

Abstract
Hematological measures are important intermediate clinical phenotypes for many acute and chronic diseases and are highly heritable. Although genomewide association studies (GWAS) have identified thousands of loci containing trait-associated variants, the causal genes underlying these associations are often uncertain. To better understand the underlying genetic regulatory mechanisms, we performed a transcriptome-wide association study (TWAS) to systematically investigate the association between genetically predicted gene expression and hematological measures in 54,542 Europeans from the Genetic Epidemiology Research on Aging cohort. We found 239 significant gene-trait associations with hematological measures; we replicated 71 associations at p < 0.05 in a TWAS meta-analysis consisting of up to 35,900 Europeans from the Women’s Health Initiative, Atherosclerosis Risk in Communities Study, and BioMe Biobank. Additionally, we attempted to refine this list of candidate genes by performing conditional analyses, adjusting for individual variants previously associated with hematological measures, and performed further fine-mapping of TWAS loci. To facilitate interpretation of our findings, we designed an R Shiny application to interactively visualize our TWAS results by integrating them with additional genetic data sources (GWAS, TWAS from multiple reference panels, conditional analyses, known GWAS variants, etc.). Our results and application highlight frequently overlooked TWAS challenges and illustrate the complexity of TWAS fine-mapping.
Keywords: fine-mapping, hematological traits, R Shiny, TWAS
1 |. INTRODUCTION
Genome-wide association studies (GWAS) have identified thousands of loci containing hematological traitassociated variants (i.e., variants associated with red cell, white cell, and platelet traits), and previous Mendelian randomization and phenome-wide association study analyses have highlighted the likely causal role of hematological trait-associated genetic variants in a variety of disorders, including autoimmune conditions and coronary heart disease (Astle et al., 2016; Chen et al., 2020; Vuckovic et al., 2020). Unfortunately, these individual single nucleotide polymorphism (SNP)-based GWAS make it difficult to identify regulatory variants with small effect sizes which in aggregate impact the same gene, even in very large sample sizes, and they identify regions of associated variants whose biological function is often not clear (Gamazon et al., 2015).
A transcriptome-wide association study (TWAS) is one gene-based method that systematically investigates the association between genetically predicted gene expression and phenotypes of interest, which can increase the power to identify novel trait-associated loci and may elucidate mechanisms of biological function (Gamazon et al., 2015; Gusev et al., 2018; Hu et al., 2019; Zhou et al., 2020). However, many challenges associated with TWAS, such as loci containing multiple associated genes, correlated gene expression, and bias with expression panels (Wainberg et al., 2019) can complicate TWAS results interpretation, particularly for well-studied traits with many identified genetic loci. Here we performed a large-scale TWAS of hematological measures using the PrediXcan method (Gamazon et al., 2015) to analyze data from 54,542 individuals of European ancestry from the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort (our discovery data set) (Banda et al., 2015; Kvale et al., 2015). Following the initial TWAS analysis, we explored several complimentary strategies (including conditional analysis, TWAS meta-analysis, TWAS using secondary gene expression reference panels, and fine-mapping tools) to improve TWAS results interpretation. Hematological phenotypes are particularly good candidates for exploring TWAS analysis interpretation due to the availability of large RNA-sequencing datasets in a relevant tissue type, high heritability across traits, and a large number of known genetic associations, most with poorly understood mechanisms and target genes.
In addition to TWAS, we performed conditional analyses to evaluate if TWAS-identified genes represented novel statistical signals or were primarily driven by variants known from GWAS (Vuckovic et al., 2020); we replicated our significant set of gene-trait associations in a meta-analyzed sample of TWAS results containing 18,100 individuals from the Women’s Health Initiative (WHI), 9345 individuals from the Atherosclerosis Risk in Communities Study (ARIC), and 8455 individuals from Mount Sinai BioMe Biobank (BioMe), all of European ancestry (Table S1); and we compared the TWAS results between primary and secondary gene expression reference panels to determine if relevant tissues with smaller sample sizes support our primary TWAS findings.
We further employed several strategies to improve our understanding and interpretation of complex genomic regions containing multiple TWAS-identified genes. First, we used FOCUS (fine-mapping of causal gene sets (Mancuso et al., 2019)) to seek to identify a set of causal genes within genomic loci containing multiple significant TWAS gene-trait associations. FOCUS is a software used to fine-map TWAS statistics at genomic risk regions, while accounting for linkage dis-equilibrium (LD) among variants and predicted expression correlation among genes at those risk regions. Second, we developed a novel web-based tool (called LocusXcanR) for integrating and visualizing TWAS and GWAS results, as well as results from multiple expression reference datasets. Taking the results of each analysis into consideration, we highlight frequently overlooked challenges of TWAS interpretation, such as failure to consider the number of proximal genes which cannot be accurately imputed with a given reference panel, but which may still be influenced by variants identified in GWAS studies. Our results illustrate the complexity of TWAS interpretation and fine-mapping efforts and provide one resource for clarifying likely gene targets for hematological trait-related genetic loci. Consideration of additional annotation resources and TWAS limitations is necessary for confident identification of gene targets.
2 |. METHODS
2.1 |. Initial TWAS analysis
We applied the PrediXcan method (detailed in Online Supporting Information) to identify expression-trait associations using individual-level genotype and phenotype data from the GERA non-Hispanic White ethnic group. The GERA cohort includes over 100,000 adults who are members of the Kaiser Permanente Medical Care Plan, Northern California Region. Genotyping was completed as previously described (Kvale et al., 2015) and genotype data were phased without external reference and imputed to 1000 Genomes Phase 3 v5. Principal components analysis was used to characterize genetic structure in this European ancestry sample (Banda et al., 2015). Hematological measures were extracted from medical records, and the first visit was used for each participant in most cases. In total, 54,542 non-Hispanic White individuals with hematological measures were included in the analysis (see Online Supporting Information for further details).
Variants were filtered for imputation quality (R2 > 0.3). Ten hematological measures were analyzed including platelet count (PLT), red blood cell counts (red blood cell count [RBC], hematocrit [HCT], hemoglobin [HGB], mean corpuscular volume [MCV], and red cell distribution width [RDW] indices), and white blood cell counts (white blood cell count [WBC], monocyte count [MONO], neutrophil count [NEUTRO], and lymphocyte count [LYMPH]) indices). Because the Depression Genes and Networks (DGN) cohort has the largest single whole blood RNA-seq data set (Battle et al., 2014; genes = 11,538, n = 922), we used DGN gene expression panel weights from PredictDB (see URLs) to predict gene expression levels in GERA.
2.2 |. Conditional analysis of significant TWAS genes on known GWAS variants
To determine if TWAS results from GERA were driven by any previously reported genome-wide significant variant, we performed conditional analysis. For each statistically significant TWAS gene-trait association, the effect of predicted gene expression was conditioned on a set of previously reported GWAS sentinel variants (Vuckovic et al., 2020) meeting the following criteria: (1) the sentinel variant fell within a 1 Mb region of the TWAS gene, (2) the trait with which the GWAS variant was associated matched the TWAS analytical trait or was within the same trait category as the analytical trait (PLTs, red blood cell indices [HCT, HGB, MCV, RBC, and RDW], white blood cell indices [WBC, NEUTRO, MONO, and LYMPH]), and (3) the GWAS variant met an imputation quality threshold of R2 > 0.3. We used a modified version of the cpgen R package (see Online Supporting Information) to perform the conditional analysis, accounting for a PLINK KING-robust kinship matrix (Manichaikul et al., 2010), which used only genotyped variants and excluded variants with minor allele frequency less than 5% and individuals missing more than 1% of SNPs.
2.3 |. Replication of conditional TWAS results in meta-analyzed cohorts
To replicate the conditionally significant gene-trait associations in GERA, we compared each gene-trait association to a TWAS meta-analysis including ARIC, WHI, and BioMe cohorts (brief cohort summaries follow and are detailed in Online Supporting Information). All TWAS analyses were limited to self-reported white or European ancestry participants for easy comparability of the DGN European ancestry eQTL panel and with the largest single-ancestry blood cell trait GWAS (also conducted in European ancestry participants). We analyzed 10 hematological phenotypes (as noted above) across all cohorts; inverse normalized phenotypes were analyzed with appropriate covariate adjustments for demographic characteristics and principal components.
Each cohort anlayzed in this study included only participants of European ancestry with hematological measures. Atherosclerosis Risk in Communities Study (ARIC) included 9345 participants (“The Atherosclerosis Risk in Communities [ARIC] Study: design and objectives,” 1989; Women’s Health Initiative (WHI) included 18,100 women (The Women’s Health Initiative Study Group, 1998); and BioMe (the Mount Sinai BioMe Biobank) included 8455 participants.
