

Summary
Bulk and single-cell RNA sequencing do not provide full characterization of tissue spatial diversity in cancer samples, and currently available in situ techniques (multiplex immunohistochemistry and imaging mass cytometry) allow for only limited analysis of a small number of targets. The current study represents the first comprehensive approach to spatial transcriptomics of high-grade serous ovarian carcinoma using intact tumor tissue. We selected a small cohort of patients with highly annotated high-grade serous ovarian carcinoma, categorized them by response to neoadjuvant chemotherapy (poor or excellent), and analyzed pre-treatment tumor tissue specimens. Our study uncovered extensive differences in tumor composition between the poor responders and excellent responders to chemotherapy, related to cell cluster organization and localization. This in-depth characterization of high-grade serous ovarian carcinoma tumor tissue from poor and excellent responders showed that spatial interactions between cell clusters may influence chemo-responsiveness more than cluster composition alone.
Subject areas: Oncology, Pathology, Omics
Graphical abstract
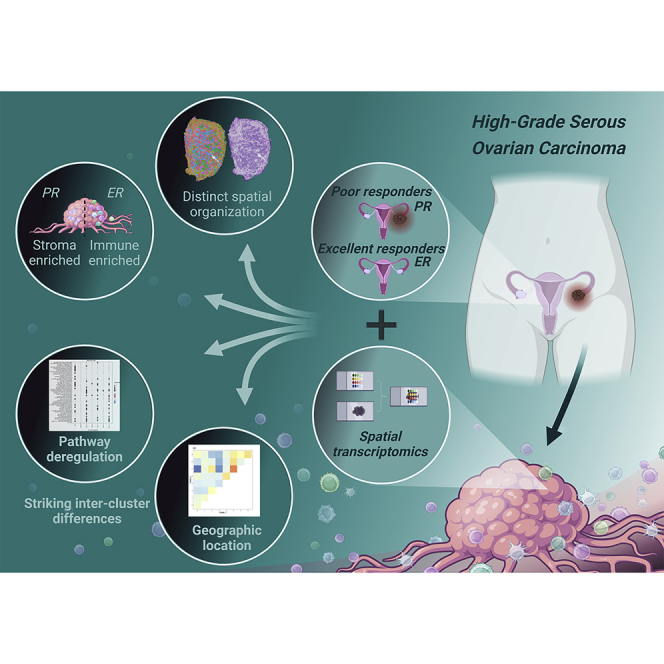
Highlights
-
•
Excellent and poor responders show different composition of tumor microenvironment
-
•
Stromal-derived cell populations dominate poorly responsive tumors
-
•
Variable regulation of inter-cluster cancer-related genes and functional pathways
-
•
Geographical location of cell populations defines cell-to-cell communication
Oncology; Pathology; Omics
Introduction
Epithelial ovarian cancer is a heterogeneous disease, with the most common subtype being high-grade serous ovarian carcinoma (HGSC) (Matulonis et al., 2016). Even within the same histologic subtype, clinical response to therapy (e.g., platinum-based chemotherapy), either before or after debulking surgery, is highly variable. For example, 44%–66% of patients (depending on stage at diagnosis) become long-term survivors with no evidence of recurrence after 10 years of observation (Rubin et al., 1999), whereas others experience disease progression within a much shorter time after first-line treatment (Davis et al., 2014; Jayson et al., 2014). Mechanisms underlying such diversity of response to treatment are not well understood (Peres et al., 2020).
We previously attempted a comprehensive multi-omics approach based on bulk transcriptomic, genomic, and proteomic analyses aimed at identifying molecular and cellular differences between responders and non-responders to neoadjuvant chemotherapy (NACT) in a highly clinically annotated dataset (Lee et al., 2020, 2021). Although differences in copy number variation and T cell infiltration were present between the two groups, these differences were not as extensive as we had anticipated. These findings suggested that important molecular differences between such tumors cannot be fully identified by bulk molecular analyses. Thus, we carried out an in-depth spatial analysis of both epithelial and stromal components of tumor tissue to explore the heterogeneity of HGSC and determine how the dynamic behavior between the tumor and the tumor microenvironment varies between poor and excellent responders to NACT. To generate these data, we performed deep spatial transcriptomic profiling in situ (Cable et al., 2021).
