

Abstract
Various adenosine receptor nucleoside-like ligands were found to modulate ATP hydrolysis by the multidrug transporter ABCG2. Both ribose-containing and rigidified (N)-methanocarba nucleosides (C2-, N6- and 5’-modified), as well as adenines (C2-, N6-, and deaza modified), were included. 57 compounds out of 63 tested either stimulated (50) or inhibited (7) basal ATPase activity. Structure-activity analysis showed a separation of adenosine receptor and ABCG2 activities. The 7-deaza modification had favorable effects in both (N)-methanocarba nucleosides and adenines. Adenine 37c (MRS7608) and (N)-methanocarba 7-deaza-5’-ethyl ester 60 (MRS7343) were found to be potent stimulators of ABCG2 ATPase activity with EC50 values of 13.2 ± 1.7 and 13.2 ± 2.2 nM, respectively. Both had affinity in the micromolar range for A3 adenosine receptor and lacked the 5’-amide agonist-enabling group (37c was reported as a weak A3 antagonist, Ki 6.82 μM). Compound 60 significantly inhibited ABCG2 substrate transport (IC50 0.44 μM). Docking simulations predicted the interaction of 60 with 21 residues in the drug-binding pocket of ABCG2.
Keywords: ABC transporter, ABCG2, A3 adenosine receptor, ATP hydrolysis, drug transport, multidrug resistance
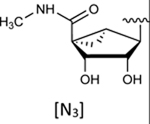
Graphical Abstract
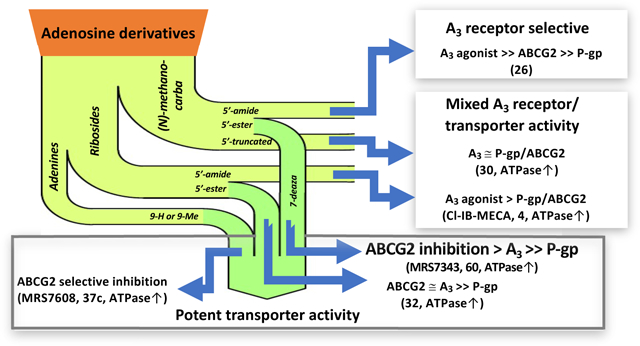
1. Introduction
Cancer patients sometimes face the difficulty of continuing therapy due to the development of resistance to chemotherapeutic drugs. One of the main causes of multidrug resistance (MDR) is the efflux action of ATP-binding cassette (ABC) transporters that are expressed on the surface of cancer cells. The transporters pump anti-cancer drugs out of cells using the energy of ATP hydrolysis [1, 2]. Three main ABC transporters contribute to the development of MDR: ABCB1 (P-glycoprotein, P-gp), ABCG2, and ABCC1 [3]. Many chemically dissimilar anticancer drugs are substrates of these transporters. Also, P-gp and ABCG2 are expressed by normal cells, such as epithelial cells found in the intestine, kidney, liver, and placenta, and by endothelial cells of the blood-brain barrier in order to export toxic compounds from the body [1, 2, 4].
To understand the mechanism of ABC transporters and find ways to overcome MDR, the structures of ABC transporters have been thoroughly analyzed, especially those of P-gp and ABCG2. ABC transporters have a transmembrane domain (TMD) that contains substrate-binding sites, typically consisting of 12 transmembrane helices [5]. Other important structural features are the nucleotide-binding domains (NBDs), which contain the conserved Walker A and Walker B motifs, and a signature region upstream of the Walker B site [5]. The structure allowing communication between the TMD and the NBD is called the intracellular domain. Human P-gp is a full transporter with six TMDs and two NBDs [2, 6, 7], whereas ABCG2 is a half-transporter that needs to form a homodimer to be functional [8, 9]. Despite extensive structural analysis, the mechanism of the transport function of ABC transporters is not yet well understood.
Understanding the interaction between drug substrates and ABC transporters is considered important to determine the mechanism of transport function [10]. Recently, we investigated whether A3 adenosine receptor agonists can interact with ABC transporters. A3 adenosine receptor agonists have been developed for the treatment of chronic diseases such as rheumatoid arthritis, psoriasis, chronic pain, and hepatocellular carcinoma [11–14]. In one clinical study, an agonist that is highly potent and selective for the A3 receptor demonstrated efflux in a CACO-2 cell bidirectional transport model [12], suggesting a possible interaction with ABC transporters in the intestinal epithelium. In our previous study, we synthesized new nucleoside derivatives of A3 adenosine ligands designed for P-gp interactions, indicated by a structure-activity relationship (SAR) analysis, and investigated the interaction of P-gp with some derivatives [15]. Those results suggested that A3 adenosine receptor ligands modulate the function of P-gp.
In this study, we investigated whether A3 adenosine receptor ligands, both adenine nucleoside and nucleobase derivatives, interact with ABCG2, another important ABC transporter, which modulates the absorption of drugs in the intestine. The interaction of A3 receptor ligands with ABCG2 either as substrates or modulators can affect their bioavailability. We performed an analysis of the effect of these A3 adenosine receptor ligands on the ATPase activity of ABCG2, with a total of 63 compounds. Of these, compound 60, a 7-deaza-5’-ester derivative with low adenosine receptor affinity, was identified as a high-affinity ligand for ABCG2. This compound was also found to significantly inhibit substrate transport by ABCG2. Furthermore, we used docking simulations to study how compound 60 binds to the ABCG2 drug-binding site. Adenine derivative 37c was also a potent transport inhibitor. In this way, we were able to identify certain A3 adenosine receptor ligands as potential modulators of human ABCG2. These results could be useful for further study of the interactions between ABCG2 and drug substrates.
2. Results
2.1. SAR studies measuring ATPase activity reveal potential A3 adenosine ligands as ABCG2 modulators
We investigated the interaction of A3 adenosine ligands with the human multidrug transporter ABCG2 by determining their effect on the ATPase activity of the transporter. In this work, 63 compounds were tested at various concentrations (e.g., 100 nM, 1.0, 2.5, and 10 μM) using total membranes (membrane vesicles isolated after hypotonic lysis) from High-Five insect cells expressing ABCG2. Membranes of intracellular organelles and the plasma membrane are included. All adenosine ligands were tested for their ability to either stimulate or inhibit the ATPase activity of ABCG2. We tested this because most compounds that inhibit basal ATPase activity are most likely able to also inhibit its transport function. In addition, we reported all the results obtained in this section as a percentage of the ATPase activity of ABCG2, with values below or equal to 30% considered as having no effect on ATPase activity, which suggests no interaction with ABCG2.
In order to study their structure-activity relationships (SARs), A3 adenosine receptor ligands were grouped into four structural categories: 1) N6 and C2-phenylethynyl modifications of (N)-methanocarba 2-arylethynyl-purine derivative (5’-amides and 5’-alcohols) (Table 1), 2) N6, C2 and riboside modifications of adenosine derivatives, including both (N)-methanocarba and ribose-containing ligands (Table 2), 3) N6 and C2 modifications of adenine derivatives (Table 3), and 4) N6 and C2 modification of (N)-methanocarba 5’-ester derivatives (Table 4). The (N)-methanocarba substitution of the ribose moiety is a rigid, fused ring system (bicyclo[3.1.0]hexane) that is intended to constrain this moiety conformationally, to enhance affinity and selectivity at the A3 adenosine receptor [12–14]. Unlike the 5’-amide agonists, adenine derivatives interact with adenosine receptors as antagonists [16].
Table 1.
