

Abstract
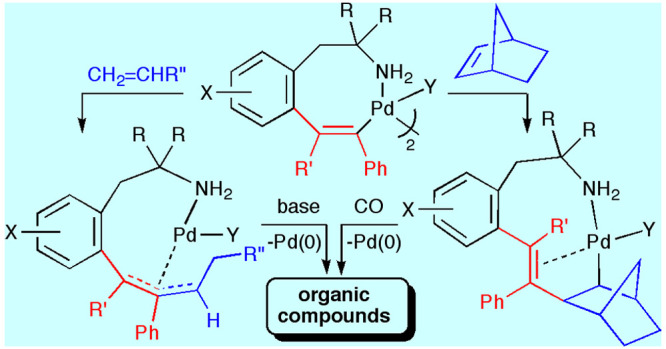
The eight-membered metallacycles arising from the insertion of 1 equiv of alkyne into the Pd–C bond of ortho-metalated homoveratrylamine and phentermine can further react with alkenes to give two different types of mononuclear complexes depending on the nature of the olefin. When terminal alkenes (styrene and ethyl acrylate) are used, a mixture of the anti/syn η3-allyl Pd(II) complexes are isolated, which evolve slowly to the syn isomers by heating the mixtures appropriately. These η3-allyl Pd(II) complexes do not react with CO or weak bases, but when they are treated with a strong base, such as KOtBu, they afford Pd(0) and the functionalized starting phenethylamines containing a 1,3-butadienyl substituent in an ortho position. When 2-norbornene was used instead of terminal alkenes, the strained olefin inserts into the alkenyl Pd(II) complex to afford a 10-membered norbornyl palladium(II) complex, in which the new C,N-chelate ligand is coordinated to the metal through an additional double bond, occupying three coordination positions. The reactivity of these norbornyl complexes depends on the substituents on the inserted alkenyl fragment, and thus they can further react with (1) KOtBu, to give Pd(0) and a tetrahydroisoquinoline nucleus containing a tricyclo[3.2.1]octyl ring, or (2) CO and TlOTf, to afford Pd(0) and amino acid derivatives or the corresponding lactones arising from an intramolecular Michael addition of the CO2H group to the α,β-unsaturated ester moiety. Crystal structures of every type of compound have been determined by X-ray diffraction studies.
Introduction
Palladium is one of the most versatile transition metals in organic synthesis, given its well-known ability to catalyze the formation of C–C and C–heteroatom bonds.1,2 Nevertheless, the extraordinarily high variability of applications of palladium relies just on a few elementary steps inherent to its reactivity, and among them, the migratory insertion reaction of an unsaturated ligand into a Pd–C bond stands out.3−5 Thus, a myriad of methods to functionalize alkenes,3,6 alkynes,7 or allenes8 through Pd catalysis have been reported so far.
Multicomponent reactions, where several reagents assemble sequentially giving rise to complex structures, are valuable protocols, since they represent an effective modular approach to green organic synthesis.9 Nevertheless, transition-metal-catalyzed processes involving the use of several unsaturated coupling partners in the reaction mixture are challenging fields due to the difficulty in controlling the order of reactivity: that is, the control of regioselectivity of the migratory insertion sequence for the different unsaturated reagents.10−12 Some successful examples of these types of reactions are the copolymerization of alkenes and CO (Scheme 1a)13 and the copolymerization of olefins with and without polar groups,14 among others.15−17 Moreover, impressive progress has been made on intramolecular cascade reactions where different unsaturated moieties are involved (Scheme 1b).5,18 In the latter case, however, the selectivity is usually determined by the structure of the substrate itself.
Scheme 1. Examples of Reactions Involving Sequential Migratory Insertion into Pd–C Bonds13,18c.

While catalytic conditions make difficult the aforementioned control of the selectivity, stoichiometric reactions can offer the advantage of stepwise insertion paths for the synthesis of valuable organic products.19−23
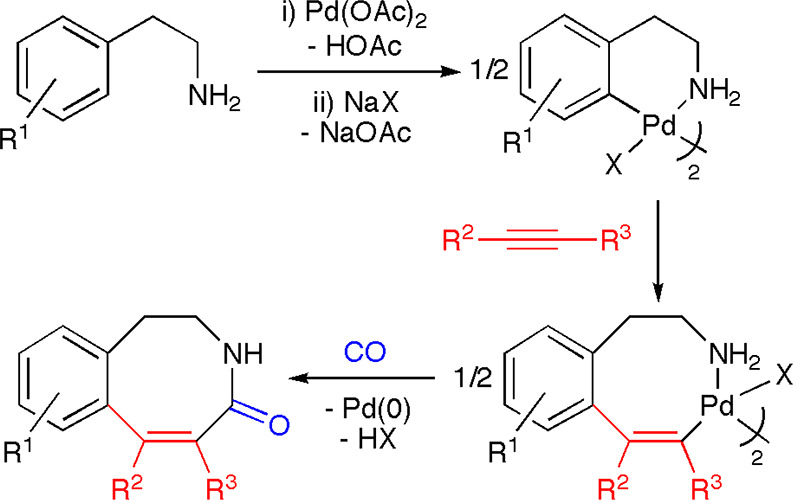
Our research group has previously reported the functionalization of primary phenethylamines through the isolation of ortho-palladated intermediates.24−26 These types of arylalkylamines constitute the core of many relevant biomolecules, such as neurotransmitters (i.e., dopamine), amino acids (i.e., phenylalanine), or marketed psychoactive drugs. The catalytic functionalization of these scaffolds has proven challenging, due to the strong coordination ability of the primary amine group to Pd(II).27 Hence, the study of the stoichiometric functionalization of primary phenethylamines can provide alternative routes to modify these interesting molecules. For instance, the sequential insertion of alkynes and CO or of alkynes and isocyanides into ortho-palladated derivatives allowed us to synthesize medium-sized rings such as eight-membered benzazocinones (Scheme 2).20,21
Scheme 2. Synthesis of Benzazocinones through the Sequential Insertion of Alkynes and CO into the Pd–C Bond of Six-Membered Palladacycles.
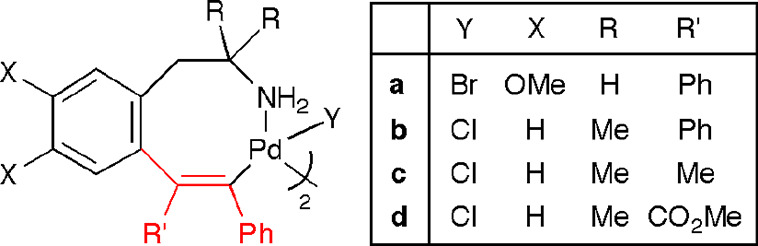
We have previously reported the synthesis and isolation of stable eight-membered C,N-palladacycles arising from the insertion of one molecule of alkyne into the Pd–C bond of ortho-palladated phentermine and homoveratrylamine (Chart 1).20 We present here the results of a study on the insertion of alkenes and alkenes/CO into these eight-membered alkenyl palladacycles. We have studied the new organometallic species generated upon each insertion, as well as the final functionalized organic products, formed through depalladation of the final organopalladium compounds. The overall processes rendering the final organopalladium complexes involve the sequential insertion of (a) alkynes and alkenes or (b) alkynes, alkenes, and CO into the Pd–C bond of the ortho-metalated derivatives from the primary phenethylamines.
Chart 1. Previously Reported Eight-Membered Alkenyl Palladacycles Used as Starting Materials.
Results and Discussion
Insertion of Styrene or Ethyl Acrylate into the Pd–C Bond
Synthesis and Structure of η3-Allyl Palladium(II) Complexes
The reaction of the previously described alkenyl palladacycle a or b, arising from the monoinsertion of diphenylacetylene into the ortho-palladated homoveratrylamine and phentermine derivative A or B, with 2 equiv of CH2=CHR′ (molar ratio Pd/alkene = 1/1; R = Ph (1), CO2Et (2)) in CH2Cl2 at room temperature (16 h) gave a mixture of two complexes, which were identified as the anti and syn isomers of the η3-allyl Pd(II) complex 1 or 2 (Scheme 3).
Scheme 3. Reactions of Eight-Membered Alkenyl Palladacycles with Terminal Alkenes.
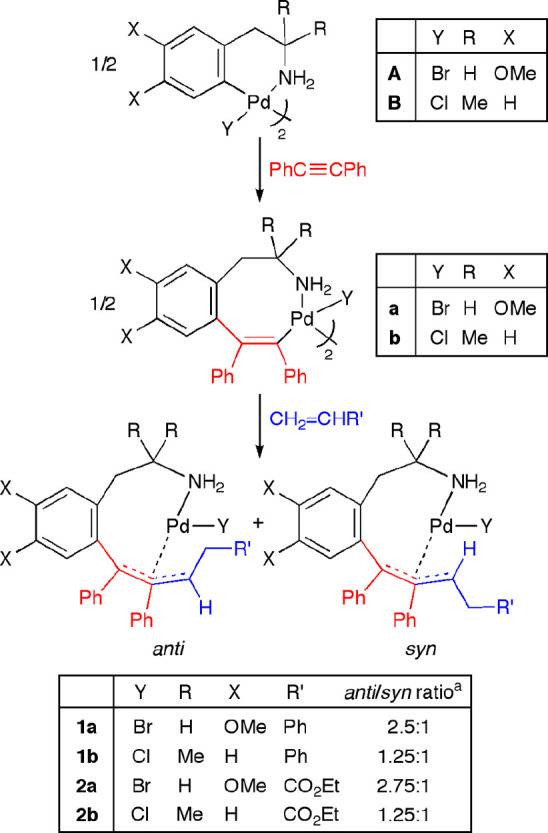
The ratios correspond to the isolated solids and were estimated by the integrals of the CH allylic signals of both isomers in the 1H NMR spectra of the mixtures.
After workup, a mixture of η3-allyl Pd(II) complexes anti-/syn-1a (2.5/1) or anti-/syn-2a (5/1) was isolated. Attempts to separate both isomers by fractional crystallization were unsuccessful. However, single crystals of anti-1a or anti-2a, suitable for X-ray diffraction studies, were obtained by slow diffusion of n-pentane into a solution of the mixture of anti-/syn-1a or anti-/syn-2a in CH2Cl2. The ratio between isomers, for both 1a and 2a, practically did not change after 48 h in CDCl3 solution at room temperature (by 1H NMR). However, the conversion to the syn isomers was complete after heating CDCl3 solutions of the mixtures at 60 °C for 10 days (syn-1a) or 48 h (syn-2a; by 1H NMR; Figure 1 and Table 1).
Figure 1.
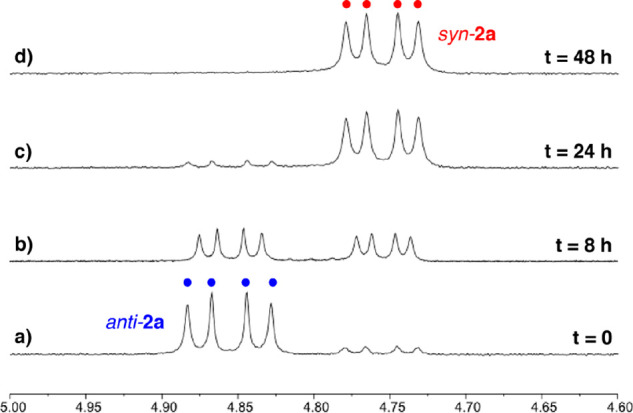
1H NMR spectra (4.6–5.0 ppm) of complex 2a (CDCl3): (a) anti-enriched mixture of complex 2a at room temperature (300 MHz) and (b) after heating at 60 °C for 8 h (400 MHz), (c) 24 h (300 MHz) and (d) 48 h (300 MHz).
Table 1. Isomerization Ratios for anti-/syn-1 and anti-/syn-2.
|
anti/syn ratio |
|||||
|---|---|---|---|---|---|
| temperature | time (h) | 1a | 1b | 2a | 2b |
| RT | 0 | 2.5/1 | 2.5/1 | 5/1 | 1.25/1 |
| 12 | 2.5/1 | 4.2/1 | |||
| 16 | 1.25/1 | ||||
| 24 | 2.5/1 | 4.2/1 | |||
| 48 | 2.5/1 | 2/1 | 3.3/1 | 1.25/1 | |
| 60 °C | 0 | 2.5/1 | 2.5/1 | 5/1 | 1.25/1 |
| 8 | 1.7/1 | 1/1 | |||
| 12 | 1/5 | ||||
| 24 | 1.1/1 | 1/10 | |||
| 48 | 1/1.25 | 1/3.3 | 0/1 | 1/20 | |
| 120 | 1/10 | ||||
| 240 | 0/1 | ||||
Enriched mixtures of both anti and syn isomers of 1b and 2b could be obtained due to the higher solubility of one of the two isomers in Et2O. When a solution of the mixture anti-/syn-1b or anti-/syn-2b (1b, ratio 1.25/1, 0.161 mmol; 2b, ratio 1.25/1, 0.260 mmol) in CH2Cl2 (1b, 3 mL; 2b, 2 mL) was treated with Et2O (15 mL), a mixture enriched in the anti (1b) or syn (2b) isomer precipitated (ratio anti/syn: 1b, 3/1; 2b: 1/3). When the mother liquors were concentrated (1b: 3 mL; 2b: 5 mL) and treated with n-pentane (1b, 20 mL) or cooled to 0 °C in an ice bath (2b), a mixture enriched in the syn (1b) or anti (2b) isomer precipitated (ratio anti/syn: 1b, 1/3; 2b: 3/1). Almost spectroscopically pure samples of anti- and syn-1b and anti- and syn-2b could be obtained by growing single crystals from the enriched mixtures.
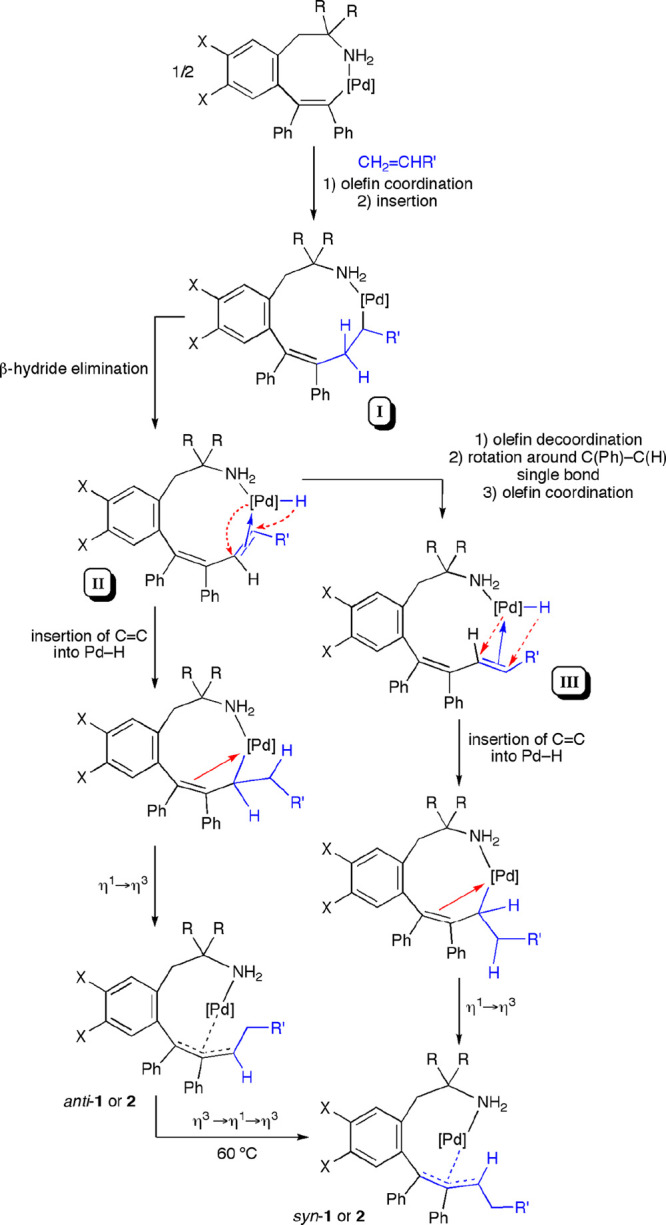
Since anti-1 and anti-2 complexes practically did not isomerize to the syn-1 and syn-2 derivatives at room temperature (Table 1), both isomers should be formed during the reaction. We proposed that the formation of anti-/syn-1 or anti-/syn-2 could be envisioned by the mechanism depicted in Scheme 4. Insertion of one molecule of the alkene into the Pd–C bond of the starting palladacycle (a or b) would afford the 10-membered alkyl palladacycle I. For this intermediate, we propose that the R′ group is at the carbon atom bonded to Pd(II), because (1) this is the regiochemistry that explains the formation of the allyl moiety coordinated to Pd(II) in complexes 1a,b and 2a,b and (2) this is the most frequent regioisomer found in the insertion of electron-poor alkenes into the Pd–C bonds of neutral complexes.22,28 Intermediate I could undergo β-hydride elimination to give the cisoid-(diene)PdH species II. Syn addition of Pd–H to the alkene moiety with regioselectivity in contrast with its previous elimination results in the formation of the η3-allyl complex anti-1 or anti-2 (Scheme 4), which seems to be formed preferentially as the kinetic product. On the other hand, the transoid-(diene)PdH intermediate III could be generated from II upon decoordination and rotation of the tertiary olefin moiety, prior to the Pd–H addition step, hence giving rise to the formation of the syn isomer. The thermal isomerization of the complexes anti-1 and anti-2 to the syn isomers at 60 °C (Table 1) proceeded by the usual η3–η1–η3 rearrangement.
Scheme 4. Formation of anti and syn Isomers of η3-Allyl Pd(II) Complexes.
Complexes 1 and 2 were fully characterized by elemental analyses and NMR and IR spectroscopic techniques. The 1H NMR spectra of all the allyl complexes showed the diastereotopic nature of the hydrogen atoms of the NH2 and CH2 groups, as well as both of Me groups of the CMe2 moiety for 1b and 2b. The most characteristic signal was that corresponding to the methine group of the inserted fragment, which appeared as a doublet of doublets in the range 4.85–5.16 ppm for the anti isomers and 4.75–4.84 ppm for the syn isomers. The two coupling constants 3JHH (anti, 11.2 ≤ 3JHH ≤ 12.3 Hz, average value 11.7 Hz; syn, 9.6 ≤ 3JHH ≤ 11.0 Hz, average value 10.3 Hz; anti, 4.5 ≤ 3JHH ≤ 5.2 Hz, average value 4.7 Hz; syn, 3.7 ≤ 3JHH ≤ 4.2 Hz, average value 4.0 Hz) were slightly smaller for the syn isomers than for the anti isomers.
The crystal structures of the η3-allyl complexes anti-1a, anti-1b, syn-1b, anti-2a·CH2Cl2, anti-2b·CHCl3, and syn-2b were determined by X-ray diffraction (see the Supporting Information). The X-ray thermal ellipsoid plots of anti-1a and syn-2b are depicted in Figures 2 and 3, respectively. In both cases, the palladium atoms adopted a square-planar geometry (mean deviation from the plane Y–Pd(1)–X(1)–N(1) 0.0194 Å, where Y = centroid from C(7), C(8), and C(9) atoms). The halogeno ligand and the amino group of the metalated fragment were coordinated in cis positions. The other two coordination sites were occupied by the allyl moiety, which was η3-bonded via C(7), C(8), and C(9), with C(7)–C(8)–C(9) angles of 119.4 and 122.5° for anti-1a and syn-2b, respectively.
