

Introduction
Historic perspective on cellular senescence
Cellular senescence, the non-dividing altered state into which many vertebrate cells enter when stressed, was first formally described by Hayflick and Moorhead in the early 1960s1,2. Their finding -- that normal cells eventually cease dividing (in culture) -- challenged the long-held idea initiated by Alexis Carrel in the early 1900s that normal cells were intrinsically ‘immortal’ (in culture)3. And, whereas Carrel suggested that organismal mortality might be a consequence of multi-cellularity, Hayflick and Moorhead suggested that the eventual cessation of cell division (subsequently termed cellular senescence) reflected organismal aging. They also noted that cells derived from malignant tumors did not undergo this form of senescence, suggesting that the senescent state existed to suppress the development of cancer. Many years later, Sager and colleagues showed that cellular senescence was a response to potential tumor-inducing stimuli, and thus formally postulated that the senescence response was a potent anti-cancer mechanism4,5. Since the 1960s, our understanding of cellular senescence, including its physiological and pathological roles, has exploded. In addition, it is now apparent that senescent cells both experience and cause many aspects of metabolic reprogramming, some aspects of which we discuss below.
Cell autonomous and non-autonomous effects of senescent cells
At the core of the cellular senescence response is an essentially irreversible arrest of cell proliferation (growth). It is now clear -- from multiple lines of evidence -- that this arrest constitutes a potent, cell autonomous anti-cancer mechanism in vivo6,7. Consequently, most humans and mice harboring mutations that prevent or suppress the senescence growth arrest (e.g., p53 or pRB inactivating or dominant negative mutations8,9) are cancer-prone and generally die an early death due to malignant tumors.
In addition, certain senescent cells become moderately resistant to cell death. In some senescent cells, this cell autonomous resistance is due to increased expression of anti-apoptotic, compared to pro-apoptotic, proteins, characteristics shared by several cancer cells. This property has led to the repurposing of certain (mostly failed) anti-cancer drugs that were initially designed to kill tumor cells, but now are being tested as senolytics -- that is, drugs that can selectively kill senescent cells, but not non-senescent counterparts10–12. However, it is not yet clear how pervasive cell death resistance is among different senescent cell types. For example, senescent endothelial cells are in general more prone to spontaneous apoptosls than senescent fibroblasts, although even fibroblasts can undergo apoptosis upon senescence when faced with certain types of DNA damage (e.g., transcription blocking lesions)13. In any case, the mechanisms of cell death resistance in senescent cells remains incompletely understood.
Finally, all senescent cells thus far examined develop a complex, multi-component senescent-associated secretory phenotype (SASP). The SASP acts cell non-autonomously to alter the behavior of neighboring cells and the tissue microenvironment. The SASP is strikingly variable and plastic. It depends on the cell type and senescence inducer (discussed below), and is dynamic, changing characteristics over time14,15. A striking, but not sole, feature of the SASP is the preponderance of pro-inflammatory molecules, including cytokines, chemokines, bioactive lipids and damage-associated molecular patterns (DAMPs; also termed Alarmins). Chronic inflammation is, of course, a major risk factor for many age-related diseases, including late-life cancer; hence the term ‘inflammaging’ has been used to describe the chronic inflammation that is a common attribute of aged tissues and might be at least partially due to the accumulation of senescent cells in aged tissues 16–18.
In recent years, it has become apparent that the phenotypes of senescent cells, in addition to the SASP, are remarkably variable, heterogeneous and plastic19. For example, for many -- but not all20 -- senescent cells, the growth arrest is initiated and/or enforced by the cyclin dependent kinase and cell cycle inhibitors p16INK4a and p21CIP1 (encoded by the CDKN2A and CDKN1A genes). Likewise, other widely used senescence markers, such as the senescence-associated beta-galactosidase (SA-Bgal)21, reduced expression of the nuclear lamina protein lamin B1 (LMNB1)22, and the relocalization and secretion of the nuclear protein HMGB1 (where, upon secretion, it acts as a DAMP)23, are neither invariable nor universal senescence markers. Thus, assessing the ability of specific stimuli to induce a senescence response -- and assessing the burden of senescent cells in tissues in vivo -- requires the use of multiple markers24.
Adaptive vs maladaptive effects of senescent cells
The senescence response can be beneficial or deleterious, depending on the physiological context. This dualism is consistent with the evolutionary theory of antagonistic pleiotropy. Antagonistic pleiotropy postulates that traits selected to ensure the survival of young organisms in natural environments, in which life spans are short, can become deleterious in modern protected environments, in which life spans are significantly longer25. Thus, aging is likely a consequence of the declining force of natural selection with age.
With regard to the beneficial effects of cellular senescence, as noted above, the senescence growth arrest protects young organisms from developing cancer. In addition, SASP factors can optimize the morphogenesis of certain structures in the embryo26,27, and initiate parturition in the placenta28,29. Finally, senescent cells occur transiently at sites of tissue damage where they contribute to wound healing, tissue repair and regeneration, most likely through specific SASP factors30–34.
In contrast, senescent cells increase with age in most mammalian tissues, where they appear to persist. Whether this increase is due to increased production or decreased clearance, for example by the immune system, is unclear. More importantly, experiments using human cells and tissues, transgenic mouse models and pharmacological interventions in cells and mice strongly implicate senescent cells in a large number of age-related pathologies, ranging from neurodegeneration to, ironically, age-related cancer6,35–38. Most of the detrimental effects of senescent cells can be attributed to the SASP, which, as noted above, is rich in pro-inflammatory molecules.
What initiates a senescence response? Little is known about how senescent cells are induced in vivo, particularly during aging. Known inducers of senescence responses -- at least in cultured cells and mouse models -- include, among others, DNA damage, activated oncogenes and mitochondrial dysfunction (discussed below). Because senescent cells are rare, even in old and diseased tissue, it has been difficult to determine how they were induced to senesce in vivo. However, single cell profiling, at both the trancriptomic and proteomic levels, promise to help identify the major drivers of senescence during natural aging and in age-related pathologies. In all cases, senescent cells must undergo metabolic reprogramming in order to maintain their viable growth arrested state and express the genes and proteins needed to sustain the highly complex, dynamic and heterogenous SASP14,39. The causes and consequences of these metabolic shifts are discussed below.
Metabolic drivers of senescence
Several forms of metabolic stress can both drive senescence and influence the SASP (Figure 1). Here we describe some of these and discuss them in the context of aging and disease.
Figure 1. Relationships between metabolism and cellular senescence.
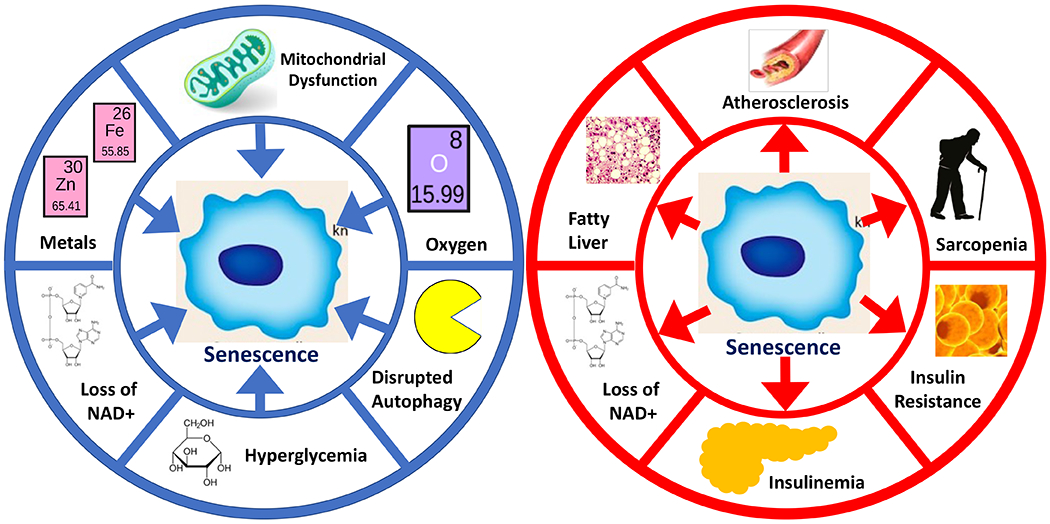
Left: Metabolic drivers of senescence. Mitochondrial dysfunction can drive senescence through disruption of cytosolic NAD+/NADH ratios, production of reactive oxygen species, and potentially other mechanisms. Accumulation of excess metals (especially transition metals) also promotes senescence. Loss of NAD+ results in senescence through loss of sirtuin or PARP activities and changes in cellular redox states. Hyperglycemia can drive senescence, though mechanistic detail is still needed. Disrupted autophagy can drive senescence in some contexts, but also prevent it in others. Non-physiological oxygen levels influence the development of senescent cells, with higher oxygen generally favoring senescence. Right: Senescent cells as drivers of metabolic disease. Senescent cells and/or the SASP can drive both formation of atherosclerotic plaques as well as plaque instability. In the liver, senescent cells can promote steatosis. The SASP also activates macrophages, which elevate CD38 and lower tissue NAD+ levels. In the pancreas, senescent beta cells promote hyperinsulinemia, but as beta cells are attacked by the immune system, this can become hypoinsulinemia. In peripheral tissues (e.g., fat) senescent cells can promote insulin resistance – so senescent cells can drive diabetes and metabolic disease in multiple ways. Finally, senescent cells promote sarcopenia in muscle tissue, which can influence basal metabolism, activity levels, and frailty.
Mitochondrial dysfunction
Mitochondria are major regulators of age-related pathology. Mice that accumulate mitochondrial DNA (mtDNA) mutations at an accelerated rate age prematurely40,41, whereas overexpression of mitochondrially-targeted catalase (mCAT) preserves mitochondrial function and extends lifespan in mice42. It is therefore not surprising that senescence and the SASP are similarly responsive to the function of mitochondria within the cell.
Many drivers of mitochondrial dysfunction also result in cellular senescence. These drivers include mtDNA depletion and mutations, inhibitors of the electron transport chain, loss of the mitochondrial chaperone protein HSPA9, loss of the mitochondrial protein deacetylase SIRT3 and the de-acylase SIRT543, and disruptions in complex I assembly44. This mitochondrial dysfunction-associated senescence (MiDAS) phenotype lacks several of the proinflammatory parts of the SASP, but instead includes its own distinct set of SASP factors. MiDAS is primarily driven by the accumulation of cytosolic NADH, which is normally oxidized by the mitochondria to NAD+ (Figure 2). This lowering of the NAD+/NADH ratio inhibits the key glycolytic enzyme, GAPDH, resulting in ATP depletion, AMPK activation and cell cycle arrest43.
Figure 2. NAD Metabolism and Cellular Senescence.
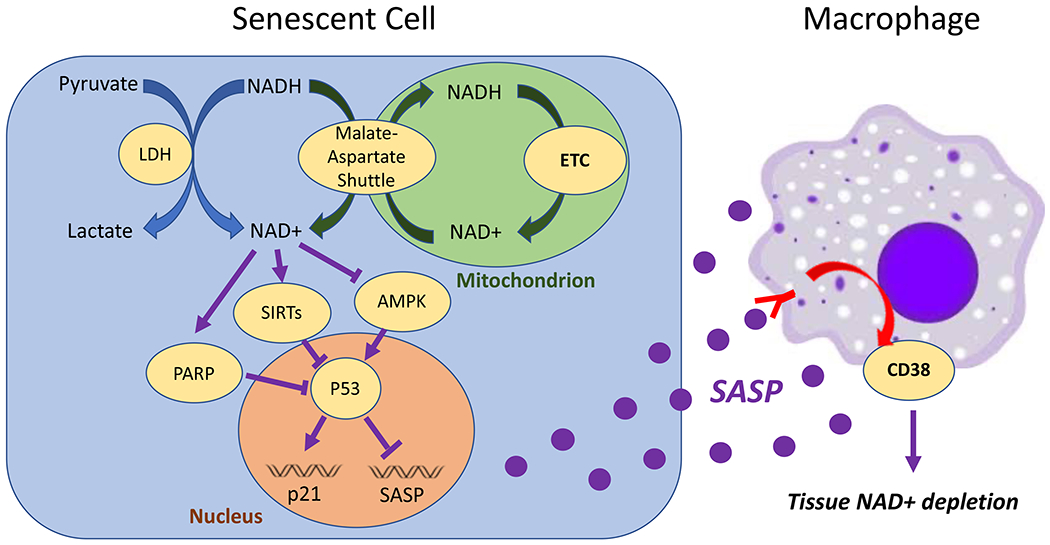
NAD+ levels and NAD+/NADH ratios are controlled by multiple pathways during senescence. The NAD+/NADH ratio is maintained by conversion of pyruvate to lactate in the cytosol, or by transfer of reducing equivalent of NADH to the mitochondrion by the malate-aspartate shuttle. NADH is then oxidized back to NAD+ in the mitochondrion by the activity of Complex I of the electron transport chain (ETC). Disruption of any of these processes leads to increased cytosolic NADH, AMPK activation, and senescence. NAD+ acts through PARPs and sirtuins to prevent genotoxic stress and p53 activation, which promotes senescence, but antagonizes the SASP. Once released by senescent cells, SASP factors can bind their cognate receptors on macrophages, which then elevate CD38. CD38 then lowers tissue levels of NAD+ in the surrounding tissue as part of cyclic ADP ribose (cADPR) synthesis.
