

SUMMARY
Metabolism of cancer cells is geared towards biomass production and proliferation. Since the metabolic resources within the local tissue are finite, this can lead to nutrient depletion and accumulation of metabolic waste. To maintain growth in these conditions, cancer cells employ a variety of metabolic adaptations, the nature of which is collectively determined by the physiology of their cell-of-origin, the identity of transforming lesions and the tissue in which cancer cells reside. Furthermore, select metabolites not only serve as substrates for energy and biomass generation, but can also regulate gene and protein expression and influence the behavior of non-transformed cells in the tumor vicinity. As they grow and metastasize, tumors can also affect and be affected by the nutrient distribution within the body. In this hallmarks update, recent advances are incorporated into a conceptual framework that may help guide further research efforts in exploring cancer cell metabolism.
eTOC BLURB
Pavlova et al. review the recent discoveries and emerging paradigms in cancer cell metabolism. New hallmarks, including the tumor metabolic diversity, the role of electron acceptors and oxidative stress protection, and the cross-talk between the tumor and whole-body metabolism are added to list of metabolic hallmarks cancer cells can exhibit.
INTRODUCTION
Unicellular organisms make decisions to build biomass and proliferate based on the availability and quality of nutrient sources in their environment. In contrast, growth and proliferation of individual cells within a metazoan organism is regulated in a non-cell-autonomous manner via a combination of tissue-specific soluble growth factors and biophysical cues. Integration of these signaling inputs allows cells to upregulate the import of necessary nutrients, use these nutrients to produce the necessary biomass components required to duplicate themselves into two daughter cells (Chandel, 2021; Lunt and Vander Heiden, 2011). In normal cells, the extent and the duration of these signals is limited by the homeostatic needs of the tissue. In contrast, transformed cells accumulate select genetic and epigenetic alterations that allow them to escape the tissue-based controls over their proliferation by maintaining major pro-survival and pro-proliferative signals in a continuous “on” state. This, in turn, leads to the establishment of a perpetual pro-anabolic state of metabolism in affected cells and enables uncontrollable accumulation of transformed cells and tumor expansion.
In a resting cell, imported nutrients are used primarily to generate energy. This process involves progressive oxidation of nutrient-sourced carbon atoms. The electrons extracted from these oxidative reactions are deposited onto the electron carrier molecules NAD+ and FAD to generate NADH and FADH2, then passed on to the components of the mitochondrial electron transport chain (ETC) and, eventually, to molecular oxygen. The continuous passage of electrons through the ETC allows a cell to maintain adenosine triphosphate (ATP) production, while also regenerating NAD+ and FAD carriers. In contrast, cells that have been instructed to proliferate not only import more nutrients from the surrounding environment, but also refocus their metabolic networks to direct the nutrient surplus into producing building blocks for biomass accumulation.
While pro-proliferative signals themselves can be maintained in an “on” state indefinitely in transformed cells, the demand for nutrients required for the sustained tumor expansion eventually outstrips the nutrient resources that the local tissue is capable of providing. The depletion of nutrients from the extracellular tumor microenvironment is often accompanied by a concurrent rise in the levels of the byproducts of anabolic metabolism. To sustain themselves, transformed cells rely on a diverse set of metabolic adaptations to withstand the limitations in nutrient supply. Thus, some cancer cells resort to the engulfment and catabolism of extracellular macromolecules and even entire cells as the source of the missing nutrients. Furthermore, some types of transformed cells adapt to the depletion of the reduced nitrogen-containing nutrients (which are in high demand in proliferating cells due to the role of nitrogen in building both proteins and nucleic acids) from their environment by scavenging of free ammonium, as well as by remodeling their metabolism to prioritize the use of nitrogen donors for nucleotide and non-essential amino acid synthesis.
In addition to structural building blocks, several key biosynthetic reactions require a source of a reducing power in a form of an electron donor NADPH. Regeneration of NADPH from NADP+ is powered by oxidation of carbon atoms in a set of dedicated reactions embedded in central carbon metabolism. In addition to NADPH, several metabolic reactions critical to tumor cell survival and/or growth are fueled by either oxidative or reductive power provided by NAD+ or NADH cofactors accordingly. In anabolically active cells, NADH/NAD+ redox pair is used not only to power oxidative phosphorylation, but also to support biosynthesis, particularly in glycolysis and the tricarboxylic (TCA) cycle, where NAD+ is required as an electron acceptor to maintain pathway flux. Thus, the balance between the reduced and oxidized forms of these cofactors is critical. Indeed, while an increased influx of oxidizable sources of carbon into cells due to the activation of pro-growth signaling favors the NADH generation, too high of a NADH/NAD+ ratio may interfere with NAD+-requiring reactions. In particular, an oversupply of NADH may overwhelm the NAD+-regenerating capacity of the ETC, especially when oxygen, the ultimate electron acceptor, becomes limiting as tumors increase in size. Thus, transformed cells must rely on robust oxidative stress defense mechanisms to mitigate the damage caused by free electron radicals to cellular structures.
While the import and utilization of nutrients by cells is controlled by signaling pathways, it has become clear that some metabolic intermediates themselves act as potent signaling modulators. Thus, changes in levels of select metabolites can enact sweeping changes to the cellular gene and protein expression landscape by controlling epigenetic changes to the cellular DNA, RNA and histones, and even modulating protein production directly. In addition, it has been demonstrated that as a tumor expands, the altered metabolite composition of its milieu acts as a potent signal that regulates the behavior of diverse non-transformed cell types in its vicinity, which further helps promote tumor expansion. Finally, there is a growing appreciation that tumors engage not only in a metabolic cross-talk with the cells in their immediate environment, but can also influence the metabolic economy of the entire organism.
The connection between tumorigenesis and deregulated metabolism was first described a century ago by the German biochemist Otto Warburg. In the past twenty years, equipped with new and detailed understanding of the genetic and epigenetic mechanisms that underlie cell transformation, as well as with the modern experimental technologies, the field of cancer metabolism has entered a prolific and exciting new era. While five years ago, we suggested there were several emerging concepts (Pavlova and Thompson, 2016), there has been an emergence of new, paradigm-shifting discoveries in cancer cell metabolism. The original conception of the hallmarks of cancer metabolism has been expanded to incorporate these new discoveries as described below.
DEREGULATED UPTAKE OF GLUCOSE AND AMINO ACIDS
Glucose is a principal carbon source consumed by mammalian cells. Catabolism of glucose through glycolysis and the TCA cycle not only fuels ATP generation, but also produces carbon intermediates to support the biosynthesis of macromolecules. In mammalian cells, glucose uptake is regulated non-cell-autonomously through growth factor signaling and positional cues (Thompson, 2011). Thus, normal cells can only acquire enough glucose to support growth and proliferation when stimulated by cell type-specific growth factors such as insulin, platelet-derived growth factor (PDGF), or epidermal growth factor (EGF). Growth factor stimulation triggers the activation of downstream signaling events including the receptor tyrosine kinase (RTK) – phosphoinositide 3-kinase (PI3K) – Akt1 (also known as protein kinase B) cascade. The RTK-PI3K-Akt1 axis promotes the expression of glucose transporter 1 (GLUT1) and its translocation from intracellular membranes to the cell surface to facilitate glucose uptake (Barthel et al., 1999; Wieman et al., 2007). In addition, Akt1 activation increases the activity of hexokinase such that the imported glucose can be phosphorylated and captured for use in glycolysis and downstream metabolic pathways (Rathmell et al., 2003) (Figure 1). In addition to soluble growth factors, signaling inputs from mechanical cues – including those provided by the cellular attachments to the extracellular matrix (ECM) and by the biophysical properties of the matrix – also serve as gatekeepers of glucose uptake and utilization in normal cells (Park et al., 2020; Schafer et al., 2009).
Figure 1. Deregulated uptake of glucose and amino acids.
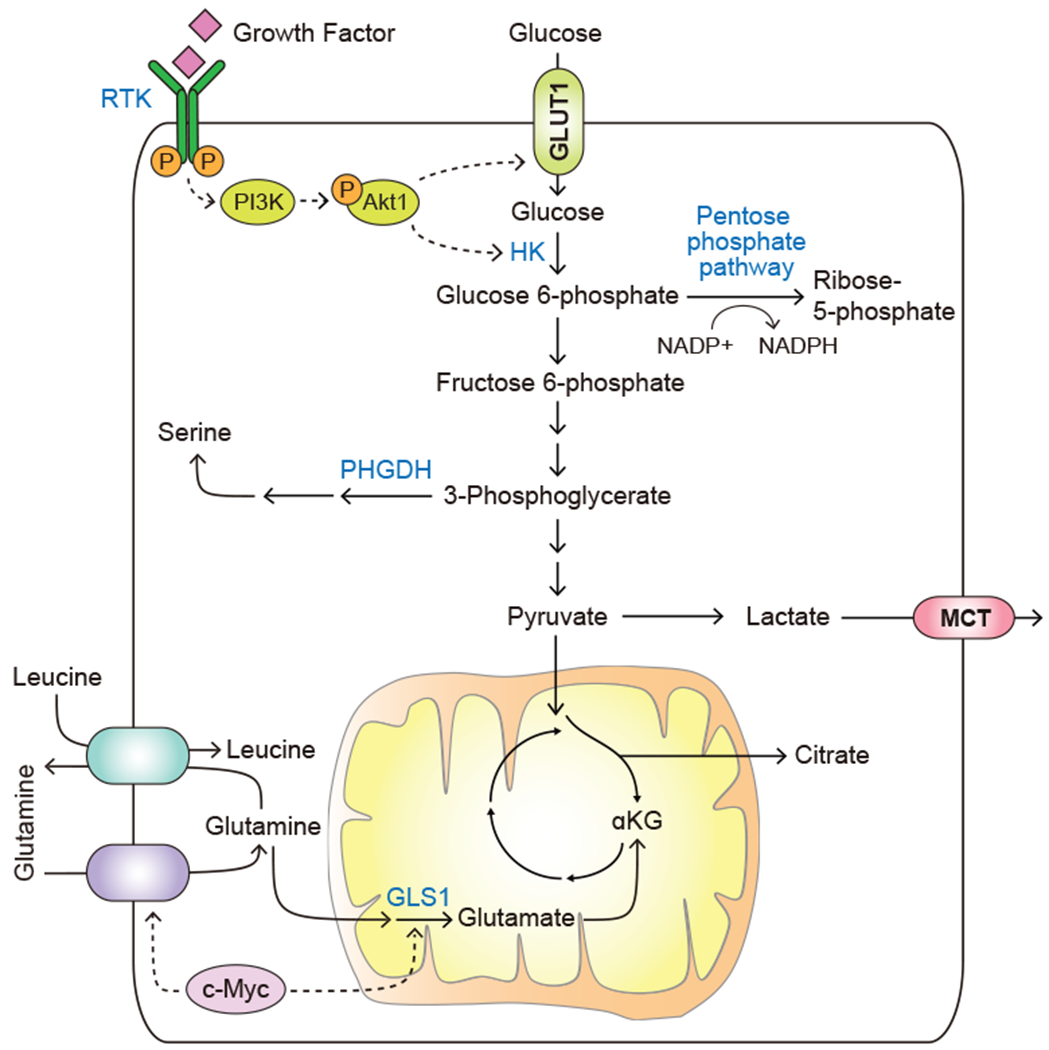
Growth factor stimulation or oncogenic activation triggers the uptake of glucose and amino acids. Solid arrows depict metabolite movement or metabolic reactions. Dashed arrows depict regulatory effects of signal transduction components. RTK, receptor tyrosine kinase; PI3K, phosphoinositide 3-kinase; HK, hexokinase; GLUT1, glucose transporter, also known as SLC2A1; PHGDH, phosphoglycerate dehydrogenase; MCT, monocarboxylate transporter; GLS1, glutaminase, also known as GLS.
As both direct and indirect consequence of oncogenic mutations, cancer cells often display enhanced ability to take up glucose from the extracellular environment. Genomic amplifications of the RTK-encoding genes such as EGF receptor (EGFR), erb-b2 receptor (ERBB2) and c-Met are frequently observed in human cancers (Lawrence et al., 2014). Similarly, oncogenic mutations in PI3K or genetic loss of its negative regulators, PTEN and INPP4B, are often identified as driver events in tumorigenesis (Lawrence et al., 2014). These genetic alterations converge upon the activation of the PI3K-Akt1 signaling cascade in a manner independent of signals provided by the external signaling inputs, allowing cell-autonomous glucose uptake in quantities that allow sustained growth and proliferation.
Adoption of the invasive phenotype by transformed cells elevates the cell’s energy requirements to maintain the actin remodeling required to support cell motility and therefore elevates the needs for glucose uptake and catabolism even further (Zanotelli et al., 2019). In turn, invasion-associated degradation of one of the abundant ECM components, hyaluronic acid, has been recently found to increase cellular glucose uptake and upregulate glycolysis, which helps produce ATP in quantities sufficient to support cancer cell invasion (Sullivan et al., 2018). Furthermore, reduced local cell density in itself can elevate the expression of GLUT1 transporter and increased glucose uptake and utilization (Kondo et al., 2021).
A shift to anabolic metabolism allows the allocation of a large fraction of the carbon surplus into biosynthetic pathways. Thus, the glycolytic intermediate glucose-6-phosphate can be diverted into the pentose phosphate pathway to support the production of ribose used in nucleotide biosynthesis. In this process, glucose skeleton becomes progressively oxidized, allowing cells to regenerate a reduced form of nicotinamide adenine dinucleotide phosphate (NADPH) from its oxidized form (NADP+) to support reductive metabolic reactions in the cytosol, such as those required for fatty acid biosynthesis (Boros et al., 1998; Patra and Hay, 2014). Other glycolytic intermediates further downstream also serve as anabolic precursors. These include fructose-6-phosphate, which gives rise to glucosamine-6-phosphate - a building block for glucosaminoglycan synthesis, and dihydroxyacetone phosphate (DHAP), which gives rise to glycerol – a backbone for the di- and triglyceride assembly. In addition, the 3-phosphoglycerate produced in the lower glycolytic pathway can be diverted into serine biosynthesis via a phosphoglycerate dehydrogenase (PHGDH)-catalyzed reaction (Figure 1). Serine is required by cancer cells not only as a building block for proteins, but also for phosphatidylserine-containing phospholipids for the plasma membrane assembly, as a carbon donor for nucleotide production and as a source of electrons for mitochondrial NADPH production (Fan et al., 2014). As a result, genomic amplifications of PHGDH are often observed in human malignancies including breast cancer and melanoma, where they are required to promote tumorigenesis (Locasale et al., 2011; Possemato et al., 2011).
In addition to glucose, commitment to anabolic metabolism also increases the import of the amino acid glutamine. Specifically, pro-proliferative stimuli trigger the upregulation of the plasma glutamine transporters ASCT2/SLC1A5 and SN2/SLC38A5. Expression of both transporters is positively regulated by the master proliferation-driving transcription factor c-myc (Wise et al., 2008); moreover, ASCT2 expression is also facilitated by another proliferation-associated transcription factor, E2F-3 (Reynolds et al., 2014). Furthermore, EGFR signaling facilitates the plasma membrane localization of ASCT2 protein (Avissar et al., 2008). Besides participating in protein synthesis, glutamine fulfills a number of diverse anabolic roles in the cell. Specifically, diverse cytosol-localized biosynthetic enzymes use the amide group of glutamine to fuel the generation of nucleotides, hexosamine units and asparagine, while the α-amine group of glutamine is incorporated into a number of other de novo-synthesized non-essential amino acids. Glutamine can also be catabolized in the mitochondria by the enzyme glutaminase 1 (GLS1), which is also positively regulated by c-myc (Gao et al., 2009) (Figure 1). GLS1 is crucial for providing a mitochondrial source of α-ketoglutarate for the TCA cycle (Wise et al., 2008); in addition, GLS1 and its cytosol-localized counterpart GLS2 are involved in bolstering cellular oxidative stress defenses via providing glutamate for glutathione production (Daemen et al., 2018; Suzuki et al., 2010). Besides supporting biosynthesis and bioenergetics, the export of intracellular glutamine facilitates the uptake of a number of essential amino acids from the extracellular space via the SLC5A7/SLC3A2 heterodimeric bidirectional transporter (Nicklin et al., 2009).
USE OF CENTRAL CARBON METABOLISM TO SUPPORT BIOSYNTHESIS
The TCA cycle is traditionally thought of as a catabolic process, in which carbon substrates are oxidized to generate energy. In fact, as recent quantitative analysis of glucose fate in vivo has revealed, in most adult tissues the major TCA cycle substrate that is oxidized to support oxidative phosphorylation is lactate, which is present in extracellular fluids at approximately 1 mM (Hui et al., 2017). Anabolically active cells also use TCA intermediates as precursors for the synthesis of macromolecules (Figure 2A). In part stimulated by an increased level of glucose uptake and intracellular production of pyruvate, the resulting carbon surplus allows the TCA cycle intermediates to depart central carbon metabolism to be consumed in various biosynthetic reactions. This, in turn, not only provides a source of new biomass, but also allows an anabolically active cell to avoid overproducing NADH beyond the capacity of ETC to convert it back to NAD+. Accordingly, the ability to vent some of the TCA carbon into supporting biosynthesis can be regarded as a bona fide metabolic necessity for a cell receiving pro-growth signaling stimuli and therefore experiencing a surplus of oxidizable nutrients.
Figure 2. Use of central carbon metabolism to support biosynthesis.
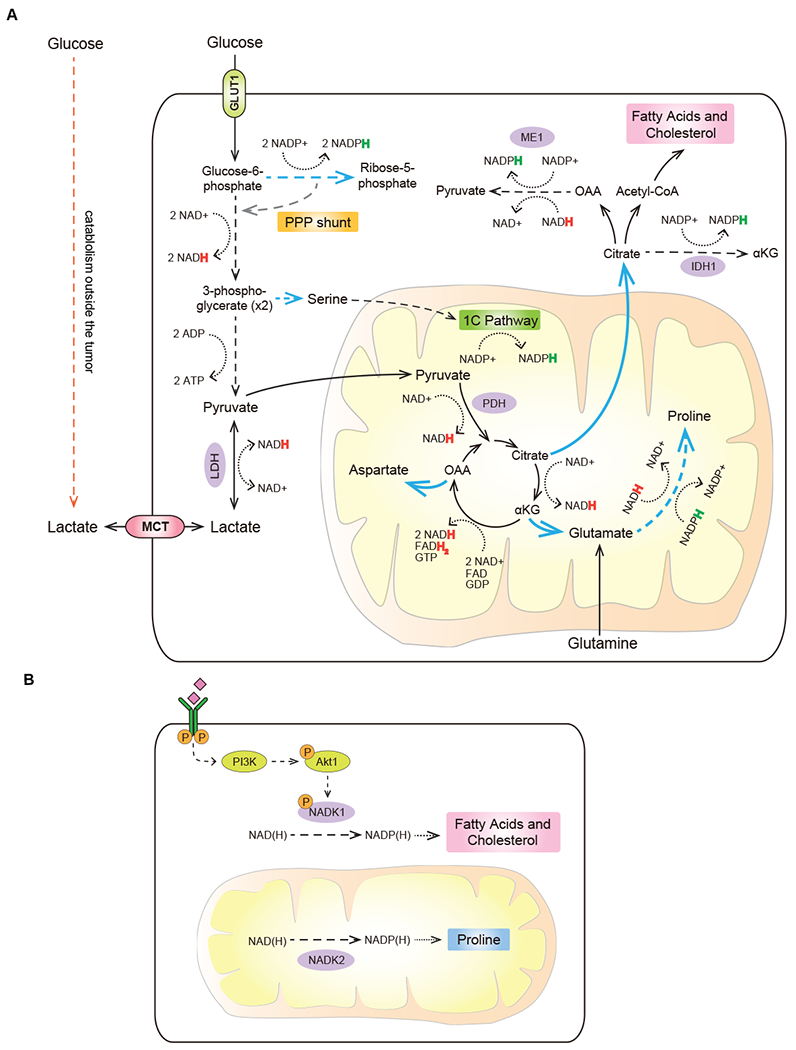
(A) Multiple central carbon metabolism intermediates serve as structural building blocks and/or donors of reducing power to support the deregulated biomass production by transformed cells. (B) Total NADPH pools can be expanded in both cytosol and mitochondria to increase reducing power in a compartment-specific manner. Blue arrows depict anabolic reactions and reaction sequences. Dashed arrows depict sequences of reactions condensed for brevity. Dotted arrows represent redox cofactor utilization. Colored “H” in NADPH and NADH represents a hydride anion carrying an extra electron. PPP, pentose phosphate pathway; LDH, lactate dehydrogenase; ME1, malic enzyme 1; IDH1, isocitrate dehydrogenase 1; PDH, pyruvate dehydrogenase; 1C pathway, one-carbon (folate) pathway; NADK1, NAD kinase 1; NADK2, NAD kinase 2.
