

Abstract

A new series of benzimidazole, 1,2,4-triazole, and 1,3,5-triazine derivatives were designed and synthesized using a microwave irradiation synthetic approach utilizing 2-phenylacetyl isothiocyanate (1) as a key starting material. All the new analogues were evaluated as anticancer agents against a panel of cancer cell lines utilizing doxorubicin as a standard drug. Most of the tested derivatives exhibited selective cytotoxic activity against MCF-7 and A-549 cancer cell lines. Furthermore, the new target compounds 5, 6, and 7 as the most potent antiproliferative agents have been assessed as in vitro EGFRWT and EGFRT790M inhibitors compared to the reference drugs erlotinib and AZD9291. They represented more potent suppression activity against the mutated EGFRT790M than the wild-type EGFRWT. Moreover, the compounds 5, 6, and 7 down-regulated the oncogenic parameter p53 ubiquitination. A docking simulation of compound 6b was carried out to correlate its molecular structure with its significant EGFR inhibition potency and its possible binding interactions within the active site of EGFRWT and the mutant EGFRT790M.
1. Introduction
Cancer disease is a terrible health epidemic that kills millions of people all over the world in both developed and developing countries.1 Despite the great progress accomplished in cancer therapy, several limitations still present. Examples are the selectivity for cancer cells, adverse effects, as well as the multiple-drug resistance acquisition by the cancer cells leading them to be unresponsive to conventional therapeutic agents.2,3 Accordingly, the innovation of new small molecules that are both potent and selective is still a serious challenge in the field of medicinal chemistry. The alteration of different protein expressions and the activity of various receptor tyrosine kinases (RTKs) are considered the main causes of many cancer types since they are responsible for the regulation of different cellular pathways such as proliferation, differentiation, migration, and angiogenesis.4,5 The epidermal growth factor receptor (EGFR) is a member of tyrosine kinases (TKs).6 It is a trans-membrane protein belonging to the erbB/HER-family and plays a pivotal role in governing cellular transduction or communication signaling through the phosphorylation of tyrosine residues in the protein domain.7,8 EGFR is one of the main tumor markers in many cancer types (such as colon, lung, liver, cervical, ovarian, breast, prostate, and bladder cancers), where its signaling in tumors, as opposed to normal cells, becomes dysregulated, resulting in EGFR overexpression and/or obtaining a gain-of-function mutation.9−13 This act is considered the main cause of tumor cell proliferation, invading the surrounding tissues and resulting in an increased angiogenesis.14 Accordingly, interrupting EGFR communicating signals is considered to be one of the prime targets to invade tumors caused by its mis-regulation.9−14 Targeted drugs inhibiting EGFR can selectively attack the cancer cells rather than normal ones, thus producing a good safety profile and less harm to the body with more patient comfortability.15 Multiple EGFR suppressors have been developed and classified into different generations. The first generation was gefitinib (Iressa), erlotinib (Tarceva), and icotinib (Conmana).16−20 Studies revealed that acquired drug resistance to the first-generation EGFR-TKIs was revealed due to T790M ″gatekeeper″ and L858R mutations in EGFR about 9–14 months after clinical treatment.21−23 The emergence of resistance paved the way toward the development of the second-generation inhibitors (EGFR TKI) (afatinib, dacomitinib, neratinib, and canertinib) that have exhibited a 60–70% objective response rate.17 The drugs related to this class contain an electrophilic acrylamide side chain that interacts irreversibly with cysteine CYS797 forming covalent complexes, thus overcoming the obstacle of resistance mediated by EGFRT790M or EGFRT790M/L858R mutation.24−27 On the other hand, due to the high reactivity of the acrylamide moiety, it interacts non-selectively with the cysteine residue in untargeted proteins, leading to toxic side effects such as diarrhea and skin rash that limited their clinical use.28−32 Recently, to solve these undesirable side effects, several third-generation inhibitors have been discovered, such as WZ4002,29 osimertinib (AZD9291) (Tagrisso),33 olmutinib (Olita),31 and rociletinib (CO1686).34 These inhibitors do not only produce good anti-tumor activity but also produce good selectivity to EGFRT790M and EGFRT790M/L858R kinases.29,35 Rociletinib and osimertinib were considered as breakthrough therapies in the mutant NSCLC treatment by the US FDA in 2014.36 Studies showed that osimertinib’s efficacy is marred by various side effects such as grade 3 venous thromboembolism and pneumonia. Its toxicity was attributed to AZ5104, which is its main metabolite, lacking selectivity between the mutant and WT EGFR.29,35−38 Accordingly, much efforts are still needed to discover new EGFR inhibitors of high selectivity to EGFRT790M and EGFRT790M/L858R kinase with low side effects (Figure 1).
Figure 1.
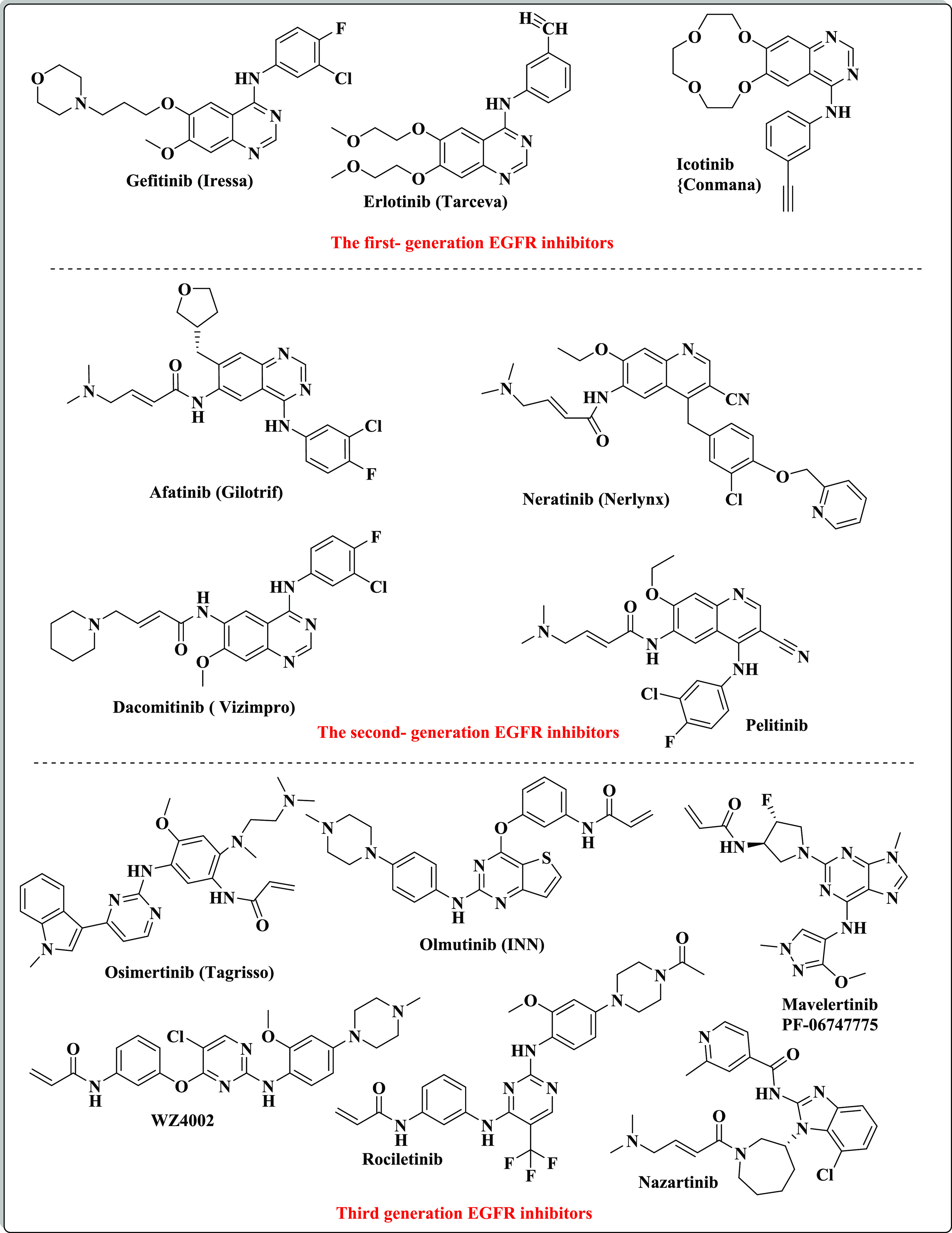
Examples of the first, second and third generations of EGFR inhibitors.
Many nitrogen-heterocyclic ring systems have recently been discovered to introduce surprisingly complex biological properties, making them one of the most significant groups in medicinal chemistry. They constitute a basic scaffold in numerous drugs due to their capabilities to imitate and interact with different biological molecules, leading to remarkable pharmacological properties.39−42 The benzimidazole scaffold participates in various compounds producing a wide range of biological activities such as antimicrobial, antiparasitic, antihistaminic, antiallergic, anticancer, and antioxidant.43−49 The benzimidazoles could be considered as auxiliary isosters of nucleotides having a potential for chemotherapeutic applications.46 In addition, the orally available third-generation EGFR inhibitor nazartinib bears a benzimidazole nucleus.50 Moreover, the triazole nucleus plays a vital role in the field of drug discovery. The triazole ring is characterized by significant stability and excellent pharmacological potency due to its electron-rich characteristics and the occurrence of an unsaturated hydrocarbon ring structure. These properties support the triazole structure to interact with various receptors (enzymes) through H-bonding that endows it with significant pharmacological actions.51−53 Currently, triazole derivatives are used to treat a wide variety of diseases specially cancer disease.51−53 Numerous anticancer drugs bearing the 1,2,4-triazole moiety are available in the market such as anastrozole,54,55 letrozole,56 and vorozole.57 Furthermore, the s-triazine (1,3,5-triazine) scaffold constitutes a basic template for the design and synthesis of various bioactive compounds with widespread applications in medicinal chemistry.58 The s-triazine core has three functionalized branches at positions 2, 4, and 6, a property that leads to easily modulating the physicochemical and biological activities of s-triazine derivatives.59 Many studies investigated the notable progress in the design, synthetic approaches, and evaluation of numerous s-triazine candidates with great promising antitumor activity acting via the inhibition of different protein kinases such as CDK2, PI3Kα/mTOR, CA, human topoisomerase IIα, hDHFR, EGFR (EGFRWT and EGFRT790M), and tubulin polymerization.60−67 There are various anticancer drugs containing the s-triazine motif that are FDA-approved such as tretamine,68 gedatolisib,69 HL 010183,70 enasidenib,71 altretamine,72 and KY-0403173 (Figure 2).
Figure 2.
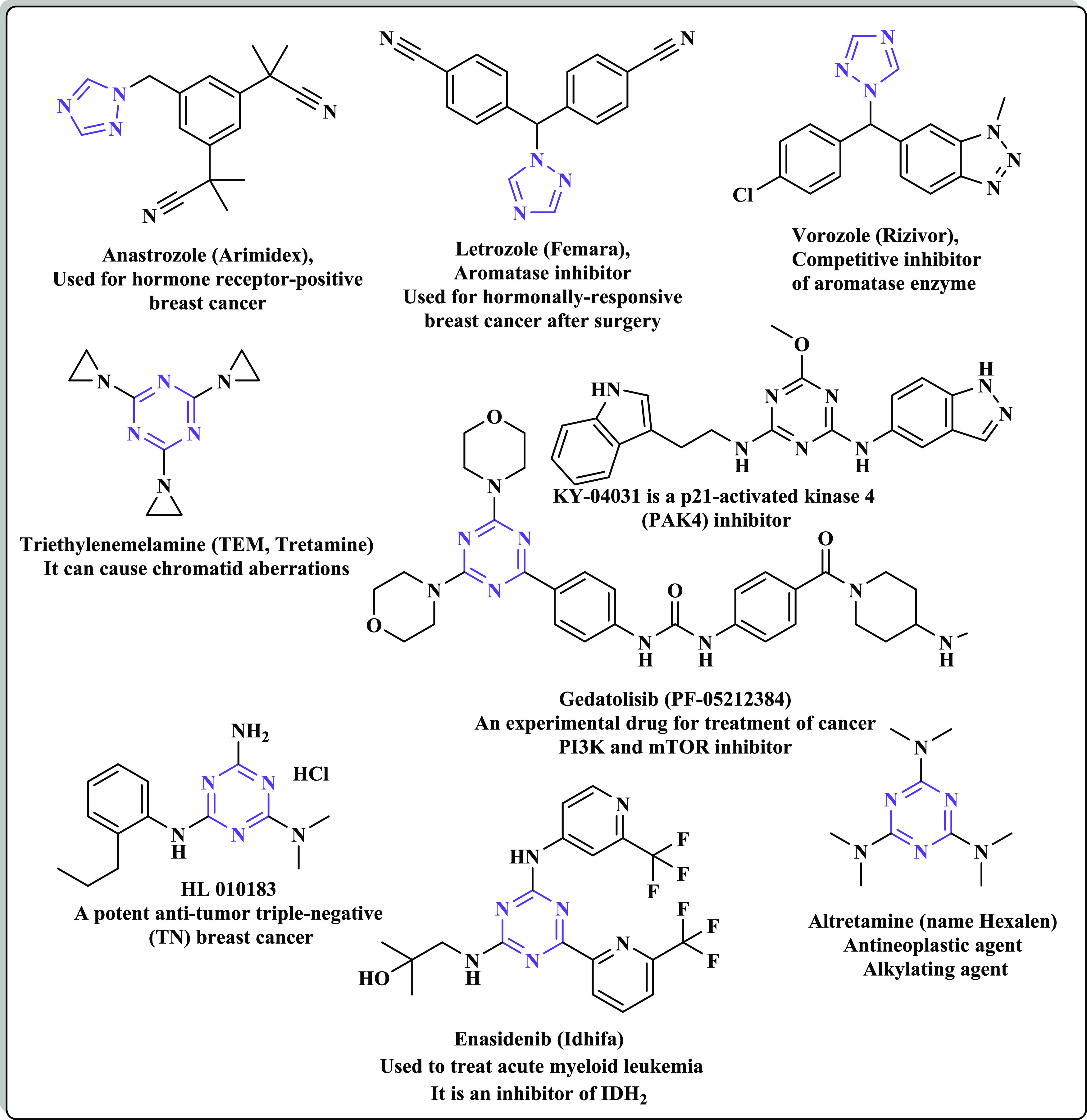
Examples of various marketed anticancer drugs bearing 1,2,4-triazole and s-triazine scaffolds.
Microwave-assisted organic synthesis (MAOS) has been widely used in green chemistry in recent years.74 Microwave irradiation is an eco-friendly approach without hazardous solvents. It helps in the synthesis of various heterocyclic compounds rapidly and in high yields.75,76 In addition, MAOS has a great contribution in chemical selectivity, catalyst-free conditions, and the absence of side products during the synthesis processes of various aromatic and heterocyclic compounds.77,78 Based on the above-mentioned knowledge and in continuation of our previous efforts in the field of design and generation of new bioactive heterocyclic compounds,79−84 this study deals with the design and microwave-assisted organic synthesis of a new set of benzimidazole, 1,2,4-triazole, and s-triazine derivatives targeting the wild-type EGFR-TK (EGFRWT) and the mutant EGFR-TK (EGFRT790M).
