

Abstract
A paradox of eukaryotic cells is that while some species assemble a complex actin cytoskeleton from a single ortholog, other species utilize a greater diversity of actin isoforms. The physiological consequences of using different actin isoforms, and the molecular mechanisms by which highly conserved actin isoforms are segregated into distinct networks, are poorly known. Here, we sought to understand how a simple biological system, composed of a unique actin and a limited set of actin‐binding proteins, reacts to a switch to heterologous actin expression. Using yeast as a model system and biomimetic assays, we show that such perturbation causes drastic reorganization of the actin cytoskeleton. Our results indicate that defective interaction of a heterologous actin for important regulators of actin assembly limits certain actin assembly pathways while reinforcing others. Expression of two heterologous actin variants, each specialized in assembling a different network, rescues cytoskeletal organization and confers resistance to external perturbation. Hence, while species using a unique actin have homeostatic actin networks, actin assembly pathways in species using several actin isoforms may act more independently.
Keywords: actin‐binding proteins, actin cytoskeleton, actin isoforms, biomimetism, Saccharomyces cerevisiae
Subject Categories: Cell Adhesion, Polarity & Cytoskeleton
Swapping of diverse actin genes into yeast allows dissection of how different actin variants are functionally segregated into distinct cellular networks.
Introduction
A fundamental characteristic of eukaryotic cells is the existence of an organized actin cytoskeleton. Dynamic actin filaments are assembled into diverse architectures which coexist within one cytoplasm, each of which is involved in the exertion of forces for various cellular functions (Blanchoin et al, 2014). Key partners are families of actin‐binding proteins (ABPs), which interact with actin monomers and filaments to regulate cytoskeletal organization and dynamics (Moseley & Goode, 2006; Pollard, 2016). Actin sequence is highly conserved across most eukaryotes, but while some cell types only express a single actin (e.g., yeasts), other cell types can express several similar actin isoforms (e.g., nonmuscle mammalian cells express beta‐ and gamma‐actins, which are 99% identical), or even very different actin isoforms (e.g., Chlamydomonas reinhardtii expresses two actins, IDA5 and NAP1, which are only 65% identical) (Gunning et al, 2015; Boiero Sanders et al, 2020). An extreme case is plants, which can express a multitude of actin isoforms (e.g., Zea mays and Arabidopsis thaliana express 21 and 8 actin isoforms, respectively). Adding to this complexity, some actins can undergo partial posttranslational modifications (PTMs), such as arginylation or acetylation, which modify their biochemical properties (Kashina, 2014; A et al, 2020; Boiero Sanders et al, 2020).
Hence, while a number of organisms are able to assemble a complex actin cytoskeleton from one (or a limited number) of actin isoforms, other organisms require the presence of multiple actin isoforms to generate such variability. In line with this idea, segregation of actin isoforms is observed in vivo. Results from different mammalian cell lines have found that beta‐actin was located mainly in the contractile ring, stress fibers, filopodia, and cell–cell contacts while gamma‐actin was localized primarily in the cortex and lamellipodia (Dugina et al, 2009; Chen et al, 2017). In Arabidopsis thaliana, the main vegetative actin isoforms organize into different structures in epidermal cells (Kijima et al, 2018). However, it should be noted that expression in mice of a beta‐coded gamma‐actin, where the nucleotide sequence of beta‐actin is modified minimally to express gamma‐actin, led to viable mice with no detectable change in behavior (Vedula et al, 2017). This result indicates that at least in some cases, the absence of an actin isoform can be compensated by the expression of a similar isoform.
A particular challenge for the field is to understand how small differences at the molecular level lead to a major segregation of actin isoforms at the cellular level. To decipher the underlying mechanisms, it is natural to postulate that actin isoforms bear small yet significant biochemical differences. Our knowledge of the distinctions between actins is limited to a small number of actin orthologs (mainly S. cerevisiae Act1p, rabbit muscle actin, to a lesser extent beta‐ and gamma‐actins, S. pombe Act1p and plant actins). Nonetheless, these studies reveal notable differences in their biochemical properties (Nefsky & Bretscher, 1992; Buzan & Frieden, 1996; Kim et al, 1996; Bryan & Rubenstein, 2005; Takaine & Mabuchi, 2007; Kijima et al, 2016), in their mechanical properties (Orlova et al, 2001; McCullough et al, 2011), and their ability to interact with the different actin‐binding proteins (Nefsky & Bretscher, 1992; Eads et al, 1998; Takaine & Mabuchi, 2007; Ezezika et al, 2009; McCullough et al, 2011; Kang et al, 2014; Kijima et al, 2016), including nucleation factors of actin assembly (Ti & Pollard, 2011; Chen et al, 2017). How such differences account for spatial segregation of actin isoforms on a cellular scale remains unclear.
In this work, we investigated, from a general perspective, the molecular principles by which actin isoforms can be addressed to different networks. Analysis in a model system, that exploits at least two actins to perform various actin functions, would explain a particular mechanism in a relevant physiological context. However, the importance of actin renders genetic manipulations difficult, and the inter‐connection of actin networks in such models complicates cellular analysis. Mammalian systems in particular express many ABP isoforms, which makes interpretation of molecular mechanisms combinatorially challenging. Furthermore, coexpression of multiple actin isoforms makes endogenous purification as a single species difficult, although new powerful protocols have been developed in recent years for their expression and purification (Hatano et al, 2018, 2020). To overcome these limitations, we decided to adopt an alternative strategy, by determining the consequences of heterologous actin expression in a system normally using a single actin. With this approach, we aimed at measuring the consequences of a perturbation caused by the use of a different actin at the level of the cell and its cytoskeleton. We decided to use the well‐studied organism, budding yeast, for the simplicity of its genetics. Another advantage of budding yeast is that actin assembles predominantly into two well‐defined structures. These structures are actin patches, which are sites of endocytosis and where actin filaments are short and branched by the Arp2/3 complex, and actin cables, which are central for maintenance of cell polarity and intra‐cellular trafficking, and where actin filaments are nucleated by the formin family of proteins (Moseley & Goode, 2006). Lastly, budding yeast allows for clean purification of ABPs in a defined organismal context.
Our results demonstrate that actin functions are regulated both at the nucleotide level where defects in actin expression leads to cell growth defects, and at the amino acid level where expression of heterologous actins induces a massive reorganization of the actin cytoskeleton. We demonstrate that actin isoforms are used with different efficiencies by the distinct actin assembly pathways, resulting in their targeting to particular actin structures. Finally, dissection of the underlying molecular mechanisms allows us to propose an explanation of our results, and a general model of the molecular mechanisms enabling segregation of actin isoforms in cells.
Results
Generation of a library of yeast strains expressing a variety of actin orthologs
We created a library of S. cerevisiae strains that express different actin orthologs to evaluate the consequences of actin variation on yeast actin cytoskeleton assembly. In order to ensure that defects were not due to potential misfolding or nonfunctional actin, we selected a diversity of actins from other species rather than using directed mutations. This approach guarantees that the actin orthologs are functional in a biologically relevant context, and maintain key physiological properties such as polymerization, depolymerization, nucleotide binding, and hydrolysis.
We chose 126 different actins from species covering the entire eukaryotic and archeal phylogenetic tree for analysis (Appendix Table S1 and Appendix Fig S1A). We also computationally predicted ancestral sequences to extend the range of actin variant possibilities. Because the actin protein sequence is highly conserved across species, ancestral sequence reconstructions score with high confidence (Appendix Fig S1B). We obtained in total 227 actin sequences (including 101 ancestral actins), from which we selected 19 for analysis. These actin orthologs were chosen to cover a spectrum from the most similar to wild‐type S. cerevisiae’s actin (Act1p, called here Act_Sc) to very divergent actin orthologs, which represent a wide range of identities (from 99 to 60%) (Fig 1A, Table EV1 and Appendix Fig S1B and C), and to display differences across all domains of the actin fold (Fig 1B and C and Appendix Fig S1C).
Figure 1. Variety of actins selected for this study and analysis strategies.
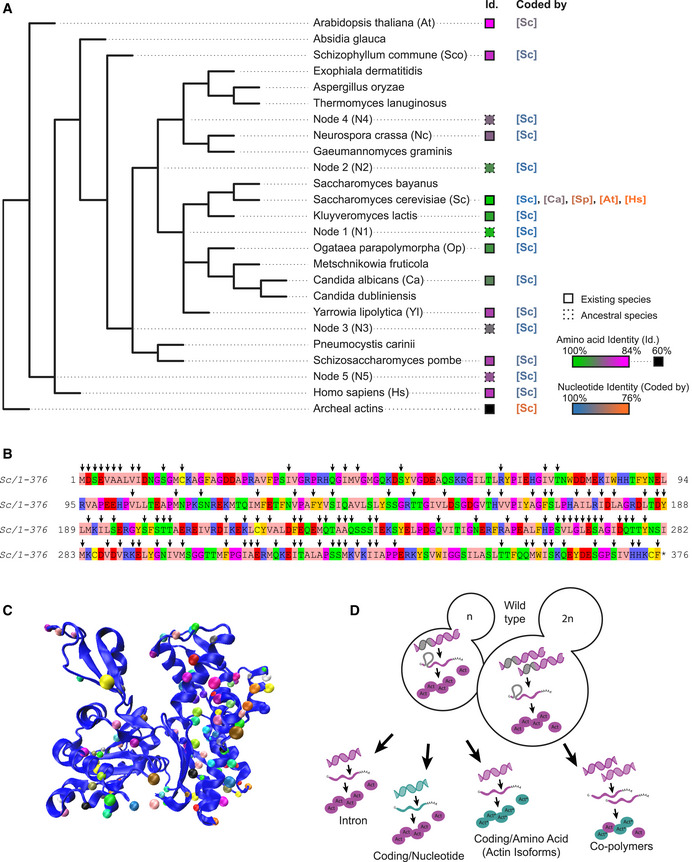
- Simplified phylogenetic tree showing mainly the Dikarya subkingdom and including the external branches Homo sapiens (Hs) and Arabidopsis thaliana (At). The Id. column indicates amino acid sequences percentage identities, ranging from 100% (green) to 84% (magenta) identity for eukaryotic actins to S. cerevisiae’s actin, and 60–62% (black) for archaeal actins. Squares’ outlines are solid or dotted for sequences deriving from existing species or ancestral reconstruction, respectively. The “coded by” column indicates, which coding sequences were originally used to code genes of interest. Nucleotide sequence identities are ranging from 100% (blue) to 76% (orange) compared to S. cerevisiae’s actin coding sequence.
- Amino acid sequence of Saccharomyces cerevisiae actin. Arrows denote all the positions that are mutated in at least one of the actin variants tested in this study.
- Schematic representation of S. cerevisiae actin 3D structure (1YAG; Vorobiev et al, 2003), showing that mutations cover all regions of the protein. Dots indicate where mutations are located, using a different color code for all actins studied here.
- Schematic showing the mutagenesis strategies applied in this study, enabling to question respectively the importance of actin’s intron, the nucleotide sequence, the amino acid sequence, and the effect of expressing copolymers. Green color indicates whether modifications are brought in the coding sequence (leading to expression of wild‐type Act1 protein (pink) or in the amino acid sequence (leading to expression of an Act1* actin ortholog).
We synthesized the actin nucleotide sequences and subcloned them in a plasmid created specifically for rapid and robust actin gene replacement under endogenous promoter control in S. cerevisiae (Appendix Fig S1D). Homologous recombination was performed on diploid cells so that the presence of a wild‐type actin copy would favor viability of the sickest strains. Despite this, we were unable to generate viable strains that could express four highly divergent archeal actins, corresponding to those of phyla Lokiarchaeota, Odinarchaeota, Thorarchaeota, and Heimdallarchaeota, which all share 60–62% of sequence identity with budding yeast actin. All mutants expressing eukaryotic actins could be generated, and were sporulated in order to study haploid cells expressing only the new actin variant. With this strategy, we created an extended library of yeast strains, from which we systematically studied the effect of deleting the actin intron in haploid cells, changing the nucleotide sequence without modifying the final actin protein in haploid cells, switching actin protein variants in haploid cells, and expressing copolymers of actin in diploid cells (Fig 1D).
Previous studies have demonstrated that the yeast actin intron is not essential for actin gene transcription and for normal cell growth (Ng et al, 1985). Indeed, our analysis found that an act1 gene construct without the intron in S. cerevisiae S288C (ScNI) does affect neither cell growth (Appendix Fig S2A and B) nor actin expression (Appendix Fig S2C and D). Fixation and phalloidin‐labeling of the actin cytoskeleton reveals that the two main structures of actin filaments in yeast, actin patches and actin cables, are well‐organized in yeast strains expressing actin in the absence of the intron and indistinguishable from wild‐type cells (Sc) (Appendix Fig S2E–G). Therefore, all experiments presented in the following sections of this study were conducted on actins expressed in the absence of an intron.
Cell fitness tolerates reduced wild‐type actin expression above a threshold
We were concerned that small changes to the actin nucleotide sequence might have consequences on actin expression levels and cell viability (Hoekema et al, 1987; Zhou et al, 2016). In mammals, for instance, nucleotide sequence was shown to differentiate beta and gamma actin functions (Vedula et al, 2017). Therefore, we expressed wild‐type actin from a range of different nucleotide sequences. We used coding sequences from other organisms, which we modified minimally so that the final product remained S. cerevisiae’s actin at the protein level (Table EV1 and Appendix Fig S2H). Western blot analysis showed that silent mutations affect wild‐type actin’s expression level to various extents (Fig 2A and B), with correlation between actin expression and the level of conservation of the nucleotide sequence (Fig EV1A). RNAseq analysis showed, on the contrary, that genes encoding actin regulators are not significantly differentially expressed (Fig EV1B). These data also revealed that a sizeable drop of actin expression (e.g., Act_Sc[Ca], derived from C. albicans’ actin gene, is expressed at 46% of normal level) has no effect on cell viability (Figs 2C and D, and EV1C and Appendix Fig S2I–J). We analyzed the organization of the cytoskeleton of phalloidin‐labeled cells by measuring the total intensity of patches and cables (Appendix Fig S2K), their numbers (Appendix Fig S2L), as well as the overall balance between these two structures whose assembly is interdependent (Burke et al, 2014) by calculating a deviation index (Antkowiak et al, 2019) (Fig 2F). Sc[Ca] cells expressing less actin also have a less bright cytoskeleton, but keep a normal distribution between actin patches and cables, and normal cell polarity (Fig 2H). However, a more drastic drop of actin expression (e.g., Act_Sc[At], derived from A. thaliana’s ACT8 gene, is expressed at 24% of normal level) affects visibly cell viability (Fig 2C and D and Appendix Fig S2I–J), the organization (Fig 2E–G and Appendix Fig S2K–L), and the polarization (Fig 2H) of the actin cytoskeleton. Unexpectedly, none of these strains expressing wild‐type actin showed a significant change in monomeric‐to‐filamentous (G/F) actin ratios (Fig EV1D). Expressing actin from a gene derived from the nucleotide sequence of H. sapiens ActB (Act_Sc[Hs]), whose nucleotide sequence is even less conserved, is lethal for cells. From these observations, we concluded that expression levels of actin orthologs should be controlled carefully in this study, but that half variations in actin expression have negligible effect on cell behavior.
