

Abstract
Cigarette smoke (CS) is known to cause impaired mitophagy and mitochondrial dysregulation in the pathogenesis of chronic obstructive pulmonary disease (COPD)/emphysema. Mitochondrial complexes and dynamics are affected by acute CS exposure in lung epithelium and mouse lung. We hypothesize that chronic CS exposure (4 months) will induce lung mitochondrial dysregulation and abnormal mitophagy. In this study, we employed the mitoQC reporter mice, a mitochondrial reporter strain, which can reflect the mitophagy based on the fluorescence-tagged mitochondria. Chronic CS exposure induced lung inflammatory cell infiltration, airspace enlargement, and lung cellular senescence. We showed the higher occurrence of mitophagy in the lung cells by CS exposure, associated with more mitochondrial fluorescence signals (GFP+/mCherry+). After chronic CS exposure, the mitochondrial complexes and function related genes were inhibited, while protein levels of complexes I and III slightly changed. Additionally, chronic CS exposure down-regulated most of the mitochondrial dynamic markers at gene expression level, included mitochondrial fusion/fission and mitochondrial translocate/transfer markers. For the markers related to mitophagy, Pink1 and Parkin, decreased gene and protein levels of Parkin, and decreased gene expression of Pink1, were identified in the CS exposure group. Hence, CS-induced mitophagy is mediated by Pink1-Parkin independent mechanism. Thus, we have shown that chronic CS exposure induced mitophagy, which is observed using a state-of-the art mitoQC reporter mice, as well as the dysregulated mitochondrial complexes and dynamics. Our results suggested that dysregulated mitochondrial function and dynamics are associated with CS-induced lung injury and phenotypic development of chronic lung diseases, such as COPD/ emphysema.
Keywords: Mitochondrial dysregulation, Mitophagy, Cigarette Smoke exposure, MitoQC reporter
Graphical Abstract
Introduction
Cigarette smoke (CS) is one of the main etiological risks of inducing lung diseases, such as chronic obstructive pulmonary disease (COPD), emphysema, asthma, interstitial lung disease, and lung cancer (Centers for Disease et al., 2010). COPD is one of the most common diseases developed by chronic exposure of CS with high morbidity and mortality, affecting more than 10% of the population globally (Blanco et al., 2019). It is well known that CS contains thousands of toxic chemicals responsible for lung inflammation, oxidative stress, and tissue damage (Bowler et al., 2004; Nyunoya et al., 2014; Patel et al., 2008). We and others, have shown that CS-induced lung injury was associated with mitochondrial dysfunction which manifested due to excessive generation of reactive oxygen species (ROS) in mitochondria, inhibition of mitochondrial potential, dysregulated mitophagy, and dysfunctional mitochondrial complexes (Banzet et al., 1999; Hara et al., 2013; Lerner et al., 2016; Sundar et al., 2019). However, the detailed mechanisms underlying CS-induced dysregulated mitochondrial dynamics, and mitochondrial quality control are still lacking.
Mitochondria are involved in cellular energy maintenance, regulation of oxidative stress, and cellular senescence (Ahmad et al., 2015; Jiang et al., 2017). Tissue injury due to mitochondrial dysfunction is usually associated with altered mitochondrial dynamics (mitochondrial fission and fusion) (Blackstone and Chang, 2011; Chauhan et al., 2014). Mitochondrial fusion, which is evident as elongated mitochondria inhibits mitochondrial degradation by allowing mitochondrial component interaction, thereby diminishing mitochondrial damage while also allowing consumption of less mitochondrial ATP and promoting cell survival. (Gomes et al., 2011; Youle and van der Bliek, 2012). Previous studies have shown elongated mitochondria in lung epithelia from smokers and fibroblasts from COPD patients (Ahmad et al., 2015; Hara et al., 2013; Hoffmann et al., 2013a). Eventually, the damaged mitochondria are removed via mitophagy, via autophagosomes followed by lysosomal degradation (Ashrafi and Schwarz, 2013; Zhu et al., 2013). The PTEN-induced kinase 1 (Pink1) - Parkin pathway plays an important role in mitophagy: stabilized Pink1 on mitochondrial outer membrane of damaged mitochondria recruits Parkin from the cytosol, which is then recognized by autophagosomes to initiate mitophagy (Ashrafi and Schwarz, 2013; Chen and Dorn, 2013; Scarffe et al., 2014; Zhu et al., 2013). While basal levels of mitophagy are observed even in Parkin knockout (KO) mice, demonstrating some Pink1-Parkin independent mitophagy (McWilliams et al., 2018), mitochondrial dysfunction and impaired mitophagy due to CS exposure is associated with the Pink1-Parkin pathway, leading to CS induced necroptosis and COPD pathogenesis (Ito et al., 2015; Mizumura et al., 2014). Hence, there is imperative to determine the mechanism underlying impaired mitophagy and dysregulated mitochondrial quality control in the pathophysiology of COPD. In this study, we determine the effects of chronic CS exposure (4 months) on mitophagy in the lungs using the mitoQC transgenic mice and provide a model to understand CS induced mitophagy and its effects in the pathogenesis of lung diseases.
We employed a transgenic mouse model: mitoQC mice, which contained fluorescent reporter on mitochondria that enable visualization of mitochondrial quality and the occurrence of mitophagy (McWilliams et al., 2016). The mitoQC reporter is a fused protein tag expressed by constuct knockin into the Rosa26 locus which allows the reporter system produced in all mammalian tissues (Soriano, 1999). We hypothesize that chronic CS exposure will induce mitophagy and dysregulated mitochondrial function. We tested the hypothesis by exposing the mitoQC mice to chronic CS for 4 months and determined the mechanism of mitophagy in a COPD mouse model.
Methods
1. Animal and Cigarette smoke exposure
MitoQC mice (C57BL/6 background) were obtained from Ian G. Ganley. Ph.D (University of Dundee, UK) which was generated by Taconic Artemis, and maintained at University of Rochester Medical Center, USA (Material Transfer Agreement on 17th April, 2018; C57BL/6-Gt(ROSA)26SorGanlH EM:11343). Genotype was confirmed using genomic DNA isolated from tails as mentioned in the previous publication (McWilliams et al., 2016). MitoQC mice (both male and female, 4–6 months old) were exposed to CS for 4 months, 5 days/week, with 2 hrs/day. The total particular matter (TPM, mg/m3) was around 300, using the Baumgartner Jaeger mainstream smoke machine. Air exposed control group mice were housed separately until sacrifice. Following 4-months of exposure, mice were sacrificed 24 hr after the last exposure, and lungs were either snap-frozen for protein/RNA characterization or inflated for histological sectioning. Lung lobes inflated with agarose were fixed with formalin (10%) for Hematoxylin and Eosin (H&E) and Senescence-Associated beta-Galactosidase (SA-β-Gal) staining, lung lobes inflated with OCT performed with frozen sectioning were used for fluorescence imaging to detect mitophagy based on mitoQC reporter system.
2. H&E staining and Lung morphometry
The H&E staining and lung morphometry are described previously by us (Wang et al., 2020). Briefly, lung sections (5 μm) were immersed in xylene (5 minutes × 3 times) for deparafinization, followed by rehydration with 100%, 95%, and 70% ethanol (3 minutes each). Slides were quickly washed in water, and stained with hematoxylin for 1 minute. Slides were washed in tap water for 5 minutes after hematoxylin staining and then immersed in 7% ammonia water for 10 seconds. Sections were washed in running water for another 5 minutes. The cleaned slides were soaked in Eosin for 1 minute, and then washed with 95% ethanol for 1 minute. The slides were then dehydrated with 95% and 100% ethanol (3 minutes × 2 times), and xylene (5minutes × 3 times). Finally, slides were mounted in Permount mounting media for microscopy, and the H&E stained sections were imaged under 20x magnifications to measure airspace enlargement (mean linear intercept, Lm). Lm measurements were performed by MetaMorph software (Molecular Devices). A technique named Measure Mean Airspace Diameter created by MetaMorph was used for airspace measurement. Briefly, individual image was loaded, and set the threshold in between 100–160, then, put the parameters, respectively: Vertical Lines: 0; Horizontal Lines: 6; Line Thickness: 1; Line Color: 255. The airspace value will be recorded into an Excel sheet. At least ten images were used to measure the Lm, and all the Lm values reflected in each image were averaged as one sample value.