Effects of N6 and C2-phenylethynyl modifications of (N)-methanocarba 2-arylethynyl-purine derivatives (5’-amides and 5’-alcohols) on the ATPase activity of human ABCG2
| ||||
|---|---|---|---|---|
| No. | R1 or R3 | R2 | ATPase activity of ABCG2 % Stimulation or inhibition | |
| 2.5 μMb | 10 μMb | |||
| 6-Benzylamino and 6-Alkyl derivatives | ||||
| 5 |
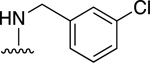
|
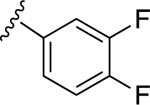
|
74.4±5.2 ↑ @ 0.5 μM | 56.8±9.9 ↑ @ 5 μM |
| 6 | H |
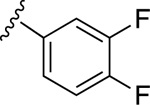
|
NE | 64.2±11.5 ↑ |
| 7 | CH3 |
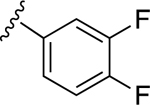
|
49.6±9.0 ↑ | 72.6±15.1 ↑ |
| 8 |
|
|
51.3±19.1 ↑ | NE |
| 9 |
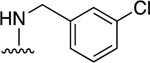
|
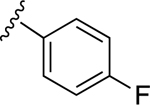
|
60.3±8.4 ↑ | 49.3±15.7 ↑ |
| 10 |
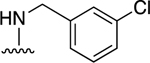
|
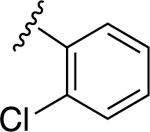
|
49.3±12.8 ↑ | NE |
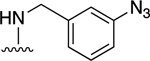
| 11 |
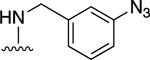
|
|
55.5±11.9 ↑ @ 0.5 μM | 78.3±8.7 ↑ @ 5 μM |
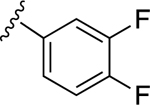
| 12 |
|
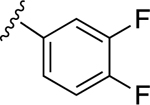
|
61.9±9.8 ↑ | NE |
| 6-Phenylcyclopropylamino derivatives | ||||
| 13 |
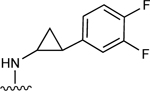
|
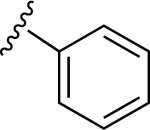
|
110.6±12.1 ↑ | 46.7±5.9 ↑ |
| 14 |
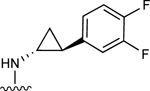
|
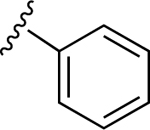
|
74.8±1.9 ↑ | ND |
| 15 |
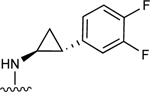
|
|
100.7±26.0 ↑ | ND |
| Triazole-Extended 6-Benzylamino derivatives | ||||
| 16 |
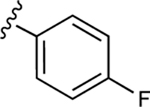
|
|
54.4±6.1 ↓ | 74.7±6.3 ↓ |
| 17 |
|
|
43.5±5.0 ↓ | 76.9±6.4 ↓ |
| 18 |
|
|
44.7±7.2 ↓ | 49.4±0.9 ↓ |
| 19 |
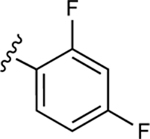
|
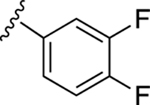
|
NE | 57.1±6.3↓ |
| 20 |
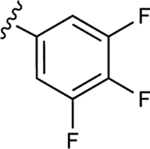
|
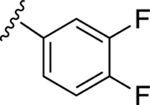
|
38.9±0.5 ↓ | 48.9±4.7 ↓ |
| 21 |
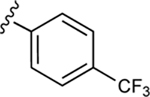
|
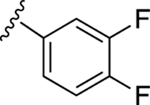
|
NE | 44.5±8.5 ↓ |
| 22 |
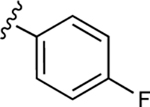
|
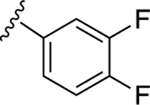
|
NE | NE |
| 23 |
|
|
NE | NE |
Modulation of the basal ATPase activity of ABCG2 by purine analogs showing percentage stimulation or inhibition. The values shown in the table indicate stimulation (upward arrow) or inhibition (downward arrow) of basal ATPase activity. All the values represent mean ± SD of three independent experiments and basal ATPase activity in this study is considered as 0%. ATPase activities were determined as described in Materials and Methods. ATPase assays were performed on total membranes prepared from High-Five insect cells expressing human ABCG2. Membranes (5 μg protein/100 μL) were incubated with indicated concentrations of adenosine derivatives, in the presence and absence of sodium orthovanadate (0.3 mM), in ATPase assay buffer as previously described [33]. The effects on the basal ATPase activity below or equal to 30% are indicated as NE, no effect. ND indicates not determined.
Concentrations were 2.5 and 10 μM, unless otherwise indicated.
Table 2.
Effect of N6, C2 and riboside modifications of adenosine derivatives (both (N)-methanocarba and ribose-containing) on the ATPase activity of ABCG2
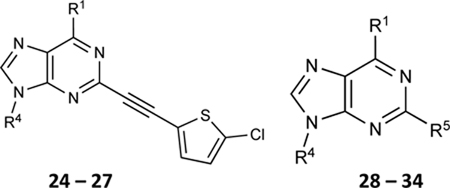
| ||||
|---|---|---|---|---|
| No. | R1 | R4 [R5] | ATPase activity of ABCG2 % Stimulation or inhibition | |
| 2.5 μMb | 10 μMb | |||
| (N)-Methanocarba derivatives | ||||
| 24 | H |
|
NE | NE |
| 25 | NHCH3 |
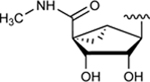
|
59.2±5.9 ↑ | NE |
| 26 | NH(CH2)2CH3 |
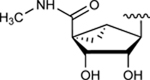
|
77.9±3.3 ↑ | 40.0±7.3 ↑ |
| 27 | NHCH3 |
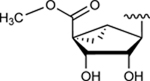
|
89.1±2.1 ↑ | 79.0±8.1 ↑ |
| 28 |
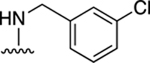
|
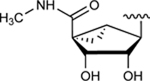
|
NE | 36.6±0.9 ↓ |
| 29 |
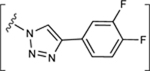
|
|
97.5±23.1 ↑ | 92.9±15.1 ↑ |
| 30 |
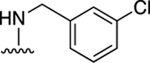
|
|
75.2±13.8 ↑ | 54.1±8.6 ↑ |
| Ribose derivatives | ||||
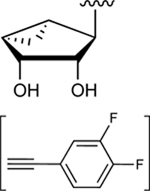
| 31 |
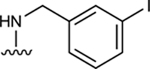
|
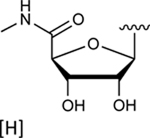
|
66.2±7.3 ↑ | 98.2±5.4 ↑ |
| 4 |
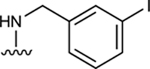
|
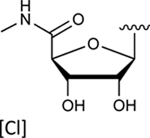
|
100.8±5.8 ↑ | 69.9±6.0 ↑ |
| 32 | NHCH3 |
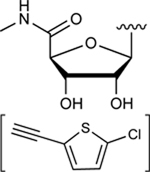
|
111.9±16.8 ↑ | 121.7±10.5 ↑ |
| 33 | NH(CH2)2CH3 |
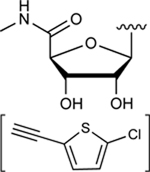
|
124.5±3.4 ↑ | 121.7±21.2 ↑ |
| 34 | NHCH3 |
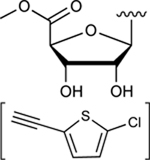
|
NE @ 100 nM | 52.2±8.7↑ @ 1 μM |
Modulation of the basal ATPase activity of ABCG2 by A3 adenosine receptor ligands showing percentage stimulation or inhibition. Details are the same as given in the legend to Table 1.
Concentrations were 2.5 and 10 μM, unless otherwise indicated.
Table 3.
Effect of N6 and C2 modifications of adenine and 7-deazaadenine derivatives on the ATPase activity of ABCG2. R4 is either H or CH3
| ||||
|---|---|---|---|---|
| No. | R1 | R2 or R5 | ATPase activity of ABCG2 % Stimulation or inhibition | |
| 100 nMc | 1 μMc | |||
| R4 = H | ||||
| 35 |
|
|
165.2±10.1↑ @ 2.5 μM | 163.5±4.9↑@ 10 μM |
| 36 |
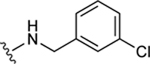
|
|
40.5±3.8 ↑ | 63.7±2.3 ↑ |
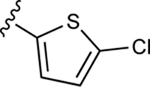
| 37a |
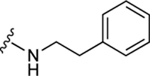
|
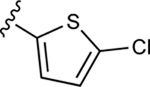
|
80.8±7.9 ↑ | 133.1±22.9 ↑ |
| 37b |
|
|
95.9±8.1 ↑ | 110.5±6.7 ↑ |
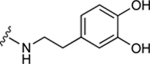
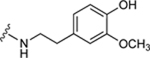
| 37c |
|
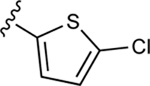
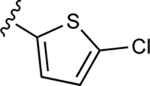
|
111.2±13.2 ↑ | 40.0±7.3 ↑ |
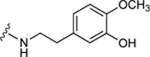
| 38 |
|
| | 79.6±9.6 ↑ | 142.4±7.3 ↑ |
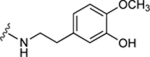
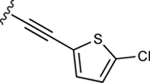
| 39 |
|
|
104.1±12.5 ↑ | 158.8±4.1 ↑ |
| R4 = CH3 | ||||
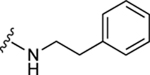
| 40 |
|
|
92.1±13.9 ↑ | 108.3±1.4 ↑ |
Modulation of the basal ATPase activity of ABCG2 by A3 adenosine receptor ligands showing percentage stimulation. Details are the same as given in the legend to Table 1.
Concentrations were 100 nM and 1 μM, unless otherwise indicated.
Table 4.
