

Abstract
K2P (KCNK) potassium channels form “back-ground” or “leak” currents that have critical roles in cell excitability control in the brain, cardiovascular system, and somatosensory neurons. Similar to many ion channel families, studies of K2Ps have been limited by poor pharmacology. Of six K2P subfamilies, the thermo- and mechanosensitive TREK subfamily comprising K2P2.1 (TREK-1), K2P4.1 (TRAAK), and K2P10.1 (TREK-2) are the first to have structures determined for each subfamily member. These structural studies have revealed key architectural features that underlie K2P function and have uncovered sites residing at every level of the channel structure with respect to the membrane where small molecules or lipids can control channel function. This polysite pharmacology within a relatively small (~70 kDa) ion channel comprises four structurally defined modulator binding sites that occur above (Keystone inhibitor site), at the level of (K2P modulator pocket), and below (Fenestration and Modulatory lipid sites) the C-type selectivity filter gate that is at the heart of K2P function. Uncovering this rich structural landscape provides the framework for understanding and developing subtype-selective modulators to probe K2P function that may provide leads for drugs for anesthesia, pain, arrhythmia, ischemia, and migraine.
Keywords: K2P channel, TREK subfamily, Ruthenium red, ML335/ML402, Fluoxetine, PIP2
4.1. Introduction
Ion channel proteins facilitate the flow of bioelectricity that underlies the physiology of thought, movement, mood, and sensation [1]. The K2P (KCNK) potassium channel family comprises a set of 15 members (Fig. 4.1a) of the voltage-gated ion channel (VGIC) superfamily [2] that have central roles in controlling cell excitability by producing “leak” potassium currents that are largely time and voltage-independent [3–5]. The 15 K2P subtypes comprise six subfamilies (TREK, TWIK, TASK, TALK, THIK, and TRESK) that within each subfamily are related by sequence similarities and regulation by shared types of signals [6] (Fig. 4.1a). A diverse range of stimuli affect K2Ps depending on the subfamily type and include physical gating commands such as pressure and temperature (TREK) [4, 7], external and internal pH (TREK, TASK, TALK, and TWIK) [4, 8], chemicals such as volatile anesthetics (TREK, TASK, THIK, TRESK) and antidepressants (TREK) [4, 9], lipids and polyunsaturated fatty acids (PUFAs) (TREK, THIK) [4, 9], and protein–protein interactions with partners such as 14–3–3, G-proteins, Protein Kinase A, and Protein Kinase C (TREK, TASK, TALK, TWIK, TRESK) [4, 9].
Fig. 4.1.
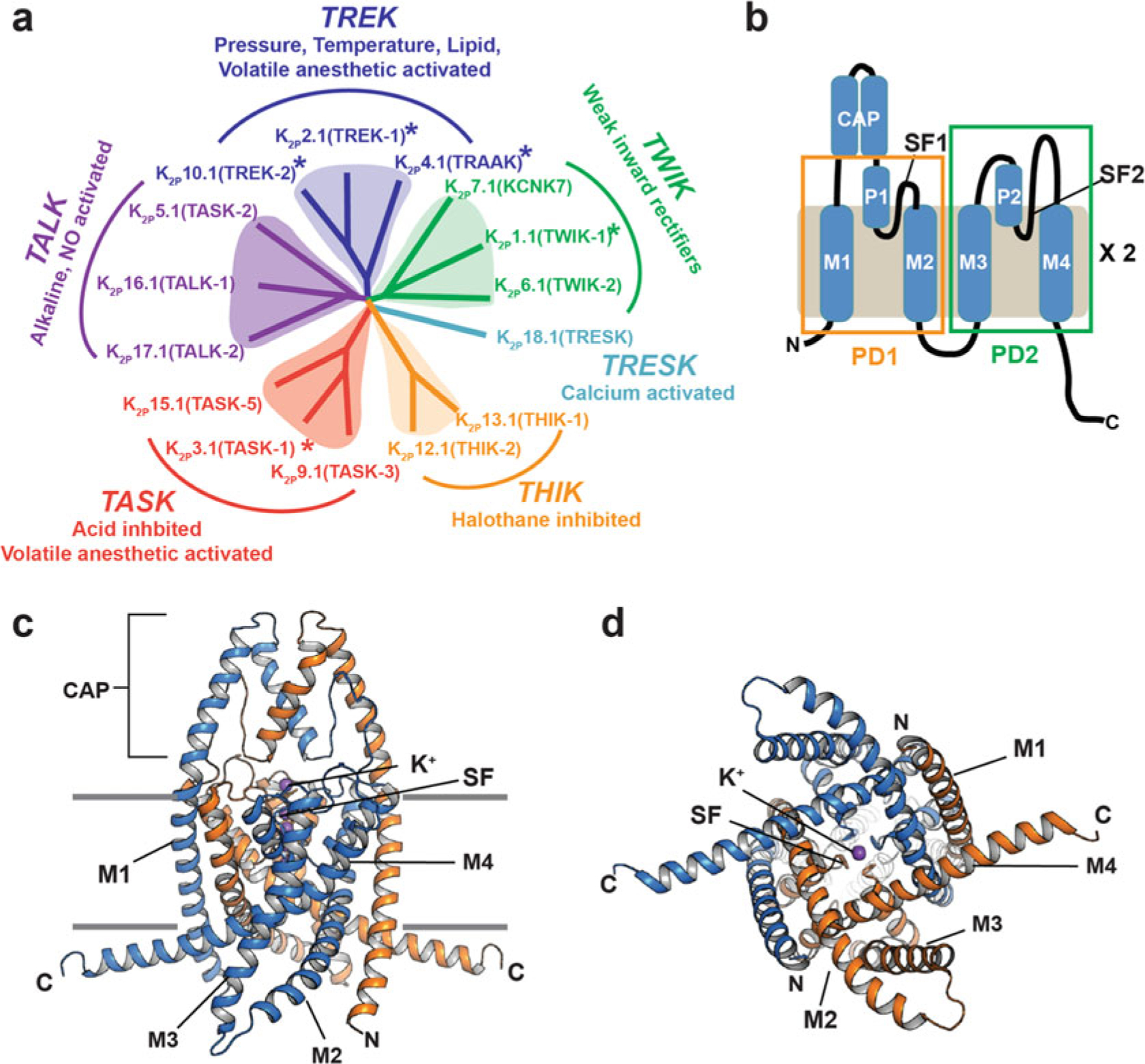
K2P channel relationships and architecture (a) K2P channel dendrogram. Subfamilies are indicated. Asterisks indicate structurally characterized K2Ps. (b) K2P subunit diagram. Pore domains 1 and 2 (PD1 and PD2), transmembrane helices (M1–M4), pore helices (P1 and P2), selectivity filters (SF1 and SF2), and CAP domain are indicated. (c and d) Cartoon diagram of the K2P2.1 (TREK-1) structure (PDB:6CQ6) [11]. Chains are colored marine and orange. Potassium ions are purple. (c), side view, (d), cytoplasmic view. Channel elements are labeled as in “b”
K2Ps are named for their unique architecture. Each subunit bears two pore-forming domains, PD1 and PD2, each comprising two transmembrane helices (M1–M2 and M3–M4) bridged by a pore helix (P1 and P2) and selectivity filter (SF1 and SF2) (Fig. 4.1b). K2P subunits dimerize to create a channel in which the pore is intrinsically heterotetrameric due to sequence differences between PD1 and PD2. Structures have been determined for exemplars of five of the fifteen K2Ps subtypes (Fig. 4.1a), K2P1.1 (TWIK-1), [10] K2P2.1 (TREK-1), [11] K2P3.1 (TASK-1), [12] K2P4.1 (TRAAK), [13] and K2P10.1 (TREK-2) [14] (Fig. 4.1c, d). These structures reveal a common protein scaffold that defines the K2P family.
Similar to other VGICs [15, 16], the two transmembrane segments of the pore domains form outer (M1 and M3) and inner (M2 and M4) helices that define the pore and support the pore helices and selectivity filter [10–14]. The pore helices and selectivity filter coordinate a set of four potassium ions on the channel central axis. K2Ps have a unique structural feature, the CAP domain (Fig. 4.1b, c). This extracellular structural element forms an arch directly over the channel pore and creates the bifurcated extracellular ion pathway (EIP) from which the ions exit the channel after passing through the selectivity filter [10, 13]. The M1 helix is domain-swapped between the two subunits, but how this structural intertwining of the subunits impacts function, assembly, or biogenesis is not clear. Each subunit also bears sequences at both the N- and C-termini that are likely to be unstructured on their own but that provide sites for protein–protein interactions that impact function [4, 9].
