

Abstract
Two decades of research have established that Nuclear Factor-κB (NF-κB) signaling plays a critical role in reprogramming the fat cell transcriptome towards inflammation in response to overnutrition and metabolic stress. Several groups have suggested that inhibition of NF-κB signaling could have metabolic benefits for obesity-associated adipose tissue inflammation. However, two significant problems arise with this approach. The first is how to deliver general NF-κB inhibitors into adipocytes without allowing these compounds to disrupt normal functioning in cells of the immune system. The second issue is that general inhibition of canonical NF-κB signaling in adipocytes will likely lead to a massive increase in adipocyte apoptosis under conditions of metabolic stress, leading full circle into a secondary inflammation (However, this problem may not be true for non-canonical NF-κB signaling.). This review will focus on the research that has examined canonical and non-canonical NF-κB signaling in adipocytes, focusing on genetic studies that examine loss-of-function of NF-κB specifically in fat cells. Although the development of general inhibitors of canonical NF-κB signaling seems unlikely to succeed in alleviating adipose tissue inflammation in humans, the door remains open for more targeted therapeutics. In principle, these would include compounds that interrogate NF-κB DNA binding, protein-protein interactions, or post-translational modifications that partition NF-κB activity towards some genes and away from others in adipocytes. I also discuss the possibility for inhibitors of non-canonical NF-κB signaling to realize success in mitigating fat cell dysfunction in obesity. To plant the seeds for such approaches, much biochemical “digging” in adipocytes remains; this includes identifying—in an unbiased manner–NF-κB direct and indirect targets, genomic DNA binding sites for all five NF-κB subunits, NF-κB protein-protein interactions, and post-translational modifications of NF-κB in fat cells.
Keywords: NF-kappaB, adipocytes, obesity, inflammation
INTRODUCTION
Obesity continues to represent a significant health epidemic worldwide. Chronic overnutrition is associated with various metabolic disorders, including metabolic syndrome, Type 2 diabetes, cardiovascular disease, hypertension, and epithelial cancers, especially breast cancer [1–19]. An increasingly recognized adipose tissue immunometabolic function is at the epicenter of these diseases. Most now believe that overnutrition-related metabolic stress on the adipocyte causes it to send out distress signals in the form of inflammatory chemokines. These chemokines, in turn, are thought to recruit a variety of immune cells into the area of distress, which secrete “heavy-hitting” inflammatory cytokines of their own, such as TNF-α. These hormones, along with a slew of fatty acids released from dead and dying adipocytes, can travel in the blood to other tissues and precipitate a state of chronic insulin resistance. If left unresolved, this can lead to metabolic disorders such as Type 2 diabetes, hypertension, and heart disease.
Although the research community has made significant progress in understanding what factors are responsible for initiating and maintaining inflammatory processes in white adipose tissue (WAT), particularly at the transcription level, major unanswered questions remain. As discussed extensively in this review, approaches targeting the canonical NF-κB pathway are unlikely to be successful, as evidenced by the fact that high-fat diet (HFD)-induced inflammation is worsened (or at least not alleviated) in several adipose or adipocyte-specific NF-κB loss-of-function models. In addition, we can only assume that a general NF-κB inhibitor could have dire consequences for the innate immune system. Thus, efforts should continue to investigate additional mechanistic actions of NF-κB activity in adipocytes to gain insights that could be useful in developing precision-targeted therapeutics.
In a previous review [20], I likened the adipocyte as representing the auxiliary power unit (APU) on a jet aircraft, whereas macrophages represent the main turbofan engines. As in the case of aviation technology, a transient firing of inflammatory gene expression in adipocytes may be all it takes to spool up the jet engines of the immune cells and enter into cruising mode. Therefore, it is imperative to understand precisely how adipocytes turn over their inflammatory APUs. Two major signaling pathways seem to promote the expression of immunometabolic loci in adipocytes, the Nuclear Factor-κB (NF-κB) pathway and the NLRP3 pathway. Both pathways seem to be indispensable for the expression of chemokines and cytokines in adipocytes as well as immune cell recruitment and infiltration; however, this review focuses primarily on NF-κB. Specifically, this review summarizes and evaluates the research that has been invested into understanding the function and regulation of NF-κB signaling specifically within adipocytes.
The thesis of this review is, therefore, that (1) events occurring in adipocytes prime adipose tissue inflammation in obesity (as opposed to cells of the innate immune system), and this is mediated mainly through NF-κB (2) we likely canʼt abolish metabolic inflammation through the use of general pharmacological inhibitors of canonical NF-κB signaling without causing increased apoptosis of adipocytes, which would only worsen inflammation (3) therefore, much more research is needed to understand NF-κB function in adipocytes, including (a) identifying direct and indirect transcriptional/functional targets of each individual NF-κB subunit in adipocytes; (b) identifying genomic targets of each individual NF-κB subunit through ChIP-Seq studies; and (c) identifying various post-translational modifications and protein interaction partners of NF-κB proteins in adipocytes.
OVERVIEW OF NF-κB SIGNALING
The term “NF-κB” refers to five protein subunits in mice and man: p65/Rel-A, RelB, p50 (Nfkb1), p52 (Nfkb2), and c-Rel [21,22]. Rel-A, RelB, and c-Rel function as transcriptional activators, whereas p50 and p52 act as transcriptional repressors. The latter two proteins are synthesized as precursors (p105 and p100, respectively) which are subsequently cleaved into their active forms. In the canonical or “classic” NF-κB pathway, p65 and p50 exist as a heterodimer in a cytosolic complex with an inhibitor protein, IκBα, in latent cells, thereby preventing their nuclear localization and transcriptional activation (Figure 1). A variety of stimuli leads to phosphorylation of IKKβ, which exits as part of another cytoplasmic complex with its sister protein IKKα and a regulatory subunit, NEMO (Note: we will discuss these stimuli in detail in a subsequent section.). Once phosphorylated, IKKβ phosphorylates IκBα, leading to its ubiquitination and destruction and consequent release of p65/p50 into the nucleus. Meanwhile, a pool of p50 protein molecules exists in the nuclei of unstimulated cells; p50 forms homodimers that generally repress transcription on their own [23–26]. However, the canonical p65/p50 heterodimer potently activates transcription in most cases.
Figure 1. Canonical NF-κB Action in Adipocytes.

Several stimuli can activate NF-κB in adipocytes, including TLR ligation, ER stress, and hypoxia. This leads to activation of IKKβ, which forms part of a trimeric complex consisting of IKKα and NEMO as partners. IKKβ proceeds to phosphorylate IκBα, which normally sequesters NF-κB proper (p65/Rel-A and p50) in the cytosol. Once phosphorylated, IκBα is ubiquitinated and destroyed, freeing p65/p50 to translocate into the nucleus and bind near target genes. For the purposes of this review, there are two major classes of NF-κB target genes in adipocytes: inflammatory (chemokine and cytokine) genes, and apoptosis/cell cycle-related genes (although by no means is this a comprehensive list). Arrows indicate the fact that we don’t know if or precisely where NF-κB binds to these genes in live cells.
An alternative (non-canonical) pathway also exists in which RelB and p52 regulate a subset of target genes in some cell types, particularly in the immune system (Figure 2). A few key differences separate the canonical from the non-canonical pathway (reviewed by S-C Sun in 2017 [27]). In the latter, RelB physically associates with a precursor form of p52, p100 (encoded by NFKB2 in humans). As an IκBα family member, p100 functions to sequester RelB in the cytoplasm of latent cells. Upon stimulation through ligand binding to its receptor, NF-κB inducing kinase (NIK) phosphorylates IKKα, which exists as a dimeric complex that lacks a NEMO equivalent. Phosphorylated IKKα then phosphorylates p100, leading to the selective ubiquitin/proteasome-mediated proteolytic cleavage of the latter, discarding the C-terminal inhibitory domain. RelB/p52 heterodimers can thus translocate into the nucleus and activate target genes. Only a few stimuli are known to activate the alternative pathway, such as lymphotoxin LTα1β2 [28], BAFF (B-Cell Activating Factor, [29,30], and RANKL [31]. The non-canonical NF-κB pathway mainly functions in lymphoid organ development and B cell maturation and, more generally, innate and adaptive immunity [27]. As we shall discuss in detail later, the pathway also operates in mature adipocytes, with adipocyte-specific Relb−/− animals presenting with a striking metabolic phenotype [32].
Figure 2. Non-canonical NF-κB Signaling in Adipocytes.
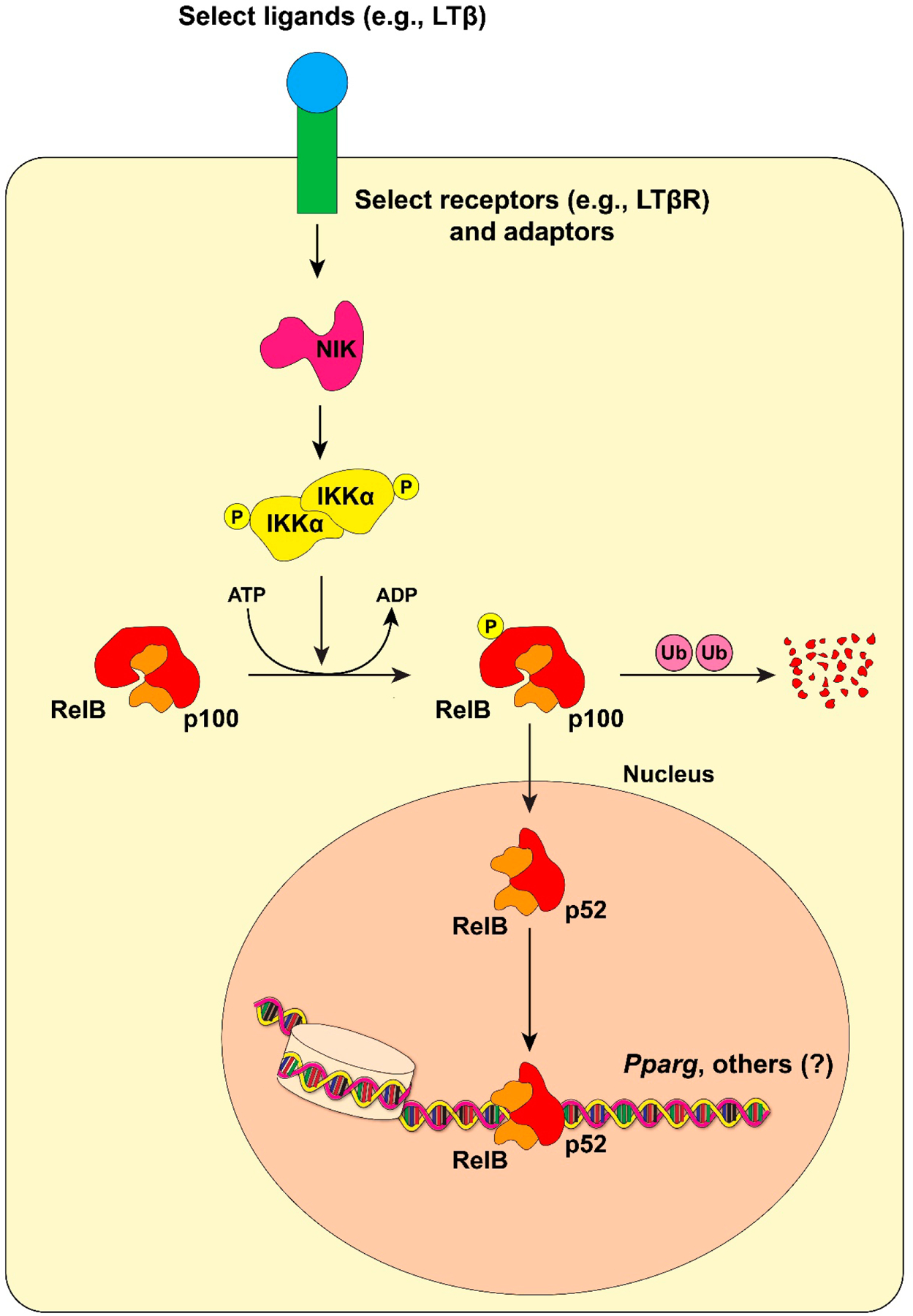
The process begins through ligation of specific receptors (e.g., LTβR), which activates receptor-associated adaptor proteins to activate NIK (NF-κB-Inducing Kinase). NIK then phosphorylates IKKα, which in turn phosphorylates p100. p100 functions as a de facto IκBα protein for RelB; upon phosphorylation, p100 is ubiquitinated and partially degraded. This leaves behind p52 and RelB to translocate into the nucleus and regulate target genes. The identify of these direct RelB/p52 target genes is largely unknown, except for one study demonstrating binding of RelB to the Pparg2 promoter in adipocytes through ChIP analysis [32].
ACTIVATORS OF CANONICAL NF-κB SIGNALING IN ADIPOCYTES
There are only three well-established general stimuli for classical NF-κB signaling in adipocytes (Figure 1), with varying degrees of understanding for each. In this section, I briefly discuss all of them.
Toll-Like Receptors—
TLRs are pattern recognition receptors that respond to a variety of foreign agents such as Lipopolysaccharide (LPS), CpG DNA, and Flagellin, depending on the specific receptor (reviewed in [33]). Some TLRs engage a protein complex consisting of MyD88, IRAKs, and TRAF6, which, when activated, stimulate the phosphorylation of IKKβ and subsequent activation of p65/p50. Other TLRs are MyD88-independent, but TRIF-dependent [33]. Adipocytes express several TLR proteins, with TLR2 and especially TLR4 boasting the highest expression [34–37]. The bacterial endotoxin LPS binds avidly to TLR4 [38–40], as do free fatty acids (FFAs), although the latter agent binds to the receptor indirectly [41,42]. Free fatty acids can also ligand with TLR2 [43].
Of significant interest, global deletion of Tlr4 protects mice from high fat diet-induced inflammation [44]; mice harboring a natural loss-of-function germline mutation in Tlr4 [45] can also consume a high-fat diet with relative impunity [45]. However, various tissues express Tlr4 in mice [40], and we don’t know with certainty what, if any, contribution Tlr4-deficient adipocytes had made to the phenotypes observed in these studies. A true adipocyte-specific Tlr4−/− mouse, driven by the established Adiponectin promoter-Cre (Adipoq-Cre) transgene (discussed in more detail later), might be incredibly useful in examining the immunometabolic significance of this pathway in adipose tissue in vivo.
Hypoxia—
the role of low pO2 in adipocyte dysfunction during obesity and its ability to promote inflammation in adipose tissue has captured much attention in recent years. The reason for the hypoxia may be relatively simple: during periods of high overnutrition, the rate of adipocyte enlargement outpaces the ability of the vasculature to service them (reviewed in [46]), resulting in a state of chronic hypoxia. Furthermore, two decades of research has established a link between obesity and inflammation, and a link between obesity and hypoxia in adipose tissue, at least in mice (note: studies of oxygenation in human WAT have produced contradictory results, reviewed in [47]), so perhaps this implies a causal relationship between obesity-induced hypoxia and inflammation in adipocytes.
Indeed, a slew of articles in the last few years have demonstrated that hypoxia can activate NF-κB in adipocytes [48–51], although the exact details of how low pO2 leads to putative phosphorylation and activation of IKKβ (an obligate step to release p65/p50) remain elusive. An obvious first responder to hypoxia is Hypoxia-Inducible Factor-1α (HIF-1α), and while we can reasonably posit that ubiquitination of HIF-1α ceases in O2-starved adipocytes, thus activating the transcription factor [52], it is not clear if this causes activation of NF-κB or whether NF-κB activation occurs in an HIF-1α-independent manner. While one cannot immediately draw a pathway from HIF-1α-driven changes in adipocyte transcriptional programming to nuclear translocation of p65/p50, several studies have unambiguously demonstrated NF-κB activation under hypoxic conditions. (Note: while HIF-1α parks itself on the Rela promoter in O2-deprived adipocytes [52], presumably driving its expression, this still doesn’t provide a full explanation of how the translated p65 protein finds its way into the nucleus under these conditions.) In 2014, He et al. demonstrated that intermittent hypoxia induces “something” in 3T3-L1 nuclear extracts to bind to κB-containing nucleotides in Electrophoretic Mobility Shift Assays (EMSAs), although it wasnʼt clear which, if any, specific NF-κB subunits occupied those sites [48]. Expression of TNF-α and IL-6 (both of which are established NF-κB targets, at least in other cell types ([53–57]), were increased in adipocytes exposed to severe IH, as was the expected increase in expression and protein levels of HIF-1α.