Additionally, mitochondria are a source of reactive oxygen species (ROS). Loss of mitochondrial superoxide dismutase (SOD2) in mice drives cellular senescence45. A transgenic cell culture model of mitochondrial elimination by mitophagy protects against senescence and the expression of many pro-inflammatory SASP factors; this protection is associated with reductions in both reactive oxygen species (ROS) and nuclear DNA damage foci, especially telomere-associated foci46. Similarly, mitochondrial ROS can activate the JUN N-terminal kinase, which in turn promotes release of cytosolic chromatin fragments and activation of proinflammatory components of the SASP47. These and many other studies suggest that mitochondria-derived ROS may be a key driver of cellular senescence.
Notably, despite evidence that senescent cells drive aging and limit both health span and median lifespan48,49, few interventions that directly antagonize ROS extend lifespan50. Moreover, none of these interventions have demonstrated a reduction in age-related accumulation of senescent cells. Rather, studies that target mitochondria and prevent senescence or SASP modules in vivo also target other aspects of mitochondrial biology. Thus, it is an open question as to whether mitochondrial ROS are actually causal for the accumulation of senescent cells with age. For example, PGC-1B depletion lowers mitochondrial biogenesis, and rapamycin increases mitophagy46. Although both manipulations decrease ROS, they also alter many other aspects of mitochondrial metabolism and function. Additionally, ROS are essential signaling molecules for several processes, such as cell proliferation51, innate immunity52, stem cell renewal53,54, differentiation55 and survival56. The relationship between ROS, senescence, and aging in vivo is therefore likely to be more complex than culture models currently indicate.
More recent studies also showed that lowering ER-mitochondria contact by ablating the Itpr2 calcium channel in mice lowers the number of age-related senescent cells and the levels of several SASP factors, resultinf in an extension of health span and lifespan57. However, it is unclear if these effects are due to reduced mitochondrial calcium overload, diminished mitochondrial-ER contacts, or some combination of both. These strategies demonstrate a definitive role for mitochondria in the age-related accumulation of senescent cells, but are not specifically informative about whether ROS, per se, are to blame. The role mitochondrial function in cellular senescence and its associated phenotypes is also the subject of several recent reviews58–61.
An important caveat to many cell culture studies, especially those using mouse cells, is that while ROS - including mitochondrial ROS - can certainly induce senescence in culture, a majority of these studies were performed in atmospheric, rather than physiological, oxygen concentrations, which alters senescence responses62. Thus, many of the observed phenomena are potential artifacts of a hyperoxic state. Next, we discuss the role of oxygen levels in the development of senescence.
Oxygen
Phenotypes of cellular senescence are highly dependent on the levels of oxygen available to the cell. This dependence is often inversely correlated, as more oxygen tends to accelerate senescence. With over 60 oxygen-consuming enzymes in the mammalian genome63, it is not surprising that oxygen levels are a key modulator of many biological processes, including senescence. Murine cells are more sensitive to oxygen levels, as murine fibroblasts cultured at atmospheric oxygen levels undergo rapid senescence, but, due to endogenous telomerase activity, will proliferate extensively if oxygen levels are lowered to 3% - closer to levels observed in the stroma in vivo62. Extension of replicative lifespan and stemness are also observed in a subset of human cell lines, such as human amniotic fluid stem cells64. Additionally, atmospheric O2 levels are required for senescence reinforcement following treatment with drugs that activate cell cycle arrest, such as nutlin-3a65,66. Furthermore, lower O2 levels are required for a human-like SASP in cultured murine fibroblasts67, and this SASP is closer to that observed with age. Despite these findings, sub-physiological levels of oxygen (i.e., hypoxia) can also activate AMPK, which can suppress the SASP by mTOR inhibition68. More broadly, these data signal the potential for artifacts when studying senescence (and likely many other conditions) using atmospheric rather than physiological oxygen levels. Oxygen concentrations vary strongly depending on the tissue and species69, so physiological levels for relevant cell types is an important consideration when studying senescence. Several recent articles review the role of oxygen in diverse biological phenomena, including senescence63,69–71.
Unfortunately, hyperoxia is a medical necessity under certain conditions. For example, premature infants, whose lungs have not yet developed, often require supplemental oxygen – but later experience increased risks of respiratory conditions, such as asthma, in early childhood72. Similarly, individuals that experience the severe symptoms of COVID-19 often require supplemental oxygen73. Treatment of fetal human airway smooth muscle cells and lung fibroblasts with moderate hyperoxia (40% O2) increased markers of senescence in as short as 7 days72,74, suggesting that senescent cells may play a role in these disorders. Conversely, hyperbaric 100% oxygen was recently found to lower markers of senescence in specific populations of peripheral blood mononuclear cells75, so cell type may play an important role in senescent cell responses to oxygen. Additionally, a murine cardiac ischemia-reperfusion model accumulates senescent cells, and elimination of these cells improves recovery76, indicating that fluctuations in oxygen can also drive senescence and pathology. Oxygen level is therefore an important consideration both in cell culture settings, and potentially for the formation of senescence under specific circumstances in animal models and humans.
Disrupted NAD+ metabolism
NAD+ plays a major role in the regulation of both the cell cycle arrest and the SASP during senescence (Figure 2). NAD+ is a major cofactor for both poly-ADP-ribose polymerase (PARP) and sirtuin family proteins (SIRTs). PARP antagonizes senescence by protecting against genotoxic stress77, but also promotes NF-kB activation and secretory phenotypes in senescent cells78. Indeed, loss of PARP1 during epithelial cell senescence is associated with increased levels of unrepaired single strand breaks, which in turn reinforce cell cycle arrest through p38MAPK activation79. Similarly, inhibition of nicotinamide phosphoribosyltransferase (NAMPT) – which prevents NAD salvage and lowers total NAD+ levels – both promotes a senescence arrest and suppresses proinflammatory aspects of the SASP43,80; this effect resembles the phenotype of MiDAS. Importantly, NAD+ levels decline with age in many tissues, and this decline is associated with multiple degenerative conditions, including age-related muscle loss and diabetes81–83.
Several sirtuins play key roles in senescence response and the SASP. Beyond SIRT3 and SIRT5, which antagonize MiDAS43, SIRTs 1, 2, and 6 have known roles in senescence. SIRT2 deacetylates and stabilizes the mitotic checkpoint kinase BUBR1, which declines during aging84, and protects against aneuploidy - a potent driver of senescence85. Notably, supplementation with the NAD+ precursor NMN extends the median lifespan of progeric BUBR1 hypomorphic mice86. SIRT6 knockout mice show premature aging and a hyperinflammatory state that is driven at least in part by de-repression of retrotransposons87, which also occurs with senescence88. Furthermore, overexpression of SIRT6 prevents the loss of homologous recombination observed during senescence89. Finally, SIRT1 is degraded by autophagy during senescence90. Loss of SIRT1 activity is associated with multiple aspects of senescence, including SASP activation91 and cell cycle arrest92, as well as several senescence-associated degenerative pathologies including neurodegeneration, cachexia, fatty liver and atherosclerosis93,94. Together, the sirtuins and PARP demonstrate the importance of NAD+-consuming enzymes in senescence and help explain why loss of NAD+ levels with age can be so deleterious to a tissue.
NAD+ is not always beneficial in the context of senescence. We hypothesized that since loss of NAD+ antagonizes the SASP, increased NAD+ might have the opposite effect and promote segments of the SASP39. This proved to be the case as NAMPT and NAD+ are elevated in senescent cells. This rise is associated with decreased AMPK-and p53 activation, leading to increased p38MAPK and NF-kB activity and the secretion of proinflammatory SASP factors80. Thus, NAD+ may antagonize senescence, but once senescence is established, NAD+ increases may instead promote degenerative pathologies through the SASP.
Cellular senescence also plays a role in tissue-level NAD+ metabolism. The SASP promotes CD38 activation in macrophages. CD38 consumes NAD+ during the synthesis of cyclic-ADP ribose (cADPR)95,96, and is a primary driver of NAD+ depletion with age97. Therefore, senescent cells may drive NAD+ loss, which in turn could lead to more senescent cells (Fig 1). This degenerative feedback loop could therefore drive multiple NAD+ and senescence-dependent age-related conditions (e.g. diabetes82). In contrast, eliminating senescent cells or suppressing the SASP might protect NAD+ homeostasis, and CD38 inhibition in turn might be protective and prevent increased senescence with age.
As noted above, MiDAS results from lowered cytosolic NAD+/NADH ratios43, but reduced NAD+/NADH ratios can occur without mitochondrial dysfunction. For example, inhibition of the malate-aspartate shuttle, which transports reducing equivalents of NADH from the cytosol to the mitochondria, by aminooxyacetate (AOA) drives senescence43. Loss of the shuttle enzyme malate dehydrogenase 1 (MDH1) has a similar phenotype98. Similar results occur when malic enzymes, which catalyze NAD+-dependent conversion malate to pyruvate in the cytosol (ME1) and mitochondria (ME2) are inhibited99. These enzymes are reciprocally regulated by p53; depletion of either drives p53-dependent senescence, but p53 in turn represses malic enzymes99. These studies show that the interplay between NAD+/NADH ratios and p53 is a key regulator of senescence under conditions of metabolic stress.
Hyperglycemia
Numerous studies demonstrate that culturing cells in high levels of glucose accelerates cellular senescence100,101. However, because multiple pathways by which this acceleration occurs have been identified, no unifying model currently explains all forms of hyperglycemia-associated senescence. Most studies focus on endothelial cells, including retinal endothelial cells, but fibroblasts and renal epithelial cells have also been studied100–104. Nonetheless, the few mechanisms ascribed to senescence induction seem unlikely to be common to multiple cell lineages. For example, arginase 1 (ARG1) expression and activity are induced in retinal endothelial cells cultured in high glucose, resulting in reduced nitric oxide (NO) and elevated oxidative stress101. Retinal endothelial cells in a mouse model of streptozotocin (STZ)-induced diabetic retinopathy undergo senescence in an ARG1-dependent manner105, and inhibition of ARG1 activity prevents high glucose-induced senescence in cultured retinal epithelial cells and diabetes-induced retinal senescence in mice. Ironically, prior reports in human umbilical vein endothelial cells (HUVECs) showed that arginase activity declined during high glucose-induced senescence, and this decline was linked to decreased NO synthesis106. Supplementation with arginine prevented these effects and antagonized senescence in high glucose-treated HUVECs. Generally, NO antagonizes endothelial cell senescence106, so the key effector of glucose-induced senescence in these cells is likely related to the level of cellular NO production. This mechanism is unlikely to be universal because ARG1 is expressed in only a few cell types, and NO is not produced at appreciable levels by all cells.
High glucose also drives senescence in fibroblasts100,104, and glucose restriction can extend replicative lifespan107. Although fibroblasts produce NO, they do not express ARG1, so it is unclear whether the phenotypes observed with endothelial cells translate to this cell type. For example, inhibition of nitric oxide synthases (NOS) with N(G)-nitro-L-arginine methyl ester (L-NAME) drives senescence in endothelial cells106, but can antagonize senescence when fibroblasts are co-cultured with macrophages108. Thus, the role of NO in the senescence responses of different cell types requires further investigation.