Figure 2. Effects of silent mutations on actin expression levels, cell viability, and cytoskeletal organization.
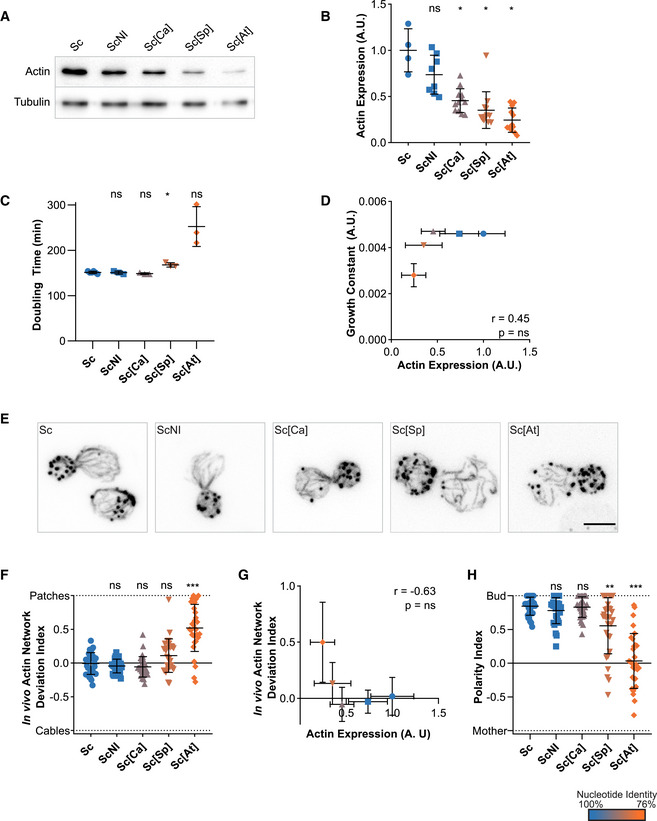
In this figure, the shape of the dots allows to identify the strains on the different graphs (circles for Sc, squares for ScNI, triangles for Sc[Ca], inversed triangles for Sc[Sp] and diamonds for Sc[At]). The color of the dots indicates the percentage of identity of the nucleotide sequences to the actin gene of S. cerevisiae, ranging from 100% (blue) to 76% (orange).
- Actin expression levels shown by western blotting for strains expressing S. cerevisiae’s actin protein from various coding sequences, with tubulin (Tub1p) as a loading control.
- Quantification of actin expression levels, showing a decrease when more silent mutations are present. Data are presented as mean ± SD (n = 4 for Sc, n = 8 for ScNI; n = 12 for Sc[Ca], Sc[Sp], and Sc[At]; 2 biological replicates with n/2 technical replicates each). *P < 0.05 (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
- Doubling times of yeast strains cultures, grown at 25°C in YPD medium. Data are presented as mean ± SD (n = 6 for Sc, n = 3 for ScNI, Sc[Ca], Sc[Sp] and Sc[At]; technical replicates). *P < 0.05 (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
- Level of actin expression as a function of growth constant does not show any clear correlation. Rather, there is an apparent level of actin expression (0.25 < expression < 0.35) below which growth rates drastically reduce. Data are presented as mean ± SD (for actin expression values, n = 4 for Sc, n = 8 for ScNI; n = 12 for Sc[Ca], Sc[Sp] and Sc[At]; 2 biological replicates with n/2 technical replicates each; for growth constants, n = 6 for Sc, n = 3 for ScNI, Sc[Ca], Sc[Sp] and Sc[At]; technical replicates). r is a Pearson correlation coefficient considered nonsignificant if its two‐tailed P‐value is > 0.05.
- Phalloidin staining depicting F‐actin organization. Images are maximum intensity projections of 3D stacks. Scale bar: 3 µm.
- In vivo actin network deviation indexes, defined to evaluate the patch‐cable balance compared to S. cerevisiae haploid cells (value is 0 in S. cerevisiae’s cells, 1 when cells contain only actin patches and −1 when cells contain only cables). Data are presented as mean ± SD (n = 30 for all conditions). ***P < 0.001 (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
- In vivo actin network deviation indexes as a function of actin expression levels does not show any clear correlation. Rather, we observe a threshold of actin expression levels (0.25 < expression < 0.35) below which actin cytoskeleton organization is affected. Data are presented as mean ± SD (for actin expression values, n = 4 for Sc, n = 8 for ScNI; n = 12 for Sc[Ca], Sc[Sp] and Sc[At]; 2 biological replicates with n/2 technical replicates each; for indexes, n = 30 for all conditions). Pearson correlation coefficient r is considered nonsignificant if P > 0.05.
- Polarity indexes, defined to assess whether cell polarity is normal or affected (value is 1 when all patches of medium to large budded cells are present in the bud, and −1 refers when all patches are in the mother cell). Data are presented as mean ± SD (n = 30 for all conditions). **P < 0.01, ***P < 0.001 (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
Data information: Abbreviations: ns ‐ nonsignificant, Sc ‐ wild‐type S. cerevisiae cells, ScNI – S. cerevisiae cells where the actin gene has been replaced with the wild‐type gene but without the intron, Sc[X] – S. cerevisiae cells where the actin gene has been replaced with a gene carrying silent mutations based on the sequences from species X (for the list of species and coding, see Table EV1 or Fig 1).
Source data are available online for this figure.
Figure EV1. Effects of silent mutations on gene expression, cell growth, and monomeric‐to‐filamentous actin ratio, related to Fig 2.
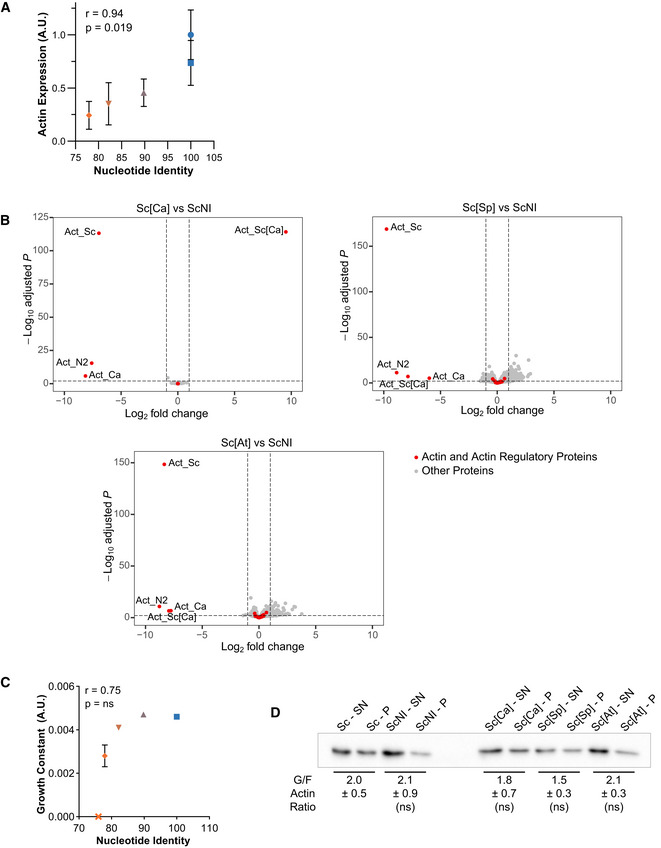
- Actin expression levels, relative to wild‐type, as a function of nucleotide conservation, showing that increased number of silent mutations lowers actin expression. Data are presented as mean ± SD (n = 4 for Sc, n = 8 for ScNI; n = 12 for Sc[Ca], Sc[Sp] and Sc[At]; 2 biological replicates with n/2 technical replicates each). Pearson correlation coefficient r is considered nonsignificant if P > 0.05.
- Differential gene expression of Sc[Ca], Sc[Sp] and Sc[At] strains compared to ScNI strain. Y‐axis represents the adjusted P‐value of FDR (False Discovery Rate) calculated with Benjamini and Hochberg method, and X‐axis fold‐changes. Red dots highlight proteins of interest (actin and 32 regulatory proteins) and grey dots represent all the other proteins identified by RNA‐seq.
- Growth constant as a function of nucleotide identity, showing a threshold of nucleotide conservation (78% < id < 82%) below which growth rates drastically reduce. Data are presented as mean ± SD (n = 6 for Sc, n = 3 for ScNI, Sc[Ca], Sc[Sp] and Sc[At]; technical replicates). Pearson correlation coefficient r is considered nonsignificant if P > 0.05.
- Evaluation of monomeric‐to‐filamentous actin ratios. Data are presented as mean ± SD (n = 12 for Sc and ScNI and 4 for Sc[Ca], Sc[Sp] and Sc[At]; 2 biological replicates with n/2 technical replicates each). (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
Actin amino acid sequence variations affect cell fitness and imbalance the linear‐to‐branched actin network ratio
We next focused our attention on the consequences of expressing 15 heterologous eukaryotic actin orthologs in yeast haploid cells. Actin genes were designed based on S. cerevisiae’s act1 sequence by making point mutations using yeast codon usage. Overall, all coding sequences used in this section are more than 90% identical to that of S. cerevisiae, which, according to the previous section, lowers the risk that actin expression is reduced excessively. Only 8 actin orthologs led to viable conditions. Their expression level varied, and appeared not to be correlated with the evolutionary relationship (Fig 3A and B). For example, Act_N1 was only expressed at 39% despite having a 98.4% identity to wild‐type actin and showed normal viability and cytoskeletal organization (Fig 3A–G). For two strains studied in detail in this article, N2 and Ca, we also verified by RNAseq that genes encoding actin regulators, including the Arp2/3 complex and the two formins Bni1 and Bnr1, are not significantly differentially expressed (Fig EV2A).
Figure 3. Effects of swapping actin for different variants on cell viability and cytoskeletal organization.
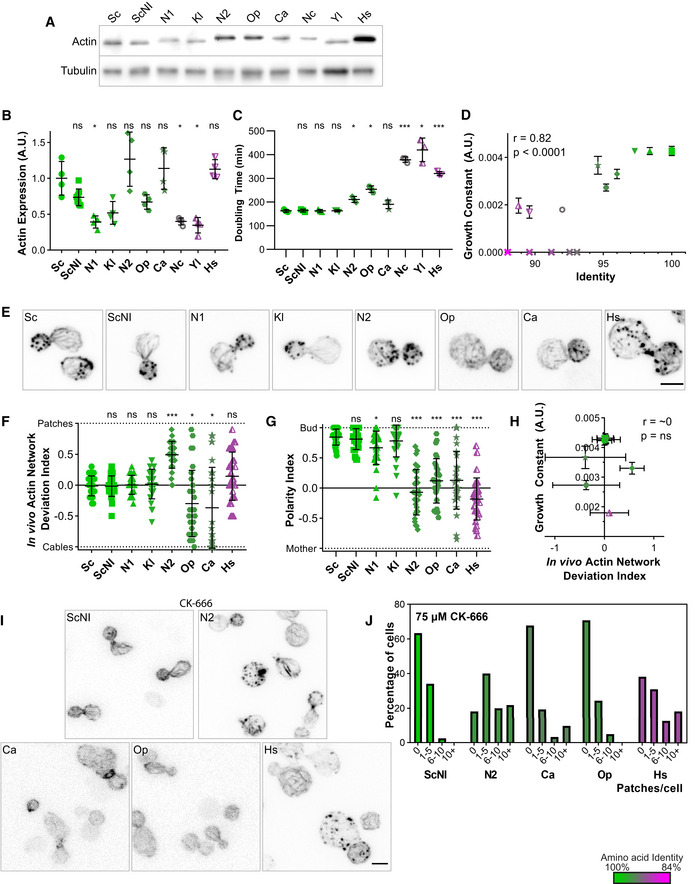
In this figure, the shape of the dots allows to identify the strains on the different graphs (closed circles for Sc, closed squares for ScNI, closed triangles for N1, inversed closed triangles for Kl, closed diamonds for N2, closed pentagons for Op, stars for Ca, open circles for Nc, half‐open triangles for Yl, half‐open inversed triangles for Hs and crosses for nonviable strains). The color of the dots indicates the percentage of identity of the amino acid sequences to S. cerevisiae’s actin, ranging from 100% (green) to 84% (magenta).
- Actin expression levels shown by western blotting for strains expressing S. cerevisiae’s actin or other actins, with tubulin (Tub1p) as a loading control.
- Quantification of actin expression levels showing varying levels of expression that do not correlate with evolutionary relationship. Data are presented as mean ± SD (n = 8 for ScNI, n = 4 for all other strains; 2 biological replicates with n/2 technical replicates each). *P < 0.05 (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
- Doubling times of yeast strain cultures grown at 25°C in YPD medium. Data are presented as mean ± SD (n = 3 for all conditions; technical replicates). *P < 0.05, ***P < 0.001 (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
- Growth constant as a function of percentage identity of the actin variant, showing clear correlation (n = 3 for all conditions; technical replicates). Data are presented as mean ± SD. r corresponds to the Pearson correlation coefficient with a two‐tailed P value and a confidence interval of 95%.
- Phalloidin staining of F‐actin organization. Images are maximum intensity projections of 3D stacks and contrasts were adapted due to the fact that phalloidin labeling had a very different efficiency depending on the actin ortholog expressed. Micrographs of Sc and Sc_NI cells are reproduced from Fig 2E. Scale bar: 3 µm.