Replication of the conditionally significant GERA gene-trait associations was performed using meta-analyzed TWAS results from ARIC, WHI, and BioMe. As described above, PrediXcan was used to facilitate gene expression imputation and association in each cohort separately, and the meta-analysis association test was conducted using METAL (Willer et al., 2010). Seventeen gene-trait associations remained statistically significant after conditional analysis; thus, for this set of genes, we defined a Bonferroni-corrected statistically significant replication threshold at p-value < 2.94 × 10−3.
2.4 |. Characterization of TWAS-identified fine-mapping loci
We next performed fine-mapping of TWAS-identified loci. Fine-mapping loci refers to fine-mapping analysis of trait-specific genomic locations that contain, and are centered at sentinel TWAS genes. That is, we took the set of trait-specific statistically significant GERA TWAS genes, selected the most significant gene in the set (the sentinel gene), and assigned it to a locus along with any other statistically significant TWAS genes within a 1 Mb window of the sentinel gene. We then selected the next most significant TWAS gene which had not yet been assigned to a locus and continued in this fashion until all statistically significant TWAS genes had been assigned to a locus.
We then defined locus categories based on whether the locus contained a single gene or multiple genes and whether the locus replicated in TWAS meta-analysis at either a lenient or strict threshold. For this fine-mapping analysis, the statistical significance of replicated genes was qualified based on two different thresholds – a stringent threshold Bonferroni-corrected for all 239 statistically significant TWAS gene-trait associations at p-value < 2.09 × 10−4, and a more lenient threshold at p-value < 0.05. Thus, locus categories were defined as follows: 1 = single gene locus, strict replication (p < 2.09E−04); 2 = single gene locus, replication (p < 0.05); 3 = single gene locus, no replication; 4 = multi gene locus, strict replication (p < 2.09E−04); 5 = multi gene locus, replication (p < 0.05); 6 = multi gene locus, no replication.
2.5 |. TWAS fine-mapping strategies
To facilitate TWAS fine-mapping and allow for better interpretation of TWAS results, we employed several different strategies including the use of R Shiny, FOCUS, and secondary TWAS reference panels. Details of each method follow.
2.5.1 |. R Shiny application
We used R’s convenient Shiny package (version 1.5.0, implemented in R 4.0.3) to produce a web application (LocusXcanR) that displays our GERA TWAS results. All GERA TWAS results were produced using PrediXcan as described above. We also included GERA GWAS results in the R Shiny app; analysis was performed using Bolt LMM as implemented in rvtests (Zhan et al., 2016). GERA conditional analysis results were produced using cpgen. Known GWAS sentinel variants were obtained from (Vuckovic et al., 2020). Model weights and model variants were taken from our primary DGN reference panel from PredictDB or secondary reference panels from PredictDB (Genotype-Tissue Expression [GTEx] Project; The Genotype-Tissue Expression GTEx project, 2013; whole blood [GWB] and Epstein-Barr virus [EBV] transformed lymphocytes [GTL]; and Multi-Ethnic Study of Atherosclerosis [MESA] [Liu et al., 2013] monocytes [MSA]. These are considered secondary reference panels due to their smaller sample sizes compared with DGN). Supplementary Methods contains further details relevant to R Shiny data and visualizations.
2.5.2 |. Fine-mapping Of CaUsal gene Sets (FOCUS)
We used the Fine-mapping Of CaUsal gene Sets (FOCUS) (Mancuso et al., 2019) software to fine-map TWAS statistics at genomic risk regions. As input, we used GERA GWAS summary data along with eQTL weights from PredictDB Depression Genes and Networks whole blood data, and the European LD reference panel from 1000 Genomes Phase 3. The software outputs a credible set of genes at each locus which can be used to explain observed genomic risk.
2.5.3 |. TWAS secondary reference panels
We used a set of secondary gene expression reference panels relevant to whole blood to assist with fine-mapping by assessing consistency between our primary TWAS results from DGN and TWAS results from secondary reference panels. Thus, in addition to DGN weights, weights for gene expression using RNA sequencing data were obtained from the Genotype-Tissue Expression project (version 7) (Zhang & Lin, 2013) (whole blood, genes = 6208; and EBV transformed lymphocytes, genes = 3000), and Multi-Ethnic Study of Atherosclerosis (Europeans only, monocytes, genes = 4647) (Mogil et al., 2018).
2.6 |. Ethics statement
We performed secondary data analysis on deidentified data only (exempt research). All individual studies included were approved by relevant local institutional review boards, and participants provided written informed consent.
3 |. RESULTS
3.1 |. Initial TWAS analysis
We applied the PrediXcan method to identify expression-trait associations using individual-level genotype and phenotype data from the GERA non-Hispanic White ethnic group. Analyzed blood cell traits included PLT, RBC, HCT, HGB, MCV, RDW, WBC, MONO, NEUTRO, and LYMPH indices. We used DGN whole blood expression panel weights from PredictDB (a database of weights provided by PrediXcan; see URLs) to predict gene expression levels in GERA. Among the 11,538 genes in the DGN expression panel, 11,438 genes were predicted in GERA and 51% of those genes achieved DGN model R2 > 0.05 (see Table S2 for model R2 values for significant genes and Table S3 for genes included in DGN but not predicted in GERA). The number of GERA variants used for prediction was equal to the number of variants included in the prediction model (i.e., complete variant matching) for 74% of the predicted genes; the remaining genes used fewer variants from GERA than were included in the prediction models. We tested each of the 11,438 predicted genes individually for association with each of the 10 hematological measures, resulting in a Bonferroni-corrected p-value threshold of p < 4.37 × 10−7. Through the subsequent study analyses, we will refer to these results as “marginal TWAS.”
Overall, we identified 295 statistically significant marginal TWAS associations (p < 4.37 × 10−7), with each hematological measure having at least one significant association (Table S2). Among these, 47 marginal TWAS associations fell into the major histocompatibility complex (MHC) or HLA region (GRCh37; chr 6: 28,477,797–33,448,354) and were not considered in subsequent analyses (Table S4); disentangling a set of causal genes within the MHC region is exceptionally difficult due to the highly polymorphic genetic loci and complex LD in the region. Another nine significant associations included genes which contained only a single variant in the prediction model. These associations were also not included in subsequent analyses (Table S4). The remaining 239 significant associations included genes predicted from 2 to 112 variants, with a median of 21 variants used in predictive models. Among this set of 239 associations, we replicated 71 at p < 0.05 with same direction of effect for the blood cell trait in TWAS meta-analysis.
3.2 |. Conditional analysis of significant TWAS genes on known GWAS variants
To determine whether any of the 239 remaining significant gene-trait associations were novel signals, not driven by any previously reported genome-wide significant variant, we performed conditional analysis. Since we performed TWAS with individual-level data, we conditioned the predicted gene expression value of each statistically significant marginal TWAS gene on the set of nearby (within ± 1 Mb of the gene) sentinel GWAS variants within the hematological category (RBC, WBC, and PLT) from the largest current European ancestry focused GWAS for hematological measures (Vuckovic et al., 2020). We found that 222 (93%) of all marginal TWAS significant associations were attenuated by known GWAS variants and became nonsignificant (p > 4.37 × 10−7) upon conditional analysis. Another 15 of the associations remained significant after conditional analysis, and the remaining two associations did not have GWAS-reported variants within a 1 Mb window of the gene (Table S5).
3.3 |. Replication of conditional TWAS results in meta-analyzed cohorts
For confirmation of robust and/or novel signal, we attempted to replicate these 17 conditionally significant findings in a TWAS meta-analysis which included up to 32,036 European ancestry individuals from three cohorts: ARIC, WHI, and BioMe. Two of the 17 conditionally significant gene-trait associations (HIST1H2BO-HGB and HIST1H2BO-RDW) included in the replication set met the stringent significance threshold (p < 2.94 × 10−3). However, HIST1H2BO is situated within 1 Mb of the MHC region already excluded above (GRCh37; chr 6: 28,477,797–33,448,354), with this signal potentially reflecting long-range LD with the MHC region, and has poor model R2 = 0.016. Additionally, OR2B6 associated with HGB, MCV, and RWD; ZNF192 associated with HGB, MCV, and RDW; and ZSCAN12 associated with MCV meet a more lenient significance threshold (p < 0.05). Yet, OR2B6, ZNF192, and ZSCAN12 are also located on chromosome 6 within approximately 500 kb of the MHC region and all have poor model R2 < 0.015. The remaining seven gene-trait associations did not meet any replication criteria.