Figure 2.
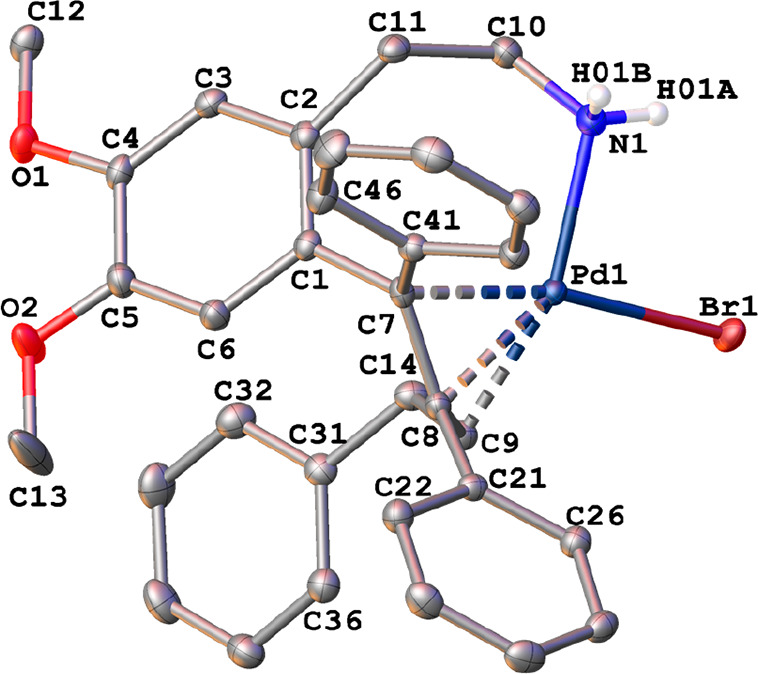
X-ray thermal ellipsoid plot (50% probability) of anti-1a along with the labeling scheme. The hydrogen atoms bonded to carbon have been omitted for clarity. Selected bond lengths (Å) and angles (deg): Pd(1)–Br(1) = 2.4959(3), Pd(1)–N(1) = 2.1124(15), Pd(1)–C(7) = 2.1295(15), Pd(1)–C(8) = 2.1047(15), Pd(1)–C(9) = 2.1181(16), C(7)–C(8) = 1.458(2), C(8)–C(9) = 1.427(2); Br(1)–Pd(1)–N(1) = 92.82(4), C(7)–C(8)–C(9) = 119.41(14), C(8)–C(9)–C(14) = 131.14(14).
Figure 3.
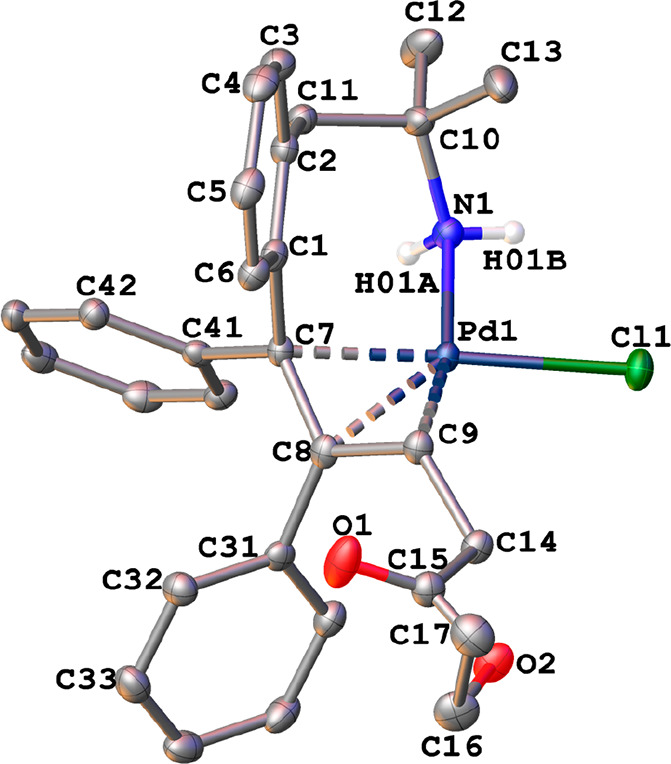
X-ray thermal ellipsoid plot (50% probability) of syn-2b along with the labeling scheme. The hydrogen atoms bonded to carbon have been omitted for clarity. Selected bond lengths (Å) and angles (deg): Pd(1)–Cl(1) = 2.3864(5), Pd(1)–N(1) = 2.1257(18), Pd(1)–C(7) = 2.1198(19), Pd(1)–C(8) = 2.1400(19), Pd(1)–C(9) = 2.1167(19), C(7)–C(8) = 1.441(3), C(8)–C(9) = 1.421(3); Cl(1)–Pd(1)–N(1) = 89.81(5), C(7)–C(8)–C(9) = 117.38(18), C(8)–C(9)–C(14) = 122.49(18).
Some catalytic transformations involving the sequential migratory insertion of alkynes and alkenes into a Pd–C11,12,17,29 or a Pd–H15 bond have been reported in the literature. In these processes, substituted 1,3-butadienes are formed. The mechanism proposed for those catalytic transformations does not consider the formation of η3-allyl intermediates. In our case, however, the presence of a coordinating group, such as NH2, may favor the isomerization of the 1,3-butadiene to form a stabilized allyl Pd(II) complex. As far as we are aware, this is the first stoichiometric study where the sequential migratory insertion of an alkyne and an alkene has been studied.
Reactivity of the η3-Allyl Complexes
The nucleophilic attack to η3-allyl Pd(II) intermediates to give functionalized alkenes is a well-known strategy in organic synthesis.30 However, most of the times the key η3-allyl Pd(II) species are generated through the oxidative addition of allyl halides, pseudohalides or acetates to Pd(0). We wondered if the η3-allyl complexes 1 and 2 could undergo the attack of a suitable nucleophile to render an organic derivative. Hence, we performed a range of reactions with several nucleophiles, such as KCN, [Tl(acac)], p-toluidine, and the phosphorus ylide Ph3PCHCOMe. There was no reaction at room temperature with any of these nucleophiles. When the reaction mixture was heated in refluxing toluene, complicated mixtures were produced. The reaction of complex 1b and dimethyl malonate in the presence of Cs2CO3 (CHCl3, room temperature) afforded an anti/syn mixture of isomers of a new complex containing the η3-allyl moiety and a coordinated malonate, whose structures were not studied further. When a solution of this complex in CHCl3 was heated at 60 °C, again a complicated mixture was obtained. In order to facilitate the nucleophilic attack, we performed the reaction of the η3-allyl complex 1b with p-toluidine in the presence of TlOTf, which would generate a cationic complex by replacing the Cl– ligand by OTf– in the coordination sphere of Pd(II). In this case, the stable cationic η3-allyl complex 3b was isolated, which contained a coordinated p-toluidine ligand (Scheme 5). This complex was stable in refluxing toluene and did not decompose to the expected organic product.
Scheme 5. Reactions of η3-Allyl Complex 1b with p-Toluidine.
The 1H NMR of complex 3b at room temperature showed some broad signals and seemed to correspond to a mixture of two isomers with a fluxional behavior (Figure 4). Perhaps the most significant resonances are those attributed to the Me groups: three sharp singlets at 1.45 (3 H), 1.42 (3 H), and 1.26 ppm (2.2 H) and two broad singlets at 2.28 (5.4 H) and 0.94 ppm (2.2 H). When the spectrum was measured at −40 °C, the broad singlet below 1 ppm become sharper, and the signal at 2.28 ppm split into two new singlets with the relative intensities 3:2.2. According to these data, we assumed that both isomers, anti-/syn-3b, are formed in a 1.33/1 ratio, one of which presented a fluxional behavior in solution. The less stable isomer should be anti-3b due to steric hindrance of the toluidine ligand and the substituent on the terminal allylic carbon, and for it, it was reasonable to suppose that the neutral ligand could be coming in and out of the metal coordination sphere.
Figure 4.
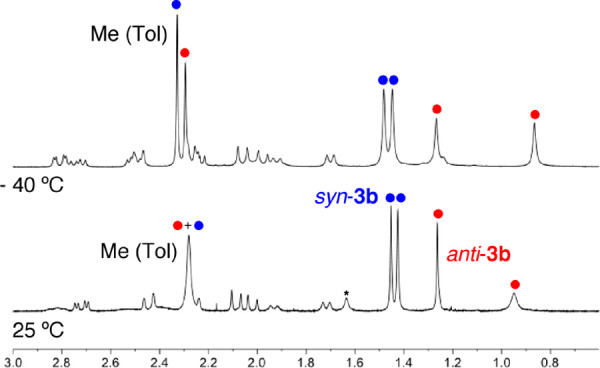
1H NMR spectra (0.6–3.0 ppm) of complex 3b (400 MHz) in CDCl3 at 25 °C (bottom) and −40 °C (top). The asterisk indicates the signal corresponding to H2O. The blue and red circles correspond to the syn and anti isomers, respectively.
The crystal structure of the η3-allyl complex syn-3b·CH2Cl2 was determined by X-ray diffraction (Figure 5), showing that the phenyl and benzyl substituents at the meso and terminal carbon atoms of the allylic unit occupied mutually syn positions and indicating that this was, in fact, the most stable isomer. For this complex there were two independent molecules in the asymmetric unit (A and A′). For molecule A, the palladium atom exhibited a square-planar geometry (mean deviation from the plane: X–Pd(1)–N(1)–N(2) 0.0067 Å, where X = centroid from C(7), C(8), and C(9) atoms). Both amino groups were coordinated in cis positions. The other two coordination sites were occupied by the allyl moiety, which was η3-bonded via C(7), C(8), and C(9), with a C(7)–C(8)–C(9) angle of 116.9(2)°. The allyl plane, defined by atoms C(7), C(8), and C(9), formed a dihedral angle of 118.1° with the Pd(II) coordination plane. The aromatic rings formed angles of 71.3° (metalated ring), 65.5° (phenyl ring at C7), 71.9° (phenyl ring at C8), 73.3° (phenyl ring at C14), and 92.4° (toluidine ring) with respect to the Pd coordination plane, to avoid steric hindrance. Hydrogen bond interactions were observed between the cationic palladium moiety and the OTf– anion (see the Supporting Information).
Figure 5.
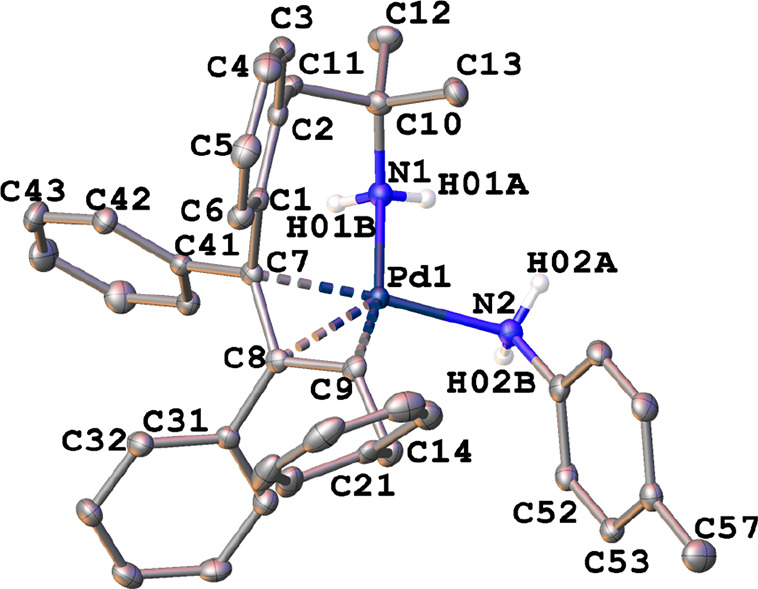
Thermal ellipsoid plot (50% probability) of the cation of one (A) of the two independent molecules of the complex syn-3b·CH2Cl2 showing the labeling scheme. The solvent molecule and the hydrogen atoms bonded to carbon have been omitted for clarity. Selected bond lengths (Å) and angles (deg) are given for both independent molecules (A and A′). For A: Pd(1)–N(1) = 2.131(2), Pd(1)–N(2) = 2.161(2), Pd(1)–C(7) = 2.100(2), Pd(1)–C(8) = 2.138(2), Pd(1)–C(9) = 2.165(2), C(7)–C(8) = 1.454(3), C(8)–C(9) = 1.408(3); N(1)–Pd(1)–N(2) = 90.26(9), C(7)–C(8)–C(9) = 116.9(2), C(8)–C(9)–C(14) = 124.9(2). For A′: Pd(1′)–N(1′) = 2.130(2), Pd(1′)–N(2′) = 2.163(2), Pd(1′)–C(7′) = 2.117(2), Pd(1′)–C(8′) = 2.134(2), Pd(1′)–C(9′) = 2.165(2), C(7′)–C(8′) = 1.452(3), C(8′)–C(9′) = 1.413(3); N(1′)–Pd(1′)–N(2′) = 88.94(8), C(7′)–C(8′)–C(9′) = 117.5(2), C(8)–C(9)–C(14) = 124.0(2).
Additionally, we studied the behavior of the anti-/syn-1a,b complexes toward a strong base. The reaction of these complexes with KOtBu in toluene under a N2 atmosphere afforded Pd(0) and the functionalized phenethylamines containing a 1,3-butadienyl substituent in an ortho position (4a,b; Scheme 6). We did not observe intramolecular cyclization arising from the possible nucleophilic attack of the NH2 group to the η3-allyl moiety. The compound 4a was a colorless solid and was easily isolated, but 4b was obtained as a liquid. In order to get a more easily isolable derivative, triflic acid was added to a solution of compound 4b in diethyl ether (Scheme 6). The corresponding ammonium triflate 5b precipitated in the reaction mixture and was obtained as a white powder in moderate yield (50%).
Scheme 6. Synthesis of Functionalized Phenethylamine Derivatives Containing 1,3-Butadienyl Substituents.
Miura et al.12 described the intermolecular three-component coupling of aryl iodides, diarylacetylenes, and alkenes in the presence of palladium acetylacetonate and silver acetate as catalysts, to give the corresponding 1:1:1 and 1:2:1 coupling products (1,3-butadiene and 1,3,5-hexatriene derivatives, respectively). The mechanism proposed for these catalytic transformations followed the sequential steps analogous to those of the synthesis of compounds 4 reported here, (1) formation of an aryl Pd(II) complex (either by oxidative addition or C–H activation), (2) alkyne insertion into the Pd–C bond to form an alkenyl complex, (3) alkene insertion into the Pd–C bond to form an alkyl intermediate, and (4) β-H elimination, assisted by a base, to afford the final diene. In our case, and due to the particular nature of the initial aryl group, we have been able to isolate all of the organometallic intermediates involved in the process.
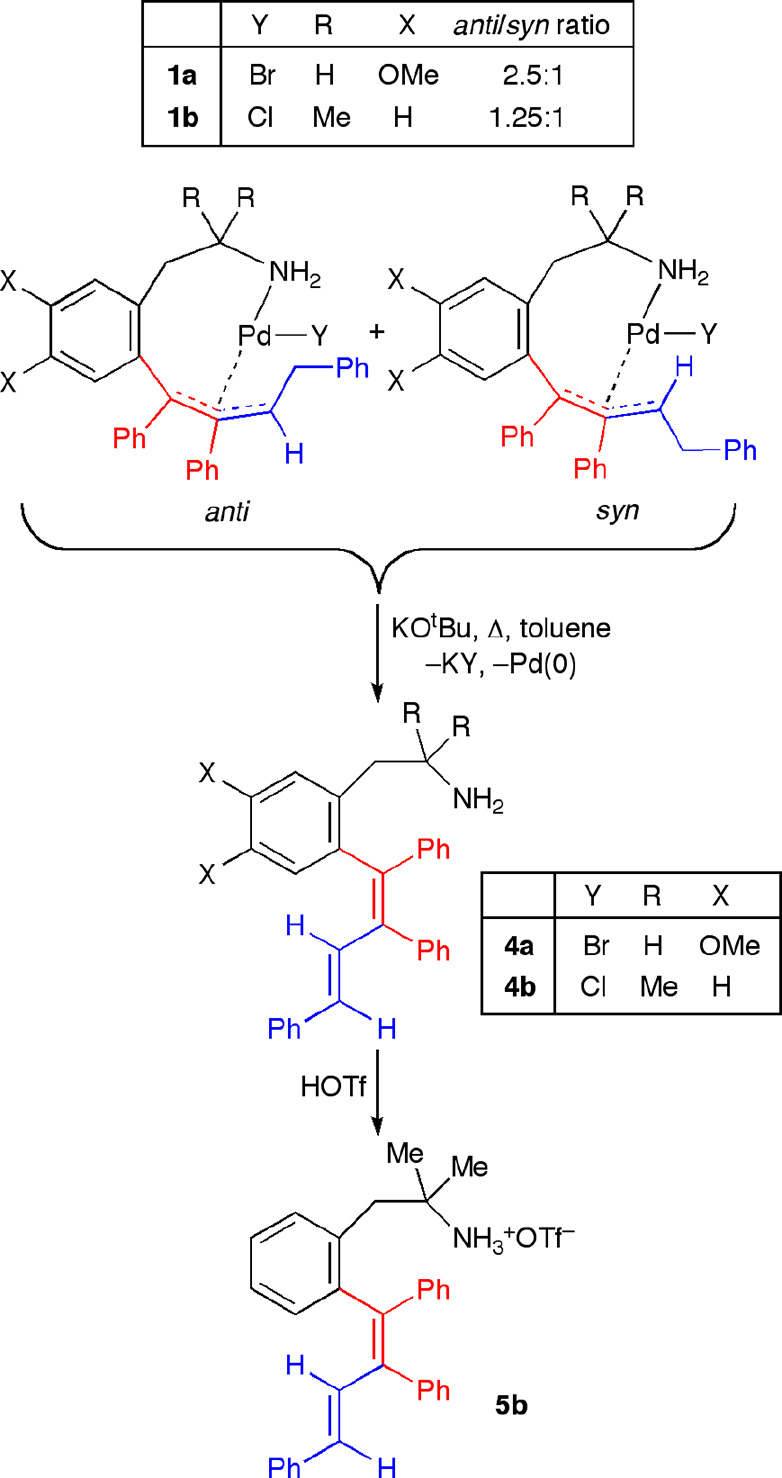
The crystal structure of the ammonium triflate 5b·H2O was solved by X-ray diffraction studies (Figure 6) and showed a slightly distorted planar diene skeleton (mean deviation from the plane: C(7)–C(8)–C(9)–C(1)–C(14)–C(21)–C(31)–C(41) = 0.041 Å) with an E,E geometry. Within the butadiene fragment, the average C–C double-bond distance (C7–C8, C9–C14) was 1.35(2) Å, being slightly longer than that corresponding to the most substituted unsaturated unit, and the average single-bond value (C1–C7, C7–C41, C8–C9, C8–C31, C14–C21) was 1.48(2) Å. The aryl rings formed angles of 62.5, 20.8, 69.1, and 48.0° with respect to the diene plane, adopting a helical conformation and releasing steric hindrance.
Figure 6.
