Karns R, Shaw KA, Cutler DJ, Okou DT, Dodd A, Quinn K, Mondal K, Aronow BJ, Haberman Y, Linn A, Price A, Bezold R, Lake K, Jackson K, Walters TD, Griffiths A, Baldassano RN, Noe JD, Hyams JS, Crandall WV, Kirschner BS, Heyman MB, Snapper S, Guthery SL, Dubinsky MC, Leleiko NS, Otley AR, Xavier RJ, Stevens C, Daly MJ, Zwick ME, Kugathasan S, Clinical and genomic correlates of neutrophil reactive oxygen species production in pediatric patients with Crohn’s disease. Gastroenterology 154, 2097–2110 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Marks DJ, Miyagi K, Rahman FZ, Novelli M, Bloom SL, Segal AW, Inflammatory bowel disease in CGD reproduces the clinicopathological features of Crohn’s disease. Am. J. Gastroenterol 104, 117–124 (2009). [DOI] [PubMed] [Google Scholar]
- 23.Dalal SR, Chang EB, The microbial basis of inflammatory bowel diseases. J. Clin. Invest 124, 4190–4196 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Manichanh C, Borruel N, Casellas F, Guarner F, The gut microbiota in IBD. Nat. Rev. Gastroenterol. Hepatol 9, 599–608 (2012). [DOI] [PubMed] [Google Scholar]
- 25.Frank DN, Amand AL St., Feldman RA, Boedeker EC, Harpaz N, Pace NR, Molecular-phylogenetic characterization of microbial community imbalances in human inflammatory bowel diseases. Proc. Natl. Acad. Sci. U.S.A 104, 13780–13785 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Sokol H, Pigneur B, Watterlot L, Lakhdari O, Bermúdez-Humarán LG, Gratadoux J-J, Blugeon S, Bridonneau C, Furet J-P, Corthier G, Grangette C, Vasquez N, Pochart P, Trugnan G, Thomas G, Blottiere HM, Doré J, Marteau P, Seksik P, Langella P, Faecalibacterium prausnitzii is an anti-inflammatory commensal bacterium identified by gut microbiota analysis of Crohn disease patients. Proc. Natl. Acad. Sci. U.S.A 105, 16731–16736 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Darfeuille-Michaud A, Boudeau J, Bulois P, Neut C, Glasser A-L, Barnich N, Bringer M-A, Swidsinski A, Beaugerie L, Colombel J-F, High prevalence of adherent-invasive Escherichia coli associated with ileal mucosa in Crohn’s disease. Gastroenterology 127, 412–421 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Martin HM, Campbell BJ, Hart CA, Mpofu C, Nayar M, Singh R, Englyst H, Williams HF, Rhodes JM, Enhanced Escherichia coli adherence and invasion in Crohn’s disease and colon cancer. Gastroenterology 127, 80–93 (2004). [DOI] [PubMed] [Google Scholar]
- 29.Gevers D, Kugathasan S, Denson LA, Vázquez-Baeza Y, Van Treuren W, Ren B, Schwager E, Knights D, Song SJ, Yassour M, Morgan XC, Kostic AD, Luo C, Gonzalez A, McDonald D, Haberman Y, Walters T, Baker S, Rosh J, Stephens M, Heyman M, Markowitz J, Baldassano R, Griffiths A, Sylvester F, Mack D, Kim S, Crandall W, Hyams J, Huttenhower C, Knight R, Xavier RJ, The treatment-naive microbiome in new-onset Crohn’s disease. Cell Host Microbe 15, 382–392 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Rath HC, Herfarth HH, Ikeda JS, Grenther WB, Hamm TE Jr., Balish E, Taurog JD, Hammer RE, Wilson KH, Sartor RB, Normal luminal bacteria, especially Bacteroides species, mediate chronic colitis, gastritis, and arthritis in HLA-B27/human beta2 microglobulin transgenic rats. J. Clin. Invest 98, 945–953 (1996). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kim SC, Tonkonogy SL, Albright CA, Tsang J, Balish EJ, Braun J, Huycke MM, Sartor RB, Variable phenotypes of enterocolitis in interleukin 10-deficient mice monoassociated with two different commensal bacteria. Gastroenterology 128, 891–906 (2005). [DOI] [PubMed] [Google Scholar]
- 32.Garrett WS, Gallini CA, Yatsunenko T, Michaud M, DuBois A, Delaney ML, Punit S, Karlsson M, Bry L, Glickman JN, Gordon JI, Onderdonk AB, Glimcher LH, Enterobacteriaceae act in concert with the gut microbiota to induce spontaneous and maternally transmitted colitis. Cell Host Microbe 8, 292–300 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Pauleau A-L, Murray PJ, Role of nod2 in the response of macrophages to toll-like receptor agonists. Mol. Cell. Biol 23, 7531–7539 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Shiloh MU, MacMicking JD, Nicholson S, Brause JE, Potter S, Marino M, Fang F, Dinauer M, Nathan C, Phenotype of mice and macrophages deficient in both phagocyte oxidase and inducible nitric oxide synthase. Immunity 10, 29–38 (1999). [DOI] [PubMed] [Google Scholar]