1.1. Rational and Design
Computational studies represented that the ATP active pocket of EGFR-TK possesses mainly five regions, as follows: (1) an adenine binding pocket bearing the key amino acid residues that can interact with the adenine ring via hydrogen bond formation, (2) a sugar zone (hydrophilic ribose pocket), (3) hydrophobic zone I (this area is not used by ATP but displays a pivotal role in the inhibitor selectivity), (4) hydrophobic region II (this area is also not used by ATP and can be used to determine the inhibitor specificity), and (5) a phosphate binding area that is important for improving the characteristics of inhibitor pharmacokinetics85,86 (Figure 3).
Figure 3.

The structure of the ATP-binding site of EGFR-TK.
Additionally, various studies demonstrated that the main pharmacophoric features shared by multiple EGFR-TKIs are four common areas: (i) a flat hetero-aromatic ring system, fitting in the adenine binding pocket and that can participate in H-bonding interactions with different amino acids such as Met793, Thr854, and Thr790 residues; (ii) a terminal hydrophobic head that occupies the hydrophobic zone I; (iii) an imino moiety (NH– spacer) that can participate in generating hydrogen bonds with different amino acid residues present in the linker region; and (iv) a hydrophobic tail that fits in the hydrophobic region II87−89 (Figure 4).
Figure 4.
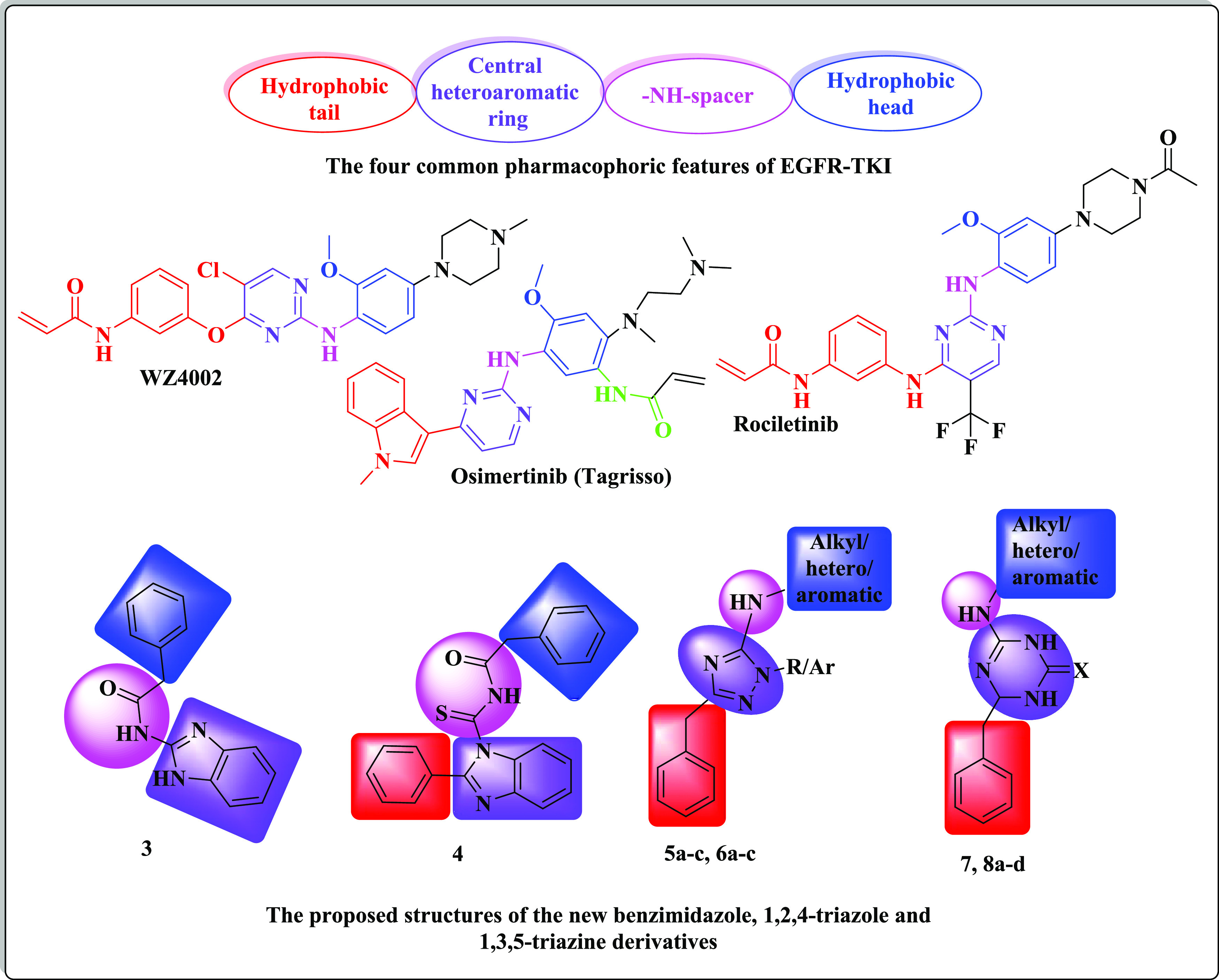
The designed molecular structures of new benzimidazole, 1,2,4-triazole, and 1,3,5-triazine derivatives.
Since benzimidazole, 1,2,4-triazole, and s-triazine are bio-isosteric, this work deals with the design and MAOS synthesis of new sets bearing one of the previously mentioned heterocyclic nuclei possessing the essential pharmacophoric features of EGFR-TKIs. The first position was the benzimidazole moiety (as compounds 3 and 4), and this scaffold was replaced by 1,2,4-triazole (as compounds 5 and 6) and 1,3,5-triazine nuclei (as compounds 7 and 8) to fit in the adenine binding pocket, where the heterocyclic nitrogen atoms act as hydrogen-bond acceptors leading to excellent EGFR-TK potency.90 The second position was the terminal benzyl moiety (hydrophobic head) (as compounds 3 and 4), which might be replaced with aliphatic, heterocyclic, or substituted phenyl structures (as compounds 5–8). The third position was the NH linker, as a site for the creation of different hydrogen bonds. The used linkers may be an imino group as in compounds 5–8 or a carbonothioyl-acetamide linker (as in compound 4). The fourth position was the phenyl group (hydrophobic tail), where a phenyl ring was incorporated at position-2 of the benzimidazole nucleus as in compound 4 or replaced by a benzyl ring at positions-5/3 of the 1,2,4-triazole nucleus or position-6 of the triazine ring to occupy the hydrophobic zone II of the ATP binding site. The fourth position of the benzimidazole 3 was left unsubstituted to find out the impact of the phenyl substitution on the target activity (Figure 4).
All the newly synthesized compounds were screened for their anti-proliferative activities against a panel of human cancer cell lines. Furthermore, the most promising compounds as cytotoxic agents were assessed as EGFRWT, EGFRT790M, and p53 ubiquitination inhibitors. Since compound 6b represented the most promising cytotoxic activity as well as EGFRWT and EGFRT790M inhibition activity, it was selected as a representative example to emphasize its possible binding patterns in the active pockets of EGFRWT and EGFRT790M via a molecular docking study.
2. Results and Discussion
2.1. Chemistry
The new target compounds 3–8 were synthesized in short reaction times and with high yields utilizing a microwave irradiation process (Schemes 1 and 2, Table 1). The chemical structures of the new compounds were elucidated using microanalytical and spectral data (IR, 1H, 13C NMR, MS). 2-Phenylacetyl isothiocyanate (1) was utilized as a key starting material and treated with different aliphatic, heterocyclic, and/or aromatic amines, namely, isopropylamine, 2-aminothiazole, o-phenylenediamine, m-aminophenol, and o-chloro-p-nitroaniline, in acetonitrile at room temperature91 to afford the corresponding thiourea derivatives 2a–e, respectively. IR spectra of 2a–e represented characteristic absorption bands at the regions 3370–3135, 1690–1660, and 1173–1146 cm–1 due to NH, C=O, and C=S groups, respectively. 1H NMR spectra of the new compounds 2a–e revealed singlet signals at the range δ 3.43–3.82 ppm representing the two methylene protons of CH2-ph. In addition, the expected signals of the aromatic protons appeared at the corresponding region δ 7.20–8.61 ppm, while the two NH protons appeared as D2O exchangeable signals at the range δ 12.11–13.0 ppm. Compound 2a exhibited a doublet signal at δ 1.06 ppm and another multiplet at δ 3.80–3.84 ppm that is an evidence of the presence of the −CH(CH3)2 group. Compounds 2c and 2d exhibited two additional D2O exchangeable signals at δ 5.01 and 9.66 ppm due to NH2 and OH groups, respectively.
Scheme 1. Synthesis of New Substituted Benzimidazole Derivatives Utilizing the Microwave Irradiation Synthetic Approach.

Scheme 2. Synthesis of New Substituted 1,2,4-Triazole and 1,3,5-Triazine Derivatives Utilizing the Microwave Irradiation Synthetic Approach.

Table 1. The Reaction Times and the Yields of the Newly Synthesized Compounds Using the Microwave Irradiation Technique.
| microwave
irradiation method |
||
|---|---|---|
| compound no. | time (min) | yield (%) |
| 3 | 2 | 80 |
| 4 | 3 | 96 |
| 5a | 2 | 98 |
| 5b | 5 | 96 |
| 5c | 7 | 86 |
| 6a | 6 | 95 |
| 6b | 9 | 85 |
| 6c | 10 | 89 |
| 7 | 5 | 97 |
| 8a | 4 | 96 |
| 8b | 9 | 97 |
| 8c | 7 | 92 |
| 8d | 10 | 98 |
Microwave irradiation of the thiourea derivative 2c in DMF for 2 min led to its cyclization, forming the corresponding N-(1H-benzo[d]imidazol-2-yl)-2-phenylacetamide benzimidazole (3), while its microwave irradiation with benzoyl chloride afforded92,93 the corresponding N-(2-phenyl-1H-benzo[d]imidazole-1-carbonothioyl) benzamide (4) (Scheme 1). IR spectra of the later derivatives 3 and 4 exhibited absorption bands at the ranges 3220–3128 and 1670–1699 cm–1 correlated to NH and C=O groups, respectively. Furthermore, 4 represented an additional band at 1266 cm–1 due to its C=S moiety. 1H NMR spectra of compounds 3 and 4 exhibited singlet signals at the region δ 3.70–3.91 due to CH2-ph and D2O exchangeable signals at the range δ 11.74–12.78 ppm representing NH groups, while the aromatic-Hs appeared as multiplet signals at their expected up-field region δ 7.36–8.24 ppm. Moreover, 13C NMR spectra of both 3 and 4 showed signals at the corresponding regions δ 40.06 and 40.32 ppm referring to CH2-ph and δ 173.30 and 167.0 due to C=O groups as well as various signals at the range δ 114.05–166.53 ppm representing the aromatic carbons.
On the other hand, treatment of compounds 2a, b, d, and e with hydrazine hydrate or phenyl hydrazine under microwave irradiation afforded the corresponding 1,3,4-triazole derivatives 5a–c and 6a–c, respectively, as the reported methods.91−94 IR spectral data of the latter triazole derivatives were devoid of any absorption bands correlated to C=O or C=S groups; instead, they exhibited absorption bands at 3288–3199 cm–1 contributing to NH groups, 3328 cm–1 referring to OH of compound 5c, and 1664–1639 cm–1 due to C=N groups. Furthermore, 1H NMR data of compounds 5 and 6 showed singlet signals at the region δ 4.23–3.50 ppm representing the presence of the two methylene protons of CH2-ph and D2O exchangeable signals at the range δ 7.21–12.31 ppm due to NH and OH protons, in addition to the expected multiplet signals at δ 6.60–8.38 ppm representing the aromatic protons. The −CH(CH3)2 residue of 5a was confirmed by the presence of doublet–multiplet signals at δ 1.06 and 3.39 ppm. 13C NMR spectra of compounds 5 and 6 represented singlet signals at the range δ 38.83–42.07 ppm ascribed to CH2-ph and different signals at the region δ 102.52–170.51 ppm referring to the aromatic carbons. Also, the isopropyl residue of 5a appeared as two additional singlets at δ 22.86 and 39.84 ppm.
Furthermore, cyclization of the thiazolyl derivative 2b with urea using microwave irradiation gave the corresponding 1,3,5-triazin-2-one derivative 7. On the other hand, microwave irradiation of compounds 2a, b, d, and e with thiourea led to the formation of the corresponding 1,3,5-triazin-2-thione analogues 8a–d, respectively (Scheme 2), following the reported reactions.91−94 The IR spectrum of compound 7 showed a characteristic absorption band at 1685 cm–1 characteristic for the carbonyl group of the triazine ring, in addition to an absorption band at 3206 cm–1 due to NH moieties. IR spectra of compound 8 exhibited the C=S group as an absorption band at the region1177–1127 cm–1. Moreover, 1H NMR data of compound 8 exhibited CH2-ph protons as a singlet signal at δ 3.51–3.82 ppm, the aromatic protons as multiplet signals in the down field expected region δ 6.65–7.76 ppm, and NH and OH protons as D2O exchangeable singlets at the region δ 7.96–12.35 ppm. The isopropyl protons of compound 8a appeared as a doublet–multiplet signal at δ 1.05 and 3.36 ppm. In addition, 13C NMR spectra showed signals at δ 40.06–42.15 ppm due to CH2-ph, 22.85 and 40.15 due to −CH(CH3)2 of compound 8a, and 113.65–170.75 related to the aromatic carbons. Further support for the suggested structures of the new compounds was gained by their mass spectra, which were in accordance with the proposed structures representing their correct molecular ion peaks beside some other important peaks (cf. Experimental Section).
2.2. Biological Activity
2.2.1. In Vitro Evaluation of Cytotoxic Potentials of the Newly Prepared Derivatives
The newly synthesized compounds 2–8 were investigated for their potential cytotoxic activities against a panel of four different human cancer cell lines—hepatocellular carcinoma (HepG-2), prostate carcinoma (PC-3), breast adenocarcinoma (MCF-7), and non-small cell lung cancer cells (A-549)—and the normal peripheral blood mononuclear cells (PBMCs) using an MTT assay.95 Doxorubicin and erlotinib served as reference standards. The concentrations of the tested derivatives that induced 50% inhibition of the cell viability (IC50, μM) were determined and tabulated in Table 2.