Results
Spatial transcriptomics to explore the tumor microenvironment of HGSC
We assessed gene expression of 12 chemotherapy-naive high-grade serous ovarian carcinoma samples with full spatial orientation. These 12 samples were classified into one of two groups: six were considered excellent responders to NACT (ER) and six were considered poor responders to NACT (PR) (Lee et al., 2020) (Figure 1A). After initial analysis, one sample in the PR group was excluded owing to poor tissue quality (high necrotic fraction).
Figure 1.

Spatial transcriptomics to explore the tumor microenvironment of high-grade serous ovarian carcinoma (HGSC)
(A) Schematic representation of the study for tissue collection in patients with HGSC. R0, no gross residual disease. Created with BioRender.com.
(B) Representative image of a hematoxylin-and-eosin-stained section (top) and unsupervised clustering (bottom).
(C) Heatmap of putative cell composition of each cluster in the excellent responder (ER) and poor responder (PR) groups.
(D) Dot-plot representing differential gene expression of known ovarian cancer genes (COSMIC) in bulk RNA sequencing and in spatial transcriptomics of the main clusters (0–4). Black boxes are shown around the point when the gene set enrichment analysis adjusted p value is <0.1.
For the first step, we used an unsupervised clustering approach to generate expression profiles of all samples grouped together, and we identified nine distinct clusters. We opted for the unsupervised approach to define clusters of cells with similar transcript patterns and determine the distribution of these clusters inside the tissue (Figure 1B). In the second step, we examined the distribution of the defined clusters in the clinically defined ER and PR groups. We observed that not all clusters were identified between the two groups or in all samples within each group. For example, clusters 0–4 were largely represented in both groups, whereas cluster seven was exclusive to the PR group and cluster eight was exclusive to the ER group (Figure S1A, and Table S1). Also, the clusters were represented in different proportions in each group (Figure S1B). The false-discovery rates (FDR) of clusters 6–8 (Table S1) show a reduced number of genes with significantly differentially expressed genes (DEGs) at the cluster definition. Clusters 0–4 are the largest physical clusters and also the ones with the most DEGs with significant FDR. Overlaying the cluster perimeters onto the hematoxylin-and-eosin-stained images, we found that distinct patterns of stromal and epithelial areas identified through gene expression mirrored the stromal and epithelial areas defined by morphology (Figure 1B). This approach demonstrates the effectiveness of using an unsupervised approach in spatial transcriptomics to detect cell populations, which has not been previously described in attempts at deep sequencing coupled with spatial distribution.
In the next step, we aimed to identify the cell types present in these tissues (Figure 1C). For this, we used well-defined markers of 20 cell types and examined larger cell areas, named stromal and immunity CD45. We identified all 20 proposed cell types in both the stromal and immunity subgroups of cells. It is important to highlight that many cell types were identified within the same cluster; this shows once again the potential of resolved spatial profiling for characterizing more cell types than in conventional histology and RNA sequencing.
A comparison of the cell populations enriched in the two response groups identified more stromal-dominated cell groups, largely formed by myofibroblasts rather than conventional cancer-associated fibroblasts (as defined by the cell markers used for cell identification), in the PR group (Figure S2). This could be explained by poor identification of cancer-associated fibroblasts in previous studies, which have been mainly based on imaging techniques that use a limited number of nonspecific markers. This transcriptomic-based approach allows the use of multiple markers at the same time, which ensures better identification of cell types. However, ER tumors contained more immune-related areas; more than half of the clusters presented a high proportion of immune cells, including T cells, B cells, and natural killer cells (Figure S2). This finding supports the well-established role of cytotoxic immune cells in strengthening chemotherapy response (Wang et al., 2018) and better progression-free survival and overall survival (Zhang et al., 2003) (Figures 1A and S2). Conventional immunohistochemistry or immunofluorescence techniques for studying the tumor microenvironment require high-quality and intact tissue with viable expression of the protein of interest (often destroyed by chemicals and tissue archiving processes); also, these techniques are highly operator-dependent. With the spatial transcriptomics approach, we identified cell types and subtypes using only a fragment of tissue RNA, without requiring complete and intact cell structure. We observed epithelial-mesenchymal-transition-like cells in most of the clusters in the PR group, in contrast with the ER group (Figure 1C). This confirms the prominent role of myofibroblasts and mesenchymal cells, rather than cancer-associated fibroblasts, in promoting poor response to therapy.