- 35.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, Littman DR, Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139, 485–498 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Chassaing B, Srinivasan G, Delgado MA, Young AN, Gewirtz AT, Vijay-Kumar M, Fecal lipocalin 2, a sensitive and broadly dynamic non-invasive biomarker for intestinal inflammation. PLOS ONE 7, e44328 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Stidham RW, Lee TCH, Higgins PDR, Deshpande AR, Sussman DA, Singal AG, Elmunzer BJ, Saini SD, Vijan S, Waljee AK, Systematic review with network meta-analysis: The efficacy of anti-TNF agents for the treatment of Crohn’s disease. Aliment. Pharmacol. Ther 39, 1349–1362 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Robertson BR, O’Rourke JL, Neilan BA, Vandamme P, On SLW, Fox JG, Lee A, Mucispirillum schaedleri gen. nov., sp. nov., a spiral-shaped bacterium colonizing the mucus layer of the gastrointestinal tract of laboratory rodents. Int. J. Syst. Evol. Microbiol 55, 1199–1204 (2005). [DOI] [PubMed] [Google Scholar]
- 39.Kamada N, Sakamoto K, Seo S-U, Zeng MY, Kim Y-G, Cascalho M, Vallance BA, Puente JL, Nuñez G, Humoral immunity in the gut selectively targets phenotypically virulent attaching-and-effacing bacteria for intraluminal elimination. Cell Host Microbe 17, 617–627 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Sareila O, Kelkka T, Pizzolla A, Hultqvist M, Holmdahl R, NOX2 complex-derived ROS as immune regulators. Antioxid. Redox Signal 15, 2197–2208 (2011). [DOI] [PubMed] [Google Scholar]
- 41.DeLeo FR, Renee J, McCormick S, Nakamura M, Apicella M, Weiss JP, Nauseef WM, Neutrophils exposed to bacterial lipopolysaccharide upregulate NADPH oxidase assembly. J. Clin. Invest 101, 455–463 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Kim Y-G, Kamada N, Shaw MH, Warner N, Chen GY, Franchi L, Nuñez G, The Nod2 sensor promotes intestinal pathogen eradication via the chemokine CCL2-dependent recruitment of inflammatory monocytes. Immunity 34, 769–780 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Davis KM, Nakamura S, Weiser JN, Nod2 sensing of lysozyme-digested peptidoglycan promotes macrophage recruitment and clearance of S. pneumoniae colonization in mice. J. Clin. Invest 121, 3666–3676 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xu L, Lochhead P, Ko Y, Claggett B, Leong RW, Ananthakrishnan AN, Systematic review with meta-analysis: Breastfeeding and the risk of Crohn’s disease and ulcerative colitis. Aliment. Pharmacol. Ther 46, 780–789 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Mikhailov TA, Furner SE, Breastfeeding and genetic factors in the etiology of inflammatory bowel disease in children. World J. Gastroenterol 15, 270–279 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Koch MA, Reiner GL, Lugo KA, Kreuk LSM, Stanbery AG, Ansaldo E, Seher TD, Ludington WB, Barton GM, Maternal IgG and IgA antibodies dampen mucosal T helper cell responses in early life. Cell 165, 827–841 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Hurley WL, Theil PK, Perspectives on immunoglobulins in colostrum and milk. Nutrients 3, 442–474 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Brandtzaeg P, Mucosal immunity: Integration between mother and the breast-fed infant. Vaccine 21, 3382–3388 (2003). [DOI] [PubMed] [Google Scholar]
- 49.Chen J, Trounstine M, Alt FW, Young F, Kurahara C, Loring JF, Huszar D, Immunoglobulin gene rearrangement in B cell deficient mice generated by targeted deletion of the JH locus. Int. Immunol 5, 647–656 (1993). [DOI] [PubMed] [Google Scholar]
- 50.Roopenian DC, Christianson GJ, Sproule TJ, Brown AC, Akilesh S, Jung N, Petkova S, Avanessian L, Choi EY, Shaffer DJ, Eden PA, Anderson CL, The MHC class I-like IgG receptor controls perinatal IgG transport, IgG homeostasis, and fate of IgG-Fc-coupled drugs. J. Immunol 170, 3528–3533 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Horowitz JE, Warner N, Stapes J, Crowley E, Murchie R, Van Hout C, King AK, Fiedler K, Reid JG, Overton JD, Shuldiner AR, Baras A; Geisinger-Regeneron DiscovEHR Collaboration, Dewey FE, Griffiths A, Gottesman O, Muise AM, Gonzaga-Jauregui C, Mutation spectrum of NOD2 reveals recessive inheritance as a main driver of early onset Crohn’s disease. bioRxiv 098574 (2017). [DOI] [PMC free article] [PubMed]