Table 2. In Vitro Cytotoxic Potency of the Newly Synthesized Compounds 2–8 against Various Human Cancer Cell Lines and Normal Cells Representing SI of the Most Active Derivativesa.
| IC50 (mean ± SEM) (μM) |
|||||
|---|---|---|---|---|---|
| compd. no. | HepG-2 | PC-3 | MCF-7 | A-549 | PBMC |
| 2a | 57.85 ± 0.07 | 26.82 ± 2.21 | 37.73 ± 0.08 | 34.57 ± 0.06 | 121.34 ± 11.35 |
| 2b | 60.85 ± 0.06 | 44.50 ± 3.51 | 55.44 ± 0.04 | 57.48 ± 0.05 | 133.30 ± 13.67 |
| 2c | 47.66 ± 0.04 | 25.44 ± 0.04 | 21.76 ± 0.08 | 27.81 ± 0.04 | 144.56 ± 14.89 |
| 2d | 34.77 ± 0.03 | 39.32 ± 0.0 | 24.74 ± 0.08 | 20.84 ± 0.04 | 157.78 ± 16.35 |
| 2e | 37.76 ± 0.05 | 40.56 ± 0.05 | 25.64 ± 0.08 | 25.96 ± 0.05 | 168.54 ± 17.36 |
| 3 | 37.73 ± 0.02 | 17.29 ± 0.03 | 6.59 ± 0.07 | 10.42 ± 0.05 | 179.25 ± 18.75 |
| 4 | 56.65 ± 0.04 | 25.41 ± 0.05 | 8.32 ± 0.04 | 15.61 ± 0.06 | 186.68 ± 19.36 |
| 5a | 35.66 ± 0.04 | 16.49 ± 0.05 | 5.42 ± 0.05 | 10.37 ± 0.04 | 174.90 ± 18.24 |
| SI = 4.90 | SI = 10.60 | SI = 32.26 | SI = 16.86 | ||
| 5b | 38.49 ± 0.02 | 18.22 ± 0.03 | 4.18 ± 0.03 | 8.27 ± 0.03 | 165.76 ± 17.36 |
| SI = 4.30 | SI = 9.09 | SI = 39.65 | SI = 20.04 | ||
| 5c | 27.59 ± 0.04 | 18.44 ± 0.04 | 4.33 ± 0.04 | 12.30 ± 0.04 | 146.32 ± 16.15 |
| SI = 5.23 | SI = 7.93 | SI = 33.79 | SI = 11.89 | ||
| 6a | 10.48 ± 0.03 | 16.30 ± 0.09 | 4.30 ± 0.04 | 5.20 ± 0.03 | 157.45 ± 16.89 |
| SI = 15.02 | SI = 9.65 | SI = 36.61 | SI = 30.27 | ||
| 6b | 5.47 ± 0.02 | 13.21 ± 0.06 | 1.29 ± 0.03 | 3.18 ± 0.03 | 178.23 ± 17.69 |
| SI = 32.58 | SI = 13.49 | SI = 138.16 | SI = 56.04 | ||
| 6c | 7.73 ± 0.04 | 26.41 ± 0.05 | 2.51 ± 0.06 | 5.80 ± 0.05 | 187.67 ± 19.76 |
| SI = 24.27 | SI = 7.10 | SI = 74.76 | SI = 32.35 | ||
| 7 | 29.75 ± 0.03 | 17.90 ± 0.03 | 4.65 ± 0.07 | 7.43 ± 0.04 | 165.23 ± 18.05 |
| 8a | 59.84 ± 0.05 | 36.55 ± 0.03 | 13.75 ± 0.08 | 25.46 ± 0.05 | 173.45 ± 18.95 |
| SI = 2.91 | SI = 4.74 | SI = 12.61 | SI = 6.81 | ||
| 8b | 60.48 ± 0.05 | 42.56 ± 0.06 | 17.95 ± 0.04 | 37.21 ± 0.03 | 196.67 ± 20.67 |
| SI = 3.25 | SI = 4.62 | SI = 10.95 | SI = 5.28 | ||
| 8c | 45.49 ± 0.04 | 32.48 ± 0.04 | 10.31 ± 0.04 | 8.29 ± 0.03 | 188.89 ± 19.86 |
| SI = 4.15 | SI = 5.81 | SI = 18.32 | SI = 22.78 | ||
| 8d | 47.86 ± 0.05 | 39.55 ± 0.08 | 14.77 ± 0.05 | 9.50 ± 0.06 | 179.09 ± 18.96 |
| SI = 3.74 | SI = 4.52 | SI = 12.12 | SI = 18.85 | ||
| DOX | 4.51 ± 0.26 | 8.11 ± 0.05 | 4.17 ± 0.2 | 8.20 ± 0.08 | 250.00 ± 26.56 |
| SI = 55.43 | SI = 30.82 | SI = 59.95 | SI = 30.48 | ||
| erlotinib | 8.19 ± 0.4 | 8.89 ± 0.6 | 4.16 ± 0.2 | 3.76 ± 0.2 | 45.75 ± 26.56 |
| SI = 5.58 | SI = 5.14 | SI = 10.99 | SI = 12.16 | ||
DOX: doxorubicin; IC50: compound concentration required to inhibit the cell viability by 50%; SEM: standard error mean; each value is the mean of three independent determinations; SI: selectivity index.
Based on the resultant data, the examined compounds showed versatile antiproliferative activities against the tested cell lines. It could be noted that the benzimidazole derivatives 3 and 4, the triazole derivatives 5 and 6, and the triazine derivatives 7 and 8 elicited superior cytotoxicity against MCF-7 and A-549 cell lines. The N-phenyl-1,2,4-triazole compounds 6a–c exhibited the most potent cytotoxic activity against MCF-7 of IC50 values ranging from 1.29 to 4.30 μM that were evidently near those of the reference drugs (doxorubicin and erlotinib) of IC50 4.17 and 4.16 μM, respectively. Furthermore, the latter derivatives reduced the viability of A-549 cells with 1.3–2.6-folds more potency than doxorubicin and approximately equivalent potency to erlotinib, exhibiting IC50’s ranging from 3.18 to 5.80 μM and IC50doxorubicin, erlotinib of 8.20 and 3.76 μM, respectively. The 3-OH-phenyl derivative 6b was 3.2-folds more potent than doxorubicin and erlotinib against MCF-7 cells and 2.6-folds more potent than doxorubicin against the A-549 cell line. The oxygen atom of the hydroxyl group might produce an additional H-binding interaction with the target protein. In addition, the HepG-2 cell line exhibited promising sensitivity against 6a–c that was slightly higher than its sensitivity against erlotinib but slightly less than that against doxorubicin, exhibiting IC50’s of 5.47–10.48 μM and IC50doxorubicin, erlotinib of 4.51 and 8.19 μM.
Both MCF-7 and A-549 cell lines displayed an equipotent or a slightly less sensitivity against the 1,2,4-triazole analogues 5a–c than that against the reference drugs, displaying IC50’s ranging from 4.18 to 5.42 and 7.43 to 12.30 μM, respectively. Moreover, the 1,3,5-triazinone 7 was nearly equivalent to doxorubicin against MCF-7 and A-549 cell lines with IC50’s of 4.65 and 7.43 μM, but it showed nearly 2-folds less potency against A-549 compared to erlotinib.
With the exception of compounds 8c and 8d that were as potent as doxorubicin against the A-549 cell line, a detectable drop in the cytotoxic activity was observed by the 1,3,5-triazin-2-thione derivatives 8 against both MCF-7 and A-549 cell lines with IC50 values of 10.31–25.46 and 37.21–25.46 μM, respectively. On the other hand, the tested compounds showed moderate to weak antiproliferative activities against hepatocellular carcinoma (HepG-2) and prostate carcinoma (PC-3) cell lines. On the other hand, all the tested derivatives produced low cytotoxicity against the normal PBMC cell line with IC50 values <100 μM, confirming the safety margin of the newly synthesized derivatives.
2.2.2. In Vitro Inhibition of EGFRWT and EGFRT790M Activity
Following the primary screening for cytotoxic potentials, the most active congeners 5a–c, 6a–c, and 7 that revealed the most promising antiproliferative activities were further investigated for their possible mechanism of actions against cancer cells. They were assessed in terms of in vitro kinase inhibitory efficiencies against the wild-type EGFRWT and the mutant form EGFRT790M using a homogeneous time resolved fluorescence (HTRF) assay.96,97 The results are summarized as IC50 values (μM) in Table 3 using erlotinib and AZD9291 as positive controls.
Table 3. Kinase Inhibitory Assay of the Newly Synthesized Derivatives 5–7 in Comparison with Erlotinib and AZD9291 against EGFRWT and Mutant EGFRT790Ma.
| IC50 (mean ± SEM) (μM) |
||
|---|---|---|
| compound no. | EGFRWT | EGFRT790M |
| erlotinib | 0.09 ± 0.05 | 0.55 ± 0.10 |
| AZD9291 | 0.52 ± 0.03 | 0.03 ± 0.01 |
| 5a | 0.25 ± 0.01 | 0.17 ± 0.05 |
| 5b | 0.22 ± 0.15 | 0.13 ± 0.11 |
| 5c | 0.24 ± 0.30 | 0.14 ± 0.50 |
| 6a | 0.18 ± 0.10 | 0.12 ± 0.18 |
| 6b | 0.08 ± 0.05 | 0.09 ± 0.01 |
| 6c | 0.15 ± 0.02 | 0.13 ± 0.07 |
| 7 | 0.22 ± 0.05 | 0.18 ± 0.11 |
IC50: compound concentration required to inhibit the enzymes’ activities by 50%; SEM: standard error mean; each value is the mean of three independent values.
Excellent inhibitory activities were obtained by the examined compounds 5 and 6 against EGFRWT, which were about 2–6.5 times more potent than AZD9291 representing IC50 values ranging from 0.08 to 0.25 μM and IC50AZD9291 of 0.52 μM. On the other hand, erlotinib (IC50 of 0.095 μM) represented about 2.7–1.6-folds more potency against the wild form of EGFR compared with 5a–c and 6a and c. Interestingly, compound 6b appeared to be a 1-fold more potent EGFRWT inhibitor than erlotinib with an IC50 value of 0.08 μM. Additionally, the resultant data investigated that compounds 5a–c, 6a–c, and 7 were 6.1–3.2-folds more active against the mutated form of EGFRT790M than the reference drug erlotinib, exhibiting IC50’s ranging from 0.09 to 0.18 μM and IC50 erlotinib of 0.55 μM. Reversely, the tested analogues 5, 6, and 7 appeared to be less potent EGFRT790M suppressors compared to the reference drug AZD9291 with IC50 of 0.03 μM. It is evident that the 3-hydroxyphenyl derivative 6b represented the most promising suppression activity against the wild and the mutant form T790M of EGFR compared with the reference standards erlotinib and AZD9291 (Figure 4). The obtained results were in agreement with the data of cytotoxicity evaluation. The docking study correlated the enhanced activity of 6b to its hydroxyl oxygen that was a site for H-bonding in the active regions of EGFRWT and EGFRT790M, while the N-phenyl moiety increased the hydrophobic interaction with the target enzyme.
Moreover, it could be detected that all the compounds exhibited more potent inhibitory activity against the mutant form EGFRT790M over the wild-type form EGFRWT, which can overcome the resistance problem to EGFR-TKIs that develops due to the T790M mutation of the EGFR gene.
2.2.3. In Vivo Inhibition of p53 Ubiquitination
The p53 protein plays a crucial role in the regulation of cancer development through its action as a suppressing molecule that binds to E3 ubiquitin ligase, thus inhibiting its role as a transcription activator.98,99 Therefore, interfering with p53 binding on E3 ligase can interfere with tumor development and progression. Following the reported methodology,99 the obtained results exhibited that the compounds 5–7 showed moderate inhibitory actions toward in vivo p53 ubiquitination compared to the reference diphenyl imidazole (DPI). According to Table 4, compounds 5a–c have recorded IC50 values greater than the reference drug of IC50’s of 0.68, 0.62, and 0.60 nM and IC50DPI of 0.26 ± 0.005 nM. A higher potency was reported by the compounds 6a–c and 7 affording IC50 values ranging from 0.59 to 0.48 nM. Accordingly, these results revealed that the tested analogues can still act as p53 ubiquitination inhibitors and thus can intervene with cancer cell growth and development.
Table 4. IC50 Values Obtained Due to In Vivo p53 Ubiquitination Inhibition of MCF-7 Cells.
| IC50 (mean ± SEM) (nM) | |
|---|---|
| compound no. | p53 ubiquitination |
| DPI | 0.26 ± 0.005 |
| 5a | 0.68 ± 0.01 |
| 5b | 0.62 ± 0.05 |
| 5c | 0.60 ± 0. 03 |
| 6a | 0.59 ± 0. 01 |
| 6b | 0.50 ± 0.05 |
| 6c | 0.48 ± 0.02 |
| 7 | 0.48 0 ± 0.05 |
2.3. Molecular Modeling Study on EGFRWT and Mutant EGFRT790M
In the current docking simulation, the potent kinase inhibitors 5–7 were selected based on the potency and scaffold type to correlate the structure–activity relationship with their behavior and the possible binding interactions within the active sites of EGFRWT and mutant EGFRT790M. Thus, the domains of EGFRWT and mutant EGFRT790M kinase complexed with erlotinib and AZD9291 (PDB ID: 1M17 and 6JX0)100,101 were downloaded from the Protein Data Bank. The docking calculations were done using MOE-Dock (Molecular Operating Environment) software version 2014.0901.102,103 At the beginning, redocking of the native ligands (erlotinib and AZD9291) was achieved within their own binding sites of EGFRWT and EGFRT790M, giving energy scores −11.40 and −12.66 kcal/mol with RMDS values (root mean square deviation) of 0.91 and 1.02 Å, respectively. It was noted that the compounds 5–7 approximately displayed similar binding poses with promising energy scores that are depicted in Tables 5 and 6.
