

Abstract
Native mass spectrometry (MS) is aimed at preserving and determining the native structure, composition, and stoichiometry of biomolecules and their complexes from solution after they are transferred into the gas phase. Major improvements in native MS instrumentation and experimental methods over the past few decades have led to a concomitant increase in the complexity and heterogeneity of samples that can be analyzed, including protein-ligand complexes, protein complexes with multiple coexisting stoichiometries, and membrane protein-lipid assemblies. Heterogeneous features of these biomolecular samples can be important for understanding structure and function. However, sample heterogeneity can make assignment of ion mass, charge, composition, and structure very challenging due to the overlap of tens or even hundreds of peaks in the mass spectrum. In this review, we cover data analysis, experimental, and instrumental advances and strategies aimed at solving this problem, with an in-depth discussion of theoretical and practical aspects of the use of available deconvolution algorithms and tools. We also reflect upon current challenges and provide a view of the future of this exciting field.
Graphical Abstract
1. INTRODUCTION
1.1. HOW DOES HETEROGENEITY ARISE IN NATIVE MASS SPECTROMETRY?
Native mass spectrometry (MS) enables preservation of noncovalent interactions and thus study of intact biomolecular complexes.1-3 With this technique analytes are gently ionized from aqueous solution into the gas phase, and the mass-to-charge ratio (m/z) is measured. Instrumental parameters are carefully controlled to produce ion populations with low charge states and minimally-perturbed structures, in line with the general goal of native MS to preserve native-like structure (i.e., as close to structures present in the condensed phase as possible). This is most commonly achieved using electrospray ionization (ESI) from approximately micron-diameter capillaries (“nanoelectrospray ionization”, nESI).4 Volatile buffer salts (e.g., ammonium acetate), which disproportionate into volatile neutral molecules that evaporate during nESI, are often used in native MS to produce adequate ionic strength (~100 mM or greater) to maintain biomolecular folds in solution rather than common non-volatile biochemical buffer salts (e.g., sodium chloride). This is due to the propensity of the latter to condense onto the biomolecular ions in essentially random stoichiometries, spread the signal of interest into many peaks, and reduce resolution, as well as to suppress ionization and signal of analytes of interest.5-11 Though it is possible to use other ionization methods, such as Matrix-Assisted Laser Desorption Ionization (MALDI)12,13 or “Inlet Ionization”,14 to transfer native-like ions to the gas phase, this review focuses on approaches to heterogeneity in native nESI-MS.
Since the introduction of biomolecular ESI in 1989 by Fenn and coworkers15 and subsequent pioneering work in the study of intact biomolecular complexes,16-34 the capabilities of native MS have rapidly advanced. While a comprehensive treatment of the history of this field2,35-39 is beyond the scope of this review, we highlight major advancements in instrumentation in the 1990s and early 2000s, including the extension of quadrupole m/z ranges, improvements in transmission of large complexes and mass resolution, and development and commercialization of quadrupole-time-of-flight (Q-TOF), ion mobility-mass spectrometry (IM-MS), and Orbitrap instruments.40-54 These early improvements in turn enabled native MS investigation of samples of ever-increasing size and complexity, including intact viruses and MDa-size complexes.27,29,55,56 Landmark achievements in the mid-2000s and 2010s expanded the use of native MS to membrane proteins embedded in detergent micelles,57,58 lipid Nanodiscs,59-61 and other membrane mimetics,62-64 as well as proteins with numerous proteoforms and extensive glycosylation.65,66 These advancements together with the advantages offered over classic techniques, such as easily-changed solution conditions, minimal sample requirements, and experiment speed, have led to a rapid rise in the use of native MS as a valuable tool in structural biology.3,37,39,67-72
However, the expansion of native MS to study more complex samples has introduced concomitant challenges in interpreting their often highly complicated mass spectra. Ions of large biomolecules and their complexes produced by nESI typically exhibit a distribution of charge states, owing in part to the stochastic nature of the number of charges in the late nESI droplet at the time the biomolecule/complex is ionized.7-9 For relatively homogeneous ion populations, this charge state distribution is often approximately Gaussian,73 and the presence of a non-Gaussian charge state distribution may indicate heterogeneity. In either case, the signal of each biomolecule/complex is thus spread out across the m/z spectrum at several peaks, and a basic goal of native MS is to determine the mass and charge state of each ion from this peak distribution. How, then, does heterogeneity arise in native MS? For the purposes of this review, we define a “heterogeneous” ion population to be one composed of multiple ions that differ in ways beyond their isotopic composition, charge state, or the identity of the charge carrier. Heterogeneity can arise from the biology that produces the individual biomolecules in question (e.g., proteoforms of a protein), distributed association of these biomolecules into complexes (e.g., different stoichiometries of the protein or other biomolecule monomers in related complexes), the presence of multiple conformations or topologies of the same biomolecule/complex in solution, binding of small molecules and ligands (such as lipids, polysaccharides, or other cofactors), adduction of metals and salts present in the aqueous buffer solution, or even from artifacts of the nESI process, such as unwanted activation and dissociation of otherwise homogeneous complexes.9,74-76 In some extreme cases, the mass spectrum for a heterogeneous native ion population may even superficially resemble that of a polydisperse long-chain polymer ion population produced by ESI, with tens or even hundreds of overlapped peaks in the mass spectrum and a wide distribution of charge states.77-80
It has therefore long been recognized that combatting heterogeneity is essential for the success of MS in accurately characterizing native biomolecular samples. Much discussion in the literature to date,3,38,81,82 especially early in the history of this field, has focused on approaches that aim to reduce complexity and heterogeneity at the sample preparation stage with additional or refined purification steps and chromatographic separations or through gas-phase fragmentation/dissociation (such as in tandem MS,19,32,83-86 native top-down MS,87-90 and other methods19,21,28,44,82,91-114) of the heterogeneous subunits. However, advancement in structural biology relies fundamentally upon accurate understanding of biomolecular structure and function in physiologically relevant states, and examples illustrating the importance of heterogeneous features, such as different proteo- and glycoforms, stoichiometries and identities of bound ligands and other cofactors, and multiple coexisting stoichiometries or conformations, in both functional and disease-associated systems abound.71,81,115-122
Thus, this review focuses on approaches which do not seek to rid biomolecular samples of their inherent heterogeneity and instead aim to facilitate interpretation and analysis of their complicated spectra. Strategies of this kind include use of software tools and deconvolution algorithms to directly analyze all mass spectral data as recorded by the instrument (data post-processing and analysis, which are the topics of §2.1 and §2.2), instrumental and experimental approaches aimed at separating ion signals online with mass and charge measurements (§2.3), and manipulation of ion populations to spread out otherwise overlapped signals with solution additives or ion/ion reactions (§2.3). Strategies for extracting composition and structural information without complete analysis of mass spectral data are described in §2.4, including a classification of common heterogeneity types. The order in which algorithms and computational methods are presented should not be taken to imply strict chronology or judgment of value. We also note that the first three algorithms described (§2.1.1-2.1.3) were developed originally for interpretation of denatured ion mass spectra but have also been applied to intact native MS data. We begin discussion of algorithms with MaxEnt (§2.1.1), though this algorithm is not the earliest described here, because it is still widely available and used today through commercial implementations and because its underlying theory predates even the application of ESI to the study of biomolecular complexes.
Below, we highlight some of the most important and widely-used data analysis, experimental, and instrumentation-based approaches to tackling the problem of heterogeneity in native MS, dating from the 1990s to the present and focusing on developments in the last decade. We note that, while available deconvolution algorithms and data analysis tools have been discussed in other reviews,123-126 these have largely focused on their applications, rather than their theoretical basis, benefits, and potential drawbacks. In the interest of filling this gap in the literature, we devote a majority of our discussion to the sections describing data analysis approaches to heterogeneity, as a major objective of this review is to educate potential users on both theoretical and practical aspects of the available tools. While the focus of this review is not on the applications of these algorithms and other methods, we provide references of this kind for interested readers. We follow this with discussion of other (instrumental and experimental) approaches which, in parallel to data analysis algorithms, aim to facilitate interpretation of heterogeneous mass spectra through data simplification and reduction while preserving heterogeneity instead of through sample preparations and/or dissociation methods that result in loss of information. In these sections we draw upon the numerous comprehensive reviews of these specific topics. Although many online solution-phase separation approaches (such as online chromatography methods127-138 and capillary zone electrophoresis139-145) have been introduced to help solve this problem, our discussion is confined to instrumentation and techniques commonly available within mass spectrometers themselves or with small modifications. As this review is written from an academic viewpoint, we refer readers not only to recent native MS work on biotherapeutics127,146-153 but also to many recent efforts and helpful perspectives on this topic from industry scientists representing a variety of biopharmaceutical companies, drawing upon these insights where possible.81,87,131,154-163 Importantly, though implementation of the methods we describe below faces unique challenges in industry (namely, rigorous standardization and commercialization),154 we hold that educating potential users on the theoretical aspects of these approaches is important and beneficial to all who utilize native MS, regardless of background. We conclude by reflecting upon the progress of native MS with respect to the problem of heterogeneity, discussing remaining challenges and future strategies for the field, and providing our view of optimal approaches to facilitate analysis and interpretation of the complicated mass spectra of heterogeneous biomolecules, which is paramount for continued growth of this technique as a tool in structural biology.
1.2. “CHARGE-STATE-SPECIFIC” AND “ZERO-CHARGE” MASS SPECTRA
In principle, every mass spectrum can be decomposed into separate mass spectra for ions of each particular charge state in the observed ion population. For a completely monodisperse ion population, in which all ions are identical but for their isotopic contents, charge state, and charge carriers, these “charge-state-specific” mass spectra will each contain essentially the same information, varying only in abundance according to the charge state distribution. However, for an ion population whose composition varies as a function of charge state, extracting charge-state-specific mass spectra from the observed mass spectrum may make these composition differences much clearer and inform further investigation of the possible physiological or other relevance of these differences. At other times, it can be useful to compile the mass and charge information from all identifiable charge states into a single plot of abundance versus mass (i.e., not m/z), possibly after subtracting the mass of any charge carriers. Such a plot is called a “zero-charge” mass spectrum and is akin to other mass distribution measurements, as in size exclusion chromatography, multi-angle light scattering, and analytical ultracentrifugation, albeit with the typically much greater sensitivity and mass resolution offered by mass spectrometry.2
Beyond producing either zero-charge and/or charge-state-specific mass spectra after deconvolution (§2.1), it can often be useful to accurately determine the mass of the repeated subunit within a polydisperse sample or the mass which is conserved across all members of the polydisperse ion populations (§2.2). Some software tools also enable deduction of the subunit topology of complexes (§2.2), which provides additional useful information in understanding biological structure and function. Instrumental and experimental methods, such as those which separate ions in dimensions other than m/z and those which manipulate ion charge states, add to the arsenal of information which can be gleaned from otherwise complicated mass spectra of heterogeneous samples (§2.3). Even with these state-of-the-art methods, it is sometimes not possible to fully analyze the mass spectrum, but useful information can often be obtained from a more coarse-grained or global perspective (§2.4).
In the following section, we discuss how these different types of information can be obtained from native mass spectra of heterogeneous samples using state-of-the-art computational, instrumental, and experimental methods. These strategies enable uncovering a plethora of valuable information important for proper understanding of structure and function as well as for quality control of manufactured biotherapeutics for extremely challenging samples, such as membrane proteins, lipid Nanodiscs, polymers, antibodies, viruses, and other large biomolecular complexes.81,164-168 This includes, for example, identities and stoichiometries of lipids, detergents, and other small molecules bound, profiling of glyo- and proteoforms, determination of subunit composition and topology, and characterization of the conformation and polydispersity of large complexes.
2. STATE-OF-THE-ART APPROACHES TO HETEROGENEITY IN NATIVE MASS SPECTROMETRY
2.1. DECONVOLUTION AND CONSTRUCTION OF A ZERO-CHARGE MASS SPECTRUM
2.1.1. MaxEnt
One of the oldest and most widely-used approaches to deconvolution of biomolecular mass spectra, especially those exhibiting multiple charge states for each ion, is the maximum-entropy or “MaxEnt” method introduced by Skilling in 1984.169 Although application to deconvolution of mass spectra and commercialization was not achieved until the 1990s, this algorithm traces its roots back much earlier to work170-172 by Shannon, Shore and Johnson, and Tikochinsky, Tishby, and Levine, who developed the concept of information entropy relating to the probability of observing various noisy data sets based on a hypothetical underlying (i.e., noiseless) data set. After initially applying the algorithm to challenges in image processing,169-172 Skilling, Ferrige and coworkers recognized the potential for applying it more generally to other signal processing problems, including deconvolution of electrospray mass spectra.173-175 MaxEnt is still employed today in deconvolution of mass spectra, including as recently as 2021 in which this method was used to characterize structural glycoform heterogeneity of the SARS-CoV-2 spike protein receptor-binding domain.176 Figure 1 illustrates use of MaxEnt for intact antibody samples and glycan-mediated heterogeneity.177 Additional examples of applications of MaxEnt deconvolution to investigation of various intact noncovalent complexes throughout the past several decades are provided in the references.176-196
Figure 1.

Example of MaxEnt mass spectral deconvolution for an intact antibody exhibiting multiple glycoforms (A) and after de-glycosylation (B). Insets show mass spectra used for deconvolution. Reprinted in part with permission from ref. 177. © 2008 Bentham Science Publishers Ltd.
Broadly, the MaxEnt algorithm attempts to explain observed data in the mass spectrum by 1) generating a hypothetical zero-charge spectrum, 2) dividing the hypothetical masses by each of the charge states assumed to be present in the ion distribution (with a correction for the charge carrier mass), and 3) adding the resulting m/z distributions with a charge-state-specific abundance scaling together in the mass spectrum and comparing these to the observed data.173-175 Ideally, mismatches (i.e., “error”) between the hypothetical and observed mass spectra should be randomly distributed over all m/z values. Mathematically, this means that the plausibility for a particular hypothetical mass spectrum given an observed mass spectrum is greatest when the “evidence”, defined as − Σm/z/p(ε(m/z)) log(p(ε(m/z))), with ε(m/z) the error at a given m/z value and p(ε(m/z)) its probability, is maximized.169,173 In other words, a “good” fit to an experimental mass spectrum should not have error piled up into just a few m/z values, rather the error should be spread out over all m/z values. Marshall and coworkers introduced an implementation of MaxEnt in 1997 in which the distribution of charge states is assumed to be “smooth” for electrospray mass spectra, i.e., there is an “evidence” penalty for abrupt discontinuities in the charge state distribution assigned to each peak in the zero-charge spectrum.197
Practically, MaxEnt requires specification of the mass range for the reconstructed zero-charge spectrum (which automatically determines the range of possible charge states based on the m/z range of the experimental mass spectrum) as well as a target full-width-at-half-maximum of the peaks (assumed symmetrical) expected in the zero-charge spectrum.173,174 Because MaxEnt software assumes symmetrical Gaussian peak shapes in fitting, determination of the accurate mass of ions with asymmetric peak shapes (e.g., those with adducts) can be difficult or even prohibitive, as noted in early ESI-MS studies of large (~310-2.2 kDa) biological oligomeric complexes from bacteria and crabs.180,181 It is possible to run MaxEnt with an intentionally very broad zero-charge mass range and concomitant charge state range, but better results are obtained the more closely the user can restrict the mass range (and thus also charge state range) to those actually present in the ion population. This belies a fundamental pitfall of the MaxEnt method in analyzing experimental data, namely that it does not inherently distinguish between noise and true signal and thus will attempt to explain all data used as input, including any baseline or noise left in the experimental mass spectrum, by forcing it into a mass bin in the zero-charge spectrum. This can result in numerous artifactual peaks in the deconvolved zero-charge mass spectrum, occasionally with intensities matching that of the true average mass distribution which may complicate interpretation and analysis, as exemplified through comparison of deconvolution of empty MSP1D1 Nanodisc sample spectra acquired using three different mass spectrometer platforms.196
Thus, MaxEnt often performs better with an initial background subtraction (such as a low-order polynomial that excludes ~30% of the raw experimental data), noise thresholding, and/or smoothing of the input data.173,175 Two major artifacts that can be caused by these mitigating steps include unwanted exclusion of low-abundance peaks and a reconstructed spectrum that can in some cases be highly dependent on the background subtraction and denoising/smoothing used. If an overly broad or narrow charge state or mass range is specified, additional artifact peak distributions can arise.181 Commercially available MaxEnt algorithms173,175,198-201 (such as those available from mass spectrometer manufacturing companies such as Waters Corporation, Bruker Corporation, Agilent Technologies, Thermo Fisher Scientific, and SCIEX) for deconvolving mass spectra do not report mass distributions specific to each charge state, thus charge-state-specific information is largely lost. Finally, the assumed noise statistics used for calculating the “evidence” of a hypothetical zero-charge spectrum and for iterating the algorithm may be different in different commercial implementations of the algorithm, e.g., Gaussian noise statistics in the Micromass/Waters implementation173,189,199 and Poisson noise statistics in that from SCIEX.200 Thus, even if convergence of the algorithm is achieved (which may not even happen if the “evidence space” does not have a single, large extremum), different final zero-charge mass spectra may be obtained using the same input parameters but different commercial implementations of MaxEnt.
2.1.2. Fenn Averaging and Deconvolution Algorithms
In 1989 Mann, Meng, and Fenn introduced two simple methods for determining the mass and charge state belonging to a sequence of well-resolved peaks in protein electrospray mass spectra.73 The first of these methods, which they call an “averaging algorithm”, begins by assigning charge states to all of the ostensibly related peaks in the mass spectrum by assuming that the mass of the charge carrier is known. (If the adduct mass is not known, its effect on charge state assignments is mitigated by fortuitous cancellation of some of the adduct mass terms.) Although the mass of the protein can then be calculated directly from the observed m/z values and assigned charge states (for example, by averaging the mass values calculated for each charge state), a simple way to “tune” the mass of the ion to improve the fit to experimental data is also described in detail.73 This method accounts to some extent for instrument calibration error as well as inaccuracies in determining a nominal mass for each peak in the observed sequence. Ion abundances play no role in this algorithm.
The second algorithm described in the same paper calculates the sum of abundances in the mass spectrum for all m/z values that can be associated with a trial protein mass, a charge carrier mass, and a set of assigned charge states. For a simple sequence of well-resolved peaks with identical abundances, the algorithm produces a “deconvolved” spectrum with large peaks at the protein mass (and multiples thereof), as well as a sequence of smaller peaks that can be used to confirm the highest charge state present in the ion population (see Figure 2).73,202-209 The algorithm produces poorer results for experimental mass spectra with lower resolution and/or with different abundances for each charge state, and an early comparison of the commercial implementation of this algorithm and that of MaxEnt found the latter to be superior especially when the signal-to-noise ratio (S/N) is poor.189 Additionally, as seen in Figure 2, the deconvolved spectrum produced using Fenn’s algorithm is prone to a large, increasing baseline and high-intensity sidebands relative to the true protein mass.73 Charge state assignments can also have large uncertainties for large proteins and complexes due to their typically poor desolvation, as has been previously discussed.210 While these algorithms are not widely used today as they were in the decade following their introduction,203-209 they illustrate fundamental mathematical relationships between the spacings of peaks in biomolecular electrospray mass spectra that set the stage for powerful algorithms introduced later on that can be used to analyze much more complex mass spectra. An article by Hagen and Monnig202 compares this method to Reinhold and Reinhold’s implementation of MaxEnt211 as well as their own “multiplicative correlation” algorithm (MCA), in which signals at expected m/z values for a given mass and charge assignment are multiplied rather than added. This method can be less prone to outputting a large baseline or artifact peaks.
Figure 2.
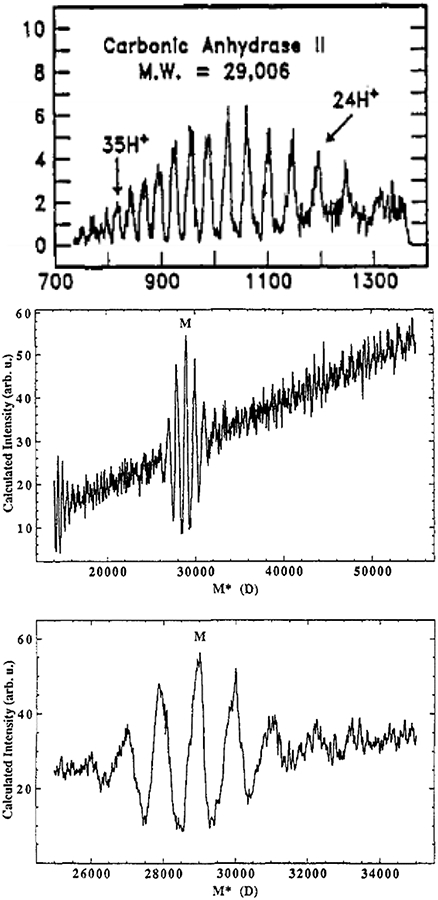
Deconvolution of carbonic anhydrase II ESI spectrum using Fenn’s deconvolution algorithm. Mass spectrum (top), zero-charge mass spectrum (middle), and “zoom” of zero-charge mass spectrum near the determined accurate mass showing sidebands (bottom). Adapted with permission from ref. 73. © 1989 American Chemical Society.
2.1.3. ZScore
Following their work improving results from application of MaxEnt to ESI mass spectra,197 in 1998 Zhang and Marshall introduced a deconvolution method,212 ZScore, in the MagTran data analysis package that fits broadly into the category of “onion-peeling” algorithms (see also Massign,213 discussed in §2.1.5). In such algorithms, one attempts to identify and computationally remove a signal that dominates the spectrum, leaving only less dominant signals. The removed signal is normalized for charge and added to a zero-charge spectrum to which more signals will be successively added. One repeats this process until only uninterpretable data and noise remain in the mass spectrum, and all assigned signals have been added to the zero-charge spectrum. Two essential characteristics of ZScore are that the onion-peeling starts with the highest-abundance peak in the mass spectrum and proceeds through successively lower-abundance peaks, and that, to be considered “interpretable”, a peak must have a set of “partner” peaks corresponding to adjacent charge states and/or isotopomer peaks. This latter characteristic is determined from the “ZScore” value, which is related to either 1) the logarithmic sum of the S/N in the mass spectrum at m/z values where an interpretable peak and all its expected partners should be located (for mass spectra), or 2) the (resolution-weighted) sum of the reciprocals of the differences in the expected and measured m/z of an interpretable peak and its partners (for centroided or “stick” spectra).212
Advantages of ZScore include the ability to work with either raw mass spectra or centroided data over a wide range of mass spectral resolution. The ZScore itself for a hypothetical peak assignment tends to increase as more partner peaks for it are found, thus the accuracy of the algorithm in assigning charge states increases the more partner peaks are present. Because each set of partner of peaks need not be related to any other set of partner peaks in composition, ZScore can often straightforwardly deconvolve mixtures of ions of interest and/or contaminants (see Figure 3),212 such as protein mixtures, peptide digests (including in hydrogen/deuterium exchange experiments), and protein fragments by gas-phase dissociation, as illustrated in the literature.214-219 Zscore has also been utilized in MS analysis of binding sites of the chemotherapeutic cisplatin to native proteins.220,221 The algorithm requires no user input parameters, tends to run very quickly on modern computers, and is fully automated. However, difficulties can arise for ions with overlapping sets of partner peaks, as can often be the case for biomolecular complexes with different oligomeric states.212 Furthermore, the accuracy of the algorithm tends to decrease for lower-abundance partner peak sets, as artifacts leftover from “peeling away” previous peak sets begin to dominate the mass spectrum. Ojha and coworkers later introduced an algorithm222 similar to the component of ZScore for low-resolution spectra212 but which incorporates charge state assignments based on Reinhold and Reinhold’s entropy-based algorithm,211 selected after comparison with Hagen and Monnig’s MCA algorithm202 (see §2.1.2). They found the entropy-based algorithm to be relatively insensitive to overestimation of charge state maxima and to be an improvement over Zscore through allowance of single m/z values to correspond to more than one charge state distribution. Kelleher and coworkers combined ZScore with filtering of high-frequency data, arguing that such data are very likely to be noise and that filtering them out before processing with ZScore can result in cleaner zero-charge mass spectra.217
Figure 3.
Deconvolution (right) of both low (top) and high (bottom) resolution ESI mass spectra (left) for a mixture of three proteins using Zscore. Reprinted with permission from ref. 212. © 1998 American Society for Mass Spectrometry.
2.1.4. SOMMS
As the ability of mass spectrometers to ionize and detect more complex distributions of analyte ions improved,42,79 many researchers began to realize that methods originally developed for interpretation of denatured mass spectra such as MaxEnt and others described above could often be insufficient for native mass spectra with highly-overlapped peaks. In 2006, van Breukelen, van den Heuvel, and coworkers introduced SOMMS223 (SOlving complex Macromolecular Mass Spectra) as an adjuvant method to assist interpretation with other algorithms like MaxEnt, especially in cases where a heterogeneous mixture of protein complexes and subcomplexes is present in the ion population, as exemplified in the literature.120,223-227 In contrast to MaxEnt, SOMMS has the user input as much information as the user knows ahead of time about the expected sample composition: subunit masses, charge state distribution, and likely complex stoichiometries. Using either a user-suggested charge state range or one calculated based on the Rayleigh charge limit, as well as a multinomial distribution (building off previous work using binomial distributions42) of all possible subcomplexes, the algorithm first identifies all m/z at which overlaps of signals from more than one ion composition are expected. Data at these m/z are ignored, and the remaining “unique” signals are then assigned to a composition and charge state based on the table of calculated possible m/z values for the intact complex or subcomplexes. “Partner” peaks belonging to the same ion composition are then identified by scanning over charge states, and the charge state distribution thereby found is fit to a Gaussian intensity distribution. After this process is repeated for each set of partner peaks, a reconstructed mass spectrum is calculated using the identified (sub)complex masses and fitted charge-state distributions. The reconstructed mass spectrum can be compared visually to experimental data to confirm proper assignment of ions in the mass spectrum and locate unidentified peaks. The program CHAMP by Benesch and coworkers228 builds off the tools in SOMMS for many of the same goals, as discussed further in §2.2.4.2.
2.1.5. Massign
This program, introduced by Morgner and Robinson,213 is an “onion-peeling” algorithm (see also Zscore,212 §2.1.3) in which readily-identified peak series are assigned and computationally “removed” from the experimental mass spectrum, leaving behind more challenging peaks. The algorithm is designed to handle overlapped peaks (for example, a dimer with twice the charge of a corresponding monomer) by assuming the charge state distribution for each ion is Gaussian. This process is iterated until essentially only noise and uninterpretable peaks remain, and the output is a “stack” of reconstructed mass spectra for each identified complex as well as the experimental and summed, reconstructed mass spectrum. As with programs like SOMMS223 and CHAMP,228 the user can input information about component protein masses and possible complex stoichiometries to identify and eliminate as many peaks series as possible before unknowns are addressed. The composition of unknown series of peaks identified by this algorithm are assigned, if possible, by Massign based on user-input subunit masses and composition constraints.
Charge states for “partner” peaks belonging to the same ion composition can be assigned either automatically or with some user intervention, based on the fit between experimental peak maxima and predicted peak positions for the series based on an assumed charge state assignment. In addition to specifying trial peak widths, the user can also adjust a “broadening factor” to account for non-Gaussian peak shapes caused by, e.g., unresolved non-specific adducts. Similarly to CHAMP228 (see §2.2.4.2), Massign can also incorporate an empirical mass correction representing non-specific adduction of buffer and water molecules that is based on the expected surface area of globular proteins as a function of sequence mass. Morgner and Robinson demonstrated, using a small number of topological constraints based on condensed-phase data, almost unique assignment of several subcomplexes produced by collisional activation of native rotary ATPase from E. hirae, which contains 9 different protein subunit types and 19-26 total subunits in its intact form (see Figure 4).213
Figure 4.

Assignment of peaks in native mass spectrum (black trace) for subcomplexes (colored traces) for rotary ATPase from Enterococcus hirae using Massign. Reprinted in part with permission from ref. 213. © 2012 American Chemical Society.
Massign can perform well for even large, multi-component complexes (such as membrane proteins) when subunit mass, stoichiometry, and topological constraints are supplied (Figure 4), as has been demonstrated for many different sample types in the literature.117,229-247 Fundamentally, the complexity of the problem in the absence of these constraints is superexponential (i.e., factorial) in the number of subunits, thus Massign performs best when a large amount of user-supplied information from prior mass measurements or condensed-phase structural data is available.
2.1.6. PeakSeeker
Sometimes native mass spectra contain series of peaks with similar masses and charge states that are not well resolved. In such situations, the resulting mass spectral peaks may have multiple local maxima or shoulders representing different ion masses. PeakSeeker,229 introduced by Lu et al., uses two main strategies to identify all the overlapped peaks under a “complex” experimental peak by 1) identification of all readily apparent peak maxima, optionally with the use of Mexican-hat wavelet-based noise filtering and 2) a subsequent shoulder detection algorithm that uses the second derivative of the (smoothed) mass spectrum. The first level of peak identification can be based on either local maxima exceeding an absolute or intensity-adjusted signal-to-noise ratio, or on the presence of local maxima after convolution with a Mexican-hat wavelet (which ideally sharpens the component peaks). The second level of peak identification relies on the fact that the second derivative of a smooth shoulder peak has a characteristic number of zero-crossings that indicate its presence. Though PeakSeeker’s shoulder peak detection is adapted from Massign213 (§2.1.5), its deconvolution algorithm differs in that up to five simulated charge state series can be fit to the experimental mass spectrum at a time using least squares regression, rather than “onion-peeling”. Figure 5 illustrates use of PeakSeeker to interpret a native mass spectrum of a ~1 MDa protein complex,229 and it has also been used to investigate chromatin.248,249
Figure 5.

Peak fitting and deconvolution of native mass spectrum for human TCP-1 ring complex (pictured in (e)) using PeakSeeker. Reprinted with permission from ref. 229. © 2015 American Society for Mass Spectrometry.
2.1.7. Bayesian Deconvolution: UniDec and PMI Intact
As native MS sample preparation and instruments improved, the 2010s saw the advent of highly polydisperse native analytes,250 such as lipoprotein Nanodiscs (with108,196,234,251-254 and without59,78,196,255-258 embedded membrane proteins) or membrane proteins embedded in detergent micelles.259-261 Mass spectra of these complexes can be extremely challenging to analyze due to their relatively broad charge states distributions and overlapped adduct (detergent, lipid, glycan, or other small molecule) distributions, resulting in tens or possibly hundreds of peaks spanning a few thousand m/z (or even hundreds of thousands of peaks, as expected for glycoproteins with extremely varied glycoforms262). Adduct distributions are often not identical for different charge states, in part because ESI tends to add more charges to native-like larger ions (i.e., with more bound ligands), and also because gas-phase collisional activation of the ions to remove solvent can often dislodge some of the adducts. MaxEnt173-175 (§2.1.1) and other relatively simple deconvolution algorithms may perform poorly for these types of samples, owing to the flatness of the probability surface, the challenge of accurately guessing input charge and mass parameters, and other factors. In this section we describe UniDec and PMI Intact, both Bayesian deconvolution algorithms developed for interpreting heterogeneous native MS data.
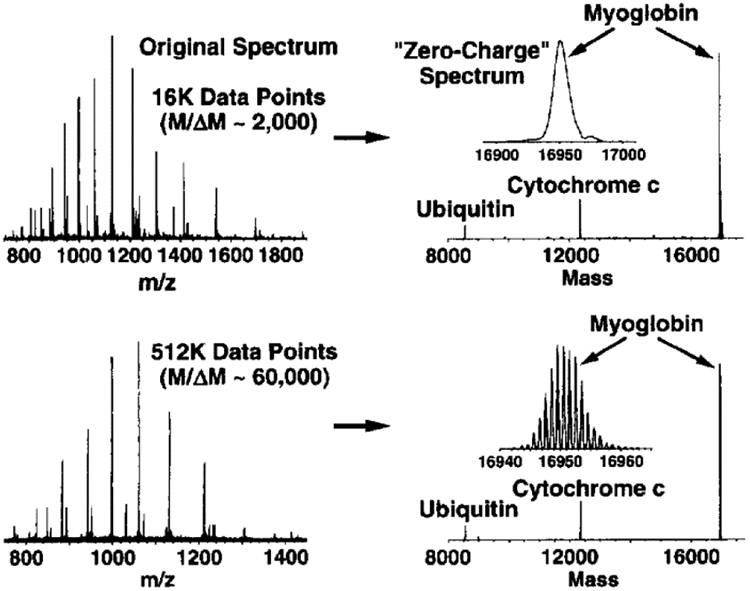
Following on Marty, Gross, and Sligar’s use of a maximum entropy-like algorithm255 for deconvolving “empty” lipoprotein Nanodisc native mass spectra, Marty and Robinson introduced the Bayesian analysis suite “UniDec” in 2015.263,264 The UniDec algorithm begins by conceiving of the information in an experimental mass spectrum as being decomposed into a rectangular matrix with m/z and charge state as its axes and a peak profile with a user-selected shape and width. The matrix is initialized as a uniform distribution. Three steps are iterated to achieve a final matrix: 1) smoothing of the charge state distribution to avoid “orphan” masses at a particular charge state that have no corresponding peaks at adjacent charge states, 2) summation of the matrix along the charge state axis and convolution with the chosen peak shape to produce a simulated m/z spectrum, and 3) adjustment of the matrix entries to reflect the mismatch between the simulated and experimental m/z spectrum. Once convergence of the algorithm is achieved, a final zero-charge spectrum is produced by multiplying each m/z trace in the matrix assigned a particular charge state by that charge state, correcting for charge carrier mass, summing the resulting data for all charge states, and convolving with a user-chosen peak shape function. UniDec requires an input charge state range (either a default range or user-specified) and allows the user to input a subunit mass filter for multiply-adducted species such as lipids or detergents. Outputs include charge-state-specific mass spectra (see Figure 6), zero-charge deconvolved spectra, and heat maps of m/z versus charge, all of which can be highly useful in interpreting native MS data for heterogeneous samples.80,115,167,258,265-282
Figure 6.

Native mass spectrum (black trace) of native oligomeric state distribution for polydisperse αB-crystallin, with deconvolved charge-state-specific mass spectra (colored traces), (A). Dependence of subunit stoichiometry distribution on collisional activation (B, C) as revealed using UniDec. Reprinted with permission from ref. 263. © 2015 American Chemical Society.
Marty has added numerous tools for analyzing the output, including macromolecular mass defect analysis108 (to identify, e.g., peptide stoichiometry inside lipoprotein Nanodiscs80,253,254; see §2.2.3.2) and proteomics tools to identify post-translational modifications and protein isoforms. A set of scoring algorithms to evaluate the plausibility of the reconstructed spectrum and peak assignments is available in UniDec.283,284 Batch processing capabilities to facilitate, e.g., adduct binding kinetics and thermodynamics measurements have been added in a modified version of UniDec called “MetaUniDec”.285 Marty also introduced a tunable “SoftMax” function to reduce the likelihood of producing artifactual peaks at multiples (i.e., harmonics) of true peaks in the zero-charge spectrum (see Figure 7).286 UniDec and MetaUniDec are both freely available open-source Python programs, and a recent preprint manuscript describes all of UniDec’s features in depth.264
Figure 7.