Spatial analysis of HGSC unravels characteristics of the tumor microenvironment
Next, we examined whether our approach, as compared with blending all RNA information from cells (i.e., bulk sequencing), could reveal differences in expression of specific genes. We used previously published bulk RNA-sequencing data from the same samples (Lee et al., 2020), as well as the spatial transcriptomics data, used as individual clusters and combined, imitating bulk sequencing. We observed that the blending phenomenon hides deeper profiling of the tumor, in addition to destroying the geographic organization of the tissue. First, we used the genes reported in the Cancer Gene Census (https://cancer.sanger.ac.uk/census) as a Catalogue Of Somatic Mutations In Cancer (COSMIC) whose aberrant expression has been causally implicated in cancer and deregulated from previous dataset analyses. The comparison of expression of these genes between the groups mentioned above highlighted profound differences, which often reached statistical significance (Figure 1D). Most prominently, genes such as NF1, MSH6, CTNNB1, BARD1, AKT1, and AKT2 showed inverse expression levels in several clusters compared with that shown in bulk sequencing for the PR group compared with the ER group. For example, BARD1 showed lower expression in the PR group by bulk sequencing, but it had higher expression in the PR group by in situ sequencing (when considering all clusters together). This results from its higher expression in the PR group (cluster 0), which represents the biggest physical size-wise cluster of the PR samples (Figure 1D). This finding is quite important because it signifies higher definition of the spatial technique, which allows for a more accurate analysis due to the preservation of the original cell localization. Similarly, other DNA repair genes, such as ATR, ARID2, and ARID1B, had significantly higher expression in PR samples when all clusters were considered together, as compared with results from bulk sequencing (lower expression in PR samples). This finding suggests that tumors from the PR group had better DNA surveillance and higher cell survival compared with the ER group.
In additional analyses, we used gene set enrichment analysis to compare the PR group with the ER group (Figure 2A and Table S2). We observed that specific enriched pathways from bulk sequencing were also enriched in the combined cluster analysis following the same pattern of deregulation (e.g., GOBP_Cell Population Proliferation, Hallmark_Angiogenesis, Hallmark_WNT Beta Catenin Signaling, Hallmark_Interferon Gamma Response, and Hallmark_Interferon Gamma Response). Conversely, we also observed pathways that were enriched with both techniques, but in the opposite direction (e.g., Hallmark_UV response, Hallmark_TGF Beta signaling, and Hallmark_Oxidative phosphorylation). When looking specifically into single clusters, we observed a large variation in patterns. Some pathways were statistically enriched in specific clusters of the ER group compared with the PR group, proving that a real and deep characterization of tumor heterogeneity can be achieved only when exploring specific clusters of cells. We clearly observed how different clusters showed opposite trends of upregulation and downregulation of pathways between the two groups, and these opposite trends were not apparent in the bulk analysis. This was clearer when analyzing immune-related pathways, in which we observed downregulation of multiple pathways in bulk sequencing but differential upregulation in specific clusters from our spatial transcriptomic analysis (e.g., GOBP_Activation of immune response, GOBP_Activation of innate immune response, and GOBP_Immune response-regulating signaling pathway; Figure S1C). By combining all clusters together, we confirmed in the pathway analysis our previous finding of reduced representation of immune cells in PR tissues, and that pathways of cell activation to immune response, adaptive immune response, and regulation of innate response were downregulated in the PR group when all clusters were considered together. Single-cluster analysis instead identified cluster 4 as consistently showing upregulation of immune pathways, mostly the innate immune pathway, in the PR group. In the cell classification analyses, this cluster showed higher representation of macrophages, dendritic cells, and neutrophils, consistent with prominent activation of the innate response.
Figure 2.