3. SA-β-gal staining
The frozen sections (10μm) prepared from OCT inflated lung lobes were stained with SA-β-gal (Cat#: K802, BioVision, Milpitas, CA) according to manufacturer’s protocol. Briefly, frozen sections were warmed at room temperature for 15mins, followed by washing (PBS/10mins × 2times) to remove the OCT. Sections were fixed with 4% paraformaldehyde (10mins). followed by washing in PBS 3 times. Freshly prepared staining solution containing 1mg/mL X-gal substrate was incubated with sections overnight at 37°C. After overnight incubation, the staining solution was removed followed by washing once with PBS. Stained slides were mounted in mounting media containing 70% glycerol (Xylene-free mounting media) for microscopy. The positive staining with SA-β-gal was showing as blue color.
4. Fluorescence imaging
The frozen sections (10μm) prepared from OCT inflated lung lobes were warmed at room temperature for 15mins and washed with PBS (10 minutes × 2 times) to remove the OCT, then mounted with ProLong™ Gold Antifade Mountant (contained DAPI) (Cat#: P36931, Thermo Fisher Scientific, Waltham, MA). Mounted slides were stored overnight at 4°C and visualized in fluorescence microscope (Olympus, BX51) using green (FITC, Ex: 470±40) and mCherry (APC, Ex: 595±40) fluorescence filters. Normal mitochondria appear as GFP+/mCherry+, while mitochondria undergoing mitophagy appear GFP−/mCherry+. The images were obtained using 40x magnification with the same exposure times for all samples (400ms for both GFP and mCherry, 10ms for DAPI). Total fluorescence intensity in the whole imaje was measured using image J; with at least five images measured.
5. Mitochondrial isolation
Mitochondria were isolated from snap-frozen lung tissue using the Mitochondria Isolation Kit (for Tissue) (Cat#: 89801, Thermo Fisher Scientific, Waltham, MA) according to the manufacturer’s protocol. Briefly, around 100mg lung tissues were washed in PBS 3 times and minced in pieces in 800 μL PBS. Mildly homogenize it for 5 sec by a polytron grinder to disrupt the tissues but not lysis the cells. Centrifuge the mixtures at 1000 g for 3 mins at 4°C, then remove the supernatant. Resuspend the pellet in 800μL of BSA/reagent A solution and vortex for 5sec, then incubate on ice for 2 mins. After 2 mins incubation, add 10 μL isolation reagent B incubate another 5 mins on ice, vortex the mixtures every minute. Finally, another 800μL reagent C was added and mixed thoroughly, then spun down for 10 mins at 4°C, 700g. Transfer the supernatant to a new tube, and spin down 12000 g for 15mins at 4°C. Discard the supernatant and resuspend the pellets as mixtures of normal mitochondrial and lysosome contained mitochondrial fragments (mitophagy). Isolated Mitochondrial were then lysed with RIPA buffer to characterize the mitochondrial proteins.
6. Protein isolation and western blot
Total mitochondrial protein was isolated by lysing in RIPA buffer with protease inhibitor (Cat#: 78440; Thermo Fisher Scientific, Waltham, MA) and quantitated using Pierce™ BCA Protein Assay Kit (Cat#: 23225, Thermo Fisher Scientific, Waltham, MA). Protein (20μg) was run on a 10% SDS-polyacrylamide electrophoresis (SDS-PAGE) followed by transfer onto nitrocellulose membranes (Cat#: 1620112, Bio-Rad, Hercules, CA) in Tris-glycine buffer with 20% methanol at 4°C overnight. The membranes were blocked with 5% BSA in TBS-T (TBS with 0.025% TritonX-100) for 1 hr at room temperature and then probed with the specific primary antibodies in blocking buffer overnight at 4°C, primary antibody (Rabbit host) used in this study included: Total OXPHOS Rodent WB Antibody Cocktail (1: 1000, ab110413, Abcam, Waltham, MA), Anti-Opa1 (1:1000, 80471S, Cell Signaling technology, Danvers, MA), Anti-Pink1 (1:1000, ab23707, Abcam, Waltham, MA), Anti-Drp1 (1:1000, 8570S, Cell signaling technology, Danvers, MA), Anti-Parkin (1:1000, 2132S, Cell signaling technology, Danvers, MA), Anti-VDAC (HRP) (1:1000, ab185063, Abcam, Waltham, MA). The, membranes were washed with TBS-T 4 times, 10minutes each, to remove the primary antibody and then probed with goat anti-rabbit secondary antibody (1:5000, 1706515; Bio-Rad, Hercules, CA). The membranes were washed again with TBS-T 4 times, 10 minutes each, and then developed with Pierce ECL Western Blotting Substrate (Cat# 32106; Thermo Fisher Scientific, Waltham, MA). The developed membranes were imaged via Bio-Rad ChemiDoc MP imaging system (Biorad, Hercules, CA), and bands were quantitated by densitometry. Results were expressed as fold change normalized VDAC endogenous control. Unedited full-blot is presented as supplementary information (Suppl Fig 1).
7. RNA isolation and Nanostring measurement
Snap frozen lung samples (~30mg) were mechanically homogenized in 300μL Qiazol reagent (Cat#:79306; Qiagen, Germantown, MD) for RNA isolation. Add another 700μL of Qiazol reagent to the homogenates. The mixtures were then mixed with chloroform thoroughly and spin down at 15000 rpm for 30mins at 4°C. Transfer the aqueous (upper) phase (~500μL) into a new tube, add 500 μL isopropanol, mix thoroughly, and incubate at −20°C for 2 hrs. Then, the mixtures were centrifuged at 15000 rpm for 30mins at 4°C, then discarded the supernatant. Wash the RNA pellets with 75% ethanol prepared in Rnase free water. Centrifuge again at 15000 rpm for 30 mins at 4°C. Remove the ethanol, resuspend the RNA in Rnase free water. RNA quality and quantity was determined using NanoDrop - Microvolume Spectrophotometer (Thermo Fisher Scientific, Waltham, MA). Total 50ng RNA was used in NanoString profiling. The customized panel from NanoString contained all the mitochondrial related genes was used in this study. The NanoString profiling was conducted according to the manufacturer’s protocol. Briefly, the RNA was incubated with hybridization codeset reagent from NanoString for 16hr at 65°C, then loaded into the nCounter Cartridge and proceeded the profiling in nCounter SPRINT profiler (Nanostring, Seattle, WA). All the gene expressions were analyzed by the nSovler 4.0 software. Endogenous gene used for normalization included: Abcf1, Gusb, Hprt, Ldha, Polr1b, and Rplp0. The RNA transcript levels showed in this study are normalized counts. The RNA transcript levels were measured by NanoString Technology, which have been confirmed by qRT-PCR in our previous publication (Wang et al., 2021).
8. Statistics
All the statistical analysis and graph preparations were done by via GraphPad Prism software (version 9.0, GraphPad Prism, San Diego, CA). The results were presented as dot blot with mean ± SEM, and the 2-tailed student’s t-test was conducted in this study. The significant difference was considered as P < 0.05.
Results
1. Chronic CS exposure induced airspace enlargement, lung inflammation, and cellular senescence
To characterize the CS exposure induced lung injury, H&E, and SA-β-Gal stained lung sections were applied. We observed airspace enlargement and infiltration of inflammatory cells into the airspace following CS exposure compared to lung sections from air exposed controls which showed normal airspace and no inflammatory cell infiltration (Fig. 1A). Lung sections from CS-exposed mice also demonstrated increased SA-β-Gal positive staining (Fig. 1B). Interestingly, we also showed a significant accumulation of SA-β-Gal in the infiltrated inflammatory cells (Fig. 1B). Together, our results demonstrate that chronic CS exposure in mitoQC mice induces lung inflammation, airspace enlargement, and cellular senescence, in line with our previous observations (Sundar et al., 2018b). Hence, we further determined the mitochondrial dysregulation and mitophagy in mitoQC reporter mice after chronic CS exposure.
Figure 1. Chronic CS induced senescence, airspace enlargement and inflammation in mitoQC mice.