Unlike other potassium channels, the K2P principal gate is the selectivity filter “C-type” gate [11, 17–22]. In line with this mechanism of control, the structures of K2P1.1 (TWIK-1), [10] K2P2.1 (TREK-1) [11], K2P4.1 (TRAAK), [13] and K2P10.1 (TREK-2) [14] show that these channels lack the inner gate that is present in most other VGIC superfamily members (Fig. 4.1d). The K2P structures have shown that the M4 helix is mobile and can adopt conformations that range between an “up” state and a “down” state [13, 14, 23–25]. The “down” state creates a fenestration just below the P2 pore helix that is open to the center of the membrane bilayer [10, 13, 24, 25]. These M4 conformational changes are linked to C-type gate control [17, 18, 25–27] but do not impede access from the intracellular side [26]. Intriguingly, the recent K2P3.1 (TASK-1) [12] structure shows a “down” state in which M4 creates an intracellular barrier, termed the “X-gate” that appears to be a special feature of the TASK subfamily [12] and that highlights structural diversification of the M4 segment within the K2P family.
K2Ps have roles in a multitude of physiological responses and pathological conditions such as action potential propagation [28, 29], anesthetic responses [30, 31], microglial surveillance [32], sleep duration [33], pain, [34–36] arrhythmia [37], ischemia [30, 38, 39], cardiac fibrosis [40], depression [41], migraine [42], intraocular pressure regulation [43], pulmonary hypertension [44], acute respiratory distress syndrome [45], and cancer [46]. Despite these clear physiological roles, the pharmacology of K2Ps is generally poor [9, 47] and has been a barrier to understanding K2P function. The paucity of reagents to probe K2P activity has motivated multiple efforts that have begun to define new K2P modulators [11, 36, 48–52] and key structural aspects of K2P channel pharmacology [11, 14, 53, 54].
K2Ps are thought to be potential therapeutic targets for pain [47, 55, 56], anesthesia [9, 47], arrhythmia [57, 58], ischemia [59], depression [60], and migraine [9, 61]. Although there are no approved drugs that target K2Ps specifically, recent advances in the discovery of new classes of a variety of K2P modulators should enable elaboration of a suite of new pharmacological tools directed at this channel family [11, 12, 36, 48, 49, 51, 62–64].
4.2. The TREK Subfamily: Model Polymodal Ion Channels
The TREK subfamily comprising K2P2.1 (TREK-1), K2P10.1 (TREK-2), and K2P4.1 (TRAAK) is both the most extensively studied K2P family and the only subfamily for which structures of each subtype are known [11, 13, 14] (Fig. 4.1a). TREK channels are polymodal ion channels that respond to diverse physical and chemical gating cues including temperature, pressure, pH, and modulatory lipids [7, 65]. The sensors for these signals reside in different parts of the channel. The intracellular C-terminal tail is key to modulation by temperature [17, 18, 66–68], pressure [18, 69, 70], intracellular pH [70, 71], responses to lipids such as phosphoinositol [4, 5] bis-phosphate (PIP2) [68, 72, 73], and control by phosphorylation [17, 74, 75]. The sensor for extracellular pH is a histidine [19, 76] located in the loop that connects the P1 helix to the CAP domain [11]. Gating cues from the extracellular pH sensor [19, 76] and the C-terminal tail [17, 18] converge on the selectivity filter C-type gate and make this channel element the nexus of signal integration and functional control [11, 17, 18, 21, 22].
TREK subfamily channels are found throughout the central and peripheral nervous system [4, 28, 29, 34, 47, 77], the eye [43], and the heart [58]. Since their discovery, TREK subfamily channels have been implicated as therapeutic targets for pain, ischemia, and depression [47, 60, 65, 78]. K2P2.1 (TREK-1) and K2P10.1 (TREK-2) share ~65% sequence similarity, whereas K2P2.1 (TREK-1) and K2P4.1 (TRAAK) share only ~40% similarity [79–81]. Most of this sequence divergence is embedded in the N- and C-terminal cytoplasmic regions. In line with the high degree of conservation in the core elements of the channel, the structures of homodimers of each of the TREK subfamily members are similar to each other [11]. The three subtypes also have been shown to heterodimerize and provide further functional diversity from this subfamily [42, 82–84]. An important consequence of heterodimerization is that it creates a channel in which the PD1 and PD2 domains are all different from each other. How these differences manifest in functional diversification remains to be defined. Understanding how the similarities and differences within the TREK subfamily contribute to function is of critical importance for developing pharmacological tools and potential therapeutics targeted toward this complex subfamily.
Both activation and inhibition of TREK channels have proposed therapeutic benefits. The ability of TREK channels to stabilize the membrane potential and reduce excitability together with their high expression in sensory neurons gives activators of this K2P subfamily the potential to function as analgesics or anesthetics [35, 47, 85] and as agents against migraine. [42] Interestingly, the original studies of K2P2.1 (TREK-1) knockout mice indicated that inhibition of this channel might have a role in mitigating depression [41]. Pharmacologically relevant antipsychotics have also been reported to inhibit K2P2.1 (TREK-1) [86]. Consequently, there has been an effort to explore K2P2.1 (TREK-1) inhibitors, such as the peptide spadin [87, 88] and small-molecule norfluoxetine [60] as new directions for treating depression. Inhibiting K2P2.1 (TREK-1) with such agents would facilitate membrane depolarization, but how such effects could result in modulation of the mental disease remains unclear and harder to understand than the links between TREK channel activation and pain suppression.
Because TREK subfamily channels are readily studied in a variety of experimental systems from potassium transport deficient yeast [48] to Xenopus oocytes [11, 22, 52, 54, 75, 80], to transfected mammalian cells [11, 22, 75, 80, 81, 89], to reconstitution assays using purified channels [13, 27, 51, 90], this K2P subfamily has been a key model for understanding K2P channel biophysics in general and is leading the way in demonstrating the potential of K2Ps as pharmacological targets [47].
4.3. The Polysite Pharmacology of TREK Channels
Structural studies of TREK K2Ps have revealed a strikingly rich structural landscape for functional control, especially given their modest size (~70 kDa). Binding sites for small molecules are found at every layer of the protein starting from its extracellular side through the portion that interacts with the membrane bilayer inner leaflet (Fig. 4.2). This polysite pharmacology comprises four defined binding sites for small molecules or lipids: the Keystone inhibitor site [54], the K2P modulator pocket, [11] the Fenestration site [14, 53], and the Modulatory lipid site [11]. Each offers a distinct structural environment and mechanism for controlling K2P function.
Fig. 4.2.