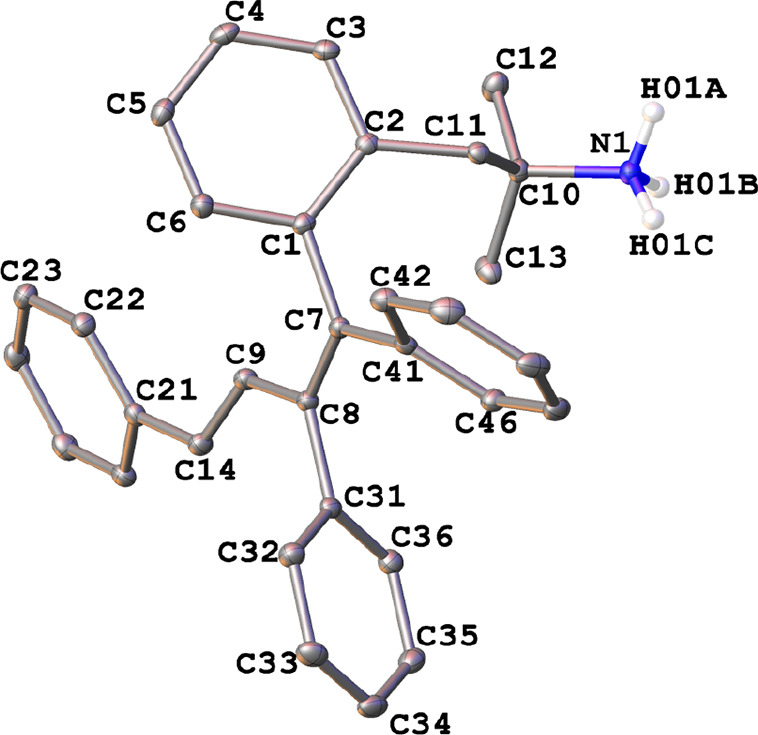
Thermal ellipsoid plot (50% probability) of the cation of compound 5b·H2O along with the labeling scheme. The solvent molecule and the hydrogen atoms bonded to carbon have been omitted for clarity. Selected bond lengths (Å) and angles (deg): C(1)–C(7) = 1.503(2), C(7)–C(8) = 1.359(2), C(8)–C(9) = 1.461(2), C(9)–C(14) = 1.341(2), C(14)–C(21) = 1.466(2), C(8)–C(31) = 1.496(2), C(7)–C(41) = 1.487(2); C(1)–C(7)–C(41) = 115.93(15), C(1)–C(7)–C(8) = 120.51(16), C(7)–C(8)–C(9) = 120.85(16), C(8)–C(9)–C(14) = 127.20(17), C(9)–C(8)–C(31) = 116.96(15).
We also attempted to insert a third unsaturated molecule into the Pd–C bonds of the η3-allyl complexes by bubbling CO through a solution of 1b in CH2Cl2 (room temperature, 4 h). Nevertheless, no reaction was observed, probably because these complexes were too stable. A similar reaction, with addition of TlOTf and use of THF as the solvent, led to a cationic complex with no CO inserted.
Insertion of 2-Norbornene into the Pd–C Bond
Synthesis and Structure of 10-Membered Norbornyl Palladium(II) Complexes
In contrast to styrene or ethyl acrylate, when 1 equiv of 2-norbornene was added to a solution of the alkenyl complex b (arising from insertion of one molecule of diphenylacetylene into the Pd–C bond of palladacycle B; see Scheme 7 for its structural formula), no insertion reaction was observed, neither at room temperature nor on heating the mixture to 65 °C in CHCl3. We also attempted the insertion reactions using as starting materials the eight-membered palladacycles c and d, arising from the insertion of methyl phenylpropiolate and 1-phenyl-1-propyne, respectively, into the Pd–C bond of complex B. When palladacycle d was used, the reaction at room temperature led to a new species that was tentatively assigned to the norbornene coordination monomer species 6d (Scheme 7), which could not be fully characterized, since it evolved easily to the starting dimeric complex d when we tried to crystallize it. Nevertheless, when these reaction mixtures (palladacycle c or d and 2-norbornene) were heated to 65 °C, we could successfully isolate the complexes 7c,d, where the insertion of the 2-norbornene moiety into the alkenyl Pd(II) complex had taken place (Scheme 7). These complexes have a monomeric nature, since the Pd center completes its coordination sphere with the intramolecular olefin moiety.
Scheme 7. Sequential Insertion of Alkyne/2-Norbornene into the Pd–C Bond of Six-Membered Palladacycle B.
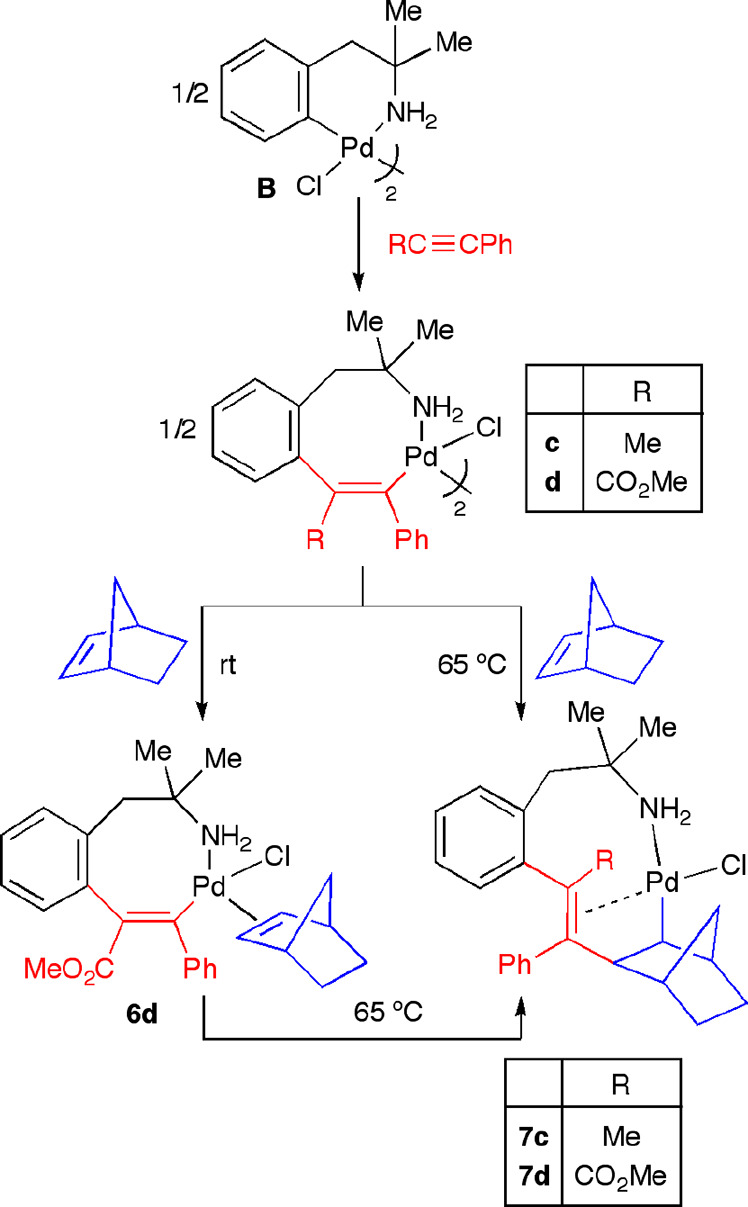
The crystal structures of complexes 7c·CHCl3 and 7d·1/2CHCl3 were solved by X-ray diffraction studies (see Figure 7 and the Supporting Information), and both showed similar features. For the complex 7c·CHCl3, the atoms Cl(1), N(1), and C(1) together with the midpoint of the C(3)–C(4) double bond form a square plane around the palladium atom (mean deviation from the plane: Pd(1)–C(1)–N(1)–Cl(1)–X 0.0230 Å, where X = centroid from C(3) and C(4) atoms). The norbornene unit adopted an exo conformation, arising from the syn addition of the Pd–C bond to the exo face of the olefin, as expected. The C(3)–C(4) double bond forms an angle of 61.8° with the palladium coordination plane. The methyl and phenyl substituents are mutually trans, which requires the isomerization of the first inserted alkyne. This arrangement reduces the steric hindrance between the substituents and is the normal behavior for di-inserted derivatives containing cyclometalated amines.31
Figure 7.
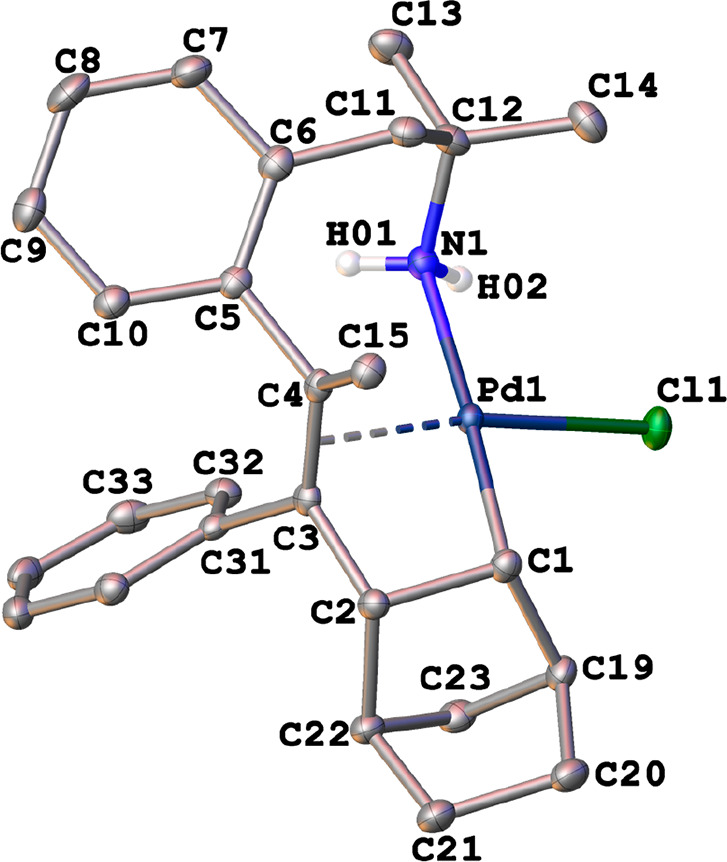
Thermal ellipsoid plot (50% probability) of the complex 7c·CHCl3 along with the labeling scheme. The solvent molecule and the hydrogen atoms bonded to carbon have been omitted for clarity. Selected bond lengths (Å) and angles (deg): Pd(1)–N(1) = 2.2185(19), Pd(1)–Cl(1) = 2.3589(5), Pd(1)–C(1) = 2.047(2), Pd(1)–C(3) = 2.159(2), Pd(1)–C(4) = 2.203(2), Pd(1)–X = 2.065, C(1)–C(2) = 1.553(3), C(2)–C(3) = 1.542(3), C(3)–C(4) = 1.403(3), C(4)–C(5) = 1.512(3); N(1)–Pd(1)–Cl(1) = 88.03(5), Cl(1)–Pd(1)–C(1) = 93.88(6), C(1)–Pd(1)–X = 76.7, X–Pd(1)–N(1) = 101.3, C(2)–C(3)–C(4) = 117.17(17), C(3)–C(4)–C(5) = 122.65(18). X represents the midpoint of the double bond C(3)–C(4).
We performed an analogous experiment reversing the order of the addition of the unsaturated reagents. That is, we studied the reactivity of the previously reported norbornyl derivative e (arising from the insertion of 2-norbornene into the six-membered palladacyle B) toward alkynes (Scheme 8). A suspension of the norbornyl Pd(II) complex e in CHCl3 was heated to 65 °C in the presence of methyl phenylpropiolate. The complex 7d, which arose from a reverse insertion order of the reagent addition, was isolated in 20% yield from the reaction mixture. In this reaction, palladacycle b was also formed. The formation of 7d could be explained through the deinsertion of the 2-norbornene fragment in e to give the starting palladacycle B, which in turn would undergo the sequential insertion of the alkyne and 2-norbornene. The ability of this cyclic olefin to insert reversibly into Pd–aryl bonds is well-known. This behavior has promoted the use of norbornene as an essential ligand in the functionalization of haloarenes: for instance, in the versatile Catellani type reaction.2 We tried to apply this strategy to obtain the diphenylacetylene/2-norbornene insertion derivative. Nevertheless, the reaction of the norbornyl complex e with diphenylacetylene gave rise mainly to the stable eight-membered palladacycle b, arising from monoinsertion of the alkyne (Scheme 8). That is, the olefin did not insert into the Pd–C bond of b, as noted above.
Scheme 8. Sequential Insertion of Norbornene/Alkynes into the Pd–C Bond of Palladacycle B.
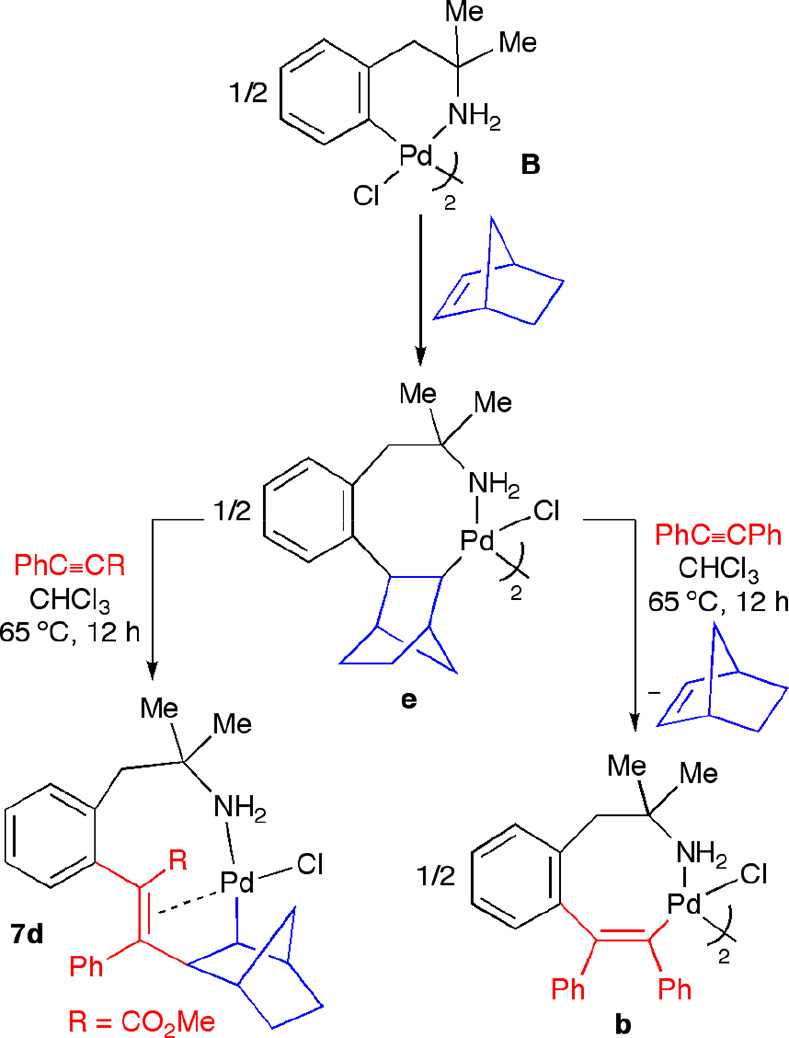
The synthesis of alkenyl palladacycles arising from the insertion of one molecule of alkyne into the Pd–C bond of a starting complex is not always a simple task, due to the possibility of di- and tri-insertion processes. This aspect is especially relevant when a very electrophilic alkyne, such as dimethyl acetylenedicarboxylate (DMAD), is used. We have previously tried to isolate the palladacycle f arising from monoinsertion of DMAD into the Pd–C bond of B; nevertheless, mixtures of mono-, di-, and tri-inserted products were obtained (Scheme 9). We thought that performing the reaction of B and DMAD in the presence of 2-norbornene could drive the reaction to the formation of a sequential DMAD/norbornene insertion product, given the fact that norbornene seems to react readily with other monoinserted alkyne derivatives. Indeed, when a solution of the palladacycle B in CHCl3 was heated to 65 °C in the presence of 1 equiv of DMAD and 1 equiv of 2-norbornene, the alkyne/alkene sequential insertion product 7f was isolated in good yield (Scheme 9). The crystal structure of complex 7f was also solved by X-ray diffraction studies (see the Supporting Information) and showed features similar to those discussed for 7c·CHCl3.
Scheme 9. Sequential Insertion of Norbornene/DMAD into the Pd–C Bond of Palladacycle B.
Reactivity of the Norbornyl Complexes
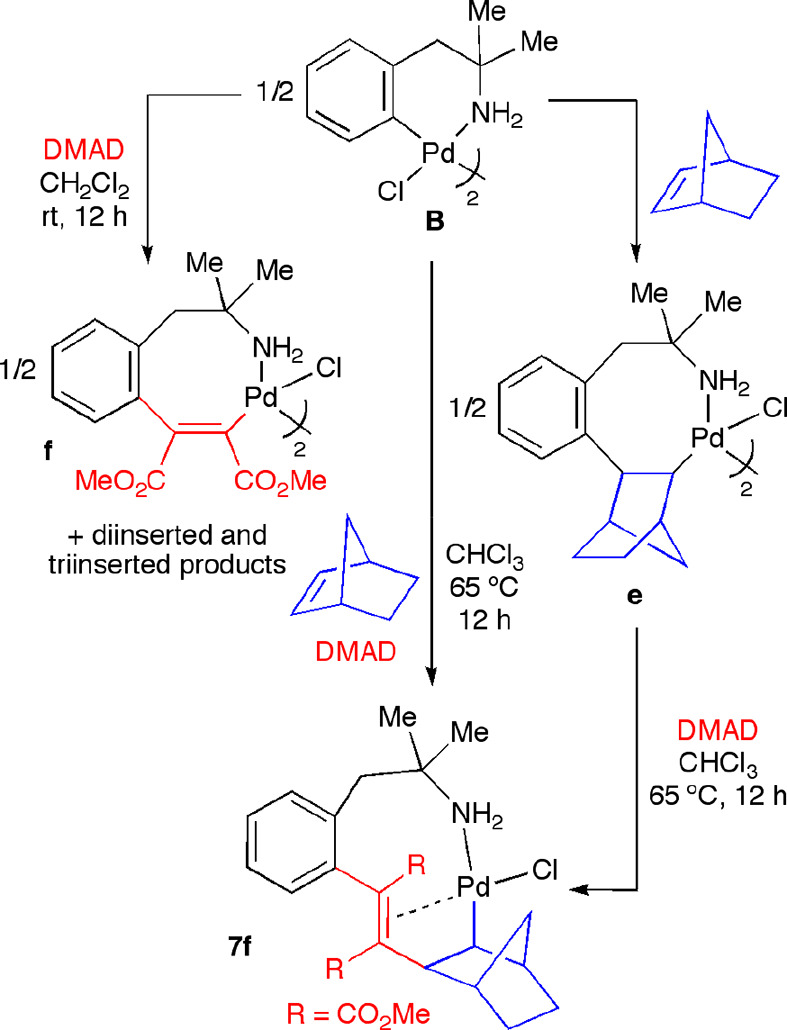
We explored the reactivity of the complex 7f toward KOtBu in MeCN at 78 °C. From the reaction mixture, we could isolate the tetrahydroisoquinoline derivative 8f by treatment of the crude product with HOTf: that is, from the protonation of V (Scheme 10). A possible path to explain the formation of 8f would involve the intramolecular carbopalladation of the alkene moiety, giving rise to a cyclopropyl ring. The new organometallic intermediate IV would then undergo a C–N coupling process with reductive elimination of Pd(0). Several Pd-catalyzed procedures to promote the cyclopropanation of 2-norbornene have been reported in the literature.32 Some of these procedures rely on the migratory insertion of norbornene into a Pd alkynyl33 or Pd alkenyl34 complex, and some organometallic intermediates similar to complexes 7 have been proposed, where the alkenyl moiety is coordinated intramolecularly to Pd.