In another study, Taylor et al. showed that human adipocytes are exquisitely sensitive to intermittent hypoxia (IH), with the magnitude of NF-κB activation to this stimulus being significantly greater than for human primary microvascular pulmonary endothelial cells or human primary small airway epithelial cells [49]. In this study, the authors observed IH-dependent increases in the expression and secretion of IL-6, TNF-α, and another previously identified NF-κB target, IL-8 [58,59]. Here, an IH-induced increase in p65 nuclear translocation was indicated in human primary adipocyte nuclear extracts, similar to that induced by TNF-α. In addition, DNA binding and phosphorylation of p65 in response to both IH and TNF-α were significantly increased in human primary adipocytes. Although these studies suggest that hypoxia induces the expression of specific cytokines in adipocytes through NF-κB, the use of studies in which NF-κB-deficient adipocytes (whether through genetic or pharmacological means) are exposed to IH could provide additional information on the role of this stress on inflammation in adipocytes. (Although one recent study showed that pharmacological inhibition of NF-κB in adipocytes exposed to IH improves glucose uptake [51], more extensive studies with genetic deletion of IKKβ and individual NF-κB subunits in O2-starved adipocytes could be fruitful.) Several questions also remain, including (1) How do we make the putative connection between the observed increase in HIF-1α levels and the putative increase in IκBα phosphorylation in O2-starved adipocytes (although it is formally possible that hypoxia activates p65 in an IκBα-independent manner); (2) do we also see hypoxia-induced increases in the expression of a slew of small NF-κB-dependent chemokines such as RANTES [60], CCL5 [61], and MCP-1 [62–65] that we believe act as chemoattractants for immune cells in human adipose tissue?
In another study, hypoxia reduced NF-κB signaling in response to TNF-α [66], although conditions used may have been somewhat unphysiological as the investigators dropped O2 concentrations to 1% for up to 24 h (most other studies mentioned above used O2 concentrations in the 5–10% range or intermittently or for shorter periods). In that study, TNF-α-induced phosphorylation of p65 and expression of IL-6 were also severely blunted in the face of hypoxia. Further studies should evaluate the compound effects of multiple NF-κB stimuli in adipocytes to determine if their effects are additive, synergistic, or antagonistic.
One of the most recent—and perhaps enlightening–studies on this topic has demonstrated that the Receptor for Advanced Glycation End Products (RAGE) mediates the activation of NF-κB by hypoxia in adipocytes; lack of RAGE significantly blunts hypoxia-induced NF-κB activation. In addition, pharmacological inhibition of NF-κB reduced the hypoxia-induced increase in RAGE expression [51]. These results might imply that activation of NF-κB occurs independently of activation of HIF-1α in O2-starved adipocytes; that is to say, HIF-1α and NF-κB reprogram the adipocyte transcriptome through parallel pathways. On the other hand, HIF-1α directly binds to and regulates the expression of the RAGE gene [67], and other results suggest that HIF-1α activates NF-κB directly and through increased expression of RAGE [68]. An experiment involving NF-κB activation in HIF-1α-deficient, hypoxia-exposed adipocytes could lay this argument to rest.
Of note, lack of HIF-1α in adipocytes of mice decreases the expression of numerous chemokines and cytokines in adipose tissue, including several known NF-κB target genes [52]. The lack of HIF-1α in adipocytes also affords the animals significant protection from HFD-induced insulin resistance and adipose tissue inflammation. Clearly, nature has built-in some level of cross-talk between HIF-1α and NF-κB in adipocytes, with RAGE possibly assuming an intermediary role. However, the connection between hypoxia and a presumed increase in the appearance of advanced glycation end products in adipocytes remains unclear. (While we must note that hypoxia increases AGE production, most of that research has focused on tumor cells [69,70]). In any case, hypoxia-mediated damage to hypertrophied adipocytes seems plausible, and AGEs released from damaged cells could ligand with RAGE on neighboring cells and trigger NF-κB activation. Based on limited data available, it is tempting to speculate that the order of events occurring in O2-starved adipocytes includes (1) hypoxia-induced increase in HIF-1α activity, which drives an adaptation to anaerobic glycolysis and an initial wave of chemokine and RAGE expression (2) the pursuant increase in glucose utilization leads to an increase in the production of AGEs and perhaps other ligands of RAGE in hypoxic adipocytes (3) ligation of RAGE and subsequent activation of NF-κB, resulting in a second wave of chemokine and cytokine production.
Inflammatory Cytokines—
A few inflammatory protein hormones can activate NF-κB in adipocytes, most notably TNF-α [32,49,71–74]. In perhaps the seminal study of this question, Tourniaire et al. demonstrated that TNF-α dramatically and dose-dependently induce the expression of numerous NF-κB-dependent chemokines in human primary adipocytes as well as differentiated 3T3-L1 adipocytes [72]. This investigation followed the pioneering work of the much earlier Ruan study of TNF-α action in 3T3-L1 cells [74]. As evidence that TNF-α mediates these inductions through NF-κB in vivo, overexpression of p65/Rel-A (using aP2-p65 transgenic mice) in adipose tissue mimicked the effect of TNF-α treatment on chemokine expression in the primary cells, whereas TNF-α-induced expression of most of the same chemokines was abolished in Rela−/− Mouse Embryonic Fibroblasts (MEFs) [72] (However, we must note that since aP2 is also highly expressed in macrophages [75–77], and since the authors used whole adipose tissue and not fractionated adipose tissue, it is unknowable from this study which cell type is primarily responsible for the observed increase in chemokine expression.). Further, Gene Set Enrichment Analysis (GSEA) indicated that κB sites were the most overrepresented transcription factor binding sites associated with loci responding to TNF-α in both human primary adipocytes and 3T3-L1 adipocytes. Finally, pharmacological inhibition of NF-κB obliterated TNF-α-stimulated chemokine gene expression in adipocytes [72]. Of note, the expression of several NF-κB-dependent chemokines correlated strongly with expression of TNF-α in human obese adipose tissue, suggesting that the pathway operates in a physiologically-relevant manner in vivo [72] (Note: as alluded to earlier, the term “NF-κB-dependent chemokines” refers to the fact that others had shown these loci to represent NF-κB targets in other cell types, not necessarily adipocytes.).
ER Stress—
other stimuli, such as ER stress and increased ROS, can also activate NF-κB signaling, at least in several non-adipocyte cell types (reviewed in [78]). Several human and animal studies have demonstrated an obesity-linked increase in ER stress in adipocytes and adipose tissue [79–84]. As for the specific link between ER stress and NF-κB, one group has shown that the ATF6 (Activating Transcription Factor-6) branch of the Unfolded Protein Response (UPR) pathway can activate NF-κB in rat renal tubular epithelial cells [85]; it remains to be determined if ATF6 can do the same in adipocytes. In any case, as most of this work has focused on the IRE-JNK-IRS-1 signaling pathway for mediating ER-induced inflammation and insulin resistance, it may be worthwhile to induce ER stress in NF-κB-deficient adipocytes (i.e., IKKβ, p65/Rel-A, or RelB KOs) to determine the dispensability of NF-κB for ER-induced metabolic inflammation.
NF-κB DURING ADIPOGENESIS
We begin our discussion proper of NF-κB activity in adipocytes by considering expression patterns of individual NF-κB subunits during adipogenesis, the process through which fibroblast-like preadipocytes assume gene expression profiles of mature fat cells and acquire their defining functional and morphological features. In a 2004 study—one of the first to specifically examine NF-κB activity in adipocytes—Berg et al. demonstrated that protein levels of several NF-κB subunits (p52, p65, and RelB) increase during adipogenesis in 3T3-L1 cells [86]. The basal activity of an NF-κB reporter plasmid also increased during adipogenesis, as did activity stimulated by LPS and TNF-α. However, whereas TNF-α potently stimulated NF-κB activity in both preadipocytes and adipocytes, the magnitude of LPS-induced NF-κB transcriptional activity diminished in mature adipocytes. Analogous results were obtained when the authors looked at LPS- and TNF-α-induced- NF-κB DNA binding through EMSAs, except that NF-κB DNA binding was actually decreased in adipocytes upon LPS treatment. The group used p65 antibodies to demonstrate that the gel shift complexes contained p65, and Western blotting confirmed LPS-induced nuclear translocation of p65 and RelB in preadipocytes and p65 in mature adipocytes. RelB and p52 nuclear levels appeared to remain high in mature adipocytes regardless of whether the cells received LPS treatment [86].
The authors then went on to show that although mature 3T3-L1 adipocytes are resistant to LPS-stimulation of classical NF-κB activity (a reflection of high constitutive NF-κB activity), LPS still significantly induced the expression of the NF-κB target serum amyloid A3 (SAA3 [86]). This induction occurred through increased binding of the transcription factor SEF (SAA3 Enhancer Factor) to an EMSA probe with an SEF binding site, a protein already known to regulate SAA3 expression. The authors also showed that LPS stimulation results in an ~80-fold increase in IL-6 secretion in both preadipocytes and adipocytes, although it should be noted that IL-6 secretion by an equivalent number of J774 macrophages scored more than an order of magnitude higher (which would give credence to the generally accepted view that Adipose Tissue Macrophages [ATMs] and other immune cells represent the primary source of circulating cytokines in obesity). Curiously, though, in contrast to LPS, TNF-α robustly stimulated NF-κB transcriptional activity in both preadipocytes and adipocytes throughout the study [86].
One possible explanation for these results (specifically, the blunted NF-κB response to LPS in mature fat cells) would be background ligation of adipocyte Tlr4 with LPS or FFAs present in the cell culture serum [39,40,42,87,88]. Although some studies suggest similar expression of TLR4/Tlr4 in preadipocytes and adipocytes [36,89–91], at least two studies demonstrated a significant increase in expression of protein levels of Tlr4 during the process of 3T3-L1 adipogenesis [37,87]. Differing expression levels of Tlr4 could account for the vastly different “deltas” of NF-κB activation upon LPS stimulation in preadipocytes and adipocytes, at least in the 3T3-L1 model. Another intriguing possibility is background ligation of TLR4/Tlr4 with Fetuin A/FFAs in adipocytes but not preadipocytes. Fetuin-A functions as a secreted glycoprotein that is believed to act as a direct ligand for TLR4 in the presence of FFAs [42] and is believed to represent a link between obesity and metabolic inflammation [41]. In support of this explanation, protein levels of Fetuin A are nearly undetectable in 3T3-L1 preadipocytes but dramatically induced in mature adipocytes [92]. In other words, LPS-induced activation of NF-κB may have been reduced in adipocytes because they were already responding to Fetuin A/FFAs in the serum, whereas the response to FFAs in preadipocytes would have been minimal due to lack of Fetuin A expression. In any case, evidence suggests that 3T3-L1 preadipocytes can respond to LPS but not FFAs/Fetuin A, whereas mature adipocytes can respond to both. It remains to be determined if a similar paradigm exists in human preadipocytes and adipocytes, although it is certainly possible that in an in vivo environment, Fetuin-A released from mature adipocytes can complex with extracellular FFAs to activate TLR4 in nearby preadipocytes.
STUDIES OF INDIVIDUAL NF-κB SUBUNITS IN ADIPOCYTES IN VITRO AND IN VIVO
This section provides a brief overview of research conducted on NF-κB in adipocytes per individual subunit. Some of the studies discussed here are elaborated upon in subsequent sections.
p65/Rel-A
p65/Rel-A protein levels are nearly undetectable in adipocyte precursors but rise rapidly during differentiation [86]. p65/Rel-A has enjoyed the most research attention of all the NF-κB subunits in adipocytes or adipose tissue and shall be discussed extensively in later sections.
Rel-B (p68)
We know relatively little about alternative (non-canonical) NF-κB signaling in adipocytes. RelB is readily detectable in adipocyte precursors but is induced further upon differentiation [86], suggesting the protein has an important role to play in mature adipocytes. Adipocyte-specific RelB knockout animals (RelBFatKO) present with improved glucose and insulin tolerance despite having increased epididymal fat pad weight and adipocyte hypertrophy [32], partially consistent with other studies that link “healthy” adipose tissue expansion with improved insulin sensitivity and glucose tolerance [93–95]. However, we should note that the authors readily detected Relb expression in subcutaneous WAT; the authors speculated that nominal Relb expression in non-adipocyte cells masked an effective knockout in adipocytes. Further in vitro experiments in the same study demonstrated that LTβR (Lymphotoxin β-Receptor) signaling attenuates adipogenesis of 3T3-L1 cells; experiments in RelB-, p52-, or p50-deficient Mouse Embryonic Fibroblasts (MEFs) showed that RelB and p52 were the subunits specifically responsible for this effect. The authors then went on to demonstrate that RelB (but not Rel-A) binds to NF-κB sites in the Pparg promoter to repress the expression of the gene primarily responsible for adipogenesis. To this author’s knowledge, this manuscript represents the one and only study in the field to demonstrate binding of any NF-κB subunit to genomic DNA in adipocytes through chromatin immunoprecipitation (ChIP) analysis.
In the final series of experiments, the authors went on to show that LTβR-mediated alternative NF-κB signaling inhibits glucose uptake and expression of various adipose-specific genes, including Adipoq and Slc2a4 (encoding Glut4). This result suggests that in vivo, alternative NF-κB signaling might very well participate in insulin resistance in adipocytes. Curiously, the authors also demonstrated that TNF-α signaling (through Rel-A) works synergistically with LTβR signaling (through RelB and p52) in a time-dependent manner to inhibit Pparg expression. It thus appears that non-canonical NF-κB signaling in adipocytes would lead to a relative shutdown in mature fat cell operations, although the physiological significance of this finding is unknown.
c-Rel
c-Rel assumes the role of the most rogue member of the NF-κB posse. This author was unable to find even one peer-reviewed article examining the function of c-Rel specifically within adipocytes, and according to the website BioGPS, c-Rel expression is minimal in human and mouse adipose tissue. However, in 2002 Ruan et al. showed that c-Rel is readily detectable by western blotting in both TNF-α-treated and untreated 3T3-L1 adipocytes [74]. Furthermore, several years ago, my lab readily reverse-transcribed and amplified c-Rel cDNA from 3T3-L1 adipocytes, indicating that its expression is anything but silent in mouse fat cells. To date, the mystery of any putative function of c-Rel in adipocytes remains unsolved. Some discovery-driven pilot studies could prove prudent here, such as Crel shRNA knockdowns combined with RNA-seq in 3T3-L1 adipocytes. Follow-up studies involving adipocyte-specific Crel−/− animals have the potential to reveal new biology that may or may not be related to metabolic inflammation in adipocytes.
Nfkb1 (p50)
Little attention has been paid to p50 (Nfkb1) function per se in adipocytes. For instance, there appear to be no studies in which researchers hit p50-deficient adipocytes with inflammatory agents and measure the expression of chemokine genes. About all we know is that p50 levels are readily detectable in preadipocytes and remain stable during adipogenesis [86]. Furthermore, nascent adipocytes can make do without p50; Nfkb1−/− MEFs demonstrate no issues with differentiation [32].