3.4 |. Characterization of TWAS-Identified fine-mapping loci
Based on the conditional analysis, previously reported GWAS sentinel variants were at least partly responsible for and attenuated 93% of the significant marginal TWAS signals. Thus, we next examined if TWAS aided fine-mapping and identification of regulatory mechanisms at these loci. To better contextualize if fine-mapping in GERA was consistent in additional cohorts, we also examined replication of TWAS significant genes in these known loci. The 239 marginal TWAS associations resided in 120 trait-specific, physically nonoverlapping cis loci (i.e., the cis region of each locus is ± 1 Mb of the locus’s TWAS sentinel gene start and end positions). Over half (57%) of these loci contained only a single significant gene, while another 19% contained two significant genes. The remaining 24% of nonoverlapping loci contained three or more significant genes, with up to 11 significant genes at a single locus. These 120 loci contained 87 unique index genes (defined as the most significant TWAS gene within the locus). Most loci did not contain a TWAS gene that replicated in meta-analysis (67% total; i.e., 47% of all loci were single-gene loci that did not replicate plus 20% of all loci were multi-gene loci that did not contain any gene that replicated, even at a marginal level). Ten percent of all loci were single-gene loci that met a marginal replication threshold (p < 0.05), and 20% of all loci were multi-gene loci that met this marginal threshold for at least one gene at the locus. The remaining four loci (3%) contained multiple TWAS genes with at least one gene meeting a strict replication threshold (p < 2.09 × 10−4) (Table S2). These included the following index gene-phenotype associations: TRIM68-MCV, USP49-MCV, PSMD3-NEUTRO, and PSMD3-WBC.
3.5 |. TWAS fine-mapping strategies
To facilitate TWAS fine-mapping and allow for better interpretation of whether a given TWAS-identified gene was truly likely to associate with hematological variation, or whether it was likely to be a spurious association due to correlation of expression with nearby genes or other factors, we created an R Shiny application (LocusXcanR) to interactively visualize TWAS sentinel genes in context, one locus at a time. LocusXcanR allowed us to integrate multiple sources of information from our primary TWAS analysis, including gene expression prediction models, TWAS meta-analysis, TWAS using secondary reference panels (whole blood and EBV transformed lymphocytes from GTEx, and monocytes from MESA), GWAS analysis of all hematological measures, and correlation among genetic variants (i.e., LD) and among predicted gene expression levels. We highlight several loci to demonstrate the utility of the application, showcase some of the challenges that arise when TWAS identifies multiple significant genes at a single locus, and illustrate some of the complexities that are inherent in TWAS fine-mapping. In the sections that follow, we feature TWAS genes that fall into loci with a low, intermediate, or high level of complexity. All the figures in the following sections originate from LocusXcanR (http://shiny.bios.unc.edu/gera-twas/), which could be readily adapted to future TWAS analyses for other complex traits.
3.5.1 |. HK1locus
The HK1 gene is known to be associated with several red blood cell traits including HGB, MCV, HCT, mean corpuscular hemoglobin, RBC, and RDW in GWAS analyses (Vuckovic et al., 2020) and is a Mendelian gene for hemolytic anemia [MIM 142600]. Our TWAS results confirmed previously reported HK1 GWAS associations with HCT and MCV (assigned based on nearest gene for lead GWAS variants). The marginal TWAS tests for association between HK1 and HCT (p = 3.84 × 10−8) and MCV (p = 1.05 × 10−7) were statistically significant (Figure 1); associations were all but eliminated by conditional analysis on known GWAS sentinel variants (HCT p = 2.58 × 10−1; MCV p = 4.36 × 10−2); HK1 with HCT replicated in meta-analysis (p = 4.63 × 10−2); and HK1 was the most significant TWAS gene among only two other genes (HKDC1 and TSPAN15) implicated by GWAS at these loci, with the other two genes showing no TWAS signal. Thus, results point simply to HK1 as the most likely causal gene at this locus.
FIGURE 1.
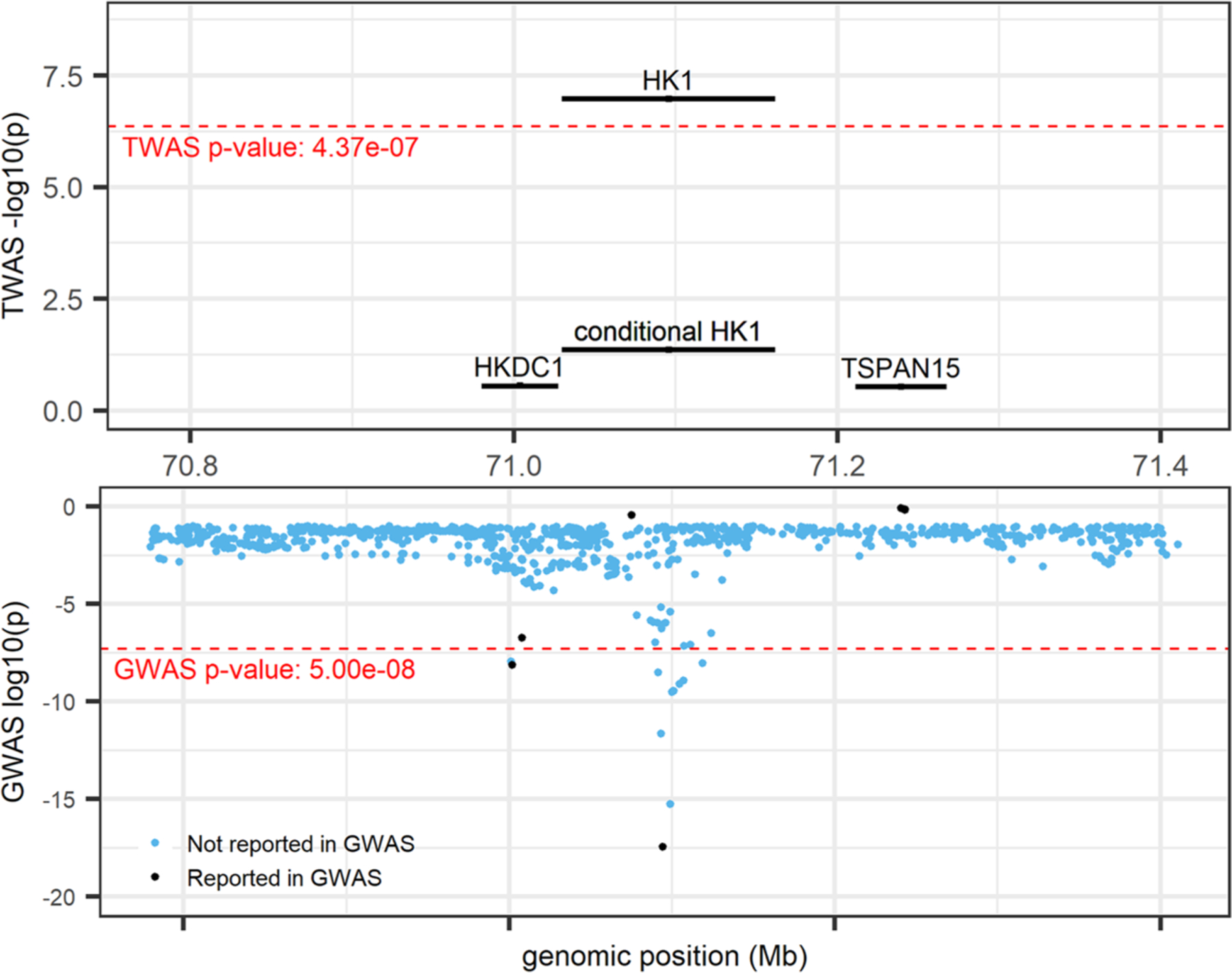
HK1locus (locus 60; chr 10: 70,029,740–72,161,638; trait = MCV) from R Shiny. TWAS results (top panel) and GWAS results (bottom panel). Marginal and conditional results for HK1 are presented in the top panel. Black-colored genes and variants denote those previously reported by UK Biobank and BCX GWAS (Vuckovic et al., 2020), blue variants denote those not previously reported as UK Biobank and BCX GWAS sentinel variants. GWAS, genome-wide association studies; TWAS, transcriptome-wide association study
3.5.2 |. CREB5locus
The marginal TWAS tests for association between CREB5 and NEUTRO (p = 1.41 × 10−12) and WBC (p = 4.01 × 10−10) were the only TWAS significant associations at this locus (Figure 2a), and associations were essentially eliminated by conditional analysis on known GWAS sentinel variants (NEUTRO p = 9.04 × 10−1; WBC p = 3.97 × 10−1). However, at this locus, both CREB5 and JAZF1 (TWAS NEUTRO p = 5.26 × 10−3, WBC p = 2.42 × 10−3) had previously been annotated as being the nearest and/or assigned gene for one or more GWAS sentinel variants. Predicted gene expression for CREB5 and JAZF1 was not highly correlated (r2 between 0.0–0.2), and the genes appeared to share only a single, nonsignificant predictive model variant (Figure 2b). CREB5 and JAZF1 both replicated at a lenient significance threshold for NEUTRO (p = 1.25 × 10−2, p = 8.98 × 10−3, respectively), and CREB5 also replicated at a lenient threshold for WBC (p = 2.61 × 10−2) but JAZF1-WBC did not replicate (p = 0.11). Both genes appeared in the GTEx whole blood (GWB) and MESA monocyte (MSA) secondary reference panels, but neither gene met the significance threshold for either reference panel. Importantly, Human Protein Atlas (Uhlén et al., 2015) reported that CREB5 was enhanced in blood and brain tissues and was specifically cell type enriched for NEUTRO (Human Protein Atlas., 2020b). JAZF1 on the other hand had low tissue specificity (Human Protein Atlas., 2020d). Together the TWAS, GWAS, and Human Protein Atlas results point to CREB5 as the most likely, and most biologically plausible, gene over JAZF1 at this locus.
