

Abstract
Influenza A virus (IAV) infection alters lung epithelial cell metabolism in vitro by promoting a glycolytic shift. We hypothesized that this shift benefits the virus rather than the host and that inhibition of glycolysis would improve infection outcomes. A/WSN/33 IAV-inoculated C57BL/6 mice were treated daily from 1 day post-inoculation (d.p.i.) with 2-deoxy-D-glucose (2-DG) to inhibit glycolysis and with the pyruvate dehydrogenase kinase (PDK) inhibitor dichloroacetate (DCA) to promote flux through the TCA cycle. To block OXPHOS, mice were treated every other day from 1 d.p.i. with the Complex I inhibitor rotenone (ROT). 2-DG significantly decreased nocturnal activity, reduced respiratory exchange ratios, worsened hypoxemia, exacerbated lung dysfunction, and increased humoral inflammation at 6 d.p.i. DCA and ROT treatment normalized oxygenation and airway resistance and attenuated IAV-induced pulmonary edema, histopathology, and nitrotyrosine formation. None of the treatments altered viral replication. These data suggest that a shift to glycolysis is host-protective in influenza.
Keywords: Acute respiratory distress syndrome, Metabolism, Nitrotyrosine, Oxidative phosphorylation
Introduction
Seasonal influenza A virus (IAV) morbidity and mortality rates remain high despite the use of vaccinations and antiviral medications. Severe primary influenza can progress to acute respiratory distress syndrome (ARDS), which has a mortality rate of approximately 40%1. We have shown that mice with influenza A/WSN/33 (H1N1) infection develop essential features of severe ARDS by 6 days post inoculation (d.p.i.), including decreases in arterial oxygenation, static lung compliance, and alveolar fluid clearance rate, which are accompanied by pulmonary edema and inflammation2. This model serves as an ideal tool to study metabolic aberrations within the host during IAV infection at both organismal and cellular levels.
Viruses such as IAV depend on host cells for their replication and assembly and therefore require both a readily available energy source and molecular building blocks including amino acids, carbohydrates, and lipids. In addition, the host immune response to viral infection requires large amounts of energy and intermediate metabolites. Following IAV infection, there is evidence that respiratory epithelial cell metabolism shifts from using OXPHOS as the primary means of ATP generation to using glycolysis3,4. Previous studies have shown that IAV increases glucose uptake, glucose consumption, glycolytic rate, and ATP production in epithelial cells while glycolysis inhibition may reduce viral replication5. However, the majority of these studies used continuously growing cancer cell lines, which often preferentially generate ATP by glycolysis even in the absence of infection6. Hence, the impact of this glycolytic shift on host lung function remains unclear.
Since it provides the virus with ATP and intermediate metabolites, we hypothesized that this glycolytic shift is beneficial to viral replication but detrimental to the host. Hence, we predicted that inhibition of glycolysis would impede viral replication, promote a shift in ATII cell metabolism towards OXPHOS, and attenuate clinical features of ARDS. Conversely, we expected that systemic blockade of either entry of pyruvate into the TCA cycle or OXPHOS would exacerbate the metabolic aberrations in IAV-infected mice and increase ARDS severity. Surprisingly, we found the opposite to be the case. When mice were treated systemically with 2-deoxy-D-glucose (2-DG) to inhibit hexokinase (the first step in glycolysis), ARDS severity increased. Conversely, treatment of mice with dichloroacetate (DCA), which inhibits pyruvate dehydrogenase kinase (PDK) and thereby promotes conversion of pyruvate to acetyl CoA, reduced IAV-induced hypoxemia and lung dysfunction. Likewise, treatment with rotenone (ROT), which inhibits Complex I of the electron transport chain, significantly attenuated IAV effects. These data indicate that a shift to glycolysis is a host-protective response in influenza and suggest that modulators of host metabolism can improve influenza outcomes.
Methods
Mice.
Adult C57BL/6 mice (Mus musculus) of both sexes were purchased from Charles River Laboratories (Wilmington, MA). Mice were provided with ad libitum food and water. All activities were approved by the Institutional Animal Care and Use Committee of The Ohio State University.
Mouse infection.
Mice were inoculated intranasally with 10,000 plaque-forming units (pfu) of Mycoplasma- and endotoxin-free influenza A/WSN/33 in 50 μl PBS with 0.1% BSA under ketamine/xylazine anesthesia, as in previous studies7. Mock controls were inoculated intranasally with 50 μl PBS with 0.1% BSA. Data for each experimental group were derived from a minimum of two independent mock or IAV infections.
Mouse treatments.
To inhibit glycolysis, mice were treated daily from 1 to 5 d.p.i. with 0.76 g/kg 2-deoxy-D-glucose (2-DG; Sigma-Aldrich, St. Louis, MO) in 50 μl sterile saline by intraperitoneal (i.p.) injection8. Treatment controls received 50 μl sterile saline i.p. daily. To block PDH kinase, mice were treated daily with 50 mg/kg dichloroacetate (DCA; 50 mg/kg) in 70 μl sterile saline i.p.9 Treatment controls received 70 μl sterile saline i.p. daily. To inhibit OXPHOS, mice were injected every other day from 1 to 5 d.p.i. with 0.8 mg/kg rotenone (ROT; Abcam, Cambridge, MA) in 10 μl 100% dimethyl sulfoxide (DMSO) i.p.10. Controls received 10 μl DMSO at the same treatment intervals.
Measurement of whole-body metabolism.
Open circuit calorimetry and measurement of mouse activity were performed simultaneously using the Oxymax/CLAMS metabolic chamber system (Columbus Instruments, Columbus, OH)11. At 5 d.p.i., mice were fasted and placed in individual CLAMS chambers at room temperature: an untreated mock-infected mouse was included as a control in all CLAMS experiments. Data were collected over the following 24 hours (12 hours light, 12 hours dark). Dark period V̇O2, V̇CO2, and respiratory exchange ratio (RER) were calculated from area under curve (AUC) values.
Histopathology and scoring of severity.
Hematoxylin- and eosin-stained thin sections were generated from formalin-inflated, paraffin-embedded lungs by standard methods. Each section was evaluated in a blinded fashion and scored based on the overall extent of peribronchial, peribronchiolar, interstitial, and intra-alveolar leukocyte infiltrates throughout both lungs. A score of 1 reflected normal histology for each parameter. Mild, moderate, and severe pathology were scored as 2, 3, and 4, respectively, for each parameter. A mean overall score was then calculated by dividing the total lesion score by 412.
Immunohistochemistry:
Deparaffinized lung tissue sections were subjected to antigen retrieval then immunostained with a murine anti-nitrotyrosine monoclonal antibody (Abcam, ab7048, Cambridge, UK) or a nonspecific isotype control antibody (Abcam ab18469). Bound antibody was detected with Mouse on Mouse Polymer HRP (BioCare Medical, Pacheco, CA) and DAB+ Chromogen (Agilent Dako, Santa Clara, CA). Slides were counterstained with hematoxylin. Additional details are provided in an online data supplement.
Other methods.
Measurements of cardiopulmonary function in conscious mice, lung mechanics in mechanically ventilated anesthetized mice, total lung water content, and viral titers were performed as previously described, as was analysis of bronchoalveolar lavage fluid (BALF) composition. Additional details are provided in an online data supplement.
Statistical analyses.
Descriptive statistics were calculated using Instat 3.05 (GraphPad Software, San Diego, CA). Gaussian data distribution was verified by the method of Kolmogorov and Smirnov. Differences between group means were analyzed by t-test for two-group comparisons and by ANOVA for multi-group comparisons, with a post hoc Tukey-Kramer multiple comparison post-test for ANOVA. Correlations were identified from Pearson correlation coefficients. P<0.05 was considered statistically significant.
Results
IAV infection reduces dark period activity and alters whole body metabolism in mice.
We used the CLAMS system to determine the impact of IAV infection and metabolic inhibitors on activity and whole-body metabolism in mice. We found that IAV infection resulted in a significant decline in nocturnal activity in fasted mice at 6 d.p.i. (Fig. 1A). Treatment with the glycolytic inhibitor 2-DG further decreased nocturnal activity. However, treatment with the PDK inhibitor DCA or the Complex I inhibitor ROT had no such effect. None of the treatments reduced activity in mock-infected mice.
Figure 1. Influenza A/WSN/33 (H1N1) virus infection reduces dark period activity and alters whole body metabolism in mice.