- 52.Asquith M, Powrie F, An innately dangerous balancing act: Intestinal homeostasis, inflammation, and colitis-associated cancer. J. Exp. Med 207, 1573–1577 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Han X, Uchida K, Jurickova I, Koch D, Willson T, Samson C, Bonkowski E, Trauernicht A, Kim M-O, Tomer G, Dubinsky M, Plevy S, Kugathsan S, Trapnell BC, Denson LA, Granulocyte-macrophage colony-stimulating factor autoantibodies in murine ileitis and progressive ileal Crohn’s disease. Gastroenterology 136, 1261–1271.e3 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Loy A, Pfann C, Steinberger M, Hanson B, Herp S, Brugiroux S, Gomes Neto JC, Boekschoten MV, Schwab C, Urich T, Ramer-Tait AE, Rattei T, Stecher B, Berry D, Lifestyle and horizontal gene transfer-mediated evolution of Mucispirillum schaedleri, a core member of the murine gut microbiota. mSystems 2, e00171–16 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lipinski S, Till A, Sina C, Arlt A, Grasberger H, Schreiber S, Rosenstiel P, DUOX2-derived reactive oxygen species are effectors of NOD2-mediated antibacterial responses. J. Cell Sci 122, 3522–3530 (2009). [DOI] [PubMed] [Google Scholar]
- 56.Lupfer CR, Anand PK, Liu Z, Stokes KL, Vogel P, Lamkanfi M, Kanneganti T-D, Reactive oxygen species regulate caspase-11 expression and activation of the non-canonical NLRP3 inflammasome during enteric pathogen infection. PLOS Pathog 10, e1004410 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Deshmukh HS, Hamburger JB, Ahn SH, McCafferty DG, Yang SR, Fowler VG Jr., Critical role of NOD2 in regulating the immune response to Staphylococcus aureus. Infect. Immun 77, 1376–1382 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Marks DJ, Harbord MW, MacAllister R, Rahman FZ, Young J, Al-Lazikani B, Lees W, Novelli M, Bloom S, Segal AW, Defective acute inflammation in Crohn’s disease: A clinical investigation. Lancet 367, 668–678 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Smith AM, Rahman FZ, Hayee B, Graham SJ, Marks DJB, Sewell GW, Palmer CD, Wilde J, Foxwell BMJ, Gloger IS, Sweeting T, Marsh M, Walker AP, Bloom SL, Segal AW, Disordered macrophage cytokine secretion underlies impaired acute inflammation and bacterial clearance in Crohn’s disease. J. Exp. Med 206, 1883–1897 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Alnabhani Z, Hugot J-P, Montcuquet N, Le Roux K, Dussaillant M, Roy M, Leclerc M, Cerf-Bensussan N, Lepage P, Barreau F, Respective roles of hematopoietic and nonhematopoietic Nod2 on the gut microbiota and mucosal homeostasis. Inflamm. Bowel Dis 22, 763–773 (2016). [DOI] [PubMed] [Google Scholar]
- 61.Campbell C, Dikiy S, Bhattarai SK, Chinen T, Matheis F, Calafiore M, Hoyos B, Hanash A, Mucida D, Bucci V, Rudensky AY, Extrathymically generated regulatory T cells establish a niche for intestinal border-dwelling bacteria and affect physiologic metabolite balance. Immunity 48, 1245–1257.e9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Petnicki-Ocwieja T, Hrncir T, Liu Y-J, Biswas A, Hudcovic T, Tlaskalova-Hogenova H, Kobayashi KS, Nod2 is required for the regulation of commensal microbiota in the intestine. Proc. Natl. Acad. Sci. U.S.A 106, 15813–15818 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Rehman A, Sina C, Gavrilova O, Hasler R, Ott S, Baines JF, Schreiber S, Rosenstiel P, Nod2 is essential for temporal development of intestinal microbial communities. Gut 60, 1354–1362 (2011). [DOI] [PubMed] [Google Scholar]