- In vivo actin network deviation indexes. Data are presented as mean ± SD (n = 30 for all conditions). *P < 0.05, ***P < 0.001 (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
- Polarity indexes. Data are presented as mean ± SD (n = 30 for all conditions). *P < 0.05, ***P < 0.001 (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
- Growth constant as a function of the in vivo actin network deviation index. Data are presented as mean ± SD (for growth constants, n = 3 for all conditions; technical replicates; for indexes, n = 30 for all conditions). r is a Pearson correlation coefficient considered nonsignificant if its two‐tailed P‐value is > 0.05.
- Effect of CK‐666 (75 µM) on the organization of the actin cytoskeleton. Cells were stained with phalloidin after 30 min incubation with CK‐666. Images are maximum intensity projections of 3D stacks. Scale bar: 3 µm.
- Quantification of actin patch resistance to CK‐666 treatment. Bar graphs represent the percentage of cells with a given number of visible actin patches after CK‐666 treatment. (n = 41 for ScNI, 55 for N2, 31 for Ca, 41 for Op, 55 for Hs).
Data information: Abbreviations: ns—nonsignificant, Sc – wild‐type S. cerevisiae cells, ScNI – S. cerevisiae cells where the actin gene has been replaced with the wild‐type gene but without the intron, the other abbreviations correspond to cells expressing actins from other species (for the list of species see Table EV1 or Fig 1).
Source data are available online for this figure.
Figure EV2. Effects of swapping actin for different variants on gene expression and monomeric‐to‐filamentous actin ratio, related to Fig 3.
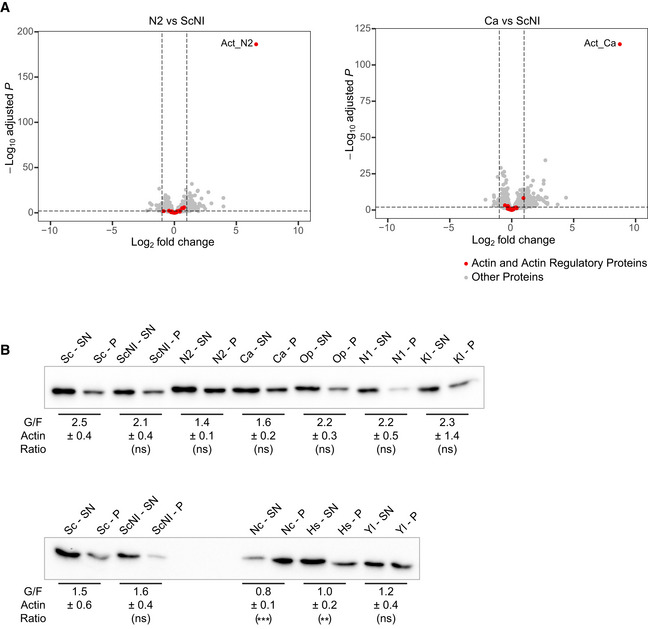
- Differential gene expression of Ca and N2 strains compared to ScNI strain. Y‐axis represents the adjusted P‐value of FDR (False Discovery Rate) calculated with Benjamini and Hochberg method, and X‐axis fold‐changes. Red dots highlight proteins of interest (actin and 32 regulatory proteins) and grey dots represent all the other proteins identified by RNA‐seq.
- Evaluation of monomeric‐to‐filamentous actin ratios. Control strains and strains expressing the most (resp. the least) conserved actin variants are analyzed in the upper (resp. the lower) western blot. Data are presented as mean ± SD (n = 12 for Sc and ScNI and n = 4 for all other conditions; 2 biological replicates with n/2 technical replicates each). **P < 0.01, ***P < 0.001 (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
Source data are available online for this figure.
While yeast strains expressing heterologous actin orthologs similar to S. cerevisiae wild‐type actin (identity > 97%) grew well (Fig 3C and D and Appendix Fig S3D and E), had normal cytoskeletal organization (Fig 3E and F and Appendix Fig S3F and G), and were polarized normally (Fig 3G), yeast strains expressing more distant actins (identity < 97%) showed moderate to severe defects (Fig 3C–H). The strength of the growth phenotypes correlated with the degree of conservation of the actin orthologs (Fig 3D). Of the 8 strains expressing actin orthologs, only 6 could be labeled with phalloidin at varying levels of intensity, which can be attributed, apart from Act_N1, to different phalloidin binding sites from Act_Sc (Appendix Fig S3F and H). Interestingly, consequences on the organization of the actin cytoskeleton were not the same for all mutants. While cells expressing Act_N2 assembled, on average, an abnormally high number of actin patches, strains expressing Act_Op or Act_Ca assembled, on the contrary, a higher number of actin cables and few actin patches (Fig 3E, F and H, and Appendix Fig S3F–G). Though, the overall G/F actin ratio was maintained in these strains (Fig EV2B). Considering that actin networks do not assemble independently in cells, but that homeostatic actin networks share a limited monomeric actin pool (Burke et al, 2014), these observations suggest an imbalanced assembly between branched‐ and linear‐actin structures from the use of different actin variants. We hypothesized that the cellular machinery cannot use Act_N2 efficiently to assemble actin cables, and cannot use Act_Op or Act_Ca to assemble actin patches, thus leading to an overproduction of patches in N2 cells and an overproduction of cables in Op and Ca cells. It is also possible that patch or cable assembly is boosted by the use of a particular actin ortholog, although this hypothesis seems less likely, since it is generally easier to disrupt a function than make it more efficient. In strains expressing the three most divergent actin orthologs Act_Nc, Act_Yl, and Act_Hs, phenotypes were even stronger, but cells also failed to maintain a normal G/F actin ratio (Fig 3C and D, EV2B and Appendix Fig S3D and E). Fewer F‐actin was assembled, suggesting that both actin assembly pathways may be affected by the use of these actin isoforms.
In wild‐type cells where actin networks are in homeostasis, addition of the small molecule inhibitor CK‐666 shifts efficiently actin assembly from patches to cables (Burke et al, 2014). We tested whether such property was conserved in the strains studied here. We observed that a strain over‐assembling actin patches, such as N2, showed acute resistance to Arp2/3 perturbations with a persistence of actin patches on treatment with CK‐666 (Fig 3I and J, and Appendix Fig S3I–J). Conversely, Op and Ca strains were more sensitive to CK‐666. These results indicate that strains with increased branched network are buffered against Arp2/3 perturbations. To further understand the mechanism of resistance to CK‐666, we attempted to compare the density of actin networks in individual patches of Sc and N2 cells. Actin patches from N2 cells are 1.6‐fold brighter for both phalloidin‐actin and Arc15‐GFP labeling (Appendix Fig S3 K–M). However, the higher phalloidin labeling intensity of N2 cells (Appendix Fig S3F) for similar amount of expressed (Fig 3A and B) and assembled (Fig EV2B) actin as wild‐type cells suggests a higher affinity of phalloidin for Act_N2, and therefore an overestimation of the amount of actin assembled in N2 cells. Taken together, these crude estimations suggest that individual actin patches of N2 cells are more densely branched than those of wild‐type cells.
A biomimetic assay recapitulates actin ortholog preference for branched‐ or linear‐network assembly
We then aimed to understand the molecular principles that allow different actin ortholog to be assembled specifically to certain actin networks, and hypothesized that heterologous actin orthologs may bind defectively to certain ABPs of S. cerevisiae. Because actin assembly into patches and cables involves a large number of proteins in cells, we adopted a reductionist approach based on a reconstituted assay. We considered the subset of ABPs that are most essential for actin patch or cable assembly in vivo. Beyond formins and the Arp2/3 complex, these proteins include profilin, a small globular protein that favors formin assembly, capping protein, a heterodimer that binds to barbed ends, ADF/cofilin, a small protein that promotes the disassembly of actin filaments, and tropomyosin, a helical coiled‐coil protein that binds and stabilizes linear‐actin filaments nucleated by formins (Moseley & Goode, 2006; Pollard, 2016).
In addition to wild‐type actin, we purified Act_N2 and Act_Ca from cultures of the corresponding yeast strains. We reconstituted in vitro, in a common experimental environment, branched‐ and linear‐actin network assembly, respectively, from WASp‐ and formin‐coated beads (Antkowiak et al, 2019). We used in these experiments a small fraction of Alexa‐568‐labeled rabbit muscle actin (1%), which integrates equally well to Act_N2 and Act_Ca actin filaments (Appendix Fig S4A and B). First, we assessed the capabilities of the different actin orthologs to assemble into such networks. Act_Ca assembled only into linear‐actin networks (Fig 4A), providing explanation for the inability of Act_Ca cells to assemble actin patches. Act_N2 assembled both into branched‐ and linear‐actin networks, although noticeably shorter but brighter Arp2/3 comet tails were observed for an overall similar amount of assembled actin, in striking similarity with what we have estimated for actin patches in N2 cells.
Figure 4. In vitro reconstitution of branched‐ and linear‐actin networks assembly from purified actins.
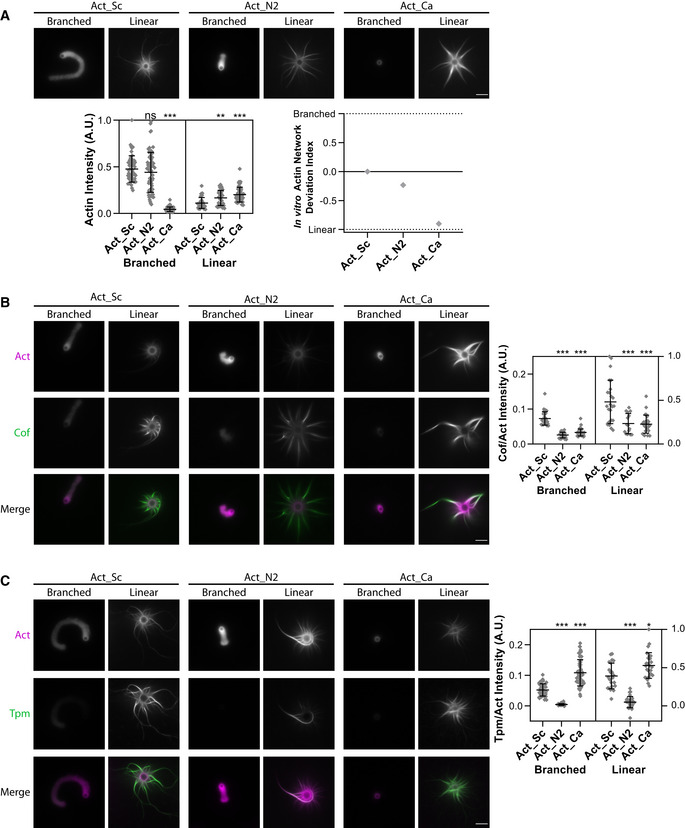
Standard conditions include Las17‐ (branched) and Bni1‐ (linear) coated beads, 8 µM F‐actin assembled from different purified variants and 1% labeled with Alexa‐568 rabbit muscle actin, 15 µM of profilin, 1 µM of capping protein, 500 nM of Arp2/3 complex and 600 nM of ADF/cofilin. Snapshots of representative actin networks were taken after 30 min. Scale bars: 6 µm.
- (Top) Snapshots of actin networks assembled from three different actins sources: Act_Sc, Act_N2, and Act_Ca. (Bottom left) Quantification of actin fluorescence on beads. Data are presented as mean ± SD (n = 52 for Act_Sc branched, n = 70 for Act_N2 branched, n = 81 for Act_Ca branched, n = 32 for Act_Sc linear, n = 34 for Act_N2 linear and n = 47 for Act_Ca linear). **P < 0.01, ***P < 0.001 (Kruskal–Wallis test, with multiple comparisons). (Bottom right) In vitro actin network deviation indexes.
- (Left) Snapshots of representative actin networks assembled in the presence of 600 nM Alexa‐488‐labeled ADF/cofilin (replacement of unlabeled ADF/cofilin). (Right) Quantification of ADF/cofilin’s fluorescence intensity relative to actin. Data are presented as mean ± SD (n = 34 for Act_Sc branched, n = 28 for Act_N2 branched, n = 43 for Act_Ca branched, n = 24 for Act_Sc linear, n = 15 for Act_N2 linear and n = 27 for Act_Ca linear). ***P < 0.001 (Kruskal–Wallis test, with multiple comparisons).
- (Left) Snapshots of representative actin networks assembled in the presence 1 µM Alexa 488‐tropomyosin. (Right) Quantification of tropomyosin’s fluorescence intensity relative to actin. Data are presented as mean ± SD (n = 45 for Act_Sc branched, n = 63 for Act_N2 branched, n = 57 for Act_Ca branched, n = 22 for Act_Sc linear, n = 32 for Act_N2 linear and n = 27 for Act_Ca linear). *P < 0.05, ***P < 0.001 (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
Data information: For all microscopy images, intensity levels were adapted for images of branched‐ and linear‐actin networks separately as their brightness is different. To obtain a correct representation of the amount of ADF/cofilin and tropomyosin bound to the two networks, the intensity levels of ADF/cofilin and tropomyosin were matched similarly to those of the corresponding actin networks. Abbreviations: ns—nonsignificant, Act_Sc—purified S. cerevisiae actin, Act_N2—purified Node 2 actin, Act_Ca—purified C. albicans actin (for more details see Table EV1, Fig 1 and Appendix Fig S1B and C).
Source data are available online for this figure.
As this simplest protein system could not explain the impossibility of yeast to assemble cables from Act_N2, we hypothesized that an additional ABP, involved in the stabilization or disassembly of one of those actin networks, may bind abnormally to Act_N2. We labeled ADF/cofilin, which is known to promote branched‐network disassembly by inducing Arp2/3 debranching while stabilizing linear networks (Michelot et al, 2007; Chan et al, 2009). ADF/cofilin bound to linear‐actin networks with higher affinity than to the branched‐actin networks (Fig 4B), as previously reported (Gressin et al, 2015). However, ADF/cofilin bound similarly to both actin variants, albeit with reduced affinity compared to wild‐type actin (Fig 4B). We next labeled tropomyosin, which inhibits branched‐network assembly and promotes linear‐network stabilization (Appendix Fig S4C; Bernstein & Bamburg, 1982; Pruyne et al, 1998; Blanchoin et al, 2001; DesMarais et al, 2002; Antkowiak et al, 2019). Tropomyosin bound with higher efficiency to linear‐actin networks, as expected (Fig 4C). Its binding to Act_Sc and Act_Ca was similar; however, tropomyosin was almost completely absent from actin networks assembled from Act_N2 (Fig 4C). This inability of tropomyosin to bind to Act_N2 provides a likely explanation for why actin patch assembly is favored in N2 cells.