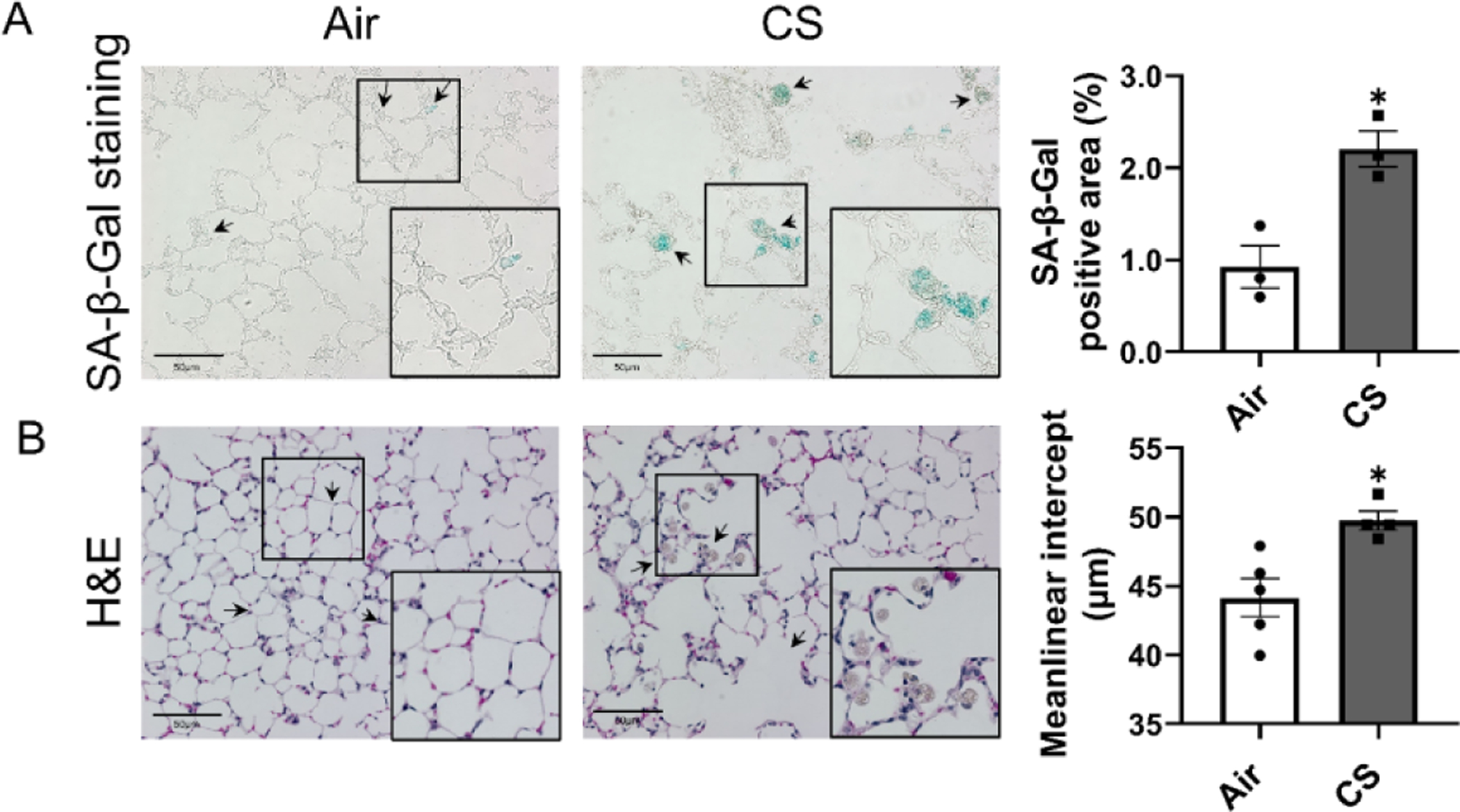
MitoQC mice were exposed to CS for 4 month, and lungs were sectioned for (A) senescence-associated beta-galactosidase (SA-β-Gal) staining, and (B) H&E staining. Represented pictures (20x objective) were taken by light microscope (Nikon Eclipse Ni-U microscope). Positive stained SA-β-Gal area was calculated as percentage compared to lung tissue area, and pointed by black arrows. Airspace enlargement and Inflammation cell infiltration caused by CS was determined in H&E stained sections and pointed by black arrows. A higher magnification image from the black square was listed in the right bottom corner. Data was presented as mean ± SEM (n=3 for SA-β-Gal staining, and n=4–5 for H&E staining; * P < 0.05 vs. air; scale bar = 50μm).
2. Chronic CS exposure induced mitophagy observed in MitoQC reporter mice
The mitoQC reporter mice express pH-sensitive GFP/mCherry fluorophore where healthy mitochondria will show both green and mCherry fluorescence (GFP+/mCherry+), while the mitochondria undergoing mitophagy will show only mCherry fluorescence puncta (GFP−/mCherry+) since GFP is deactivated by low pH in the lysosome. Our images of frozen lung sections, show GFP−/mCherry+ puncta only in lungs from CS-exposed mitoQC mice (Fig 2), whereas the air group showed only GFP+/mCherry+ across lung sections. The fluorescence ratio of mCherry/GFP was significantly up-regulated in the CS group, indicating more mitophagy occurred after CS exposure in the lungs. Interestingly, we also showed more mitochondrial signals (GFP+/mCherry+) in infiltrated inflammatory cells in airspace, and the GFP−/mCherry+ puncta were observed mostly within the area that contained inflammatory cells (Fig 2).
Figure 2. Chronic CS induced mitophagy in lung alveolar epithelium in mitoQC mice.
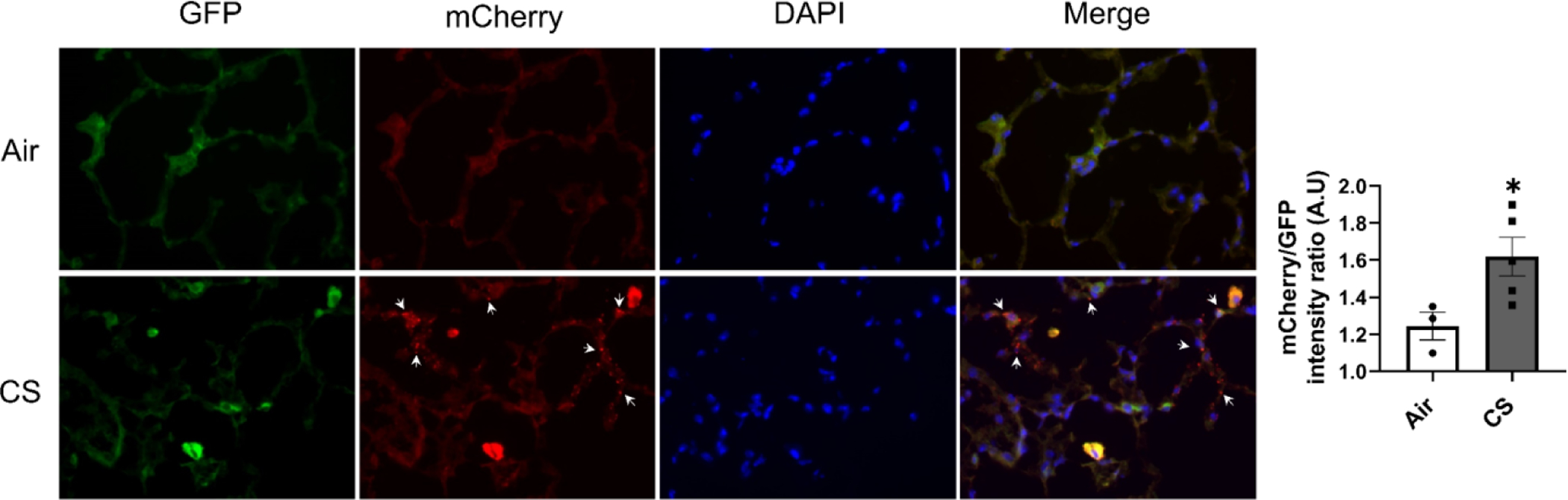
MitoQC mice were exposed to CS for 4 month, and lungs were fixed by OCT and cryo-sectioned. Represented pictures were taken by fluorescence microscope (Olympus BX51) under 40x objective. The GFP+/mCherry+ signal indicates the cytosolic mitochondria, while both the mCherry only signal (GFP−/mCherry+) and the ratio of mCherry/GFP intensity was used to calculated the occurred mitophagy. Mitophagy observed in CS exposure group was pointed by white arrows. Data was presented as mean ± SEM (n=3 for air group, and n=5 for CS group; * P < 0.05 vs. air).
3. Dysregulated Mitochondrial fraction occurred after chronic CS exposure
Given our observation that CS induces mitophagy in lungs, we wanted to determine how CS dysregulates the mitochondrial complexes and functions. We isolated mitochondrial from lung homogenates and characterized the OXPHOS complex abundance. We found changes in protein abundance in these complxes with complex I (Ndufb8) showing a slight increase in while complex III (Uqcrc2) showed a decrease in protein abundance (Fig 3) in smoke exposed mice versus air exposed controls. Other complexes (Complex II (Sdhb), IV (Mtco), and V (Atp5a)) showed no significant differences between air and CS groups.