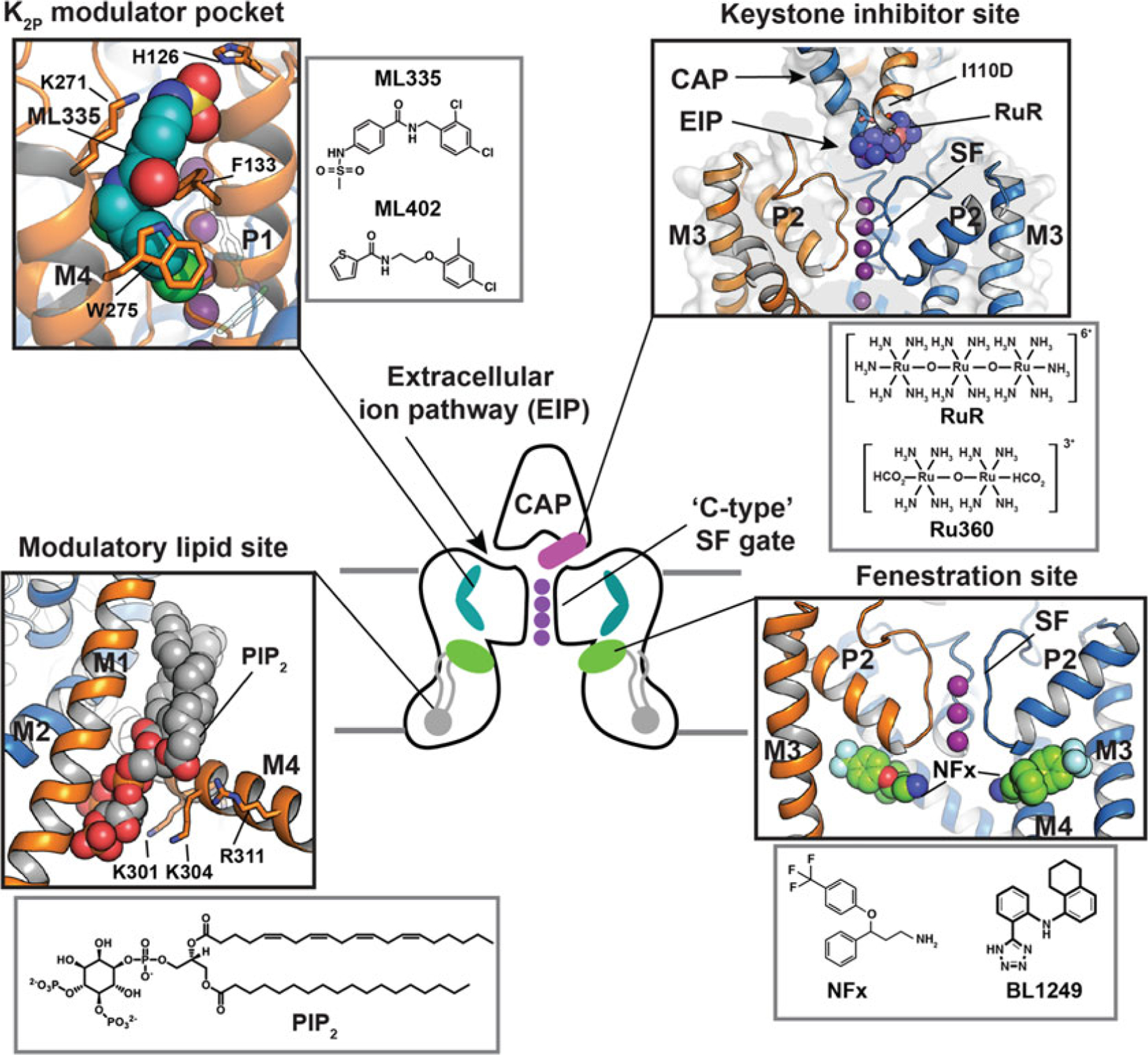
Polysite model of TREK subfamily modulation. Central cartoon shows the locations of structurally defined K2P small molecule binding sites including the Keystone inhibitor site (magenta), K2P modulator pocket (cyan), fenestration site (green), and modulatory lipid site (grey). CAP and “C-type” SF gates are indicated. Potassium ions are shown (purple). Grey lines denote the lipid bilayer. Black boxes show the details of the individual sites. Grey boxes show modulator chemical structures
4.3.1. The Keystone Inhibitor Site: Block by Polynuclear Ruthenium Amines
The trinuclear oxo-bridged ruthenium amine ruthenium red (RuR) [91] is a polycation with many biological applications [92], including a ~50 year legacy of use as an inhibitor of diverse ion channels. RuR has been shown to inhibit three K2P channels, two from the TREK subfamily, K2P4.1 (TRAAK) [93, 94] and K2P10.1 (TREK-2) [93], as well as K2P9.1 (TASK-3) [95–97]. Functional studies showed that a negatively charged residue at the base of the K2P CAP domain comprises a key RuR sensitivity determinant in the natively RuR sensitive channels K2P9.1 (TASK-3) [95–97] and K2P10.1 (TREK-2). Further, placing a negatively charged residue at the CAP base is sufficient for rendering a non-RuR sensitive K2P responsive to RuR inhibition [54, 93]. Hence, this negative residue is both necessary and sufficient for RuR sensitivity in the context of a K2P channel.
Structural studies of a RuR-sensitive K2P2.1 (TREK-1) mutant, I110D [54], revealed that RuR inhibits K2Ps in a 1:1 stoichiometry matching functional studies [93, 94] and places one ruthenium amine moiety directly over the channel pore while the remainder of the RuR molecule occupies one of the two branches of the extracellular ion pathway (EIP). This “finger in the dam” mechanism provides both, and even an electrostatic and physical barrier that prevents the flow of potassium ions through the selectivity filter.
RuR interacts directly with the negatively charged residues that form the RuR-sensitivity determinant and that constitute the “Keystone inhibitor site” at the base of the CAP domain. The principal mode of binding is through a multipronged interaction made by the two acidic residues at the Keystone inhibitor site with multiple RuR elements. This sort of direct engagement of RuR by multiple acidic sidechains is likely to contribute to RuR block of other classes of RuR-sensitive channels where the binding site is thought to be rich in acidic residues such as TRP channels [98–105], the mitochondrial calcium uniporter (MCU) [106–109], CALHM calcium channels [110–112], ryanodine receptors [113, 114], and Piezo channels [115, 116]. The dinuclear ruthenium amine, Ru360 [117], an inhibitor of the mitochondrial calcium uniporter [106, 118, 119] not previously known to affect potassium channels also binds to the Keystone inhibitor site in a similar way, although due to its reduced electrostatic interactions relative to RuR, Ru360 is a weaker blocker (IC50 = 0.287 vs. 11.3 μM, for RuR and Ru360, respectively) [54] (Fig. 4.2).
Once sites of modulator action are known, it is possible to use the structural information to alter the protein or the ligand to create molecules having new properties. Using a structure-based protein engineering approach, our lab-created RuR super-responder K2P2.1 (TREK-1) mutants having IC50s in the low nanomolar range by placing acidic resides at Asn147 site at the external mouth of the selectivity filter in conjunction with the I110D mutation (IC50 = 12.7 nM) [54]. Because of the shared pore architecture among K2Ps, this strategy is generalizable to other K2P members to create subtypes endowed with a high-affinity RuR sensitivity and could provide a means for exploring their functions. The demonstration that compounds such as RuR and Ru360 can block K2P function by reaching through the EIP raises the possibility of identifying other classes of molecules that could work similarly. Two interesting directions for making subtype-selective modulators directed at the Keystone inhibitor site would be to capitalize on the renewed interest in synthesizing novel polyruthenium amine derivatives [120] or to design compounds having moieties that interact with the Keystone inhibitor site but that also make specific contacts to non-conserved features of CAP exterior. Biologics, such as nanobodies, may be particularly suited to this type of molecular recognition mode as one can envision that a long variable loop from the nanobody could reach through the EIP to block the pore while other parts of the protein recognize subtype-specific features of the CAP exterior and EIP entryway.
4.3.2. The K2P Modulator Pocket: A Cryptic Small Molecule Binding Site for K2P Control
The K2P modulator pocket (Fig. 4.2) is unrelated to any previously known small molecule binding pocket in the VGIC superfamily and was discovered in structural studies of K2P2.1 (TREK-1) with two novel activators, ML335 (N-[(2,4-dichlorophenyl)methyl]-4-(methanesulfonamido) benzamide) and ML402 (N-[2-(4-chloro-2-methylphenoxy)ethyl]thiophene-2-carboxamide (Fig. 4.2) [11]. This L-shaped pocket is found in the P1–M4 interface, an intersubunit interface involved in C-type gating [17, 18]. Both compounds bind in similar ways and act as molecular wedges that stabilize the P1–M4 interface and directly activate the channel selectivity filter C-type gate [11, 22]. In the unliganded structure, the K2P modulator pocket is occluded by P1–M4 interface interactions that require small movements of few residues to open, making this pocket a cryptic site that relies on conformational change similar to cryptic sites described for soluble proteins [121]. Rigidification of the P1–M4 interface is central to channel activation [11, 22]. The observation that these two compounds stabilize this intersubunit interface highlights the general importance of intersubunit interfaces as sites of channel control.
ML335 and ML402 are remarkably selective, activating K2P2.1 (TREK-1) and K2P10.1 (TREK-2) but not K2P4.1 (TRAAK) [11] (Fig. 4.3). This strong subtype selectivity originates from a single lysine residue on the N-terminal end of M4 that engages in a cation–π interaction with the upper ring of each of the activators (Fig. 4.2). The equivalent residue in K2P4.1 (TRAAK) is glutamine and exchanging K→Q in K2P2.1 (TREK-1) and Q→K in K2P4.1 (TRAAK) at this position is sufficient for rendering the former insensitive to ML335 and ML402 activation and the latter sensitive to activation by both compounds [11]. The importance of a single amino acid difference in an otherwise conserved small molecule binding pocket underscores the potential for exploiting local differences and structural knowledge to develop subtype-selective K2P modulators.
Fig. 4.3.