Geographical location of specific cell populations define cell-to-cell communication
(A) Dot-plot of gene set enrichment analysis comparing bulk RNA sequencing and spatial transcriptomics of the main clusters (0–4). Black boxes are shown around the point when the gene set enrichment analysis adjusted p value is < 0.1.
(B) Heatmap indicating the physical distance between clusters of the excellent responder (ER) and poor responder (PR) groups, where each square represents a cluster-cluster comparison, the blue range of colors indicates longer distance between clusters, and the red range of colors indicates shorter physical distance between clusters.
(C) Heatmap of ligand-receptor (LR) analysis showing patterns of the top co-expressed LRs between clusters of ER (left) and PR (right).
Geographical location of specific cell populations defines cell-to-cell communication
Next, we analyzed the spatial localization of the identified cell clusters by measuring the physical distance between them inside the tissues (Figure 2B). We observed that cluster distribution largely differed between groups. For example, in the PR group, the clusters tended to be physically larger and distributed throughout the whole tissue area, which translates into a larger distance between spots from the same cluster in the heatmap (blue areas). By comparison, in the ER tissues, clusters were smaller and more compact, and the distance between spots in the same cluster was smaller (yellow areas).
When analyzing the distances between clusters, we found that some clusters were often located close to each other in one group and significantly farther from each other in the other group. For example, clusters 2 and 5 were closer to each other in the ER group, whereas clusters 3 and 8 were closer to each other in the PR group. This indicates that different cell-to-cell contacts are found in different groups, showing that the disposition of a cell type, more so than its abundance, could explain different mechanisms of response to therapy. For example, cluster 8 (represented mostly by epithelial-mesenchymal-transition-like cells, mesenchymal cells, endothelial cells, and myofibroblasts) had more physical interactions in tissues from the PR group than in the ER group, and the most frequent interaction was between clusters 5 and 2, which are highly dominated by immune cells.
To explore the relationship between cells of neighboring clusters, we performed a ligand-receptor (LR) analysis to identify the expression of an LR couple (identified as the product of L in one cluster and R in the neighboring cluster) in cluster combinations. To explore the extent of LR expression, we chose five cluster combinations: 5–2, 8–1, 8–2, 8–3, and 8–4, which are differentially located from each other in ER and PR tissues (Figure 2B).
The heatmap showing co-expression patterns defines two distinct patterns in the ER and PR tissues (Figure 2C). We found that co-expression of specific LR pairings exists depending on their geographic localization. Specifically, at clusters 5 and 2 of the ER group (geographically close), there was stronger co-expression of GPC3-IGF1R (score 0.45) compared with clusters 8 and 2 in the same ER group (geographically distant; score 0.18). The same pattern is observed when looking in cluster 8–2 (geographically close in PR), where the same LR (GPC3-CD81) had strong co-expression patterns (score 0.66) compared with clusters 5 and 2 (geographically distant; score 0.41- Table S3). These findings indicate that the co-expression pattern of specific LRs depends on the geographic localization in the tissue; closer interactions lead to stronger co-expression patterns.
Discussion
Single-cell RNA sequencing has greatly improved detailed analysis of the tumor microenvironment. However, single-cell RNA sequencing methods mostly analyze suspensions of dissociated cells, which completely erase the complexity of the physical organization of cells and provide partial information concerning cell-cell interactions. To overcome such limitations, some investigators have coupled single-cell RNA sequencing on microdissected tissues with staining for multiple targets and imaging mass cytometry (Zhu et al., 2021), or employed bioinformatic tools to profile physically interacting cells in ”multiplets” derived from partially undissociated tissues (Andrews et al., 2021). However, such techniques are not able to produce high-resolution RNA sequencing applied directly on intact tissues through a simple and scalable pipeline. To the best of our knowledge, the current manuscript is the first application of an in situ RNA-sequencing approach to study cell-cell communication in HGSC tissues from poor and excellent responders to NACT.