FIGURE 2.
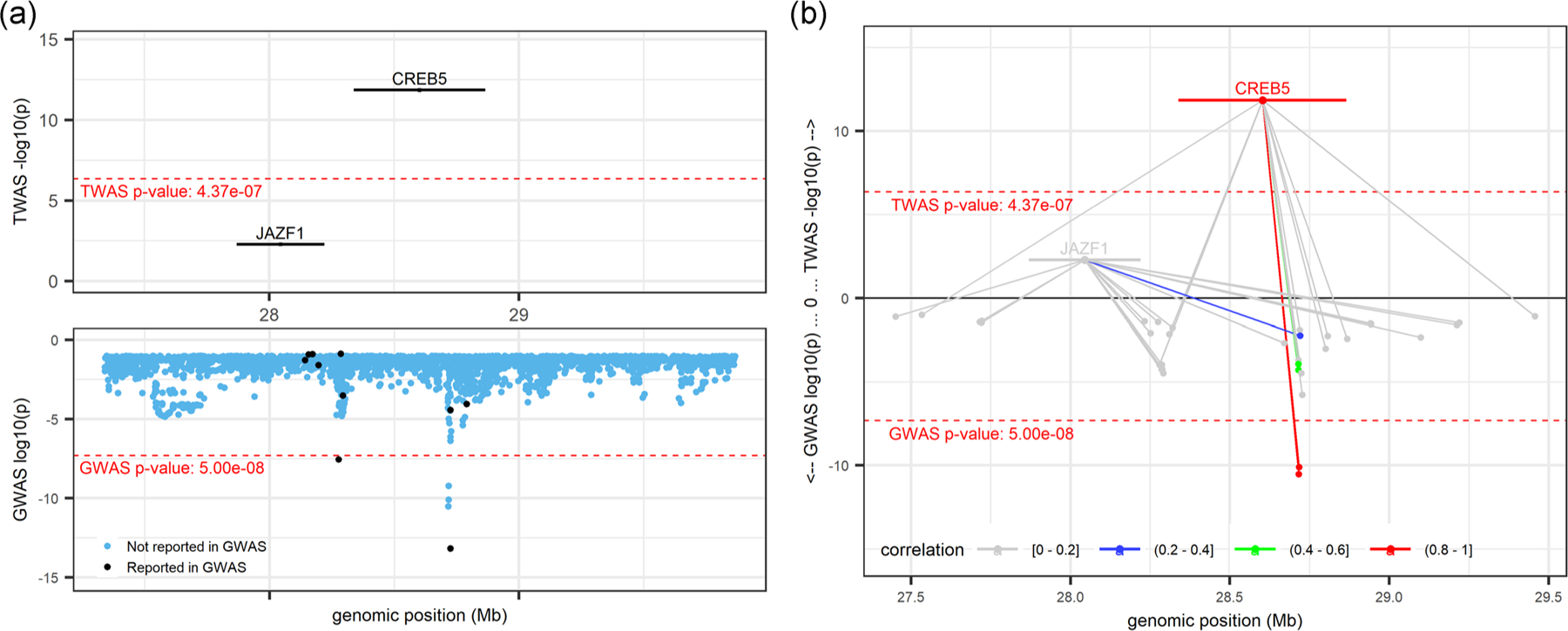
CREB5locus (locus 40; chr 7: 27,338,940–29,865,511; trait = NEUTRO) from R Shiny. TWAS results (top panels) and GWAS results (bottom panels). Marginal TWAS result displayed in (a), with Black-colored genes and variants denoting those previously reported by GWAS, blue variants denote those not previously reported as GWAS sentinel variants. (b) Mirrored-Manhattan locus-zoom plot displaying genes connected to their predictive model variants. Color scale, increasing from light gray to red, indicates the predicted expression correlation (r2) between the index TWAS gene and all other genes in the locus and the LD between the index variant and all other variants in the locus. GWAS, genome-wide association studies; TWAS, transcriptome-wide association study
3.5.3 |. CD164locus
The CD164 gene is known to play a role in hematopoiesis (Watt et al., 1998; Zannettino et al., 1998) and has been associated with several blood cell indices in GWAS analyses (Vuckovic et al., 2020). Our TWAS results prioritized CD164 over other genes at the locus as being significantly associated with MCV (p = 2.54 × 10−12) and categorized it into a multi-gene locus along with MICAL1 (p = 4.20 × 10−7) (Figure 3a). Conditional analysis on sentinel GWAS variants all but eliminated the TWAS signal for both CD164 (p = 8.61 × 10−2) and MICAL1 (p = 1.26 × 10−1). Interestingly, Figure 3b shows that CD164 and MICAL1 were not highly correlated in their predicted gene expression (r2: 0.2–0.4) and did not share any predictive model variants. We also note that both genes replicated in meta-analysis at a lenient threshold (CD164 p = 1.56 × 10−3; MICAL1 p = 7.06 × 10−3; Figure 3c). Additionally, while MICAL1 is not available in secondary reference panels, CD164 met the TWAS significance threshold for its association with MCV in GTEx whole blood and MESA monocytes (GTEx p = 2.61 × 10−9 and MESA p = 4.09 × 10−8). Thus, the evidence at this locus suggests that expression of CD164 and MICAL1 are both reasonable candidates for being regulated by red cell-associated genetic variants although we note that Human Protein Atlas reports low tissue specificity for MICAL1 (Human Protein Atlas., 2020f).
FIGURE 3.