Scheme 10. Reaction of Complex 7f with KOtBu.
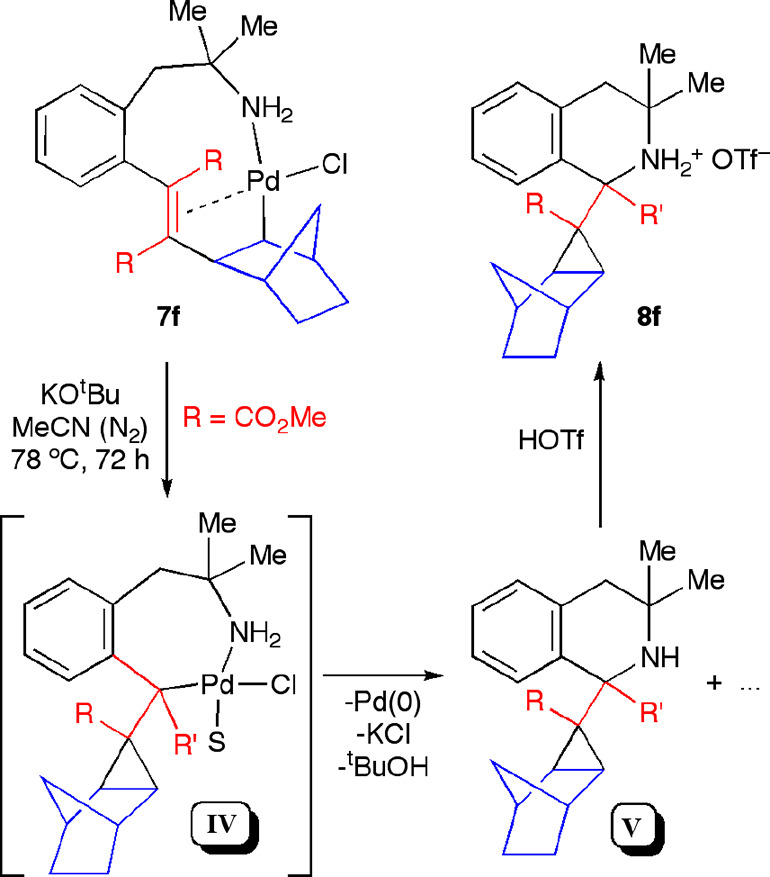
The crystal structure of 8f was solved by X-ray diffraction studies (Figure 8) and showed the isoquinoline nucleus derived from phentermine substituted at C1 with a methoxycarbonyl group and a tricyclo[3.2.1]octyl ring. It is worth noting that highly strained hydrocarbons such as this cyclopropane-containing norbornyl moiety have been tested as new highly energetic materials for liquid-fueled propulsion systems.35
Figure 8.
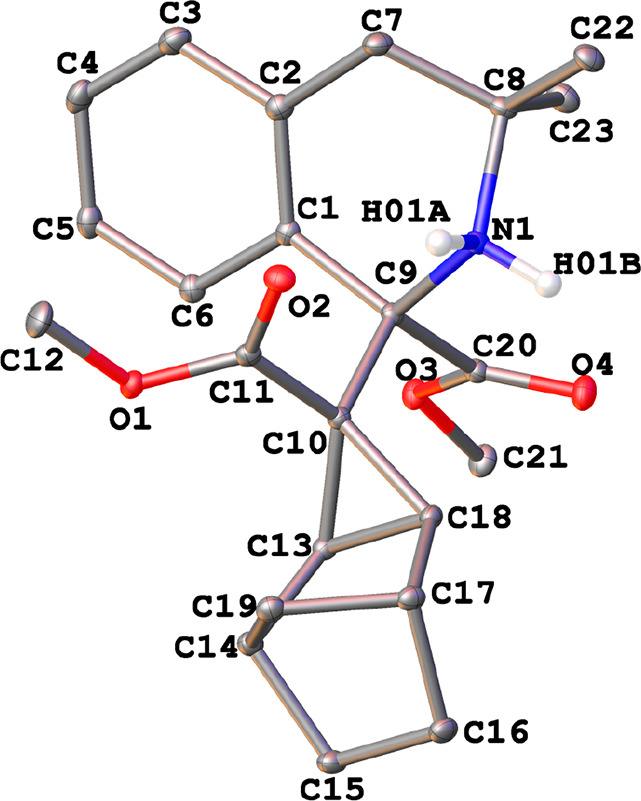
Thermal ellipsoid plot (50% probability) of the cation of compound 8f along with the labeling scheme. The hydrogen atoms bonded to carbon have been omitted for clarity. Selected bond lengths (Å) and angles (deg): N(1)–C(8) = 1.5233(17), N(1)–C(9) = 1.5134(16), C(9)–C(10) = 1.5595(18), C(10)–C(13) = 1.5071(17), C(10)–C(18) = 1.5259(18), C(13)–C(18) = 1.5261(17); C(1)–C(9)–N(1) = 111.04(10), N(1)–C(9)–C(20) = 105.93(10), C(20)–C(9)–C(10) = 110.53(10), C(10)–C(18)–C(13) = 59.18(8), C(18)–C(13)–C(10) = 60.40(8), C(13)–C(10)–C(18) = 60.42(8).
We studied whether a further insertion of an unsaturated molecule could be carried out in the complexes 7c,d,f, obtained after alkyne/norbornene insertion. Complex 7f did not react when it was stirred in CHCl3 in the presence of CO at room temperature for 5 h. When the reaction mixture was heated to 65 °C, a complicated mixture was formed. In order to facilitate the insertion of CO, we performed the reaction in the presence of TlOTf in acetone, hence generating a cationic Pd(II) intermediate in situ. In this case, the reaction afforded the lactone 9f and Pd(0) (Scheme 11). The formation of 9f could be explained as the result of the hydrolysis of the acyl Pd(II) intermediate generated upon CO insertion into the Pd–C bond present in 7f. Under those conditions, the carboxylic acid derivative underwent an intramolecular Michael addition of the CO2H group to the α,β-unsaturated ester moiety.
Scheme 11. Reactions of Complexes 7 with CO.
When the cationic derivatives of complexes 7c,d (generated in situ) reacted with CO at room temperature, the amino acid derivatives 10c,d were obtained (Scheme 11). In the case of 10d, intramolecular cyclization to give 9d took place after heating at 65 °C in CHCl3 for 7 days. As expected, the derivative 10c was stable upon heating, since it was not activated for the Michael addition step, as occurred in the cases of the alkenyl complexes bearing a CO2Me substituent.
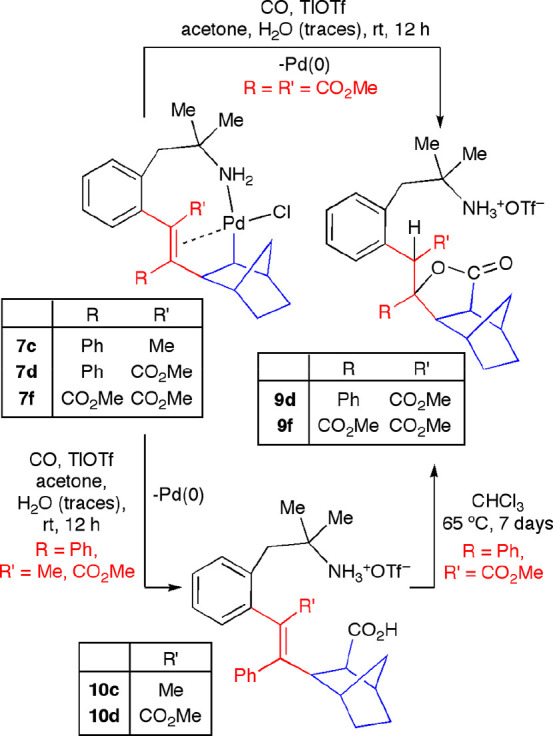
The crystal structure of 9f·Et2O has been solved by X-ray diffraction studies (Figure 9) and showed an ammonium salt derived from the starting phentermine containing a bicyclo[2.2.1]heptane lactone substituent. The hexahydro-4,7-methanoisobenzofuran-1(3H)-one core is a known compound that has attracted interest because of its potential use as a prostaglandin/thromboxane receptor antagonist.36 The most frequent routes for its synthesis involved (a) the cycloaddition reaction of furan-2(5H)-one and cyclopentadiene37 and (b) the oxidation of cis-2,3-bis(hydroxymethyl)bicyclo[2.2.1]heptane, which could be performed in a stoichiometric way with standard organic oxidants38 or by catalysis, with the use of enzymes39 or transition metals.40
Figure 9.
Thermal ellipsoid plot (50% probability) of the cation of 9f·Et2O along with the labeling scheme. The solvent molecule and the hydrogen atoms bonded to carbon have been omitted for clarity. Selected bond lengths (Å) and angles (deg): N(1)–C(8) = 1.5209(19), C(11)–C(12) = 1.522(2), C(11)–C(14) = 1.557(2), C(14)–O(4) = 1.4423(18), C(17)–O(3) = 1.2010(19), C(17)–O(4) = 1.3514(19), C(16)–C(17) = 1.499(2); C(1)–C(11)–C(12) = 107.19(12), C(11)–C(14)–O(4) = 109.88(12), C(14)–O(4)–C(17) = 111.86(11), O(4)–C(17)–C(16) = 111.77(13), C(17)–C(16)–C(15) = 104.53(12), C(16)–C(15)–C(14) = 104.02(12), C(15)–C(14)–O(4) = 106.07(11).
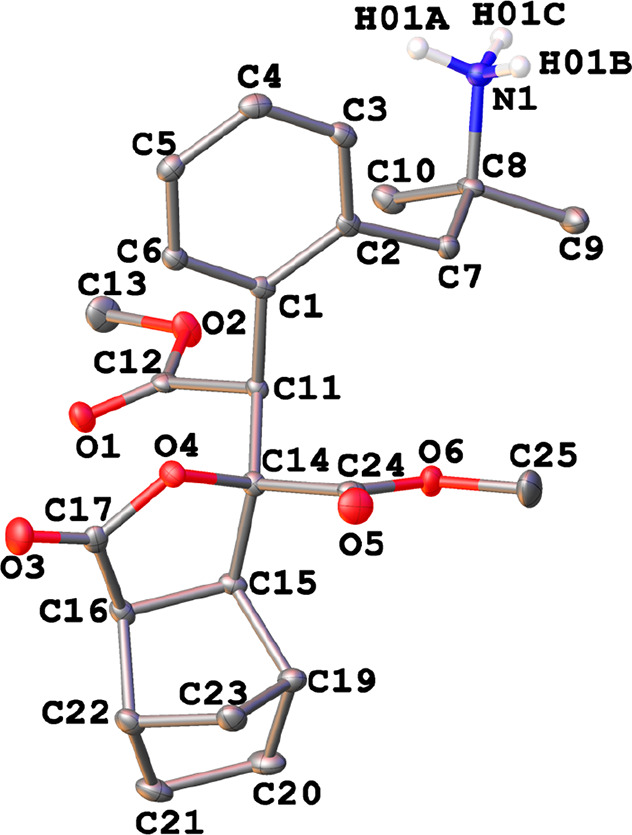
The crystal structure of 10c has also been determined by X-ray diffraction studies (Figure 10), and it showed the ammonium salt derived from the sequential insertion of alkyne/norbornene/CO into the initial phentermine palladacycle. Both substituents (the carboxylate and the alkenyl groups) at the norbornadienyl moiety were in an exo disposition.
Figure 10.
Thermal ellipsoid plot (50% probability) of the cation of 10c·H2O along with the labeling scheme. The solvent molecule and the hydrogen atoms bonded to carbon have been omitted for clarity. Selected bond lengths (Å) and angles (deg): N(1)–C(8) = 1.518(2), C(1)–C(11) = 1.504(3), C(11)–C(14) = 1.342(3), C(14)–C(15) = 1.532(2), C(15)–C(16) = 1.582(2), C(16)–C(17) = 1.506(3), C(17)–O(1) = 1.218(2), C(17)–O(2) = 1.328(2); N(1)–C(8)–C(7) = 104.46(14), C(1)–(11)–C(14) = 121.73(17), C(11)–C(14)–C(15) = 121.56(17), C(14)–C(15)–C(16) = 118.26(15), C(15)–C(16)–C(17) = 116.58(15), C(16)–C(17)–O(1) = 126.22(17), C(16)–C(17)–O(2) = 111.65(16).
Conclusion
In summary, the stoichiometric sequential insertion of alkynes and alkenes into the Pd–C bond of metalated phenethylamines has been studied. The result of these reactions depends on the nature of the olefin used. The insertion of styrene or ethyl acrylate into the eight-membered palladacycle arising from monoinsertion of diphenylacetylene affords the η3-allyl species 1 or 2, via sequential β-H elimination/hydropalladation steps. However, the insertion of 2-norbornene leads to norbornyl Pd(II) complexes with an alkenyl moiety intramolecularly coordinated to the metal center.
The treatment with a strong base of the organometallic complexes obtained upon the sequential insertion of alkyne/alkene (1, 2, and 7) also led to different results. While the η3-allyl complexes 1 and 2 afforded 1,3-butadienyl-substituted phenethylamines (4b and 5b), the norbornyl Pd derivative 7f gave a tetrahydroisoquinoline derivative upon cyclopropanation of the norbornene fragment and C–N coupling (8f).
The η3-allyl complexes 1 and 2 were unreactive toward CO insertion. In contrast, the norbornyl derivatives 7c,d,f afforded interesting amino acid or lactone derivatives upon CO insertion into the Pd–C bond and subsequent hydrolysis, under the appropriate conditions.
Overall, we have shown that the isolation of the organometallic intermediates obtained upon stepwise insertion of alkynes and alkenes into palladated phenethylamines allows the synthesis of functionalized primary phenethylamines. These stoichiometric routes avoid the problems associated with substrates that make difficult the development of catalytic processes (such as primary alkylamines) and allow the control of the regioselective insertion of the different unsaturated coupling partners.
Experimental Section
Caution! Special precautions should be taken in handling thallium(I) compounds, which are toxic.
General Procedures
Infrared spectra were recorded on a PerkinElmer 16F-PC-FT spectrometer. C, H, N, and S analyses were carried out with a LECO CHNS-932 microanalyzer. Conductance measurements and melting point determinations were carried out as described elsewhere.41 Unless stated otherwise, NMR spectra were recorded in CDCl3 with Bruker Avance 300, 400, and 600 spectrometers. Chemical shifts are referenced to TMS (1H and 13C{1H}). Signals in the 1H and 13C NMR spectra of all compounds were assigned with the help of APT, HMQC, and HMBC experiments. High-resolution electrospray ionization mass spectra (ESI-MS) were recorded on an Agilent 6220 Accurate-Mass time-of-flight (TOF) LC/MS. Reactions were carried out at room temperature without special precautions against moisture unless specified otherwise. The groups C6H4 and C6H2 are denoted by Ar. Free and inserted 2-norbornene are denoted by C7H10 (free), CH(C5H8)CH (inserted), and nor (both, NMR signals).
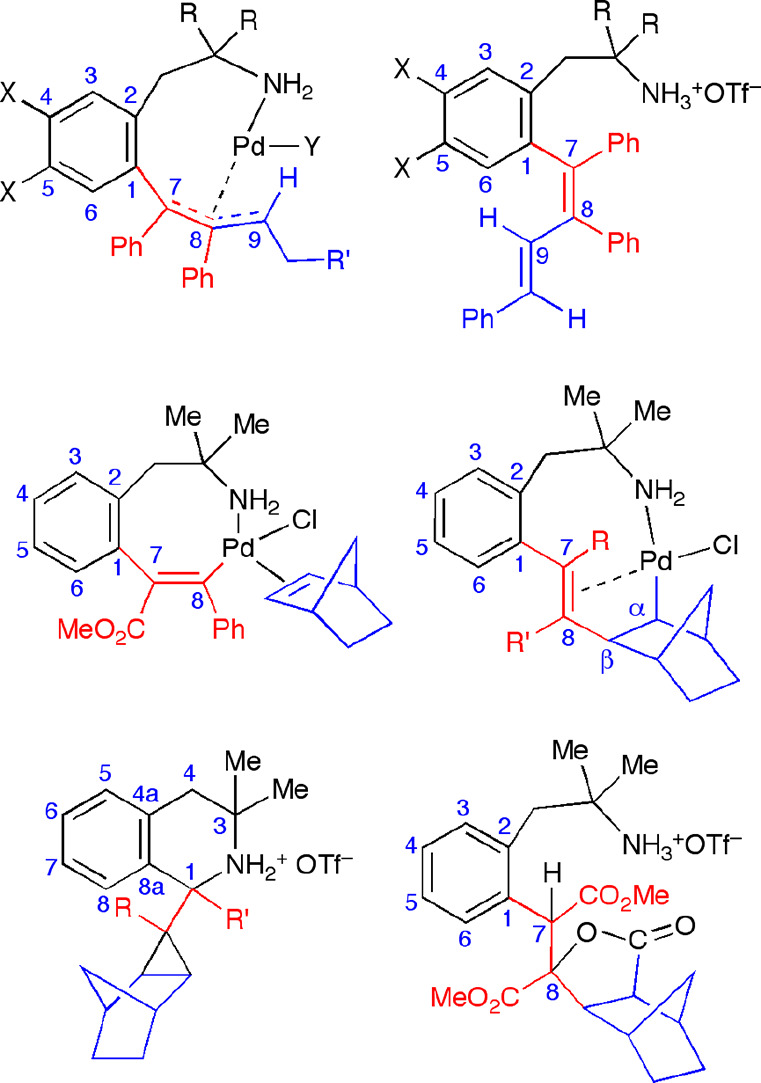
Styrene, ethyl acrylate, 2-norbornene, dimethyl acetylenedicarboxylate (DMAD), methyl phenylpropiolate, p-toluidine (Tol), KOtBu, and HOTf (HO3SCF3) were used as received from comertial sources. The palladacycles [Pd{C,N-C6H2(CH2CH2NH2)-2-(OMe)2-4,5}(μ-Br)]2 (A),26 [Pd{C,N-C6H4(CH2CMe2NH2)-2}(μ-Cl)]2 (B),24 [Pd{C,N-C(Ph)=C(Ph)C6H2(CH2CH2NH2)-2-(OMe)2-4,5}(μ-Br)]2 (a), [Pd{C,N-C(Ph)=C(R)C6H4(CH2CMe2NH2)-2}(μ-Cl)]2 (R = Ph (b), Me (c), CO2Me (d)),20 and [Pd{C,N-CH(C5H8)CHC6H4(CH2CMe2NH2)-2}(μ-Cl)]2 (e)19 were prepared as previously reported. TlOTf was prepared by the reaction of Tl2CO3 and HO3SCF3 (1/2) in water and recrystallized from acetone/Et2O. Chart 2 gives the numbering schemes for the new organometallic and organic derivatives.
Chart 2. Numbering Schemes for the New Palladium(II) Complexes and the Organic Derivatives.