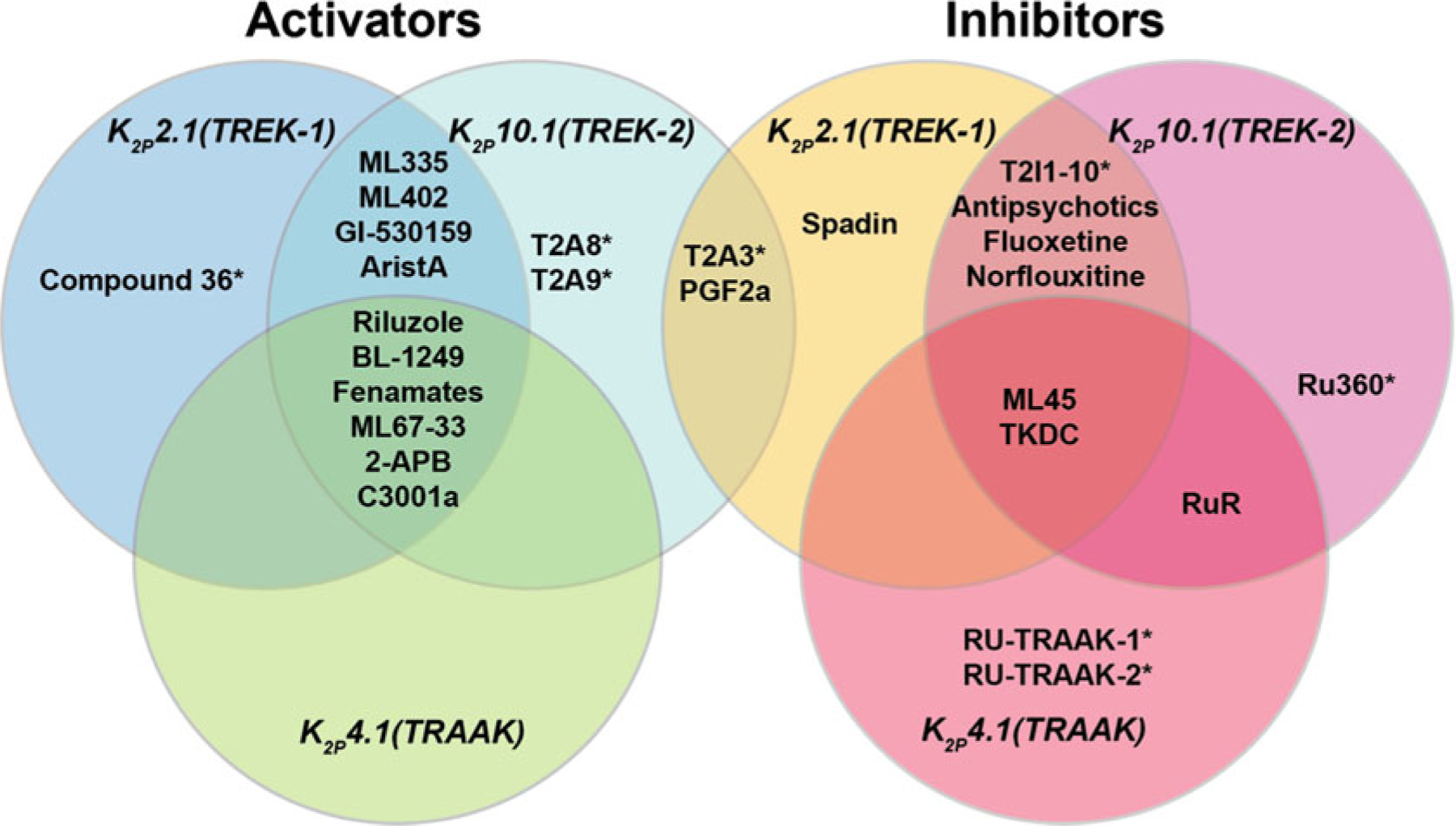
Selectivity profiles of TREK subfamily small molecule activators and inhibitors. Asterisks indicate modulators lacking a complete profile of subtype selectivity
The K2P modulator pocket is unique to K2Ps [11]. Currently, there are no known natural ligands for this site, but it seems unlikely that such a well-defined binding site is only recognized by two unnatural small molecules. Stabilization of the P1–M4 interface is central for integrating gating cues that arise in other parts of the protein, particularly the C-terminal tail [11, 17, 18, 22]. Because the K2P modulator pocket is in the center of this interface, it seems very likely that there are natural compounds such as lipids, metabolites, signaling molecules, or regulatory proteins that target this site. Hence, understanding the normal function of this part of the channel and whether Nature has exploited natural compounds to affect K2P activity through the K2P modulator pocket remains an important direction for future study.
4.3.3. The Fenestration Site: A Binding Site for Activators and Inhibitors
The K2P M4 transmembrane helix is a key moving element and serves as a means to convey gating cues from temperature [17, 18, 25, 26], pressure [18, 26, 27], and phosphorylation [17] to the C-type gate. Structural studies of TREK subfamily channels have defined two extreme positions of the M4 helix termed “up” and “down” [13, 14, 23–25]. The “down “ state creates a fenestration just below the P2 pore helix that is open to the center of the membrane bilayer [10, 13, 24, 25], the “Fenestration site” (Fig. 4.2). Structural studies of K2P10.1 (TREK-2) have shown that this site binds to the K2P inhibitors fluoxetine and norfluoxetine [14]. These compounds bind to a site defined by the lower part of the P2 pore helix and M4 (Fig. 4.2) and require the M4 helix to adopt the “down” position.
Remarkably, crystal structures of a K2P10.1 (TREK-2) complex with a brominated version of an activator, the fenamate BL-1249, although not defining the entire compound, strongly indicate that this molecule and perhaps other activators bind to the Fenestration site created by the “down” M4 position [53]. How can the binding of a small molecule to the same site yield opposite functional outcomes of inhibition and activation? Clearly, the answer cannot be in the stabilization of the M4 “down” state over the “up” state as the binding of both inhibitors and activators to the Fenestration site requires an M4 “down” conformation [14, 53]. Interestingly, it is suggested that activators such as BL-1249 use their tetrazole moiety to create a binding site for potassium ions in the central cavity and thereby stabilize the selectivity filter C-type gate [53]. Given this type of mechanism, it is notable that the norfluoxetine structure places the norfluoxetine amine just below the selectivity filter where its expected positive charge could provide an unfavorable modification to the potassium ion conduction pathway that would lead to channel inhibition (Fig. 4.2). The fenestration site is commonly found in the VGIC superfamily of which K2Ps are members and serves as the site of action for multiple types of activators of different classes of potassium channels [53]. Understanding the relationship between the occupation of this site, effects on the selectivity filter C-type gate, and the relationship between the properties of activators and inhibitors that can inhabit this site is an important challenge for further development of K2P modulators.
4.3.4. The Modulatory Lipid Site: PIP2 and the C-Terminal Tail
PIP2 is an important modulatory lipid for TREK subfamily channels [68, 72, 73]. The likely site of PIP2 action has been located in a series of K2P2.1 (TREK-1) structures [11, 22]. These show the presence of a phospholipid that co-purified with the channel and that was bound to a groove at the M1/M2/M4 interface (Fig. 4.2). The phosphoinositol headgroup contacts an electro-positive patch on the C-tail comprising five residues implicated in PIP2 modulation (Arg297, Lys301, Lys302, Lys304, and R311) [68, 72] (Fig. 4.2). This same stretch of the C-terminal tail also contains the intracellular proton sensor site, Glu306 [71], and inhibitory phosphorylation site, Ser300 [74]. The key PIP2-interacting residues are in a portion of the channel that is most affected by movements of M4 between the “up” and “down” positions. Hence, it seems likely that regulatory impacts of the modulatory lipid, intracellular pH sensor, and phosphorylation site on the C-terminal tail are all tightly intertwined with M4 motions [122]. Further study is needed to unravel these interactions, to understand whether other lipids reported to impact TREK channel function, such as phosphatidyl serine and phosphatidic acid [68, 123], compete with PIP2 at this site, whether this site can be targeted by small molecules, and to define how changes in this lower part of the channel impact the dynamics and function of the C-type gate.
4.4. Subtype Specific Modulators in the TREK Subfamily
The growing progress in developing modulators for the TREK subfamily has been well-reviewed recently elsewhere [9, 47] and is, therefore, not reiterated here. As new modulators are uncovered, one key question is whether it will be possible to create subtype-specific modulators that can not only distinguish among the various K2P subfamilies but also among the different subtypes within a subfamily. Such a high level of target selectivity would provide powerful tools for delineating the biological functions of these channels and could provide starting points for the development of subtype-selective pharmacology for these therapeutically relevant targets.
With respect to subtype selectivity, currently characterized TREK subfamily activators mostly fall into two categories. There are many examples of molecules that affect all three members of the TREK subfamily such as Riluzole [89, 124], BL-1249 [52], fenamates [36], ML67–33 [48], 2-Aminoethoxydiphenyl Borate (2-APB) [125, 126], and C3001a [85]. Some of these, such as BL-1249 [52] show limited selectivity for K2P2.1 (TREK-1) and K2P10.1 (TREK-2) over K2P4.1 (TRAAK). The second category of compounds activate K2P2.1 (TREK-1) and K2P10.1 (TREK-2) but not K2P4.1 (TRAAK) and include ML335 [11], ML402 [11], GI-530159 [127], and aristolochic acid (AristA) [128]. Given that K2P2.1 (TREK-1) and K2P10.1 (TREK-2) have sequences that are more similar to each other than they are to K2P4.1 (TRAAK) (Fig. 4.1a), it is not surprising that K2P4.1 (TRAAK) exhibits different responses to some modulators. There is a report of a K2P2.1 (TREK-1) selective activator, Compound 36 [36], based on studies of less selective caffeic acid ester activators [36, 56, 63], but a detailed understanding of this high degree of selectivity remains to be defined. The compounds T2A8 and T2A9 are also reported to activate K2P10.1 (TREK-1) with some selectivity [62]. The fact that there are already compounds showing some degree of selectivity provides an encouraging sign that developing activators having better subtype selectivity is a goal that can be reached.