In another study, high glucose promoted senescence in fibroblasts by lowering the levels of SIRT3, and transgenic overexpression of SIRT3 prevented this high glucose-induced senescence104. Loss of SIRT3 can drive MiDAS43, so lowering of SIRT3 by excess glucose is an appealing link between these processes. Furthermore, high glucose accelerates the loss of sirtuin expression in endothelial cells103, and this loss is also associated with increased senescence. Since SIRT3 deacetylates mitochondrial proteins, and high-sugar diets can increase mitochondrial protein acetylation109,110 and decrease NAD+/NADH ratios111, it is possible that hyperglycemia drives senescence by MiDAS or a MiDAS-like phenotype.
Hyperglycemia can also result in the formation of advanced glycation end-products (AGEs), which can drive senescence in some models. For example, treatment of kidney proximal tubule epithelial cells with exogenous AGEs (e.g., glycated albumin) triggered increased expression of p21112 or p16113, coupled to activation of SA-Bgal, in a receptor for advanced glycation end-products receptor (RAGE)-dependent manner. Notably, these studies focused only on effects observed 48 hours after treatment, so chronic exposure and effects of AGE removal were not considered. Further, these effects may not extend to other cell types. Conversely, activation of RAGE extends the replicative lifespan of murine preadipocytes via binding to p53 and antagonizing p21 expression114. More research is therefore required to fully elucidate the relationship between AGEs and cellular senescence. Furthermore, studies linking high glucose to senescence have not addressed the SASP, and therefore the consequences of this form of senescence are still somewhat unclear. Despite these needs, the links between diabetes and senescence are growing, and increases in senescent cells at sites of diabetic complications indicate that this is an important area for future research.
Metabolism of senescent cells
Senescent cells show several alterations in their metabolic states (Figure 3). Here we review some of these alterations and their significance in the development of senescence phenotypes.
Figure 3. Altered metabolic states of senescent cells.
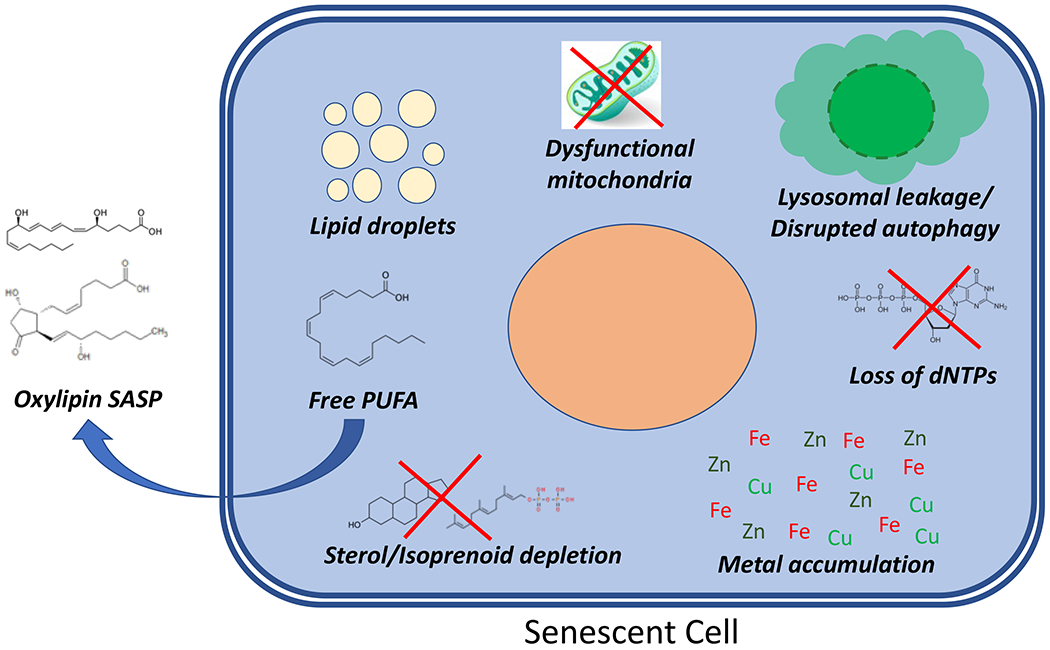
Senescent cells have increased free cytosolic polyunsaturated fatty acids (PUFAs), which in turn can be converted into oxylipins as part of a lipid-based SASP. Fatty acids also accumulate in lipid droplets during senescence. Sterols preferentially accumulate in the ER of some senescent cells in a p53-dependent manner, inhibiting sterol-response element binding protein 2 (SREBP2) activation and lowering sterol synthesis in the cell as a whole. Senescent cells often have increased mitochondrial mass or mtDNA, but this is coupled to altered membrane potential and ROS production. Lysosomes of senescent cells can become permeable, acidifying the cytosol and disrupting some forms of autophagy. This can lead to accumulation of transition metals inside senescent cells. Finally, senescent cells can have lower dNTP levels due to loss of ribonucleotide reductase 2. *Note that senescence is a complex and varied phenomenon, and it is likely that individual metabolic alterations may be present in some, but not all, forms of senescence.
Lipid metabolism
A growing body of evidence indicates that senescent cells alter lipid metabolism. One of the first observations along these lines came from the finding that the sphingomyelin/ceramide pathway, and especially sphingomyelinase, was significantly more active in senescent cells115. Addition of ceramides at concentrations that approximate those found in senescent cells lower markers of cell proliferation and increase markers of senescence in both fibroblasts115 and endothelial cells116. Overall, ceramide and sphingosine promote senescence, whereas sphingosine-1-phosphate (S1P) promotes cell proliferation via its cognate receptors. Ceramide-activated protein phosphatases (PP1 and PP2A) can promote a senescence arrest by dephosphorylating CDK2 and specifically up-regulating p21117. Sphingosine has similar effects, but the mechanisms are less understood118. The relationships among ceramides, sphingolipids and cellular senescence were recently reviewed119.
Additional studies indicate that senescent cells upregulate beta-oxidation - the metabolic breakdown of lipids120 - and that this activity is necessary for the secretion of many proinflammatory components of the SASP in oncogene-induced senescence. In addition, fatty acid synthase (FASN) was elevated in senescent cells and required for both the cell cycle arrest and parts of the SASP121. The expression of several enzymes related to both fatty acid synthesis (e.g. FASN) and oxidation (e.g. ACADS, ACADL, ACADSB, ACADVL) increase in senescent cells122, whereas stearoyl-CoA desaturase – which converts saturated fatty acids into monounsaturated fatty acids (MUFAs) - declines123. These data suggest that saturated fatty acids might be an important substrate for energy production in senescent cells.
Beyond energy use and storage, fatty acids play additional roles in senescence responses. Notably, lipid droplets increase and accumulate with at least some forms of cellular senescence. Exogenous lipids are preferentially incorporated into triacylglyerols, which form the lipid droplets in senescent cells123. Furthermore, free polyunsaturated fatty acid (PUFA) levels increase in senescent cells124, likely via p38-dependent activation of phospholipase A2, which cleaves PUFAs from cellular membranes125. Senescent cells incorporate higher levels of PUFAs into triglycerides126, which ultimately accumulate in lipid droplets. Notably, animal models of obesity show increased numbers of senescent cells in the brain, and these cells show increased numbers of lipid droplets. Importantly, clearance of senescent cells lowered the accumulation of these droplets127. Lowering exogenous lipid levels in cell culture media prevented both lipid droplet accumulation and the upregulation of many SASP factors127. These data suggest that lipid droplets, or at least the presence of lipids, is required for some key aspects of cellular senescence and the SASP.
The release of free PUFAs from the plasma membrane is associated with multiple forms of senescence124,126. Additionally, levels of several PUFAs, such as eicosapentanoic acid (EPA), arachidonic acid (AA), dihomo-gamma-linolenic acid (DGLA) and docosohexanoic acid (DHA), are highly elevated during MiDAS124, above other forms of senescence. This elevation may reflect an attempt by cells to preserve the NAD redox balance by using PUFA desaturation to convert NADH into NAD+, as described128. Beyond accumulating as triglycerides in lipid droplets, free PUFAs are also converted to oxygenated signaling molecules collectively known as oxylipins124. Oxylipins associated with cellular senescence include leukotrienes125,129 and prostaglandins, including dihomo-prostaglandins124,130. Senescence-associated leukotriene release drives profibrotic features of pulmonary fibrosis, and eliminating senescent cells ameliorates these effects125,131. The synthesis of prostaglandins is essential for reinforcing the senescence-associated cell cycle arrest, as inhibitors of the inducible prostaglandin synthase 2 (PTGS2 or COX-2) extend the replicative lifespan of cultured cells132 and allow increased numbers of dividing cells in response to genotoxic stress-induced and oncogene-induced senescence124.
Surprisingly, it does not appear that prostaglandin synthesis promotes senescence through interactions with prostaglandin receptors. Rather, senescent cells upregulate expression of the prostaglandin transporter (PGT/SLCO2A1), but without a commensurate elevation in expression of prostaglandin dehydrogenase (PGDH), which inactivates prostaglandins by converting them to 15-keto derivatives. These metabolic changes result in the intracellular accumulation of 15-deoxy prostaglandins, especially 15d-PGJ2 and senescence-associated dihomo-15d-PGJ2. These electrophilic prostaglandins have multiple activities inside cells. Primarily, they chage the structure of proteins by Michael addition to exposed cysteines133 -- that is, the nucleophilic addition of a carbanion or other nucleophile to an α,β-unsaturated carbonyl compound containing an electron withdrawing group.
The intracellular accumulation of dihomo-15d-PGJ2 in senescent cells promotes the activation of endogenous HRAS, thereby reinforcing senescence in a manner analogous to oncogene activation124. Notably, this property of senescent cells allows the detection of senolysis because dihomo-15d-PGJ2 is only ~343 Da in size and thus escapes the cytosol during apoptosis. Consequently, it can be detected in culture media or biological fluids upon senolysis124. Together, these studies demonstrate that senescence and lipid metabolism are intricately linked (Figure 4), but also highlight the importance of future work, as many areas of lipid metabolism are still underexplored in the context of senescence.
Figure 4. Lipid metabolism in senescent cells.
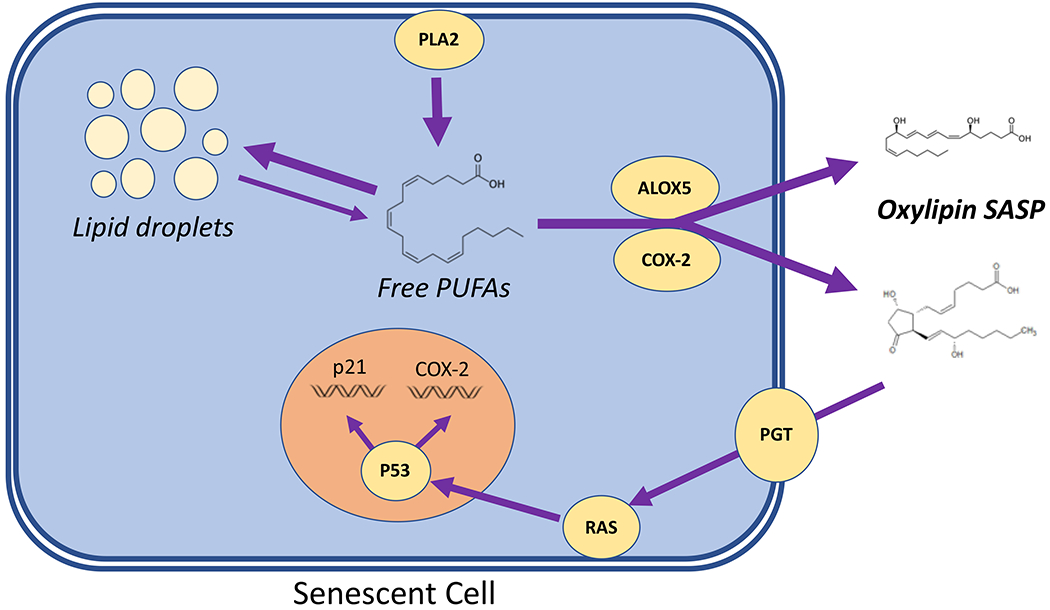
Activation of phospholipase A2 (PLA2) frees polyunsaturated fatty acids (PUFAs) from the plasma membrane, which then accumulate in the form of triglycerides in lipid droplets but are also used as substrates for oxylipin synthases such as arachidonate 5-lipoxygenase (ALOX5) and cyclooxygenase 2 (COX-2), resulting in release of an oxylipin SASP. Prostaglandin transporter (PGT) imports prostaglandins into the cytosol, where cyclopentenone prostaglandins activate RAS, leading to p53 activation, leading to both cell cycle arrest p21 and elevation of COX-2, reinforcing oxylipin biosynthesis.