Structural analysis provides plausible explanation of defective interactions
We searched for a structural understanding of why Act_N2 and Act_Ca do not interact properly with specific ABPs of S. cerevisiae. Based on the structural information available of the interactions of actin with its binding partners, we identified actin residues that are within 5 Å at the actin–actin interface in a filament, or at the interface between G‐ or F‐actin and the ABPs used in our biomimetic assay (Winn et al, 2011), with the exception of the Arp2/3 mother filament, which were within 10 Å since the coordinates were not released when this study was performed (Fäßler et al, 2020; Fig 5A).
Figure 5. ABPs interfaces with actin.
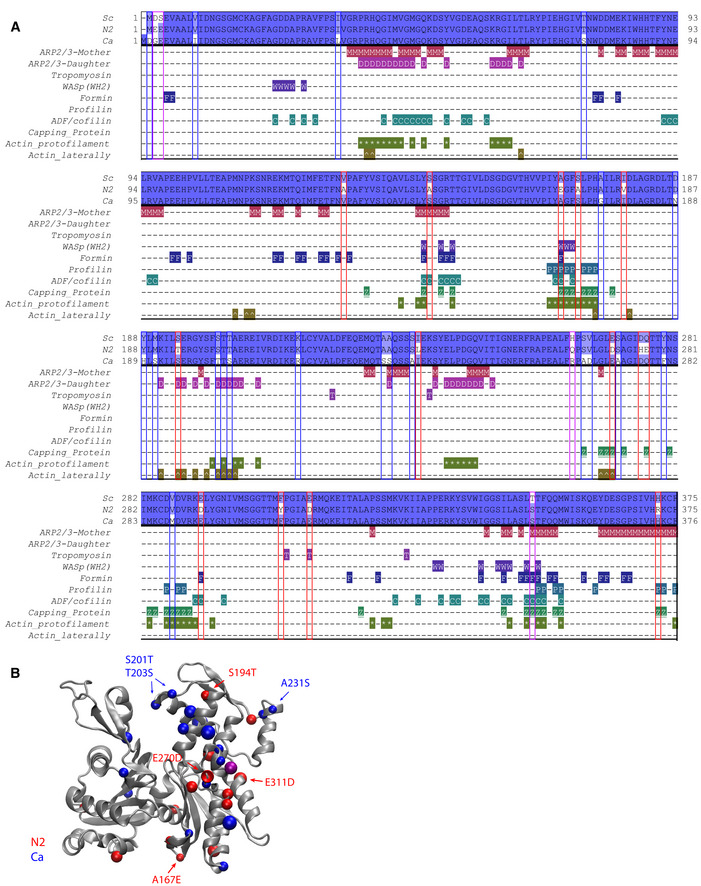
- Sequence alignment of three actins (Act_Sc, Act_N2, and Act_Ca; deep blue indicates conserved residues, light blue and white indicates nonconserved), indicating contacts between proteins used in the biomimetic assay (with Arp2/3 complex at the mother filament interface (M), with Arp2/3 complex at the daughter filaments interface (D), with tropomyosin (T), with WASP’s WH2 (W), with formin (F), with profilin (P), with ADF/cofilin (C), with capping protein (Z), at the protofilament interface (*) and laterally (^).
- Schematic representation of actin 3D structure (1YAG; Vorobiev et al, 2003). Color dots correspond to positions where Act_Sc has different residues compared to Act_N2 (red) and Act_Ca (blue). Purple dots correspond to positions where both Act_N2 and Act_Ca have different residues compared to Act_Sc.
Data information: Abbreviations: Sc—S. cerevisiae, N2—Node 2, Ca—C. albicans (for more details, see Table EV1, Fig 1, and Appendix Fig S1B and C).
At protomer:protomer interfaces, wild‐type actin differed by one residue (Val287Met) and two residues (Ala167Glu and Ser170Ala) relative to Act_Ca and Act_N2, respectively (Fig 5B). In particular, the Ala167Glu substitution has been shown to affect actin filament stiffness (Kang et al, 2012; Hocky et al, 2016; Scipion et al, 2018). Furthermore, four differences were observed in inter‐strand contacts relative to Act_Sc (Ser194Thr and Glu270Asp for Act_N2) and (Ser201Thr and Thr203Ser for Act_Ca) (Fig 5B). Together, these substitutions may subtly alter the relative filament plasticity, which in turn may have an influence on the association or activity of filament binding and filament nucleating proteins (McCullough et al, 2011; von der Ecken et al, 2015). In addition, we identified 15 nonconserved residues of Act_N2 or Act_Ca that are surface exposed on the actin protomer structures and contact a binding partner (Table EV2). Tropomyosin is likely to be particularly susceptible to small changes in the actin filaments, since it loosely associates with the actin filament surface via shape and charge complementarity (Popp & Robinson, 2012; von der Ecken et al, 2016). Particularly, Act_N2 filament Asp311 potentially places the negative charge at ~1.5 Å closer to the actin, relative to the Act_Sc and Act_Ca filaments (glutamic acid), which may be inappropriate for tropomyosin binding. Act_N2 has substitutions in interfaces with all the proteins used in the in vitro assays, including Arp2/3 and formin interfaces, which could have impaired the activities of these filament nucleating complexes. Act_Ca has fewer substitutions in the actin regulating proteins, with the notable exception of Arp2/3. In particular, substitutions in the actin interfaces with Arp2/3 subunits in the daughter filament may indicate that the nucleation process of the daughter filament is impaired for Act_Ca with S. cerevisiae’s Arp2/3.
Dual expression of a patch and a cable‐favoring actin rescues cell viability and cytoskeletal organization
The identification of heterologous actin orthologs favoring the specific assembly of actin patches or cables suggested that actin functions could be separated from the use of two carefully selected actin variants (Fig 6A). To test this possibility, we switched to a diploid yeast cell background. We verified first that both N2/N2 and Ca/Ca cells display similar phenotypes to their haploid equivalents, with slow growth and unbalanced actin patch and cable assembly (Fig 6B–D and Appendix Fig S5A–C). We then crossed strains to generate cells (N2/Ca and Ca/N2) expressing a total amount of actin comparable to control conditions (Fig EV3A), and shared between the two variants of interest (Fig EV3B). We also controlled that the level of expression of actin regulators is maintained in cells expressing Act_N2 and Act_Ca (Fig EV3C). Strikingly, cell growth (Fig 6B and Appendix Fig S5A and B), actin cytoskeleton organization (Figs 6C–E and EV3D and Appendix Fig S5C), and cell polarity (Fig 6E) were rescued in diploid cells expressing both Act_N2 and Act_Ca. However, verification that each of the actin structures was enriched by each of the variants is difficult to do in the absence of specific antibodies; nevertheless, our results indicate that defective actin functions in cells carrying a single actin variant were carried out more normally when the other actin variant was simultaneously expressed.
Figure 6. Effect of a dual expression of actins on cell viability and cytoskeletal organization.
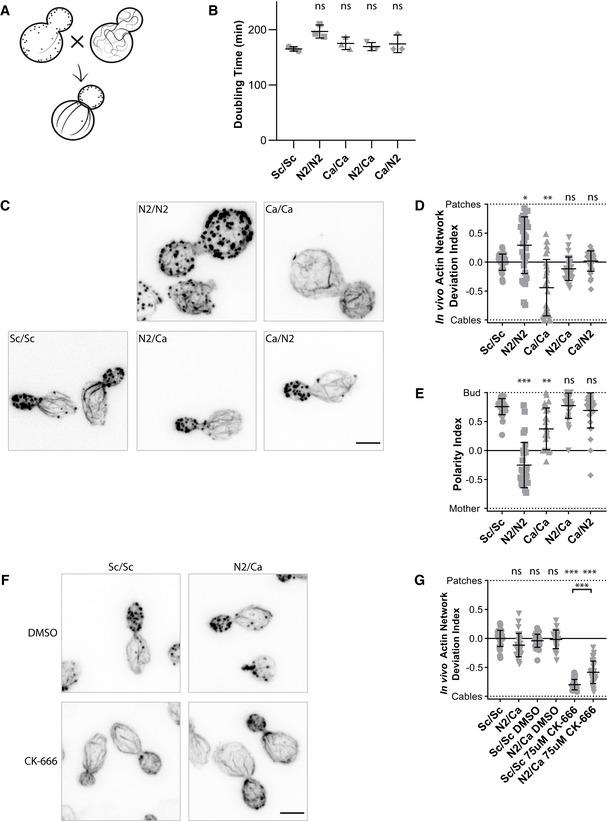
- Schematic of the experiment performed in a diploid yeast background.
- Doubling times of diploid yeast strains cultures, grown at 25°C in YPD medium. N2/Ca and Ca/N2 cells express the same actins but markers used for selection are exchanged. Data are presented as mean ± SD (n = 3 for all conditions; technical replicates). (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
- Phalloidin staining depicting F‐actin organization. Images are maximum intensity projections of 3D stacks. Scale bar: 3 µm.
- In vivo actin network deviation indexes. Data are presented as mean ± SD (n = 30 for all conditions). *P < 0.05, **P < 0.01 (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
- Polarity Indexes. Data are presented as mean ± SD (n = 30 for all conditions). **P < 0.01, ***P < 0.001 (Kruskal–Wallis test, with multiple comparisons).
- Effect of CK‐666 (75 µM) on the organization of the actin cytoskeleton. Cells were stained with phalloidin after 30‐min incubation with CK‐666. Images are maximum intensity projections of 3D stacks. Scale bar: 3 µm.
- In vivo actin network deviation indexes of cells treated with DMSO or CK‐666. Data are presented as mean ± SD (n = 30 for all conditions). ***P < 0.001 (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
Data information: Each strain is represented by a dot of specific shape in all panels. Abbreviations: ns—nonsignificant, Sc/Sc—wild‐type diploid S. cerevisiae cells, N2/N2 –diploid S. cerevisiae cells expressing only N2 actin, Ca/Ca—diploid S. cerevisiae cells expressing only C. albicans actin, N2/Ca and Ca/N2—diploid S. cerevisiae cells expressing N2 actin and C. albicans actin at the same time (for more details, see Table EV1, Fig 1, and Appendix Fig S1B and C).
Source data are available online for this figure.
Figure EV3. Effect of a dual expression of actins on protein expression and monomeric‐to‐filamentous actin ratio, related to Fig 6.
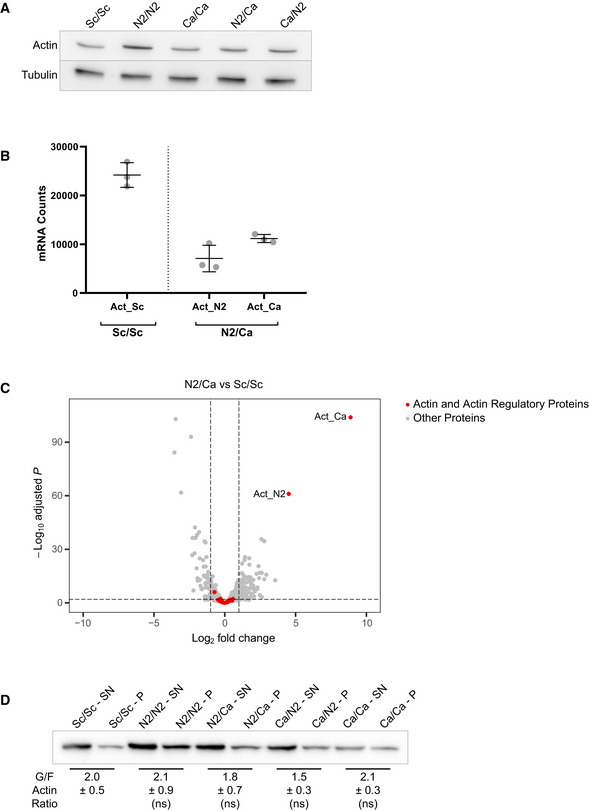
- Total actin expression levels shown by western blotting for diploid strains expressing one or two actin variants, with tubulin (Tub1p) as a loading control.
- Comparison of actin gene expression levels in Sc/Sc and Ca/N2 strains. Data are presented as mean ± SD (n = 3 for all conditions; biological replicates).
- Differential gene expression of N2/Ca strain compared to Sc/Sc strain. Y‐axis represents the adjusted P‐value of FDR (False Discovery Rate) calculated with Benjamini and Hochberg method, and X‐axis fold‐changes. Red dots highlight proteins of interest (actin and 32 regulatory proteins) and grey dots represent all the other proteins identified by RNA‐seq.
- Evaluation of monomeric‐to‐filamentous actin ratios. Data are presented as mean ± SD (n = 4 for all conditions; 2 biological replicates with 2 technical replicates each). (Brown–Forsythe and Welch ANOVA tests, with Dunnett’s T3 multiple comparisons tests).
Source data are available online for this figure.
F‐actin network homeostasis is affected in a two‐actin system
Generation of yeast strains with partially separated actin functions enabled us to question some differences between species sharing a single actin for multiple cellular functions, and species using different actin variants. We were especially curious to know what the physiological consequences would be on actin network homeostasis for wild‐type diploid (Sc/Sc) cells and N2/Ca cells, which share the same ratio of branched and linear network but possess different actin variants. As expected, addition of CK‐666 in wild‐type cells resulted in the disappearance of actin patches and an increase of actin cables (Fig 6F–G). On the contrary, addition of CK‐666 to N2/Ca cells had a weaker effect, as a large number of actin patches could still be observed (Fig 6F–G). Together, these results show that while F‐actin network homeostasis is preserved in a yeast strain using a single actin ortholog, actin re‐distribution from one network to another is less effective in the context of a yeast strain expressing two different actin variants.