TREK subfamily inhibitors show slightly more selectivity than activators and follow the same pattern having broadly acting inhibitors and inhibitors showing some selectivity. ML45 [48] and TKDC [129] inhibit all three TREK subfamily channels. There are a set of molecules that inhibit K2P2.1 (TREK-1) and K2P10.1 (TREK-2) but not K2P4.1 (TRAAK), namely, antipsychotics [86], fluoxetine [84], and norfluoxetine [130]. A series of inhibitors (T2I1–10) unable to discriminate between K2P2.1 (TREK-1) and K2P10.1 (TREK-2) have also been reported [62], but whether these compounds affect K2P4.1 (TRAAK) remains to be established. The polyruthenium blockers RuR and Ru360 show an unusual inhibition profile. RuR and Ru360 inhibit K2P10.1 (TREK-2) but not K2P2.1 (TREK-1) [54, 93, 94]. K2P4.1 (TRAAK) is sensitive to RuR but the mechanism of inhibition must be different from the “finger in the dam” mechanism as K2P4.1 (TRAAK) lacks the defining acidic residue in the Keystone inhibitor site [54]. Ru-TRAAK-1 and Ru-TRAAK-2 inhibit K2P4.1 (TRAAK) as well as K2Ps from other subfamilies, such as K2P1.1 (TWIK-1), K2P3.1 (TASK-1), and K2P18.1 (TRESK) [51]. The action of these compounds on other TREK subfamily members has not been reported, but given their ability to inhibit K2Ps outside of the TREK subfamily, it would be surprising if they did not also show some activity against the more closely related K2P2.1 (TREK-1) or K2P10.1 (TREK-2). The peptide spadin has been reported to act as a K2P2.1 (TREK-1)-selective inhibitor [87, 88], but its mechanism of action remains unclear. T2A3, T2A8, T2A9, and the bioactive lipid 11-deoxyprostaglandin-F2α form an unusual class K2P2.1 (TREK-1) inhibitors that are reported to also act as K2P10.1 (TREK-2) activators [62]. How such dual action occurs is not known, but has been proposed to involve a short part of the P2–M4 loop [62]. As with the activators, the growing examples of subtype-selective inhibitors indicate that developing better and more selective inhibitors of the TREK subfamily should also be feasible, especially as more structural data about how such molecules interact with K2Ps becomes available.
So far, the origins of subtype selectivity for TREK modulators are understood for only two sites at the level of the atomic interactions, the Keystone inhibitor site, and the K2P modulator pocket. For the Keystone inhibitor site, the negative charge at the Keystone inhibitor site is the principal determinant controlling RuR and Ru360 inhibition [54] (see Sect. 1.3.1). How RuR affects K2P4.1 (TRAAK) remains unknown, as this channel lacks the negative residue at the Keystone inhibitor site and is inhibited with a stoichiometry higher than the 1:1 interaction of the Keystone inhibitor site [93, 94]. The basis of the subtype selectivity of the ML335 and ML402 activators [11] arises from a single lysine in the K2P modulator pocket that controls subtype selectivity (see Sect. 1.3.2). Although not yet mapped in atomic detail, BL-1249 shows a ~10-fold selectivity for K2P2.1 (TREK-1) and K2P10.1 (TREK-2) over K2P4.1 (TRAAK) that originates from differences in the M2/M3 helix interface [52]. Understanding the molecular origins of the subtype specificity for this compound as well as GI-530159 [127], aristolochic acid (AristA) [128], and C3001a [85] will require further studies that combines both structural and functional approaches.
4.5. Perspectives on K2P Channel Polysite Pharmacology
Structural data for complexes of K2P2.1 (TREK-1) and K2P10.1 (TREK-2) with various modulators have uncovered a surprisingly large number of unique sites for small molecule control present at every level of the channel structure with respect to the membrane. These sites are arranged above (Keystone inhibitor site [54]), at the level of (K2P modulator pocket [11]), and below (fenestration [14] and modulatory lipid [11] sites) the structure that is at the heart of channel function, the C-type gate [17, 18, 20–22]. It seems likely that there is a fifth site within the channel cavity, as there is good functional evidence that this architectural feature is targeted in TREK channels by alkylammonium pore blockers [9, 53] and this shared K2P architectural element is the site of crystallographically defined small molecule block of K2P3.1 (TASK-1) [12]. All of these sites have functions that are crucial for the normal functioning and modulation of K2Ps. The intersection of small molecule modulators and key sites of channel modulation emphasizes the importance of building an integrated understanding of modulator action and the basic mechanisms of channel function.
The structural pharmacology of the Keystone inhibitor site, K2P modulator pocket, and Fenestration site has been defined by crystal structures of K2Ps complexed with nonnatural compounds that exert powerful effects on channel function. These findings highlight two key outstanding questions: To what extent has Nature has capitalized on these control points to influence TREK channel activity? and What are the naturally occurring modulators that target these sites? Answering such questions will be important for developing a better understanding of the roles of K2Ps in physiological responses. One known natural modulator of great physiological interest whose site remains to be defined on the TREK subfamily is the site of action of the activator arachidonic acid [75, 131]. From a structural perspective, although K2P4.1 (TRAAK) was the first member of the TREK subfamily structurally characterized [13] and received its name due to its sensitivity to arachidonic acid [131], it remains the only channel in the TREK subfamily lacking any modulator-bound structures. Given the fact that this channel stands apart with respect to the selectivity of many modulators (Fig. 4.3), defining the site of arachidonic acid action in the TREK subfamily along with obtaining structural data for small molecule:K2P4.1 (TRAAK) complexes will provide important guides for unraveling natural mechanisms of channel modulation and better templates for subtype-selective modulator discovery.
The clear crosstalk between various K2P moving parts and the C-type gate [17, 18, 52] raises the question of whether the action of modulators at the various sites influence each other. The strength of polyruthenium amine block at the Keystone inhibitor site is not influenced by C-type gate stabilization by compounds occupying the K2P modulator pocket [54], but the extent to which there may be crosstalk among the other sites remains to be evaluated. Understanding such interactions could provide insight into how to boost the efficacy of current modulators and will refine our understanding of how signals from various parts of the channel impinge on the C-type gate.
The current structural knowledge of modulator sites provides a framework to discover a new chemical matter that can affect K2P function in novel ways. Such molecules may be engineered to block or enhance the consequences of various physical and chemical stimuli or to modify the channel chemically so that its biogenesis, distribution, and interaction with other cellular components can be followed in complex cell types and tissues. Besides further crystallographic studies of new K2P: modulator complexes, an obvious key advance will be to understand structural consequences of heterodimer formation [42, 82–84] and to image K2Ps in lipid membrane environments using cryo-electron microscopy (cryo-EM) so that interactions between the channel and bilayer can be better understood. The initial burst of activity surrounding K2P structural pharmacology is the first of many waves that will fill out our understanding of this important ion channel family and should lead to new and novel therapeutic strategies for a host of brain, cardiac, and nervous system diseases.
Acknowledgments
We thank F. C. Chatelain for comments on the manuscript and P. Deal for help with figure preparation. This work was supported by grant NIH-R01-MH093603 to D.L.M.
Contributor Information
Lianne Pope, Cardiovascular Research Institute, University of California San Francisco, CA, US.
Daniel L. Minor, Jr, Cardiovascular Research Institute, University of California San Francisco, CA, US; Departments of Biochemistry and Biophysics, and Cellular and Molecular Pharmacology, University of California, San Francisco, CA, USA; California Institute for Quantitative Biomedical Research, University of California, San Francisco, CA, USA; Kavli Institute for Fundamental Neuroscience, University of California, San Francisco, CA, USA; Molecular Biophysics and Integrated Bio-imaging Division, Lawrence Berkeley National Laboratory, Berkeley, CA, USA.