Lysosomes and autophagy
Autophagy, the process by which a cell breaks down its organelles and macromolecules, is dysregulated in senescent cells. Specific factors show selective microautophagic degradation in senescent cells, whereas macroautophagy appears to be less active in senescent cells134,135. Inhibition of autophagy by depletion of ATG7, ATG12 or LAMP2 induces senescence136, implying that loss of autophagic homeostasis during senescence may reinforce the senescence arrest and maintain the senescent state. However, activation of autophagy is associated with oncogene-induced senescence, and overexpression of ULK3 also promotes senescence137. The complex nature of the relationship between senescence and autophagy are the subject of recent reviews135,138.
Notably, many senescent cells have dysfunctional lysosomes. Indeed, SA-Bgal is a lysosomal beta-galactosidase that is markedly upregulated in senescent cells139,140, and SA-Bgal may reflect lysosomal dysfunction. Recent findings show that lysosomal permeability increases in senescent cells. This increase results in the previously-observed loss of lysosomal acidity141,142, but also elevates cytosolic acidification143. To combat this increase, senescent cells upregulate glutaminolysis by increasing glutaminase 1 (GLS1) expression, which increases ammonia production and neutralizes the cytosolic acidity143. Inhibition of GLS1 kills senescent cells both in culture and in vivo and ameliorates several indicators of age-related organ dysfunction143. Therefore, lysosomal dysfunction, while potentially deleterious, is an exploitable weakness for targeted elimination of senescent cells.
Disruption of transition metal homeostasis
Transition metals – divalent cations that occupy a central block in the periodic table - play a key role in several cellular processes, but the role of these metals in senescence is only beginning to be understood. Surprisingly, senescent cells were found to accumulate transition metals at much higher levels than non-senescent or immortalized cells144. These transition metals included iron, manganese and zinc. Later, other transition metals, such as copper145, were found to accumulate in senescent cells.
Iron
Iron levels are tightly controlled in most cells. Iron is required for oxygen transport, respiration and oxidative phosphorylation146. However, too much unbound or “labile” iron can catalyze the production of ROS and peroxidized lipids, which can drive toxic cell processes, including a form of iron-dependent cell death known as ferroptosis147. Iron overload is antagonized by the protein ferritin, which binds and sequesters iron in an inactive form. Senescent cells import increased levels of iron by elevating the transferrin receptor, which then endocytoses iron-bound transferrin148. Ferritin levels increase in senescent cells, sequestering much of the imported iron in an inactive state148. Because iron is released from ferritin by targeted autophagy (ferritinophagy), and senescent cells display compromised autophagy and lysosomal dysfunction, senescent cells likely accumulate iron at least in part due to failure to degrade ferritin. Activation of autophagy by rapamycin partially restored cellular iron levels148. While iron metabolizing protein levels suggest that labile iron may actually decrease during senescence148, no reports of direct measurement of the labile iron pool in senescent cells exist to date. If the labile iron pool does indeed decrease during senescence, this finding might explain the observation that senescent cells are resistant to ferroptosis, as lack of labile iron would prevent iron-catalyzed lipid peroxidation. However, rapamycin had no effect on ferroptosis resistance in senescent cells, emphasizing the need for more research in this area.
Copper
Copper is an essential trace element required for the activity of multiple enzymes, including superoxide dismutase, neurotransmitter synthases, amine oxidases, ceruloplasmin and lysyl oxidase149. Excess copper can promote senescence in human fibroblasts150 and human glioblastoma cells by downregulating the polycomb protein Bmi-1151, although both copper and oxygen levels were supraphysiological in these studies. Similar to iron, copper accumulates in senescent cells145, and, much like iron, this accumulation appears due to defective lysosomal autophagic degradation of copper chaperones. However, since copper-dependent antioxidant enzymes (e.g. superoxide dismutase, glutathione reductase) are elevated during senescence145, and prevent senescence152, this elevation may be a compensatory mechanism to counteract the more oxidative environment observed in senescent cells. Much like iron, rapamycin partially abrogates copper accumulation in senescent cells145, suggesting regulation by autophagy, but the consequences of this elevation are unclear.
Zinc
Zinc is notable for its essential use by many organisms. One reason for this use is that, unlike iron and copper, zinc does not catalyze the formation of radicals from lipid peroxides153. Thus, zinc is less toxic and more redox inactive than iron and copper. Nonetheless, zinc levels are inversely associated with lifespan in C. elegans154. Zinc levels also increase in senescent cells, and are generally higher than iron or copper levels144. Excess zinc promotes senescence in cultured vascular smooth muscle cells155, endothelial cells156 and fibroblasts157. Despite these studies showing an elevation of transition metals during senescence, no functional significance has yet been ascribed to the iron, copper or zinc accumulation in senescent cells. Since overload of these metals drives senescence, this accumulation may enforce a cell cycle arrest program that maintains senescent cells in an essentially irreversible growth-arrested state.
Senescence and dNTP synthesis
Since senescent cells do not divide, their principal uses for dNTPs are for DNA repair and mitochondria DNA synthesis. Ribose-linked ATP, GTP and CTP are converted to dNTPs by ribonucleotide reductase (RRM2). Unsurprisingly, RRM2 declines during senescence158. However, when senescence is induced by oncogenic RAS, RRM2 depletion actually precedes the growth arrest and occurs during the hyperproliferative phase prior to cell cycle arrest158. Increased proliferation in the absence of dNTPs causes replication fork collapse, DNA double strand breaks and cell cycle arrest. Addition of exogenous dNTPs allows continued proliferation in this model158. Eliminating p53 or p16 allows cells to bypass oncogenic RAS-induced senescence by increasing nucleotide synthesis via shunting metabolites through the pentose phosphate pathway159,160. Notably, RAS activation antagonizes the hippo pathway transducers YAP/TAZ, and activation of this pathway restores RRM2 expression and allows escape from RAS-induced senescence161. These findings suggest that senescent cells integrate external cues to control DNA metabolism.
Interventions that reciprocally target senescence and metabolism.
As described above, senescence and metabolism and intricately linked. It would therefore follow that interventions that target metabolism might influence the accumulation of senescent cells - or properties of senescent cells, such as the SASP. Conversely, targeting senescent cells with, for example, senolytic drugs, should influence the development of metabolic disorders, as shown in Figure 1 (right). In this section, we explore some of these links.
Senolytics as regulators of metabolism
Transgenic mice and drugs that allow the selective killing of senescent cells (senolysis/senolytic drugs) reveal that senescent cells promote many age-related conditions, thereby limiting both lifespan and health span48,49. Senolysis prevents several pathologies, including chronic kidney disease48,162, osteopenia163, and osteoarthritis164. Senescent cells also promote tumorigenesis48,165, including both cancer relapse and several side effects of anti-cancer chemotherapy166,167. It is therefore not surprising that senescent cells also promote aspects of metabolic disease.
Senescent cells promote both diabetes and the degenerative complications of diabetes. Diabetes can be driven by pancreatic cell senescence, promoting insulitis and Type I diabetes168. Diabetes can also be driven by peripheral senescent cells in obese mice, which drive insulin resistance and Type 2 diabetes169. Surprisingly, increasing the presence of senescent cells by transgenically overexpressing p16INK4a in pancreatic beta cells of mice actually increases insulin secretion170, so the development of senescence may reflect metabolic compensation for high blood glucose levels. Similarly, insulin resistance can drive hyperinsulinemia and promote beta cell senescence171, implicating peripheral senescent cells as feedback drivers of senescence in the pancreas. Furthermore, complications of diabetes, including diabetic kidney disease172,173 and diabetic retinopathy174, potentially result from senescent cells driven by increased blood glucose levels, as described above. In each case, senolytic therapies improve health outcomes in terms of either blood glucose levels168,169,171 or development of diabetes-driven complications172,174. Together, these studies implicate senescent cells as a major nexus for interventions into diabetes and its associated disorders.
Dietary interventions and senescence
Multiple dietary interventions can limit the accumulation of senescent cells with age. Calorie restriction (CR) was one of the first lifespan-extending interventions to be identified175. Thus, when it was found that senescent cells can limit lifespan48, it seemed that links between CR and senescence were likely. CR lowers markers of senescence in the colons of mice and humans176 and in inguinal white adipose tissue177. CR prevents the development of senescence in the kidney of aged animals, whereas high calorie diets increase senescence178.
Other dietary interventions can similarly extend health span and either or both median and maximum lifespan. These interventions include methionine restriction179 and ketogenic diets180,181. Methionine restriction lowers markers of senescence and the SASP182, while injection of mice with beta-hydroxybutyrate, a major ketone body produced by ketogenic diets, lowers markers of senescence in vascular smooth muscle and endothelial cells183. These results indicate that dietary interventions that create a favorable metabolic state that can limit the accumulation of senescent cells.
Metabolic interventions that influence senescent cells
As described above, hyperglycemia induces senescence. It therefore reasons that diabetes could be pro-senescence, and interventions that lower blood sugar should antagonize the formation of senescent cells. Indeed, kidney proximal tubule cells become senescent within 4 weeks of induction of diabetes in mice, and this rise in senescent cells can be prevented by lowering glucose levels with insulin or inhibiting glucose transport into the cells by inhibiting the activity of the sodium-glucose co-transporter SGLT2102. Similarly, acarbose - which lowers blood sugar by antagonizing intestinal carbohydrate absorption – extends lifespan in mice184 and prevents diet-induced atherosclerosis-associated senescence in rabbits185.
Another antidiabetic medication, metformin, extends both lifespan and health span in mice186, and is a drug being studied for the first clinical trial to monitor general aspects of aging (under review; see https://www.afar.org/tame-trial). Metformin antagonizes the development of senescence in response to ceramide in immortal mouse myoblast cells187 and extends the replicative lifespan of human fibroblasts and mesenchymal stem cells188. Additionally, metformin lowers several SASP factor levels, including many proinflammatory cytokines, by interfering with NF-kB activation189. Furthermore, metformin protects against the development of senescence in murine models of intervertebral disc degeneration190 and chronic kidney disease191. Metformin therefore interferes with senescence at several levels and is perhaps the best-characterized metabolic intervention that antagonizes senescence and the SASP.
Atherosclerosis, statins, and senescence
Statins, such as simvastatin, are commonly used among older adults as preventives for elevated cholesterol levels and associated atherosclerosis-related complications, such as stroke and heart attack. They prevent cholesterol synthesis by inhibiting HMG-CoA reductase, the rate-limiting enzyme in the mevalonate pathway that leads to cholesterol biosythesis192. Statins prevent the senescence growth arrest, with lowered markers of senescence, in endothelial progenitor cells193., which is dependent on lowered levels of farnesylpyrophosphate and geranylgeranylpyrophosphate193. Statins also inhibit many aspects of the proinflammatory SASP of senescent fibroblasts, but, as with endothelial progenitor senescence, this effect is independent of the cholesterol-lowering properties of the drug194. Rather, HMG-CoA reductase inhibition lowers farnesyl-CoA and geranylgeranyl-CoA levels, which in turn are required for membrane localization and activation of rho-type GTPases, including RAS and RAC, which are required for the SASP. Since p16-positive (presumably senescent) foamy intimal cells promote deleterious features of atherosclerosis195, it is possible that statins may have anti-atherogenic properties beyond regulating cholesterol. However, statins can also act as radiosensitizers – increasing cancer cell senescence in response to irradiation196,. So, the effects of statins are likely to be context-specific. Surprisingly, p53 activation during senescence limits both the mevalonate and cholesterol synthesis pathways197, and therefore may be a mechanism by which p53 activation limits aspects of the SASP15,66. The relationship between senescence, cholesterol and atherosclerosis is therefore likely to be more complex than previously thought.
Exercise as an intervention for senescence
While numerous studies highlight the health benefits of exercise and its improvement of health span with age, few studies to date have demonstrated lifespan extension due to exercise in mice198,199; just one late life exercise study showed lifespan extension200. There is evidence that exercise can prevent senescence and segments of the proinflammatory SASP in the sera and hearts of aged mice199. More notably, exercise offsets diet-induced senescence and the SASP in mouse adipose and liver tissues201, suggesting that the most beneficial aspects of exercise may not pertain to already healthy animals, but rather those likely to develop metabolic disease. In humans, exercise and activity are associated with reduced markers of endothelial and leukocyte cell senescence202,203. These data suggest that exercise can offset deleterious aspects of senescence brought about by poor diet and sedentary lifestyles.