Figure 3. Chronic CS induced dysregulated mitochondrial complexes in mitoQC mice.
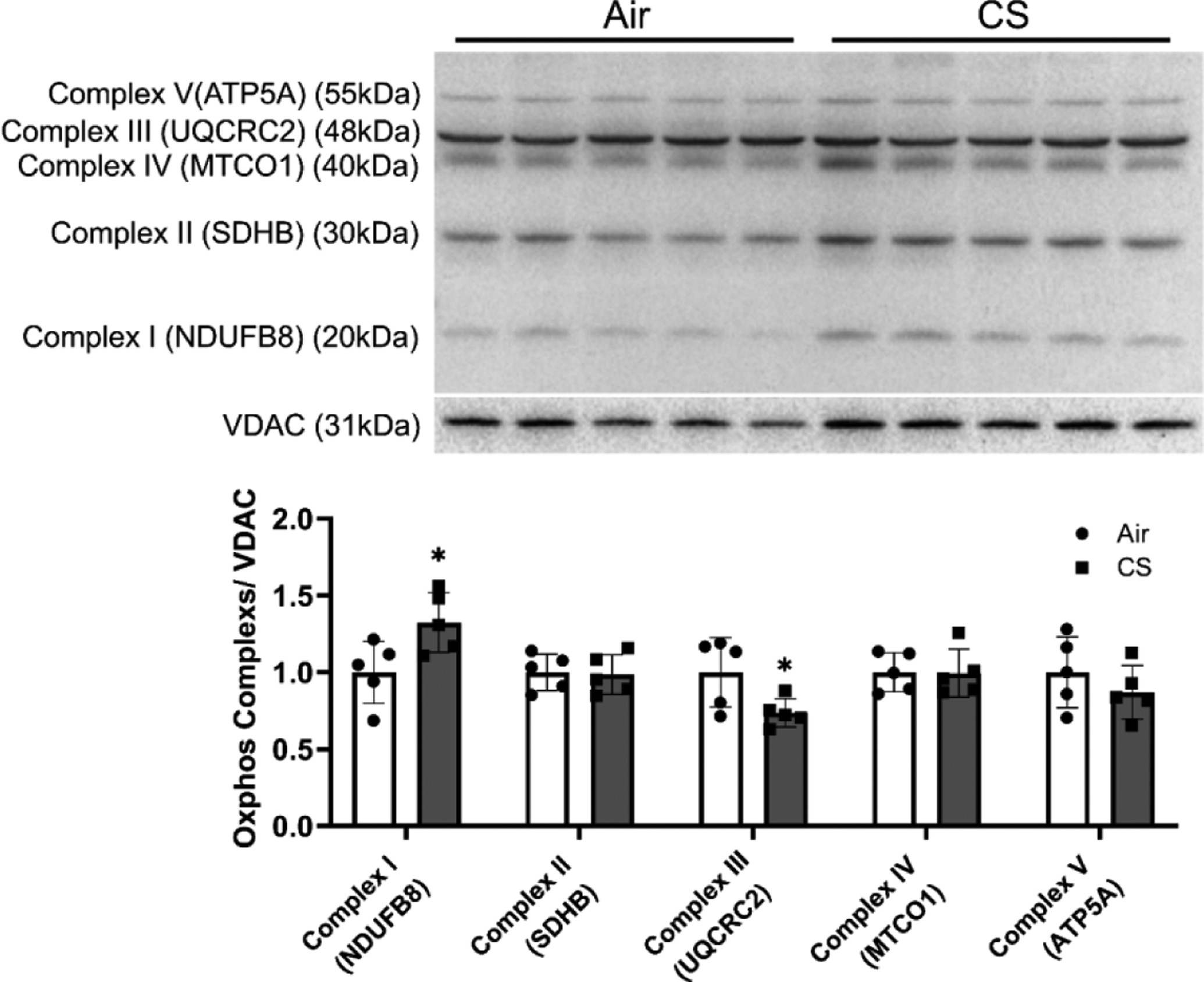
MitoQC mice were exposed to CS for 4 month, and lungs were snap frozen for mitochondrial protein isolation. Proteins from mitochondrials were isolated and mitochondrial complexes abundances were measured by immunoblot. Represented bands were shown, and densitometry was used to evaluate protein expression change folds for complex 1–5. VDAC was used as endogenous control for mitochondrial proteins for normalization. Data was presented as mean ± SEM (n=5; * P < 0.05).
In addition to determining the protein abundance of mitochondrial complexes, we also determined the effects on RNA transcript levels of genes related to mitochondrial complexes and its functions in lungs after 4 month CS exposure. Employing our customized NanoString panels, we characterized mitochondrial gene profiles between the air exposed control and chronic CS exposure groups. Most ATP synthases gene expressions were inhibited by CS and other mitochondrial complexes related to transcript levels (Fig. 4A, Supplement Table S1). Especially, Atp5a1, Atp5b, and Atp5d, were significantly decreased after chronic CS exposure while Atp6v0d2 was statistically up-regulated in the CS group compared to air controls (Fig. 4B). Similarly, we observed that gene transcript levels related to mitochondrial complexes I, II, and III, were all inhibited. For complex I-related genes, Ndufa10, Ndufb8, and Ndufv1 were down-regulated after chronic CS exposure (Fig 4B); gene markers related to complex II: Sdha, also showed decreased gene expression level in the CS group compared to air controls (Fig 4B). Gene expressions related to complex III: Cox4i2 and Cyc1 were also inhibited after chronic CS exposure (Fig 4B).
Figure 4. Chronic CS induced dysregulated mitochondrial complexes dysregulation in mitoQC mice.
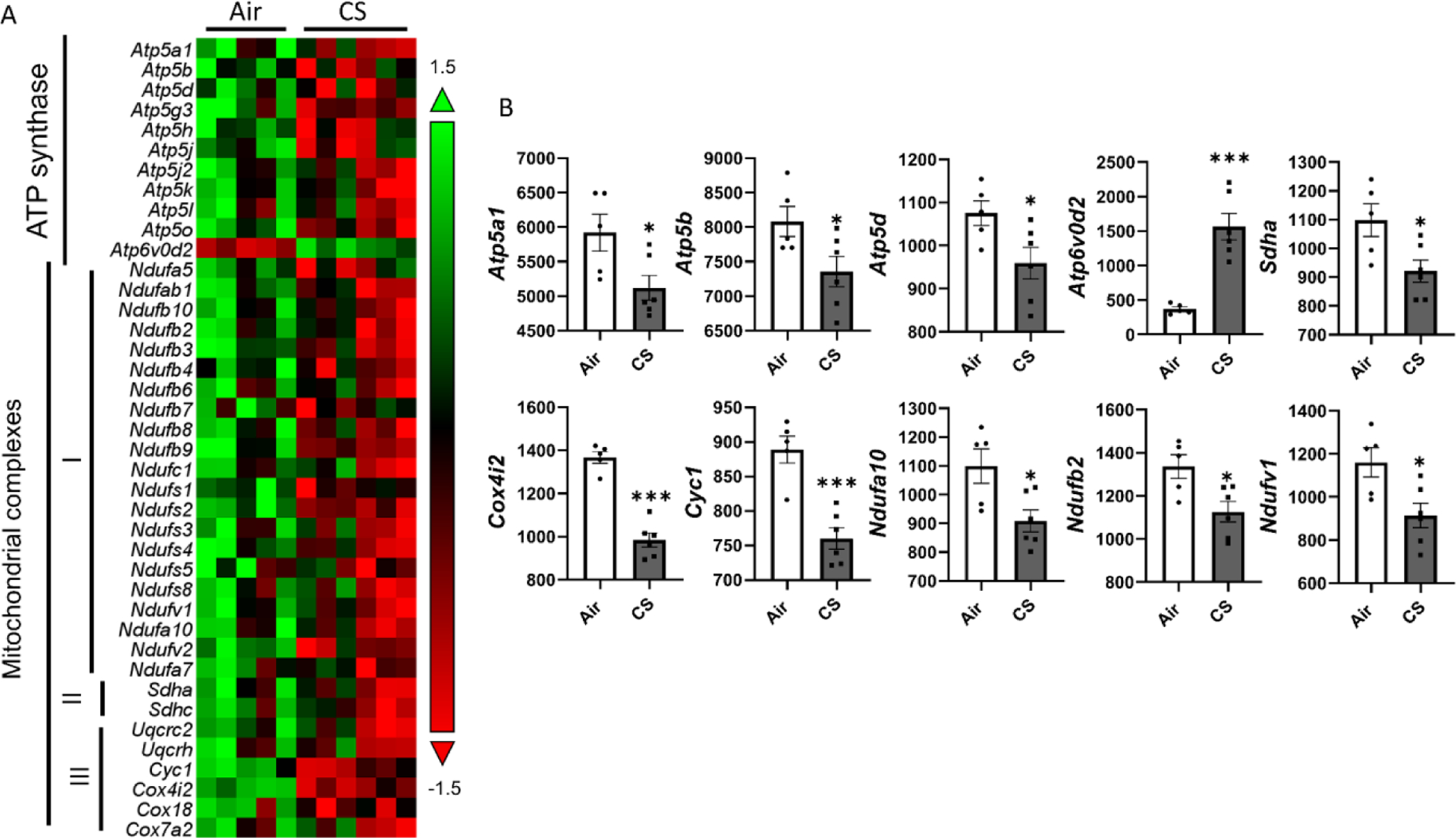
MitoQC mice were exposed to CS for 4 month, and lungs were snap frozen for transcript level measurements. RNA was isolated from lung homogenates and expression levels were measured by nanostring based on customized panel. Genes related to mitochondrial complexes and ATP synthesis were measured, and the abundances of transcripts were showed as normalized counts. Data was presented as mean ± SEM (n=5; * P < 0.05, *** P < 0.001).