Two important findings emerged from our analyses: one is the assessed importance of the stromal component as a potential driver of poor response to chemotherapy; the second is the identification of differential clusters (distribution and composition) in ER compared with PR tissues, both at a cohort level and at a single patient level. The existence of such clusters could be recognized only by the in situ technique. Focusing on the epithelial-mesenchymal transition pathway, known to contribute to tumor progression, metastasis, and resistance to therapy (Ashrafizadeh et al., 2021; Liu et al., 2021; Smith and Bhowmick, 2016), we noticed that among different areas of the same tumor tissue, as well as in specific areas of the tissue within the whole group of patients (in this case, the PR group), the epithelial-mesenchymal transition pathway was subject to large variation in downregulation or upregulation. This finding means that specific populations of cells may contribute to the poor response to therapy.
The physical location of the cell clusters in the TME is important to understand response to therapy. For example, we identified cluster 8, a mainly mesenchymal cluster, as a cluster with multiple physical connections to all the other clusters, mostly in the PR group. This underlines the role of crosstalk between the mesenchymal compartment and other cell clusters in potentially sustaining resistance. Moreover, the tight physical connection between clusters 2 and 5 (both represented mostly by immune cells) presented homogeneously in the ER tissues, suggesting a fundamental role of the immune response in enhancing the cytotoxic effect of chemotherapy. In conclusion, comparison of the in situ technique with bulk RNA sequencing allowed characterization of specific cell subpopulations with defined molecular traits. These clusters, with their specific set of overexpressed genes, might be important determinants of lack of response to therapy, representing the ideal target for effective therapeutic approaches.
Our work overcomes many of the limitations of previous approaches. Each sequenced spot of 50-μm diameter allows analysis of the transcriptome of a limited number of cells ranging between 3 and 5. Despite this low resolution, one strength of this technique is the ability to perform the analysis on undissociated tissues. Dissociation techniques can often cause major environmental stresses that could impact the transcriptional machinery (Denisenko et al., 2020; van den Brink et al., 2017). When this technique is used, tissues do not undergo extensive manipulations before RNA sequencing and are homogeneously treated. It is likely that further refinement of the bioinformatics tools might address the limitations of deconvolution and assess each cell type at a higher resolution for cell identification and transcriptomics analyses.
In conclusion, our work provides a new understanding of the tumor microenvironment and chemotherapy response in ovarian cancer. We identified spatially defined cell clusters, in particular, the ones represented by mesenchymal-derived cells, in tumor samples that were resistant to chemotherapy.
Limitations of the study
The small sample size in this project represents a potential limitation; we are evaluating an expanded cohort. Moreover, the current techniques allow for a resolution of 5–15 cells; ongoing efforts are aimed at further increasing the resolution of spatial transcriptomics to a single cell level.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Biological samples | ||
| Patients with HGSC (tumor tissue) | MDACC | N/A |
| Critical commercial assays | ||
| Dual Index Kit TT Set A | 10X Genomics | 1000215 |
| Visium Spatial Gene Expression Slide & Reagents Kit | 10X Genomics | 1000187 |
| Visium Spatial Gene Expression Starter Kit | 10X Genomics | 1000200 |
| Visium Spatial Tissue Optimization Slide & Reagents Kit | 10X Genomics | 1000193 |
| Deposited data | ||
| RNA Sequencing data | Lee et al., 2020 | EGA: EGAD00001005240 |
| Spatial Transcriptomics sequencing data | Own data | GSE189843 |
| Software and algorithms | ||
| spaceranger v1.1.0 | 10X Genomics | |
| R package Seurat v4.0.1 | Hafemeister and Satija, 2019 | |
| R package GSVA v1.36.3 | Hanzelmann et al., 2013 | |
| R package fgsea v1.14.0 | Gennady Korotkevich et al., 2021 | |
| SingleCellSignalR | Cabello-Aguilar et al., 2020 | |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Anil K. Sood (asood@mdanderson.org).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Patients
High-grade serous ovarian carcinoma biopsy samples from 12 female patients (age range 45–78 years old) were collected under a protocol approved by the Institutional Review Board of The University of Texas MD Anderson Cancer Center, and all patients provided written informed consent. The fresh frozen tumor tissues were obtained from the MD Anderson Gynecologic Tumor Bank. Patients underwent a standardized therapeutic approach following chemotherapy and surgery protocols established in the Department of Gynecology Oncology & Reproductive Medicine at MD Anderson.