Global knockout of p50 in mice results in a severe reduction in NF-κBʼs transcriptional activity, particularly in B cells. However, no metabolic phenotype was reported in this original Nfkb1−/− study [96], possibly because the animals were not challenged with a HFD. However, the more recent Tang study showed that Nfkb1−/− mice displayed a 50% decrease in fat mass at 12 weeks of age; an increase in energy expenditure more than offset increased food intake [97]. In addition, inflammatory gene expression increased in the adipose tissue of Nfkb1−/− mice, including IL-6, which could help explain the increase in energy expenditure. Within the adipose tissue, these results might be explained through an increase in the expression of genes that would normally be repressed by p50 in the latent state, an increase in p65 homodimerization and transcriptional activation at loci that would normally be activated by p65/p50 heterodimers in the stimulated state, or both.
As with other NF-κB subunits, the field could be well-served by examining any metabolic phenotype of true adipocyte-specific Nfkb1−/− animals. This genetic model would allow us to evaluate the function of p50 specifically in adipocytes without the confounding effects of loss of p50 in other tissues.
Nfkb2 (p52)
Like p50, little research in the adipocyte biology field has focused on p52 per se. Unlike p50, however—whose expression is boringly unchanged from fibroblast to fat cell during 3T3-L1 adipogenesis—p52 is dramatically induced during this process [86]. In addition, LTβR (a stimulator of the alternative NF-κB pathway) potently inhibits adipogenesis of Mouse Embryonic Fibroblasts (MEFs) in a p52-dependent manner [32]. Protein levels of p100 are also under the control of TNF-α, as TNF-α treatment markedly increases the concentration of p100 in 3T3-L1 nuclear extracts [32]. Having received the honor for “most induced NF-κB subunit during adipogenesis [86]”, further studies involving p52 and its precursor in adipocytes are certainly warranted.
In Vitro Studies
This section discusses in vitro studies mostly involving genetic or pharmacological inhibition of NF-κB in adipocytes. These studies are summarized in Table 1, including the investigations that examined previously-discussed NF-κB activation pathways in adipocytes (e.g., hypoxia). The discovery in the late 1990s by the Hotamisligil group that TNF-α mediates insulin resistance in mice [98–101] resulted in a flurry of activity to examine this pathway specifically within adipocytes. At around the same time, several groups discovered that adipocytes avidly express Toll-Like Receptors (TLRs) and respond to some of their ligands (i.e., LPS and free fatty acids) through the NF-κB pathway. In one of the first studies of its kind, Ruan et al. demonstrated that TNF-α alters the transcriptional profile of adipocytes into a devolving pattern resembling preadipocytes and that this occurs mainly through NF-κB [74]. Shortly thereafter, the same group showed that this effect can be potently inhibited by TZDs (Thiazolidinediones), pointing towards an antagonism between NF-κB and PPARγ [102]; the authors speculated that competition for coactivators could explain this mutual inhibition. However, several years later, another group demonstrated that nuclear p65 can physically interact with PPARγ, but only in the absence of ligand, at least in rat kidney cells [103], suggesting that p65 directly inhibits PPARγ. In any case, the bulk of in vitro evidence would suggest that inhibiting canonical NF-κB activity in adipocytes with a drug would likely mimic the action of TZDs to some extent and afford some level of metabolic benefit. However, as described in the next section, general inhibition of NF-κB in adipocytes in vivo appears to precipitate apoptosis and worsen inflammation.
Table 1.
In vitro Studies of NF-κB in Adipocytes.
| Authors | Title | Year published | Major findings | Comments |
|---|---|---|---|---|
| Ruan et al. [74] | “Tumor necrosis factor-alpha suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: nuclear factor-κB activation by TNF-alpha is obligatory” | 2002 | • TNF-α increases expression of preadipocyte-specific genes and suppresses insulin signaling and metabolic gene expression in 3T3-L1 adipocytes • Many chemokines and cytokine-induced proteins were also increased by TNF-α • TNF-α largely mediates these effects through NF-κB • TNF-α treatment induces rapid nuclear accumulation of p65/Rel-A • Inhibition of NF-κB function in TNF-α-treated cells results in increased cell death, suggesting that NF-κB action in adipocytes inhibits apoptosis |
• One of the first papers (if not the first) to show TNF-α-dependent activation of NF-κB in adipocytes |
| Ruan et al. [102] | “Troglitazone antagonizes tumor necrosis factor-alpha-induced reprogramming of adipocyte gene expression by inhibiting the transcriptional regulatory functions of NF-κB” | 2003 | • TZDs largely block the suppressive effects of TNF-α on numerous adipocyte genes • TGZ does not affect TNF-α-induced NF-κB nuclear translocation or DNA binding • p65/Rel-A and PPARγ mutually inhibit each other’s gene regulatory activity in 3T23-L1 cells • p65 can inhibit PPARγ-dependent gene expression from a promoter lacking a functional NF-κB binding site, and PPARγ can inhibit p65-mediated gene expression from a promoter lacking a PPARγ site • p65 may inhibit PPARγ activity, in part, by interacting non-productively with critical PPARγ coactivator proteins |
• Paper demonstrates that TZD-mediated improvements in insulin sensitivity and inflammation occur—at least in part--through antagonizing TNF-α-induced NF-κB transcriptional activity |
| Berg, A et al. [86] | “Adipocyte differentiation induces dynamic changes in NF-κB expression and activity” | 2004 | • Several NF-κB subunits are induced during differentiation • Nfkb2 (p52) showed the largest increase in protein levels during differentiation • Relatively low sensitivity to LPS in differentiated adipocytes • One of the first papers to show secretion of inflammatory cytokines from adipocytes and subsequent activation of macrophages in vitro |
• Paper demonstrates that multiple NF-κB subunits are alive and well in differentiated adipocytes • Could FFAs and/or LPS in serum have activated TLR4 and contributed to “constitutive” NF-κB activation in this study? |
| Laurencikiene, J et al. [71] | “NF-κB is important for TNF-alpha-induced lipolysis in human adipocytes” | 2007 | • Inhibition of NF-κB activity partially blocks TNF-α-induced lipolysis in human adipocytes • NF-κB activity is required for full TNF-α-stimulated induction of HSL and Perilipin expression in adipocytes • This effect of an NF-κB inhibitor is mediated, at least in part, through attenuation of TNF-α-mediated repression of Hsl and Plin expression |
• Proof-of-principle that NF-κB inhibitors might be useful for attenuating adipocyte lipolysis in obesity-associated inflammation • In vivo studies are needed however |
| Suganami, T et al. [88] | “Role of the Toll-like receptor 4/NF-κB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages” | 2007 | • Pharmacological inhibition of NF-κB inhibits increases in MCP-1 and TNF-α expression and secretion that occur when adipocytes and macrophages are co-cultured • Co-culture of the two cell types activates NF-κB in both • TNF-α increases MCP-1 expression and secretion in adipocytes through NF-κB • Pharmacological inhibition of NF-κB inhibits coculture-induced lipolysis in adipocytes but not TNF-α-induced lipolysis |
• Study suggests that NF-κB is not required for TNF-α-induced lipolysis in 3T3-L1 adipocytes • May contrast with Laurencikiene et al. [71], who showed that NF-κB inhibition only partially blocks TNF-α-induced lipolysis in human adipocytes |
| Vitseva, O.I. et al. [35] | “Inducible Toll-like receptor and NF-κB regulatory pathway expression in human adipose tissue” | 2008 | • LPS increases expression of RELA as well as phosphorylation and nuclear translocation of p65 in cultured human adipocytes • LPS induces secretion of TNF-α and IL-6 in cultured human adipocytes • TLR2 and TLR4 expression high in human adipose tissue and inducible with TLR ligands |
• One of the first papers to demonstrate high expression of TLR4 in human adipose tissue • Study was conducted on adipose tissue and isolated adipocytes from obese subjects • Study also demonstrates that NF-κB activation can induce TLR4 expression in adipocytes |
| Schaeffler, A et al. [87] | “Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-κB pathway in adipocytes links nutritional signaling with innate immunity” | 2009 | • Treatment with several fatty acids (stearic, palmitic, and palmitoleic) induces nuclear translocation of p65 in 3T3-L1 adipocytes • Pharmacological inhibition of NF-κB blocks fatty acid-induced increases in the secretion of MCP-1 and resistin |
• Another paper reinforcing the paradigm that fatty acid ligation of TLRs increases secretion of inflammatory cytokines through NF-κB |
| Zoico et al. [104] | “The effects of Adiponectin on interleukin-6 and MCP-1 secretion in lipopolysaccharide-treated 3T3-L1 adipocytes: role of the NF-κB pathway” | 2009 | • LPS increases the expression of IL-6 and MCP-1 in differentiated adipocytes • This induction is largely blocked by pre-treatment with Adiponectin • Adiponectin targets NF-κB in its inhibition of inflammation in adipocytes |
• Study provides a mechanism by which adiponectin inhibits the expression of inflammatory cytokines in adipocytes • Highlights functional antagonism between NF-κB and PPARγ |
| Hoareau et al. [105] | “Signaling pathways involved in LPS induced TNF-alpha production in human adipocytes” | 2010 | • Main finding is that both the NF-κB pathway and p38 MAPK pathway are involved in LPS-stimulated expression and secretion of TNF-α in primary human adipocytes • Primary human macrophages exhibit greater sensitivity to LPS stimulation (in terms of TNF-α output) than do primary human adipocytes • But adipocytes secrete more TNF-α at high concentrations of LPS (1 μg/mL) |
• Results would call into question the efficacy of using NF-κB drug inhibitors to block the production of TNF-α in adipocytes, since the pathway also depends up on p38 MAPK • Study may contradict another one showing that TNF-α expression in adipocytes is minimal compared to that in macrophages [88] |
| Turner, J.J. et al. [106] | “Investigation of nuclear factor-κB inhibitors and interleukin-10 as regulators of inflammatory signaling in human adipocyte” | 2010 | • Pharmacological inactivation of NF-κB, or • overexpression of dominant-negative iκBα or IKK2 proteins, strongly inhibits LPS stimulation of IL-6 and MCP-1 induction in primary human adipocytes |
• This paper simply adds more evidence to the importance of NF-κB in mediating the expression of inflammatory chemokines and cytokines in human adipocytes. |
| Krinninger, P. et al. [107] | “Role of the adipocyte-specific NF-κB activity in the regulation of IP-10 and T cell migration” | 2011 | • Expression and secretion of CXC chemokine interferon-γ-induced protein 10 kDa (IP-10) is significantly greater in adipocytes than in preadipocytes • Induced expression and secretion of IP-10 and murine and human adipocytes is largely dependent upon NF-κB • Conditioned media from induced adipocytes in which NF-κB activity has been abolished fails to result in increased T-celf migration |
• One of the first papers (if not the first) to link NF-κB activation in adipocytes to T-cell migration |
| Zhong, L et al. [108] | “Effect of NF-κB decoy on insulin resistance of adipocytes from patients with type 2 diabetes mellitus” | 2011 | • Differentiated adipocytes from patients with T2DM have greater levels of NF-κB and inflammatory gene expression than those from non-diabetic patients • Interference with NF-κB activity via sequestration of NF-κB subunits with κB site-containing oligonucleotides improves insulin signaling and impairs inflammatory gene expression |
• The observation that NF-κB activity is higher in fat cells from patients with diabetes provides proof that this pathway is important for metabolic inflammation |
| Lira, F. S. et al. [109] | “Both Adiponectin and interleukin-10 inhibit LPS-induced activation of the NF-κB pathway in 3T3-L1 adipocytes” | 2012 | • IL-10 and/or Adiponectin can inhibit LPS-stimulated NF-κB nuclear activity, at least at certain time points • Curiously, treatment with IL-10 and/or Adiponectin actually increases LPS-stimulated IL-6 secretion at early time points but inhibits IL-6 secretion at later time points |
• Study provides an important counterpoint to other studies, i.e., hormones with known anti-inflammatory activity act to potently inhibit NF-κB in adipocytes and correlates this with reduced cytokine secretion |
| Harte, A. et al. [110] | “NF-κB as a potent regulator of inflammation in human adipose tissue, influenced by depot, adiposity, T2DM status, and TNF-alpha” | 2013 | • NF-κB activity is higher in human omental WAT than in subcutaneous WAT in both lean and obese subjects (consistent with the idea that visceral fat tends to become much more inflamed than subcutaneous fat) • Much higher NF-κB activity in subcutaneous WAT from patients with Type 2 diabetes than in patients without diabetes • Just the opposite is true for JNK activity • TNF-α is a potent inducer of NF-κB in human primary adipocytes, less so for JNK |
• Significant finding is that NF-κB, not JNK, appears to be the dominant inflammatory signaling pathway in human adipocytes • May contrast with prior mouse models that suggest JNK is a predominant factor influencing diabetes • Provides further justification for pursuing NF-κB therapeutic interventions as treatment for obesity-associated diabetes |
| Tourniaire et al. [72] | “Chemokine Expression in Inflamed Adipose Tissue Is Mainly Mediated by NF-κB” | 2013 | • TNF-α induces mouse and human adipocytes to secrete multiple chemokines in a dose-dependent manner • NF-κB inactivation completely abolished TNF-α-induced increases in expression of select chemokines in human adipocytes • Much higher levels of chemokine gene expression in WAT from obese vs. non-obese human subjects • High levels of chemokine gene expression in ap2-p65 mice as compared to WT controls • TNF-induced chemokine gene expression abolished in p65-deficient MEFs |
• This manuscript arguably represents a landmark paper in the adipocyte NF-κB field • Tied several concepts together to demonstrate NF-κB-dependent expression of chemokines in adipocytes • Used genetic gain- and loss-of-function models and pharmacological approaches to demonstrate a critical role of NF-κB in human adipocytes |
| He, Q. et al. [48] | “Effects of varying degrees of intermittent hypoxia on proinflammatory cytokines and adipokines in rats and 3T3-L1 adipocytes” | 2014 | • Intermittent hypoxia (IM) induces NF-κB DNA binding in nuclear extracts from 3T3-L1 adipocytes • IH also increases mRNA and protein levels of two NF-κB targets (TNF-α and IL-6) in cellular and animal models |
• One of the first studies to demonstrate that NF-κB can be activated in 3T3-L1 adipocytes by IH |
| Taylor, C et al. [49] | “Human adipocytes are highly sensitive to intermittent hypoxia-induced NF-κB activity and subsequent inflammatory gene expression” | 2014 | • Intermittent hypoxia (IH) induces iκBα phosphorylation and p65 DNA binding activity in human adipocytes • This activity tended to be much higher than in human primary microvascular pulmonary endothelial cells and human primary small airway epithelial cells • Significant IH-induced expression and secretion of inflammatory gene expression in human adipocytes |
• Manuscript shows that transient hypoxia can lead to activation of NF-κB in human subcutaneous and visceral adipocytes • Suggests that hypoxia could represent a major mechanism triggering inflammatory signaling through NF-κB in vivo |
| Weidermann A, et al. [32] | “Classical and alternative NF-κB signaling cooperate in regulating adipocyte differentiation and function” | 2016 | • Alternative NF-κB pathway activation attenuates adipogenesis • RelB (and probably p52) directly regulates Pparg2 promoter upon stimulation • Alternative NF-κB signaling impairs adipogenesis and insulin action in mature adipocytes while stimulating lipolysis • Canonical NF-κB signaling primes non-canonical signaling in adipocytes by increasing expression of RelB and p100 |
• One and only paper in the field to examine non-canonical NF-κB activity in adipocytes • Suggests non-canonical NF-κB signaling impairs insulin signaling in adipocytes • Both classical and alternative NF-κB signaling are involved in full repression of PPARγ in adipocytes • Paper suggests that inhibiting non-canonical NF-κB signaling in adipocytes could have beneficial metabolic effects |
| Tang, Y et al. [51] | “RAGE/NF-κB pathway mediates hypoxia-induced insulin resistance in 3T3-L1 adipocytes” | 2020 | • Impaired activation of NF-κB by hypoxia in RAGE-deficient adipocytes • An NF-κB inhibitor blunts hypoxia-induced increase in RAGE expression in adipocytes |
• Study suggests hypoxia induces NF-κB activity indirectly through RAGE Also suggests a positive feedback loop in adipocytes, wherein NF-κB activity boosts RAGE expression during hypoxia |
Most of the other in vitro studies in the adipocyte NF-κB field have focused on several keywords: TNF-α, p65/Rel-A, TLRs, and hypoxia. These studies started in the 3T3-L1 cell line and quickly moved to human adipocytes. A common theme emerged in these studies, basically regressing to (1) a variety of stimuli activate NF-κB in adipocytes (discussed in an earlier section) (2) this activation involves nuclear translocation and/or phosphorylation of Rel-A/p65; (3) the activating stimuli increase the expression of numerous cytokine and chemokine genes but not in the absence of functional NF-κB. One limitation of some of these studies is that NF-κB activation was assessed through DNA binding to synthetic probes without identifying bona fide genomic binding sites for these proteins. This caveat is an important consideration; while a powerful quantitative tool, EMSA studies only tell us that transcription factors are present in nuclear extracts and competent to bind to DNA. They provide no information as to whether these proteins occupy particular genomic sites in living cells. As I shall elaborate upon later, I believe this represents a significant knowledge gap in the field.