Conclusions
As noted throughout this article, the interplay between senescent cells and systemic metabolism is dynamic. Senescent cells can disrupt metabolic homeostasis in multiple tissues, and these changes in turn can promote senescence, creating a feedback loop that can drive pathology. This loop occurs in the context of NAD+, where the SASP promotes CD38-dependent NAD+ breakdown by immune cells95,96, while lowered NAD+ levels in turn can promote senescence39 (Figure 2). Similarly, peripheral senescence can result in insulin resistance169, while insulin resistance in turn drives stress and senescence in pancreatic beta cells171. This degenerative cycle leads to elevated blood glucose and senescence in the eye and kidney, ultimately driving diabetic complications172,174 (Figure 5). These feedback loops require interventions that break the cycle in order to be efficacious.
Figure 5. Multiple roles for senescence in diabetes and its complications.
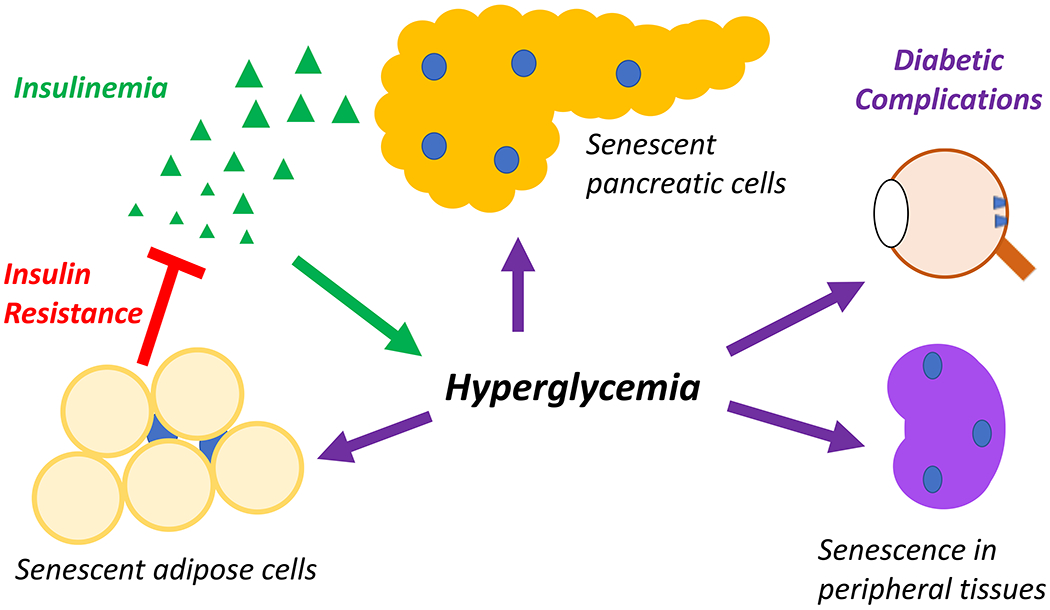
Senescent adipose cells can result in insulin resistance, which results in hyperglycemia, which can in turn promote additional adipose tissue senescence. Hyperglycemia also forces pancreatic beta cells to over-produce insulin. This stress results in beta cell senescence, leading to insulinemia, resulting in further hyperglycemia. Hyperglycemia also results in senescence in peripheral tissues such as the retina and the kidney, fueling diabetic complications.
There are three types of interventions that target senescent cells and their degenerative pathologies. First, we can slow the formation of senescent cells, as observed during dietary restriction and similar interventions. Second, we can allow senescent cells to accumulate, but prevent them from causing harm, as observed during metformin-mediated SASP suppression or after CD38 inhibition. Finally, we can use senolysis to remove senescent cells. Unlike the first two interventions, senolysis can be used intermittently - allowing for a “hit and run” approach that might be easier to implement in humans - whereas dietary and suppressive drug regimens require regular adherence to maintain benefits.
The metabolic changes observed in senescent cells also imply that additional disorders might be driven by senescent cells. For example, it was recently shown that senescent cells occur in the chorioamnionic membrane prior to birth in mammals, and that these cells send signals to promote parturition28. Given that parturition is promoted by prostaglandins E2 (PGE2) and F2α (PGF2α), it may be that senescence-associated prostaglandins are part of normal (and potentially premature) labor in humans. Prostaglandins also have additional roles, such as the essential role of PGD2 in sleep initiation204,205. Given that many people experience increased sleep disruption with age206, it is tempting to speculate that senescent cells may produce additional PGD2 that drives age-related sleep disruption.
Finally, age is a major outcome of human nutrition, while nutritional interventions can prevent many age-associated diseases207. Much is known about the role of macronutrient stress in longevity and senescence, but less is known about how malnutrition of key micronutrients controls the development of senescence. For example, choline deficiency is common in the elderly in the United States207, and is a driver of hepatic steatosis – a condition also promoted during age by senescent cells208. The relationship between nutrition and senescence is therefore fertile ground for future exploration and will undoubtedly be the subject of multiple studies moving forward.
Clearly, many aspects of the interplay between metabolism and cellular senescence need additional research. Encouragingly, however, emerging tools of modern biology, including single cell and systems biology and artificial intelligence, are making it easier to obtain deep insights into this complex interplay, which will undoubtedly lead to more efficacious interventions into age-related metabolic pathologies in the future.
Synopsis:
Cellular senescence entails a permanent proliferative arrest, coupled to multiple phenotypic changes. Among these changes is the release of a numerous biologically active molecules collectively known as the senescence-associated secretory phenotype, or SASP. A growing body of literature indicates that both senescence and the SASP are sensitive to cellular and organismal metabolic states, which in turn can drive phenotypes associated with metabolic dysfunction. Here, we review the current literature linking senescence and metabolism, with an eye toward findings at the cellular level, including both metabolic inducers of senescence and alterations in cellular metabolism associated with senescence. Additionally, we consider how interventions that target either metabolism or senescent cells might influence each other and mitigate some of the pro-aging effects of cellular senescence. We conclude that the most effective interventions will likely break a degenerative feedback cycle by which cellular senescence promotes metabolic diseases, which in turn promote senescence.
Footnotes
Conflict of interest
Dr. Campisi is a scientific founder of Unity Biotechnology, which develops senolytic therapies. Drs. Wiley and Campisi hold patents for induction and detection of senolysis using metabolic targets.
References
- 1.Hayflick L The limited in vitro lifetime of human diploid cell strains. Exp Cell Res 37, 614–636 (1965). [DOI] [PubMed] [Google Scholar]
- 2.Hayflick L & Moorhead PS The serial cultivation of human diploid cell strains. Exp Cell Res 25, 585–621 (1961). [DOI] [PubMed] [Google Scholar]
- 3.Carrel A On the permanent life of tissues outside of the organism. J Exp Med 15, 516–528 (1912). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Sager R, Tanaka K, Lau CC, Ebina Y & Anisowicz A Resistance of human cells to tumorigenesis induced by cloned transforming genes. Proc Natl Acad Sci USA 80, 7601–7605 (1984). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Sager R Senescence as a mode of tumor suppression. Environ Health Persp 93, 59–62 (1991). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Campisi J Aging, cellular senescence, and cancer. Annu Rev Physiol 75, 685–705 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Collado M, Blasco MA & Serrano M Cellular senescence in cancer and aging. Cell 130, 223–233 (2007). [DOI] [PubMed] [Google Scholar]
- 8.de Keizer PL, Laberge RM & Campisi J p53: Pro-aging or pro-longevity? Aging 2, 377–379 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Chicas A et al. Dissecting the unique role of the retinoblastoma tumor suppressor during cellular senescence. Cancer Cell 17, 376–387 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Chang J et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nature Med 22, 78–83 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Yosef R et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nature Comm 7, 11190 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Zhu Y et al. Identification of a novel senolytic agent, navitoclax, targeting the Bcl-2 family of anti-apoptotic factors. Aging Cell 15, 428–435 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Kim DE et al. Deficiency in the DNA repair protein ERCC1 triggers a link between senescence and apoptosis in human fibroblasts and mouse skin. Aging Cell 19, e13072 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Basisty N et al. A proteomic atlas of senescence-associated secretomes for aging biomarker development. PLoS Biol 18, e3000599 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Coppe JP et al. Senescence-associated secretory phenotypes reveal cell non-automous functions of oncogenic RAS and the p53 tumor suppressor. PLoS Biol 6, 2853–2868 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Cevenini E, Monti D & Franceschi C Inflamm-ageing. Curr Opin Clin Nutr Metab Care 16, 14–20 (2013). [DOI] [PubMed] [Google Scholar]
- 17.Franceschi C & Campisi J Chronic inflammation (inflammaging) and its potential contribution to age-associated diseases. J Gerontol 69, 54–59 (2014). [DOI] [PubMed] [Google Scholar]
- 18.Furman D et al. Chronic inflammation in the etiology of disease across the life span. Nature Med 25, 1822–1832 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Hernandez-Segura A et al. Unmasking transcriptional heterogeneity in senescent cells. Curr Biol 27, 2652–2660 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Alimonti A et al. A novel type of cellular senescence that can be enhanced in mouse models and human tumor xenografts to suppress prostate tumorigenesis. J Clin Invest 120, 681–693 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dimri GP et al. A novel biomarker identifies senescent human cells in culture and in aging skin in vivo. Proc Natl Acad Sci USA 92, 9363–9367 (1995). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Freund A, Laberge RM, Demaria M & Campisi J Lamin B1 loss is a senescence-associated biomarker. Molec Biol Cell 23, 2066–2075 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Davalos AR et al. p53-dependent release of Alarmin HMGB1 is a central mediator of senescent phenotypes. J Cell Biol 201, 613–629 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Gorgoulis V et al. Cellular Senescence: Defining a Path Forward. Cell 179, 813–827, doi: 10.1016/j.cell.2019.10.005 (2019). [DOI] [PubMed] [Google Scholar]
- 25.Williams GC Pleiotropy, natural selection, and the evolution of senescence. Evolution 11, 398–411 (1957). [Google Scholar]
- 26.Muñoz-Espín D et al. Programmed cell senescence during mammalian embryonic development. Cell 155, 1104–1118 (2013). [DOI] [PubMed] [Google Scholar]
- 27.Storer M et al. Senescence is a developmental mechanism that contributes to embryonic growth and patterning. Cell 155, 1119–1130 (2013). [DOI] [PubMed] [Google Scholar]
- 28.Menon R, Richardson LS & Lappas M Fetal membrane architecture, aging and inflammation in pregnancy and parturition. Placenta 79, 40–45, doi: 10.1016/j.placenta.2018.11.003 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Menon R et al. Placental membrane aging and HMGB1 signaling associated with human parturition. Aging 8, 216–230 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Chu X, Wen J & Raju RP Rapid senescence-like response after acute injury. Aging Cell 19, e13201 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Demaria M et al. An essential role for senescent cells in optimal wound healing through secretion of PDGF-AA. Developmental cell 31, 722–733, doi: 10.1016/j.devcel.2014.11.012 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Kortlever RM & Bernards R Senescence, wound healing and cancer: the PAI-1 connection. Cell Cycle 5, 2697–2703 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Ritschka B et al. The senescence-associated secretory phenotype induces cellular plasticity and tissue regeneration. Genes Dev 31, 172–183 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Sarig R et al. Transient p53-mediated regenerative senescence in the injured heart. Circulation 139, 2491–2494 (2019). [DOI] [PubMed] [Google Scholar]
- 35.Adams PD Healing and hurting: molecular mechanisms, functions and pathologies of cellular senescence. Molec Cell 36, 2–14 (2009). [DOI] [PubMed] [Google Scholar]
- 36.Baker DJ & Sedivy JM Probing the depths of cellular senescence. J Cell Biol 202, 11–13 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Childs BG et al. Senescent cells: an emerging target for diseases of ageing. Nature Rev Drug Disc 16, 718–735 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Tominaga K The emerging role of senescent cells in tissue homeostasis and pathophysiology. Pathobiol Aging & Age-related Dis 5, 27743 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Wiley CD & Campisi J From Ancient Pathways to Aging Cells-Connecting Metabolism and Cellular Senescence. Cell Metab 23, 1013–1021, doi: 10.1016/j.cmet.2016.05.010 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Trifunovic A et al. Premature ageing in mice expressing defective mitochondrial DNA polymerase. Nature 429, 417–423, doi: 10.1038/nature02517 (2004). [DOI] [PubMed] [Google Scholar]