In cells committed to anabolic metabolism, citrate produced in the TCA cycle can be exported out of the mitochondria and used to generate building blocks for the synthesis of fatty acids and cholesterol in the cytosol. As lipids are a primary constituent of the cellular membranes, activation of the lipogenic transcriptional program, which is coordinated by the SREBP1 transcription factor, is a key part of the cellular pro-anabolic program orchestrated by the mTORC1 activation (Porstmann et al., 2008). In addition, conversion of citrate to oxaloacetate and acetyl-CoA in an ATP-citrate lyase (ACLY)-catalyzed reaction – an initiating step in the de novo production of both fatty acyl chains as well as cholesterol - is positively regulated by Akt1 (Bauer et al., 2005). Furthermore, the principal fatty acid synthesis enzyme, fatty acid synthase (FASN) is frequently upregulated and is essential for tumorigenesis (Menendez and Lupu, 2007).
Lipogenesis is a highly NADPH-consuming process – for instance, building one molecule of palmitic acid requires 14 molecules of NADPH. To help balance these requirements with the cellular NADPH supply, oxaloacetate generated in the ACLY-mediated reaction can be converted to malate, which can re-enter the mitochondria or become oxidized to pyruvate in a malic enzyme 1 (ME1)-catalyzed reaction, producing NADPH. NADPH can also be produced from the conversion of cytosolic citrate to isocitrate and then, via cytosol-localized isocitrate dehydrogenase (IDH1), to α-ketoglutarate. Highlighting the role that these NADPH-producing reactions play in enabling de novo lipogenesis, depletion of either ME1 or IDH1 in various cellular contexts was shown to inhibit the de novo lipogenesis and attenuate tumor growth in vivo (Calvert et al., 2017; Fernandes et al., 2018; Shao et al., 2020).
In addition, not only the capacity to recharge NADP+ back into NADPH, but the total size of the cellular NADPH pool can be dynamically regulated in accordance with the cell’s metabolic needs (Figure 2B). Two NAD kinase isoforms – a cytosol-localized NADK1 and a mitochondria-localized NADK2 – allow cells to convert NAD+ to NADP+ in their respective cellular compartments. The activity of NADK1 is directly stimulated by Akt1-mediated phosphorylation (Hoxhaj et al., 2019), which may allow cells to tune their capacity for performing NADPH-driven cytosolic biosynthetic reactions, such as de novo lipogenesis, in accordance with the PI3K/Akt1-driven signaling inputs. Similarly, NADK2 was found to be essential for the NADPH-dependent de novo biosynthesis of proline, which takes place in the mitochondrial compartment (Tran et al., 2021; Zhu et al., 2021).
Another TCA intermediate that cells use to divert a portion of the TCA carbon away from full oxidation is α-ketoglutarate, which can give rise to an amino acid glutamate. Glutamate, in turn, can serve as a precursor for several other non-essential amino acids, including proline. Notably, in addition to acting as a vent for the TCA cycle carbon, production of one molecule of proline also consumes ATP, NADH and NADPH, dampening the mitochondrial electron load even further. In support of the vent hypothesis, blocking either citrate export or proline synthesis increases oxidative stress levels (Schworer et al., 2020). Finally, TCA intermediate oxaloacetate can also be vented away from the TCA cycle into the synthesis of aspartate - a precursor of pyrimidine bases and asparagine (Birsoy et al., 2015; Sullivan et al., 2015).
Though glucose uptake is increased by pro-proliferative stimuli, a relatively small fraction of its carbon enters the mitochondria for oxidation. To moderate the entry of pyruvate carbon into the TCA cycle, the gatekeeper enzyme pyruvate dehydrogenase (PDH), which converts pyruvate into the acetyl-CoA, is heavily negatively regulated - both allosterically via its products as well as through inhibitory phosphorylation by one of the four pyruvate dehydrogenase kinases (PDKs). Notably, deregulation of PDH activity via either PDK1 overexpression or PDH phosphatase (PDP2) depletion triggers oxidative stress and an onset of senescence in KRAS-transformed cells (Kaplon et al., 2013).
As an alternative to being oxidized in the mitochondria, glycolysis-derived pyruvate can be reduced to lactate in a cytosol-localized lactate dehydrogenase (LDH)-driven reaction. Importantly, producing lactate regenerates one NAD+ equivalent from NADH. Furthermore, because lactate equilibrates rapidly with the extracellular space through the monocarboxylate (MCT) transporters, this metabolic route renders the entire glycolytic cascade red ox-neutral. With no NADH consumed or generated, metabolism of glucose to lactate bypasses the need for ETC and for molecular oxygen altogether, while still sustaining a modest energy yield of 2 molecules of ATP.
Metabolism of glucose to lactate becomes a necessity under hypoxic conditions, in which the ETC’s capacity to unload NADH of electrons is limited by the low oxygen availability. To this end, a master hypoxia response coordinator hypoxia-inducible factor 1α (HIF1α) enables this route by simultaneously upregulating glucose transporter GLUT1, lactate dehydrogenase LDH and the PDH-inhibitory kinase PDK1 (Kierans and Taylor, 2021).
Hypoxia is not the only scenario in which cells preferentially convert glucose to lactate. Indeed, nearly a century ago, Otto Warburg discovered that tumors consume glucose in excess of normal tissue and preferentially convert it to lactate instead of oxidizing it in mitochondria - even when oxygen is present in abundance (Warburg et al., 1927). Since opting out of the mitochondrial route of glycolytic carbon oxidation forfeits almost 95% of energy locked in glucose carbons, Warburg had theorized that such truncated form of glycolysis reflects a carcinogen-inflicted damage to mitochondria, hypothesizing it to be the root cause of cancer (Warburg, 1956). Though Warburg’s finding has become foundational for the field of cancer metabolism, his own explanation of this effect had since been refuted. Indeed, most cancer cells continue to oxidize carbon in mitochondria and require the functional ETC for growth (Cavalli et al., 1997; Martinez-Reyes et al., 2020; Weinberg et al., 2010). Moreover, non-transformed cells – including lymphocytes (Frauwirth et al., 2002) and endothelial cells (Fitzgerald et al., 2018) – also exhibit Warburg effect when stimulated with pro-proliferative signals while still retaining the need for oxidative phosphorylation (Diebold et al., 2019; Sena et al., 2013), altogether revealing the Warburg effect to be a general metabolic strategy that accompanies cell proliferation.
The adaptive utility of the Warburg effect continues to be debated to this day (DeBerardinis and Chandel, 2020; Liberti and Locasale, 2016). First, aerobic glycolysis may allow cells to regenerate ATP faster than does the TCA cycle (Epstein et al., 2017). Second, accumulation of lactate and the concomitant acidification of the extracellular milieu play an important role in establishing a favorable microenvironment for tumorigenesis (Boedtkjer and Pedersen, 2020). Third, a preferential conversion of pyruvate to lactate not only diverts glucose-derived pyruvate away from being oxidized in the mitochondria but also helps to alleviate the electron load by directly regenerating NAD+ from NADH (Luengo et al., 2021). Fourth, a switch to aerobic glycolysis allows a cell to increase the capacity to generate the glycolytic intermediates for supporting biomass accumulation safely and without the risk of overwhelming the ETC with the surplus of electrons (Vander Heiden et al., 2009).
A series of recent studies have challenged the long-standing view of lactate as a byproduct of tumor metabolism. Indeed, tracing the conversion of the systemically injected 13C isotope-labeled glucose and lactate in cancer patient volunteers and genetically engineered mouse tumor models has revealed that circulating lactate is an important contributor to the tumor TCA cycle. When patients with lung and brain tumors received a systemic infusion of 13C isotope-labeled glucose, contribution of glucose-derived (13C ) carbons to the pools of lactate and the TCA cycle intermediate citrate in tumors was found to exceed their contribution to the pools of upper glycolytic intermediates, such as 3-phosphoglycerate (Courtney et al., 2018; Hensley et al., 2016). Such labeling pattern is consistent with these tumor types not only utilizing glucose directly, but also taking up and metabolizing lactate produced elsewhere in the body. Furthermore, when similar infusions were performed with 13C-labeled lactate, lactate carbons were found to contribute to the pools of TCA cycle intermediates in both tumors and in non-transformed tissues (Faubert et al., 2017; Hui et al., 2017). The mathematical framework for evaluating the extent to which lactate-sourced carbons contribute to the TCA cycle in various tissues continues to be a topic of a vigorous debate (Liu et al., 2020); furthermore, the applicability of the lactate paradigm across different tumor types (Ghergurovich et al., 2021), as well as the identities of lactate-producing and lactate-consuming cell and tissue types continue to be actively investigated (Jang et al., 2019; TeSlaa et al., 2021).
Given that lactate-transporting MCT1 transporters are relatively ubiquitously expressed, these recent discoveries position the role of lactate in the whole-body carbon economy as that of a constitutively available source of oxidizable carbon that is maintained homeostatically at the organismal level (Rabinowitz and Enerback, 2020). Glucose uptake, in contrast, is a highly regulated event that is closely linked to the proliferative state of the cell. The potential for extracellular lactate to provide a source of oxidizable carbon might be especially adaptive when considered in the context of a glucose-poor microenvironment of advanced tumors. In this context, utilization of lactate as well as of potential other non-glucose sources of oxidizable carbon can, while placing a higher burden on ETC-mediated NAD+ regeneration from NADH, provide a cell with a critical advantage of allocating the limited supply of glucose towards supporting biomass production. While the full spectrum of the heterogeneity of carbon source choice by tumor cells continues to be actively investigated and debated, these studies highlight the complexity of interlinked metabolic tradeoffs that cells must contend with as they carry out their anabolic programs within a metabolically challenging environment of a tumor.
USE OF OPPORTUNISTIC MODES OF NUTRIENT ACQUISITION
Besides the principal low-molecular weight nutrients – i.e., glucose and amino acids - cancer cells are able to utilize a wide spectrum of alternative nutrient sources, the need for which can be driven by the specific metabolic circumstances. Autophagy involves capture and lysosome-mediated degradation of intracellular proteins or entire intracellular structures (such as ribosomes, mitochondria and parts of the endoplasmic reticulum). Nutrients supplied via autophagy are critical for cell survival when the uptake of low-molecular weight metabolic substrates is compromised (Lum et al., 2005). In addition to serving as an emergency nutrient supply, deployment of organelle-specific autophagy can help cull damaged intracellular structures (Chourasia et al., 2015), and even change the biophysical properties of the cell’s interior by reducing molecular crowding (Delarue et al., 2018).
Despite its outsize and multilayered importance in cell physiology, one thing autophagy cannot do is provide sufficient material for building new biomass for cell proliferation. However, mammalian cells were also found to use their lysosomes to digest non-discriminately scavenged macromolecules of extracellular origin in a manner reminiscent of unicellular eukaryotes. This is achieved via the extension of membrane protrusions and formation of macropinosomes in a process termed macropinocytosis. Macromolecules engulfed in this manner are subsequently trafficked to lysosomes for degradation. Positioned downstream of growth factor signaling, PI3K activation is required to initiate membrane ruffling and macropinosome closure. Genetic alterations observed in cancers such as oncogenic mutant forms of Ras GTPases increase the rate and volume at which macropinocytosis occurs, independent of growth factor stimulation (Bar-Sagi and Feramisco, 1986; Commisso, 2019; Commisso et al., 2013). As a result, cancer cells harboring oncogenic Ras mutations display an increased ability to utilize extracellular protein through macropinocytosis such that they are able to maintain cell survival and proliferation in amino acid depleted tumor microenvironment (Commisso et al., 2013; Kamphorst et al., 2015) (Figure 3B).
Figure 3. Use of opportunistic modes of nutrient acquisition.
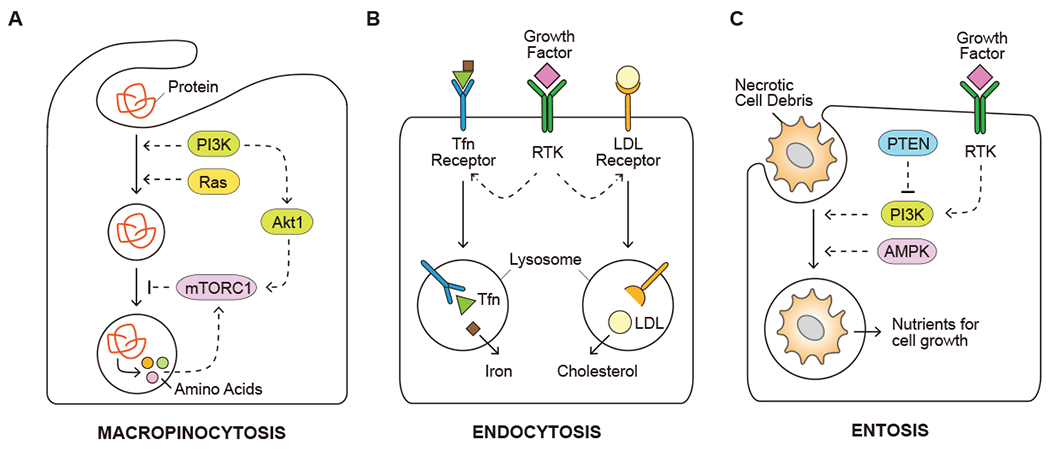
(A) Capture of extracellular proteins by macropinocytosis to recover amino acids. (B) Uptake of insoluble nutrients, such as iron and cholesterol, through receptor-mediated endocytosis. (C) Utilization of dying cells and/or necrotic cell debris for nutrient acquisition. Solid arrows depict movement of metabolites. Dashed arrows depict regulatory effects of signal transduction components. Tfn, transferrin; LDL, low-density lipoprotein; mTORC1, mechanistic target of rapamycin, complex 1; PTEN, phosphatase and tensin homolog; AMPK, AMP-activated protein kinase.
The internalized macropinosomes are subject to additional regulations as they deliver cargos to lysosomes for degradation. Active mTORC1 complex suppresses the catabolism of extracellular proteins as an amino acid source; accordingly inhibition of mTORC1 activity in cells facing nutrient-depleted conditions facilitates the survival and growth of Ras-transformed cells in amino acid-depleted conditions in presence of extracellular albumin (Nofal et al., 2017; Palm et al., 2015). In agreement, Akt1 activation of mTORC1 suppresses the utilization of internalized albumin when free amino acids are abundant (Figure 3A). By contrast, the small GTPase Rac1 and phospholipase C (PLC) downstream of PI3K activity can promote cell growth that depends on the catabolism of extracellular proteins through macropinocytosis (Palm et al., 2017).
While the use of serum albumin by macropinocytosis as a source of amino acids has been studied the most, other extracellular macromolecules can also be taken up by macropinocytosis. Pancreatic ductal adenocarcinoma (PDAC) cells often reside in a desmoplastic microenvironment that contains a dense collagen network. It was reported that PDAC cells are able to utilize extracellular collagen in glucose- or glutamine-limited conditions, partly through macropinocytosis (Olivares et al., 2017). Proline is a major amino acid component of collagen proteins. Accordingly, collagen internalized by PDAC cells can provide a source of proline to cells, which could be further catabolized by proline dehydrogenase (PRODH) as a source of electrons for energy production and as the TCA cycle substrate (Olivares et al., 2017). In addition to the utilization of proteins, macropinocytosis was also found to participate in scavenging other extracellular components such as exosomes and lysophospholipids (Kamphorst et al., 2013; Nakase et al., 2015).
Of note, scavenging of extracellular lipids becomes elevated under hypoxic conditions, where it serves to help restore the balance of saturated (SFA) to monounsaturated (MUFA) fatty acids in cellular membranes. Indeed, the introduction of a double bond into an acyl chain, which converts an SFA molecule into its MUFA counterpart, is driven by the enzyme stearoyl-CoA desaturase 1 (SCD1), which requires molecular oxygen as an electron acceptor. Low-oxygen conditions inhibit SCD1, thereby altering the SFA/MUFA ratio in favor of saturated fatty acids. This, in turn, alters the biophysical properties of cellular membranes, making them less fluid and pliable. The endoplasmic reticulum membranes appear to be particularly affected by the SFA/MUFA imbalance, in part due to the intricate topography of the ER membrane compared to other cellular membrane structures (Rong et al., 2013). In fact, hypoxia-associated SCD1 inhibition was found to trigger ER stress in cells with a hyperactive mTORC1 and, consequently, a high translational burden (Young et al., 2013). Consequently, adaptations such as uptake of extracellular lipids, or, in other contexts, release of lipids from internal lipid storage depots was found to maintain the fatty acid balance when the cell’s fatty acid desaturation capacity becomes compromised by oxygen deficit (Ackerman et al., 2018; Kamphorst et al., 2013).
In addition to a bulk mode of nutrient scavenging via macropinocytosis, cells can internalize some macromolecules from the extracellular environment via a selective, receptor-mediated endocytosis. For instance, low-density lipoprotein (LDL) is the major carrier of extracellular cholesterol. Mammalian cells capture LDL through plasma membrane LDL receptors, which are trafficked to endolysosomal compartments to release cholesterol required for cellular membrane assembly (Brown and Goldstein, 1979) (Figure 3B). Aberrant activation of the RTK-PI3K-Akt1 signaling in cancer cells was found to upregulate LDL receptor in part through mTORC1 and sterol regulatory element binding protein (SREBP) activity (Edinger and Thompson, 2002; Porstmann et al., 2008; Streicher et al., 1996). In addition, exogenous cholesterol can be imported into cells in its high-density lipoprotein (HDL) form via scavenger receptor B1 (SCARB1). Clear cell renal cell carcinomas in particular were found to be cholesterol auxotrophs and rely primarily on SCARB1-mediated HDL import as a source of cholesterol (Riscal et al., 2021). Furthermore, iron is essential for various metabolic activities such as biosynthesis of the heme prosthetic group and iron-sulfur clusters (Rouault, 2015). Endocytosis of the extracellular iron carrier, transferrin (Tfn), by Tfn receptors is the major source of intracellular iron, and is critical during the regulation of iron-dependent ferroptotic cell death (see below) (De Domenico et al., 2008; Jiang et al., 2021).
Finally, cells are able to engulf entire living cells and/or dying cells in a non-phagocytic process referred to as entosis, which can be induced upon metabolic stress including glucose starvation (Hamann et al., 2017; Overholtzer et al., 2007). Similarly, PTEN-deficient prostate cancer cells were reported to scavenge necrotic cell debris through macropinocytosis in an AMPK-dependent manner (Kim et al., 2018) (Figure 3C). Similarly to the engulfment of soluble proteins, nutrients from the engulfed living or dead cells can be recovered from the lysosomal digestive system to support cell proliferation (Hamann et al., 2017; Kim et al., 2018).