4. Chronic CS exposure dysregulated the mitochondrial dynamics
In addition to quantifying the mitochondrial functions related markers, we also measured the mitochondrial dynamics related markers (protein and gene expressions). As shown in Fig 5, we measured the mitochondrial dynamic markers from the protein isolated from the mitochondrial only and not total cellular protein. We showed a significant increase in OPA1 Mitochondrial Dynamin Like GTPase (Opa1) protein levels and decreased Parkin protein expression in the CS group compared to the air group (Fig 5). While the protein levels of Dynamin-1-like protein (Drp1, gene symbol: Dnml1) did demonstrate a decreased trend in the CS group compared to air control the data was not statistically significant (Fig 5). There is no difference in protein levels of Pink1 between the air and CS groups (Fig 5).
Figure 5. Chronic CS induced dysregulated mitochondrial dynamics in mitoQC mice.
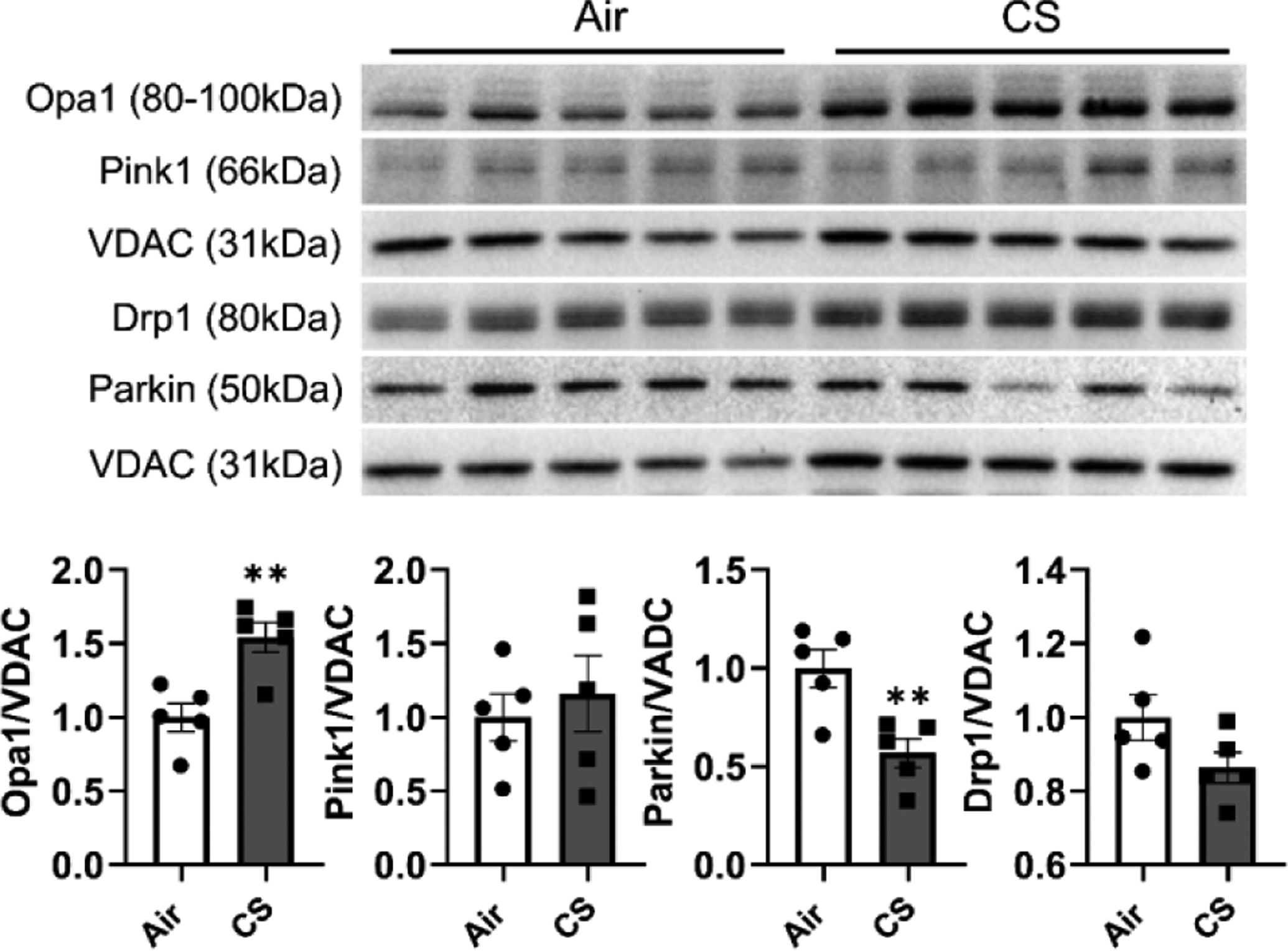
MitoQC mice were exposed to CS for 4 month, and lungs were snap frozen for mitochondrial protein isolation. Proteins from mitochondrials were isolated and related mitochondrial dynamic markers were measured by immunoblot. Represented bands were shown, and densitometry was used to evaluate protein expression change folds for Opa-1, Pink1, Drp-1, and Parkin. VDAC was used as endogenous control for mitochondrial proteins for normalization. Data was presented as mean ± SEM (n=5; * P < 0.05, ** P <0.01).
With gene profiling of mitochondrial dynamics-related markers, we observed that markers related to mitochondrial fusion and fission, mitophagy, mitochondrial transportation, and translocation, as well as oxidative stress, were dysregulated (Fig 6A, and supplement table S1). Specifically, the mitochondrial fission and fusion-related gene expression levels: Mitofusin-1 (Mfn1), Mitofusin-2 (Mfn2), and Dnm1l, are decreased after chronic CS exposure (Fig 6B). Gene expressions of Mitochondrial Rho GTPase 1 (Rhot1) and Mitochondrial Rho GTPase 2 (Rhot2), which are responsible for the mitochondrial transport, are decreased significantly in the CS group (Fig 6B). The mitophagy-related markers: Pink1 and Prkn, showed down-regulation after CS exposure (Fig 6B). The oxidative stress marker, Superoxide dismutase 2, mitochondrial (Sod2), was increased in CS exposed mouse lungs compared to the air group (Fig 6B). However, the gene expression for Opa1 (Fig 5) showed no significant difference between CS and air groups (Supplement Table S1).
Figure 6. Chronic CS induced dysregulated mitochondrial dynamic dysregulation in mitoQC mice.