Native mass spectrum of aquaporin Z (AqpZ) with bound palmitoyloleoylphosphatidylcholine (POPC) lipids (A), corresponding zero-charge mass spectrum from UniDec showing artefactual “satellite” peaks (B), and zero-charge mass spectrum after application of UniDec’s SoftMax function to suppress satellite peaks (C). Reprinted in part with permission from ref. 286. © 2019 American Society for Mass Spectrometry.
Expanding the capabilities of their Byonic peptide and protein identification software originally introduced to the market primarily for use in proteomics in 2011,287 Bern and coworkers at Protein Metrics, Inc. separately developed a new program, Protein Metrics Intact288 (“PMI Intact”) that also utilizes Bayesian inference. The heart of the PMI Intact algorithm is a matrix of intensity values that are a function of both m/z and assigned charge state and are iteratively corrected by comparing the simulated m/z spectrum obtained from the matrix with the experimental m/z spectrum. PMI Intact identifies candidate charge states for an experimental spectrum using a “parsimonious algorithm” that attempts to explain all zero-charge mass spectrum data with as few charge states as possible.288 Peak-sharpening algorithms are subsequently used on the deconvolved data to resolve remaining overlapped features in the zero-charge spectrum. PMI includes a “comb filter” to identify peak series equally spaced in m/z, such as those arising from polydisperse adduction of a subunit, which can greatly improve analysis of mass spectra representing samples of this type, including those of great interest in the biopharmaceutical industry154,155 such as highly disperse antibody-drug conjugates.122,163,168,289-293 PMI is coded and compiled in C++ for increased speed, has batch processing capabilities, allows the user to easily select different liquid chromatography-MS (LC-MS) retention data to analyze, and can be used to automatically assign peaks based on protein sequence data or other user-supplied information. It is also vendor-neutral and can produce user-friendly, customizable reports for non-MS users, features important for its use in industry.154 Users can also input expected mass differences (arising, e.g., from known ligand masses) to bias charge state assignments toward those consistent with these mass differences.293 Figure 8 shows a comparison of results from PMI and other deconvolution algorithms (Agilent’s PMod, two implementations of MaxEnt, iFAMS, and UniDec) for a 40 kDa PEGylated protein.163 This example illustrates the superior performance of more sophisticated and recent deconvolution algorithms which utilize Bayesian (UniDec, PMI Intact) or Fourier Transform (iFAMS, see §2.1.9) over earlier tools, as indicated by their faithful reproduction of the reference mass distribution observed for the singly-charged ions using MALDI-MS.
Figure 8.
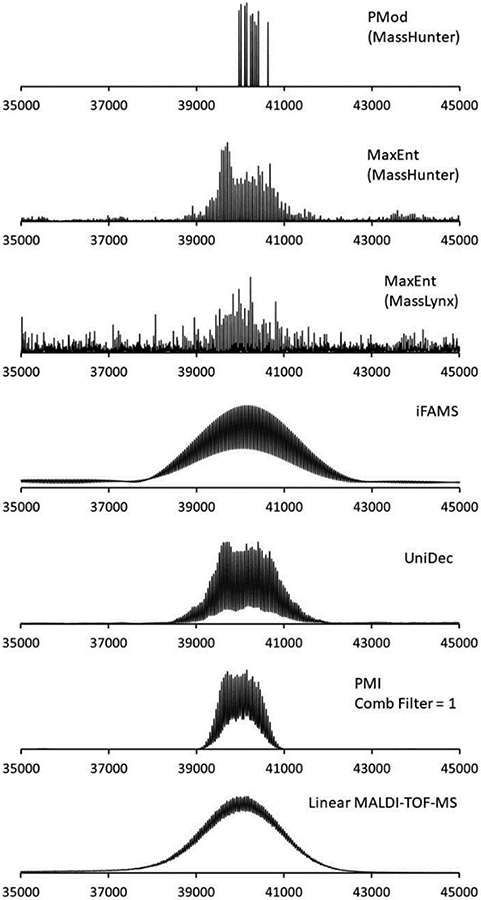
Comparison of deconvolution of ESI mass spectra for PEGylated granulocyte colony stimulating factor protein using different deconvolution algorithms (top 6 traces) and MALDI-TOF 1+ charge state mass spectrum (bottom trace). Reprinted with permission from ref. 163. © 2019 American Chemical Society.
2.1.8. Game-Theoretic Approach: AutoMass
Assigning mass and charge to peaks in native mass spectra for charge state distributions of different ions at their “boundaries”, i.e., at the extreme high m/z end of one distribution where it overlaps with the extreme low m/z end of a different distribution, can be especially problematic. This can occur, for example, when two different stoichiometries of a complex are present in the ion population, or for different symmetries of a viral capsid. Peak assignments in boundary regions can be very challenging due to the presence of “overassigned” peaks (i.e., peaks consistent with more than one mass and charge assignment) and low-intensity peaks. Peng and coworkers introduced AutoMass294 to combat this challenge and also to achieve accurate mass and charge assignments with minimal input from the user, building off ideas introduced in their earlier tool for the same purpose, LeastMass.295 AutoMass treats charge and mass assignment of the peaks in a mass spectrum as “competitors” in a zero-sum game and seeks a game theoretic solution that simultaneously minimizes the maximum “loss” for mass assignment (the standard deviation of the m/z discrepancy for observed peaks given a particular set of m/z assignments) and maximizes the minimum “loss” for charge assignment (the shift in charge for the observed peaks given a particular set of m/z assignments). AutoMass applies this algorithm after smoothing, Gaussian baseline subtraction, and thresholding of the mass spectrum. In this manner, the boundaries between peak distributions can be determined automatically, enabling peak assignment for mass spectra containing many tens of overlapped peaks. Peng and coworkers demonstrated application of AutoMass to assignment of intact 3-4 MDa hepatitis B viral capsids with T=3 and T=4 symmetries and also to the tens of products with different protein stoichiometries produced upon collision-induced dissociation (CID) of isolated T=3 and T=4 ions.294
2.1.9. Fourier Transform Approaches: iFAMS
Many analytes of interest in native MS, or in ESI-MS more generally, differ primarily in the polydispersity of one or more constituent subunits. For example, long-chain homopolymers and copolymers contain many identical monomer subunits in varying stoichiometry, and challenging samples such as lipoprotein Nanodiscs or detergent micelles containing membrane proteins are polydisperse in the number of constituent lipid detergent molecules. In these and many other cases, the ESI mass spectrum often contains tens or even hundreds of overlapped peaks due to the charge distribution and polydisperse subunit distribution. However, the regular spacing between peaks in the mass spectrum for a given charge state due to the varying number of repeated subunits forms a pattern with a “frequency” that can be analyzed using Fourier Transform (FT). In 2004, Prebyl and Cook introduced the use of FT to analyze electrosprayed polymer mass spectra,296 noting that a much simpler set of peaks is present in the Fourier spectrum than in the mass spectrum itself, with each peak occurring at some integer multiple of the reciprocal of the monomer mass (its characteristic frequency, 1/ms, where ms is the monomer/subunit mass). This provided a straightforward way of measuring the subunit mass (from the spacing of the Fourier spectrum) and determining which charge states may be present in the ion population (from the integer multiples of the characteristic frequency at which a peak is present).
Cleary and Prell expanded this concept in 2016 to analysis of Nanodiscs, heavily metal ion-adducted native proteins, and polymers, producing an open-source Python program called iFAMS (interactive Fourier-Transform Analysis for Mass Spectrometry).78 This program automates computation of the FT of an input mass spectrum (by treating it as a composite of three functions, see Figure 9),256 identification of Fourier-domain peaks, and determination of the subunit mass and charge states in the ion population. Signal for individual charge states in the Fourier spectrum can be readily extracted and inverse Fourier-Transformed to reconstruct individual charge-state-specific mass spectra as well as a zero-charge spectrum in iFAMS (as compared with other deconvolution methods in Figure 8).163 One disadvantage of using FT for some samples is that the ion population may not be sufficiently polydisperse to yield well-resolved peaks in the Fourier domain, although this can be mitigated somewhat by using harmonic peaks,256 which are spaced more widely (see Figure 10). Another disadvantage is the possibility for two or more types of ion to have overlapping Fourier-domain frequencies, e.g., two heavily sodiated proteins of different masses but similar charge states.
Figure 9.

Schematic of iFAMS Fourier Transform-based algorithm, showing decomposition of mass spectrum into “comb”, “peak shape”, and “peak envelope” functions (left) and their corresponding functions in the Fourier spectrum (right). * indicates convolution, and × indicates pointwise multiplication. Reprinted in part with permission from ref. 256. © 2018 American Society for Mass Spectrometry.
Figure 10.

Deconvolution of native mass spectrum of dimyristoylphosphatidylcholine MSP1D1 Nanodiscs using iFAMS Fourier Transform-based algorithm (left), and corresponding Fourier spectrum (right), illustrating the use of higher-harmonic data (inset). Colored traces in mass spectrum correspond to reconstructed peak envelope functions for the charge states indicated with the same color in the inset. Reprinted in part with permission from ref. 256. © 2018 American Society for Mass Spectrometry.
To address these problems, Cleary and Prell introduced the use of Gábor Transform (GT),297 which is a type of “windowed” or “short-time” Fourier Transform in which the intensities of “local” frequencies in the mass spectrum are plotted against the mass spectrum itself, into iFAMS.298 GT and FT analysis with iFAMS of heterogeneous mass spectra of α-hemolysin complexes in detergent micelles enabled separation of the overlapped frequency signals of two oligomeric states, as well as determination of detergent stoichiometries and reconstruction of zero-charge mass spectra,299 and iFAMS has also been used to characterize functionalized polymer constructs for protein conjugation.300 In many cases, GT can readily overcome pitfalls of FT analysis due to separation of frequency signal from different types of ions according to their m/z. Another advantage is that salt cluster ions, which typically increase in mass as their charge state increases, can be distinguished at a glance from native biomolecular ions, which typically change little in mass over their charge state envelope and give rise to a “negatively chirped” GT signal (see §2.4.2.3).298 Similar to FT, a disadvantage of GT is that low polydispersity samples may give rise to overlapped GT signals for different charge states, although even the isotope pattern may be sufficient for GT analysis in mass spectra where isotopes are resolved. Both FT and GT analysis can serve as ideal “notch filters”, dispensing with nearly all chemical noise as well as white noise, though windowing artifacts can sometimes show up as “ringing” near the baseline of reconstructed spectra.256 Further capabilities of FT analysis for distinguishing between different compositional heterogeneity types77 are discussed in §2.4.1.3.
2.1.10. MetaOdysseus
Some metals of physiological and human health relevance (e.g. zinc and platinum) have complex isotope patterns compared to those of common organic atoms, thus it is important for the study of metalloproteins and other metal-containing analytes to develop deconvolution algorithms that can handle these challenging isotope patterns. In 2021 Peris-Díaz, Krężel, and coworkers published the most recent of the deconvolution algorithms reviewed here: MetaOdysseus,284 a software suite written in R. MetaOdysseus can be used for analysis of native, bottom-up, and native top-down mass spectra. After spectra are smoothed with one of three included algorithms, convolution with a Mexican hat wavelet can optionally be performed to help identify peaks. The three main features of MetaOdysseus are charge state deconvolution, mass assignment, and statistical scoring. Two algorithms can be used for charge state deconvolution: 1) an algorithm similar to that of ZScore212 (see §2.1.3) for peak assignment for high-resolution, low-charge mass spectra, and 2) an “onion-peeling” algorithm based on fitting simulated spectra to experiment and which can account for peak broadening often observed in native MS due to adducts. Mass assignment is achieved through cross-correlation with a generated expected mass pattern based on the amino acid sequence of the protein of interest as well as a library of common labeling reagents and metal isotope distributions. MetaOdysseus incorporates the UniScore283 scoring schemes developed by Marty which can be used to evaluate results from deconvolution and mass assignment.
2.2. DATA REDUCTION
2.2.1. Monomer Mass
In many chemical applications, it may sometimes be necessary to determine the accurate masses of repeated subunits in polydisperse samples, i.e., the sample components whose stoichiometry varies in the sample. This can be especially important when samples are prepared from mixing reagents with similar masses or when bulk average measurements fail to properly distinguish successfully made products from leftover reagents, conditions which apply to lipid Nanodiscs, polymers, antibodies, and other biotherapeutics.77,81,163,164,166,168,301,302 Several approaches have been demonstrated to address this challenge.
2.2.1.1. FT Methods.
Prebyl and Cook noted in their 2004 Fourier Transform-based algorithm for analyzing electrospray mass spectra of polymers that the characteristic spacing between fundamental peaks in the Fourier spectrum (which represent the charge states present) is the inverse of the monomer mass.296 From simulated spectra of polydisperse 40-kDa polymers with a charge state distribution spanning 15-22+ and exact monomer mass of 160.0 Da, they found that the accuracy of the monomer mass determined from peak spacing in the Fourier spectrum decreases with S/N of the mass spectrum. However, reasonable agreement (7% error) is achieved even for a very low S/N of 5:1 and with very poor resolution of the mass spectrum, which exhibits a high, curved baseline due to the overlapping tails of tens of adjacent peaks. Applying their method to ESI spectra of sodium poly(styrenesulfonate) with a nominal average mass of 4.6 kDa, they determined a monomer mass 1.2% lower than the expected monomer mass. However, they were able to confidently assign all of the charge states present in the ion population and attributed some of the monomer mass inaccuracy to substitutions of protons with sodium ions during ESI that could be mitigated by adjusting the pH of the ESI solution (to 0.1-0.3% monomer mass error).
Cleary and Prell demonstrated automated determination of subunit mass from nESI mass spectra using a similar algorithm in iFAMS for sodiated and potassiated ubiquitin, long-chain poly(ethylene glycol), and lipoprotein Nanodisc samples.78 They found typically less than 0.2% root mean squared deviation of determined subunit masses from their known exact masses for the metal adducts, ethylene glycol monomer unit, and Nanodisc phospholipids. The precision of the monomer adduct mass for poly(ethylene glycol) was sufficient to distinguish the Fourier spectrum signal from that potentially arising from sodium metal adduction.
2.2.1.2. “Double FT”.
Because fundamental peaks in the Fourier spectrum are spaced by the reciprocal of the repeated subunit mass, another approach to determining the subunit mass is to apply another (forward) Fourier Transform to the Fourier spectrum itself. This results in a “double FT spectrum” in which a peak is expected at the mass of the subunit. Marty demonstrated that the mass of the phospholipid subunit in Nanodiscs can be recovered in this way without directly analyzing the initial Fourier spectrum (see Figure 11).302 This method was also recently employed to distinguish mixtures of poorly-resolved lipid head groups attached to protein ions.303
Figure 11.

UniDec-based “double FT” analysis of native mass spectrum of Nanodiscs containing mixtures of POPC and either PO-phosphatidylserine (POPS) or PO-phosphatidylglycerol (POPG) lipids. Example pure POPC Nanodisc native mass spectrum (A), corresponding Fourier spectrum (B) and “double FT” spectrum (C), revealing apparent average lipid mass (yellow dot). Measured apparent average lipid masses for different bulk lipid compositions (D, E) and reconstructed Nanodisc lipid composition versus bulk lipid composition (F). Reprinted with permission from ref. 302. © 2016 American Chemical Society.
The double FT spectrum can be similar in appearance to the spectra produced by Fenn’s deconvolution method73 (see §2.1.2), but with the major peaks at multiples of the repeated subunit rather than the total ion mass. However, a large baseline is often present in the double FT spectrum, and numerous other signals can be present, potentially making the method difficult to use when mass spectral resolution is too low. Intriguingly, this method can also be used to approximate the bulk fraction of two different types of lipids in mixed-lipid Nanodiscs (see §2.4.1.2 and 2.4.1.3). Further discussion of samples this type of analysis is well-suited for, as well as of potential caveats, is included in §2.4.1.3.
2.2.2. Base Mass or “De-adducting” Measurements
For many samples, the complementary problem to determination of accurate monomer/repeated subunit masses (§2.2.1), i.e., measurement of the “base mass,” or mass conserved across many or all members of a polydisperse ion population, may be of interest. For example, in studying ligand-bound proteins, non-specific adduction of sodium and potassium or other common metal ions may obfuscate the relative abundances of other proteoforms or of ligands bound to the protein. Recently-introduced methods computationally remove these nuisance adducts to reveal the underlying base masses of interest (e.g., the abundance distribution of a protein and its ligand-bound complexes).
2.2.2.1. SWARM.
Klassen and coworkers introduced the SWARM (“Sliding Window Adduct Removal Method”) algorithm in 2019 to effectively remove patterns of adducts from mass spectra and reveal more clearly the peaks belonging to base masses of interest.304 This is achieved by first smoothing the experimental spectrum, with optional baseline subtraction. It is assumed that the user knows the mass of the protein and ligands in the sample and is interested primarily in determining the relative abundances of different ligand states. It is further assumed that identical non-specific adduction occurs for each base mass. A region of the mass spectrum is then selected to represent the pattern of non-specific adducts expected for each base mass in the ion population. This region must be well-separated from peaks associated with other base masses; often a region from the native mass spectrum of the ligand-free protein is used. Within the selected region, the low-m/z side of the base mass peak is ignored, and the remaining portion is called the “template window” (see Figure 12).304 The template window is then scaled vertically and horizontally according to the pre-assigned charge state and abundance of each target base mass in the spectrum and slid over to the base mass peak m/z value. The resulting scaled and translated template is subtracted from the smoothed mass spectrum, and this process is repeated for all target base masses. The resulting SWARM-processed spectrum thus reveals the abundances of each base mass with non-specific adducts removed. Klassen and coworkers originally demonstrated the utility of this algorithm in studying equilibrium and kinetics between ligand states of carbonic anhydrase, lysozyme, and the C-terminal portion of human galectin-3 with glycan ligands in the presence of non-specific sodium and potassium adducts.304 They recently showed the facility with which the abundances of a library of glycans attached to CUPRA linkers can be determined from highly-overlapped mass spectra and also demonstrated utility for quantifying weak protein-glycan interactions.305,306 Marty and coworkers applied a similar algorithm to deconvolve base masses of interest for zinc- and lipid-bound rhodopsin.265
Figure 12.

Native mass spectrum of human galectin-3 C-terminal domain without (A) and with (B) extensive sodium adduction (black traces), and final de-adducted base mass spectra from application of SWARM (red traces). Blue insets illustrate de-adducting template that includes oligosaccharide and sodium adduct profiles. Reprinted with permission from ref. 304. © 2019 American Society for Mass Spectrometry.
2.2.3. Mass Defect Analysis
In their FT-based analysis of ESI-MS of polymers described above (§2.2.1.1),296 Prebyl and Cook also pointed out that, in principle, the phase information in the Fourier spectrum could be used to determine the average total mass of the end groups on polymers (plus the mass of any non-covalent adducts) modulo the monomer mass. This procedure would effectively be analogous to Kendrick Mass Defect307 analysis common in polymer mass spectrometry as well as to Macromolecular Mass Defect108,253 analysis in native MS, both described below.
2.2.3.1. Kendrick Mass Defect Analysis.
In 1963 Kendrick introduced a method for characterizing polymer mass spectra based on the difference in mass defect between an ion and a chosen molecular fragment (e.g., a monomer).307 Part of the original motivation for this method was to reduce the size of mass spectral datasets for more efficient storage and although the method is not typically used in its original form in native MS, it illustrates key concepts that are used in the related Macromolecular Mass Defect method, which has utility in native MS (§2.2.3.2). First, a molecular fragment of interest is chosen, typically one present in varying stoichiometry within the analyte population. The “Kendrick mass” of each analyte ion is defined as the product of its measured accurate mass and the nominal (nearest-integer) mass of the molecular fragment, divided by the exact mass of the molecular fragment. The “Kendrick mass defect” is then defined as the nominal mass of an analyte minus its Kendrick mass. Thus, if an analyte has a mass that is an exact multiple of the molecular fragment mass, it will have a Kendrick mass defect of exactly 0. Typically, the Kendrick mass defects of each analyte in an ion population are plotted against their Kendrick masses. In such a plot, analytes belonging to a “family,” such as linear polymers with the same end groups but differing monomer numbers, will fall along horizontal lines corresponding to the same total end group mass. Data falling along lines of non-zero slope can indicate the presence of analytes with a different repeated subunit other than the chosen molecular fragment. Kendrick mass defect analysis has become a major tool in polymer analysis for the ease with which researchers can make judgments about sample composition from visual analysis of the plot, and it has also been adapted for native ESI-MS investigation of polymers, gangliosides, and other analytes.308-312 For example, the number of different horizontal groupings in the Kendrick mass defect plot can reveal how many different combinations of end groups are present. This is analogous to modular arithmetic, in which numbers are considered equivalent if they have the same remainder after division by a chosen natural number. Kendrick mass analysis readily reveals which “remainders” are present in the polymer ion population as well as what the mass of the end groups are in a given ion, modulo the chosen molecular fragment mass. Although it is therefore possible for different combinations of end groups to yield the same Kendrick mass defect, the researcher can often make unique assignments for a given Kendrick mass defect based on additional information about the sample.
2.2.3.2. Macromolecular Mass Defect Analysis.
Marty extended the ideas of Kendrick mass analysis to the study of polydisperse native biomolecular ion complexes, in particular, lipoprotein Nanodiscs containing varying numbers of lipids and embedded membrane proteins.108,253,254,313 In this case, the molecular fragment mass used in the “macromolecular mass defect” (MMD) analysis is the known molecular mass of the lipid.108 After the native mass spectrum is deconvolved to a zero-charge mass spectrum, it is computationally divided into strips starting and ending at consecutive integer multiples of the lipid mass. The strips are then overlaid and the intensities summed to produce a plot of intensity versus MMD, the x-axis of which is simply the remainder obtained upon dividing each ion’s mass by the mass of the lipid (as shown in Figure 13). This method has the advantage of providing a global-average distribution of the MMD over all lipidation states, effectively increasing the S/N of each MMD. MMD analysis is available in UniDec, with options for making 2-dimensional plots of MMD versus mass and applying Richardson-Lucy peak sharpening to assist in determining MMD values. Marty and coworkers illustrated that this method can be extremely useful for determining peptide and small membrane protein incorporation into Nanodiscs as a function of bulk peptide/protein composition in the Nanodisc assembly mixture (see Figure 13), which can reveal stability and specificity (i.e., preference for particular oligomeric states and/or lipid interactions) of the inserted molecules in lipid environments of varying compositions.253,254,302,313
Figure 13.

Native mass spectra of charge-reduced Nanodisc-embedded melittin for different bulk melittin: Nanodisc concentrations (A), corresponding zero-charge spectra deconvolved using UniDec (B), MMD profiles reconstructed using MetaUniDec and sharpened with the Richardson-Lucy algorithm with peak label numbers indicating stoichiometry of incorporated melittin (C), and variation of melittin incorporation as a function of bulk melittin:Nanodisc concentration for DMPG and DMPC lipids (D, E). Reprinted with permission from ref. 253. © 2019 American Chemical Society.
2.2.4. Modeling Complex Topologies
Reconciling observed masses for protein complexes with reasonable complex stoichiometries and topologies is important in determining quaternary structure using data from native IM-MS,67,74,91,123,124,314-334 surface-induced dissociation (SID),313,335-344 and other complementary methods.39,72,83,88,91,92,99,102,335,336,341,345-348 SOMMS,223 CHAMP,228 and SUMMIT349 include algorithms for this purpose and are described below. Although a detailed analysis of these and other quaternary structure modeling programs is beyond the scope of this review,72,314,326,340,345,350-352 possible structures determined from these programs can provide tight constraints for modeling atomistic structures and interpreting complementary information from other structural methods. We briefly highlight these capabilities below.
2.2.4.1. SOMMS.
In addition to its mass spectrum deconvolution algorithm, SOMMS223 (see also §2.1.4) can be useful in analyzing multi-protein complexes with two or more different types of protein subunits by calculating hypothetical spectra a priori based on combinations of known subunit masses and user-input charge state ranges. SOMMS performs best with high-quality prior measurements of the subunit masses and may not be optimal for identifying unknown components or for analyzing experimental mass spectra in which many unknown contaminant ions are present.
2.2.4.2. CHAMP.
Benesch and coworkers introduced an algorithm, CHAMP228 (Calculating Heterogeneous Assembly and Mass spectra of Proteins), that shares similarities with SOMMS223 and uses a more sophisticated approach than previous efforts off which it builds42,79,353 to charge state distribution assignment as well as a χ2-based optimization algorithm for the reconstructed mass spectrum. Empirical relationships between mass and the native charge state envelope as well as a mass adjustment factor based on the estimated surface area of each putative complex are used to more realistically predict m/z distributions based on user input. A three-stage optimization algorithm based on the χ2 statistic for the difference between the reconstructed and experimental mass spectra is used to tune the fitting parameters; this three-stage optimization was found to avoid local-minimum “traps” in the fitting parameter surface and converge faster than a simpler steepest-descent approach.228 High-quality fits of calculated spectra to poorly resolved mass spectra representing very polydisperse ion populations, such as oligomers of small heat shock proteins228 and αB-crystallins,354 and to investigate selectivity of lipid binding to membrane proteins were obtained using CHAMP.119,354 Like SOMMS, CHAMP performs best when the user can supply as much input information about the component proteins as possible.
2.2.4.3. SUMMIT.
Taking a structure-based approach to elucidating heterogeneous multi-protein complexes, in 2008 Robinson and coworkers introduced SUMMIT (SUMming Masses for Interaction Topology) to generate protein interaction networks and, in some cases, atomic model structures.349 This program uses a multi-technique approach in which subcomplexes are deliberately formed using solution-phase chemical cross-linking,355,356 gas-phase dissociation of the intact complex and subcomplexes, and gel electrophoresis. Both native and denaturing MS are used to assign the identities and masses of the subunits, and overlapping information for different subcomplexes is used to generate a “protein interaction network,” which is a map of likely subunit interfaces.349,351,357-366 The interaction network can be used along with other computational approaches, such as homology modeling, to build 3-dimensional models of the intact complex that are consistent with the experimental data (see Figure 14). In addition to other uses to SUMMIT to reveal the architecture and interactions of subunits within complexes,351,357-366 the Robinson group has demonstrated the utility of this powerful combined approach for assigning the 3-dimensional structure of the 19S proteasome lid, which contains 9 distinct protein subunits, and the yeast exosome complex, which contains 10 distinct subunits. A major advantage of this method is that the number of subcomplexes with overlapping information is maximized, vastly reducing the number of possible structures consistent with all of the structural data.
Figure 14.

Representative native mass spectra of intact yeast exosome and its subcomplexes formed in solution or generated by collision-induced dissociation (middle), interaction networks generated by SUMMIT from native MS and solution-phase data (bottom, A-C), subcomplex map (top), and final proposed 3-dimensional model topology of the intact exosome (center). Reprinted with permission from ref. 349. © 2008 American Chemical Society.
2.3. INSTRUMENTAL AND EXPERIMENTAL APPROACHES
2.3.1. Charge Detection of Single Particles
Especially for very large ions approaching the MDa size range, native ESI mass spectra can exhibit very poor resolution due to adduction of buffer salts and other small cosolute molecules in addition to heterogeneity resulting from the presence of multiple isoforms.210,367 The resolving power and sensitivity of TOF, Orbitrap, and FTICR instruments tend to decrease at very high m/z due to a number of instrumental factors368 including space-charge repulsion, further complicating mass spectral analysis.369 An experimental alternative to (nearly) simultaneous detection of multiple ions per scan, as is the case for these instrument types, is charge detection mass spectrometry (CDMS), in which individual ions are trapped and their accurate masses measured one at a time or in very small groups.369,370 Initially introduced by Benner in the mid-1990s,371,372 innovative work done to increase speed and sensitivity373-378 and, more recently, to enable performance of these experiments in Orbitrap instruments,379 has made CDMS an exciting addition to the arsenal of native MS techniques that aims to circumvent many of these challenges of conventional biomolecular ESI-MS. We provide a brief overview of CDMS innovation and recent exciting applications to challenging, heterogeneous samples below and encourage interested readers to the many in-depth reviews and landmark publications available in the literature.55,164,372-375,377-389
2.3.1.1. Benner Trap.
Improving on earlier instrumentation for determining masses of aerosol and cosmic dust particles,390,391 Benner designed a mass spectrometer consisting of an ESI source, an electrostatic ion gate, and two electrostatic ion mirrors on either side of a cylindrical inductive pick-up electrode.371,372 The pick-up electrode is connected to a field-effect transistor assembly that transmits signal to an external amplifier and detector. Single ions that pass through the gate and trigger a response in the detector assembly are trapped by rapidly switching on the ion mirrors, which cause the ion to oscillate back and forth repeatedly through the pick-up electrode (on the order of tens of passes in a few milliseconds). Each time the ion exits or enters the pick-up electrode, a characteristic spike and dip in the voltage on the pick-up electrode are digitally recorded. By adjusting the potentials on the ion mirrors, only ions within a certain range of kinetic energies are trapped, enabling kinetic energy selection. Because the magnitude of the voltage spikes on the detector is proportional to the charge of the ion, and the time required for the ion to traverse the pick-up electrode is related to its m/z, the detector read-out can be used to determine both the charge state and the mass of the single trapped ion. The mass distribution for a sample can be reconstructed by superimposing results from many such individual measurements. Benner initially illustrated the use of CDMS to measure the mass and charge of the pBR322 plasmid (2.88 MDa) carrying ~690 charges in positive ion mode.371 This technology was soon applied to the analysis of viruses and viral capsids55 and large, heterogeneous DNA samples.389,392
Jarrold370,377,383-388,393-395 and, separately, Williams374-376,396-398 later showed how FT analysis of the detector signal can be used to rapidly assign ion mass and charge, even when a small number of ions are simultaneously trapped. The incorporation of multiple pick-up electrodes arranged in a row results in faster signal acquisition and greater sensitivity. In addition, ion kinetic energy uncertainty can be reduced to ~0.45% using a hemispherical electrostatic energy selector prior to trapping,393 and the ratio of the time ions spend outside and inside the pick-up electrodes can also be used to more accurately determine ion kinetic energy.397 Jarrold and coworkers demonstrated detection of pyruvate kinase aggregates up to 40-mers (2.43 MDa) using CDMS and noted that the larger aggregates are typically 5-6% more massive than predicted simply based on the native tetramer mass, indicating significant adduction of salts, solvents, and other cosolutes at this size.388 Plots of the measured m/z of these ions versus their mass illustrate that, even in the absence of space-charge repulsion and other effects common to conventional ESI-MS, the mass spectrum would exhibit extreme overlap of ion signals.388 Williams has used CDMS to track solvent evaporation from multi-MDa ions produced by ESI and as a method for determining their collision cross sections from CDMS measurements,398 a finding with promising implications in the future study of very large biomolecular complexes.
2.3.1.2. CDMS in Orbitrap Instruments.
Recently, Kelleher399-402 and Heck and Makarov379 demonstrated that CDMS can be performed in modified Orbitrap instruments. For these experiments, only a few (~100) ions are trapped in the Orbitrap per scan. Their transient signals are collected, and the charge of each ion is deduced from the current it induces on the detection electrodes as measured against a calibration curve. This results in simultaneous measurement of m/z and charge (see Figure 15) and greatly improves mass accuracy due to the reduction in space-charge repulsion owing to the small number of trapped ions. Kelleher showed that this method can drastically improve identification of 0-30 kDa proteoforms from extremely complex mixtures, such as fractions of human cell extracts, even using direct infusion ESI.399 This method can be extremely useful in distinguishing otherwise overlapping m/z signals for different oligomers of the same species, as shown for immunoglobulin-M in Figure 15, and for very large complexes, such as Adeno-associated virus capsids with (4.91 MDa) and without (3.74 MDa) genome cargo.379
Figure 15.

Two-dimensional histogram of single-ion signals measured using Orbitrap CDMS of immunoglobulin-M oligomers (A), m/z of histograms of single-particle centroids illustrating m/z overlap (B), and corresponding mass histograms (C). Reprinted with permission from ref. 379. © 2020 Springer Nature America, Inc.
2.3.2. Cutting-Edge IM-MS Instrumentation
Ion mobility separation is a technique that can be integrated into mass spectrometer instruments to provide complementary information through separation of ions based on size and shape,325 and IM-MS instruments have been commercially available since 2004.49 In addition to providing some structural information via collision cross section (CCS) measurements, which can be useful for characterizing structural heterogeneity, for distinguishing between different possible conformations, and for classifying ions (see §2.4.2.1-2.4.2.2), IM-MS can serve as a filter to enable isolation of heterogeneous species which may overlap in the mass spectral domain,322,324 as illustrated in the literature.66,121,299,321,329,403-414 We direct readers to many excellent reviews on the principles and history of IM-MS and aim to provide an overview of exciting innovation in this field to improve resolution and separation capabilities, a critical development as samples amenable to study with native MS become increasingly more complex.67,315,322-325,328-331,333,415-422
Beyond conventional drift-tube IM separation,423 in which resolving power increases with the square root of drift tube length and presents rapidly diminishing returns in instrument design, a number of promising alternative IM technologies have been introduced and rapidly developed over the last two decades. Improved IM separation and S/N can be helpful in studying heterogeneous mixtures because analytes with identical m/z that are not separated in the mass spectrometry step can in many cases be separated by IM based on shape and size. The resulting mobility-selected mass spectra are often better resolved and simpler than the full mass spectrum, resulting in more facile assignment of peaks. However, while IM resolving power in commercial instruments can be relatively high for small ions, such as peptides, lipids, and oligonucleotides (typically ~50-300 CCS/ΔCCS with many currently available instruments417), resolving power for larger proteins and biomolecular complexes is often substantially lower. Much recent effort has gone into developing instrumentation to address this challenge while minimizing signal loss and ion heating/unfolding for native ions.
In Traveling Wave Ion Mobility (TWIM) instruments,415,418 such as Waters’ Synapt series,50,51 a high degree of ion separation (up to ~40:1 for small biomolecules and somewhat less for native proteins)424 using a relatively short (on the order of ~10-25 cm) IM cell can be achieved. Waters’ recently-introduced Cyclic Ion Mobility cell design425 effectively turns the TWIM cell into a circular path, in which multi-pass separations and much higher resolution are possible (~750 for isobaric 491-Da peptides in 100 passes around the cyclic cell; see Figure 16), although at present only a few results for native protein complexes have been published.425-429 Separation of the reverse-sequence peptides as shown in Figure 16 represents an important milestone for the utility of IM-based separation of isobaric heterogeneous analytes. Because they have the exact same mass and amino acid composition, differences in their mobility should ultimately be due solely to conformational differences. This and other key work done to achieve separation of isomers and mixtures of small molecules420,430-439 provide an exciting glimpse to the future separation of larger, heterogeneous samples with continued improvements to IM-MS instrumentation, though these capabilities have utility even now as native MS expands to the investigation of endogenous small molecule ligands bound to biomolecular complexes.167,440
Figure 16.