Table 5. Docking Study of Compounds 5–7 within EGFRWT (PDB Code: 1M17) Using MOE Software Version 2014.0901.
| compd. no. | docking score (kcal/mol) | amino acid residues (bond length Å) | atoms of compound | type of bond |
|---|---|---|---|---|
| erlotinib | –11.40 | Met769(2.70) | N1(quinazoline) | H-acc |
| 5a | –10.15 | Lys721(2.92) | N-4(1,2,4-triazole) | H-acc |
| Met769(2.75) | N(thiazole) | H-acc | ||
| 5b | –10.32 | Lys721(2.65) | N-4(1,2,4-triazole) | H-acc |
| Met769(2.95) | N(linker NH) | H-acc | ||
| 5c | –9.85 | Lys702 | phenol | arene-cation |
| Lys721(2.75) | N-4(1,2,4-triazole) | H-acc | ||
| Met769(2.60) | O(OH) | H-acc | ||
| 6a | –10.50 | Lys721(3.22) | N-2(1,2,4-triazole) | H-acc |
| Met769(2.88) | N(thiazole) | H-acc | ||
| 6b | –10.88 | Val702 | phenol | arene-cation |
| Lys721(3.63) | N-2(1,2,4-triazole) | H-acc | ||
| Met769(3.27) | O(OH) | H-acc | ||
| 6c | –10.25 | Lys721(3.20) | N-2(1,2,4-triazole) | H-acc |
| Met769(3.60) | O(NO2) | H-acc | ||
| 7 | –10.36 | Lys721(2.70) | N-3(1,3,5-triazine) | H-acc |
| Met769(2.80) | N(thiazole) | H-acc |
Table 6. Docking Study of Compounds 5–7 within EGFRT790M (PDB Code: 6JX0) Using MOE Software Version 2014.0901.
| compd. no. | docking score (kcal/mol) | amino acid residues (bond length Å) | atoms of compound | type of bond |
|---|---|---|---|---|
| AZD9291 | –12.66 | Val726 | indole | arene-cation |
| Met793(2.88) | N-1(pyrimidine) | H-acc | ||
| Asp800(3.27) | N(N(CH3)2) | H-don | ||
| 5a | –11.20 | Leu718 | thiazole | arene-cation |
| Met793(2.95) | N-1(1,2,4-triazole) | H-don | ||
| Met793(2.80) | N(linker NH) | H-don | ||
| 5b | –10.74 | Met793(3.15) | N-1(1,2,4-triazole) | H-don |
| Met793(3.00) | N(linker NH) | H-don | ||
| 5c | –10.70 | Leu718 | phenol | arene-cation |
| Met793(2.77) | N-1(1,2,4-triazole) | H-don | ||
| Met793(2.60) | N(linker NH) | H-don | ||
| 6a | –11.45 | Leu718 | thiazole | arene-cation |
| Met793(3.55) | N-4(1,2,4-triazole) | H-acc | ||
| Met793(3.26) | N(linker NH) | H-don | ||
| 6b | –11.75 | Leu718 | phenol | arene-cation |
| Met793(3.65) | N-4(1,2,4-triazole) | H-acc | ||
| Met793(3.06) | N(linker NH) | H-don | ||
| 6c | –11.35 | Leu718 | 2-Cl-4-NO2-C6H3 | arene-cation |
| Met793(3.22) | N-4(1,2,4-triazole) | H-acc | ||
| Met793(2.90) | N(linker NH) | H-don | ||
| 7 | –11.20 | Leu718 | thiazole | arene-cation |
| Met793(3.15) | N-1(1,3,5-triazine) | H-don | ||
| Met793(2.85) | N(linker NH) | H-don |
All the screened derivatives 5–7 afforded H-bonding with the key amino acids Met769 and Met793 within the active sites of EGFRWT and mutant EGFRT790M kinases like the original ligands erlotinib and AZD9291, respectively. Furthermore, the existence of 1,2,4-triazoles in compounds 5 and 6, in addition to the 1,3,5-triazine moiety in compound 7, potentiates fixation within the binding pockets of EGFRWT and EGFRT790M enzymes through extra H-bonding with Lys721 and Met793, respectively.
By focusing upon compound 6b as the most active inhibitor, it fulfilled the key interactions in the active site of EGFRWT with energy score −10.88 kcal/mol, where hydrogen bonding was established between N-2 of the 1,2,4-triazole moiety and the side chain of Lys721 (distance: 3.63 Å), as well as the Pi-cation interaction of the phenolic ring with the Val702 residue. The presence of the H-bond acceptor between the hydroxyl oxygen and the backbone of the key amino acid Met769 improved the fitting within the active site of the enzyme (distance: 3.27 Å) (Figure 5).
Figure 5.
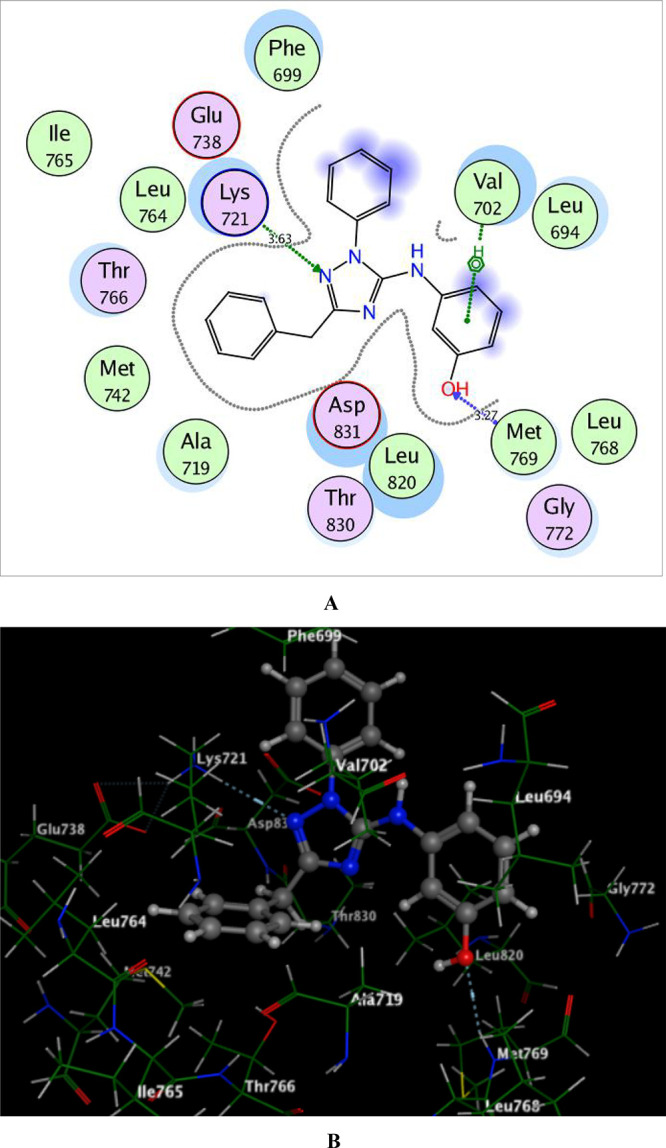
2D and 3D schematic binding interactions (A and B) of compound 6b into EGFRWT (PDB code: 1M17) using the MOE software.
Regarding the docking of 6b within the ATP-binding pocket of EGFRT790M allowing energy score −13.27 kcal/mol, it was found that N-2 of the 1,2,4-triazole scaffold and the NH linker at position-5 played a vital role in the binding through a bidentate hydrogen-bonded interaction with the backbone of the hinge Met793 (distance: 3.65 and 3.06 Å, respectively). Moreover, the phenolic ring shared fixation through Pi-cation interaction with Leu718 (Figure 6).
Figure 6.
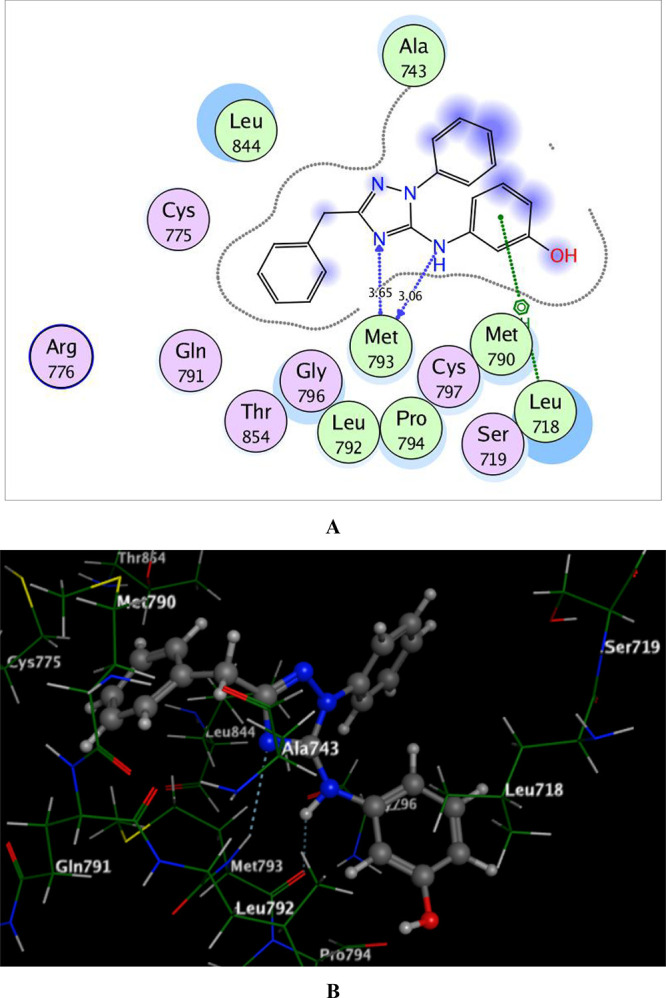
2D and 3D schematic binding interactions (A and B) of compound 6b into EGFRT790M (PDB code: 6JX0) using the MOE software.
The analysis of the docking results demonstrated that compound 6b with the highest EGFRWT and EGFRT790M inhibitory activities adopted good binding mode through its characterized structure of the 1,2,4-triazole core and the phenolic ring linked via the NH group forming hydrophilic and hydrophobic interactions.
3. Conclusions
A new set of benzimidazole, 1,2,4-triazole, and 1,3,5-triazine derivatives was designed and synthesized using microwave irradiation. The cytotoxic activity of all the new analogues was evaluated against a panel of four human cancer cell lines—HepG-2, PC-3, MCF-7, and A-549—in addition to the normal peripheral blood mononuclear cells (PBMCs) using doxorubicin and erlotinib as standard drugs. The gained results represented the significant selective cytotoxicity of some of the examined derivatives against MCF-7 and A-549 cell lines. The most potent cytotoxic activity against MCF-7 cells was revealed by the N-phenyl-1,2,4-triazole analogues 6a–c, exhibiting IC50 values ranging from 1.29 to 4.30 μM that were evidently near those of the reference compounds (doxorubicin and erlotinib) of IC50 of 4.17 and 4.16 μM, respectively. Moreover, A-549 cancer cells represented about 1.3–2.6-folds more sensitivity against the latter derivatives than that against doxorubicin and approximately equal sensitivity to that obtained against erlotinib exhibiting IC50’s ranging from 3.18 to 5.80 μM and IC50; doxorubicin, erlotinib of 8.20 and 3.76 μM, respectively. On the other hand, both MCF-7 and A-549 cell lines displayed an equipotent or a slightly less sensitivity against the 1,2,4-triazole analogues 5a–c and the 1,3,5-triazinone 7 than that against the reference drugs displaying IC50’s ranging from 4.18 to 5.42 and 7.43 to 12.30 μM, respectively. With the exception of compounds 8c and 8d that were as potent as doxorubicin against the A-549 cell line, an observable decrease in the cytotoxic activity was detected in the 1,3,5-triazin-2-thione derivatives 8 against both MCF-7 and A-549 cell lines with IC50 values of 10.31–25.46 and 37.21–25.46 μM, respectively. Moreover, moderate to weak antiproliferative activity against hepatocellular carcinoma (HepG-2) and prostate carcinoma (PC-3) cell lines was detected by the tested compounds. All the tested derivatives represented low cytotoxicity against the normal PBMC cell line with IC50 values <100 μM, confirming the safety margin of the new derivatives.
Furthermore, the new target compounds showing the most promising anticancer activity (5, 6, and 7) were evaluated as in vitro EGFRWT and EGFRT790M inhibitors compared to the reference drugs erlotinib and AZD9291. Generally, the target derivatives represented a promising inhibitory effect against EGFRWT and EGFRT790M with more potency against the mutant form EGFRT790M, which is a good property to overcome the EGFR-TKI resistance problem. Also, derivative 6b represented the most potent suppression effect against both EGFRWT and EGFRT790M.
Moreover, compounds 5–7 down-regulated the oncogenic parameter p53 ubiquitination, representing approximately an equivalent suppression potency to the reference diphenyl imidazole (DPI). The docking simulation study was performed for the promising inhibitors 5–7, giving energy scores of −11.40 and −12.66 kcal/mol with RMDS values of 0.91 and 1.02 Å, respectively. Compound 6b was chosen as a representative example to find out the binding modes of the compound in the active pocket of EGFRWT and the mutant EGFRT790M. It adopted promising binding interactions with the active sites of the tested proteins through its 1,2,4-triazole scaffold and the phenolic ring linked via the NH group forming various hydrophilic and hydrophobic interactions in the active pocket of the wild EGFRWT and its mutated form EGFRT790M.
As an overview on the obtained results, it has been investigated that 6b is a new potent antitumor agent exhibiting a safety profile against the normal cells as well as a promising inhibitory impact against EGFRWT and EGFRT790M. These advantages together indicated that 6b could be considered as an auspicious lead compound for the future evolution of new more potent anticancer candidates inhibiting EGFR mutations.
4. Experimental Section
4.1. Chemistry
The instruments used for measuring the melting points, spectral data (IR, mass, 1H NMR and 13C NMR, X-ray) and elemental analysis are provided in detail in the Supplementary Information.
4.1.1. Synthesis of the Thiourea Derivatives 2a–e
A mixture of 2-phenylacetyl isothiocyanate (1) (0.01 mol) and different amine derivatives, namely, isopropylamine, 2-aminothiazole, o-phenylenediamine, m-aminophenol, and o-chloro-p-nitroaniline (0.01 mol) in dry acetonitrile (20 mL), was stirred at room temperature for 3 h. The solid product obtained was filtered and recrystallized from ethanol to give the corresponding thiourea derivatives 2a–e, respectively.
4.1.1.1. N-(Isopropylcarbamothioyl)-2-phenylacetamide (2a)
Pale yellow crystals; yield 98%, m.p. 70–72 °C. IR (KBr) (υ, cm–1): 3287, 3192 (NH), 3064 (CHarom), 2974, 2930, 2875 (CHaliph), 1660 (C=O), 1639 (C=N), 1173 (C=S); 1H NMR (DMSO-d6) δ: 1.06 (d, 6H, CH3), 3.43 (s, 2H, CH2-ph), 3.80–3.84 (m, 1H, CH(CH3)2), 7.20–7.33 (m, 5H, Ar-H, J = 7.27 Hz), 7.96 (br.s, 1H, NH, D2O exchangeable), 11.35 (br.s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 22.85, 42.89, 46.86, 126.70, 128.63, 129.33, 137.11, 169.56, 179.12; MS (70 eV) m/z (%): 236 (M+, 19). Anal. calcd for C12H16N2Os (236.33): C, 60.99; H, 6.82; N, 11.85. Found: C, 60.86; H, 7.12; N, 11.67.
4.1.1.2. 2-Phenyl-N-(thiazol-2-ylcarbamothioyl) Acetamide (2b)
Pale brown crystals; yield 90%, m.p. 242–244 °C; IR (KBr) (υ, cm–1): 3267, 3171 (NH), 3080 (CHarom), 2947, 2893 (CHaliph), 1685 (C=O), 1567 (C=N), 1166 (C=S); 1H NMR (DMSO-d6) δ: 3.78 (s, 2H, CH2-ph), 7.20–7.26 (d, 2H, thiazole-H4, H5), 7.34–7.63 (m, 5H, Ar-H, J = 7.67 Hz), 8.01 (br.s, 1H, NH, D2O exchangeable), 12.37 (br.s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 42.12, 113.96, 130.36, 138.06, 127.27, 128.88, 129.70, 135.47, 158.46, 169.62; MS (70 eV) m/z (%): 277 (M+, 18). Anal. calcd for C12H11N3OS2 (277.36): C, 51.97; H, 4.00; N, 15.15. Found: C, 51.62; H, 3.87; N, 14.86.