Discussion
Budding yeast tolerates reduced expression of actin
Here, we investigated, at the cellular and at the molecular level, the consequences of perturbing a simple system, which uses a single actin ortholog and a limited number of regulatory proteins to assemble an organized actin cytoskeleton. We first found that a sizeable drop of actin expression in S. cerevisiae has no or little effect on the cell. For example, it is clear that Act_N1, which differs from wild‐type actin only by 5 amino acids (all located far from the binding site of the antibody used in this study), is expressed at 39% but that cells are indistinguishable from wild type at the sensitivity of our experiments. This result is surprising, and even seems contradictory with the fact that actin networks compete for a limited monomeric actin pool. Our measurements of G/F actin ratios in Sc and N1 strains do not reveal any significant difference. However, we would like to point out that these measurements do not distinguish actin monomers and filaments from small oligomers. Several studies report the presence of important amount of actin oligomers in yeast and other eukaryotic cell types (Okreglak & Drubin, 2010; Smith et al, 2013; Qu et al, 2015; Raz‐Ben Aroush et al, 2017); their function is not clear, but it is possible that they are not only intermediate species between filamentous and monomeric actin during actin disassembly. They may contribute to actin assembly (Okreglak & Drubin, 2010; Smith et al, 2013), but we would like to put forward the possibility that they could serve to buffer actin level variations in order to maintain constant levels of monomeric actin. This hypothesis could explain how a strain like N1 manages to maintain limiting but constant levels of monomeric actin despite different overall actin levels. A verification of this hypothesis would deserve a complete study, which is beyond the scope of this paper, but the strains generated here could help the community to address this question.
Identification of actin variants that favor branched‐ or linear‐actin networks assembly
Then, we demonstrated that small variations in the actin amino acid sequence are sufficient to induce a global reorganization of the actin cytoskeleton. This finding highlights the fact that, despite the remarkably high sequence conservation of actin orthologs across species, there are sufficient differences in sequence for cells to segregate multiple actin variants into diverse actin networks. It also highlights how careful one has to be in performing biochemical assays with actin binding proteins and actin from different species or cell biology studies where actin orthologs from other species are introduced.
Generally, we found that mutant cells expressing a heterologous actin assemble an abnormal distribution of actin patches and cables. This result is coherent with the literature, which shows particularly in yeast that homeostatic actin networks compete for a limited pool of actin monomers (Burke et al, 2014; Fig 7 top and bottom left). In this context, it is rational to postulate that the inability of an actin variant to assemble efficiently in a given actin network leads to an expansion of the other actin networks, provided that those can use this actin normally (Fig 7 top middle). This is the case at least with the most conserved of these actin variants, where conservation of G/F actin ratios suggests that actin is well redistributed toward these structures. It may not be the case for the less well‐conserved actin variants, which show higher G/F actin ratios. In these strains, it is possible that both assembly pathways are perturbed, leading to an incomplete actin redistribution and increased pool of monomeric actin.
Figure 7. Schematic model of the differences between a cell expressing one or two actins to perform two cellular functions.
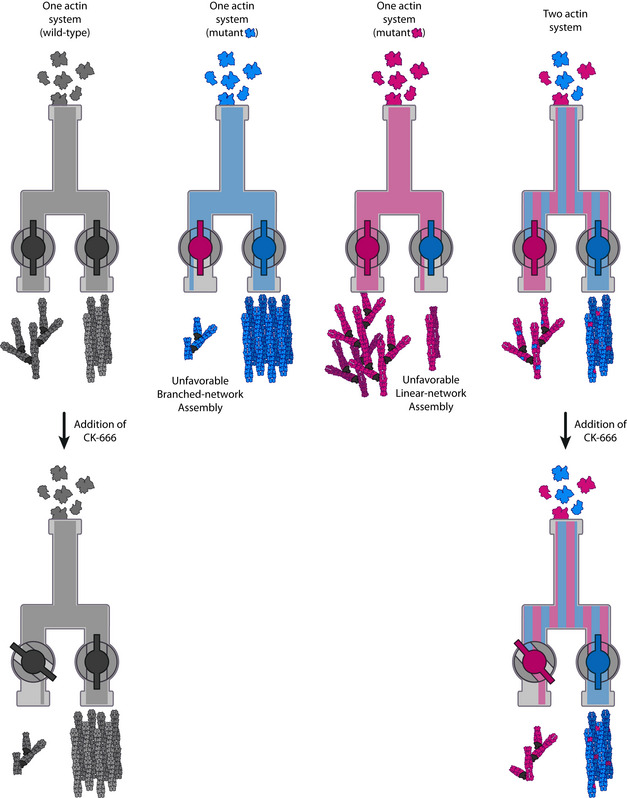
(Top) A model of the molecular mechanisms by which two actin isoforms may segregate to different actin networks. On the left, a system carrying wild‐type actin is able to generate both the branched and linear‐actin networks. On the two central panels, defective interactions of an actin isoform with one or several ABPs affect branched‐ or linear‐network assembly. On the right, combining these two actin variants in one cell should trigger a natural segregation of actins and rescues the wild‐type actin organization. (Bottom) Effect of perturbing an actin assembly pathway for cells using one or two actin variants. On the left, when one actin is shared for two actin functions, the inhibition of one actin assembly pathway (e.g., branched networks with CK‐666) leads to a reinforcement of the other actin assembly pathway. On the right, when two actin variants are used for two different actin functions, this effect is limited as both actin networks assemble more independently. In other words, having a system with two actin variants can buffer against the addition of the drug.
The possibility to rescue yeast cell viability with the simultaneous expression of patch‐favoring and cable‐favoring actin variants reinforces the possibility that actin isoforms compensate for the other actin’s lack of efficiency to form a certain structure (Fig 7 top right). The lack of specific probes that can differentiate between these actin variants prevented us from localizing them in cells and from verifying the extent of segregation. We expect the integration of an actin variant within a particular network to be dependent on its innate ability to assemble in such a network, and also to be affected by relative efficiencies of other coexpressed actin variants to integrate within branched‐ and linear‐actin networks. Such a hypothesis is purely speculative and should be formally tested in the future. Nevertheless, our observations strongly suggest that we were successful in performing a partial separation of function, and in transforming yeast from being an organism that uses a single actin into an organism using two actin variants to perform several actin‐based functions.
Molecular subtleties guide actins to appropriate networks
This study was originally motivated to provide a systematic description of the molecular principles by which different actin isoforms could become spatially segregated into different networks. Experiments performed here show that a biomimetic system, using a reduced set of essential proteins for patch and cable assembly, is sufficient to provide basic molecular explanation of differences observed in cells. While one actin (Act_Ca) seems inefficient in nucleating or assembling into branched‐actin networks, the other actin (Act_N2) seems to assemble in both types of actin networks. However, Act_N2 is defective in binding to tropomyosin, which is an essential component for cable stability in cells, as it protects them from the action of disassembly factors such as ADF/cofilin. This result highlights that the segregation of actin isoforms can be influenced after filament nucleation. Although actin nucleators tend to be in the spotlight, it must be stressed that most ABPs have different effects on branched‐ and linear‐actin networks, and their influence needs to be taken into account (Rotty et al, 2015; Suarez & Kovar, 2016; Antkowiak et al, 2019). Proteins like tropomyosin and ADF/cofilin stabilize linear networks of actin filaments, while enhancing disassembly of branched networks. In this context, any actin variant with defective binding to ADF/cofilin or tropomyosin will naturally be more present within branched networks, while absent from linear ones.
Overall, the principles outlined above should be valid regardless of the mechanism by which variation is brought to the specific actin, whether it is through changes in the peptide sequence or through posttranslational modifications. Also, from an evolutionary perspective, the proposed mechanism appears to be efficient in allowing the emergence of new actin isoforms associated with discrete actin functions. Our model implies that a simple actin gene duplication, followed by minimal mutation in one actin copy, which impairs an essential interaction with an ABP, could be sufficient to trigger a global reorganization of the actin cytoskeleton, whereby each actin network becomes enriched in one actin isoform or the other.
We now have precise structural information on how actin interacts with many ABPs (Baek et al, 2008; Eads et al, 1998; Fedorov et al, 1997; Otomo et al, 2005; Pollard, 2016; Shaaban et al, 2020; Tanaka et al, 2018; Thompson et al, 2013; Urnavicius et al, 2015; von der Ecken et al, 2015). Careful analysis of actin–actin and actin–ABPs interactions can be a powerful tool to predict which ABPs affect the roles of specific actin isoforms in discrete actin networks. For example, such analysis indicates that most Act_Ca substitutions affect its interface with the Arp2/3 complex, providing potential explanation for defective assembly into branched‐actin networks. In parallel, our knowledge of the molecular principles involved in the assembly of the different actin networks of the cell allows us to anticipate the consequences of varying the affinity between actin and an ABP.
Consequences of multiple functions deriving from a single or multiple actin isoforms
We also analyzed in this work the effect of the Arp2/3 inhibitor CK‐666 on strains carrying different actin isoforms. We found that strains over‐assembling actin patches were more resistant to the effect of the drug, while strains over‐assembling actin cables were less resistant. In the case of N2 cells, which are more resistant to CK‐666 than Sc cells, we have estimated that actin patches were denser than in Sc cells. These estimates are crude, complicated by the fact that most actin variants have different phalloidin binding sites. Nevertheless, in the case of N2 cells, these observations are consistent with the idea that a greater flow of monomeric actin to branched networks may promote a more efficient actin nucleation by the Arp2/3 complex.
Finally, the generation of a yeast strain carrying two different actin variants allowed us to question the main differences between systems using the same actin and systems using several actin isoforms to perform various actin‐based functions. We showed that addition of CK‐666 in strains expressing both actins Act_Ca and Act_N2 did not lead to similar cytoskeletal reorganization as in wild‐type strains (Fig 7 bottom). While cells expressing wild‐type actin can easily shift actin use from patches to cables, the mechanism was less efficient for a two‐actin system, indicating a perturbed homeostasis of actin networks. This observation brings additional evidence that assembly of both actin networks is more independent in a two‐actin system. Therefore, it is possible that an important difference highlighted here is that organisms using a single actin for multiple actin functions have the possibility for global reorganization of the actin cytoskeleton, where increased assembly of a specific actin network occurs at the expense of others. Conversely, for organisms using multiple actin isoforms, the various actin assembly pathways may be modulated separately, allowing for more autonomous actin networks and functions.
Materials and Methods
Reconstruction of ancestral protein sequences and selection of actins
Actin amino acid sequences from 126 different species, selected from different branches of the eukaryotic and archeal tree of life to cover a wide range of variations, were collected from annotated and reviewed UniProtKB/Swiss‐Prot entries. For species encoding more than one actin, the cytoplasmic actin with the most similar sequence to S. cerevisiae actin was selected. The resulting 126 selected sequences from different species were aligned using the Multiple Sequence Alignment program Clustal Omega (Madeira et al, 2019) in Pearson/FASTA format. The phylogenetic tree of the 126 species was created based on the NCBI taxonomy using the phylogenetic tree generator phyloT (https://phylot.biobyte.de/). Ancestral actin sequence reconstruction was performed from multiple sequence alignment and phylogenetic tree inputs using FastML (Ashkenazy et al, 2012). Ninety nine percent of the amino acids in the ancestral sequences are predicted with an accuracy > 95% and uncertain residues correspond to conservative substitutions (Grantham score < 100; Grantham, 1974).
Generation of plasmids for efficient and rapid actin gene replacement
We generated two plasmid backbones, which carried in succession a sequence upstream of the yeast actin promoter (−804 to −467 from act1 gene) as a first site for homologous recombination, a first selection marker (URA3 or HIS3), the yeast actin promoter (−473 to 0), the act1 coding sequence, a short sequence downstream of the actin gene (+1,437 to +1,703), a second selection marker (LEU2 or KanMX3), and lastly a sequence downstream of the yeast actin gene as a second site for homologous recombination (+1,543 to +2,071). The advantage of having two different selection markers within the same plasmid is to easily select correct insertions of DNA fragments from partial insertions, which are more frequent when targeting an essential gene like actin. These plasmids also contain four unique restriction sites: PacI and XbaI, on each side of the actin gene, allowed to subclone easily new actin coding sequences in the plasmid; Bsu36I and AatII before and after the two sites for homologous recombination, allowed to obtain linear DNA fragments for yeast transformation.
The new actin genes used in this study were obtained commercially from whole gene synthesis techniques (Synbio Technologies). For analysis of S. cerevisiae’s actin expression effects from various nucleotide sequences, we selected multiple actin nucleotide sequences from the specified species and we point mutated the corresponding codons so that the translation product is S. cerevisiae’s actin. For analysis of exogenous actin expression effects, we manually changed the coding sequence of S. cerevisiae’s actin gene (act1) by changing the specific codons that correspond to amino acid mutations respecting the budding yeast codon usage. All plasmids generated for this study are listed in Appendix Table S2 and the actin sequences are given in Appendix Fig S1C.
Yeast strain generation
Actin gene replacement was performed in diploid cells. S. cerevisiae were transformed using the LiAc/SS carrier DNA/PEG method (Gietz & Schiestl, 2007) and grown on dual selection media. Correct insertion of DNA fragments was verified by PCR for all strains and sequenced. Strains were stored as heterozygous diploids and haploid mutant strains were isolated by tetrad analysis for study. Appendix Table S3 lists all the haploid and diploid yeast strains used in this study.
Yeast growth assays
For the measurement of doubling times, yeast cells were grown overnight in YPD (2% bacto‐peptone, 1% yeast extract, 2% dextrose) at 25°C. Cultures were diluted to optical density (OD) 0.1, and growth was followed by measuring the OD every hour. Growth curves were fitted with the formula:
where N represents the number of cells at a given time t and N 0 represents the initial number of cells. The doubling time is calculated as follows:
For yeast growth assays on plate, yeast cells were grown in YPD overnight at 25°C. Equal amounts of cells, calculated from OD600 measurements of log phase growing cultures, were serially diluted and spotted onto YPD plates. Pictures of plates were taken after 2 days of growth at 25°C.
Actin cytoskeleton organization in yeast
Yeast cell phalloidin staining and imaging
Log phase cultures in YPD medium at 25°C were fixed with 4% formaldehyde for 2 h. For CK‐666‐sensitivity assays, cells were treated with the indicated concentration of CK‐666 (Sigma‐Aldrich SML0006) for 30 min before fixation. After fixation, cells were washed twice in PBS and stained overnight with 250‐nM Phalloidin‐Alexa568 (Invitrogen, ref. A12380) at 25°C. Samples were washed twice with PBS, resuspended in PBS‐70% glycerol and directly mounted for imaging. Cells were imaged using a Leica TCS SP8 X White Light Laser confocal microscope equipped with an HC PL APO CS2 100×/1.4NA Oil objective and a hybrid detector. Z‐stack images were collected every 0.3 µm with Las X 3.5.5.19976 software.