EXPANDED NEED FOR ELECTRON ACCEPTORS
Carbon metabolism via glycolysis and the TCA cycle requires NAD+ as an electron acceptor. Enhanced glycolytic activity of transformed cells makes them dependent on the constant regeneration of NAD+. The Warburg effect is one prominent adaptation that anabolically active cells use to regenerate NAD+ from NADH via the LDH-mediated reaction. As a consequence, lactate secretion is characteristic of cancer cells that display high rates of glycolysis (Vander Heiden et al., 2009). Cytosolic electrons in a form of NADH can also be transported into the mitochondria through dedicated electron shuttles, such as the malate-aspartate shunt and the glycerol phosphate shunt, to facilitate the regeneration of NAD+ in the electron transport chain (ETC) (Figure 4). Recently, MCART1 (encoded by SLC25A51) was identified as a mitochondrial NAD+ transporter that could also function to communicate NAD+ and NADH pools between the cytosolic and mitochondrial compartments (Kory et al., 2020; Luongo et al., 2020).
Figure 4. Expanded need for electron acceptors.
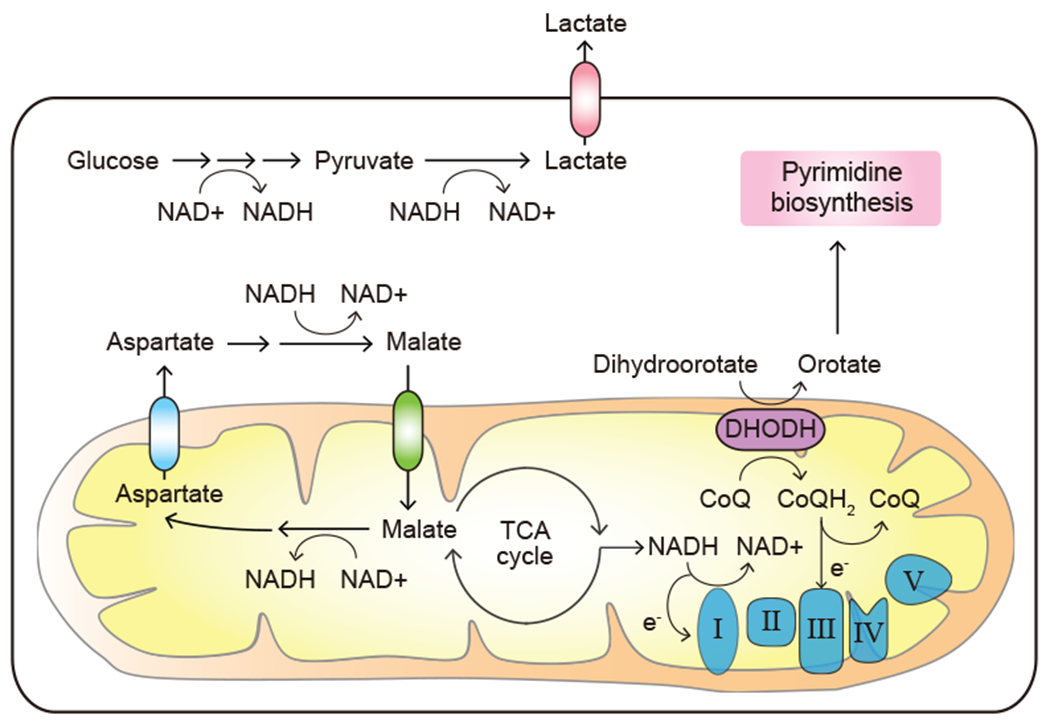
Proliferating cancer cells display a high demand for the regeneration of electron acceptors including NAD+. DHODH, dihydroorotate dehydrogenase; CoQ, ubiquinone; CoQH2, ubiquinol; e−, electron.
Dissecting the specific roles of mitochondrial respiration in supporting anabolic metabolism and cell proliferation revealed that one essential function of the ETC was to regenerate NAD+ to support aspartate biosynthesis (Birsoy et al., 2015; Sullivan et al., 2015). Indeed, aspartate production could be compromised by inhibiting either Complex I or Complex III of the ETC. Reciprocally, supplementation of electron acceptor substrates, such as pyruvate or α-ketobutyrate, helped increase the NAD+/NADH ratio, restored aspartate biosynthesis and enabled cell proliferation even in cells in which ETC was inhibited (Birsoy et al., 2015; Sullivan et al., 2015). The availability of aspartate product asparagine was also compromised by the ETC inhibitors (Krall et al., 2021). Interestingly, when ETC inhibitors were applied at low doses, asparagine supplementation sufficed to restore nucleotide synthesis and cell proliferation without rescuing cellular aspartate abundance (Krall et al., 2021).
Electron flux along the ETC not only helps regenerate NAD+ and FAD, but also directly connects the energy-producing capacity of mitochondria to pyrimidine biosynthesis through the activity of dihydroorotate dehydrogenase (DHODH). To this end, oxidation of ubiquinol by the ETC Complex III regenerated ubiquinone to serve as a critical electron acceptor for supporting the DHODH function and hence pyrimidine synthesis in proliferating cells (Martinez-Reyes et al., 2020) (Figure 4). Taken together, proliferating cancer cells have a perpetual high demand for the regeneration of electron acceptors to generate precursors for both protein and nucleic acid biosynthesis.
ELEVATED RELIANCE ON OXIDATIVE STRESS PROTECTION MECHANISMS
Deregulated anabolic metabolism coupled to the limitations associated with the tumor microenvironment exposes transformed cells to increased levels of reductive stress (for example, high NADH, low O2). Indeed, as proliferating cancer cells import increased quantities of carbon sources, the NADH supply may increase beyond what the ETC has the capacity to handle, particularly if either O2 or ADP become limiting. A number of pathways that reduce NADH/NAD+ ratio to avoid reductive stress are outlined above. Cancer cells are also subjected to oxidative stress. In the presence of environmental oxidants such as H2O2, NO, or even O2, macromolecular oxidation has the capacity to damage intracellular macromolecules such as lipids, which, in turn, can lead to loss of cellular integrity. Cancer cells rely on a number of cellular antioxidant defense mechanisms, including the glutathione (GSH) system and the thioredoxin (TRX) system, to protect themselves from such oxidative damage. As a result, genetic and metabolic alterations that facilitate these protection mechanisms are often found in tumors.
The mutant forms of NRF2 (encoded by NFE2L2) transcription factor as well as of its E3 ubiquitin ligase KEAP1 are found in solid tumors, and are most prevalent in lung cancer, which is characterized with elevated levels of oxidative stress in part as a result of high levels of extracellular oxygen present in lung tissue (Martincorena et al., 2017). Both NRF2 and KEAP1 mutations impair the binding of KEAP1 to NRF2 protein to target the latter for degradation. This, in turn, increases NRF2 protein levels and promotes the NRF2-driven transcriptional program, resulting in an increased expression of enzymes involved in GSH biosynthesis (Figure 5). Increased production of GSH requires a greater supply of its limiting substrate, cysteine. Accordingly, NRF2-driven transcriptional response also increases xCT (encoded by SLC7A11) expression, which promotes the import of the oxidized form of cysteine, cystine, into cells (Habib et al., 2015; Ye et al., 2014). Consumption of intracellular cysteine stores upon oxidative stress triggers a decline in its levels, which is sensed by the GCN2 kinase. GCN2, in turn, triggers the accumulation of activating transcription factor 4 (ATF4). ATF4 increase further promotes xCT expression, facilitating cystine uptake (Sato et al., 2004; Ye et al., 2014). When the levels of extracellular cystine are depleted, as is often seen in tumor microenvironment (Kamphorst et al., 2015; Sullivan et al., 2019), ATF4 can also upregulate enzymes involved in the de novo cysteine synthesis from methionine via the transsulfuration pathway (Zhu et al., 2019) (Figure 5).
Figure 5. Elevated reliance on oxidative stress protection mechanisms.
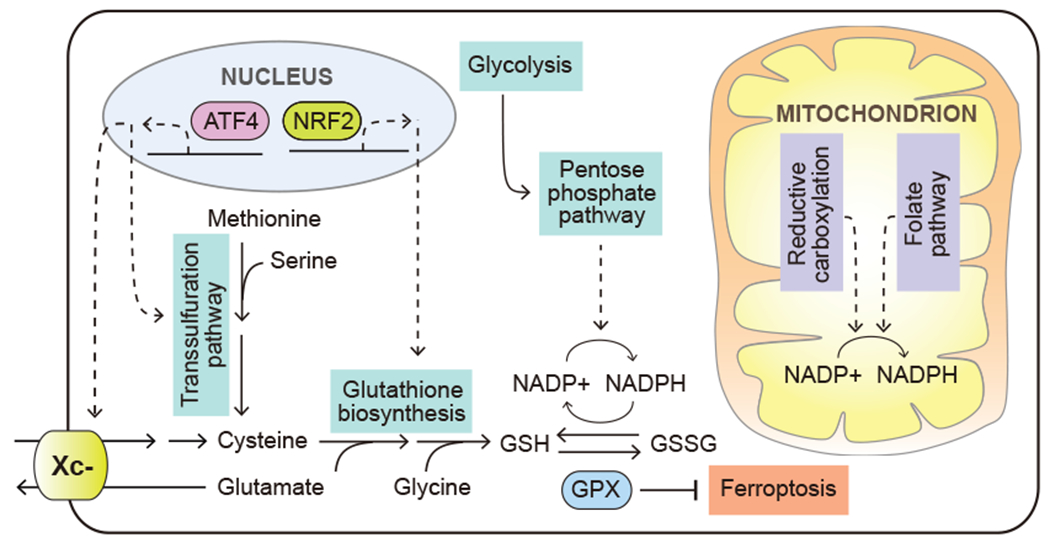
Cancer cells depend on a variety of metabolic mechanisms to defend oxidative stress, including upregulated glutathione biosynthesis, increased NADPH regeneration and suppression of ferroptosis. Solid arrows depict metabolite movement or metabolic reactions. Dashed arrows depict regulatory effects of signal transduction components. ATF4, activating transcription factor 4; NRF2, nuclear factor erythroid 2-related factor 2, also known as NFE2L2; Xc-, system Xc-, cystine/glutamate antiporter; GPX, glutathione peroxidase.
In addition to increasing the production of GSH, transformed cells often respond to oxidative stress by altering their metabolism to maintain the antioxidant capacity of these ROS-scavenging molecules. A challenge that cancer cells often encounter during tumor initiation is the oxidative stress induced by detachment from the extracellular matrix. Activation of receptor tyrosine kinase and the downstream PI3K pathway was found to facilitate anchorage-independent growth by increasing glucose uptake and its utilization in the pentose phosphate pathway to support NADPH regeneration from NADP+ (Schafer et al., 2009) (Figure 5).
RTK-driven tyrosine phosphorylation of IDH1 can also regulate the shuttling of NADPH between the cytosol and the mitochondria. Tyrosine phosphorylation of IDH1 has been reported to favor the reductive flow of glutamine-derived α-ketoglutarate into citrate, which consumes cytosolic NADPH. Citrate production is followed by its retrograde transport into the mitochondria, where the IDH1’s counterpart, IDH2, can convert the imported citrate back into α-ketoglutarate, thus regenerating NADPH in the mitochondrial compartment (Jiang et al., 2016). A reverse cycle, in which IDH2 consumes mitochondrial NADPH to catalyze reductive carboxylation of α-ketoglutarate into citrate in cells with genetically impaired ETC, has also been reported (Mullen et al., 2014).
As metastasizing cells enter the circulation, exposure to a more oxygenated environment may further elevate the oxidative stress burden. One adaptation that circulating tumor cells employ to counteract oxidative stress relies on the propensity of circulating tumor cells to cluster together (Aceto et al., 2014). Thus, cells clustering together were found to create a hypoxic pocket in the core of the cluster, which, through HIF1α accumulation, may limit the oxidative metabolism of carbon and favor the redox-neutral glycolytic route, as well as trigger mitophagy to cull oxidatively damaged mitochondria (Labuschagne et al., 2019).
The importance of robust ROS defense in facilitating metastasis is further supported by studies from in vivo tumorigenesis models. In a melanoma model, circulating tumor cells and cells from metastatic nodules were found to experience higher levels of oxidative stress than the subcutaneous tumors (Piskounova et al., 2015); accordingly, both a robust flux through the one-carbon pathway as well as an increased uptake of lactate were found to be among the metabolic determinants of successful metastatic growth (Piskounova et al., 2015; Tasdogan et al., 2020). Both of these adaptations can help augment the cell’s NADPH production capacity – one via the one-carbon pathway-mediated NADPH production, while the second one by sparing glucose carbons to be used in the oxidative branch of the PPP pathway (Figure 5). In contrast to these findings, studies from other tumorigenic contexts have revealed that elevated levels of ROS may, in fact, facilitate metastasis. Studies from a mouse pancreatic cancer model have shown that the loss of TIGAR expression, which compromises the cell’s ability to use PPP as a NADPH source, increased cellular ROS levels, promoted cell invasiveness and facilitated metastatic colonization of the lung (Cheung et al., 2020). Whether the difference in level or location of the ROS produced, or the cell lineage context itself contributes to these disparate outcomes, remains to be further elucidated.
Oxidative stress, and in particular, oxidative damage to the cellular lipid components, can lead to ferroptotic cell death. Recent studies have revealed a sophisticated regulation of ferroptosis by cellular metabolic activities, including iron storage and release, homeostasis of selenium, phospholipid peroxidation, and cysteine and GSH availability (Jiang et al., 2021; Zheng and Conrad, 2020). Interestingly, the transit through the lymphatic system prior to entering the blood environment was found to facilitate melanoma metastasis by equipping the melanoma cells with better ability to withstand an increased risk of ferroptosis associated with the iron- and oxygen-rich environment of the blood (Ubellacker et al., 2020). Multiple mechanisms are likely to underlie the protective effect of such transit through the lymphatic environment, including the access to ferroptosis-protective agents oleic acid and glutathione, both of which are particularly abundant in lymph (Ubellacker et al., 2020). The mechanism behind the protective role of oleic acid in ferroptosis is a topic of active investigation, but might at least in part be due to oleic acid, a MUFA, competing with, and thereby reducing the presence of PUFA species in the cellular membranes (Magtanong et al., 2019). Indeed, as PUFA species contain multiple double bonds in their chemical structure, they can sustain the most profound oxidative damage from ferroptosis, negatively affecting membrane function and cell integrity and survival.
Targeting metabolic mechanisms that protect cells from ferroptosis have emerged as promising therapeutic interventions in pre-clinical studies. For example, blocking sterol regulatory element-binding protein (SREBP)-mediated lipogenesis was found to synergize with perturbations of the glutathione peroxidase 4 (GPX4) defense mechanism to induce ferroptosis in PI3K-mutated cancer cells (Yi et al., 2020). In addition to the GPX4-dependent ferroptosis defense system, the oxidoreductase ferroptosis suppressor protein 1 (FSP1) was recently found to produce ubiquinol from ubiquinone to serve as a ROS scavenger, thereby revealing a glutathione-independent ferroptosis defense mechanism (Bersuker et al., 2019; Doll et al., 2019). Accordingly, DHODH-mediated ubiquinol production was reported as a targetable vulnerability in cancer cells with low GPX4 expression (Mao et al., 2021). Taken together, the process of tumorigenesis often exposes cancer cells to elevated oxidative stress that has the potential to be exploited therapeutically in the future.
INCREASED DEMAND FOR NITROGEN
Carbon sources can serve as structural intermediates for biosynthesis or become oxidized to generate energy or support NADPH production. In contrast, reduced nitrogen is used for macromolecular synthesis or converted to waste. Increased nitrogen demand is uniquely associated with proliferation. Consequently, deregulated proliferation of cancer cells preferentially depletes the microenvironment of the preferred growth-supporting nitrogen donors – most prominently, glutamine. To maintain growth and proliferation even after exhausting the supply of preferred exogenous sources of reduced nitrogen, cancer cells therefore must rely on adaptations that allow them to use - and reuse - the alternative nitrogen sources at their disposal.
Glutamine is a preferred nitrogen-providing nutrient in animals (Bott et al., 2019a). Given a uniquely high demand for it, glutamine levels are maintained at an approximately 600-700 μM in circulation – almost an order of magnitude higher than other amino acids (Marchesini et al., 1983). During an injury or a systemic infection, circulating glutamine levels become depleted; furthermore, a local depletion of glutamine levels is observed in both solid tumors and in healing wounds. For example, a recent study has found the levels of free glutamine in the interstitial fluid of mammary tumor allografts to be as low as 100 μM (Edwards et al., 2021). However, not all tumors are equally glutamine-limited – for instance, in some genetically engineered models of murine cancers, glutamine concentration in the tumor interstitial fluid has been reported to be the same as in circulation (Sullivan et al., 2019). Tumors have also been reported to produce glutamine de novo from glutamate and free ammonium via glutamine synthetase (also known as glutamate-ammonia ligase, or GLUL)-driven reaction (Figure 6A). A number of tumorigenesis-associated transcription factors, including c-myc (Bott et al., 2015) and Yap (Cox et al., 2016) can upregulate GLUL expression in various cellular contexts. In addition to being regulated at the level of gene expression, glutamine depletion also triggers GLUL accumulation at the protein level (Nguyen et al., 2016). Indeed, tumor-specific deletion of GLUL was shown to severely delay tumor formation in a genetically engineered mouse model (Bott et al., 2019b).
Figure 6. Increased demand for nitrogen.
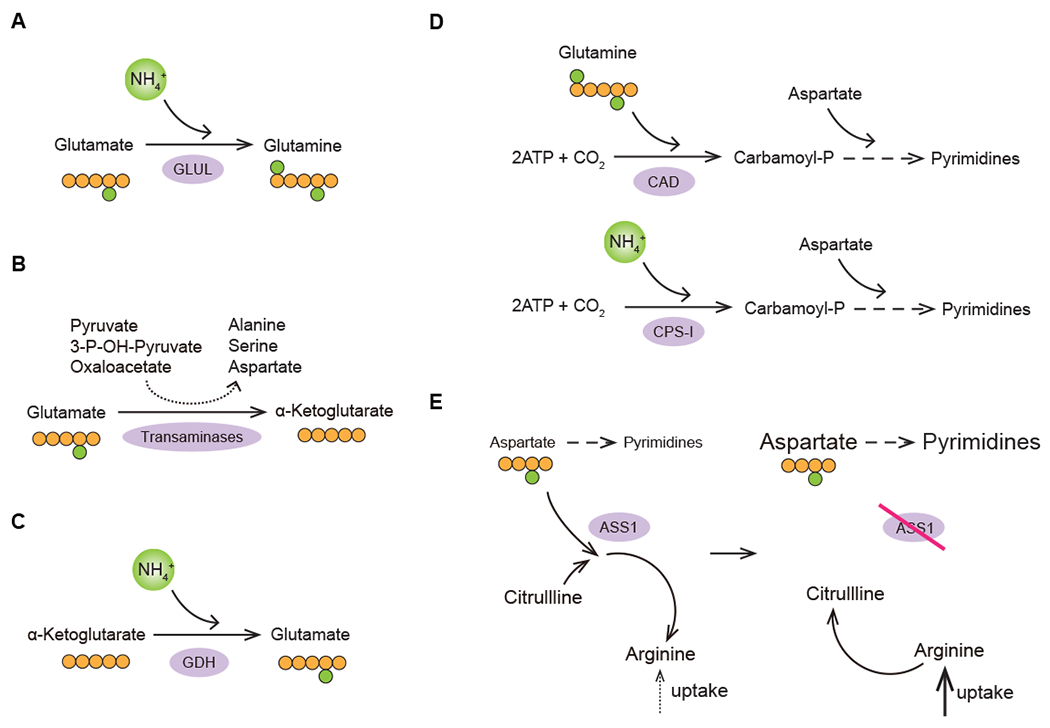
Nitrogen-saving strategies used by transformed cells to mitigate the deficit of reduced nitrogen carriers in the tumor microenvironment. (A) De novo glutamine synthesis; (B) Preference for transamination of glutamate to α-ketoglutarate as a strategy to maximize non-essential amino acid synthesis in proliferating cells; (C) De novo glutamate synthesis; (D) Preference for CPS-I-mediated route of carbamoyl-phosphate production in LKB1-deificient tumors; (E) Increased availability of aspartate for pyrimidine synthesis coupled to an increased reliance on arginine import in ASS1-deficient tumors. GLUL; glutamate-ammonia ligase (glutamine synthetase); GDH1, glutamate dehydrogenase 1; CAD, carbamoyl-phosphate synthetase 2, aspartate transcarbamylase, and dihydroorotase; CPS-I, carbamoyl phosphate synthetase I; ASS1, argininosuccinate synthetase 1.