Arrival time distribution of reverse-sequence peptides SDGRG and GRGDS as a function of number of passes around cyclic TWIM cell (A) and corresponding plot of IM resolution as a function of number of passes (inset corresponds to arrival time distribution after 100 passes). Reprinted with permission from ref. 425. © 2019 American Chemical Society.
Trapped Ion Mobility Spectrometry (TIMS)419 devices,404,419,441-447 such as those present in Bruker’s timsTOF series,443,448 are a recent addition to the native IM-MS arsenal, building upon development of this technique in 2011 by Park, Fernandez-Lima, and coworkers.441,449 These TIMS mass spectrometers offer the advantage of performing many different types of tandem experiments online after IM separation. IM resolving power up to ~400:1 is possible on these instruments for small molecules,450 and “microheterogeneity” (multiple conformations of a protein for a single mass and charge state) has been observed using TIMS.404 More recently, Bleiholder and coworkers have introduced a setup utilizing two TIMS cells (tandem TIMS)442 which enables collisional activation of mobility-selected ions and subsequent mobility-based structural analysis of their new conformations and/or fragments, expanding native MS capabilities in structural biology. 451
Although integrated IM separation is not yet available commercially for Orbitrap instruments from Thermo Fisher Scientific, prototype instruments coupling drift-tube IM to Orbitrap mass analyzers have recently been demonstrated.452-455 Due to the relatively slow scan rate of the Orbitrap, sensitivity and resolution can be poor if ion packets are introduced into the drift tube only after all ions from the previous scan have exited the drift tube (amounting to a small duty cycle). To increase the duty cycle, FT methods,456-459 in which many packets of ions are released into the drift tube in rapid succession using precisely-controlled gating at a single or chirped frequency, have been introduced by Clowers, Laganowsky, and Russell (see Figure 17).454,457,460,461 This combination of the superior resolution and activation capabilities of the Orbitrap with additional separation of ions in the mobility dimension represents an exciting new development in the field of native MS toward analyzing large, extremely polydisperse samples. Advantages and disadvantages of these gating methods have been compared to other high-duty cycle gating methods, such as Hadamard Transform.462
Figure 17.

Schematic of entrance and exit gate synchronous chirped pulsing for FT-drift tube ion mobility-MS experiments (A). FT-IM-MS data for native cytochrome C obtained on a prototype FT-IM-Orbitrap instrument (B) and Fourier Transform of extracted ion chromatograms from FT-IM-MS data converted to arrival time distributions (C). Reprinted with permission from ref. 454. © 2018 American Chemical Society.
Very high sensitivity can be achieved in prototype IM-MS instruments based on Structures for Lossless Ion Manipulations (SLIM) developed by Richard Smith,463-466 printed circuit board ion optics with small cross sectional areas that maximize ion transmission. IM separation for native-like protein ions in a ~46 cm SLIM-TOF instrument was found to have a resolution of ~13-42, with CCSs nearly identical to those measured using drift-tube MS.466 In these development-stage instruments, effective drift tube path lengths of ~540 m based on multiple passes around a serpentine SLIM board have resulted in resolving power of >1,800:1, although trapping times for the ions can be so long that substantial unfolding and even chemical reactions with trace reactive background gases often occur on the timescale of the separation.463 Ever-increasing complexity of samples could lead to not only signal overlap in m/z but also in ion mobility, thus improved separation in more dimensions than m/z may be very advantageous.
2.3.3. Ion Reactions for Improved Separation
A common problem in native MS for complex samples is that nESI-generated ions of different charge states can sometimes overlap at the same m/z. Since the early days of native MS, researchers have utilized various methods to manipulate the charge states of ions with minimal perturbation to their native-like structures and thus shift their signal in m/z to facilitate analysis.467-476 Increasing charge states can move mass spectral peaks to lower m/z, where instrument resolving power, trapping, and transmission efficiencies are often higher. Decreasing charge states (as in Charge Reduction Electrospray Mass Spectrometry developed more than 20 years ago by Lloyd Smith and coworkers473,475,476) can be advantageous because the spacing between mass spectral peaks increases, reducing peak overlap. These effects on charge state can be achieved through addition of chemical reagents to sample solutions and through gas-phase ion/ion reactions performed in the instrument. In this section, we describe recent developments in this area, made possible by early pioneering work utilizing ion/ion reactions, supercharging reagents, superbases and proton sponges, α-particle emitters, and corona discharge for charge manipulation.301,468-483
2.3.3.1. Charge Manipulation.
Native Supercharging ESI, a modification of denaturing supercharging electrospray ionization, uses small, polar chemical additives with high boiling points to encourage attachment of unusually high numbers of charges to native-like ions during the ESI droplet evaporation process.481,484-486 In most cases, this results in extensive unfolding of the ions, even to the point that nearly linear structures are produced.480,484,487-499 However, in some cases, such as ions with very strong noncovalent interactions, less unfolding is observed,481 and native stoichiometries are preserved, as for anthrax lethal toxin ((PA63)8(LFN)4, ~630 kDa) in its pore form.484 Many reagents have shown excellent supercharging properties, including some that are very economical.80,480,488,498,500 In addition to moving mass spectral peaks into m/z regions with higher resolving power, supercharging reagents can improve the efficiency of top-down dissociation methods, such as CID and electron capture/transfer dissociation (ECD, ETD).94,481,501-503
Manipulation of peaks to shift the opposite direction in m/z and thus spread out overlapped peaks can be done via charge reduction, which dates back to work done to measure gas-phase basicities of reagents and the addition of superbases and proton sponges to samples.301,473,475-478 Numerous experiments have demonstrated charge reduction of native biomolecular complexes upon addition of strong bases or their salts,470,504,505 such as triethylammonium salts,120,504,506-508 trimethylamine N-oxide (TMAO),509 polyamines,460 and imidazole derivatives,272,479 to native ESI buffers. Laganowsky showed that charge reduction can be especially useful in revealing multiple lipid adduction states for membrane proteins embedded in lipid-detergent micelles which were not accessible without addition of TMAO (see Figure 18).509 In addition to separating overlapping peaks in native mass spectra, charge reduction can also improve the ability of methods such as SID to maintain compact ion structures upon dissociation, useful for determining quaternary structure and subunit interactions in complexes.344,510-513
Figure 18.

Native mass spectrum of Escherichia coli ammonia channel B trimers with PO-phosphatidylethanolamine adducts and no TMAO (A) and with TMAO (B), illustrating charge reduction by TMAO and associated increase in the number of observable lipid adducts. Reprinted with permission from ref. 509. © 2019 American Society for Mass Spectrometry.
2.3.3.2. Ion/Ion Reactions.
Gas-phase ion/ion reactions469,514 performed inside the mass spectrometer are another common route to simplify otherwise complicated mass spectra. Building upon the use of corona discharge in ESI pioneered by Lloyd Smith,473,477 Cation to Anion Proton Transfer Reactions (CAPTR), introduced by Bush, is a method for reducing the charge of native protein cations in the gas phase by reacting them with small reagent anions that abstract protons.515,516 The reagent anions are produced from flowing fluorocarbons through a corona discharge in the ion source, after which they are trapped inside the instrument. The polarity of the ion source is rapidly switched to enable introduction of native protein ions produced by ESI, and these ions are combined with the reagent anions, to which they transfer protons, thereby reducing their charge state (see Figure 19). This method (as well as charge reduction and manipulation in general) can facilitate analysis of heterogeneous samples, as illustrated for the overlapped peak in the native mass spectrum in Figure 19A corresponding to charge states of two different proteins and subsequent separation of each species’ peaks with CAPTR in Figure 19D. Advantages of CAPTR for native biomolecular complexes can be similar to those of charge reduction reagents. A similar effect on charge states in native mass spectra can be obtained using a form of ETD, in which the multiply-charged protein ions capture an electron produced by a cathode or transferred from a radical anion reagent in the gas phase without subsequent dissociation (“ETnoD” or nondissociative electron transfer), as illustrated by results from the Barran and Sobott groups.107,517,518 Kaltashov and coworkers have also utilized transfer of electrons from ETD reagents (such as fluoranthrene radical anions) to protein ions isolated in small m/z windows to reduce mass spectral complexity and spread the resulting reduced charge states along the m/z axis (“limited charge reduction”).147,519,520
Figure 19.

Native mass spectrum of bovine serum albumin (BSA, blue) and yeast enolase dimer (green) (A), CAPTR of BSA15+ in the absence of enolase (B) and enolase21+ in the absence of BSA (C), and CAPTR of BSA15+/enolase21+ peak (D,E) from the red box in (A). Reprinted with permission from ref. 515. © 2015 American Society for Mass Spectrometry.
In 2020 McLuckey, a long-time innovator in gas-phase ion/ion chemistry,467-469,483,514,521 introduced a method similar to CAPTR, but instead using a modified source in which two electrospray sources are used, one in positive ion mode (for the ion of interest) and the other in negative ion mode.522 The anionic reagent is a protein ion with multiple negative charges, such as (insulin chain A)5-6− or (holomyoglobin)~10−, which, due to its physical size and large negative charge, has a large adduction cross section for recombination with the positively-charged native biomolecular ion of interest. This process can result in multiple adductions of the reagent protein to the ion of interest, reducing its net charge and producing a characteristic train of peaks at high m/z that can be used to identify it and determine its charge state based on the known mass of the reagent protein (see Figure 20).515 Additionally, proton transfer charge reduction capabilities are now possible on commercial instruments, such as Thermo Fisher Scientific’s Orbitrap Eclipse Tribrid mass spectrometer,523 and we refer interested readers to a recent review covering the latest developments in gas-phase ion/ion reactions in MS.514
Figure 20.

Post-ion attachment mass spectrum of E. coli 70S ribosome-related subunits showing charge reduction upon gas-phase attachment of holo-myoglobin10– and associated resolution of non-isobaric components of the native ion distribution (green box). Reprinted with permission from ref. 522. © 2020 American Chemical Society.
2.4. GLOBAL APPROACHES TO HETEROGENEITY
2.4.1. Average Composition
In some cases of extreme compositional heterogeneity, such as that in random copolymers or mixed-lipid Nanodiscs,77,258,302,524-526 it may be very difficult, even with the above state-of-the-art methods, to determine the complete mass and stoichiometry distributions for the entire ion population. However, a number of methods have been introduced to determine global average composition information for these types of samples.
2.4.1.1. Inference from Fragmentation Data.
Although Prebyl and Cook hinted at the possibility of using FT-based analysis methods to determine the monomer ratio of copolymers in the original article describing their FT-based algorithm (see §2.2.1.1),296 ultimately they demonstrated an alternative method relying on CID.527,528 In this method, it is assumed that the monomer composition of the end regions of the polymer ions is representative of the composition of the polymers as a whole. CID is used to fragment the polymer ion population formed using electrospray ionization, and low-mass fragment ions produced by this process are assigned based on the known monomer masses. The ratio of the total intensity of low-mass ions associated with each monomer is calculated and assumed to represent the global average composition of the copolymer sample. These authors examined the accuracy and precision of this approach at various collision voltages, including whether it is better to use only intact monomer fragments versus include secondary fragments.527 Intriguingly, for ~20 kDa poly(styrene sulfonate)-co-(maleic acid) samples, in which the styrene sulfonate (SS) monomers are significantly more acidic than the maleic acid (MA) monomers, observed fragment ratios were typically a factor of at least 3 times the monomer ratios provided by the manufacturer. The authors attributed this to a difference in ionization efficiency between ions with unusually high SS content and those with low SS content owing to the high acidity of this monomer. While the accuracy and precision of the method can be excellent, a calibration curve is necessary to reconcile the bias of the method toward the higher-acidity monomer.528 Importantly, recent work on the gas-phase behavior of lipids has demonstrated that deducing the average composition of such polydisperse analytes as membrane proteins and lipid Nanodiscs from observed lipid loss upon activation can be unreliable.80 108,303
2.4.1.2. “Double FT” Approach.
FT-based methods have enabled analysis of average composition directly from mass spectra of intact large ions, instead of relying on fragmentation data. As described above in §2.2.1.2, the “double FT" approach can be used to approximate the composition of Nanodiscs containing two different types of lipid. Marty illustrated use of this method to determine the composition of Nanodiscs formed from MSP1D1(−) and binary mixtures of palmitoyloleoylphosphatidylcholine (POPC) and either PO-phosphatidylserine (POPS) or PO-phosphatidylglycerol (POPG).302 Overall, excellent agreement was observed between the double FT-based lipid composition and the bulk lipid composition used to assemble the Nanodiscs. For these determinations, it was assumed that the average lipid mass found by double FT of the mass spectrum is a simple bulk population-weighted average of the lipid monomer masses. Double FT was recently used to determine the identity and relative abundances of lipid head groups adducted to native proteins and found excellent agreement with expected masses as well as abundances anticipated from the appearance of the raw mass spectra.303
2.4.1.3. Distinguishing Compositional Heterogeneity Types Using FT-Based Methods.
Cleary and Prell showed that FT-based approaches, such as those implemented in iFAMS, can be used not only to characterize the bulk composition of ion populations formed from two types of subunits, but also to reveal what type of heterogeneity is present in the sample (see Figure 21).77 They introduced a classification scheme for different common types of sample heterogeneity: “superpositions”/simple mixtures (Class I), mixtures satisfying a “mean-proportional-variance condition” (Class II), and mixtures following a multinomial subunit distribution (Class III). Class I includes mixtures of analytes that contain exclusively one type of subunit, such as a mixture of homopolymers or single-lipid Nanodiscs. Class II includes analytes for which incorporation of different subunits is essentially random, and the distribution of the entire population is well described by a convolution of separate distributions, one for each type of subunit, as may be the case for Nanodiscs made from pre-mixed non-interacting lipids or for random copolymers. Class III includes ion populations for which the incorporation of a particular type of subunit follows a multinomial distribution, such as different isotopes of a particular atom in an ion or different protein isoforms into a protein complex whose stability is not affected by the identity of the isoform.
Figure 21.

Schematic of different classes of compositional heterogeneity for analyte mixtures (left) and representative mass spectra and corresponding Fourier spectra for model ion populations representing each heterogeneity class. Reprinted with permission from ref. 77. © 2020 Royal Society of Chemistry.
Class I and II populations can often be clearly distinguished by their corresponding Fourier spectra, even when the bulk compositions of the mixtures are identical. This work also provided mathematical justification for Marty’s “double FT” approach302 (see §2.4.1.2) in analyzing binary phospholipid Nanodiscs. For the double FT approach to work, the mean number of each subunit type incorporated into the ion population must be proportional to the variance in the distribution for that subunit across the whole population (the “mean-proportional-variance condition”).77 Under other conditions, the result from the double FT approach can be inaccurate. Fortunately, the mean-proportional-variance condition likely holds for many common types of assembly mechanisms for copolymers, mixed-detergent micelles, Nanodiscs, and other membrane mimetics.
The FT approach of iFAMS can also be used to infer information about the assembly mechanism for heterogeneous ion populations based on their apparent membership in the various classes described above. For example, from their Class II FT spectra, it was deduced that phospholipids incorporate into Nanodiscs without extensive equilibration of their composition after Nanodisc assembly is arrested by complete removal of detergent,529,530 in agreeance with other experiments showing that lipid exchange between fully-formed Nanodiscs is very slow.524,531,532 This distinction, which is only possible through analysis of the Class behavior of the FT spectra because extensive equilibration would result in no change in bulk composition, illustrates the utility of compositional analysis even for poorly-resolved heterogeneous ion populations.
2.4.2. Assigning Biomolecular Ions to Chemical and Structural Class
For some samples, complete analysis of their mass spectra to achieve the level of detailed interpretation in many of the aforementioned strategies may not be possible. However, various features, including charge state, CCS, and/or Fourier frequency information, can still enable a coarse-grained level of characterization of the ion’s chemical or structural class, which we detail below.
2.4.2.1. Small Biomolecular Ions.
Although calling the structures of small biomolecular ions “native-like” may be inappropriate in many cases (for example, isolated phospholipid ions may have structures rather different from those when they are packed into cell membranes in vivo), structural classification and prediction based on electrospray IM-MS data provide key insights into how this approach might be used more generally for larger native biomolecular ions in the future. This type of classification could be particularly relevant for heterogeneous biomolecular complexes involving many small molecules (either bound or free in clusters) for which the identities are unknown and/or for which there are coincident masses. Because IM separation in the low-field limit reflects the “size” (CCS) to charge ratio of ions,533 some approaches for classifying ions according to their chemical structure take advantage of different typical densities belonging to each class. For example, over a wide range of masses, lipids tend to have lower densities (thus higher CCS) in the gas phase than do nearly isobaric carbohydrates, which tend to have high density owing to their very high number of internal hydrogen bonds.534-538 Peptides tend to fall somewhere in between, and small drug-like molecules tend to be lower in mass than the other three classes, yet span a wider range of CCS/z ratio (see Figure 22).537-539 McLean537,538 and Xu536,539 have demonstrated reliable and reproducible classification of lipids, sugars and polysaccharides, nucleotides, peptides, and small drug-like molecules into different regions within electrospray ion mobility-mass spectra, with CCS measured in nitrogen on both drift-tube and traveling-wave type ion mobility-mass spectrometers. Subclassification of phospholipids according to head group type has also been demonstrated, although isobaric lipids with different head groups types often differ in CCS by only a few percent,540 illustrating the necessity of increased separation and resolving power as described in §2.3.2. Zhu,535 McLean,537,538 Baker,534 and Xu536 have introduced efforts to build large, publicly available online compendia of CCS information for metabolites to improve database- and Machine Learning-based prediction of CCS using structural information as well as prediction of structure (from biomolecule type to more detailed Lewis structure) using IM-MS data. In combination with gas-phase isolation and fragmentation, remarkably detailed structures can be predicted using these approaches. This foundational work toward classification of small molecules using IM-MS data holds great promise with future interpretation of increasingly complex, larger, and heterogeneous samples, especially as IM-MS instrumentation continues to improve and native IM-MS is applied to investigate endogenous and/or unknown bound small molecules and ligands (see §2.3.2).142,167,440,541
Figure 22.

Composite nitrogen drift tube IM-MS data for singly-charged biological molecules illustrating separation into different m/z vs. CCS regions according to structure type. Reprinted in part with permission from ref. 534. © 2017 Royal Society of Chemistry.
2.4.2.2. Classification of Large Biomolecular Ion Conformation Using Native IM-MS Data.
Quantitative correlations of mass with charge state35,495,542-548 and with CCS320,495,549,550 have long been noted in IM-MS research. Based on a simplistic picture of the electrospray process, the charge state for globular ions is expected to follow a mass1/2 dependence due to the Rayleigh criterion for fission of charge droplets (the “Charge Residue Model”).7,543,551 By contrast, extended, quasi-linear structures478 should follow the relationship [(z−1) ln(z−1)] ∝ mass, where z is the charge of the ion.495 Structures in between these extremes, such as mostly globular native protein ions with unfolded or disordered regions, may follow intermediate behavior (see Figure 23).548 Likewise, CCS is expected to scale with mass2/3 for globular ions320,495,549,550 and mass1 for quasi-linear structures.488,495 Because solvent accessible surface area (SASA) can be computed very quickly by many molecular visualization and dynamics programs, some researchers have used SASA for modeled protein structures in place of experimental or computed CCS values.547,548,552 Empirical mass scaling exponents for charge, CCS, and SASA have been measured for a wide variety of proteins with structures ranging from intrinsically disordered or semi-disordered to compact globular. Using these expected scaling relationships, it is often possible to assign protein and protein complex ions (even with the same m/z) to different structural classes by examining IM-MS data. This simple approach can be very useful in determining whether a given set of solution and/or instrumental conditions produces a structurally homogeneous vs. heterogeneous ion population, and whether these structures are likely compact, partially unfolded, or extensively unfolded, especially important in cases where native MS reveal species not previously identified by other structural methods.299,379,406,553,554 Oligomers can also be classed into linear, compact, and other topologies based on expected CCS scaling with mass, even for samples containing mixtures of these topologies.548,552
Figure 23.
Representative native mass spectra of globular proteins (A: chicken-egg lysozyme, B: bovine β-lactoglobulin, C: human transferrin) and intrinsically disordered proteins (IDPs; D: Sic1-KID from Saccharomyces cerevisiae residues 215-284, E: human stathmin-4, F: murine ataxin-3 residues 1-291), illustrating relatively low charge states for globular proteins and multimodal distributions for IDPs. Reprinted with permission from ref. 548. © 2017 American Society for Mass Spectrometry.
2.4.2.3. Gábor-Transform Isolation of Biomolecular Ion Signal from High-Salt Background Signal.
One limitation in the use of native MS in structural biology is the reliance upon volatile buffer salts such as ammonium acetate rather than those which more closely resemble physiological conditions, due to the tendency of nonvolatile salts to complicate mass spectra and suppress ionization of the analyte of interest.10,11,483,555 Efforts to circumvent these challenges include the use of submicron emitter tips5,306,556-558 as well as improvements to data analysis methods (see SWARM, §2.2.2.1). Gábor Transform (see §2.1.9) of highly congested native mass spectra in iFAMS was used to characterize the masses of monomeric protein ions electrosprayed from buffers containing a relatively high concentration of salt (100 mM NaCl in Tris or HEPES buffer).298 Despite signal from large salt cluster ions dominating the mass spectrum, GT enabled isolation of signal arising from the protein. As illustrated in Figure 24, signal from protein ions (which tend to follow a negatively chirped pattern) can be visually distinguished in the GT spectrogram from interfering/overlapping salt cluster signal (which appear as horizontal stripes or positively chirped patterns). This difference arises from the essentially constant mass of the protein ions as a function of charge state, whereas the charge state of salt cluster ions and clusters of small molecules such as lipids tends to increase with mass. By the same reasoning, protein ions of similar m/z but different masses can in principle be readily distinguished in GT spectra upon visual inspection due to their different chirp patterns, as was demonstrated for α-hemolysin hexameric and heptameric complexes which were overlapped in both the FT spectrum and the mass spectrum.299 As seen in Figure 24, higher charge states (indicative of some unfolding) can often be easier to detect in a high salt cluster background than fully-folded native ions of lower charge states, but the chirp pattern established by these higher-signal peaks can facilitate visual identification of lower-signal native peaks.298
Figure 24.

