

Abstract
Co‐evolution between hosts’ and parasites’ genomes shapes diverse pathways of acquired immunity based on silencing small (s)RNAs. In plants, sRNAs cause heterochromatinization, sequence degeneration, and, ultimately, loss of autonomy of most transposable elements (TEs). Recognition of newly invasive plant TEs, by contrast, involves an innate antiviral‐like silencing response. To investigate this response’s activation, we studied the single‐copy element EVADÉ (EVD), one of few representatives of the large Ty1/Copia family able to proliferate in Arabidopsis when epigenetically reactivated. In Ty1/Copia elements, a short subgenomic mRNA (shGAG) provides the necessary excess of structural GAG protein over the catalytic components encoded by the full‐length genomic flGAG‐POL. We show here that the predominant cytosolic distribution of shGAG strongly favors its translation over mostly nuclear flGAG‐POL. During this process, an unusually intense ribosomal stalling event coincides with mRNA breakage yielding unconventional 5’OH RNA fragments that evade RNA quality control. The starting point of sRNA production by RNA‐DEPENDENT‐RNA‐POLYMERASE‐6 (RDR6), exclusively on shGAG, occurs precisely at this breakage point. This hitherto‐unrecognized “translation‐dependent silencing” (TdS) is independent of codon usage or GC content and is not observed on TE remnants populating the Arabidopsis genome, consistent with their poor association, if any, with polysomes. We propose that TdS forms a primal defense against EVD de novo invasions that underlies its associated sRNA pattern.
Keywords: RDR6, ribosome stalling, small RNAs, translation, transposons
Subject Categories: Chromatin, Transcription & Genomics; Plant Biology; RNA Biology
Analyzing the initiation of RNA silencing of the Arabidopsis transposon EVADE (EVD) reveals a ribosome stalling event during EVD translation that correlates with the production of small RNAs involving RNA‐dependent RNA polymerase 6 (RDR6).
Introduction
Transposable elements (TEs) colonize and threaten the integrity of virtually all genomes (Huang et al, 2012). Chromosomal rearrangements caused by their highly repetitive nature (Fedoroff, 2012) are usually circumvented by cytosine methylation and/or histone‐tail modifications at their loci of origin. The ensuing heterochromatic DNA is not conducive to transcription by RNA Pol II, bringing TEs into an epigenetically silent transcriptional state (Allshire & Madhani, 2018). This “transcriptional gene silencing” (TGS) is observed at the majority of TE loci in plants, including the model species Arabidopsis thaliana, and causes, over evolutionary times, accumulating mutations resulting in mostly degenerated, non‐autonomous entities (Vitte & Bennetzen, 2006; Civáň et al, 2011). Nonetheless, the genome invasiveness of these remnants remains evident by their methyl cytosine‐marked DNA, which is perpetuated over generations by METHYL‐TRANSFERASE 1 (MET1), among other factors. MET1 reproduces symmetrical methylation sites from mother to daughter strands during DNA replication (Kankel et al, 2003) aided by the (hetero)chromatin remodeler DEFICIENT IN DNA METHYLATION 1 (DDM1) (Saze et al, 2003; Zemach et al, 2013).
Loss of MET1 or DDM1 functions in Arabidopsis leads to genome‐wide demethylation, transcriptional reactivation of many TE remnants, and mobilization of a small portion of intact, autonomous TEs (Mirouze et al, 2009; Tsukahara et al, 2010). Their proliferation together with genome‐wide deposition of aberrant epigenetic marks likely explains why met1 and ddm1 mutants accumulate increasingly severe genetic and phenotypic burdens over inbred generations (Vongs et al, 1993). However, such secondary events can be avoided by backcrossing the first homozygous generation of ddm1‐ or met1‐derived mutants with wild‐type plants, upon which continuous selfing of F2 plants creates “epigenetic recombinant inbred lines” (epiRILs). These harbor only mosaics of de‐methylated DNA while maintaining wild‐type (WT) MET1 and DDM1 functions (Reinders et al, 2009; Teixeira et al, 2009). One such met1 epiRIL, epi15, endows epigenetic reactivation of the autonomous, long terminal repeat (LTR) retroelement EVADÉ (EVD) in theTy1/Copia family, which is one of the most proliferative families in plants (Vitte & Panaud, 2005). Of the two EVD copies in the Arabidopsis Col‐0 genome, only one is reactivated in epi15 (Marí‐Ordóñez et al, 2013). By providing a proxy for a de novo genomic invasion, this reactivation granted a unique opportunity to grasp how, over multiple inbred generations, newly invasive TEs might be detected and eventually epigenetically silenced (Marí‐Ordóñez et al, 2013).
We found that EVD is initially confronted to post‐transcriptional gene silencing (PTGS) akin to that mounted against plant viruses (Voinnet, 2005; Marí‐Ordóñez et al, 2013). Antiviral RNA‐DEPENDENT RNA POLYMERASE 6 (RDR6) produces cytosolic, long double‐stranded (ds)RNAs from EVD‐derived transcripts, which are then processed by DCL4 or DCL2, two of the four Arabidopsis Dicer‐like RNase‐III enzymes, into populations of respectively 21‐ and 22‐nt small interfering (si)RNAs. However, despite their loading into the antiviral PTGS effectors ARGONAUTE1 and ARGONAUTE2 (AGO1/2), they do not suppress expression of EVDs increasingly more abundant genomic copies. This ultimately gives way to DCL3, instead of DCL4/2, to process the RDR6‐made long dsRNAs into 24‐nt siRNAs. In association with AGO4‐clade AGOs, these species guide RNA‐directed DNA methylation (RdDM) of EVD copies. Initially localized within the EVD gene body, it later spreads into the LTRs to eventually shut down the expression of EVD genome‐wide via TGS (Marí‐Ordóñez et al, 2013).
A key, unsolved question prompted by this proposed suite of events pertains to the mechanisms whereby RDR6 is initially recruited onto EVD, and more generally on newly invasive TEs, during the primary antiviral‐like silencing phase. “Homology‐” or “identity”‐based silencing entails sequence complementarity between TE transcripts and host‐derived small RNAs. Loaded into AGOs, they likely attract RDR6 concomitantly to silencing execution. One such type of PTGS occurs with TEs reactivated in ddm1/met1 mutants, which, by displaying complementarity mostly to host‐encoded microRNAs, spawn “epigenetically activated siRNAs” (easiRNAs) in an AGO1‐dependent manner (Creasey et al, 2014). easiRNA production likely entails substantial co‐evolution between host and TE genomes (Sarazin & Voinnet, 2014) because miRNAs usually target short and highly conserved TE regions, including the primer‐binding sites required for retroelements’ reverse transcription (RT; Šurbanovski et al, 2016; Borges et al, 2018). Another form of acquired immunity underlying identity‐based silencing is conferred by siRNAs derived from relics of previous genome invasions by the same or sequence‐related TE(s) (Fultz & Slotkin, 2017).
New intruder TEs are unlikely to engage either form of identity‐based silencing, as indeed noted for EVD (Creasey et al, 2014). Thus, RDR6‐dependent PTGS initiation should involve intrinsic features of the TEs themselves (Sarazin & Voinnet, 2014). In the yeast Cryptococcus neoformans, stalled spliceosomes on suboptimal TE introns provide an opportunity for an RDR‐containing complex to co‐transcriptionally initiate such innate PTGS (Dumesic et al, 2013). Studies of transgene silencing in plants (Luo & Chen, 2007; Thran et al, 2012) have advocated other possible mechanisms, though none has yet been linked to epigenetically reactivated TEs. These studies describe how uncapped, prematurely terminated or non‐polyadenylated transcripts might stimulate RDR activities when they evade or overwhelm RNA quality control (RQC) pathways that normally degrade these “aberrant” RNAs (Herr et al, 2006; Gy et al, 2007; Parent et al, 2015). A recent model also contends that widespread translation‐coupled RNA degradation as a consequence of suboptimal codon usage and low GC content might trigger RDR‐dependent silencing in plants (Kim et al, 2021).
Initiation of innate PTGS in the context of EVD likely ties in with an unusual process of splicing‐coupled premature cleavage and polyadenylation (PCPA) shared by Ty1/Copia retroelements to optimize protein expression from their compact genomes (Oberlin et al, 2017). On the one hand, an unspliced and full‐length (fl) GAG‐POL isoform codes for a polyprotein processed into protease, integrase/reverse‐transcriptase RNase, and GAG nucleocapsid components. On the other hand, a spliced and prematurely terminated short (sh) GAG subgenomic isoform is solely dedicated to GAG production. Though less abundant than the flGAG‐POL mRNA, shGAG is substantially more translated (Oberlin et al, 2017). This presumably results in a molar excess of structural GAG for viral‐like particle (VLP) formation compared to Pr‐IN‐RT‐RNase required for reverse transcription (RT) and, ultimately, mobilization (Oberlin et al, 2017; Lee et al, 2020). Supporting the notion that genome expression of Ty1/Copia elements influences PTGS initiation, EVD‐derived RDR6‐dependent siRNAs do not map onto the unspliced flGAG‐POL mRNA, but instead specifically onto the spliced shGAG transcript of which, intriguingly, they only cover approximately the 3’ half (Oberlin et al, 2017).
Here, we show that differential subcellular distribution of the two mRNA isoforms due to splicing‐coupled PCPA accounts for the peculiar EVD siRNA distribution and activity patterns. While the flGAG‐POL isoform remains largely nuclear, the shGAG mRNA is enriched in the cytosol and endows vastly disproportionate translation over flGAG‐POL. However, a previously uncharacterized innate PTGS process accompanies active shGAG translation, manifested as a discrete and unusually intense ribosome stalling event independent of codon usage or GC content, among other tested parameters. Ribosome stalling coincides precisely with the starting point of shGAG siRNA production and maps to the 5’ ends of discrete, shGAG‐derived RNA breakage fragments. These harbor unconventional 5’OH termini that prevent their RQC‐based degradation via 5’P‐dependent XRN4 action (Stevens, 2001; Peach et al, 2015). Based on the well‐documented substrate competition between XRN4 and RDR6 (Gazzani, 2004; Gy et al, 2007; Gregory et al, 2008; Moreno et al, 2013; Martínez‐de‐Alba et al, 2015), we suggest that the 5’OH status of breakage fragments contributes to their conversion into dsRNA by RDR6, thereby initiating PTGS of EVD. We further show that splicing‐coupled PCPA suffices to recapitulate this “translation‐dependent silencing” (TdS) in reporter‐gene settings. Given that Ty1/Copia retroelements share a PCPA‐based genome expression strategy (Oberlin et al, 2017), the phenomenon discovered here with EVD might constitute a more generic primal defense that shapes the siRNA patterns initially associated with Ty1/Copia TEs.
Results
shGAG is the main source and target of EVD‐derived siRNAs
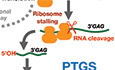
Arabidopsis lines constitutively overexpressing an LTR‐deficient but otherwise intact form of EVD driven by the 35S promoter (35S:EVDwt ) recapitulate the restriction of EVD siRNA to the 3’ part of the shGAG sequence (Marí‐Ordóñez et al, 2013; Oberlin et al, 2017; Fig 1A and B). We explored EVD transcripts levels in 35S:EVDwt in WT (siRNA‐proficient) as opposed to rdr6 (siRNA‐deficient) background (Fig 1B, Appendix Fig S1A). Both in RNA blot and qRT‐PCR analyses, the spliced shGAG mRNA levels were increased in rdr6 compared to WT, whereas those of unspliced flGAG‐POL were globally unchanged (Fig 1C and D). Accordingly, accumulation of the GAG protein—mainly produced via shGAG translation (Oberlin et al, 2017)—was higher in rdr6 compared to WT background (Fig 1E). Essentially identical results were obtained upon epigenetic reactivation of endogenous EVD in non‐transgenic Arabidopsis with the ddm1 single‐ versus ddm1 rdr6 double‐mutant background (Appendix Fig S1B–E). Following EVD mobilization from an early (F8) to a more advanced (F11) epi15 inbred generation (Marí‐Ordóñez et al, 2013) revealed that its progressively increased copy number correlates with progressively higher steady‐state levels of EVD‐derived transcripts and EVD‐derived siRNAs (Appendix Fig S1F and G). Again, these siRNAs disproportionately target the shGAG relative to flGAG‐POL mRNA from F8 to F11 (Appendix Fig S1H). Collectively, these results indicate that PTGS activated de novo by EVD is both triggered by, and targeted against, the spliced shGAG mRNA. Therefore, features associated with shGAG, but not flGAG‐POL, likely stimulate RDR6 recruitment, which we explored by testing current models for PTGS initiation from TEs and transgenes.
Figure 1. EVD shGAG is both a trigger and a target of RDR6‐dependent but miRNA‐independent siRNAs.
-
AEVD flGAG‐POL and spliced shGAG mRNAs are distinguishable using specific PCR primer sets (arrows) for quantification and northern analysis. (35S) Cauliflower Mosaic Virus 35S promoter, (Pr) protease, (IN) integrase, (RT‐RNase) reverse‐transcriptase RNase; red squares: stop codons.
-
BsRNA‐seq reads profile of EVD expressed from 35S:EVDWT in WT (black) or rdr6 (red). (RPM) Reads per million. Positions are indicated in nucleotides (nt) from the start of the 35S sequence. Dashed vertical lines: shGAG and GAG‐POL 3’ ends.
-
CNorthern analysis of EVD RNA isoforms using a probe for the GAG region or for ACTIN2 (ACT2) as a loading control.
-
DqPCR quantification of shGAG and flGAG‐POL normalized to ACT2 and to GLYCERALDEHYDE‐3‐PHOSPHATE DEHYDROGENASE C SUBUNIT (GAPC) levels. qPCR was performed on n = 3 biological replicates; bars: standard error. **P < 0.01 (two‐sided t‐test between indicated values).
-
EWestern analysis of GAG and RDR6 with Coomassie (coom.) staining as a loading control. Arrow indicates cognate RDR6 protein band.