Other studies in Table 1 highlighted potential species differences in NF-κB action in adipocytes; for instance, whereas pharmacological inhibition of NF-κB partially blocks TNF-α-induced lipolysis in human adipocytes [71], pharmacological inhibition of NF-κB in 3T3-L1 cells (mouse) doesnʼt block TNF-α-induced lipolysis at all [88] (It should be noted, however, that different methods of pharmacological inhibition could also explain these differences.). These studies provide a robust base from which to launch future studies, especially discovery-driven studies such as shRNA or CRISPR-mediated loss of function studies in adipocytes, combined with microarrays or RNA-seq, ChIP-Seq, and identifying novel NF-κB interaction partners or post-translational modifications in adipocytes.
TRUE ADIPOCYTE-SPECIFIC NF-κB LOSS-OF-FUNCTION STUDIES IN VIVO REVEAL A SECONDARY, APOPTOSIS-DRIVEN AND NF-κB-INDEPENDENT INFLAMMATION
A few notable loss-of-function studies have specifically examined NF-κB activity in adipocytes in vivo, presented in Table 2. These studies represent true adipocyte-specific knockouts, in which the investigators used the Adipoq promoter/control region to drive Cre recombinase expression for the excision of floxed alleles in mature adipocytes. As described in a seminal paper by Lee et al. [118], the Adipoq promoter affords the best and most adipocyte-specific method of manipulating gene expression in vivo, with minimal “leakage” of expression in other tissues. Most prior studies had used the aP2 (Fabp4) promoter, which is considered “leaky” with substantial expression in macrophages and other cell types. Therefore, studies of NF-κB manipulation that employ the aP2-Cre transgene cannot be regarded as genuinely adipocyte-specific, with macrophage alterations in NF-κB activity a major confounding variable. Therefore, Table 2 does not include all gain- or loss-of-function studies of NF-κB in vivo, as they either used aP2-Cre to drive expression of Cre or examined NF-κB activity or related gene expression whole adipose tissue without fractionation into mature adipocytes vs the stromovascular fraction. Thus, one cannot determine from which cell type (adipocytes vs macrophages) the metabolic phenotypes arise. Those studies are presented in Table 3.
Table 2.
True Adipocyte-Specific NF-κB Knockout Studies.
| Authors | Title | Year published | Major findings | Comments |
|---|---|---|---|---|
| Kwon et al. [111] | “Adipocyte-specific IKKbeta signaling suppresses adipose tissue inflammation through an IL-13-dependent paracrine feedback pathway” | 2014 | • HFD-induced inflammation is worsened in adipocyte-specific IKKβ KO mice in epidydimal WAT, but not subcutaneous WAT • Expression and secretion of IL-13, an anti-inflammatory cytokine, is decreased in epididymal WAT of adipocyte-specific IKKβ-KO mice |
• First paper to demonstrate that lack of NF-κB in adipocytes does not improve measures of inflammation • Reduced expression and secretion of IL-13 may play a causative role in this phenomenon |
|
| ||||
| Park, S. et al. [112] | “IKKbeta Is Essential for Adipocyte Survival and Adaptive Adipose Remodeling in Obesity” | 2016 | • Adipocyte-specific IKKβ KO mice present with a severe diabetic phenotype on an HFD • Defect appears to be in adipocyte remodeling, lipodystrophy of visceral fat • Inflammation, lipolysis worsened in adipose tissue despite lack of NF-κB signaling • IKKβ appears to protect against adipocyte apoptosis |
• Counterintuitively, adipocyte-specific IKKβ KO animals have insulin resistance and are not protected against inflammation in WAT. Results are consistent with Gao et al. in the aspect of apoptosis but inconsistent since IKKβ KO in adipocytes did not increase energy expenditure. |
| Weidemann A, et al. [32] | “Classical and alternative NF-κB signaling cooperate in regulating adipocyte differentiation and function” | 2016 | • Adipocyte-specific Relb KO animals protected from insulin resistance despite increased adipocyte hypertrophy • An activator of non-canonical NF-κB signaling can induce systemic insulin resistance, but not in adipocyte-specific Relb-KO mice |
• Manuscript demonstrates that non-canonical NF-κB signaling in adipocytes promotes impaired systemic insulin sensitivity and glucose tolerance in vivo • Specific gene targets of RelB/p52 in adipocytes that mediate these effects are unknown |
Table 3.
In vivo Studies Implicating NF-κB in Adipocyte Immunometabolism.
| Authors | Title | Year published | Major findings | Comments |
| Zamboni et al. [113] | “Adiponectin gene expression and adipocyte NF-κB transcriptional activity in elderly overweight and obese women: inter-relationships with fat distribution, hs-CRP, leptin, and insulin resistance” | 2007 | • iκBα gene expression is negatively correlated with abdominal adiposity in humans • This indirectly implies that abdominal adiposity is associated with greater activity of NF-κB itself |
• A limitation of this study was that whole adipose tissue was used, unknowable which cell types are responsible for various correlations |
| Chiang et al. [114] | “The protein kinase IKKepsilon regulates energy balance in obese mice.” | 2009 | • HFD consumption results in a 5-fold increase in NF-κB activity in visceral and subcutaneous WATs • Expression of IKKε increased dramatically in adipocytes from mice on HFD (far more so than in cells of the SVF) • Smaller, more numerous adipocytes in Ikbke-/- mice; animals exhibited increased energy expenditure and less HFD-induced weight gain (despite eating more calories) • Dramatically increased Ucp1 expression in WAT of Ikbke-/- mice • Improved glucose, lipid homeostasis, and insulin signaling in the HFD-fed KO animals • Transfection of an IKKε vector into 3T3-L1 adipocytes reduces glucose uptake • Protection of Ikbke-/- mice from hepatic steatosis • Significantly reduced chronic inflammation seen in multiple tissues of HFD-fed Ikbke-/- mice |
• Manuscripts identified a major new player in adipose tissue inflammation • IKKε, an established target of classical NF-κB signaling, promotes insulin resistance and impairs adipogenesis in adipose tissues • Loss-of-function results suggest that IKKε mediates major actions of NF-κB in WAT • Effects appear to be largely due to IKKε deficiency in adipocytes, but an adipocyte-specific model could still be warranted |
| Tang, T et al. [97] | “Uncoupling of inflammation and insulin resistance by NF-κB in transgenic mice through elevated energy expenditure.” | 2010 | • Significant increase in expression of NF-κB target genes in WAT of aP2-p65 transgenic mice on a normal diet (expected result) • aP2-Rel-A/p65 transgenic animals display elevated energy expenditure and decreased epididymal fat mass. • p65/Rel-A inhibits adipogenesis • Resistance of aP2-p65 mice to diet-induced obesity • Enhanced insulin sensitivity of aP2-p65 mice on HFD • Similar phenotype in global Nfkb1-/-(p50) KO mice |
• Rela overexpression model in adipocytes and macrophages • Energy expenditure could be an artifactual result (since aP2-p65 KO mice also displayed increased energy expenditure [115]) • Enhanced insulin sensitivity is surprising but possibly a result of increased energy expenditure and lower fat pad mass |
| Jiao, P et al. [116] | “Constitutive activation of IKKβ in adipose tissue prevents diet-induced obesity in mice” | 2012 | • Body weights of aP2-IKKβ transgenic animals are reduced on both normal and high-fat diets • Reduced fat pad weights in the Tg mice • Expression of IL-6 and MCP-1 increased in aP2-IKKβ WAT on both chow and HFD as well as in aP2-IKKβ isolated adipocytes • Macrophage infiltration may have been enhanced in aP2-IKKβ Tg mice (not quantified in this study) • Increased food intake and energy expenditure in aP2-IKKβ Tg mice • Increased expression of fatty acid oxidation and thermogenesis-related genes in BAT and muscle • Circulating levels of IL-6 and TNF-α increased in aP2-IKKβ Tg mice • Improved insulin sensitivity • Animals largely phenocopy aP2-p65 Tg mice |
• Results corroborate Tang et al. [97] with very similar phenotypes • Increased energy expenditure appears to be linked to elevated IL-6 in both models • Suggests that overall energy balance can override local inflammation in WAT • Also suggests that local inflammation in WAT cannot drive systemic insulin resistance unless there is a chronic positive energy balance |
| Gao, Z et al. [117] | “p65 inactivation in adipocytes and macrophages attenuates adipose inflammatory response in lean but not in obese mice” | 2015 | • Adipocyte/macrophage p65-κO reduces adipose inflammation in lean mice • Surprisingly, the same knockout scheme worsened inflammation in obese mice • Adipocyte apoptosis was increased in the KO mice, possibly leading to a secondary inflammation • KO obese mice displayed increased energy expenditure, possibly explaining lack of signs of insulin resistance despite increased WAT inflammation |
• What transcription factors are involved in stimulating apoptosis-induced inflammation in the absence of p65? • Would similar effects be observed using a true adipocyte-specific knockout scheme (Adipoq-Cre?) |
With that caveat in mind, only two studies examined Adipoq-Cre-driven deletion of canonical NF-κB pathway components in vivo; both of these involved IKKβ [111,112]. The most surprising findings in both studies were that HFD-induced WAT inflammation was actually worsened in the face of IKKβ deficiency. In the first study, the effect (specifically, an increase in HFD-induced expression of inflammatory cytokines) was seen only in visceral WAT [111], suggesting that either IKKβ is dispensable for inflammation in subcutaneous WAT or that this tissue did not “see” any inflammatory stimuli under the experimental conditions. Curiously, whereas whole epididymal adipose tissue expression of cytokines was accentuated in adipocyte-specific IKKβ-KO animals, an expected decrease in cytokine-stimulated TNF-α, IL-6, and IL-13 expression was observed in differentiated adipocytes from these animals. This, of course, suggests a level of compensation occurring in non-adipose cells in vivo, a phenomenon discussed in more detail later. Of note, there was no sign of cell-autonomous apoptosis occurring in the differentiated but unstimulated adipocytes from these animals, leading the authors to passively conclude that the worsened inflammation in visceral fat was not the result of increased fat cell death per se. In contrast, while the study by Park et al. also demonstrated worsened inflammation in epidydimal WAT of adipocyte-specific IKKβ-KO mice, there was a marked impairment of adipocyte remodeling and a significant increase in caspase-mediated fat cell death [112]. It is important to note that the worsened inflammation in both models occurred independently of any changes in food intake or body weight.
In the Kwon study, it is quite possible that the observed increase in inflammatory cytokine expression was mainly coming from cells of the stromovascular fraction; the authors of this study evidently did not fractionate the WAT prior to their analysis. Adipocyte death was not quantified directly in that study, but the high degree of macrophage infiltration and crown-like structures suggests that this parameter did increase. Previous studies had shown that NF-κB regulates many apoptosis-related genes, including Bax [115], Bcl-2 [119], Bim [120], XIAP [121], CIDEA [122], and many others, at least in non-fat cells. In the Park study, fat cell apoptosis was unequivocally increased in epididymal WAT of adipocyte-specific IKKβ-KO mice, as evidenced by elevated levels of activated (cleaved) Caspase-3 and Poly (ADP-ribose) proteins [112]. Likewise, HFD-induced macrophage infiltration was also increased in epididymal WAT of adipocyte-specific IKKβ-KO mice, consistent with the previous observation that genetically-forced apoptosis of fat cells also results in extensive macrophage infiltration [123]. These results clearly indicate that macrophage infiltration can occur in adipose tissue devoid of classical NF-κB signaling in adipocytes. Notably, classical NF-κB signaling in macrophages is also dispensable for infiltration in adipose tissue, as Fabp4 promoter-driven deletion of IKKβ resulted in a very similar phenotype [112].
The only other study using the Adipoq promoter to drive deletion of an NF-κB subunit was Weidemann et al., who used the system to delete RelB from adipocytes [32], as discussed earlier. They showed the peculiar result of increased insulin sensitivity and glucose tolerance in the face of increased adipocyte hypertrophy, accompanied by increased expression of adipocyte-specific markers in epidydimal WAT. These changes occurred without any alterations in food intake or body weight (although there was a significant increase in the mass of epidydimal WAT). The implication that alternative (non-canonical) NF-κB signaling impairs overall glucose tolerance and insulin sensitivity was corroborated by the fact that an agonistic α-LTβR antibody failed to impair insulin sensitivity or glucose tolerance in the adipocyte-specific RelB-KO mice. This manuscript stands alone as the sole paper to examine the function of non-canonical NF-κB signaling in adipocytes in an in vivo context and suggests that p65/p50 is not the only game in town for promoting metabolic dysfunction in fat. This also implies that targeting non-canonical NF-κB signaling in adipocytes could represent a viable alternative to inhibiting canonical signaling, especially since there is no evidence—at least so far—that non-canonical NF-κB signaling regulates apoptosis in adipocytes.
Taken together, the bulk of the evidence from the best adipocyte-specific studies suggests a “dammed if you do, dammed if you donʼt” quandary when it comes to canonical NF-κB activation in adipocytes: having normal NF-κB activity in adipocytes will readily promote HFD-induced expression of inflammatory cytokines and macrophage infiltration through direct mechanisms, whereas inhibiting NF-κB will have a similar effect through an indirect route (increased fat cell apoptosis). Thus, if canonical NF-κB is to remain an effective therapeutic target for metabolic inflammation in obesity, we may have to find a way to specifically target only a subset of canonical NF-κB target genes in adipocytes. We should also determine what, if any, non-NF-κB dependent signals are released from apoptotic adipocytes that promote local macrophage infiltration and inflammation. Possible alternative means of inflammatory signaling in adipose tissue devoid of NF-κB in adipocytes are discussed in a subsequent section.
NON-TRULY ADIPOCYTE-SPECIFIC NF-κB MANIPULATION STUDIES IN VIVO
Several [highly valuable] studies have examined NF-κB function in WAT in vivo that either did not use truly adipocyte-specific deletion or examined parameters such as chemokine expression in whole WAT without separation of adipocytes from non-adipocytes. The genetic or pharmacological manipulations used in these studies affected NF-κB function or activity in adipocytes or adipose tissue and resulted in a significant metabolic phenotype. These studies are presented in Table 3 and discussed briefly below. This list includes some human studies that clearly link adipose tissue inflammation to increased NF-κB activity.
In one of the significant manuscripts to examine NF-κB-related parameters in human WAT, Zamboni et al. showed in 2007 a negative correlation between IκBα gene expression and abdominal obesity [113]. This observation makes sense from the standpoint of lower IκBα levels implying higher NF-κB activity in visceral WAT, but since IκBα is itself a target of p65/p50 [124–126], this fact calls into question the direction of causality in this study. In another landmark paper in 2009, Chiang et al. demonstrated that germline lack of IKKε-protects mice from HFD-induced insulin resistance, with dramatic reductions in inflammatory gene expression in WAT [114]. However, it is unclear what contribution the loss of IKKε in adipocytes made to this phenotype, especially since the animals displayed increased energy expenditure. (Increased energy expenditure would tend to reduce metabolic stress on adipocytes, which would, in turn, reduce inflammatory pressure to begin with). As an NF-κB-related protein with clear implications in adipose inflammation, the reader should, at this point, guess what the authorʼs suggested strategy to interrogate this question further would be.