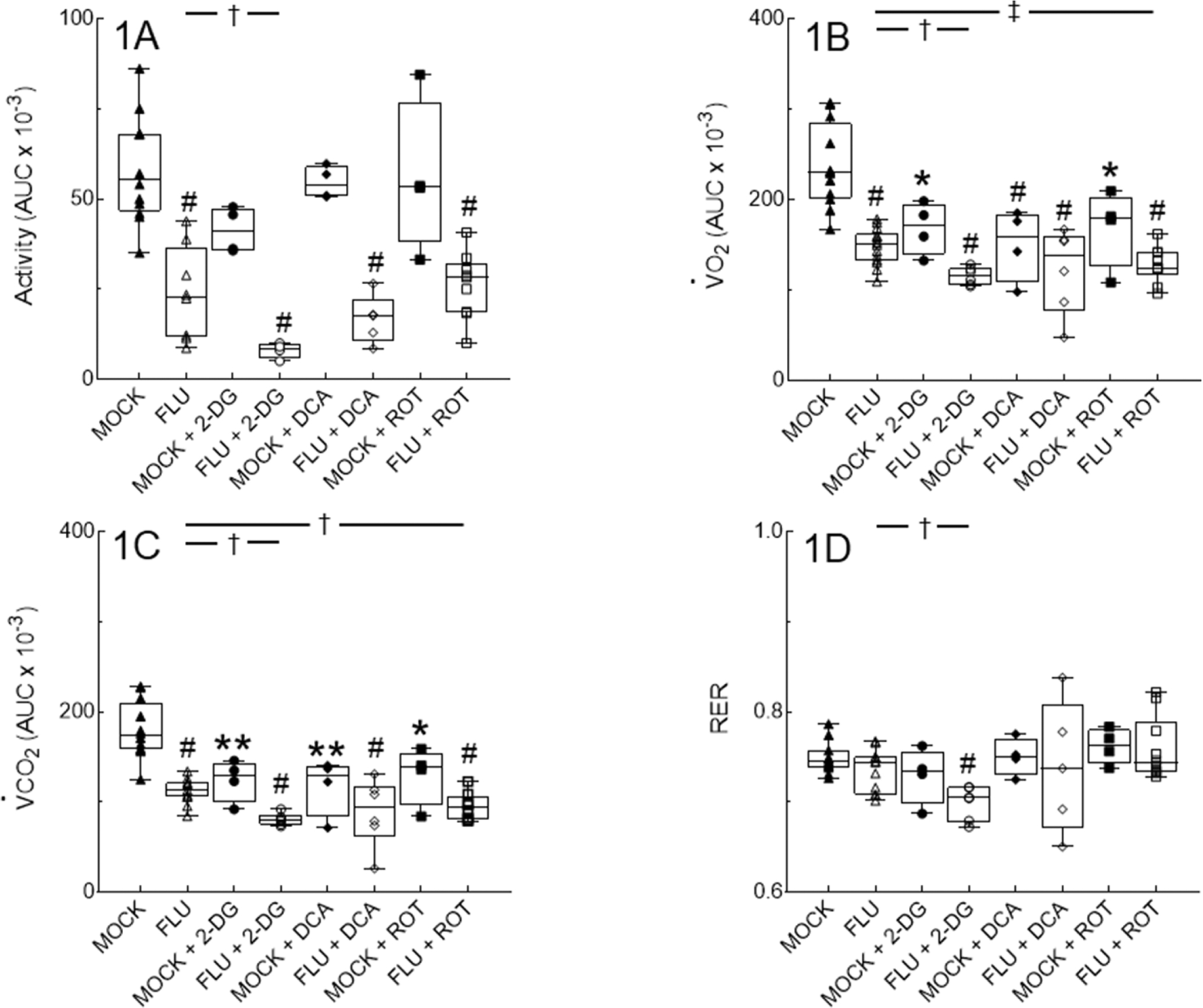
Effect of mock and IAV infection (FLU) and treatment with 2-deoxy-D-glucose (2-DG; 0.76 g/kg, daily), dichloroacetate (DCA; 50 mg/kg, daily), or rotenone (ROT; 0.8 mg/kg. every other day) from 1–5 days post-inoculation (d.p.i.) on: (A) Nocturnal activity (expressed as area under curve [AUC]); (B) O2 consumption (V̇O2; expressed as AUC); (C) CO2 production (V̇CO2; expressed as AUC); and (D) Respiratory exchange ratio (RER). All measurements were made from 5–6 d.p.i. n=12, 8, 4, 4, 4, 5, 4, and 9 per group (from 12 individual mock or IAV infections). Data in box represent first quartile, median, and third quartile for each experimental group. Whiskers indicate highest and lowest sample values within the group. *: P<0.05, **: P<0.005, #: P<0.001, vs. mock-infected mice. †: P<0.05, ‡: P<0.005, vs. IAV-infected mice.
IAV infection resulted in a significant decrease in dark period O2 consumption (V̇O2; Fig. 1B) and CO2 production rate (V̇CO2; Fig. 1C) in fasted mice at 6 d.p.i. Interestingly, treatment with all three metabolic inhibitors had comparable effects on V̇O2 and V̇CO2 in mock-infected mice to those of IAV infection. V̇O2 in 2-DG-treated, IAV-infected mice was significantly lower than V̇O2 in both 2-DG-treated mock-infected mice and untreated IAV-infected mice. Effects of DCA and ROT treatment on V̇O2 were more variable, but overall V̇O2 in DCA- or ROT-treated, IAV-infected mice did not differ from mock-infected, inhibitor-treated controls.
There was a significant correlation between activity and V̇O2 (P=0.0122, r2=0.3014) and activity and V̇CO2 (P=0.0025, r2 =0.4063) in untreated mock- and influenza-infected mice. For 2-DG-treated mock- and influenza-infected mice, these correlations persist (P=0.0035, r2=0.7833 for V̇O2 and P=0.0032, r2=0.7855 for V̇CO2). Likewise, there was a significant correlation between activity and V̇O2 (P=0.0002, r2=0.7277) and activity and V̇CO2 (P=0.005, r2=0.5617) in rotenone-treated mock- and influenza-infected mice. In contrast, there was no significant correlation between activity and V̇O2 or V̇CO2 in DCA-treated mock- and influenza-infected mice.
Respiratory exchange ratio (RER) can be used as a surrogate for the respiratory quotient (RQ), which is an indicator of whether energy supply is primarily derived from carbohydrates, proteins, or fats. If only carbohydrates are being metabolized, then the RQ is 1.0; for proteins only the RQ is 0.8, and for lipids only the RQ is 0.713. RER values in untreated mock-infected mice were consistent with mixed, but lipid-dominant, substrate utilization (mean RER 0.75; Fig. 1D). Because all three metabolic inhibitors had comparable effects on both V̇O2 and V̇CO2 in mock-infected mice, RER values were not significantly altered in this group. Likewise, IAV infection alone did not significantly change RERs. However, 2-DG-treated IAV infected mice demonstrated significantly lower RER values at 6 d.p.i., indicative of a shift towards predominately lipid catabolism for energy production. In contrast, RER values did not differ from those of mock-infected controls in either DCA- or ROT treated-mice.
Inhibition of PDK and Complex I but not glycolysis improves cardiopulmonary function in IAV-infected mice without altering viral replication.
As in our previous studies, IAV infection resulted in significant hypoxemia (Fig. 2A) and bradycardia (Fig. 2B) at 6 d.p.i. Interestingly, treatment with the glycolysis inhibitor 2-DG significantly worsened IAV-induced hypoxemia, although its effects on cardiac function were more variable. In contrast, blockade of PDK with DCA significantly attenuated IAV-induced hypoxemia and inhibition of Complex I with ROT restored carotid SaO2 saturation almost to normal levels. ROT treatment also attenuated IAV-induced bradycardia. None of the treatments caused hypoxemia or bradycardia in mock-infected mice.
Figure 2. Inhibition of PDK and Complex I but not glycolysis improves cardiopulmonary function in IAV-infected mice at 6 d.p.i. without altering viral replication.