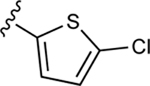
Effect of N6 and C2 modifications of (N)-methanocarba 5’-ester derivatives on the ATPase activity of ABCG2. R2 is 2-chloro-thien-5-yl, unless noted. X, Y, Z = N, unless otherwise noted
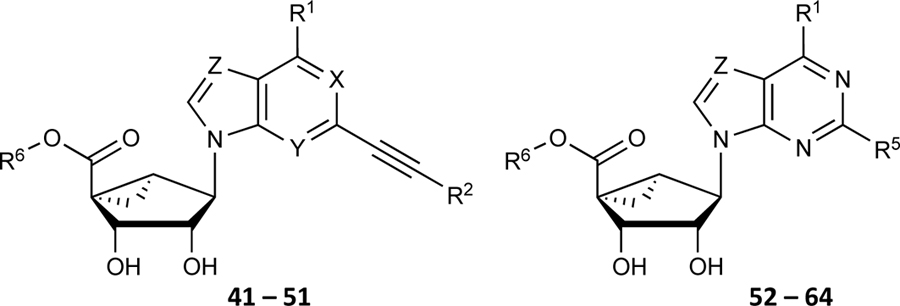
| ||||
|---|---|---|---|---|
| No. | R1 | R6 (and other) | ATPase activity of ABCG2 % Stimulation or Inhibition | |
| 100 nM | 1 μM | |||
| X, Y, Z as indicated | ||||
| 41 | NHCH3 | CH2CH3 | NE | 92.9±12.4 ↑ |
| 42 | NHCH3 | CH2CH3
(X = CH) |
NE | 50.3±6.1 ↑ |
| 43 | NHCH3 | CH2CH3
(Y = CH) |
NE | 62.2±7.5 ↑ |
| 44 | NHCH3 | CH2CH3
(Z = CH) |
41.0±11.2 ↑ | 90.3±2.2 ↑ |
| R6 as indicated, X, Y, Z = N | ||||
| 45 | NHCH3 | (CH2)2CH3 | 33.9±4.7 ↑ | 83.5±12.6 ↑ |
| 46 | NHCH3 | CH(CH3)2 | NE | 51.7±6.9 ↑ |
| 47 | NHCH3 | (CH2)3CH3 | NE | 92.0±7.9 ↑ |
| 48 | NHCH3 | (CH2)2-CH(CH3)2 | NE | 50.9±3.1 ↑ |
| 49 | NHCH3 | (CH2)2-c-Hex | NE | 50.8±9.6 ↑ |
| 50 | NHCH3 | CH2Ph | 50.3±0.8 ↑ | 105.2±10.3 ↑ |
| 51 | NHCH3 | (CH2)2Ph | NE | 64.6±6.9 ↑ |
| 52 | NHCH3 | (CH2)3Ph | 38.8±1.3 ↑ | 61.6±3.8 ↑ |
| R6 = CH2CH3, X, Y, Z = N | ||||
| 53 | NHCH3 |
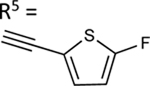
|
NE | 54.5±1.7 ↑ |
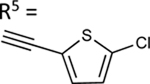
| 54 |
|
|
64.3±13.2 ↑ | 96.4±5.6 ↑ |
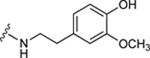
| 55 |
|
|
59.3±2.9 ↑ | NE |
| R6 = CH2CH3, Z = CH | ||||
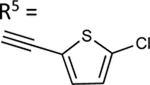
| 56 | NHCH3 | R5 = I | NE | NE |
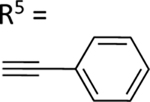
| 57 | NHCH3 |
|
NE | NE |
| 58 |
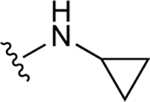
|
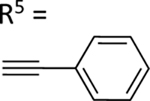
|
NE | 73.0±10.0 ↑ |
| 59 |
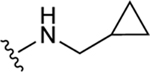
|
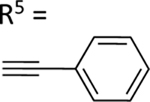
|
NE | 79.8±3.1 ↑ |
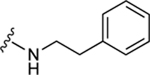
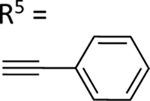
| 60 |
|
|
77.3±10.9 ↑ | 104.5±10.1 ↑ |
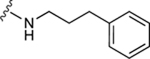
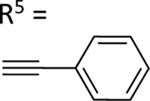
| 61 |
|
|
37.9±5.1 ↑ | 82.8±11.1 ↑ |
| 62 |
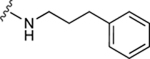
|
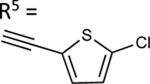
|
45.5±10.4 ↑ | 64.3±11.5 ↑ |
| 63 |
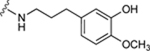
|
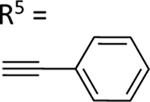
|
55.9±10.7 ↑ | 84.9±11.3 ↑ |
| 64 |
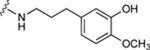
|
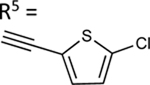
|
74.2±9.1 ↑ | 79.7±7.5 ↑ |
Modulation of the basal ATPase activity of ABCG2 by A3 adenosine analogs showing percentage stimulation. Details are the same as given in the legend to Table 1.
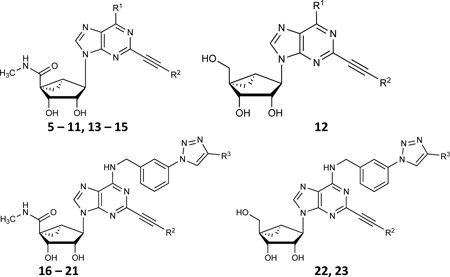
The synthetic routes to the newly prepared compounds are similar to our published nucleoside synthesis [12–20] and are shown in: Scheme 1A (16, 18 – 21, prepared by a click reaction [19] of a nucleoside azide with an arylacetylene), Scheme 1B (12, 22 and 23, were prepared by sequential nucleophilic substitution reaction with 3-azido-benzyl amine, followed by Sonogashira coupling with C2-arylethynyl group acidic hydrolysis of the isopropylidene group and a click reaction of the nucleoside azide derivatives with an arylacetylene). Scheme 2 (61 – 64, prepared from compound 70 [17] by nucleophilic amination with 3-phenylpropan-1-amine derivatives to give compounds 71–72 with good yield, which upon Sonogashira coupling (73 – 76) and subsequent acid hydrolysis afforded the desired nucleoside derivatives (61 – 64). Scheme S1, Supporting Information (38, 39, prepared by sequential nucleophilic substitution reaction with an amine, installation of a C2-arylethynyl group).
Scheme 1.

Synthesis of (N)-methanocarba nucleoside derivatives 12, 16 and 18 – 23. A) Reagents and conditions: (i) phenylacetylene, sodium ascorbate, CuSO4.5H2O, t-BuOH-H2O, rt, 87–90%. B) (i) 3-azidobenzylamine, DIPEA, 2-propanol, 87%, (ii) TBAF, THF, rt, 91%; (iii) 3,4-di-F-phenylacetylene, PdCl2(Ph3P)2, CuI, Et3N, DMF, 68%; (iv) 10% TFA, MeOH, 70°C, 89%; (v) phenylacetylene, sodium ascorbate, CuSO4.5H2O, t-BuOH-H2O, rt, 87–88%.
Scheme 2.

Synthesis of 7-deaza (N)-methanocarba nucleoside 5’-esters 61 – 64. Reagents and conditions: (i) Ph(CH2)3NH2, DIPEA, 2-propanol, 69–72%; (ii) arylalkyne, PdCl2(Ph3P)2, CuI, Et3N, DMF, rt, 67–72%; (iii) 10% TFA, MeOH, 70 °C, 88–91%.
First, we tested (N)-methanocarba nucleoside compound 5, which was found in our earlier study to stimulate the ATPase activity of P-gp (ABCB1) [15]. This ligand consists of R1: N6-(3-chlorobenzyl) and R2: 3,4-difluorophenyl [15]. As shown in Table 1, compound 5 was observed to stimulate the basal ATPase activity of ABCG2 at 0.5 and 5 μM concentrations. The extent of the stimulation was determined by testing concentrations ranging from 0.05 to 5 μM, to provide an EC50 value of 0.08 ± 0.01 μM (Figure 1A). We then decided to design derivatives of compound 5 with the goal of improving their affinity towards the transporter. For this reason, compounds 6 and 7 were synthesized by retaining the parent structure of compound 5 but replacing the R1: N6-(3-chlorobenzyl) with a hydrogen atom 6 or methyl group 7. These adenosine ligands were observed to stimulate the ATPase activity of ABCG2 at higher concentrations, and the results indicate that the N6-(3-chlorobenzyl) group substantially increases the affinity of the adenosine receptor ligand toward ABCG2.
Fig. 1.

Modulation of ABCG2 ATPase activity by A3 adenosine receptor ligands. Results for four representative ligands, the potent A3 agonist 5 (A), triazole-extended 6-benzylamino derivative 17 (B), adenine (A3 antagonist) derivative 37c (C), and (N)-methanocarba-7-deazaadenosine 5’-ester derivative 60 (D) are shown. Each experiment was performed at least three times in duplicate. The EC50 and IC50 values were calculated using GraphPad Prism (GraphPad Software, San Diego, CA, USA).