Several other studies manipulating NF-κB components in both adipocytes and macrophages demonstrated a metabolic phenotype with significant alternations in food intake and/or energy expenditure. Two of these involved the overexpression of two NF-κB components (IKKβ and p65/Rel-A), and one involved the deletion of p65/Rel-A. Curiously, both overexpression of p65/Rel-A [97] and KO of p65/Rel-A [117] results in a similar phenotype of elevated energy expenditure. It is not clear why energy expenditure was increased in both models; however, in the Tang study, circulating levels of IL-6 were increased by more than 20-fold, and IL-6 has long been recognized as a potent central regulator of energy expenditure [127–133]. aP2-IKKβ transgenic animals have a similar metabolic phenotype as the aP2-p65 mice, with resistance to diet-induced obesity, enhanced insulin sensitivity, and increased energy expenditure [116]. Circulating levels of IL-6 were also significantly up-regulated in the aP2-IKKβ study [116]. In the Gao study—in which the authors deleted p65 from adipocytes and macrophages—the expression of IL-6 doubled in apoptosis-laden adipose tissue, although circulating IL-6 levels remained unchanged [117]. Thus, an explanation for the increased energy expenditure in adipocyte/macrophage-specific p65/Rel-A-KO animals remains elusive (although the timing of serum cytokine measurements or other experimental parameters might have played a role).
Of note, adipocyte-specific IKKβ-KO mice present with nominal energy expenditure. This result implies either that the increase in energy expenditure seen in aP2-p65 transgenic mice represents an artifactual finding or that the increase in circulating IL-6 seen in those animals mainly came from macrophages. As circulating IL-6 was not measured in either adipocyte-specific IKKβ-KO study, it is difficult to infer the specific roles of IKKβ vs p65 in macrophages and adipocytes and their contributions to overall energy expenditure.
Both the aP2-p65 transgenic model [97] and the aP2-p65-κO model [117] also presented with substantial HFD-induced local adipose tissue inflammation. As discussed earlier, the increase in inflammation in the KO study corresponded to an increase in adipocyte and macrophage apoptosis, as demonstrated by increased caspase 7 cleavage in both cell types [117]. This result was consistent with the adipocyte-specific IKKβ-KO models discussed earlier and confirmed that the apoptosis-inhibiting effects of IKKβ are mediated, at least in part, through p65. The increase in WAT inflammation in the transgenic models [97,116] was arguably expected, although perhaps somewhat unphysiological. As mentioned earlier, an increase in circulating cytokines (especially IL-6), as well as increased expression of fatty acid oxidation and thermogenesis genes in BAT and muscle, could explain the increased energy expenditure observed in models of constitutive NF-κB activity. These results indicate an uncoupling between the well-accepted notion of overnutrition-induced WAT inflammation being causally related to insulin resistance in obesity. While interesting in their own right, targeted studies involving adipocyte-specific deficiencies of each NF-κB subunit could yield more powerful and physiologically-relevant information.
SECONDARY APOPTOSIS-DRIVEN NF-κB-INDEPENDENT INFLAMMATION IN WAT
It remains unclear at this time how the expression of inflammatory cytokines or infiltration of immune cells within adipose tissue are maintained when NF-κB function is taken out of action in adipocytes. A possible explanation is that adipocyte apoptosis activated ATMs (Adipose Tissue Macrophages) and other immune cells not through increased secretion of inflammatory cytokines but through other mechanisms. An intriguing candidate for this would be the NLR Family Pyrin Domain Containing 3 (NLRP3) inflammasome pathway [134–138]. Like TLRs, NLRP3 is a cytoplasmic pattern recognition receptor in macrophages and other cell types that detects products of damaged (endogenous) cells (Danger-Associated Molecular Patterns, or DAMPs) such as lysophosphatidylcholine and uric acid. Once activated, NLRP3 oligomerizes to form an “inflammasome”, an intracellular signaling module that activates pro-Caspase-1. Caspase-1 then cleaves pro-interleukin-1β and pro-IL-18 into their biologically active forms. The increase in IL-1β can then lead to infiltration of immune cells. Inflammasome activation can also lead to an inflammatory cell death known as pyroptosis [139,140].
In one of the landmark papers examining the role of the Nlrp3 inflammasome in obesity-associated insulin resistance, Nlrp3 components were expressed in macrophages but not adipocytes [134]. However, later studies demonstrated inducible expression of Nlrp3 components in fat cells [141–144]. Thus, it seems like an attractive model to explain the fact that inflammation is worsened (as measured by an increase in macrophage infiltration or an increase in expression of inflammatory cytokines) in true NF-κB KO models is that adipocyte apoptosis increases the production of DAMPs, which then ligands with macrophages to activate the Nlrp3 inflammasome and induce production of IL-1β and IL-18. These, in turn, could recruit additional immune cells and lead to a secondary wave of cytokines such as IL-6 and others that account for the significant increases in these hormones seen in the Park study of IKKβ deficiency in adipocytes [112] (Of note, the LPS-induced increase in the expression of some of these is reduced in Nlrp3-deficient human visceral adipocytes [144], suggesting that NF-κB cannot maintain their expression without the Nlrp3 inflammasome in place.). Since both studies looked at inflammatory gene expression in whole adipose tissue, not fractionated cells, we cannot determine the primary origin of these cytokines, although it is safe to say that they are not coming from dead adipocytes. Therefore, it is likely that most of the increased WAT cytokine expression seen in NF-κB knockdown studies in vivo comes from surviving adipocytes and macrophages (in an Nlrp3-dependent manner), other immune cells that featured intact NF-κB activity, or both. Another critical limitation of in vitro studies is that NF-κB inactivation appears to be insufficient to induce apoptosis in this setting; in an in vivo setting, apoptosis in the adipocyte-specific NF-κB knockout was only seen in epididymal WAT and only when an HFD was consumed [112].
Several groups have reported that components of the Nlrp3 inflammasome are under transcriptional control of NF-κB, most notably IL-1β and Nlrp3 itself [145–147]. In the canonical model of Nlrp3 activation, its expression must first be “primed” through NF-κB activation. Thus, a two-step model has been proposed, in which various factors (LPS, TNF-α, etc.) activate NF-κB to stimulate transcription of chemokines and cytokines as well as the components of the inflammasome themselves [147]. Should the inflammation continue to the point of damage to nearby cells, the second stage becomes activated (through ligation of DAMP receptors), culminating in increased secretion of IL-1β and IL-18. If true, this provides an argument against the idea that a compensatory increase in Nlrp3 inflammasome mediates the increase in expression of inflammatory cytokines in WAT of aP2-p65 KO mice [117] since the first priming step for Nlrp3 activation presumably would have failed in both adipocytes and macrophages. However, an alternative pathway has also been proposed in which basal levels of Nlrp3 components are sufficient to mediate inflammasome activation independently of new protein synthesis or NF-κB [148–150]. Any future studies involving truly adipocyte-specific knockout of NF-κB subunits would do well to examine the activity of the Nlrp3 inflammasome pathway in adipocytes as well as the remaining immune cells.
2020 AND BEYOND: “OMICS” RESEARCH INVOLVING NF-κB IN ADIPOSE TISSUE
The recent turn of the decade saw a continuation of research that involved NF-κB in adipocytes—somewhat serendipitously—but did not necessarily focus on NF-κB from the outset. I will briefly discuss two of the most salient articles in this section. The first study by Mohallem et al. employed an unbiased, integrated quantitative proteomics study to identify novel proteins involved in mediating TNF-α-induced insulin resistance in 3T3-L1 adipocytes [151]. Perhaps surprisingly, only one NF-κB subunit protein was upregulated (in amount) in this study, p52 (which is not to say that the other subunits were not transcriptionally activated upon TNF-α treatment); however, the expression of numerous NF-κB-dependent gene products was unsurprisingly increased. The upregulation of the Nfkb2 gene product was consistent with the results of Wiedemann et al., who also demonstrated an increase in p100 levels in adipocytes upon TNF-α treatment [32]. Those authors proposed a model in which TNF-α “primes” Relb and Nfkb2 expression, whereas ligation of the LTβR triggers the activation of RelB/p52. Since the authors of the proteomics study did not treat the cells with ligands that can directly activate non-canonical NF-κB, the functional significance of the results is unknown. One secreted gene product that was upregulated in the proteomics study was galetin-3-binding protein, a glycoprotein that had previously been shown to be involved in macrophage recruitment [152].
There was a notable lack of several “high-profile” NF-κB-dependent chemokines that were differentially regulated by TNF-α in the Mohallem study, such as Il-6, Ccl5, Rantes, and others, although there were still a number of upregulated secreted proteins that had previously been shown to be NF-κB dependent, such as Csf1 [153,154], C3 [155], Col1a2 [156,157], and S100A6 [158]. One possible explanation would be the time frame measured for these secreted proteins, which occurred between Day 4 and Day 8 of TNF-α treatment; in the seminal Tourniaire study [72], the authors observed significant increases in the expression of chemokines such as CCL2 and CCL5 in human adipocytes after a 24-hour TNF-α treatment. Other studies that were previously discussed herein–also using shorter TNF-α treatment times–corroborate these results [74,86]. It is thus possible that a transient secretory burst of these chemokines in the Mohallem study [151] had already come and gone and was not apparent through prolonged TNF-α treatment. It is also possible that species differences could account for these discrepancies. Further studies involving full time courses of TNF-α treatment in both mouse and human adipocytes could shed light on this issue.
The second study unveiled a fascinating relationship between the circadian clock and NF-κB activity in human omental fat [159]. The authors made the serendipitous discovery that BMAL1, a core clock activator, competes with NF-κB for control of some targets (including PER2, a critical clock repressor gene), with NF-κB motifs being overrepresented in close proximity to BMAL1 motifs in human Omental Adipose Precursors (OAPs). Significantly, the greater amount of NF-κB activity at or near BMAL1 bindings sites in OAPs from obese individuals differentiated them from OAPs derived from non-obese individuals. A causal relationship was demonstrated by the fact that inhibition of NF-κB in OAPs restored normal clock function in these cells. Another example of a “disputed” gene with regards to BMAL1/NF-κB antagonism was the well-known chemokine MCP-1 (encoded by Ccl2), which was repressed by BMAL1 but activated when BMAL1 was displaced by p65/REL-A. To demonstrate the physiological significance of these findings in vivo, the authors inhibited NF-κB pharmacologically and through a genetic approach (Tamoxifen-dependent IKKβ-KO mice). Both models demonstrated a reduction in PER2 expression, and the genetic model also showed reduced CCL2 expression, proving that NF-κB can antagonize normal clock activity in adipocytes in vivo. One limitation of this study, however, was the fact that only human OAPs were used, rather than mature human adipocytes; it may be interesting to study those, especially since several NF-κB subunits are much higher in expression in preadipocytes as compared to adipocytes, at least in mouse 3T3-L1 cells [86]. In any case, this study adds an exciting new temporal dimension to NF-κB biology in adipocytes and is perfectly in line with the mechanistically-focused studies that I would like to see more of in the future.
UNBIASED, DISCOVERY-DRIVEN APPROACHES FOR FUTURE STUDIES OF NF-κB BIOCHEMISTRY IN ADIPOCYTES
Although we have made significant progress in understanding the adipocyte-specific role of NF-κB in metabolic inflammation, much work remains. This is especially true if we are to realistically follow through with notions of developing NF-κB inhibitors as a treatment or prophylactic for obesity-associated metabolic inflammation. The bulk of evidence would suggest that general inhibitors of the canonical NF-κB pathway are a “no-go” due to their putative adipocyte apoptosis-inducing effects as well as likely detrimental effects on the immune system. However, all may not be lost if one believes that it is possible to intervene at some NF-κB target loci in adipocytes but not others. One hypothetical example of how we could achieve this would be to develop a drug that blocks protein-protein interactions between NF-κB and an adipocyte-specific transcription factor that only interacts with NF-κB at chemokine (but apoptosis-related) genes. In principle, this would allow normal NF-κB activity at all target loci in non-adipocyte immune cells, thereby preserving the functioning of the immune system. Of course, this will require discovery-driven experiments to identify such factors and to prove, perhaps with recombinant peptides fragments, that disrupting their interactions with NF-κB produces the desired outcome. The ideal unbiased approaches for finding novel NF-κB-interacting proteins in adipocytes—such as Tandem Affinity Purification (TAP) or Stable Isotope Labeling by Amino Acids in Cell Culture (SILAC)—would also prove useful in identifying post-translational modifications of all proteins involved [160–166]. Utilizing these approaches with and without an inflammatory stimulus would constitute an especially powerful approach for uncovering new NF-κB biology in adipocytes. A recent in silico study identifying functional NF-κB interaction partners in adipose tissue represents an excellent first step in the quest to identify new NF-κB biological networks involved in obesity-associated inflammation [167].
The idea of inhibiting transcription factors as a treatment for disease is not new. Indeed, inhibition of specific transcription factors has gained traction in recent years as a more general therapeutic option. Once thought as “undruggable” (mainly due to lack of an enzyme active site), efforts have focused mainly on inhibiting interactions with other transcription factors or proteins or inhibiting the enzymes that post-translationally modify transcription factors. After all, transcription factors are only as good as the proteins they interact with; thus, inhibiting specific protein-protein interactions might also represent a viable approach, and it may even be possible to modulate specific genomic targets of NF-κB but not others (For some sense of the veracity of this approach, refer to reference [168], where Zhao et al. demonstrated that in lymphoblastoid B cells, the transcription factor FOXM1 both co-occupies nearly 50% of genomic NF-κB binding sites and physically interacts with p65/Rel-A. They too pointed towards the idea of a “selective blockade” for NF-κB based on these interactions, especially since most adult tissues do not express FOXM1.).
As it turns out, to our delight, most of the research on transcription factor inhibition has focused on disrupting protein-protein interactions, including NF-κB as a prime target of interest (reviewed in [169]). For instance, several groups have developed compounds that inhibit NF-κB translocation, DNA binding, and transactivation, many of which focus on inhibiting the enzymes that install post-translational modifications into NF-κB proteins [170,171]. In another proof-of-concept example, other groups have developed compounds to inhibit c-Myc/Max dimerization, and several of these have shown functional success in vitro [172,173].
Two more pieces of information would prove helpful in selectively targeting NF-κB-regulated loci in adipocytes. The first piece is a catalog of messages (including siRNAs) whose expression is NF-κB dependent in adipocytes, which we could achieve by combining genetic knockdowns with RNA-seq. The second piece is a catalog of genomic binding sites for each NF-κB subunit in the presence and absence of an inflammatory stimulus; this would also allow us to infer which transcriptional targets are likely to be direct and whether binding sites for other transcription factors, especially those shown to physically interact with NF-κB subunits, are placed in close proximity. There is a relative paucity of studies examining genomic binding sites for NF-κB in live cells; although likely to prove technically challenging (especially in adipocytes), several groups have realized success in that endeavor in recent years [168,174–177].
Meanwhile, future in vivo studies should focus on documenting the metabolic phenotypes of true adipocyte-specific knockouts (driven by Adiponectin-Cre transgenic mice) of all five NF-κB subunits. To this author’s knowledge, except for RelB [32], no research labs have systematically analyzed true adipocyte-specific knockouts of individual NF-κB subunits. Although conducting such experiments might seem to represent somewhat of a tautology (some would argue that demonstrating, for instance, that adipocyte-specific p65 KO mice have reduced obesity-associated inflammation yields no new biology), the Gao study, in which such mice demonstrated increased inflammation [117], underscores the need to prosecute such studies in order to uncover new and unexpected biological outcomes. One possible way to eliminate all classical NF-κB activity in adipocytes would be to engineer Adiponectin-IκBα-mut transgenic mice, in which mature adipocytes would express a dominant-negative IκBα mutant protein under the control of Adipoq control elements. Such an approach might represent the most parsimonious way to reduce the functional activity of all five NF-κB subunits specifically within adipocytes in an in vivo context.