-
F, GsRNA‐seq profiles from EVD de‐repressed in the ddm1 (F) or ddm1 dcl1 (G) backgrounds. Different siRNA size categories are stacked. Nomenclature as in (B).
Source data are available online for this figure.
shGAG siRNA production is miRNA‐independent
Though unlikely (Creasey et al, 2014; Sarazin & Voinnet, 2014), we first considered that production of RDR6‐dependent siRNAs from shGAG might require its cleavage by miRNAs via the easiRNA pathway (Creasey et al, 2014). Arabidopsis miRNA biogenesis depends on DCL1 and the dsRNA‐binding protein HYL1, among other factors (Brodersen & Voinnet, 2006). Analyses of publicly available sRNA‐seq data (Creasey et al, 2014) showed, however, that epigenetically reactivated EVD spawns qualitatively and quantitatively identical shGAG‐only siRNAs in both ddm1 single and ddm1 dcl1 double mutants (Fig 1F and G). Moreover, levels of shGAG siRNA, shGAG mRNA, and GAG protein remained unchanged in 35S:EVDwt plants with either the WT, hypomorphic dcl1‐11, or loss‐of‐function hyl1‐2 background (Appendix Fig S2A–D). By contrast and as expected, production of trans‐acting (ta)siRNAs, which is both miRNA and RDR6 dependent, was dramatically reduced and the levels of tasiRNA precursors and target transcripts enhanced in both mutant backgrounds (Appendix Fig S2A–I). Therefore, RDR6 recruitment to the spliced shGAG mRNA is unlikely to involve endogenous miRNAs via an identity‐based mechanism. We then explored known innate processes of PTGS initiation instead.
Splicing‐coupled premature cleavage and polyadenylation suffices to generate EVD‐like siRNA accumulation and activity patterns
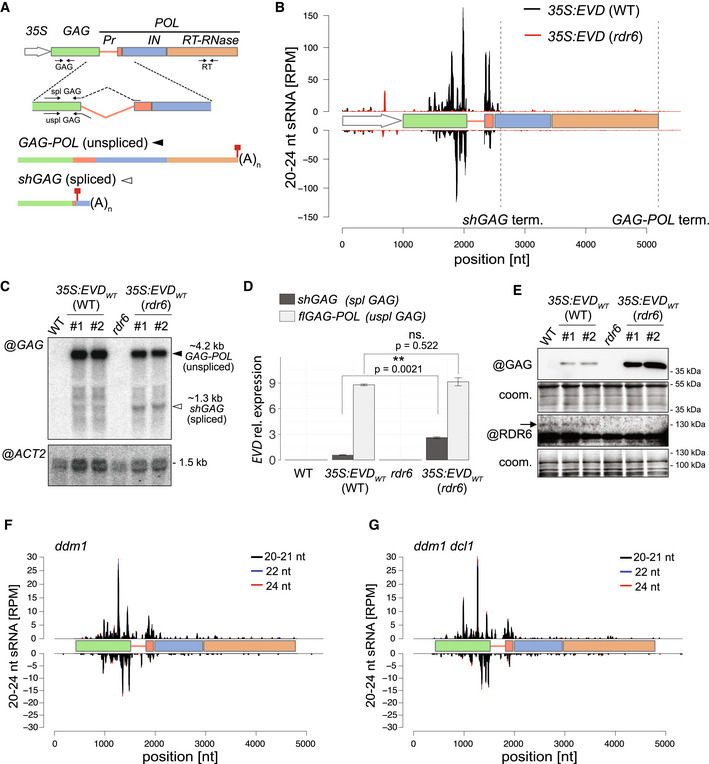
Some cases of transgene‐induced PTGS correlate with a lack of polyadenylation due to aberrant RNA transcription (Luo & Chen, 2007). We ruled out that this feature underlies EVD‐derived siRNA production because shGAG displays no overt polyadenylation defects regardless of the onset of PTGS (Appendix Fig S3A–E). Next, we considered splicing defects, such as inaccurate splicing or spliceosome stalling, and premature transcriptional termination as possible PTGS triggers, two processes previously independently linked to innate, RDR‐dependent siRNA production in plants and fungi (Dumesic et al, 2013; Dalakouras et al, 2019). Ty1/Copia elements have introns that are significantly longer than those of Arabidopsis genes. Moreover, shGAG undergoes atypical splicing‐coupled PCPA (Oberlin et al, 2017). When engineered between the GFP and GUS sequences of a translational fusion, the shGAG intron and proximal PCPA signal spawn unspliced flGFP‐GUS and spliced GFP‐only (shGFP) mRNAs in the Arabidopsis line 35S:GFP‐EVDint/ter ‐GUS (Oberlin et al, 2017) (Fig 2A and B; Appendix Fig S4A). Since this artificial system recapitulates the production of respectively flGAG‐POL and shGAG, we asked whether an EVD‐like siRNA pattern was likewise reproduced.
Figure 2. The EVD intron and terminator suffice to initiate PTGS.
- The 35S:GFP‐EVDint/ter‐GUS fusion was made by introducing the EVD intron and proximal shGAG terminator (including the premature cleavage and polyadenylation site; PCAP) between the GFP and GUS coding sequence. Like EVD, it spawns full‐length unspliced and short‐spliced mRNAs. Red squares: stop codons.
- Expression levels of shGFP (spliced) and GFP‐EVDint/ter‐GUS (unspliced) transcripts, relative to ACT2 and AT4G26410 (RHIP1), in the WT or rdr6 background. qPCR was performed on three biological replicates and error bars represent the standard error on. *P < 0.05 (two‐sided t‐test against corresponding controls).
- sRNA‐seq profile mapped on the genomic 35S:GFP‐EVDint/ter‐GUS locus. (RPM) Reads per million. Positions indicated in nucleotides (nt) from the start of the 35S sequence. Dashed vertical lines: shGFP and GFP‐GUS 3’ ends.
- Low‐molecular‐weight RNA analysis of the GFP‐ and GUS‐spanning regions. tasiRNA255 is a control for the rdr6 mutation and miR159 provides a loading control.
Source data are available online for this figure.
The majority of RDR6‐dependent 21‐nt siRNAs mapped to the GFP, but not the GUS region downstream of the PCPA signal (Fig 2C and D) suggesting that, just like shGAG in EVD, the spliced shGFP mRNA is the main source of siRNAs in GFP‐EVDint/ter ‐GUS. Accordingly, and similar to EVD (Oberlin et al, 2017), many siRNAs spanned the exon–exon junction of GFP‐EVDint/ter ‐GUS (Appendix Fig S4B). Moreover, shGFP, unlike flGFP‐GUS, over‐accumulated in GFP‐EVDint/ter ‐GUS plants with the rdr6 background (Fig 2B, Appendix Fig S4A), indicating that only shGFP is efficiently targeted by PTGS (Fig 2B). Therefore, in the reconstituted setting, the intron and PCPA signal found in shGAG suffice to spawn RDR6‐dependent siRNAs displaying accumulation and activity patterns resembling those generated in the authentic EVD context (Fig 1A–D).
Neither splicing nor intron‐retention per se initiate RDR6 recruitment
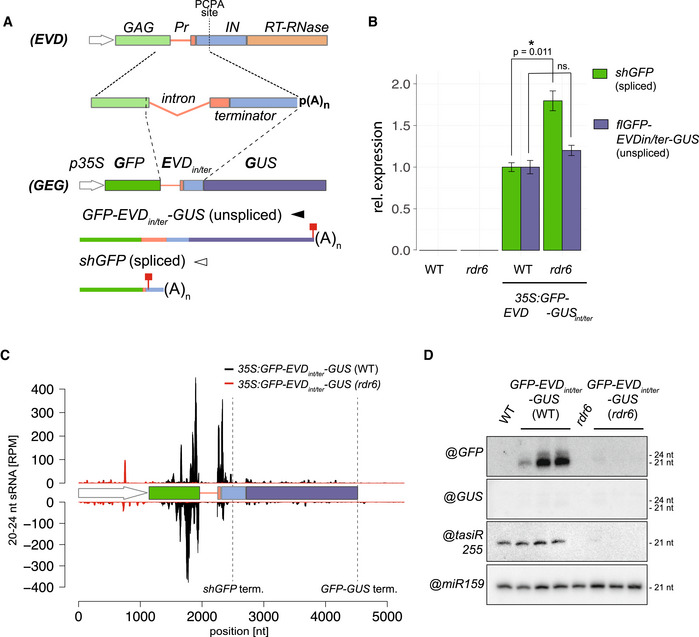
The above result prompted us to investigate a potential facilitating role for splicing in shGAG siRNA biogenesis or, conversely, a role for intron retention in inhibiting RDR6 recruitment to flGAG‐POL. We used previously engineered Arabidopsis EVD‐overexpression lines with a point‐mutated U1 snRNP‐binding site (35S:EVDmU1 ) or a fully deleted intron (35S:EVDΔi ) (Oberlin et al, 2017; Fig 3A–C). 35S:EVDΔi spawns fully matured shGAG transcripts that do not associate with the spliceosome, leading exclusively to prematurely terminated and polyadenylated mRNA species with a stop codon (Oberlin et al, 2017; Fig 3B,D,E, Appendix Fig S5A). However, lack of the intron, and hence splicing, did not prevent RDR6‐dependent siRNA production from 35S:EVDΔi , which was comparable to that of 35S:EVDwt (Fig 3E). Moreover, the shGAG mRNA and GAG protein levels from 35S:EVDΔi were higher in an rdr6 compared to WT background (Fig 3D–F), indicating that EVD’s unconventional splicing is unlikely to underpin shGAG siRNA production.
Figure 3. Impact of splicing and premature termination on EVD silencing.
-
A–CConstructs and isoforms transcribed from 35S:EVD wt (A), 35S:EVD Δintron (B) and 35S:EVD mU1 (C). Probes for northern analysis of GAG exon 1 (GAG‐ex1), intron (GAG‐in), exon2 (GAG‐ex2), and the POL region are depicted with black lines. Red squares: stop codons.
-
DRelative expression levels of spliced and unspliced transcripts in the three EVD constructs relative to ACT2. qPCR was performed on three biological replicates and error bars represent the standard error. (ns.) = non‐significant, *P < 0.05, **P < 0.01, (two‐sided t‐test between indicated samples/targets).
-
EHigh‐ and low‐molecular‐weight RNA analysis of EVD GAG (GAG‐ex1) and EVD intron (GAG‐in) in two independent T1 bulks from each indicated line. The filled arrows on the right‐hand side or with an asterisk on the blots correspond to the transcripts depicted in (A‐C). ACT2: loading control for mRNAs; tasiR255, miR173, and U6: loading controls for sRNAs. Hybridizations for GAG‐ex2 and POL probes are found in Appendix Fig S5A.
-
FWestern blot analysis of the GAG protein with Coomassie (coom.) staining as a loading control.
Source data are available online for this figure.
To test the alternative possibility that intron‐retention or specific sequences within the EVD intron prevent siRNA biogenesis from flGAG‐POL, we analyzed the siRNAs from 35S:EVDmU1 . Impeding U1 binding and its inhibitory action on PCPA causes a complete lack of splicing in EVDmU1 (Fig 3C and D). This generates short unspliced transcripts, alternatively terminated at the cognate shGAG terminator or at an intronic cryptic site previously mapped by 3’ RACE (Oberlin et al, 2017), both detected here by northern analysis (Fig 3C–E, Appendix Fig S5A). Both alternatively terminated transcripts likely undergo translation, albeit largely unproductively (Fig 3F), because low levels of cryptic GAG translation products were detectable in rdr6 compared to WT (Appendix Fig S5B). EVDmU1 bestowed RDR6‐dependent siRNA production expanding—as expected from its non‐spliceable nature—into the retained intron sequence (Fig 3E). The near‐complete lack of siRNAs downstream of the intron (Appendix Fig S5A), by contrast, suggested that both cryptically terminated shGAG transcripts are mainly involved in recruiting RDR6. Therefore, even though the shGAG intron and PCPA signal suffice to trigger PTGS from EVD and GFP‐EVDint/ter ‐GUS (Figs 1 and 2), neither splicing nor intron‐retention per se seem to initiate PTGS. This suggests that splicing‐coupled PCPA does not co‐transcriptionally condition the sensitivity of shGAG to RDR6 but, rather, downstream in the gene expression pathway.
RDR6 recruitment onto shGAG likely requires translation
Splicing‐coupled PCPA, conserved among Arabidopsis Ty1/Copia elements, correlates with the over‐representation of shGAG on polysomes as opposed to the paradoxically more abundant flGAG‐POL (Oberlin et al, 2017). However, among the ddm1‐ or met1‐reactivated Ty1/Copia elements sharing the same genome expression strategy, only EVD spawns detectable RDR6‐dependent shGAG siRNAs (Oberlin et al, 2017), prompting us to explore the basis for this difference. Polysome association, independently of translation efficiency, is the most decisive prerequisite for any given RNA to engage the translation machinery. For instance, many non‐coding RNAs are mostly nuclear (Khanduja et al, 2016), and aberrant (e.g., uncapped and/or poly(A)−) mRNAs are actively degraded by RQC, both of which explain their general absence from polysomes (Doma & Parker, 2007). We conducted genome‐wide correlation analyses between steady‐state transcript accumulation, polysome association, and siRNA levels of reactivated TEs in the ddm1 versus ddm1 rdr6 background by calculating the ratio of polysome‐associated versus total mRNA levels. The same approach was applied to Arabidopsis protein‐coding compared with non‐coding RNAs used as references (Oberlin et al, 2017). This analysis revealed two distinct TE populations according to the levels of associated RDR6‐dependent siRNAs. On the one hand, approximately ¾ of ddm1 de‐repressed TEs (530/674) display varying degrees of polysome association, some within the range of protein‐coding genes (Fig 4A, quartiles 1–3). However, RDR6‐dependent siRNA production does not accompany their reactivation presumably because of their low expression levels (Fig 4B, quartiles 1–3). The remaining ¼ (144/674) of TEs spawn RDR6‐dependent siRNAs, correlating with higher RNA expression levels (Fig 4A and B quartile 4). Nonetheless, unlike those of quartiles 1‐3, these TEs, almost exclusively composed of degenerated LTR/Gypsy elements (i.e., elements shorter than‐full‐length reference ORFs; Fig 4C), resemble non‐coding RNAs in being poorly associated with polysomes, if at all (Fig 4A, quartile 4). By contrast, EVD is the sole LTR/Copia element within quartile 4, in which it is one of the most strongly polysome‐associated elements that concurrently spawn 21–22‐nt siRNAs. Furthermore, when the two EVD isoforms are considered separately, shGAG emerges as a clear outlier by being associated with polysomes to the same extent as protein‐coding mRNAs, (Fig 4A, quartile 4, inlay). flGAG‐POL, by contrast, displays low polysome association albeit higher than most degenerated LTR/Gypsy elements populating quartile 4. In summary, shGAG, compared to flGAG‐POL, is both vastly overrepresented on polysomes (Oberlin et al, 2017) and is the major, if not unique source of EVD‐derived siRNAs (Figs 1 and 3, and Appendix Fig S1). This analysis suggests, therefore, that translation is the step stimulated by splicing‐coupled PCPA of shGAG, upon which RDR6 is recruited specifically onto this mRNA isoform.