Synthesis of anti-/syn-1a
Styrene (23 μL, 0.202 mmol) was added to a solution of palladacycle a (110 mg, 0.100 mmol) in CH2Cl2 (10 mL), and the mixture was stirred for 20 h. The resulting mixture was filtered through a plug of Celite, the filtrate was concentrated to ca. 1 mL, and Et2O (15 mL) was added. The mixture was cooled to 0 °C, the resulting suspension was filtered, and the pale yellow solid was air-dried to give a mixture of anti-/syn-1a. Yield: 86 mg, 0.132 mmol, 66%. Mp: 143 °C. Anal. Calcd for C32H32BrNO2Pd (648.936): C, 59.23; H, 4.97; N, 2.16. Found: C, 59.32; H, 5.10; N, 2.19. IR (cm–1): ν(NH) 3319 m, 3258 w, 3206 m, 3135 w. The NMR spectra of this complex showed two sets of signals in approximately a 2.5/1 ratio (determined by 1H integration), corresponding to the mixture anti-/syn-1a. Data for anti-1a (extracted from the mixture) are as follows. 1H NMR (300.1 MHz): δ 1.63–1.70 (m, 1 H, NH2), 1.88 (m, 1 H, CH2Ar), 2.56 (br dd, 1 H, CH2Ar, 2JHH = 15.3, 3JHH = 7.8 Hz), 2.76–2.85 (m, partially obscured by the resonance of MeO, 1 H, CH2Ph), 2.86 (s, 3 H, MeO), 2.97–3.02 (br d, 1 H, CH2N, 2JHH = 12.6 Hz), 3.22–3.35 (m, 2 H, 1 H of CH2N + 1 H of NH2), 2.38 (br dd, 1 H, CH2Ph, 2JHH = 17.4, 3JHH = 4.5 Hz), 3.87 (s, 3 H, MeO), 5.11 (dd, 1 H, CH, 3JHH = 12.3, 3JHH = 4.5 Hz), 6.47 (s, 1 H, H6), 6.64 (s, 1 H, H3), 6.80–7.27 (m, 15 H, Ph). 13C{1H} NMR (75.5 MHz): δ 35.2 (CH2Ar), 39.1 (CH2Ph), 42.9 (CH2N), 54.7 (MeO), 55.9 (MeO), 85.0 (CH), 93.8 (C7), 111.9 (CH6), 116.1 (CH3), 123.6 (C8), 125.8 (CH, Ph), 126.8 (CH, Ph), 127.0 (CH, Ph), 127.3 (CH, Ph), 128.0 (CH, Ph), 128.4 (CH, Ph), 128.5 (CH, Ph), 128.6 (CH, Ph), 129.6 (CH, Ph), 132.2 (C1), 135.1 (i-C, Ph), 140.4 (i-C, Ph), 141.1 (i-C, Ph), 143.6 (C2), 146.4 (C5), 148.3 (C4).
anti/syn Isomerization of 1a
An NMR tube was charged with a 2.5:1 mixture of anti-/syn-1a (20 mg) and CDCl3 (0.6 mL), and the solution was heated at 60 °C. The sample was checked by 1H NMR periodically, until complete conversion. After 10 days at 60 °C, a spectroscopically pure sample of syn-1a was obtained. Data for syn-1a are as follows. 1H NMR (400.9 MHz): δ 1.61–1.67 (m, 1 H, NH2), 1.89 (br dd, 1 H, CH2Ar, 2JHH = 15.3, 3JHH = 10.8 Hz), 2.47 (br dd, 1 H, CH2Ar, 2JHH = 15.4, 3JHH = 8.0 Hz), 2.86–2.90 (m, 1 H, CH2Ph), 3.23 (dd, partially obscured by the resonance of NH2, 1 H, CH2N, 2JHH = 12.0, 3JHH = 8.0 Hz), 3.02 (br d, 1 H, NH2, 2JHH = 12.0 Hz), 3.42 (s, 3 H, MeO), 3.46 (br dd, 1 H, CH2Ph, 2JHH = 14.2, 3JHH = 3.5 Hz), 3.87 (s, 3 H, MeO), 4.80 (dd, 1 H, CH, 3JHH = 11.0, 3JHH = 3.7 Hz), 6.39 (s, 1 H, H6), 6.60 (s, 1 H, H3), 6.19 (br d, 1 H, Ph, 3JHH = 7.0 Hz), 6.80 (br d, 2 H, Ph, 3JHH = 7.0 Hz), 6.90–7.23 (m, 10 H, Ph), 7.40 (m, 1 H, Ph), 7.85 (br d, 1 H, Ph, 3JHH = 6.0 Hz). 13C{1H} NMR (75.5 MHz): δ 34.8 (CH2Ar), 35.8 (CH2Ph), 43.6 (CH2N), 55.6 (MeO), 60.0 (MeO), 84.5 (CH), 90.8 (C7), 110.0 (CH6), 116.2 (CH3), 124.0 (C8), 126.0 (CH, Ph), 127.0 (CH, Ph), 127.3 (CH, Ph), 127.7 (CH, Ph), 128.4 (CH, Ph), 128.5 (CH, Ph), 129.0 (CH, Ph), 131.8 (C1), 133.2 (i-C, Ph), 133.7 (CH, Ph), 135.1 (i-C, Ph), 140.5 (i-C, Ph), 142.4 (C2), 147.5 (C5), 148.2 (C4).
Synthesis of anti-/syn-1b
Method A
Styrene (52 μL, 0.453 mmol) was added to a solution of palladacycle b (200 mg, 0.213 mmol) in CH2Cl2 (10 mL). The solution was stirred for 12 h, the solvent was concentrated to ca. 5 mL, Et2O (5 mL) was added, and the resulting suspension was filtered through a plug of Celite. The filtrate was concentrated to ca. 1 mL, and n-pentane (20 mL) was added. The suspension was filtered, and the pale yellow solid was washed with n-pentane (2 × 5 mL) and air-dried to give a mixture of anti-/syn-1b (ratio ca. 1.25:1 by 1H NMR). Yield: 155 mg, 0.270 mmol, 63%. Dec pt: 252 °C. Anal. Calcd for C32H32ClNPd (572.485): C, 67.14; H, 5.63; N, 2.44. Found: C, 67.19; H, 5.60; N, 2.39. IR (cm–1): ν(NH) 2295 w, 3238 w.
Method B
In a different preparation, it was possible to obtain two different crops by fractional crystallization, each of them enriched in one of the isomers. Styrene (30 μL, 0.262 mmol) was added to a solution of palladacycle b (120 mg, 0.128 mmol) in CH2Cl2 (10 mL). The mixture was stirred for 12 h and filtered through a plug of Celite. The filtrate was concentrated to ca. 3 mL, and Et2O (15 mL) was added. The resulting suspension was filtered, and the solid was air-dried to afford 57 mg of an anti-enriched mixture of both isomers (anti/syn = 3/1). The mother liquors were concentrated to ca. 3 mL, and n-pentane (20 mL) was added. The resulting suspension was filtered, and the solid was air-dried to afford 25 mg of a syn-enriched mixture of both isomers (anti/syn = 1/3). Almost spectroscopically pure samples of anti- and syn-1b could be obtained by growing single crystals from the enriched mixtures. Data for anti-1b are as follows. 1H NMR (300.1 MHz): δ 1.34 (s, 6 H, CMe2), 1.86 (br d, 1 H, NH2, 2JHH = 10.8 Hz), 2.11 (d, 1 H, CH2Ar, 2JHH = 15.0 Hz), 2.38 (br d, 1 H, CH2Ar, 2JHH = 14.7 Hz), 2.59 (dd, 1 H, CH2Ph, 2JHH = 16.5, 3JHH = 11.7 Hz), 3.18 (br d, 1 H, NH2, 2JHH = 10.8 Hz), 3.60 (dd, 1 H, CH2Ph, 2JHH = 16.5, 3JHH = 4.5 Hz), 5.16 (dd, 1 H, CH, 3JHH = 11.7, 3JHH = 4.5 Hz), 6.86–6.91 (m, 3 H, Ph + Ar), 7.04–7.34 (m, 16 H, Ph + Ar). 13C{1H} NMR (75.5 MHz): δ 27.8 (Me, CMe2), 36.0 (Me, CMe2), 39.0 (CH2Ph), 46.8 (CH2Ar), 52.3 (CMe2), 88.3 (CH), 89.4 (C7), 122.9 (C8), 125.9 (CH), 126.3 (CH), 126.7 (CH), 127.0 (CH), 127.5 (CH), 127.8 (CH, Ph), 127.9 (CH, Ph), 128.3 (br s, CH, Ph), 129.2 (CH), 129.4 (CH), 128.5 (CH, Ph), 132.3 (CH), 134.9 (CH3), 137.6 (C2), 139.8 (i-C, Ph), 140.4 (C1), 141.0 (i-C, Ph), 143.2 (i-C, Ph). Data for syn-1b are as follows. 1H NMR (400.9 MHz): δ 1.29 (s, 3 H, Me, CMe2), 1.30 (s, 3 H, Me, CMe2), 1.85 (br d, 1 H, NH2, 2JHH = 10.0 Hz), 2.09 (d, 1 H, CH2Ar, 2JHH = 14.8 Hz), 2.28 (dd, 1 H, CH2Ar, 2JHH = 14.8, 4JHH = 1.2 Hz), 3.17 (dd, 1 H, CH2Ph, 2JHH = 14.8, 3JHH = 10.6 Hz), 3.29 (br d, 1 H, NH2, 2JHH = 10.4 Hz), 3.40 (dd, 1 H, CH2Ph, 2JHH = 15.2, 3JHH = 4.0 Hz), 4.84 (dd, 1 H, CH, 3JHH = 10.4, 3JHH = 4.0 Hz), 6.50 (br s, 1 H, Ph), 6.87–6.91 (m, 4 H, Ph), 6.99–7.04 (m, 5 H, H3 + H6 + 3 H of Ph), 7.08 (td, 1 H, H5, 3JHH = 7.6, 4JHH = 1.6 Hz), 7.11–7.20 (m, 6 H, Ph), 7.22 (td, 1 H, H4, 3JHH = 7.2, 4JHH = 1.6 Hz), 7.36 (br s, 1 H, Ph), 7.84 (br s, 1 H, Ph). 13C{1H} NMR (100.8 MHz): δ 27.2 (Me, CMe2), 35.1 (CH2Ph), 36.1 (Me, CMe2), 46.3 (CH2Ar), 53.1 (CMe2), 84.7 (CH), 88.0 (C7), 124.5 (C8), 125.8 (CH, Ph), 126.2 (CH6), 126.9 (CH5), 127.1 (CH, Ph), 127.28 (CH4 or Ph), 127.29 (CH4 or Ph), 127.5 (CH, Ph), 127.9 (br s, CH, Ph), 128.21 (CH, Ph), 128.25 (CH, Ph), 128.4 (CH, Ph), 128.7 (CH, Ph), 131.4 (CH, Ph), 133.6 (br s, CH, Ph), 134.9 (i-C, Ph), 135.1 (CH3), 138.4 (C2), 139.7 (i-C, Ph), 140.0 (C1), 142.2 (i-C, Ph).
anti/syn Isomerization of 1b
An NMR tube was charged with a 2.5/1 mixture of anti-/syn-1b (20 mg) and CDCl3 (0.6 mL), and the solution was heated at 60 °C. The sample was checked by 1H NMR periodically, until no change was observed. After 5 days at 60 °C, a 1/10 mixture of anti-/syn-1b was obtained.
Synthesis of anti-/syn-2a
Ethyl acrylate (40 μL, 0.367 mmol) was added to a solution of palladacycle a (200 mg, 0.183 mmol) in CH2Cl2 (10 mL), and the mixture was stirred for 48 h. The resulting mixture was filtered through a plug of Celite, the filtrate was concentrated to ca. 1 mL, and Et2O (20 mL) was added. The suspension was filtered, and the pale yellow solid was washed with Et2O (2 × 5 mL) and air-dried to give an anti-/syn-2a mixture (171 mg; ratio ca. 5/1 by 1H NMR). The filtrate was concentrated to ca. 1 mL, and n-pentane (20 mL) was added. The resulting suspension was filtered, and the solid was air-dried to afford a anti-/syn-2a mixture (30 mg; ratio ca. 1/5 by 1H NMR). Yield: 201 mg, 0.312 mmol, 85%. Mp: 169 °C. Anal. Calcd for C29H32BrNO4Pd (644.901): C, 54.01; H, 5.00; N, 2.17. Found: C, 54.25; H, 5.00; N, 2.49. IR (cm–1): ν(NH) 3293 w, 3214 br; ν(CO) 1727 s, 1711 s. Data for anti-2a are as follows. 1H NMR (400.9 MHz): δ 1.19 (t, 3 H, MeCH2, 3JHH = 7.2 Hz), 1.61 (br d, 1 H, NH2, 2JHH = 12.0 Hz), 1.90 (dd, 1 H, CH2Ar, 2JHH = 10.8, 3JHH = 4.8 Hz), 2.35 (dd, 1 H, CH2CO2Et, 2JHH = 17.3, 3JHH = 11.6 Hz), 2.58 (br dd, 1 H, CH2Ar, 2JHH = 15.6, 4JHH = 6.8 Hz), 2.93 (m, 1 H, CH2N), 3.26–3.33 (m, 3 H, 1 H of NH2 + 1 H of CH2N + 1 H of CH2CO2Et), 3.86 (s, 3 H, MeO), 3.91 (s, 3 H, MeO), 4.01 (m, 1 H, CH2O), 4.15 (m, 1 H, CH2O), 4.85 (dd, 1 H, CH, 3JHH = 11.6, 3JHH = 4.8 Hz), 6.68 (s, 1 H, H3), 6.87 (s, 1 H, H6), 6.99–7.10 (m, 8 H, Ph), 7.11–7.25 (m, 2 H, Ph). 13C{1H} NMR (100.8 MHz): δ 14.1 (MeCH2), 35.1 (CH2Ar), 39.4 (CH2CO2Et), 42.9 (CH2N), 56.0 (MeO), 56.0 (MeO), 60.8 (CH2O), 79.1 (CH), 93.8 (C7), 112.5 (CH6), 116.4 (CH3), 123.9 (C8), 126.8 (CH), 127.2 (CH), 127.3 (CH), 127.9 (CH), 128.0 (CH), 129.6 (CH), 132.1 (C1), 132.6 (C2), 140.7 (i-C, Ph), 143.1 (i-C, Ph), 147.0 (C5), 148.6 (C4), 170.0 (CO).
anti/syn Isomerization of 2a
An NMR tube was charged with a 5/1 anti-/syn-2a mixture (20 mg) and CDCl3 (0.6 mL), and the solution was heated at 60 °C. The sample was checked by 1H NMR periodically, until complete conversion. After 48 h at 60 °C, a spectroscopically pure sample of syn-2a was obtained. Data for syn-2a are as follows. 1H NMR (300.1 MHz): δ 1.10 (t, 3 H, MeCH2, 3JHH = 7.2 Hz), 1.67 (br d, 1 H, NH2, 2JHH = 10.8 Hz), 1.88 (dd, 1 H, CH2Ar, 2JHH = 15.6, 3JHH = 10.8 Hz), 2.49 (dd, 1 H, CH2Ar, 2JHH = 15.3, 3JHH = 6.6 Hz), 2.80 (dd, 1 H, CH2CO2Et, 2JHH = 18.0, 3JHH = 10.2 Hz), 2.90 (m, partially obscured by the resonance of CH2CO2Et, 1 H, CH2N), 3.25–3.32 (m, 2 H, 1 H of CH2N + 1 H of CH2CO2Et), 3.37 (br d, 1 H, NH2, 2JHH = 9.9 Hz), 3.89 (s, 3 H, MeO), 3.93–3.96 (m, partially obscured by the resonance of MeO, 1 H, CH2O), 3.96–4.05 (m, partially obscured by the resonance of MeO, 1 H, CH2O), 3.99 (s, 3 H, MeO), 4.75 (dd, 1 H, CH, 3JHH = 10.2, 3JHH = 4.2 Hz), 6.63 (s, 1 H, H3), 6.87–6.91 (m, partially obscured by the resonance of H6, 2 H, Ph), 6.93 (s, 1 H, H6), 7.01–7.17 (m, 6 H, Ph), 7.27–7.34 (m, 1 H, Ph), 7.65 (br d, 1 H, Ph, 3JHH = 6.0 Hz). 13C{1H} NMR (75.5 MHz): δ 14.1 (MeCH2), 34.7 (CH2Ar), 34.9 (CH2CO2Et), 43.7 (CH2N), 56.0 (MeO), 56.2 (MeO), 60.4 (CH2O), 75.2 (CH), 92.2 (C7), 110.1 (CH6), 116.2 (CH3), 125.6 (C8), 127.1 (CH), 127.3 (CH), 127.6 (CH), 127.8 (CH), 128.8 (CH), 131.5 (C1), 133.0 (CH), 133.1 (C2), 134.6 (i-C, Ph), 142.2 (i-C, Ph), 147.8 (C5), 148.4 (C4), 170.8 (CO).
Synthesis of anti-/syn-2b
Method A
Ethyl acrylate (50 μL, 0.460 mmol) was added to a solution of palladacycle b (200 mg, 0.213 mmol) in CH2Cl2 (10 mL). The solution was stirred for 12 h and then concentrated to ca. 5 mL. Et2O (5 mL) was added, and the resulting suspension was filtered through a plug of Celite. The filtrate was concentrated to ca. 1 mL, and n-pentane (20 mL) was added. The suspension was filtered, and the pale yellow solid was washed with n-pentane (2 × 5 mL) and air-dried to give as anti-/syn- mixture (ratio ca. 1.25/1 by 1H NMR). Yield: 197 mg, 0.346 mmol, 81%. Mp: 127 °C. Anal. Calcd for C29H32ClNO2Pd (568.451): C, 61.27; H, 5.67; N, 2.46. Found: C, 61.38; H, 5.42; N, 2.46. IR (cm–1): ν(NH) 3293 w, 3214 br; ν(CO) 1727 s, 1711 s.