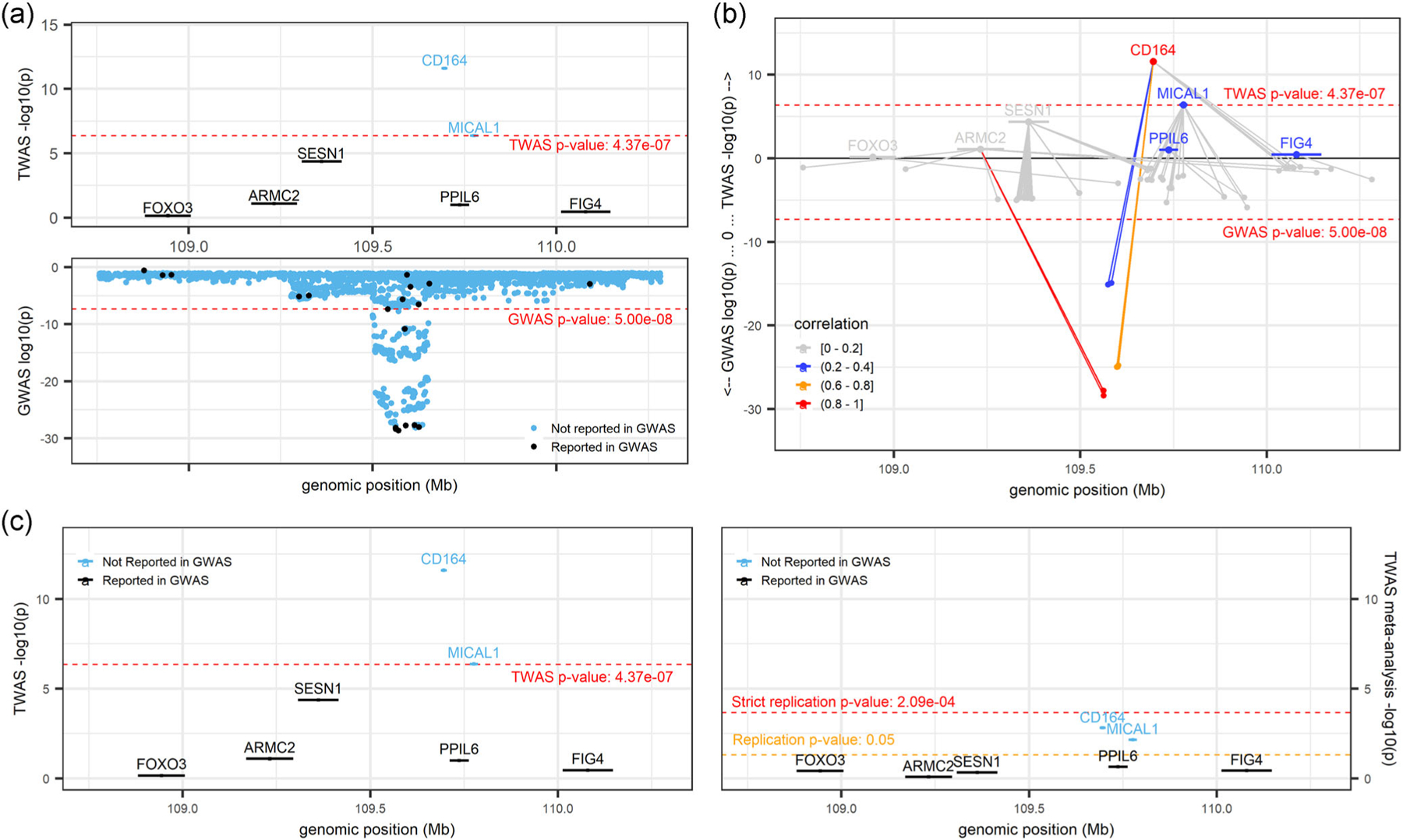
CD164locus (locus 36; chr 6: 108,687,717–110,703,762; trait = MCV) from R Shiny. (a) Marginal TWAS results in the top panel and GWAS results in the bottom panel. Black-colored genes and variants denote those previously implicated by GWAS, and blue-colored genes and variants denote those not previously implicated by GWAS. (b) Mirrored-Manhattan locus-zoom plot displaying genes connected to their predictive model variants. TWAS results in the top panel, GWAS results in the bottom panel. Color scale, increasing from light gray to red, indicates the predicted expression correlation (r2) between the index TWAS gene and all other genes in the locus and the LD between the index variant and all other variants in the locus. (c) Comparison of marginal TWAS (left panel) and TWAS meta-analysis (right panel). Black-colored genes denote those previously implicated by GWAS sentinel variants, and blue genes denote those not previously implicated by GWAS sentinel variants. GWAS, genome-wide association studies; TWAS, transcriptome-wide association study
3.5.4 |. PSMD3locus
The PSMD3 locus contained a much higher level of complexity because it fell into a region containing many marginal TWAS genes, had a complex gene-gene correlation and LD pattern, and included a combination of genes previously reported by GWAS as well as genes that had not been reported by GWAS. Thus, TWAS results did not clearly pinpoint the most likely causal gene. While PSMD3 appeared as the index TWAS gene associated with WBC (Figure 4a), eight other genes were also TWAS significant at this locus. Five of those genes (IKZF3, GSDMB, ORMDL3, MED24, and CCR7) replicated at a lenient significance threshold (p < 0.05), and PSMD3 replicated at a more stringent threshold (p < 2.09 × 10−4) (Figure 4c). We saw a complex network of shared model variants and correlation/LD patterns in Figure 4b, notably with MED24 and CCR7 (the next most significant genes at this locus) being only slightly correlated (r2 between 0.2 and 0.4) with PSMD3. The FOCUS fine-mapping results (Figure 4d) aligned to the TWAS results and indicated PSMD3 and MED24 as the most likely causal genes at the locus, each having posterior inclusion probabilities (PIPs) equal to 1.0. PIPs for all other genes at this locus, including CCR7, were less than 0.021 (Figure 4d).
FIGURE 4.
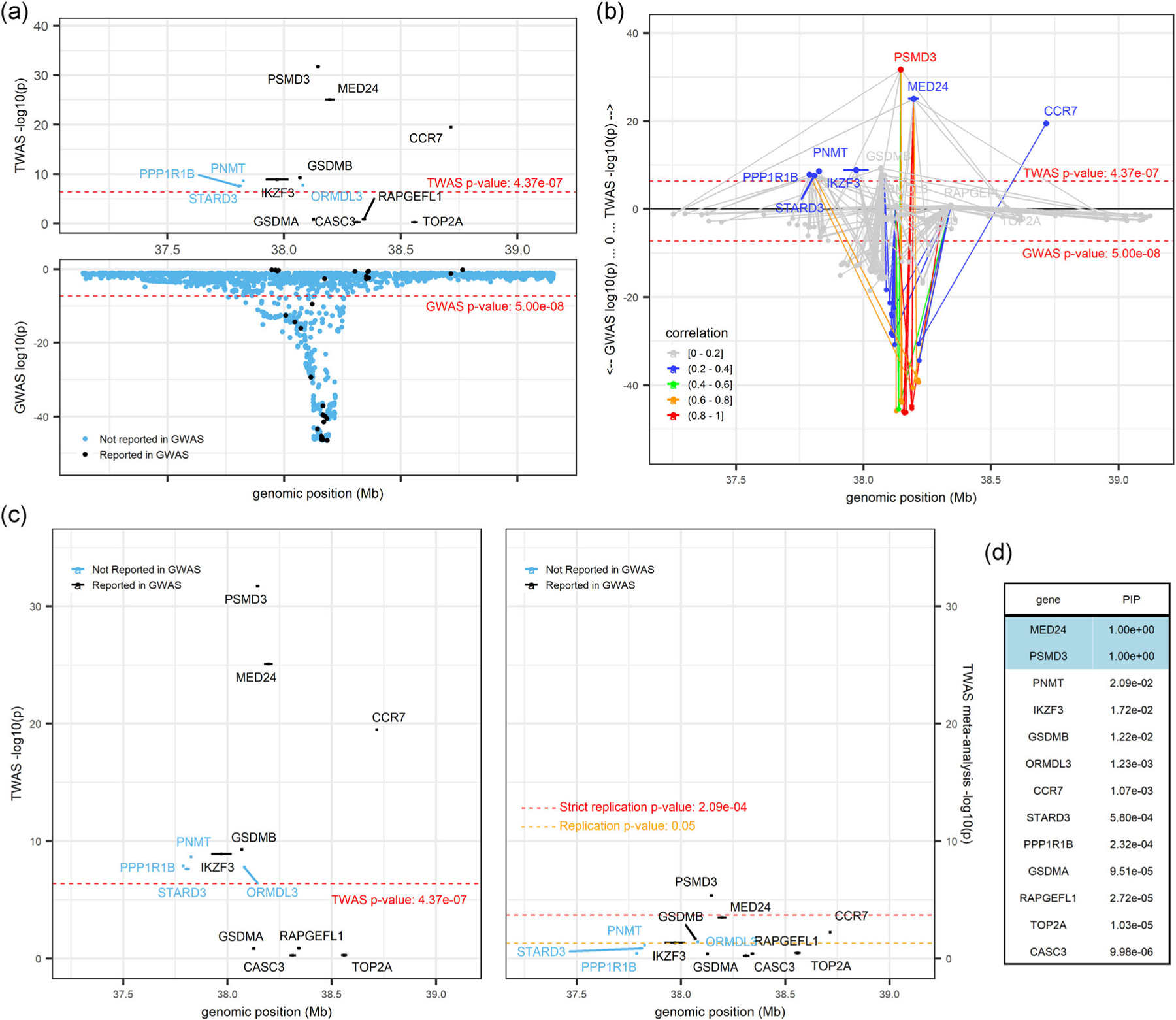
PSMD3locus (locus 101; chr 17: 37,137,050–39,154,213; trait = white blood cell count). (a) displays marginal TWAS results (top panel) and GWAS results (bottom panel), with genes and variants colored in blue and black to denote those not reported by GWAS and those reported by GWAS, respectively. (b) is a mirrored-Manhattan locus-zoom plot displaying genes connected to their predictive model variants with TWAS results (top panel) and GWAS results (bottom panel). Color scale, increasing from light gray to red, indicates the predicted expression correlation (r2) between the index TWAS gene and all other genes in the locus and the LD between the index variant and all other variants in the locus. (c) presents marginal TWAS results (left panel) and meta-analysis TWAS results (right panel), with genes colored in blue and black to denote those not reported by GWAS and those reported by GWAS, respectively. (d) displays the FOCUS posterior inclusion probabilities (PIPs) for each gene at this locus. FOCUS, Fine-mapping Of CaUsal gene Sets; GWAS, genome-wide association studies; TWAS, transcriptome-wide association study
4 |. DISCUSSION
Our follow-up fine-mapping, replication, and conditional analyses for a large-scale TWAS of 10 hematological measures demonstrates that results from marginal TWAS analyses alone cannot illuminate causal genes at loci for these traits.