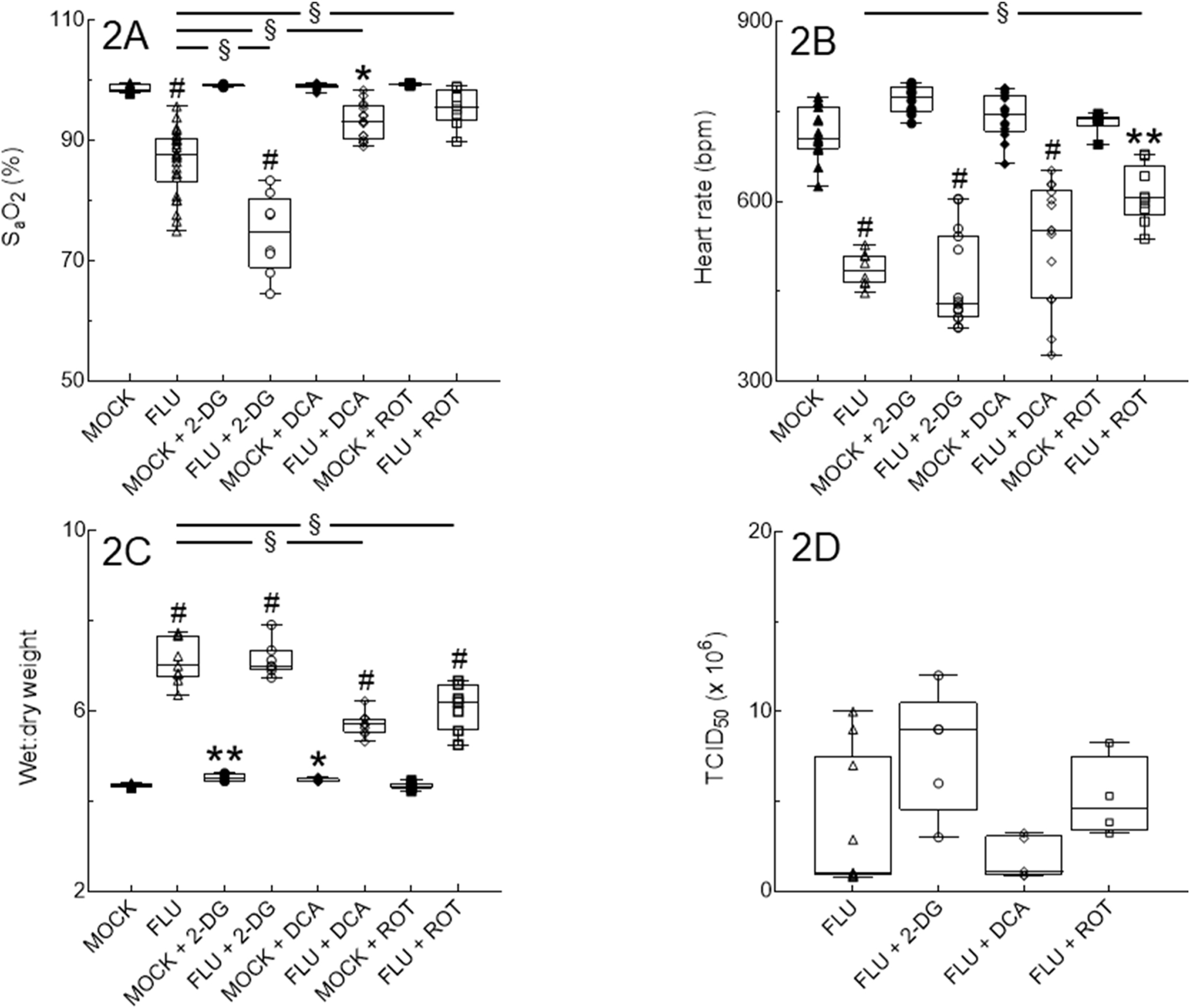
Effect of mock and IAV infection (FLU) and treatment with 2-DG, DCA, or ROT from 1–5 d.p.i. on: (A) Carotid arterial O2 saturation (SaO2; n=9, 26, 5, 8, 10, 16, 6, and 8 per group, from 16 independent mock or IAV infections); (B) Heart rate (n=9, 26, 5, 8, 10, 16, 6, and 8 per group, from 16 independent mock or IAV infections); (C) Lung water content (wet:dry weight ratio; n=6, 9, 5, 7, 5, 10, 8, and 7 per group, from 16 independent mock or IAV infections); and (D) Viral replication (n=5/group, from 2 independent IAV infections per group). Data in box represent first quartile, median, and third quartile for each experimental group. Whiskers indicate highest and lowest sample values within the group. *: P<0.05, **: P<0.005, #: P<0.001, vs. untreated mock-infected mice. §: P<0.001, vs. IAV-infected mice.
IAV infection resulted in significant pulmonary edema (increased lung wet:dry weight ratio) at 6 d.p.i. (Fig. 2C). Treatment with 2-DG or DCA modestly increased lung wet:dry weights in mock-infected mice, but only the latter significantly attenuated pulmonary edema in IAV-infected animals. Treatment with ROT had no effect in mock-infected mice but resulted in a significant decrease in edema in IAV-infected animals. However, neither DCA nor ROT treatment restored lung wet:dry weights to normal levels. Interestingly, none of the inhibitors had any significant effect on viral replication in IAV-infected mice at 6 d.p.i. (Fig 2D).
Detrimental effects of IAV infection on lung function are prevented by PDK and Complex I inhibition and exacerbated by inhibition of glycolysis.
Relative to mock controls, pulmonary resistance increased (Fig. 3A) and static lung compliance decreased (Fig. 3B) after IAV infection. 2-DG treatment had no additional impact on airway resistance but caused a further significant decrease in static lung compliance compared to IAV at 6 d.p.i. In contrast, DCA and ROT treatment reduced pulmonary resistance and increased static lung compliance to mock levels.
Figure 3. Detrimental effects of IAV infection on lung function at 6 d.p.i. are prevented by PDK and Complex I inhibition and exacerbated by inhibition of glycolysis.
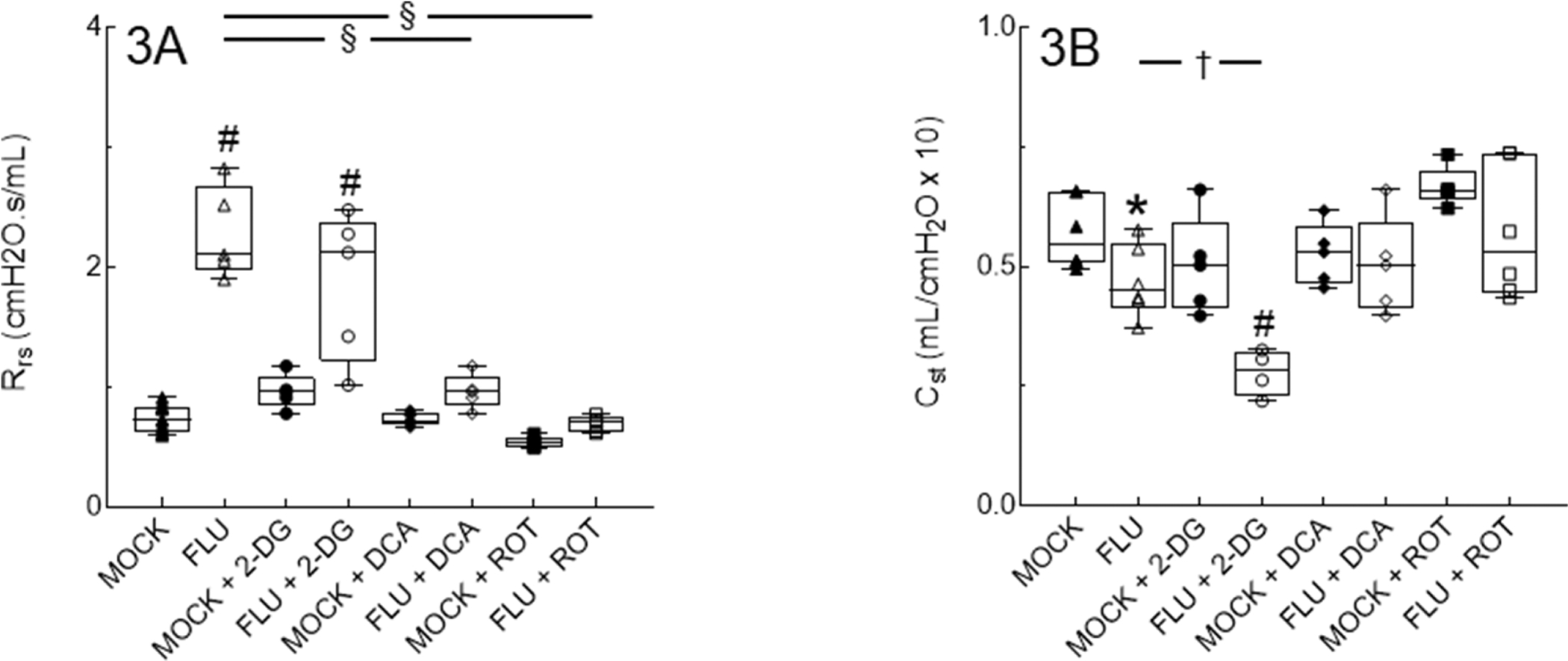
Effect of mock and IAV infection (FLU) and treatment with 2-DG, DCA, or ROT on: (A) Total respiratory system resistance (Rrs; cm H2O · s/ml); and (B) Static lung compliance (Cst; ml/cm H2O). n = 5/group (from 2 independent mock or IAV infections per group). Data in box represent first quartile, median, and third quartile for each experimental group. Whiskers indicate highest and lowest sample values within the group. *: P<0.05, #: P<0.001, vs. mock-infected mice. †: P<0.05, §: P<0.001, vs. IAV-infected mice.
Inhibition of PDK in IAV-infected mice decreases BALF inflammatory infiltrates.