Method B
In a different preparation, it was possible to obtain two different crops by fractional crystallization, each of them enriched in one of the isomers. Ethyl acrylate (40 μL, 0.262 mmol) was added to a solution of palladacycle b (150 mg, 0.160 mmol) in CH2Cl2 (10 mL). The mixture was stirred for 12 h and then filtered through a plug of Celite. The filtrate was concentrated to ca. 2 mL, and Et2O (15 mL) was added. The resulting suspension was filtered, and the solid was washed with Et2O (2 × 5 mL) and air-dried to afford 58 mg of a syn-enriched mixture of both isomers (anti/syn = 1/3). The mother liquors were concentrated to ca. 5 mL and cooled to 0 °C. A suspension formed, which was filtered, and the solid was air-dried to afford 60 mg of an anti-enriched mixture of both isomers (anti/syn = 3/1). Almost spectroscopically pure samples of anti- and syn-2b could be obtained by growing single crystals from the enriched mixtures. Data for anti-2b are as follows. 1H NMR (400.9 MHz): δ 1.22 (t, 3 H, MeCH2, 3JHH = 7.2 Hz), 1.27 (s, 3 H, Me, CMe2), 1.32 (s, 3 H, Me, CMe2), 1.81 (br d, 1 H, NH2, 2JHH = 10.0 Hz), 2.09 (d, 1 H, CH2Ar, 2JHH = 14.8 Hz), 2.21 (dd, 1 H, CH2CH, 2JHH = 17.2, 3JHH = 11.2 Hz), 2.36 (dd, 1 H, CH2Ar, 2JHH = 14.8, 4JHH = 1.6 Hz), 3.13 (br d, partially obscured by the resonance of CH2CH, 1 H, NH2, 2JHH = 9.6 Hz), 3.16 (dd, 1 H, CH2CH, 2JHH = 17.2, 3JHH = 5.2 Hz), 4.11 (m, 2 H, CH2O), 4.94 (dd, 1 H, CH, 3JHH = 11.2, 3JHH = 5.2 Hz), 6.86–6.90 (m, 2 H, Ph), 7.03–7.11 (m, 6 H, 5 H of Ph + H6), 7.13 (td, partially obscured by the resonance of H6, 1 H, H4, 3JHH = 7.6, 4JHH = 1.2 Hz), 7.21 (m, 2 H, o-H, Ph), 7.27 (m, partially obscured by the resonance of CHCl3, 1 H, p-H, Ph), 7.30 (dd, 1 H, H3, 3JHH = 7.6, 4JHH = 1.2 Hz), 7.34 (td, 1 H, H5, 3JHH = 7.6, 4JHH = 1.6 Hz). 13C{1H} NMR (100.8 MHz): δ 14.2 (MeCH2), 27.7 (Me, CMe2), 36.0 (Me, CMe2), 39.0 (CH2CH), 46.6 (CH2Ar), 52.3 (CMe2), 60.7 (CH2O), 81.0 (CH), 90.0 (C7), 123.5 (C8), 126.5 (CH5), 126.8 (p-CH, Ph), 127.1 (o-CH, Ph), 127.6 (m-CH, Ph), 127.9 (m-CH, Ph), 128.2 (CH4), 128.3 (m-CH, Ph), 128.9 (CH6), 129.1 (o-CH, Ph), 129.5 (o-CH, Ph), 132.2 (p-CH, Ph), 135.0 (CH3), 137.8 (C2), 140.49 (C1), 140.55 (i-C, Ph), 142.8 (i-C, Ph), 169.4 (CO). Data for syn-2b are as follows. 1H NMR (300.1 MHz): δ 1.16 (t, 3 H, MeCH2, 3JHH = 7.2 Hz), 1.28 (s, 3 H, Me, CMe2), 1.30 (s, 3 H, Me, CMe2), 1.82 (br d, 1 H, NH2, 2JHH = 9.9 Hz), 2.09 (d, 1 H, CH2Ar, 2JHH = 14.7 Hz), 2.29 (dd, 1 H, CH2Ar, 2JHH = 14.4, 4JHH = 1.8 Hz), 2.76 (dd, 1 H, CH2CH, 2JHH = 18.3, 3JHH = 9.9 Hz), 3.17 (dd, 1 H, CH2CH, 2JHH = 18.3, 3JHH = 4.2 Hz), 3.22 (br d, partially obscured by the resonance of CH2CH, 1 H, NH2, 2JHH = 9.3 Hz), 4.01 (m, 2 H, CH2O), 4.79 (dd, 1 H, CH, 3JHH = 9.6, 3JHH = 4.2 Hz), 6.87–7.36 (m, 13 H, Ph + Ar), 7.65 (br s, 1 H, CH, Ph or Ar). 13C{1H} NMR (75.5 MHz): δ 14.1 (MeCH2), 27.2 (Me, CMe2), 34.4 (CH2CH), 36.1 (Me, CMe2), 46.3 (CH2Ar), 53.4 (CMe2), 60.5 (CH2O), 76.7 (CH), 88.9 (C7), 125.5 (C8), 126.6 (CH), 127.1 (CH), 127.3 (CH), 127.5 (CH), 127.6 (CH), 127.9 (CH), 128.3 (CH), 128.5 (br s, CH), 128.8 (CH), 129.5 (CH), 131.2 (CH), 133.0 (br s, CH), 134.5 (i-C, Ph), 135.1 (CH3), 138.3 (C2), 139.8 (C1), 142.0 (i-C, Ph), 170.4 (CO).
anti/syn Isomerization of 2b
An NMR tube was charged with a 1.25/1 anti-/syn-1b mixture (20 mg) and CDCl3 (0.6 mL), and the solution was heated at 60 °C. The sample was checked by 1H NMR periodically, until no change was observed. After 48 days at 60 °C, a 1/20 anti-/syn-2b mixture was obtained.
Synthesis of anti-/syn-3b
TlOTf (56 mg, 0.158 mmol) was added to a suspension of complex 1b (90 mg, 0.157 mmol) in acetone (10 mL), and the resulting mixture was stirred for 2 h. The solvent was removed, and CH2Cl2 (15 mL) was added. The suspension was filtered through a plug of Celite, p-toluidine (17 mg, 0.159 mmol) was added to the filtrate, and the mixture was stirred for another 30 min. The solvent was concentrated to ca. 1 mL, and n-pentane was added. The suspension was filtered, and the pale yellow solid was washed with cold n-pentane (2 × 5 mL) and air-dried to give an anti-/syn-3b mixture (ratio ca. 1/1.33 by 1H NMR). Yield: 60.0 mg, 0.076 mmol, 48%. Mp: 120 °C. ΛM (Ω–1 mol–1 cm2): 106 (c = 2.7 × 10–4 M). Anal. Calcd for C40H41F3N2O3PdS (793.237): C, 60.57; H, 5.21; N, 3.53; S, 4.04. Found: C, 60.38; H, 5.17; N, 3.30; S, 4.04. IR (cm–1): ν(NH) 3295 w, 3251 m, 3159 w. Data for anti-3b (minor isomer, extracted from the mixture) are as follows. δ 0.86 (s, 3 H, Me, CMe2), 1.27 (s, 3 H, Me, CMe2), 1.92 (br d, 1 H, NH2, 2JHH = 12.6 Hz), 1.98 (d, 1 H, CH2, 2JHH = 15.2 Hz), 2.20–2.29 (m, 1 H, CH2, overlapped with one H of CH2 of the major isomer), 2.29 (s, 3 H, Me, Tol), 2.44–2.55 (m, 1 H, CH2, overlapped with one H of CH2 of the major isomer), 2.73 (dd, 1 H, CH2, 2JHH = 14.8, 3JHH = 9.2 Hz), 4.13 (br d, 1 H, NH2, 2JHH = 11.2 Hz), 4.74 (dd, 1 H, CH, 3JHH = 8.8, 3JHH = 6.0 Hz), 5.33 (br d, 1 H, NH2, 2JHH = 11.2 Hz), 5.54 (br d, 1 H, NH2, 2JHH = 10.4 Hz), 6.32–7.55 (m, 23 H, Ph + Ar, overlapped with the aromatic protons of the major isomer). Data for syn-3b (major isomer, extracted from the mixture) are as follows. 1H NMR (400.9 MHz, −40 °C): δ 1.45 (s, 3 H, Me, CMe2), 1.48 (s, 3 H, Me, CMe2), 1.70 (br d, 1 H, NH2, 2JHH = 10.8 Hz), 2.06 (d, 1 H, CH2, 2JHH = 15.2 Hz), 2.20–2.29 (m, 1 H, CH2, overlapped with one H of CH2 of the minor isomer), 2.33 (s, 3 H, Me, Tol), 2.44–2.55 (m, 1 H, CH2, overlapped with one H of CH2 of the minor isomer), 2.81 (dd, 1 H, CH2Ar, 2JHH = 16.4, 3JHH = 4.0 Hz), 3.92 (d, 1 H, NH2, 2JHH = 9.6 Hz), 4.24 (dd, 1 H, CH, 3JHH = 10.0, 3JHH = 4.0 Hz), 5.02 (br d, 1 H, NH2, 2JHH = 11.2 Hz), 6.30 (br d, 1 H, NH2, 2JHH = 10.0 Hz), 6.32–7.55 (m, 23 H, Ph + Ar, overlapped with the aromatic protons of the minor isomer). Data for both isomers are as follows. 13C{1H} NMR (100.8 MHz, −40 °C): δ 20.7 (Me, Tol, major), 20.8 (Me, Tol, minor), 25.9 (Me, CMe2, minor), 27.4 (Me, CMe2, major), 33.8 (CH2, minor), 34.4 (Me, CMe2, major), 34.8 (Me, CMe2, minor), 41.4 (CH2, major), 46.5 (CH2, minor), 47.2 (CH2, major), 52.6 (CMe2, major), 53.0 (CMe2, minor), 84.6 (CH, minor), 86.6 (C, major), 87.1 (C, minor), 93.7 (CH, major), 188.8 (CH, major), 119.8 (CH, minor), 119.9 (q, CF3, 1JCF = 318.8 Hz), 124.1 (C, major), 126.3 (CH), 126.4 (CH), 126.5 (CH), 126.7, 126.8 (C, minor), 127.1 (CH), 127.2 (CH), 127.3 (CH), 127.4 (CH), 127.6 (CH), 127.7 (CH), 127.8 (CH), 127.8 (CH), 127.9 (CH), 128.0 (CH), 128.1 (CH), 128.1 (CH), 128.4 (CH), 128.6 (CH), 128.7 (CH), 129.0 (CH), 129.5 (CH), 129.7 (CH), 129.8 (CH), 130.0 (CH), 131.5 (CH), 131.9 (CH), 132.7 (CH), 133.1 (C, major), 133.6 (C, minor), 134.1 (C, minor), 134.9 (CH), 135.0 (CH), 137.7 (C, major), 138.2 (C, minor), 138.5 (C, minor), 138.9 (C, minor), 139.0 (C, major), 139.4 (C, minor), 139.6 (C, major), 139.7 (C, major), 139.9 (C, major), 140.8 (C, minor), 141.2 (C, major).
Synthesis of 4a·1/2H2O
In a Carius tube, KOtBu (190 mg, 1.55 mmol) was added to a solution of anti-/syn-1a (100 mg, 0.154 mmol) in dry toluene (10 mL), under a nitrogen atmosphere. The mixture was heated at 100 °C for 12 h. Decomposition to metallic palladium was observed. The solvent was removed, and CH2Cl2 (20 mL) was added to the residue. The resulting suspension was filtered through a plug of Celite, the filtrate was concentrated to ca. 1 mL, n-pentane (20 mL) was added, and the mixture was cooled to 0 °C. The resulting suspension was filtered, and the solid was air-dried to give 4a·1/2H2O as a colorless solid. Yield: 55 mg, 0.117 mmol, 76%. Mp: 129 °C. Anal. Calcd for C32H31NO2·1/2H2O (470.611): C, 81.67; H, 6.85; N, 2.97. Found: C, 81.84; H, 6.60; N, 3.05. ESI-HRMS (m/z): exact mass calcd for C32H32NO2, 462.2433 [(M + H)+]; found, 462.2433. IR (cm–1): ν(NH) 3375 w. 1H NMR (300.1 MHz): δ 1.22 (br s, 3 H, NH2 + 1 H of H2O), 2.50–2.61 (m, 2 H, CH2Ar), 2.63–2.70 (m, 1 H, CH2N), 2.70–2.83 (m, 1 H, CH2N), 3.86 (s, 3 H, MeO), 3.95 (s, 3 H, MeO), 6.23 (d, 1 H, CH = , 3JHH = 15.9 Hz), 6.80 (s, 1 H, H3), 6.81 (s, 1 H, H6), 6.86–6.90 (m, 2 H, Ph), 6.93 (d, 1 H, CH = , 3JHH = 15.9 Hz), 6.97–7.00 (m, 3 H, Ph), 7.12–7.24 (m, 7 H, Ph), 7.27–7.35 (m, 3 H, Ph). 13C{1H} NMR(100.8 MHz): δ 37.4 (CH2Ar), 42.8 (CH2N), 55.8 (MeO), 56.1 (MeO), 111.9 (CH3), 112.6 (CH6), 126.3 (CH, Ph), 126.4 (CH, Ph), 126.9 (CH, Ph), 127.3 (CH, Ph), 127.4 (CH, Ph), 128.1 (CH, Ph), 128.5 (CH, Ph), 130.5 (CH, Ph), 130.9 (CH = ), 131.2 (C2), 131.3 (CH, Ph), 132.3 (CH = ), 133.8 (C1), 137.5 (i-C, Ph), 139.7 (i-C, Ph), 139.7 (i-C, Ph), 141.3 (C7), 141.8 (C8), 147.0 (C4), 148.2 (C5).
Synthesis of 5b·H2O
In a Carius tube, KOtBu (250 mg, 2.04 mmol) was added to a solution of anti-/syn-1b (120 mg, 0.209 mmol) in dry toluene (10 mL), under a nitrogen atmosphere. The mixture was heated at 100 °C for 12 h. Decomposition to metallic palladium was observed. The solvent was removed, and Et2O (20 mL) was added to the residue. The suspension was filtered through a plug of Celite, and the solvent was removed from the filtrate to give crude 4b as an oily residue, which was was characterized by 1H NMR. Data for 4b are as follows. 1H NMR (400.9 MHz): δ 0.99 (br s, 2 H, NH2), 1.04 (s, 3 H, Me, CMe2), 1.09 (s, 3 H, Me, CMe2), 2.34, 2.54 (AB system, 2 H, CH2Ar, 2JAB = 13.6 Hz), 6.20 (d, 1 H, CH = , 3JHH = 15.6 Hz), 6.81–6.85 (m, 2 H), 6.96–7.00 (m, 3 H), 7.03 (d, 1 H, CH=, 3JHH = 16.0 Hz), 7.10–7.47 (m, 14 H). Crude 4b was dissolved in CH2Cl2 (5 mL), HOTf (0.05 mL, 0.565 mmol) was added, and the resulting solution was stirred for 30 min. The solvent was concentrated to ca. 1 mL, Et2O (20 mL) was added, and the mixture was cooled to 0 °C. The resulting suspension was filtered, and the solid was washed with Et2O (2 × 5 mL) and air-dried to give the salt 5b·H2O as a colorless solid. Yield: 63 mg, 0.105 mmol, 50%. Mp: 131 °C. ΛM (Ω–1 mol–1 cm2): 108.5 (c = 5.09 × 10–4 M). Anal. Calcd for C33H32F3NO3S·H2O (597.696): C, 66.31; H, 5.73; N, 2.34; S, 5.36. Found: C, 66.02; H, 5.59; N, 2.43; S, 5.59. ESI-HRMS (m/z): exact mass calcd for C32H32N, 430.2529 [(M – CF3SO3)+]; found, 430.2533. IR (cm–1): ν(NH) 3479 w. 1H NMR (400.9 MHz): δ 1.23 (s, 3 H, Me, CMe2), 1.25 (s, 3 H, Me, CMe2), 2.55, 2.62 (AB system, 2 H, CH2Ar, 2JAB = 14.0 Hz), 3.00 (br s, 2 H, H2O), 6.22 (d, 1 H, CH=, 3JHH = 16.0 Hz), 6.75–6.95 (m, 4 H, Ph), 6.94–6.97 (m, 2 H, Ph), 6.99 (d, 1 H, CH = , 3JHH = 16.0 Hz), 7.09–7.21 (m, 6 H, Ph), 7.22–7.27 (m, 4 H, 3 H of Ph + H3), 7.33 (td, 1 H, H4, 3JHH = 7.6, 4JHH = 1.2 Hz), 7.38 (td, 1 H, H5, 3JHH = 7.2, 4JHH = 1.2 Hz), 7.50 (dd, 1 H, H6, 3JHH = 7.2, 4JHH = 1.2 Hz), 7.70 (br s, 3 H, NH3). 13C{1H} NMR (100.8 MHz): δ 24.7 (Me, CMe2), 26.4 (Me, CMe2), 42.4 (CH2Ar), 57.0 (CMe2), 119.7 (q, CF3, 1JCF = 318.7 Hz), 126.5 (CH, Ph), 127.0 (CH, Ph), 127.3 (CH5), 127.6 (CH, Ph), 128.1 (CH, Ph), 128.5 (CH, Ph), 128.6 (CH4), 130.3 (CH, Ph), 130.6 (CH = ), 130.9 (CH, Ph), 132.2 (CH3), 133.4 (CH = ), 133.9 (C2), 134.2 (CH6), 137.3 (i-C, Ph), 139.4 (i-C, Ph), 140.5 (C8), 141.26 (i-C, Ph), 141.34 (C7), 142.2 (C1).
Synthesis of [Pd{C,N-C(Ph)=C(CO2Me)C6H4CH2CMe2NH2-2}Cl(C7H10)] (6d)
2-Norbornene (C7H10; 25 mg, 0.265 mmol) was added to a solution of palladacycle d (65 mg, 0.072 mmol) in CH2Cl2 (10 mL), and the yellow solution was stirred for 2 h. The solvent was removed under vacuum at room temperature (to prevent the evolution of the complex), and Et2O (10 mL) was added. The resulting suspension was filtered, and the solid was washed with Et2O (2 × 5 mL) and air-dried to afford complex 6d as a colorless solid. Yield: 58 mg, 0.106 mmol, 73%. Mp: 182 °C dec. Anal. Calcd for C27H32ClNO2Pd (544.429): C, 59.56; H, 5.92; N, 2.57. Found: C, 59.37; H, 6.01; N, 2.41. IR (cm–1): ν(NH) 3323 w, 3267 w; n(CO) 1725 s. 1H NMR (400.9 MHz): δ 0.66 (br d, 1 H, CH, nor, 3JHH = 8.0 Hz), 0.97 (br d, 1 H, CH2, nor, 2JHH = 9.6 Hz), 1.04 (br m, 1 H, CH2, nor), 1.11 (br m, 1 H, CH2, nor), 1.35 (s, 3 H, Me, CMe2), 1.38 (m, partially obscured by the resonance of Me group, 2 H, CH2, nor), 1.40 (s, 3 H, Me, CMe2), 1.79 (br s, 1 H, CH, nor), 2.05 (br d, 1 H, CH2, nor, 2JHH = 10.0 Hz), 2.49 (d, 1 H, CH2Ar, 2JHH = 15.6 Hz), 2.67 (d, 1 H, CH2Ar, 2JHH = 15.2 Hz), 3.08 (br s, 1 H, CH, nor), 3.26 (s, 3 H, MeO), 4.36 (d, 1 H, CH, nor, 3JHH = 7.6 Hz), 7.16 (dd, 1 H, H3, 3JHH = 7.6, 4JHH = 1.2 Hz), 7.23–7.35 (m, 6 H, Ar + Ph), 7.83 (d, 1 H, H6, 3JHH = 7.2 Hz), 7.95 (m, 1 H, o-H, Ph). The 1H resonance corresponding to the NH2 group was not observed. 13C{1H} NMR (75.5 MHz): δ 18.0 (CH, nor), 27.6 (Me, CMe2), 27.8 (CH2, nor), 28.9 (CH2, nor), 36.5 (Me, CMe2), 36.9 (CH2, nor), 40.4 (CH, nor), 40.6 (CH, nor), 47.1 (CH2Ar), 51.1 (CH, nor), 52.7 (MeO), 54.9 (CMe2), 99.8 (C7), 123.9 (CH6), 126.8 (CH, Ph), 127.0 (CH, Ph), 127.3 (CH, Ph), 127.4 (CH, Ph), 127.5 (CH5), 127.9 (CH4), 134.0 (C1), 134.1 (o-CH, Ph), 135.1 (CH3), 137.2 (i-C or C8), 138.4 (C2), 170.0 (CO).