4.1 |. TWAS discovery analyses
While 17 of our 239 marginal gene-trait associations did remain significant after conditional analysis or contained no known GWAS sentinel variants within a 1 Mb region of the gene, we found no substantive evidence from meta-analysis nor secondary reference panels to support these associations as novel discoveries for hematological traits. Conditional analyses suggested that nearly all our TWAS findings were driven at least in part by GWAS sentinel variants from the largest recent European-focused GWAS analysis for hematological measures (Vuckovic et al., 2020). This is perhaps not surprising given the greater statistical power for this GWAS analysis, which was conducted in 563,085 participants (vs. 54,542 participants in our analysis). However, for 61 gene-trait associations (26%), some residual signal (p < 0.05) remained after conditioning on GWAS. For example, although JAK2 is a well-known blood cell-associated signal from GWAS (Vuckovic et al., 2020) and the Mendelian disease literature for platelet disorders [MIM 147796], its association with platelet count remained statistically significant after conditional analysis. Thus, our TWAS results also suggested that there are likely additional regulatory variants at the JAK2 locus which are not tagged by current GWAS single variants. Similarly, for other gene-trait associations retaining some residual significance after conditional analysis, our results suggest that additional small-effect regulatory variants remain to be discovered for these genes which associate with blood cell indices, illustrating the power advantages from aggregate tests like TWAS.
4.2 |. TWAS Fine-Mapping
For TWAS fine-mapping, we grouped the 239 TWAS-wide significant gene-trait associations into 120 loci. To effectively interpret these results, we introduced LocusXcanR, an R Shiny application that integrates TWAS and GWAS information into locus-specific, interactive visualizations which we use to assist with TWAS fine-mapping and TWAS results interpretation. We showed the utility of LocusXcanR by highlighting the varying levels of complexity at several TWAS loci and demonstrating where TWAS aligns with or provides advantages over GWAS. For example, the HK1-MCV locus showed a very simple genomic locus in which we found that TWAS confirmed what we already knew from GWAS. Coding variants in HK1 are known to be associated with hemolytic anemia due to hexokinase deficiency [MIM 142600], providing a clear link to red blood cell-related traits.
The CREB5 locus further demonstrated one of the advantages of TWAS over GWAS in that the TWAS results provided clarity regarding the likely causal gene at the locus. At this locus, CREB5 and JAZF1 had both been implicated by GWAS, likely assigned based on their physical proximity to the GWAS sentinel variant. However, CREB5 showed a stronger TWAS signal, replicated in the much smaller meta-analysis sample, and Human Protein Atlas provided clear evidence of enrichment in blood (specifically neutrophils) (Human Protein Atlas., 2020b, 2020d; Uhlén et al., 2015). These results in aggregate supported CREB5 as the likely causal gene at this locus, even though CREB5 may not be the closest gene in proximity to all sentinel GWAS variants within the region.
4.3 |. TWAS challenges
We further highlight the challenges, particularly at multi-gene loci, which should be considered when interpreting TWAS findings, including total and/or predicted expression correlation, shared predictive model variants, the relevance of reference tissue panel, biological plausibility, and so forth and demonstrated the importance of interpreting TWAS results in context. Although TWAS is useful for prioritizing candidate causal genes, researchers should guard against the hasty conclusion that the most significant gene is the only causal gene or even the most likely causal gene. For example, the conclusion at the CD164 locus is not evident from TWAS results. While TWAS points to CD164 as the causal gene, as does existing knowledge of the gene’s biological function, taking the full context of this locus into consideration, it is not out of the realm of possibilities that both CD164 and MICAL1 are causal at this locus.
Furthermore, at the PSMD3 locus we see potentially misleading TWAS results when marginal TWAS statistics are interpreted alone. The PSMD3-WBC association appears as the sentinel gene at this locus. However, several pieces of evidence support other genes, including CCR7, as the most likely biologically plausible causal gene at the locus. First, PSMD3 and MED24 have no immune cell specificity (Human Protein Atlas., 2020e, 2020g; Uhlén et al., 2015). Second, CCR7 was also TWAS significant, it replicated at a lenient threshold in meta-analysis, and is enriched for expression in blood and lymphoid tissues, especially T-cells (Human Protein Atlas., 2020a). However, CCR7 was not highly correlated with nor does it appear to share model variants with lead gene PSMD3, and the FOCUS results show a posterior inclusion probability of only 0.001. Finally, CCR7 is known to be involved in the migration of neutrophils to lymph nodes (Beauvillain et al., 2011). While it is certainly possible at multi-gene TWAS loci for multiple genes to be contributing to trait regulation, it is also possible for spurious or nonrelevant genes to be identified based on shared eQTLs across tissues that are not relevant to a given trait or correlation of gene expression.
Moreover, proximal genes which cannot be accurately imputed with a given reference panel, but which may still be influenced by variants identified by GWAS studies, must also be considered. For example, the gene colony-stimulating factor 3 (CSF3), which has a known key role in the production, differentiation, and function of granulocytes [MIM 138970], is also situated within the PSMD3 locus. However, this gene has very low constitutive expression in whole blood (Human Protein Atlas., 2020c), and it is not depicted in Figure 4 (or LocusXcanR) because a predictive model could not be fit for this gene in the DGN reference panel (likely due to very low expression); therefore, CFS3 cannot be detected as a possible target gene at this locus (Supplementary Table 7 contains CSF3, along with other genes that have been assigned by one or more GWAS variants but are not included in DGN). This genomic region is extremely complex and highly pleiotropic, and any interpretation of this locus using TWAS results alone is likely to be overly simplistic. This complex locus shows the importance of considering statistical evidence from TWAS, GWAS, and FOCUS fine-mapping as well as trait biology in the interpretation of TWAS findings.
5 |. LIMITATIONS
While we have used PrediXcan and pre-calculated PredictDB weights for our analysis, we note a limitation in doing so. The variants included in PredictDB were not always available in our analytical cohort (generally due to poor imputation quality), so some predictive models did not use all PredictDB weights. We note that 70% of our TWAS significant genes were predicted with complete variant matching (i.e. used all model variants) and 85% of TWAS significant genes used at least 90% of model variants; we have included this information in Table S2 for transparency, and these details should be taken into account when interpreting TWAS results.
The cohorts that we have included in our TWAS meta-analysis also pose some limitations on our ability to replicate GERA TWAS sentinel genes. The smaller sample sizes of the meta-analyzed cohorts are likely the primary reason why GERA TWAS sentinel genes fail to replicate. Additionally, it may be the case that major contributing variants exhibit differential allele frequencies across cohorts; although this is less likely than in multi-ethnic analyses because all cohorts are of European ancestry, it could still contribute to poorer power for replication. Furthermore, differences in imputation quality across cohorts could also explain the failure to replicate TWAS sentinel genes in meta-analysis.
Although FOCUS, in some cases, helps to identify a set of the most likely causal genes at a locus, we have shown that it does not always provide enough evidence above and beyond TWAS to fully identify a putative causal gene set at a complex locus. Additionally, FOCUS performs a summary statistics-based TWAS method and then proceeds to fine-mapping the TWAS results from this method. However, we performed TWAS using PrediXcan, and thus, the fine-mapping results from FOCUS may not exactly match our PrediXcan TWAS results. In future, the FOCUS software could be extended to take pre-calculated TWAS results as input (using the TWAS method of the researcher’s choosing), bypassing the need to use GWAS summary statistics or to recompute predicted gene expression.
Our analysis is primarily conducted using whole blood TWAS weights only, with supplemental TWAS results available in our app for a few other blood-related tissues (whole blood and EBV transformed lymphocytes from GTEx and monocytes from MESA); we felt this was the most prudent approach to limit false positives and reduce needed multiple testing correction, versus an approach using TWAS weights in, for example, all GTEx tissues. However, this choice could be inappropriate if the main relevant tissue at some blood cell-related loci is not in fact whole blood, and it limits our ability to use FOCUS fine-mapping to overcome the choice of tissue for TWAS training. Joint/multiple tissue TWAS approaches such as UTMOST (Hu et al., 2019) and MR-JTI (Zhou et al., 2020) could be employed in the future to assess the relevance of other tissues at blood-cell-related loci.