- 41.Kujoth GC et al. Mitochondrial DNA mutations, oxidative stress, and apoptosis in mammalian aging. Science 309, 481–484, doi: 10.1126/science.1112125 (2005). [DOI] [PubMed] [Google Scholar]
- 42.Schriner SE et al. Extension of murine life span by overexpression of catalase targeted to mitochondria. Science 308, 1909–1911, doi: 10.1126/science.1106653 (2005). [DOI] [PubMed] [Google Scholar]
- 43.Wiley CD et al. Mitochondrial Dysfunction Induces Senescence with a Distinct Secretory Phenotype. Cell Metab 23, 303–314, doi: 10.1016/j.cmet.2015.11.011 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Miwa S et al. Low abundance of the matrix arm of complex I in mitochondria predicts longevity in mice. Nat Commun 5, 3837, doi: 10.1038/ncomms4837 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Velarde MC, Flynn JM, Day NU, Melov S & Campisi J Mitochondrial oxidative stress caused by Sod2 deficiency promotes cellular senescence and aging phenotypes in the skin. Aging (Albany NY) 4, 3–12, doi: 10.18632/aging.100423 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Correia-Melo C et al. Mitochondria are required for pro-ageing features of the senescent phenotype. EMBO J 35, 724–742, doi: 10.15252/embj.201592862 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Vizioli MG et al. Mitochondria-to-nucleus retrograde signaling drives formation of cytoplasmic chromatin and inflammation in senescence. Genes Dev 34, 428–445, doi: 10.1101/gad.331272.119 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Baker DJ et al. Naturally occurring p16(Ink4a)-positive cells shorten healthy lifespan. Nature 530, 184–189, doi: 10.1038/nature16932 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Xu M et al. Senolytics improve physical function and increase lifespan in old age. Nat Med 24, 1246–1256, doi: 10.1038/s41591-018-0092-9 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Perez VI et al. The overexpression of major antioxidant enzymes does not extend the lifespan of mice. Aging Cell 8, 73–75, doi: 10.1111/j.1474-9726.2008.00449.x (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Diebold L & Chandel NS Mitochondrial ROS regulation of proliferating cells. Free Radic Biol Med 100, 86–93, doi: 10.1016/j.freeradbiomed.2016.04.198 (2016). [DOI] [PubMed] [Google Scholar]
- 52.West AP, Shadel GS & Ghosh S Mitochondria in innate immune responses. Nat Rev Immunol 11, 389–402, doi: 10.1038/nri2975 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Morimoto H et al. ROS are required for mouse spermatogonial stem cell self-renewal. Cell Stem Cell 12, 774–786, doi: 10.1016/j.stem.2013.04.001 (2013). [DOI] [PubMed] [Google Scholar]
- 54.Juntilla MM et al. AKT1 and AKT2 maintain hematopoietic stem cell function by regulating reactive oxygen species. Blood 115, 4030–4038, doi: 10.1182/blood-2009-09-241000 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Tormos KV et al. Mitochondrial complex III ROS regulate adipocyte differentiation. Cell Metab 14, 537–544, doi: 10.1016/j.cmet.2011.08.007 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Maryanovich M & Gross A A ROS rheostat for cell fate regulation. Trends Cell Biol 23, 129–134, doi: 10.1016/j.tcb.2012.09.007 (2013). [DOI] [PubMed] [Google Scholar]
- 57.Ziegler DV et al. Calcium channel ITPR2 and mitochondria-ER contacts promote cellular senescence and aging. Nat Commun 12, 720, doi: 10.1038/s41467-021-20993-z (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Vasileiou PVS et al. Mitochondrial Homeostasis and Cellular Senescence. Cells 8, doi: 10.3390/cells8070686 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Chapman J, Fielder E & Passos JF Mitochondrial dysfunction and cell senescence: deciphering a complex relationship. FEBS Lett 593, 1566–1579, doi: 10.1002/1873-3468.13498 (2019). [DOI] [PubMed] [Google Scholar]
- 60.Korolchuk VI, Miwa S, Carroll B & von Zglinicki T Mitochondria in Cell Senescence: Is Mitophagy the Weakest Link? EBioMedicine 21, 7–13, doi: 10.1016/j.ebiom.2017.03.020 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Ziegler DV, Wiley CD & Velarde MC Mitochondrial effectors of cellular senescence: beyond the free radical theory of aging. Aging Cell 14, 1–7, doi: 10.1111/acel.12287 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Parrinello S et al. Oxygen sensitivity severely limits the replicative lifespan of murine fibroblasts. Nat Cell Biol 5, 741–747, doi: 10.1038/ncb1024 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Nakazawa MS, Keith B & Simon MC Oxygen availability and metabolic adaptations. Nat Rev Cancer 16, 663–673, doi: 10.1038/nrc.2016.84 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Casciaro F et al. Prolonged hypoxia delays aging and preserves functionality of human amniotic fluid stem cells. Mech Ageing Dev 191, 111328, doi: 10.1016/j.mad.2020.111328 (2020). [DOI] [PubMed] [Google Scholar]
- 65.Leontieva OV et al. Hypoxia suppresses conversion from proliferative arrest to cellular senescence. Proc Natl Acad Sci U S A 109, 13314–13318, doi: 10.1073/pnas.1205690109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Wiley CD et al. Small-molecule MDM2 antagonists attenuate the senescence-associated secretory phenotype. Sci Rep 8, 2410, doi: 10.1038/s41598-018-20000-4 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Coppe JP et al. A human-like senescence-associated secretory phenotype is conserved in mouse cells dependent on physiological oxygen. PLoS One 5, e9188, doi: 10.1371/journal.pone.0009188 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.van Vliet T et al. Physiological hypoxia restrains the senescence-associated secretory phenotype via AMPK-mediated mTOR suppression. Mol Cell 81, 2041–2052 e2046, doi: 10.1016/j.molcel.2021.03.018 (2021). [DOI] [PubMed] [Google Scholar]
- 69.Ast T & Mootha VK Oxygen and mammalian cell culture: are we repeating the experiment of Dr. Ox? Nat Metab 1, 858–860, doi: 10.1038/s42255-019-0105-0 (2019). [DOI] [PubMed] [Google Scholar]
- 70.Baik AH & Jain IH Turning the Oxygen Dial: Balancing the Highs and Lows. Trends Cell Biol 30, 516–536, doi: 10.1016/j.tcb.2020.04.005 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.van Vliet T, Casciaro F & Demaria M To breathe or not to breathe: Understanding how oxygen sensing contributes to age-related phenotypes. Ageing Res Rev 67, 101267, doi: 10.1016/j.arr.2021.101267 (2021). [DOI] [PubMed] [Google Scholar]
- 72.Parikh P et al. Hyperoxia-induced Cellular Senescence in Fetal Airway Smooth Muscle Cells. Am J Respir Cell Mol Biol 61, 51–60, doi: 10.1165/rcmb.2018-0176OC (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Daher A et al. Clinical course of COVID-19 patients needing supplemental oxygen outside the intensive care unit. Sci Rep 11, 2256, doi: 10.1038/s41598-021-81444-9 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.You K et al. Moderate hyperoxia induces senescence in developing human lung fibroblasts. Am J Physiol Lung Cell Mol Physiol 317, L525–L536, doi: 10.1152/ajplung.00067.2019 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hachmo Y et al. Hyperbaric oxygen therapy increases telomere length and decreases immunosenescence in isolated blood cells: a prospective trial. Aging (Albany NY) 12, 22445–22456, doi: 10.18632/aging.202188 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Dookun E et al. Clearance of senescent cells during cardiac ischemia-reperfusion injury improves recovery. Aging Cell 19, e13249, doi: 10.1111/acel.13249 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fleury H et al. Exploiting interconnected synthetic lethal interactions between PARP inhibition and cancer cell reversible senescence. Nat Commun 10, 2556, doi: 10.1038/s41467-019-10460-1 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Ohanna M et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev 25, 1245–1261, doi: 10.1101/gad.625811 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Nassour J et al. Defective DNA single-strand break repair is responsible for senescence and neoplastic escape of epithelial cells. Nat Commun 7, 10399, doi: 10.1038/ncomms10399 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Nacarelli T et al. NAD(+) metabolism governs the proinflammatory senescence-associated secretome. Nat Cell Biol 21, 397–407, doi: 10.1038/s41556-019-0287-4 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Gomes AP et al. Declining NAD(+) induces a pseudohypoxic state disrupting nuclear-mitochondrial communication during aging. Cell 155, 1624–1638, doi: 10.1016/j.cell.2013.11.037 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Yoshino J, Mills KF, Yoon MJ & Imai S Nicotinamide mononucleotide, a key NAD(+) intermediate, treats the pathophysiology of diet- and age-induced diabetes in mice. Cell Metab 14, 528–536, doi: 10.1016/j.cmet.2011.08.014 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Zhang H et al. NAD(+) repletion improves mitochondrial and stem cell function and enhances life span in mice. Science 352, 1436–1443, doi: 10.1126/science.aaf2693 (2016). [DOI] [PubMed] [Google Scholar]
- 84.Baker DJ et al. Increased expression of BubR1 protects against aneuploidy and cancer and extends healthy lifespan. Nature Cell Biol 15, 96–102 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Andriani GA et al. Whole chromosome instability induces senescence and promotes the SASP. Science Rep 6, 35218 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.North BJ et al. SIRT2 induces the checkpoint kinase BubR1 to increase lifespan. EMBO J 33, 1438–1453, doi: 10.15252/embj.201386907 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Simon M et al. LINE1 Derepression in Aged Wild-Type and SIRT6-Deficient Mice Drives Inflammation. Cell Metab 29, 871–885 e875, doi: 10.1016/j.cmet.2019.02.014 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.De Cecco M et al. L1 drives IFN in senescent cells and promotes age-associated inflammation. Nature 566, 73–78, doi: 10.1038/s41586-018-0784-9 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Mao Z et al. Sirtuin 6 (SIRT6) rescues the decline of homologous recombination repair during replicative senescence. Proc Natl Acad Sci U S A 109, 11800–11805, doi: 10.1073/pnas.1200583109 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Xu C et al. SIRT1 is downregulated by autophagy in senescence and ageing. Nat Cell Biol 22, 1170–1179, doi: 10.1038/s41556-020-00579-5 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Hayakawa T et al. SIRT1 suppresses the senescence-associated secretory phenotype through epigenetic gene regulation. PLoS One 10, e0116480, doi: 10.1371/journal.pone.0116480 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ota H et al. Sirt1 inhibitor, Sirtinol, induces senescence-like growth arrest with attenuated Ras-MAPK signaling in human cancer cells. Oncogene 25, 176–185, doi: 10.1038/sj.onc.1209049 (2006). [DOI] [PubMed] [Google Scholar]
- 93.Bonkowski MS & Sinclair DA Slowing ageing by design: the rise of NAD(+) and sirtuin-activating compounds. Nat Rev Mol Cell Biol 17, 679–690, doi: 10.1038/nrm.2016.93 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Covarrubias AJ, Perrone R, Grozio A & Verdin E NAD(+) metabolism and its roles in cellular processes during ageing. Nat Rev Mol Cell Biol 22, 119–141, doi: 10.1038/s41580-020-00313-x (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Covarrubias AJ et al. Senescent cells promote tissue NAD(+) decline during ageing via the activation of CD38(+) macrophages. Nat Metab 2, 1265–1283, doi: 10.1038/s42255-020-00305-3 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Chini CCS et al. CD38 ecto-enzyme in immune cells is induced during aging and regulates NAD(+) and NMN levels. Nat Metab 2, 1284–1304, doi: 10.1038/s42255-020-00298-z (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Camacho-Pereira J et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab 23, 1127–1139, doi: 10.1016/j.cmet.2016.05.006 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lee SM et al. Cytosolic malate dehydrogenase regulates senescence in human fibroblasts. Biogerontology 13, 525–536, doi: 10.1007/s10522-012-9397-0 (2012). [DOI] [PubMed] [Google Scholar]