Figure 4. Expression, but not translation, is associated with RDR6 activity on most ddm1‐reactivated TEs except EVD .
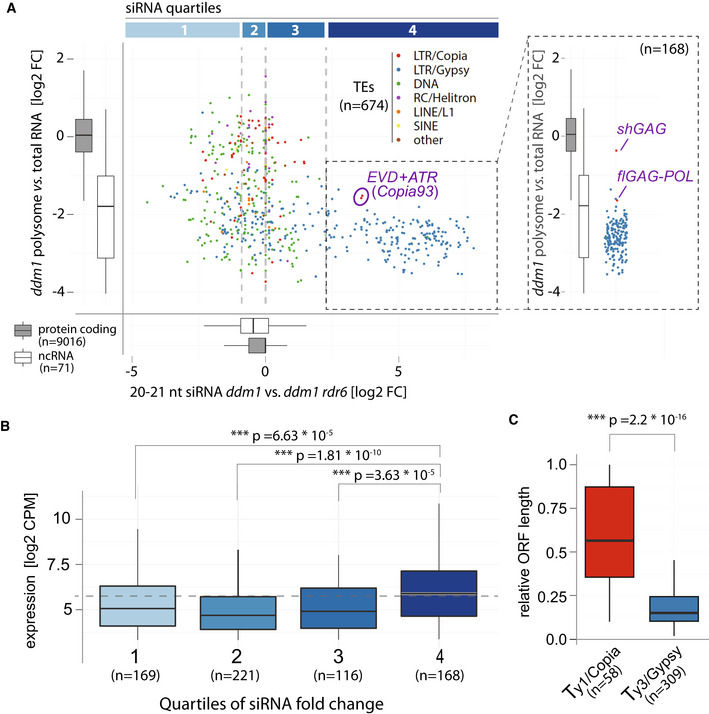
- Scatter plot comparing polysome association score (defined as fold‐change between abundance in polysome libraries vs. total RNA) and RDR6‐dependent siRNA levels of TEs found de‐repressed in ddm1 (brief description of RDR6 dependency). Quartiles of siRNA levels are confined by gray vertical lines. For comparison and reference, polysome association and RDR6‐dependent siRNA levels of protein coding and non‐coding transcripts are displayed as boxplots. Copia93 elements: EVD (AT5G17125) + ATR (AT1G34967), are circled. Inlet: Polysome association score of TEs in quartile 4, EVD mRNA isoforms are displayed separately.
- Boxplots of RNA expression levels of TEs in ddm1 from the quartiles in (A).
- ORF length of Ty1/Copia and Ty3/Gypsy elements expressed in ddm1 relative to their genomic length.
Data information: In all panels: ***P < 0.001, (Wilcoxon rank‐sum test against labeled controls or protein coding gene cohort). For all boxplots, the central band represents the median, boxes are range from the first to third quartile and whiskers range to the largest value within 1.5 times the interquartile range.
Splicing‐coupled PCPA promotes selective translation and PTGS initiation from shGAG‐like mRNA isoforms
To test whether differential translation due to splicing‐coupled PCPA indeed underlies siRNA production from shGAG as opposed to flGAG‐POL, we used GFP‐EVDint/ter ‐GUS, from which the two EVD RNA isoforms and associated siRNA production/activity patterns are recapitulated (Fig 2). Of the shGAG‐like shGFP‐ and flGAG‐POL‐like flGFP‐GUS‐ mRNAs, only the former produced a detectable protein under the form of free GFP (Appendix Fig S4C and D) despite accumulation of both mRNAs (Fig 2A, Appendix Fig S4A). Free GFP levels were increased in the rdr6 background (Appendix Fig S4C), coinciding with increased shGFP‐ but unchanged flGFP‐GUS‐ mRNA levels (Fig 2B). The lack of detectable GFP‐GUS fusion protein—the expected product of flGFP‐GUS—in either WT or rdr6 backgrounds (Fig 2D, Appendix Fig S4C and D) was not due to intrinsically poor translatability. Indeed, GFP‐GUS was the sole protein detected in independent lines undergoing RDR6‐dependent PTGS of 35S:GFP‐GUS, a construct identical to 35S:GFP‐EVDint/ter ‐GUS, save the shGAG intron and PCPA signal (Fig 5A and B). As expected, the GFP‐GUS fusion protein and GFP‐GUS mRNA levels were strongly enhanced in the rdr6 versus WT background (Fig 5A and B). Yet, in contrast to GFP‐EVDint/ter ‐GUS, from which siRNAs are restricted to shGFP, the siRNAs from GFP‐GUS encompassed both the GFP and GUS sequences (Fig 5A). These results therefore indicate that splicing‐coupled PCPA promotes selective translation of, and PTGS initiation from, shGAG‐like as opposed to flGAG‐POL‐like mRNA isoforms.
Figure 5. Splicing promotes translation and siRNA biogenesis from short‐spliced mRNAs by influencing nucleocytoplasmic distribution of RNA isoforms.
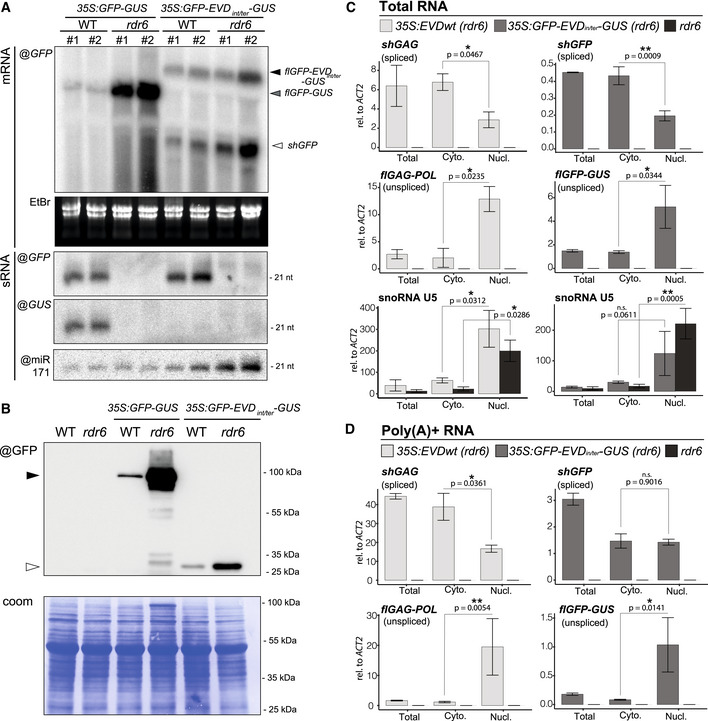
- Comparison of RNA isoforms and sRNA patterns generated by 35S:GFP‐GUS and 35S:GFP‐EVDint/ter‐GUS. High‐ and low‐molecular‐weight RNA analysis using a GFP or GUS probe in two independent transgenic lines from each construct in the WT or rdr6 background. mRNA isoforms are indicated with arrows and correspond to the transcripts depicted in Fig 2A. EtBr staining of the agarose gel and miR171 probe serve as loading control for mRNAs and sRNAs, respectively.
- Western blot analysis of the translation products from GFP and GFP‐GUS transcripts. Coomassie (coom.) staining as a loading control. Black arrow: GFP‐GUS fusion protein; white arrow: GFP protein.
- Nucleo‐cytosolic distribution of 35S:EVD and 35S:GFP‐EVDint/ter‐GUS RNA isoforms in rdr6 relative to that of ACT2 analyzed by qPCR. RNA extracted from total, nuclear (Nucl) and cytoplasmic (Cyto) fractions was reverse‐transcribed with random hexamers and oligo(dT). snoRNA U5 is shown as a nuclear‐only RNA control.
- Same as in (C) but using exclusively oligo(dT) to reverse transcribe poly(A)+ RNAs.
Data information: Both in (C) and in (D), qPCR was performed on n = 3 biological replicates; bars: standard error. *P < 0.05, **P < 0.01 (two‐sided t‐test between indicated samples).
Source data are available online for this figure.
Intron retention causes selective nuclear seclusion of flGAG‐POL‐like mRNAs
What mechanism linked to splicing‐coupled PCPA might underpin the differential translation of shGAG‐like versus flGAG‐POL‐like mRNAs? Noteworthy, splicing generally enhances mRNA nuclear export and translation (Valencia et al, 2008; Sørensen et al, 2017). Conversely, polyadenylated, unspliced mRNAs are retained in the nucleus in Arabidopsis and only exported to the cytoplasm upon splicing (Jia et al, 2020). Moreover, 5’ splice motifs and U1 snRNP‐binding promote chromatin tethering of long non‐coding RNAs in animal cells (Lee et al, 2015; Yin et al, 2020). We thus tested if intron‐retention might promote nuclear sequestration of the unspliced flGFP‐GUS and flGAG‐POL or if, conversely, splicing might favor export of shGFP and shGAG to the cytoplasm, thereby selectively promoting their translation. We performed nucleo‐cytosolic fractionation (Appendix Fig S5C) to analyze the relative distributions of EVD‐derived RNA isoforms produced in 3S:EVDwt or 35S:GFP‐EVDint/ter ‐GUS plants, using spliced/unspliced isoform‐specific PCR amplification. Additionally, unspliced isoforms were selectively analyzed using qPCR primer sets designed to amplify sequences located near the 3’ end of flGAG‐POL or flGFP‐GUS, and absent from shGAG and shGFP (Figs 1A and 2A). A similar approach was used to differentiate the unspliced versus spliced ACTIN mRNA (Appendix Fig S5D). Finally, the nuclear‐only snoRNA U5 (Fig 5C) was used as a control to assess the quality of nuclear enrichments. To optimize accumulation of both types of RNA isoforms, the experiments were all conducted in the PTGS‐deficient rdr6 background.
The analysis revealed strikingly distinct nucleo‐cytosolic distribution patterns for the full‐length versus short‐spliced mRNAs from both systems. Indeed, while the spliced shGFP and shGAG were found predominantly in the cytosol (Fig 5C), flGAG‐POL and flGFP‐GUS were strongly enriched in nuclear fractions (Fig 5C, Appendix Fig S5D). To validate that nuclear unspliced full‐length transcripts are bona fide poly(A)+ mRNAs as opposed to nascent transcripts or splicing intermediates, cDNA from the same RNA samples was synthesized using exclusively oligo‐dT to capture polyadenylated RNAs only. This approach generated comparable results (Fig 5D), indicating that nuclear full‐length transcripts are properly terminated mRNAs. Corresponding results were obtained in epi15 F11 plants displaying endogenous EVD reactivation (Appendix Fig S5E and F). Collectively, these findings suggest that the unique splicing behavior of EVD—which is recapitulated in GFP‐EVDint/ter ‐GUS—not only allows production of the GAG‐encoding shGAG subgenomic mRNA, but simultaneously promotes nuclear retention of flGAG‐POL. This is likely contributing to the disproportionate translation of shGAG over flGAG‐POL, although we do not exclude the involvement of other processes. For instance, in animal cells, exon–junction complex (EJC) deposition enhances translation of mRNAs even when tethered to intron‐less transcripts (Nott et al, 2004). This could also contribute to enhance translation of the splicing‐dependent shGAG isoform. Under these premises, splicing‐coupled PCPA likely predisposes shGAG, as opposed to flGAG‐POL, to one or several co‐translational processes which, in turn, signal(s) RDR6 recruitment.
Saturation of co‐translational mRNA decay is unlikely to trigger shGAG siRNA production
In plants and fungi, decapping coupled to 5’‐>3’ exonucleolytic activity operated by cytosolic XRN proteins regulate the intrinsic half‐life of most actively translated transcripts by degrading decapped mRNAs after the last translating ribosome (Kastenmayer & Green, 2000; Hu et al, 2009; Pelechano et al, 2015; Yu et al, 2016). Of the three Arabidopsis XRNs, XRN2 and XRN3 are nuclear, whereas XRN4 is cytosolic and, hence, mediates co‐translational mRNA decay (Gregory et al, 2008; Yu et al, 2016; Kurihara, 2017). Remarkably, transcripts undergoing improper decapping and/or XRN4‐mediated exonucleolysis constitute competing substrates for RDR6 in Arabidopsis (Gazzani, 2004; Gy et al, 2007; Gregory et al, 2008; Moreno et al, 2013; Martínez‐de‐Alba et al, 2015) (Appendix Fig S6A). For instance, loss‐of‐RDR6 function suppresses the lethality of decapping mutants by preventing production of undesirable siRNAs from hundreds of endogenous mRNAs (Martínez‐de‐Alba et al, 2015). Conversely, loss of XRN4 activity enhances RDR6‐dependent PTGS (Gy et al, 2007; Gregory et al, 2008; Moreno et al, 2013). These observations strongly suggest that RDR6‐dependent PTGS takes over co‐translational mRNA decay when this process becomes saturated by highly abundant and/or highly translated mRNAs.