6 |. SUMMARY
In summary, we found that TWAS results enrich our understanding of GWAS, can help to explain trait variation, and are superior to merely selecting the nearest gene. We have shown that the gene, or genes, implicated in TWAS, in some cases, clearly overlap with what is known in GWAS and from prior knowledge of important genes in hematopoietic processes. However, while we showed that TWAS may help in some cases to pinpoint likely causal genes, we emphasize the need for investigators not to interpret TWAS results alone and out of context. We introduced an R Shiny application and demonstrated its utility in assisting researchers in this endeavor by leveraging the TWAS and GWAS information available from the analytical cohort and interactively visualizing results one locus at a time. The results of this analysis are accessible online (http://shiny.bios.unc.edu/gera-twas/), and we also made the layout of this application available for others to import and analyze their own TWAS results in the LocusXcanR R package, available on GitHub (https://github.com/amanda-tapia/LocusXcanR). Together with a clearer understanding of the relationship between TWAS and GWAS results, biological insight, and subject matter expertise, TWAS results can help us formulate mechanistic hypotheses for functional experimental validation.
Supplementary Material
ACKNOWLEDGMENTS
The data and materials included in this report result from collaboration between multiple studies and organizations. The authors thank the staff and participants of GERA. The authors also thank the WHI investigators and staff for their dedication and the study participants for making the program possible. A listing of WHI investigators can be found at: https://www.whi.org/researchers/Documents%20%20Write%20a%20Paper/WHI%20Investigator%20Long%20List.pdf. The authors thank the staff and participants of the ARIC study for their important contributions. The authors also thank all participants in the Mount Sinai Biobank all our recruiters who have assisted and continue to assist in data collection and management and are grateful for the computational resources and staff expertise provided by Scientific Computing at the Icahn School of Medicine at Mount Sinai. GERA: Genotyping of the GERA cohort was funded by a grant from the National Institute on Aging, National Institute of Mental Health, and National Institute of Health Common Fund (RC2 AG036607). WHI: Funding support for the “Exonic variants and their relation to complex traits in minorities of the WHI” study is provided through the NHGRI PAGE program (NIHU01HG007376). The WHI program is funded by the National Heart, Lung, and Blood Institute, National Institutes of Health, U.S. Department of Health and Human Services through contracts HHSN268201100046C, HHSN268201100001C, HHSN268201100002C, HHSN26 8201100003C, HHSN268201100004C, and HHSN271201 100004C. ARIC: The Atherosclerosis Risk in Communities study has been funded in whole or in part with Federal funds from the National Heart, Lung, and Blood Institute, National Institutes of Health, Department of Health and Human Services (contract numbers HHSN268201700001I, HHSN268201700002I, HHSN26 8201700003I, HHSN268201700004I, and HHSN2682017 00005I), R01HL087641, R01HL059367, and R01HL0866 94; National Human Genome Research Institute contract U01HG004402; and National Institutes of Health contract HHSN268200625226C. Infrastructure was partly supported by Grant Number UL1RR025005, a component of the National Institutes of Health and NIH Roadmap for Medical Research. BioMe: The Mount Sinai BioMe Biobank has been supported by The Andrea and Charles Bronfman Philanthropies and in part by Federal funds from the NHLBI and NHGRI (U01HG00638001; U01HG007417; X01HL134588). This study was funded by R01HL129132 and R01HL146500. C.K and L.M.R. were funded by R01HG010297. C.K. and S.B. were also funded by U01HG007376. LMR was additionally funded by T32 HL129982, and KL2TR002490. Y.L. was further supported by R01GM105785. R.J.F.L. was funded by R01DK110113, R01DK107786, R01HL142302, and R01 DK124097. H.C. and E.J. were supported by grants from the National Eye Institute (R01 EY027004), the National Institute of Diabetes and Digestive and Kidney Diseases (R01 DK116738), and from the National Cancer Institute (R01 CA2416323). B.R. was supported by the National Science Foundation Graduate Research Fellowship under Grant No. DGE-1650116.
Funding information
National Institute of Diabetes and Digestive and Kidney Diseases, Grant/Award Numbers: R01 DK116738, R01 DK124097, R01DK107786; National Institutes of Health, Grant/Award Numbers: R01DK110113, R01HL129132; National Science Foundation, Grant/Award Number: DGE-1650116; National Eye Institute, Grant/Award Number: EY027004; National Cancer Institute, Grant/Award Number: R01 CA2416323; North Carolina Translational and Clinical Sciences Institute, University of North Carolina at Chapel Hill, Grant/Award Number: KL2TR002490; National Heart, Lung, and Blood Institute, Grant/Award Numbers: HL129982, R01HL142302; National Human Genome Research Institute, Grant/Award Numbers: R01HG010297, U01HG007376
Footnotes
URLS
cpgen: https://github.com/cheuerde/cpgen
FOCUS: https://github.com/bogdanlab/focus
Human Protein Atlas: https://www.proteinatlas.org/
LocusXcanR R package for R Shiny application: https://github.com/amanda-tapia/LocusXcanR
LocusXcanR R Shiny application for GERA results: http://shiny.bios.unc.edu/gera-twas/
METAL: https://genome.sph.umich.edu/wiki/METAL_Documentation
Online Mendelian Inheritance in Man (OMIM): https://www.omim.org/
PLINK KING kinship matrix: https://www.cog-genomics.org/plink/2.0/distance#make_king
PredictDB: http://predictdb.org/
PrediXcan: https://github.com/hakyimlab/PrediXcan
R Shiny: https://shiny.rstudio.com/
CONFLICT OF INTERESTS
The authors declare that there are no conflict of interests.
SUPPORTING INFORMATION
Additional supporting information may be found in the online version of the article at the publisher’s website.
DATA AVAILABILITY STATEMENT
Data are available through dbGaP (GERA- phs000674, ARIC- phs000557, WHI- phs000386, and BioMe- phs000925), or by application to study coordinating centers with approved manuscript proposals (ARIC-https://sites.cscc.unc.edu/aric/, WHI https://www.whi.org/, and BioMe-https://icahn.mssm.edu/research/ipm/programs/biome-biobank/, GERA https://divisionofresearch.kaiserpermanente.org/genetics/rpgeh/biobank/).