For the next series of compounds, we decided to synthesize methanocarba derivatives with R1 retaining N6-(3-chlorobenzyl) but replacing the R2 group (3,4-difluorophenyl) with a phenyl group 8, a 4-fluorophenyl group 9, and a 2-chlorophenyl group 10. These compounds all stimulated the basal ATPase activity of ABCG2 at a 2.5 μM concentration, confirming our previous observation that N6-(3-chlorobenzyl) helps to generate high affinity substrates of ABCG2. Furthermore, we synthesized compound 11 by retaining the parent structure of 5 but replacing R1 (N6-(3-chlorobenzyl)) with N6-(3-azido-benzyl)), and R2 remained 3,4-difluorophenyl. Our results showed stimulation of ATPase activity of ABCG2 by 11 at low concentrations, similar to compound 5. However, when we modified methanocarba derivative 11 by replacing the 5’-amide with an adenosine-like 5’-hydroxyl group to obtain compound 12, there was no change in the stimulatory effect. To increase the stimulatory effect of compound 5, we synthesized compounds 13, 14, and 15 by replacing the N6-(3-chlorobenzyl) at position R1 with (3,4-difluorophenyl) cyclopropylamino and replacing the initial R2 3,4-difluorophenyl with a phenyl group. These compounds showed stimulation of basal ATPase activity of ABCG2 at concentrations in the nanomolar range, with EC50 values of 16.0 ± 3.0 nM, 13.0 ± 3.0 nM, and 14.0 ± 5.0 nM, respectively (Table 5). Compounds 13, 14 and 15 only differ structurally in terms of their stereochemistry.
Table 5.
EC50 and IC50 values for ABCG2 ATPase activity and inhibitory effect on mitoxantrone efflux by A3 adenosine receptor ligands
| Compounds | ATPase activity | Inhibition of mitoxantrone efflux | |
|---|---|---|---|
| EC50 or IC50 (nM) | % of pcDNA control cells a, at 10 μM of compounds | IC50 (μM)b | |
| 5 | EC50 = 80.0 ± 10.0 | 99.2 ± 12.0 | NCc |
| 13 | EC50 = 16.0 ± 3.0 | 100.0 ± 6.3 | NC |
| 14 | EC50 = 13.0 ± 3.0 | 97.4 ± 12.1 | NC |
| 15 | EC50 = 14.0 ± 5.0 | 97.2 ± 11.9 | NC |
| 16 | IC50 = 2590 ± 40 | 101.7 ± 17.0 | NC |
| 17 | IC50 = 2570 ± 30 | 92.5 ± 13.5 | NC |
| 20 | IC50 = 4010 ± 20 | 105.3 ± 3.0 | NC |
| 35 | EC50 = 50.0 ± 10.0 | 81.4 ± 11.5 | NC |
| 37c | EC50 = 13.2 ± 1.7 | 110.4 ± 15.8 | 4.98 ± 1.69 |
| 60 | EC50 = 13.2 ± 2.2 | 92.6 ± 9.2 | 0.44 ± 0.06 |
| 64 | EC50 = 24.3 ± 4.9 | 109.1 ± 16.4 | 0.60 ± 0.08 |
Fluorescence intensity of pcDNA control cells was taken as a 100% inhibition, and percentage of mitoxantrone efflux inhibition in R-5 cells by 10 μM of A3 adenosine receptor ligands was calculated compared to pcDNA control cells.
Compound concentration that produces 50% inhibition of mitoxantrone efflux in R-5 cells is given as IC50 value.
NC, Not calculated.
In our previous work, we identified triazole derivative 17 as an adenosine receptor ligand that partially inhibited the basal ATPase activity of P-gp [15]. This (N)-methanocarba nucleoside was designed with the same parent structure of compound 5, although at position R1 the compound had an extended 3-substituted benzyl, and at R2, a 3,4-difluorophenyl. For this reason, we decided to select compound 17 and its derivatives 16, 18, 19, 20, and 21 to investigate their effect on the basal ATPase activity of ABCG2 in total membranes of High-Five insect cells. Triazole compounds 16, 17, 18, 19, 20, and 21 all have an extended 3-substituted benzyl at the R1 position, but all differ structurally at the R3 position: 16 (3-fluorophenyl), 17 (3,4-difluorophenyl), 18 (3,5-difluorophenyl), 19 (2,4-difluorophenyl), 20 (2,3,4-trifluorophenyl), and 21 (4-trifluoromethylphenyl). Compounds 16, 17, and 20 all inhibit the basal ATPase activity of ABCG2, with IC50 values of 2590 ± 40 nM, 2570 ± 30 nM, and 4010 ± 20 nM, respectively (Figure S1A, Figure 1B, and Table 5). In addition, compound 18, which has 3,5-difluorophenyl at the R3 position shows partial inhibition of the ATPase activity of ABCG2. Compound 19 with 2,4-difluorophenyl at position R3 has no effect on the ATPase activity at 2.5 μM. Furthermore, we synthesized compound 21 with trifluoromethylphenyl at the R3 position, which exhibited partial inhibition of basal ATPase activity at a 10 μM concentration. To increase the inhibitory effect of compounds 16 and 17, we decided to modify the ligands by replacing the 5’-CONHCH3 on the methanocarba ring with CH2OH to obtain compounds 22 and 23. We speculated that modifying the ring could increase the hydrogen bonding with residues in the drug-binding pocket of ABCG2. However, compounds 22 and 23 had no effect on the basal ATPase activity of ABCG2.
Subsequently, we decided to synthesize additional adenosine ligands with the goal of improving the interaction with ABCG2 by modifying the analogs found to stimulate the ATPase activity. In Table 2 we show the effect of N6, C2 and riboside modifications of adenosine derivatives having either (N)-methanocarba or ribose rings on the ATPase activity of ABCG2. In this SAR study, 6-amino-truncated (N)-methanocarba compound 24, with structural features similar to those of compound 6, was designed by replacing the 3,4-difluorophenyl R2 substituent with 2-chloro-thien-5-yl. This group was added to the structure in an effort to increase the potential ionic bonding, non-covalent sulfur interactions and π-π interactions of the nucleoside derivative with the drug-binding pocket of ABCG2. Compound 24 was observed to have no effect on the basal ATPase activity of ABCG2, indicating no interaction with ABCG2 (see Table 2). Furthermore, compounds 25 and 26 were designed by replacing the R1, initially a hydrogen atom (24), with 25 (methylamino), and 26 (propylamino). The presence of these amino groups at the R1 position increased the affinity of the ligands toward ABCG2 by stimulating the ATPase activity at a 2.5 μM concentration. In addition, we modified compound 25, by replacing 5’-CONHCH3 on the methanocarba ring with a COOMe to obtain compound 27 and observed an increase in affinity of the adenosine ligand towards ABCG2. Compound 28 was later synthesized with structural features similar to those of compound 5, but by using click chemistry we converted the alkyne at position R5 to a 1,2,3-triazole. This structural change at the R5 position of compound 28 was observed to switch the effect of ATPase activity from stimulation (compound 5) to partial inhibition at a 10 μM concentration. Additionally, we designed compound 29 by the introduction of an azide at the R5 position. This resulted in stimulation of the basal ATPase activity of ABCG2 at a 2.5 μM concentration. Compound 30 was then synthesized with structural features like those of compound 5, but with truncation of the 5’-CONHCH3 group at the R4 position on the methanocarba ring system (i.e., replacement with a hydrogen atom). The results in Table 2 indicate that most of the adenosine derivatives stimulate the ATPase activity at lower concentrations, and we can switch the affinity of the ligand towards ABCG2 by replacing the alkyne at position R5 with a 1,2,3-triazole (see for example compound 28).
In the last two decades, Food and Drug Administration guidelines for drug design have recommended the testing of investigational drugs to determine whether they are substrates of the ABCG2 multidrug transporter. For this reason, we tested N6-(3-iodobenzyl) compounds 31 and 4, which are currently in phase 2 and 3 clinical trials [21]. These potent A3 adenosine receptor agonists contain a modified ribose moiety at the R4 position rather than an (N)-methanocarba ring system as in compound 5, and either a hydrogen (31) or chloro (4) at the R5 position rather than a 3,4-difluorophenylethynyl group. These compounds were observed to stimulate the basal ATPase activity of ABCG2, with EC50 values of 70 ± 30 nM (31) and 1420 ± 190 nM (4) (Figures S1B and S1C). To investigate the effect of the ribose group on the interaction of the adenosine ligands with ABCG2, we designed compounds 32, 33, and 34 by replacing the methanocarba ring system of compounds 25, 26, and 27 with a ribose group at position R4. These adenosine derivatives with a ribose-containing group all stimulated the ATPase activity of ABCG2 at lower concentrations, indicating that a methanocarba ring that enforces a North conformation was not required for interaction with this transporter (see Table 2).
To further identify ligands with apparent high affinity based on their ability to stimulate the ATPase activity of ABCG2 at low nanomolar concentrations, we decided to select known adenine derivatives recently reported to have moderate binding affinity toward the human A3 adenosine receptor [16]. These ligands were synthesized based on the N6 and C2 modifications of adenine derivatives, and their effect on the ATPase activity of ABCG2 was determined, and results can be seen in Table 3. All adenine derivatives, which are compounds 35, 36, 37a–c, 38, 39, and 40, were observed to stimulate the basal ATPase activity of ABCG2 at 100 nM and 1 μM concentrations, unless otherwise indicated in the table legend. To investigate the extent of their stimulation, we decided to select compounds 35 and 37c and test whether these ligands can stimulate the ATPase activity of ABCG2 in the nanomolar range. For this reason, we tested concentrations ranging from 1 nM to 250 nM, with EC50 values of 50.0 ± 10.0 nM for 35, and EC50 value of 13.2 ± 1.7 nM for 37c (Figure 1C).