Likewise, we reasoned that intense translation might overwhelm XRN4‐mediated co‐translational decay of shGAG and thereby concurrently promote RDR6 action (Appendix Fig S6A). This would predict an accumulation of RNA degradation fragments (reflecting XRN4 activity) coinciding with siRNA accumulation. PARE (parallel amplification of RNA ends) and related methods map mostly XRN4 products associated with co‐translational decay as well as non‐translational RNA cleavage events, e.g., miRNA‐mediated slicing of non‐coding RNAs (Gregory et al, 2008; Schon et al, 2018). We therefore conducted nanoPARE analyses, which capture both capped and uncapped RNA fragments (Schon et al, 2018), in ddm1 vs WT Arabidopsis (Fig 6A, Appendix Fig S6B). Simultaneously, mRNA‐seq (i.e., SMART‐seq2) was conducted on the same RNA to monitor gene expression (Schon et al, 2018; Fig 6A). Analysis of TAS1c, which undergoes miR173‐mediated slicing, confirmed that the ensuing 3’ RNA cleavage fragment, a common substrate of XRN4 (Schon et al, 2018), was readily detected in both backgrounds, despite spawning vast amounts of RDR6‐dependent siRNAs (Fig 6A). Analyzing EVD upon its reactivation in ddm1 revealed a low level of RNA degradation fragments spanning the entirety of EVD despite the siRNAs being exclusively derived from shGAG (Fig 6A). Had RNA degradation contributed to siRNA biogenesis, these species would be expected to be distributed along the entirety of EVD, encompassing both shGAG and flGAG‐POL. Inspection of the housekeeping ACT2 locus revealed a similar ORF‐spanning degradation pattern, albeit at substantially higher levels (~10‐fold), presumably reflecting the higher transcript abundance. However, ACT2 does not spawn siRNAs (Fig 6A). These observations therefore reveal no overt correlation between abundance of RNA degradation products, siRNA production, and/or polysome association.
Figure 6. Intense, discrete ribosome stalling on shGAG correlates with RDR6‐dependent siRNA accumulation.
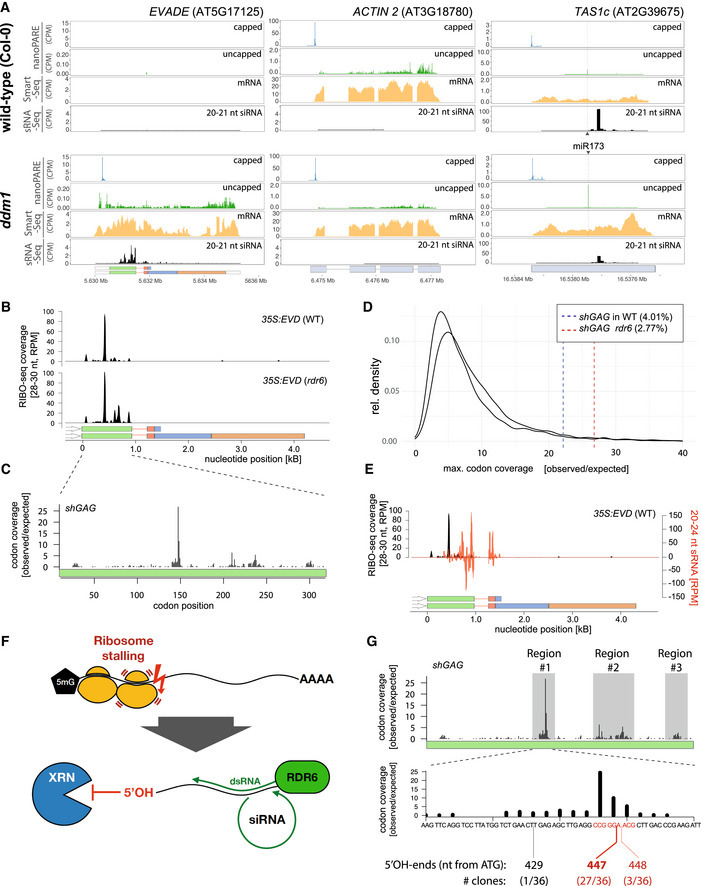
- EVD, ACT2, and TAS1c capped and uncapped 5’ ends from nanoPARE and Smart‐seq2 libraries along 20–21 nt siRNA in WT and ddm1.
- RIBO‐seq coverage profiles from 35S:EVD in WT or rdr6. RPM: Reads per million.
- Ribosomal footprints on shGAG in rdr6 displaying codon occupancy at P‐sites to calculate codon coverage. The coverage observed at each codon position was divided by the expected mean coverage along the entire GAG coding sequence.
- Maximal individual codon coverage over the expected coverage for all translated transcripts of Arabidopsis. Vertical lines indicate the strength of stalling sites of shGAG in the WT or rdr6 background. Percentages specify the proportion of transcripts with more pronounced stalling events than shGAG.
- Overlay between 35S:EVD siRNAs (red) and RIBO‐seq profiles (black) in the WT background.
- Schematic representation of putative ribosome stalling‐linked mRNA breakage generating 5’OH ends. Lack of 5’PO4 prevents XRN 5’‐>3’ exonucleolytic activity (see Fig 6A), granting the RNA to be used as template by RDR6.
- Overlap between ribosomal footprints and mapping of 5’OH ends from 35S:EVD in rdr6 cloned through RtbC ligation. Regions investigated are highlighted in gray. 5’OH ends were only successfully cloned from region #1. Alignment of sequenced clones to EVD is displayed in Appendix Fig S9.
Source data are available online for this figure.
The above results did not formally exclude the possibility that at least some EVD‐associated degradation products identified by nanoPARE might contribute to siRNA biogenesis via competing RDR6 vs XRN4 activities. This would be genetically diagnosed by an increased accumulation of shGAG siRNAs in xrn4 in contrast to their loss in rdr6 (Gy et al, 2007; Gregory et al, 2008). To test this idea without the potential complication of EVD overexpression artificially saturating XRN4 activity in 35S:EVDwt , we introgressed the xrn4 null mutation into epi15 at the early F8 inbred generation, when PTGS of EVD is commonly initiated (Marí‐Ordóñez et al, 2013). As negative controls, we used loss‐of‐function alleles of nuclear XRN2 and XRN3, which, by not contributing to co‐translational mRNA decay, should not influence siRNA production. Finally, the rdr6 mutation was introgressed in parallel, to prevent shGAG siRNA biogenesis. We analyzed two‐to‐three independent lineages with WT versus homozygous mutant backgrounds isolated from segregating F2s. However, neither xrn4 nor xrn2/xrn3 differed from the WT background with regard to EVD expression, copy number, or shGAG siRNA levels (Appendix Fig S6C–N). In contrast, EVD expression and copy numbers were increased in rdr6, coinciding with reduced shGAG siRNA levels (Appendix Fig S6F–H). We conclude from these collective results that saturation of XRN4‐dependent co‐translational mRNA decay (Appendix Fig S6A) is unlikely to underlie shGAG siRNA production. siRNAs are, instead, abruptly spawned from the middle up to the 3’ end of shGAG, as if their production coincided with a discrete co‐translational event (Fig 1B). A similar rationale should apply to the discrete shGFP‐centric siRNA pattern spawned from GFP‐EVDint/ter ‐GUS (Fig 2C).
The initiation of RDR6 activity coincides with isolated and intense ribosome stalling events
To overcome the caveat of EVD cell‐specific expression (Marí‐Ordóñez et al, 2013) and simultaneously investigate which co‐translational event(s) might trigger the siRNA patterns in both shGAG and shGFP, we generated RIBO‐seq datasets (Ingolia et al, 2009) from 35S:EVDwt and 35S:GFP‐EVDint/ter ‐GUS. This resulted in high‐quality ribosome footprints (RFPs) displaying the characteristic triplet periodicity (Appendix Fig S7A–E). We found that EVD RFPs in the 35S:EVDwt background map near‐exclusively onto shGAG, underscoring its preferential translation (Figs 4A and 6B). However, a strong and isolated footprint peak was detected near the middle of the shGAG ORF (Fig 6B), suggesting intense ribosome stalling at this position. This stalling peak was also found within the shGAG coding sequence of endogenous EVD in one public ddm1 RIBO‐seq library (Kim et al, 2021) (Appendix Fig S8A). By specifying codon occupancy of ribosome P‐sites—the sites of peptidyl transfer activity—reflecting the codon dwell time, we found that > 35% of shGAG translating ribosomes are located on two consecutive codons (pos.148–149) coinciding with this peak (Fig 6C). Having normalized these proportions to ORF lengths, we compared them to those of actively translated Arabidopsis mRNAs. To exclude artifacts from transcripts with low coverage, we restricted our analysis to the most abundant mRNA isoforms with coverage available for more than 70% of ORFs, as described (Sabi & Tuller, 2015). We found that shGAG ranks among the top 4.01 and 2.77% (in WT and rdr6 backgrounds, respectively) of Arabidopsis transcripts displaying the most intense stalling events (Fig 6D). Remarkably, overlaying siRNAs and codon coverage intensity revealed that the intense stalling position coincides nearly exactly with the 5’ starting point of the RDR6‐dependent EVD siRNA pattern (Fig 6E). Stalling is not a consequence of RDR6 recruitment, because it also occurs in the rdr6 background (Fig 6B).
To explore further a possible link between discrete, intense ribosome stalling and RDR6 activity, RFPs were conducted in the 35S:GFP‐EVDint/ter ‐GUS background. As seen above for shGAG versus flGAG‐POL in the EVD context, the analysis confirmed the vastly disproportional translation of shGFP versus flGFP‐GUS (Appendix Fig S8B). It also identified a major stalling site only in the shGFP ORF (whose detection was enhanced in the rdr6 background) in which two prominently covered and consecutive codons (pos. 235–236) accounted for ~40% of footprints (Appendix Fig S8C). Similarly to shGAG, shGFP ranked among the top 3.21–4.14% Arabidopsis transcripts displaying the most intense stalling events (Appendix Fig S8D). Furthermore, this stalling site was located between major peaks of shGFP siRNAs, in this case, in both the 5’ and 3’ directions within GFP‐EVDint/ter ‐GUS (Appendix Fig S8E).
A recent model advocates a possible link between suboptimal codon usage and PTGS initiation in plants (Kim et al, 2021). However, overlaying the Arabidopsis codon adaptation index with the codon coverage of ribosomes on shGAG and shGFP did not reveal any overt correlation between ribosome stalling and codon suboptimality (Appendix Fig S9). The cited study showed that codon optimization by increasing the CG3 content (CG content on the 3rd codon position) in a region corresponding, surprisingly, to the shGAG 3’UTR enhanced translation of a linked luciferase ORF (Kim et al, 2021). Yet, our analysis shows that neither CG nor CG3 content overtly influences ribosome association along shGAG or shGFP, let alone the intense stalling event detected on either mRNA (Appendix Fig S9). Two consecutive codons at the stalling site identified on shGAG code for proline and glycine (Appendix Fig S9A) and, interestingly, single prolines (P) and/or glycines (G) at P‐sites correlate with ribosome stalling in animals and fungi (Artieri & Fraser, 2014; Sabi & Tuller, 2015; Zhao et al, 2021). To assess whether the consecutive proline‐glycine amino acids influence translation and siRNA biogenesis, P148 and G149 were mutated to serine (S) and alanine (A), respectively, in the EVD construct (Appendix Fig S10A). In several bulks of T1 transformants, neither siRNA‐ nor Gag‐ levels were altered compared to those produced from the unmodified construct (Appendix Fig S10C and E). Consistent with this negative result, > 45% codon occupancy on shGFP occurs on consecutive glutamate and leucine, not proline/glycine, codons (Appendix Fig S9B).
In addition to the identity of some codons, secondary RNA structures have been correlated with ribosome stalling (Doma & Parker, 2006; Yan et al, 2015; Bao et al, 2020), including G‐quadruplexes (Song et al, 2016; Fay et al, 2017). In particular, sites of “ribothrypsis”—a ribosome stalling‐induced process recently described in metazoans—are positively correlated with such occurrences within ORFs (Ibrahim et al, 2018). By forming secondary structures, G‐quadruplexes are thought to act as “roadblocks” hampering proper ribosome progression during elongation (Song et al, 2016). G‐quadruplex scoring along the shGAG and shGFP mRNA did reveal potential hot spots of such motifs (Appendix Fig S9). While they are localized far upstream or downstream of shGAG ribosome stalling sites (Appendix Fig S9A), a putative G‐quadruplex is indeed located immediately downstream of the main shGFP stalling site (Appendix Fig S9B), which we deleted in the shGFP construct (Appendix Fig S10B). However, this did not have any overt effect on siRNA and Gag levels (Appendix Fig S10D and F). Therefore, EVD and GFP‐EVDint/ter ‐GUS transcripts display similar behavior, whereby RDR6‐dependent production of shGAG‐ or shGFP‐only siRNAs coincides with highly localized and unusually intense ribosome stalling events. While these stalling events have likely distinct albeit as‐yet‐unidentified causes for each transcript, both appear to stimulate co‐translational processing of RNA intermediates that, in turn, serve as RDR6 substrates.
Ribosome stalling correlates with production of 5’‐hydroxy 3’‐cleavage fragments that possibly serve as RDR6 substrates
As described above, nanoPARE in ddm1 did not reveal any discrete RNA products with 5’ ends mapping consistently at, or near, the stalling site in shGAG. We also failed to detect such products using classic 5’ RACE (Llave et al, 2002). Noteworthy, this technique relies on a 5’ monophosphate (5’P) for RNA ligation of 5’ adaptors (Silber et al, 1972; Wang & Fang, 2015). Intriguingly, 5’P was reported to be absent from various 3’ cleavage RNA fragments produced co‐translationally in budding yeast, including upon ribosome stalling (Peach et al, 2015; Navickas et al, 2020). A lack of 5’P is also strongly suspected for the 3’ cleavage products of ribothrypsis (Ibrahim et al, 2018). Since siRNA production from EVD initiates just downstream of the major stalling site (codons 148–149; Fig 6E), we thus considered the possibility that discrete 3’ cleavage RNA fragments devoid of a 5’P—and thus akin to the 5‘OH RNA associated with the above‐mentioned processes (Peach et al, 2015; Ibrahim et al, 2018; D’Orazio et al, 2019; Navickas et al, 2020)—might constitute RDR6 templates (Fig 6F).