- 64.Robertson SJ, Zhou JY, Geddes K, Rubino SJ, Cho JH, Girardin SE, Philpott DJ, Nod1 and Nod2 signaling does not alter the composition of intestinal bacterial communities at homeostasis. Gut Microbes 4, 222–231 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Shanahan MT, Carroll IM, Grossniklaus E, White A, von Furstenberg RJ, Barner R, Fodor A, Henning SJ, Sartor RB, Gulati AS, Mouse Paneth cell antimicrobial function is independent of Nod2. Gut 63, 903–910 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Ramanan D, Tang MS, Bowcutt R, Loke P, Cadwell K, Bacterial sensor Nod2 prevents inflammation of the small intestine by restricting the expansion of the commensal Bacteroides vulgatus. Immunity 41, 311–324 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Bunker JJ, Flynn TM, Koval JC, Shaw DG, Meisel M, McDonald BD, Ishizuka IE, Dent L, Wilson PC, Jabri B, Antonopoulos DA, Bendelac A, Innate and adaptive humoral responses coat distinct commensal bacteria with immunoglobulin A. Immunity 43, 541–553 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Dieleman LA, Palmen MJHJ, Akol H, Bloemena E, Peña AS, Meuwissen SGM, Van Rees EP, Chronic experimental colitis induced by dextran sulphate sodium (DSS) is characterized by Th1 and Th2 cytokines. Clin. Exp. Immunol 114, 385–391 (1998). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Franchi L, Kamada N, Nakamura Y, Burberry A, Kuffa P, Suzuki S, Shaw MH, Kim Y-G, Nuñez G, NLRC4-driven production of IL-1β discriminates between pathogenic and commensal bacteria and promotes host intestinal defense. Nat. Immunol 13, 449–456 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kozich JJ, Westcott SL, Baxter NT, Highlander SK, Schloss PD, Development of a dual-index sequencing strategy and curation pipeline for analyzing amplicon sequence data on the MiSeq Illumina sequencing platform. Appl. Environ. Microbiol 79, 5112–5120 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Schloss PD, Westcott SL, Ryabin T, Hall JR, Hartmann M, Hollister EB, Lesniewski RA, Oakley BB, Parks DH, Robinson CJ, Sahl JW, Stres B, Thallinger GG, Van Horn DJ, Weber CF, Introducing mothur: Open-source, platform-independent, community-supported software for describing and comparing microbial communities. Appl. Environ. Microbiol 75, 7537–7541 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Bloom SM, Bijanki VN, Nava GM, Sun L, Malvin NP, Donermeyer DL, Dunne WM Jr., Allen PM, Stappenbeck TS, Commensal Bacteroides species induce colitis in host-genotype-specific fashion in a mouse model of inflammatory bowel disease. Cell Host Microbe 9, 390–403 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Hehemann J-H, Kelly AG, Pudlo NA, Martens EC, Boraston AB, Bacteria of the human gut microbiome catabolize red seaweed glycans with carbohydrate-active enzyme updates from extrinsic microbes. Proc. Natl. Acad. Sci. U.S.A 109, 19786–19791 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S2. Kinetics of intestinal inflammation in doubly deficient Nod2/Cybb mice.
Fig. S3. Cellular infiltrates in the large intestine of Tac- and Jax-fostered mice.
Fig. S1. Skip lesions in older Tac-fostered Nod2−/−Cybb−/− mice.
Fig. S5. Characterization of Tac-fostered doubly deficient Nod2/Cybb mice before the development of colitis.
Fig. S6. Analysis of the gut microbiota in Tac- and Jax-fostered doubly deficient Nod2/Cybb mice and quantification of mucus-dwelling bacteria in fostered mice.
Fig. S4. CD-like inflammation in Nod2−/−Cybb−/− mice is characterized by TH1 immune responses.
Fig. S8. ROS production after microbial stimulation.
Fig. S7. Increased abundance of Mucispirillum in inflamed colonic areas in Nod2−/−Cybb−/− mice.
Fig. S9. Immunoglobulin production in fostered mice.
Fig. S11. Mucispirillum-specific IgA and IgG cooperate in conferring protection against colitis in doubly deficient Nod2/Cybb mice.
Fig. S10. Mucispirillum-specific colonization and Ig production in JH−/−, IgA−/− and FcRn−/− mice
Fig. S12. Specificity of Mucispirillum-induced intestinal inflammation in doubly deficient Nod2/Cybb mice.
Fig. S13. Efficiency of B cell depletion in doubly deficient Nod2/Cybb mice.
Table S1. The abundances and LEfSe data of individual OTUs in Jax- and Tac-fostered mice. File S1. Data in tabular format.
Data Availability Statement
All data needed to evaluate the conclusions in the paper are present in the paper and/or the Supplementary Materials. The microbiota FASTQ files have been deposited in the NCBI database under the accession numbers PRJNA489470 and PRJNA488108.