4.1.1.3. N-((2-Aminophenyl)carbamothioyl)-2-phenylacetamide (2c)
Yellow crystals; yield 94%; m.p. 188–190 °C; IR (KBr) (υ, cm–1): 3370, 3327, 3135 (NH, NH2), 3030 (CHarom), 2950, 2830 (CHaliph), 1690 (C=O), 1670 (C=N), 1167 (C=S); 1H NMR (DMSO-d6) δ: 3.82 (s, 2H, CH2-ph), 5.01 (br.s, 2H, NH2, D2O exchangeable), 6.57–7.80 (m, 9H, Ar-H, J = 7.10 Hz), 11.63 (br.s, 1H, NH, D2O exchangeable), 12.13 (br.s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 42.80, 109.96, 116.32, 127.40, 127.67, 128.87, 129.90, 132.80, 134.45, 143.70, 173.30, 180.67; MS (70 eV) m/z (%): 285 (M+, 14). Anal. calcd for C15H15N3Os (285.37): C, 63.13; H, 5.30; N, 14.73. Found: C, 63.09; H, 5.27; N, 14.69.
4.1.1.4. N-((3-Hydroxyphenyl)carbamothioyl)-2-phenylacetamide (2d)
Beige powder; yield 98%, m.p. 260–262o C; IR (KBr) (υ, cm–1): 3304 (OH), 3246, 3199 (NH), 3063 (CHarom), 2906, 2820 (CHaliph), 1684 (C=O), 1609 (C=N), 1146 (C=S); 1H NMR (DMSO-d6) δ: 3.83 (s, 2H, CH2-ph), 6.67–7.36 (m, 9H, Ar-H, J = 7.05 Hz), 9.66 (br.s, 1H, OH, D2O exchangeable), 11.67 (br.s, 1H, NH, D2O exchangeable), 12.41 (br.s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 39.94, 111.28, 113.86, 114.90, 127.48, 128.93, 129.95, 134.74, 139.13, 157.98, 173.75, 178.85; MS (70 eV) m/z (%): 286 (M+, 39). Anal. calcd for C15H14N2O2S (286.35): C, 62.92; H, 4.93; N, 9.78. Found: C, 61.21; H, 5.21; N, 9.67.
4.1.1.5. N-((2-Chloro-4-nitrophenyl)carbamothioyl)-2-phenylacetamide (2e)
Yellow powder; yield 97%, m.p. 105–107o C; IR (KBr) (υ, cm–1): 3275, 3198 (NH), 3091, 3009 (CHarom), 2921, 2815 (CHaliph), 1684 (C=O), 1625 (C=N), 1147 (C=S); 1H NMR (DMSO-d6) δ: 3.86 (s, 2H, CH2-ph), 6.84–7.35 (m, 5H, Ar-H, J = 7.69 Hz), 7.94–8.63 (m, 3H, Ar-H, J = 7.31, 7.63 Hz), 12.11 (br.s, 1H, NH, D2O exchangeable), 12.85 (br.s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 42.77, 120.43, 122.98, 125.13, 126.92, 130.02, 134.44, 136.36, 145.35, 151.83, 173.98, 180.07; MS (70 eV) m/z (%): 349 (M+, 29). Anal. calcd for C15H12ClN3O3S (349.79): C, 51.51; H, 3.46; N, 12.01. Found: C, 51.13; H, 3.67; N, 11.69.
4.1.2. Synthesis of N-(1H-Benzo[d]imidazol-2-yl)-2-phenylacetamide (3)
A solution of compound 2c (2.85 g; 0.01 mol) in ethyl alcohol (5 mL) was irradiated under MW for 2 min at 80 °C. After cooling at room temperature, the precipitate was filtered and recrystallized from ethanol to give the corresponding compound 3.
White crystals; yield (80%); m.p. 206–208 °C; IR (KBr) (υ, cm–1): 3200, 3128 (NH), 3067, 3028 (CHarom), 2910, 2820 (CHalkyl), 1670 (C=O), 1512 (C=N); 1H NMR (DMSO-d6) δ: 3.76 (s, 2H, CH2-ph), 6.57–7.80 (m, 9H, Ar-H, J = 7.28, 7.68 Hz), 11.74 (br. s, 1H, NH, D2O exchangeable), 12.11 (br.s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 40.06, 116.32, 116.50, 127.43, 128.87, 128.90, 129.88, 130.01, 134,70, 143.66, 173.30; MS (70 eV) m/z (%): 251 (M+, 5). Anal. calcd for C15H13N3O (251.29): C, 71.70; H, 5.21; N, 16.72. Found: C, 71.53; H, 5.03; N, 16.48.
4.1.3. Synthesis of 2-Phenyl-N-(2-phenyl-1H-enzo[d]imidazole-1-carbonothioyl)acetamide (4)
A mixture of compound 2c (2.85 g; 0.01 mol) and benzoyl chloride (0.01 mol) in ethyl alcohol (5 mL) was irradiated under MW radiation for 3 min at 80 °C, and then it was treated with cold water. The formed solid was filtered, washed with water, dried, and recrystallized from ethanol to give compound 4. Yellow crystals; yield 96%, m.p. 260–280 °C; IR (KBr) (υ, cm–1): 3219 (NH), 3063, 3026 (CHarom), 2960, 2820 (CHaliph), 1699 (C=O), 1625 (C=N), 1266 (C=S); 1H NMR (DMSO-d6) δ: 3.91 (s, 2H, CH2-ph),7.36–8.24 (m, 14H, Ar-H, J = 7.48, 7.94 Hz), 12.78 (br.s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 40.32, 114.05, 125.02, 128.92, 129.09, 129.37, 129.60, 129.87, 129.98, 131.99, 134.18, 144.71, 166.53, 167.0; MS (70 eV) m/z (%): 371 (M+, 68). Anal. calcd for C22H17N3OS (371.46): C, 71.14; H, 4.61; N, 11.31. Found: C, 71.11; H, 4.58; N, 11.28.
4.1.4. Synthesis of 1,2,4-Triazole Derivatives 5a–c and 6a–c
A mixture of 2a, b, and d (0.01 mol) and hydrazine hydrate or phenyl hydrazine (0.01 mol) was heated in MW for 2–10 min at 120–150 °C in the presence of DMF as a solvent. After cooling, the reaction mixture was poured into ice water. The obtained precipitate was filtered and recrystallized from ethanol to give corresponding products 5a–c and 6a–c, respectively.
4.1.4.1. 5-Benzyl-N-isopropyl-4H-1,2,4-triazol-3-amine (5a)
White needles, yield 98%, m.p. 84–86 °C; IR (KBr) (υ, cm–1): 3288 (NH), 3065, 3029 (CHarom), 2975, 2930, 2874 (CHaliph), 1639 (C=N); 1H NMR (DMSO-d6) δ: 1.06 (d, 6H, CH3), 3.39 (m, 1H, CH), 3.82 (s, 2H, CH2-ph), 7.21 (br. s, 1H, NH, D2O exchangeable), 7.22–7.31 (m, 5H, Ar-H, J = 7.26, 7.30 Hz), 7.95 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 22.86, 39.84, 42.90, 126.69, 128.62, 129.34, 137.13, 155.40, 169.51; MS (70 eV) m/z (%): 216 (M+, 69). Anal. calcd for C12H16N4 (216.28): C, 66.64; H, 7.46; N, 25.90. Found: C, 66.60; H, 7.40; N, 25.88.
4.1.4.2. N-(5-Benzyl-4H-1,2,4-triazol-3-yl)thiazol-2-amine (5b)
White powder; yield 96%, m.p. 220–222 °C; IR (KBr) (υ, cm–1): 3199 (NH), 3056, 3020 (CHarom), 2922, 2850 (CHaliph), 1663 (C=N); 1H NMR (DMSO-d6) δ: 3.84 (s, 2H, CH2-ph), 7.16, 7.25 (2d, 2H, thiazole-H4, H5), 7.30–7.32 (m, 5H, Ar-H, J = 7.17, 7.23, 7.30 Hz), 7.45 (br. s, 1H, NH, D2O exchangeable), 12.31 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 42.07, 114.09, 127.39, 128.95, 129.64, 135.26, 138.04, 158.37, 159.04, 169.86; MS (70 eV) m/z (%): 257 (M+, 18). Anal. calcd for C12H11N5S (257.31): C, 56.01; H, 4.31; N, 27.22. Found: C, 55.97; H, 4.29; N, 26.98.
4.1.4.3. 3-((5-Benzyl-4H-1,2,4-triazol-3-yl)amino)phenol (5c)
Pale brown crystals; yield 86%, m.p. 110–112 °C; IR (KBr) (υ, cm–1): 3328 (OH), 3270, 3145 (NH), 3086, 3027 (CHarom), 2964, 2840 (CHaliph), 1662 (C=N). 1H NMR (DMSO-d6) δ: 3.50 (s, 2H, CH2-ph), 6.62–7.31 (m, 9H, Ar-H, J = 7.37, 7.74 Hz), 8.10 (br. s, 1H, NH, D2O exchangeable), 9.30 (br. s, 1H, NH, D2O exchangeable), 9.60 (br. s, 1H, OH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 38.83, 102.52, 110.41, 115.98, 125.06, 128.07, 130.15, 136.49, 143.80, 156.12, 159.54, 160.08, 165.13; MS (70 eV) m/z (%): 266 (M+, 81). Anal. calcd for C15H14N4O (266.30): C, 67.65.17; H, 5.30; N, 21.04. Found: C, 67.40; H, 5.18; N, 20.98.
4.1.4.4. N-(3-Benzyl-1-phenyl-1H-1,2,4-triazol-5-yl)thiazol-2-amine (6a)
Brown powder, yield 95%, m.p. 170–172 °C; IR (KBr) (υ, cm–1): 3285 (NH), 3084, 3026 (CHarom), 2919 (CHaliph), 1664 (C=N). 1H NMR (DMSO-d6) δ: 3.50 (s, 2H, CH2-ph), 6.67, 7.10 (2d, 2H, thiazole-H4, H5), 7.26–7.74 (m, 10H, Ar-H, J = 7.10, 7.37 Hz), 10.23 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 40.08, 112.49, 126.99, 128.53, 128.71, 128.75, 129.12, 129.48, 129.83, 136.38, 149.75, 170.43; MS (70 eV) m/z (%): 333 (M+, 10). Anal. calcd for C18H15N5S (333.41): C, 64.84; H, 4.53; N, 21.01. Found: C, 64.81; H, 4.50; N, 20.97.
4.1.4.5. 3-((3-Benzyl-1-phenyl-1H-1,2,4-triazol-5-yl)amino)phenol (6b)
White powder, yield 85%, m.p. 158–160 °C; IR (KBr) (υ, cm–1): 3285 (OH), 3206 (NH), 3084, 3027 (CHarom), 2915 (CHaliph), 1664 (C=N); 1H NMR (DMSO-d6) δ: 3.51 (s, 2H, CH2-ph), 6.66–7.73 (m, 14H, Ar-H, J = 6.68, 7.16 Hz), 9.09 (br. s, 1H, NH, D2O exchangeable), 9.90 (br. s, 1H, OH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 40.94, 112.52, 118.98, 127.02, 128.77, 129.15, 129.49, 136.36, 149.73, 170.51; MS (70 eV) m/z (%): 342 (M+, 25). Anal. calcd for C21H18N4O (342.39): C, 73.67; H, 5.30; N, 16.36. Found: C, 73.96; H, 5.15; N, 15.96.
4.1.4.6. 3-Benzyl-N-(2-chloro-4-nitrophenyl)-1-phenyl-1H-1,2,4-triazol-5-amine (6c)
White powder, yield 89%, m.p. 180–182 °C; IR (KBr) (υ, cm–1): 3284 (NH), 3084, 3026 (CHarom), 2915, 2840 (CHaliph), 1664 (C=N); 1H NMR (DMSO-d6) δ: 4.23 (s, 2H, CH2-ph), 6.67–7.60 (m, 10H, Ar-H, J = 8.18 Hz), 8.22–8.38 (d, 2H, Ar-H, J = 7.59 Hz), 9.22 (s, 1H, Ar-H), 9.90 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 40.39, 118.43, 122.26, 124.83, 126.12, 128.04, 136.14, 138.36, 139.25, 151.83, 158.32, 162.15; MS (70 eV) m/z (%): 406 (M+, 49). Anal. calcd for C21H16ClN5O2 (405.84): C, 62.15; H, 3.97; N, 17.26. Found: C, 62.11; H, 3.95; N, 17.24.
4.1.5. Synthesis of 4-Benzyl-6-(thiazol-2-ylamino)-1,3,5-triazin-2(5H)-one (7)
A mixture of 2b (2.77 g; 0.01 mol) and urea (0.60 g; 0.01 mol) in DMF (5 mL) was heated under MW irradiation for 5 min at 130 °C. After cooling to room temperature, the reaction mixture was poured onto ice. The obtained precipitate was filtered and recrystallized from ethanol to give the corresponding compound 7.
Pale brown crystals, yield 97%, m.p. 242–244 °C IR (KBr) (υ, cm–1): 3206 (NH), 3081 (CHarom), 2950, 2885 (CHaliph), 1686 (C=O), 1626 (C=N); 1H NMR (DMSO-d6) δ: 3.77 (s, 2H, CH2-ph), 6.93–7.47 (m, 7H, Ar-H, J = 7.32 Hz), 11.19 (br. s, 1H, NH, D2O exchangeable), 12.35 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 40.13, 113.96, 127.27, 128.88, 129.69, 135.47, 138.10150.41, 158.44, 169.63; MS (70 eV) m/z (%): 285 (M+, 32). Anal. calcd for C13H11N5OS (285.32): C, 54.72; H, 3.89; N, 24.55. Found: C, 54.70; H, 3.86; N, 24.51.
4.1.6. Synthesis 1,3,5-Triazin-2-thione Derivatives 8a–d
An equimolar ratio of 2a, b, d, and e (0.01 mol) and thiourea (0.74 g; 0.01 mol) in DMF (5 mL) was heated in MW for 2–10 min at 130–150 °C. After cooling to room temperature, the reaction mixture was poured onto ice. The obtained precipitate was filtered and recrystallized from ethanol to give corresponding products 8a–d, respectively.
4.1.6.1. 6-Benzyl-4-(isopropylimino)-3,4-dihydro-1,3,5-triazine-2(1H)-thione (8a)
Pale brown crystals, yield 96%, m.p. 260–262 °C; IR (KBr) (υ, cm–1): 3289, 3110 (NH), 3065, 3030 (CHarom), 2975, 2930, 2875 (CHaliph), 1639 (C=N), 1175 (C=S); 1H NMR (DMSO-d6) δ: 1.05 (d, 6H, CH3), 3.36 (m, 1H, CH), 3.82 (s, 2H, ph-CH2), 7.21–7.30 (m, 5H, Ar-H, J = 7.12 Hz), 7.96 (br. s, 1H, NH, D2O exchangeable), 11.35 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 22.85, 40.15, 42.88, 126.71, 128.60, 128.64, 129.27, 129.33, 132.66, 137.11, 169.56; MS (70 eV) m/z (%): 260 (M+, 29). Anal. calcd for C13H16N4S (260.36): C, 59.97; H, 6.19; N, 21.52. Found: C, 59.93; H, 6.15; N, 21.49.