Based on the results obtained concerning the interaction of compound 37c with ABCG2, we then decided to design adenosine ligands with the goal of improving their affinity towards the transporter. Compounds 41, 42, 43, and 44 were all modified by replacing the methyl group at the R6 position of compound 27 with an ethyl group, yielding compound 41. Compounds 42 (1-deaza), 43 (3-deaza), and 44 (7-deaza) are structurally like 41 but have a CH in place of a nitrogen atom on the ring system (see Table 4). As shown in Table 4, compounds 41, 42, and 43 all stimulated the basal ATPase activity of ABCG2 at a 1 μM concentration, although we observed no interaction with the transporter at a 100 nM concentration. In the case of compound 44, we observed partial stimulation of ATPase activity at a 100 nM concentration. Compounds 45 - 52 were all designed by replacing the ethyl group at position R6 of 41 with (CH2)2CH3 (45), CH(CH3)2 (46), (CH2)3CH3 (47), (CH2)2-CH(CH3)2 (48), (CH2)2-c-Hex (49), CH2Ph (50), (CH2)2Ph (51), and (CH2)3Ph (52). Most of these compounds had no effect on the basal ATPase activity of human ABCG2 at a concentration of 100 nM, suggesting that these adenosine ligands have low affinity towards ABCG2. On the other hand, compounds 45, 50, and 52 all showed partial stimulation of the ATPase activity of ABCG2. Moreover, compound 53 was synthesized by replacing the 2-chloro-thien-5-yl at the R5 position of (compound 27) with 2-fluoro-thien-5-yl, and the methyl group at the R6 position with ethyl, in an effort to increase its ABCG2 affinity. However, the results shown in Table 4 indicate that compound 53 had no effect on the ATPase activity of ABCG2 at a 100 nM concentration.
In the previous series of adenosine ligands tested, we identified compound 37c to be a high affinity ligand that stimulated the basal ATPase assay of ABCG2 in the nanomolar range with an EC50 value of 13.2 ± 1.7 nM. To improve on the affinity of ligands toward ABCG2, we decided to structurally design compound 54 by replacing the hydrogen atom at position R4 of 37c with the methanocarba ring system. Compound 54 was observed to stimulate the basal ATPase activity of ABCG2 at both 100 nM and 1 μM concentrations. Afterward, we synthesized compound 55 which is structurally almost identical to 54, differing only by the substitution of the hydroxyl group and methoxy group on the phenyl ring of position R1. We observed stimulation of basal ATPase activity of ABCG2 at 100 nM (see results in Table 4). Furthermore, compounds 56 and 57 were synthesized by replacing the 2-fluoro-thien-5-yl-ethynyl group at position R5 of 44 with iodo in the case of 56, and with a phenyl ethynyl group in 57. These compounds all showed no interaction with the protein at a 100 nM concentration. Compounds 58 and 59 on the other hand, which had structural features similar to those of 57, differing only at position R1, also showed no effect on the ATPase activity of ABCG2.
The aim of structurally modifying these ligands is to identify adenosine analogs which can interact with ABCG2 at the nanomolar range. For this reason, compound 60 was designed by replacing the N6-cyclopropylmethylamino group of compound 59 at position R1 with 2-phenylethylamino. Compound 60 was observed to stimulate the ATPase activity of ABCG2 at both 100 nM and 1 μM, with a percentage stimulation of 77% at a 100 nM concentration (Table 4). We then tested a range of concentrations of 60 from 1 nM to 250 nM and obtained an EC50 value of 13.2 ± 2.2 nM, as shown in the graph in Figure 1D. To improve on the affinity, compound 61 was synthesized by replacing the phenylethylamino group of 60 at position R1 with 3-phenylpropylamino. Subsequently, compound 62 was later designed by replacing the phenyl ring at position R2 of 61 with a 2-chloro-thien-5-yl moiety to increase the ionic bonding or π-π interactions between the adenosine ligand and residues in the drug binding pocket of ABCG2. Compounds 61 and 62 were observed to partially stimulate the ATPase activity of ABCG2 at a 100 nM concentration. In addition, compound 63, which is structurally like 55 but differs by replacing the 2-chloro-thien-5-yl at position R5 of 55 with a phenyl ring and adding an extra carbon atom on the chain of the functional group at position R1 of compound 55. Furthermore, we designed compound 64, which is structurally almost identical to 63 except that the phenyl ring of 63 was replaced with 2-chloro-thien-5-yl at position R2 of 64. From the results shown in Table 4, compound 63 was observed to partially stimulate the activity of ABCG2 at a 100 nM concentration. Compound 64, on the other hand, had a percentage stimulation of 74% at 100 nM, and we later tested the concentration range from 1 nM to 250 nM, with an EC50 value of 24.3 ± 4.9 nM, as shown in Table 5. The SAR results suggest that compounds 37c and 60 are most likely high affinity ligands for ABCG2 based on modulation of ATPase activity at low nanomolar concentrations and could be useful candidates for pharmacological probes of the human A3 adenosine receptor.
2.2. Selected A3 adenosine ligands inhibit Mitoxantrone transport mediated by ABCG2
Based on structural-activity analysis, we selected A3 adenosine receptor ligands that have a high affinity with ABCG2 for substrate transport assays to determine if these compounds affect the transport function of ABCG2. To reveal this, substrate transport assays were performed using flow cytometry. For these assays, R-5 cells expressing ABCG2 and pcDNA control cells were used. Mitoxantrone was used as a fluorescent substrate of ABCG2, as described in Materials and Methods.
As shown in Figure 2A, the fluorescence intensity of R-5 cells (blue) was lower than that of pcDNA cells (red) indicating mitoxantrone was pumped out by ABCG2 transporters expressed on the R-5 cell surface. Adding an A3 adenosine ligand to R-5 incubating cells increased the fluorescence intensity, indicating that the compound inhibited mitoxantrone efflux by ABCG2. All selected A3 adenosine ligands, as shown in Table 5, were able to inhibit mitoxantrone efflux completely (more than 80% inhibition) at a concentration of 10 μM. Of these compounds, compound 60 demonstrated the most significant inhibitory effect, with an IC50 value of 0.44 ± 0.06 μM, as shown in Figures 2A and 2B. Therefore, compound 60 was determined to be a lead compound for further analysis.
Fig. 2.

Compound 60 inhibited mitoxantrone transport mediated by ABCG2. (A) Representative histogram showing the effect of compound 60 on mitoxantrone transport at indicated concentrations. Fluorescence intensity of pcDNA control cells is shown in red and R-5 cells expressing ABCG2 are shown in blue. The reversal effect of compound 60 for R-5 cells at 0.25, 0.5 and 1 μM are shown in orange, light green, and green, respectively. (B) Concentration-dependent inhibition of mitoxantrone efflux by compound 60. Mitoxantrone efflux by R-5 cells in the absence of compound 60 was taken as 100%, and the percentage of efflux at the indicated concentrations of compound 60 was calculated. Data points were plotted as the mean ± S.D. (n = 3). Compound concentration that produces 50% inhibition of mitoxantrone transport in R-5 cells is given as IC50 value, calculated using GraphPad Prism.
2.3. Interaction of compound 60 with residues in the drug-binding pocket of ABCG2
To determine whether the lead A3 adenosine receptor ligand (compound 60) binds to the drug-binding pocket of human ABCG2, docking analysis was done using the cryo-EM structure of a human ABCG2 mutant bound to a substrate (PDBID: 6HCO)[22]. In this structure, a single molecule of estrone-3-sulfate (E1S), an endogenous substrate of ABCG2, is bound in the central cavity of the drug-binding pocket. In our docking simulations we set 26 residues from each monomer in the binding cavity to have flexible side chains and docked the structure of compound 60. We obtained 18 different docking poses with docking scores ranging from −12.9 kcal/mol to −11.9 kcal/mol. In the lowest energy pose, we identified 21 residues within 4.5 Å from the ligand, belonging to both monomers. Among these, eleven were found within 4.5 Å of E1S (Figure 3), suggesting an overlap between the binding of both compound 60 and E1S. The mostly hydrophobic residues in close proximity are predicted to interact favorably with the hydrophobic substituents of 60, including its R1 and R5 groups. No polar interactions were observed. Compound 60 might also bind to the NBDs of ABCG2. However, based on its stimulatory effect on the ATPase activity of ABCG2, that would be very unlikely. This needs to be explored further.
Fig. 3.

Docking of (N)-methanocarba-7-deazaadenosine 5’-ester derivative 60 in the drug-binding pocket of human ABCG2. The cryo-EM structure of a human ABCG2 mutant bound to E1S (PDBID: 6HCO) was used as a template for docking. On the left side, the structure of ABCG2 depicts the two monomers of (G2 and G2’) in cyan and yellow cartoons and the binding site for the compound 60 is boxed. The close-up in the right panel depicts the expanded region showing the lowest energy docking pose for compound 60 and the residues in the drug-binding pocket interacting with it. A total of 21 residues were identified to be within 4.5 Å of compound 60, and eleven of these (highlighted in yellow) were also within 4.5 Å of substrate E1S.