The demand for glutamine can rise even further in hypoxic conditions, when molecular oxygen levels fall below the level necessary to maintain the flow of electrons through the ETC to regenerate NAD+ from NADH (Metallo et al., 2011). In this metabolic scenario, glutamine-derived α–ketoglutarate can provide the cancer cell with a source of citrate to support de novo lipogenesis, making use of the reversibility of the IDH1-catalyzed reaction – a phenomenon termed reductive carboxylation. Mutant EGFR-driven non-small cell lung carcinoma cell lines have been reported to engage in a reductive flux from α-ketoglutarate to citrate even in the presence of oxygen as a result of EGFR-driven phosphorylation of the IDH1’s Y42 and Y391 tyrosine residues (Chen et al., 2019a; Chen et al., 2019b). Such an adaptation, while elevating the demand for glutamine, may help reduce TCA cycle-generated reductive stress.
A cell has more than one option at its disposal for converting glutamate to α-ketoglutarate. One of these options involves oxidative deamination of glutamate into α-ketoglutarate via the glutamate dehydrogenase (GDH)-catalyzed reaction, which releases free ammonium. Alternatively, α-ketoglutarate can be produced via transamination, in which the amino group of glutamate is transferred via a transaminase enzyme onto one of the available ketoacid acceptors, generating non-essential amino acids such as aspartate, serine and alanine (Figure 6B). Notably, proliferating mammary epithelial cells were found to favor the transamination route over the GDH-driven deamination, while quiescent cells opted for the GDH option (Coloff et al., 2016). Such an adaptation may allow proliferating cells to maximize the use of available amino group reserves to optimally support non-essential amino acid synthesis.
TCA cycle-generated α-ketoglutarate can also give rise to glutamate using the reversibility of the GDH-catalyzed reaction. Ammonium often accumulates in the tumor microenvironment, and can reach up to 3 mM. At this level of ammonium accumulation, cells were found to utilize the reverse direction of the GDH-driven reaction, which allows them to produce glutamate and use it to fuel the synthesis of other non-essential amino acids (Figure 6C) (Spinelli et al., 2017).
A unique ammonium-scavenging adaptation that is characteristic to transformed cells harboring a specific oncogenic lesion involves a non-canonical route of carbamoyl phosphate synthesis - the initiating step for building pyrimidine bases (Kim et al., 2017). Proliferating cells produce carbamoyl phosphate synthesis via the multifunctional CAD enzyme in a reaction that requires glutamine as an amide donor. However, non-small cell lung carcinoma cells that harbor the combination of a mutant KRAS allele and the loss of the tumor suppressor LKB1 were found to express a different carbamoyl phosphate-producing enzyme, carbamoyl phosphate synthetase 1 (CPS1). Notably, CPS1 produces carbamoyl phosphate in a more economical manner than the CAD enzyme: while CAD requires glutamine as a source of nitrogen, CPS1 can use free ammonium ion instead (Figure 6D). Normally, CPS1 expression is restricted to the liver, where it works to facilitate the ammonium clearance via the urea cycle. However, in the context of LKB1 loss, CPS1 becomes aberrantly upregulated and is used by cancer cells to fulfill a critical anabolic role (Kim et al., 2017). Opting out of a costly CAD-catalyzed reaction in favor of a more economical CPS1-catalyzed route allows a cell to eliminate a portion of its glutamine requirement and freeing up remaining glutamine for other biosynthetic needs.
Some tumor cells can also increase their pyrimidine synthesis by reducing the flux of aspartate into the urea cycle (Figure 6E). Most normal cells can utilize aspartate to initiate the clearance of excess reduced nitrogen in the urea cycle in a reaction catalyzed by the enzyme argininosuccinate synthase 1 (ASS1). However, many solid tumors, including pancreatic cancer, prostate cancer and melanoma, lose the ASS1 expression, predominantly due to its epigenetic silencing (Delage et al., 2010). The hypoxia-associated acidification of the extracellular milieu can further contribute to the ASS1 downregulation in a non-cell-autonomous manner (Silberman et al., 2019). Without aspartate being consumed to initiate the clearance of excess reduced nitrogen, proliferating cancer cells can afford to divert more of the available aspartate supply towards pyrimidine biosynthesis (Rabinovich et al., 2015). However, the loss of ASS1 expression comes at a cost as it blocks the ability of cells to regenerate arginine from citrulline in the urea cycle, making them vulnerable to the exogenous depletion of arginine. Accordingly, arginine-depleting therapeutic strategies, such as PEGylated recombinant arginase enzyme are being explored as a potential metabolic therapy intervention in these tumor contexts (Zou et al., 2019). Furthermore, cancer cells with downregulated ASS1 or other urea cycle enzymes display a characteristically imbalanced pyrimidine-to-purine ratio, which, in turn, is associated with a higher mutational burden in this subset of cancers – a feature that makes transformed cells more “visible” to the antitumor immune attack and thus susceptible to immune checkpoint inhibitor therapeutics (Lee et al., 2018).
HETEROGENEITY OF METABOLIC ADAPTATIONS
Cancer can arise from a variety of tissues and cell types with distinct physiologic differences in metabolism; moreover, tumors may harbor different combinations of transforming oncogenic lesions. A combination of these factors can help create tumors with distinct metabolic characteristics. As cancer cells leave the tissue of origin and colonize other organs they can be potentially constrained by the tissue microenvironment of the organ to which they metastasize. As a result, the diversity of cancer-associated metabolic portraits is shaped both by factors intrinsic to cancer cells as well as the specifics of the metabolic terrain they encounter (Figure 7).
Figure 7. Heterogeneity of metabolic adaptations.
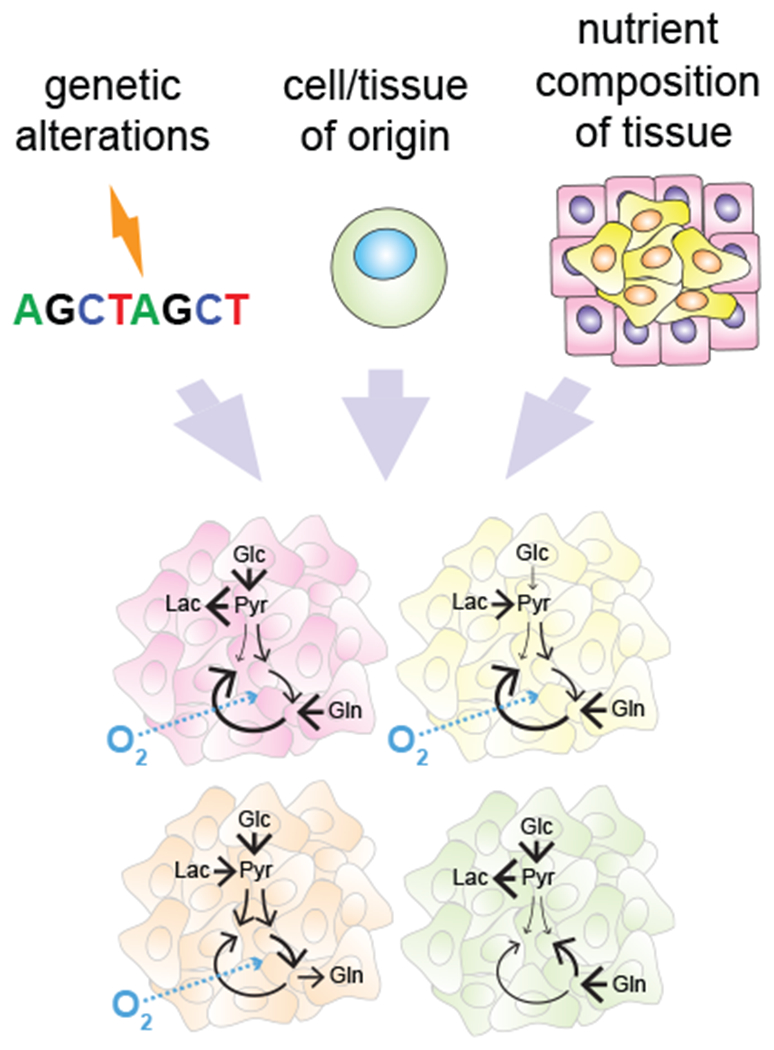
Tumor metabolic identity is shaped by both cell-intrinsic factors such as the identity of transforming genetic lesions and the metabolic phenotype of the cell-of-origin, and by the availability of metabolites in a specific tissue environment. Glc, glucose; Pyr; pyruvate; Lac, lactate; Gln, glutamine.
Some oncogenic lesions harbored by cancer cells are directly responsible for modulating specific metabolic pathways in a manner that facilitates tumorigenesis. Several clear examples have emerged, including the pseudohypoxic state established by the HIF1α and HIF2α transcription factor stabilization in Von Hippel Lindau factor (VHL)-deficient renal cell carcinomas (Bratslavsky et al., 2007), an increased reliance on glutaminolysis-dependent glutamate supply for powering the import of cystine in tumors that have lost the KEAP1 tumor suppressor expression (Romero et al., 2017) and constitutively activated reductive metabolism of α-ketoglutarate in a subset of EGFR-transformed non-small cell lung carcinomas (Chen et al., 2019b). Even within the same tissue-of-origin context, expression of different driver oncogenes can sometimes lead to not only different but even opposite metabolic outcomes – for instance, c-Met receptor tyrosine kinase-transformed liver tumors were shown to upregulate GLUL and accumulate glutamine, whereas c-myc-transformed tumors of the same origin were found to deplete glutamine form their environment (Yuneva et al., 2012).
The tissue of origin is another important factor that determines how cancer cells engage their metabolic pathways and support growth. As mentioned above, liver tumors driven by the transgenic c-myc expression consume glutamine; in contrast, c-myc-driven lung tumorigenesis is associated with GLUL expression and glutamine production (Yuneva et al., 2012). Similarly, lung tumorigenesis driven by a combination of Kras oncogene and Trp53 inactivation rely on catabolism of branched-chain amino acids (BCAAs) as nutrients, whereas pancreatic tumors driven by the same transforming genetic combination do not catabolize BCAAs (Mayers et al., 2016).
Evidence from genetically engineered animal tumor models is further corroborated by the transcriptomic analysis of human tumors and corresponding normal tissues. These studies reveal that transformed cells retain the distinguishing metabolic gene expression patterns of their tissue of origin (Gaude and Frezza, 2016; Hu et al., 2013). Thus, despite the accumulation of genetic perturbations, tumors retain the pre-existing metabolic features of their native tissue. However, during transformation, these pre-existing metabolic properties are repurposed to support anabolic growth. In some tumors, these pre-existing metabolic phenotypes become amplified genetically. For instance, amplification of PHGDH, a limiting enzyme for de novo serine biosynthesis is frequently found in tumors that arise from basal-like mammary epithelial cell subtypes with characteristically high native expression of PHGDH (Gromova et al., 2015); similarly, amplifications of NARPT, an enzyme that drives the de novo synthesis of NAD+, are detected in cancers that arise from tissues with high native NARPT expression (Chowdhry et al., 2019). Conversely, some metabolic enzymes act as bona fide tumor suppressors in select tissues. This is exemplified by the uniform loss of the rate-limiting gluconeogenic enzyme fructose-1,6-bisphosphatase 1 (FBP1) expression in tumors that originate from liver and kidney – two professional gluconeogenic tissues. Loss of FBP1 appears to exert a wide range of tumor-promoting effects, among which are promotion of aerobic glycolysis, activation of HIF1α transcription factor as well as the ability to foster tumor-promoting characteristics in other cells in the tumor vicinity - such as in hepatic stellate cells (Li et al., 2014; Li et al., 2020).
One prominent example of heterogeneity of metabolic adaptations is the preference for a particular anaplerotic substrate for the TCA cycle. Two contrasting anaplerotic strategies predominate in transformed cells – one in which the refilling of the TCA cycle takes place at the point of α-ketoglutarate, which is derived from the catabolism of glutamine, and the second one in which the pool of the TCA intermediate oxaloacetate is replenished through the carboxylation of pyruvate in a pyruvate carboxylase (PC)-driven reaction.
Though glutamine-mediated anaplerosis is near-universal in cultured cells, cells can reorient their carbon flow and switch to PC-driven anaplerosis when glutamine is depleted (Cheng et al., 2011). Interestingly, in vivo studies with 13C carbon isotope-containing glucose infusions demonstrate that the preference for a particular anaplerotic route depends on a tumor type. Indeed, pancreatic and lung tumors were found to favor the PC-mediated anaplerotic route and required it for growth (Lau et al., 2020; Sellers et al., 2015). In contrast, 13C carbon infusion studies in xenografts of colorectal origin have revealed that these tumors utilized glutamine as their anaplerotic source (Zhao et al., 2019). The dependence on a particular anaplerotic strategy may further point to the link between the metabolic character of the tissue of origin and the tumors arising from it. For instance, neither lung tumors nor normal lung were found to be net glutamine consumers (Davidson et al., 2016), a pattern that is also consistent with the role the lung tissue plays in supplying de novo synthesized glutamine for the body in conditions when circulating levels of glutamine are compromised (Labow and Souba, 2000).
Resumption of growth in the context of a newly colonized organ requires metastatic cancer cells to adapt to a different nutrient microenvironment compared to their native tissue. Depending on the context, such adaptations may involve taking advantage of the substrates that are abundant at a new site while adapting to the limited quantities of other nutrients. For instance, central nervous system is relatively depleted of nutrients such as lipids, compared to other metastatic niches. Accordingly, tumors that metastasize to the brain rely on the de novo synthesis of fatty acids (Ferraro et al., 2021). In addition, the ability to synthesize asparagine de novo was shown to be required for the metastasis of mammary carcinoma cells to the lung, but not for the growth of primary tumors (Knott et al., 2018).
Not only the limitation, but the surplus of select nutrients can contribute to shaping the metabolic character of cancer cells that colonize it. For example, mammary tumors that metastasize to either lung or bone are highly dependent on PGC1α expression and the mitochondrial route of carbon metabolism (Andrzejewski et al., 2017), whereas their liver-bound counterparts are, in contrast, highly glycolytic and rely on high PDK1 expression, which reduces the entry of glucose carbon into the mitochondria (Dupuy et al., 2015). The highly glycolytic nature of liver metastases may reflect the cells taking advantage of the abundant supply of glucose in liver tissue. In contrast, colorectal tumors that metastasize to liver display an ability to metabolize another sugar that is abundant in this nutrient-rich organ – fructose. To this end, liver metastatic clones from colorectal tumors are selected to express high levels of aldolase B (ALDOB), which allows them to use fructose as a glycolytic substrate (Bu et al., 2018). Similarly, mammary tumor cells that metastasize to the lung elevate their use of pyruvate as an anaplerotic substrate as compared to primary tumors – an adaptation that allows them to take advantage of high pyruvate levels in the lung (Christen et al., 2016). This finding reveals that not only primary lung tumors (Sellers et al., 2015), but also tumors that metastasize to the lung can take advantage of the increased pyruvate supply in the lung tissue, further underscoring the role of the nutrient composition of the surrounding milieu in shaping the tumor metabolic character.
ALTERATIONS IN METABOLITE-DRIVEN SIGNALING EVENTS
The activity of a variety of regulatory pathways can be influenced by the availability and abundance of particular metabolites. For example, cells use the AMP/ATP ratio to inform signaling events through kinases such as AMPK (Hardie, 2011). Glutamine and the glycolytic intermediate, fructose-6-phosphate, are essential substrates for the hexosamine biosynthetic pathway. The levels of these metabolites directly influence the production of UDP-N-acetylglucosamine (UDP-GlcNAc) that is essential for protein glycosylation and the surface expression of certain growth factor receptors (Wellen et al., 2010). In addition, the O-linked GlcNAc transferase (OGT) is often upregulated in a variety of cancer types and can sustain oncogenic signaling pathways through stable glycosylation on serine/threonine sites of proteins involved in signal transduction (Lynch et al., 2012; Ma et al., 2013).
Nutrient availability also regulates gene expression epigenetically through its effects on DNA and histone modifications. Acetyl-CoA and S-adenosylmethionine (SAM) are required substrates of acetyl- and methyl-transferases respectively, which add post-translational modifications to histone proteins and modulate transcription of the associated DNA. Other metabolites found to modify histone and non-histone proteins include succinyl-CoA, crotonyl-CoA and lactate (Tan et al., 2011; Zhang et al., 2019; Zhang et al., 2011), although their physiological significance remains to be further characterized. During histone deacetylation, NAD+ is a required reactant to support the enzymatic action of the Sirtuin family deacetylases. Similarly, histone and DNA methylation marks can be reversed; α-ketoglutarate is required for the function of dioxygenases including ten eleven translocation (TET) DNA hydroxylases/demethylases and JmjC domain-containing (JMJD) histone demethylases (Figure 8). Perturbations of these metabolite-dependent chromatin modifying processes can contribute to tumorigenesis. Owing to the structural similarity to α-ketoglutarate, succinate and fumarate act as competitive inhibitors of the α-ketoglutarate-dependent dioxygenases. Mutations in succinate dehydrogenase (SDH) occur frequently in familial paragangliomas, and fumarate hydratase (FH) is a tumor suppressor frequently lost in leiomyomas and renal cell cancer. Cancer-associated mutations in these genes can lead to the accumulation of succinate and fumarate respectively and the inhibition of DNA and histone demethylases. Similarly, oncogenic mutations in isocitrate dehydrogenase 1 (IDH1) or IDH2 are prevalent in glioma, acute myeloid leukemia (AML), cholangiocarcinoma and chondrosarcoma. Mutant IDH converts α-ketoglutarate to D-2-hydroxyglutarate (D-2-HG), which competitively inhibits α-ketoglutarate-dependent dioxygenases (Figure 8). As a result, these cancer types often display a hypermethylated genome that represses the cellular differentiation program and promotes tumorigenesis. A similar pattern of dioxygenase inhibition and associated with it hypermethylated epigenetic landscape can be triggered by the accumulation of the D-2-HG enantiomer L-2-hydroxyglutarate (L-2-HG). Accumulation of L-2-HG occurs independently of isocitrate dehydrogenase enzyme activity and is instead driven by the low pH-associated changes in substrate specificity of at least two dehydrogenase enzymes (specifically, lactate and malate dehydrogenase), as well as, in the context of renal cell carcinoma cells, loss of L-2-HG dehydrogenase (L2HGDH), which is required to decrease L-2-HG (Intlekofer et al. 2015; Shim et al., 2014).
Figure 8. Alterations in metabolite-driven signaling events.