4.1.6.2. 6-Benzyl-4-(thiazol-2-ylimino)-3,4-dihydro-1,3,5-triazine-2(1H)-thione (8b)
Brown crystals, yield 97%, m.p. 151–153 °C; IR (KBr) (υ, cm–1): 3220 (NH), 3072, 3028 (CHarom), 2918, 2862 (CHaliph), 1620 (C=N), 1164 (C=S); 1H NMR (DMSO-d6) δ: 3.77 (s, 2H, CH2-ph), 7.20–7.28 (m, 5H, Ar-H, J = 7.34 Hz), 7.35–7.48 (2d, 2H, thiazole-H4, H5, J = 7.20, 7.27 Hz), 8.33 (br. s, 1H, NH, D2O exchangeable), 12.35 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 40.06, 113.97, 127.28, 128.88, 129.69, 135.47, 138.11, 158.42, 169.62; MS (70 eV) m/z (%): 301 (M+, 42). Anal. calcd for C13H11N5S2 (301.39): C, 51.81; H, 3.68; N, 23.24. Found: C, 51.78; H, 3.79; N, 23.22.
4.1.6.3. 6-Benzyl-4-((3-hydroxyphenyl)imino)-3,4-dihydro-1,3,5-triazine-2(1H)-thione (8c)
Brown crystals, yield 92%, m.p. 147–149 °C. IR (KBr) (υ, cm–1): 3357 (OH), 3216 (NH), 3050, 3022 (CHarom), 2925, 2815 (CHaliph), 1603(C=N), 1177 (C=S); 1H NMR (DMSO-d6) δ: 3.51 (s, 2H, ph-CH2), 6.65–7.76 (m, 9H, Ar-H, J = 6.68, 7.28 Hz), 8.73 (br. s, 1H, NH, D2O exchangeable), 9.09 (br. s, 1H, NH, D2O exchangeable), 9.90 (br. s, 1H, OH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 41.90, 112.20, 113.65, 114.65, 127.40, 128.95, 129.90, 134.74, 139.13, 150.42, 156.90, 170.75; MS (70 eV) m/z (%): 310 (M+, 32). Anal. calcd for C16H14N4OS (310.37): C, 61.92; H, 4.55; N, 18.05. Found: C, 61.89; H, 4.52; N, 18.03.
4.1.6.4. 6-Benzyl-4-((2-chloro-4-nitrophenyl)imino)-3,4-dihydro-1,3,5-triazine-2(1H)-thione (8d)
Yellow powder, yield 98%, m.p. 70–72 °C. IR (KBr) (υ, cm–1): 3198 (NH), 3098, 3066 (CHarom), 2923, 2850 (CHaliph), 1625 (C=N), 1127 (C=S); 1H NMR (DMSO-d6) δ: 3.60 (s, 2H, CH2-ph), 6.66–7.74 (m, 8H, Ar-H, J = 7.01 Hz), 8.73 (br. s, 1H, NH, D2O exchangeable), 9.09 (br. s, 1H, NH, D2O exchangeable); 13C NMR (DMSO-d6) δ: 42.15, 112.28, 113.80, 114.78, 127.80, 128.90, 129.70, 135.04, 138.83, 147.60, 156.75, 169.95 ; MS (70 eV) m/z (%): 374 (M+, 19). Anal. calcd for C16H12ClN5O2S (373.82): C, 51.41; H, 3.24; N, 18.74. Found: C, 51.39; H, 3.21; N, 18.70.
4.2. Biological Activity
4.2.1. In Vitro Cytotoxic Assay (MTT)
The potential cytotoxic properties of the prepared compounds 2–8 were evaluated against a panel of four human cancer cell lines, including hepatocellular carcinoma (HepG-2), prostate carcinoma (PC-3), breast adenocarcinoma (MCF-7), and non-small cell lung cancer cells (A-549), and the normal peripheral blood mononuclear cells (PBMCs) using the MTT assay95 depending on the development of purple formazan crystals by mitochondrial dehydrogenases. More details were provided in Supporting Information.
4.2.2. In Vitro Inhibition Assay of EGFRWT and Mutant EGFRT790M Activities
The compounds that exhibited the most potent cytotoxic activity were further examined for their inhibitory activities against both EGFRWT and EGFRT790M. A homogeneous time resolved fluorescence (HTRF) assay96,97 was applied in this test with EGFRWT and EGFRT790M (Sigma). More details were provided in Supplementary Information.
4.2.3. In Vivo Determination of p53 Ubiquitination
The potential of different prepared derivatives as potent p53 ubiquitination inhibitors was evaluated using the standard procedure and protocol previously applied.98,99 Briefly, cells were allowed to grow for 24 h to reach 50% confluency. Thereafter, of 1 μg p53, 4 μg MDM2 and 1 μg HIS-ubiquitin were transfected with the Gene Juice reagent, and then cells were grown for another 20 h. More details were provided in Supplementary Information.
4.3. Molecular Modeling Study on EGFRWT and Mutant EGFRT790M
The domains of EGFRWT and mutant EGFRT790M kinase complexed with erlotinib and AZD9291 (PDB ID: 1M17 and 6JX0)100,101 were downloaded from the Protein Data Bank. The docking calculations were done using MOE-Dock (Molecular Operating Environment) software version 2014.0901.88,89 More details were provided in the Supplementary Information.
Acknowledgments
The authors are grateful to the Deanship of Scientific Research, King Saud University, for funding through the Vice Deanship of Scientific Research Chairs.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acsomega.1c06836.
Experimental section; chemistry, in vitro cytotoxic assay (MTT), in vitro inhibition assay of EGFRWT and mutant EGFRT790M activities, in vivo determination of p53 ubiquitination, molecular modeling study on EGFRWT and mutant EGFRT790M, 1H-NMR spectra of the new compounds, 13C-NMR spectra of the new compounds, IR spectra of the new compounds, and mass spectra of the new compounds (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Ewes W. A.; Elmorsy M. A.; El-Messery S. M.; Nasr M. N. A. Synthesis, biological evaluation and molecular modeling study of [1,2,4]-Triazolo[4,3-c] quinazolines: New class of EGFR-TK inhibitors. Bioorg. Med. Chem. 2020, 28, 115373. 10.1016/j.bmc.2020.115373. [DOI] [PubMed] [Google Scholar]
- Shafei A.; El-Bakly W.; Sobhy A.; Wagdy O.; Reda A.; Aboelenin O.; Marzouk A.; El Habak K.; Mostafa R.; Ali M. A.; Ellithy M. A review on the efficacy and toxicity of different doxorubicin nanoparticles for targeted therapy in metastatic breast cancer. Biomed. Pharmacother. 2017, 95, 1209–1218. 10.1016/j.biopha.2017.09.059. [DOI] [PubMed] [Google Scholar]
- Hassan G. S.; Georgey H. H.; Mohammed E. Z.; George R. F.; Mahmoud W. R.; Omar F. A. Mechanistic selectivity investigation and 2D-QSAR study of some new antiproliferative pyrazoles and pyrazolopyridines as potential CDK2 inhibitors. Eur. J. Med. Chem. 2021, 218, 113389. 10.1016/j.ejmech.2021.113389. [DOI] [PubMed] [Google Scholar]
- Lemmon M. A.; Schlessinger J. Cell signaling by receptor tyrosine kinases. Cell 2010, 141, 1117–1134. 10.1016/j.cell.2010.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim M.; Baek M.; Kim D. J. Protein Tyrosine signaling and its potential therapeutic implications in carcinogenesis. Curr. Pharm. Des. 2017, 23, 4226–4246. 10.2174/1381612823666170616082125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bansal R.; Malhotra A. Therapeutic progression of quinazolines as targeted chemotherapeutic agents. Eur. J. Med. Chem. 2021, 211, 113016. 10.1016/j.ejmech.2020.113016. [DOI] [PubMed] [Google Scholar]
- Mokhtar A. M.; El-Messery S. M.; Ghaly M. A.; Hassan G. S. Targeting EGFR tyrosine kinase: Synthesis, in vitro antitumor evaluation, and molecular modeling studies of benzothiazole-based derivatives. Bioorg. Chem. 2020, 104, 104259. 10.1016/j.bioorg.2020.104259. [DOI] [PubMed] [Google Scholar]
- Zhang H.; Berezov A.; Wang Q.; Zhang G.; Drebin J.; Murali R.; Greene M. I. ErbB receptors: from oncogenes to targeted cancer therapies. J. Clin. Invest. 2007, 117, 2051–2058. 10.1172/JCI32278. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Othman I. M. M.; Alamshany Z. M.; Tashkandi N. Y.; Gad-Elkareem M. A. M.; Anwar M. M.; Nossier E. S. New pyrimidine and pyrazole-based compounds as potential EGFR inhibitors: synthesis, anticancer, antimicrobial evaluation and computational studies. Bioorg. Chem. 2021, 114, 105078. 10.1016/j.bioorg.2021.105078. [DOI] [PubMed] [Google Scholar]
- Ahmed M. F.; Santali E. Y.; El-Deen E. M. M.; Naguib I. A.; El-Haggar R. Development of Pyridazine Derivatives as Potential EGFR inhibitors and Apoptosis Inducers: Design, Synthesis, Anticancer Evaluation, and Molecular Modeling Studies. Bioorg. Chem. 2021, 106, 104473. 10.1016/j.bioorg.2020.104473. [DOI] [PubMed] [Google Scholar]
- Capdevila J.; Elez E.; Macarulla T.; Ramos F. J.; Ruiz-Echarri M.; Tabernero J. Anti-epidermal growth factor receptor monoclonal antibodies in cancer treatment. Cancer Treat. Rev. 2009, 35, 354–363. 10.1016/j.ctrv.2009.02.001. [DOI] [PubMed] [Google Scholar]
- Abd El-Meguid E. A.; Moustafa G. O.; Awad H. M.; Zaki E. R.; Nossier E. S. Novel benzothiazole hybrids targeting EGFR: Design, synthesis, biological evaluation and molecular docking studies. J. Mol. Struct. 2021, 1240, 130595. 10.1016/j.molstruc.2021.130595. [DOI] [Google Scholar]
- Seshacharyulu P.; Ponnusamy M. P.; Haridas D.; Jain M.; Ganti A. K.; Batra S. K. Targeting the EGFR signaling pathway in cancer therapy. Expert Opin. Ther. Targets 2012, 16, 15–31. 10.1517/14728222.2011.648617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kannaiyan R.; Mahadevan D. A comprehensive review of protein kinase inhibitors for cancer therapy. Expert Rev. Anticancer Ther. 2018, 18, 1249–1270. 10.1080/14737140.2018.1527688. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Petrelli A.; Giordano S. From single- to multi-target drugs in cancer therapy: When a specificity becomes an advantage. Curr. Med. Chem. 2008, 15, 422–432. 10.2174/092986708783503212. [DOI] [PubMed] [Google Scholar]
- Hawata M. A.; El-Sayed W. A.; Nossier E. S.; Abdel-Rahman A. A. H. Synthesis and Cytotoxic Activity of New Pyrimido [1, 2-c] quinazolines,[1, 2, 4] triazolo [4, 3-c] quinazolines and (quinazolin-4-yl)-1H-pyrazoles Hybrids. Biointerface Res. Appl. Chem. 2022, 12, 5217–5233. [Google Scholar]
- Xiao Z.; Zhou Z.; Chu C.; Zhang Q.; Zhou L.; Yang Z.; Li X.; Yu L.; Zheng P.; Xu S.; Zhu W. Design, synthesis and antitumor activity of novel thiophene-pyrimidine derivatives as EGFR inhibitors overcoming T790M and L858R/T790M mutations. Eur. J. Med. Chem. 2020, 203, 112511. 10.1016/j.ejmech.2020.112511. [DOI] [PubMed] [Google Scholar]
- Barker A. J.; Gibson K. H.; Grundy W.; Godfrey A. A.; Barlow J. J.; Healy M. P.; Woodburn J. R.; Ashton S. E.; Curry B. J.; Scarlett L.; Henthorn L.; Richards L. Studies leading to the identification of ZD1839 (Iressa): an orally active, selective epidermal growth factor receptor tyrosine kinase inhibitor targeted to the treatment of cancer. Bioorg. Med. Chem. Lett. 2001, 11, 1911–1914. 10.1016/S0960-894X(01)00344-4. [DOI] [PubMed] [Google Scholar]
- Khattab R. R.; Alshamari A. K.; Hassan A. A.; Elganzory H. H.; El-Sayed W. A.; Awad H. M.; Nossier E. S.; Hassan N. A. Click chemistry-based synthesis, cytotoxic activity and molecular docking of novel triazole-thienopyrimidine hybrid glycosides targeting EGFR. J. Enzyme Inhib. Med. Chem. 2021, 36, 504–516. 10.1080/14756366.2020.1871335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pao W.; Miller V. A.; Politi K. A.; Riely G. J.; Somwar R.; Zakowski M. F.; Kris M. G.; Varmus H. Acquired resistance of lung adenocarcinomas to gefitinib or erlotinib is associated with a second mutation in the EGFR kinase domain. PLoS Med. 2005, 2, 225–235. 10.1371/journal.pmed.0020073. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ko B.; Paucar D.; Halmos B. EGFR T790M: Revealing the secrets of a gatekeeper. Lung Cancer 2017, 8, 147–159. 10.2147/LCTT.S117944. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. A.; Arcila M. E.; Rekhtman N.; Sima C. S.; Zakowski M. F.; Pao W.; Kris M. G.; Miller V. A.; Ladanyi M.; Riely G. J. Analysis of tumor specimens at the time of acquired resistance to EGFR-TKI therapy in 155 patients with EGFR-mutant lung cancers. Clin. Cancer Res. 2013, 19, 2240–2247. 10.1158/1078-0432.CCR-12-2246. [DOI] [PMC free article] [PubMed] [Google Scholar]
- El-Serwy W. S.; Mohamed N. A.; El-Serwy W. S.; Nossier E. S.; Mahmoud K. Synthesis, molecular modeling studies and biological evaluation of novel pyrazole derivatives as antitumor and EGFR inhibitors. Int. J. Pharm. Technol. 2016, 8, 25192–25209. [Google Scholar]
- Xiao Q.; Qu R.; Gao D.; Yan Q.; Tong L.; Zhang W.; Ding J.; Xie H.; Li Y. Discovery of 5-(methylthio) pyrimidine derivatives as L858R/T790M mutant selective Epidermal Growth Factor Receptor (EGFR) inhibitors. Bioorg. Med. Chem. 2016, 24, 2673–2680. 10.1016/j.bmc.2016.04.032. [DOI] [PubMed] [Google Scholar]