In sum, the last few decades have shown significant progress in understanding NF-κB activity in adipocytes. However, the prospects of developing a general drug inhibitor for canonical NF-κB signaling as a treatment for obesity-associated metabolic inflammation seem dim, mainly due to a likely increase in adipocyte apoptosis. However, selective targeting of specific NF-κB-regulated loci in adipocytes through precision drugs could remain a viable option. To achieve that end, we should commit to conducting many unbiased, discovery-driven investigations in adipocytes, including generating a comprehensive list of direct and indirect targets of NF-κB in adipocytes, mapping the NF-κB cistromes (for all five NF-κB subunits), and identifying novel protein interaction partners and post-translational modifications for NF-κB. These approaches should complement physiological, in vivo studies using truly adipocyte-specific NF-κB subunit loss-of-function models. Developing drug inhibitors of non-canonical NF-κB signaling may also hold some promise for treating obesity-associated inflammation.
ACKNOWLEDGEMENT
This work was supported by grant R15 DK119792 “Transcriptional Regulation Of Adipocyte Inflammation By Early B-Cell Factor-1 (Ebf1).”
Footnotes
CONFLICTS OF INTEREST
The author declares that he has no conflict of interest.
REFERENCES
- 1.Antuna-Puente B, Feve B, Fellahi S, Bastard JP. Adipokines: the missing link between insulin resistance and obesity. Diabetes Metab. 2008;34(1):2–11. [DOI] [PubMed] [Google Scholar]
- 2.Batista ML Jr, Olivan M, Alcantara PS, Sandoval R, Peres SB, Neves RX, et al. Adipose tissue-derived factors as potential biomarkers in cachectic cancer patients. Cytokine. 2013;61(2):532–9. [DOI] [PubMed] [Google Scholar]
- 3.Dorresteijn JA, Visseren FL, Spiering W. Mechanisms linking obesity to hypertension. Obes Rev. 2012;13(1):17–26. [DOI] [PubMed] [Google Scholar]
- 4.Fuentes E, Fuentes F, Vilahur G, Badimon L, Palomo I. Mechanisms of chronic state of inflammation as mediators that link obese adipose tissue and metabolic syndrome. Mediators Inflamm. 2013;2013:136584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Goran MI, Alderete TL. Targeting adipose tissue inflammation to treat the underlying basis of the metabolic complications of obesity. Nestle Nutrition Institute workshop series. 2012;73:49–60; discussion p1–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Guilherme A, Virbasius JV, Puri V, Czech MP. Adipocyte dysfunctions linking obesity to insulin resistance and type 2 diabetes. Nat Rev Mol Cell Biol. 2008;9(5):367–77. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Hefetz-Sela S, Scherer PE. Adipocytes: impact on tumor growth and potential sites for therapeutic intervention. Pharmacol Ther. 2013;138(2):197–210. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Heilbronn LK, Campbell LV. Adipose tissue macrophages, low grade inflammation and insulin resistance in human obesity. Curr Pharm Des. 2008;14(12):1225–30. [DOI] [PubMed] [Google Scholar]
- 9.Lysaght J, van der Stok EP, Allott EH, Casey R, Donohoe CL, Howard JM, et al. Pro-inflammatory and tumour proliferative properties of excess visceral adipose tissue. Cancer Lett. 2011;312(1):62–72. [DOI] [PubMed] [Google Scholar]
- 10.Matafome P, Santos-Silva D, Sena CM, Seica R. Common mechanisms of dysfunctional adipose tissue and obesity-related cancers. Diabetes Metab Res Rev. 2013;29(4):285–95. [DOI] [PubMed] [Google Scholar]
- 11.Mlinar B, Marc J. New insights into adipose tissue dysfunction in insulin resistance. Clin Chem Lab Med. 2011. Sep 6;49(12):1925–35. [DOI] [PubMed] [Google Scholar]
- 12.Nieman KM, Romero IL, Van Houten B, Lengyel E. Adipose tissue and adipocytes support tumorigenesis and metastasis. Biochim Biophys Acta. 2013;1831(10):1533–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Rasouli N, Kern PA. Adipocytokines and the metabolic complications of obesity. J Clin Endocrinol Metab. 2008;93(11 Suppl 1):S64–73. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Romeo GR, Lee J, Shoelson SE. Metabolic syndrome, insulin resistance, and roles of inflammation--mechanisms and therapeutic targets. Arterioscler Thromb Vasc Biol. 2012;32(8):1771–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Sun S, Ji Y, Kersten S, Qi L. Mechanisms of inflammatory responses in obese adipose tissue. Annu Rev Nutr. 2012;32:261–86. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Tanti JF, Ceppo F, Jager J, Berthou F. Implication of inflammatory signaling pathways in obesity-induced insulin resistance. Front Endocrinol. 2012;3:181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Tilg H, Moschen AR. Inflammatory mechanisms in the regulation of insulin resistance. Mol Med. 2008;14(3–4):222–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Trinchieri G Cancer and inflammation: an old intuition with rapidly evolving new concepts. Annu Rev Immunol. 2012;30:677–706. [DOI] [PubMed] [Google Scholar]
- 19.Xu H, Barnes GT, Yang Q, Tan G, Yang D, Chou CJ, et al. Chronic inflammation in fat plays a crucial role in the development of obesity-related insulin resistance. J Clin Invest. 2003;112(12):1821–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Griffin MJ. Nipping Adipocyte Inflammation in the Bud. Immunometabolism. 2021;3(2):e210012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Hayden MS, Ghosh S. Shared principles in NF-kappaB signaling. Cell. 2008;132(3):344–62. [DOI] [PubMed] [Google Scholar]
- 22.Baker RG, Hayden MS, Ghosh S. NF-kappaB, inflammation, and metabolic disease. Cell Metab. 2011;13(1):11–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhong H, May MJ, Jimi E, Ghosh S. The phosphorylation status of nuclear NF-kappa B determines its association with CBP/p300 or HDAC-1. Mol Cell. 2002;9(3):625–36. [DOI] [PubMed] [Google Scholar]
- 24.Grundstrom S, Anderson P, Scheipers P, Sundstedt A. Bcl-3 and NFkappaB p50-p50 homodimers act as transcriptional repressors in tolerant CD4+ T cells. J Biol Chem. 2004;279(9):8460–8. [DOI] [PubMed] [Google Scholar]
- 25.Carmody RJ, Ruan Q, Palmer S, Hilliard B, Chen YH. Negative regulation of toll-like receptor signaling by NF-kappaB p50 ubiquitination blockade. Science. 2007;317(5838):675–8. [DOI] [PubMed] [Google Scholar]
- 26.Elsharkawy AM, Oakley F, Lin F, Packham G, Mann DA, Mann J. The NF-kappaB p50:p50:HDAC-1 repressor complex orchestrates transcriptional inhibition of multiple pro-inflammatory genes. J Hepatol. 2010;53(3):519–27. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Sun SC. The non-canonical NF-kappaB pathway in immunity and inflammation. Nat Rev Immunol. 2017;17(9):545–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Dejardin E, Droin NM, Delhase M, Haas E, Cao Y, Makris C, et al. The lymphotoxin-beta receptor induces different patterns of gene expression via two NF-kappaB pathways. Immunity. 2002;17(4):525–35. [DOI] [PubMed] [Google Scholar]
- 29.Claudio E, Brown K, Park S, Wang H, Siebenlist U. BAFF-induced NEMO-independent processing of NF-kappa B2 in maturing B cells. Nat Immunol. 2002;3(10):958–65. [DOI] [PubMed] [Google Scholar]
- 30.Kayagaki N, Yan M, Seshasayee D, Wang H, Lee W, French DM, et al. BAFF/BLyS receptor 3 binds the B cell survival factor BAFF ligand through a discrete surface loop and promotes processing of NF-kappaB2. Immunity. 2002;17(4):515–24. [DOI] [PubMed] [Google Scholar]
- 31.Novack DV, Yin L, Hagen-Stapleton A, Schreiber RD, Goeddel DV, Ross FP, et al. The IkappaB function of NF-kappaB2 p100 controls stimulated osteoclastogenesis. J Exp Med. 2003;198(5):771–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Weidemann A, Lovas A, Rauch A, Andreas N, von Maltzahn J, Riemann M, et al. Classical and alternative NF-kappaB signaling cooperate in regulating adipocyte differentiation and function. Int J Obes. 2016;40(3):452–9. [DOI] [PubMed] [Google Scholar]
- 33.Fitzgerald KA, Kagan JC. Toll-like Receptors and the Control of Immunity. Cell. 2020;180(6):1044–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bes-Houtmann S, Roche R, Hoareau L, Gonthier MP, Festy F, Caillens H, et al. Presence of functional TLR2 and TLR4 on human adipocytes. Histochem Cell Biol. 2007;127(2):131–7. [DOI] [PubMed] [Google Scholar]
- 35.Vitseva OI, Tanriverdi K, Tchkonia TT, Kirkland JL, McDonnell ME, Apovian CM, et al. Inducible Toll-like receptor and NF-kappaB regulatory pathway expression in human adipose tissue. Obesity. 2008;16(5):932–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Kopp A, Buechler C, Neumeier M, Weigert J, Aslanidis C, Scholmerich J, et al. Innate immunity and adipocyte function: ligand-specific activation of multiple Toll-like receptors modulates cytokine, adipokine, and chemokine secretion in adipocytes. Obesity. 2009;17(4):648–56. [DOI] [PubMed] [Google Scholar]
- 37.Poulain-Godefroy O, Le Bacquer O, Plancq P, Lecoeur C, Pattou F, Fruhbeck G, et al. Inflammatory role of Toll-like receptors in human and murine adipose tissue. Mediators Inflamm. 2010;2010:823486. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Hoshino K, Takeuchi O, Kawai T, Sanjo H, Ogawa T, Takeda Y, et al. Pillars Article: Cutting Edge: Toll-Like Receptor 4 (TLR4)-Deficient Mice Are Hyporesponsive to Lipopolysaccharide: Evidence for TLR4 as the Lps Gene Product. J. Immunol. 2016;197(7):2563–6. [PubMed] [Google Scholar]
- 39.Poltorak A, He X, Smirnova I, Liu MY, Van Huffel C, Du X, et al. Defective LPS signaling in C3H/HeJ and C57BL/10ScCr mice: mutations in Tlr4 gene. Science. 1998;282(5396):2085–8. [DOI] [PubMed] [Google Scholar]
- 40.Qureshi ST, Lariviere L, Leveque G, Clermont S, Moore KJ, Gros P, et al. Endotoxin-tolerant mice have mutations in Toll-like receptor 4 (Tlr4). J Exp Med. 1999;189(4):615–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Trepanowski JF, Mey J, Varady KA. Fetuin-A: a novel link between obesity and related complications. Int J Obes. 2015;39(5):734–41. [DOI] [PubMed] [Google Scholar]
- 42.Pal D, Dasgupta S, Kundu R, Maitra S, Das G, Mukhopadhyay S, et al. Fetuin-A acts as an endogenous ligand of TLR4 to promote lipid-induced insulin resistance. Nat Med. 2012;18(8):1279–85. [DOI] [PubMed] [Google Scholar]
- 43.Snodgrass RG, Huang S, Choi IW, Rutledge JC, Hwang DH. Inflammasome-mediated secretion of IL-1beta in human monocytes through TLR2 activation; modulation by dietary fatty acids. J Immunol. 2013;191(8):4337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Shi H, Kokoeva MV, Inouye K, Tzameli I, Yin H, Flier JS. TLR4 links innate immunity and fatty acid-induced insulin resistance. J Clin Invest. 2006;116(11):3015–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Suganami T, Mieda T, Itoh M, Shimoda Y, Kamei Y, Ogawa Y. Attenuation of obesity-induced adipose tissue inflammation in C3H/HeJ mice carrying a Toll-like receptor 4 mutation. Biochem Biophys Res Commun. 2007;354(1):45–9. [DOI] [PubMed] [Google Scholar]
- 46.Trayhurn P Hypoxia and adipose tissue function and dysfunction in obesity. Physiol Rev. 2013;93(1):1–21. [DOI] [PubMed] [Google Scholar]
- 47.Lempesis IG, van Meijel RLJ, Manolopoulos KN, Goossens GH. Oxygenation of adipose tissue: A human perspective. Acta Physiol. 2020;228(1):e13298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.He Q, Yang QC, Zhou Q, Zhu H, Niu WY, Feng J, et al. Effects of varying degrees of intermittent hypoxia on proinflammatory cytokines and adipokines in rats and 3T3-L1 adipocytes. PLoS One. 2014;9(1):e86326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Taylor CT, Kent BD, Crinion SJ, McNicholas WT, Ryan S. Human adipocytes are highly sensitive to intermittent hypoxia induced NF-kappaB activity and subsequent inflammatory gene expression. Biochem Biophys Res Commun. 2014;447(4):660–5. [DOI] [PubMed] [Google Scholar]
- 50.Lee MY, Wang Y, Mak JC, Ip MS. Intermittent hypoxia induces NF-kappaB-dependent endothelial activation via adipocyte-derived mediators. Am J Physiol Cell Physiol. 2016;310(6):C446–55. [DOI] [PubMed] [Google Scholar]
- 51.Tang Y, Wang J, Cai W, Xu J. RAGE/NF-κB pathway mediates hypoxia-induced insulin resistance in 3T3-L1 adipocytes. Biochem Biophys Res Commun. 2020;521(1):77–83. [DOI] [PubMed] [Google Scholar]
- 52.Lee YS, Kim JW, Osborne O, Oh DY, Sasik R, Schenk S, et al. Increased adipocyte O2 consumption triggers HIF-1alpha, causing inflammation and insulin resistance in obesity. Cell. 2014;157(6):1339–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Collart MA, Baeuerle P, Vassalli P. Regulation of tumor necrosis factor alpha transcription in macrophages: involvement of four kappa B-like motifs and of constitutive and inducible forms of NF-kappa B. Mol Cell Biol. 1990;10(4):1498–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Libermann TA, Baltimore D. Activation of interleukin-6 gene expression through the NF-kappa B transcription factor. Mol Cell Biol. 1990;10(5):2327–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Shakhov AN, Collart MA, Vassalli P, Nedospasov SA, Jongeneel CV. Kappa B-type enhancers are involved in lipopolysaccharide-mediated transcriptional activation of the tumor necrosis factor alpha gene in primary macrophages. J Exp Med. 1990;171(1):35–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Shakhov AN, Kuprash DV, Azizov MM, Jongeneel CV, Nedospasov SA. Structural analysis of the rabbit TNF locus, containing the genes encoding TNF-beta (lymphotoxin) and TNF-alpha (tumor necrosis factor). Gene. 1990;95(2):215–21. [DOI] [PubMed] [Google Scholar]
- 57.Son YH, Jeong YT, Lee KA, Choi KH, Kim SM, Rhim BY, et al. Roles of MAPK and NF-kappaB in interleukin-6 induction by lipopolysaccharide in vascular smooth muscle cells. J Cardiovasc Pharmacol. 2008;51(1):71–7. [DOI] [PubMed] [Google Scholar]
- 58.Kunsch C, Rosen CA. NF-kappa B subunit-specific regulation of the interleukin-8 promoter. Mol Cell Biol. 1993;13(10):6137–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Kang HB, Kim YE, Kwon HJ, Sok DE, Lee Y. Enhancement of NF-kappaB expression and activity upon differentiation of human embryonic stem cell line SNUhES3. Stem Cells Dev. 2007;16(4):615–23. [DOI] [PubMed] [Google Scholar]