To explore such a connection and simultaneously characterize and map the 5’ ends of putative shGAG 3’ cleavage fragments, we used the RtcB RNA ligase. RtcB contributes to tRNAs splicing by ligating RNAs with 3’P ends (or 2’,3’‐cyclic phosphate) to 5’OH ends and was used previously to map co‐translational RNA cleavage fragments in yeast (Desai & Raines, 2012; Peach et al, 2015). A 5’ RNA adaptor with a 3’P end was therefore RtcB‐ligated to total RNA extracted from plants expressing 35S:EVD or non‐transgenic controls, both in the rdr6 background. Use of rdr6 prevented conversion of potential RDR6 templates into dsRNA as well as the accumulation of confounding cleavage fragments potentially caused by the ensuing secondary siRNAs. The ligated RNA was then subjected to reverse transcription using EVD‐specific primers surrounding the major stalling site (Fig 6G; Region #1), amplified through PCR, and cloned following standard RACE procedures. Based on the EVD ribosome footprint profile (Fig 6C), we also investigated two additional regions more covered with ribosomes than expected (Fig 6G; Regions #2 & 3). Only region #1 yielded detectable amplification products within the expected size range. Nonetheless, gel excision within the anticipated size ranges followed by cloning was performed for all regions in all genotypes (Appendix Fig S11A–C). Sanger sequencing revealed that 30 out of 36 fragments cloned from region #1 displayed 5’OH ends consistently mapping at nucleotides 447–448, strikingly defining the intense ribosome stalling site on shGAG (Fig 6G, Appendix Fig S11D) from which siRNA production is initiated (Fig 6E). By contrast, the clones obtained from regions #2 and #3 were either devoid of EVD sequences or empty. These results are consistent with the notion that the intense ribosome stalling event correlates with breakage of the shGAG RNA and that the ensuing 5’OH fragments serve as templates for RDR6 to initiate dsRNA production and downstream siRNA processing. Given that XRNs require a 5‐P for their 5’‐>3’ exonucleolytic activities (Stevens, 2001; Schon et al, 2018), this could explain the insensitivity of shGAG‐derived siRNA accumulation to any xrn mutation and to xrn4 in particular (Appendix Fig S6). Being linked to 3’ cleavage fragments inaccessible to the competing activity of XRN4, ribosome stalling might thus optimize the recruitment of RDR6 on shGAG for PTGS initiation. We note that nanoPARE, while being indiscriminative of RNA 5’‐ends (including 5’OH) requires a 3’ polyA tail to generate cDNA. Thus, the fact that the technique failed to detect the shGAG 5’OH cleavage fragments could indicate that they are indeed mostly poly(A)−. Preliminary PAGE‐based analyses as conducted for the 3’ ends of ribothrypsis products in mammalian cells (Ibrahim et al, 2018) suggested that discrete shGAG‐derived 3’ cleavage fragments are found in the poly(A)‐ fraction isolated from 35S:EVD in the rdr6 background (Appendix Fig S12A). Lack‐of‐poly(A) could further optimize RDR6 recruitment because the enzyme is inhibited in vitro by 3’ adenosine stretches (Baeg et al, 2017).
Discussion
Translation as an initiator of PTGS and epigenetic silencing
Protein synthesis is commonly merely seen as a target of PTGS by reducing the amount of available RNA and/or interfering with translation. Our study adds to a growing body of work identifying translation also as a trigger for PTGS (Sun et al, 2020; Iwakawa et al, 2021). This became evident after epigenetic reactivation of EVD, from which splicing‐coupled PCPA generates separate RNA isoforms from a single transcription unit. Of the two, the shorter subgenomic shGAG RNA undergoes disproportionate translation over flGAG‐POL as an indispensable feature of Ty1/Copia biology because this likely provides the stochiometric protein balance necessary for efficient amplification and mobilization of the element. This process, however, concomitantly correlates with RDR6 activity. shGAG translation efficacy per se is within the range of moderately translated Arabidopsis mRNAs and is unlikely to explain this effect, nor do GAG expression or abundance. Rather, an exceptionally intense and highly discrete ribosome stalling event coincides with RDR6‐dependent PTGS of shGAG. Our data also suggest how intron‐retention in combination with active splicing accounts for the mostly nuclear versus cytosolic localization of flGAG‐POL versus shGAG, respectively. Their asymmetrical subcellular distribution concurrently rationalizes (i) the disproportionate translation efficacies of each mRNA, (ii) the shGAG‐centric distribution of translation‐dependent EVD‐derived siRNAs, and consequently, (iii) the contrasted sensitivity of each isoform to cytosolic PTGS. Splicing‐coupled PCPA probably underlies most, if not all, of features (i–iii) because they were recapitulated with the GFP‐EVDint/ter ‐GUS construct containing the shGAG intron and proximal PCPA signal (Figs 2 and 5, and Appendix Fig S4). Since splicing‐coupled PCPA is at the very core of the Ty1/Copia genome expression strategy (Oberlin et al, 2017), the process described here for EVD is likely to be broadly applicable.
Being mostly nuclear, flGAG‐POL, the template for RT required for mobilization, is neither a potent trigger nor a target of PTGS, likely explaining why increasing amounts of shGAG siRNAs have little impact on EVD’s genomic proliferation over successive epi15 inbred generations (Marí‐Ordóñez et al, 2013). Previously attributed to GAG‐mediated protection of flGAG‐POL as part of VLPs (Marí‐Ordóñez et al, 2013), we now consider flGAG‐POL nuclear retention as an additional and perhaps major contributor to this shielding effect. The ensuing rise in EVD genomic copies causes increasing levels of RDR6‐dependent shGAG dsRNA over generations. We previously suggested that these levels eventually saturate DCL4/DCL2 activities in the highly cell‐specific expression domain of EVD, acting as a prerequisite to DCL3 recruitment and RdDM, ultimately causing LTR methylation and TGS of all EVD copies (Marí‐Ordóñez et al, 2013). This proposed saturation‐coupled PTGS‐to‐TGS switch invariably occurs in epi15 and other EVD‐reactivating epiRILs when the EVD copy number reaches 40–50 (Marí‐Ordóñez et al, 2013), causing only sporadic and minor developmental defects even in advanced generations (Mirouze et al, 2009; Marí‐Ordóñez et al, 2013; Quadrana et al, 2016). By contrast, EVD copy number increases well beyond 80 in rdr6 mutants already in F2s (Appendix Fig S6H) displaying loss of fertility (Appendix Fig S12B) likely solely ascribable to enhanced EVD proliferation. These data attest to a central role for RDR6 in controlling EVD’s mobilization and perhaps that of other autonomous TEs, at the level of translation. At least in the multi‐generational context of epi15, our results also establish a hitherto‐unrecognized role for translation in not only PTGS, but also, ultimately, epigenetic silencing and TGS.
Translation‐dependent silencing as a sensor for de novo invading, foreign genetic elements
The vast majority of ddm1‐reactivated TEs that spawn RDR6‐dependent siRNAs is composed of LTR/Gypsy elements (Fig 4), which is the family most prominently associated with easiRNA production (Creasey et al, 2014; Borges et al, 2018). Arabidopsis LTR/Gypsy elements generally display significantly shorter‐than‐full‐length ORFs as compared to the other main classes of Arabidopsis TEs, including the LTR/Copia family to which EVD belongs (Oberlin et al, 2017; Fig 4C). Although they likely constitute, therefore, degenerated transcription units, a substantial fraction of such LTR/Gypsy is nonetheless highly expressed as a possible source of abundant aberrant RNAs (Fig 4B). Thus, alternatively or concurrently to easiRNA production, some of these LTR/Gypsy remnants might also enter the RDR6 pathway by saturating RQC either co‐transcriptionally or post‐transcriptionally. While this process possibly underlies a previously documented expression‐dependent form of innate TE silencing (Panda et al, 2016; Fultz & Slotkin, 2017), such loci might in turn autonomously produce siRNAs and become sources of identity‐based silencing. Regardless, the combined action of all these silencing pathways likely explains why most siRNA‐generating TEs in ddm1 do not actively engage translation, as evidenced by their conspicuous underrepresentation on polysomes (Fig 4A; quartile 4; Oberlin et al, 2017). In fact, siRNA‐generating TEs are equally or even less polysome‐associated than are non‐coding RNAs (Fig 4A), indicating that there is no general correlation between siRNA production and translation. Conversely, numerous TEs are translated, yet do not spawn siRNAs (Fig 4A). These data contradict recent claims advocating a general correlation between siRNA production and translation based on the untested premise that most siRNA‐generating TEs are translated (Kim et al, 2021), whereas they are, in fact, absent from polysomes (Oberlin et al, 2017; Fig 4A). Based on our experimental findings, we argue, on the contrary, that the process of “translation‐dependent silencing” (TdS) described here is an attribute of only a handful of evolutionary young TEs. These chiefly include EVD, which concurrently undergoes productive translation (mostly of shGAG) and spawns RDR6‐dependent siRNAs (Figs 1 and 4, and Appendix Fig S1).
EVD is among the few autonomously transposing LTR/TEs in the Arabidopsis Col‐0 genome (Mirouze et al, 2009; Reinders et al, 2009; Tsukahara et al, 2010; Gilly et al, 2014) and, as such, is unlikely controlled by identity‐based mechanisms. TdS might enable the plant to detect its activity as the first line of defense against de novo invasions, for instance upon horizontal transfer of active TEs. TdS may likewise underpin silencing triggered upon experimental transfer of “exogenous” TEs between species separated by millions of years of evolution. For instance, similarly to EVD, the epigenetic silencing of two tobacco retrotransposons, Tnt1 and Tto1, is copy‐dependent when they are horizontally transferred into Arabidopsis by transgenesis (Hirochika et al, 2000; Fultz & Slotkin, 2017). Although the translation dynamics of Tnt1 and Tto1 in Arabidopsis has not been investigated, TdS might also contribute to their initial recognition in addition to a transcriptional‐level of regulation as proposed forTto1 (Fultz & Slotkin, 2017).
Viruses divert a substantial fraction of the host translational apparatus to their highly compact and TE‐like genomic and subgenomic RNAs (Gao et al, 2003; Dreher & Miller, 2006; Sztuba‐Solińska et al, 2011), which might also predispose them to TdS. In all these circumstances, a key feature of TdS is an innate ability to detect transcripts by virtue of their foreign—as opposed to aberrant—nature, independently of any sequence homology to the host genome. We propose that foreignness is perceived by anomalies manifested during active translation, which likely include abnormal ribosome stalling. Despite extensive reverse genetics in EVD and shGFP, we failed to identify possible causes of stalling (Appendix Figs S9 and S10), having additionally ruled out any contribution of codon usage and CG/CG3 content (Appendix Fig S9). This is in sharp contrast with a recent model proposed by Kim and coworkers (Kim et al, 2021), which contends that PTGS via RDR6 might be caused by a multitude of presumptive translation stalling events. These were allegedly ascribed to pervasive suboptimal codons and low content in CG and CG3 along the ORFs of certain mRNAs, including TE‐derived RNAs. This interpretation is neither compatible with the highly discrete nature of the stalling events experimentally detected in our study nor with the noticeable absence, from polysomes, of the TE RNAs used by Kim et al. to build their model, EVD excepted (Fig 4A). Possible causes of ribosome stalling, including mRNA‐extrinsic ones, are further evoked in Appendix Discussion and Appendix Fig S13. In particular, we argue why a recently proposed model of RISC‐mediated stalling‐coupled RDR6 recruitment (Iwakawa et al, 2021) during tasiRNA biogenesis unlikely applies to EVD, leading us to consider a potential causal role for stalling‐coupled 5’‐OH RNA fragment generation in stimulating RDR6 activity. Since we were unable, however, to genetically impede stalling (Appendix Fig S10), we can only speculate in the model described in the next section since, at this stage, 5’OH RNA fragments might be mere byproducts of ribosome stalling.
Biogenesis of 5’OH RNA fragments and their putative link to RDR6 recruitment
Independently of their possible role in TdS, a first question pertains to how 5’OH RNA fragments might be generated. In budding yeast, the metal‐independent endonuclease Cue2 cleaves, within the colliding ribosome’s A site, mRNAs undergoing stalling‐induced no‐go decay, which generates 5’OH 3’ RNA fragments (D’Orazio et al, 2019). The mammalian homolog, N4BP2, additionally contains a polynucleotide kinase domain, which might directly couple endonucleolysis with the 5’P‐dependent XRN‐licensing step evoked below (D’Orazio et al, 2019). We failed, however, to identify a plant Cue2/ N4BP2 ortholog such that other mechanisms might underlie what we conservatively refer to as “translation‐linked mRNA breakage” here.
Intense stalling is usually resolved on the protein side by ubiquitin‐mediated proteolysis (Joazeiro, 2019). Translation‐decoupled RNA degradation is involved on the RNA side (Ikeuchi et al, 2018), including XRN‐mediated exonucleolysis operated in processing (P) bodies (Maldonado‐Bonilla, 2014). Yet, 5’OH RNA is not directly accessible to XRN action, which requires 5‐P termini (Stevens, 2001). In budding yeast, the Trl1 kinase phosphorylates these fragments to license their degradation by XRNs (Navickas et al, 2020), which also likely occurs during ribothrypsis in mammalian cells (D’Orazio et al, 2019). Alternatively/additively, the mouse and fission yeast DXO/Rai1, which removes incomplete 5’‐mRNA caps, catalyzes the removal of 5’OH ends, exposing 5’P for subsequent 5’‐>3’ exoribonuclease activity (Doamekpor et al, 2020). In contrast to RNAi‐deficient budding yeast or RNAi‐proficient mammalian cells, plants display RDR activities (Stein et al, 2003; Drinnenberg et al, 2009). We suggest that in these organisms, 5’OH termini would not only disqualify XRN4 action, but concurrently optimize that of RDR6, which is known to compete with XRN4 for substrates, including those evading co‐translational decay (Gy et al, 2007; Gregory et al, 2008). RDR6 action in TdS is possibly further facilitated by the striking physical proximity of P‐bodies—where unresolved 5’OH RNA fragments should primarily accumulate—with the so‐called “siRNA bodies” involved in RDR6‐dependent tasiRNA processing (Martínez‐de‐Alba et al, 2015). In principle, RDR6 could also pick up a multitude of RNA cleavage fragments predictably produced via siRNA‐guided cleavage of shGAG by RISCs. However, RISC‐mediated slicing produces 5’P termini (Martinez & Tuschl, 2004), qualifying these RNAs as XRN4‐, as opposed to RDR6‐, substrates unlikely, therefore, to contribute prominently to shGAG siRNA production.