Inhibitor treatment had no effect on BALF macrophage counts in mock-infected mice, which remained low, and did not induce any neutrophil response in this group (Figs. 4A and 4B, respectively). 2-DG and ROT treatment had no effect on BALF infiltrates in IAV-infected mice at 6 d.p.i. However, DCA treatment significantly decreased BALF macrophages and neutrophils at this timepoint.
Figure 4. Inhibition of PDK in IAV-infected mice decreases BALF inflammatory infiltrates.
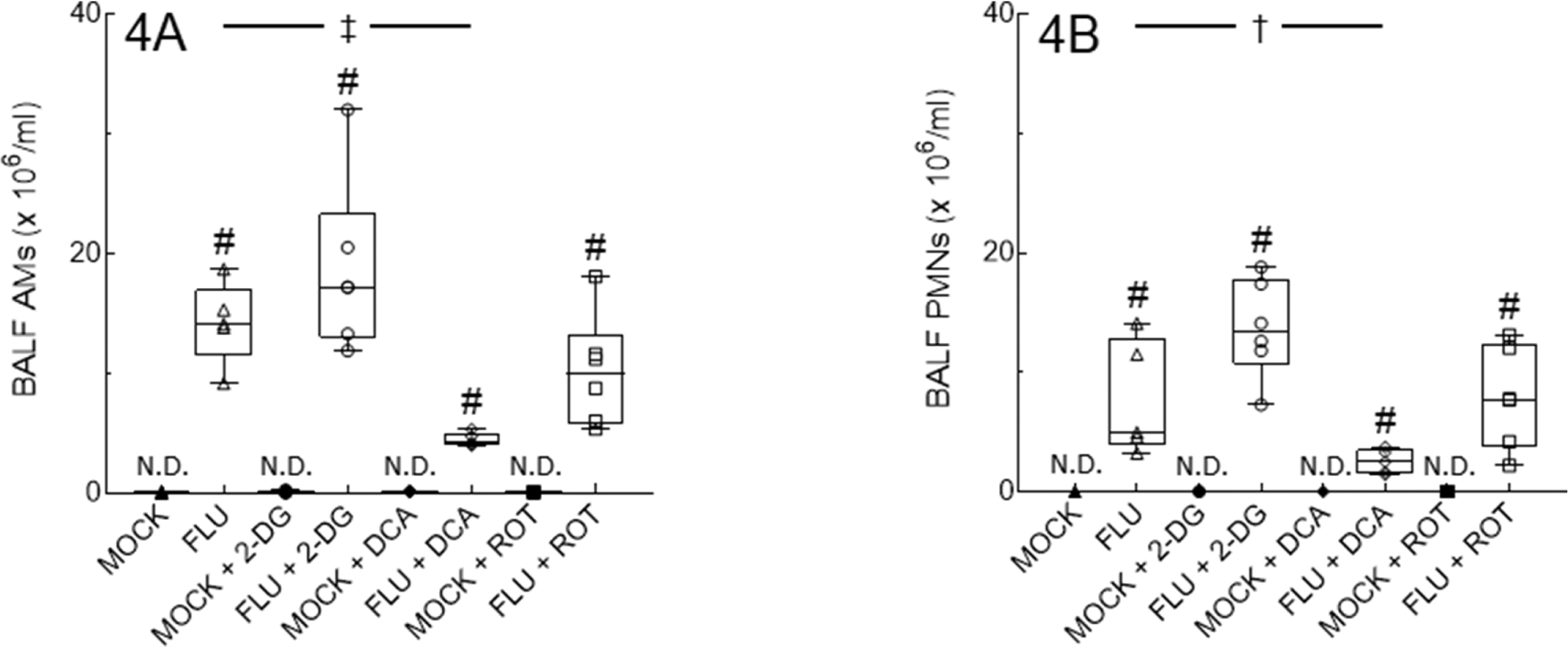
Effect of mock and IAV infection (FLU) and treatment with 2-DG, DCA, or ROT on day 6 BALF: (A) Alveolar macrophages (AMs); and (B) Neutrophils (PMNs). All n = 5/group (from 2 independent mock or IAV infections per group). Data in box represent first quartile, median, and third quartile for each experimental group. Whiskers indicate highest and lowest sample values within the group. N.D.: none detected. #: P<0.001, vs. mock-infected mice. †: P<0.05, ‡: P<0.005, vs. IAV-infected mice.
2-DG and DCA treatment significantly alter BALF cytokine and chemokine responses to IAV infection.
IAV infection significantly increased BALF levels of the cytokines IFN-γ, IL-6, and IL-10, and the chemokine KC/CXCL-1 at 6 d.p.i., although effects on IL-10 were relatively modest (Figs. 5A–5D). 2-DG treatment significantly increased BALF IL-6 and KC and decreased IL-10. DCA treatment caused a significantly greater increase in BALF IL-6 than that induced by 2-DG, but did not impact the IFN-γ, IL-10, and KC responses to IAV infection. Treatment with ROT had no impact on any of these mediators.
Figure 5. 2-DG and DCA treatment significantly alter BALF cytokine and chemokine responses to IAV infection.
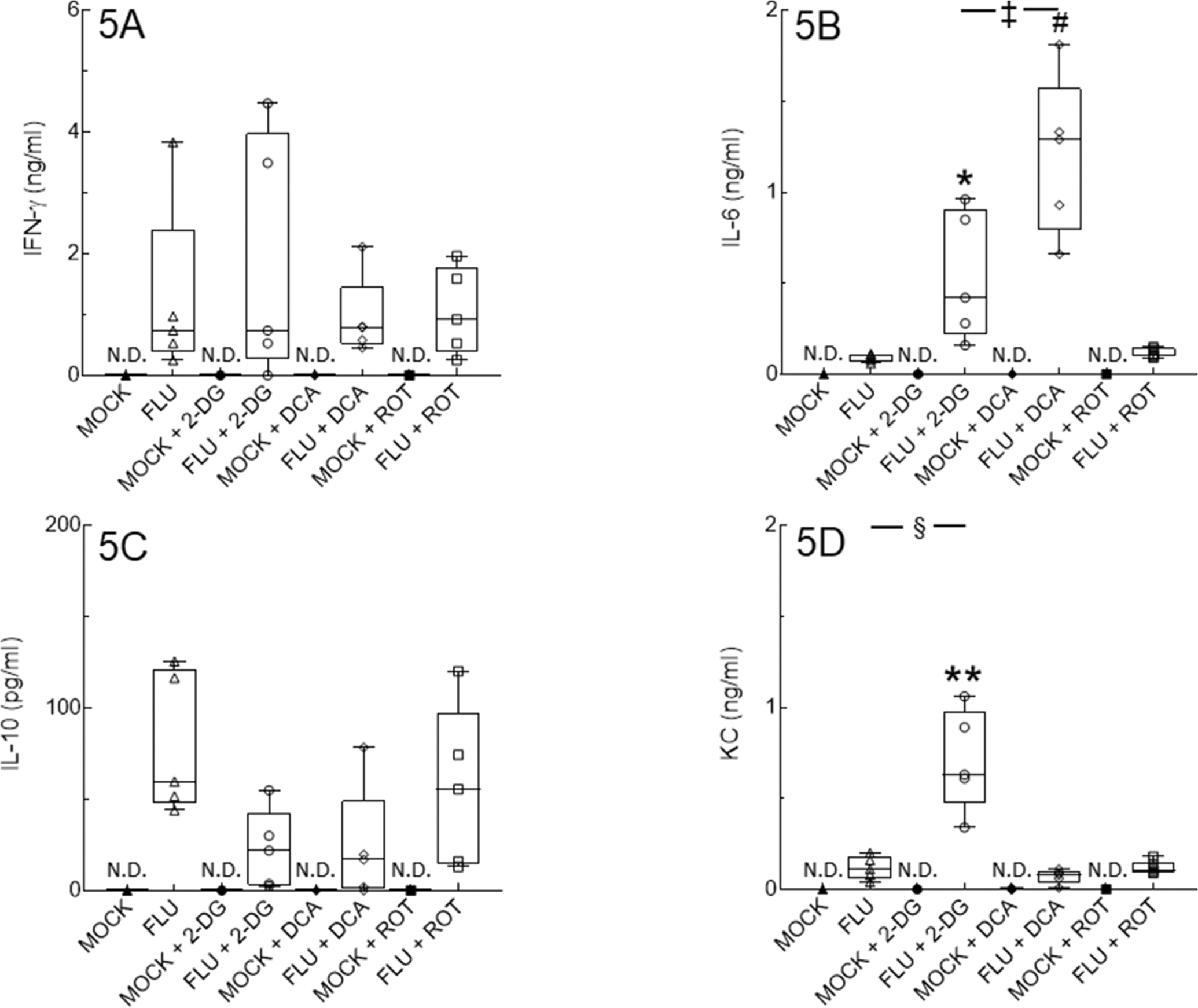
Effect of mock and IAV infection (FLU) and treatment with 2-DG, DCA, or ROT on day 6 BALF levels of: (A) IFN-γ (ng/ml); (B) IL-6 (ng/ml); (C) IL-10 (pg/ml); and (D) KC/CXCL-1 (ng/ml). All n = 5/group (from 2 independent mock or IAV infections per group). Data in box represent first quartile, median, and third quartile for each experimental group. Whiskers indicate highest and lowest sample values within the group. N.D.: none detected. *: P<0.05, **: P<0.005, #: P<0.001, vs. IAV-infected mice. ‡: P<0.005, §: P<0.001, vs. 2-DG-treated, IAV-infected mice.