Mass spectrum of native-like anthrax toxin Lethal Factor N-terminal domain electrosprayed from Tris/sodium chloride buffer, (top red trace), corresponding Fourier spectrum (right red trace), and Gabor spectrogram (heat map), illustrating separation of negatively-chirped protein signal (labeled according to charge state in inset) and sodium chloride clusters (horizontal bands). Note that cluster ion and protein ions signals are strongly overlapped in both m/z and frequency but are easy to identify in the Gabor spectrogram. Reprinted with permission from ref. 298. © 2019 Wiley-VCH Verlag GmbH & Co.
3. CONCLUSIONS AND OUTLOOK
3.1. CURRENT STATE OF THE FIELD
Above we have provided an overview of past and state-of-the-art approaches toward overcoming the problem of heterogeneity in native MS, with a specific focus on strategies which enable preservation of inherent heterogeneity of samples important for understanding biological structure and function and aim to facilitate analysis and interpretation. Initial efforts in this area focused on accurate assignment of charge states and masses to relatively simple native mass spectra representing few ions and with ample resolution of individual charge states.29,559-565 Of course, as instrumentation rapidly improved and landmark achievements were made, samples of ever-increasing complexity have become routine to investigate with this powerful technique.
Today, it is possible to analyze mass spectra representing highly polydisperse ion populations, with broad charge state distributions and tens or even hundreds of overlapping peaks, and researchers have a plentiful buffet of programs and algorithms from which to select. Automation and batch processing has continued to improve to the point that some published articles in the field of native MS now reflect many tens to hundreds of individual mass spectra80,254,524 that might be effectively hopeless to analyze by hand, an improvement that parallels software development in “omics” fields.566,567 Adjuvant strategies for separating complex ion mixtures using chemical reactions or labeling during the electrospray process or within the mass spectrometer have further expanded the range of challenging samples that can be addressed. Deconvolution approaches now span the range from game theory to Bayesian inference to Fourier/Gábor Transform methods from signal processing theory. This plethora of “orthogonal” deconvolution methods offers the promise of cross-validation, although to date this has been rarely implemented in the literature.163,202 Furthermore, for the past 25 years or so, the use of volatile salt “buffers”568 (such as ammonium acetate) has been nearly universal in native IM-MS due to the adverse effects of salt adduction when using more physiologically-relevant buffers (Tris, HEPES, phosphate buffers, etc.). Modern deconvolution methods, including Gábor Transform and SWARM, as well as the recent use of submicron nESI emitters5,6,306,555-558 may finally liberate native IM-MS from dependence on volatile salts and artifacts arising from their use.568 Other current efforts toward better understanding of detergent/lipid properties and their influence on membrane protein behavior, as well as engineered and tailored membrane scaffold proteins and lipids for Nanodisc construction and other membrane mimetics, also present exciting avenues for the future of native MS.257,258,569-575
Additionally, continual improvements and innovation to instrumentation, including increasing mass resolution and separation capabilities and implementing various techniques including ion/ion reaction capabilities into high-performing mass analyzer instrument platforms, demonstrates the rapid, ever-evolving state of this field. Thanks to these advancements, native MS investigation of extremely large, heterogeneous samples, such as intact viral capsids, multimeric protein complexes, and membrane proteins, is now in many laboratories routine. Recent work in combining native MS with other techniques, such as cryo-electron microscopy (cryo-EM) and omics approaches,440,576-581 and in analyzing samples directly from native environments, lipid vesicles, and/or crude cell lysates266,271,541,573,582-591 constitutes the very exciting, hybrid future of structural biology and of the role of native MS within it.592,593
3.2. REMAINING CHALLENGES
3.2.1. Recalcitrant Features of Heterogeneity
Despite major improvements in theory, software, sample preparation, and instrumentation, it remains very challenging to quantitate heterogeneous mixtures with very different component intensities, although solving this problem would be highly beneficial for drug development, fundamental biochemistry, and related fields. For example, this problem arises when large and small peaks overlap in the mass spectrum or Fourier/Gábor spectrum, in which case it can be extremely challenging to decide whether the small peak is present. Curiously, resolution generally improves in Fourier/Gábor spectra with higher polydispersity in the corresponding mass spectrum. Thus the complementarity of this method with other methods operating on the m/z domain suggests that combining both approaches may provide an optimal path forward in mixture quantitation. For both types of approach, however, it is still very challenging in general to analyze polydisperse multi-subunit ion populations if the subunit masses are not near-multiples of one another.
Another outstanding question pervading native IM-MS is whether measured ion abundances do in fact quantitatively reflect those present in solution, let alone under what conditions native-like ions may be relevant for understanding structure and function. Recent results555 using submicron nESI emitters indicate that biomolecular ions formed from solutions containing higher concentrations of physiological salts (such as sodium chloride) can be stabilized in more compact conformations, consistent with what has long been known about effects on protein stability from different ions first described by Hofmeister in 1888.594,595 Thus experimental methods which enable ionization of biomolecules from physiological buffers5,6,555,556 and deconvolution methods298,304 that can eliminate remaining background salt cluster signal and/or accurately account for salt adduction to biomolecular ions will be especially important for approaching this question for heterogeneous mixtures.
3.2.2. Is There a “Complexity Limit” in Native Mass Spectrometry?
All of the data analysis methods described in our review are ultimately limited by the resolution of the mass spectra, which typically decreases as the ion population grows more heterogeneous.367 Although the resolving power and sensitivity of modern mass spectrometers continue to improve, researchers will inevitably need to understand yet larger, more complex and heterogeneous samples. It is therefore imperative to continue developing methods that anticipate these future advances or which can work together synergistically to combat the problem. For example, many current deconvolution methods can be and are regularly used without the luxury of isotopic resolution, which somewhat paradoxically can simplify the deconvolution process and interpretation of the resulting data. How will these algorithms perform if and when much higher resolution is readily available? It is plausible that unique assignments of peaks for complex isotope distributions of overlapped ions representing different species will be very challenging within the m/z domain, and high m/z resolution may lead to extensive harmonic overlap in FT/GT approaches, complicating deconvolution. Perhaps methods like SWARM could be combined with Bayesian, game theoretical, or FT/GT methods, for example, to first “de-isotope” the mass spectrum before further processing. Alternatively, charge manipulation or CDMS methods might be combined with deconvolution approaches to handle highly heterogeneous samples that suffer from space-charge repulsion or other resolution-reducing phenomena that occur with conventional mass spectrometry instrumentation. Continued investigation of these theoretical challenges in advance of improvements in instrumental resolution is therefore highly desirable.
3.2.3. Education Barriers
The variety of methods described in this review for approaching heterogeneous native samples with IM-MS is both exciting and daunting. Are there now too many options to choose from when deconvolving a complex mass spectrum? How should a researcher go about deciding which one to use? Many of the deconvolution methods here involve a substantial dose of mathematics, probability theory, signal processing, and facility with programming that many researchers may not have encountered in their training. Thus developers in this field face a major challenge of educating potential users on both theoretical aspects of how these approaches work as well as their practical use. A number of the data analysis tools described in this review have been made deliberately open-source so that users around the world can adapt the code to their own purposes, but doing so can be very intimidating for many new users. Fortunately, modern software sharing platforms, such as GitHub and GitLab, online science communities like Zenodo, and video sharing platforms (YouTube, Vimeo, and many others) offer researchers new and innovative ways to share their developments with others in ways beyond the written page, including through step-by-step video tutorials. Workshops at conferences aimed at training new users on the theory and best practices for using these programs are increasingly common. It is our view that increased training of undergraduate and graduate students in practical scientific programming and modern data analysis methods will be highly beneficial in preparing the next generation of scientists to use these methods to their full potential. In parallel, we recommend that developers of these methods make a concerted effort to use online tools such as those mentioned above to lower the barrier for access to these powerful programs. Several good models for these recommendations exist already in both industry and academia. Protein Metrics Inc., for example, hosts regular user meetings and webinars for their software, which includes PMI Intact discussed here, as well as other tools for omics research.287,288 The National Resource for Native MS-Guided Structural Biology,596 funded by the National Institutes of Health since 2018, hosts regular workshops led by algorithm developers, instrumentation innovators, and technique pioneers with a goal to educate potential users.
3.3. FUTURE STRATEGIES AND BEST PRACTICES
In our view, future advances to overcome the heterogeneity problem in native MS should embrace and preserve the inherent heterogeneity of samples rather than requiring researchers to mitigate it or make samples more homogenous, as was typically the focus of early efforts and much related discussion in the literature to date. Sustained growth of native MS as a tool in structural biology in many ways depends upon this strategy, as these heterogeneous features, such as bound small molecules, multiple coexisting stoichiometries, etc., continue to be revealed as important for understanding biological structure and function. Specifically with regard to optimal deconvolution methods, critical, necessary features include: ease and flexibility/customization of use (both practically and with regard to different kinds of samples/information amenable), availability of resources and education materials for users including both practical and theoretical aspects as well as cautions about potential artifacts, ability to output information in formats digestible for both native MS experts and novices, compatibility with different mass spectrometer platforms and data types, and minimal requirements for user input which may ultimately bias results and lead to errors. Ease of interpretation is especially important for integration of these tools into industry settings, in addition to rigorous validation and automation of these methods.154 We also envision a future in which multiple different tools can be integrated onto the same platform to provide complementary and/or supporting information, including development of field-wide scoring metrics, which has been the focus of some recent efforts already.283,597,598
Based on our above analysis, we believe that a number of strategies exist that can be immediately undertaken to address the challenges outlined in §3.2. For example, streamlining existing software programs based on user feedback will greatly increase their widespread utility and application, thus continued conference and online workshops aimed at training users on and improving software through direct interaction will be very useful.596 Convergence on a small number of universal data formats amenable to multiple software platforms will provide a path towards improved reproducibility and cross-platform validation, as will inclusion of metadata needed for reproduction of analysis results in public data repositories.123 Continued development of cross-platform validation methods (such as comparing results from “orthogonal” approaches, e.g., Bayesian and FT/GT methods, or even feeding them into each other163,202,256) and standardization of quality scores283,284 for results produced from them will help users identify artifacts and better characterize uncertainties. For example, FT/GT methods can greatly facilitate identification of charge states for distinct, highly overlapped peaks in mass spectra,299 thus inputting the range of charge states thereby identified may greatly reduce artifacts of other deconvolution methods that perform best when the charge state range is confined to correct values. Experimental and instrumental improvements possible in the near future include development of robust inlet-based separation beyond liquid chromatography (such as capillary zone electrophoresis139-141), next-generation nESI tip design (including reliable production of submicron emitters5,6,118,306,555,556,558 and theta-glass emitters for rapid mixing of samples during the ESI process599-601), and more efficient in-source desolvation.210,602
In the more distant future, we also envision theoretical and instrument developments that reveal new types of information in native IM-MS data. For example, field alignment of biomolecular ions in IM-MS instruments may be used to separate ions based on structural differences not easily observed in experiments on current low-field IM-MS instruments.328 Further theoretical investigation into the relationship between observed heterogeneity/polydispersity and assembly mechanisms and kinetics may reveal information that is very challenging to deduce by other means.77,524 Continued improvement in modeling of dissociation, unfolding, and labeling kinetics and energetics will also allow researchers to design experimental protocols that can unveil subtle structural details and possibly differences not resolved by conventional native IM-MS.92,111,317,326,398,501,513,515,516,603-616 Finally, using the data analysis tools described in this review, streamlining the interface between native IM-MS and complementary state-of-the-art structural methods, such as cryo-EM and coherent diffractive imaging, will likely provide unprecedented insight into composition, structure, and behavior of highly heterogeneous biomolecular systems.
ACKNOWLEDGMENTS
A.D.R. is supported by the National Institute of General Medical Sciences (Award 2T32GM00759) and Peter O’Day Fellowship in Biological Sciences and Office of the Vice President for Research and Innovation at the University of Oregon, and she is an ARCS Scholar supported by the ARCS Oregon Chapter. Research highlighted in this review was supported by the National Institute for Allergy and Infectious Diseases (Award R21AI125804 to J.S.P.) and the National Science Foundation (Award CHE-1752994 to J.S.P.).
LIST OF ABBREVIATIONS USED
- CAPTR
Cation to Anion Proton Transfer Reactions
- CCS
collision cross section
- CDMS
charge detection mass spectrometry
- CHAMP
Calculating Heterogeneous Assembly and Mass spectra of Proteins
- CID
collision-induced dissociation
- cryo-EM
cryogenic electron microscopy
- ECD
electron capture dissociation
- ESI
electrospray ionization
- ETD
electron transfer dissociation
- ETnoD
nondissociative electron transfer
- FT
Fourier Transform
- GT
Gábor Transform
- iFAMS
interactive Fourier-Transform Analysis for Mass Spectrometry
- IM-MS
ion mobility-mass spectrometry
- LC-MS
liquid chromatography-mass spectrometry
- m/z
mass-to-charge ratio
- MA
maleic acid
- MALDI
Matrix-Assisted Laser Desorption Ionization
- MCA
Multiplicative Correlation Algorithm
- MMD
Macromolecular Mass Defect
- MS
mass spectrometry
- nESI
nanoelectrospray ionization
- PMI Intact
Protein Metrics Intact
- POPC
palmitoyloleoylphosphatidylcholine
- POPG
palmitoyloleoylphosphatidylglycerol
- POPS
palmitoyloleoylphosphatidylserine
- Q-TOF
quadrupole-time-of-flight
- S/N
signal-to-noise ratio
- SASA
solvent accessible surface area
- SID
surface-induced dissociation
- SLIM
Structures for Lossless Ion Manipulations
- SOMMS
SOlving complex Macromolecular Mass Spectra
- SS
styrene sulfonate
- SUMMIT
SUMming Masses for Interaction Topology
- SWARM
Sliding Window Adduct Removal Method
- TIMS
Trapped Ion Mobility Spectrometry
- TMAO
trimethylamine N-oxide
- TWIM
Traveling Wave Ion Mobility
Biography
Amber D. Rolland graduated summa cum laude from the University of Central Arkansas with a B.S. in Chemistry and joined the PhD program in Biochemistry at the University of Oregon in 2016. Her PhD thesis focuses on improving native IM-MS methodology to characterize protein structure and its dependence on environment, including physiological membranes. Rolland is an ARCS Foundation scholar and recipient of a Peter O’Day Fellowship in Biological Sciences, John R. Moore Scholarship, two Doctoral Student Service Awards, and the Doctoral Research Fellowship from the University of Oregon.
James S. Prell graduated summa cum laude from Washington University in St. Louis in 2005 with a B.A. in Mathematics, Chemistry, and German and minors in Religious Studies and Music. He received a PhD in Chemistry in 2011 from the University of California, Berkeley, in the group of Prof. Evan R. Williams and was a postdoctoral scholar in the group of Prof. Stephen R. Leone from 2011-2014. He joined the Department of Chemistry and Biochemistry at the University of Oregon in 2014, where his group studies fundamentals and applications of native IM-MS, focusing on theory, analytical software, and experimental method development to characterize biomolecule structures and interactions. He received a National Science Foundation CAREER Award and an American Society for Mass Spectrometry Research Award in 2018.
References
- (1).van den Heuvel RHH; Heck AJR Native Protein Mass Spectrometry: From Intact Oligomers to Functional Machineries. Curr. Opin. Chem. Biol 2004, 8, 519–526. [DOI] [PubMed] [Google Scholar]
- (2).Leney AC; Heck AJR Native Mass Spectrometry: What Is in the Name? J. Am. Soc. Mass Spectrom 2017, 28, 5–13. [DOI] [PubMed] [Google Scholar]
- (3).Barth M; Schmidt C Native Mass Spectrometry—a Valuable Tool in Structural Biology. J. Mass Spectrom 2020, 55, e4578. [DOI] [PubMed] [Google Scholar]
- (4).Wilm M; Mann M Analytical Properties of the Nanoelectrospray Ion Source. Anal. Chem 1996, 68, 1–8. [DOI] [PubMed] [Google Scholar]
- (5).Susa AC; Xia Z; Williams ER Small Emitter Tips for Native Mass Spectrometry of Proteins and Protein Complexes from Nonvolatile Buffers That Mimic the Intracellular Environment. Anal. Chem 2017, 89, 3116–3122. [DOI] [PubMed] [Google Scholar]
- (6).Susa AC; Xia Z; Williams ER Native Mass Spectrometry from Common Buffers with Salts That Mimic the Extracellular Environment. Angew. Chem. Int. Ed 2017, 56, 7912–7915. [DOI] [PubMed] [Google Scholar]
- (7).Felitsyn N; Peschke M; Kebarle P Origin and Number of Charges Observed on Multiply-Protonated Native Proteins Produced by Esi. Int. J. Mass Spectrom 2002, 219, 39–62. [Google Scholar]
- (8).Kebarle P; Verkerk UH Electrospray: From Ions in Solution to Ions in the Gas Phase, What We Know Now. Mass Spectrom. Rev 2009, 28, 898–917. [DOI] [PubMed] [Google Scholar]
- (9).Peschke M; Verkerk UH; Kebarle P Features of the ESI Mechanism That Affect the Observation of Multiply Charged Noncovalent Protein Complexes and the Determination of the Association Constant by the Titration Method. J. Am. Soc. Mass Spectrom 2004, 15, 1424–1434. [DOI] [PubMed] [Google Scholar]
- (10).Iavarone AT; Udekwu OA; Williams ER Buffer Loading for Counteracting Metal Salt-Induced Signal Suppression in Electrospray Ionization. Anal. Chem 2004, 76, 3944–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Metwally H; McAllister RG; Konermann L Exploring the Mechanism of Salt-Induced Signal Suppression in Protein Electrospray Mass Spectrometry Using Experiments and Molecular Dynamics Simulations. Anal. Chem 2015, 87, 2434–2442. [DOI] [PubMed] [Google Scholar]
- (12).Chen F; Gülbakan B; Weidmann S; Fagerer SR; Ibáñez AJ; Zenobi R Applying Mass Spectrometry to Study Non-Covalent Biomolecule Complexes. Mass Spectrom. Rev 2016, 35, 48–70. [DOI] [PubMed] [Google Scholar]
- (13).Tanaka K; Waki H; Ido Y; Akita S; Yoshida Y; Yoshida T; Matsuo T Protein and Polymer Analyses up to m/z 100000 by Laser Ionization Time-of-Flight Mass Spectrometry. Rapid Commun. Mass Spectrom 1988, 2, 151–153. [Google Scholar]
- (14).Trimpin S "Magic" Ionization Mass Spectrometry. J. Am. Soc. Mass Spectrom 2016, 27, 4–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).Fenn J; Mann M; Meng C; Wong S; Whitehouse C Electrospray Ionization for Mass Spectrometry of Large Biomolecules. Science 1989, 246, 64–71. [DOI] [PubMed] [Google Scholar]
- (16).Chowdhury SK; Katta V; Chait BT An Electrospray-Ionization Mass Spectrometer with New Features. Rapid Commun. Mass Spectrom 1990, 4, 81–87. [DOI] [PubMed] [Google Scholar]
- (17).Katta V; Chait BT Observation of the Heme-Globin Complex in Native Myoglobin by Electrospray-Ionization Mass Spectrometry. J. Am. Chem. Soc 1991, 113, 8534–8535. [Google Scholar]
- (18).Ganem B; Li YT; Henion JD Detection of Noncovalent Receptor-Ligand Complexes by Mass Spectrometry. J. Am. Chem. Soc 1991, 113, 6294–6296. [Google Scholar]
- (19).Light-Wahl KJ; Loo JA; Edmonds CG; Smith RD; Witkowska HE; Shackleton CHL; Wu C-SC Collisionally Activated Dissociation and Tandem Mass Spectrometry of Intact Hemoglobin B-Chain Variant Proteins with Electrospray Ionization. Biol. Mass Spectrom 1993, 22, 112–120. [DOI] [PubMed] [Google Scholar]
- (20).Miranker A; Robinson C; Radford S; Aplin R; Dobson C Detection of Transient Protein Folding Populations by Mass Spectrometry. Science 1993, 262, 896–900. [DOI] [PubMed] [Google Scholar]
- (21).Light-Wahl KJ; Schwartz BL; Smith RD Observation of the Noncovalent Quaternary Associations of Proteins by Electrospray Ionization Mass Spectrometry. J. Am. Chem. Soc 1994, 116, 5271–5278. [DOI] [PubMed] [Google Scholar]
- (22).Mann M; Wilm M Electrospray Mass Spectrometry for Protein Characterization. Trends Biochem. Sci 1995, 20, 219–224. [DOI] [PubMed] [Google Scholar]
- (23).Fitzgerald MC; Chernushevich I; Standing KG; Whitman CP; Kent SB Probing the Oligomeric Structure of an Enzyme by Electrospray Ionization Time-of-Flight Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A 1996, 93, 6851–6856. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Mirza UA; Chait BT Do Proteins Denature During Droplet Evolution in Electrospray Ionization? Int. J. Mass Spectrom. Ion Processes 1997, 162, 173–181. [Google Scholar]
- (25).Benjamin DR; Robinson CV; Hendrick JP; Hartl FU; Dobson CM Mass Spectrometry of Ribosomes and Ribosomal Subunits. Proc. Natl. Acad. Sci. U. S. A 1998, 95, 7391–7395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Rostom AA; Robinson CV Detection of the Intact Groel Chaperonin Assembly by Mass Spectrometry. J. Am. Chem. Soc 1999, 121, 4718–4719. [Google Scholar]
- (27).van Berkel WJ; van den Heuvel RH; Versluis C; Heck AJ Detection of Intact Megadalton Protein Assemblies of Vanillyl-Alcohol Oxidase by Mass Spectrometry. Protein Sci. 2000, 9, 435–439. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Rostom AA; Fucini P; Benjamin DR; Juenemann R; Nierhaus KH; Hartl FU; Dobson CM; Robinson CV Detection and Selective Dissociation of Intact Ribosomes in a Mass Spectrometer. Proc. Natl. Acad. Sci. U. S. A 2000, 97, 5185–5190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (29).Tito MA; Tars K; Valegard K; Hajdu J; Robinson CV Electrospray Time-of-Flight Mass Spectrometry of the Intact MS2 Virus Capsid. J. Am. Chem. Soc 2000, 122, 3550–3551. [Google Scholar]
- (30).Tito MA; Miller J; Walker N; Griffin KF; Williamson ED; Despeyroux-Hill D; Titball RW; Robinson CV Probing Molecular Interactions in Intact Antibody: Antigen Complexes, an Electrospray Time-of-Flight Mass Spectrometry Approach. Biophys. J 2001, 81, 3503–3509. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (31).Hernández H; Robinson CV Dynamic Protein Complexes: Insights from Mass Spectrometry. J. Biol. Chem 2001, 276, 46685–46688. [DOI] [PubMed] [Google Scholar]
- (32).Sobott F; Robinson CV Characterising Electrosprayed Biomolecules Using Tandem-MS—the Noncovalent GroEL Chaperonin Assembly. Int. J. Mass Spectrom 2004, 236, 25–32. [Google Scholar]
- (33).Ilag LL; Ubarretxena-Belandia I; Tate CG; Robinson CV Drug Binding Revealed by Tandem Mass Spectrometry of a Protein–Micelle Complex. J. Am. Chem. Soc 2004, 126, 14362–14363. [DOI] [PubMed] [Google Scholar]
- (34).Hernández H; Robinson CV Determining the Stoichiometry and Interactions of Macromolecular Assemblies from Mass Spectrometry. Nat. Protoc 2007, 2, 715. [DOI] [PubMed] [Google Scholar]
- (35).Heck AJR; van den Heuvel RHH Investigation of Intact Protein Complexes by Mass Spectrometry. Mass Spectrom. Rev 2004, 23, 368–389. [DOI] [PubMed] [Google Scholar]
- (36).Loo JA Studying Noncovalent Protein Complexes by Electrospray Ionization Mass Spectrometry. Mass Spectrom. Rev 1997, 16, 1–23. [DOI] [PubMed] [Google Scholar]
- (37).Marcoux J; Robinson, Carol V. Twenty Years of Gas Phase Structural Biology. Structure 2013, 21, 1541–1550. [DOI] [PubMed] [Google Scholar]
- (38).Hilton GR; Benesch JLP Two Decades of Studying Non-Covalent Biomolecular Assemblies by Means of Electrospray Ionization Mass Spectrometry. J. R. Soc. Interface 2012, 9, 801–816. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (39).Boeri Erba E; Petosa C The Emerging Role of Native Mass Spectrometry in Characterizing the Structure and Dynamics of Macromolecular Complexes. Protein Sci. 2015, 24, 1176–1192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (40).Winger BE; Light-Wahl KJ; Ogorzalek Loo RR; Udseth HR; Smith RD Observation and Implications of High Mass-to-Charge Ratio Ions from Electrospray Ionization Mass Spectrometry. J. Am. Soc. Mass Spectrom 1993, 4, 536–545. [DOI] [PubMed] [Google Scholar]
- (41).Collings BA; Douglas DJ An Extended Mass Range Quadrupole for Electrospray Mass Spectrometry. Int. J. Mass Spectrom. Ion Processes 1997, 162, 121–127. [Google Scholar]
- (42).Sobott F; Benesch JLP; Vierling E; Robinson CV Subunit Exchange of Multimeric Protein Complexes: Real-Time Monitoring of Subunit Exchange between Small Heat Shock Proteins by Using Electrospray Mass Spectrometry. J. Biol. Chem 2002, 277, 38921–38929. [DOI] [PubMed] [Google Scholar]
- (43).van den Heuvel RHH; van Duijn E; Mazon H; Synowsky SA; Lorenzen K; Versluis C; Brouns SJJ; Langridge D; van der Oost J; Hoyes J, et al. Improving the Performance of a Quadrupole Time-of-Flight Instrument for Macromolecular Mass Spectrometry. Anal. Chem 2006, 78, 7473–7483. [DOI] [PubMed] [Google Scholar]
- (44).Olsen JV; Macek B; Lange O; Makarov A; Horning S; Mann M Higher-Energy C-Trap Dissociation for Peptide Modification Analysis. Nat. Methods 2007, 4, 709–712. [DOI] [PubMed] [Google Scholar]
- (45).Rose RJ; Damoc E; Denisov E; Makarov A; Heck AJR High-Sensitivity Orbitrap Mass Analysis of Intact Macromolecular Assemblies. Nat. Methods 2012, 9, 1084–1086. [DOI] [PubMed] [Google Scholar]
- (46).Tang X-J; Fred Brewer C; Saha S; Chernushevich I; Ens W; Standing KG; Chait BT Investigation of Protein-Protein Noncovalent Interactions in Soybean Agglutinin by Electrospray Ionization Time-of-Flight Mass Spectrometry. Rapid Commun. Mass Spectrom 1994, 8, 750–754. [DOI] [PubMed] [Google Scholar]
- (47).Verentchikov AN; Ens W; Standing KG Reflecting Time-of-Flight Mass Spectrometer with an Electrospray Ion Source and Orthogonal Extraction. Anal. Chem 1994, 66, 126–133. [DOI] [PubMed] [Google Scholar]
- (48).Morris HR; Paxton T; Dell A; Langhorne J; Berg M; Bordoli RS; Hoyes J; Bateman RH High Sensitivity Collisionally-Activated Decomposition Tandem Mass Spectrometry on a Novel Quadrupole/Orthogonal-Acceleration Time-of-Flight Mass Spectrometer. Rapid Commun. Mass Spectrom 1996, 10, 889–896. [DOI] [PubMed] [Google Scholar]
- (49).Thalassinos K; Slade SE; Jennings KR; Scrivens JH; Giles K; Wildgoose J; Hoyes J; Bateman RH; Bowers MT Ion Mobility Mass Spectrometry of Proteins in a Modified Commercial Mass Spectrometer. Int. J. Mass Spectrom 2004, 236, 55–63. [Google Scholar]
- (50).Giles K; Pringle SD; Worthington KR; Little D; Wildgoose JL; Bateman RH Applications of a Travelling Wave-Based Radio-Frequency-Only Stacked Ring Ion Guide. Rapid Commun. Mass Spectrom 2004, 18, 2401–2414. [DOI] [PubMed] [Google Scholar]
- (51).Pringle SD; Giles K; Wildgoose JL; Williams JP; Slade SE; Thalassinos K; Bateman RH; Bowers MT; Scrivens JH An Investigation of the Mobility Separation of Some Peptide and Protein Ions Using a New Hybrid Quadrupole/Travelling Wave IMS/Oa-TOF Instrument. Int. J. Mass Spectrom 2007, 261, 1–12. [Google Scholar]
- (52).Hu Q; Noll RJ; Li H; Makarov A; Hardman M; Cooks RG The Orbitrap: A New Mass Spectrometer. J. Mass Spectrom 2005, 40, 430–443. [DOI] [PubMed] [Google Scholar]
- (53).Makarov A Electrostatic Axially Harmonic Orbital Trapping: A High-Performance Technique of Mass Analysis. Anal. Chem 2000, 72, 1156–1162. [DOI] [PubMed] [Google Scholar]
- (54).Hardman M; Makarov AA Interfacing the Orbitrap Mass Analyzer to an Electrospray Ion Source. Anal. Chem 2003, 75, 1699–1705. [DOI] [PubMed] [Google Scholar]
- (55).Fuerstenau SD; Benner WH; Thomas JJ; Brugidou C; Bothner B; Siuzdak G Mass Spectrometry of an Intact Virus. Angew. Chem. Int. Ed 2001, 40, 541–544. [DOI] [PubMed] [Google Scholar]
- (56).Siuzdak G; Bothner B; Yeager M; Brugidou C; Fauquet CM; Hoey K; Change C-M Mass Spectrometry and Viral Analysis. Chem. Biol 1996, 3, 45–48. [DOI] [PubMed] [Google Scholar]
- (57).Barrera NP; Di Bartolo N; Booth PJ; Robinson CV Micelles Protect Membrane Complexes from Solution to Vacuum. Science 2008, 321, 243–246. [DOI] [PubMed] [Google Scholar]
- (58).Laganowsky A; Reading E; Hopper JTS; Robinson CV Mass Spectrometry of Intact Membrane Protein Complexes. Nat. Protoc 2013, 8, 639–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (59).Marty MT; Zhang H; Cui WD; Blankenship RE; Gross ML; Sligar SG Native Mass Spectrometry Characterization of Intact Nanodisc Lipoprotein Complexes. Anal. Chem 2012, 84, 8957–8960. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (60).Hopper JTS; Yu YTC; Li DF; Raymond A; Bostock M; Liko I; Mikhailov V; Laganowsky A; Benesch JLP; Caffrey M, et al. Detergent-Free Mass Spectrometry of Membrane Protein Complexes. Nat. Methods 2013, 10, 1206–1208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (61).Leney AC; Fan X; Kitova EN; Klassen JS Nanodiscs and Electrospray Ionization Mass Spectrometry: A Tool for Screening Glycolipids against Proteins. Anal. Chem 2014, 86, 5271–5277. [DOI] [PubMed] [Google Scholar]
- (62).Leney AC; McMorran LM; Radford SE; Ashcroft AE Amphipathic Polymers Enable the Study of Functional Membrane Proteins in the Gas Phase. Anal. Chem 2012, 84, 9841–9847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (63).Watkinson TG; Calabrese AN; Giusti F; Zoonens M; Radford SE; Ashcroft AE Systematic Analysis of the Use of Amphipathic Polymers for Studies of Outer Membrane Proteins Using Mass Spectrometry. Int. J. Mass Spectrom 2015, 391, 54–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (64).Keener JE; Zhang G; Marty MT Native Mass Spectrometry of Membrane Proteins. Anal. Chem 2021, 93, 583–597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (65).Rosati S; van den Bremer ETJ; Schuurman J; Parren PWHI; Kamerling JP; Heck AJR In-Depth Qualitative and Quantitative Analysis of Composite Glycosylation Profiles and Other Micro-Heterogeneity on Intact Monoclonal Antibodies by High-Resolution Native Mass Spectrometry Using a Modified Orbitrap. mAbs 2013, 5, 917–924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (66).Olivova P; Chen W; Chakraborty AB; Gebler JC Determination of N-Glycosylation Sites and Site Heterogeneity in a Monoclonal Antibody by Electrospray Quadrupole Ion-Mobility Time-of-Flight Mass Spectrometry. Rapid Commun. Mass Spectrom 2008, 22, 29–40. [DOI] [PubMed] [Google Scholar]
- (67).Konijnenberg A; Butterer A; Sobott F Native Ion Mobility-Mass Spectrometry and Related Methods in Structural Biology. Biochim. Biophys. Acta Proteins Proteom 2013, 1834, 1239–1256. [DOI] [PubMed] [Google Scholar]
- (68).Liko I; Allison TM; Hopper JTS; Robinson CV Mass Spectrometry Guided Structural Biology. Curr. Opin. Struct. Biol 2016, 40, 136–144. [DOI] [PubMed] [Google Scholar]