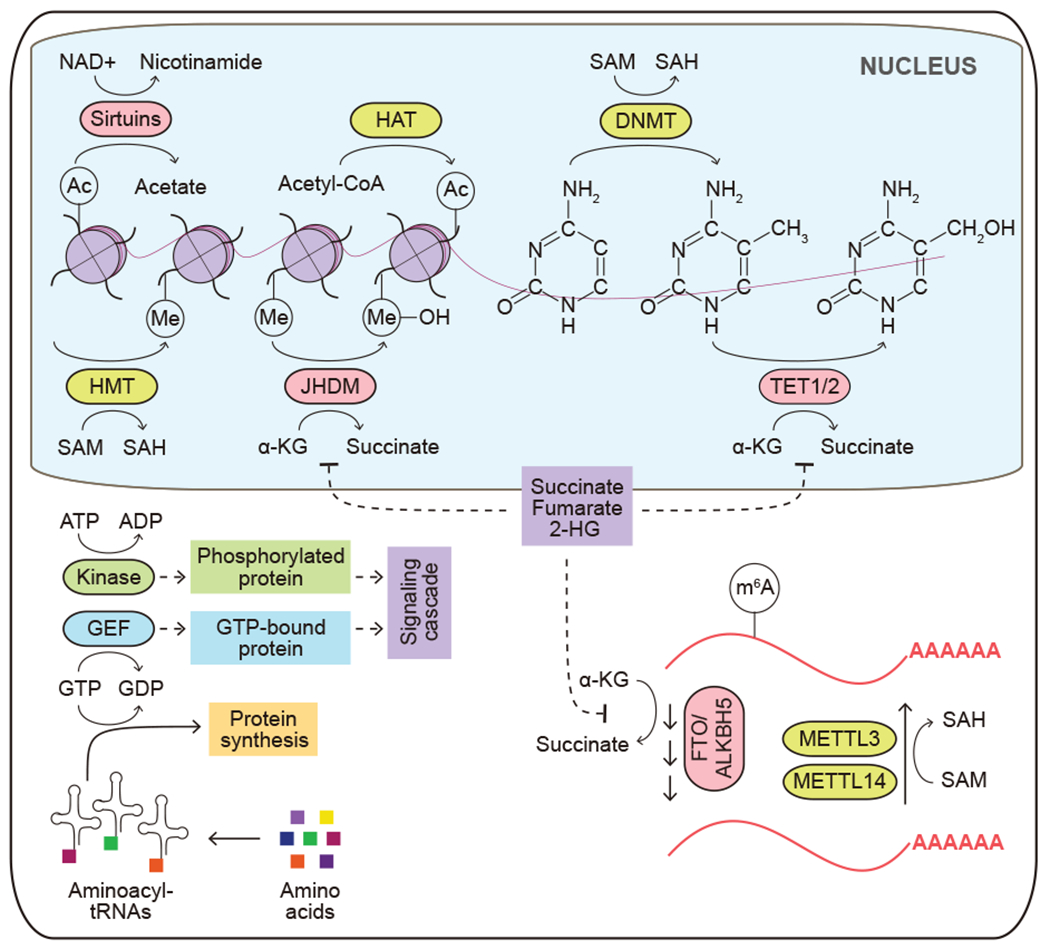
The availability and abundance of metabolites regulate signaling events and gene expression. Solid arrows depict metabolite movement or metabolic reactions. Dashed arrows depict regulatory effects of signal transduction components. HAT, histone acetyltransferase; Ac, an acetyl mark; Me, a methyl mark; HMT, histone methyltransferase; DNMT, DNA methyltransferase; JHDM, Jumonji domain-containing histone demethylase; SAM, S-adenosylmethionine; SAH, S-adenosylhomocysteine; TET1/2, ten-eleven translocation methylcytosine dioxygenase 1/2; GEF, guanine nucleotide exchange factor; FTO, fat mass and obesity-associated protein; ALKBH5, alkb homolog 5; METTL3, methyltransferase 3; METTL14, methyltransferase 14; m6A, N6-methyladenosine; α-KG, α-ketoglutarate; 2-HG, 2-hydroxyglutarate.
Metabolite-driven regulation of gene expression extends to the modification of RNA. Reduction in the SAM to S-adenosylhomocysteine (SAM/SAH) ratio following methionine restriction reduces the activity of the N6-adenosine methyltransferase METTL16, which methylates mRNA and promotes translation. In contrast, the activation of mTORC1 downstream of growth factor or oncogenic signaling can stimulate m6A mRNA methylation via Wilms’ tumor 1-associating protein (WTAP) expression and SAM synthesis, which in turn promotes tumor cell growth by enhancing mRNA translation (Cho et al., 2021; Villa et al., 2021). Interestingly, like their DNA and protein counterparts, the RNA m6A demethylases, fat mass and obesity-associated protein (FTO) and alkB homolog 5 (ALKBH5), are α-ketoglutarate-dependent dioxygenases, and their activities are promoted by the presence of α-ketoglutarate, iron and oxygen, and are inhibited by 2-hydroxyglutarate produced by mutant IDH enzymes (Kim and Lee, 2021; Su et al., 2018) (Figure 8).
Protein synthesis can be also regulated by nutrient supply independently of post-translational mRNA modifications. In amino acid-depleted environments, glutamine-specific tRNA isoacceptors became selectively uncharged, while other tRNAs retain charging of their cognate amino acids (Pavlova et al., 2020). Reduced availability of charged tRNAGln preferentially triggers the depletion of proteins containing polyglutamine tracts, which are overrepresented among the components of the gene transcription machinery, and therefore may alter cellular transcriptional output (Pavlova et al., 2020). Taken together, nutrient status and metabolite abundance regulate intracellular signaling cascade and gene expression at multiple levels, and as a result can contribute to tumorigenesis.
METABOLIC INTERACTIONS WITH THE TUMOR MICROENVIRONMENT
Metazoan tissues consist not only of cells that carry out the primary tissue function, but also of a variety of accessory, or stromal, cell types, including fibroblasts, immune cells and endothelial cells. Stromal cells play supportive roles that help maintain the tissue homeostasis, as well as coordinate tissue repair in an event of an injury. Transformed cells do not cut ties with this support network - on the contrary, their pro-proliferative state can influence stromal cell behavior, often co-opting stromal cells to engage in tissue repair activities that can promote cancer cell survival and expansion (Dvorak, 1986; Foster et al., 2018). While a complex messaging system consisting of numerous growth factors and cytokines has been implicated in this cross-talk (Tlsty and Coussens, 2006), there is a growing appreciation that the metabolic factors within the tumor microenvironment (TME) also play an important role in establishing and maintaining the wound healing-like state observed in growing tumors (Morandi et al., 2016; Schworer et al., 2019) (Figure 9).
Figure 9. Metabolic interactions with the tumor microenvironment.
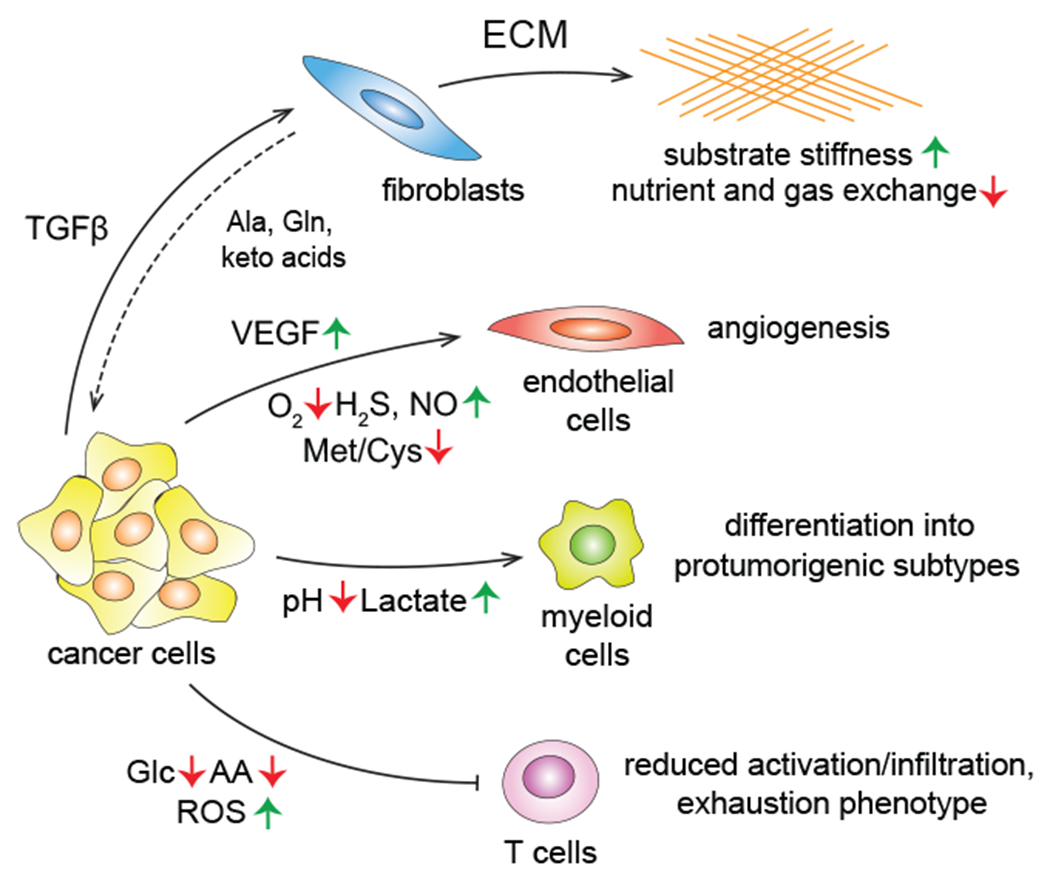
Tumor cells reshape the behavior of the surrounding stromal compartment in a cell type-specific manner using both signaling and metabolic influences. TGFβ, transforming growth factor β; ECM, extracellular matrix; Ala, alanine; VEGF, vascular endothelial growth factor; Met, methionine; Cys, cysteine; ROS, reactive oxygen species.
Tissue repair is an immense anabolic event in itself, and transformed cells are not the only cells of the TME that have an active anabolic metabolism. In fact, in some tumorigenic contexts, transformed cells represent a minority of cells within the growing tumor mass (Neesse et al., 2011). Consequently, the metabolic activity of the stromal component can be a potent shaping force in the TME. However, while the metabolic objective of transformed cells is to build more of themselves, the metabolic phenotypes of the tumor-associated stromal cells are dictated by the more circumscribed roles they play in tissue homeostasis. For instance, many solid tumor types are populated by abundant cancer-associated fibroblasts (CAFs) that are recruited from the local tissue fibroblast populations and synthesize ample quantities of extracellular matrix (ECM). Interestingly, matrix proteins are highly enriched in proline and glycine (Schworer et al., 2020), and the production of tumor matrix consumes large quantities of these amino acids. TGFβ, a potent inducer of ECM production in fibroblasts, was shown to coordinate a robust anabolic program in these cells which allows them to increase the uptake of both glucose and glutamine, promoting the synthesis of glycine from glucose via the serine synthetic pathway as well as production of proline from glutamine (Schworer et al., 2020). Thus, the anabolic metabolism of CAFs can contribute to the glucose and glutamine depletion observed in many growing tumors.
Under some circumstances, tissue-specific fibroblasts can also provide nutritional support to tumors through synthesis and/or release of non-essential amino acids or the keto acids of essential amino acids. For instance, ovarian cancer-associated fibroblasts can synthesize and release glutamine into the TME (Yang et al., 2016), while the pancreatic cancer-associated fibroblasts, or stellate cells, were found to supply the TME with alanine (Sousa et al., 2016) and the keto acids of BCAAs (Zhu et al., 2020). To support the production and/or recycling of amino acids and keto acids into the TME, it appears that some fibroblasts in growing tumors engage in the macropinocytosis of soluble extracellular proteins to help augment amino acid supplies for their production of matrix and release of surplus amino acids and keto acids into the TME (Zhang et al., 2021).
The deposition of the dense lattices of ECM can also alter the biophysical properties of the TME, resulting in the stiffening of the substrates on which cells anchor themselves to (Kalli and Stylianopoulos, 2018). Encountering a stiff, rather than a soft, substratum acts as a physical cue that alters the properties of the embedded cancer cells (Bertero et al., 2019). The excess ECM build-up can lead to the collapse of the existing capillary network within the TME (Stylianopoulos et al., 2013), which could further exacerbate the limited solute and gas exchange between the tumor and the circulation. Oxygen availability in particular is lower across a spectrum of solid tumors compared to the surrounding tissue (Muz et al., 2015) , and is a parameter that is particularly important for shaping the metabolic characteristics of the TME. Among its many effects, insufficient oxygen availability promotes enhanced glucose consumption and its conversion to lactate, increased consumption of glutamine as a result of the reliance on glutamine-dependent reductive carboxylation (Metallo et al., 2011), acidification of the tumor extracellular milieu, and resultant epigenetic changes (Intlekofer et al., 2015).
Hypoxic niches rich in lactate can induce resident innate immune cells such as type II macrophages to release VEGF, a growth factor that stimulates the endothelial cell recruitment and new capillary growth and branching (Carmona-Fontaine et al., 2017; Colegio et al., 2014). In addition to VEGF, endothelial cells integrate a number of TME-associated metabolic stimuli into their decision-making. Among angiogenesis-promoting metabolic stimuli is the buildup of gasotransmitters such as arginine-derived nitric oxide (Gallo et al., 1998) and cysteine-derived hydrogen sulfide, as well as amino acid depletion in the TME (Longchamp et al., 2018). Taken together, the diversity of the TME-shaping stimuli and their effects therefore result in a complex and dynamic picture, in which the activities of various TME-associated factors either help deplete or restore supplies of nutrients to the tumor.
For effector T cells - the immune cell population at the forefront of the antitumor immune attack - the nutrient-depleted metabolic terrain of the TME is particularly restricting. Upon antigen stimulation, T lymphocytes switch from quiescence to a robustly anabolic state – an adaptation that fuels their proliferation and differentiation into tumor-killing effector cells. As a result, nutrient depletion can derail T cell activation and function more easily than other cell types. For example, T cells are markedly dependent on the exogenous uptake of non-essential amino acids – such as serine (Ma et al., 2017) and alanine (Ron-Harel et al., 2019) in lieu of an ability to synthesize these nutrients de novo. Moreover, T cell activation is also exquisitely sensitive to the depletion of glutamine and glucose (Ho et al., 2015). In addition, effector T cells are sensitive to increased levels of oxidative stress in the TME, which can trigger a so-called exhaustion phenotype (Vardhana et al., 2020). Moreover, transformed cells and their non-transformed TME associates, such as type II macrophages, further help suppress the T cell populations by engaging in elimination of nutrients required to sustain T cell effector function. To this end, both cancer cells and tumor-associated macrophages were shown to secrete amino acid-catabolic enzymes indoleamine 2,3-dioxygenase 1 (IDO1) that degrade tryptophan, which further hampers T cell effector function and, similarly to glucose depletion, leads to the emergence of the immunosuppressive Treg population (McGaha et al., 2012).
Given that tumors with low effector T cell populations fail to respond to immune checkpoint inhibitor therapies (Mariathasan et al., 2018), normalizing the metabolic state of the TME to counteract the tumor-promoting cell populations and enhance the tumoricidal activities of the T cells has potential to enhance the success and the durability of immunotherapy. Thus, a recent study has shown that blocking glutamine utilization by mammary tumor cells via GLS1 deletion can increase glutamine levels in the TME by almost an order of magnitude, thereby promoting the T cell antioxidant capacity and enhancing their infiltration into the tumor (Edwards et al., 2021). Similarly, tumor-inhibitory effects have been reported with inhibitors of IDO1-mediated tryptophan degradation (Tang et al., 2021).
Taken together, the TME represents a complex metabolic ecosystem in which multiple metabolically active cell types contribute to the emergence of a tumor-specific metabolic milieu that in turn, modulates the behavior of its cellular components and facilitates tumor expansion. Though numerous metabolic interactions between the individual components of the tumor have been described, the complete rulebook of the TME metabolic economy continues to be elucidated. For instance, in vivo metabolic tracer experiments in a mouse model of a colorectal carcinoma have recently revealed that TME-residing cells do not utilize nutrients in equal measure – that is, transformed cells were found to be the dominant consumers of glutamine but the lion’s share of glucose went to cells of the myeloid lineage, such as macrophages (Reinfeld et al., 2021). Finally, new cell types continue to be added to the cast of characters within the TME that support the anabolic metabolism of transformed cells. Among such cell types are cancer-associated adipocytes, which can serve as providers of fatty acids in melanoma (Zhang et al., 2018), as well as tumor-associated neurons, which were found to act as an unexpected source of serine for the pancreatic cancer cells (Banh et al., 2020).
INTEGRATION INTO THE WHOLE-BODY METABOLIC ECONOMY
There is increasing evidence that carcinogenesis is influenced by organismal metabolic regulation. Thus, metabolic changes associated with obesity and type II diabetes appear to play an important role in facilitating tumorigenesis. In addition, an aging-associated increase in serum methylmalonic acid has recently been implicated in directly promoting the aggressive behavior of cancer cells (Gomes et al., 2020). Furthermore, some tumors display an ability to influence the metabolic state of an entire organism through mechanisms that are only just beginning to be unraveled (Figure 10). Determining the mechanisms underlying these observations may help instruct strategies for cancer prevention, early detection and novel therapeutic interventions.
Figure 10. Integration into the whole-body metabolic economy.
Tumors co-opt the signals from systemic metabolic regulatory molecules and release a variety of soluble factors to modulate the nutrient storage and release across a spectrum of metabolic organs. IGF-I, insulin-like growth factor I. Created with BioRender.
Compelling epidemiologic evidence indicates that obesity and type II diabetes are prominent risk factors for numerous types of cancer (Arnold et al., 2015; Gallagher and LeRoith, 2015). Fasting plasma glucose levels are characteristically elevated (up to 7 mM from a healthy 4-5 mM) in individuals with type II diabetes. By itself, this increase is unlikely to provide cancer cells with extra glucose. This is because GLUT1, the major glucose transporter expressed by cancer cells, already operates at a maximal capacity even at physiologic levels of glucose (Gorovits and Charron, 2003). Thus, the access of cancer cells to glucose is unlikely to be directly impacted by abnormally elevated plasma glucose.
Even at high levels of extracellular glucose, the influx of glucose into most cells is controlled primarily by the ligand-mediated activation of cell- and tissue-specific receptor tyrosine kinases, which in turn, regulate the relative expression and cell surface localization of GLUT1. While tissue-specific metabolism is controlled by locally produced growth factors, such as EGF, PDGF, or FGF, nutrient utilization on the organismal scale is governed by systemically circulating factors that act upon a variety of tissues at the same time. These factors include insulin, which is produced almost exclusively by the pancreas, as well as insulin-like growth factor (IGF-I), which is produced by the liver as well as a variety of local tissue sites and influenced by central nervous system production of growth hormone (Pollak, 2012).
As the ability of insulin to facilitate glucose uptake into insulin-dependent tissues such as muscle, becomes impaired in individuals with metabolic syndrome and type II diabetes, circulating levels of insulin and IGF-I undergo a compensatory increase. Compelling genetic and pharmacologic evidence indicates that systemic increases in IGF-I in particular play a prominent role in tumorigenesis. Indeed, though they are rarely mutated, receptors for IGF1 are frequently overactive in tumors – e.g. up to 50% of breast cancers were found to show signs of increased IGF-I receptor activity, which is associated with poor survival (Law et al., 2008). Moreover, the growth of orthotopically transplanted and carcinogen-induced tumors was found to be enhanced in mice with elevated IGF-I levels (de Ostrovich et al., 2008; Wu et al., 2002). Loss-of-function genetic evidence from both animal models and human populations also lends further support for the involvement of the systemic IGF-I signaling in tumorigenesis. Thus, a homozygous deletion of IGF-I receptor or a dual ablation of its proximal effectors IRS1 and IRS2 were shown to markedly delay Kras-driven tumorigenesis in mice (Klinakis et al., 2009; Xu et al., 2018). Evidence from human population studies also implicates IGF-I signaling in tumorigenesis. Indeed, cancer is all but absent among individuals with congenital IGF-I deficiency syndromes, such as Laron syndrome (Steuerman et al., 2011).