Inhibition of PDK or Complex I activity, but not glycolysis, reduces lung pathology severity.
Lungs from mock-infected mice were histologically normal. Lungs from all infected groups contained peribronchial and perivascular mononuclear cell and neutrophil infiltrates at 6 d.p.i., although lesion severity varied from moderate to marked depending on the location within in lung parenchyma (Figs. 6A–6H). Subjectively, alveolar and peribronchial inflammation appeared to be attenuated in both DCA- and ROT-treated mice. This was reflected in significantly lower mean lesion scores in both groups, relative to untreated IAV-infected mice (Fig. 7). .
Figure 6. Inhibition of PDK or Complex I activity, but not glycolysis, reduces lung pathology severity.
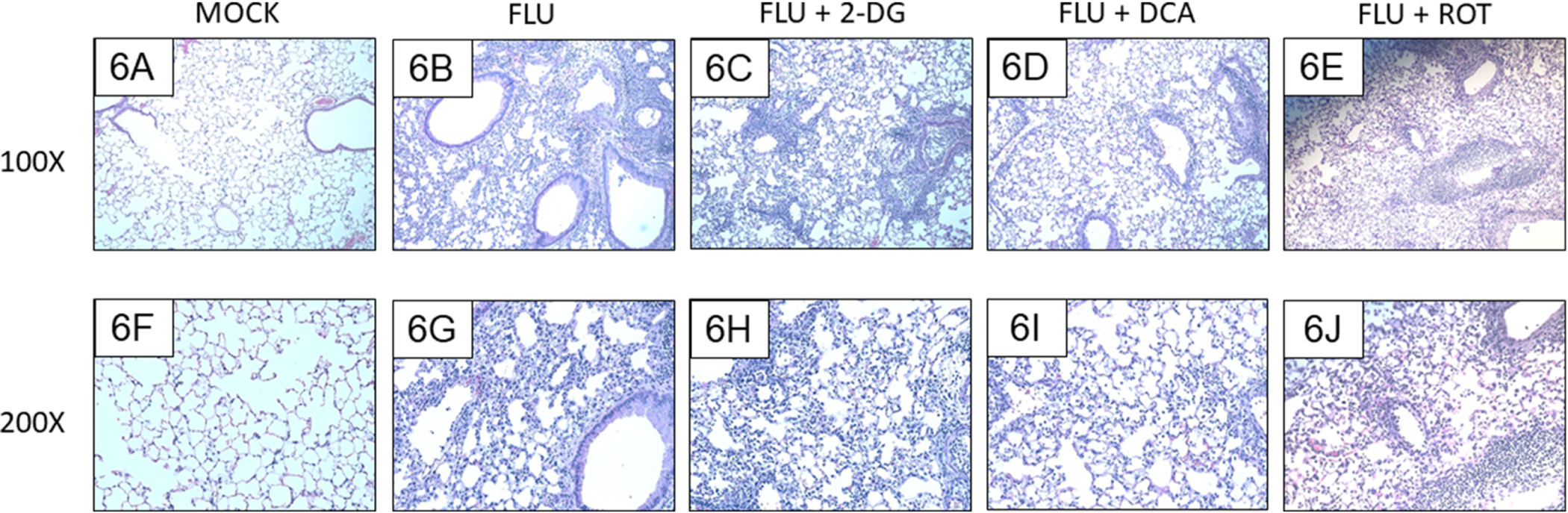
Effect of mock and IAV infection (FLU) and treatment with 2-DG, DCA, or ROT on day 6 lung histology in: (A and F) Mock-infected mice; (B and G) IAV-infected mice; (C and H) 2-DG-treated, IAV-infected mice; (D and I) DCA-treated, IAV-infected mice; and (E and J) ROT-treated, IAV-infected mice. All n = 4/group (from 3 independent mock or IAV infections per group). Magnifications, 100X (A-E) and 200X (F-J). Representative photomicrographs are shown. Brightness and contrast were adjusted equally for all images to improve visibility.
Figure 7. Inhibition of PDK or Complex I activity, but not glycolysis, reduces lung lesion scores.
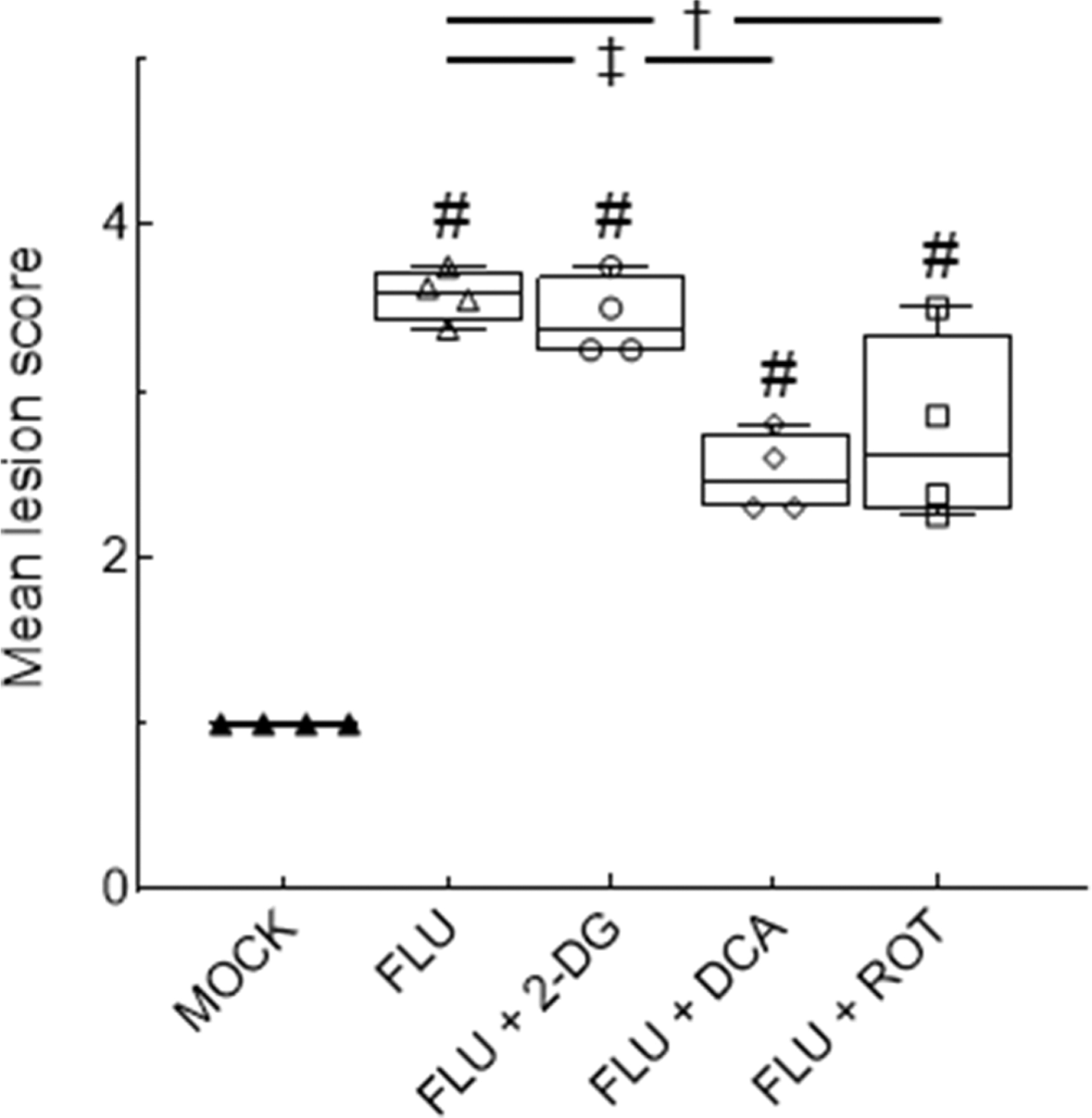
Effect of mock and IAV infection (FLU) and treatment with 2-DG, DCA, or ROT on mean histopathologic lesion scores. All n = 4/group (from 3 independent mock or IAV infections per group). #: P<0.001, vs. mock-infected mice. †: P<0.05, ‡: P<0.005, vs. IAV-infected mice.