References
- 1.Hille B (2001) Ion channels of excitable membranes, 3rd edn. Sinauer Associates, Inc., Sunderland, MA [Google Scholar]
- 2.Yu FH, Yarov-Yarovoy V, Gutman GA, Catterall WA (2005) Overview of molecular relationships in the voltage-gated ion channel superfamily. Pharmacol Rev 57:387–395 [DOI] [PubMed] [Google Scholar]
- 3.Enyedi P, Czirjak G (2010) Molecular background of leak K+ currents: two-pore domain potassium channels. Physiol Rev 90:559–605 [DOI] [PubMed] [Google Scholar]
- 4.Feliciangeli S, Chatelain FC, Bichet D, Lesage F (2014) The family of K2P channels: salient structural and functional properties. J Physiol 10.1113/jphysiol.2014.287268 [DOI] [PMC free article] [PubMed]
- 5.Renigunta V, Schlichthorl G, Daut J (2015) Much more than a leak: structure and function of K(2)p-channels. Pflugers Arch 467:867–894 [DOI] [PubMed] [Google Scholar]
- 6.Goldstein SA et al. (2005) International Union of Pharmacology. LV Nomenclature and molecular relationships of two-P potassium channels. Pharmacol Rev 57:527–540 [DOI] [PubMed] [Google Scholar]
- 7.Douguet D, Honore E (2019) Mammalian mechanoelectrical transduction: structure and function of forcegated ion channels. Cell 179:340–354 [DOI] [PubMed] [Google Scholar]
- 8.Sepulveda FV, Pablo Cid L, Teulon J, Niemeyer MI (2015) Molecular aspects of structure, gating, and physiology of pH-sensitive background K2P and Kir K+-transport channels. Physiol Rev 95:179–217 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Sterbuleac D (2019) Molecular determinants of chemical modulation of two-pore domain potassium channels. Chem Biol Drug Des 94:1596–1614 [DOI] [PubMed] [Google Scholar]
- 10.Miller AN, Long SB (2012) Crystal structure of the human two-pore domain potassium channel K2P1. Science 335:432–436 [DOI] [PubMed] [Google Scholar]
- 11.Lolicato M et al. (2017) K2P2.1 (TREK-1)-activator complexes reveal a cryptic selectivity filter binding site. Nature 547:364–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rödström KEJ et al. (2020) A lower X-gate in TASK channels traps inhibitors within the vestibule. Nature 582:443–447 [DOI] [PubMed] [Google Scholar]
- 13.Brohawn SG, del Marmol J, MacKinnon R (2012) Crystal structure of the human K2P TRAAK, a lipid- and mechano-sensitive K+ ion channel. Science 335:436–441 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Dong YY et al. (2015) K2P channel gating mechanisms revealed by structures of TREK-2 and a complex with Prozac. Science 347:1256–1259 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Payandeh J, Minor DL Jr (2014) Bacterial Voltage-Gated Sodium Channels (BacNas) from the soil, sea, and salt lakes enlighten molecular mechanisms of electrical signaling and pharmacology in the brain and heart. J Mol Biol 10.1016/j.jmb.2014.08.010 [DOI] [PMC free article] [PubMed]
- 16.Catterall WA, Wisedchaisri G, Zheng N (2017) The chemical basis for electrical signaling. Nat Chem Biol 13:455–463 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bagriantsev SN, Clark KA, Minor DL Jr (2012) Metabolic and thermal stimuli control K(2P)2.1 (TREK-1) through modular sensory and gating domains. EMBO J 31:3297–3308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Bagriantsev SN, Peyronnet R, Clark KA, Honore E, Minor DL Jr (2011) Multiple modalities converge on a common gate to control K2P channel function. EMBO J 30:3594–3606 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Cohen A, Ben-Abu Y, Hen S, Zilberberg N (2008) A novel mechanism for human K2P2.1 channel gating. Facilitation of C-type gating by protonation of extracellular histidine residues. J Biol Chem 283:19448–19455 [DOI] [PubMed] [Google Scholar]
- 20.Piechotta PL et al. (2011) The pore structure and gating mechanism of K2P channels. EMBO J 30:3607–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Schewe M et al. (2016) A non-canonical voltage-sensing mechanism controls gating in K2P K(+) channels. Cell 164:937–949 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lolicato M et al. (2020) K2P channel C-type gating involves asymmetric selectivity filter order-disorder transitions. bioRxiv 10.1101/2020.03.20.000893 [DOI] [PMC free article] [PubMed]
- 23.Brohawn SG, Campbell EB, MacKinnon R (2013) Domain-swapped chain connectivity and gated membrane access in a Fab-mediated crystal of the human TRAAK K+ channel. Proc Natl Acad Sci U S A 110:2129–2134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brohawn SG, Campbell EB, MacKinnon R (2014) Physical mechanism for gating and mechanosensitivity of the human TRAAK K+ channel. Nature 516:126–130 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Lolicato M, Riegelhaupt PM, Arrigoni C, Clark KA, Minor DL Jr (2014) Transmembrane helix straightening and buckling underlies activation of mechanosensitive and thermosensitive K (2P) channels. Neuron 84:1198–1212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McClenaghan C et al. (2016) Polymodal activation of the TREK-2 K2P channel produces structurally distinct open states. J Gen Physiol 147:497–505 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Aryal P et al. (2017) Bilayer-mediated structural transitions control mechanosensitivity of the TREK-2 K2P channel. Structure 25:708–718. e702 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Kanda H et al. (2019) TREK-1 and TRAAK are principal K(+) channels at the nodes of ranvier for rapid action potential conduction on mammalian myelinated afferent nerves. Neuron 10.1016/j.neuron.2019.08.042 [DOI] [PMC free article] [PubMed]
- 29.Brohawn SG et al. (2019) The mechanosensitive ion channel TRAAK is localized to the mammalian node of Ranvier. eLife 8 [DOI] [PMC free article] [PubMed]
- 30.Heurteaux C et al. (2004) TREK-1, a K+ channel involved in neuroprotection and general anesthesia. EMBO J 23:2684–2695 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lazarenko RM et al. (2010) Anesthetic activation of central respiratory chemoreceptor neurons involves inhibition of a THIK-1-like background K(+) current. J Neurosci 30:9324–9334 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Madry C et al. (2018) Microglial ramification, surveillance, and interleukin-1beta release are regulated by the two-pore domain K(+) channel THIK-1. Neuron 97:299–312. e296 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Yoshida K et al. (2018) Leak potassium channels regulate sleep duration. Proc Natl Acad Sci U S A 115:E9459–E9468 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Alloui A et al. (2006) TREK-1, a K+ channel involved in polymodal pain perception. EMBO J 25:2368–2376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Devilliers M et al. (2013) Activation of TREK-1 by morphine results in analgesia without adverse side effects. Nat Commun 4:2941. [DOI] [PubMed] [Google Scholar]
- 36.Vivier D et al. (2017) Development of the first two-pore domain potassium channel TREK-1 (TWIK-Related K+ Channel 1)-selective agonist possessing in vivo anti-nociceptive activity. J Med Chem 10.1021/acs.jmedchem.6b01285 [DOI] [PubMed]
- 37.Decher N et al. (2017) Sodium permeable and “hyper-sensitive” TREK-1 channels cause ventricular tachycardia. EMBO Mol Med 9:403–414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Laigle C, Confort-Gouny S, Le Fur Y, Cozzone PJ, Viola A (2012) Deletion of TRAAK potassium channel affects brain metabolism and protects against ischemia. PLoS One 7:e53266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wu X et al. (2013) Involvement of TREK-1 activity in astrocyte function and neuroprotection under simulated ischemia conditions. J Mol Neurosci 49:499–506 [DOI] [PubMed] [Google Scholar]
- 40.Abraham DM et al. (2018) The two-pore domain potassium channel TREK-1 mediates cardiac fibrosis and diastolic dysfunction. J Clin Invest 128:4843–4855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Heurteaux C et al. (2006) Deletion of the background potassium channel TREK-1 results in a depression-resistant phenotype. Nat Neurosci 9:1134–1141 [DOI] [PubMed] [Google Scholar]
- 42.Royal P et al. (2019) Migraine-associated TRESK mutations increase neuronal excitability through alternative translation initiation and inhibition of TREK. Neuron 101:232–245. e236 [DOI] [PubMed] [Google Scholar]
- 43.Yarishkin O et al. (2018) TREK-1 channels regulate pressure sensitivity and calcium signaling in trabecular meshwork cells. J Gen Physiol 150:1660–1675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Lambert M et al. (2018) Loss of KCNK3 is a hallmark of RV hypertrophy/dysfunction associated with pulmonary hypertension. Cardiovasc Res 114:880–893 [DOI] [PubMed] [Google Scholar]