- (69).van Duijn E Current Limitations in Native Mass Spectrometry Based Structural Biology. J. Am. Soc. Mass Spectrom 2010, 21, 971–978. [DOI] [PubMed] [Google Scholar]
- (70).Benesch JLP; Ruotolo BT Mass Spectrometry: Come of Age for Structural and Dynamical Biology. Curr. Opin. Struct. Biol 2011, 21, 641–649. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (71).Struwe WB; Robinson CV Relating Glycoprotein Structural Heterogeneity to Function – Insights from Native Mass Spectrometry. Curr. Opin. Struct. Biol 2019, 58, 241–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (72).Boeri Erba E; Signor L; Petosa C Exploring the Structure and Dynamics of Macromolecular Complexes by Native Mass Spectrometry. J. Proteom 2020, 222, 103799. [DOI] [PubMed] [Google Scholar]
- (73).Mann M; Meng CK; Fenn JB Interpreting Mass-Spectra of Multiply Charged Ions. Anal. Chem 1989, 61, 1702–1708. [Google Scholar]
- (74).Tong W; Wang G How Can Native Mass Spectrometry Contribute to Characterization of Biomacromolecular Higher-Order Structure and Interactions? Methods 2018, 144, 3–13. [DOI] [PubMed] [Google Scholar]
- (75).Wang W; Kitova EN; Klassen JS Nonspecific Protein–Carbohydrate Complexes Produced by Nanoelectrospray Ionization. Factors Influencing Their Formation and Stability. Anal. Chem 2005, 77, 3060–3071. [DOI] [PubMed] [Google Scholar]
- (76).Miller ZM; Zhang JD; Donald WA; Prell JS Gas-Phase Protonation Thermodynamics of Biological Lipids: Experiment, Theory, and Implications. Anal. Chem 2020, 92, 10365–10374. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (77).Cleary SP; Prell JS Distinct Classes of Multi-Subunit Heterogeneity: Analysis Using Fourier Transform Methods and Native Mass Spectrometry. Analyst 2020, 145, 4688–4697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (78).Cleary SP; Thompson AM; Prell JS Fourier Analysis Method for Analyzing Highly Congested Mass Spectra of Ion Populations with Repeated Subunits. Anal. Chem 2016, 88, 6205–6213. [DOI] [PubMed] [Google Scholar]
- (79).Aquilina JA; Benesch JLP; Bateman OA; Slingsby C; Robinson CV Polydispersity of a Mammalian Chaperone: Mass Spectrometry Reveals the Population of Oligomers in αΒ-Crystallin. Proc. Natl. Acad. Sci. U. S. A 2003, 100, 10611–10616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (80).Keener JE; Zambrano DE; Zhang G; Zak CK; Reid DJ; Deodhar BS; Pemberton JE; Prell JS; Marty MT Chemical Additives Enable Native Mass Spectrometry Measurement of Membrane Protein Oligomeric State within Intact Nanodiscs. J. Am. Chem. Soc 2019, 141, 1054–1061. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (81).Wohlschlager T; Scheffler K; Forstenlehner IC; Skala W; Senn S; Damoc E; Holzmann J; Huber CG Native Mass Spectrometry Combined with Enzymatic Dissection Unravels Glycoform Heterogeneity of Biopharmaceuticals. Nat. Commun 2018, 9, 1713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (82).Benesch JLP; Robinson CV Mass Spectrometry of Macromolecular Assemblies: Preservation and Dissociation. Curr. Opin. Struct. Biol 2006, 16, 245–251. [DOI] [PubMed] [Google Scholar]
- (83).Benesch JLP; Aquilina JA; Ruotolo BT; Sobott F; Robinson CV Tandem Mass Spectrometry Reveals the Quaternary Organization of Macromolecular Assemblies. Chem. Biol 2006, 13, 597–605. [DOI] [PubMed] [Google Scholar]
- (84).McLuckey SA; Goeringer DE Slow Heating Methods in Tandem Mass Spectrometry. J. Mass Spectrom 1997, 32, 461–474. [Google Scholar]
- (85).Shepherd DA; Marty MT; Giles K; Baldwin AJ; Benesch JLP Combining Tandem Mass Spectrometry with Ion Mobility Separation to Determine the Architecture of Polydisperse Proteins. Int. J. Mass Spectrom 2015, 377, 663–671. [Google Scholar]
- (86).Loo JA; Edmonds CG; Smith RD Tandem Mass Spectrometry of Very Large Molecules: Serum Albumin Sequence Information from Multiply Charged Ions Formed by Electrospray Ionization. Anal. Chem 1991, 63, 2488–2499. [DOI] [PubMed] [Google Scholar]
- (87).Donnelly DP; Rawlins CM; DeHart CJ; Fornelli L; Schachner LF; Lin Z; Lippens JL; Aluri KC; Sarin R; Chen B, et al. Best Practices and Benchmarks for Intact Protein Analysis for Top-Down Mass Spectrometry. Nat. Methods 2019, 16, 587–594. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (88).Zhou M; Lantz C; Brown KA; Ge Y; Paša-Tolić L; Loo JA; Lermyte F Higher-Order Structural Characterisation of Native Proteins and Complexes by Top-Down Mass Spectrometry. Chem. Sci 2020, 11, 12918–12936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (89).Schachner LF; Tran DP; Lee AS; McGee JP; Jooss K; Durbin KR; Seckler HS; Adams L; Cline EN; Melani RD, et al. Reassembling Protein Complexes after Controlled Disassembly by Top-Down Mass Spectrometry in Native Mode. Int. J. Mass Spectrom 2021, 465, 116591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (90).Li HL; Wolff JJ; Van Orden SL; Loo JA Native Top-Down Electrospray Ionization-Mass Spectrometry of 158 kDa Protein Complex by High-Resolution Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem 2014, 86, 317–320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (91).Wang H; Eschweiler J; Cui W; Zhang H; Frieden C; Ruotolo BT; Gross ML Native Mass Spectrometry, Ion Mobility, Electron-Capture Dissociation, and Modeling Provide Structural Information for Gas-Phase Apolipoprotein E Oligomers. J. Am. Soc. Mass Spectrom 2019, 30, 876–885. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (92).Zhou M; Liu W; Shaw JB Charge Movement and Structural Changes in the Gas-Phase Unfolding of Multimeric Protein Complexes Captured by Native Top-Down Mass Spectrometry. Anal. Chem 2020, 92, 1788–1795. [DOI] [PubMed] [Google Scholar]
- (93).Zubarev RA Electron-Capture Dissociation Tandem Mass Spectrometry. Curr. Opin. Biotechnol 2004, 15, 12–16. [DOI] [PubMed] [Google Scholar]
- (94).Zubarev RA; Horn DM; Fridriksson EK; Kelleher NL; Kruger NA; Lewis MA; Carpenter BK; McLafferty FW Electron Capture Dissociation for Structural Characterization of Multiply Charged Protein Cations. Anal. Chem 2000, 72, 563–573. [DOI] [PubMed] [Google Scholar]
- (95).Zubarev RA; Kelleher NL; McLafferty FW Electron Capture Dissociation of Multiply Charged Protein Cations. A Nonergodic Process. J. Am. Chem. Soc 1998, 120, 3265–3266. [Google Scholar]
- (96).Coon JJ; Shabanowitz J; Hunt DF; Syka JEP Electron Transfer Dissociation of Peptide Anions. J. Am. Soc. Mass Spectrom 2005, 16, 880–882. [DOI] [PubMed] [Google Scholar]
- (97).Holden DD; Brodbelt JS Ultraviolet Photodissociation of Native Proteins Following Proton Transfer Reactions in the Gas Phase. Anal. Chem 2016, 88, 12354–12362. [DOI] [PubMed] [Google Scholar]
- (98).Holden DD; McGee WM; Brodbelt JS Integration of Ultraviolet Photodissociation with Proton Transfer Reactions and Ion Parking for Analysis of Intact Proteins. Anal. Chem 2016, 88, 1008–1016. [DOI] [PubMed] [Google Scholar]
- (99).O’Brien JP; Li W; Zhang Y; Brodbelt JS Characterization of Native Protein Complexes Using Ultraviolet Photodissociation Mass Spectrometry. J. Am. Chem. Soc 2014, 136, 12920–12928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (100).Reilly JP Ultraviolet Photofragmentation of Biomolecular Ions. Mass Spectrom. Rev 2009, 28, 425–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (101).Marzluff EM; Campbell S; Rodgers MT; Beauchamp JL Collisional Activation of Large Molecules Is an Efficient Process. J. Am. Chem. Soc 1994, 116, 6947–6948. [Google Scholar]
- (102).Sever AIM; Yin V; Konermann L Interrogating the Quaternary Structure of Noncanonical Hemoglobin Complexes by Electrospray Mass Spectrometry and Collision-Induced Dissociation. J. Am. Soc. Mass Spectrom 2021, 32, 270–280. [DOI] [PubMed] [Google Scholar]
- (103).Borysik AJ; Robinson CV Formation and Dissociation Processes of Gas-Phase Detergent Micelles. Langmuir 2012, 28, 7160–7167. [DOI] [PubMed] [Google Scholar]
- (104).Brodbelt JS Photodissociation Mass Spectrometry: New Tools for Characterization of Biological Molecules. Chem. Soc. Rev 2014, 43, 2757–2783. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (105).Brodbelt JS; Wilson JJ Infrared Multiphoton Dissociation in Quadrupole Ion Traps. Mass Spectrom. Rev 2009, 28, 390–424. [DOI] [PubMed] [Google Scholar]
- (106).Cotham VC; Shaw JB; Brodbelt JS High-Throughput Bioconjugation for Enhanced 193 nm Photodissociation Via Droplet Phase Initiated Ion/Ion Chemistry Using a Front-End Dual Spray Reactor. Anal. Chem 2015, 87, 9396–9402. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (107).Lermyte F; Williams JP; Brown JM; Martin EM; Sobott F Extensive Charge Reduction and Dissociation of Intact Protein Complexes Following Electron Transfer on a Quadrupole-Ion Mobility-Time-of-Flight Ms. J. Am. Soc. Mass Spectrom 2015, 26, 1068–1076. [DOI] [PubMed] [Google Scholar]
- (108).Marty MT; Hoi KK; Gault J; Robinson CV Probing the Lipid Annular Belt by Gas-Phase Dissociation of Membrane Proteins in Nanodiscs. Angew. Chem. Int. Edit 2016, 55, 550–554. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (109).Nesatyy VJ; Laskin J Dissociation of Noncovalent Protein Complexes by Triple Quadrupole Tandem Mass Spectrometry: Comparison of Monte Carlo Simulation and Experiment. Int. J. Mass Spectrom 2002, 221, 245–262. [Google Scholar]
- (110).Popa V; Trecroce DA; McAllister RG; Konermann L Collision-Induced Dissociation of Electrosprayed Protein Complexes: An All-Atom Molecular Dynamics Model with Mobile Protons. J. Phys. Chem. B 2016, 120, 5114–5124. [DOI] [PubMed] [Google Scholar]
- (111).Rabuck-Gibbons JN; Keating JE; Ruotolo BT Collision Induced Unfolding and Dissociation Differentiates ATP-Competitive from Allosteric Protein Tyrosine Kinase Inhibitors. Int. J. Mass Spectrom 2018, 427, 151–156. [Google Scholar]
- (112).Schwartz BL; Bruce JE; Anderson GA; Hofstadler SA; Rockwood AL; Smith RD; Chilkoti A; Stayton PS Dissociation of Tetrameric Ions of Noncovalent Streptavidin Complexes Formed by Electrospray Ionization. J. Am. Soc. Mass Spectrom 1995, 6, 459–465. [DOI] [PubMed] [Google Scholar]
- (113).Shaw JB; Li W; Holden DD; Zhang Y; Griep-Raming J; Fellers RT; Early BP; Thomas PM; Kelleher NL; Brodbelt JS Complete Protein Characterization Using Top-Down Mass Spectrometry and Ultraviolet Photodissociation. J. Am. Chem. Soc 2013, 135, 12646–12651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (114).Syka JEP; Coon JJ; Schroeder MJ; Shabanowitz J; Hunt DF Peptide and Protein Sequence Analysis by Electron Transfer Dissociation Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A 2004, 101, 9528–9533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (115).Wu D; Struwe WB; Harvey DJ; Ferguson MAJ; Robinson CV N-Glycan Microheterogeneity Regulates Interactions of Plasma Proteins. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 8763–8768. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (116).Gupta K; Donlan JAC; Hopper JTS; Uzdavinys P; Landreh M; Struwe WB; Drew D; Baldwin AJ; Stansfeld PJ; Robinson CV The Role of Interfacial Lipids in Stabilizing Membrane Protein Oligomers. Nature 2017, 541, 421–424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (117).Bechara C; Noell A; Morgner N; Degiacomi MT; Tampe R; Robinson CV A Subset of Annular Lipids Is Linked to the Flippase Activity of an ABC Transporter. Nat. Chem 2015, 7, 255–262. [DOI] [PubMed] [Google Scholar]
- (118).Agasid MT; Sørensen L; Urner LH; Yan J; Robinson CV The Effects of Sodium Ions on Ligand Binding and Conformational States of G Protein-Coupled Receptors—Insights from Mass Spectrometry. J. Am. Chem. Soc 2021, 143, 4085–4089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (119).Laganowsky A; Reading E; Allison TM; Ulmschneider MB; Degiacomi MT; Baldwin AJ; Robinson CV Membrane Proteins Bind Lipids Selectively to Modulate Their Structure and Function. Nature 2014, 510, 172–175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (120).van de Waterbeemd M; Snijder J; Tsvetkova IB; Dragnea BG; Cornelissen JJ; Heck AJR Examining the Heterogeneous Genome Content of Multipartite Viruses Bmv and Ccmv by Native Mass Spectrometry. J. Am. Soc. Mass Spectrom 2016, 27, 1000–1009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (121).Bernstein SL; Dupuis NF; Lazo ND; Wyttenbach T; Condron MM; Bitan G; Teplow DB; Shea JE; Ruotolo BT; Robinson CV, et al. Amyloid-Beta Protein Oligomerization and the Importance of Tetramers and Dodecamers in the Aetiology of Alzheimer's Disease. Nat. Chem 2009, 1, 326–331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (122).Brücher D; Franc V; Smith SN; Heck AJR; Plückthun A Malignant Tissues Produce Divergent Antibody Glycosylation of Relevance for Cancer Gene Therapy Effectiveness. mAbs 2020, 12, 1792084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (123).Allison TM; Barran P; Benesch JLP; Cianferani S; Degiacomi MT; Gabelica V; Grandori R; Marklund EG; Menneteau T; Migas LG, et al. Software Requirements for the Analysis and Interpretation of Native Ion Mobility Mass Spectrometry Data. Anal. Chem 2020, 92, 10881–10890. [DOI] [PubMed] [Google Scholar]
- (124).Allison TM; Barran P; Cianférani S; Degiacomi MT; Gabelica V; Grandori R; Marklund EG; Menneteau T; Migas LG; Politis A, et al. Computational Strategies and Challenges for Using Native Ion Mobility Mass Spectrometry in Biophysics and Structural Biology. Anal. Chem 2020, 92, 10872–10880. [DOI] [PubMed] [Google Scholar]
- (125).Peng W-P; Chou S-W; Patil AA Measuring Masses of Large Biomolecules and Bioparticles Using Mass Spectrometric Techniques. Analyst 2014, 139, 3507–3523. [DOI] [PubMed] [Google Scholar]
- (126).Sharon M How Far Can We Go with Structural Mass Spectrometry of Protein Complexes? J. Am. Soc. Mass Spectrom 2010, 21, 487–500. [DOI] [PubMed] [Google Scholar]
- (127).Tassi M; De Vos J; Chatterjee S; Sobott F; Bones J; Eeltink S Advances in Native High-Performance Liquid Chromatography and Intact Mass Spectrometry for the Characterization of Biopharmaceutical Products. J. Sep. Sci 2018, 41, 125–144. [DOI] [PubMed] [Google Scholar]
- (128).Bults P; Spanov B; Olaleye O; van de Merbel NC; Bischoff R Intact Protein Bioanalysis by Liquid Chromatography – High-Resolution Mass Spectrometry. J. Chromatogr. B 2019, 1110–1111, 155–167. [DOI] [PubMed] [Google Scholar]
- (129).Gahoual R; Heidenreich A-K; Somsen GW; Bulau P; Reusch D; Wuhrer M; Haberger M Detailed Characterization of Monoclonal Antibody Receptor Interaction Using Affinity Liquid Chromatography Hyphenated to Native Mass Spectrometry. Anal. Chem 2017, 89, 5404–5412. [DOI] [PubMed] [Google Scholar]
- (130).Ehkirch A; Hernandez-Alba O; Colas O; Beck A; Guillarme D; Cianférani S Hyphenation of Size Exclusion Chromatography to Native Ion Mobility Mass Spectrometry for the Analytical Characterization of Therapeutic Antibodies and Related Products. J. Chromatogr. B 2018, 1086, 176–183. [DOI] [PubMed] [Google Scholar]
- (131).Haberger M; Heidenreich A-K; Hook M; Fichtl J; Lang R; Cymer F; Adibzadeh M; Kuhne F; Wegele H; Reusch D, et al. Multiattribute Monitoring of Antibody Charge Variants by Cation-Exchange Chromatography Coupled to Native Mass Spectrometry. J. Am. Soc. Mass Spectrom 2021, DOI: 10.1021/jasms.1020c00446. [DOI] [PubMed] [Google Scholar]
- (132).Haberger M; Leiss M; Heidenreich A-K; Pester O; Hafenmair G; Hook M; Bonnington L; Wegele H; Haindl M; Reusch D, et al. Rapid Characterization of Biotherapeutic Proteins by Size-Exclusion Chromatography Coupled to Native Mass Spectrometry. mAbs 2016, 8, 331–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (133).Yan Y; Xing T; Wang S; Daly TJ; Li N Online Coupling of Analytical Hydrophobic Interaction Chromatography with Native Mass Spectrometry for the Characterization of Monoclonal Antibodies and Related Products. J. Pharm. Biomed. Anal 2020, 186, 113313. [DOI] [PubMed] [Google Scholar]
- (134).Yan Y; Liu AP; Wang S; Daly TJ; Li N Ultrasensitive Characterization of Charge Heterogeneity of Therapeutic Monoclonal Antibodies Using Strong Cation Exchange Chromatography Coupled to Native Mass Spectrometry. Anal. Chem 2018, 90, 13013–13020. [DOI] [PubMed] [Google Scholar]
- (135).Yan Y; Xing T; Wang S; Daly TJ; Li N Coupling Mixed-Mode Size Exclusion Chromatography with Native Mass Spectrometry for Sensitive Detection and Quantitation of Homodimer Impurities in Bispecific IgG. Anal. Chem 2019, 91, 11417–11424. [DOI] [PubMed] [Google Scholar]
- (136).Ma F; Raoufi F; Bailly MA; Fayadat-Dilman L; Tomazela D Hyphenation of Strong Cation Exchange Chromatography to Native Mass Spectrometry for High Throughput Online Characterization of Charge Heterogeneity of Therapeutic Monoclonal Antibodies. mAbs 2020, 12, 1763762. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (137).Füssl F; Trappe A; Cook K; Scheffler K; Fitzgerald O; Bones J Comprehensive Characterisation of the Heterogeneity of Adalimumab Via Charge Variant Analysis Hyphenated on-Line to Native High Resolution Orbitrap Mass Spectrometry. mAbs 2019, 11, 116–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (138).Füssl F; Cook K; Scheffler K; Farrell A; Mittermayr S; Bones J Charge Variant Analysis of Monoclonal Antibodies Using Direct Coupled pH Gradient Cation Exchange Chromatography to High-Resolution Native Mass Spectrometry. Anal. Chem 2018, 90, 4669–4676. [DOI] [PubMed] [Google Scholar]
- (139).Jooß K; McGee JP; Melani RD; Kelleher NL Standard Procedures for Native CZE-MS of Proteins and Protein Complexes up to 800 kDa. Electrophoresis 2021, 42, 1050–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (140).Dykstra AB; Flick TG; Lee B; Blue LE; Angell N Chip-Based Capillary Zone Electrophoresis Mass Spectrometry for Rapid Resolution and Quantitation of Critical Quality Attributes in Protein Biotherapeutics. J. Am. Soc. Mass Spectrom 2021, 32, 1952–1963. [DOI] [PubMed] [Google Scholar]
- (141).Shen X; Liang Z; Xu T; Yang Z; Wang Q; Chen D; Pham L; Du W; Sun L Investigating Native Capillary Zone Electrophoresis-Mass Spectrometry on a High-End Quadrupole-Time-of-Flight Mass Spectrometer for the Characterization of Monoclonal Antibodies. Int. J. Mass Spectrom 2021, 462, 116541. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (142).Jooß K; Schachner LF; Watson R; Gillespie ZB; Howard SA; Cheek MA; Meiners MJ; Sobh A; Licht JD; Keogh M-C, et al. Separation and Characterization of Endogenous Nucleosomes by Native Capillary Zone Electrophoresis–Top-Down Mass Spectrometry. Anal. Chem 2021, 93, 5151–5160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (143).Le-Minh V; Tran NT; Makky A; Rosilio V; Taverna M; Smadja C Capillary Zone Electrophoresis-Native Mass Spectrometry for the Quality Control of Intact Therapeutic Monoclonal Antibodies. J. Chromatogr. A 2019, 1601, 375–384. [DOI] [PubMed] [Google Scholar]
- (144).Gstöttner C; Nicolardi S; Haberger M; Reusch D; Wuhrer M; Domínguez-Vega E Intact and Subunit-Specific Analysis of Bispecific Antibodies by Sheathless CE-MS. Anal. Chim. Acta 2020, 1134, 18–27. [DOI] [PubMed] [Google Scholar]
- (145).Han M; Rock BM; Pearson JT; Rock DA Intact Mass Analysis of Monoclonal Antibodies by Capillary Electrophoresis—Mass Spectrometry. J. Chromatogr. B 2016, 1011, 24–32. [DOI] [PubMed] [Google Scholar]
- (146).Eschweiler JD; Kerr R; Rabuck-Gibbons J; Ruotolo BT Sizing up Protein–Ligand Complexes: The Rise of Structural Mass Spectrometry Approaches in the Pharmaceutical Sciences. Annu. Rev. Anal. Chem 2017, 10, 25–44. [DOI] [PubMed] [Google Scholar]
- (147).Bobst CE; Sperry J; Friese OV; Kaltashov IA Simultaneous Evaluation of a Vaccine Component Microheterogeneity and Conformational Integrity Using Native Mass Spectrometry and Limited Charge Reduction. J. Am. Soc. Mass Spectrom 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (148).Yang Y; Du Y; Kaltashov IA The Utility of Native MS for Understanding the Mechanism of Action of Repurposed Therapeutics in COVID-19: Heparin as a Disruptor of the SARS-CoV-2 Interaction with Its Host Cell Receptor. Anal. Chem 2020, 92, 10930–10934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (149).Kaltashov IA; Bobst CE; Pawlowski J; Wang G Mass Spectrometry-Based Methods in Characterization of the Higher Order Structure of Protein Therapeutics. J. Pharm. Biomed. Anal 2020, 184, 113169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (150).Sacco MD; Ma C; Lagarias P; Gao A; Townsend JA; Meng X; Dube P; Zhang X; Hu Y; Kitamura N, et al. Structure and Inhibition of the SARS-CoV-2 Main Protease Reveal Strategy for Developing Dual Inhibitors against Mpro and Cathepsin L. Sci. Adv 2020, 6, eabe0751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (151).Mehaffey MR; Lee J; Jung J; Lanzillotti MB; Escobar EE; Morgenstern KR; Georgiou G; Brodbelt JS Mapping a Conformational Epitope of Hemagglutinin A Using Native Mass Spectrometry and Ultraviolet Photodissociation. Anal. Chem 2020, 92, 11869–11878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (152).Zhu W; Li M; Zhang J Integrating Intact Mass Analysis and Middle-Down Mass Spectrometry Approaches to Effectively Characterize Trastuzumab and Adalimumab Structural Heterogeneity. J. Proteome Res 2021, 20, 270–278. [DOI] [PubMed] [Google Scholar]
- (153).Liu M; Van Voorhis WC; Quinn RJ Development of a Target Identification Approach Using Native Mass Spectrometry. Sci. Rep 2021, 11, 2387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (154).Campuzano IDG; Sandoval W Denaturing and Native Mass Spectrometric Analytics for Biotherapeutic Drug Discovery Research: Historical, Current, and Future Personal Perspectives. J. Am. Soc. Mass Spectrom 2021, 32, 1861–1885. [DOI] [PubMed] [Google Scholar]
- (155).Kellie JF; Tran JC; Jian W; Jones B; Mehl JT; Ge Y; Henion J; Bateman KP Intact Protein Mass Spectrometry for Therapeutic Protein Quantitation, Pharmacokinetics, and Biotransformation in Preclinical and Clinical Studies: An Industry Perspective. J. Am. Soc. Mass Spectrom 2021, 32, 1886–1900. [DOI] [PubMed] [Google Scholar]
- (156).Terral G; Beck A; Cianferani S Insights from Native Mass Spectrometry and Ion Mobility-Mass Spectrometry for Antibody and Antibody-Based Product Characterization. J. Chromatogr. B 2016, 1032, 79–90. [DOI] [PubMed] [Google Scholar]
- (157).Kazarian AA; Barnhart W; Campuzano IDG; Cabrera J; Fitch T; Long J; Sham K; Wu B; Murray JK Purification of Guanine-Quadruplex Using Monolithic Stationary Phase under Ion-Exchange Conditions. J. Chromatogr. A 2020, 1634, 461633. [DOI] [PubMed] [Google Scholar]
- (158).Skeene K; Khatri K; Soloviev Z; Lapthorn C Current Status and Future Prospects for Ion-Mobility Mass Spectrometry in the Biopharmaceutical Industry. Biochim. Biophys. Acta Proteins Proteom 2021, 140697. [DOI] [PubMed] [Google Scholar]
- (159).Deslignière E; Ehkirch A; Duivelshof BL; Toftevall H; Sjögren J; Guillarme D; D’Atri V; Beck A; Hernandez-Alba O; Cianférani S State-of-the-Art Native Mass Spectrometry and Ion Mobility Methods to Monitor Homogeneous Site-Specific Antibody-Drug Conjugates Synthesis. Pharmaceuticals 2021, 14, 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (160).Chen Z; Kellie JF; Hottenstein CS; Szapacs ME Native High-Resolution Mass Spectrometry Analysis of Noncovalent Protein Complexes up to 450 kDa. Bioanalysis 2020, 12, 1353–1362. [DOI] [PubMed] [Google Scholar]
- (161).Zhang Z; Hug C; Tao Y; Bitsch F; Yang Y Solving Complex Biologics Truncation Problems by Top-Down Mass Spectrometry. J. Am. Soc. Mass Spectrom 2021. [DOI] [PubMed] [Google Scholar]
- (162).Shaw JB; Liu W; Vasil’ev YV; Bracken CC; Malhan N; Guthals A; Beckman JS; Voinov VG Direct Determination of Antibody Chain Pairing by Top-Down and Middle-Down Mass Spectrometry Using Electron Capture Dissociation and Ultraviolet Photodissociation. Anal. Chem 2020, 92, 766–773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (163).Campuzano IDG; Robinson JH; Hui JO; Shi SDH; Netirojjanakul C; Nshanian M; Egea PF; Lippens JL; Bagal D; Loo JA, et al. Native and Denaturing Ms Protein Deconvolution for Biopharma: Monoclonal Antibodies and Antibody–Drug Conjugates to Polydisperse Membrane Proteins and Beyond. Anal. Chem 2019, 91, 9472–9480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (164).O'Connor DM; Lutomski C; Jarrold MF; Boulis NM; Donsante A Lot-to-Lot Variation in Adeno-Associated Virus Serotype 9 (AAV9) Preparations. Hum. Gene Ther. Methods 2019, 30, 214–225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (165).Hammerschmid D; van Dyck JF; Sobott F; Calabrese AN Interrogating Membrane Protein Structure and Lipid Interactions by Native Mass Spectrometry. In Biophysics of Membrane Proteins: Methods and Protocols, Vol. Postis VLG; Goldman A, Eds.; Springer; US, 2020; pp 233–261. [DOI] [PubMed] [Google Scholar]
- (166).Patrick JW; Laganowsky A Probing Heterogeneous Lipid Interactions with Membrane Proteins Using Mass Spectrometry. In Lipid-Protein Interactions: Methods and Protocols, Vol. Kleinschmidt JH; Ed. Springer; New York, 2019; pp 175–190. [DOI] [PubMed] [Google Scholar]
- (167).Gault J; Donlan JAC; Liko I; Hopper JTS; Gupta K; Housden NG; Struwe WB; Marty MT; Mize T; Bechara C, et al. High-Resolution Mass Spectrometry of Small Molecules Bound to Membrane Proteins. Nat. Methods 2016, 13, 333–336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (168).Čaval T; Tian W; Yang Z; Clausen H; Heck AJR Direct Quality Control of Glycoengineered Erythropoietin Variants. Nat. Commun 2018, 9, 3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (169).Skilling J Data Analysis: The Maximum Entropy Method. Nature 1984, 309, 748–749. [Google Scholar]
- (170).Shannon CE A Mathematical Theory of Communication. Bell Sys. Tech. J 1948, 27, 379–423. [Google Scholar]
- (171).Shore J; Johnson R Axiomatic Derivation of the Principle of Maximum Entropy and the Principle of Minimum Cross-Entropy. IEEE Trans. Inf. Theory 1980, 26, 26–37. [Google Scholar]
- (172).Tikochinsky Y; Tishby NZ; Levine RD Consistent Inference of Probabilities for Reproducible Experiments. Phys. Rev. Lett 1984, 52, 1357–1360. [Google Scholar]
- (173).Ferrige AG; Seddon MJ; Green BN; Jarvis SA; Skilling J; Staunton J Disentangling Electrospray Spectra with Maximum Entropy. Rapid Commun. Mass Spectrom 1992, 6, 707–711. [Google Scholar]
- (174).Ferrige AG; Seddon MJ; Jarvis S; Skilling J; Aplin R Maximum Entropy Deconvolution in Electrospray Mass Spectrometry. Rapid Commun. Mass Spectrom 1991, 5, 374–377. [Google Scholar]
- (175).Ferrige AG; Seddon MJ; Jarvis S; Skilling J; Welch J The Application of Maxent to Electrospray Mass Spectrometry. In Maximum Entropy and Bayesian Methods: Seattle, 1991, Vol. Smith CR; Erickson GJ; Neudorfer PO, Eds.; Springer; Netherlands, 1992; pp 327–335. [Google Scholar]
- (176).Roberts DS; Mann M; Melby JA; Larson EJ; Zhu Y; Brasier AR; Jin S; Ge Y Structural O-Glycoform Heterogeneity of the SARS-CoV-2 Spike Protein Receptor-Binding Domain Revealed by Top-Down Mass Spectrometry. J. Am. Chem. Soc 2021, 143, 12014–12024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (177).Beck A; Wagner-Rousset E; Bussat M-C; Lokteff M; Klinguer-Hamour C; Haeuw J-F; Goetsch L; Wurch T; Van Dorsselaer A; Corvaia N Trends in Glycosylation, Glycoanalysis and Glycoengineering of Therapeutic Antibodies and Fc-Fusion Proteins. Curr. Pharm. Biotechnol 2008, 9, 482–501. [DOI] [PubMed] [Google Scholar]
- (178).Yu Y; Liu H; Yu Z; Witkowska HE; Cheng Y Stoichiometry of Nucleotide Binding to Proteasome AAA+ ATPase Hexamer Established by Native Mass Spectrometry. Mol. Cell. Proteom 2020, 19, 1997–2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (179).Thompson NJ; Rosati S; Rose RJ; Heck AJR The Impact of Mass Spectrometry on the Study of Intact Antibodies: From Post-Translational Modifications to Structural Analysis. Chem. Commun 2013, 49, 538–548. [DOI] [PubMed] [Google Scholar]
- (180).Sanglier S; Leize E; van Dorsselaer A; Zal F Comparative ESI-MS Study of ~2.2 MDa Native Hemocyanins from Deep-Sea and Shore Crabs: From Protein Oligomeric State to Biotope. J. Am. Soc. Mass Spectrom 2003, 14, 419–429. [DOI] [PubMed] [Google Scholar]
- (181).Sanglier S; Ramström H; Haiech J; Leize E; van Dorsselaer A Electrospray Ionization Mass Spectrometry Analysis Revealed a ~310 kDa Noncovalent Hexamer of HPr Kinase/Phosphatase from Bacillus Subtilis. Int. J. Mass Spectrom 2002, 219, 681–696. [Google Scholar]
- (182).Zal F; Green BN; Lallier FH; Toulmond A Investigation by Electrospray Ionization Mass Spectrometry of the Extracellular Hemoglobin from the Polychaete Annelid Alvinella Pompejana: An Unusual Hexagonal Bilayer Hemoglobin. Biochemistry 1997, 36, 11777–11786. [DOI] [PubMed] [Google Scholar]
- (183).Robinson CV; Chung EW; Kragelund BB; Knudsen J; Aplin RT; Poulsen FM; Dobson CM Probing the Nature of Noncovalent Interactions by Mass Spectrometry. A Study of Protein–CoA Ligand Binding and Assembly. J. Am. Chem. Soc 1996, 118, 8646–8653. [Google Scholar]
- (184).Lengqvist J; Svensson R; Evergren E; Morgenstern R; Griffiths WJ Observation of an Intact Noncovalent Homotrimer of Detergent-Solubilized Rat Microsomal Glutathione Transferase-1 by Electrospray Mass Spectrometry. J. Biol. Chem 2004, 279, 13311–13316. [DOI] [PubMed] [Google Scholar]
- (185).Ashcroft AE; Brinker A; Coyle JE; Weber F; Kaiser M; Moroder L; Parsons MR; Jager J; Hartl UF; Hayer-Hartl M, et al. Structural Plasticity and Noncovalent Substrate Binding in the Groel Apical Domain: A Study Using Electrospray Ionization Mass Spectrometry and Fluorescence Binding Studies. J. Biol. Chem 2002, 277, 33115–33126. [DOI] [PubMed] [Google Scholar]
- (186).Pinkse MWH; Heck AJR; Rumpel K; Pullen F Probing Noncovalent Protein—Ligand Interactions of the cGMP-Dependent Protein Kinase Using Electrospray Ionization Time of Flight Mass Spectrometry. J. Am. Soc. Mass Spectrom 2004, 15, 1392–1399. [DOI] [PubMed] [Google Scholar]
- (187).Bolgar MS; Gaskell SJ Determination of the Sites of 4-Hydroxy-2-Nonenal Adduction to Protein by Electrospray Tandem Mass Spectrometry. Anal. Chem 1996, 68, 2325–2330. [Google Scholar]
- (188).Bennett KL; Smith SV; Lambrecht RM; Truscott RJW; Sheil MM Rapid Characterization of Chemically-Modified Proteins by Electrospray Mass Spectrometry. Bioconjugate Chem. 1996, 7, 16–22. [DOI] [PubMed] [Google Scholar]
- (189).De BK; Woolfitt AR; Barr JR; Daneshvar MI; Sampson JS; Ades EW; Carlone GM Analysis of Recombinant Acylated Pneumococcal Surface Adhesin A of Streptococcus Pneumoniae by Mass Spectrometry. Arch. Biochem. Biophys 2003, 419, 147–157. [DOI] [PubMed] [Google Scholar]
- (190).White HD; Ashcroft AE Real-Time Measurement of Myosin–Nucleotide Noncovalent Complexes by Electrospray Ionization Mass Spectrometry. Biophys. J 2007, 93, 914–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (191).Tahallah N; van den Heuvel RHH; van den Berg WAM; Maier CS; van Berkel WJH; Heck AJR Cofactor-Dependent Assembly of the Flavoenzyme Vanillyl-Alcohol Oxidase. J. Biol. Chem 2002, 277, 36425–36432. [DOI] [PubMed] [Google Scholar]
- (192).Lamkemeyer T; Zeis B; Decker H; Jaenicke E; Waschbüsch D; Gebauer W; Markl J; Meissner U; Rousselot M; Zal F, et al. Molecular Mass of Macromolecules and Subunits and the Quaternary Structure of Hemoglobin from the Microcrustacean Daphnia Magna. FEBS J. 2006, 273, 3393–3410. [DOI] [PubMed] [Google Scholar]
- (193).Brockwell D; Yu L; Cooper S; Mccleland S; Cooper A; Attwood D; Gaskell SJ; Barber J Physicochemical Consequences of the Perdeuteriation of Glutathione S-Transferase from S. Japonicum. Protein Sci 2001, 10, 572–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (194).Green BN; Sannes-Lowery KA; Loo JA; Satterlee JD; Kuchumov AR; Walz DA; Vinogradov SN Electrospray Ionization Mass Spectrometric Study of the Multiple Intracellular Monomeric and Polymeric Hemoglobins of Glycera Dibranchiata. J. Protein Chem 1998, 17, 85–97. [DOI] [PubMed] [Google Scholar]
- (195).Said N; Gahoual R; Kuhn L; Beck A; François Y-N; Leize-Wagner E Structural Characterization of Antibody Drug Conjugate by a Combination of Intact, Middle-up and Bottom-up Techniques Using Sheathless Capillary Electrophoresis – Tandem Mass Spectrometry as NanoESI Infusion Platform and Separation Method. Anal. Chim. Acta 2016, 918, 50–59. [DOI] [PubMed] [Google Scholar]
- (196).Campuzano IDG; Li H; Bagal D; Lippens JL; Svitel J; Kurzeja RJM; Xu H; Schnier PD; Loo JA Native Ms Analysis of Bacteriorhodopsin and an Empty Nanodisc by Orthogonal Acceleration Time-of-Flight, Orbitrap and Ion Cyclotron Resonance. Anal. Chem 2016, 88, 12427–12436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (197).Zhang Z; Guan S; Marshall AG Enhancement of the Effective Resolution of Mass Spectra of High-Mass Biomolecules by Maximum Entropy-Based Deconvolution to Eliminate the Isotopic Natural Abundance Distribution. J. Am. Soc. Mass Spectrom 1997, 8, 659–670. [Google Scholar]
- (198).Bruker Daltonik GmbH. Dataanalysis 5.0. 2017. (accessed August 19, 2021). [Google Scholar]
- (199).Cottrell JC; Green BN Maxent: An Essential Maximum Entropy Based Tool for Interpreting Multiply-Charged Electrospray Data. 1998. https://www.waters.com/webassets/cms/library/docs/an212.pdf (accessed August 19, 2021).
- (200).SCIEX. Bio Tool Kit: A Complete Set of Tools for Biomolecule Characterization. 2017. https://www.sciex.com/content/dam/SCIEX/pdf/tech-notes/all/BioToolKit-peakview.pdf (accessed August 19, 2021).
- (201).Agilent Technologies. Agilent Masshunter Bioconfirm Software, version B.04.00; Agilent Technologies: Santa Clara, CA, 2012. [Google Scholar]
- (202).Hagen JJ; Monnig CA Method for Estimating Molecular Mass from Electrospray Spectra. Anal. Chem 1994, 66, 1877–1883. [Google Scholar]