We selected six patients with poor response to NACT with carboplatin and paclitaxel and six patients with excellent response to NACT. The definition of poor response was stable or progressive disease after three to four cycles according to radiologic evaluation and/or suboptimal interval cytoreduction after NACT according to the Response Evaluation Criteria in Solid Tumors (RECIST 1.1). Excellent responders were those with complete response or only microscopic disease left at the time of interval surgery and/or pathologic analysis after interval surgery (Lee et al., 2020).
Method details
Spatial transcriptomics
Tissue optimization
For spatial transcriptomics, we used the Visium Spatial Gene Expression Reagent Kit (10X Genomics). Tissue optimization was done using the Visium Spatial Tissue Optimization Slide & Reagent kit. The optimization, as well as the analysis, was performed using tissue at 10-μm thickness per the manufacturer’s protocol. The selected permeabilization time was 20 min. Bright-field images of the tissue were captured according to the requirements of the manufacturer, using a Nikon Eclipse Ni microscope and Nikon DS-Ri1 8-bit camera. The objective was a Nikon Plan Apo with 10× magnification and 0.45 numerical aperture. We used Nikon Elements software with a resolution of 0.91 μm/pixel. Camera settings were as follows: format 1280 × 1024 (fine), exposure 10.0 ms (±0.0 EV), analog gain 1.00.
Library construction
The Visium Spatial Gene Expression Slide & Reagent kit was used for library construction. One section per patient (10 μm) was mounted into each capture area. Each capture area on a Visium for FFPE slide has an array containing capture probes that bind to RNA. The probe pairs are extended to incorporate complements of the spatial barcodes, and sequencing libraries are prepared. The libraries are then sequenced, and data visualized to determine which genes are expressed and where, as well as in what quantity. A capture area is defined as a 6.5 × 6.5 mm area that contains 5,000 barcoded spots that are 55 μm in diameter and 100 μm center to center between spots, providing an average resolution of 1–10 cells (10X Genomics). All of the following steps (cDNA synthesis and amplification, library construction, and post-library construction quality control) were done according to the manufacturer’s protocol. The libraries were sequenced on a NovaSeq 6000 (Illumina).
Quantification and statistical analysis
The Visium sequencing data were processed by spaceranger v1.1.0 with commands spaceranger mkfastq and spaceranger count. The count matrix output and image data were further analyzed by R package Seurat v4.0.1. Specifically, the matrices of each sample were loaded individually and the function SCTransform was used to perform data transformation (Hafemeister and Satija, 2019). To remove batch effects, the list of data from individual samples was processed by functions SelectIntegrationFeatures, PrepSCTIntegration, and FindIntegrationAnchors, and finally integrated into a single Seurat object by the function IntegrateData. The integrated data underwent principal component analysis, and 30 principal components were used for Uniform Manifold Approximation and Projection. Cluster-specific cell type scores were calculated by performing gene set variation analysis on the average expression matrix of clusters using R package GSVA v1.36.3 (Hanzelmann et al., 2013). The pairwise distances between two clusters were calculated by median Euclidean distances of each spot pair on the slides in the two clusters. The differential gene expression analyses between two clusters were performed by the function FindMarkers, and the resulting ranks were used for gene set enrichment analyses by R package fgsea v1.14.0(Gennady Korotkevich et al., 2021). The ligand receptor interaction (SingleCellSignalR) scores were calculated by multiplying the average expression of the ligand and the receptor in the respective clusters and further normalized to [0,1], as performed in previous studies (Cabello-Aguilar et al., 2020). The codes used to perform this analysis are in Data S1.