Both PDK and Complex I inhibition decrease alveolar epithelial 3-nitrotyrosine in IAV-infected mice.
Mock infection did not induce formation of 3-nitrotyrosine in alveolar macrophages or epithelial cells (Figs. 8F and 8K), although non-specific immunoreactivity was observed in pulmonary vessels. Both intra-alveolar macrophages and type I and II alveolar epithelial cells exhibited strong immunoreactivity at 6 d.p.i., which was increased by 2-DG treatment (Figs. 8G, 8H, 8L, and 8M). Interestingly, both DCA and ROT treatment significantly decreased 3-nitrotyrosine staining in the alveolar epithelium and 3-nitrotyrosine reactivity appeared to be confined primarily to intra-alveolar macrophages and occasional ATII cells in these groups (Figs. 8I and 8N, and 8J and 8O, respectively). No 3-nitrotyrosine immunoreactivity was detectable in sections immunostained with an isotype control antibody (Figs. 8A–8E).
Figure 8. Both PDK and Complex I inhibition decrease alveolar epithelial 3-nitrotyrosine in IAV-infected mice.
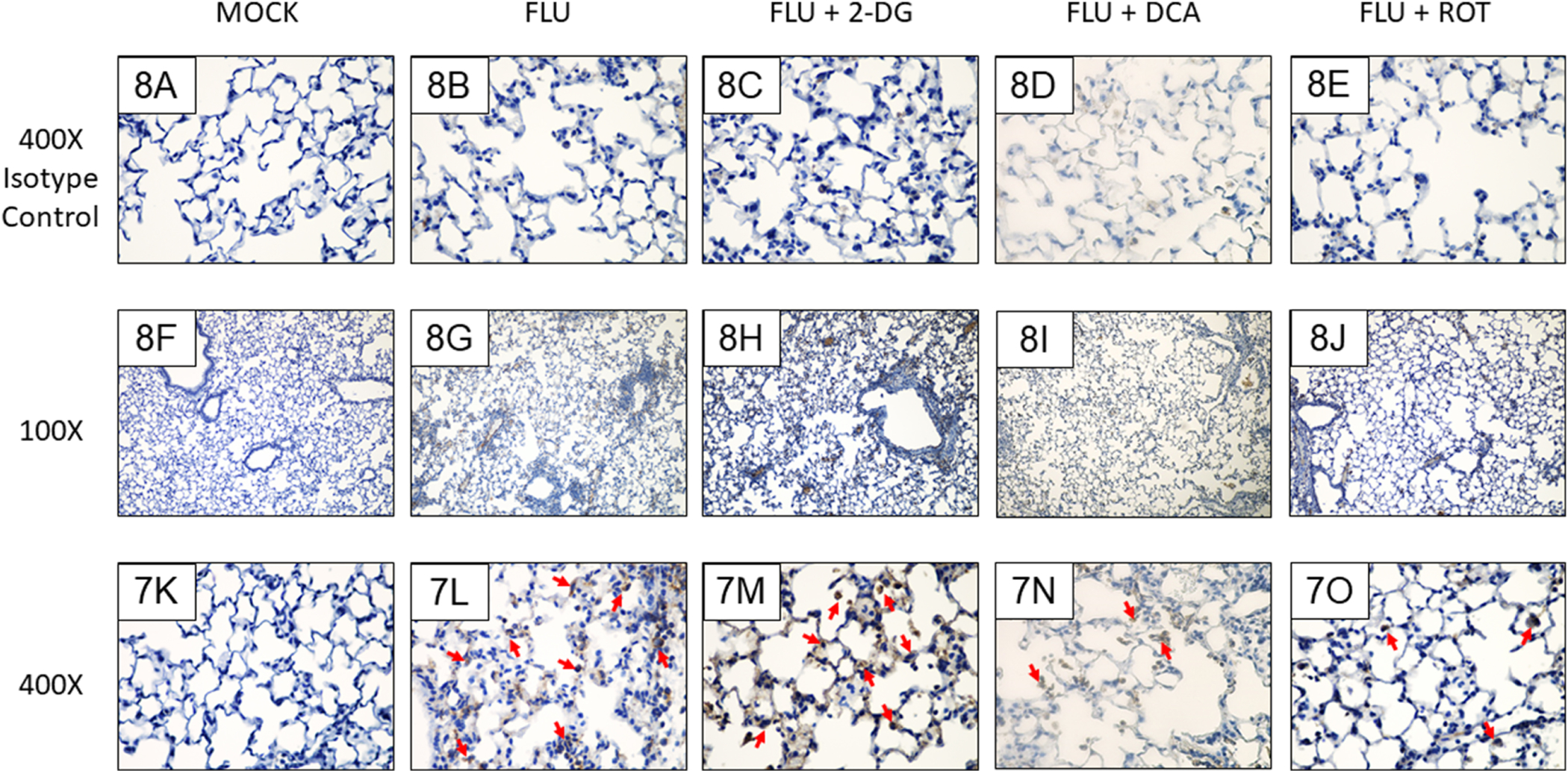
Effect of mock and IAV infection (FLU) and treatment with 2-DG or ROT on day 6 3-nitrotyrosine formation in: (F and K) Mock-infected mice; (G and L) IAV-infected mice; (H and M) 2-DG-treated, IAV-infected mice; (I and N) DCA-treated, IAV-infected mice; and (J and O) ROT-treated, IAV-infected mice. (A-E) Isotype controls. All n = 3/group (from 2 independent mock or IAV infections per group). Magnifications, 400X (A-E and K-O), 100X (F-J). Representative photomicrographs are shown. Brightness and contrast were adjusted equally for all images to improve visibility. Red arrowheads indicate 3-nitrotyrosine-positive cells.
Discussion
Each year, 3–5 million cases of severe influenza occur with >300,000 deaths worldwide14. Acute respiratory distress syndrome is a potentially fatal consequence of severe IAV infection, with few effective treatments currently available. Viral replication, alveolar epithelial cell dysfunction, and an overwhelming inflammatory response contribute to disease pathogenesis. In the present study, inhibition of the glycolytic pathway exacerbated disease severity while blockade of PDK or Complex I improved clinical features. This provides insight into IAV pathogenesis as this glycolytic shift may allow the host to adapt to infection while PDK and OXPHOS pathway may prove to be potential therapeutic targets.
As in our previous studies, we found that influenza infection resulted in both hypoxemia and bradycardia, which were accompanied by reduced metabolic activity (V̇O2 and V̇CO2). In contrast, human influenza infection results in tachycardia and increased V̇O2 and V̇CO2. This is a significant difference between murine influenza models and human influenza, and a limitation of the former. While we do not know the reason for this discrepancy, we speculate that, because mice have such high resting metabolic rates (and hence very high energy demands), the reduction in food consumption associated with sickness (and reflected in decreased nocturnal activity) results in starvation and an inability to generate enough ATP to keep V̇O2, V̇CO2, and heart rate up at normal levels: this effect is reflected by the very rapid weight loss observed in influenza-infected mice.
Viruses require readily available energy sources and molecular building blocks to replicate and assemble progeny. IAV rapidly induces increased glucose uptake, glycolysis and lactic acid production in epithelial cell lines5 and primary normal human bronchial epithelial (NHBE) cells15. In vivo, IAV has been shown to increase glucose uptake by non-immune cells in the lungs of a pediatric patient15 and infiltrating macrophages in a murine model16. However, there is conflicting evidence regarding IAV’s dependence on glycolysis for successful propagation and no information regarding the functional consequences of this glycolytic shift for the host lung. Kohio and Adamson showed that supplementation of the culture media with glucose enhanced H1N1 IAV (A/PR/8/34) replication in MDCK cells4. Moreover, pre-treatment of cultures with 2-DG prior to IAV inoculation suppressed viral replication, which these authors attributed to reduced viral entry due to inhibition of the glucose-dependent vacuolar-type H+ ATPase pump. In contrast, Zhao et al. showed that ATII cell-specific deletion of HIF-1α, which promotes glycolysis, enhanced A/PR/8 (H1N1) IAV replication in vitro and in vivo and increased pulmonary inflammation in mice17. Finally, Wang et al. found that, despite increasing disease severity, 2-DG administration did not alter A/WSN/33 (H1N1) IAV replication in mice18. We likewise found that 2-DG treatment increased IAV severity: 2-DG-treated IAV-infected mice were significantly less active and developed more severe hypoxemia and pulmonary edema than vehicle-treated IAV-infected controls at 6 d.p.i. Additionally, static lung compliance, which is an index of lung stiffness, was significantly lower in these mice: this may reflect an inhibitory effect on surfactant production secondary to inhibition of glycolysis. These detrimental effects of 2-DG were associated with increased levels of IL-6 and KC, and reduced amounts of IL-10 in BALF, suggesting an immune effect. Interestingly, our whole-body metabolism experiments indicated that 2-DG treatment also provoked a shift to lipid catabolism as a source of energy production (decreased RER). Since this metabolic shift did not alter viral replication, this indicates that IAV has the capacity to exercise flexibility when it comes to using different energy sources for its replication. It is important to note, however, that inhibition of such an early step in such a complex metabolic pathway can have indirect downstream knock-on effects. Unless a compensatory increase in fatty acid oxidation occurs, inhibition of glycolysis by treatment with 2-DG will probably reduce availability of acetyl CoA for entry into the TCA cycle. In turn, this could lead to decreased generation of reducing intermediates and thereby a decline in total ATP generation. Currently, we cannot exclude the possibility that this phenomenon, rather than a direct effect on glycolysis could account for the severe detrimental effects of this inhibitor in IAV-infected mice.