- 99.Jiang P, Du W, Mancuso A, Wellen KE & Yang X Reciprocal regulation of p53 and malic enzymes modulates metabolism and senescence. Nature 493, 689–693, doi: 10.1038/nature11776 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Blazer S et al. High glucose-induced replicative senescence: point of no return and effect of telomerase. Biochem Biophys Res Commun 296, 93–101, doi: 10.1016/s0006-291x(02)00818-5 (2002). [DOI] [PubMed] [Google Scholar]
- 101.Patel C et al. Arginase as a mediator of diabetic retinopathy. Front Immunol 4, 173, doi: 10.3389/fimmu.2013.00173 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Kitada K et al. Hyperglycemia causes cellular senescence via a SGLT2- and p21-dependent pathway in proximal tubules in the early stage of diabetic nephropathy. J Diabetes Complications 28, 604–611, doi: 10.1016/j.jdiacomp.2014.05.010 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mortuza R, Chen S, Feng B, Sen S & Chakrabarti S High glucose induced alteration of SIRTs in endothelial cells causes rapid aging in a p300 and FOXO regulated pathway. PLoS One 8, e54514, doi: 10.1371/journal.pone.0054514 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zhang B et al. SIRT3 overexpression antagonizes high glucose accelerated cellular senescence in human diploid fibroblasts via the SIRT3-FOXO1 signaling pathway. Age (Dordr) 35, 2237–2253, doi: 10.1007/s11357-013-9520-4 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Shosha E et al. Mechanisms of Diabetes-Induced Endothelial Cell Senescence: Role of Arginase 1. Int J Mol Sci 19, doi: 10.3390/ijms19041215 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Hayashi T et al. Endothelial cellular senescence is inhibited by nitric oxide: implications in atherosclerosis associated with menopause and diabetes. Proc Natl Acad Sci U S A 103, 17018–17023, doi: 10.1073/pnas.0607873103 (2006). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Jin J & Zhang T Effects of glucose restriction on replicative senescence of human diploid fibroblasts IMR-90. Cell Physiol Biochem 31, 718–727, doi: 10.1159/000350090 (2013). [DOI] [PubMed] [Google Scholar]
- 108.Sohn JJ et al. Macrophages, nitric oxide and microRNAs are associated with DNA damage response pathway and senescence in inflammatory bowel disease. PLoS One 7, e44156, doi: 10.1371/journal.pone.0044156 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Zhang Y et al. The pivotal role of protein acetylation in linking glucose and fatty acid metabolism to beta-cell function. Cell Death Dis 10, 66, doi: 10.1038/s41419-019-1349-z (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Meyer JG et al. Temporal dynamics of liver mitochondrial protein acetylation and succinylation and metabolites due to high fat diet and/or excess glucose or fructose. PLoS One 13, e0208973, doi: 10.1371/journal.pone.0208973 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Wu J, Jin Z, Zheng H & Yan LJ Sources and implications of NADH/NAD(+) redox imbalance in diabetes and its complications. Diabetes Metab Syndr Obes 9, 145–153, doi: 10.2147/DMSO.S106087 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Liu J et al. Receptor for advanced glycation end-products promotes premature senescence of proximal tubular epithelial cells via activation of endoplasmic reticulum stress-dependent p21 signaling. Cell Signal 26, 110–121, doi: 10.1016/j.cellsig.2013.10.002 (2014). [DOI] [PubMed] [Google Scholar]
- 113.Liu J et al. Impact of ER stress-regulated ATF4/p16 signaling on the premature senescence of renal tubular epithelial cells in diabetic nephropathy. Am J Physiol Cell Physiol 308, C621–630, doi: 10.1152/ajpcell.00096.2014 (2015). [DOI] [PubMed] [Google Scholar]
- 114.Chen CY, Abell AM, Moon YS & Kim KH An advanced glycation end product (AGE)-receptor for AGEs (RAGE) axis restores adipogenic potential of senescent preadipocytes through modulation of p53 protein function. J Biol Chem 287, 44498–44507, doi: 10.1074/jbc.M112.399790 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Venable ME, Lee JY, Smyth MJ, Bielawska A & Obeid LM Role of ceramide in cellular senescence. J Biol Chem 270, 30701–30708, doi: 10.1074/jbc.270.51.30701 (1995). [DOI] [PubMed] [Google Scholar]
- 116.Venable ME & Yin X Ceramide induces endothelial cell senescence. Cell Biochem Funct 27, 547–551, doi: 10.1002/cbf.1605 (2009). [DOI] [PubMed] [Google Scholar]
- 117.Lee JY, Bielawska AE & Obeid LM Regulation of cyclin-dependent kinase 2 activity by ceramide. Exp Cell Res 261, 303–311, doi: 10.1006/excr.2000.5028 (2000). [DOI] [PubMed] [Google Scholar]
- 118.Chao R, Khan W & Hannun YA Retinoblastoma protein dephosphorylation induced by D-erythro-sphingosine. J Biol Chem 267, 23459–23462 (1992). [PubMed] [Google Scholar]
- 119.Trayssac M, Hannun YA & Obeid LM Role of sphingolipids in senescence: implication in aging and age-related diseases. J Clin Invest 128, 2702–2712, doi: 10.1172/JCI97949 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Quijano C et al. Oncogene-induced senescence results in marked metabolic and bioenergetic alterations. Cell Cycle 11, 1383–1392, doi: 10.4161/cc.19800 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Fafian-Labora J et al. FASN activity is important for the initial stages of the induction of senescence. Cell Death Dis 10, 318, doi: 10.1038/s41419-019-1550-0 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Flor AC, Wolfgeher D, Wu D & Kron SJ A signature of enhanced lipid metabolism, lipid peroxidation and aldehyde stress in therapy-induced senescence. Cell Death Discov 3, 17075, doi: 10.1038/cddiscovery.2017.75 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Maeda M, Scaglia N & Igal RA Regulation of fatty acid synthesis and Delta9-desaturation in senescence of human fibroblasts. Life Sci 84, 119–124, doi: 10.1016/j.lfs.2008.11.009 (2009). [DOI] [PubMed] [Google Scholar]
- 124.Wiley CD et al. Oxylipin biosynthesis reinforces cellular senescence and allows detection of senolysis. Cell Metab Epub Ahead of Print, doi: 10.1016/j.cmet.2021.03.008 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Wiley CD et al. Secretion of leukotrienes by senescent lung fibroblasts promotes pulmonary fibrosis. JCI Insight 4, doi: 10.1172/jci.insight.130056 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Lizardo DY, Lin YL, Gokcumen O & Atilla-Gokcumen GE Regulation of lipids is central to replicative senescence. Mol Biosyst 13, 498–509, doi: 10.1039/c6mb00842a (2017). [DOI] [PubMed] [Google Scholar]
- 127.Ogrodnik M et al. Obesity-Induced Cellular Senescence Drives Anxiety and Impairs Neurogenesis. Cell Metab 29, 1233, doi: 10.1016/j.cmet.2019.01.013 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kim W et al. Polyunsaturated Fatty Acid Desaturation Is a Mechanism for Glycolytic NAD(+) Recycling. Cell Metab 29, 856–870 e857, doi: 10.1016/j.cmet.2018.12.023 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Catalano A, Rodilossi S, Caprari P, Coppola V & Procopio A 5-Lipoxygenase regulates senescence-like growth arrest by promoting ROS-dependent p53 activation. EMBO J 24, 170–179, doi: 10.1038/sj.emboj.7600502 (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Martien S et al. Cellular senescence involves an intracrine prostaglandin E2 pathway in human fibroblasts. Biochim Biophys Acta 1831, 1217–1227, doi: 10.1016/j.bbalip.2013.04.005 (2013). [DOI] [PubMed] [Google Scholar]
- 131.Schafer MJ et al. Cellular senescence mediates fibrotic pulmonary disease. Nat Commun 8, 14532, doi: 10.1038/ncomms14532 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Han JH et al. Selective COX-2 inhibitor, NS-398, inhibits the replicative senescence of cultured dermal fibroblasts. Mech Ageing Dev 125, 359–366, doi: 10.1016/j.mad.2004.02.002 (2004). [DOI] [PubMed] [Google Scholar]
- 133.Shibata T et al. 15-deoxy-delta 12,14-prostaglandin J2. A prostaglandin D2 metabolite generated during inflammatory processes. J Biol Chem 277, 10459–10466, doi: 10.1074/jbc.M110314200 (2002). [DOI] [PubMed] [Google Scholar]
- 134.Kang C & Elledge SJ How autophagy both activates and inhibits cellular senescence. Autophagy 12, 898–899, doi: 10.1080/15548627.2015.1121361 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Kwon Y, Kim JW, Jeoung JA, Kim MS & Kang C Autophagy Is Pro-Senescence When Seen in Close-Up, but Anti-Senescence in Long-Shot. Mol Cells 40, 607–612, doi: 10.14348/molcells.2017.0151 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Kang HT, Lee KB, Kim SY, Choi HR & Park SC Autophagy impairment induces premature senescence in primary human fibroblasts. PLoS One 6, e23367, doi: 10.1371/journal.pone.0023367 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Young AR et al. Autophagy mediates the mitotic senescence transition. Genes Dev 23, 798–803, doi: 10.1101/gad.519709 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rajendran P et al. Autophagy and senescence: A new insight in selected human diseases. J Cell Physiol 234, 21485–21492, doi: 10.1002/jcp.28895 (2019). [DOI] [PubMed] [Google Scholar]
- 139.Kurz DJ, Decary S, Hong Y & Erusalimsky JD Senescence-associated (beta)-galactosidase reflects an increase in lysosomal mass during replicative ageing of human endothelial cells. J Cell Sci 113 ( Pt 20), 3613–3622 (2000). [DOI] [PubMed] [Google Scholar]
- 140.Lee BY et al. Senescence-associated beta-galactosidase is lysosomal beta-galactosidase. Aging Cell 5, 187–195, doi: 10.1111/j.1474-9726.2006.00199.x (2006). [DOI] [PubMed] [Google Scholar]
- 141.Han L et al. Senescent Stromal Cells Promote Cancer Resistance through SIRT1 Loss-Potentiated Overproduction of Small Extracellular Vesicles. Cancer Res 80, 3383–3398, doi: 10.1158/0008-5472.CAN-20-0506 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar] [Retracted]
- 142.Patschan S et al. Mapping mechanisms and charting the time course of premature cell senescence and apoptosis: lysosomal dysfunction and ganglioside accumulation in endothelial cells. Am J Physiol Renal Physiol 294, F100–109, doi: 10.1152/ajprenal.00261.2007 (2008). [DOI] [PubMed] [Google Scholar]
- 143.Johmura Y et al. Senolysis by glutaminolysis inhibition ameliorates various age-associated disorders. Science 371, 265–270, doi: 10.1126/science.abb5916 (2021). [DOI] [PubMed] [Google Scholar]
- 144.Killilea DW, Atamna H, Liao C & Ames BN Iron accumulation during cellular senescence in human fibroblasts in vitro. Antioxid Redox Signal 5, 507–516, doi: 10.1089/152308603770310158 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Masaldan S et al. Copper accumulation in senescent cells: Interplay between copper transporters and impaired autophagy. Redox Biol 16, 322–331, doi: 10.1016/j.redox.2018.03.007 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Winter WE, Bazydlo LA & Harris NS The molecular biology of human iron metabolism. Lab Med 45, 92–102, doi: 10.1309/lmf28s2gimxnwhmm (2014). [DOI] [PubMed] [Google Scholar]
- 147.Dixon SJ et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell 149, 1060–1072, doi: 10.1016/j.cell.2012.03.042 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Masaldan S et al. Iron accumulation in senescent cells is coupled with impaired ferritinophagy and inhibition of ferroptosis. Redox Biol 14, 100–115, doi: 10.1016/j.redox.2017.08.015 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Festa RA & Thiele DJ Copper: an essential metal in biology. Curr Biol 21, R877–883, doi: 10.1016/j.cub.2011.09.040 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Matos L, Gouveia A & Almeida H Copper ability to induce premature senescence in human fibroblasts. Age (Dordr) 34, 783–794, doi: 10.1007/s11357-011-9276-7 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Li Y et al. Copper induces cellular senescence in human glioblastoma multiforme cells through downregulation of Bmi-1. Oncol Rep 29, 1805–1810, doi: 10.3892/or.2013.2333 (2013). [DOI] [PubMed] [Google Scholar]