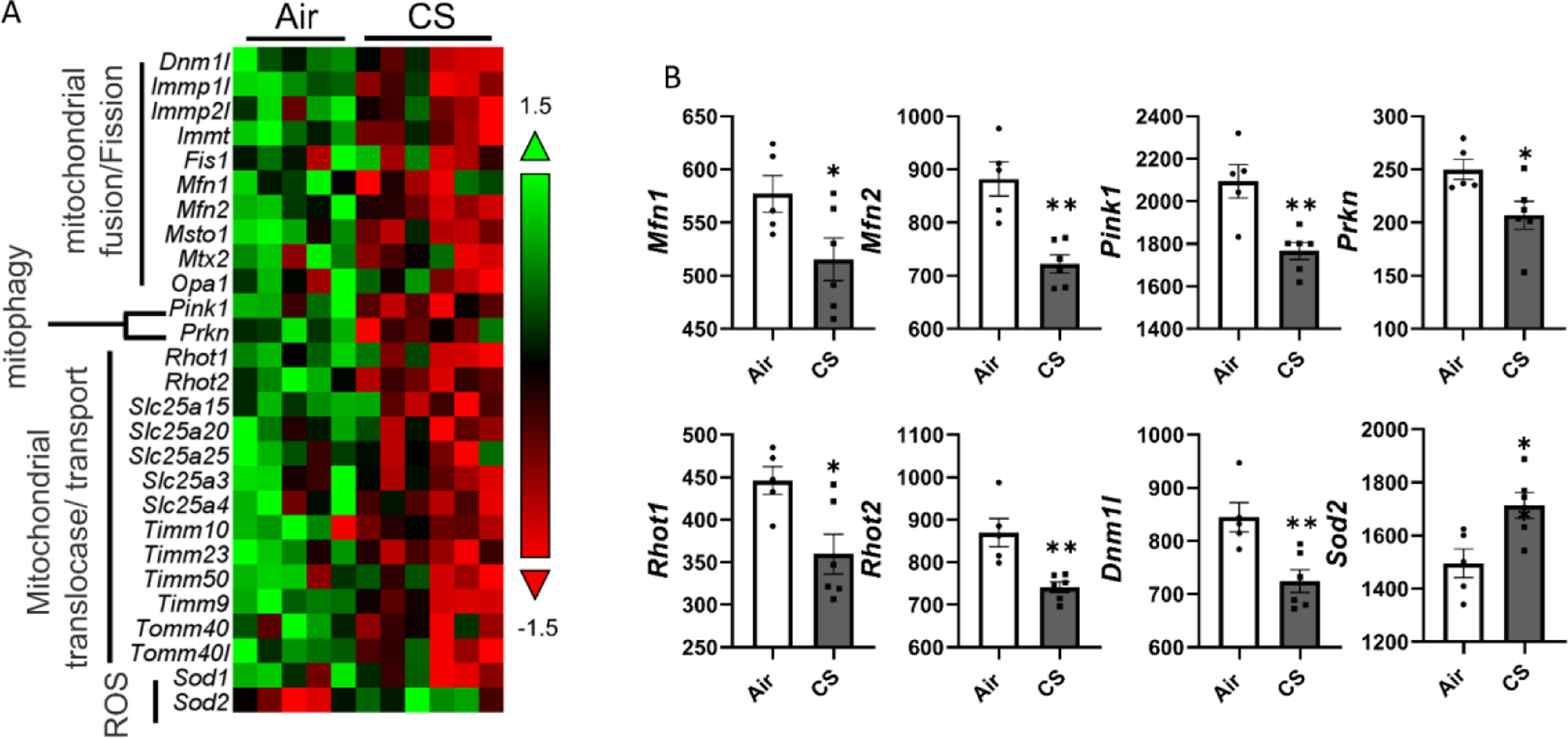
MitoQC mice were exposed to CS for 4 month, and lungs were snap frozen for transcript level measurements. RNA was isolated from lung homogenates and expression levels were measured by nanostring based on customized panel. Genes related to mitochondrial dynamics including mitochondrial translocase/transport, mitophagy, mitochondrial fussion/fission, and mitochondrial stress were measured, and the abundances of transcripts were showed as normalized counts. Data was presented as mean ± SEM (n=5; * P < 0.05, ** P < 0.01).
Discussion
CS exposure induces lung inflammation and premature lung cellular senescence leading to diseased pathologies, such as COPD/emphysema, upon chronic CS exposure (Sundar et al., 2018b). Previous publications have shown that CS exposure induces mitochondrial dysregulation and dysfunction in lung epithelium (Banzet et al., 1999; Hara et al., 2013; Hoffmann et al., 2013a; Solanki et al., 2018; Sundar et al., 2019). In our earlier study, we have shown that ten days CS exposure induced inhibition of gene markers related to mitochondrial fission and fusion (Maremanda et al., 2019). In the same study, mitophagy was observed based on the mitoQC reporter system (Maremanda et al., 2019). This study found that four months CS exposure induced lung inflammation, airspace enlargement, and cellular senescence in mitoQC reporter mice. We also observed augmented mitophagy, dysregulated mitochondrial complexes and dynamics after chronic CS exposure.
It is well known that CS-induced lung inflammation promotes airspace enlargement and premature senescence leading to development of COPD/emphysema phenotype (Sundar et al., 2018b). We have shown that CS exposure causes lung remodeling and infiltration of inflammatory cells into airspace, with increased macrophage, neutrophil, and T-cells identified in the bronchoalveolar lavage fluid (BALF) and lung sections (Sundar et al., 2018a; Wang et al., 2021; Yao et al., 2012). This study is in line with our previous observation (Sundar et al., 2018b) and confirms that chronic CS exposure induces lung inflammation, airspace enlargement, and cellular senescence in mitoQC mice. Both acute and chronic CS exposure has been shown to promote mitochondrial dysregulation, as well as the mitophagy progression (Ahmad et al., 2015; Araya et al., 2019; Maremanda et al., 2019; Sundar et al., 2019). In line with previous reports, we showed augmented mitophagy after CS exposure, especially around the alveolar epithelial cells in the inflammatory regions. Parkin-PINK1 plays an important role in mediating mitophagy, and Parkin depletion augmented accumulation of injured mitochondria in lung epithelial cells from mice exposed to CS (Araya et al., 2019). Our previous study also showed that Parkin overexpression ameliorates mitophagy dysregulation and attenuates the accumulation of damaged mitochondria (Ahmad et al., 2015). We observed that a decreased Parkin protein abundance in mitochondrial, which partially aligns with the previous publication that CS induces accumulation of damaged mitochondrial, and Parkin overexpression helps restore the normal mitophagy process (Ahmad et al., 2015; Araya et al., 2019). However, the higher incidence of mitophagy observed in this study might suggest that a role for Pink1-Parkin independent mitophagy process occurred during chronic CS exposure (McWilliams et al., 2018). The increased mitochondrial fluorescence signal (GFP+/mCherry+) could be associated with elongated mitochondria and mitochondrial accumulation induced by CS exposure (Hoffmann et al., 2013b; Sundar et al., 2019). The elongated increase ATP levels and prevent mitochondrial degradation (Gomes et al., 2011), suggesting that CS induced elongated mitochondria might be one of the protection mechanisms during the stress.
In this study, we observed dysregulation of mitochondrial complex gene expression levels. In line with our previous reports, gene levels related to all five mitochondrial complexes were decreased in CS exposed group (Maremanda et al., 2019; Sundar et al., 2019). A previous report had shown that complexes I, II, and V were downregulated in lungs from smokers and GOLD2 stage COPD patients compared to never smokers (Haji et al., 2020). Interestingly, the same report also showed decreased mitochondrial complexes in muscle tissue from COPD patients (Haji et al., 2020), while another report described the same trend in skeletal muscle from COPD patients (Leermakers et al., 2018). Interestingly, COPD phenotypes induced by ozone exposure also showed decreased protein levels of mitochondrial complexes I, III, and V (Wiegman et al., 2015). Taken together, these data suggest that inhibition of mitochondrial complexes in COPD patients might happened at a very early stage of COPD, with a progressive decline in mitochondrial function as COPD phenotype develops/progresses.
We also showed the inhibited gene expression related to mitochondrial dynamics and increased oxidative stress genes after CS exposure. Increased Sod2 gene level was found in CS exposed mitoQC mice, which aligned with the previous study (Gannon et al., 2012), and oxidative stress was usually initiated after CS exposure and responsible for the cellular injury (Wiegman et al., 2015; Yao et al., 2010). We also observed the decreased mRNA and protein levels of Parkin, and Pink1 in CS exposure group. Parkin and Pink1 play well-known roles in mediating mitophagy, and loss of Parkin exacerbates the accumulation of damaged mitochondria after CS exposure, with concomitant increase in oxidative stress (Araya et al., 2019). In the same report, increased Pink1 and impaired mitophagy were associated with Parkin deficiency, suggesting that Parkin and Pink1 were equally essential in mediating mitophagy (Araya et al., 2019). Decreased Parkin levels observed in this study might promote the delayed clearance of damaged mitochondria and promote the Pink1-Parkin independent mitophagy process.