Synthesis of [Pd{C,N-CH(C5H8)CHC(Ph)=C(Me)C6H4CH2CMe2NH2-2}Cl]·CHCl3 (7c·CHCl3)
In a Carius tube, 2-norbornene (100 mg, 1.06 mmol) was added to a solution of palladacycle c (250 mg, 0.308 mmol) in CHCl3 (15 mL), and the mixture was heated at 65 °C for 12 h. The resulting solution was filtered through a plug of Celite, the solvent was removed from the filtrate, and Et2O (20 mL) was added. The resulting suspension was filtered, and the solid was washed with Et2O (2 × 5 mL) and air-dried to give complex 7c·CHCl3 as a yellow solid. Yield: 258 mg, 0.416 mmol, 67%. Dec pt: 223 °C. Anal. Calcd for C26H32ClNPd·CHCl3 (619.796): C, 52.32; H, 5.37; N, 2.26. Found: C, 52.57; H, 5.48; N, 2.29. IR (cm–1): ν(NH) 3313 w, 3250 w. 1H NMR (300.1 MHz): δ 0.82 (br d, 1 H, NH2, 2JHH = 9.6 Hz), 0.95 (br d, 1 H, CH2, nor, 2JHH = 10.2 Hz), 1.07 (br d, 1 H, Hα, 3JHH = 7.5 Hz), 1.12 (s, 3 H, Me, CMe2), 1.20–1.27 (m, 2 H, CH2, nor), 1.37–1.50 (m, 2 H, CH2, nor), 1.64 (s, 3 H, Me, CMe2), 2.05 (br d, 1 H, CH2, nor, 2JHH = 9.9 Hz), 2.14 (br s, 1 H, CH, nor), 2.20 (br d, 1 H, NH2, 2JHH = 9.6 Hz), 2.35 (s, 3 H, MeC=), 2.57 (d, 1 H, CH2Ar, 2JHH = 12.9 Hz), 3.09 (br s, 1 H, CH, nor), 3.65 (d, 1 H, Hβ, 3JHH = 7.8 Hz), 3.96 (d, 1 H, CH2Ar, 2JHH = 12.9 Hz), 6.67 (d, 1 H, H6, 3JHH = 7.8 Hz), 6.76 (m, 1 H, H5), 7.01 (m, 2 H, H4 + H3), 7.18–7.25 (br m, partially obscured by the resonance of CHCl3, 3 H, p-H + m-H, Ph), 2.26 (s, 1 H, CHCl3), 7.42 (br d, 2 H, o-H, Ph, 3JHH = 7.8 Hz). 13C{1H} NMR (75.5 MHz): δ 21.5 (CHα), 27.9 (MeC=), 28.5 (CH2, nor), 28.9 (CH2, nor), 30.7 (Me, CMe2), 30.9 (Me, CMe2), 37.3 (CH2, nor), 40.0 (CH, nor), 41.1 (CH, nor), 48.1 (CH2Ar), 52.3 (CMe2), 54.6 (CHβ), 77.8 (C8), 107.7 (C7), 126.3 (CH5), 126.7 (CH4), 126.8 (p-CH, Ph), 131.0 (br s, CH6 or o-CH of Ph), 131.1 (CH6 or o-CH of Ph), 132.7 (CH3), 135.8 (C2), 138.6 (i-C, Ph), 142.6 (C1). The 13C resonance corresponding to m-CH of Ph was overlapped with the signal of CH6 or o-CH.
Synthesis of [Pd{C,N-CH(C5H8)CHC(Ph)=C(CO2Me)C6H4CH2CMe2NH2-2}Cl]·1/2CHCl3 (7d·1/2CHCl3)
Method A
In a Carius tube, methyl phenylpropiolate (48 μL, 0.324 mmol) was added to a solution of palladacycle e (120 mg, 0.156 mmol) in CHCl3 (10 mL), and the mixture was heated at 65 °C for 2 h. The yellow solution was concentrated to ca. 1 mL, and Et2O was added (20 mL). The suspension was filtered, and the solid was washed with Et2O (2 × 5 mL) and air-dried to give complex 7d·1/2CHCl3 as a pale yellow solid. Yield: 37 mg, 0.061 mmol, 20%. The filtrate was contentrated to ca. 2 mL, and n-pentane (20 mL) was added. The suspension was filtered, and the yellow solid was air-dried to give a ca. 1/0.7 mixture of complex 7d and palladacycle d (80 mg).
Method B
In a Carius tube, 2-norbornene (50 mg, 0.531 mmol) was added to a solution of palladacycle d (200 mg, 0.223 mmol) in CHCl3 (15 mL), and the mixture was heated at 65 °C for 4 h. The resulting solution was filtered through a plug of Celite, the filtrate was concentrated to ca. 1 mL, and Et2O (20 mL) was added. The suspension was filtered, and the solid was washed with Et2O (2 × 5 mL) and air-dried to give complex 7d·1/2CHCl3 as a pale yellow solid. Yield: 198 mg, 0.328 mmol, 73%. Dec pt: 204 °C. Anal. Calcd for C27H32ClNO2Pd·1/2CHCl3 (604.117): C, 54.68; H, 5.42; N, 2.32. Found: C, 54.64; H, 5.42; N, 2.29. IR (cm–1): ν(NH) 3320 w, 3266 w; ν(CO) 1716 s. 1H NMR (400.9 MHz): δ 0.83 (br d, 1 H, NH2, 2JHH = 9.6 Hz), 0.95 (br d, 1 H, CH2, nor, 2JHH = 10.0 Hz), 1.05 (br d, 1 H, CH2, nor, 2JHH = 10.4 Hz), 1.13 (s, 3 H, Me, CMe2), 1.19–1.23 (m, 2 H, Hα + 1 H of CH2 of nor), 1.35–1.45 (m, 2 H, CH2, nor), 1.64 (s, 3 H, Me, CMe2), 1.93 (d, 1 H, CH2, nor, 2JHH = 9.6 Hz), 2.15 (br s, 1 H, CH, nor), 2.37 (br d, 1 H, NH2, 2JHH = 9.6 Hz), 2.45 (d, 1 H, CH2Ar, 2JHH = 13.2 Hz), 3.18 (br s, 1 H, CH, nor), 3.61 (d, 1 H, Hβ, 3JHH = 8.0 Hz), 3.76 (s, 3 H, MeO), 4.35 (d, 1 H, CH2Ar, 2JHH = 12.8 Hz), 6.83 (m, 1 H, H5), 6.88 (br d, 1 H, H6, 3JHH = 7.2 Hz), 7.01 (br d, 1 H, H3, 3JHH = 7.2 Hz), 7.08 (td, 1 H, H4, 3JHH = 7.6, 4JHH = 1.2 Hz), 7.28 (br m, partially obscured by the resonance of CHCl3, 3 H, p-H + m-H, Ph), 7.48 (br s, 2 H, o-H, Ph). 13C{1H} NMR (100.8 MHz): δ 23.8 (CHα), 28.3 (CH2, nor), 28.4 (CH2, nor), 30.7 (Me, CMe2), 30.75 (Me, CMe2), 37.0 (CH2, nor), 39.8 (CH, nor), 41.2 (CH, nor), 47.4 (CH2Ar), 52.2 (CMe2), 53.3 (MeO), 54.5 (CHβ), 101.5 (C7), 126.6 (CH5), 127.5 (p-CH, Ph), 127.7 (br s, m-CH, Ph), 127.9 (CH4), 130.5 (o-CH, Ph), 132.5 (CH3), 133.2 (CH6), 135.1 (C1), 136.2 (i-C, Ph), 137.6 (C2), 169.1 (CO). The resonance corresponding to C8 was not observed.
Synthesis of [Pd{C,N-CH(C5H8)CHC(CO2Me)=C(CO2Me)C6H4CH2CMe2NH2-2}Cl] (7f)
Method A
In a Carius tube, dimethyl acetylenedicarboxylate (82 μL, 0.667 mmol) was added to a suspension of palladacycle e (240 mg, 0.312 mmol) in CHCl3 (15 mL), and the mixture was heated at 65 °C for 8 h. The resulting solution was filtered through a plug of Celite, the filtrate was concentrated to ca. 1 mL, and Et2O (20 mL) was added. The suspension was filtered, and the solid was washed with Et2O (2 × 5 mL) and air-dried to give the complex 7f as a bright yellow solid. Yield: 247 mg, 0.469 mmol, 75%.
Method B
In a Carius tube, a solution of dimethyl acetylenedicarboxylate (65 μL, 0.529 mmol) and 2-norbornene (50 mg, 0.531 mmol) in CHCl3 (5 mL) was added to a solution of palladacycle B (150 mg, 0.312 mmol) in CHCl3 (15 mL), and the mixture was heated at 65 °C for 8 h. The resulting solution was filtered through a plug of Celite, the filtrate was concentrated to ca. 2 mL, and Et2O (20 mL) was added. The suspension was filtered, and the solid was washed with Et2O (2 × 5 mL) and air-dried to give complex 7f as a bright yellow solid. Yield: 90 mg, 0.171 mmol, 33%. Dec pt: 188 °C. Anal. Calcd for C23H30ClNO4Pd (526.368): C, 52.48; H, 5.74; N, 2.66. Found: C, 52.21; H, 6.11; N, 2.67. IR (cm–1): ν(NH) 3326 m, 3267 m; ν(CO) 1731 s, 1716 s. 1H NMR (400.9 MHz): δ 1.08 (br d, partially obscured by the resonance of Me, 1 H, Hα, 3JHH = 8.0 Hz), 1.10 (s, 3 H, Me, CMe2), 1.11 (m, partially obscured by the resonance of Me, 1 H, CH2, nor), 1.21 (m, 1 H, CH2, nor), 1.33 (br d, 1 H, CH2, nor, 2JHH = 10.4 Hz), 1.44 (m, 1 H, CH2, nor), 1.51 (s, 3 H, Me, CMe2), 1.58 (m, 1 H, CH2, nor), 1.62 (br d, 1 H, NH2, 2JHH = 10.4 Hz), 2.07 (br d, 1 H, NH2, 2JHH = 10.4 Hz), 2.26 (br d, 1 H, CH2, nor, 2JHH = 10.4 Hz), 2.48 (dd, 1 H, CH2Ar, 2JHH = 13.2, 4JHH = 1.2 Hz), 2.70 (br d, 1 H, CH, nor, 3JHH = 3.6 Hz), 3.21 (br d, 1 H, CH, nor, 3JHH = 4.0 Hz), 3.60 (d, partially obscured by the resonance of MeO, 1 H, Hβ, 3JHH = 8.4 Hz), 3.61 (s, 3 H, MeO), 3.75 (s, 3 H, MeO), 4.12 (d, 1 H, CH2Ar, 2JHH = 13.2 Hz), 7.13 (d, 1 H, H3, 3JHH = 7.6 Hz), 7.20 (m, 2 H, H6 + H5), 7.28–7.33 (m, 1 H, H4). 13C{1H} NMR (100.8 MHz): δ 25.6 (CHα), 27.6 (CH2, nor), 28.3 (CH2, nor), 30.4 (Me, CMe2), 30.6 (Me, CMe2), 38.1 (CH2, nor), 39.2 (CH, nor), 42.4 (CH, nor), 47.7 (CH2Ar), 52.2 (CMe2), 52.5 (MeO), 53.3 (MeO), 53.4 (CHβ), 68.3 (C8), 102.9 (C7), 127.4 (CH5), 128.9 (CH4), 130.4 (CH6), 133.1 (CH3), 135.2 (C1), 137.2 (C2), 166.9 (CO), 167.7 (CO).
Synthesis of 8f
In a Carius tube, KOtBu (50 mg, 0.445 mmol) was added to a solution of complex 7f (120 mg, 0.228 mmol) in dry CH3CN, under an N2 atmosphere (10 mL), and the mixture was heated at 78 °C for 3 days. Decomposition to metallic palladium was observed. The solvent was removed, n-pentane (20 mL) was added, and the mixture was filtered through a plug of Celite. The solvent was removed from the filtrate, the residue was dissolved in Et2O (10 mL), and HOTf (0.01 mL, 0.113 mmol) was added. The resulting suspension was filtered, and the solid was washed with Et2O (2 × 2 mL) and air-dried to give compound 8f as a colorless solid. Yield: 23 mg, 0.043 mmol, 19%. Mp: 197 °C. ΛM (Ω–1 mol–1 cm2): 104.0 (c = 3.82 × 10–4 M). Anal. Calcd for C24H30F3NO7S (533.566): C, 54.02; H, 5.67; N, 2.62, S; 6.00. Found: C, 53.88; H, 6.09; N, 2.69; S: 5.97. The hydrogen content found in the elemental analysis was slightly outside the accepted range (6.09 vs 5.67%; Δ = 0.42). This could be attributed to the presence of small traces of Et2O in the sample (see the Supporting Information), which remained in spite of the compound being dried under vacuum. ESI-HRMS (m/z): exact mass calcd for C23H30NO4, 384.2169 [(M – CF3SO3)+]; found, 384.2174. IR (cm–1): ν(CO) 1740 (s), 1683 (s). 1H NMR (300.1 MHz): δ 0.57 (br d, 1 H, CH2, nor, 2JHH = 12.6 Hz), 0.84 (d, 1 H, CH2, nor, 2JHH = 12.3 Hz), 1.28 (s, 3 H, Me, CMe2), 1.29–1.55 (m, 4 H, CH2, nor), 1.56 (d, partially obscured by the resonance of CH2 of nor, 1 H, Hα or Hβ, 3JHH = 7.2 Hz), 1.83 (s, 3 H, Me, CMe2), 2.01 (d, 1 H, Hα or Hβ, 3JHH = 7.5 Hz), 2.62 (br s, 1 H, CH, nor), 2.75 (d, partially obscured by the resonance of CH of nor, 1 H, CH2Ar, 2JHH = 16.8 Hz), 2.76 (br s, 1 H, CH, nor), 3.02 (d, 1 H, CH2Ar, 2JHH = 17.1 Hz), 3.10 (s, 3 H, MeO), 3.90 (s, 3 H, MeO), 7.07 (m, 1 H, H5), 7.37 (m, 2 H, H7 + H6), 7.85 (m, 1 H, H8), 7.99 (br d, 1 H, NH2, 2JHH = 11.4 Hz), 9.62 (br d, 1 H, NH2, 2JHH = 11.4 Hz). 13C{1H} NMR (75.5 MHz): δ 22.4 (Me, CMe2), 27.3 (CHα or CHβ), 28.5 (Me, CMe2), 28.6 (CH2, nor), 29.0 (CH2, nor), 29.6 (CHα or CHβ), 30.1 (CH2, nor), 35.5 (CH, nor), 35.7 (C(CO2Me)CH), 37.6 (CH, nor), 40.0 (CH2Ar), 52.6 (MeO), 54.6 (MeO), 57.8 (CMe2), 69.7 (C1), 120.4 (q, CF3SO3, 1JCF = 319.5 Hz), 126.2 (C8a), 126.7 (CH7), 128.5 (CH5), 129.7 (CH6), 130.9 (C4a), 131.1 (CH8), 165.3 (CO), 174.1 (CO).
Synthesis of 9f
TlOTf (84 mg, 0.237 mmol) was added to a solution of complex 7f (125 mg, 0.237 mmol) in acetone (15 mL), and the mixture was stirred at room temperature for 12 h under an CO atmosphere, using a toy balloon. Decomposition to metallic palladium was observed. The suspension was filtered through a plug of Celite, the filtrate was concentrated to ca. 1 mL, and Et2O (20 mL) was added. The suspension was filtered, and the solid was washed with Et2O (2 × 2 mL) and air-dried to give the compound 9f as a colorless solid. Yield: 103 mg, 0.177 mmol, 75%. Mp: 223 °C. ΛM (Ω–1 mol–1 cm2): 101.4 (c = 5.04 × 10–4 M). Anal. Calcd for C25H32F3NO9S (579.592): C, 51.80; H, 5.56; N, 2.41, S; 5.53. Found: C, 51.56; H, 5.87; N, 2.50; S: 5.37. ESI-HRMS (m/z): exact mass calcd for C24H32NO6, 430.2230 [(M – CF3SO3)+]; found, 430.2225. IR (cm–1): ν(CO) 1771 vs, 1733 vs, 1626 s. 1H NMR (400.9 MHz, acetone-d6): δ 1.10 (m, 1 H, CH2, nor), 1.21 (m, 1 H, CH2, nor), 1.31 (m, 2 H, CH2, nor), 1.48 (s, 3 H, Me, CMe2), 1.54 (m, 2 H, CH2, nor), 1.58 (s, 3 H, Me, CMe2), 2.28 (br s, 1 H, CH, nor), 2.50 (br s, 1 H, CH, nor), 2.73 (d, 1 H, Hα, 3JHH = 8.0 Hz), 2.88 (d, 1 H, Hβ, 3JHH = 8.0 Hz), 3.10 (d, 1 H, CH2Ar, 2JHH = 14.4 Hz), 3.51 (s, 3 H, MeO), 3.66 (s, 3 H, MeO), 3.69 (d, 1 H, CH2Ar, 2JHH = 14.4 Hz), 4.99 (s, 1 H, H7), 7.26–7.33 (m, 2 H, H4 + H5), 7.37 (m, 1 H, H3), 7.63 (m, 1 H, H6), 7.70 (br s, 3 H, NH3). The resonance corresponding to the OH group was not observed. 13C{1H} NMR (100.8 MHz, acetone-d6): δ 25.3 (Me, CMe2), 26.9 (Me, CMe2), 27.6 (CH2, nor), 28.5 (CH2, nor), 35.1 (CH2, nor), 40.9 (CH, nor), 41.1 (CH, nor), 41.7 (CH2Ar), 51.1 (CHα), 51.3 (CHβ), 52.2 (MeO), 53.7 (MeO), 53.0 (CHAr), 57.4 (CMe2), 90.2 (C8), 128.2 (CH5), 128.7 (CH4), 131.4 (CH6), 133.1 (CH3), 134.5 (C1), 135.0 (C2), 166.3 (CO2Me), 171.2 (CO2Me), 177.6 (CO2H).