- 60.Moriuchi H, Moriuchi M, Fauci AS. Nuclear factor-kappa B potently up-regulates the promoter activity of RANTES, a chemokine that blocks HIV infection. J Immunol. 1997;158(7):3483–91. [PubMed] [Google Scholar]
- 61.Wickremasinghe MI, Thomas LH, OʼKane CM, Uddin J, Friedland JS. Transcriptional mechanisms regulating alveolar epithelial cell-specific CCL5 secretion in pulmonary tuberculosis. J Biol Chem. 2004;279(26):27199–210. [DOI] [PubMed] [Google Scholar]
- 62.Ueda A, Okuda K, Ohno S, Shirai A, Igarashi T, Matsunaga K, et al. NF-kappa B and Sp1 regulate transcription of the human monocyte chemoattractant protein-1 gene. J Immunol. 1994;153(5):2052–63. [PubMed] [Google Scholar]
- 63.Ueda A, Ishigatsubo Y, Okubo T, Yoshimura T. Transcriptional regulation of the human monocyte chemoattractant protein-1 gene. Cooperation of two NF-kappaB sites and NF-kappaB/Rel subunit specificity. J Biol Chem. 1997;272(49):31092–9. [DOI] [PubMed] [Google Scholar]
- 64.Teferedegne B, Green MR, Guo Z, Boss JM. Mechanism of action of a distal NF-kappaB-dependent enhancer. Mol Cell Biol. 2006;26(15):5759–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Xing L, Remick DG. Promoter elements responsible for antioxidant regulation of MCP-1 gene expression. Antioxid Redox Signal. 2007;9(11):1979–89. [DOI] [PubMed] [Google Scholar]
- 66.Famulla S, Horrighs A, Cramer A, Sell H, Eckel J. Hypoxia reduces the response of human adipocytes towards TNFalpha resulting in reduced NF-kappaB signaling and MCP-1 secretion. Int J Obes. 2012;36(7):986–92. [DOI] [PubMed] [Google Scholar]
- 67.Pichiule P, Chavez JC, Schmidt AM, Vannucci SJ. Hypoxia-inducible factor-1 mediates neuronal expression of the receptor for advanced glycation end products following hypoxia/ischemia. J Biol Chem. 2007;282(50):36330–40. [DOI] [PubMed] [Google Scholar]
- 68.Tafani M, Schito L, Pellegrini L, Villanova L, Marfe G, Anwar T, et al. Hypoxia-increased RAGE and P2X7R expression regulates tumor cell invasion through phosphorylation of Erk1/2 and Akt and nuclear translocation of NF-{kappa}B. Carcinogenesis. 2011;32(8):1167–75. [DOI] [PubMed] [Google Scholar]
- 69.Taneja S, Vetter SW, Leclerc E. Hypoxia and the Receptor for Advanced Glycation End Products (RAGE) Signaling in Cancer. Int J Mol Sci. 2021;22(15):8153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Khan MI, Rath S, Adhami VM, Mukhtar H. Hypoxia driven glycation: Mechanisms and therapeutic opportunities. Semin Cancer Biol. 2018;49:75–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Laurencikiene J, van Harmelen V, Arvidsson Nordström E, Dicker A, Blomqvist L, Näslund E, et al. NF-kappaB is important for TNF-alpha-induced lipolysis in human adipocytes. J Lipid Res. 2007;48(5):1069–77. [DOI] [PubMed] [Google Scholar]
- 72.Tourniaire F, Romier-Crouzet B, Lee JH, Marcotorchino J, Gouranton E, Salles J, et al. Chemokine Expression in Inflamed Adipose Tissue Is Mainly Mediated by NF-kappaB. PLoS One. 2013;8(6):e66515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Qi R, Huang J, Wang Q, Liu H, Wang R, Wang J, et al. MicroRNA-224–5p regulates adipocyte apoptosis induced by TNFalpha via controlling NF-kappaB activation. J Cell Physiol. 2018;233(2):1236–46. [DOI] [PubMed] [Google Scholar]
- 74.Ruan H, Hacohen N, Golub TR, Van Parijs L, Lodish HF. Tumor necrosis factor-alpha suppresses adipocyte-specific genes and activates expression of preadipocyte genes in 3T3-L1 adipocytes: nuclear factor-kappaB activation by TNF-alpha is obligatory. Diabetes. 2002;51(5):1319–36. [DOI] [PubMed] [Google Scholar]
- 75.Fu Y, Luo N, Lopes-Virella MF, Garvey WT. The adipocyte lipid binding protein (ALBP/aP2) gene facilitates foam cell formation in human THP-1 macrophages. Atherosclerosis. 2002;165(2):259–69. [DOI] [PubMed] [Google Scholar]
- 76.Makowski L, Boord JB, Maeda K, Babaev VR, Uysal KT, Morgan MA, et al. Lack of macrophage fatty-acid-binding protein aP2 protects mice deficient in apolipoprotein E against atherosclerosis. Nat Med. 2001;7(6):699–705. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Fu Y, Luo N, Lopes-Virella MF. Oxidized LDL induces the expression of ALBP/aP2 mRNA and protein in human THP-1 macrophages. J Lipid Res. 2000;41(12):2017–23. [PubMed] [Google Scholar]
- 78.Schmitz ML, Shaban MS, Albert BV, Gokcen A, Kracht M. The Crosstalk of Endoplasmic Reticulum (ER) Stress Pathways with NF-kappaB: Complex Mechanisms Relevant for Cancer, Inflammation and Infection. Biomedicines. 2018;6(2):58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kawasaki N, Asada R, Saito A, Kanemoto S, Imaizumi K. Obesity-induced endoplasmic reticulum stress causes chronic inflammation in adipose tissue. Sci Rep. 2012;2:799. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Jiao P, Ma J, Feng B, Zhang H, Diehl JA, Chin YE, et al. FFA-induced adipocyte inflammation and insulin resistance: involvement of ER stress and IKKbeta pathways. Obesity. 2011;19(3):483–91. [DOI] [PubMed] [Google Scholar]
- 81.Sharma NK, Das SK, Mondal AK, Hackney OG, Chu WS, Kern PA, et al. Endoplasmic reticulum stress markers are associated with obesity in nondiabetic subjects. J Clin Endocrinol Metab. 2008;93(11):4532–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Boden G, Duan X, Homko C, Molina EJ, Song W, Perez O, et al. Increase in endoplasmic reticulum stress-related proteins and genes in adipose tissue of obese, insulin-resistant individuals. Diabetes. 2008;57(9):2438–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Gregor MF, Hotamisligil GS. Thematic review series: Adipocyte Biology. Adipocyte stress: the endoplasmic reticulum and metabolic disease. J Lipid Res. 2007;48(9):1905–14. [DOI] [PubMed] [Google Scholar]
- 84.Ozcan U, Cao Q, Yilmaz E, Lee AH, Iwakoshi NN, Ozdelen E, et al. Endoplasmic reticulum stress links obesity, insulin action, and type 2 diabetes. Science. 2004;306(5695):457–61. [DOI] [PubMed] [Google Scholar]
- 85.Yamazaki H, Hiramatsu N, Hayakawa K, Tagawa Y, Okamura M, Ogata R, et al. Activation of the Akt-NF-kappaB pathway by subtilase cytotoxin through the ATF6 branch of the unfolded protein response. J Immunol. 2009;183(2):1480–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Berg AH, Lin Y, Lisanti MP, Scherer PE. Adipocyte differentiation induces dynamic changes in NF-kappaB expression and activity. Am J Physiol Endocrinol Metab. 2004;287(6):E1178–88. [DOI] [PubMed] [Google Scholar]
- 87.Schaeffler A, Gross P, Buettner R, Bollheimer C, Buechler C, Neumeier M, et al. Fatty acid-induced induction of Toll-like receptor-4/nuclear factor-kappaB pathway in adipocytes links nutritional signalling with innate immunity. Immunology. 2009;126(2):233–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Suganami T, Tanimoto-Koyama K, Nishida J, Itoh M, Yuan X, Mizuarai S, et al. Role of the Toll-like receptor 4/NF-kappaB pathway in saturated fatty acid-induced inflammatory changes in the interaction between adipocytes and macrophages. Arterioscler Thromb Vasc Biol. 2007;27(1):84–91. [DOI] [PubMed] [Google Scholar]
- 89.Pietsch J, Batra A, Stroh T, Fedke I, Glauben R, Okur B, et al. Toll-like receptor expression and response to specific stimulation in adipocytes and preadipocytes: on the role of fat in inflammation. Ann N Y Acad Sci. 2006;1072:407–9. [DOI] [PubMed] [Google Scholar]
- 90.Batra A, Pietsch J, Fedke I, Glauben R, Okur B, Stroh T, et al. Leptin-dependent toll-like receptor expression and responsiveness in preadipocytes and adipocytes. Am J Pathol. 2007;170(6):1931–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Schaffler A, Scholmerich J, Salzberger B. Adipose tissue as an immunological organ: Toll-like receptors, C1q/TNFs and CTRPs. Trends Immunol. 2007;28(9):393–9. [DOI] [PubMed] [Google Scholar]
- 92.Jialal I, Devaraj S, Bettaieb A, Haj F, Adams-Huet B. Increased adipose tissue secretion of Fetuin-A, lipopolysaccharide-binding protein and high-mobility group box protein 1 in metabolic syndrome. Atherosclerosis. 2015;241(1):130–7. [DOI] [PubMed] [Google Scholar]
- 93.Kim JY, van de Wall E, Laplante M, Azzara A, Trujillo ME, Hofmann SM, et al. Obesity-associated improvements in metabolic profile through expansion of adipose tissue. J Clin Invest. 2007;117(9):2621–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Virtue S, Vidal-Puig A. Adipose tissue expandability, lipotoxicity and the Metabolic Syndrome--an allostatic perspective. Biochim Biophys Acta. 2010;1801(3):338–49. [DOI] [PubMed] [Google Scholar]
- 95.Ghaben AL, Scherer PE. Adipogenesis and metabolic health. Nat Rev Mol Cell Biol. 2019;20(4):242–58. [DOI] [PubMed] [Google Scholar]
- 96.Sha WC, Liou HC, Tuomanen EI, Baltimore D. Targeted disruption of the p50 subunit of NF-kappa B leads to multifocal defects in immune responses. Cell. 1995;80(2):321–30. [DOI] [PubMed] [Google Scholar]
- 97.Tang T, Zhang J, Yin J, Staszkiewicz J, Gawronska-Kozak B, Jung DY, et al. Uncoupling of inflammation and insulin resistance by NF-kappaB in transgenic mice through elevated energy expenditure. J Biol Chem. 2010;285(7):4637–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Hotamisligil GS, Murray DL, Choy LN, Spiegelman BM. Tumor necrosis factor alpha inhibits signaling from the insulin receptor. Proc Natl Acad Sci U S A. 1994;91(11):4854–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Hotamisligil GS, Peraldi P, Budavari A, Ellis R, White MF, Spiegelman BM. IRS-1-mediated inhibition of insulin receptor tyrosine kinase activity in TNF-alpha- and obesity-induced insulin resistance. Science. 1996;271(5249):665–8. [DOI] [PubMed] [Google Scholar]
- 100.Peraldi P, Hotamisligil GS, Buurman WA, White MF, Spiegelman BM. Tumor necrosis factor (TNF)-alpha inhibits insulin signaling through stimulation of the p55 TNF receptor and activation of sphingomyelinase. J Biol Chem. 1996;271(22):13018–22. [DOI] [PubMed] [Google Scholar]
- 101.Uysal KT, Wiesbrock SM, Marino MW, Hotamisligil GS. Protection from obesity-induced insulin resistance in mice lacking TNF-alpha function. Nature. 1997;389(6651):610–4. [DOI] [PubMed] [Google Scholar]
- 102.Ruan H, Pownall HJ, Lodish HF. Troglitazone antagonizes tumor necrosis factor-alpha-induced reprogramming of adipocyte gene expression by inhibiting the transcriptional regulatory functions of NF-kappaB. J Biol Chem. 2003;278(30):28181–92. [DOI] [PubMed] [Google Scholar]
- 103.Wen X, Li Y, Liu Y. Opposite action of peroxisome proliferator-activated receptor-gamma in regulating renal inflammation: functional switch by its ligand. J Biol Chem. 2010;285(39):29981–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Zoico E, Garbin U, Olioso D, Mazzali G, Fratta Pasini AM, Di Francesco V, et al. The effects of adiponectin on interleukin-6 and MCP-1 secretion in lipopolysaccharide-treated 3T3-L1 adipocytes: role of the NF-kappaB pathway. Int J Mol Med. 2009;24(6):847–51. [DOI] [PubMed] [Google Scholar]
- 105.Hoareau L, Bencharif K, Rondeau P, Murumalla R, Ravanan P, Tallet F, et al. Signaling pathways involved in LPS induced TNFalpha production in human adipocytes. J Inflamm. 2010;7:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Turner JJ, Foxwell KM, Kanji R, Brenner C, Wood S, Foxwell BM, et al. Investigation of nuclear factor-kappaB inhibitors and interleukin-10 as regulators of inflammatory signalling in human adipocytes. Clin Exp Immunol. 2010;162(3):487–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Krinninger P, Brunner C, Ruiz PA, Schneider E, Marx N, Foryst-Ludwig A, et al. Role of the adipocyte-specific NF-kappaB activity in the regulation of IP-10 and T cell migration. Am J Physiol Endocrinol Metab. 2011;300(2):E304–11. [DOI] [PubMed] [Google Scholar]
- 108.Zhong L, Luo Y, Huang C, Liu L. Effect of NF-κB decoy on insulin resistance of adipocytes from patients with type 2 diabetes mellitus. Diabetes Metab. 2011;37(6):520–6. [DOI] [PubMed] [Google Scholar]
- 109.Lira FS, Rosa JC, Pimentel GD, Seelaender M, Damaso AR, Oyama LM, et al. Both adiponectin and interleukin-10 inhibit LPS-induced activation of the NF-κB pathway in 3T3-L1 adipocytes. Cytokine. 2012;57(1):98–106. [DOI] [PubMed] [Google Scholar]
- 110.Harte AL, Tripathi G, Piya MK, Barber TM, Clapham JC, Al-Daghri N, et al. NFkappaB as a potent regulator of inflammation in human adipose tissue, influenced by depot, adiposity, T2DM status, and TNFalpha. Obesity. 2013;21(11):2322–30. [DOI] [PubMed] [Google Scholar]
- 111.Kwon H, Laurent S, Tang Y, Zong H, Vemulapalli P, Pessin JE. Adipocyte-specific IKKbeta signaling suppresses adipose tissue inflammation through an IL-13-dependent paracrine feedback pathway. Cell Rep. 2014;9(5):1574–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Park SH, Liu Z, Sui Y, Helsley RN, Zhu B, Powell DK, et al. IKKbeta Is Essential for Adipocyte Survival and Adaptive Adipose Remodeling in Obesity. Diabetes. 2016;65(6):1616–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Zamboni M, Di Francesco V, Garbin U, Fratta Pasini A, Mazzali G, Stranieri C, et al. Adiponectin gene expression and adipocyte NF-kappaB transcriptional activity in elderly overweight and obese women: inter-relationships with fat distribution, hs-CRP, leptin and insulin resistance. Int J Obes. 2007;31(7):1104–9. [DOI] [PubMed] [Google Scholar]
- 114.Chiang SH, Bazuine M, Lumeng CN, Geletka LM, Mowers J, White NM, et al. The protein kinase IKKepsilon regulates energy balance in obese mice. Cell. 2009;138(5):961–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Grimm T, Schneider S, Naschberger E, Huber J, Guenzi E, Kieser A, et al. EBV latent membrane protein-1 protects B cells from apoptosis by inhibition of BAX. Blood. 2005;105(8):3263–9. [DOI] [PubMed] [Google Scholar]
- 116.Jiao P, Feng B, Ma J, Nie Y, Paul E, Li Y, et al. Constitutive activation of IKKβ in adipose tissue prevents diet-induced obesity in mice. Endocrinology. 2012;153(1):154–65. [DOI] [PubMed] [Google Scholar]