Data analysis for live imaging
Branched‐ and linear‐actin network assembly in medium budded cells was assessed from the intensity of actin patches and cables, respectively. Total cell cable intensities were calculated from maximum intensity z‐stack projections using Fiji v.1.53a. For total cell endocytic patch intensities, patches were identified using the TrackMate plugin of Fiji (Tinevez et al, 2017; Planade et al, 2019). Patch detection was corrected manually using the spot editing tool, and the integrated intensity for all patches was calculated from the analysis table of TrackMate.
Fluorescence intensity of phalloidin labeling varied between strains expressing different actin variants. For this reason, the contrast of images showed in the figures was adapted from strain to strain so that both actin structures remained clearly visible. In addition, rather than reporting total intensities, we compared the relative assembly of branched‐ and linear‐actin networks for each strain. This choice is also motivated by the fact that actin networks do not assemble independently but compete for a limited pool of monomeric actin (Burke et al, 2014). We calculated an in vivo actin network deviation index, defined as in Antkowiak et al (2019):
where (resp. ) is the total patch (respectively cable) fluorescence intensity of the cell of interest, and (resp. ) is the mean total intensity of actin patches (resp. cables) in wild‐type S. cerevisiae’s cells. This branched‐to‐linear actin network ratio was calculated for each cell, and compared to in vivo actin network deviation indexes of wild‐type S. cerevisiae’s cells.
For cell polarity, number of visible patches in the bud ( and in the mother cell ( were taken into account to calculate a polarity index:
Quantification of actin expression levels
A mouse anti‐Actin C4 primary antibody (Fisher Scientific, ref. 08691002; 1:10,000 dilution) was selected to recognize actin. Its epitope is located around amino acids 50–70, which corresponds to a highly conserved region across all actins used in this study, with the exception of position 70 (a lysine in Act_Hs; an arginine for all other actins). To test the antibody’s sensitivity to this amino acid variability, different amounts of purified rabbit muscle actin, which contains a lysine in position 70 and purified budding yeast actin, which contains an arginine in position 70, were loaded on a 12% gel. After protein migration, gels were transferred to a nitrocellulose membrane, incubated with mouse anti‐Actin C4 primary antibody (Fisher Scientific, ref. 08691002; 1:10,000 dilution) overnight at 4°C, then incubated with a goat anti‐mouse HRP antibody (Jackson ImmunoResearch, ref. 115‐035‐146; 1:10,000 dilution) for 1 h at 25°C and revealed with Western lightning plus ECL reagent (PerkinElmer, Inc., ref. NEL104001EA). After the Western blots were imaged, membranes were incubated with Ponceau S for 10 min and imaged again. Immunostaining signals were compared relative to Ponceau staining signals. The value of 12 measurements indicated on average a 1.48‐fold stronger signal for rabbit muscle actin compared to S. cerevisiae’s actin. This value was used afterwards as a normalization factor when comparing the expression of Act_Hs and the expression of other actins in yeast.
Total protein samples from S. cerevisiae strains were prepared by trichloroacetic acid precipitation as described in Reid and Schatz (1982). Twelve percent of SDS‐PAGE gels were loaded with 15 µg of total protein sample, and transferred to a nitrocellulose membrane after protein migration. Actin was recognized by anti‐Actin C4 primary antibody (Fisher Scientific, ref. 08691002; 1:10,000 dilution). For our loading control, we selected a rabbit anti‐alpha tubulin primary antibody (Abcam, ref. ab184970; 1:20,000 dilution). Western blots were incubated with goat anti‐mouse HRP (Jackson ImmunoResearch, ref. 115‐035‐146; 1:10,000 dilution) and goat Anti‐rabbit IgG H&L (HRP) (Abcam, ref. ab205718; 1:20,000 dilution) secondary antibodies. Western blots were revealed with Western Lightning Plus ECL reagent (PerkinElmer, Inc., ref. NEL104001EA), on a ChemiDoc MP imaging system (BioRad). We verified the linearity of results obtained with this method over a range of 3 µg to 30 µg of total extract loaded in the gels. Bands’ intensities were calculated using the Image Lab 6.0.1 software. Actin signals were normalized to tubulin signals. The same control sample was loaded in all membranes, and all values were normalized to this lane.
Quantification of filamentous‐to‐monomeric actin ratios
Yeast cells were grown overnight in YPD at 25°C. Cell cultures were diluted to OD 0.3 and grown for 2 h at 25°C. Two ODs of cells were harvested, sedimented by low‐speed centrifugation, and resuspended in G‐Buffer (2 mM of Tris pH 8.0, 0.2 of mM ATP, 0.5 of mM DTT, 0.1 mM of CaCl2, 1 mM of Na‐azide) containing 20 µM of latrunculin A and 20 µM of phalloidin and protease inhibitors (Protease Inhibitor Cocktail Set IV, Calbiochem, reference 539136). Cells were lysed with acid‐washed glass beads (425–600 μm, Sigma‐Aldrich) in a Precellys 24 Tissue Homogenizer. Samples were centrifuged at 240,000 g for 1 h at 4°C. Supernatant was collected and the pellet was resuspended in equal amount of G‐Buffer. Western Blot analysis of actin in supernatants and pellets was performed as described above.
Yeast RNA purification and sequencing
Budding yeast cells (2 ODs) were harvested from log‐phase cultures growing in YPD at 25°C. Cells were lysed mechanically using glass beads (425–600 μm, Sigma‐Aldrich) in Precellys 24 Tissue Homogenizer. Samples were cleared on QIAshredder columns (Qiagen) and RNA was extracted using a RNeasy Mini Kit (Qiagen) following manufacturer’s instructions. Sample quality was checked first by their absorbance values (A260/A280 ≈ 2.2 and A260/A230 > 1.8), then by using a Bioanalyzer (Model 2100, Agilent) (RNA Integrity Number > 8). Library preparation (for Stranded mRNA‐seq/standard quantity, Ligation) and sequencing (Hiseq 4000 sequencing 1 × 50 bases) was performed by the GenomEast Platform (http://genomeast.igbmc.fr/).
RNA sequencing analysis
The quality of the raw reads was assessed using FastQC v0.11.9 toolkit (https://www.bioinformatics.babraham.ac.uk/projects/fastqc/). Adapters and low‐quality reads were trimmed using Trimmomatic v0.39 (Bolger et al, 2014). The reads were aligned with HiSat2 v2.2.1 (Kim et al, 2015) using default options. Gene counts were quantified using feature Counts from the Subread v2.0.1 package (Liao et al, 2019). Alignment and gene counts were generated against the reference genome Saccharomyces cerevisiae S288C (assembly R64). The low expressed genes, which did not have more than one count per million reads (1CPM) in at least three samples within each dataset, were removed from further analysis. Gene counts were then normalized and used for differential expression testing using DESeq2 v1.28.0 (Love et al, 2014). For differential RNA‐seq analysis, genes with a Fold change between −2 to 2 and an adjusted P‐value (P adj) > 0.01 were not considered as significantly expressed. Volcanoplot was generated with the software EnhancedVolcano v1.8.0.
Protein purification and labeling
Actins
Strains expressing Act1, Act_N2, and Act_Ca were used to purify the respective actins. Large‐scale cultures were prepared at 25°C in YPD and harvested by centrifugation. Pellets were frozen in liquid nitrogen and ground in a steel blender (Waring, Winsted, CT, USA; Michelot & Drubin, 2014). Actins were affinity‐purified on a DNAse I column (Goode, 2002). Yeast powder was resuspended in G1 buffer (10 mM of Tris–HCl, 0.5 mM of ATP, 0.2 mM of DTT, 0.2 mM of CaCl2) containing protease inhibitors (Protease Inhibitor Cocktail Set IV, Calbiochem, reference 539136) and centrifuged for 30 min at 160,000 g at 4°C. The lysate was passed through a DNase I column. Bound actin was purified and eluted with G1 buffer supplemented with 50% formamide, and dialyzed against G1 buffer with less calcium (10 mM of Tris–HCl, 0.5 mM of ATP, 0.2 mM of DTT, 0.1 mM of CaCl2) overnight. Rabbit muscle actin was purified from standard procedures (Spudich and Watt, 1971).
Actin labeling
G‐actin from rabbit muscle was dialyzed against 25 mM of Hepes pH 7.5, 50 mM of KCl, 0.1 mM of CaCl2, 0.2 mM of ATP at 4°C for 12 h. A 6‐fold excess of Alexa Fluor 568 succinimidyl ester dye was added and incubated overnight. F‐actin was then centrifugated at 390,000 x g for 40 min, pellet was resuspended and dialyzed against G buffer (2 mM of Tris pH 8, 0.2 mM of ATP, 0.5 mM of DTT, 0.1 mM of CaCl2, and 1 mM of Na‐Azide) for 2 h at 4°C. Labeled actin was centrifugated at 390,000 x g for 40 min to remove insoluble components, and labeled actin was eventually loaded into a G25 column to remove unbound fluorophore.
Formin
Saccharomyces cerevisiae cells (MATa, leu2, ura3‐52, trp1, prb1‐1122, pep4‐3, pre1‐451) were transformed with a plasmid designed for formin overexpression (Gst‐Bni1(1215‐Cter)‐TEV‐9xHis) under the control of a GAL1 promoter; Antkowiak et al, 2019). The expression was induced with 2% galactose for 12 h at 30°C. The resulting cultures were centrifuged and cells were frozen in liquid nitrogen and ground in a steel blender. For protein purification, 5 g of yeast powder was thawed on ice with 45 ml of HKI10 buffer (20 mM of Hepes, pH 7.5, 200 mM of KCl, 10 mM of imidazole, pH 7.5), supplemented with 50 μl of Protease Inhibitor Cocktail Set IV and centrifugated at 160,000 g for 30 min. The supernatant was collected and then incubated with 500 μl of Nickel‐Sepharose 6 Fast Flow (GE Healthcare Life Sciences, Piscataway, NJ, USA) for 2 h at 4°C. Protein bound to Nickel‐Sepharose beads was washed with HKI20 buffer (20 mM of Hepes, pH 7.5, 200 mM of KCl, 20 mM of imidazole, pH 7.5) and cleaved from the beads by a 1‐h incubation with TEV at room temperature. The protein was concentrated with an Amicon Ultra 4‐ml device (Merck4Biosciences), dialyzed against HKG buffer (20 mM of Hepes, pH 7.5, 200 mM of KCl, 6% glycerol), flash frozen, and stored at −80°C.
Arp2/3 complex
Saccharomyces cerevisiae Arp2/3 complex was purified from commercially purchased baker’s yeast (L’Hirondelle) based on a protocol modified from (Nolen & Pollard, 2008; Doolittle et al, 2013; Antkowiak et al, 2019). Yeast powder was prepared by flash freezing droplets of liquid yeast culture in liquid nitrogen and grinding them in a steel blender. Two hundred and thirty grams of yeast powder were resuspended in a lysis buffer (20 mM of Tris–HCl, pH 7.5, 150 mM of NaCl, 2 mM of EDTA, 1 mM of DTT) supplemented with Protease Inhibitor Cocktail Set IV. The mixture was centrifuged at 160,000 g for 30 min and the supernatant was fractioned by a 50% ammonium sulfate cut. The insoluble fraction was dissolved, dialyzed in HKME buffer (25 mM of Hepes, pH 7.5, 50 mM of KCl, 1 mM of EGTA, 3 mM of MgCl2, 1 mM of DTT, 0.1 mM of ATP) overnight at 4°C and loaded onto a 2‐ml Glutathione‐Sepharose 4B (GE Healthcare Life Sciences, Piscataway, NJ, USA) column precharged with GST‐N‐WASp‐VCA (Nolen & Pollard, 2008; Doolittle et al, 2013, 3; Antkowiak et al, 2019). The column was washed with HKME buffer and bound Arp2/3 was eluted with 20 mM of Tris–HCl pH 7.5, 25 mM of KCl, 200 mM of MgCl2, 1 mM of EGTA, and 1 mM of DTT. The presence of protein was detected by using the Bradford reagent, fractions containing protein were pooled, concentrated with an Amicon Ultra 4‐ml device (Merck4Biosciences, Darmstadt, Germany), and dialyzed against HKG buffer. Concentrated Arp2/3 was flash frozen in liquid nitrogen and kept at −80°C.
WASp (Las17), Capping Protein, ADF/cofilin and Profilin
Rosetta 2(DE3)pLysS cells were transformed with a plasmid designed for S. cerevisiae Las17 (Gst‐Las17(375‐Cter)‐6xHis) overexpression. Bacterial cells were collected by centrifugation and then lysed in 20 mM of Tris–HCl, pH 7.5, 1 mM of DTT, 1 mM of EDTA, 200 mM of NaCl, 0.1% Triton X‐100, 5% glycerol and protease inhibitors (Complete Protease Inhibitor Cocktail, Roche). The lysate was centrifuged at 160,000 g for 20 min, the supernatant incubated with Glutathione‐Sepharose beads, and the protein was purified from the extract. Bound proteins were then eluted with 100 mM of l‐glutathione reduced and subjected to a second purification by addition of Nickel‐Sepharose beads 6 Fast Flow (GE Healthcare Life Sciences, Piscataway, NJ, USA). The protein was eluted with HKI500 buffer (20 mM of Hepes, pH 7.5, 200 mM of KCl, 500 mM of imidazole, pH 7.5), concentrated with an Amicon Ultra 4‐ml device and dialyzed against HKG buffer. Protein was flash frozen in liquid nitrogen and kept at −80°C.
Saccharomyces cerevisiae capping protein, ADF/cofilin and profilin, were purified as in Gressin et al (2015). Briefly, proteins were overexpressed in Rosetta 2(DE3)pLysS cells. Cultures were lysed and centrifuged, and supernatants were incubated with Nickel‐Sepharose beads 6 Fast Flow in HKI20 buffer (20 mM of Hepes, pH 7.5, 200 mM of KCl, 20 mM of imidazole, pH 7.5, 0,1% Triton X‐100, 10% glycerol). Proteins were eluted with HKI500 buffer and dialyzed against HKG buffer. They were then flash frozen in liquid nitrogen and kept at −80°C.