- (203).Hopkins CE; O'Connor PB; Allen KN; Costello CE; Tolan DR Chemical-Modification Rescue Assessed by Mass Spectrometry Demonstrates That Gamma-Thia-Lysine Yields the Same Activity as Lysine in Aldolase. Protein Sci. 2002, 11, 1591–1599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (204).Bunk DM; Welch MJ Electrospray Ionization Mass Spectrometry for the Quantitation of Albumin in Human Serum. J. Am. Soc. Mass Spectrom 1997, 8, 1247–1254. [Google Scholar]
- (205).Upmacis RK; Hajjar DP; Chait BT; Mirza UA Direct Observation of Nitrosylated Heme in Myoglobin and Hemoglobin by Electrospray Ionization Mass Spectrometry. J. Am. Chem. Soc 1997, 119, 10424–10429. [Google Scholar]
- (206).Read SM; Currie G; Bacic A Analysis of the Structural Heterogeneity of Laminarin by Electrospray-Ionisation-Mass Spectrometry. Carbohydr. Res 1996, 281, 187–201. [DOI] [PubMed] [Google Scholar]
- (207).Lei QP; Cui X; Kurtz DM; Amster IJ; Chernushevich IV; Standing KG Electrospray Mass Spectrometry Studies of Non-Heme Iron-Containing Proteins. Anal. Chem 1998, 70, 1838–1846. [DOI] [PubMed] [Google Scholar]
- (208).Donald LJ; Chernushevich IV; Zhou J; Verentchikov A; Poppe-Schriemer N; Hosfield DJ; Westmore JB; Ens W; Duckworth HW; Standing KG Preparation and Properties of Pure, Full-Length Ic1r Protein of Escherichia Coli. Use of Time-of-Flight Mass Spectrometry to Investigate the Problems Encountered. Protein Sci. 1996, 5, 1613–1624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (209).Labowsky M; Whitehouse C; Fenn JB Three-Dimensional Deconvolution of Multiply Charged Spectra. Rapid Commun. Mass Spectrom 1993, 7, 71–84. [Google Scholar]
- (210).Snijder J; Rose RJ; Veesler D; Johnson JE; Heck AJR Studying 18 MDa Virus Assemblies with Native Mass Spectrometry. Angew. Chem. Int. Ed 2013, 52, 4020–4023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (211).Reinhold BB; Reinhold VN Electrospray Ionization Mass Spectrometry: Deconvolution by an Entropy-Based Algorithm. J. Am. Soc. Mass Spectrom 1992, 3, 207–215. [DOI] [PubMed] [Google Scholar]
- (212).Zhang ZQ; Marshall AG A Universal Algorithm for Fast and Automated Charge State Deconvolution of Electrospray Mass-to-Charge Ratio Spectra. J. Am. Soc. Mass Spectrom 1998, 9, 225–233. [DOI] [PubMed] [Google Scholar]
- (213).Morgner N; Robinson CV Massign: An Assignment Strategy for Maximizing Information from the Mass Spectra of Heterogeneous Protein Assemblies. Anal. Chem 2012, 84, 2939–2948. [DOI] [PubMed] [Google Scholar]
- (214).Ro SY; Schachner LF; Koo CW; Purohit R; Remis JP; Kenney GE; Liauw BW; Thomas PM; Patrie SM; Kelleher NL, et al. Native Top-Down Mass Spectrometry Provides Insights into the Copper Centers of Membrane-Bound Methane Monooxygenase. Nat. Commun 2019, 10, 2675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (215).Johnson P; Philo M; Watson A; Mills ENC Rapid Fingerprinting of Milk Thermal Processing History by Intact Protein Mass Spectrometry with Nondenaturing Chromatography. J. Agric. Food Chem 2011, 59, 12420–12427. [DOI] [PubMed] [Google Scholar]
- (216).Honarvar E; Venter AR Ammonium Bicarbonate Addition Improves the Detection of Proteins by Desorption Electrospray Ionization Mass Spectrometry. J. Am. Soc. Mass Spectrom 2017, 28, 1109–1117. [DOI] [PubMed] [Google Scholar]
- (217).Meng F; Cargile BJ; Patrie SM; Johnson JR; McLoughlin SM; Kelleher NL Processing Complex Mixtures of Intact Proteins for Direct Analysis by Mass Spectrometry. Anal. Chem 2002, 74, 2923–2929. [DOI] [PubMed] [Google Scholar]
- (218).Ariza A; Garzon D; Abánades DR; de los Ríos V; Vistoli G; Torres MJ; Carini M; Aldini G; Pérez-Sala D Protein Haptenation by Amoxicillin: High Resolution Mass Spectrometry Analysis and Identification of Target Proteins in Serum. J. Proteom 2012, 77, 504–520. [DOI] [PubMed] [Google Scholar]
- (219).Gallagher ES; Hudgens JW Chapter Fourteen - Mapping Protein–Ligand Interactions with Proteolytic Fragmentation, Hydrogen/Deuterium Exchange-Mass Spectrometry. In Methods Enzymol., Vol. 566; Kelman Z, Ed. Academic Press, 2016; pp 357–404. [DOI] [PubMed] [Google Scholar]
- (220).Zhao T; King FL Direct Determination of the Primary Binding Site of Cisplatin on Cytochrome c by Mass Spectrometry. J. Am. Soc. Mass Spectrom 2009, 20, 1141–1147. [DOI] [PubMed] [Google Scholar]
- (221).Zhao T; King FL Mass-Spectrometric Characterization of Cisplatin Binding Sites on Native and Denatured Ubiquitin. J. Biol. Inorg. Chem 2011, 16, 633–639. [DOI] [PubMed] [Google Scholar]
- (222).Zheng H; Ojha PC; McClean S; Black ND; Hughes JG; Shaw C Heuristic Charge Assignment for Deconvolution of Electrospray Ionization Mass Spectra. Rapid Commun. Mass Spectrom 2003, 17, 429–436. [DOI] [PubMed] [Google Scholar]
- (223).van Breukelen B; Barendregt A; Heck AJR; van den Heuvel RHH Resolving Stoichiometries and Oligomeric States of Glutamate Synthase Protein Complexes with Curve Fitting and Simulation of Electrospray Mass Spectra. Rapid Commun. Mass Spectrom 2006, 20, 2490–2496. [DOI] [PubMed] [Google Scholar]
- (224).Thompson NJ; Merdanovic M; Ehrmann M; van Duijn E; Heck AJR Substrate Occupancy at the Onset of Oligomeric Transitions of DegP. Structure 2014, 22, 281–290. [DOI] [PubMed] [Google Scholar]
- (225).Poliakov A; van Duijn E; Lander G; Fu C-Y; Johnson JE; Prevelige PE; Heck AJR Macromolecular Mass Spectrometry and Electron Microscopy as Complementary Tools for Investigation of the Heterogeneity of Bacteriophage Portal Assemblies. J. Struct. Biol 2007, 157, 371–383. [DOI] [PubMed] [Google Scholar]
- (226).Snijder J; van de Waterbeemd M; Damoc E; Denisov E; Grinfeld D; Bennett A; Agbandje-McKenna M; Makarov A; Heck AJR Defining the Stoichiometry and Cargo Load of Viral and Bacterial Nanoparticles by Orbitrap Mass Spectrometry. J. Am. Chem. Soc 2014, 136, 7295–7299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (227).Weiss VU; Bereszcazk JZ; Havlik M; Kallinger P; Gösler I; Kumar M; Blaas D; Marchetti-Deschmann M; Heck AJR; Szymanski WW, et al. Analysis of a Common Cold Virus and Its Subviral Particles by Gas-Phase Electrophoretic Mobility Molecular Analysis and Native Mass Spectrometry. Anal. Chem 2015, 87, 8709–8717. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (228).Stengel F; Baldwin AJ; Bush MF; Hilton GR; Lioe H; Basha E; Jaya N; Vierling E; Benesch JLP Dissecting Heterogeneous Molecular Chaperone Complexes Using a Mass Spectrum Deconvolution Approach. Chem. Biol 2012, 19, 599–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (229).Lu J; Trnka MJ; Roh S-H; Robinson PJJ; Shiau C; Fujimori DG; Chiu W; Burlingame AL; Guan S Improved Peak Detection and Deconvolution of Native Electrospray Mass Spectra from Large Protein Complexes. J. Am. Soc. Mass Spectrom 2015, 26, 2141–2151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (230).Marcoux J; Wang SC; Politis A; Reading E; Ma J; Biggin PC; Zhou M; Tao H; Zhang Q; Chang G, et al. Mass Spectrometry Reveals Synergistic Effects of Nucleotides, Lipids, and Drugs Binding to a Multidrug Resistance Efflux Pump. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 9704–9709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (231).Morgner N; Schmidt C; Beilsten-Edmands V; Ebong I-O; Patel NA; Clerico EM; Kirschke E; Daturpalli S; Jackson SE; Agard D, et al. Hsp70 Forms Antiparallel Dimers Stabilized by Post-Translational Modifications to Position Clients for Transfer to Hsp90. Cell Rep. 2015, 11, 759–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (232).Schmidt C; Zhou M; Marriott H; Morgner N; Politis A; Robinson CV Comparative Cross-Linking and Mass Spectrometry of an Intact F-Type ATPase Suggest a Role for Phosphorylation. Nat. Commun 2013, 4, 1985. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (233).Schiffrin B; Calabrese AN; Devine PWA; Harris SA; Ashcroft AE; Brockwell DJ; Radford SE Skp Is a Multivalent Chaperone of Outer-Membrane Proteins. Nat. Struct. Mol. Biol 2016, 23, 786–793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (234).Henrich E; Peetz O; Hein C; Laguerre A; Hoffmann B; Hoffmann J; Dötsch V; Bernhard F; Morgner N Analyzing Native Membrane Protein Assembly in Nanodiscs by Combined Non-Covalent Mass Spectrometry and Synthetic Biology. eLife 2017, 6, e20954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (235).Krichel B; Falke S; Hilgenfeld R; Redecke L; Uetrecht C Processing of the SARS-CoV pp1a/ab nsp7–10 Region. Biochem. J 2020, 477, 1009–1019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (236).Sauer PV; Timm J; Liu D; Sitbon D; Boeri-Erba E; Velours C; Mücke N; Langowski J; Ochsenbein F; Almouzni G, et al. Insights into the Molecular Architecture and Histone H3-H4 Deposition Mechanism of Yeast Chromatin Assembly Factor 1. eLife 2017, 6, e23474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (237).Dunne M; Leicht S; Krichel B; Mertens HDT; Thompson A; Krijgsveld J; Svergun DI; Gomez-Torres N; Garde S; Uetrecht C, et al. Crystal Structure of the CTP1L Endolysin Reveals How Its Activity Is Regulated by a Secondary Translation Product. J. Biol. Chem 2016, 291, 4882–4893. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (238).Ebong I-O; Beilsten-Edmands V; Patel NA; Morgner N; Robinson CV The Interchange of Immunophilins Leads to Parallel Pathways and Different Intermediates in the Assembly of Hsp90 Glucocorticoid Receptor Complexes. Cell Discov. 2016, 2, 16002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (239).Macek P; Kerfah R; Erba EB; Crublet E; Moriscot C; Schoehn G; Amero C; Boisbouvier J Unraveling Self-Assembly Pathways of the 468-kDa Proteolytic Machine Tet2. Sci. Adv 2017, 3, e1601601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (240).Nitsche J; Josts I; Heidemann J; Mertens HD; Maric S; Moulin M; Haertlein M; Busch S; Forsyth VT; Svergun DI, et al. Structural Basis for Activation of Plasma-Membrane Ca2+-ATPase by Calmodulin. Commun. Biol 2018, 1, 206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (241).Wallen JR; Zhang H; Weis C; Cui W; Foster BM; Ho CMW; Hammel M; Tainer JA; Gross ML; Ellenberger T Hybrid Methods Reveal Multiple Flexibly Linked DNA Polymerases within the Bacteriophage T7 Replisome. Structure 2017, 25, 157–166. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (242).Zhang H; Harrington LB; Lu Y; Prado M; Saer R; Rempel D; Blankenship RE; Gross ML Native Mass Spectrometry Characterizes the Photosynthetic Reaction Center Complex from the Purple Bacterium Rhodobacter Sphaeroides. J. Am. Soc. Mass Spectrom 2017, 28, 87–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (243).Bernal I; Börnicke J; Heidemann J; Svergun D; Horstmann JA; Erhardt M; Tuukkanen A; Uetrecht C; Kolbe M Molecular Organization of Soluble Type III Secretion System Sorting Platform Complexes. J. Mol. Biol 2019, 431, 3787–3803. [DOI] [PubMed] [Google Scholar]
- (244).Wittig S; Haupt C; Hoffmann W; Kostmann S; Pagel K; Schmidt C Oligomerisation of Synaptobrevin-2 Studied by Native Mass Spectrometry and Chemical Cross-Linking. J. Am. Soc. Mass Spectrom 2019, 30, 149–160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (245).Prajapati S; Haselbach D; Wittig S; Patel MS; Chari A; Schmidt C; Stark H; Tittmann K Structural and Functional Analyses of the Human PDH Complex Suggest a “Division-of-Labor” Mechanism by Local E1 and E3 Clusters. Structure 2019, 27, 1124–1136. [DOI] [PubMed] [Google Scholar]
- (246).Lu Y; Goodson C; Blankenship RE; Gross ML Primary and Higher Order Structure of the Reaction Center from the Purple Phototrophic Bacterium Blastochloris viridis: A Test for Native Mass Spectrometry. J. Proteome Res 2018, 17, 1615–1623. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (247).Wittig S; Songailiene I; Schmidt C Formation and Stoichiometry of CRISPR-Cascade Complexes with Varying Spacer Lengths Revealed by Native Mass Spectrometry. J. Am. Soc. Mass Spectrom 2020, 31, 538–546. [DOI] [PubMed] [Google Scholar]
- (248).Li S; Yen L; Pastor WA; Johnston JB; Du J; Shew CJ; Liu W; Ho J; Stender A; Clark AT, et al. Mouse MORC3 Is a GHKL ATPase That Localizes to H3K4me3 Marked Chromatin. Proc. Natl. Acad. Sci. U. S. A 2016, 113, E5108–E5116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (249).Larson AG; Elnatan D; Keenen MM; Trnka MJ; Johnston JB; Burlingame AL; Agard DA; Redding S; Narlikar GJ Liquid Droplet Formation by HP1α Suggests a Role for Phase Separation in Heterochromatin. Nature 2017, 547, 236–240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (250).Mishra S; Chandler SA; Williams D; Claxton DP; Koteiche HA; Stewart PL; Benesch JLP; McHaourab HS Engineering of a Polydisperse Small Heat-Shock Protein Reveals Conserved Motifs of Oligomer Plasticity. Structure 2018, 26, 1116–1126. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (251).Hebling CM; Morgan CR; Stafford DW; Jorgenson JW; Rand KD; Engen JR Conformational Analysis of Membrane Proteins in Phospholipid Bilayer Nanodiscs by Hydrogen Exchange Mass Spectrometry. Anal. Chem 2010, 82, 5415–5419. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (252).Marty MT; Wilcox KC; Klein WL; Sligar SG Nanodisc-Solubilized Membrane Protein Library Reflects the Membrane Proteome. Anal. Bioanal. Chem 2013, 405, 4009–4016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (253).Walker LR; Marzluff EM; Townsend JA; Resager WC; Marty MT Native Mass Spectrometry of Antimicrobial Peptides in Lipid Nanodiscs Elucidates Complex Assembly. Anal. Chem 2019, 91, 9284–9291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (254).Walker LR; Marty MT Revealing the Specificity of a Range of Antimicrobial Peptides in Lipid Nanodiscs by Native Mass Spectrometry. Biochemistry 2020, 59, 2135–2142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (255).Marty MT; Zhang H; Cui WD; Gross ML; Sligar SG Interpretation and Deconvolution of Nanodisc Native Mass Spectra. J. Am. Soc. Mass Spectrom 2014, 25, 269–277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (256).Cleary SP; Li H; Bagal D; Loo JA; Campuzano IDG; Prell JS Extracting Charge and Mass Information from Highly Congested Mass Spectra Using Fourier-Domain Harmonics. J. Am. Soc. Mass Spectrom 2018, 29, 2067–2080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (257).Reid DJ; Keener JE; Wheeler AP; Zambrano DE; Diesing JM; Reinhardt-Szyba M; Makarov A; Marty MT Engineering Nanodisc Scaffold Proteins for Native Mass Spectrometry. Anal. Chem 2017, 89, 11189–11192. [DOI] [PubMed] [Google Scholar]
- (258).Kostelic MM; Ryan AM; Reid DJ; Noun JM; Marty MT Expanding the Types of Lipids Amenable to Native Mass Spectrometry of Lipoprotein Complexes. J. Am. Soc. Mass Spectrom 2019, 30, 1416–1425. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (259).Cong X; Liu Y; Liu W; Liang X; Russell DH; Laganowsky A Determining Membrane Protein–Lipid Binding Thermodynamics Using Native Mass Spectrometry. J. Am. Chem. Soc 2016, 138, 4346–4349. [DOI] [PubMed] [Google Scholar]
- (260).Reading E; Liko I; Allison TM; Benesch JLP; Laganowsky A; Robinson CV The Role of the Detergent Micelle in Preserving the Structure of Membrane Proteins in the Gas Phase. Angew. Chem. Int. Ed 2015, 54, 4577–4581. [DOI] [PubMed] [Google Scholar]
- (261).Reading E; Walton TA; Liko I; Marty MT; Laganowsky A; Rees DC; Robinson CV The Effect of Detergent, Temperature, and Lipid on the Oligomeric State of MscL Constructs: Insights from Mass Spectrometry. Chem. Biol 2015, 22, 593–603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (262).Tjondro HC; Ugonotti J; Kawahara R; Chatterjee S; Loke I; Chen S; Soltermann F; Hinneburg H; Parker BL; Venkatakrishnan V, et al. Hyper-Truncated Asn355- and Asn391-Glycans Modulate the Activity of Neutrophil Granule Myeloperoxidase. J. Biol. Chem 2021, 296, 100144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (263).Marty MT; Baldwin AJ; Marklund EG; Hochberg GKA; Benesch JLP; Robinson CV Bayesian Deconvolution of Mass and Ion Mobility Spectra: From Binary Interactions to Polydisperse Ensembles. Anal. Chem 2015, 87, 4370–4376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (264).Kostelic MM; Marty MT Deconvolving Native and Intact Protein Mass Spectra with Unidec. ChemRxiv 2020, DOI: 10.26434/chemrxiv.13417118.v13417111 (submitted December 21, 2020). [DOI] [PubMed] [Google Scholar]
- (265).Norris CE; Keener JE; Perera SMDC; Weerasinghe N; Fried SDE; Resager WC; Rohrbough JG; Brown MF; Marty MT Native Mass Spectrometry Reveals the Simultaneous Binding of Lipids and Zinc to Rhodopsin. Int. J. Mass Spectrom 2021, 460, 116477. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (266).Chorev DS; Tang H; Rouse SL; Bolla JR; von Kügelgen A; Baker LA; Wu D; Gault J; Grünewald K; Bharat TAM, et al. The Use of Sonicated Lipid Vesicles for Mass Spectrometry of Membrane Protein Complexes. Nat. Protoc 2020, 15, 1690–1706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (267).Ahdash Z; Lau AM; Byrne RT; Lammens K; Stüetzer A; Urlaub H; Booth PJ; Reading E; Hopfner K-P; Politis A Mechanistic Insight into the Assembly of the HerA–NurA Helicase–Nuclease DNA End Resection Complex. Nucleic Acids Res. 2017, 45, 12025–12038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (268).Jia M; Sen S; Wachnowsky C; Fidai I; Cowan JA; Wysocki VH Characterization of [2Fe–2S]-Cluster-Bridged Protein Complexes and Reaction Intermediates by Use of Native Mass Spectrometric Methods. Angew. Chem. Int. Ed 2020, 59, 6724–6728. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (269).Campuzano IDG; Nshanian M; Spahr C; Lantz C; Netirojjanakul C; Li H; Wongkongkathep P; Wolff JJ; Loo JA High Mass Analysis with a Fourier Transform Ion Cyclotron Resonance Mass Spectrometer: From Inorganic Salt Clusters to Antibody Conjugates and Beyond. J. Am. Soc. Mass Spectrom 2020, 31, 1155–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (270).Chen J; Chiu C; Gopalkrishnan S; Chen AY; Olinares PDB; Saecker RM; Winkelman JT; Maloney MF; Chait BT; Ross W, et al. Stepwise Promoter Melting by Bacterial RNA Polymerase. Mol. Cell 2020, 78, 275–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (271).VanAernum ZL; Busch F; Jones BJ; Jia M; Chen Z; Boyken SE; Sahasrabuddhe A; Baker D; Wysocki VH Rapid Online Buffer Exchange for Screening of Proteins, Protein Complexes and Cell Lysates by Native Mass Spectrometry. Nat. Protoc 2020, 15, 1132–1157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (272).Townsend JA; Keener JE; Miller ZM; Prell JS; Marty MT Imidazole Derivatives Improve Charge Reduction and Stabilization for Native Mass Spectrometry. Anal. Chem 2019, 91, 14765–14772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (273).Kieuvongngam V; Olinares PDB; Palillo A; Oldham ML; Chait BT; Chen J Structural Basis of Substrate Recognition by a Polypeptide Processing and Secretion Transporter. eLife 2020, 9, e51492. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (274).Mickolajczyk KJ; Olinares PDB; Niu Y; Chen N; Warrington SE; Sasaki Y; Walz T; Chait BT; Kapoor TM Long-Range Intramolecular Allostery and Regulation in the Dynein-Like AAA Protein Mdn1. Proc. Natl. Acad. Sci. U. S. A 2020, 117, 18459–18469. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (275).Moghadamchargari Z; Huddleston J; Shirzadeh M; Zheng X; Clemmer DE; M. Raushel F; Russell DH; Laganowsky A, Intrinsic GTPase Activity of K-Ras Monitored by Native Mass Spectrometry. Biochemistry 2019, 58, 3396–3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (276).Lin C-W; McCabe JW; Russell DH; Barondeau DP Molecular Mechanism of ISC Iron–Sulfur Cluster Biogenesis Revealed by High-Resolution Native Mass Spectrometry. J. Am. Chem. Soc 2020, 142, 6018–6029. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (277).Wu D; Li J; Struwe WB; Robinson CV Probing N-Glycoprotein Microheterogeneity by Lectin Affinity Purification-Mass Spectrometry Analysis. Chem. Sci 2019, 10, 5146–5155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (278).Patrick JW; Boone CD; Liu W; Conover GM; Liu Y; Cong X; Laganowsky A Allostery Revealed within Lipid Binding Events to Membrane Proteins. Proc. Natl. Acad. Sci. U. S. A 2018, 115, 2976–2981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (279).Matthews SJ; Pacholarz KJ; France AP; Jowitt TA; Hay S; Barran PE; Munro AW MhuD from Mycobacterium tuberculosis: Probing a Dual Role in Heme Storage and Degradation. ACS Infect. Dis 2019, 5, 1855–1866. [DOI] [PubMed] [Google Scholar]
- (280).Cruz AR; den Boer MA; Strasser J; Zwarthoff SA; Beurskens FJ; de Haas CJC; Aerts PC; Wang G; de Jong RN; Bagnoli F, et al. Staphylococcal Protein A Inhibits Complement Activation by Interfering with IgG Hexamer Formation. Proc. Natl. Acad. Sci. U. S. A 2021, 118, e2016772118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (281).Ma C; Sacco MD; Hurst B; Townsend JA; Hu Y; Szeto T; Zhang X; Tarbet B; Marty MT; Chen Y, et al. Boceprevir, GC-376, and Calpain Inhibitors II, XII Inhibit SARS-CoV-2 Viral Replication by Targeting the Viral Main Protease. Cell Res. 2020, 30, 678–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (282).Delfosse V; Huet T; Harrus D; Granell M; Bourguet M; Gardia-Parège C; Chiavarina B; Grimaldi M; Le Mével S; Blanc P, et al. Mechanistic Insights into the Synergistic Activation of the RXR–PXR Heterodimer by Endocrine Disruptor Mixtures. Proc. Natl. Acad. Sci. U. S. A 2021, 118, e2020551118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (283).Marty MT A Universal Score for Deconvolution of Intact Protein and Native Electrospray Mass Spectra. Anal. Chem 2020, 92, 4395–4401. [DOI] [PubMed] [Google Scholar]
- (284).Peris-Díaz MD; Guran R; Zitka O; Adam V; Krężzel A Mass Spectrometry-Based Structural Analysis of Cysteine-Rich Metal-Binding Sites in Proteins with MetaOdysseus R Software. J. Proteome Res 2021, 20, 776–785. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (285).Reid DJ; Diesing JM; Miller MA; Perry SM; Wales JA; Montfort WR; Marty MT MetaUniDec: High-Throughput Deconvolution of Native Mass Spectra. J. Am. Soc. Mass Spectrom 2018, 30, 118–127. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (286).Marty MT Eliminating Artifacts in Electrospray Deconvolution with a SoftMax Function. J. Am. Soc. Mass Spectrom 2019, 30, 2174–2177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (287).Bern M; Kil YJ; Becker C Byonic: Advanced Peptide and Protein Identification Software. Curr. Protoc. Bioinformatics 2012, 40, 13.20.11–13.20.14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (288).Bern M; Caval T; Kil YJ; Tang W; Becker C; Carlson E; Kletter D; Sen KI; Galy N; Hagemans D, et al. Parsimonious Charge Deconvolution for Native Mass Spectrometry. J. Proteome Res 2018, 17, 1216–1226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (289).Campuzano IDG; Netirojjanakul C; Nshanian M; Lippens JL; Kilgour DPA; Van Orden S; Loo JA Native-MS Analysis of Monoclonal Antibody Conjugates by Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Anal. Chem 2018, 90, 745–751. [DOI] [PubMed] [Google Scholar]
- (290).Leblanc Y; Faid V; Lauber MA; Wang Q; Bihoreau N; Chevreux G A Generic Method for Intact and Subunit Level Characterization of mAb Charge Variants by Native Mass Spectrometry. J. Chromatogr. B 2019, 1133, 121814. [DOI] [PubMed] [Google Scholar]
- (291).Lin Y-H; Zhu J; Meijer S; Franc V; Heck AJR Glycoproteogenomics: A Frequent Gene Polymorphism Affects the Glycosylation Pattern of the Human Serum Fetuin/α-2-HS-Glycoprotein. Mol. Cell. Proteom 2019, 18, 1479–1490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (292).Neviani V; van Deventer S; Wörner TP; Xenaki KT; van de Waterbeemd M; Rodenburg RNP; Wortel IMN; Kuiper JK; Huisman S; Granneman J, et al. Site-Specific Functionality and Tryptophan Mimicry of Lipidation in Tetraspanin CD9. FEBS J. 2020, 287, 5323–5344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (293).Busch F; VanAernum ZL; Ju Y; Yan J; Gilbert JD; Quintyn RS; Bern M; Wysocki VH Localization of Protein Complex Bound Ligands by Surface-Induced Dissociation High-Resolution Mass Spectrometry. Anal. Chem 2018, 90, 12796–12801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (294).Tseng Y-H; Uetrecht C; Yang S-C; Barendregt A; Heck AJR; Peng W-P Game-Theory-Based Search Engine to Automate the Mass Assignment in Complex Native Electrospray Mass Spectra. Anal. Chem 2013, 85, 11275–11283. [DOI] [PubMed] [Google Scholar]
- (295).Tseng Y-H; Uetrecht C; Heck AJR; Peng W-P Interpreting the Charge State Assignment in Electrospray Mass Spectra of Bioparticles. Anal. Chem 2011, 83, 1960–1968. [DOI] [PubMed] [Google Scholar]
- (296).Prebyl BS; Cook KD Use of Fourier Transform for Deconvolution of the Unresolved Envelope Observed in Electrospray Ionization Mass Spectrometry of Strongly Ionic Synthetic Polymers. Anal. Chem 2004, 76, 127–136. [Google Scholar]
- (297).Gábor D Theory of Communication. J. Inst. Electr. Eng 1946, 93, 429–460. [Google Scholar]
- (298).Cleary SP; Prell JS Liberating Native Mass Spectrometry from Dependence on Volatile Salt Buffers by Use of Gábor Transform. ChemPhysChem 2019, 20, 519–523. [DOI] [PubMed] [Google Scholar]
- (299).Wilson JW; Rolland AD; Klausen GM; Prell JS Ion Mobility-Mass Spectrometry Reveals That α-Hemolysin from Staphylococcus aureus Simultaneously Forms Hexameric and Heptameric Complexes in Detergent Micelle Solutions. Anal. Chem 2019, 91, 10204–10211. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (300).Yang SH; Chen B; Wang J; Zhang K Characterization of High Molecular Weight Multi-Arm Functionalized PEG–Maleimide for Protein Conjugation by Charge-Reduction Mass Spectrometry Coupled to Two-Dimensional Liquid Chromatography. Anal. Chem 2020, 92, 8584–8590. [DOI] [PubMed] [Google Scholar]
- (301).Bagal D; Zhang H; Schnier PD Gas-Phase Proton-Transfer Chemistry Coupled with TOF Mass Spectrometry and Ion Mobility-MS for the Facile Analysis of Poly(ethylene glycols) and PEGylated Polypeptide Conjugates. Anal. Chem 2008, 80, 2408–2418. [DOI] [PubMed] [Google Scholar]
- (302).Hoi KK; Robinson CV; Marty MT Unraveling the Composition and Behavior of Heterogeneous Lipid Nanodiscs by Mass Spectrometry. Anal. Chem 2016, 88, 6199–6204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (303).Donor MT; Wilson JW; Shepherd SO; Prell JS Lipid Head Group Adduction to Soluble Proteins Follows Gas-Phase Basicity Predictions: Dissociation Barriers and Charge Abstraction. Int. J. Mass Spectrom 2021, 469, 116670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (304).Kitov PI; Han L; Kitova EN; Klassen JS Sliding Window Adduct Removal Method (SWARM) for Enhanced Electrospray Ionization Mass Spectrometry Binding Data. J. Am. Soc. Mass Spectrom 2019, 30, 1446–1454. [DOI] [PubMed] [Google Scholar]
- (305).Kitov PI; Kitova EN; Han L; Li Z; Jung J; Rodrigues E; Hunter CD; Cairo CW; Macauley MS; Klassen JS A Quantitative, High-Throughput Method Identifies Protein–Glycan Interactions Via Mass Spectrometry. Commun. Biol 2019, 2, 268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (306).Báez Bolivar EG; Bui DT; Kitova EN; Han L; Zheng RB; Luber EJ; Sayed SY; Mahal LK; Klassen JS Submicron Emitters Enable Reliable Quantification of Weak Protein–Glycan Interactions by ESI-MS. Anal. Chem 2021, 93, 4231–4239. [DOI] [PubMed] [Google Scholar]
- (307).Kendrick E A Mass Scale Based on CH2 = 14.0000 for High Resolution Mass Spectrometry of Organic Compounds. Anal. Chem 1963, 35, 2146–2154. [Google Scholar]
- (308).Fouquet TNJ The Kendrick Analysis for Polymer Mass Spectrometry. J. Mass Spectrom 2019, 54, 933–947. [DOI] [PubMed] [Google Scholar]
- (309).Hughey CA; Hendrickson CL; Rodgers RP; Marshall AG; Qian K Kendrick Mass Defect Spectrum: A Compact Visual Analysis for Ultrahigh-Resolution Broadband Mass Spectra. Anal. Chem 2001, 73, 4676–4681. [DOI] [PubMed] [Google Scholar]
- (310).Lee H; An HJ; Lerno LA Jr; German JB; Lebrilla CB Rapid Profiling of Bovine and Human Milk Gangliosides by Matrix-Assisted Laser Desorption/Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometry. Int. J. Mass Spectrom 2011, 305, 138–150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (311).Nakamura S; Fouquet T; Sato H Molecular Characterization of High Molecular Weight Polyesters by Matrix-Assisted Laser Desorption/Ionization High-Resolution Time-of-Flight Mass Spectrometry Combined with on-Plate Alkaline Degradation and Mass Defect Analysis. J. Am. Soc. Mass Spectrom 2019, 30, 355–367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (312).Shi Y; Bajrami B; Yao X Passive and Active Fragment Ion Mass Defect Labeling: Distinct Proteomics Potential of Iodine-Based Reagents. Anal. Chem 2009, 81, 6438–6448. [DOI] [PubMed] [Google Scholar]
- (313).Harvey SR; VanAernum ZL; Kostelic MM; Marty MT; Wysocki VH Probing the Structure of Nanodiscs Using Surface-Induced Dissociation Mass Spectrometry. Chem. Commun 2020, 56, 15651–15654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (314).Eschweiler JD; Frank AT; Ruotolo BT Coming to Grips with Ambiguity: Ion Mobility-Mass Spectrometry for Protein Quaternary Structure Assignment. J. Am. Soc. Mass Spectrom 2017, 28, 1991–2000. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (315).Ben-Nissan G; Sharon M The Application of Ion-Mobility Mass Spectrometry for Structure/Function Investigation of Protein Complexes. Curr. Opin. Chem. Biol 2018, 42, 25–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (316).Bleiholder C Structure Elucidation from Ion Mobility-Mass Spectrometry Data: Are Detailed Structures Amenable?, Proceedings of Advancing Mass Spectrometry for Biophysics and Structural Biology Meeting, Ann Arbor, MI, July 28-August 1, 2017; Advancing Mass Spectrometry: 2017. [Google Scholar]
- (317).Canzani D; Laszlo KJ; Bush MF Ion Mobility of Proteins in Nitrogen Gas: Effects of Charge State, Charge Distribution, and Structure. J. Phys. Chem. A 2018, 122, 5625–5634. [DOI] [PubMed] [Google Scholar]
- (318).Jurneczko E; Barran PE How Useful Is Ion Mobility Mass Spectrometry for Structural Biology? The Relationship between Protein Crystal Structures and Their Collision Cross Sections in the Gas Phase. Analyst 2011, 136, 20–28. [DOI] [PubMed] [Google Scholar]
- (319).Wyttenbach T; Bowers MT Structural Stability from Solution to the Gas Phase: Native Solution Structure of Ubiquitin Survives Analysis in a Solvent-Free Ion Mobility–Mass Spectrometry Environment. J. Phys. Chem. B 2011, 115, 12266–12275. [DOI] [PubMed] [Google Scholar]
- (320).Bush MF; Hall Z; Giles K; Hoyes J; Robinson CV; Ruotolo BT Collision Cross Sections of Proteins and Their Complexes: A Calibration Framework and Database for Gas-Phase Structural Biology. Anal. Chem 2010, 82, 9557–9565. [DOI] [PubMed] [Google Scholar]
- (321).Bleiholder C; Dupuis NF; Wyttenbach T; Bowers MT Ion Mobility–Mass Spectrometry Reveals a Conformational Conversion from Random Assembly to β-Sheet in Amyloid Fibril Formation. Nat. Chem 2011, 3, 172–177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (322).Dodds JN; Baker ES Ion Mobility Spectrometry: Fundamental Concepts, Instrumentation, Applications, and the Road Ahead. J. Am. Soc. Mass Spectrom 2019, 30, 2185–2195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (323).Advances in Ion Mobility-Mass Spectrometry: Fundamentals, Instrumentation and Applications; Donald WA; Prell JS, Eds.; Comprehensive Analytical Chemistry: Vol. 83; Elsevier, 2019. ISBN: 978-0-444-64154-0 [Google Scholar]
- (324).Ewing MA; Glover MS; Clemmer DE Hybrid Ion Mobility and Mass Spectrometry as a Separation Tool. J. Chromatogr. A 2016, 1439, 3–25. [DOI] [PubMed] [Google Scholar]
- (325).Gabelica V; Marklund E Fundamentals of Ion Mobility Spectrometry. Curr. Opin. Chem. Biol 2018, 42, 51–59. [DOI] [PubMed] [Google Scholar]
- (326).Hall Z; Politis A; Robinson CV Structural Modeling of Heteromeric Protein Complexes from Disassembly Pathways and Ion Mobility-Mass Spectrometry. Structure 2012, 20, 1596–1609. [DOI] [PubMed] [Google Scholar]
- (327).Lanucara F; Holman SW; Gray CJ; Eyers CE The Power of Ion Mobility-Mass Spectrometry for Structural Characterization and the Study of Conformational Dynamics. Nat. Chem 2014, 6, 281–294. [DOI] [PubMed] [Google Scholar]
- (328).Larriba-Andaluz C; Prell JS Fundamentals of Ion Mobility in the Free Molecular Regime. Interlacing the Past, Present and Future of Ion Mobility Calculations. Int. Rev. Phys. Chem 2020, 39, 569–623. [Google Scholar]
- (329).Maurer MM; Donohoe GC; Valentine SJ Advances in Ion Mobility-Mass Spectrometry Instrumentation and Techniques for Characterizing Structural Heterogeneity. Analyst 2015, 140, 6782–6798. [DOI] [PubMed] [Google Scholar]
- (330).May JC; McLean JA Ion Mobility-Mass Spectrometry: Time-Dispersive Instrumentation. Anal. Chem 2015, 87, 1422–1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (331).Morris CB; Poland JC; May JC; McLean JA Fundamentals of Ion Mobility-Mass Spectrometry for the Analysis of Biomolecules. In Ion Mobility-Mass Spectrometry : Methods and Protocols, Vol. Paglia G; Astarita G, Eds.; Springer; US, 2020; pp 1–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (332).Saikusa K; Fuchigami S; Takahashi K; Asano Y; Nagadoi A; Tachiwana H; Kurumizaka H; Ikeguchi M; Nishimura Y; Akashi S Gas-Phase Structure of the Histone Multimers Characterized by Ion Mobility Mass Spectrometry and Molecular Dynamics Simulation. Anal. Chem 2013, 85, 4165–4171. [DOI] [PubMed] [Google Scholar]
- (333).Wyttenbach T; Pierson NA; Clemmer DE; Bowers MT Ion Mobility Analysis of Molecular Dynamics. Annu. Rev. Phys. Chem 2014, 65, 175–196. [DOI] [PubMed] [Google Scholar]