In contrast to the detrimental effects of 2-DG, post-inoculation DCA treatment resulted in significant attenuation of IAV-induced hypoxemia and pulmonary edema. Interestingly, despite the fact that lung water content remained elevated, DCA treatment also restored normal lung function (pulmonary resistance and static lung compliance) in IAV-infected mice at 6 d.p.i. The most likely explanation for this discrepancy is that the increase in pulmonary wet:dry weights seen in influenza-infected mice is a reflection of peribronchial edema: this would have a greater impact on airway resistance and less effect on static lung compliance than would alveolar interstitial edema and/or impaired surfactant production (which we have shown is a component of influenza infection in our model19). However, DCA had no effect on VĊO2, V̇O2, and RER. DCA was the only metabolic inhibitor tested that reduced BALF alveolar macrophage and neutrophil counts. DCA also induced a dramatic IL-6 response, which we have found to be associated with improved outcomes in our influenza model. While Yamane et al., showed similar effects in mice infected with A/PR/8 IAV treated with a different PDK inhibitor (diisopropylamine dichloroacetate)20, these authors did not measure metabolic rate and found that PDK inhibition reduced IAV replication, which could explain its beneficial effects in their model. In contrast, we did not observe any antiviral effect of DCA in A/WSN/33 IAV-infected mice.
In the absence of an antiviral effect, it is difficult to explain how DCA treatment attenuates IAV-induced hypoxemia and lung injury. Activation of PDK promotes formation of lactate from pyruvate (aerobic glycolysis) and its inhibition enhances flux from glycolysis into to the TCA cycle by increasing activity of the pyruvate dehydrogenase complex, which catalyzes oxidative decarboxylation of pyruvate to acetyl CoA. Increased TCA cycle activity leads to increased generation of reducing intermediates (NADH and FADH2), which are essential for synthesis of fatty acids, nucleotides, and some amino acids (e.g., glutamate), as well as for reduction of oxidized antioxidants and OXPHOS. Enhanced flux from glucose into the TCA cycle will also increase generation of acetyl CoA for fatty acid synthesis. Hence, by promoting synthesis of antioxidants and metabolic intermediates to replace those consumed by viral replication, DCA treatment may facilitate host cell survival.
It is not clear why systemic inhibition of PDK with DCA results in reduced pulmonary infiltrates. We have little evidence that DCA treatment reduces leukocyte recruitment to the lung, since it has no effect on BALF levels of either the neutrophil chemoattractant KC or the monocyte chemoattractant MCP-1 (data not shown). Hence, reduced leukocyte infiltration into the lungs of DCA-treated, IAV-infected mice probably reflects an alteration in leukocyte TCA cycle metabolism. A recent report from Woods et al. showed that, in influenza, tissue-resident and infiltrating monocyte-derived macrophages are metabolically distinct16. Tissue-resident macrophages appear to primarily generate energy by oxidative phosphorylation irrespective of influenza infection status, while infiltrating monocyte-derived macrophages upregulate glycolysis in response to infection. Conceivably, therefore, DCA treatment could impact both cell types while 2-DG and ROT only affect one subset or the other. However, it will be necessary to further characterize BALF macrophage subsets in our model to determine the differential effects of each inhibitor on both resident and infiltrating cells.
Post-inoculation ROT treatment also had significant beneficial effects on arterial oxygen saturation, airway resistance, and static lung compliance in IAV-infected mice. In addition, ROT significantly reduced pulmonary edema. However, it did not impact IAV-induced pulmonary inflammation. Although ROT was shown to be toxic when administered to rats at very high doses for prolonged periods21, this inhibitor is documented to be generally safe when used in vivo10,21,22. Indeed, we found no evidence of weight loss, hypoxemia, pulmonary edema or lung pathology in mock-infected mice treated with ROT at the same dose and frequency as infected animals. To our knowledge, this is the first study to demonstrate a beneficial effect of ROT in IAV-infected mice, although other investigators have found that treatment of IAV-infected mice with the Complex I inhibitor metformin attenuates disease23. However, metformin also inhibits AMP kinase, so could be modulating influenza severity independently of its effects on Complex I. Nevertheless, there is evidence that ROT treatment is protective in other inflammatory diseases. For example, ROT suppressed caspase activation, TNF-α production, and hepatocellular apoptosis in a murine model of LPS-induced hepatitis, resulting in improved survival22. Likewise, myeloperoxidase activity, neutrophil recruitment, and TNF-α levels significantly decreased after ROT treatment in an ischemia-reperfusion model of intestinal mucosal damage24. Finally, in mice with LPS-induced lung injury, ROT treatment decreased wet-to-dry ratios, BALF neutrophil counts, and pulmonary myeloperoxidase concentrations thereby decreasing disease severity10. However, in our model, ROT did not attenuate the host inflammatory response to IAV infection.
Increased oxidative stress is a primary mechanism that underpins IAV-induced acute lung injury25. In normal cells, mitochondrial electron transport chain Complexes I and III constitutively produce ROS, including hydroxyl radicals and superoxide anions. During IAV infection, alveolar epithelial cells and macrophages express inducible nitric oxide synthase (Nos2) and generate large quantities of nitric oxide and ROS26–28. Mitochondrial superoxide can react rapidly with cytosolic nitric oxide to form peroxynitrite29 which is a principal mediator of oxidative stress-dependent lung injury25. Peroxynitrite is highly reactive and can damage DNA, RNA, proteins, lipids, and metal centers. 3-nitrotyrosine is a stable marker of peroxynitrite formation and increased 3-nitrotyrosine formation is associated with greater influenza severity30,31. We found that both chronic DCA and ROT treatment decreased formation of 3-nitrotyrosine in the lung, particularly in alveolar epithelial cells. This finding indicates that ROS generated by Complex I in the mitochondria of alveolar epithelial cells may contribute more to the development of severe lung injury in IAV-infected mice than macrophage-derived ROS. This is in agreement with an earlier study showing that treatment with the mitochondrion-targeted anti-oxidant MitoTEMPO improved IAV outcomes32. However, MitoTEMPO treatment also reduced viral replication, which could underlie its observed protective effect. Additionally, while we cannot exclude the possibility that ROT effects are secondary to allosteric inhibition of TCA cycle enzymes resulting from reduced electron flow through Complex I, the fact that DCA treatment, which should accelerate the TCA cycle, also improves influenza outcomes suggests that this is not the case. Finally, it should be noted that ROT can also disrupt microtubules33. Given the important role of these organelles in influenza viral entry and replication within cells34, we cannot exclude the possibility that ROT effects are independent of its impact on Complex I. Nevertheless, we believe that the lack of impact of ROT treatment on viral replication makes this last possibility less likely.
In conclusion, our data show that treatment of IAV-infected mice with DCA (a PDK inhibitor) or ROT (which blocks Complex I of the electron transport chain) attenuated clinical features of ARDS. In contrast, inhibition of glycolysis with 2-DG resulted in a shift in whole-body metabolism towards lipid catabolism and increased ARDS severity. Although further research is necessary to better understand the mechanisms underlying these effects, our data provide novel insight into influenza pathogenesis and suggest that PDK and Complex I may serve as promising therapeutic targets for host-directed treatment of IAV infection. Likewise, should further research indicate that PDK and/or Complex I-derived ROS play a similar role in the pathogenesis of COVID-19, ROT and related agents might prove to be a valuable part of our armamentarium against this devastating pandemic.
Supplementary Material
Research highlights.