- 152.Zhang Y et al. A new role for oxidative stress in aging: The accelerated aging phenotype in Sod1(-/)(−) mice is correlated to increased cellular senescence. Redox Biol 11, 30–37, doi: 10.1016/j.redox.2016.10.014 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 153.Oteiza PI Zinc and the modulation of redox homeostasis. Free Radic Biol Med 53, 1748–1759, doi: 10.1016/j.freeradbiomed.2012.08.568 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 154.Kumar J et al. Zinc Levels Modulate Lifespan through Multiple Longevity Pathways in Caenorhabditis elegans. PLoS One 11, e0153513, doi: 10.1371/journal.pone.0153513 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 155.Salazar G, Huang J, Feresin RG, Zhao Y & Griendling KK Zinc regulates Nox1 expression through a NF-kappaB and mitochondrial ROS dependent mechanism to induce senescence of vascular smooth muscle cells. Free Radic Biol Med 108, 225–235, doi: 10.1016/j.freeradbiomed.2017.03.032 (2017). [DOI] [PubMed] [Google Scholar]
- 156.Malavolta M et al. Changes in Zn homeostasis during long term culture of primary endothelial cells and effects of Zn on endothelial cell senescence. Exp Gerontol 99, 35–45, doi: 10.1016/j.exger.2017.09.006 (2017). [DOI] [PubMed] [Google Scholar]
- 157.Rudolf E & Cervinka M Stress responses of human dermal fibroblasts exposed to zinc pyrithione. Toxicol Lett 204, 164–173, doi: 10.1016/j.toxlet.2011.04.028 (2011). [DOI] [PubMed] [Google Scholar]
- 158.Aird KM et al. Suppression of nucleotide metabolism underlies the establishment and maintenance of oncogene-induced senescence. Cell Rep 3, 1252–1265, doi: 10.1016/j.celrep.2013.03.004 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Aird KM et al. ATM couples replication stress and metabolic reprogramming during cellular senescence. Cell Rep 11, 893–901, doi: 10.1016/j.celrep.2015.04.014 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Buj R et al. Suppression of p16 Induces mTORC1-Mediated Nucleotide Metabolic Reprogramming. Cell Rep 28, 1971–1980 e1978, doi: 10.1016/j.celrep.2019.07.084 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 161.Santinon G et al. dNTP metabolism links mechanical cues and YAP/TAZ to cell growth and oncogene-induced senescence. EMBO J 37, doi: 10.15252/embj.201797780 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Baar MP et al. Targeted Apoptosis of Senescent Cells Restores Tissue Homeostasis in Response to Chemotoxicity and Aging. Cell 169, 132–147 e116, doi: 10.1016/j.cell.2017.02.031 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Farr JN et al. Targeting cellular senescence prevents age-related bone loss in mice. Nat Med 23, 1072–1079, doi: 10.1038/nm.4385 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 164.Jeon OH et al. Local clearance of senescent cells attenuates the development of post-traumatic osteoarthritis and creates a pro-regenerative environment. Nat Med 23, 775–781, doi: 10.1038/nm.4324 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Alimirah F et al. Cellular Senescence Promotes Skin Carcinogenesis through p38MAPK and p44/42MAPK Signaling. Cancer Res 80, 3606–3619, doi: 10.1158/0008-5472.CAN-20-0108 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Demaria M et al. Cellular Senescence Promotes Adverse Effects of Chemotherapy and Cancer Relapse. Cancer Discov 7, 165–176, doi: 10.1158/2159-8290.CD-16-0241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Wiley CD et al. SILAC Analysis Reveals Increased Secretion of Hemostasis-Related Factors by Senescent Cells. Cell Rep 28, 3329–3337 e3325, doi: 10.1016/j.celrep.2019.08.049 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Thompson PJ et al. Targeted Elimination of Senescent Beta Cells Prevents Type 1 Diabetes. Cell Metab 29, 1045–1060 e1010, doi: 10.1016/j.cmet.2019.01.021 (2019). [DOI] [PubMed] [Google Scholar]
- 169.Palmer AK et al. Targeting senescent cells alleviates obesity-induced metabolic dysfunction. Aging Cell 18, e12950, doi: 10.1111/acel.12950 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Helman A et al. p16(Ink4a)-induced senescence of pancreatic beta cells enhances insulin secretion. Nat Med 22, 412–420, doi: 10.1038/nm.4054 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 171.Aguayo-Mazzucato C et al. Acceleration of beta Cell Aging Determines Diabetes and Senolysis Improves Disease Outcomes. Cell Metab 30, 129–142 e124, doi: 10.1016/j.cmet.2019.05.006 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Kim SR et al. Increased renal cellular senescence in murine high-fat diet: effect of the senolytic drug quercetin. Transl Res 213, 112–123, doi: 10.1016/j.trsl.2019.07.005 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 173.Verzola D et al. Accelerated senescence in the kidneys of patients with type 2 diabetic nephropathy. Am J Physiol Renal Physiol 295, F1563–1573, doi: 10.1152/ajprenal.90302.2008 (2008). [DOI] [PubMed] [Google Scholar]
- 174.Crespo-Garcia S et al. Pathological angiogenesis in retinopathy engages cellular senescence and is amenable to therapeutic elimination via BCL-xL inhibition. Cell Metab, doi: 10.1016/j.cmet.2021.01.011 (2021). [DOI] [PubMed] [Google Scholar]
- 175.Heilbronn LK & Ravussin E Calorie restriction and aging: review of the literature and implications for studies in humans. Am J Clin Nutr 78, 361–369, doi: 10.1093/ajcn/78.3.361 (2003). [DOI] [PubMed] [Google Scholar]
- 176.Fontana L et al. The effects of graded caloric restriction: XII. Comparison of mouse to human impact on cellular senescence in the colon. Aging Cell 17, e12746, doi: 10.1111/acel.12746 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 177.Mau T, O’Brien M, Ghosh AK, Miller RA & Yung R Life-span Extension Drug Interventions Affect Adipose Tissue Inflammation in Aging. J Gerontol A Biol Sci Med Sci 75, 89–98, doi: 10.1093/gerona/glz177 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 178.Cui J et al. Mitochondrial autophagy involving renal injury and aging is modulated by caloric intake in aged rat kidneys. PLoS One 8, e69720, doi: 10.1371/journal.pone.0069720 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 179.Miller RA et al. Methionine-deficient diet extends mouse lifespan, slows immune and lens aging, alters glucose, T4, IGF-I and insulin levels, and increases hepatocyte MIF levels and stress resistance. Aging Cell 4, 119–125, doi: 10.1111/j.1474-9726.2005.00152.x (2005). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 180.Newman JC et al. Ketogenic Diet Reduces Midlife Mortality and Improves Memory in Aging Mice. Cell Metab 26, 547–557 e548, doi: 10.1016/j.cmet.2017.08.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 181.Roberts MN et al. A Ketogenic Diet Extends Longevity and Healthspan in Adult Mice. Cell Metab 26, 539–546 e535, doi: 10.1016/j.cmet.2017.08.005 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Wang SY et al. Methionine restriction delays senescence and suppresses the senescence-associated secretory phenotype in the kidney through endogenous hydrogen sulfide. Cell Cycle 18, 1573–1587, doi: 10.1080/15384101.2019.1618124 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 183.Han YM et al. beta-Hydroxybutyrate Prevents Vascular Senescence through hnRNP A1-Mediated Upregulation of Oct4. Mol Cell 71, 1064–1078 e1065, doi: 10.1016/j.molcel.2018.07.036 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 184.Harrison DE et al. Acarbose improves health and lifespan in aging HET3 mice. Aging Cell 18, e12898, doi: 10.1111/acel.12898 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 185.Chan KC et al. Pleiotropic effects of acarbose on atherosclerosis development in rabbits are mediated via upregulating AMPK signals. Sci Rep 6, 38642, doi: 10.1038/srep38642 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 186.Martin-Montalvo A et al. Metformin improves healthspan and lifespan in mice. Nat Commun 4, 2192, doi: 10.1038/ncomms3192 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 187.Jadhav KS, Dungan CM & Williamson DL Metformin limits ceramide-induced senescence in C2C12 myoblasts. Mech Ageing Dev 134, 548–559, doi: 10.1016/j.mad.2013.11.002 (2013). [DOI] [PubMed] [Google Scholar]
- 188.Fang J et al. Metformin alleviates human cellular aging by upregulating the endoplasmic reticulum glutathione peroxidase 7. Aging Cell 17, e12765, doi: 10.1111/acel.12765 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 189.Moiseeva O et al. Metformin inhibits the senescence-associated secretory phenotype by interfering with IKK/NF-kappaB activation. Aging Cell 12, 489–498, doi: 10.1111/acel.12075 (2013). [DOI] [PubMed] [Google Scholar]
- 190.Chen D et al. Metformin protects against apoptosis and senescence in nucleus pulposus cells and ameliorates disc degeneration in vivo. Cell Death Dis 7, e2441, doi: 10.1038/cddis.2016.334 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 191.Kim H et al. Metformin inhibits chronic kidney disease-induced DNA damage and senescence of mesenchymal stem cells. Aging Cell 20, e13317, doi: 10.1111/acel.13317 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 192.Simons J The $10 billion pill. Fortune 147, 58–62, 66, 68 (2003). [PubMed] [Google Scholar]
- 193.Assmus B et al. HMG-CoA reductase inhibitors reduce senescence and increase proliferation of endothelial progenitor cells via regulation of cell cycle regulatory genes. Circ Res 92, 1049–1055, doi: 10.1161/01.RES.0000070067.64040.7C (2003). [DOI] [PubMed] [Google Scholar]
- 194.Liu S et al. Simvastatin suppresses breast cancer cell proliferation induced by senescent cells. Sci Rep 5, 17895, doi: 10.1038/srep17895 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 195.Childs BG et al. Senescent intimal foam cells are deleterious at all stages of atherosclerosis. Science 354, 472–477, doi: 10.1126/science.aaf6659 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 196.Efimova EV et al. HMG-CoA Reductase Inhibition Delays DNA Repair and Promotes Senescence After Tumor Irradiation. Mol Cancer Ther 17, 407–418, doi: 10.1158/1535-7163.MCT-17-0288 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 197.Moon SH et al. p53 Represses the Mevalonate Pathway to Mediate Tumor Suppression. Cell 176, 564–580 e519, doi: 10.1016/j.cell.2018.11.011 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Garcia-Valles R et al. Life-long spontaneous exercise does not prolong lifespan but improves health span in mice. Longev Healthspan 2, 14, doi: 10.1186/2046-2395-2-14 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Nilsson MI et al. Lifelong aerobic exercise protects against inflammaging and cancer. PLoS One 14, e0210863, doi: 10.1371/journal.pone.0210863 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 200.Schafer MJ et al. Late-life time-restricted feeding and exercise differentially alter healthspan in obesity. Aging Cell 18, e12966, doi: 10.1111/acel.12966 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 201.Schafer MJ et al. Exercise Prevents Diet-Induced Cellular Senescence in Adipose Tissue. Diabetes 65, 1606–1615, doi: 10.2337/db15-0291 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 202.Rossman MJ et al. Endothelial cell senescence with aging in healthy humans: prevention by habitual exercise and relation to vascular endothelial function. Am J Physiol Heart Circ Physiol 313, H890–H895, doi: 10.1152/ajpheart.00416.2017 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 203.Werner C et al. Physical exercise prevents cellular senescence in circulating leukocytes and in the vessel wall. Circulation 120, 2438–2447, doi: 10.1161/CIRCULATIONAHA.109.861005 (2009). [DOI] [PubMed] [Google Scholar]
- 204.Cherasse Y et al. The Leptomeninges Produce Prostaglandin D2 Involved in Sleep Regulation in Mice. Front Cell Neurosci 12, 357, doi: 10.3389/fncel.2018.00357 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Urade Y & Hayaishi O Prostaglandin D2 and sleep/wake regulation. Sleep Med Rev 15, 411–418, doi: 10.1016/j.smrv.2011.08.003 (2011). [DOI] [PubMed] [Google Scholar]
- 206.Mander BA, Winer JR & Walker MP Sleep and Human Aging. Neuron 94, 19–36, doi: 10.1016/j.neuron.2017.02.004 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 207.Roberts SB et al. Healthy Aging-Nutrition Matters: Start Early and Screen Often. Adv Nutr, doi: 10.1093/advances/nmab032 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Ogrodnik M et al. Cellular senescence drives age-dependent hepatic steatosis. Nat Commun 8, 15691, doi: 10.1038/ncomms15691 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]