Acknowledgments
We thank E. Goodoff (Research Medical Library, The University of Texas MD Anderson Cancer Center, Houston, Texas) for editorial assistance. This research was performed in collaboration with the Pathology and Histology Core, which is supported in part by the P30 Cancer Center Support Grant (NCI-CA125123) and by the Genomic and RNA Profiling Core at Baylor College of Medicine with funding from the NIH S10 grant (1S10OD023469). This research was supported, in part, by the MD Anderson Ovarian Cancer SPORE (P50 CA217685) and grants R35 CA209904 and U01 CA213759, P30 CA016672, the MD Anderson Ovarian Cancer Moon Shot, by the American Cancer Society, the Ovarian Cancer Research Alliance, the Dunwoody Fund, the Gordon Fund, and by the Frank McGraw Memorial Chair in Cancer Research. E.S. is supported by Ovarian Cancer Research Alliance (OCRA number FP00006137). S.C. is funded by the Foundation Amy Krouse Rosenthal Ovarian Cancer Early Detection Research Grant through The Foundation for Women’s Cancer 2020/2021 (AWD00005708). Graphical abstract adapted from “New strategies for treating cancer”, by BioRender.com (2022). Retrieved from https://app.biorender.com/biorender-templates
Author contributions
Conceptualization, Methodology, Investigation, Visualization, Writing (Original Draft/Review and Editing): E.S., S.C.
Conceptualization, Review and Editing, Supervision, Funding Acquisition and Project Administration: A.K.S.
Data collection and curation: E.S., S.C., S.L., E.R., D.K., J.L., and P.C.
Formal Analysis, Data Curation, Software: M.X., K.C., and J.Z.
Declaration of interests
Anil K. Sood is a shareholder of BioPath and is a consultant for Merck, Astra Zeneca, GSK, and Kiyatec. The other authors declare no competing interests.
Published: March 18, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.103923.
Supplemental information
Data and code availability
-
•
Spatial transcriptomics data have been deposited at GEO and are publicly available. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Andrews N., Serviss J.T., Geyer N., Andersson A.B., Dzwonkowska E., Sutevski I., Heijboer R., Baryawno N., Gerling M., Enge M. An unsupervised method for physical cell interaction profiling of complex tissues. Nat. Methods. 2021;18:912–920. doi: 10.1038/s41592-021-01196-2. [DOI] [PubMed] [Google Scholar]
- Ashrafizadeh M., Mirzaei S., Hashemi F., Zarrabi A., Zabolian A., Saleki H., Sharifzadeh S.O., Soleymani L., Daneshi S., Hushmandi K., et al. New insight towards development of paclitaxel and docetaxel resistance in cancer cells: EMT as a novel molecular mechanism and therapeutic possibilities. Biomed. Pharmacother. 2021;141:111824. doi: 10.1016/j.biopha.2021.111824. [DOI] [PubMed] [Google Scholar]
- Cabello-Aguilar S., Alame M., Kon-Sun-Tack F., Fau C., Lacroix M., Colinge J. SingleCellSignalR: inference of intercellular networks from single-cell transcriptomics. Nucleic Acids Res. 2020;48:e55. doi: 10.1093/nar/gkaa183. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cable D.M., Murray E., Zou L.S., Goeva A., Macosko E.Z., Chen F., Irizarry R.A. Robust decomposition of cell type mixtures in spatial transcriptomics. Nat. Biotechnol. 2021 doi: 10.1038/s41587-021-00830-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Davis A., Tinker A.V., Friedlander M. Platinum resistant" ovarian cancer: what is it, who to treat and how to measure benefit? Gynecol. Oncol. 2014;133:624–631. doi: 10.1016/j.ygyno.2014.02.038. [DOI] [PubMed] [Google Scholar]
- Denisenko E., Guo B.B., Jones M., Hou R., De Kock L., Lassmann T., Poppe D., Clement O., Simmons R.K., Lister R., Forrest A.R.R. Systematic assessment of tissue dissociation and storage biases in single-cell and single-nucleus RNA-seq workflows. Genome Biol. 2020;21:130. doi: 10.1186/s13059-020-02048-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gennady Korotkevich V.S., Budin N., Shpak B., Artyomov M.N., Sergushichev A. Fast gene set enrichment analysis. bioRxiv. 2021 doi: 10.1101/060012. Preprint at. [DOI] [Google Scholar]