- Blair J. A.; Rauh D.; Kung C.; Yun C. H.; Fan Q. W.; Rode H.; Zhang C.; Eck M. J.; Weiss W. A.; Shokat K. M. Structure-guided development of affinity probes for tyrosine kinases using chemical genetics. Nat. Chem. Biol. 2007, 229–238. 10.1038/nchembio866. [DOI] [PubMed] [Google Scholar]
- Zhou W.; Liu X.; Tu Z.; Zhang L.; Ku X.; Bai F.; Zhao Z.; Xu Y.; Ding K.; Li H. Discovery of pteridin-7(8H)-one-based irreversible inhibitors targeting the epidermal growth factor receptor (EGFR) kinase T790M/L858R mutant. J. Med. Chem. 2013, 56, 7821–7837. 10.1021/jm401045n. [DOI] [PubMed] [Google Scholar]
- Li J.; An B.; Song X.; Zhang Q.; Chen C.; Wei S.; Fan R.; Li X.; Zou Y. Design, synthesis and biological evaluation of novel 2,4-diaryl pyrimidine derivatives as selective EGFRL858R/T790M inhibitors. Eur. J. Med. Chem. 2021, 212, 113019. 10.1016/j.ejmech.2020.113019. [DOI] [PubMed] [Google Scholar]
- Giaccone G.; Wang Y. Strategies for overcoming resistance to EGFR family tyrosine kinase inhibitors. Cancer Treat. Rev. 2011, 37, 456–464. 10.1016/j.ctrv.2011.01.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou W.; Ercan D.; Chen L.; Yun C. H.; Li D.; Capelletti M.; Cortot A. B.; Chirieac L.; Iacob R. E.; Padera R.; Engen J. R.; Wong K. K.; Eck M. J.; Gray N. S.; Jänne P. A. Novel mutant-selective EGFR kinase inhibitors against EGFR T790M. Nature 2009, 462, 1070–1074. 10.1038/nature08622. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ward R. A.; Anderton M. J.; Ashton S.; Bethel P. A.; Box M.; Butterworth S.; Colclough N.; Chorley C. G.; Chuaqui C.; Cross D. A. E.; Dakin L. A.; Debreczeni J. É.; Eberlein C.; Finlay M. R. V.; Hill G. B.; Grist M.; Klinowska T. C. M.; Lane C.; Martin S.; Orme J. P.; Smith P.; Wang F.; Waring M. J. Structure-and reactivity-based development of covalent inhibitors of the activating and gatekeeper mutant forms of the epidermal growth factor receptor (EGFR). J. Med. Chem. 2013, 56, 7025–7048. 10.1021/jm400822z. [DOI] [PubMed] [Google Scholar]
- Kim E. S. Olmutinib: first global approval. Drugs 2016, 76, 1153–1157. 10.1007/s40265-016-0606-z. [DOI] [PubMed] [Google Scholar]
- Song J.; Jang S.; Lee J. W.; Jung D.; Lee S.; Min K. H. Click chemistry for improvement in selectivity of quinazoline-based kinase inhibitors for mutant epidermal growth factor receptors. Bioorg. Med. Chem. Lett. 2019, 29, 477–480. 10.1016/j.bmcl.2018.12.020. [DOI] [PubMed] [Google Scholar]
- Wang Z.; Ye W.; Qin Y.; You H.; Zhang S.; Fan F.; Wang Y.; Zheng L. Development and validation of a UPLC–MS/MS method for quantification of C-005, a novel third-generation EGFR TKI, and its major metabolite in plasma: Application to its first-in-patient study. J. Chromatogr., B 2021, 1162, 122475. 10.1016/j.jchromb.2020.122475. [DOI] [PubMed] [Google Scholar]
- Sequist L. V.; Soria J. C.; Goldman J. W.; Wakelee H. A.; Gadgeel S. M.; Varga A.; Papadimitrakopoulou V.; Solomon B. J.; Oxnard G. R.; Dziadziuszko R.; Aisner D. L.; Doebele R. C.; Galasso C.; Garon E. B.; Heist R. S.; Logan J.; Neal J. W.; Mendenhall M. A.; Nichols S.; Piotrowska Z.; Wozniak A. J.; Raponi M.; Karlovich C. A.; Jaw-Tsai S.; Isaacson J.; Despain D.; Matheny S. L.; Rolfe L.; Allen A. R.; Camidge D. R. Rociletinib in EGFR-mutated non–small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1700–1709. 10.1056/NEJMoa1413654. [DOI] [PubMed] [Google Scholar]
- Cross D. A.; Ashton S. E.; Ghiorghiu S.; Eberlein C.; Nebhan C. A.; Spitzler P. J.; Orme J. P.; Finlay M. R. V.; Ward R. A.; Mellor M. J.; Hughes G.; Rahi A.; Jacobs V. N.; Brewer M. R.; Ichihara E.; Sun J.; Jin H.; Ballard P.; al-Kadhimi K.; Rowlinson R.; Klinowska T.; Richmond G. H. P.; Cantarini M.; Kim D. W.; Ranson M. R.; Pao W. AZD9291, an irreversible EGFR TKI, overcomes T790M-mediated resistance to EGFR inhibitors in lung cancer. Cancer Discovery 2014, 4, 1046–1061. 10.1158/2159-8290.CD-14-0337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cui J. J. A New Challenging and Promising Era of Tyrosine Kinase Inhibitors. ACS Med. Chem. Lett. 2014, 5, 272–274. 10.1021/ml500091p. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou; Chen G.; Gao M.; Wu J. Design, synthesis and evaluation of the osimertinib analogue (C-005) as potent EGFR inhibitor against NSCLC. Bioorg. Med. Chem. 2018, 26, 6135–6145. 10.1016/j.bmc.2018.10.018. [DOI] [PubMed] [Google Scholar]
- Jänne P. A.; Yang J. C. H.; Kim D. W.; Planchard D.; Ohe Y.; Ramalingam S. S.; Ahn M. J.; Kim S. W.; Su W. C.; Horn L.; Haggstrom D.; Felip E.; Kim J. H.; Frewer P.; Cantarini M.; Brown K. H.; Dickinson P. A.; Ghiorghiu S.; Ranson M. AZD9291 in EGFR inhibitor-resistant non-small-cell lung cancer. N. Engl. J. Med. 2015, 372, 1689–1699. 10.1056/NEJMoa1411817. [DOI] [PubMed] [Google Scholar]
- Moreno L. M.; Quiroga J.; Abonia R.; Lauria A.; Martorana A.; Insuasty H.; Insuasty B. Synthesis, biological evaluation, and in silico studies of novel chalcone- and pyrazoline-based 1,3,5-triazines as potential anticancer agents. RSC Adv. 2020, 10, 34114. 10.1039/D0RA06799G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martins P.; Jesus J.; Santos S.; Raposo L. R.; Roma-Rodrigues C.; Baptista P. V.; Fernandes A. R. Heterocyclic Anticancer Compounds: Recent Advances and the Paradigm Shift towards the Use of Nanomedicine’s Tool Box. Molecules 2015, 20, 16852–16891. 10.3390/molecules200916852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Facchetti G.; Rimoldi I. Anticancer platinum (II) complexes bearing N-heterocycle rings. Bioorg. Med. Chem. Lett. 2019, 29, 1257–1263. 10.1016/j.bmcl.2019.03.045. [DOI] [PubMed] [Google Scholar]
- Roskoski R. Jr. Properties of FDA-approved small molecule protein kinase inhibitors. Pharmacol. Res. 2019, 144, 19–50. 10.1016/j.phrs.2019.03.006. [DOI] [PubMed] [Google Scholar]
- Liu H.-B.; Gao W.-W.; Tangadanchu V. K. R.; Zhou C.-H.; Geng R.-X. Novel aminopyrimidinyl benzimidazoles as potentially antimicrobial agents: Design, synthesis and biological evaluation. Eur. J. Med. Chem. 2018, 143, 66–84. 10.1016/j.ejmech.2017.11.027. [DOI] [PubMed] [Google Scholar]
- Alp M.; Göker H.; Brun R.; Yıldız S. Synthesis and antiparasitic and antifungal evaluation of 2′-arylsubstituted-1H,1′H-[2,5′]bisbenzimidazolyl-5-carboxamidines. Eur. J. Med. Chem. 2009, 44, 2002–2008. 10.1016/j.ejmech.2008.10.003. [DOI] [PubMed] [Google Scholar]
- Hu Z.; Ou L.; Li S.; Yang L. Synthesis and biological evaluation of 1-cyano-2-amino benzimidazole derivatives as a novel class of antitumor agents. Med. Chem. Res. 2014, 23, 3029–3038. 10.1007/s00044-013-0888-6. [DOI] [Google Scholar]
- Akhtar M. J.; Siddiqui A. A.; Khan A. A.; Ali Z.; Dewangan R. P.; Pasha S.; Yar M. S. Design, synthesis, docking and QSAR study of substituted benzimidazole linked oxadiazole as cytotoxic agents, EGFR and erbB2 receptor inhibitors. Eur. J. Med. Chem. 2017, 126, 853–869. 10.1016/j.ejmech.2016.12.014. [DOI] [PubMed] [Google Scholar]
- Demirel S.; Kilcigil G. A.; Kara Z.; Güven B.; Beşikci A. O. Synthesis and Pharmacologic Evaluation of Some Benzimidazole Acetohydrazide Derivatives as EGFR Inhibitors. Turk. J. Pharm. Sci. 2017, 285–289. 10.4274/tjps.24008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cheong J. E.; Zaffagni M.; Chung I.; Xu Y.; Wang Y.; Jernigan F. E.; Zetter B. R.; Sun L. Synthesis and anticancer activity of novel water soluble benzimidazole carbamates. Eur. J. Med. Chem. 2018, 144, 372–385. 10.1016/j.ejmech.2017.11.037. [DOI] [PubMed] [Google Scholar]
- Ayhan-Kilcigil G.; Kuş C.; Çoban T.; Özdamar E. D.; Can-Eke B. Identification of a Novel Series of N -Phenyl-5-[(2-phenylbenzimidazol-1-yl)methyl]-1,3,4-oxadiazol-2-amines as Potent Antioxidants and Radical Scavengers. Arch. Pharm. 2014, 347, 276–282. 10.1002/ardp.201300324. [DOI] [PubMed] [Google Scholar]
- Celik İ.; Ayhan-Kılcıgil G.; Guven B.; Kara Z.; Gurkan-Alp A. S.; Karayel A.; Onay-Besikci A. Design, synthesis and docking studies of benzimidazole derivatives as potential EGFR inhibitors. Eur. J. Med. Chem. 2019, 173, 240–249. 10.1016/j.ejmech.2019.04.012. [DOI] [PubMed] [Google Scholar]
- Kaur P.; Chawla A. 1,2,4-triazole: a review of pharmacological activities. Int. Res. J. Pharm. 2017, 8, 10–29. 10.7897/2230-8407.087112. [DOI] [Google Scholar]
- Rose C.; Vtoraya O.; Pluzanska A.; Davidson N.; Gershanovich M.; Thomas R.; Johnson S.; Caicedo J. J.; Gervasio H.; Manikhas G.; Ayed F. B.; Burdette-Radoux S.; Chaudri-Ross H. A.; Lang R. An open randomized trial of second-line endocrine therapy in advanced breast cancer: comparison of the aromatase inhibitors letrozole and anastrozole. Eur. J. Cancer 2003, 39, 2318–2327. 10.1016/S0959-8049(03)00630-0. [DOI] [PubMed] [Google Scholar]
- Dahmani R.; Manachou M.; Belaidi S.; Chtita S.; Boughdiri S. Structural characterization and QSAR modeling of 1,2,4-triazole derivatives as α-glucosidase inhibitors. New J. Chem. 2021, 45, 1253–1261. 10.1039/D0NJ05298A. [DOI] [Google Scholar]
- Lønning P.; Pfister C.; Martoni A.; Zamagni C. Pharmacokinetics of third-generation aromatase inhibitors. Semin. Oncol. 2003, 30, 23–32. 10.1016/S0093-7754(03)00305-1. [DOI] [PubMed] [Google Scholar]
- Lønning P. E.; Geisler J.; Dowsett M. Pharmacological and clinical profile of anastrozole. Breast Cancer Res. Treat. 1998, 49, S53–S57. 10.1023/A:1006000806630. [DOI] [PubMed] [Google Scholar]
- Njar V. C. O.; Brodie A. M. H. Comprehensive pharmacology and clinical efficacy of aromatase inhibitors. Drugs 1999, 58, 233–255. 10.2165/00003495-199958020-00003. [DOI] [PubMed] [Google Scholar]
- Goss P. E. Pre-clinical and clinical review of vorozole, a new third generation aromatase inhibitor. Cancer Res. Treat. 1998, 49, S59–S65. 10.1023/A:1006052923468. [DOI] [PubMed] [Google Scholar]
- El-Faham A.; Farooq M.; Almarhoon Z.; Abd Alhameed R.; Wadaan M. A. M.; de la Torre B. G.; Albericio F. Di-and tri-substituted s-triazine derivatives: Synthesis, characterization, anticancer activity in human breast-cancer cell lines, and developmental toxicity in zebrafish embryos. Bioorg. Chem. 2020, 94, 103397. 10.1016/j.bioorg.2019.103397. [DOI] [PubMed] [Google Scholar]
- Cascioferro S.; Parrino B.; Spanò V.; Carbone A.; Montalbano A.; Barraja P.; Diana P.; Cirrincione G. 1,3,5-Triazines: A promising scaffold for anticancer drugs development. Eur. J. Med. Chem. 2017, 142, 523–549. 10.1016/j.ejmech.2017.09.035. [DOI] [PubMed] [Google Scholar]
- Nie Z.; Perretta C.; Erickson P.; Margosiak S.; Lu J.; Averill A.; Almassy R.; Chu S. Structure-based design and synthesis of novel macrocyclic pyrazolo[1,5-a] [1,3,5] triazine compounds as potent inhibitors of protein kinase CK2 and their anticancer activities. Bioorg. Med. Chem. Lett. 2008, 18, 619–623. 10.1016/j.bmcl.2007.11.074. [DOI] [PubMed] [Google Scholar]
- Zhang B.; Zhang Q.; Xiao Z.; Sun X.; Yang Z.; Gu Q.; Liu Z.; Xie T.; Jin Q.; Zheng P.; Xu S.; Zhu W. Design, synthesis and biological evaluation of substituted 2-(thiophen-2-yl)-1,3,5-triazine derivatives as potential dual PI3Kα/mTOR inhibitors. Bioorg. Chem. 2020, 95, 103525. 10.1016/j.bioorg.2019.103525. [DOI] [PubMed] [Google Scholar]
- Lolak N.; Akocak S.; Bua S.; Sanku R. K. K.; Supuran C. T. Discovery of new ureido benzenesulfonamides incorporating 1,3,5-triazine moieties as carbonic anhydrase I, II, IX and XII inhibitors. Bioorg. Med. Chem. 2019, 27, 1588–1594. 10.1016/j.bmc.2019.03.001. [DOI] [PubMed] [Google Scholar]
- Havránková E.; Csöllei J.; Vullo D.; Garaj V.; Pazdera P.; Supuran C. T. Novel sulfonamide incorporating piperazine, aminoalcohol and 1,3,5-triazine structural motifs with carbonic anhydrase I, II and IX inhibitory action. Bioorg. Chem. 2018, 77, 25–37. 10.1016/j.bioorg.2017.12.034. [DOI] [PubMed] [Google Scholar]