The labeling of ADF/cofilin was performed using an ADF/cofilin D34C mutant (Gressin et al, 2015). Yeast ADF/cofilin D34C mutant was bound to Nickel‐Sepharose beads 6 Fast Flow as described above for the wild‐type protein. A 5‐fold excess of Alexa Fluor 488 C5‐maleimide (Thermo Fisher Scientific) was added overnight at 4°C. Bound protein was cleared from unbound fluorophore before elution in HKI500 buffer, dialyzed against HKG buffer, flash frozen, and kept at −80°C.
Tropomyosin
Rosetta 2(DE3)pLysS cells were transformed with a plasmid designed for S. cerevisiae tropomyosin Tpm1p overexpression. This tropomyosin was modified to contain an Ala‐Ser extension at the N‐terminal, which mimics its acetylation, and was purified based on a protocol modified from Skau et al (2009). Briefly, bacteria overexpressing tropomyosin were lysed by sonication in a buffer (50 mM of imidazole‐HCl, pH 6.9, 300 mM of KCl, 5 mM of MgCl2, 0.3 mM of phenylmethylsulfonyl fluoride) supplemented with protease inhibitors (Complete Protease Inhibitor Cocktail, Roche)). Cells were then boiled for 10 min, and the resulting mixture was centrifugated at 300,000 g for 20 min. The supernatant, which contains pure tropomyosin, was dialyzed overnight at 4°C against a dialysis buffer (50 mM of KCl, 10 mM of Tris–HCl, pH 7.5, and 0.5 mM of DTT). Tpm1p labeling was performed with the same strategy used for fission yeast tropomyosin Cdc8 labeling (Christensen et al, 2017). We mutated tpm1's histidine 114 by site‐directed mutagenesis to introduce a cysteine (H114C). Immediately after tropomyosin Ala‐Ser‐Tpm1p H114C purification, the protein was labeled by incubation with a 5‐fold excess Alexa Fluor 488 C5‐maleimide over tropomyosin overnight at 4°C, and separated on a Sephadex G‐25 gel filtration column. The purified fluorescent protein was flash frozen in liquid nitrogen and kept at −80°C.
Branched‐ and linear‐actin network assembly from microbeads
Functionalization of beads
Polystyrene microspheres (2 µm diameter, 2.5% solid (w/v) aqueous suspension, Polysciences, Inc) were washed with HK buffer (20 mM of Hepes, pH 7.5, 150 mM of KCl), diluted 10 times, and incubated with 1 µM Las17 for 30 min on ice. Beads were saturated with 1% bovine serum albumin (BSA) for 15 min, washed, and stored on ice in HK buffer supplemented with 0.1% BSA. Similarly, glutathione‐coated particles (4.37‐µm diameter, 0.5% solid (w/v) aqueous suspension, Spherotech, Inc.) were coated with GST‐Bni1 (1 µM) and then saturated with 1% BSA, washed, and stored in HK 0.1% BSA.
Branched and linear network reconstitution
Unlabeled and fluorescent actins were mixed to reach a final concentration of 40 µM and a labeling percentage of 1%. Actin polymerization was induced by the addition of G‐Buffer and 1x KMEI (50 mM of KCl, 1 mM of MgCl2, 1 mM of EGTA, 10 mM of imidazole Fluorescence Blank pH 7.8) for 1 h at RT. Las17‐ and Bni1‐coated beads were incubated with F‐actin and a minimal set of proteins in a motility buffer (50 mM of KCl, 5 mM of Hepes, 2.4 mM of MgCl2, 4 mM of DTT, 1 mM of ATP, 0.36% methylcellulose 1500 cP, and 1.5% BSA), which triggers actin assembly. Standard optimal protein concentrations were 8 µM of F‐actin, 15 µM of profilin, 1 µM of capping protein, 500 nM of Arp2/3 complex, and 600 nM of ADF/cofilin. When fluorescent proteins were used, their concentrations were 600 nM for Alexa 488‐ADF/cofilin (in which case no black ADF/cofilin was added) and 1 µM for Alexa 488‐tropomyosin.
Image acquisition, processing, and analysis
Images of several beads were acquired 30 min after the initiation of the experiment on a Zeiss Axio Observer Z1 equipped with a 100×/1.4NA Oil Ph3 Plan‐Apochromat objective and a Hamamatsu ORCA‐Flash 4.0LT camera. Images were acquired with Zen 2.3 blue edition using the same light intensity and exposure time.
Data quantification for biomimetic assays
Fluorescence intensity of actin networks and fluorescent ABPs was quantified using Fiji (Version 1.52p), and fluorescence of the background was subtracted. To avoid any measurement bias, all actin networks present around the beads are quantified, independently of their apparent density. Similarly to the in vivo actin network deviation index, a linear‐to‐branched ratio was calculated to compare the efficiency of actin assembly in vitro for both branched‐ and linear‐actin networks of actin filaments for a given biochemical condition. This index measures how actin assembly between branched and linear networks deviates from the values obtained when S. cerevisiae actin is used. It is defined as follows:
where is the average intensity of the branched network for all Las17 beads with a given actin, is the same value for S. cerevisiae actin, is the average intensity of the linear network for all Bni beads with a given actin, and is the same value for S. cerevisiae actin.
Single filament preparation and imaging
Monomeric actins purified from strains expressing Act_Sc, Act_N2, Act_Ca were precleared by centrifugation at 156,000 g for 1 h at 4°C, and mixed with Alexa568‐labeled monomeric rabbit muscle actin to reach a final labeling percentage of 1%. The top 2/3 fraction was kept for the experiment and 3 µM of actin was polymerized for 1 h by addition of 1x KMEI, in the presence of 3 µM of phalloidin. Filaments were then carefully diluted 300 times in Fluorescence Buffer (10 mM of imidazole–HCl, pH 7.0, 50 mM of KCl, 1 mM of EGTA, 1 mM of MgCl2, 70 mM of DTT, 2.5 mg/ml of glucose, 15 µg/ml of catalase, 70 µg/ml of glucose oxidase, 0.1% of BSA, and 0.3% of methylcellulose 1500 cP) and loaded onto plasma‐cleaned coverslips. Filaments were observed using a Nikon Eclipse Ti microscope, equipped for TIRFM (Total Internal Reflection Fluorescence Microscopy) with a 60× NA 1,49 objective, a Prime 95B scientific CMOS camera (Photometrics), and using Metamorph software. For image analysis, filaments were segmented using the Fiji plugin JFilament, and their mean fluorescence intensity was measured at 3 pixel width.
Actin‐ABP contact analysis
The amino acid positions of the substitutions between Saccharomyces cerevisiae (Act_Sc), Node 2 (Act_N2), and Candida albicans (Act_Ca) actin sequences were inspected within high‐resolution X‐ray crystal and cryoEM structures of complexes containing actin. The PDB accession codes are: G‐actin (1YAG) (Vorobiev et al, 2003), F‐actin (6DJN) (Chou & Pollard, 2019), ADF/cofilin (5YU8 and 1CFY) (Fedorov et al, 1997; Tanaka et al, 2018), Arp2/3 daughter filament (6W17) (Shaaban et al, 2020), profilin (1YPR and 3CHW) (Eads et al, 1998; Baek et al, 2008); CP/Arp1 (5ADX) (Urnavicius et al, 2015), WH2 (5YPU), formin (1Y64 and 4EAH) (Otomo et al, 2005; Thompson et al, 2013), and tropomyosin (5JLF) (von der Ecken et al, 2016). Actin residues that are within 5 Å of the binding protein were identified in the CCP4 program CONTACT (Winn et al, 2011), with the exception of the Arp2/3 mother filament, which were within 10 Å since the coordinates were not released when this study was performed (Fäßler et al, 2020). All contacts were visually inspected in COOT (Emsley et al, 2010).
Data reproducibility
All experiments were repeated at least two times. For each mutant, a minimum of three independent clones were generated and compared for reproducibility of phenotypes (biological replicates). The results presented in this study are those obtained for one of these mutants, which is kept in our collection (i.e., technical replicates). Western blot analyses show technical replicates from two different biological replicates, and RNAseq data are presented from three different biological replicates. For the colony area measurements, colonies were measured from 2 plates, results were normalized to control and pooled together. For cell doubling time measurement, experiments were repeated 3 times in different days.
Author contributions
Micaela Boiero Sanders: Conceptualization; Data curation; Formal analysis; Validation; Investigation; Visualization; Methodology; Writing—original draft; Writing—review and editing. Christopher P Toret: Conceptualization; Formal analysis; Investigation; Methodology; Writing—review and editing. Audrey Guillotin: Formal analysis; Investigation. Adrien Antkowiak: Formal analysis; Investigation. Thomas Vannier: Formal analysis; Validation; Investigation; Visualization. Robert C Robinson: Conceptualization; Resources; Data curation; Formal analysis; Funding acquisition; Investigation; Methodology; Writing—original draft; Writing—review and editing. Alphée Michelot: Conceptualization; Resources; Data curation; Formal analysis; Supervision; Funding acquisition; Validation; Investigation; Visualization; Methodology; Writing—original draft; Project administration; Writing—review and editing.
In addition to the CRediT author contributions listed above, the contributions in detail are:
Conceptualization, Methodology, and Writing‐review and editing: MBS, CPT, RCR, and AM. Writing‐original draft: MBS, RCR, and AM. Investigation and Formal analysis: MBS, CPT, AG, AA, TV, RCR, and AM. Validation and Visualization: MBS, TV, and AM. Data curation: MBS, RCR, and AM. Funding acquisition and Resources: RCR and AM. Supervision and Project administration: AM.
Supporting information
Appendix
Expanded View Figures PDF
Table EV1
Table EV2
Source Data for Expanded View/Appendix
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 4
Source Data for Figure 6
Acknowledgements
The authors thank Isabelle Sagot and Emilia Mauriello for their invaluable advice on the project; Sarah Würbel and Ahmed Fatmi for their technical help. This project has received funding from the European Research Council (ERC) under the European Union's Horizon 2020 research and innovation program (grant agreement n° 638376/Segregactin) to A.M., from the Labex INFORM (ANR‐11‐LABX‐0054, funded by the “Investissements d’Avenir French Government program”), and from the Fondation pour la Recherche Médicale (FRM) to M.B.S. under the program Fin de thèse (ref. FDT201904008021). R.C.R. thanks Vidyasirimedhi Institute of Science and Technology (VISTEC), RIIS and JSPS (KAKENHI grant number JP20H00476) for support. We acknowledge the France‐BioImaging infrastructure supported by the Agence Nationale de la Recherche (ANR‐10‐INSB‐04‐01). Sequencing was performed by the GenomEast platform, a member of the “France Génomique” consortium (ANR‐10‐INSB‐0009).
Disclosure and competing interests statement
The authors declare that they have no conflict of interest.
The EMBO Journal (2022) 41: e107982.