Additionally, we also found dysregulated mitochondrial fission and fusion markers. The increased Opa1 protein abundance was observed in the CS exposure group, which agree with the previous reports that Opa1 showed up-regulation after CSE treatment and in COPD patients (Hoffmann et al., 2013a; Leermakers et al., 2018). Loss of Opa1 induced mitochondrial fragmentation while overexpression of Opa1 helped to increase the mitochondrial inner membrane surface and mitochondrial elongation (Cipolat et al., 2004; Hoffmann et al., 2013a). Dysregulated Opa1 and mitochondrial structure could result in impaired mitophagy (Cipolat et al., 2004; Hoffmann et al., 2013a). Other mitochondrial dynamic markers, such as Mfn1, Mfn1, and Dnm1l (protein code: Drp1) showed were also decreased in the CS exposure group compared to air control, which was in line with our and other’s previous studies (Gannon et al., 2013; Maremanda et al., 2019; Song et al., 2017). Mfn1 and Mfn2 are responsible for mitochondrial fusion of the outer mitochondrial membrane, while Drp1 and Fis1 are involved in mitochondrial fission (Nam et al., 2017). Decreased Mfn1, Mfn2, and Drp1 reflect aberrant mitochondrial dynamics and attenuated recycling of damaged mitochondrial (Mitophagy) (Manevski et al., 2020; Westermann, 2010). Additionally, Rhot1 and Rhot2 genes, responsible for mitochondrial trafficking and transfer, were decreased after CS exposure. Rhot1 and Rhot2 help restore the healthy mitochondria and ameliorate the CS-induced stress (Liu et al., 2021; Sundar et al., 2019), further reflecting mitochondrial dysfunction by CS utilizing these mice.
Conclusion
In summary, we show that chronic CS exposure induces mitophagy, dysregulates mitochondrial complex abundance, and mitochondrial dynamics. Down-regulation of mitochondrial complex genes inhibits the mitochondrial biogensis to replace the damaged mitochondria. Decreased levels of Parkin after CS exposure can promote accumulation of damaged mitochondria with consequent disruption in mitochondrial turnover. CS induced impaired mitophagy, observed from mitoQC reporter mice might be Pink1-Parkin independent and increased mitochondrial fluorescence signal (GFP+/mCherry+) is due to the mitochondrial accumulation induced by CS exposure. Detailed signaling mechanisms involved in mitophagy, mitochondrial accumulation and mitochondrial elongation require further investigation to determine other alternate Pink-Parkin independent mitophagy pathways induced by CS and its impact on the development and progression of COPD.
Supplementary Material
Supplementary Figure 1 Unedited full-blot/gel listed in the supplementary information.
Supplementary Table S1 Gene expression change fold Ratio (CS/Air)
Research Highlights.
Cigarette smoke (CS) exposure induces mitochondrial dysregulation and abnormal mitophagy.
We employed mitoQC reporter mice, a mitochondrial reporter strain as mitophagy marker using GFP+/mCherry+.
Mitochondrial complexes and function related genes were inhibited with alterations in complexes I and III were observed
Mitochondrial dynamic markers, mitochondrial fusion/fission, and mitochondrial translocate/transfer markers were altered.
Thus, mitoQC reporter mouse model can be employed to assess the dysregulated mitochondrial complexes and dynamics.
Acknowledgments:
The authors would like to acknowledge Lakshmi Chakrapaani, PhD for their scientific inputs. MitoQC mice (C57BL/6 background) were obtained from Ian G. Ganley. Ph.D (University of Dundee, UK) which was generated by Taconic Artemis under a Material Transfer Agreement.
Funding:
The National Institutes of Health (NIH) HL147715, HL137738, HL133404, ES032159, and HL135613 supported this work.
List of Abbreviations
- COPD
Chronic Obstructive Pulmonary disease
- IPF
Idiopathic Pulmonary disease
- BALF
Bronchoalveolar Lavage fluid
- mQC
MitoQC
- CS
Cigarette smoke
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Conflicts of Interest: The authors have declared that no competing interests exist.
References
- Ahmad T, Sundar IK, Lerner CA, Gerloff J, Tormos AM, Yao H, Rahman I, 2015. Impaired mitophagy leads to cigarette smoke stress-induced cellular senescence: implications for chronic obstructive pulmonary disease. FASEB J 29, 2912–2929. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Araya J, Tsubouchi K, Sato N, Ito S, Minagawa S, Hara H, Hosaka Y, Ichikawa A, Saito N, Kadota T, Yoshida M, Fujita Y, Utsumi H, Kobayashi K, Yanagisawa H, Hashimoto M, Wakui H, Ishikawa T, Numata T, Kaneko Y, Asano H, Yamashita M, Odaka M, Morikawa T, Nishimura SL, Nakayama K, Kuwano K, 2019. PRKN-regulated mitophagy and cellular senescence during COPD pathogenesis. Autophagy 15, 510–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashrafi G, Schwarz TL, 2013. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ 20, 31–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banzet N, François D, Polla BS, 1999. Tobacco smoke induces mitochondrial depolarization along with cell death: effects of antioxidants. Redox Rep 4, 229–236. [DOI] [PubMed] [Google Scholar]
- Blackstone C, Chang CR, 2011. Mitochondria unite to survive. Nat. Cell Biol 13, 521–522. [DOI] [PubMed] [Google Scholar]
- Blanco I, Diego I, Bueno P, Casas-Maldonado F, Miravitlles M, 2019. Geographic distribution of COPD prevalence in the world displayed by Geographic Information System maps. Eur. Respir. J 54. [DOI] [PubMed] [Google Scholar]
- Bowler RP, Barnes PJ, Crapo JD, 2004. The role of oxidative stress in chronic obstructive pulmonary disease. Copd 1, 255–277. [DOI] [PubMed] [Google Scholar]
- Centers for Disease, C., Prevention, National Center for Chronic Disease, P., Health, P., Office on, S., Health, 2010. Publications and Reports of the Surgeon General, How Tobacco Smoke Causes Disease: The Biology and Behavioral Basis for Smoking-Attributable Disease: A Report of the Surgeon General Centers for Disease Control and Prevention (US), Atlanta (GA). [PubMed] [Google Scholar]
- Chauhan A, Vera J, Wolkenhauer O, 2014. The systems biology of mitochondrial fission and fusion and implications for disease and aging. Biogerontology 15, 1–12. [DOI] [PubMed] [Google Scholar]
- Chen Y, Dorn GW 2nd, 2013. PINK1-phosphorylated mitofusin 2 is a Parkin receptor for culling damaged mitochondria. Science 340, 471–475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cipolat S, Martins de Brito O, Dal Zilio B, Scorrano L, 2004. OPA1 requires mitofusin 1 to promote mitochondrial fusion. Proc Natl Acad Sci U S A 101, 15927–15932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gannon AM, Stämpfli MR, Foster WG, 2012. Cigarette smoke exposure leads to follicle loss via an alternative ovarian cell death pathway in a mouse model. Toxicol. Sci 125, 274–284. [DOI] [PubMed] [Google Scholar]
- Gannon AM, Stämpfli MR, Foster WG, 2013. Cigarette Smoke Exposure Elicits Increased Autophagy and Dysregulation of Mitochondrial Dynamics in Murine Granulosa Cells1. Biol. Reprod 88. [DOI] [PubMed] [Google Scholar]
- Gomes LC, Di Benedetto G, Scorrano L, 2011. During autophagy mitochondria elongate, are spared from degradation and sustain cell viability. Nat. Cell Biol 13, 589–598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haji G, Wiegman CH, Michaeloudes C, Patel MS, Curtis K, Bhavsar P, Polkey MI, Adcock IM, Chung KF, on behalf of the, C.c., 2020. Mitochondrial dysfunction in airways and quadriceps muscle of patients with chronic obstructive pulmonary disease. Respir. Res 21, 262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hara H, Araya J, Ito S, Kobayashi K, Takasaka N, Yoshii Y, Wakui H, Kojima J, Shimizu K, Numata T, Kawaishi M, Kamiya N, Odaka M, Morikawa T, Kaneko Y, Nakayama K, Kuwano K, 2013. Mitochondrial fragmentation in cigarette smoke-induced bronchial epithelial cell senescence. Am. J. Physiol. Lung Cell Mol. Physiol 305, L737–746. [DOI] [PubMed] [Google Scholar]