- 45.Schwingshackl A (2016) The role of stretch-activated ion channels in acute respiratory distress syndrome: finally a new target? Am J Physiol Lung Cell Mol Physiol 311:L639–L652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Petho Z, Najder K, Bulk E, Schwab A (2019) Mechanosensitive ion channels push cancer progression. Cell Calcium 80:79–90 [DOI] [PubMed] [Google Scholar]
- 47.Mathie A, Veale EL, Cunningham KP, Holden RG, Wright PD (2020) Two-pore domain potassium channels as drug targets: anesthesia and beyond. Annu Rev Pharmacol Toxicol 10.1146/annurev-pharmtox-030920-111536 [DOI] [PubMed]
- 48.Bagriantsev SN et al. (2013) A high-throughput functional screen identifies small molecule regulators of temperature- and mechano-sensitive K2P channels. ACS Chem Biol 8:1841–1851 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Tian F et al. (2019) A small-molecule compound selectively activates K2P Channel TASK-3 by acting at two distant clusters of residues. Mol Pharmacol 96:26–35 [DOI] [PubMed] [Google Scholar]
- 50.Wright PD et al. (2019) Pranlukast is a novel small molecule activator of the two-pore domain potassium channel TREK2. Biochem Biophys Res Commun 10.1016/j.bbrc.2019.09.093 [DOI] [PubMed]
- 51.Su ZW, Brown EC, Wang WW, MacKinnon R (2016) Novel cell-free high-throughput screening method for pharmacological tools targeting K+ channels. Proc Natl Acad Sci USA 113:5748–5753 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Pope L et al. (2018) Protein and chemical determinants of BL-1249 action and selectivity for K2P channels. ACS Chem Neurosci 9:3153–3165 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Schewe M et al. (2019) A pharmacological master key mechanism that unlocks the selectivity filter gate in K (+) channels. Science 363:875–880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Pope L, Lolicato M, Minor DL Jr (2020) Polynuclear ruthenium amines inhibit K2P channels via a “Finger in the Dam” mechanism. Cell Chem Biol 10.1016/j.chembiol.2020.01.011 [DOI] [PMC free article] [PubMed]
- 55.Gada K, Plant LD (2019) Two-pore domain potassium channels: emerging targets for novel analgesic drugs: IUPHAR Review 26. Br J Pharmacol 176:256–266 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Vivier D, Bennis K, Lesage F, Ducki S (2016) Perspectives on the two-pore domain potassium channel TREK-1 (TWIK-Related K(+) Channel 1). A novel therapeutic target? J Med Chem 59:5149–5157 [DOI] [PubMed] [Google Scholar]
- 57.Hancox JC, James AF, Marrion NV, Zhang H, Thomas D (2016) Novel ion channel targets in atrial fibrillation. Expert Opin Ther Targets 20:947–958 [DOI] [PubMed] [Google Scholar]
- 58.Decher N, Kiper AK, Rinne S (2017) Stretch-activated potassium currents in the heart: focus on TREK-1 and arrhythmias. Prog Biophys Mol Biol 130:223–232 [DOI] [PubMed] [Google Scholar]
- 59.Bagal SK et al. (2013) Ion channels as therapeutic targets: a drug discovery perspective. J Med Chem 56:593–624 [DOI] [PubMed] [Google Scholar]
- 60.Borsotto M et al. (2015) Targeting two-pore domain K(+) channels TREK-1 and TASK-3 for the treatment of depression: a new therapeutic concept. Br J Pharmacol 172:771–784 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Enyedi P, Czirjak G (2015) Properties, regulation, pharmacology, and functions of the K(2)p channel, TRESK. Pflugers Arch 467:945–958 [DOI] [PubMed] [Google Scholar]
- 62.Dadi PK et al. (2016) Selective small molecule activators of TREK-2 channels stimulate DRG c-fiber nociceptor K2P currents and limit calcium influx. ACS Chem Neurosci 10.1021/acschemneuro.6b00301 [DOI] [PMC free article] [PubMed]
- 63.Rodrigues N et al. (2014) Synthesis and structure-activity relationship study of substituted caffeate esters as antinociceptive agents modulating the TREK-1 channel. Eur J Med Chem 75:391–402 [DOI] [PubMed] [Google Scholar]
- 64.Liao P et al. (2019) Selective activation of TWIK-related acid-sensitive K(+) 3 subunit-containing channels is analgesic in rodent models. Sci Transl Med 11 [DOI] [PubMed] [Google Scholar]
- 65.Honore E (2007) The neuronal background K2P channels: focus on TREK1. Nat Rev Neurosci 8:251–261 [DOI] [PubMed] [Google Scholar]
- 66.Maingret F et al. (2000) TREK-1 is a heat-activated background K(+) channel. EMBO J 19:2483–2491 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Kang D, Choe C, Kim D (2005) Thermosensitivity of the two-pore domain K+ channels TREK-2 and TRAAK. J Physiol 564:103–116 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Chemin J et al. (2005) A phospholipid sensor controls mechanogating of the K+ channel TREK-1. EMBO J 24:44–53 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Maingret F, Fosset M, Lesage F, Lazdunski M, Honoré E (1999) TRAAK is a mammalian neuronal mechano-gated K+ channel. J Biol Chem 274:1381–1387 [DOI] [PubMed] [Google Scholar]
- 70.Maingret F, Patel AJ, Lesage F, Lazdunski M, Honore E (1999) Mechano- or acid stimulation, two interactive modes of activation of the TREK-1 potassium channel. J Biol Chem 274:26691–26696 [DOI] [PubMed] [Google Scholar]
- 71.Honore E, Maingret F, Lazdunski M, Patel AJ (2002) An intracellular proton sensor commands lipid- and mechano-gating of the K(+) channel TREK-1. EMBO J 21:2968–2976 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Chemin J et al. (2007) Up- and down-regulation of the mechano-gated K(2P) channel TREK-1 by PIP (2) and other membrane phospholipids. Pflugers Arch 455:97–103 [DOI] [PubMed] [Google Scholar]
- 73.Lopes CM et al. (2005) PIP2 hydrolysis underlies agonist-induced inhibition and regulates voltage gating of two-pore domain K+ channels. J Physiol 564:117–129 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Murbartian J, Lei Q, Sando JJ, Bayliss DA (2005) Sequential phosphorylation mediates receptor- and kinase-induced inhibition of TREK-1 background potassium channels. J Biol Chem 280:30175–30184 [DOI] [PubMed] [Google Scholar]
- 75.Patel AJ et al. (1998) A mammalian two pore domain mechano-gated S-like K+ channel. EMBO J 17:4283–4290 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Sandoz G, Douguet D, Chatelain F, Lazdunski M, Lesage F (2009) Extracellular acidification exerts opposite actions on TREK1 and TREK2 potassium channels via a single conserved histidine residue. Proc Natl Acad Sci U S A 106:14628–14633 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Acosta C et al. (2014) TREK2 expressed selectively in IB4-binding C-fiber nociceptors hyperpolarizes their membrane potentials and limits spontaneous pain. J Neurosci 34:1494–1509 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Waxman SG, Zamponi GW (2014) Regulating excitability of peripheral afferents: emerging ion channel targets. Nat Neurosci 17:153–163 [DOI] [PubMed] [Google Scholar]
- 79.Bang H, Kim Y, Kim D (2000) TREK-2, a new member of the mechanosensitive tandem-pore K+ channel family. J Biol Chem 275:17412–17419 [DOI] [PubMed] [Google Scholar]
- 80.Fink M et al. (1996) Cloning, functional expression and brain localization of a novel unconventional outward rectifier K+ channel. EMBO J 15:6854–6862 [PMC free article] [PubMed] [Google Scholar]
- 81.Lesage F, Maingret F, Lazdunski M (2000) Cloning and expression of human TRAAK, a polyunsaturated fatty acids-activated and mechano-sensitive K(+) channel. FEBS Lett 471:137–140 [DOI] [PubMed] [Google Scholar]
- 82.Lengyel M, Czirjak G, Enyedi P (2016) Formation of functional heterodimers by TREK-1 and TREK-2-two-pore domain potassium channel subunits. J Biol Chem 291:13649–13661 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Levitz J et al. (2016) Heterodimerization within the TREK channel subfamily produces a diverse family of highly regulated potassium channels. Proc Natl Acad Sci U S A 113:4194–4199 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Blin S et al. (2016) Mixing and matching TREK/TRAAK subunits generate heterodimeric K2P channels with unique properties. Proc Natl Acad Sci U S A 113:4200–4205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Qiu Y et al. (2020) TREK Channel family activator with a well-defined structure-activation relationship for pain and neurogenic inflammation. J Med Chem 63:3665–3677 [DOI] [PubMed] [Google Scholar]
- 86.Thummler S, Duprat F, Lazdunski M (2007) Antipsychotics inhibit TREK but not TRAAK channels. Biochem Biophys Res Commun 354:284–289 [DOI] [PubMed] [Google Scholar]