- (334).Zhou M; Politis A; Davies RB; Liko I; Wu K-J; Stewart AG; Stock D; Robinson CV Ion Mobility–Mass Spectrometry of a Rotary ATPase Reveals ATP-Induced Reduction in Conformational Flexibility. Nat. Chem 2014, 6, 208–215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (335).Blackwell AE; Dodds ED; Bandarian V; Wysocki VH Revealing the Quaternary Structure of a Heterogeneous Noncovalent Protein Complex through Surface-Induced Dissociation. Anal. Chem 2011, 83, 2862–2865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (336).Ma X; Zhou M; Wysocki VH Surface Induced Dissociation Yields Quaternary Substructure of Refractory Noncovalent Phosphorylase B and Glutamate Dehydrogenase Complexes. J. Am. Soc. Mass Spectrom 2014, 25, 368–379. [DOI] [PubMed] [Google Scholar]
- (337).Jones CM; Beardsley RL; Galhena AS; Dagan S; Cheng G; Wysocki VH Symmetrical Gas-Phase Dissociation of Noncovalent Protein Complexes Via Surface Collisions. J. Am. Chem. Soc 2006, 128, 15044–15045. [DOI] [PubMed] [Google Scholar]
- (338).Ma X; Loo JA; Wysocki VH Surface Induced Dissociation Yields Substructure of Methanosarcina thermophila 20S Proteasome Complexes. Int. J. Mass Spectrom 2015, 377, 201–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (339).Quintyn RS; Zhou M; Yan J; Wysocki VH Surface-Induced Dissociation Mass Spectra as a Tool for Distinguishing Different Structural Forms of Gas-Phase Multimeric Protein Complexes. Anal. Chem 2015, 87, 11879–11886. [DOI] [PubMed] [Google Scholar]
- (340).Seffernick JT; Harvey SR; Wysocki VH; Lindert S Predicting Protein Complex Structure from Surface-Induced Dissociation Mass Spectrometry Data. ACS Cent. Sci 2019, 5, 1330–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (341).Song Y; Nelp MT; Bandarian V; Wysocki VH Refining the Structural Model of a Heterohexameric Protein Complex: Surface Induced Dissociation and Ion Mobility Provide Key Connectivity and Topology Information. ACS Cent. Sci 2015, 1, 477–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (342).Wysocki VH; Joyce KE; Jones CM; Beardsley RL Surface-Induced Dissociation of Small Molecules, Peptides, and Non-Covalent Protein Complexes. J. Am. Soc. Mass Spectrom 2008, 19, 190–208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (343).Zhou M; Jones CM; Wysocki VH Dissecting the Large Noncovalent Protein Complex Groel with Surface-Induced Dissociation and Ion Mobility–Mass Spectrometry. Anal. Chem 2013, 85, 8262–8267. [DOI] [PubMed] [Google Scholar]
- (344).Zhou M; Wysocki VH Surface Induced Dissociation: Dissecting Noncovalent Protein Complexes in the Gas Phase. Acc. Chem. Res 2014, 47, 1010–1018. [DOI] [PubMed] [Google Scholar]
- (345).Seffernick JT; Lindert S Hybrid Methods for Combined Experimental and Computational Determination of Protein Structure. J. Chem. Phys 2020, 153, 240901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (346).Gault J; Robinson CV Cracking Complexes to Build Models of Protein Assemblies. ACS Cent. Sci 2019, 5, 1310–1311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (347).Liu XR; Zhang MM; Gross ML Mass Spectrometry-Based Protein Footprinting for Higher-Order Structure Analysis: Fundamentals and Applications. Chem. Rev 2020, 120, 4355–4454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (348).Macias LA; Santos IC; Brodbelt JS Ion Activation Methods for Peptides and Proteins. Anal. Chem 2020, 92, 227–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (349).Taverner T; Hernández H; Sharon M; Ruotolo BT; Matak-Vinković D; Devos D; Russell RB; Robinson CV Subunit Architecture of Intact Protein Complexes from Mass Spectrometry and Homology Modeling. Acc. Chem. Res 2008, 41, 617–627. [DOI] [PubMed] [Google Scholar]
- (350).Russel D; Lasker K; Webb B; Velázquez-Muriel J; Tjioe E; Schneidman-Duhovny D; Peterson B; Sali A Putting the Pieces Together: Integrative Modeling Platform Software for Structure Determination of Macromolecular Assemblies. PLoS Biol. 2012, 10, e1001244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (351).Politis A; Park AY; Hyung S-J; Barsky D; Ruotolo BT; Robinson CV Integrating Ion Mobility Mass Spectrometry with Molecular Modelling to Determine the Architecture of Multiprotein Complexes. PLoS One 2010, 5, e12080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (352).Seffernick JT; Canfield SM; Harvey SR; Wysocki VH; Lindert S Prediction of Protein Complex Structure Using Surface-Induced Dissociation and Cryo-Electron Microscopy. Anal. Chem 2021, 93, 7596–7605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (353).Baldwin Andrew J.; Lioe H; Hilton Gillian R.; Baker Lindsay A.; Rubinstein John L.; Kay Lewis E.; Benesch Justin L. P. The Polydispersity of αB-Crystallin Is Rationalized by an Interconverting Polyhedral Architecture. Structure 2011, 19, 1855–1863. [DOI] [PubMed] [Google Scholar]
- (354).Hilton GR; Hochberg GKA; Laganowsky A; McGinnigle SI; Baldwin AJ; Benesch JLP C-Terminal Interactions Mediate the Quaternary Dynamics of αB-Crystallin. Philos. Trans. R. Soc. B 2013, 368, 20110405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (355).Sinz A; Arlt C; Chorev D; Sharon M Chemical Cross-Linking and Native Mass Spectrometry: A Fruitful Combination for Structural Biology. Protein Sci. 2015, 24, 1193–1209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (356).Yu C; Huang L Cross-Linking Mass Spectrometry: An Emerging Technology for Interactomics and Structural Biology. Anal. Chem 2018, 90, 144–165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (357).Zhou M; Sandercock AM; Fraser CS; Ridlova G; Stephens E; Schenauer MR; Yokoi-Fong T; Barsky D; Leary JA; Hershey JW, et al. Mass Spectrometry Reveals Modularity and a Complete Subunit Interaction Map of the Eukaryotic Translation Factor eIF3. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 18139–18144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (358).Schreiber A; Stengel F; Zhang Z; Enchev RI; Kong EH; Morris EP; Robinson CV; da Fonseca PCA; Barford D Structural Basis for the Subunit Assembly of the Anaphase-Promoting Complex. Nature 2011, 470, 227–232. [DOI] [PubMed] [Google Scholar]
- (359).Sharon M; Mao H; Boeri Erba E; Stephens E; Zheng N; Robinson CV Symmetrical Modularity of the COP9 Signalosome Complex Suggests Its Multifunctionality. Structure 2009, 17, 31–40. [DOI] [PubMed] [Google Scholar]
- (360).Chorev DS; Moscovitz O; Geiger B; Sharon M Regulation of Focal Adhesion Formation by a Vinculin-Arp2/3 Hybrid Complex. Nat. Commun 2014, 5, 3758. [DOI] [PubMed] [Google Scholar]
- (361).Casañal A; Kumar A; Hill CH; Easter AD; Emsley P; Degliesposti G; Gordiyenko Y; Santhanam B; Wolf J; Wiederhold K, et al. Architecture of Eukaryotic mRNA 3′-End Processing Machinery. Science 2017, 358, 1056–1059. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (362).Hernández H; Makarova OV; Makarov EM; Morgner N; Muto Y; Krummel DP; Robinson CV Isoforms of U1-70k Control Subunit Dynamics in the Human Spliceosomal U1 snRNP. PLoS One 2009, 4, e7202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (363).Politis A; Schmidt C; Tjioe E; Sandercock AM; Lasker K; Gordiyenko Y; Russel D; Sali A; Robinson CV Topological Models of Heteromeric Protein Assemblies from Mass Spectrometry: Application to the Yeast eIF3:eIF5 Complex. Chem. Biol 2015, 22, 117–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (364).Shakeel S; Rajendra E; Alcón P; O’Reilly F; Chorev DS; Maslen S; Degliesposti G; Russo CJ; He S; Hill CH, et al. Structure of the Fanconi anaemia Monoubiquitin Ligase Complex. Nature 2019, 575, 234–237. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (365).Lane LA; Nadeau OW; Carlson GM; Robinson CV Mass Spectrometry Reveals Differences in Stability and Subunit Interactions between Activated and Nonactivated Conformers of the (Aβγδ)4 Phosphorylase Kinase Complex. Mol. Cell. Proteom 2012, 11, 1768–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (366).Kleiner D; Shmulevich F; Zarivach R; Shahar A; Sharon M; Ben-Nissan G; Bershtein S The Interdimeric Interface Controls Function and Stability of Ureaplasma urealiticum Methionine S-Adenosyltransferase. J. Mol. Biol 2019, 431, 4796–4816. [DOI] [PubMed] [Google Scholar]
- (367).Lössl P; Snijder J; Heck AJR Boundaries of Mass Resolution in Native Mass Spectrometry. J. Am. Soc. Mass Spectrom 2014, 25, 906–917. [DOI] [PubMed] [Google Scholar]
- (368).Chen X; Westphall MS; Smith LM Mass Spectrometric Analysis of DNA Mixtures: Instrumental Effects Responsible for Decreased Sensitivity with Increasing Mass. Anal. Chem 2003, 75, 5944–5952. [DOI] [PubMed] [Google Scholar]
- (369).Bruce JE; Cheng X; Bakhtiar R; Wu Q; Hofstadler SA; Anderson GA; Smith RD Trapping, Detection, and Mass Measurement of Individual Ions in a Fourier Transform Ion Cyclotron Resonance Mass Spectrometer. J. Am. Chem. Soc 1994, 116, 7839–7847. [Google Scholar]
- (370).Keifer DZ; Pierson EE; Jarrold MF Charge Detection Mass Spectrometry: Weighing Heavier Things. Analyst 2017, 142, 1654–1671. [DOI] [PubMed] [Google Scholar]
- (371).Benner WH A Gated Electrostatic Ion Trap to Repetitiously Measure the Charge and m/z of Large Electrospray Ions. Anal. Chem 1997, 69, 4162–4168. [Google Scholar]
- (372).Fuerstenau SD; Benner WH Molecular Weight Determination of Megadalton DNA Electrospray Ions Using Charge Detection Time-of-Flight Mass Spectrometry. Rapid Commun. Mass Spectrom 1995, 9, 1528–1538. [DOI] [PubMed] [Google Scholar]
- (373).Draper BE; Jarrold MF Real-Time Analysis and Signal Optimization for Charge Detection Mass Spectrometry. J. Am. Soc. Mass Spectrom 2019, 30, 898–904. [DOI] [PubMed] [Google Scholar]
- (374).Elliott AG; Harper CC; Lin H-W; Susa AC; Xia Z; Williams ER Simultaneous Measurements of Mass and Collisional Cross-Section of Single Ions with Charge Detection Mass Spectrometry. Anal. Chem 2017, 89, 7701–7708. [DOI] [PubMed] [Google Scholar]
- (375).Harper CC; Elliott AG; Oltrogge LM; Savage DF; Williams ER Multiplexed Charge Detection Mass Spectrometry for High-Throughput Single Ion Analysis of Large Molecules. Anal. Chem 2019, 91, 7458–7465. [DOI] [PubMed] [Google Scholar]
- (376).Harper CC; Williams ER Enhanced Multiplexing in Fourier Transform Charge Detection Mass Spectrometry by Decoupling Ion Frequency from Mass to Charge Ratio. J. Am. Soc. Mass Spectrom 2019, 30, 2637–2645. [DOI] [PubMed] [Google Scholar]
- (377).Keifer DZ; Shinholt DL; Jarrold MF Charge Detection Mass Spectrometry with Almost Perfect Charge Accuracy. Anal. Chem 2015, 87, 10330–10337. [DOI] [PubMed] [Google Scholar]
- (378).Todd AR; Jarrold MF Dramatic Improvement in Sensitivity with Pulsed Mode Charge Detection Mass Spectrometry. Anal. Chem 2019, 91, 14002–14008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (379).Wörner TP; Snijder J; Bennett A; Agbandje-McKenna M; Makarov AA; Heck AJR Resolving Heterogeneous Macromolecular Assemblies by Orbitrap-Based Single-Particle Charge Detection Mass Spectrometry. Nat. Methods 2020, 17, 395–398. [DOI] [PubMed] [Google Scholar]
- (380).Botamanenko DY; Jarrold MF Ion-Ion Interactions in Charge Detection Mass Spectrometry. J. Am. Soc. Mass Spectrom 2019, 30, 2741–2749. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (381).Keifer DZ; Jarrold MF Single-Molecule Mass Spectrometry. Mass Spectrom. Rev 2017, 36, 715–733. [DOI] [PubMed] [Google Scholar]
- (382).Doussineau T; Mathevon C; Altamura L; Vendrely C; Dugourd P; Forge V; Antoine R Mass Determination of Entire Amyloid Fibrils by Using Mass Spectrometry. Angew. Chem. Int. Ed 2016, 55, 2340–2344. [DOI] [PubMed] [Google Scholar]
- (383).Contino NC; Pierson EE; Keifer DZ; Jarrold MF Charge Detection Mass Spectrometry with Resolved Charge States. J. Am. Soc. Mass Spectrom 2013, 24, 101–108. [DOI] [PubMed] [Google Scholar]
- (384).Keifer DZ; Motwani T; Teschke CM; Jarrold MF Acquiring Structural Information on Virus Particles with Charge Detection Mass Spectrometry. J. Am. Soc. Mass Spectrom 2016, 27, 1028–1036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (385).Keifer DZ; Pierson EE; Hogan JA; Bedwell GJ; Prevelige PE; Jarrold MF Charge Detection Mass Spectrometry of Bacteriophage P22 Procapsid Distributions above 20 MDa. Rapid Commun. Mass Spectrom 2014, 28, 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (386).Miller LM; Barnes LF; Raab SA; Draper BE; El-Baba TJ; Lutomski CA; Robinson CV; Clemmer DE; Jarrold MF Heterogeneity of Glycan Processing on Trimeric SARS-CoV-2 Spike Protein Revealed by Charge Detection Mass Spectrometry. J. Am. Chem. Soc 2021, 143, 3959–3966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (387).Pierson EE; Keifer DZ; Asokan A; Jarrold MF Resolving Adeno-Associated Viral Particle Diversity with Charge Detection Mass Spectrometry. Anal. Chem 2016, 88, 6718–6725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (388).Pierson EE; Keifer DZ; Contino NC; Jarrold MF Probing Higher Order Multimers of Pyruvate Kinase with Charge Detection Mass Spectrometry. Int. J. Mass Spectrom 2013, 337, 50–56. [Google Scholar]
- (389).Schultz JC; Hack CA; Benner WH Mass Determination of Megadalton-DNA Electrospray Ions Using Charge Detection Mass Spectrometry. J. Am. Soc. Mass Spectrom. 1998, 9, 305–313. [DOI] [PubMed] [Google Scholar]
- (390).Philip MA; Gelbard F; Arnold S An Absolute Method for Aerosol Particle Mass and Charge Measurement. J. Colloid Interface Sci 1983, 91, 507–515. [Google Scholar]
- (391).Hars G; Tass Z Application of Quadrupole Ion Trap for the Accurate Mass Determination of Submicron Size Charged Particles. J. Appl. Phys 1995, 77, 4245–4250. [Google Scholar]
- (392).Halim MA; Bertorelle F; Doussineau T; Antoine R Direct Determination of Molecular Weight Distribution of Calf-Thymus Dnas and Study of Their Fragmentation under Ultrasonic and Low-Energy Infrared Irradiations. A Charge Detection Mass Spectrometry Investigation. Rapid Commun. Mass Spectrom 2019, 33, 35–39. [DOI] [PubMed] [Google Scholar]
- (393).Contino NC; Jarrold MF Charge Detection Mass Spectrometry for Single Ions with a Limit of Detection of 30 Charges. Int. J. Mass Spectrom 2013, 345-347, 153–159. [Google Scholar]
- (394).Dunbar CA; Rayaprolu V; Wang JCY; Brown CJ; Leishman E; Jones-Burrage S; Trinidad JC; Bradshaw HB; Clemmer DE; Mukhopadhyay S, et al. Dissecting the Components of Sindbis Virus from Arthropod and Vertebrate Hosts: Implications for Infectivity Differences. ACS Infect. Dis 2019, 5, 892–902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (395).Mabbett SR; Zilch LW; Maze JT; Smith JW; Jarrold MF Pulsed Acceleration Charge Detection Mass Spectrometry: Application to Weighing Electrosprayed Droplets. Anal. Chem 2007, 79, 8431–8439. [DOI] [PubMed] [Google Scholar]
- (396).Elliott AG; Merenbloom SI; Chakrabarty S; Williams ER Single Particle Analyzer of Mass: A Charge Detection Mass Spectrometer with a Multi-Detector Electrostatic Ion Trap. Int. J. Mass Spectrom 2017, 414, 45–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (397).Elliott AG; Harper CC; Lin H-W; Williams ER Mass, Mobility and MSn Measurements of Single Ions Using Charge Detection Mass Spectrometry. Analyst 2017, 142, 2760–2769. [DOI] [PubMed] [Google Scholar]
- (398).Harper CC; Elliott AG; Lin H-W; Williams ER Determining Energies and Cross Sections of Individual Ions Using Higher-Order Harmonics in Fourier Transform Charge Detection Mass Spectrometry (FT-CDMS). J. Am. Soc. Mass Spectrom 2018, 29, 1861–1869. [DOI] [PubMed] [Google Scholar]
- (399).Kafader JO; Melani RD; Durbin KR; Ikwuagwu B; Early BP; Fellers RT; Beu SC; Zabrouskov V; Makarov AA; Maze JT, et al. Multiplexed Mass Spectrometry of Individual Ions Improves Measurement of Proteoforms and Their Complexes. Nat. Methods 2020, 17, 391–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (400).Kafader JO; Durbin KR; Melani RD; Des Soye BJ; Schachner LF; Senko MW; Compton PD; Kelleher NL Individual Ion Mass Spectrometry Enhances the Sensitivity and Sequence Coverage of Top-Down Mass Spectrometry. J. Proteome Res 2020, 19, 1346–1350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (401).McGee JP; Melani RD; Yip PF; Senko MW; Compton PD; Kafader JO; Kelleher NL Isotopic Resolution of Protein Complexes up to 466 kDa Using Individual Ion Mass Spectrometry. Anal. Chem 2021, 93, 2723–2727. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (402).Kafader JO; Melani RD; Senko MW; Makarov AA; Kelleher NL; Compton PD Measurement of Individual Ions Sharply Increases the Resolution of Orbitrap Mass Spectra of Proteins. Anal. Chem 2019, 91, 2776–2783. [DOI] [PubMed] [Google Scholar]
- (403).Bernstein SL; Liu D; Wyttenbach T; Bowers MT; Lee JC; Gray HB; Winkler JR α-Synuclein: Stable Compact and Extended Monomeric Structures and pH Dependence of Dimer Formation. J. Am. Soc. Mass Spectrom 2004, 15, 1435–1443. [DOI] [PubMed] [Google Scholar]
- (404).Ridgeway ME; Silveira JA; Meier JE; Park MA Microheterogeneity within Conformational States of Ubiquitin Revealed by High Resolution Trapped Ion Mobility Spectrometry. Analyst 2015, 140, 6964–6972. [DOI] [PubMed] [Google Scholar]
- (405).Uetrecht C; Barbu IM; Shoemaker GK; van Duijn E; Heck AJR Interrogating Viral Capsid Assembly with Ion Mobility–Mass Spectrometry. Nat. Chem 2011, 3, 126–132. [DOI] [PubMed] [Google Scholar]
- (406).Young LM; Cao P; Raleigh DP; Ashcroft AE; Radford SE Ion Mobility Spectrometry–Mass Spectrometry Defines the Oligomeric Intermediates in Amylin Amyloid Formation and the Mode of Action of Inhibitors. J. Am. Chem. Soc 2014, 136, 660–670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (407).Snijder J; Uetrecht C; Rose RJ; Sanchez-Eugenia R; Marti GA; Agirre J; Guerin DMA; Wuite GJL; Heck AJR; Roos WH Probing the Biophysical Interplay between a Viral Genome and Its Capsid. Nat. Chem 2013, 5, 502–509. [DOI] [PubMed] [Google Scholar]
- (408).El-Baba TJ; Lutomski CA; Wang B; Trimpin S Characterizing Synthetic Polymers and Additives Using New Ionization Methods for Mass Spectrometry. Rapid Commun. Mass Spectrom 2014, 28, 1175–1184. [DOI] [PubMed] [Google Scholar]
- (409).Loo JA; Berhane B; Kaddis CS; Wooding KM; Xie Y; Kaufman SL; Chernushevich IV Electrospray Ionization Mass Spectrometry and Ion Mobility Analysis of the 20S Proteasome Complex. J. Am. Soc. Mass Spectrom 2005, 16, 998–1008. [DOI] [PubMed] [Google Scholar]
- (410).Marcoux J; Champion T; Colas O; Wagner-Rousset E; Corvaïa N; Van Dorsselaer A; Beck A; Cianférani S Native Mass Spectrometry and Ion Mobility Characterization of Trastuzumab Emtansine, a Lysine-Linked Antibody Drug Conjugate. Protein Sci. 2015, 24, 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (411).Österlund N; Moons R; Ilag LL; Sobott F; Gräslund A Native Ion Mobility-Mass Spectrometry Reveals the Formation of β-Barrel Shaped Amyloid-β Hexamers in a Membrane-Mimicking Environment. J. Am. Chem. Soc 2019, 141, 10440–10450. [DOI] [PubMed] [Google Scholar]
- (412).Kłoniecki M; Jabłonowska A; Poznański J; Langridge J; Hughes C; Campuzano I; Giles K; Dadlez M Ion Mobility Separation Coupled with MS Detects Two Structural States of Alzheimer's Disease Aβ1–40 Peptide Oligomers. J. Mol. Biol 2011, 407, 110–124. [DOI] [PubMed] [Google Scholar]
- (413).Woods LA; Radford SE; Ashcroft AE Advances in Ion Mobility Spectrometry–Mass Spectrometry Reveal Key Insights into Amyloid Assembly. Biochim. Biophys. Acta Proteins Proteom 2013, 1834, 1257–1268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (414).Young LM; Saunders JC; Mahood RA; Revill CH; Foster RJ; Tu L-H; Raleigh DP; Radford SE; Ashcroft AE Screening and Classifying Small-Molecule Inhibitors of Amyloid Formation Using Ion Mobility Spectrometry–Mass Spectrometry. Nat. Chem 2015, 7, 73–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (415).Shvartsburg AA; Smith RD Fundamentals of Traveling Wave Ion Mobility Spectrometry. Anal. Chem 2008, 80, 9689–9699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (416).Pukala TL; Ruotolo BT; Zhou M; Politis A; Stefanescu R; Leary JA; Robinson CV Subunit Architecture of Multiprotein Assemblies Determined Using Restraints from Gas-Phase Measurements. Structure 2009, 17, 1235–1243. [DOI] [PubMed] [Google Scholar]
- (417).Dodds JN; May JC; McLean JA Correlating Resolving Power, Resolution, and Collision Cross Section: Unifying Cross-Platform Assessment of Separation Efficiency in Ion Mobility Spectrometry. Anal. Chem 2017, 89, 12176–12184. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (418).Richardson K; Langridge D; Giles K Fundamentals of Travelling Wave Ion Mobility Revisited: I. Smoothly Moving Waves. Int. J. Mass Spectrom 2018, 428, 71–80. [Google Scholar]
- (419).Michelmann K; Silveira JA; Ridgeway ME; Park MA Fundamentals of Trapped Ion Mobility Spectrometry. J. Am. Soc. Mass Spectrom 2015, 26, 14–24. [DOI] [PubMed] [Google Scholar]
- (420).Wu C; Siems WF; Klasmeier J; Hill HH Separation of Isomeric Peptides Using Electrospray Ionization/High-Resolution Ion Mobility Spectrometry. Anal. Chem 2000, 72, 391–395. [DOI] [PubMed] [Google Scholar]
- (421).Clowers BH; Hill HH Mass Analysis of Mobility-Selected Ion Populations Using Dual Gate, Ion Mobility, Quadrupole Ion Trap Mass Spectrometry. Anal. Chem 2005, 77, 5877–5885. [DOI] [PubMed] [Google Scholar]
- (422).Eldrid C; Thalassinos K Developments in Tandem Ion Mobility Mass Spectrometry. Biochem. Soc. Trans 2020, 48, 2457–2466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (423).Bohrer BC; Merenbloom SI; Koeniger SL; Hilderbrand AE; Clemmer DE Biomolecule Analysis by Ion Mobility Spectrometry. Annu. Rev. Anal. Chem 2008, 1, 293–327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (424).Giles K; Williams JP; Campuzano I Enhancements in Travelling Wave Ion Mobility Resolution. Rapid Commun. Mass Spectrom 2011, 25, 1559–1566. [DOI] [PubMed] [Google Scholar]
- (425).Giles K; Ujma J; Wildgoose J; Pringle S; Richardson K; Langridge D; Green M A Cyclic Ion Mobility-Mass Spectrometry System. Anal. Chem 2019, 91, 8564–8573. [DOI] [PubMed] [Google Scholar]
- (426).Richardson K; Langridge D; Dixit SM; Ruotolo BT An Improved Calibration Approach for Traveling Wave Ion Mobility Spectrometry: Robust, High-Precision Collision Cross Sections. Anal. Chem 2021, 93, 3542–3550. [DOI] [PubMed] [Google Scholar]
- (427).Sanda M; Morrison L; Goldman R N- and O-Glycosylation of the SARS-CoV-2 Spike Protein. Anal. Chem 2021, 93, 2003–2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (428).Sisley EK; Ujma J; Palmer M; Giles K; Fernandez-Lima FA; Cooper HJ Lesa Cyclic Ion Mobility Mass Spectrometry of Intact Proteins from Thin Tissue Sections. Anal. Chem 2020, 92, 6321–6326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (429).Eldrid C; Ujma J; Kalfas S; Tomczyk N; Giles K; Morris M; Thalassinos K Gas Phase Stability of Protein Ions in a Cyclic Ion Mobility Spectrometry Traveling Wave Device. Anal. Chem 2019, 91, 7554–7561. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (430).Hofmann J; Hahm HS; Seeberger PH; Pagel K Identification of Carbohydrate Anomers Using Ion Mobility–Mass Spectrometry. Nature 2015, 526, 241–244. [DOI] [PubMed] [Google Scholar]
- (431).Pagel K; Harvey DJ Ion Mobility–Mass Spectrometry of Complex Carbohydrates: Collision Cross Sections of Sodiated N-Linked Glycans. Anal. Chem 2013, 85, 5138–5145. [DOI] [PubMed] [Google Scholar]
- (432).Kyle JE; Zhang X; Weitz KK; Monroe ME; Ibrahim YM; Moore RJ; Cha J; Sun X; Lovelace ES; Wagoner J, et al. Uncovering Biologically Significant Lipid Isomers with Liquid Chromatography, Ion Mobility Spectrometry and Mass Spectrometry. Analyst 2016, 141, 1649–1659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (433).Helmer PO; Nordhorn ID; Korf A; Behrens A; Buchholz R; Zubeil F; Karst U; Hayen H Complementing Matrix-Assisted Laser Desorption Ionization-Mass Spectrometry Imaging with Chromatography Data for Improved Assignment of Isobaric and Isomeric Phospholipids Utilizing Trapped Ion Mobility-Mass Spectrometry. Anal. Chem 2021, 93, 2135–2143. [DOI] [PubMed] [Google Scholar]
- (434).Dodds JN; May JC; McLean JA Investigation of the Complete Suite of the Leucine and Isoleucine Isomers: Toward Prediction of Ion Mobility Separation Capabilities. Anal. Chem 2017, 89, 952–959. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (435).Kliman M; May JC; McLean JA Lipid Analysis and Lipidomics by Structurally Selective Ion Mobility-Mass Spectrometry. Biochim. Biophys. Acta Mol. Cell Biol. Lipids 2011, 1811, 935–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (436).Groessl M; Graf S; Knochenmuss R High Resolution Ion Mobility-Mass Spectrometry for Separation and Identification of Isomeric Lipids. Analyst 2015, 140, 6904–6911. [DOI] [PubMed] [Google Scholar]
- (437).Deng L; Ibrahim YM; Baker ES; Aly NA; Hamid AM; Zhang X; Zheng X; Garimella SVB; Webb IK; Prost SA, et al. Ion Mobility Separations of Isomers Based Upon Long Path Length Structures for Lossless Ion Manipulations Combined with Mass Spectrometry. ChemistrySelect 2016, 1, 2396–2399. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (438).Jeanne Dit Fouque K; Ramirez CE; Lewis RL; Koelmel JP; Garrett TJ; Yost RA; Fernandez-Lima F Effective Liquid Chromatography–Trapped Ion Mobility Spectrometry–Mass Spectrometry Separation of Isomeric Lipid Species. Anal. Chem 2019, 91, 5021–5027. [DOI] [PubMed] [Google Scholar]
- (439).Pu Y; Ridgeway ME; Glaskin RS; Park MA; Costello CE; Lin C Separation and Identification of Isomeric Glycans by Selected Accumulation-Trapped Ion Mobility Spectrometry-Electron Activated Dissociation Tandem Mass Spectrometry. Anal. Chem 2016, 88, 3440–3443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (440).Gault J; Liko I; Landreh M; Shutin D; Bolla JR; Jefferies D; Agasid M; Yen H-Y; Ladds MJGW; Lane DP, et al. Combining Native and ‘Omics’ Mass Spectrometry to Identify Endogenous Ligands Bound to Membrane Proteins. Nat. Methods 2020, 17, 505–508. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (441).Fernandez-Lima F; Kaplan DA; Suetering J; Park MA Gas-Phase Separation Using a Trapped Ion Mobility Spectrometer. Int. J. Ion Mobil. Spectrom 2011, 14, 93–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (442).Liu FC; Ridgeway ME; Park MA; Bleiholder C Tandem Trapped Ion Mobility Spectrometry. Analyst 2018, 143, 2249–2258. [DOI] [PubMed] [Google Scholar]
- (443).Liu FC; Kirk SR; Bleiholder C On the Structural Denaturation of Biological Analytes in Trapped Ion Mobility Spectrometry – Mass Spectrometry. Analyst 2016, 141, 3722–3730. [DOI] [PubMed] [Google Scholar]
- (444).Silveira JA; Ridgeway ME; Park MA High Resolution Trapped Ion Mobility Spectrometery of Peptides. Anal. Chem 2014, 86, 5624–5627. [DOI] [PubMed] [Google Scholar]
- (445).Jeanne Dit Fouque K; Fernandez-Lima F Recent Advances in Biological Separations Using Trapped Ion Mobility Spectrometry – Mass Spectrometry. Trends Anal. Chem 2019, 116, 308–315. [Google Scholar]
- (446).Ridgeway ME; Lubeck M; Jordens J; Mann M; Park MA Trapped Ion Mobility Spectrometry: A Short Review. Int. J. Mass Spectrom 2018, 425, 22–35. [Google Scholar]
- (447).Ridgeway ME; Bleiholder C; Mann M; Park MA Trends in Trapped Ion Mobility – Mass Spectrometry Instrumentation. Trends Anal. Chem 2019, 116, 324–331. [Google Scholar]
- (448).Borotto NB; Graham KA Fragmentation and Mobility Separation of Peptide and Protein Ions in a Trapped-Ion Mobility Device. Anal. Chem 2021, 93, 9959–9964. [DOI] [PubMed] [Google Scholar]
- (449).Fernandez-Lima FA; Kaplan DA; Park MA Note: Integration of Trapped Ion Mobility Spectrometry with Mass Spectrometry. Rev. Sci. Instrum 2011, 82, 126106–126106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (450).Adams KJ; Montero D; Aga D; Fernandez-Lima F Isomer Separation of Polybrominated Diphenyl Ether Metabolites Using NanoESI-TIMS-MS. Int. J. Ion Mobil. Spectrom 2016, 19, 69–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (451).Liu FC; Cropley TC; Ridgeway ME; Park MA; Bleiholder C Structural Analysis of the Glycoprotein Complex Avidin by Tandem-Trapped Ion Mobility Spectrometry-Mass Spectrometry (Tandem-TIMS/MS). Anal. Chem 2020, 92, 4459–4467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (452).Ibrahim YM; Garimella SVB; Prost SA; Wojcik R; Norheim RV; Baker ES; Rusyn I; Smith RD Development of an Ion Mobility Spectrometry-Orbitrap Mass Spectrometer Platform. Anal. Chem 2016, 88, 12152–12160. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (453).Poltash ML; McCabe JW; Patrick JW; Laganowsky A; Russell DH Development and Evaluation of a Reverse-Entry Ion Source Orbitrap Mass Spectrometer. J. Am. Soc. Mass Spectrom 2019, 30, 192–198. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (454).Poltash ML; McCabe JW; Shirzadeh M; Laganowsky A; Clowers BH; Russell DH Fourier Transform-Ion Mobility-Orbitrap Mass Spectrometer: A Next-Generation Instrument for Native Mass Spectrometry. Anal. Chem 2018, 90, 10472–10478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (455).Poltash ML; McCabe JW; Shirzadeh M; Laganowsky A; Russell DH Native IM-Orbitrap MS: Resolving What Was Hidden. Trends Anal. Chem 2020, 124, 115533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (456).Knorr FJ; Eatherton RL; Siems WF; Hill HH Fourier Transform Ion Mobility Spectrometry. Anal. Chem 1985, 57, 402–406. [DOI] [PubMed] [Google Scholar]
- (457).Morrison KA; Siems WF; Clowers BH Augmenting Ion Trap Mass Spectrometers Using a Frequency Modulated Drift Tube Ion Mobility Spectrometer. Anal. Chem 2016, 88, 3121–3129. [DOI] [PubMed] [Google Scholar]
- (458).Clowers BH; Siems WF; Yu Z; Davis AL A Two-Phase Approach to Fourier Transform Ion Mobility Time-of-Flight Mass Spectrometry. Analyst 2015, 140, 6862–6870. [DOI] [PubMed] [Google Scholar]
- (459).Davis AL; Reinecke T; Morrison KA; Clowers BH Optimized Reconstruction Techniques for Multiplexed Dual-Gate Ion Mobility Mass Spectrometry Experiments. Anal. Chem 2019, 91, 1432–1440. [DOI] [PubMed] [Google Scholar]
- (460).Lyu J; Liu Y; McCabe JW; Schrecke S; Fang L; Russell DH; Laganowsky A Discovery of Potent Charge-Reducing Molecules for Native Ion Mobility Mass Spectrometry Studies. Anal. Chem 2020, 92, 11242–11249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (461).Poltash ML; Shirzadeh M; McCabe JW; Moghadamchargari Z; Laganowsky A; Russell DH New Insights into the Metal-Induced Oxidative Degradation Pathways of Transthyretin. Chem. Commun 2019, 55, 4091–4094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (462).Davis AL; Liu W; Siems WF; Clowers BH Correlation Ion Mobility Spectrometry. Analyst 2017, 142, 292–301. [DOI] [PubMed] [Google Scholar]
- (463).Deng L; Webb IK; Garimella SVB; Hamid AM; Zheng X; Norheim RV; Prost SA; Anderson GA; Sandoval JA; Baker ES, et al. Serpentine Ultralong Path with Extended Routing (Super) High Resolution Traveling Wave Ion Mobility-MS Using Structures for Lossless Ion Manipulations. Anal. Chem 2017, 89, 4628–4634. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (464).Webb IK; Garimella SVB; Tolmachev AV; Chen T-C; Zhang X; Norheim RV; Prost SA; LaMarche B; Anderson GA; Ibrahim YM, et al. Experimental Evaluation and Optimization of Structures for Lossless Ion Manipulations for Ion Mobility Spectrometry with Time-of-Flight Mass Spectrometry. Anal. Chem 2014, 86, 9169–9176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (465).Hollerbach AL; Conant CR; Nagy G; Monroe ME; Gupta K; Donor M; Giberson CM; Garimella SVB; Smith RD; Ibrahim YM Dynamic Time-Warping Correction for Shifts in Ultrahigh Resolving Power Ion Mobility Spectrometry and Structures for Lossless Ion Manipulations. J. Am. Soc. Mass Spectrom 2021, 32, 996–1007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (466).Allen SJ; Eaton RM; Bush MF Analysis of Native-Like Ions Using Structures for Lossless Ion Manipulations. Anal. Chem 2016, 88, 9118–9126. [DOI] [PubMed] [Google Scholar]
- (467).Herron WJ; Goeringer DE; McLuckey SA Product Ion Charge State Determination Via Ion/Ion Proton Transfer Reactions. Anal. Chem 1996, 68, 257–262. [DOI] [PubMed] [Google Scholar]
- (468).McLuckey SA; Goeringer DE Ion/Molecule Reactions for Improved Effective Mass Resolution in Electrospray Mass Spectrometry. Anal. Chem 1995, 67, 2493–2497. [DOI] [PubMed] [Google Scholar]
- (469).Stephenson JL Jr; McLuckey SA Charge Manipulation for Improved Mass Determination of High-Mass Species and Mixture Components by Electrospray Mass Spectrometry. J. Mass Spectrom 1998, 33, 664–672. [DOI] [PubMed] [Google Scholar]
- (470).Catalina MI; van den Heuvel RHH; van Duijn E; Heck AJR Decharging of Globular Proteins and Protein Complexes in Electrospray. Chem. Eur. J 2005, 11, 960–968. [DOI] [PubMed] [Google Scholar]
- (471).Iavarone AT; Jurchen JC; Williams ER Supercharged Protein and Peptide Ions Formed by Electrospray Ionization. Anal. Chem 2001, 73, 1455–1460. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (472).Cheng X; Gale DC; Udseth HR; Smith RD Charge State Reduction of Oligonucleotide Negative Ions from Electrospray Ionization. Anal. Chem 1995, 67, 586–593. [Google Scholar]
- (473).Ebeling DD; Westphall MS; Scalf M; Smith LM Corona Discharge in Charge Reduction Electrospray Mass Spectrometry. Anal. Chem 2000, 72, 5158–5161. [DOI] [PubMed] [Google Scholar]
- (474).Frey BL; Lin Y; Westphall MS; Smith LM Controlling Gas-Phase Reactions for Efficient Charge Reduction Electrospray Mass Spectrometry of Intact Proteins. J. Am. Soc. Mass Spectrom 2005, 16, 1876–1887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (475).Scalf M; Westphall MS; Krause J; Kaufman SL; Smith LM Controlling Charge States of Large Ions. Science 1999, 283, 194–197. [DOI] [PubMed] [Google Scholar]