- Żołnowska B.; Sławiński J.; Szafrański K.; Angeli A.; Supuran C. T.; Kawiak A.; Wieczór M.; Zielińska J.; Bączek T.; Bartoszewska S. Novel 2-(2-arylmethylthio-4-chloro-5-methylbenzenesulfonyl)-1-(1,3,5-triazin-2-ylamino) guanidine derivatives: Inhibition of human carbonic anhydrase cytosolic isozymes I and II and the transmembrane tumor-associated isozymes IX and XII, anticancer activity, and molecular modeling studies. Eur. J. Med. Chem. 2018, 143, 1931–1941. 10.1016/j.ejmech.2017.11.005. [DOI] [PubMed] [Google Scholar]
- Hashem H. E.; Amr A. E.-G. E.; Nossier E. S.; Elsayed E. A.; Azmy E. M. Synthesis, antimicrobial activity and molecular docking of novel thiourea derivatives tagged with thiadiazole, imidazole and triazine moieties as potential DNA gyrase and topoisomerase IV inhibitors. Molecules 2020, 25, 2766. 10.3390/molecules25122766. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou X.; Lin K.; Ma X.; Chui W. K.; Zhou W. Design, synthesis, docking studies and biological evaluation of novel dihydro-1,3,5-triazines as human DHFR inhibitors. Eur. J. Med. Chem. 2017, 125, 1279–1288. 10.1016/j.ejmech.2016.11.010. [DOI] [PubMed] [Google Scholar]
- Narva S.; Chitti S.; Amaroju S.; Bhattacharjee D.; Rao B. B.; Jain N.; Alvala M.; Sekhar K. V. G. C. Design and synthesis of 4-morpholino-6-(1,2,3,6-tetrahydropyridin-4-yl)-N-(3,4,5-trimethoxyphenyl)-1,3,5-triazin-2-amine analogues as tubulin polymerization inhibitors. Bioorg. Med. Chem. Lett. 2017, 27, 3794–3801. 10.1016/j.bmcl.2017.06.060. [DOI] [PubMed] [Google Scholar]
- Wong J. R.; Morton L. M.; Tucker M. A.; Abramson D. H.; Seddon J. M.; Sampson J. N.; Kleinerman R. A. Risk of subsequent malignant neoplasms in long-term hereditary retinoblastoma survivors after chemotherapy and radiotherapy. Am. J. Clin. Oncol. 2014, 32, 3284. 10.1200/JCO.2013.54.7844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dehnhardt C. M.; Venkatesan A. M.; Chen Z.; Delos-Santos E.; Ayral-Kaloustian S.; Brooijmans N.; Yu K.; Hollander I.; Feldberg L.; Lucas J.; Mallon R. Identification of 2-oxatriazines as highly potent pan-PI3K/mTOR dual inhibitors. Bioorg. Med. Chem. Lett. 2011, 21, 4773–4778. 10.1016/j.bmcl.2011.06.063. [DOI] [PubMed] [Google Scholar]
- Koh M.; Lee J. C.; Min C.; Moon A. A novel metformin derivative, HL010183, inhibits proliferation and invasion of triple-negative breast cancer cells. Bioorg. Med. Chem. 2013, 21, 2305–2313. 10.1016/j.bmc.2013.02.015. [DOI] [PubMed] [Google Scholar]
- Kim E. S. Enasidenib: first global approval. Drugs. 2017, 77, 1705–1711. 10.1007/s40265-017-0813-2. [DOI] [PubMed] [Google Scholar]
- Keldsen N.; Havsteen H.; Vergote I.; Bertelsen K.; Jakobsen A. Altretamine (hexamethylmelamine) in the treatment of platinum-resistant ovarian cancer: a phase II study. Gynecol. Oncol. 2003, 88, 118–122. 10.1016/S0090-8258(02)00103-8. [DOI] [PubMed] [Google Scholar]
- Guo H.; Diao Q. P. 1, 3, 5-Triazine-azole Hybrids and their Anticancer Activity. Curr. Top. Med. Chem. 2020, 20, 1481–1492. 10.2174/1568026620666200310122741. [DOI] [PubMed] [Google Scholar]
- Daştan A.; Kulkarni A.; Török B. Environmentally benign synthesis of heterocyclic compounds by combined microwave-assisted heterogeneous catalytic approaches. Green Chem. 2012, 14, 17–37. 10.1039/C1GC15837F. [DOI] [Google Scholar]
- Kaur N.; Kishore D. Microwave-Assisted Synthesis of Six-Membered O-Heterocycles. Synth. Commun. 2014, 44, 3047–3081. 10.1080/00397911.2013.796383. [DOI] [Google Scholar]
- Kaur N. Microwave-Assisted Synthesis of Five-Membered O-Heterocycles. Synth. Commun. 2014, 44, 3483–3508. 10.1080/00397911.2013.800213. [DOI] [Google Scholar]
- Diaz-Ortiz A.; Moreno A.; Sanchez-Migallon A.; Prieto P.; Carrillo J. R.; Vazquez E.; Gomez M.; Herrero M. A. Microwave-assisted reactions in heterocyclic compounds with applications in medicinal and supramolecular chemistry. Comb. Chem. High Throughput Screening 2007, 10, 877–902. [DOI] [PubMed] [Google Scholar]
- De la Hoz A.; Loupy A. Eds. Microwaves in Organic Synthesis; 3rd ed.; Wiley-VCH: Weinheim, Germany, 2012, 127–207. [Google Scholar]
- Amr A. E.-G. E.; Mageid R. E. A.; El-Naggar M.; Naglah A. M.; Nossier E. S.; Elsayed E. A. Chiral Pyridine-3, 5-bis-(L-phenylalaninyl-L-leucinyl) Schiff Base Peptides as Potential Anticancer Agents: Design, Synthesis, and Molecular Docking Studies Targeting Lactate Dehydrogenase-A. Molecules 2020, 25, 1096. 10.3390/molecules25051096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abd El-Meguid E. A.; El-Deen E. M. M.; Nael M. A.; Anwar M. M. Novel benzimidazole derivatives as anti-cervical cancer agents of potential multi-targeting kinase inhibitory activity. Arab. J. Chem. 2020, 13, 9179–9195. 10.1016/j.arabjc.2020.10.041. [DOI] [Google Scholar]
- Srour A. M.; Ahmed N. S.; Abd El-Karim S. S.; Anwar M. M.; El-Hallouty S. M. Design, synthesis, biological evaluation, QSAR analysis and molecular modelling of new thiazol-benzimidazoles as EGFR inhibitors. Bioorg. Med. Chem. 2020, 28, 115657. 10.1016/j.bmc.2020.115657. [DOI] [PubMed] [Google Scholar]
- Othman I. M. M.; Gad-Elkareem M. A. M.; Amr A. E.-G. E.; Al-Omar M. A.; Nossier E. S.; Elsayed E. A. Novel heterocyclic hybrids of pyrazole targeting dihydrofolate reductase: design, biological evaluation and in silico studies. J. Enzyme Inhib. Med. Chem. 2020, 35, 1491–1502. 10.1080/14756366.2020.1791842. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dawood D. H.; Nossier E. S.; Ali M. M.; Mahmoud A. E. Synthesis and molecular docking study of new pyrazole derivatives as potent anti-breast cancer agents targeting VEGFR-2 kinase. Bioorg. Chem. 2020, 101, 103916. 10.1016/j.bioorg.2020.103916. [DOI] [PubMed] [Google Scholar]
- Anwar M. M.; Abd El-Karim S. S.; Mahmoud A. H.; Amr A. E.-G. E.; Al-Omar M. A. A Comparative Study of the Anticancer Activity and PARP-1 Inhibiting Effect of Benzofuran–Pyrazole Scaffold and Its Nano-Sized Particles in Human Breast Cancer Cells. Molecules 2019, 24, 2413. 10.3390/molecules24132413. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang J.; Yang P. L.; Gray N. S. Targeting cancer with small molecule kinase inhibitors. Nat. Rev. Cancer. 2009, 9, 28–39. 10.1038/nrc2559. [DOI] [PubMed] [Google Scholar]
- Khattab R. R.; Hassan A. A.; Osman D. A. A.; Abdel-Megeid F. M.; Awad H. M.; Nossier E. S.; El-Sayed W. A. Synthesis, anticancer activity and molecular docking of new triazolo [4, 5-d] pyrimidines based thienopyrimidine system and their derived N-glycosides and thioglycosides. Nucleos Nucleot Nucl. 2021, 40, 1090–1113. 10.1080/15257770.2021.1975297. [DOI] [PubMed] [Google Scholar]
- Mowafy S.; Galanis A.; Doctor Z. M.; Paranal R. M.; Lasheen D. S.; Farag N. A.; Jänne P. A.; Abouzid K. A. M. Toward discovery of mutant EGFR inhibitors; Design, synthesis and in vitro biological evaluation of potent 4-arylamino-6-ureido and thioureido-quinazoline derivatives. Bioorg. Med. Chem. 2016, 24, 3501–3512. 10.1016/j.bmc.2016.05.063. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Wu H.; Wang L.; Liu Y.; Knapp S.; Liu Q.; Gray N. S. Exploration of type II binding mode: a privileged approach for kinase inhibitor focused drug discovery ?. ACS Chem. Biol. 2014, 9, 1230–1241. 10.1021/cb500129t. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Furet P.; Caravatti G.; Lydon N.; Priestle J. P.; Sowadski J. M.; Trinks U.; Traxler P. Modelling study of protein kinase inhibitors: binding mode of staurosporine and origin of the selectivity of CGP 52411. J. Comput.-Aided Mol. Des. 1995, 9, 465–472. 10.1007/BF00124317. [DOI] [PubMed] [Google Scholar]
- Liu Y.; Gray N. S. Rational design of inhibitors that bind to inactive kinase conformations. Nat. Chem. Biol. 2006, 2, 358–364. 10.1038/nchembio799. [DOI] [PubMed] [Google Scholar]
- Abou Elmagd W. S. I.; Hemdan M. M.; Samy S. S.; Youssef A. S. A. Thiosemicarbazide Derivatives as Building Block in Synthesis of Target Heterocyclic Compounds with Their Antimicrobial Assessment. J. Heterocycl. Chem. 2017, 54, 1391–1395. 10.1002/jhet.2719. [DOI] [Google Scholar]
- Hemdan M. M.; El-Sayed A. A. E. Use of Phthalimidoacetyl Isothiocyanate as a Scaffold in the Synthesis of Target Heterocyclic Systems, and Their Antimicrobial Assessment. Chem. Pharm. Bull. 2016, 64, 483–489. 10.1248/cpb.c16-00099. [DOI] [PubMed] [Google Scholar]
- El-Sayed A. A.; Atta-allah S. R.; Hemdan M. M. Utility of 3- ( thiophen-2-yl ) prop-2-enoyl isothiocyanate in heterocyclic synthesis. J. Chem. Res. 2019, 43, 1–8. 10.1177/1747519819862176. [DOI] [Google Scholar]
- Hemdan M. M.; Abou Elmagd W. S.; Samy S. S.; Youssef A. S. Dodecanoyl thiosemicarbazide derivatives as useful synthons in the synthesis of 1, 2, 4-triazole, 1, 3, 4-thiadiazole, and 1, 3-benzothiazole derivatives. Synth. Commun. 2016, 46, 710–718. 10.1080/00397911.2016.1164865. [DOI] [Google Scholar]
- Nossier E. S.; El-Hallouty S. M.; Zaki E. R. Synthesis, anticancer evaluation and molecular modeling of some substituted thiazolidinonyl and thiazolyl pyrazole derivatives. Int. J. Pharm. Pharm. Sci. 2015, 7, 353–359. [Google Scholar]
- Kassem A. F.; Moustafa G. O.; Nossier E. S.; Khalaf H. S.; Mounier M. M.; Al-Yousef S. A.; Mahmoud S. Y. In-vitro anticancer potentiality and molecular modelling study of novel amino acid derivatives based on N1, N3-bis-(1-hydrazinyl-1-oxopropan-2-yl) isophthalamide. J. Enzyme Inhib. Med. Chem. 2019, 34, 1247–1258. 10.1080/14756366.2019.1613390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jia Y.; Quinn C. M.; Gagnon A. I.; Talanian R. Homogeneous time-resolved fluorescence and its applications for kinase assays in drug discovery. Anal. Biochem. 2006, 356, 273–281. 10.1016/j.ab.2006.05.006. [DOI] [PubMed] [Google Scholar]
- El-Sayed A. A.; Nossier E. S.; Almehizia A. A.; Amr A. E.-G. E. Design, synthesis, anticancer evaluation and molecular docking study of novel 2, 4-dichlorophenoxymethyl-based derivatives linked to nitrogenous heterocyclic ring systems as potential CDK-2 inhibitors. J. Mol. Struct. 2022, 1247, 131285. 10.1016/j.molstruc.2021.131285. [DOI] [Google Scholar]
- Amr A. E.-G. E.; Elsayed E. A.; Al-Omar M. A.; Badr Eldin H. O.; Nossier E. S.; Abdallah M. M. Design, synthesis, anticancer evaluation and molecular modeling of novel estrogen derivatives. Molecules 2019, 24, 416. 10.3390/molecules24030416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nossier E. S.; Abd El-Karim S. S.; Khalifa N. M.; El-Sayed A. S.; Hassan E. S. I.; El-Hallouty S. M. Kinase inhibitory activities and molecular docking of a novel series of anticancer pyrazole derivatives. Molecules 2018, 23, 3074. 10.3390/molecules23123074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yan X. E.; Ayaz P.; Zhu S. J.; Zhao P.; Liang L.; Zhang C. H.; Wu Y. C.; Li J. L.; Choi H. G.; Huang X.; Shan Y.; Shaw D. E.; Yun C. H. Structural basis of AZD9291 selectivity for EGFR T790M. J. Med. Chem. 2020, 63, 8502–8511. 10.1021/acs.jmedchem.0c00891. [DOI] [PubMed] [Google Scholar]
- Mohi El-Deen E. M.; Abd El-Meguid E. A.; Hasabelnaby S.; Karam E. A.; Nossier E. S. Synthesis, docking studies, and in vitro evaluation of some novel thienopyridines and fused thienopyridine–quinolines as antibacterial agents and DNA gyrase inhibitors. Molecules 2019, 24, 3650. 10.3390/molecules24203650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Abd El-Karim S. S.; Mohamed H. S.; Abdelhameed M. F.; Amr A. E.-G. E.; Almehizia A. A.; Nossier E. S. Design, synthesis and molecular docking of new pyrazole-thiazolidinones as potent anti-inflammatory and analgesic agents with TNF-α inhibitory activity. Bioorg. Chem. 2021, 111, 104827. 10.1016/j.bioorg.2021.104827. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.