2.4. Comparison of affinity for A3 adenosine and other receptors
The affinities of the analogues at the A3 adenosine receptor, the k-opioid receptor and the TSPO (translocator protein, also known as peripheral benzodiazepine receptor, on the outer mitochondrial membrane) are listed in Tables S1 – S4 (Supporting Information). Many of these receptor binding affinities were previously reported [12–14, 17–20, 23]. SAR analysis showed a separation of adenosine receptor affinity and ABCG2 activity. For example, the human A3 adenosine receptor agonists of single digit nM affinity (4, 5, 8 – 11, 15, 17, 25 – 29, 31 – 33, 45, 50) showed variable effects on ABCG2. Among them: the following compounds stimulated ATP hydrolysis at multiple concentrations: 4, 5, 26, 27, 29, 31 – 33, 45, 50; the following compounds stimulated ATP hydrolysis at 2.5 μM, but not 10 μM: 8, 10, 15 and 25. The following compounds inhibited ATP hydrolysis at both concentrations: 16 – 18 and 20, but 19, 21, and 28 inhibited only at 10 μM. Among a separated subset of compounds that achieved ≥50% ATPase stimulation at 2.5 μM (4, 5, 7 – 9, 11 – 15, 25 – 27, 29 – 33, 35, 37 – 40, 55, 63 and 64) or ≥50% inhibition at 2.5 μM (16), compounds 7, 12 and 30, 35, 37 – 40, 63 and 64 were weak in A3 adenosine receptor binding (Ki ≥99 nM).
Compounds 56 – 64, which had considerable ABCG2 activity, were 5’-ester derivatives that lacked the adenine 7 position nitrogen; thus, they are 7H-pyrrolo[2,3-d]pyrimidin-4-amine derivatives. One of the important results of this SAR study of the ABCG2 transporter is that the 7-deaza and 5’-ethyl ester analogues are preferred. It is noteworthy that a potent stimulator of ATP hydrolysis, 60, a 7-deaza-5’-ester derivative, has low A3 adenosine receptor affinity (Ki 0.483 μM for the human and >10 μM for the mouse homologues) [17]. Compound 60 was inactive at the human A1 and A2A receptors [17]. Furthermore, similar 5’-ester derivatives in the methanocarba series have low efficacy as partial agonists at the A3 adenosine receptor. This compound also bound to the human k-opioid receptor as a weak partial agonist with a binding Ki value of 0.21 μM [17].
The affinities of the adenine derivatives (Table 3) for ABCG2 were greater, in general, than for the adenosine receptor (Table S3, Supporting Information). Compound 37c, a weak adenine antagonist of the A3 receptor (Ki 6.82 μM at the human homologue) [16] was able to potently stimulate ATP hydrolysis, similar to the activity of 60. Among the adenine derivatives studied (Table 3), the 9-H and 9-methyl analogues were comparable in their ABCG2 activity (cf. 37 and 40). The adenine derivatives had highly variable human A3 receptor affinity (Table S3, Supporting Information) but were consistently weak or inactive in other adenosine receptor subtypes [16]. Compound 37c was inactive at the human A1 and A2A receptors. In this adenine series related to 37c, as reported by Yu et al. [16], four analogues were found to be inactive as the mouse A3 receptor. Thus, we found that some of the adenine derivative compounds were more selective for ABCG2 compared to adenosine receptors.
3. Discussion
Due to its involvement in the development of MDR, ABCG2 is one of the most thoroughly studied ABC transporters, similar to P-gp. Recently ABCG2 inhibitors of diverse chemical classes, including repurposed known drugs, have been reported for potential use as anti-cancer agents [24, 25]. To understand the mechanism of the transport function of ABCG2, it is important to understand interactions between the drug-binding site of ABCG2 and drug substrates [26, 27]. We previously found that A3 adenosine derivatives interact with the drug-binding site of P-gp and can be important modulators of P-gp [15]. Therefore, we decided to extend this approach to ABCG2 and examined the functional effects of an even broader range of adenosine and adenine derivatives on the ATPase activity of this transporter. Synthetic nucleosides are recognized as a good source of useful and orally bioavailable pharmaceuticals, with many derivatives approved for treatment of certain diseases. Furthermore, the approach of conformationally constraining the ribose-like moiety by means of a bicyclic methanocarba substitution has been applied to a wide range of drug targets [17, 20, 28, 29].
In this study, we employed a structural-activity relationship approach [16] as a tool to develop a pharmacophore model for the interaction of A3 adenosine receptor ligands with ABCG2. We tested the modulatory effect of the derivatives, a total of 63 compounds, on ABCG2-mediated ATP hydrolysis. Of these compounds, 57 compounds were found to modulate ABCG2 ATP hydrolysis, with 50 compounds stimulating and 7 compounds inhibiting the basal ATPase activity. These results contrast with our previous findings with P-gp, as most of the tested A3 adenosine derivatives showed neither stimulation nor inhibition of basal ATPase activity of that transporter [15]. It seems that the A3 adenosine ligands are more interactive and have greater affinity with ABCG2 than with P-gp.
Among the compounds that stimulate ABCG2 ATPase activity, adenine derivative 37c and 5’-ethyl ester derivative 60 were identified to be high-affinity ligands for ABCG2 with EC50 values of 13.2 ± 1.7 and 13.2 ± 2.2 nM, respectively (Table 5). Several other compounds stimulated ABCG2 ATPase activity at nearly the same level of potency (EC50 values, nM), i.e., adenine derivative 35 (50.0 ± 10.0) and 5’-ethyl ester derivative 64 (24.3 ± 4.9). Compound 60 contained a 2-phenylethyl group at the N6 position, while the corresponding substituent of 64 was a modified 3-phenylpropyl group. N6-(2-Phenyl-cyclopropyl) derivatives 13 – 15 also potently stimulated ATPase activity. Both adenine derivatives 35 and 37c contained the following substitutions: 9-H, N6-arylalkyl and C2-arylethynyl. Figure 4 is a schematic representation of how adenosine derivatives affect the ATPase activity of ABCG2. In summary, several compounds potently stimulated ATPase activity with weak adenosine receptor interaction (e.g., 37c and 60), while other examined nucleoside analogues were potent A3 adenosine receptor agonists and weak ABCG2 and P-gp modulators (e.g., riboside 5’-methylamide 4 and (N)-methanocarba 5’-methylamide derivative 26). Other compounds were relatively potent modulators of both ABCG2 and the A3 adenosine receptor (e.g., riboside 5’-methylamide with a C2-arylethynyl group 32, which was additionally very weak at P-gp). However, the 5’-truncated (N)-methanocarba derivative 30 inhibited the ATPase activity of ABCG2 and was additionally potent at stimulating P-gp ATPase activity (116% increase of ATPase activity at 2.5 μM [15]).
Fig. 4.

Schematic representation of the modulatory effect of tested nucleoside adenines on the ATPase activity of human ABCG2.
Substrate transport assays were performed for selected A3 adenosine receptor ligands, and all tested compounds were able to completely inhibit mitoxantrone transport by ABCG2. Compound 60 had the greatest inhibitory effect on mitoxantrone transport, with an IC50 value of 0.44 ± 0.06 μM (Figure 2 and Table 5). In addition, our docking data suggests that compound 60 can bind in the drug-binding cavity of ABCG2. Furthermore, the many residues within 4.5 Å allow extensive interactions between (N)-methanocarba 5’-ester derivative 60 and ABCG2, which seems to be essential for distinguishing substrates and inhibitors [30]. These results suggest that this lead compound strongly interacts with ABCG2 and plays an important role in the modulation of substrate transport and ATP hydrolysis.
In conclusion, we found that A3 adenosine receptor ligands can exhibit a greater modulatory effect on ABCG2 activity than on P-gp activity. We were able to identify lead compound 60 as a promising candidate modulator of ABCG2. Our results could be useful for the development of highly potent ABCG2 inhibitors.
4. Materials and Methods
4.1. Chemicals
Dimethyl sulfoxide (DMSO), sodium butyrate compounds 31 and 4 and other chemicals, except the specified, fluorescent substrates used in this work, were obtained from Sigma-Aldrich (St. Louis, MO).