- (476).Scalf M; Westphall MS; Smith LM Charge Reduction Electrospray Mass Spectrometry. Anal. Chem 2000, 72, 52–60. [DOI] [PubMed] [Google Scholar]
- (477).Campuzano IDG; Schnier PD Coupling Electrospray Corona Discharge, Charge Reduction and Ion Mobility Mass Spectrometry: From Peptides to Large Macromolecular Protein Complexes. Int. J. Ion Mobil. Spectrom 2013, 16, 51–60. [Google Scholar]
- (478).Schnier PD; Gross DS; Williams ER On the Maximum Charge State and Proton Transfer Reactivity of Peptide and Protein Ions Formed by Electrospray Ionization. J. Am. Soc. Mass Spectrom 1995, 6, 1086–1097. [DOI] [PubMed] [Google Scholar]
- (479).Sun J; Kitova EN; Klassen JS Method for Stabilizing Protein-Ligand Complexes in Nanoelectrospray Ionization Mass Spectrometry. Anal. Chem 2007, 79, 416–425. [DOI] [PubMed] [Google Scholar]
- (480).Lomeli SH; Peng IX; Yin S; Ogorzalek Loo RR; Loo JA New Reagents for Increasing ESI Multiple Charging of Proteins and Protein Complexes. J. Am. Soc. Mass Spectrom 2010, 21, 127–131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (481).Lomeli SH; Yin S; Ogorzalek Loo RR; Loo JA Increasing Charge While Preserving Noncovalent Protein Complexes for ESI-MS. J. Am. Soc. Mass Spectrom 2009, 20, 593–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (482).Smith LM Is Charge Reduction in ESI Really Necessary? J. Am. Soc. Mass Spectrom 2008, 19, 629–631. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (483).Pan P; McLuckey SA The Effect of Small Cations on the Positive Electrospray Responses of Proteins at Low pH. Anal. Chem 2003, 75, 5468–5474. [DOI] [PubMed] [Google Scholar]
- (484).Sterling HJ; Kintzer AF; Feld GK; Cassou CA; Krantz BA; Williams ER Supercharging Protein Complexes from Aqueous Solution Disrupts Their Native Conformations. J. Am. Soc. Mass Spectrom 2012, 23, 191–200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (485).Polfer NC Supercharging Proteins: How Many Charges Can a Protein Carry? Angew. Chem. Int. Ed 2017, 56, 8335–8337. [DOI] [PubMed] [Google Scholar]
- (486).Abaye DA; Agbo IA; Nielsen BV Current Perspectives on Supercharging Reagents in Electrospray Ionization Mass Spectrometry. RSC Adv. 2021, 11, 20355–20369. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (487).Cassou CA; Williams ER Desalting Protein Ions in Native Mass Spectrometry Using Supercharging Reagents. Analyst 2014, 139, 4810–4819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (488).Going CC; Williams ER Supercharging with m-Nitrobenzyl Alcohol and Propylene Carbonate: Forming Highly Charged Ions with Extended, Near-Linear Conformations. Anal. Chem 2015, 87, 3973–3980. [DOI] [PubMed] [Google Scholar]
- (489).Iavarone AT; Williams ER Mechanism of Charging and Supercharging Molecules in Electrospray Ionization. J. Am. Chem. Soc 2003, 125, 2319–2327. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (490).Sterling HJ; Cassou CA; Susa AC; Williams ER Electrothermal Supercharging of Proteins in Native Electrospray Ionization. Anal. Chem 2012, 84, 3795–3801. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (491).Sterling HJ; Cassou CA; Trnka MJ; Burlingame AL; Krantz BA; Williams ER The Role of Conformational Flexibility on Protein Supercharging in Native Electrospray Ionization. Phys. Chem. Chem. Phys 2011, 13, 18288–18296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (492).Sterling HJ; Daly MP; Feld GK; Thoren KL; Kintzer AF; Krantz BA; Williams ER Effects of Supercharging Reagents on Noncovalent Complex Structure in Electrospray Ionization from Aqueous Solutions. J. Am. Soc. Mass Spectrom 2010, 21, 1762–1774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (493).Sterling HJ; Prell JS; Cassou CA; Williams ER Protein Conformation and Supercharging with DMSO from Aqueous Solution. J. Am. Soc. Mass Spectrom 2011, 22, 1178–1186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (494).Sterling HJ; Williams ER Origin of Supercharging in Electrospray Ionization of Noncovalent Complexes from Aqueous Solution. J. Am. Soc. Mass Spectrom 2009, 20, 1933–1943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (495).Donor MT; Ewing SA; Zenaidee MA; Donald WA; Prell JS Extended Protein Ions Are Formed by the Chain Ejection Model in Chemical Supercharging Electrospray Ionization. Anal. Chem 2017, 89, 5107–5114. [DOI] [PubMed] [Google Scholar]
- (496).Zenaidee MA; Donald WA Extremely Supercharged Proteins in Mass Spectrometry: Profiling the pH of Electrospray Generated Droplets, Narrowing Charge State Distributions, and Increasing Ion Fragmentation. Analyst 2015, 140, 1894–1905. [DOI] [PubMed] [Google Scholar]
- (497).Zhang JD; Donor MT; Rolland AD; Leeming MG; Wang H; Trevitt AJ; Kabir KMM; Prell JS; Donald WA Protonation Isomers of Highly Charged Protein Ions Can Be Separated in FAIMS-MS. Int. J. Mass Spectrom 2020, 457, 116425. [Google Scholar]
- (498).Teo CA; Donald WA Solution Additives for Supercharging Proteins Beyond the Theoretical Maximum Proton-Transfer Limit in Electrospray Ionization Mass Spectrometry. Anal. Chem 2014, 86, 4455–4462. [DOI] [PubMed] [Google Scholar]
- (499).Zenaidee MA; Leeming MG; Zhang F; Funston TT; Donald WA Highly Charged Protein Ions: The Strongest Organic Acids to Date. Angew. Chem. Int. Ed 2017, 56, 8522–8526. [DOI] [PubMed] [Google Scholar]
- (500).Going CC; Xia Z; Williams ER New Supercharging Reagents Produce Highly Charged Protein Ions in Native Mass Spectrometry. Analyst 2015, 140, 7184–7194. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (501).Sterling HJ; Williams ER Real-Time Hydrogen/Deuterium Exchange Kinetics Via Supercharged Electrospray Ionization Tandem Mass Spectrometry. Anal. Chem 2010, 82, 9050–9057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (502).Yin S; Loo JA Top-Down Mass Spectrometry of Supercharged Native Protein–Ligand Complexes. Int. J. Mass Spectrom 2011, 300, 118–122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (503).Zhang J; Ogorzalek Loo RR; Loo JA Increasing Fragmentation of Disulfide-Bonded Proteins for Top-Down Mass Spectrometry by Supercharging. Int. J. Mass Spectrom 2015, 377, 546–556. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (504).Stiving AQ; Jones BJ; Ujma J; Giles K; Wysocki VH Collision Cross Sections of Charge-Reduced Proteins and Protein Complexes: A Database for Collision Cross Section Calibration. Anal. Chem 2020, 92, 4475–4483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (505).Dyachenko A; Gruber R; Shimon L; Horovitz A; Sharon M Allosteric Mechanisms Can Be Distinguished Using Structural Mass Spectrometry. Proc. Natl. Acad. Sci. U. S. A 2013, 110, 7235–7239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (506).Lemaire D; Marie G; Serani L; Laprevote O Stabilization of Gas-Phase Noncovalent Macromolecular Complexes in Electrospray Mass Spectrometry Using Aqueous Triethylammonium Bicarbonate Buffer. Anal. Chem 2001, 73, 1699–1706. [DOI] [PubMed] [Google Scholar]
- (507).Verkerk UH; Peschke M; Kebarle P Effect of Buffer Cations and of H3O+ on the Charge States of Native Proteins. Significance to Determinations of Stability Constants of Protein Complexes. J. Mass Spectrom 2003, 38, 618–631. [DOI] [PubMed] [Google Scholar]
- (508).Pacholarz KJ; Barran PE Use of a Charge Reducing Agent to Enable Intact Mass Analysis of Cysteine-Linked Antibody-Drug-Conjugates by Native Mass Spectrometry. EuPA Open Proteom. 2016, 11, 23–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (509).Patrick JW; Laganowsky A Generation of Charge-Reduced Ions of Membrane Protein Complexes for Native Ion Mobility Mass Spectrometry Studies. J. Am. Soc. Mass Spectrom 2019, 30, 886–892. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (510).Harvey SR; Liu Y; Liu W; Wysocki VH; Laganowsky A Surface Induced Dissociation as a Tool to Study Membrane Protein Complexes. Chem. Commun 2017, 53, 3106–3109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (511).Zhou M; Dagan S; Wysocki VH Impact of Charge State on Gas-Phase Behaviors of Noncovalent Protein Complexes in Collision Induced Dissociation and Surface Induced Dissociation. Analyst 2013, 138, 1353–1362. [DOI] [PubMed] [Google Scholar]
- (512).Harvey SR; VanAernum ZL; Wysocki VH Surface-Induced Dissociation of Anionic vs Cationic Native-Like Protein Complexes. J. Am. Chem. Soc 2021, 143, 7698–7706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (513).Hall Z; Politis A; Bush MF; Smith LJ; Robinson CV Charge-State Dependent Compaction and Dissociation of Protein Complexes: Insights from Ion Mobility and Molecular Dynamics. J. Am. Chem. Soc 2012, 134, 3429–3438. [DOI] [PubMed] [Google Scholar]
- (514).Foreman DJ; McLuckey SA Recent Developments in Gas-Phase Ion/Ion Reactions for Analytical Mass Spectrometry. Anal. Chem 2020, 92, 252–266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (515).Laszlo KJ; Bush MF Analysis of Native-Like Proteins and Protein Complexes Using Cation to Anion Proton Transfer Reactions (CAPTR). J. Am. Soc. Mass Spectrom 2015, 26, 2152–2161. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (516).Laszlo KJ; Munger EB; Bush MF Folding of Protein Ions in the Gas Phase after Cation-to-Anion Proton-Transfer Reactions. J. Am. Chem. Soc 2016, 138, 9581–9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (517).Jhingree JR; Beveridge R; Dickinson ER; Williams JP; Brown JM; Bellina B; Barran PE Electron Transfer with No Dissociation Ion Mobility–Mass Spectrometry (ETnoD IM-MS). The Effect of Charge Reduction on Protein Conformation. Int. J. Mass Spectrom 2017, 413, 43–51. [Google Scholar]
- (518).Lermyte F; Łącki MK; Valkenborg D; Gambin A; Sobott F Conformational Space and Stability of ETD Charge Reduction Products of Ubiquitin. J. Am. Soc. Mass Spectrom 2017, 28, 69–76. [DOI] [PubMed] [Google Scholar]
- (519).Abzalimov RR; Kaltashov IA Electrospray Ionization Mass Spectrometry of Highly Heterogeneous Protein Systems: Protein Ion Charge State Assignment Via Incomplete Charge Reduction. Anal. Chem 2010, 82, 7523–7526. [DOI] [PubMed] [Google Scholar]
- (520).Yang Y; Niu C; Bobst CE; Kaltashov IA Charge Manipulation Using Solution and Gas-Phase Chemistry to Facilitate Analysis of Highly Heterogeneous Protein Complexes in Native Mass Spectrometry. Anal. Chem 2021, 93, 3337–3342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (521).Pitts-McCoy AM; Harrilal CP; McLuckey SA Gas-Phase Ion/Ion Chemistry as a Probe for the Presence of Carboxylate Groups in Polypeptide Cations. J. Am. Soc. Mass Spectrom 2019, 30, 329–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (522).Abdillahi AM; Lee KW; McLuckey SA Mass Analysis of Macro-Molecular Analytes Via Multiply-Charged Ion Attachment. Anal. Chem 2020, 92, 16301–16306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (523).Huguet R; Mullen C; Srzentić K; Greer JB; Fellers RT; Zabrouskov V; Syka JEP; Kelleher NL; Fornelli L Proton Transfer Charge Reduction Enables High-Throughput Top-Down Analysis of Large Proteoforms. Anal. Chem 2019, 91, 15732–15739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (524).Zhang G; Keener JE; Marty MT Measuring Remodeling of the Lipid Environment Surrounding Membrane Proteins with Lipid Exchange and Native Mass Spectrometry. Anal. Chem 2020, 92, 5666–5669. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (525).Kostelic MM; Zak CK; Jayasekera H; Marty MT Assembly of Model Membrane Nanodiscs for Native Mass Spectrometry. Anal. Chem 2021, 93, 5972–5979. [DOI] [PubMed] [Google Scholar]
- (526).Weidner SM; Trimpin S Mass Spectrometry of Synthetic Polymers. Anal. Chem 2008, 80, 4349–4361. [DOI] [PubMed] [Google Scholar]
- (527).Prebyl BS; Johnson JD; Cook KD Calibration for Determining Monomer Ratios in Copolymers by Electrospray Ionization Mass Spectrometry. Int. J. Mass Spectrom 2004, 238, 207–214. [Google Scholar]
- (528).Prebyl BS; Johnson JD; Tuinman AA; Zhou SL; Cook KD Qualitative Assessment of Monomer Ratios in Putative Ionic Terpolymer Samples by Electrospray Ionization Mass Spectrometry with Collision-Induced Dissociation. J. Am. Soc. Mass Spectrom 2002, 13, 921–927. [DOI] [PubMed] [Google Scholar]
- (529).Denisov IG; Grinkova YV; Lazarides AA; Sligar SG Directed Self-Assembly of Monodisperse Phospholipid Bilayer Nanodiscs with Controlled Size. J. Am. Chem. Soc 2004, 126, 3477–3487. [DOI] [PubMed] [Google Scholar]
- (530).Denisov IG; Sligar SG Nanodiscs in Membrane Biochemistry and Biophysics. Chem. Rev 2017, 117, 4669–4713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (531).Lai G; Forti KM; Renthal R Kinetics of Lipid Mixing between Bicelles and Nanolipoprotein Particles. Biophys. Chem 2015, 197, 47–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (532).Nakano M; Fukuda M; Kudo T; Miyazaki M; Wada Y; Matsuzaki N; Endo H; Handa T Static and Dynamic Properties of Phospholipid Bilayer Nanodiscs. J. Am. Chem. Soc 2009, 131, 8308–8312. [DOI] [PubMed] [Google Scholar]
- (533).Prell JS Modelling Collisional Cross Sections. In Advances in Ion Mobility-Mass Spectrometry: Fundamentals, Instrumentation, and Applications, Comprehensive Analytical Chemistry: Vol. 83; Donald WA; Prell JS, Eds.; Elsevier, 2019; pp 1–22. [Google Scholar]
- (534).Zheng X; Aly NA; Zhou Y; Dupuis KT; Bilbao A; Paurus Vanessa L.; Orton DJ; Wilson R; Payne SH; Smith RD, et al. A Structural Examination and Collision Cross Section Database for over 500 Metabolites and Xenobiotics Using Drift Tube Ion Mobility Spectrometry. Chem. Sci 2017, 8, 7724–7736. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (535).Zhou Z; Luo M; Chen X; Yin Y; Xiong X; Wang R; Zhu Z-J Ion Mobility Collision Cross-Section Atlas for Known and Unknown Metabolite Annotation in Untargeted Metabolomics. Nat. Commun 2020, 11, 4334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (536).Ross DH; Cho JH; Xu L Breaking Down Structural Diversity for Comprehensive Prediction of Ion-Neutral Collision Cross Sections. Anal. Chem 2020, 92, 4548–4557. [DOI] [PubMed] [Google Scholar]
- (537).May JC; Morris CB; McLean JA Ion Mobility Collision Cross Section Compendium. Anal. Chem 2017, 89, 1032–1044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (538).Picache JA; Rose BS; Balinski A; Leaptrot Katrina L.; Sherrod SD; May JC; McLean JA Collision Cross Section Compendium to Annotate and Predict Multi-Omic Compound Identities. Chem. Sci 2019, 10, 983–993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (539).Hines KM; Ross DH; Davidson KL; Bush MF; Xu L Large-Scale Structural Characterization of Drug and Drug-Like Compounds by High-Throughput Ion Mobility-Mass Spectrometry. Anal. Chem 2017, 89, 9023–9030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (540).Hines KM; Herron J; Xu L Assessment of Altered Lipid Homeostasis by HILIC-Ion Mobility-Mass Spectrometry-Based Lipidomics. J. Lipid Res 2017, 58, 809–819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (541).Ben-Nissan G; Belov ME; Morgenstern D; Levin Y; Dym O; Arkind G; Lipson C; Makarov AA; Sharon M Triple-Stage Mass Spectrometry Unravels the Heterogeneity of an Endogenous Protein Complex. Anal. Chem 2017, 89, 4708–4715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (542).Tolić LP; Anderson GA; Smith RD; Brothers HM; Spindler R; Tomalia DA Electrospray Ionization Fourier Transform Ion Cyclotron Resonance Mass Spectrometric Characterization of High Molecular Mass Starburst™ Dendrimers. Int. J. Mass Spectrom. Ion Processes 1997, 165, 405–418. [Google Scholar]
- (543).Fernandez de la Mora J Electrospray Ionization of Large Multiply Charged Species Proceeds Via Dole’s Charged Residue Mechanism. Anal. Chim. Acta 2000, 406, 93–104. [Google Scholar]
- (544).Nesatyy VJ; Suter MJF On the Conformation-Dependent Neutralization Theory and Charging of Individual Proteins and Their Non-Covalent Complexes in the Gas Phase. J. Mass Spectrom 2004, 39, 93–97. [DOI] [PubMed] [Google Scholar]
- (545).Kaltashov IA; Mohimen A Estimates of Protein Surface Areas in Solution by Electrospray Ionization Mass Spectrometry. Anal. Chem 2005, 77, 5370–5379. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (546).Testa L; Brocca S; Grandori R Charge-Surface Correlation in Electrospray Ionization of Folded and Unfolded Proteins. Anal. Chem 2011, 83, 6459–6463. [DOI] [PubMed] [Google Scholar]
- (547).Testa L; Brocca S; Santambrogio C; D’Urzo A; Habchi J; Longhi S; Uversky VN; Grandori R Extracting Structural Information from Charge-State Distributions of Intrinsically Disordered Proteins by Non-Denaturing Electrospray-Ionization Mass Spectrometry. Intrinsically Disord. Proteins 2013, 1, e25068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (548).Natalello A; Santambrogio C; Grandori R Are Charge-State Distributions a Reliable Tool Describing Molecular Ensembles of Intrinsically Disordered Proteins by Native MS? J. Am. Soc. Mass Spectrom 2017, 28, 21–28. [DOI] [PubMed] [Google Scholar]
- (549).Marklund EG; Degiacomi MT; Robinson CV; Baldwin AJ; Benesch JLP Collision Cross Sections for Structural Proteomics. Structure 2015, 23, 791–799. [DOI] [PubMed] [Google Scholar]
- (550).Ruotolo BT; Benesch JLP; Sandercock AM; Hyung S-J; Robinson CV Ion Mobility-Mass Spectrometry Analysis of Large Protein Complexes. Nat. Protoc 2008, 3, 1139–1152. [DOI] [PubMed] [Google Scholar]
- (551).Dole M; Mack LL; Hines RL; Mobley RC; Ferguson LD; Alice MB Molecular Beams of Macroions. J. Chem. Phys 1968, 49, 2240–2249. [Google Scholar]
- (552).Santambrogio C; Natalello A; Brocca S; Ponzini E; Grandori R Conformational Characterization and Classification of Intrinsically Disordered Proteins by Native Mass Spectrometry and Charge-State Distribution Analysis. Proteomics 2019, 19, 1800060. [DOI] [PubMed] [Google Scholar]
- (553).Ilag LL; Videler H; McKay AR; Sobott F; Fucini P; Nierhaus KH; Robinson CV Heptameric (L12)6/L10 Rather Than Canonical Pentameric Complexes Are Found by Tandem MS of Intact Ribosomes from Thermophilic Bacteria. Proc. Natl. Acad. Sci. U. S. A 2005, 102, 8192–8197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (554).Reardon PN; Jara KA; Rolland AD; Smith DA; Hoang HTM; Prell JS; Barbar EJ The Dynein Light Chain 8 (LC8) Binds Predominantly "In-Register" to a Multivalent Intrinsically Disordered Partner. J. Biol. Chem 2020, 295, 4912–4922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (555).Xia Z; DeGrandchamp JB; Williams ER Native Mass Spectrometry Beyond Ammonium Acetate: Effects of Nonvolatile Salts on Protein Stability and Structure. Analyst 2019, 144, 2565–2573. [DOI] [PubMed] [Google Scholar]
- (556).Susa AC; Lippens JL; Xia Z; Loo JA; Campuzano IDG; Williams ER Submicrometer Emitter ESI Tips for Native Mass Spectrometry of Membrane Proteins in Ionic and Nonionic Detergents. J. Am. Soc. Mass Spectrom 2018, 29, 203–206. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (557).Panczyk EM; Gilbert JD; Jagdale GS; Stiving AQ; Baker LA; Wysocki VH Ion Mobility and Surface Collisions: Submicrometer Capillaries Can Produce Native-Like Protein Complexes. Anal. Chem 2020, 92, 2460–2467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (558).Nguyen GTH; Tran TN; Podgorski MN; Bell SG; Supuran CT; Donald WA Nanoscale Ion Emitters in Native Mass Spectrometry for Measuring Ligand–Protein Binding Affinities. ACS Cent. Sci 2019, 5, 308–318. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (559).McKay AR; Ruotolo BT; Ilag LL; Robinson CV Mass Measurements of Increased Accuracy Resolve Heterogeneous Populations of Intact Ribosomes. J. Am. Chem. Soc 2006, 128, 11433–11442. [DOI] [PubMed] [Google Scholar]
- (560).Cunniff JB; Vouros P Mass and Charge State Assignment for Proteins and Peptide Mixtures Via Noncovalent Adduction in Electrospray Mass Spectrometry. J. Am. Soc. Mass Spectrom 1995, 6, 1175–1182. [DOI] [PubMed] [Google Scholar]
- (561).Liepold L; Oltrogge LM; Suci PA; Young MJ; Douglas T Correct Charge State Assignment of Native Electrospray Spectra of Protein Complexes. J. Am. Soc. Mass Spectrom 2009, 20, 435–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (562).Maleknia SD; Downard KM Charge Ratio Analysis Method: Approach for the Deconvolution of Electrospray Mass Spectra. Anal. Chem 2005, 77, 111–119. [DOI] [PubMed] [Google Scholar]
- (563).Almudaris A; Ashton DS; Beddell CR; Cooper DJ; Craig SJ; Oliver RWA The Assignment of Charge States in Complex Electrospray Mass Spectra. Eur. J. Mass Spectrom 1996, 2, 57–67. [Google Scholar]
- (564).Tabb DL; Shah MB; Strader MB; Connelly HM; Hettich RL; Hurst GB Determination of Peptide and Protein Ion Charge States by Fourier Transformation of Isotope-Resolved Mass Spectra. J. Am. Soc. Mass Spectrom 2006, 17, 903–915. [DOI] [PubMed] [Google Scholar]
- (565).Winkler R Esiprot: A Universal Tool for Charge State Determination and Molecular Weight Calculation of Proteins from Electrospray Ionization Mass Spectrometry Data. Rapid Commun. Mass Spectrom 2010, 24, 285–294. [DOI] [PubMed] [Google Scholar]
- (566).Smith R; Mathis AD; Ventura D; Prince JT Proteomics, Lipidomics, Metabolomics: A Mass Spectrometry Tutorial from a Computer Scientist's Point of View. BMC Bioinform. 2014, 15, S9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (567).Chouinard CD; Nagy G; Smith RD; Baker ES Ion Mobility-Mass Spectrometry in Metabolomic, Lipidomic, and Proteomic Analyses. In Advances in Ion Mobility-Mass Spectrometry: Fundamentals, Instrumentation, and Applications, Comprehensive Analytical Chemistry: Vol. 83; Donald WA; Prell JS, Eds.; Elsevier, 2019; pp 123–159. [Google Scholar]
- (568).Konermann L Addressing a Common Misconception: Ammonium Acetate as Neutral pH “Buffer” for Native Electrospray Mass Spectrometry. J. Am. Soc. Mass Spectrom 2017, 28, 1827–1835. [DOI] [PubMed] [Google Scholar]
- (569).Bolla JR; Corey RA; Sahin C; Gault J; Hummer A; Hopper JTS; Lane DP; Drew D; Allison TM; Stansfeld PJ, et al. A Mass-Spectrometry-Based Approach to Distinguish Annular and Specific Lipid Binding to Membrane Proteins. Angew. Chem. Int. Edit 2020, 59, 3523–3528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (570).Urner LH; Liko I; Yen H-Y; Hoi K-K; Bolla JR; Gault J; Almeida FG; Schweder M-P; Shutin D; Ehrmann S, et al. Modular Detergents Tailor the Purification and Structural Analysis of Membrane Proteins Including G-Protein Coupled Receptors. Nat. Commun 2020, 11, 564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (571).Kundlacz T; Bender J; Schmidt C Effects of Non-Ionic and Zwitterionic Detergents on Soluble Proteins During Native Mass Spectrometry Experiments. Int. J. Mass Spectrom 2021, 468, 116652. [Google Scholar]
- (572).Han L; Xue X; Roy R; Kitova EN; Zheng RB; St-Pierre Y; Lowary TL; Klassen JS Neoglycolipids as Glycosphingolipid Surrogates for Protein Binding Studies Using Nanodiscs and Native Mass Spectrometry. Anal. Chem 2020, 92, 14189–14196. [DOI] [PubMed] [Google Scholar]
- (573).Hoi KK; Bada Juarez JF; Judge PJ; Yen H-Y; Wu D; Vinals J; Taylor GF; Watts A; Robinson CV Detergent-Free Lipodisq Nanoparticles Facilitate High-Resolution Mass Spectrometry of Folded Integral Membrane Proteins. Nano Letters 2021, 21, 2824–2831. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (574).Landreh M; Costeira-Paulo J; Gault J; Marklund EG; Robinson CV Effects of Detergent Micelles on Lipid Binding to Proteins in Electrospray Ionization Mass Spectrometry. Anal. Chem 2017, 89, 7425–7430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (575).Landreh M; Marty MT; Gault J; Robinson CV A Sliding Selectivity Scale for Lipid Binding to Membrane Proteins. Curr. Opin. Struct. Biol 2016, 39, 54–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (576).Benesch JLP; Ruotolo BT; Simmons DA; Barrera NP; Morgner N; Wang L; Saibil HR; Robinson CV Separating and Visualising Protein Assemblies by Means of Preparative Mass Spectrometry and Microscopy. J. Struct. Biol 2010, 172, 161–168. [DOI] [PubMed] [Google Scholar]
- (577).Jore MM; Lundgren M; van Duijn E; Bultema JB; Westra ER; Waghmare SP; Wiedenheft B; Pul Ü; Wurm R; Wagner R, et al. Structural Basis for CRISPR RNA-Guided DNA Recognition by Cascade. Nat. Struct. Mol. Biol 2011, 18, 529–536. [DOI] [PubMed] [Google Scholar]
- (578).Olinares PDB; Kang JY; Llewellyn E; Chiu C; Chen J; Malone B; Saecker RM; Campbell EA; Darst SA; Chait BT Native Mass Spectrometry-Based Screening for Optimal Sample Preparation in Single-Particle Cryo-EM. Structure 2021, 29, 186–195.e186. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (579).Su C-C; Lyu M; Morgan CE; Bolla JR; Robinson CV; Yu EWA ‘Build and Retrieve’ Methodology to Simultaneously Solve Cryo-EM Structures of Membrane Proteins. Nat. Methods 2021, 18, 69–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (580).Chen S; Wu D; Robinson CV; Struwe WB Native Mass Spectrometry Meets Glycomics: Resolving Structural Detail and Occupancy of Glycans on Intact Glycoproteins. Anal. Chem 2021. [DOI] [PubMed] [Google Scholar]
- (581).Wu D; Robinson CV Connecting ‘Multi-Omics’ Approaches to Endogenous Protein Complexes. Trends Chem. 2021, 3, 445–455. [Google Scholar]
- (582).Chorev DS; Baker LA; Wu D; Beilsten-Edmands V; Rouse SL; Zeev-Ben-Mordehai T; Jiko C; Samsudin F; Gerle C; Khalid S, et al. Protein Assemblies Ejected Directly from Native Membranes Yield Complexes for Mass Spectrometry. Science 2018, 362, 829–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (583).Yin Z; Huang J; Miao H; Hu O; Li H High-Pressure Electrospray Ionization Yields Supercharged Protein Complexes from Native Solutions While Preserving Noncovalent Interactions. Anal. Chem 2020, 92, 12312–12321. [DOI] [PubMed] [Google Scholar]
- (584).Saikusa K; Kato D; Nagadoi A; Kurumizaka H; Akashi S Native Mass Spectrometry of Protein and DNA Complexes Prepared in Nonvolatile Buffers. J. Am. Soc. Mass Spectrom 2020, 31, 711–718. [DOI] [PubMed] [Google Scholar]
- (585).Sakamoto W; Azegami N; Konuma T; Akashi S Single-Cell Native Mass Spectrometry of Human Erythrocytes. Anal. Chem 2021, 93, 6583–6588. [DOI] [PubMed] [Google Scholar]
- (586).Gan J; Ben-Nissan G; Arkind G; Tarnavsky M; Trudeau D; Noda Garcia L; Tawfik DS; Sharon M Native Mass Spectrometry of Recombinant Proteins from Crude Cell Lysates. Anal. Chem 2017, 89, 4398–4404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (587).Rogawski R; Rogel A; Bloch I; Gal M; Horovitz A; London N; Sharon M Intracellular Protein-Drug Interactions Probed by Direct Mass Spectrometry of Cell Lysates. Angew. Chem. Int. Ed 2021, DOI: 10.1002/anie.202104947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (588).Vimer S; Ben-Nissan G; Sharon M Direct Characterization of Overproduced Proteins by Native Mass Spectrometry. Nat. Protoc 2020, 15, 236–265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (589).Ben-Nissan G; Vimer S; Warszawski S; Katz A; Yona M; Unger T; Peleg Y; Morgenstern D; Cohen-Dvashi H; Diskin R, et al. Rapid Characterization of Secreted Recombinant Proteins by Native Mass Spectrometry. Commun. Biol 2018, 1, 213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (590).Takano K; Arai S; Sakamoto S; Ushijima H; Ikegami T; Saikusa K; Konuma T; Hamachi I; Akashi S Screening of Protein-Ligand Interactions under Crude Conditions by Native Mass Spectrometry. Anal. Bioanal. Chem 2020, 412, 4037–4043. [DOI] [PubMed] [Google Scholar]
- (591).Vimer S; Ben-Nissan G; Marty M; Fleishman SJ; Sharon M Direct-MS Analysis of Antibody-Antigen Complexes. Proteomics 2021, DOI: 10.1002/pmic.202000300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (592).Čaval T; Buettner A; Haberger M; Reusch D; Heck AJR Discrepancies between High-Resolution Native and Glycopeptide-Centric Mass Spectrometric Approaches: A Case Study into the Glycosylation of Erythropoietin Variants. J. Am. Soc. Mass Spectrom 2021, 32, 2099–2104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (593).Li H; Nguyen HH; Ogorzalek Loo RR; Campuzano IDG; Loo JA An Integrated Native Mass Spectrometry and Top-Down Proteomics Method That Connects Sequence to Structure and Function of Macromolecular Complexes. Nat. Chem 2018, 10, 139–148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (594).Tadeo X; López-Méndez B; Castaño D; Trigueros T; Millet O Protein Stabilization and the Hofmeister Effect: The Role of Hydrophobic Solvation. Biophys. J 2009, 97, 2595–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (595).Kunz W; Henle J; Ninham BW ‘Zur Lehre Von Der Wirkung Der Salze’ (About the Science of the Effect of Salts): Franz Hofmeister's Historical Papers. Curr. Opin. Colloid Interface Sci 2004, 9, 19–37. [Google Scholar]
- (596).National Resource for Native MS-Guided Structural Biology. https://nativems.osu.edu/ (accessed August 19, 2021).
- (597).Quetschlich D; Esser TK; Newport TD; Fiorentino F; Shutin D; Chen S; Davis R; Lovera S; Liko I; Stansfeld PJ, et al. Navia: A Program for the Visual Analysis of Complex Mass Spectra. Bioinformatics 2021, DOI: 10.1093/bioinformatics/btab1436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (598).Wu Z; Roberts DS; Melby JA; Wenger K; Wetzel M; Gu Y; Ramanathan SG; Bayne EF; Liu X; Sun R, et al. MASH Explorer: A Universal Software Environment for Top-Down Proteomics. J. Proteome Res 2020, 19, 3867–3876. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (599).Mortensen DN; Williams ER Theta-Glass Capillaries in Electrospray Ionization: Rapid Mixing and Short Droplet Lifetimes. Anal. Chem 2014, 86, 9315–9321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (600).Fisher CM; Kharlamova A; McLuckey SA Affecting Protein Charge State Distributions in Nano-Electrospray Ionization Via In-Spray Solution Mixing Using Theta Capillaries. Anal. Chem 2014, 86, 4581–4588. [DOI] [PubMed] [Google Scholar]
- (601).Wang H; Yong G; Brown SL; Lee HE; Zenaidee MA; Supuran CT; Donald WA Supercharging Protein Ions in Native Mass Spectrometry Using Theta Capillary Nanoelectrospray Ionization Mass Spectrometry and Cyclic Alkylcarbonates. Anal. Chim. Acta 2018, 1003, 1–9. [DOI] [PubMed] [Google Scholar]
- (602).Wilson JW; Donor MT; Shepherd SO; Prell JS Increasing Collisional Activation of Protein Complexes Using Smaller Aperture Source Sampling Cones on a Synapt Q-IM-TOF Instrument with a Stepwave Source. J. Am. Soc. Mass Spectrom 2020, 31, 1751–1754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (603).Laskin J; Futrell J On the Efficiency of Energy Transfer in Collisional Activation of Small Peptides. J. Chem. Phys 2002, 116, 4302–4310. [Google Scholar]
- (604).Ruotolo BT; Hyung S-J; Robinson PM; Giles K; Bateman RH; Robinson CV Ion Mobility–Mass Spectrometry Reveals Long-Lived, Unfolded Intermediates in the Dissociation of Protein Complexes. Angew. Chem. Int. Ed 2007, 46, 8001–8004. [DOI] [PubMed] [Google Scholar]
- (605).Hyung S-J; Robinson CV; Ruotolo BT Gas-Phase Unfolding and Disassembly Reveals Stability Differences in Ligand-Bound Multiprotein Complexes. Chem. Biol 2009, 16, 382–390. [DOI] [PubMed] [Google Scholar]
- (606).Hopper JTS; Oldham NJ Collision Induced Unfolding of Protein Ions in the Gas Phase Studied by Ion Mobility-Mass Spectrometry: The Effect of Ligand Binding on Conformational Stability. J. Am. Soc. Mass Spectrom 2009, 20, 1851–1858. [DOI] [PubMed] [Google Scholar]
- (607).Donor MT; Mroz AM; Prell JS Experimental and Theoretical Investigation of Overall Energy Deposition in Surface-Induced Unfolding of Protein Ions. Chem. Sci 2019, 10, 4097–4106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (608).Donor MT; Shepherd SO; Prell JS Rapid Determination of Activation Energies for Gas-Phase Protein Unfolding and Dissociation in a Q-IM-TOF Mass Spectrometer. J. Am. Soc. Mass Spectrom 2020, 31, 602–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (609).Cheung See Kit M; Shepherd SO; Prell JS; Webb IK Experimental Determination of Activation Energies for Covalent Bond Formation Via Ion/Ion Reactions and Competing Processes. J. Am. Soc. Mass Spectrom 2021, DOI: 10.1021/jasms.1021c00025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (610).Dixit SM; Polasky DA; Ruotolo BT Collision Induced Unfolding of Isolated Proteins in the Gas Phase: Past, Present, and Future. Curr. Opin. Chem. Biol 2018, 42, 93–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (611).Tian Y; Han L; Buckner AC; Ruotolo BT Collision Induced Unfolding of Intact Antibodies: Rapid Characterization of Disulfide Bonding Patterns, Glycosylation, and Structures. Anal. Chem 2015, 87, 11509–11515. [DOI] [PubMed] [Google Scholar]
- (612).Zhong Y; Han L; Ruotolo BT Collisional and Coulombic Unfolding of Gas-Phase Proteins: High Correlation to Their Domain Structures in Solution. Angew. Chem. Int. Ed 2014, 53, 9209–9212. [DOI] [PubMed] [Google Scholar]
- (613).Laszlo KJ; Bush MF Effects of Charge State, Charge Distribution, and Structure on the Ion Mobility of Protein Ions in Helium Gas: Results from Trajectory Method Calculations. J. Phys. Chem. A 2017, 121, 7768–7777. [DOI] [PubMed] [Google Scholar]
- (614).Konermann L; Rodriguez AD; Liu J On the Formation of Highly Charged Gaseous Ions from Unfolded Proteins by Electrospray Ionization. Anal. Chem 2012, 84, 6798–6804. [DOI] [PubMed] [Google Scholar]
- (615).Landreh M; Liko I; Uzdavinys P; Coincon M; Hopper JTS; Drew D; Robinson CV Controlling Release, Unfolding and Dissociation of Membrane Protein Complexes in the Gas Phase through Collisional Cooling. Chem. Commun 2015, 51, 15582–15584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (616).Silveira JA; Fort KL; Kim D; Servage KA; Pierson NA; Clemmer DE; Russell DH From Solution to the Gas Phase: Stepwise Dehydration and Kinetic Trapping of Substance P Reveals the Origin of Peptide Conformations. J. Am. Chem. Soc 2013, 135, 19147–19153. [DOI] [PubMed] [Google Scholar]