Concluding Remarks
Transient ribosome stalling is a normal and favorable feature of translation, enabling proper folding of nascent peptides (Rodnina, 2016). Accordingly, many mechanisms exist to resolve such instances (Buskirk & Green, 2017) including ribothrypsis in mammalian cells, an apparent widespread component of ordinary translation (Ibrahim et al, 2018). However, while its initiation strongly resembles that of mammalian ribothrypsis, TdS is unlikely to be ubiquitous in plants, since its RNA products, by directly engaging RDR6 for amplified siRNA production, would promote degradation of the entire mRNA pool independently of its stalled or even merely translated status. While this would be highly detrimental as a common form of endogenous gene regulation, the process seems particularly well suited to eliminate highly proliferating foreign RNAs such as those of viruses and TEs.
Material and Methods
Plant material and growth conditions
Plants were grown in a growth chamber on soil at 22°C for two weeks in a 12‐h/12‐h light cycle and then transferred to a 16‐h/8‐h light cycle and pools of three to five plants were sampled for inflorescence tissue. Mutant genotypes met1‐3, dcl1‐11, ddm1‐2 (seventh inbred generation), hyl1‐2, rdr6‐12, xrn2‐2, xrn3‐3, xrn4‐3 plants are all derived from the Col‐0 ecotype (Vongs et al, 1993; Peragine et al, 2004; Vazquez et al, 2004; Gy et al, 2007). Genotyping primers are described in Appendix Table S1. met1‐derived epiRIL#15 plants (epi15) were described previously (Reinders et al, 2009; Marí‐Ordóñez et al, 2013). 35S:EVDwt, 35S:EVDmU1 , 35S:EVDΔi , 35S:GFP‐GUS and 35S:GFP‐EVDint/ter ‐GUS overexpression lines were previously depicted (Marí‐Ordóñez et al, 2013; Oberlin et al, 2017).
Constructs and plasmids
All constructs are available from addgene (www.addgene.org): 35S:EVDwt (#167119), 35S:EVDmU1 (#167121), 35S:EVDΔI (#167120), 35S:GFP‐GUS (#167122) and 35S:GFP‐EVDint/ter‐GUS (#167123; Marí‐Ordóñez et al, 2013; Oberlin et al, 2017).
Cyto‐nuclear fractionations
For each sample, twice 250 mg of 3‐week‐old seedlings grown in ½ strength (2.2 g/l) Murashige and Skoog medium (#M0231, Duchefa Biochemie) was ground to fine powder in liquid nitrogen and homogenized in 575 μl of lysis buffer (10 mM Tris‐HCl pH 7.4, 150 mM NaCl, 0.15% IGEPAL (CA‐630, Merk) and 1× cOmplete protease inhibitor cocktail (Roche)). Lysates were gently mixed and incubated on ice for 10 min. before being filtered through one layer of Miracloth. 400 μl from each lysate was recovered and one set aside as Total. The second set of cell lysate were gently overlaid on top of 1 ml of cold sucrose buffer (10 mM Tris‐HCl pH 7.4, 150 mM NaCl, 24% sucrose and 1× cOmplete EDTA‐free protease inhibitor cocktail (#04693159001, Roche)) in protein low binding 1.5‐mL tubes (LoBind, Eppendorf) by slowly pipetting against the side of the tube. Samples were centrifuged at 3,500 g for 10 min. to separate nuclei (pellet) from cytoplasm (supernatant). Cytoplasmic fractions were cleared by centrifugation at 14,000 g for 1 min. in a new tube and the resulting supernatant set aside. Nuclear pellets were rinsed by inverting the tube 3–5 times without disturbing the pellet with 1 ml of 1× PBS, 0.5 mM EDTA. Nuclei were spin for 15 s at 1,300 g before gently removing the wash solution. Nuclei pellets were resuspended by pipetting in 200 μl of nuclear lysis buffer (10 mM Tris–HCl pH 7.4, 300 mM NaCl, 7.5 mM MgCl2, 0.2 mM EDTA pH8, 1 M urea, 1% IGEPAL, and 1× cOmplete protease inhibitor cocktail). For isolation of total RNA and protein from the different fractions, samples were mixed 1 volume of acid PCI (phenol/chloroform/isoamyl alcohol, #X985 Carl Roth). In addition, nuclear fractions were further homogenized after addition of PCI by passing the sample through a 21‐gauge needle with a 1‐ml syringe. All steps were carried on ice or centrifuged at 4°C. Buffers were freshly prepared in advance and chilled on ice before use.
Nucleic acid and protein extractions
RNA was extracted from frozen and ground tissue with TRIzol reagent (#93289, Sigma) and precipitated with 1× vol. of cold isopropanol. For RNA extraction from cyto‐nuclear fractionations, 20 μg of glycogen (#R0551, ThermoFisher) and 0.1× vol. of sodium acetate 3 M pH5.2 was mixed with recovered aqueous phases after PCI before RNA precipitation with 1× vol. of cold isopropanol. DNA was extracted using the DNeasy Plant Mini Kit (#69204, Qiagen) according to manufacturer’s guidelines.
Protein of frozen and ground tissue was homogenized in extraction buffer (0.7 M sucrose, 0.5 M Tris–HCl, pH 8, 5 mM EDTA, pH 8, 0.1 M NaCl, 2% β‐mercaptoethanol), and cOmplete EDTA‐free protease inhibitor cocktail (#04693159001, Roche). Water‐saturated and Tris‐buffered phenol (pH 8) was added to an equal volume and samples were agitated for 5 min. Phases were separated by 30‐min centrifugation (12,000 g at 4°C). Proteins were precipitated from the phenol phase (including those from PCI) by the addition of 5 volumes of 0.1 M ammonium acetate in methanol. Precipitated proteins were collected by centrifugation for 30 min (12,000 g at 4°C), washed twice with ammonium acetate in methanol and resuspended in resuspension buffer (3% SDS, 62.3 mM Tris‐HCl, pH 8, 10% glycerol).
RNA and protein blot analysis
For high‐molecular‐weight RNA analysis, 5–10 µg of total RNA was separated on a 1.2% agarose MOPS‐buffered gel with 2.2 M formaldehyde. RNA was partially hydrolyzed on gel with 5× gel volumes of 0.05N NaOH for 20 min. Gel was washed twice for 20 min with 20X SSC, transferred overnight by capillarity to a HyBond‐NX membrane (#RPN303, GE Healthcare) and UV‐crosslinked for fixation. For high‐molecular‐weight RNA analysis by PAGE, 1–40 µg of RNA (total, poly(A)+ or poly(A)−) was separated on a denaturing 4% polyacrylamide–urea gel, transferred to a HyBond‐NX membrane by electroblotting and UV‐crosslinked. For low‐molecular‐weight RNA analysis, 10–40 µg of total RNA was separated on a denaturing 17.5% polyacrylamide–urea gel, transferred to a HyBond‐NX membrane by electroblotting and chemically crosslinked (Pall & Hamilton, 2008). Probes from PCR products were radiolabeled using the Prime‐a‐Gene kit (#U1100, Promega) in the presence of [α‐32P]‐dCTP (Hartmann Analytic) and oligo probes were radiolabeled by incubation of PNK (#EK0031, Thermo) in the presence of [γ‐32P]‐ATP. Membranes were hybridized with these probes in PerfectHyb hybridization buffer (#H7033, Sigma) and detected on a Typhoon FLA 9500 (GE Healthcare) laser scanner. Oligonucleotides used for probe generation are listed in Appendix Table S1.
Proteins were separated on SDS‐polyacrylamide gels, transferred to Immobilon‐P PVDF membranes (#IPVH00010, Millipore) by electroblotting and incubated with antibodies in 1× PBS with 0.1% Tween‐20 and 5% nonfat dried milk. After incubation with HRP‐conjugated secondary goat antibody against rabbit or rat primary antibodies (Sigma), detection was performed with the Clarity Max Western ECL substrate (#1705062, BIO‐RAD) on a ChemiDoc Touch imaging system (BIO‐RAD). Affinity‐purified antibodies were used at the specified dilutions: GAG (1:2,000; Oberlin et al, 2017), GFP (1:5,000 Chromotek #3H9‐100), GUS (1:1,000 Sigma‐Aldrich #G5545), H3 (1:10,000 Abcam #ab1791), UGPase (1:2,000 Agrisera #AS5 086). Protein loading was confirmed by Coomassie staining of membranes.
Quantitative PCR
RNA was treated with DNaseI (#EN0521, Thermo Scientific) and cDNA was subsequently synthesized with the Maxima First‐Strand cDNA Synthesis Kit (#K1641, Thermo Scientific), or RevertAid cDNA Synthesis Kit with Oligo(dT) (#K1612, Thermo Scientific). qPCRs were run on a LightCycler480 II (Roche) or a QuantStudio5 (Applied Biosystems) machine with the SYBR FAST qPCR Kit (KAPA Biosystems). Ct values were determined by the 2nd derivative max method of minimally two technical replicates for each biological replicate. Relative expression values were computed as ratios of Ct values between targets of interest and ACT2 and/or GAPC reference mRNA unless otherwise indicated. EVD copy numbers were determined by direct qPCR on genomic DNA, comparing relative EVD and ACT2 levels, normalized by their inherent copy numbers of two and one in WT plants, respectively. Oligonucleotides used are listed in Appendix Table S1.
Separation of polyadenylated mRNA
Isolation of poly(A)+ from non‐poly(A) RNA was performed from 75 μg of Trizol‐extracted total RNA from floral buds, using the DynabeadsTM mRNA Purification Kit (Ambion Cat#.61006) following the manufacturer’s instructions. Non‐polyA RNA was precipitated from the DynabeadsTM‐unbound fraction and resuspended in the same volume (200 μl) as the poly(A)+ RNA fraction. Efficiency of the separation was confirmed by running aliquots of each fraction on a 1% agarose gel to monitor efficient depletion of rRNA in poly(A)+ fractions before downstream analysis.
Cloning and mapping of 5’OH ends
Non‐canonical cleavage sites in EVADE transcript were mapped by a modified 5’ RACE method. Total RNA isolated from a pool of 2‐ to 3‐week‐old plants extracted by standard protocols (See nucleic acid extraction section) was taken for RNA ligations after DNase I treatment ((#EN0521, Thermo scientific). RNA adapters with a 5’ inverted dT modification (see Appendix Table S1) were ligated to the DNase‐treated RNA by T4 RNA ligase 1 (#M0204S, New England Biolabs) to render the canonical cleavage products not available for subsequent ligation reaction. To map the cleavage products with a 5’ hydroxyl group, the RNA was subsequently ligated to an RNA adapter with a 3’ phosphate group by RtcB ligase (#M0458S, New England Biolabs). The ligated RNA was converted to cDNA with RevertAid first‐strand cDNA synthesis kit (#K1612, Thermo scientific) and a primer specific to the EVADE transcript (Appendix Table S1). The cDNA was amplified by nested PCR by using primers from the adapter RNA and primers located ~100 nucleotides downstream of each stalling site (all adaptor and primers sequences can be found in Appendix Table S1). The PCR products were separated on an agarose gel and the DNA fragments were extracted from the gel by GeneJET gel extraction kit (#K0691, ThermoFisher scientific). The DNA fragments were cloned in pJET1.2 vectors by using CloneJET PCR cloning kit (#K1232, ThermoFisher scientific) and ~50 colonies were screened for each potential cleavage site by Sanger sequencing technology.
Small RNA sequencing
Small RNA sequencing of 35S:EVDwt and 35S:GFP‐EVDint/ter ‐GUS was performed as follows. Total RNA was resolved on a 17.5% polyacrylamide‐urea gel and sizes between 18–30 nt were excised, eluted overnight in elution buffer (20 mM Tris–HCl (pH 7.9), 1 mM EDTA, 400 mM ammonium acetate, 0.5% (w/v) SDS), and collected by precipitation with equal volumes of isopropanol. RNA was quantified using the Qubit™ RNA HS Assay Kit (Thermo Scientific) and subsequently cloned using the Small RNA‐Seq Library Prep Kit (Lexogen). Sequencing was performed on an Illumina HiSeq 4000 machine.
RIBO‐seq
For RIBO‐seq libraries, frozen inflorescence tissue was ground in digestion buffer (100 mM Tris·HCl (pH 8), 40 mM KCl, 20 mM MgCl2, 2% (v/v) polyoxyethylene (10) tridecyl ether, 1% (v/v) de‐oxycholic acid, 1 mM DTT, 10 unit/ml DNase I (Thermo Scientific), 100 μg/ml cycloheximide). Precleared solutions were incubated with 650 U RNase I (Ambion) for 45 min at 25°C. Nuclease digestion was stopped by the addition of 10 μl SUPERase In RNase Inhibitor (Ambion). Resulting monosomes were purified by ultracentrifugation of the lysate on a sucrose cushion (1 M sucrose, 20 mM HEPES (pH 7.6), 100 mM KCl, 5 mM MgCl2, 10 μg/ml cycloheximide, 10 units/ml RiboLock (Thermo Scientific) and cOmplete protease inhibitor cocktail (Roche) for 4 h at 250,000 g in 4°C. RNA was extracted using the TRIzol RNA extraction described above and treated with 10 U PNK (Thermo Scientific) for 30 min. Ribosomal RNA depletion was performed using the RiboMinus Plant Kit (Thermo Scientific) and libraries were generated as above, except that the 25–32 nt RNA fraction was excised from the denaturing polyacrylamide gel prior to RNA ligation.
nanoPARE
NanoPARE library preparation and analysis was performed following the protocol from Schon et al (2018). Briefly, 10 ng of total RNA was isolated from inflorescences. Two biological replicates each of Col‐0 and ddm1‐2 were used for reverse transcription. After 9 cycles of PCR pre‐amplification, 5 ng aliquots of cDNA were separately tagmented and amplified using either standard Smart‐seq2 Tn5 primers or 5’‐end enrichment primers. The resulting Smart‐seq2 and nanoPARE libraries were sequenced on an Illumina HiSeq 2500 using paired‐end 50‐bp reads and single‐end 50‐bp reads, respectively.