Owing to the fact that systemic insulin and IGF-I release is governed primarily by food intake, dietary modifications can help reduce the circulating levels of both of these factors. Indeed, Peyton Rous’ experiments with underfed animals in 1914 provided early experimental evidence that reduced caloric intake has potential to attenuate tumor growth (Rous, 1914). Notably, caloric restriction-mediated attenuated tumorigenesis in animals could be readily reverted by injecting recombinant IGF-I (Dunn et al., 1997), further implicating the reduction in IGF-I levels as a key factor in diet-mediated attenuation of tumor growth. Insulin/IGF-I-normalizing dietary strategies – such as ketogenic (i.e., very low carbohydrate) diet – were shown to achieve similar tumor inhibitory effects in various mouse tumor models (Ho et al., 2011; Venkateswaran et al., 2007). On a population level, treatments to lower extracellular glucose and insulin levels, such as metformin, have been shown to reduce the incidence of cancer in diabetic patients (Noto et al., 2012). However, not all tumors are sensitive to caloric restriction or glucose reduction. In particular, tumors with activating mutations in PI-3 kinase were not affected by dietary restriction in animals, indicating that tumor genetic identity may play a key role in the extent to which tumors rely on systemic nutrient uptake signals (Kalaany and Sabatini, 2009).
In addition to co-opting the inputs from the organism-wide metabolic governance factors, some tumors seem to also influence the metabolic set point of the entire body. The most prominent manifestation of a tumor-orchestrated takeover of the whole-body metabolism is a multi-organ catabolic state of cachexia (Porporato, 2016). Cachexia is characterized with loss of insulin sensitivity across a number of tissues, progressive wasting of skeletal muscle, heart, liver, adipose and brain tissue, absence of appetite and immune system suppression. Though not an exclusively cancer-associated phenomenon – i.e., severe trauma or AIDS are also associated with cachexia – cachexia frequently accompanies cancer at its advanced stages, and is purported to be a direct cause of death from cancer in at least 20% of patients (Porporato, 2016). Cachexia can be induced by a number of circulating factors, including TNFα (originally named cachectin), inflammatory cytokines and glucocorticoid hormones. Whether these or other factors are directly or indirectly responsible for the cachexia associated with specific cancer types remains an area of active investigation.
Recently, tumors have also been shown to influence the metabolic state of an organism is by scrambling normal circadian oscillations in metabolic gene expression in the liver (Hojo et al., 2017; Masri et al., 2016) and in the adipose tissue (Tsoli et al., 2014). Whether disruption in the metabolic cycling that accompanies the diurnal cycle contributes to either cancer growth or the catabolic state exhibited by liver, muscle and fat during cancer progression remains to be determined.
CONCLUSIONS
Metabolic changes associated with tumorigenesis allow transformed cells to sustain aberrant accumulation and colonize diverse tissues by escaping tissue homeostatic defenses and co-opting both the internal signaling mechanisms as well as a spectrum of local tissue and whole-body resources. Importantly, not only transformed cells themselves, but also the stromal cells within the TME, as well as metabolic balance of the entire organism become remodeled in cancer, altogether promoting cancer cell accumulation and dissemination, reducing the ability of immune system to counteract tumor growth and directly contributing to cancer-associated lethality. Dissecting the metabolic adaptations tumors rely on to promote these changes and to sustain growth even in metabolically unfavorable environments can help instruct novel therapeutic and, potentially, dietary combinations that may synergize with existing therapeutic interventions such as chemotherapy, targeted inhibitors and immune checkpoint blockade approaches.
ACKNOWLEDGEMENTS
We thank Dr. Aggie Bielska for critical reading of the manuscript and the rest of the members of the Thompson laboratory for valuable discussions. This work was supported by the NIH Grant R01 CA248355-01 and Memorial Sloan Kettering Cancer Center Support Grant/Core Grant P30 CA008748. Image templates for Figure 10 are from BioRender (https://biorender.com).
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
DECLARATION OF INTERESTS
C.B.T. is a founder of Agios Pharmaceuticals and a member of its scientific advisory board. He is also a former member of the Board of Directors and stockholder of Merck and Charles River Laboratories. He holds patents related to cellular metabolism. He is a member of the advisory board of Cell Metabolism. N.N.P. and J.Z. declare no competing interests.
Bibliography and References
- Aceto N, Bardia A, Miyamoto DT, Donaldson MC, Wittner BS, Spencer JA, Yu M, Pely A, Engstrom A, Zhu H, et al. (2014). Circulating tumor cell clusters are oligoclonal precursors of breast cancer metastasis. Cell 158, 1110–1122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ackerman D, Tumanov S, Qiu B, Michalopoulou E, Spata M, Azzam A, Xie H, Simon MC, and Kamphorst JJ (2018). Triglycerides Promote Lipid Homeostasis during Hypoxic Stress by Balancing Fatty Acid Saturation. Cell Rep 24, 2596–2605 e2595. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Andrzejewski S, Klimcakova E, Johnson RM, Tabaries S, Annis MG, McGuirk S, Northey JJ, Chenard V, Sriram U, Papadopoli DJ, et al. (2017). PGC-1alpha Promotes Breast Cancer Metastasis and Confers Bioenergetic Flexibility against Metabolic Drugs. Cell Metab 26, 778–787 e775 [DOI] [PubMed] [Google Scholar]
- Arnold M, Pandeya N, Byrnes G, Renehan PAG, Stevens GA, Ezzati PM, Ferlay J, Miranda JJ, Romieu I, Dikshit R, et al. (2015). Global burden of cancer attributable to high body-mass index in 2012: a population-based study. Lancet Oncol 16, 36–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avissar NE, Sax HC, and Toia L (2008). In human entrocytes, GLN transport and ASCT2 surface expression induced by short-term EGF are MAPK, PI3K, and Rho-dependent. Dig Dis Sci 53, 2113–2125. [DOI] [PubMed] [Google Scholar]
- Banh RS, Biancur DE, Yamamoto K, Sohn ASW, Walters B, Kuljanin M, Gikandi A, Wang H, Mancias JD, Schneider RJ, et al. (2020). Neurons Release Serine to Support mRNA Translation in Pancreatic Cancer. Cell 183, 1202–1218 e1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bar-Sagi D, and Feramisco JR (1986). Induction of membrane ruffling and fluid-phase pinocytosis in quiescent fibroblasts by ras proteins. Science 233, 1061–1068. [DOI] [PubMed] [Google Scholar]
- Barthel A, Okino ST, Liao J, Nakatani K, Li J, Whitlock JP Jr., and Roth RA (1999). Regulation of GLUT1 gene transcription by the serine/threonine kinase Akt1. J Biol Chem 274, 20281–20286. [DOI] [PubMed] [Google Scholar]
- Bauer DE, Hatzivassiliou G, Zhao F, Andreadis C, and Thompson CB (2005). ATP citrate lyase is an important component of cell growth and transformation. Oncogene 24, 6314–6322. [DOI] [PubMed] [Google Scholar]
- Bersuker K, Hendricks JM, Li Z, Magtanong L, Ford B, Tang PH, Roberts MA, Tong B, Maimone TJ, Zoncu R, et al. (2019). The CoQ oxidoreductase FSP1 acts parallel to GPX4 to inhibit ferroptosis. Nature 575, 688–692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bertero T, Oldham WM, Grasset EM, Bourget I, Boulter E, Pisano S, Hofman P, Bellvert F, Meneguzzi G, Bulavin DV, et al. (2019). Tumor-Stroma Mechanics Coordinate Amino Acid Availability to Sustain Tumor Growth and Malignancy. Cell Metab 29, 124–140 e110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Birsoy K, Wang T, Chen WW, Freinkman E, Abu-Remaileh M, and Sabatini DM (2015). An Essential Role of the Mitochondrial Electron Transport Chain in Cell Proliferation Is to Enable Aspartate Synthesis. Cell 162, 540–551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boedtkjer E, and Pedersen SF (2020). The Acidic Tumor Microenvironment as a Driver of Cancer. Annu Rev Physiol 82, 103–126. [DOI] [PubMed] [Google Scholar]
- Boros LG, Lee PW, Brandes JL, Cascante M, Muscarella P, Schirmer WJ, Melvin WS, and Ellison EC (1998). Nonoxidative pentose phosphate pathways and their direct role in ribose synthesis in tumors: is cancer a disease of cellular glucose metabolism? Med Hypotheses 50, 55–59. [DOI] [PubMed] [Google Scholar]
- Bott AJ, Maimouni S, and Zong WX (2019a). The Pleiotropic Effects of Glutamine Metabolism in Cancer. Cancers (Basel) 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott AJ, Peng IC, Fan Y, Faubert B, Zhao L, Li J, Neidler S, Sun Y, Jaber N, Krokowski D, et al. (2015). Oncogenic Myc Induces Expression of Glutamine Synthetase through Promoter Demethylation. Cell Metab 22, 1068–1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bott AJ, Shen J, Tonelli C, Zhan L, Sivaram N, Jiang YP, Yu X, Bhatt V, Chiles E, Zhong H, et al. (2019b). Glutamine Anabolism Plays a Critical Role in Pancreatic Cancer by Coupling Carbon and Nitrogen Metabolism. Cell Rep 29, 1287–1298 e1286. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bratslavsky G, Sudarshan S, Neckers L, and Linehan WM (2007). Pseudohypoxic pathways in renal cell carcinoma. Clin Cancer Res 13, 4667–4671. [DOI] [PubMed] [Google Scholar]
- Brown MS, and Goldstein JL (1979). Receptor-mediated endocytosis: insights from the lipoprotein receptor system. Proc Natl Acad Sci U S A 76, 3330–3337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bu P, Chen KY, Xiang K, Johnson C, Crown SB, Rakhilin N, Ai Y, Wang L, Xi R, Astapova I, et al. (2018). Aldolase B-Mediated Fructose Metabolism Drives Metabolic Reprogramming of Colon Cancer Liver Metastasis. Cell Metab 27, 1249–1262 e1244. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calvert AE, Chalastanis A, Wu Y, Hurley LA, Kouri FM, Bi Y, Kachman M, May JL, Bartom E, Hua Y, et al. (2017). Cancer-Associated IDH1 Promotes Growth and Resistance to Targeted Therapies in the Absence of Mutation. Cell Rep 19, 1858–1873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carmona-Fontaine C, Deforet M, Akkari L, Thompson CB, Joyce JA, and Xavier JB (2017). Metabolic origins of spatial organization in the tumor microenvironment. Proc Natl Acad Sci U S A 114, 2934–2939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cavalli LR, Varella-Garcia M, and Liang BC (1997). Diminished tumorigenic phenotype after depletion of mitochondrial DNA. Cell Growth Differ 8, 1189–1198. [PubMed] [Google Scholar]
- Chandel NS (2021). Metabolism of Proliferating Cells. Cold Spring Harb Perspect Biol 13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Xia S, Wang M, Lin R, Li Y, Mao H, Aguiar M, Famulare CA, Shih AH, Brennan CW, et al. (2019a). Mutant and Wild-Type Isocitrate Dehydrogenase 1 Share Enhancing Mechanisms Involving Distinct Tyrosine Kinase Cascades in Cancer. Cancer Discov 9, 756–777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen PH, Cai L, Huffman K, Yang C, Kim J, Faubert B, Boroughs L, Ko B, Sudderth J, McMillan EA, et al. (2019b). Metabolic Diversity in Human Non-Small Cell Lung Cancer Cells. Mol Cell 76, 838–851 e835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheng T, Sudderth J, Yang C, Mullen AR, Jin ES, Mates JM, and DeBerardinis RJ (2011). Pyruvate carboxylase is required for glutamine-independent growth of tumor cells. Proc Natl Acad Sci U S A 108, 8674–8679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheung EC, DeNicola GM, Nixon C, Blyth K, Labuschagne CF, Tuveson DA, and Vousden H. (2020). Dynamic ROS Control by TIGAR Regulates the Initiation and Progression of Pancreatic Cancer. Cancer Cell 37, 168–182 e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cho S, Lee G, Pickering BF, Jang C, Park JH, He L, Mathur L, Kim SS, Jung S, Tang HW, et al. (2021). mTORC1 promotes cell growth via m(6)A-dependent mRNA degradation. Mol Cell 81, 2064–2075 e2068. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chourasia AH, Boland ML, and Macleod KF (2015). Mitophagy and cancer. Cancer Metab 3, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chowdhry S, Zanca C, Rajkumar U, Koga T, Diao Y, Raviram R, Liu F, Turner K, Yang H, Brunk E, et al. (2019). NAD metabolic dependency in cancer is shaped by gene amplification and enhancer remodelling. Nature 569, 570–575. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Christen S, Lorendeau D, Schmieder R, Broekaert D, Metzger K, Veys K, Elia I, Buescher JM, Orth MF, Davidson SM, et al. (2016). Breast Cancer-Derived Lung Metastases Show Increased Pyruvate Carboxylase-Dependent Anaplerosis. Cell Rep 17, 837–848. [DOI] [PubMed] [Google Scholar]
- Colegio OR, Chu NQ, Szabo AL, Chu T, Rhebergen AM, Jairam V, Cyrus N, Brokowski CE, Eisenbarth SC, Phillips GM, et al. (2014). Functional polarization of tumour-associated macrophages by tumour-derived lactic acid. Nature 513, 559–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Coloff JL, Murphy JP, Braun CR, Harris IS, Shelton LM, Kami K, Gygi SP, Selfors LM, and Brugge JS (2016). Differential Glutamate Metabolism in Proliferating and Quiescent Mammary Epithelial Cells. Cell Metab 23, 867–880. [DOI] [PubMed] [Google Scholar]
- Commisso C (2019). The pervasiveness of macropinocytosis in oncological malignancies. Philos Trans R Soc Lond B Biol Sci 374, 20180153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Commisso C, Davidson SM, Soydaner-Azeloglu RG, Parker SJ, Kamphorst JJ, Hackett S, Grabocka E, Nofal M, Drebin JA, Thompson CB, et al. (2013). Macropinocytosis of protein is an amino acid supply route in Ras-transformed cells. Nature 497, 633–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Courtney KD, Bezwada D, Mashimo T, Pichumani K, Vemireddy V, Funk AM, Wimberly J, McNeil SS, Kapur P, Lotan Y, et al. (2018). Isotope Tracing of Human Clear Cell Renal Cell Carcinomas Demonstrates Suppressed Glucose Oxidation In Vivo. Cell Metab 28, 793–800 e792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cox AG, Hwang KL, Brown KK, Evason K, Beltz S, Tsomides A, O’Connor K, Galli GG, Yimlamai D, Chhangawala S, et al. (2016). Yap reprograms glutamine metabolism to increase nucleotide biosynthesis and enable liver growth. Nat Cell Biol 18, 886–896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Daemen A, Liu B, Song K, Kwong M, Gao M, Hong R, Nannini M, Peterson D, Liederer BM, de la Cruz C, et al. (2018). Pan-Cancer Metabolic Signature Predicts Co-Dependency on Glutaminase and De Novo Glutathione Synthesis Linked to a High-Mesenchymal Cell State. Cell Metab 28, 383–399 e389. [DOI] [PubMed] [Google Scholar]
- Davidson SM, Papagiannakopoulos T, Olenchock BA, Heyman JE, Keibler MA, Luengo A, Bauer MR, Jha AK, O’Brien JP, Pierce KA, et al. (2016). Environment Impacts the Metabolic Dependencies of Ras-Driven Non-Small Cell Lung Cancer. Cell Metab 23, 517–528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- De Domenico I, McVey Ward D, and Kaplan J (2008). Regulation of iron acquisition and storage: consequences for iron-linked disorders. Nat Rev Mol Cell Biol 9, 72–81. [DOI] [PubMed] [Google Scholar]
- de Ostrovich KK, Lambertz I, Colby JK, Tian J, Rundhaug JE, Johnston D, Conti CJ, DiGiovanni J, and Fuchs-Young R (2008). Paracrine overexpression of insulin-like growth factor-1 enhances mammary tumorigenesis in vivo. Am J Pathol 173, 824–834. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeBerardinis RJ, and Chandel NS (2020). We need to talk about the Warburg effect. Nat Metab 2, 127–129. [DOI] [PubMed] [Google Scholar]
- Delage B, Fennell DA, Nicholson L, McNeish I, Lemoine NR, Crook T, and Szlosarek PW (2010). Arginine deprivation and argininosuccinate synthetase expression in the treatment of cancer. Int J Cancer 126, 2762–2772. [DOI] [PubMed] [Google Scholar]
- Delarue M, Brittingham GP, Pfeffer S, Surovtsev IV, Pinglay S, Kennedy KJ, Schaffer M, Gutierrez JI, Sang D, Poterewicz G, et al. (2018). mTORC1 Controls Phase Separation and the Biophysical Properties of the Cytoplasm by Tuning Crowding. Cell 174, 338–349 e320. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Diebold LP, Gil HJ, Gao P, Martinez CA, Weinberg SE, and Chandel NS (2019). Mitochondrial complex III is necessary for endothelial cell proliferation during angiogenesis. Nat Metab 1, 158–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doll S, Freitas FP, Shah R, Aldrovandi M, da Silva MC, Ingold I, Goya Grocin A, Xavier da Silva TN, Panzilius E, Scheel CH, et al. (2019). FSP1 is a glutathione-independent ferroptosis suppressor. Nature 575, 693–698. [DOI] [PubMed] [Google Scholar]
- Dunn SE, Kari FW, French J, Leininger JR, Travlos G, Wilson R, and Barrett JC (1997). Dietary restriction reduces insulin-like growth factor I levels, which modulates apoptosis, cell proliferation, and tumor progression in p53-deficient mice. Cancer Res 57, 4667–4672. [PubMed] [Google Scholar]
- Dupuy F, Tabaries S, Andrzejewski S, Dong Z, Blagih J, Annis MG, Omeroglu A, Gao D, Leung S, Amir E, et al. (2015). PDK1-Dependent Metabolic Reprogramming Dictates Metastatic Potential in Breast Cancer. Cell Metab 22, 577–589. [DOI] [PubMed] [Google Scholar]
- Dvorak HF (1986). Tumors: wounds that do not heal. Similarities between tumor stroma generation and wound healing. N Engl J Med 315, 1650–1659. [DOI] [PubMed] [Google Scholar]
- Edinger AL, and Thompson CB (2002). Akt maintains cell size and survival by increasing mTOR-dependent nutrient uptake. Mol Biol Cell 13, 2276–2288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Edwards DN, Ngwa VM, Raybuck AL, Wang S, Hwang Y, Kim LC, Cho SH, Paik Y, Wang Q, Zhang S, et al. (2021). Selective glutamine metabolism inhibition in tumor cells improves antitumor T lymphocyte activity in triple-negative breast cancer. J Clin Invest 131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Epstein T, Gatenby RA, and Brown JS (2017). The Warburg effect as an adaptation of cancer cells to rapid fluctuations in energy demand. PLoS One 12, e0185085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fan J, Ye J, Kamphorst JJ, Shlomi T, Thompson CB, and Rabinowitz JD (2014). Quantitative flux analysis reveals folate-dependent NADPH production. Nature 510, 298–302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Faubert B, Li KY, Cai L, Hensley CT, Kim J, Zacharias LG, Yang C, Do QN, Doucette S, Burguete D, et al. (2017). Lactate Metabolism in Human Lung Tumors. Cell 171, 358–371 e359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes LM, Al-Dwairi A, Simmen RCM, Marji M, Brown DM, Jewell SW, and Simmen FA (2018). Malic Enzyme 1 (ME1) is pro-oncogenic in Apc(Min/+) mice. Sci Rep 8, 14268. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraro GB, Ali A, Luengo A, Kodack DP, Deik A, Abbott KL, Bezwada D, Blanc L, Prideaux B, Jin X, et al. (2021). Fatty Acid Synthesis Is Required for Breast Cancer Brain Metastasis. Nat Cancer 2, 414–428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fitzgerald G, Soro-Arnaiz I, and De Bock K (2018). The Warburg Effect in Endothelial Cells and its Potential as an Anti-angiogenic Target in Cancer. Front Cell Dev Biol 6, 100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Foster DS, Jones RE, Ransom RC, Longaker MT, and Norton JA (2018). The evolving relationship of wound healing and tumor stroma. JCI Insight 3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Frauwirth KA, Riley JL, Harris MH, Parry RV, Rathmell JC, Plas DR, Elstrom RL, June CH, and Thompson CB (2002). The CD28 signaling pathway regulates glucose metabolism. Immunity 16, 769–777. [DOI] [PubMed] [Google Scholar]