- Hoffmann RF, Zarrintan S, Brandenburg SM, Kol A, de Bruin HG, Jafari S, Dijk F, Kalicharan D, Kelders M, Gosker HR, Ten Hacken NH, van der Want JJ, van Oosterhout AJ, Heijink IH, 2013a. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res 14, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoffmann RF, Zarrintan S, Brandenburg SM, Kol A, de Bruin HG, Jafari S, Dijk F, Kalicharan D, Kelders M, Gosker HR, ten Hacken NHT, van der Want JJ, van Oosterhout AJM, Heijink IH, 2013b. Prolonged cigarette smoke exposure alters mitochondrial structure and function in airway epithelial cells. Respir. Res 14, 97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ito S, Araya J, Kurita Y, Kobayashi K, Takasaka N, Yoshida M, Hara H, Minagawa S, Wakui H, Fujii S, Kojima J, Shimizu K, Numata T, Kawaishi M, Odaka M, Morikawa T, Harada T, Nishimura SL, Kaneko Y, Nakayama K, Kuwano K, 2015. PARK2-mediated mitophagy is involved in regulation of HBEC senescence in COPD pathogenesis. Autophagy 11, 547–559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang Y, Wang X, Hu D, 2017. Mitochondrial alterations during oxidative stress in chronic obstructive pulmonary disease. Int. J. Chron. Obstruct. Pulmon. Dis 12, 1153–1162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leermakers PA, Schols AMWJ, Kneppers AEM, Kelders MCJM, de Theije CC, Lainscak M, Gosker HR, 2018. Molecular signalling towards mitochondrial breakdown is enhanced in skeletal muscle of patients with chronic obstructive pulmonary disease (COPD). Sci. Rep 8, 15007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lerner CA, Rutagarama P, Ahmad T, Sundar IK, Elder A, Rahman I, 2016. Electronic cigarette aerosols and copper nanoparticles induce mitochondrial stress and promote DNA fragmentation in lung fibroblasts. Biochem Biophys Res Commun 477, 620–625. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu D, Gao Y, Liu J, Huang Y, Yin J, Feng Y, Shi L, Meloni BP, Zhang C, Zheng M, Gao J, 2021. Intercellular mitochondrial transfer as a means of tissue revitalization. Signal transduction and targeted therapy 6, 65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manevski M, Muthumalage T, Devadoss D, Sundar IK, Wang Q, Singh KP, Unwalla HJ, Chand HS, Rahman I, 2020. Cellular stress responses and dysfunctional Mitochondrial-cellular senescence, and therapeutics in chronic respiratory diseases. Redox Biol 33, 101443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maremanda KP, Sundar IK, Rahman I, 2019. Protective role of mesenchymal stem cells and mesenchymal stem cell-derived exosomes in cigarette smoke-induced mitochondrial dysfunction in mice. Toxicol Appl Pharmacol 385, 114788. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams TG, Prescott AR, Allen GF, Tamjar J, Munson MJ, Thomson C, Muqit MM, Ganley IG, 2016. mito-QC illuminates mitophagy and mitochondrial architecture in vivo. J. Cell Biol 214, 333–345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McWilliams TG, Prescott AR, Montava-Garriga L, Ball G, Singh F, Barini E, Muqit MMK, Brooks SP, Ganley IG, 2018. Basal Mitophagy Occurs Independently of PINK1 in Mouse Tissues of High Metabolic Demand. Cell Metab 27, 439–449.e435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mizumura K, Cloonan SM, Nakahira K, Bhashyam AR, Cervo M, Kitada T, Glass K, Owen CA, Mahmood A, Washko GR, Hashimoto S, Ryter SW, Choi AM, 2014. Mitophagy-dependent necroptosis contributes to the pathogenesis of COPD. J. Clin. Invest 124, 3987–4003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nam HS, Izumchenko E, Dasgupta S, Hoque MO, 2017. Mitochondria in chronic obstructive pulmonary disease and lung cancer: where are we now? Biomark. Med 11, 475–489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nyunoya T, Mebratu Y, Contreras A, Delgado M, Chand HS, Tesfaigzi Y, 2014. Molecular processes that drive cigarette smoke-induced epithelial cell fate of the lung. Am J Respir Cell Mol Biol 50, 471–482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patel RR, Ryu JH, Vassallo R, 2008. Cigarette smoking and diffuse lung disease. Drugs 68, 1511–1527. [DOI] [PubMed] [Google Scholar]
- Scarffe LA, Stevens DA, Dawson VL, Dawson TM, 2014. Parkin and PINK1: much more than mitophagy. Trends Neurosci 37, 315–324. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Solanki HS, Babu N, Jain AP, Bhat MY, Puttamallesh VN, Advani J, Raja R, Mangalaparthi KK, Kumar MM, Prasad TSK, Mathur PP, Sidransky D, Gowda H, Chatterjee A, 2018. Cigarette smoke induces mitochondrial metabolic reprogramming in lung cells. Mitochondrion 40, 58–70. [DOI] [PubMed] [Google Scholar]
- Song C, Luo B, Gong L, 2017. Resveratrol reduces the apoptosis induced by cigarette smoke extract by upregulating MFN2. PLoS One 12, e0175009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Soriano P, 1999. Generalized lacZ expression with the ROSA26 Cre reporter strain. Nat. Genet 21, 70–71. [DOI] [PubMed] [Google Scholar]
- Sundar IK, Maremanda KP, Rahman I, 2019. Mitochondrial dysfunction is associated with Miro1 reduction in lung epithelial cells by cigarette smoke. Toxicol. Lett 317, 92–101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar IK, Rashid K, Gerloff J, Li D, Rahman I, 2018a. Genetic Ablation of p16(INK4a) Does Not Protect against Cellular Senescence in Mouse Models of Chronic Obstructive Pulmonary Disease/Emphysema. Am J Respir Cell Mol Biol 59, 189–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sundar IK, Rashid K, Gerloff J, Rangel-Moreno J, Li D, Rahman I, 2018b. Genetic ablation of histone deacetylase 2 leads to lung cellular senescence and lymphoid follicle formation in COPD/emphysema 32, 4955–4971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Sundar IK, Blum JL, Ratner JR, Lucas JH, Chuang TD, Wang Y, Liu J, Rehan VK, Zelikoff JT, Rahman I, 2020. Prenatal Exposure to Electronic-Cigarette Aerosols Leads to Sex-Dependent Pulmonary Extracellular-Matrix Remodeling and Myogenesis in Offspring Mice. Am J Respir Cell Mol Biol 63, 794–805. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Q, Sundar IK, Lucas JH, Muthumalage T, Rahman I, 2021. Molecular clock REV-ERBα regulates cigarette smoke-induced pulmonary inflammation and epithelial-mesenchymal transition. JCI Insight 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Westermann B, 2010. Mitochondrial fusion and fission in cell life and death. Nat. Rev. Mol. Cell Biol 11, 872–884. [DOI] [PubMed] [Google Scholar]
- Wiegman CH, Michaeloudes C, Haji G, Narang P, Clarke CJ, Russell KE, Bao W, Pavlidis S, Barnes PJ, Kanerva J, Bittner A, Rao N, Murphy MP, Kirkham PA, Chung KF, Adcock IM, 2015. Oxidative stress-induced mitochondrial dysfunction drives inflammation and airway smooth muscle remodeling in patients with chronic obstructive pulmonary disease. J Allergy Clin Immunol 136, 769–780. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Arunachalam G, Hwang JW, Chung S, Sundar IK, Kinnula VL, Crapo JD, Rahman I, 2010. Extracellular superoxide dismutase protects against pulmonary emphysema by attenuating oxidative fragmentation of ECM. Proc Natl Acad Sci U S A 107, 15571–15576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yao H, Chung S, Hwang JW, Rajendrasozhan S, Sundar IK, Dean DA, McBurney MW, Guarente L, Gu W, Rönty M, Kinnula VL, Rahman I, 2012. SIRT1 protects against emphysema via FOXO3-mediated reduction of premature senescence in mice. J. Clin. Invest 122, 2032–2045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Youle RJ, van der Bliek AM, 2012. Mitochondrial fission, fusion, and stress. Science 337, 1062–1065. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Wang KZ, Chu CT, 2013. After the banquet: mitochondrial biogenesis, mitophagy, and cell survival. Autophagy 9, 1663–1676. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Figure 1 Unedited full-blot/gel listed in the supplementary information.
Supplementary Table S1 Gene expression change fold Ratio (CS/Air)