4.2. Chemical Synthesis
The adenosine and adenine derivatives (Tables 1 – 4) included many already published A3 adenosine receptor agonists and antagonists (Tables S1 – S4, Supporting Information) [12–20, 24]. Many of the derivatives were based on a (N)-methanocarba substitution of the native ribose moiety, which utilizes a rigid bicyclic ring to fix a North-envelope conformation of the pseudo ribose ring [12, 17, 20]. Also, various substitutions (including truncation) of the N6, 5’ and C2 positions were examined. Finally, adenine derivatives lacking the ribose moiety were also compared. The newly synthesized C2-arylethynyl compounds are: 5’-hydroxy (N)-methanocarba 12, 22 and 23; 5’-methylamide (N)-methanocarba 16, 18 – 21; and 5’-ester-7-deaza (N)-methanocarba 61 – 64 analogues. Table 1 includes C2-arylethynyl (N)-methanocarba analogues (both 5’-methylamide and 5’-hydroxy). Table 2 includes a variety of ribose and (N)-methanocarba analogues, including 4’-truncated compounds. Table 3 represents N6 substituted C2-arylethynyl adenine derivatives (both 9-H and 9-CH3), with two 7-deazaadenines are included (38, 39). Table 4 shows C2-arylethynyl (N)-methanocarba analogues containing 5’-esters. The details of the chemical synthesis of the new compounds and representative NMR and mass spectra results are provided in the Supporting Information.
4.3. Preparation of total membrane vesicles from High-Five insect cells
The membrane vesicles were prepared by hypotonic lysis of High-Five insect cells expressing the human multidrug transporter ABCG2 and differential centrifugation, as previously described [31, 32].
4.4. ATPase activity of ABCG2
Human ABCG2-mediated ATPase activity was measured using methods previously described [33]. Briefly, total membrane vesicles (10 μg protein/tube) were diluted with ATPase 2X buffer (50 mM MES-Tris (pH 6.8), 50 mM KCl, 10 mM MgCl2, 5 mM NaN3, 1 mM EGTA, 1 mM ouabain, and 2 mM DTT). One μL of an adenosine or adenine analog dissolved in dimethyl sulfoxide (DMSO) in the presence and absence of sodium orthovanadate was added to the ATPase assay mixture. DMSO was added as a control sample in this experiment. The mixtures were pre-incubated at 37°C for 2 min before the addition of 5 mM ATP to each test tube to start the reaction. After 20 min the reaction was stopped by addition of SDS, and the inorganic phosphate generated was measured colorimetrically. Three independent experiments in duplicate were carried out and the results are reported as mean ± SE.
4.5. Cell culture
Human embryonic kidney cells (HEK293) transfected with either empty pcDNA vector (named pcDNA cells) or pcDNA3 vector containing full-length ABCG2 (termed R-5 cells) were provided by Dr. Robert W. Robey, Laboratory of Cell Biology, National Cancer Institute. Cells were cultured in Eagle’s Minimum Essential Medium (Corning, New York, NY, USA) supplemented with 10% fetal bovine serum (FBS), 100 units/ml penicillin, 100 μg/ml streptomycin and 2 mg/ml G418 (Invitrogen, Carlsbad, CA, USA), as described previously [34]. High-Five insect cells were obtained from ThermoFisher Scientific (Waltham, MA).
4.6. Cell surface expression of ABCG2 and transport assay
Cell surface expression and the effect of the test compounds on the transport function of ABCG2 and P-gp was determined using flow cytometry [34–36]. The BacMam baculovirus transduction system was used to express P-gp on HeLa cells for transport function assays of P-gp, as described previously [37, 38]. To determine the cell surface expression of ABCG2, R-5 cells were incubated in Iscove’s Modified Dulbecco’s Medium (IMDM) containing 5% FBS with phycoerythrin-labeled 5D3 antibody (Thermo Fisher Scientific, Waltham, MA, USA) at 37°C for 1 hour. After incubation, cells were washed with cold IMDM and resuspended with cold PBS containing 1% bovine serum albumin (BSA). In transport assays, R-5 ABCG2-expressing cells were incubated in IMDM medium containing 5% FBS with fluorescent substrate, mitoxantrone for ABCG2 at a final concentration of 5 μM at 37°C for 45 min. Then the cells were washed with cold IMDM and resuspended with cold PBS containing 1% BSA. The cell surface expression of ABCG2 and the transport was measured by flow cytometry using a FACS CANTO II instrument with FACS Diva software (BD Biosciences, Franklin Lakes, NJ, USA). The data were analyzed using FlowJo software (Tree Star Inc., Ashland, OR USA).
4.7. In silico docking of compounds in the drug-binding pocket of ABCG2
AutoDock Vina [39] software was used to dock compound 60 in the structure of the inward-open conformation of ABCG2 bound to estrone-3-sulfate and 5D3-Fab (PDBID: 6HCO) [22]. The MGLtools software package (Scripps Research Institute, La Jolla, CA, USA) was used to prepare the pdbqt files. In our docking simulations we set 26 residues from each monomer in the biding cavity to have flexible side chains and docked the structure of compound 60. These residues were N393, N398, V401, L405, I409, T413, N424, F431, F432, T435, N436, F439, S440, V442, S443, Y538, L539, T542, I543, V546, F547, M549, I550, L554, L555. The receptor grid centered at x = 155, y = 155, and z = 149, and a box with dimensions 44 Å × 40 Å × 50 Å was used. The exhaustiveness level for both proteins was set at 100 to ensure that the global minimum of the scoring function would be found considering the large box size and the number of flexible residues. Molecular models were analyzed using PyMOL, Version 1.7, Schrödinger, LLC [40, 41] (New York, NY, USA).
Supplementary Material
Highlights.
Interaction of adenosine/adenine derivatives was more widespread with ABCG2 than P-gp
Distinct SAR for ABCG2 ATPase stimulation compared to A3 adenosine receptor affinity
7-Deaza (N)-methanocarba 5’-ester derivative 60 stimulated ATPase with EC50 13.2 nM
2-(5-Cl-thien-2-yl)ethynyl-adenine derivative 37c stimulated ATPase with EC50 13.2 nM
Acknowledgements
We thank George Leiman for editorial assistance. This work was supported by funds from the Intramural Research Program of the National Institutes of Health, National Cancer Institute, Center for Cancer Research [ZIABC010030] and the Molecular Recognition Section, Laboratory of Bioorganic Chemistry, National Institute of Diabetes and Digestive and Kidney Diseases [NIDDK, ZIADK31117]. Dr. Megumi Murakami was supported in part by a Fellowship from the Japan Society for the Promotion of Science. The computational resources of the NIH HPC Biowulf cluster (http://hpc.nih.gov) were used for docking studies. We thank John Lloyd (NIDDK) for mass spectral determinations. We thank Dr. Bryan L. Roth (Univ. North Carolina at Chapel Hill) and National Institute of Mental Health’s Psychoactive Drug Screening Program (Contract # HHSN-271-2008-00025-C) for screening data.
Abbreviations:
- ABC transporters
ATP-binding cassette transporters
- AR
adenosine receptor
- MDR
multidrug resistance
- TMD
transmembrane domain
- NBD
nucleotide-binding domain
Footnotes
Declaration of competing interests
The authors declare no competing financial interests.