- Gallagher EJ, and LeRoith D (2015). Obesity and Diabetes: The Increased Risk of Cancer and Cancer-Related Mortality. Physiol Rev 95, 727–748. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gallo O, Masini E, Morbidelli L, Franchi A, Fini-Storchi I, Vergari WA, and Ziche M (1998). Role of nitric oxide in angiogenesis and tumor progression in head and neck cancer. J Natl Cancer Inst 90, 587–596. [DOI] [PubMed] [Google Scholar]
- Gao P, Tchernyshyov I, Chang TC, Lee YS, Kita K, Ochi T, Zeller KI, De Marzo AM, Van Eyk JE, Mendell JT, et al. (2009). c-Myc suppression of miR-23a/b enhances mitochondrial glutaminase expression and glutamine metabolism. Nature 458, 762–765. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaude E, and Frezza C (2016). Tissue-specific and convergent metabolic transformation of cancer correlates with metastatic potential and patient survival. Nat Commun 7, 13041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ghergurovich JM, Lang JD, Levin MK, Briones N, Facista SJ, Mueller C, Cowan AJ, McBride MJ, Rodriguez ESR, Killian A, et al. (2021). Local production of lactate, ribose phosphate, and amino acids within human triple-negative breast cancer. Med (N Y) 2, 736–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gomes AP, Ilter D, Low V, Endress JE, Fernandez-Garcia J, Rosenzweig A, Schild T, Broekaert D, Ahmed A, Planque M, et al. (2020). Age-induced accumulation of methylmalonic acid promotes tumour progression. Nature 585, 283–287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorovits N, and Charron MJ (2003). What we know about facilitative glucose transporters - Lessons from cultured cells, animal models, and human studies. Biochem Mol Biol Edu 31, 163–172. [Google Scholar]
- Gromova I, Gromov P, Honma N, Kumar S, Rimm D, Talman ML, Wielenga VT, and Moreira JM (2015). High level PHGDH expression in breast is predominantly associated with keratin 5-positive cell lineage independently of malignancy. Mol Oncol 9, 1636–1654. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Habib E, Linher-Melville K, Lin HX, and Singh G (2015). Expression of xCT and activity of system xc(−) are regulated by NRF2 in human breast cancer cells in response to oxidative stress. Redox Biol 5, 33–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hamann JC, Surcel A, Chen R, Teragawa C, Albeck JG, Robinson DN, and Overholtzer M (2017). Entosis Is Induced by Glucose Starvation. Cell Rep 20, 201–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hardie DG (2011). AMP-activated protein kinase: an energy sensor that regulates all aspects of cell function. Genes Dev 25, 1895–1908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hensley CT, Faubert B, Yuan Q, Lev-Cohain N, Jin E, Kim J, Jiang L, Ko B, Skelton R, Loudat L, et al. (2016). Metabolic Heterogeneity in Human Lung Tumors. Cell 164, 681–694. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho PC, Bihuniak JD, Macintyre AN, Staron M, Liu X, Amezquita R, Tsui YC, Cui G, Micevic G, Perales JC, et al. (2015). Phosphoenolpyruvate Is a Metabolic Checkpoint of Anti-tumor T Cell Responses. Cell 162, 1217–1228. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ho VW, Leung K, Hsu A, Luk B, Lai J, Shen SY, Minchinton AI, Waterhouse D, Bally MB, Lin W, et al. (2011). A low carbohydrate, high protein diet slows tumor growth and prevents cancer initiation. Cancer Res 71, 4484–4493. [DOI] [PubMed] [Google Scholar]
- Hojo H, Enya S, Arai M, Suzuki Y, Nojiri T, Kangawa K, Koyama S, and Kawaoka S (2017). Remote reprogramming of hepatic circadian transcriptome by breast cancer. Oncotarget 8, 34128–34140. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoxhaj G, Ben-Sahra I, Lockwood SE, Timson RC, Byles V, Henning GT, Gao P, Selfors LM, Asara JM, and Manning BD (2019). Direct stimulation of NADP(+) synthesis through Akt-mediated phosphorylation of NAD kinase. Science 363, 1088–1092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu J, Locasale JW, Bielas JH, O’Sullivan J, Sheahan K, Cantley LC, Vander Heiden MG, and Vitkup D (2013). Heterogeneity of tumor-induced gene expression changes in the human metabolic network. Nat Biotechnol 31, 522–529. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hui S, Ghergurovich JM, Morscher RJ, Jang C, Teng X, Lu W, Esparza LA, Reya T, Le Z, Yanxiang Guo J, et al. (2017). Glucose feeds the TCA cycle via circulating lactate. Nature 551, 115–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Intlekofer AM, Dematteo RG, Venneti S, Finley LW, Lu C, Judkins AR, Rustenburg AS, Grinaway PB, Chodera JD, Cross JR, et al. (2015). Hypoxia Induces Production of L-2-Hydroxyglutarate. Cell Metab 22, 304–311. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jang C, Hui S, Zeng X, Cowan AJ, Wang L, Chen L, Morscher RJ, Reyes J, Frezza C, Hwang HY, et al. (2019). Metabolite Exchange between Mammalian Organs Quantified in Pigs. Cell Metab 30, 594–606 e593. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang L, Shestov AA, Swain P, Yang C, Parker SJ, Wang QA, Terada LS, Adams ND, McCabe MT, Pietrak B, et al. (2016). Reductive carboxylation supports redox homeostasis during anchorage-independent growth. Nature 532, 255–258. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang X, Stockwell BR, and Conrad M (2021). Ferroptosis: mechanisms, biology and role in disease. Nat Rev Mol Cell Biol 22, 266–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalaany NY, and Sabatini DM (2009). Tumours with PI3K activation are resistant to dietary restriction. Nature 458, 725–731. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalli M, and Stylianopoulos T (2018). Defining the Role of Solid Stress and Matrix Stiffness in Cancer Cell Proliferation and Metastasis. Front Oncol 8, 55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Cross JR, Fan J, de Stanchina E, Mathew R, White EP, Thompson CB, and Rabinowitz JD (2013). Hypoxic and Ras-transformed cells support growth by scavenging unsaturated fatty acids from lysophospholipids. Proc Natl Acad Sci U S A 110, 8882–8887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kamphorst JJ, Nofal M, Commisso C, Hackett SR, Lu W, Grabocka E, Vander Heiden MG, Miller G, Drebin JA, Bar-Sagi D, et al. (2015). Human pancreatic cancer tumors are nutrient poor and tumor cells actively scavenge extracellular protein. Cancer Res 75, 544–553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaplon J, Zheng L, Meissl K, Chaneton B, Selivanov VA, Mackay G, van der Burg SH, Verdegaal EM, Cascante M, Shlomi T, et al. (2013). A key role for mitochondrial gatekeeper pyruvate dehydrogenase in oncogene-induced senescence. Nature 498, 109–112. [DOI] [PubMed] [Google Scholar]
- Kierans SJ, and Taylor CT (2021). Regulation of glycolysis by the hypoxia-inducible factor (HIF): implications for cellular physiology. J Physiol 599, 23–37. [DOI] [PubMed] [Google Scholar]
- Kim J, Hu Z, Cai L, Li K, Choi E, Faubert B, Bezwada D, Rodriguez-Canales J, Villalobos P, Lin YF, et al. (2017). CPS1 maintains pyrimidine pools and DNA synthesis in KRAS/LKB1-mutant lung cancer cells. Nature 546, 168–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim J, and Lee G (2021). Metabolic Control of m(6)A RNA Modification. Metabolites 11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim SM, Nguyen TT, Ravi A, Kubiniok P, Finicle BT, Jayashankar V, Malacrida L, Hou J, Robertson J, Gao D, et al. (2018). PTEN Deficiency and AMPK Activation Promote Nutrient Scavenging and Anabolism in Prostate Cancer Cells. Cancer Discov 8, 866–883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klinakis A, Szabolcs M, Chen G, Xuan S, Hibshoosh H, and Efstratiadis A (2009). Igf1r as a therapeutic target in a mouse model of basal-like breast cancer. Proc Natl Acad Sci U S A 106, 2359–2364. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Knott SRV, Wagenblast E, Khan S, Kim SY, Soto M Wagner M, Turgeon MO, Fish L, Erard N, Gable AL, et al. (2018). Asparagine bioavailability governs metastasis in a model of breast cancer. Nature 554, 378–381. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kondo H, Ratcliffe CDH, Hooper S, Ellis J, MacRae JI, Hennequart M, Dunsby CW, Anderson KI, and Sahai E (2021). Single-cell resolved imaging reveals intra-tumor heterogeneity in glycolysis, transitions between metabolic states, and their regulatory mechanisms. Cell Rep 34, 108750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kory N, Uit de Bos J, van der Rijt S, Jankovic N, Gura M, Arp N, Pena IA, Prakash G, Chan SH, Kunchok T, et al. (2020). MCART1/SLC25A51 is required for mitochondrial NAD transport. Sci Adv 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Krall AS, Mullen PJ, Surjono F, Momcilovic M, Schmid EW, Halbrook CJ, Thambundit A, Mittelman SD, Lyssiotis CA, Shackelford DB, et al. (2021). Asparagine couples mitochondrial respiration to ATF4 activity and tumor growth. Cell Metab 33, 1013–1026 e1016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Labow BI, and Souba WW (2000). Glutamine. World J Surg 24, 1503–1513. [DOI] [PubMed] [Google Scholar]
- Labuschagne CF, Cheung EC, Blagih J, Domart MC, and Vousden KH (2019). Cell Clustering Promotes a Metabolic Switch that Supports Metastatic Colonization. Cell Metab 30, 720–734 e725. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lau AN, Li Z, Danai LV, Westermark AM, Darnell AM, Ferreira R, Gocheva V, Sivanand S, Lien EC, Sapp KM, et al. (2020). Dissecting cell-type-specific metabolism in pancreatic ductal adenocarcinoma. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Law JH, Habibi G, Hu K, Masoudi H, Wang MY, Stratford AL, Park E, Gee JM, Finlay P, Jones HE, et al. (2008). Phosphorylated insulin-like growth factor-i/insulin receptor is present in all breast cancer subtypes and is related to poor survival. Cancer Res 68, 10238–10246. [DOI] [PubMed] [Google Scholar]
- Lawrence MS, Stojanov P, Mermel CH, Robinson JT, Garraway LA, Golub TR, Meyerson M, Gabriel SB, Lander ES, and Getz G. (2014). Discovery and saturation analysis of cancer genes across 21 tumour types. Nature 505, 495–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee JS, Adler L, Karathia H, Carmel N, Rabinovich S, Auslander N, Keshet R, Stettner N, Silberman A, Agemy L, et al. (2018). Urea Cycle Dysregulation Generates Clinically Relevant Genomic and Biochemical Signatures. Cell 174, 1559–1570 e1522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, Qiu B, Lee DS, Walton ZE, Ochocki JD, Mathew LK, Mancuso A, Gade TP, Keith B, Nissim I, et al. (2014). Fructose-1,6-bisphosphatase opposes renal carcinoma progression. Nature 513, 251–255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li F, Huangyang P, Burrows M, Guo K, Riscal R, Godfrey J, Lee KE, Lin N, Lee P, Blair IA, et al. (2020). FBP1 loss disrupts liver metabolism and promotes tumorigenesis through a hepatic stellate cell senescence secretome. Nat Cell Biol 22, 728–739. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberti MV, and Locasale JW (2016). The Warburg Effect: How Does it Benefit Cancer Cells? Trends Biochem Sci 41, 211–218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu S, Dai Z, Cooper DE, Kirsch DG, and Locasale JW (2020). Quantitative Analysis of the Physiological Contributions of Glucose to the TCA Cycle. Cell Metab 32, 619–628 e621. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Locasale JW, Grassian AR, Melman T, Lyssiotis CA, Mattaini KR, Bass AJ, Heffron G, Metallo CM, Muranen T, Sharfi H, et al. (2011). Phosphoglycerate dehydrogenase diverts glycolytic flux and contributes to oncogenesis. Nat Genet 43, 869–874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Longchamp A, Mirabella T, Arduini A, MacArthur MR, Das A, Trevino-Villarreal JH, Hine C, Ben-Sahra I, Knudsen NH, Brace LE, et al. (2018). Amino Acid Restriction Triggers Angiogenesis via GCN2/ATF4 Regulation of VEGF and H2S Production. Cell 173, 117–129 e114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luengo A, Li Z, Gui DY, Sullivan LB, Zagorulya M, Do BT, Ferreira R, Naamati A, Ali A, Lewis CA, et al. (2021). Increased demand for NAD(+) relative to ATP drives aerobic glycolysis. Mol Cell 81, 691–707 e696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lum JJ, Bauer DE, Kong M, Harris MH, Li C, Lindsten T, and Thompson CB (2005). Growth factor regulation of autophagy and cell survival in the absence of apoptosis. Cell 120, 237–248. [DOI] [PubMed] [Google Scholar]
- Lunt SY, and Vander Heiden MG (2011). Aerobic glycolysis: meeting the metabolic requirements of cell proliferation. Annu Rev Cell Dev Biol 27, 441–464. [DOI] [PubMed] [Google Scholar]
- Luongo TS, Eller JM, Lu MJ, Niere M, Raith F, Perry C, Bornstein MR, Oliphint P, Wang L, McReynolds MR, et al. (2020). SLC25A51 is a mammalian mitochondrial NAD(+) transporter. Nature 588, 174–179. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lynch TP, Ferrer CM, Jackson SR, Shahriari KS, Vosseller K, and Reginato MJ (2012). Critical role of O-Linked beta-N-acetylglucosamine transferase in prostate cancer invasion, angiogenesis, and metastasis. J Biol Chem 287, 11070–11081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma EH, Bantug G, Griss T, Condotta S, Johnson RM, Samborska B, Mainolfi N, Suri V, Guak H, Balmer ML, et al. (2017). Serine Is an Essential Metabolite for Effector T Cell Expansion. Cell Metab 25, 345–357. [DOI] [PubMed] [Google Scholar]
- Ma Z, Vocadlo DJ, and Vosseller K (2013). Hyper-O-GlcNAcylation is anti-apoptotic and maintains constitutive NF-kappaB activity in pancreatic cancer cells. J Biol Chem 288, 15121–15130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Magtanong L, Ko PJ, To M, Cao JY, Forcina GC, Tarangelo A, Ward CC, Cho K, Patti GJ, Nomura DK, et al. (2019). Exogenous Monounsaturated Fatty Acids Promote a Ferroptosis-Resistant Cell State. Cell Chem Biol 26, 420–432 e429. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mao C, Liu X, Zhang Y, Lei G, Yan Y, Lee H, Koppula P, Wu S, Zhuang L, Fang B, et al. (2021). DHODH-mediated ferroptosis defence is a targetable vulnerability in cancer. Nature 593, 586–590. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marchesini G, Bianchi G, Zoli M, Dondi C, Forlani G, Melli A, Bua V, Vannini P, and Pisi E (1983). Plasma amino acid response to protein ingestion in patients with liver cirrhosis. Gastroenterology 85, 283–290. [PubMed] [Google Scholar]
- Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, Kadel EE III, Koeppen H, Astarita JL, Cubas R, et al. (2018). TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 554, 544–548. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martincorena I, Raine KM, Gerstung M, Dawson KJ, Haase K, Van Loo P, Davies H, Stratton MR, and Campbell PJ (2017). Universal Patterns of Selection in Cancer and Somatic Tissues. Cell 171, 1029–1041 e1021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martinez-Reyes I, Cardona LR, Kong H, Vasan K, McElroy GS, Werner M, Kihshen H, Reczek CR, Weinberg SE, Gao P, et al. (2020). Mitochondrial ubiquinol oxidation is necessary for tumour growth. Nature 585, 288–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Masri S, Papagiannakopoulos T, Kinouchi K, Liu Y, Cervantes M, Baldi P, Jacks T, and Sassone-Corsi P (2016). Lung Adenocarcinoma Distally Rewires Hepatic Circadian Homeostasis. Cell 165, 896–909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mayers JR, Torrence ME, Danai LV, Papagiannakopoulos T, Davidson SM, Bauer MR, Lau AN, Ji BW, Dixit PD, Hosios AM, et al. (2016). Tissue of origin dictates branched-chain amino acid metabolism in mutant Kras-driven cancers. Science 353, 1161–1165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McGaha TL, Huang L, Lemos H, Metz R, Mautino M, Prendergast GC, and Mellor AL (2012). Amino acid catabolism: a pivotal regulator of innate and adaptive immunity. Immunol Rev 249, 135–157. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Menendez JA, and Lupu R (2007). Fatty acid synthase and the lipogenic phenotype in cancer pathogenesis. Nat Rev Cancer 7, 763–777. [DOI] [PubMed] [Google Scholar]
- Metallo CM, Gameiro PA, Bell EL, Mattaini KR, Yang J, Hiller K, Jewell CM, Johnson ZR, Irvine DJ, Guarente L, et al. (2011). Reductive glutamine metabolism by IDH1 mediates lipogenesis under hypoxia. Nature 481, 380–384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morandi A, Giannoni E, and Chiarugi P (2016). Nutrient Exploitation within the Tumor-Stroma Metabolic Crosstalk. Trends Cancer 2, 736–746. [DOI] [PubMed] [Google Scholar]
- Mullen AR, Hu Z, Shi X, Jiang L, Boroughs LK, Kovacs Z, Boriack R, Rakheja D, Sullivan LB, Linehan WM, et al. (2014). Oxidation of alpha-ketoglutarate is required for reductive carboxylation in cancer cells with mitochondrial defects. Cell Rep 7, 1679–1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Muz B, de la Puente P, Azab F, and Azab AK (2015). The role of hypoxia in cancer progression, angiogenesis, metastasis, and resistance to therapy. Hypoxia (Auckl) 3, 83–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nakase I, Kobayashi NB, Takatani-Nakase T, and Yoshida T (2015). Active macropinocytosis induction by stimulation of epidermal growth factor receptor and oncogenic Ras expression potentiates cellular uptake efficacy of exosomes. Sci Rep 5, 10300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Neesse A, Michl P, Frese KK, Feig C, Cook N, Jacobetz MA, Lolkema MP, Buchholz M, Olive KP, Gress TM, et al. (2011). Stromal biology and therapy in pancreatic cancer. Gut 60, 861–868. [DOI] [PubMed] [Google Scholar]
- Nguyen TV, Lee JE, Sweredoski MJ, Yang SJ, Jeon SJ, Harrison JS, Yim JH, Lee SG, Handa H, Kuhlman B, et al. (2016). Glutamine Triggers Acetylation-Dependent Degradation of Glutamine Synthetase via the Thalidomide Receptor Cereblon. Mol Cell 61, 809–820. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nicklin P, Bergman P, Zhang B, Triantafellow E, Wang H, Nyfeler B, Yang H, Hild M, Kung C, Wilson C., et al. (2009). Bidirectional transport of amino acids regulates mTOR and autophagy. Cell 136, 521–534. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nofal M, Zhang K, Han S, and Rabinowitz JD (2017). mTOR Inhibition Restores Amino Acid Balance in Cells Dependent on Catabolism of Extracellular Protein. Mol Cell 67, 936–946 e935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Noto H, Goto A, Tsujimoto T, and Noda M (2012). Cancer risk in diabetic patients treated with metformin: a systematic review and meta-analysis. PLoS One 7, e33411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Olivares O, Mayers JR, Gouirand V, Torrence ME, Gicquel T, Borge L, Lac S, Roques J, Lavaut MN, Berthezene P, et al. (2017). Collagen-derived proline promotes pancreatic ductal adenocarcinoma cell survival under nutrient limited conditions. Nat Commun 8, 16031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Overholtzer M, Mailleux AA, Mouneimne G, Normand G, Schnitt SJ, King RW, Cibas ES, and Brugge JS (2007). A nonapoptotic cell death process, entosis, that occurs by cell-in-cell invasion. Cell 131, 966–979. [DOI] [PubMed] [Google Scholar]