REFERENCES
- Astle WJ, Elding H, Jiang T, Allen D, Ruklisa D, Mann AL, Mead D, Bouman H, Riveros-Mckay F, Kostadima MA, Lambourne JJ, Sivapalaratnam S, Downes K, Kundu K, Bomba L, Berentsen K, Bradley JR, Daugherty LC, Delaneau O … Soranzo N (2016). The allelic landscape of human blood cell trait variation and links to common complex disease. Cell, 167(5), 1415–1429. 10.1016/j.cell.2016.10.042 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Banda Y, Kvale MN, Hoffmann TJ, Hesselson SE, Ranatunga D, Tang H, Sabatti C, Croen LA, Dispensa BP, Henderson M, Iribarren C, Jorgenson E, Kushi LH, Ludwig D, Olberg D, Quesenberry CP, Rowell S, Sadler M, Sakoda LC … Risch N (2015). Characterizing race/ethnicity and genetic ancestry for 100,000 subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort. Genetics, 200(4), 1285–1295. 10.1534/genetics.115.178616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Battle A, Mostafavi S, Zhu X, Potash JB, Weissman MM, McCormick C, Haudenschild CD, Beckman KB, Shi J, Mei R, Urban AE, Montgomery SB, Levinson DF, & Koller D (2014). Characterizing the genetic basis of transcriptome diversity through RNA-sequencing of 922 individuals. Genome Research, 24(1), 14–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beauvillain C, Cunin P, Doni A, Scotet M, Jaillon S, Loiry ML, Magistrelli G, Masternak K, Chevailler A, Delneste Y, & Jeannin P (2011). CCR7 is involved in the migration of neutrophils to lymph nodes. Blood, 117(4), 1196–1204. 10.1182/blood-2009-11-254490 [DOI] [PubMed] [Google Scholar]
- Chen MH, Raffield LM, Mousas A, Sakaue S, Huffman JE, Moscati A, Trivedi B, Jiang T, Akbari P, Vuckovic D, Bao EL, Zhong X, Manansala R, Laplante V, Chen M, Lo KS, Qian H, Lareau CA, Beaudoin M … Lettre G (2020). Trans-ethnic and ancestry-specific blood-cell genetics in 746,667 individuals from 5 global populations. Cell, 182(5), 1198–1213. 10.1016/j.cell.2020.06.045 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gamazon ER, Wheeler HE, Shah KP, Mozaffari SV, Aquino-Michaels K, Carroll RJ, Eyler AE, Denny JC, GTEx C, Nicolae DL, Cox NJ, & Im HK (2015). A gene-based association method for mapping traits using reference transcriptome data. Nature Genetics, 47, 1091–1098. 10.1038/ng.3367 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gusev A, Mancuso N, Won H, Kousi M, Finucane HK, Reshef Y, Song L, Safi A, Schizophrenia Working Group of the Psychiatric Genomics, C., McCarroll S, Neale BM, Ophoff RA, O’Donovan MC, Crawford GE, Geschwind DH, Katsanis N, Sullivan PF, Pasaniuc B, & Price AL (2018). Transcriptome-wide association study of schizophrenia and chromatin activity yields mechanistic disease insights. Nature Genetics, 50(4), 538–548. 10.1038/s41588-018-0092-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu Y, Li M, Lu Q, Weng H, Wang J, Zekavat SM, Yu Z, Li B, Gu J, Muchnik S, Shi Y, Kunkle BW, Mukherjee S, Natarajan P, Naj A, Kuzma A, Zhao Y, Crane PK, Alzheimer’s Disease Genetics C,, … Zhao H (2019). A statistical framework for cross-tissue transcriptome-wide association analysis. Nature Genetics, 51(3), 568–576. 10.1038/s41588-019-0345-7 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Human Protein Atlas. (2020a). Human Protein Atlas CCR7. https://www.proteinatlas.org/ENSG00000126353-CCR7
- Human Protein Atlas. (2020b). Human Protein Atlas CREB5. https://www.proteinatlas.org/ENSG00000146592-CREB5
- Human Protein Atlas. (2020c). Human Protein Atlas CSF3. https://www.proteinatlas.org/ENSG00000108342-CSF3/tissue
- Human Protein Atlas. (2020d). Human Protein Atlas JAZF1. https://www.proteinatlas.org/ENSG00000153814-JAZF1
- Human Protein Atlas. (2020e). Human Protein Atlas MED24. https://www.proteinatlas.org/ENSG00000008838-MED24
- Human Protein Atlas. (2020f). Human Protein Atlas MICAL1. https://www.proteinatlas.org/ENSG00000135596-MICAL1/tissue
- Human Protein Atlas. (2020g). Human Protein Atlas PSMD3. https://www.proteinatlas.org/ENSG00000108344-PSMD3
- Kvale MN, Hesselson S, Hoffmann TJ, Cao Y, Chan D, Connell S, Croen LA, Dispensa BP, Eshragh J, Finn A, Gollub J, Iribarren C, Jorgenson E, Kushi LH, Lao R, Lu Y, Ludwig D, Mathauda GK, McGuire WB … Risch N (2015). Genotyping informatics and quality control for 100,000 subjects in the Genetic Epidemiology Research on Adult Health and Aging (GERA) cohort. Genetics, 200(4), 1051–1060. 10.1534/genetics.115.178905 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu Y, Ding J, Reynolds LM, Lohman K, Register TC, De La Fuente A, Howard TD, Hawkins GA, Cui W, Morris J, Smith SG, Barr RG, Kaufman JD, Burke GL, Post W, Shea S, McCall CE, Siscovick D, Jacobs DR J. r… Hoeschele I (2013). Methylomics of gene expression in human monocytes. Human Molecular Genetics, 22(24), 5065–5074. 10.1093/hmg/ddt356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lonsdale J, Thomas J, Salvatore M, Phillips R, Lo E, Shad S, Hasz R, Walters G, Garcia F, Young N, Foster B, Moser M, Karasik E, Gillard B, Ramsey K, Sullivan S, Bridge J, Magazine H, Syron J… Moore HF (2013). The Genotype-Tissue Expression (GTEx) project. Nature Genetics, 45(6), 580–585. 10.1038/ng.2653 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mancuso N, Freund MK, Johnson R, Shi H, Kichaev G, Gusev A, & Pasaniuc B (2019). Probabilistic fine-mapping of transcriptome-wide association studies. Nature Genetics, 51(4), 675–682. 10.1038/s41588-019-0367-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manichaikul A, Mychaleckyj JC, Rich SS, Daly K, Sale M, & Chen WM (2010). Robust relationship inference in genome-wide association studies. Bioinformatics, 26(22), 2867–2873. 10.1093/bioinformatics/btq559 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mogil LS, Andaleon A, Badalamenti A, Dickinson SP, Guo X, Rotter JI, Johnson WC, Im HK, Liu Y, & Wheeler HE (2018). Genetic architecture of gene expression traits across diverse populations. PLoS Genetics, 14(8), e1007586. 10.1371/journal.pgen.1007586 [DOI] [PMC free article] [PubMed] [Google Scholar]
- The Atherosclerosis Risk in Communities (ARIC) Study: design and objectives. (1989). The ARIC investigators. American Journal of Epidemiology, 129(4), 687–702. [PubMed] [Google Scholar]
- The Women’s Health Initiative Study Group. (1998). Design of the Women’s Health Initiative clinical trial and observational study. Controlled Clinical Trials, 19(1), 61–109. [DOI] [PubMed] [Google Scholar]
- Uhlén M, Fagerberg L, Hallström BM, Lindskog C, Oksvold P, Mardinoglu A, Sivertsson Å, Kampf C, Sjöstedt E, Asplund A, Olsson I, Edlund K, Lundberg E, Navani S, Szigyarto CA, Odeberg J, Djureinovic D, Takanen JO, Hober S… Pontén F (2015). Proteomics tissue-based map of the human proteome. Science, 347(6220), 1260419. 10.1126/science.1260419 [DOI] [PubMed] [Google Scholar]
- Vuckovic D, Bao EL, Akbari P, Lareau CA, Mousas A, Jiang T, Chen MH, Raffield LM, Tardaguila M, Huffman JE, Ritchie SC, Megy K, Ponstingl H, Penkett CJ, Albers PK, Wigdor EM, Sakaue S, Moscati A, Manansala R… Soranzo N (2020). The polygenic and monogenic basis of blood traits and diseases. Cell, 182(5), 1214–1231. 10.1016/j.cell.2020.08.008 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainberg M, Sinnott-Armstrong N, Mancuso N, Barbeira AN, Knowles DA, Golan D, Ermel R, Ruusalepp A, Quertermous T, Hao K, Björkegren JLM, Im HK, Pasaniuc B, Rivas MA, & Kundaje A (2019). Opportunities and challenges for transcriptome-wide association studies. Nature Genetics, 51(4), 592–599. 10.1038/s41588-019-0385-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watt SM, Bühring HJ, Rappold I, Chan JY, Lee-Prudhoe J, Jones T, Zannettino AC, Simmons PJ, Doyonnas R, Sheer D, & Butler LH (1998). CD164, a novel sialomucin on CD34(+) and erythroid subsets, is located on human chromosome 6q21. Blood, 92(3), 849–866. [PubMed] [Google Scholar]
- Willer CJ, Li Y, & Abecasis GR (2010). METAL: Fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26(17), 2190–2191. 10.1093/bioinformatics/btq340 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zannettino AC, Bühring HJ, Niutta S, Watt SM, Benton MA, & Simmons PJ (1998). The sialomucin CD164 (MGC-24v) is an adhesive glycoprotein expressed by human hematopoietic progenitors and bone marrow stromal cells that serves as a potent negative regulator of hematopoiesis. Blood, 92(8), 2613–2628. [PubMed] [Google Scholar]
- Zhan X, Hu Y, Li B, Abecasis GR, & Liu DJ (2016). RVTESTS: An efficient and comprehensive tool for rare variant association analysis using sequence data. Bioinformatics, 32(9), 1423–1426. 10.1093/bioinformatics/btw079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, & Lin X (2013). Some considerations of classification for high dimension low-sample size data. Statistical Methods in Medical Research, 22(5), 537–550. 10.1177/0962280211428387 [DOI] [PubMed] [Google Scholar]
- Zhou D, Jiang Y, Zhong X, Cox NJ, Liu C, & Gamazon ER (2020). A unified framework for joint-tissue transcriptome-wide association and Mendelian randomization analysis. Nature Genetics, 52, 1239–1246. 10.1038/s41588-020-0706-2 [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
Data are available through dbGaP (GERA- phs000674, ARIC- phs000557, WHI- phs000386, and BioMe- phs000925), or by application to study coordinating centers with approved manuscript proposals (ARIC-https://sites.cscc.unc.edu/aric/, WHI https://www.whi.org/, and BioMe-https://icahn.mssm.edu/research/ipm/programs/biome-biobank/, GERA https://divisionofresearch.kaiserpermanente.org/genetics/rpgeh/biobank/).