Seed counting
Plants germinated and grown in parallel under the same conditions were individually covered with paper bags before the maturation of siliques and harvested upon ripening. Total amount of seeds from each plant was counted twice with a C3 High Sensitive Seed Counter (Elmor).
Data analysis
Analysis of sRNA sequencing is based on the following workflow. Reads were trimmed using bbduk (BBTools: sourceforge.net/projects/bbmap/, version 38.41; ktrim=r k=23 mink=11 hdist=1) mapped against the TAIR10 Arabidopsis genome with STAR (Dobin et al, 2013) (version 2.5.2a; ‐‐outFilterMismatchNoverLmax 0.05 ‐‐outFilterMatchNmin 16 ‐‐outFilterScoreMinOverLread 0 ‐‐alignIntronMax 500 ‐‐alignIntronMin 50 ‐‐outFilterMultimapNmax 50), quantified using Rsubread (Liao et al, 2013) (version 1.20.6; allowMultiOverlap=T, largestOverlap=T, isPairedEnd=F, strandSpecific=1, countMultiMappingReads=T, fraction=T) and differential analysis using DESeq2 (Love et al, 2014; version 1.10.1). Reads were split in different lengths with Samtools (Li et al, 2009; version 0.1.19), and locus coverage among those read length was visualized using BEDtools (Quinlan & Hall, 2010; version 2.15.0) and R cran (version 3.2.5).
RIBO‐seq libraries were analyzed as follows. Reads were trimmed of adapter sequences with bbduk as above. Reads mapping to rRNA loci using Bowtie2 (Langmead & Salzberg, 2012) (version 2.2.1; ‐k 1 ‐x) were discarded from further analysis. Subsequent mapping and quantification were performed as for the sRNA sequencing analysis using STAR (Dobin et al, 2013) and Rsubread (Liao et al, 2013)as above, but reads were mapped to both Arabidopsis genome and transcriptome sequences. Quality control of the RIBO‐seq libraries was performed with the riboWaltz (Lauria et al, 2018; version 1.1.0) package. P‐site occupancies were estimated using the RiboProfiling (Popa et al, 2016; version 1.0.3) package based on 5’ read offsets determined by the coverage profile around start codons dependent on read lengths. Codon occupancies were compiled for all three possible frames to generate a single codon occupancy score. A ribosomal stalling score at each codon position was defined as the ratio of observed over expected counts, where the expectation was the mean of occupancy counts over the entire transcript. To improve quality of the assessment, only the most translated isoform per gene and only isoforms with a minimal read coverage of 70% were considered. Codon dwell time was estimated as the mean value of log‐normalized codon occupancies per individual transcript and codon usage was estimated from the subset of genes considered translated. Stop codons and stop codons containing di‐codons were excluded from the analysis. Data were visualized using R cran and the packages Gviz (Hahne & Ivanek, 2016) and ggplot2 (Wickham, 2009).
Author contributions
AM‐O, SO, and OV conceived and designed the study. SO and AM‐O performed most experiments. RR cloned and sequenced the EVD 5’OH ends, MT and VB‐B investigated EVD copy number in rdr6 and RNA isoform distribution in cyto‐nuclear fractions. MAS, AP, and MDN performed nanoPARE. LL conducted seed counting. SO and MAS performed computer analyses. SO and MT performed statistical analyses. AM‐O, SO, and OV analyzed the data and wrote the manuscript.
Supporting information
Appendix
Source Data for Appendix
Source Data for Figure 1
Source Data for Figure 2
Source Data for Figure 3
Source Data for Figure 5
Source Data for Figure 6
Acknowledgments
We thank members of the Voinnet and Marí‐Ordóñez laboratories and colleagues for critical reading of the manuscript and for discussions. This work was supported by a core grant attributed to OV by the ETH‐Zürich that covered the largest part of this study, including the PhD studentships of S.O. and A.M.O. under the Life Science Zürich Graduate School program and the “Syngenta Fonds in Honor of Heinz Imhof” – ETH Foundation Project: De novo silencing of active retrotransposons. In Arabidopsis. Part of the work was also supported by the NCCR RNA & Disease funded by the Swiss National Science Foundation, no. 4112b. This work was additionally supported by the Gregor Mendel Institute of the Austrian Academy of Sciences core funding attributed to A.M.O and M.D.N. Open access funding provided by Eidgenossische Technische Hochschule Zurich.
Conflict of interest
The authors declare that they have no conflict of interest.
EMBO reports (2022) 23: e53400.
Contributor Information
Arturo Marí‐Ordóñez, Email: arturo.mari-ordonez@gmi.oeaw.ac.at.
Olivier Voinnet, Email: voinneto@ethz.ch.
Data availability
Sequencing data generated in this study are accessible on the Gene Expression Omnibus (GEO) under the accession number GSE167484 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE167484). Data from previous studies including sRNA sequencing in ddm1 & ddm1 rdr6, ddm1 & ddm1 dcl1, isoform‐specific sequencing data of total and polysome‐associated mRNA in TE de‐repressed backgrounds are found under the accession numbers GSE41755 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE41755; Nuthikattu et al, 2013), GSE52952 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE52952; Creasey et al, 2014), GSE93584 (http://www.ncbi.nlm.nih.gov/geo/query/acc.cgi?acc=GSE93584; Oberlin et al, 2017) and PRJNA598331 (https://www.ncbi.nlm.nih.gov/bioproject/PRJNA598331; Kim et al, 2021). Every other raw data used in this study (including raw image files, qPCR data, and Sanger sequencing traces) have been deposited in Zenodo (www.zenodo.org) under the https://doi.org/10.5281/zenodo.5564305.
References
- Allshire RC, Madhani HD (2018) Ten principles of heterochromatin formation and function. Nat Rev Mol Cell Biol 19: 229–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Artieri CG, Fraser HB (2014) Accounting for biases in riboprofiling data indicates a major role for proline in stalling translation. Genome Res 24: 2011–2021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Baeg K, Iwakawa H, Tomari Y (2017) The poly(A) tail blocks RDR6 from converting self mRNAs into substrates for gene silencing. Nat Plants 3: 17036–17044 [DOI] [PubMed] [Google Scholar]
- Bao C, Loerch S, Ling C, Korostelev AA, Grigorieff N, Ermolenko DN (2020) mRNA stem‐loops can pause the ribosome by hindering A‐site tRNA binding. Elife 9: e55799 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Borges F, Parent J‐S, Ex FV, Wolff P, Martínez G, Köhler C, Martienssen RA (2018) Transposon‐derived small RNAs triggered by miR845 mediate genome dosage response in Arabidopsis . Nat Genet 50: 186–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brodersen P, Voinnet O (2006) The diversity of RNA silencing pathways in plants. Trends Genet 22: 268–280 [DOI] [PubMed] [Google Scholar]
- Buskirk AR, Green R (2017) Ribosome pausing, arrest and rescue in bacteria and eukaryotes. Philos Trans R Soc Lond B Biol Sci 372: 20160183 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Civáň P, Švec M, Hauptvogel P (2011) On the coevolution of transposable elements and plant genomes. J Bot 2011: 1–9 [Google Scholar]
- Creasey KM, Zhai J, Borges F, Ex FV, Regulski M, Meyers BC, Martienssen RA (2014) miRNAs trigger widespread epigenetically activated siRNAs from transposons in Arabidopsis . Nature 508: 411–415 [DOI] [PMC free article] [PubMed] [Google Scholar]
- D’Orazio KN, Wu CC‐C, Sinha N, Loll‐Krippleber R, Brown GW, Green R (2019) The endonuclease Cue2 cleaves mRNAs at stalled ribosomes during No Go Decay. Elife 8: e49117 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dalakouras A, Lauter A, Bassler A, Krczal G, Wassenegger M (2019) Transient expression of intron‐containing transgenes generates non‐spliced aberrant pre‐mRNAs that are processed into siRNAs. Planta 249: 457–468 [DOI] [PubMed] [Google Scholar]
- Desai KK, Raines RT (2012) tRNA ligase catalyzes the GTP‐dependent ligation of RNA with 3’‐phosphate and 5’‐hydroxyl termini. Biochemistry 51: 1333–1335 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doamekpor SK, Gozdek A, Kwasnik A, Kufel J, Tong L (2020) A novel 5’‐hydroxyl dinucleotide hydrolase activity for the DXO/Rai1 family of enzymes. Nucleic Acids Res 48: 349–358 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, Gingeras TR (2013) STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 29: 15–21 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doma MK, Parker R (2006) Endonucleolytic cleavage of eukaryotic mRNAs with stalls in translation elongation. Nature 440: 561–564 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Doma MK, Parker R (2007) RNA quality control in eukaryotes. Cell 131: 660–668 [DOI] [PubMed] [Google Scholar]
- Dreher TW, Miller WA (2006) Translational control in positive strand RNA plant viruses. Virology 344: 185–197 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Drinnenberg IA, Weinberg DE, Xie KT, Mower JP, Wolfe KH, Fink GR, Bartel DP (2009) RNAi in budding yeast. Science 326: 544–550 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dumesic P, Natarajan P, Chen C, Drinnenberg I, Schiller B, Thompson J, Moresco J, Yates J, Bartel D, Madhani H (2013) Stalled spliceosomes are a signal for RNAi‐mediated genome defense. Cell 152: 957–968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fay MM, Lyons SM, Ivanov P (2017) RNA G‐Quadruplexes in biology: principles and molecular mechanisms. J Mol Biol 429: 2127–2147 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fedoroff NV (2012) Transposable elements, epigenetics, and genome evolution. Science 338: 758–767 [DOI] [PubMed] [Google Scholar]
- Fultz D, Slotkin RK (2017) Exogenous transposable elements circumvent identity‐based silencing, permitting the dissection of expression‐dependent silencing. Plant Cell 29: 360–376 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Havecker ER, Baranov PV, Atkins JF, Voytas DF (2003) Translational recoding signals between gag and pol in diverse LTR retrotransposons. RNA 9: 1422–1430 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gazzani S (2004) A link between mRNA turnover and RNA interference in Arabidopsis . Science 306: 1046–1048 [DOI] [PubMed] [Google Scholar]
- Gilly A, Etcheverry M, Madoui M‐A, Guy J, Quadrana L, Alberti A, Martin A, Heitkam T, Engelen S, Labadie K et al (2014) TE‐Tracker: systematic identification of transposition events through whole‐genome resequencing. BMC Bioinformatics 15: 377 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gregory BD, O’Malley RC, Lister R, Urich MA, Tonti‐Filippini J, Chen H, Millar AH, Ecker JR (2008) A link between RNA metabolism and silencing affecting Arabidopsis development. Dev Cell 14: 854–866 [DOI] [PubMed] [Google Scholar]
- Gy I, Gasciolli V, Lauressergues D, Morel J‐B, Gombert J, Proux F, Proux C, Vaucheret H, Mallory AC (2007) Arabidopsis FIERY1, XRN2, and XRN3 are endogenous RNA silencing suppressors. Plant Cell 19: 3451–3461 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hahne F, Ivanek R (2016) Visualizing genomic data using gviz and bioconductor. Methods Mol Biol 1418: 335–351 [DOI] [PubMed] [Google Scholar]
- Herr AJ, Molnar A, Jones A, Baulcombe DC (2006) Defective RNA processing enhances RNA silencing and influences flowering of Arabidopsis . Proc Natl Acad Sci 103: 14994–15001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hirochika H, Okamoto H, Kakutani T (2000) Silencing of retrotransposons in Arabidopsis and reactivation by the ddm1 mutation. Plant Cell 12: 357–368 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu W, Sweet TJ, Chamnongpol S, Baker KE, Coller J (2009) Co‐translational mRNA decay in Saccharomyces cerevisiae . Nature 461: 225–229 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang CRL, Burns KH, Boeke JD (2012) Active transposition in genomes. Annu Rev Genet 46: 651–675 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ibrahim F, Maragkakis M, Alexiou P, Mourelatos Z (2018) Ribothrypsis, a novel process of canonical mRNA decay, mediates ribosome‐phased mRNA endonucleolysis. Nat Struct Mol Biol 25: 302–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ikeuchi K, Izawa T, Inada T (2018) Recent progress on the molecular mechanism of quality controls induced by ribosome stalling. Front Genet 9: 743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ingolia NT, Ghaemmaghami S, Newman JR, Weissman JS (2009) Genome‐wide analysis in vivo of translation with nucleotide resolution using ribosome profiling. Science 324: 218–223 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iwakawa H, Lam AYW, Mine A, Fujita T, Kiyokawa K, Yoshikawa M, Takeda A, Iwasaki S, Tomari Y (2021) Ribosome stalling caused by the Argonaute‐microRNA‐SGS3 complex regulates the production of secondary siRNAs in plants. Cell Rep 35: 109300 [DOI] [PubMed] [Google Scholar]
- Jia J, Long Y, Zhang H, Li Z, Liu Z, Zhao Y, Lu D, Jin X, Deng X, Xia R et al (2020) Post‐transcriptional splicing of nascent RNA contributes to widespread intron retention in plants. Nat Plants 6: 780–788 [DOI] [PubMed] [Google Scholar]