- Hafemeister C., Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. doi: 10.1186/s13059-019-1874-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hanzelmann S., Castelo R., Guinney J. GSVA: gene set variation analysis for microarray and RNA-seq data. BMC Bioinformatics. 2013;14:7. doi: 10.1186/1471-2105-14-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jayson G.C., Kohn E.C., Kitchener H.C., Ledermann J.A. Ovarian cancer. Lancet. 2014;384:1376–1388. doi: 10.1016/S0140-6736(13)62146-7. [DOI] [PubMed] [Google Scholar]
- Lee S., Zhao L., Little L.D., Westin S.N., Jazarei A.A., Fleming N.D., Zhang J., Futreal P.A., Sood A.K. Distinct T cell receptor repertoire diversity of clinically defined high-grade serous ovarian cancer treatment subgroups. iScience. 2021;24:102053. doi: 10.1016/j.isci.2021.102053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee S., Zhao L., Rojas C., Bateman N.W., Yao H., Lara O.D., Celestino J., Morgan M.B., Nguyen T.V., Conrads K.A., et al. Molecular analysis of clinically defined subsets of high-grade serous ovarian cancer. Cell Rep. 2020;31:107502. doi: 10.1016/j.celrep.2020.03.066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X., He M., Li L., Wang X., Han S., Zhao J., Dong Y., Ahmad M., Li L., Zhang X., et al. EMT and cancer cell stemness associated with chemotherapeutic resistance in esophageal cancer. Front. Oncol. 2021;11:672222. doi: 10.3389/fonc.2021.672222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Matulonis U.A., Sood A.K., Fallowfield L., Howitt B.E., Sehouli J., Karlan B.Y. Ovarian cancer. Nat. Rev. Dis. Primers. 2016;2:16061. doi: 10.1038/nrdp.2016.61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peres L.C., Sinha S., Townsend M.K., Fridley B.L., Karlan B.Y., Lutgendorf S.K., Shinn E., Sood A.K., Tworoger S.S. Predictors of survival trajectories among women with epithelial ovarian cancer. Gynecol. Oncol. 2020;156:459–466. doi: 10.1016/j.ygyno.2019.12.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rubin S.C., Randall T.C., Armstrong K.A., Chi D.S., Hoskins W.J. Ten-year follow-up of ovarian cancer patients after second-look laparotomy with negative findings. Obstet. Gynecol. 1999;93:21–24. doi: 10.1016/s0029-7844(98)00334-2. [DOI] [PubMed] [Google Scholar]
- Smith B.N., Bhowmick N.A. Role of EMT in metastasis and therapy resistance. J. Clin. Med. 2016;5:17. doi: 10.3390/jcm5020017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- van den Brink S.C., Sage F., Vertesy A., Spanjaard B., Peterson-Maduro J., Baron C.S., Robin C., van Oudenaarden A. Single-cell sequencing reveals dissociation-induced gene expression in tissue subpopulations. Nat. Methods. 2017;14:935–936. doi: 10.1038/nmeth.4437. [DOI] [PubMed] [Google Scholar]
- Wang W., Zou W., Liu J.R. Tumor-infiltrating T cells in epithelial ovarian cancer: predictors of prognosis and biological basis of immunotherapy. Gynecol. Oncol. 2018;151:1–3. doi: 10.1016/j.ygyno.2018.09.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L., Conejo-Garcia J.R., Katsaros D., Gimotty P.A., Massobrio M., Regnani G., Makrigiannakis A., Gray H., Schlienger K., Liebman M.N., et al. Intratumoral T cells, recurrence, and survival in epithelial ovarian cancer. N. Engl. J. Med. 2003;348:203–213. doi: 10.1056/NEJMoa020177. [DOI] [PubMed] [Google Scholar]
- Zhu Y., Ferri-Borgogno S., Sheng J., Yeung T.L., Burks J.K., Cappello P., Jazaeri A.A., Kim J.H., Han G.H., Birrer M.J., et al. SIO: a spatioimageomics pipeline to identify prognostic biomarkers associated with the ovarian tumor microenvironment. Cancers (Basel) 2021;13:1777. doi: 10.3390/cancers13081777. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
-
•
Spatial transcriptomics data have been deposited at GEO and are publicly available. Accession numbers are listed in the key resources table.
-
•
This paper does not report original code.
-
•
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.