- Palm W, Araki J, King B, DeMatteo RG, and Thompson CB (2017). Critical role for PI3-kinase in regulating the use of proteins as an amino acid source. Proc Natl Acad Sci U S A 114, E8628–E8636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Palm W, Park Y, Wright K, Pavlova NN, Tuveson DA, and Thompson CB (2015). The Utilization of Extracellular Proteins as Nutrients Is Suppressed by mTORC1. Cell 162, 259–270. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park JS, Burckhardt CJ, Lazcano R, Solis LM, Isogai T, Li L, Chen CS, Gao B, Minna JD, Bachoo R, et al. (2020). Mechanical regulation of glycolysis via cytoskeleton architecture. Nature 578, 621–626. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Patra KC, and Hay N (2014). The pentose phosphate pathway and cancer. Trends Biochem Sci 39, 347–354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, King B, Josselsohn RH, Violante S, Macera VL, Vardhana SA, Cross JR, and Thompson CB (2020). Translation in amino-acid-poor environments is limited by tRNA(Gln) charging. Elife 9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pavlova NN, and Thompson CB (2016). The Emerging Hallmarks of Cancer Metabolism. Cell Metab 23, 27–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piskounova E, Agathocleous M, Murphy MM, Hu Z, Huddlestun SE, Zhao Z, Leitch AM, Johnson TM, DeBerardinis RJ, and Morrison SJ (2015). Oxidative stress inhibits distant metastasis by human melanoma cells. Nature 527, 186–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pollak M (2012). The insulin and insulin-like growth factor receptor family in neoplasia: an update. Nat Rev Cancer 12, 159–169. [DOI] [PubMed] [Google Scholar]
- Porporato PE (2016). Understanding cachexia as a cancer metabolism syndrome. Oncogenesis 5, e200. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Porstmann T, Santos CR, Griffiths B, Cully M, Wu M, Leevers S, Griffiths JR, Chung YL, and Schulze A (2008). SREBP activity is regulated by mTORC1 and contributes to Akt-dependent cell growth. Cell Metab 8, 224–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Possemato R, Marks KM, Shaul YD, Pacold ME, Kim D, Birsoy K, Sethumadhavan S, Woo HK, Jang HG, Jha AK, et al. (2011). Functional genomics reveal that the serine synthesis pathway is essential in breast cancer. Nature 476, 346–350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinovich S, Adler L, Yizhak K, Sarver A, Silberman A, Agron S, Stettner N, Sun Q, Brandis A, Helbling D, et al. (2015). Diversion of aspartate in ASS1-deficient tumours fosters de novo pyrimidine synthesis. Nature 527, 379–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rabinowitz JD, and Enerback S (2020). Lactate: the ugly duckling of energy metabolism. Nat Metab 2, 566–571. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rathmell JC, Fox CJ, Plas DR, Hammerman PS, Cinalli RM, and Thompson CB (2003). Akt-directed glucose metabolism can prevent Bax conformation change and promote growth factor-independent survival. Mol Cell Biol 23, 7315–7328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinfeld BI, Madden MZ, Wolf MM, Chytil A, Bader JE, Patterson AR, Sugiura A, Cohen AS, Ali A, Do BT, et al. (2021). Cell-programmed nutrient partitioning in the tumour microenvironment. Nature 593, 282–288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reynolds MR, Lane AN, Robertson B, Kemp S, Liu Y, Hill BG, Dean DC, and Clem BF (2014). Control of glutamine metabolism by the tumor suppressor Rb. Oncogene 33, 556–566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Riscal R, Bull CJ, Mesaros C, Finan JM, Carens M, Ho ES, Xu JP, Godfrey J, Brennan P, Johansson M, et al. (2021). Cholesterol Auxotrophy as a Targetable Vulnerability in Clear Cell Renal Cell Carcinoma. Cancer Discov. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Romero R, Sayin VI, Davidson SM, Bauer MR, Singh SX, LeBoeuf SE, Karakousi TR, Ellis DC, Bhutkar A, Sanchez-Rivera FJ, et al. (2017). Keap1 loss promotes Kras-driven lung cancer and results in dependence on glutaminolysis. Nat Med 23, 1362–1368. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ron-Harel N, Ghergurovich JM, Notarangelo G, LaFleur MW, Tsubosaka Y, Sharpe AH, Rabinowitz JD, and Haigis MC (2019). T Cell Activation Depends on Extracellular Alanine. Cell Rep 28, 3011–3021 e3014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rong X, Albert CJ, Hong C, Duerr MA, Chamberlain BT, Tarling EJ, Ito A, Gao J, Wang B, Edwards PA, et al. (2013). LXRs regulate ER stress and inflammation through dynamic modulation of membrane phospholipid composition. Cell Metab 18, 685–697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rouault TA (2015). Mammalian iron-sulphur proteins: novel insights into biogenesis and function. Nat Rev Mol Cell Biol 16, 45–55. [DOI] [PubMed] [Google Scholar]
- Rous P (1914). The Influence of Diet on Transplanted and Spontaneous Mouse Tumors. J Exp Med 20, 433–451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sato H, Nomura S, Maebara K, Sato K, Tamba M, and Bannai S (2004). Transcriptional control of cystine/glutamate transporter gene by amino acid deprivation. Biochem Biophys Res Commun 325, 109–116. [DOI] [PubMed] [Google Scholar]
- Schafer ZT, Grassian AR, Song L, Jiang Z, Gerhart-Hines Z, Irie HY, Gao S, Puigserver P, and Brugge JS (2009). Antioxidant and oncogene rescue of metabolic defects caused by loss of matrix attachment. Nature 461, 109–113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schworer S, Berisa M, Violante S, Qin W, Zhu J, Hendrickson RC, Cross JR, and Thompson CB (2020). Proline biosynthesis is a vent for TGFbeta-induced mitochondrial redox stress. EMBO J 39, e103334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schworer S, Vardhana SA, and Thompson CB (2019). Cancer Metabolism Drives a Stromal Regenerative Response. Cell Metab 29, 576–591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sellers K, Fox MP, Bousamra M 2nd, Slone SP., Higashi RM., Miller DM., Wang Y., Yan J, Yuneva MO, Deshpande R, et al. (2015). Pyruvate carboxylase is critical for non-small-cell lung cancer proliferation. J Clin Invest 125, 687–698. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sena LA, Li S, Jairaman A, Prakriya M, Ezponda T, Hildeman DA, Wang CR, Schumacker PT, Licht JD, Perlman H, et al. (2013). Mitochondria are required for antigen-specific T cell activation through reactive oxygen species signaling. Immunity 38, 225–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shao C, Lu W, Du Y, Yan W, Bao Q, Tian Y, Wang G, Ye H, and Hao H (2020). Cytosolic ME1 integrated with mitochondrial IDH2 supports tumor growth and metastasis. Redox Biol 36, 101685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shim EH, Livi CB, Rakheja D, Tan J, Benson D, Parekh V, Kho EY, Ghosh AP, Kirkman R, Velu S, et al. (2014). L-2-Hydroxyglutarate: an epigenetic modifier and putative oncometabolite in renal cancer. Cancer Discov 4, 1290–1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silberman A, Goldman O, Boukobza Assayag O, Jacob A, Rabinovich S, Adler L, Lee JS, Keshet R, Sarver A, Frug J, et al. (2019). Acid-Induced Downregulation of ASS1 Contributes to the Maintenance of Intracellular pH in Cancer. Cancer Res 79, 518–533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sousa CM, Biancur DE, Wang X, Halbrook CJ, Sherman MH, Zhang L, Kremer D, Hwang RF, Witkiewicz AK, Ying H, et al. (2016). Pancreatic stellate cells support tumour metabolism through autophagic alanine secretion. Nature 536, 479–483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spinelli JB, Yoon H, Ringel AE, Jeanfavre S, Clish CB, and Haigis MC (2017). Metabolic recycling of ammonia via glutamate dehydrogenase supports breast cancer biomass. Science 358, 941–946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steuerman R, Shevah O, and Laron Z (2011). Congenital IGF1 deficiency tends to confer protection against post-natal development of malignancies. Eur J Endocrinol 164, 485–489. [DOI] [PubMed] [Google Scholar]
- Streicher R, Kotzka J, Muller-Wieland D, Siemeister G, Munck M, Avci H, and Krone W (1996). SREBP-1 mediates activation of the low density lipoprotein receptor promoter by insulin and insulin-like growth factor-I. J Biol Chem 271, 7128–7133. [DOI] [PubMed] [Google Scholar]
- Stylianopoulos T, Martin JD, Snuderl M, Mpekris F, Jain SR, and Jain RK (2013). Coevolution of solid stress and interstitial fluid pressure in tumors during progression: implications for vascular collapse. Cancer Res 73, 3833–3841. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Su R, Dong L, Li C, Nachtergaele S, Wunderlich M, Qing Y, Deng X, Wang Y, Weng X, Hu C, et al. (2018). R-2hG Exhibits Anti-tumor Activity by Targeting FTO/m(6)A/MYC/CEBPA Signaling. Cell 172, 90–105 e123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan LB, Gui DY, Hosios AM, Bush LN, Freinkman E, and Vander Heiden MG (2015). Supporting Aspartate Biosynthesis Is an Essential Function of Respiration in Proliferating Cells. Cell 162, 552–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan MR, Danai LV, Lewis CA, Chan SH, Gui DY, Kunchok T, Dennstedt EA, Vander Heiden MG, and Muir A (2019). Quantification of microenvironmental metabolites in murine cancers reveals determinants of tumor nutrient availability. Elife 8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sullivan WJ, Mullen PJ, Schmid EW, Flores A, Momcilovic M, Sharpley MS, Jelinek D, Whiteley AE, Maxwell MB, Wilde BR, et al. (2018). Extracellular Matrix Remodeling Regulates Glucose Metabolism through TXNIP Destabilization. Cell 175, 117–132 e121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suzuki S, Tanaka T, Poyurovsky MV, Nagano H, Mayama T, Ohkubo S, Lokshin M, Hosokawa H, Nakayama T, Suzuki Y, et al. (2010). Phosphate-activated glutaminase (GLS2), a p53-inducible regulator of glutamine metabolism and reactive oxygen species. Proc Natl Acad Sci U S A 107, 7461–7466. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tan M, Luo H, Lee S, Jin F, Yang JS, Montellier E, Buchou T, Cheng Z, Rousseaux S, Rajagopal N, et al. (2011). Identification of 67 histone marks and histone lysine crotonylation as a new type of histone modification. Cell 146, 1016–1028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tang K, Wu YH, Song Y, and Yu B (2021). Indoleamine 2,3-dioxygenase 1 (IDO1) inhibitors in clinical trials for cancer immunotherapy. J Hematol Oncol 14, 68. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tasdogan A, Faubert B, Ramesh V, Ubellacker JM, Shen B, Solmonson A, Murphy MM, Gu Z, Gu W, Martin M, et al. (2020). Metabolic heterogeneity confers differences in melanoma metastatic potential. Nature 577, 115–120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- TeSlaa T, Bartman CR, Jankowski CSR, Zhang Z, Xu X, Xing X, Wang L, Lu W, Hui S, and Rabinowitz JD (2021). The Source of Glycolytic Intermediates in Mammalian Tissues. Cell Metab 33, 367–378 e365. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thompson CB (2011). Rethinking the regulation of cellular metabolism. Cold Spring Harb Symp Quant Biol 76, 23–29. [DOI] [PubMed] [Google Scholar]
- Tlsty TD, and Coussens LM (2006). Tumor stroma and regulation of cancer development. Annu Rev Pathol 1, 119–150. [DOI] [PubMed] [Google Scholar]
- Tran DH, Kesavan R, Rion H, Soflaee MH, Solmonson A, Bezwada D, Vu HS, Cai F, Phillips JA 3rd, DeBerardinis RJ, et al. (2021). Mitochondrial NADP(+) is essential for proline biosynthesis during cell growth. Nat Metab 3, 571–585. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsoli M, Schweiger M, Vanniasinghe AS, Painter A, Zechner R, Clarke S, and Robertson G (2014). Depletion of white adipose tissue in cancer cachexia syndrome is associated with inflammatory signaling and disrupted circadian regulation. PLoS One 9, e92966. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ubellacker JM, Tasdogan A, Ramesh V, Shen B, Mitchell EC, Martin-Sandoval MS, Gu Z, McCormick ML, Durham AB, Spitz DR, et al. (2020). Lymph protects metastasizing melanoma cells from ferroptosis. Nature 585, 113–118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vander Heiden MG, Cantley LC, and Thompson CB (2009). Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science 324, 1029–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vardhana SA, Hwee MA, Berisa M, Wells DK, Yost KE, King B, Smith M, Herrera PS, Chang HY, Satpathy AT, et al. (2020). Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat Immunol 21, 1022–1033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Venkateswaran V, Haddad AQ, Fleshner NE, Fan R, Sugar LM, Nam R, Klotz LH, and Pollak M (2007). Association of diet-induced hyperinsulinemia with accelerated growth of prostate cancer (LNCaP) xenografts. J Natl Cancer Inst 99, 1793–1800. [DOI] [PubMed] [Google Scholar]
- Villa E, Sahu U, O’Hara BP, Ali ES, Helmin KA, Asara JM, Gao P, Singer BD, and Ben-Sahra I (2021). mTORC1 stimulates cell growth through SAM synthesis and m(6)A mRNA-dependent control of protein synthesis. Mol Cell 81, 2076–2093 e2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Warburg O (1956). On respiratory impairment in cancer cells. Science 124, 269–270. [PubMed] [Google Scholar]
- Warburg O, Wind F, and Negelein E (1927). The Metabolism of Tumors in the Body. J Gen Physiol 8, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weinberg F, Hamanaka R, Wheaton WW, Weinberg S, Joseph J, Lopez M, Kalyanaraman B, Mutlu GM, Budinger GR, and Chandel NS (2010). Mitochondrial metabolism and ROS generation are essential for Kras-mediated tumorigenicity. Proc Natl Acad Sci U S A 107, 8788–8793. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wellen KE, Lu C, Mancuso A, Lemons JM, Ryczko M, Dennis JW, Rabinowitz JD, Coller HA, and Thompson CB (2010). The hexosamine biosynthetic pathway couples growth factor-induced glutamine uptake to glucose metabolism. Genes Dev 24, 2784–2799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wieman HL, Wofford JA, and Rathmell JC (2007). Cytokine stimulation promotes glucose uptake via phosphatidylinositol-3 kinase/Akt regulation of Glut1 activity and trafficking. Mol Biol Cell 18, 1437–1446. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wise DR, DeBerardinis RJ, Mancuso A, Sayed N, Zhang XY, Pfeiffer HK, Nissim I, Daikhin E, Yudkoff M, McMahon SB, et al. (2008). Myc regulates a transcriptional program that stimulates mitochondrial glutaminolysis and leads to glutamine addiction. Proc Natl Acad Sci U S A 105, 18782–18787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu Y, Yakar S, Zhao L, Hennighausen L, and LeRoith D (2002). Circulating insulin-like growth factor-I levels regulate colon cancer growth and metastasis. Cancer Res 62, 1030–1035. [PubMed] [Google Scholar]
- Xu H, Lee MS, Tsai PY, Adler AS, Curry NL, Challa S, Freinkman E, Hitchcock DS, Copps KD, White MF, et al. (2018). Ablation of insulin receptor substrates 1 and 2 suppresses Kras-driven lung tumorigenesis. Proc Natl Acad Sci U S A 115, 4228–4233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Achreja A, Yeung TL, Mangala LS, Jiang D, Han C, Baddour J, Marini JC, Ni J, Nakahara R, et al. (2016). Targeting Stromal Glutamine Synthetase in Tumors Disrupts Tumor Microenvironment-Regulated Cancer Cell Growth. Cell Metab 24, 685–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ye P, Mimura J, Okada T, Sato H, Liu T, Maruyama A, Ohyama C, and Itoh K (2014). Nrf2- and ATF4-dependent upregulation of xCT modulates the sensitivity of T24 bladder carcinoma cells to proteasome inhibition. Mol Cell Biol 34, 3421–3434. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi J, Zhu J, Wu J, Thompson CB, and Jiang X (2020). Oncogenic activation of PI3K-AKT-mTOR signaling suppresses ferroptosis via SREBP-mediated lipogenesis. Proc Natl Acad Sci U S A 117, 31189–31197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young RM, Ackerman D, Quinn ZL, Mancuso A, Gruber M, Liu L, Giannoukos DN, Bobrovnikova-Marjon E, Diehl JA, Keith B, et al. (2013). Dysregulated mTORC1 renders cells critically dependent on desaturated lipids for survival under tumor-like stress. Genes Dev 27, 1115–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yuneva MO, Fan TW, Allen TD, Higashi RM, Ferraris DV, Tsukamoto T, Mates JM, Alonso FJ, Wang C, Seo Y, et al. (2012). The metabolic profile of tumors depends on both the responsible genetic lesion and tissue type. Cell Metab 15, 157–170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanotelli MR, Rahman-Zaman A, VanderBurgh JA, Taufalele PV, Jain A, Erickson D, Bordeleau F, and Reinhart-King CA (2019). Energetic costs regulated by cell mechanics and confinement are predictive of migration path during decision-making. Nat Commun 10, 4185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang D, Tang Z, Huang H, Zhou G, Cui C, Weng Y, Liu W, Kim S, Lee S, Perez-Neut M, et al. (2019). Metabolic regulation of gene expression by histone lactylation. Nature 574, 575–580. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang M, Di Martino JS, Bowman RL, Campbell NR, Baksh SC, Simon-Vermot T, Kim IS, Haldeman P, Mondal C, Yong-Gonzales V, et al. (2018). Adipocyte-Derived Lipids Mediate Melanoma Progression via FATP Proteins. Cancer Discov 8, 1006–1025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Recouvreux MV, Jung M, Galenkamp KMO, Li Y, Zagnitko O, Scott DA, Lowy AM, and Commisso C (2021). Macropinocytosis in Cancer-Associated Fibroblasts Is Dependent on CaMKK2/ARHGEF2 Signaling and Functions to Support Tumor and Stromal Cell Fitness. Cancer Discov 11, 1808–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Z, Tan M, Xie Z, Dai L, Chen Y, and Zhao Y (2011). Identification of lysine succinylation as a new post-translational modification. Nat Chem Biol 7, 58–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao Y, Zhao X, Chen V, Feng Y, Wang L, Croniger C, Conlon RA, Markowitz S, Fearon E, Puchowicz M, et al. (2019). Colorectal cancers utilize glutamine as an anaplerotic substrate of the TCA cycle in vivo. Sci Rep 9, 19180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng J, and Conrad M (2020). The Metabolic Underpinnings of Ferroptosis. Cell Metab 32, 920–937. [DOI] [PubMed] [Google Scholar]
- Zhu J, Berisa M, Schworer S, Qin W, Cross JR, and Thompson CB (2019). Transsulfuration Activity Can Support Cell Growth upon Extracellular Cysteine Limitation. Cell Metab 30, 865–876 e865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu J, Schworer S, Berisa M, Kyung YJ, Ryu KW, Yi J, Jiang X, Cross JR, and Thompson CB (2021). Mitochondrial NADP(H) generation is essential for proline biosynthesis. Science 372, 968–972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhu Z, Achreja A, Meurs N, Animasahun O, Owen S, Mittal A, Parikh P, Lo TW, Franco-Barraza J, Shi J, et al. (2020). Tumour-reprogrammed stromal BCAT1 fuels branched-chain ketoacid dependency in stromal-rich PDAC tumours. Nat Metab 2, 775–792. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zou S, Wang X, Liu P, Ke C, and Xu S (2019). Arginine metabolism and deprivation in cancer therapy. Biomed Pharmacother 118, 109210. [DOI] [PubMed] [Google Scholar]