- 87.Maati HMO et al. (2012) Spadin as a new antidepressant: absence of TREK-1-related side effects. Neuropharmacology 62:278–288 [DOI] [PubMed] [Google Scholar]
- 88.Mazella J et al. (2010) Spadin, a sortilin-derived peptide, targeting rodent TREK-1 channels: a new concept in the antidepressant drug design. PLoS Biol 8: e1000355. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Lesage F, Terrenoire C, Romey G, Lazdunski M (2000) Human TREK2, a 2P domain mechano-sensitive K+ channel with multiple regulations by polyunsaturated fatty acids, lysophospholipids, and Gs, Gi, and Gq protein-coupled receptors. J Biol Chem 275:28398–28405 [DOI] [PubMed] [Google Scholar]
- 90.Brohawn SG, Su Z, MacKinnon R (2014) Mechanosensitivity is mediated directly by the lipid membrane in TRAAK and TREK1 K+ channels. Proc Natl Acad Sci U S A 111:3614–3619 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Fletcher JM, Greenfield BF, Hardy CJ, Scargill D, Woodhead JL (1961) Ruthenium red. J Chem Soc:2000–2006
- 92.Clarke MJ (2002) Ruthenium metallopharmaceuticals. Coordin Chem Rev 232:69–93 [Google Scholar]
- 93.Braun G, Lengyel M, Enyedi P, Czirjak G (2015) Differential sensitivity of TREK-1, TREK-2 and TRAAK background potassium channels to the polycationic dye ruthenium red. Br J Pharmacol 172:1728–1738 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Czirjak G, Enyedi P (2002) Formation of functional heterodimers between the TASK-1 and TASK-3 two-pore domain potassium channel subunits. J Biol Chem 277:5426–5432 [DOI] [PubMed] [Google Scholar]
- 95.Musset B et al. (2006) Effects of divalent cations and spermine on the K+ channel TASK-3 and on the outward current in thalamic neurons. J Physiol 572:639–657 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Czirjak G, Enyedi P (2003) Ruthenium red inhibits TASK-3 potassium channel by interconnecting glutamate 70 of the two subunits. Mol Pharmacol 63:646–652 [DOI] [PubMed] [Google Scholar]
- 97.Gonzalez W et al. (2013) An extracellular ion pathway plays a central role in the cooperative gating of a K(2P) K+ channel by extracellular pH. J Biol Chem 288:5984–5991 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Caterina MJ et al. (1997) The capsaicin receptor: a heat-activated ion channel in the pain pathway. Nature 389:816–824 [DOI] [PubMed] [Google Scholar]
- 99.Strotmann R, Harteneck C, Nunnenmacher K, Schultz G, Plant TD (2000) OTRPC4, a nonselective cation channel that confers sensitivity to extracellular osmolarity. Nat Cell Biol 2:695–702 [DOI] [PubMed] [Google Scholar]
- 100.Guler AD et al. (2002) Heat-evoked activation of the ion channel, TRPV4. J Neurosci 22:6408–6414 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Caterina MJ, Rosen TA, Tominaga M, Brake AJ, Julius D (1999) A capsaicin-receptor homologue with a high threshold for noxious heat. Nature 398:436–441 [DOI] [PubMed] [Google Scholar]
- 102.Voets T et al. (2002) Molecular determinants of permeation through the cation channel TRPV4. J Biol Chem 277:33704–33710 [DOI] [PubMed] [Google Scholar]
- 103.Arif Pavel M et al. (2016) Function and regulation of TRPP2 ion channel revealed by a gain-of-function mutant. Proc Natl Acad Sci U S A 113:E2363–E2372 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Voets T et al. (2004) TRPM6 forms the Mg2+ influx channel involved in intestinal and renal Mg2+ absorption. J Biol Chem 279:19–25 [DOI] [PubMed] [Google Scholar]
- 105.Story GM et al. (2003) ANKTM1, a TRP-like channel expressed in nociceptive neurons, is activated by cold temperatures. Cell 112:819–829 [DOI] [PubMed] [Google Scholar]
- 106.Kirichok Y, Krapivinsky G, Clapham DE (2004) The mitochondrial calcium uniporter is a highly selective ion channel. Nature 427:360–364 [DOI] [PubMed] [Google Scholar]
- 107.Chaudhuri D, Sancak Y, Mootha VK, Clapham DE (2013) MCU encodes the pore conducting mitochondrial calcium currents. elife 2:e00704. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Rahamimoff R, Alnaes E (1973) Inhibitory action of Ruthenium red on neuromuscular transmission. Proc Natl Acad Sci U S A 70:3613–3616 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Moore CL (1971) Specific inhibition of mitochondrial Ca++ transport by ruthenium red. Biochem Biophys Res Commun 42:298–305 [DOI] [PubMed] [Google Scholar]
- 110.Ma Z et al. (2012) Calcium homeostasis modulator 1 (CALHM1) is the pore-forming subunit of an ion channel that mediates extracellular Ca2+ regulation of neuronal excitability. Proc Natl Acad Sci U S A 109: E1963–E1971 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Dreses-Werringloer U et al. (2013) CALHM1 controls the Ca(2)(+)-dependent MEK, ERK, RSK and MSK signaling cascade in neurons. J Cell Sci 126:1199–1206 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Choi W, Clemente N, Sun W, Du J, Lu W (2019) The structures and gating mechanism of human calcium homeostasis modulator 2. Nature 10.1038/s41586-019-1781-3 [DOI] [PMC free article] [PubMed]
- 113.Ma J (1993) Block by ruthenium red of the ryanodine-activated calcium release channel of skeletal muscle. J Gen Physiol 102:1031–1056 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Smith JS et al. (1988) Purified ryanodine receptor from rabbit skeletal muscle is the calcium-release channel of sarcoplasmic reticulum. J Gen Physiol 92:1–26 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Coste B et al. (2012) Piezo proteins are pore-forming subunits of mechanically activated channels. Nature 483:176–181 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Zhao QC et al. (2016) Ion permeation and mechanotransduction mechanisms of mechanosensitive piezo channels. Neuron 89:1248–1263 [DOI] [PubMed] [Google Scholar]
- 117.Ying WL, Emerson J, Clarke MJ, Sanadi DR (1991) Inhibition of mitochondrial calcium ion transport by an oxo-bridged dinuclear ruthenium ammine complex. Biochemistry 30:4949–4952 [DOI] [PubMed] [Google Scholar]
- 118.Baughman JM et al. (2011) Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 476:341–345 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Oxenoid K et al. (2016) Architecture of the mitochondrial calcium uniporter. Nature 533:269–273 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Woods JJ, Wilson JJ (2019) Inhibitors of the mitochondrial calcium uniporter for the treatment of disease. Curr Opin Chem Biol 55:9–18 [DOI] [PubMed] [Google Scholar]
- 121.Hardy JA, Wells JA (2004) Searching for new allosteric sites in enzymes. Curr Opin Struct Biol 14:706–715 [DOI] [PubMed] [Google Scholar]
- 122.Soussia IB et al. (2018) Antagonistic effect of a cytoplasmic domain on the basal activity of polymodal potassium channels. Front Mol Neurosci 11:301. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Chemin J et al. (2005) Lysophosphatidic acid-operated K+ channels. J Biol Chem 280:4415–4421 [DOI] [PubMed] [Google Scholar]
- 124.Duprat F et al. (2000) The neuroprotective agent riluzole activates the two P domain K(+) channels TREK-1 and TRAAK. Mol Pharmacol 57:906–912 [PubMed] [Google Scholar]
- 125.Zhuo RG et al. (2016) Allosteric coupling between proximal C-terminus and selectivity filter is facilitated by the movement of transmembrane segment 4 in TREK-2 channel. Sci Rep 6:21248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Beltran L, Beltran M, Aguado A, Gisselmann G, Hatt H (2013) 2-Aminoethoxydiphenyl borate activates the mechanically gated human KCNK channels KCNK 2 (TREK-1), KCNK 4 (TRAAK), and KCNK 10 (TREK-2). Front Pharmacol 4:63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Loucif AJC et al. (2017) GI-530159, a novel, selective, mechanosensitive two-pore-domain potassium (K2P ) channel opener, reduces rat dorsal root ganglion neuron excitability. Br J Pharmacol 10.1111/bph.14098 [DOI] [PMC free article] [PubMed]
- 128.Veale EL, Mathie A (2016) Aristolochic acid, a plant extract used in the treatment of pain and linked to Balkan endemic nephropathy, is a regulator of K2P channels. Br J Pharmacol 173:1639–1652 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Luo Q et al. (2017) An allosteric ligand-binding site in the extracellular cap of K2P channels. Nat Commun 8:378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Kennard LE et al. (2005) Inhibition of the human two-pore domain potassium channel, TREK-1, by fluoxetine and its metabolite norfluoxetine. Br J Pharmacol 144:821–829 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Fink M et al. (1998) A neuronal two P domain K+ channel stimulated by arachidonic acid and polyunsaturated fatty acids. EMBO J 17:3297–3308 [DOI] [PMC free article] [PubMed] [Google Scholar]