- 117.Gao Z, Zhang J, Henagan TM, Lee JH, Ye X, Wang H, et al. P65 inactivation in adipocytes and macrophages attenuates adipose inflammatory response in lean but not in obese mice. Am J Physiol Endocrinol Metab. 2015;308(6):E496–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Lee KY, Russell SJ, Ussar S, Boucher J, Vernochet C, Mori MA, et al. Lessons on conditional gene targeting in mouse adipose tissue. Diabetes. 2013;62(3):864–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Catz SD, Johnson JL. Transcriptional regulation of bcl-2 by nuclear factor kappa B and its significance in prostate cancer. Oncogene. 2001;20(50):7342–51. [DOI] [PubMed] [Google Scholar]
- 120.Wang Z, Zhang B, Yang L, Ding J, Ding HF. Constitutive production of NF-kappaB2 p52 is not tumorigenic but predisposes mice to inflammatory autoimmune disease by repressing Bim expression. J Biol Chem. 2008;283(16):10698–706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Turner DJ, Alaish SM, Zou T, Rao JN, Wang JY, Strauch ED. Bile salts induce resistance to apoptosis through NF-kappaB-mediated XIAP expression. Ann Surg. 2007;245(3):415–25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Pettersson AT, Laurencikiene J, Nordstrom EA, Stenson BM, van Harmelen V, Murphy C, et al. Characterization of the human CIDEA promoter in fat cells. Int J Obes. 2008;32(9):1380–7. [DOI] [PubMed] [Google Scholar]
- 123.Pajvani UB, Trujillo ME, Combs TP, Iyengar P, Jelicks L, Roth KA, et al. Fat apoptosis through targeted activation of caspase 8: a new mouse model of inducible and reversible lipoatrophy. Nat Med. 2005;11(7):797–803. [DOI] [PubMed] [Google Scholar]
- 124.Haskill S, Beg AA, Tompkins SM, Morris JS, Yurochko AD, Sampson-Johannes A, et al. Characterization of an immediate-early gene induced in adherent monocytes that encodes I kappa B-like activity. Cell. 1991;65(7):1281–9. [DOI] [PubMed] [Google Scholar]
- 125.Sun SC, Ganchi PA, Ballard DW, Greene WC. NF-kappa B controls expression of inhibitor I kappa B alpha: evidence for an inducible autoregulatory pathway. Science. 1993;259(5103):1912–5. [DOI] [PubMed] [Google Scholar]
- 126.de Martin R, Vanhove B, Cheng Q, Hofer E, Csizmadia V, Winkler H, et al. Cytokine-inducible expression in endothelial cells of an I kappa B alpha-like gene is regulated by NF kappa B. EMBO J. 1993;12(7):2773–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Schobitz B, Pezeshki G, Pohl T, Hemmann U, Heinrich PC, Holsboer F, et al. Soluble interleukin-6 (IL-6) receptor augments central effects of IL-6 in vivo. FASEB J. 1995;9(8):659–64. [DOI] [PubMed] [Google Scholar]
- 128.Wallenius K, Wallenius V, Sunter D, Dickson SL, Jansson JO. Intracerebroventricular interleukin-6 treatment decreases body fat in rats. Biochem Biophys Res Commun. 2002;293(1):560–5. [DOI] [PubMed] [Google Scholar]
- 129.Wallenius V, Wallenius K, Ahren B, Rudling M, Carlsten H, Dickson SL, et al. Interleukin-6-deficient mice develop mature-onset obesity. Nat Med. 2002;8(1):75–9. [DOI] [PubMed] [Google Scholar]
- 130.Benrick A, Schele E, Pinnock SB, Wernstedt-Asterholm I, Dickson SL, Karlsson-Lindahl L, et al. Interleukin-6 gene knockout influences energy balance regulating peptides in the hypothalamic paraventricular and supraoptic nuclei. J Neuroendocrinol. 2009;21(7):620–8. [DOI] [PubMed] [Google Scholar]
- 131.Schele E, Fekete C, Egri P, Fuzesi T, Palkovits M, Keller E, et al. Interleukin-6 receptor alpha is co-localised with melanin-concentrating hormone in human and mouse hypothalamus. J Neuroendocrinol. 2012;24(6):930–43. [DOI] [PubMed] [Google Scholar]
- 132.Schele E, Benrick A, Grahnemo L, Egecioglu E, Anesten F, Palsdottir V, et al. Inter-relation between interleukin (IL)-1, IL-6 and body fat regulating circuits of the hypothalamic arcuate nucleus. J Neuroendocrinol. 2013;25(6):580–9. [DOI] [PubMed] [Google Scholar]
- 133.Timper K, Denson JL, Steculorum SM, Heilinger C, Engstrom-Ruud L, Wunderlich CM, et al. IL-6 Improves Energy and Glucose Homeostasis in Obesity via Enhanced Central IL-6 trans-Signaling. Cell Rep. 2017;19(2):267–80. [DOI] [PubMed] [Google Scholar]
- 134.Vandanmagsar B, Youm YH, Ravussin A, Galgani JE, Stadler K, Mynatt RL, et al. The NLRP3 inflammasome instigates obesity-induced inflammation and insulin resistance. Nat Med. 2011;17(2):179–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Martinon F, Burns K, Tschopp J. The inflammasome: a molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol Cell. 2002;10(2):417–26. [DOI] [PubMed] [Google Scholar]
- 136.Davis BK, Wen H, Ting JP. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu Rev Immunol. 2011;29:707–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Henao-Mejia J, Elinav E, Strowig T, Flavell RA. Inflammasomes: far beyond inflammation. Nat Immunol. 2012;13(4):321–4. [DOI] [PubMed] [Google Scholar]
- 138.Swanson KV, Deng M, Ting JP. The NLRP3 inflammasome: molecular activation and regulation to therapeutics. Nat Rev Immunol. 2019;19(8):477–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Kayagaki N, Warming S, Lamkanfi M, Vande Walle L, Louie S, Dong J, et al. Non-canonical inflammasome activation targets caspase-11. Nature. 2011;479(7371):117–21. [DOI] [PubMed] [Google Scholar]
- 140.Kayagaki N, Stowe IB, Lee BL, OʼRourke K, Anderson K, Warming S, et al. Caspase-11 cleaves gasdermin D for non-canonical inflammasome signalling. Nature. 2015;526(7575):666–71. [DOI] [PubMed] [Google Scholar]
- 141.Yin Z, Deng T, Peterson LE, Yu R, Lin J, Hamilton DJ, et al. Transcriptome analysis of human adipocytes implicates the NOD-like receptor pathway in obesity-induced adipose inflammation. Mol Cell Endocrinol. 2014;394(1–2):80–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Stienstra R, Joosten LA, Koenen T, van Tits B, van Diepen JA, van den Berg SA, et al. The inflammasome-mediated caspase-1 activation controls adipocyte differentiation and insulin sensitivity. Cell Metab. 2010;12(6):593–605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Zhang SY, Dong YQ, Wang P, Zhang X, Yan Y, Sun L, et al. Adipocyte-derived Lysophosphatidylcholine Activates Adipocyte and Adipose Tissue Macrophage Nod-Like Receptor Protein 3 Inflammasomes Mediating Homocysteine-Induced Insulin Resistance. EBioMedicine. 2018;31:202–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Unamuno X, Gomez-Ambrosi J, Ramirez B, Rodriguez A, Becerril S, Valenti V, et al. NLRP3 inflammasome blockade reduces adipose tissue inflammation and extracellular matrix remodeling. Cell Mol Immunol. 2021;18(4):1045–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Boaru SG, Borkham-Kamphorst E, Van de Leur E, Lehnen E, Liedtke C, Weiskirchen R. NLRP3 inflammasome expression is driven by NF-kappaB in cultured hepatocytes. Biochem Biophys Res Commun. 2015;458(3):700–6. [DOI] [PubMed] [Google Scholar]
- 146.Hiscott J, Marois J, Garoufalis J, DʼAddario M, Roulston A, Kwan I, et al. Characterization of a functional NF-kappa B site in the human interleukin 1 beta promoter: evidence for a positive autoregulatory loop. Mol Cell Biol. 1993;13(10):6231–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 147.Bauernfeind FG, Horvath G, Stutz A, Alnemri ES, MacDonald K, Speert D, et al. Cutting edge: NF-kappaB activating pattern recognition and cytokine receptors license NLRP3 inflammasome activation by regulating NLRP3 expression. J Immunol. 2009;183(2):787–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Lin KM, Hu W, Troutman TD, Jennings M, Brewer T, Li X, et al. IRAK-1 bypasses priming and directly links TLRs to rapid NLRP3 inflammasome activation. Proc Natl Acad Sci U S A. 2014;111(2):775–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 149.Gaidt MM, Ebert TS, Chauhan D, Schmidt T, Schmid-Burgk JL, Rapino F, et al. Human Monocytes Engage an Alternative Inflammasome Pathway. Immunity. 2016;44(4):833–46. [DOI] [PubMed] [Google Scholar]
- 150.Juliana C, Fernandes-Alnemri T, Kang S, Farias A, Qin F, Alnemri ES. Non-transcriptional priming and deubiquitination regulate NLRP3 inflammasome activation. J Biol Chem. 2012;287(43):36617–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 151.Mohallem R, Aryal UK. Regulators of TNFalpha mediated insulin resistance elucidated by quantitative proteomics. Sci Rep. 2020;10(1):20878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Sano H, Hsu DK, Yu L, Apgar JR, Kuwabara I, Yamanaka T, et al. Human galectin-3 is a novel chemoattractant for monocytes and macrophages. J Immunol. 2000;165(4):2156–64. [DOI] [PubMed] [Google Scholar]
- 153.Brach MA, Henschler R, Mertelsmann RH, Herrmann F. Regulation of M-CSF expression by M-CSF: role of protein kinase C and transcription factor NF kappa B. Pathobiology. 1991;59(4):284–8. [DOI] [PubMed] [Google Scholar]
- 154.Hohensinner PJ, Kaun C, Rychli K, Niessner A, Pfaffenberger S, Rega G, et al. Macrophage colony stimulating factor expression in human cardiac cells is upregulated by tumor necrosis factor-alpha via an NF-kappaB dependent mechanism. J Thromb Haemost. 2007;5(12):2520–8. [DOI] [PubMed] [Google Scholar]
- 155.Moon MR, Parikh AA, Pritts TA, Fischer JE, Cottongim S, Szabo C, et al. Complement component C3 production in IL-1beta-stimulated human intestinal epithelial cells is blocked by NF-kappaB inhibitors and by transfection with ser 32/36 mutant IkappaBalpha. J Surg Res. 1999;82(1):48–55. [DOI] [PubMed] [Google Scholar]
- 156.Novitskiy G, Potter JJ, Rennie-Tankersley L, Mezey E. Identification of a novel NF-kappaB-binding site with regulation of the murine alpha2(I) collagen promoter. J Biol Chem. 2004;279(15):15639–44. [DOI] [PubMed] [Google Scholar]
- 157.Nieto N Ethanol and fish oil induce NFkappaB transactivation of the collagen alpha2(I) promoter through lipid peroxidation-driven activation of the PKC-PI3K-Akt pathway. Hepatology. 2007;45(6):1433–45. [DOI] [PubMed] [Google Scholar]
- 158.Joo JH, Kim JW, Lee Y, Yoon SY, Kim JH, Paik SG, et al. Involvement of NF-kappaB in the regulation of S100A6 gene expression in human hepatoblastoma cell line HepG2. Biochem Biophys Res Commun. 2003;307(2):274–80. [DOI] [PubMed] [Google Scholar]
- 159.Maury E, Navez B, Brichard SM. Circadian clock dysfunction in human omental fat links obesity to metabolic inflammation. Nat Commun. 2021;12(1):2388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 160.Puig O, Caspary F, Rigaut G, Rutz B, Bouveret E, Bragado-Nilsson E, et al. The tandem affinity purification (TAP) method: a general procedure of protein complex purification. Methods. 2001;24(3):218–29. [DOI] [PubMed] [Google Scholar]
- 161.Burckstummer T, Bennett KL, Preradovic A, Schutze G, Hantschel O, Superti-Furga G, et al. An efficient tandem affinity purification procedure for interaction proteomics in mammalian cells. Nat Methods. 2006;3(12):1013–9. [DOI] [PubMed] [Google Scholar]
- 162.Ma Z, Fung V, DʼOrso I. Tandem Affinity Purification of Protein Complexes from Eukaryotic Cells. J Vis Exp. 2017. Jan 26;(119):55236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 163.Xu X, Song Y, Li Y, Chang J, Zhang H, An L. The tandem affinity purification method: an efficient system for protein complex purification and protein interaction identification. Protein Expr Purif. 2010;72(2):149–56. [DOI] [PubMed] [Google Scholar]
- 164.Mann M Functional and quantitative proteomics using SILAC. Nat Rev Mol Cell Biol. 2006. Dec;7(12):952–8. [DOI] [PubMed] [Google Scholar]
- 165.Ong SE. The expanding field of SILAC. Anal Bioanal Chem. 2012;404(4):967–76. [DOI] [PubMed] [Google Scholar]
- 166.Ong SE, Blagoev B, Kratchmarova I, Kristensen DB, Steen H, Pandey A, et al. Stable isotope labeling by amino acids in cell culture, SILAC, as a simple and accurate approach to expression proteomics. Mol Cell Proteomics. 2002;1(5):376–86. [DOI] [PubMed] [Google Scholar]
- 167.Sabir JSM, El Omri A, Shaik NA, Banaganapalli B, Al-Shaeri MA, Alkenani NA, et al. Identification of key regulatory genes connected to NF-κB family of proteins in visceral adipose tissues using gene expression and weighted protein interaction network. PLoS One. 2019;14(4):e0214337. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Zhao B, Barrera LA, Ersing I, Willox B, Schmidt SC, Greenfeld H, et al. The NF-kappaB genomic landscape in lymphoblastoid B cells. Cell Rep. 2014;8(5):1595–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Chen A, Koehler AN. Transcription Factor Inhibition: Lessons Learned and Emerging Targets. Trends Mol Med. 2020;26(5):508–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Gilmore TD, Herscovitch M. Inhibitors of NF-kappaB signaling: 785 and counting. Oncogene. 2006;25(51):6887–99. [DOI] [PubMed] [Google Scholar]
- 171.Gupta SC, Sundaram C, Reuter S, Aggarwal BB. Inhibiting NF-kappaB activation by small molecules as a therapeutic strategy. Biochim Biophys Acta. 2010;1799(10–12):775–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 172.Berg T Small-molecule modulators of c-Myc/Max and Max/Max interactions. Curr Top Microbiol Immunol. 2011;348:139–49. [DOI] [PubMed] [Google Scholar]
- 173.Fletcher S, Prochownik EV. Small-molecule inhibitors of the Myc oncoprotein. Biochim Biophys Acta. 2015;1849(5):525–43. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Ngo KA, Kishimoto K, Davis-Turak J, Pimplaskar A, Cheng Z, Spreafico R, et al. Dissecting the Regulatory Strategies of NF-kappaB RelA Target Genes in the Inflammatory Response Reveals Differential Transactivation Logics. Cell Rep. 2020;30(8):2758–75.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 175.Borghini L, Lu J, Hibberd M, Davila S. Variation in Genome-Wide NF-kappaB RELA Binding Sites upon Microbial Stimuli and Identification of a Virus Response Profile. J Immunol. 2018;201(4):1295–305. [DOI] [PubMed] [Google Scholar]
- 176.Zhou F, Xu X, Wang D, Wu J, Wang J. Identification of novel NF-kappaB transcriptional targets in TNFalpha-treated HeLa and HepG2 cells. Cell Biol Int. 2017;41(5):555–69. [DOI] [PubMed] [Google Scholar]
- 177.Satoh J Molecular network of ChIP-Seq-based NF-kappaB p65 target genes involves diverse immune functions relevant to the immunopathogenesis of multiple sclerosis. Mult Scler Relat Disord. 2014;3(1):94–106. [DOI] [PubMed] [Google Scholar]