Influenza infection causes a glycolytic shift in respiratory epithelial cells
Effects of influenza on whole body metabolism and nocturnal activity were measured
Systemic inhibition of glycolysis exacerbates influenza severity
Blockade of pyruvate dehydrogenase kinase or Complex I improves outcomes
Complex I blockade attenuates nitrotyrosine formation in the lungs
Acknowledgments
Studies were supported by The C. Glenn Barber Fund (to L.M.D.) and The National Heart Lung and Blood Institute at the National Institutes of Health (R01-HL137090; to I.C.D. and R01-HL138738 to K.I.S.). We would like to thank Ms. Lisa Joseph for her excellent technical support and the Comparative Pathology and Mouse Phenotyping Shared Resource at OSU CVM (especially Dr. Sue Knoblaugh and Ms. Chelssie Breece for immunohistochemistry optimization and characterization). In addition, we wish to acknowledge the technical support provided by members of the Singh lab.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Credit author statement
K.E.N., L.M.D. and A.M.N. performed experiments, acquired data, and assisted with drafting of the final manuscript; L.A.B., P.K., L.E.R. and J.M.H.D. performed data analysis and interpretation, and assisted with drafting of the final manuscript; K.I.S., and H.S. designed and directed experiments and assisted with drafting of the final manuscript; I.C.D. directed experiments, performed data interpretation, and drafted the final manuscript.
The authors report no conflicts of interest.
References
- 1.Huppert LA, Matthay MA, Ware LB. Pathogenesis of acute respiratory distress syndrome. Semin Respir Crit Care Med. 2019;40(1):31–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Traylor ZP, Aeffner F, Davis IC. Influenza A H1N1 induces declines in alveolar gas exchange in mice consistent with rapid post-infection progression from acute lung injury to ARDS. Influenza Other Respir Viruses. 2013;7(3):472–479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ritter J, Wahl A, Freund S, Genzel Y, Reichl U. Metabolic effects of influenza virus infection in cultured animal cells: Intra- and extracellular metabolite profiling. BMC Systems Biol. 2010;4(1):61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Kohio HP, Adamson AL. Glycolytic control of vacuolar-type ATPase activity: A mechanism to regulate influenza viral infection. Virology. 2013;444(1):301–309. [DOI] [PubMed] [Google Scholar]
- 5.Bahadoran A, Bezavada L, Smallwood HS. Fueling influenza and the immune response: Implications for metabolic reprogramming during influenza infection and immunometabolism. Immunol Rev. 2020;295(1):140–166. [DOI] [PubMed] [Google Scholar]
- 6.Vanhove K, Graulus GJ, Mesotten L, et al. The metabolic landscape of lung cancer: new insights in a disturbed glucose metabolism. Front Oncol. 2019;9:1215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Aeffner F, Woods PS, Davis IC. Ecto-5’-nucleotidase CD73 modulates the innate immune response to influenza infection but is not required for development of influenza-induced acute lung injury. Am J Physiol Lung Cell Mol Physiol. 2015;309(11):L1313–L1322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Zheng Z, Ma H, Zhang X, et al. Enhanced glycolytic metabolism contributes to cardiac dysfunction in polymicrobial sepsis. J Infect Dis. 2017;215(9):1396–1406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kato-Weinstein J, Lingohr MK, Orner GA, Thrall BD, Bull RJ. Effects of dichloroacetate on glycogen metabolism in B6C3F1 mice. Toxicology. 1998;130(2–3):141–154. [DOI] [PubMed] [Google Scholar]
- 10.Zmijewski JW, Lorne E, Zhao X, et al. Mitochondrial respiratory Complex I regulates neutrophil activation and severity of lung injury. Am J Respir Crit Care Med. 2008;178(2):168–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Olsen MK, Johannessen H, Cassie N, et al. Steady-state energy balance in animal models of obesity and weight loss. Scand J Gastroenterol. 2017;52(4):442–449. [DOI] [PubMed] [Google Scholar]
- 12.Gibson-Corley KN, Olivier AK, Meyerholz DK. Principles for valid histopathologic scoring in research. Vet Pathol. 2013;50(6):1007–1015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Patel H, Bhardwaj A. Physiology, Respiratory Quotient. In: StatPearls. Treasure Island (FL): StatPearls Publishing. 2020 [PubMed] [Google Scholar]
- 14.Tavares LP, Teixeira MM, Garcia CC. The inflammatory response triggered by influenza virus: a two edged sword. Inflamm Res. 2017;66(4):283–302. [DOI] [PubMed] [Google Scholar]
- 15.Smallwood HS, Duan S, Morfouace M, et al. Targeting metabolic reprogramming by influenza infection for therapeutic intervention. Cell Rep. 2017;19(8):1640–1653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Woods PS, Kimmig LM, Meliton AY, et al. Tissue-resident alveolar macrophages do not rely on glycolysis for LPS-induced inflammation. Am J Respir Cell Mol Biol. 2020;62(2):243–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Zhao C, Chen J, Cheng L, Xu K, Yang Y, Su X. Deficiency of HIF-1α enhances influenza A virus replication by promoting autophagy in alveolar type II epithelial cells. Emerg Microbes Infect. 2020;9(1):691–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wang A, Huen SC, Luan HH, et al. Opposing effects of fasting metabolism on tissue tolerance in bacterial and viral inflammation. Cell. 2016;166(6):1512–1525.e1512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Woods PS, Doolittle LM, Rosas LE, Joseph LM, Calomeni EP, Davis IC. Lethal H1N1 influenza A virus infection alters the murine alveolar type II cell surfactant lipidome. Am J Physiol Lung Cell Mol Physiol. 2016;311(6):L1160–L1169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Yamane K, Indalao IL, Chida J, Yamamoto Y, Hanawa M, Kido H. Diisopropylamine dichloroacetate, a novel pyruvate dehydrogenase kinase 4 inhibitor, as a potential therapeutic agent for metabolic disorders and multiorgan failure in severe influenza. PLoS ONE. 2014;9(5):e98032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Heinz S, Freyberger A, Lawrenz B, Schladt L, Schmuck G, Ellinger-Ziegelbauer H. Mechanistic investigations of the mitochondrial Complex I inhibitor rotenone in the context of pharmacological and safety evaluation. Sci Rep. 2017;7:45465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Ai Q, Jing Y, Jiang R, et al. Rotenone, a mitochondrial respiratory complex I inhibitor, ameliorates lipopolysaccharide/D-galactosamine-induced fulminant hepatitis in mice. Int Immunopharmacol. 2014;21(1):200–207. [DOI] [PubMed] [Google Scholar]
- 23.Fedson DS. Treating influenza with statins and other immunomodulatory agents. Antiviral Res. 2013;99(3):417–435. [DOI] [PubMed] [Google Scholar]
- 24.Ichikawa H, Takagi T, Uchiyama K, et al. Rotenone, a mitochondrial electron transport inhibitor, ameliorates ischemia-reperfusion-induced intestinal mucosal damage in rats. Redox Rep. 2004;9(6):313–316. [DOI] [PubMed] [Google Scholar]
- 25.Vlahos R, Stambas J, Selemidis S. Suppressing production of reactive oxygen species (ROS) for influenza A virus therapy. Trends Pharmacol Sci. 2012;33(1):3–8. [DOI] [PubMed] [Google Scholar]
- 26.Haddad IY, Pataki G, Hu P, Galliani C, Beckman JS, Matalon S. Quantitation of nitrotyrosine levels in lung sections of patients and animals with acute lung injury. J Clin Invest. 1994;94(6):2407–2413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Vlahos R, Stambas J, Bozinovski S, Broughton BRS, Drummond GR, Selemidis S. Inhibition of Nox2 oxidase activity ameliorates influenza A virus-induced lung inflammation. PLoS Pathog. 2011;7(2):e1001271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Khomich OA, Kochetkov SN, Bartosch B, Ivanov AV. Redox biology of respiratory viral infections. Viruses. 2018;10(8). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Beckman JS, Chen J, Ischiropoulos H, Crow JP. Oxidative chemistry of peroxynitrite. Methods Enzymol. 1994;233:229–240. [DOI] [PubMed] [Google Scholar]
- 30.Liu M, Chen F, Liu T, Chen F, Liu S, Yang J. The role of oxidative stress in influenza virus infection. Microbes Infect. 2017;19(12):580–586. [DOI] [PubMed] [Google Scholar]
- 31.Akaike T, Noguchi Y, Ijiri S, et al. Pathogenesis of influenza virus-induced pneumonia: involvement of both nitric oxide and oxygen radicals. Proc Natl Acad Sci USA. 1996;93(6):2448–2453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.To EE, Erlich JR, Liong F, et al. Mitochondrial Reactive Oxygen Species Contribute to Pathological Inflammation During Influenza A Virus Infection in Mice. Antioxid Redox Signal. 2019;32(13):929–942. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Srivastava P, Panda D. Rotenone inhibits mammalian cell proliferation by inhibiting microtubule assembly through tubulin binding. FEBS J. 2007;274(18):4788–4801. [DOI] [PubMed] [Google Scholar]
- 34.Simpson C, Yamauchi Y. Microtubules in influenza virus entry and egress. Viruses. 2020;12(1):117. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.