Data availability
RNAseq data produced in this study are available in Gene Expression Omnibus (accession number GSE189312). https://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE189312
References
- A MU, Latario CJ, Pickrell LE, Higgs HN (2020) Lysine acetylation of cytoskeletal proteins: Emergence of an actin code. J Cell Biol 219. 10.1083/jcb.202006151 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Antkowiak A, Guillotin A, Boiero Sanders M, Colombo J, Vincentelli R, Michelot A (2019) Sizes of actin networks sharing a common environment are determined by the relative rates of assembly. PLoS Biol 17: e3000317 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ashkenazy H, Penn O, Doron‐Faigenboim A, Cohen O, Cannarozzi G, Zomer O, Pupko T (2012) FastML: a web server for probabilistic reconstruction of ancestral sequences. Nucleic Acids Res 40: W580–W584 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baek K, Liu X, Ferron F, Shu S, Korn ED, Dominguez R (2008) Modulation of actin structure and function by phosphorylation of Tyr‐53 and profilin binding. Proc Natl Acad Sci USA 105: 11748–11753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bernstein BW, Bamburg JR (1982) Tropomyosin binding to F‐actin protects the F‐actin from disassembly by brain actin‐depolymerizing factor (ADF). Cell Motil 2: 1–8 [DOI] [PubMed] [Google Scholar]
- Blanchoin L, Boujemaa‐Paterski R, Sykes C, Plastino J (2014) Actin dynamics, architecture, and mechanics in cell motility. Physiol Rev 94: 235–263 [DOI] [PubMed] [Google Scholar]
- Blanchoin L, Pollard TD, Hitchcock‐DeGregori SE (2001) Inhibition of the Arp2/3 complex‐nucleated actin polymerization and branch formation by tropomyosin. Curr Biol 11: 1300–1304 [DOI] [PubMed] [Google Scholar]
- Boiero Sanders M, Antkowiak A, Michelot A (2020) Diversity from similarity: cellular strategies for assigning particular identities to actin filaments and networks. Open Biol 10: 200157 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, Usadel B (2014) Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30: 2114–2120 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bryan KE, Rubenstein PA (2005) An intermediate form of ADP‐F‐actin. J Biol Chem 280: 1696–1703 [DOI] [PubMed] [Google Scholar]
- Burke TA, Christensen JR, Barone E, Suarez C, Sirotkin V, Kovar DR (2014) Homeostatic actin cytoskeleton networks are regulated by assembly factor competition for monomers. Curr Biol 24: 579–585 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Buzan JM, Frieden C (1996) Yeast actin: polymerization kinetic studies of wild type and a poorly polymerizing mutant. Proc Natl Acad Sci USA 93: 91–95 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chan C, Beltzner CC, Pollard TD (2009) Cofilin dissociates Arp2/3 complex and branches from actin filaments. Curr Biol 19: 537–545 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen A, Arora PD, McCulloch CA, Wilde A (2017) Cytokinesis requires localized β‐actin filament production by an actin isoform specific nucleator. Nat Commun 8: 1530 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chou SZ, Pollard TD (2019) Mechanism of actin polymerization revealed by cryo‐EM structures of actin filaments with three different bound nucleotides. Proc Natl Acad Sci USA 116: 4265–4274 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christensen JR, Hocky GM, Homa KE, Morganthaler AN, Hitchcock‐DeGregori SE, Voth GA, Kovar DR (2017) Competition between Tropomyosin, Fimbrin, and ADF/Cofilin drives their sorting to distinct actin filament networks. eLife 6: e23152 [DOI] [PMC free article] [PubMed] [Google Scholar]
- DesMarais V, Ichetovkin I, Condeelis J, Hitchcock‐DeGregori SE (2002) Spatial regulation of actin dynamics: a tropomyosin‐free, actin‐rich compartment at the leading edge. J Cell Sci 115: 4649–4660 [DOI] [PubMed] [Google Scholar]
- Doolittle LK, Rosen MK, Padrick SB (2013) Purification of Arp2/3 complex from Saccharomyces cerevisiae . Methods Mol Biol 1046: 251–271 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dugina V, Zwaenepoel I, Gabbiani G, Clement S, Chaponnier C (2009) β‐ and γ‐cytoplasmic actins display distinct distribution and functional diversity. J Cell Sci 122: 2980–2988 [DOI] [PubMed] [Google Scholar]
- Eads JC, Mahoney NM, Vorobiev S, Bresnick AR, Wen KK, Rubenstein PA, Haarer BK, Almo SC (1998) Structure determination and characterization of Saccharomyces cerevisiae profilin. Biochemistry 37: 11171–11181 [DOI] [PubMed] [Google Scholar]
- von der Ecken J, Heissler SM, Pathan‐Chhatbar S, Manstein DJ, Raunser S (2016) Cryo‐EM structure of a human cytoplasmic actomyosin complex at near‐atomic resolution. Nature 534: 724–728 [DOI] [PubMed] [Google Scholar]
- von der Ecken J, Müller M, Lehman W, Manstein DJ, Penczek PA, Raunser S (2015) Structure of the F‐actin–tropomyosin complex. Nature 519: 114–117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Emsley P, Lohkamp B, Scott WG, Cowtan K (2010) Features and development of Coot. Acta Crystallogr D Biol Crystallogr 66: 486–501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ezezika OC, Younger NS, Lu J, Kaiser DA, Corbin ZA, Nolen BJ, Kovar DR, Pollard TD (2009) Incompatibility with formin Cdc12p prevents human profilin from substituting for fission yeast profilin: insights from crystal structures of fission yeast profilin. J Biol Chem 284: 2088–2097 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fäßler F, Dimchev G, Hodirnau V‐V, Wan W, Schur FKM (2020) Cryo‐electron tomography structure of Arp2/3 complex in cells reveals new insights into the branch junction. Nat Commun 11: 6437 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedorov AA, Lappalainen P, Fedorov EV, Drubin DG, Almo SC (1997) Structure determination of yeast cofilin. Nat Struct Biol 4: 366–369 [DOI] [PubMed] [Google Scholar]
- Gietz RD, Schiestl RH (2007) High‐efficiency yeast transformation using the LiAc/SS carrier DNA/PEG method. Nat Protoc 2: 31–34 [DOI] [PubMed] [Google Scholar]
- Goode BL (2002) Purification of yeast actin and actin‐associated proteins. Methods Enzymol 351: 433–441 [DOI] [PubMed] [Google Scholar]
- Grantham R (1974) Amino acid difference formula to help explain protein evolution. Science 185: 862–864 [DOI] [PubMed] [Google Scholar]
- Gressin L, Guillotin A, Guérin C, Blanchoin L, Michelot A (2015) Architecture dependence of actin filament network disassembly. Curr Biol 25: 1437–1447 [DOI] [PubMed] [Google Scholar]
- Gunning PW, Ghoshdastider U, Whitaker S, Popp D, Robinson RC (2015) The evolution of compositionally and functionally distinct actin filaments. J Cell Sci 128: 2009–2019 [DOI] [PubMed] [Google Scholar]
- Hatano T, Alioto S, Roscioli E, Palani S, Clarke ST, Kamnev A, Hernandez‐Fernaud JR, Sivashanmugam L, Chapa‐Y‐Lazo B, Jones AME et al (2018) Rapid production of pure recombinant actin isoforms in Pichia pastoris. J Cell Sci 131: jcs213827 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hatano T, Sivashanmugam L, Suchenko A, Hussain H, Balasubramanian MK (2020) Pick‐ya actin ‐ a method to purify actin isoforms with bespoke key post‐translational modifications. J Cell Sci 133: jcs241406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hocky GM, Baker JL, Bradley MJ, Sinitskiy AV, De La Cruz EM, Voth GA (2016) Cations stiffen actin filaments by adhering a key structural element to adjacent subunits. J Phys Chem B 120: 4558–4567 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoekema A, Kastelein RA, Vasser M, de Boer HA (1987) Codon replacement in the PGK1 gene of Saccharomyces cerevisiae: experimental approach to study the role of biased codon usage in gene expression. Mol Cell Biol 7: 2914–2924 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Bradley MJ, McCullough BR, Pierre A, Grintsevich EE, Reisler E, De La Cruz EM (2012) Identification of cation‐binding sites on actin that drive polymerization and modulate bending stiffness. Proc Natl Acad Sci USA 109: 16923–16927 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kang H, Bradley MJ, Cao W, Zhou K, Grintsevich EE, Michelot A, Sindelar CV, Hochstrasser M, De La Cruz EM (2014) Site‐specific cation release drives actin filament severing by vertebrate cofilin. Proc Natl Acad Sci 111: 17821–17826 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kashina A (2014) Protein arginylation, a global biological regulator that targets actin cytoskeleton and the muscle. The Anatomical Record, 297: 1630–1636 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kijima ST, Hirose K, Kong S‐G, Wada M, Uyeda TQP (2016) Distinct biochemical properties of Arabidopsis thaliana actin isoforms. Plant Cell Physiol 57: 46–56 [DOI] [PubMed] [Google Scholar]
- Kijima ST, Staiger CJ, Katoh K, Nagasaki A, Ito K, Uyeda TQP (2018) Arabidopsis vegetative actin isoforms, AtACT2 and AtACT7, generate distinct filament arrays in living plant cells. Sci Rep 8: 4381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim D, Langmead B, Salzberg SL (2015) HISAT: a fast spliced aligner with low memory requirements. Nat Methods 12: 357–360 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim E, Miller CJ, Reisler E (1996) Polymerization and in vitro motility properties of yeast actin: a comparison with rabbit skeletal alpha‐actin. Biochemistry 35: 16566–16572 [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2019) The R package Rsubread is easier, faster, cheaper and better for alignment and quantification of RNA sequencing reads. Nucleic Acids Res 47: e47 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Madeira F, Park YM, Lee J, Buso N, Gur T, Madhusoodanan N, Basutkar P, Tivey ARN, Potter SC, Finn RD et al (2019) The EMBL‐EBI search and sequence analysis tools APIs in 2019. Nucleic Acids Res 47: W636–W641 [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCullough B, Grintsevich E, Chen C, Kang H, Hutchison A, Henn A, Cao W, Suarez C, Martiel J‐L, Blanchoin L et al (2011) Cofilin‐linked changes in actin filament flexibility promote severing. Biophys J 101: 151–159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Michelot A, Berro J, Guérin C, Boujemaa‐Paterski R, Staiger CJ, Martiel J‐L, Blanchoin L (2007) Actin‐filament stochastic dynamics mediated by ADF/Cofilin. Curr Biol 17: 825–833 [DOI] [PubMed] [Google Scholar]
- Michelot A, Drubin DG (2014) Dissecting principles governing actin assembly using yeast extracts. Methods Enzymol 540: 381–397 [DOI] [PubMed] [Google Scholar]
- Moseley JB, Goode BL (2006) The yeast actin cytoskeleton: from cellular function to biochemical mechanism. Microbiol Mol Biol Rev 70: 605–645 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nefsky B, Bretscher A (1992) Yeast actin is relatively well behaved. Eur J Biochem 206: 949–955 [DOI] [PubMed] [Google Scholar]
- Ng R, Domdey H, Larson G, Rossi JJ, Abelson J (1985) A test for intron function in the yeast actin gene. Nature 314: 183–184 [DOI] [PubMed] [Google Scholar]
- Nolen BJ, Pollard TD (2008) Structure and biochemical properties of fission yeast Arp2/3 complex lacking the Arp2 subunit. J Biol Chem 283: 26490–26498 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Okreglak V, Drubin DG (2010) Loss of Aip1 reveals a role in maintaining the actin monomer pool and an in vivo oligomer assembly pathway. J Cell Biol 188: 769–777 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Orlova A, Galkin VE, VanLoock MS, Kim E, Shvetsov A, Reisler E, Egelman EH (2001) Probing the structure of F‐actin: cross‐links constrain atomic models and modify actin dynamics. J Mol Biol 312: 95–106 [DOI] [PubMed] [Google Scholar]
- Otomo T, Tomchick DR, Otomo C, Panchal SC, Machius M, Rosen MK (2005) Structural basis of actin filament nucleation and processive capping by a formin homology 2 domain. Nature 433: 488–494 [DOI] [PubMed] [Google Scholar]
- Planade J, Belbahri R, Boiero Sanders M, Guillotin A, du Roure O, Michelot A, Heuvingh J (2019) Mechanical stiffness of reconstituted actin patches correlates tightly with endocytosis efficiency. PLoS Biol 17: e3000500 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollard TD (2016) Actin and actin‐binding proteins. Cold Spring Harb Perspect Biol 8: a018226 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popp D, Robinson RC (2012) Supramolecular cellular filament systems: how and why do they form? Cytoskeleton 69: 71–87 [DOI] [PubMed] [Google Scholar]
- Pruyne DW, Schott DH, Bretscher A (1998) Tropomyosin‐containing actin cables direct the Myo2p‐dependent polarized delivery of secretory vesicles in budding yeast. J Cell Biol 143: 1931–1945 [DOI] [PubMed] [Google Scholar]
- Qu Z, Silvan U, Jockusch BM, Aebi U, Schoenenberger C‐A, Mannherz HG (2015) Distinct actin oligomers modulate differently the activity of actin nucleators. FEBS J 282: 3824–3840 [DOI] [PubMed] [Google Scholar]
- Raz‐Ben Aroush D, Ofer N, Abu‐Shah E, Allard J, Krichevsky O, Mogilner A, Keren K (2017) Actin turnover in lamellipodial fragments. Curr Biol 27: 2963–2973 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reid GA, Schatz G (1982) Import of proteins into mitochondria. Yeast cells grown in the presence of carbonyl cyanide m‐chlorophenylhydrazone accumulate massive amounts of some mitochondrial precursor polypeptides. J Biol Chem 257: 13056–13061 [PubMed] [Google Scholar]
- Rotty JD, Wu C, Haynes EM, Suarez C, Winkelman JD, Johnson HE, Haugh JM, Kovar DR, Bear JE (2015) Profilin‐1 serves as a gatekeeper for actin assembly by Arp2/3‐dependent and ‐independent pathways. Dev Cell 32: 54–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scipion CPM, Ghoshdastider U, Ferrer FJ, Yuen TY, Wongsantichon J, Robinson RC (2018) Structural evidence for the roles of divalent cations in actin polymerization and activation of ATP hydrolysis. Proc Natl Acad Sci USA 115: 10345–10350 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaaban M, Chowdhury S, Nolen BJ (2020) Cryo‐EM reveals the transition of Arp2/3 complex from inactive to nucleation‐competent state. Nat Struct Mol Biol 27: 1009–1016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Skau CT, Neidt EM, Kovar DR (2009) Role of tropomyosin in formin‐mediated contractile ring assembly in fission yeast. Mol Biol Cell 20: 2160–2173 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith MB, Kiuchi T, Watanabe N, Vavylonis D (2013) Distributed actin turnover in the lamellipodium and FRAP kinetics. Biophys J 104: 247–257 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suarez C, Kovar DR (2016) Internetwork competition for monomers governs actin cytoskeleton organization. Nat Rev Mol Cell Biol 17: 799–810 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Takaine M, Mabuchi I (2007) Properties of actin from the fission yeast Schizosaccharomyces pombe and interaction with fission yeast profilin. J Biol Chem 282: 21683–21694 [DOI] [PubMed] [Google Scholar]
- Tanaka K, Takeda S, Mitsuoka K, Oda T, Kimura‐Sakiyama C, Maeda Y, Narita A (2018) Structural basis for cofilin binding and actin filament disassembly. Nat Commun 9: 1860 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson ME, Heimsath EG, Gauvin TJ, Higgs HN, Kull FJ (2013) FMNL3 FH2‐actin structure gives insight into formin‐mediated actin nucleation and elongation. Nat Struct Mol Biol 20: 111–118 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ti S‐C, Pollard TD (2011) Purification of actin from fission yeast Schizosaccharomyces pombe and characterization of functional differences from muscle actin. J Biol Chem 286: 5784–5792 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tinevez J‐Y, Perry N, Schindelin J, Hoopes GM, Reynolds GD, Laplantine E, Bednarek SY, Shorte SL, Eliceiri KW (2017) TrackMate: an open and extensible platform for single‐particle tracking. Methods 115: 80–90 [DOI] [PubMed] [Google Scholar]
- Urnavicius L, Zhang K, Diamant AG, Motz C, Schlager MA, Yu M, Patel NA, Robinson CV, Carter AP (2015) The structure of the dynactin complex and its interaction with dynein. Science 347: 1441–1446 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vedula P, Kurosaka S, Leu NA, Wolf YI, Shabalina SA, Wang J, Sterling S, Dong DW, Kashina A (2017) Diverse functions of homologous actin isoforms are defined by their nucleotide, rather than their amino acid sequence. eLife 6: e31661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vorobiev S, Strokopytov B, Drubin DG, Frieden C, Ono S, Condeelis J, Rubenstein PA, Almo SC (2003) The structure of nonvertebrate actin: implications for the ATP hydrolytic mechanism. Proc Natl Acad Sci USA 100: 5760–5765 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winn MD, Ballard CC, Cowtan KD, Dodson EJ, Emsley P, Evans PR, Keegan RM, Krissinel EB, Leslie AG, McCoy A et al (2011) Overview of the CCP4 suite and current developments. Acta Crystallogr D Biol Crystallogr 67: 235–242 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Dang Y, Zhou M, Li L, Yu C, Fu J, Chen S, Liu Y (2016) Codon usage is an important determinant of gene expression levels largely through its effects on transcription. Proc Natl Acad Sci 113: E6117–E6125 [DOI] [PMC free article] [PubMed] [Google Scholar]