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- [1].Gottesman MM, Fojo T, Bates SE, Multidrug resistance in cancer: role of ATP-dependent transporters, Nat Rev Cancer, 2 (2002) 48–58. [DOI] [PubMed] [Google Scholar]
- [2].Ambudkar SV, Dey S, Hrycyna CA, Ramachandra M, Pastan I, Gottesman MM, Biochemical, cellular, and pharmacological aspects of the multidrug transporter, Annu Rev Pharmacol Toxicol, 39 (1999) 361–398. [DOI] [PubMed] [Google Scholar]
- [3].Robey RW, Pluchino KM, Hall MD, Fojo AT, Bates SE, Gottesman MM, Revisiting the role of ABC transporters in multidrug-resistant cancer, Nat Rev Cancer, 18 (2018) 452–464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Thiebaut F, Tsuruo T, Hamada H, Gottesman MM, Pastan I, Willingham MC, Cellular localization of the multidrug-resistance gene product P-glycoprotein in normal human tissues, Proc Natl Acad Sci U S A, 84 (1987) 7735–7738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [5].Dean M, Hamon Y, Chimini G, The human ATP-binding cassette (ABC) transporter superfamily, J Lipid Res, 42 (2001) 1007–1017. [PubMed] [Google Scholar]
- [6].Gottesman MM, Pastan I, Biochemistry of multidrug resistance mediated by the multidrug transporter, Annu Rev Biochem, 62 (1993) 385–427. [DOI] [PubMed] [Google Scholar]
- [7].Kim Y, Chen J, Molecular structure of human P-glycoprotein in the ATP-bound, outward-facing conformation, Science, 359 (2018) 915–919. [DOI] [PubMed] [Google Scholar]
- [8].Lusvarghi S, Robey RW, Gottesman MM, Ambudkar SV, Multidrug transporters: recent insights from cryo-electron microscopy-derived atomic structures and animal models, F1000Res, 9 (2020) 17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [9].Polgar O, Robey RW, Bates SE, ABCG2: structure, function and role in drug response, Expert Opin Drug Metab Toxicol, 4 (2008) 1–15. [DOI] [PubMed] [Google Scholar]
- [10].Chufan EE, Sim HM, Ambudkar SV, Molecular basis of the polyspecificity of P-glycoprotein (ABCB1): recent biochemical and structural studies, Adv Cancer Res, 125 (2015) 71–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [11].Borea PA, Varani K, Vincenzi F, Baraldi PG, Tabrizi MA, Merighi S, Gessi S, The A3 adenosine receptor: history and perspectives, Pharmacol Rev, 67 (2015) 74–102. [DOI] [PubMed] [Google Scholar]
- [12].Tosh DK, Padia J, Salvemini D, Jacobson KA, Efficient, large-scale synthesis and preclinical studies of MRS5698, a highly selective A3 adenosine receptor agonist that protects against chronic neuropathic pain, Purinergic Signal, 11 (2015) 371–387. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [13].Tosh DK, Paoletta S, Chen Z, Moss SM, Gao ZG, Salvemini D, Jacobson KA, Extended N(6) substitution of rigid C2-arylethynyl nucleosides for exploring the role of extracellular loops in ligand recognition at the A3 adenosine receptor, Bioorg Med Chem Lett, 24 (2014) 3302–3306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [14].Tosh DK, Paoletta S, Phan K, Gao ZG, Jacobson KA, Truncated Nucleosides as A(3) Adenosine Receptor Ligands: Combined 2-Arylethynyl and Bicyclohexane Substitutions, ACS Med Chem Lett, 3 (2012) 596–601. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Abel B, Tosh DK, Durell SR, Murakami M, Vahedi S, Jacobson KA, Ambudkar SV, Evidence for the Interaction of A(3) Adenosine Receptor Agonists at the Drug-Binding Site(s) of Human P-glycoprotein (ABCB1), Mol Pharmacol, 96 (2019) 180–192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [16].Yu J, Mannes P, Jung Y-H, Ciancetta A, Bitant A, Lieberman DI, Khaznadar S, Auchampach JA, Gao Z-G, Jacobson KA, Structure activity relationship of 2-arylalkynyl-adenine derivatives as human A3 adenosine receptor antagonists, MedChemComm, 9 (2018) 1920–1932. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [17].Tosh DK, Ciancetta A, Mannes P, Warnick E, Janowsky A, Eshleman AJ, Gizewski E, Brust TF, Bohn LM, Auchampach JA, Gao ZG, Jacobson KA, Repurposing of a Nucleoside Scaffold from Adenosine Receptor Agonists to Opioid Receptor Antagonists, ACS Omega, 3 (2018) 12658–12678. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Tosh DK, Ciancetta A, Warnick E, O’Connor R, Chen Z, Gizewski E, Crane S, Gao ZG, Auchampach JA, Salvemini D, Jacobson KA, Purine (N)-Methanocarba Nucleoside Derivatives Lacking an Exocyclic Amine as Selective A3 Adenosine Receptor Agonists, J Med Chem, 59 (2016) 3249–3263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [19].Tosh DK, Paoletta S, Chen Z, Crane S, Lloyd J, Gao ZG, Gizewski ET, Auchampach JA, Salvemini D, Jacobson KA, Structure-Based Design, Synthesis by Click Chemistry and in Vivo Activity of Highly Selective A(3) Adenosine Receptor Agonists, Medchemcomm, 6 (2015) 555–563. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [20].Tosh DK, Salmaso V, Rao H, Campbell R, Bitant A, Gao ZG, Auchampach JA, Jacobson KA, Direct Comparison of (N)-Methanocarba and Ribose-Containing 2-Arylalkynyladenosine Derivatives as A(3) Receptor Agonists, ACS Med Chem Lett, 11 (2020) 1935–1941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [21].Jacobson KA, Tosh DK, Jain S, Gao ZG, Historical and Current Adenosine Receptor Agonists in Preclinical and Clinical Development, Front Cell Neurosci, 13 (2019) 124. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Manolaridis I, Jackson SM, Taylor NMI, Kowal J, Stahlberg H, Locher KP, Cryo-EM structures of a human ABCG2 mutant trapped in ATP-bound and substrate-bound states, Nature, 563 (2018) 426–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [23].Tosh DK, Salmaso V, Campbell R, Rao H, Bitant A, Pottie E, Stove CP, Liu N, Gavrilova O, Gao ZG, Auchampach JA, Jacobson KA, A3 adenosine receptor agonists containing dopamine moieties for enhanced interspecies affinity, Eur. J. Med. Chem, 228 (2022) 113983. (doi: 10.1016/j.ejmech.2021.113983). [DOI] [PMC free article] [PubMed] [Google Scholar]
- [24].Silbermann K, Li J, Namasivayam V, Baltes F, Bendas G, Stefan SM, Wiese M, Superior Pyrimidine Derivatives as Selective ABCG2 Inhibitors and Broad-Spectrum ABCB1, ABCC1, and ABCG2 Antagonists, J Med Chem, 63 (2020) 10412–10432. [DOI] [PubMed] [Google Scholar]
- [25].Toyoda Y, Takada T, Suzuki H, Inhibitors of Human ABCG2: From Technical Background to Recent Updates With Clinical Implications, Front Pharmacol, 10 (2019) 208. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Kowal J, Ni D, Jackson SM, Manolaridis I, Stahlberg H, Locher KP, Structural Basis of Drug Recognition by the Multidrug Transporter ABCG2, J Mol Biol, 433 (2021) 166980. [DOI] [PubMed] [Google Scholar]
- [27].Mitchell-White JI, Stockner T, Holliday N, Briddon SJ, Kerr ID, Analysis of Sequence Divergence in Mammalian ABCGs Predicts a Structural Network of Residues That Underlies Functional Divergence, Int J Mol Sci, 22 (2021) 3012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Marquez VE, The Properties of Locked Methanocarba Nucleosides in Biochemistry, Biotechnology, and Medicinal Chemistry, in: Modified Nucleosides, 2008, pp. 305–341. [Google Scholar]
- [29].Jacobson KA, Salmaso V, Ravi R, Tosh D, Expanding the repertoire of methanocarba nucleosides from purinergic signaling to diverse targets, RSC Med. Chem, 12 (2021) 1808–1825. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [30].Jackson SM, Manolaridis I, Kowal J, Zechner M, Taylor NMI, Bause M, Bauer S, Bartholomaeus R, Bernhardt G, Koenig B, Buschauer A, Stahlberg H, Altmann KH, Locher KP, Structural basis of small-molecule inhibition of human multidrug transporter ABCG2, Nat Struct Mol Biol, 25 (2018) 333–340. [DOI] [PubMed] [Google Scholar]
- [31].Kerr KM, Sauna ZE, Ambudkar SV, Correlation between steady-state ATP hydrolysis and vanadate-induced ADP trapping in Human P-glycoprotein. Evidence for ADP release as the rate-limiting step in the catalytic cycle and its modulation by substrates, J Biol Chem, 276 (2001) 8657–8664. [DOI] [PubMed] [Google Scholar]
- [32].Sauna ZE, Ambudkar SV, Evidence for a requirement for ATP hydrolysis at two distinct steps during a single turnover of the catalytic cycle of human P-glycoprotein, Proc Natl Acad Sci U S A, 97 (2000) 2515–2520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Ambudkar SV, Drug-stimulatable ATPase activity in crude membranes of human MDR1-transfected mammalian cells, Methods Enzymol, 292 (1998) 504–514. [DOI] [PubMed] [Google Scholar]
- [34].Robey RW, Honjo Y, Morisaki K, Nadjem TA, Runge S, Risbood M, Poruchynsky MS, Bates SE, Mutations at amino-acid 482 in the ABCG2 gene affect substrate and antagonist specificity, Br J Cancer, 89 (2003) 1971–1978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Robinson AN, Tebase BG, Francone SC, Huff LM, Kozlowski H, Cossari D, Lee JM, Esposito D, Robey RW, Gottesman MM, Coexpression of ABCB1 and ABCG2 in a Cell Line Model Reveals Both Independent and Additive Transporter Function, Drug Metab Dispos, 47 (2019) 715–723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Shukla S, Schwartz C, Kapoor K, Kouanda A, Ambudkar SV, Use of baculovirus BacMam vectors for expression of ABC drug transporters in mammalian cells, Drug Metab Dispos, 40 (2012) 304–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [37].Sajid A, Lusvarghi S, Chufan EE, Ambudkar SV, Evidence for the critical role of transmembrane helices 1 and 7 in substrate transport by human P-glycoprotein (ABCB1), PLoS One, 13 (2018) e0204693. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [38].Vahedi S, Chufan EE, Ambudkar SV, Global alteration of the drug-binding pocket of human P-glycoprotein (ABCB1) by substitution of fifteen conserved residues reveals a negative correlation between substrate size and transport efficiency, Biochem Pharmacol, 143 (2017) 53–64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [39].Trott O, Olson AJ, AutoDock Vina: improving the speed and accuracy of docking with a new scoring function, efficient optimization, and multithreading, J Comput Chem, 31 (2010) 455–461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [40].Janson G, Zhang C, Prado MG, Paiardini A, PyMod 2.0: improvements in protein sequence-structure analysis and homology modeling within PyMOL, Bioinformatics, 33 (2017) 444–446. [DOI] [PubMed] [Google Scholar]
- [41].Yuan S, Chan HCS, Filipek S, Vogel H, PyMOL and Inkscape Bridge the Data and the Data Visualization, Structure, 24 (2016) 2041–2042. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.