- Joazeiro CAP (2019) Mechanisms and functions of ribosome‐associated protein quality control. Nat Rev Mol Cell Biol 20: 368–383 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kankel MW, Ramsey DE, Stokes TL, Flowers SK, Haag JR, Jeddeloh JA, Riddle NC, Verbsky ML, Richards EJ (2003) Arabidopsis MET1 cytosine methyltransferase mutants. Genetics 163: 1109–1122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kastenmayer JP, Green PJ (2000) Novel features of the XRN‐family in Arabidopsis: evidence that AtXRN4, one of several orthologs of nuclear Xrn2p/Rat1p, functions in the cytoplasm. Proc Natl Acad Sci USA 97: 13985–13990 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanduja JS, Calvo IA, Joh RI, Hill IT, Motamedi M (2016) Nuclear noncoding RNAs and genome stability. Mol Cell 63: 7–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim EY, Wang L, Lei Z, Li H, Fan W, Cho J (2021) Ribosome stalling and SGS3 phase separation prime the epigenetic silencing of transposons. Nat Plants 7: 303–309 [DOI] [PubMed] [Google Scholar]
- Kurihara Y (2017) Activity and roles of Arabidopsis thaliana XRN family exoribonucleases in noncoding RNA pathways. J Plant Res 130: 25–31 [DOI] [PubMed] [Google Scholar]
- Langmead B, Salzberg SL (2012) Fast gapped‐read alignment with Bowtie 2. Nat Methods 9: 357–359 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lauria F, Tebaldi T, Bernabò P, Groen EJN, Gillingwater TH, Viero G (2018) riboWaltz: optimization of ribosome P‐site positioning in ribosome profiling data. PLoS Comput Biol 14: e1006169 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee ES, Akef A, Mahadevan K, Palazzo AF (2015) The consensus 5’ splice site motif inhibits mRNA nuclear export. PLoS One 10: e0122743 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee SC, Ernst E, Berube B, Borges F, Parent J‐S, Ledon P, Schorn A, Martienssen RA (2020) Arabidopsis retrotransposon virus‐like particles and their regulation by epigenetically activated small RNA. Genome Res 30: 576–588 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, Durbin R & Subgroup 1000 Genome Project Data Processing (2009) The sequence Alignment/Map format and SAMtools. Bioinformatics 25: 2078–2079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, Shi W (2013) The Subread aligner: fast, accurate and scalable read mapping by seed‐and‐vote. Nucleic Acids Res 41: e108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Llave C, Xie Z, Kasschau KD, Carrington JC (2002) Cleavage of Scarecrow‐like mRNA targets directed by a class of Arabidopsis miRNA. Science 297: 2053–2056 [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, Anders S (2014) Moderated estimation of fold change and dispersion for RNA‐seq data with DESeq2. Genome Biol 15: 550–621 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo Z, Chen Z (2007) Improperly terminated, unpolyadenylated mRNA of sense transgenes is targeted by RDR6‐mediated RNA silencing in Arabidopsis . Plant Cell 19: 943–958 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Maldonado‐Bonilla LD (2014) Composition and function of P bodies in Arabidopsis thaliana . Front Plant Sci 5: 201 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marí‐Ordóñez A, Marchais A, Etcheverry M, Martin A, Colot V, Voinnet O (2013) Reconstructing de novo silencing of an active plant retrotransposon. Nat Genet 45: 1029–1039 [DOI] [PubMed] [Google Scholar]
- Martinez J, Tuschl T (2004) RISC is a 5’ phosphomonoester‐producing RNA endonuclease. Genes Dev 18: 975–980 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martínez de Alba AE, Moreno AB, Gabriel M, Mallory AC, Christ A, Bounon R, Balzergue S, Aubourg S, Gautheret D, Crespi MD et al (2015) In plants, decapping prevents RDR6‐dependent production of small interfering RNAs from endogenous mRNAs. Nucleic Acids Res 43: 2902–2913 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirouze M, Reinders J, Bucher E, Nishimura T, Schneeberger K, Ossowski S, Cao J, Weigel D, Paszkowski J, Mathieu O (2009) Selective epigenetic control of retrotransposition in Arabidopsis . Nature 461: 427–430 [DOI] [PubMed] [Google Scholar]
- Moreno AB, de Alba AEM, Bardou F, Crespi MD, Vaucheret H, Maizel A, Mallory AC (2013) Cytoplasmic and nuclear quality control and turnover of single‐stranded RNA modulate post‐transcriptional gene silencing in plants. Nucleic Acids Res 41: 4699–4708 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Navickas A, Chamois S, Saint‐Fort R, Henri J, Torchet C, Benard L (2020) No‐Go Decay mRNA cleavage in the ribosome exit tunnel produces 5’‐OH ends phosphorylated by Trl1. Nat Commun 11: 122–211 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nott A, Hir HL, Moore MJ (2004) Splicing enhances translation in mammalian cells: an additional function of the exon junction complex. Gene Dev 18: 210–222 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nuthikattu S, McCue AD, Panda K, Fultz D, DeFraia C, Thomas EN, Slotkin RK (2013) The initiation of epigenetic silencing of active transposable elements is triggered by RDR6 and 21–22 nucleotide small interfering RNAs. Plant Physiol 162: 116–131 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Oberlin S, Sarazin A, Chevalier C, Voinnet O, Ordóñez AM (2017) A genome‐wide transcriptome and translatome analysis of Arabidopsis transposons identifies a unique and conserved genome expression strategy for Ty1/Copia retroelements. Genome Res 27: 1549–1562 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pall GS, Hamilton AJ (2008) Improved northern blot method for enhanced detection of small RNA. Nat Protoc 3: 1077–1084 [DOI] [PubMed] [Google Scholar]
- Panda K, Ji L, Neumann DA, Daron J, Schmitz RJ, Slotkin RK (2016) Full‐length autonomous transposable elements are preferentially targeted by expression‐dependent forms of RNA‐directed DNA methylation. Genome Biol 17: 170–219 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parent J‐S, Jauvion V, Bouché N, Beclin C, Hachet M, Zytnicki M, Vaucheret H (2015) Post‐transcriptional gene silencing triggered by sense transgenes involves uncapped antisense RNA and differs from silencing intentionally triggered by antisense transgenes. Nucleic Acids Res 43: 8464–8475 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peach SE, York K, Hesselberth JR (2015) Global analysis of RNA cleavage by 5′‐hydroxyl RNA sequencing. Nucleic Acids Res 43: gkv536‐e108 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pelechano V, Wei W, Steinmetz LM (2015) Widespread co‐translational RNA decay reveals ribosome dynamics. Cell 161: 1400–1412 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peragine A, Yoshikawa M, Wu G, Albrecht HL, Poethig RS (2004) SGS3 and SGS2/SDE1/RDR6 are required for juvenile development and the production of trans‐acting siRNAs in Arabidopsis . Genes Dev 18: 2368–2379 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Popa A, Lebrigand K, Paquet A, Nottet N, Robbe‐Sermesant K, Waldmann R, Barbry P (2016) RiboProfiling: a Bioconductor package for standard Ribo‐seq pipeline processing. F1000Research 5: 1309 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quadrana L, Silveira AB, Mayhew GF, LeBlanc C, Martienssen RA, Jeddeloh JA, Colot V (2016) The Arabidopsis thaliana mobilome and its impact at the species level. Elife 5: e15716 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quinlan AR, Hall IM (2010) BEDTools: a flexible suite of utilities for comparing genomic features. Bioinformatics 26: 841–842 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reinders J, Wulff BBH, Mirouze M, Ordóñez AM, Dapp M, Rozhon W, Bucher E, Theiler G, Paszkowski J (2009) Compromised stability of DNA methylation and transposon immobilization in mosaic Arabidopsis epigenomes. Genes Dev 23: 939–950 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodnina MV (2016) The ribosome in action: tuning of translational efficiency and protein folding. Protein Sci 25: 1390–1406 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sabi R, Tuller T (2015) A comparative genomics study on the effect of individual amino acids on ribosome stalling. BMC Genom 16(Suppl 10): S5–S12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarazin A, Voinnet O (2014) Exploring new models of easiRNA biogenesis. Nat Genet 46: 530–531 [DOI] [PubMed] [Google Scholar]
- Saze H, Scheid OM, Paszkowski J (2003) Maintenance of CpG methylation is essential for epigenetic inheritance during plant gametogenesis. Nat Genet 34: 65–69 [DOI] [PubMed] [Google Scholar]
- Schon MA, Kellner MJ, Plotnikova A, Hofmann F, Nodine MD (2018) NanoPARE: parallel analysis of RNA 5’ ends from low‐input RNA. Genome Res 28: 1931–1942 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Silber R, Malathi VG, Hurwitz J (1972) Purification and properties of bacteriophage T4‐induced RNA ligase. Proc Natl Acad Sci USA 69: 3009–3013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song J, Perreault J‐P, Topisirovic I, Richard S (2016) RNA G‐quadruplexes and their potential regulatory roles in translation. Translation 4: e1244031 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sørensen BB, Ehrnsberger HF, Esposito S, Pfab A, Bruckmann A, Hauptmann J, Meister G, Merkl R, Schubert T, Längst G et al (2017) The Arabidopsis THO/TREX component TEX1 functionally interacts with MOS11 and modulates mRNA export and alternative splicing events. Plant Mol Biol 93: 283–298 [DOI] [PubMed] [Google Scholar]
- Stein P, Svoboda P, Anger M, Schultz RM (2003) RNAi: mammalian oocytes do it without RNA‐dependent RNA polymerase. RNA 9: 187–192 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stevens A (2001) 5’‐exoribonuclease 1: Xrn1. Methods Enzymol 342: 251–259 [DOI] [PubMed] [Google Scholar]
- Sun YH, Zhu J, Xie LH, Li Z, Meduri R, Zhu X, Song C, Chen C, Ricci EP, Weng Z et al (2020) Ribosomes guide pachytene piRNA formation on long intergenic piRNA precursors. Nat Cell Biol 22: 200–212 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Šurbanovski N, Brilli M, Moser M, Si‐Ammour A (2016) A highly specific microRNA‐mediated mechanism silences LTR retrotransposons of strawberry. Plant J 85: 70–82 [DOI] [PubMed] [Google Scholar]
- Sztuba‐Solińska J, Stollar V, Bujarski JJ (2011) Subgenomic messenger RNAs: mastering regulation of (+)‐strand RNA virus life cycle. Virology 412: 245–255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teixeira FK, Heredia F, Sarazin A, Roudier François, Boccara M, Ciaudo C, Cruaud C, Poulain J, Berdasco M, Fraga MF et al (2009) A role for RNAi in the selective correction of DNA methylation defects. Science 323: 1600–1604 [DOI] [PubMed] [Google Scholar]
- Thran M, Link K, Sonnewald U (2012) The Arabidopsis DCP2gene is required for proper mRNA turnover and prevents transgene silencing in Arabidopsis . Plant J 72: 368–377 [DOI] [PubMed] [Google Scholar]
- Tsukahara S, Kobayashi A, Kawabe A, Mathieu O, Miura A, Kakutani T (2010) Bursts of retrotransposition reproduced in Arabidopsis . Nature 461: 423–426 [DOI] [PubMed] [Google Scholar]
- Valencia P, Dias AP, Reed R (2008) Splicing promotes rapid and efficient mRNA export in mammalian cells. Proc Natl Acad Sci 105: 3386–3391 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vazquez F, Vaucheret H, Rajagopalan R, Lepers C, Gasciolli V, Mallory AC, Hilbert JL, Bartel DP, Crete P (2004) Endogenous trans‐acting siRNAs regulate the accumulation of Arabidopsis mRNAs. Mol Cell 16: 69–79 [DOI] [PubMed] [Google Scholar]
- Vitte C, Bennetzen JL (2006) Analysis of retrotransposon structural diversity uncovers properties and propensities in angiosperm genome evolution. Proc Natl Acad Sci USA 103: 17638–17643 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitte C, Panaud O (2005) LTR retrotransposons and flowering plant genome size: emergence of the increase/decrease model. Cytogenet Genome Res 110: 91–107 [DOI] [PubMed] [Google Scholar]
- Voinnet O (2005) Induction and suppression of RNA silencing: insights from viral infections. Nat Rev Genet 6: 206–220 [DOI] [PubMed] [Google Scholar]
- Vongs A, Kakutani T, Martienssen R, Richards E (1993) Arabidopsis thaliana DNA methylation mutants. Science 260: 1926–1928 [DOI] [PubMed] [Google Scholar]
- Wang C, Fang J (2015) RLM‐RACE, PPM‐RACE, and qRT‐PCR: an integrated strategy to accurately validate miRNA target genes. Methods Mol Biol 1296: 175–186 [DOI] [PubMed] [Google Scholar]
- Wickham H (2009) ggplot2: elegant graphics for data analysis. New York, NY: Springer Publishing; [Google Scholar]
- Yan S, Wen J‐D, Bustamante C, Tinoco I (2015) Ribosome excursions during mRNA translocation mediate broad branching of frameshift pathways. Cell 160: 870–881 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yin Y, Lu JY, Zhang X, Shao W, Xu Y, Li P, Hong Y, Cui LI, Shan GE, Tian B et al (2020) U1 snRNP regulates chromatin retention of noncoding RNAs. Nature 580: 147–150 [DOI] [PubMed] [Google Scholar]
- Yu X, Willmann MR, Anderson SJ, Gregory BD (2016) Genome‐wide mapping of uncapped and cleaved transcripts reveals a role for the nuclear mRNA cap‐binding complex in cotranslational RNA decay in Arabidopsis . Plant Cell 28: 2385–2397 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zemach A, Kim MY, Hsieh P‐H, Coleman‐Derr D, Eshed‐Williams L, Thao K, Harmer SL, Zilberman D (2013) The Arabidopsis nucleosome remodeler DDM1 allows DNA methyltransferases to access H1‐containing heterochromatin. Cell 153: 193–205 [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao T, Chen Y‐M, Li Y, Wang J, Chen S, Gao N, Qian W (2021) Disome‐seq reveals widespread ribosome collisions that promote cotranslational protein folding. Genome Biol 22: 16–35 [DOI] [PMC free article] [PubMed] [Google Scholar]