Synthesis of 10c
In a Carius tube, TlOTf (72 mg, 0.204 mmol) was added to a suspension of the complex 7c·CHCl3 (100 mg, 0.161 mmol) in acetone (15 mL) and the mixture was stirred for 15 min. CO was bubbled through the suspension for 3 min, the pressure of CO was increased to 1 atm, the tube was sealed, and the mixture was stirred at room temperature for 20 h. Decomposition to metallic palladium was observed. The suspension was filtered through a plug of Celite, the filtrate was concentrated to ca. 1 mL, and Et2O (20 mL) was added. The suspension was filtered, the solvent was removed from the filtrate, and the solid residue was stirred in n-pentane (20 mL). The resulting suspension was filtered, and the solid was washed with n-pentane (2 × 5 mL) and air-dried to give the compound 10c as a colorless solid. Yield: 84 mg, 0.152 mmol, 94%. Mp: 160 °C. ΛM (Ω–1 mol–1 cm2): 117.1 (c = 5.00 × 10–4 M). Compound 10c was hygroscopic, and no satisfactory elemental analysis could be obtained. ESI-HRMS (m/z): exact mass calcd for C27H34NO2, 404.2590 [(M – CF3SO3)+]; found, 404.2589. IR (cm–1): ν(NH) 3452 br; ν(CO) 1704 s. 1H NMR (400.9 MHz): δ 0.98 (br d, 1 H, CH2, nor, 2JHH = 10.4 Hz), 1.07 (s, 3 H, Me, CMe2), 1.29 (m, 2 H, CH2, nor), 1.42 (m, partially obscured by the resonance of Me group, 1 H, CH2, nor), 1.45 (s, 3 H, Me, CMe2), 1.60 (m, 1 H, CH2, nor), 1.81 (br s, 1 H, CH, nor), 1.83 (br d, partially obscured by the resonance of CH of nor, 1 H, CH2, nor), 2.19 (s, 3 H, MeC=), 2.22 (d, 1 H, CH2Ar, 2JHH = 13.6 Hz), 2.60 (br d, 1 H, CH, nor, 3JHH = 4.0 Hz), 3.08 (d, 1 H, Hα, 3JHH = 10.0 Hz), 3.22 (d, 1 H, Hβ, 3JHH = 9.6 Hz), 3.52 (d, 1 H, CH2Ar, 2JHH = 13.2 Hz), 6.58 (br s, 1 H), 6.77 (d, 1 H, H3, 3JHH = 7.6 Hz), 6.90–6.99 (m, 4 H), 7.06–7.12 (m, 6 H), 7.33 (br s, 1 H, CO2H). 13C{1H} NMR (100.8 MHz): δ 22.9 (MeC=), 24.1 (Me, CMe2), 26.0 (Me, CMe2), 29.0 (CH2, nor), 30.9 (CH2, nor), 36.8 (CH2, nor), 39.7 (CH, nor), 40.3 (CH, nor), 41.5 (CH2Ar), 50.8 (CHβ), 54.1 (CHα), 57.3 (CMe2), 125.9 (CH4 or p-CH of Ph), 126.0 (CH4 or p-CH of Ph), 126.6 (CH5), 126.9 (br s, o-CH + m-CH, Ph), 129.1 (CH6), 129.4 (CH3), 131.3 (C2), 134.4 (C7), 140.1 (i-C, Ph), 140.3 (C8), 146.0 (C1), 177.6 (CO).
Synthesis of 10d and 9d
In a Carius tube, TlOTf (50 mg, 0.141 mmol) was added to a solution of complex 7d·1/2CHCl3 (75 mg, 0.124 mmol) in acetone (15 mL), and the mixture was stirred for 2 h. CO was bubbled through the suspension for 2 min, the pressure of CO was increased to 1 atm, the tube was sealed, and the mixture was stirred at room temperature for 15 h. Decomposition to metallic palladium was observed. The suspension was filtered through a plug of Celite, the solvent was removed from the filtrate, and Et2O (20 mL) was added. The mixture was filtered through a plug of Celite, the filtrate was concentrated to ca. 1 mL, and n-pentane (20 mL) was added. The resulting suspension was filtered, and the solid was washed with n-pentane (2 × 5 mL) and air-dried to give the compound 10d as a colorless solid. Yield: 60 mg, 0.100 mmol, 81%. Mp: 122 °C. ΛM (Ω–1 mol–1 cm2): 78.3 (c = 7.4 × 10–4 M). Compound 10d was hygroscopic, and no satisfactory elemental analysis could be obtained. ESI-HRMS (m/z): exact mass calcd for C28H34NO4, 448.2482 [(M – CF3SO3)+]; found: 448.2484. IR (cm–1): ν(NH) 3486 br; ν(CO) 1715 vs, 1626 s. 1H NMR (300.1 MHz): δ 1.02 (br d, 1 H, CH2, nor, 2JHH = 9.9 Hz), 1.1 (s, 3 H, Me, CMe2), 1.20–1.32 (m, 2 H, CH2, nor), 1.36–1.47 (m, 2 H, CH2, nor), 1.51 (s, 3 H, Me, CMe2), 1.79 (br d, 1 H, CH, nor, 3JHH = 2.7 Hz), 1.83 (br d, 1 H, CH2, nor, 2JHH = 9.9 Hz), 2.19 (d, 1 H, CH2Ar, 2JHH = 14.1 Hz), 2.44 (br d, 1 H, CH, nor, 3JHH = 3.6 Hz), 3.21 (d, 1 H, Hα, 3JHH = 9.3 Hz), 3.45 (d, 1 H, CH2Ar, 2JHH = 13.8 Hz), 3.57 (d, 1 H, Hβ, 3JHH = 8.4 Hz), 3.79 (s, 3 H, OMe), 6.59 (br s, 1 H), 6.80 (d, 1 H, H3, 3JHH = 7.8 Hz), 6.90–7.09 (m, 6 H), 7.11–7.20 (m, 3 H), 7.40 (br s, 1 H, CO2H). 13C{1H} NMR (75.5 MHz): δ 23.9 (Me, CMe2), 26.8 (Me, CMe2), 29.5 (CH2, nor), 30.3 (CH2, nor), 36.6 (CH2, nor), 39.7 (CH, nor), 41.3 (CH, nor), 42.1 (CH2Ar), 50.9 (CHβ), 52.6 (OMe), 55.2 (CHα), 56.8 (CMe2), 118.3 (C), 121.4 (C), 126.7 (CH), 126.9 (CH5), 128.2 (CH), 130.2 (C7 or C8), 131.1 (CH), 131.4 (CH3), 132.1 (C1 or C2), 138.3 (C1 or C2), 138.6 (C), 156.3 (C7 or C8), 169.5 (CO2Me), 177.7 (CO2H).
In a Carius tube, a solution of amino acid 10d (45 mg, 0.075 mmol) in CHCl3 (2 mL) was heated at 65 °C for 7 days. The solvent was removed from the resulting white suspension to ca. 0.5 mL, and Et2O (10 mL) was added. The suspension was filtered, and the solid was washed with Et2O (2 × 3 mL) and air-dried to afford the cyclic lactone 9d as a colorless solid. Yield: 30 mg, 0.048 mmol, 64%. Mp: 251 °C. ΛM (Ω–1 mol–1 cm2): 88.8 (c = 4.1 × 10–4 M). Anal. Calcd for C29H34F3NO7S (597.643): C, 58.28; H, 5.73; N, 2.34, S; 5.37. Found: C, 58.15; H, 5.56; N, 2.12; S: 5.42. ESI-HRMS (m/z): exact mass calcd for C28H34NO4, 448.2482 [(M – CF3SO3)+]; found, 448.2493. IR (cm–1): ν(NH) 3167 m; ν(CO) 1749 s, 1733 s. 1H NMR (400.9 MHz, DMSO-d6): δ 0.69 (m, 2 H, CH2, nor), 0.91–0.94 (m, 2 H, 1 H of CH2 + 1 H of CH2, nor), 1.19 (s, 4 H, Me of CMe2 + CH of nor), 1.23 (s, 3 H, Me, CMe2), 1.26–1.28 (m, 2 H, 1 H of CH2 + 1 H of CH2, nor), 1.69 (s, 1 H, CH, nor), 2.17 (s, 1 H, CH, nor), 2.19 (s, 1 H, CH, nor), 2.88 (d, 1 H, CH2Ar, 2JHH = 14.0 Hz), 3.21 (s, 3 H, MeO), 3.41 (d, 1 H, CH2Ar, 2JHH = 13.6 Hz), 4.86 (s, 1 H, H7), 7.28 (br t, 1 H, H5, 3JHH = 7.2 Hz), 7.32–7.50 (m, 6 H, H3 + H4 + CH of Ph), 7.51–7.60 (br s, 1 H, CH, Ph), 7.63 (d, 1 H, H6, 3JHH = 8.0 H), 7.87 (br s, 3 H, NH3). The resonance corresponding to the OH group was not observed. 13C{1H} NMR (100.8 MHz, DMSO-d6): δ 24.8 (Me, CMe2), 24.9 (Me, CMe2), 27.4 (CH2, nor), 27.8 (CH2, nor), 33.5 (CH2, nor), 39.7 (CH, nor), 39.8 (CH, nor), 41.0 (CH2Ar), 49.5 (CH, nor), 50.4 (CH, nor), 51.5 (MeO), 54.7 (CMe2), 55.6 (CH7), 89.7 (C8), 127.2 (CH5), 127.6 (CH, Ph), 127.8 (br s, CH, Ph), 128.4 (CH4), 131.1 (CH6), 131.5 (C1), 132.2 (CH3), 135.1 (C2), 138.4 (i-C, Ph), 169.4 (CO2Me), 177.4 (CO2H).
Single-Crystal X-ray Structure Determinations
Single crystals were obtained as follows: complex anti-1a, slow diffusion of n-pentane into a solution of a 2.5/1 mixture of anti/syn-1a in CH2Cl2; complex anti-1b, slow diffusion of Et2O into a solution of a 3/1 mixture of anti-/syn-1b in CHCl3; complex syn-1b, slow diffusion of n-pentane into a solution of a 1/3 mixture of anti-/syn-1b in CHCl3; complex anti-2a·CH2Cl2, slow diffusion of n-pentane into a solution of a 5/1 mixture of anti-/syn-2a in CH2Cl2; complex syn-2a, slow diffusion of n-pentane into a solution of the complex in CHCl3, complex anti-2b·CHCl3, slow diffusion of n-pentane into a solution of a 3/1 mixture of anti-/syn-2b in CHCl3; complex syn-2b: slow diffusion of n-pentane into a solution of a 1/3 mixture of anti-/syn-2b in CHCl3; complex syn-3b·CH2Cl2: ,slow diffusion of n-pentane into a solution of anti-/syn-3b in CH2Cl2; compound 5b·H2O, slow diffusion of n-pentane into a solution of the complex in CH2Cl2, compounds 7c·CHCl3, 7d·1/2CHCl3, 7f, and 8f, slow diffusion of n-pentane into a solution of the corresponding compound in CHCl3; compound 9d, slow diffusion of Et2O into a solution of the compound in acetone; compound 9f·Et2O, slow diffusion of Et2O into a solution of 9f in acetone; compound 10c·H2O, slow diffusion of n-pentane into a solution of 10c in CHCl3.
Data Collection
Crystals suitable for X-ray diffraction were mounted in inert oil on a glass fiber and transferred to a Bruker diffractometer. Data were recorded at 100(2) K, using graphite-monochromated Mo Kα radiation (λ = 0.71073 Å) and the ω-scan mode. Multiscan absorption corrections were applied for all complexes.
Structure Solution and Refinements
Crystal structures were solved by Patterson (syn-1b, anti-2b·CHCl3, and 7c·CHCl3) or direct methods (anti-1a, anti-1b, anti-2a·CH2Cl2, syn-2a, syn-2b, syn-3b·CH2Cl2, 6b·H2O, 7d·1/2CHCl3, 7f, 8f, 9d, 9f·Et2O, and 10c·H2O), and all non-hydrogen atoms were refined anisotropically on F2 using the program SHELXL-2018/3.42 Hydrogen atoms were refined as follows: complex anti-1a, NH2, free with SADI; ordered methyl, rigid group; all others, riding; compounds anti-1b, syn-1b, anti-2a·CH2Cl2, syn-2a, syn-2b, 7c·CHCl3, 7d·1/2CHCl3, 7f, 8f, and 9d, NH2 or NH3, free with SADI; methyl, rigid group; all others, riding; complex anti-2b·CHCl3, NH2, free with DFIX; methyl, rigid group; all others, riding; compounds syn-3b·CH2Cl2 and 9f·Et2O, NH2 or NH3, free; methyl, rigid group; all others, riding; compound 6b·H2O, NH3 and H2O, free with SADI; methyl, rigid group; all others, riding; compound 10c·H2O, NH3, free with SADI; H2O (crystallization water), free with DFIX; methyl, rigid group; all others, riding.
Special Features
For anti-1a, one methyl group was disordered over two positions, with a ca. 95/5 occupancy distribution. For syn-1b, one phenyl ring was disordered over two positions, with a ca. 57/43 occupancy distribution. For syn-2a, the CO2CH2CH3 fragment was disordered over two positions, with a ca. 75/25 occupancy distribution. The structure was refined as an inversion twin. Absolute structure (Flack) parameter:43 0.027(8). For anti-2b·CHCl3, the chloroform molecule was disordered over two positions, with a ca. 81/19 occupancy distribution. For 7d·1/2CHCl3, the crystallization solvent (half a molecule of chloroform), located on a crystallographic inversion center, was disordered over two positions, with a ca. 62/38 occupancy distribution.
Relevant crystallographic data, details of the refinements, and details (including symmetry operators) of hydrogen bonds for the compounds anti-1a, anti-1b, syn-1b, anti-2a·CH2Cl2, syn-2a, anti-2b·CHCl3, syn-2b, syn-3b·CH2Cl2, 6b·H2O, 7c·CHCl3, 7d·1/2CHCl3, 7f, 8f, 9d, 9f·Et2O, and 10c·H2O are given in the Supporting Information.
Acknowledgments
We thank the Ministerio de Ciencia, Innovación y Universidades (Spain), FEDER (Project PGC2018-100719-B-I00), and Fundación Séneca (19890/GERM/15) for financial support.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.organomet.0c00787.
For anti-1a, anti-1b, syn-1b, anti-2a·CH2Cl2, syn-2a, anti-2b·CHCl3, syn-2b, syn-3b·CH2Cl2, 6b·H2O, 7c·CHCl3, 7d·1/2CHCl3, 7f, 8f, 9d, 9f·Et2O, and 10c·H2O, relevant crystallographic data, details of the refinements, details of hydrogen bonds (including symmetry operators), and NMR spectra (PDF)
Accession Codes
CCDC 2047166–2047181 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- a Zeni G.; Larock R. C. Synthesis of Heterocycles via Palladium-Catalyzed Oxidative Addition. Chem. Rev. 2006, 106, 4644–4680. 10.1021/cr0683966. [DOI] [PubMed] [Google Scholar]; b He J.; Wasa M.; Chan K. S. L.; Shao Q.; Yu J.-Q. Palladium-Catalyzed Transformations of Alkyl C-H Bonds. Chem. Rev. 2017, 117, 8754–8786. 10.1021/acs.chemrev.6b00622. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Biffis A.; Centomo P.; Del Zotto A.; Zecca M. Pd Metal Catalysts for Cross-Couplings and Related Reactions in the 21st Century: A Critical Review. Chem. Rev. 2018, 118, 2249–2295. 10.1021/acs.chemrev.7b00443. [DOI] [PubMed] [Google Scholar]; d Devendar P.; Qu R.-Y.; Kang W.-M.; He B.; Yang G.-F. Palladium-Catalyzed Cross-Coupling Reactions: A Powerful Tool for the Synthesis of Agrochemicals. J. Agric. Food Chem. 2018, 66, 8914–8934. 10.1021/acs.jafc.8b03792. [DOI] [PubMed] [Google Scholar]; e Wang D.; Weinstein A. B.; White P. B.; Stahl S. S. Ligand-Promoted Palladium-Catalyzed Aerobic Oxidation Reactions. Chem. Rev. 2018, 118, 2636–2679. 10.1021/acs.chemrev.7b00334. [DOI] [PubMed] [Google Scholar]; f Dondas H. A.; Retamosa M. d. G.; Sansano J. M. Recent Development in Palladium-Catalyzed Domino Reactions: Access to Materials and Biologically Important Carbo- and Heterocycles. Organometallics 2019, 38, 1828–1867. 10.1021/acs.organomet.9b00110. [DOI] [Google Scholar]
- Della Ca’ N.; Fontana M.; Motti E.; Catellani M. Pd/Norbornene: A Winning Combination for Selective Aromatic Functionalization via C-H Bond Activation. Acc. Chem. Res. 2016, 49, 1389–1400. 10.1021/acs.accounts.6b00165. [DOI] [PubMed] [Google Scholar]
- Beller M.; Seayad J.; Tillack A.; Jiao H. Catalytic Markovnikov and anti-Markovnikov Functionalization of Alkenes and Alkynes: Recent Developments and Trends. Angew. Chem., Int. Ed. 2004, 43, 3368–3398. 10.1002/anie.200300616. [DOI] [PubMed] [Google Scholar]
- a Vlaar T.; Ruijter E.; Maes B. U. W.; Orru R. V. A. Palladium-Catalyzed Migratory Insertion of Isocyanides: An Emergin Platform in Cross-Coupling Chemistry. Angew. Chem., Int. Ed. 2013, 52, 7084–7097. 10.1002/anie.201300942. [DOI] [PubMed] [Google Scholar]; b Mehta V. P.; García-López J.-A. σ-Alkyl-PdII Species for Remote C-H Functionalization. ChemCatChem 2017, 9, 1149–1156. 10.1002/cctc.201601624. [DOI] [Google Scholar]; c Xia Y.; Qiu D.; Wang J. Transition-Metal-Catalyzed Cross-Couplings through Carbene Migratory Insertion. Chem. Rev. 2017, 117, 13810–13889. 10.1021/acs.chemrev.7b00382. [DOI] [PubMed] [Google Scholar]; d Zeidan N.; Lautens M. Migratory Insertion Strategies for Dearomatization. Synthesis 2019, 51, 4137–4146. 10.1055/s-0037-1611918. [DOI] [Google Scholar]
- Blouin S.; Blond G.; Donnard M.; Gulea M.; Suffert J. Cyclocarbopalladation as a Key Step in Cascade Reactions: Recent Developments. Synthesis 2017, 49, 1767–1784. 10.1055/s-0036-1588708. [DOI] [Google Scholar]
- a McDonald R. I.; Liu G.; Stahl S. S. Palladium(II)-Catalyzed Alkene Functionalization via Nucleopalladation: Stereochemical Pathways and Enantioselective Catalytic Applications. Chem. Rev. 2011, 111, 2981–3019. 10.1021/cr100371y. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Dhungana R. K.; Sapkota R. R.; Niroula D.; Giri R. Walking metals: catalytic difunctionalization of alkenes at nonclassical sites. Chem. Sci. 2020, 11, 9757–9774. 10.1039/D0SC03634J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Zeni G.; Larock R. C. Synthesis of Heterocycles via Palladium π-Olefin and π-Alkyne Chemistry. Chem. Rev. 2004, 104, 2285–2310. 10.1021/cr020085h. [DOI] [PubMed] [Google Scholar]; b Cacchi S.; Fabrizi G. Synthesis and Functionalization of Indoles Through Palladium-catalyzed Reactions. Chem. Rev. 2005, 105, 2873–2920. 10.1021/cr040639b. [DOI] [PubMed] [Google Scholar]; c Zhang F.; Xin L.; Yu Y.; Liao S.; Huang X. Recent Advances in Palladium-Catalyzed Bridging C-H Activation by Using Alkenes, Alkynes or Diazo Compounds as Bridging Reagents. Synthesis 2021, 53, 238–254. 10.1055/s-0040-1707268. [DOI] [Google Scholar]
- a Rodríguez A.; Albert J.; Ariza X.; Garcia J.; Granell J.; Farràs J.; La Mela A.; Nicolás E. Catalytic C-H Activation of Phenylethylamines or Benzylamines and Their Annulation with Allenes. J. Org. Chem. 2014, 79, 9578–9585. 10.1021/jo501658s. [DOI] [PubMed] [Google Scholar]; b Ye J.; Ma S. Palladium-Catalyzed Cyclization Reactions of Allenes in the Presence of Unsaturated Carbon-Carbon Bonds. Acc. Chem. Res. 2014, 47, 989–1000. 10.1021/ar4002069. [DOI] [PubMed] [Google Scholar]; c Yang B.; Qiu Y.; Bäckvall J.-E. Control of Selectivity in Palladium(II)-Catalyzed Oxidative Transformations of Allenes. Acc. Chem. Res. 2018, 51, 1520–1531. 10.1021/acs.accounts.8b00138. [DOI] [PubMed] [Google Scholar]
- a Balme G.; Bossharth E.; Monteiro N. Pd-Assisted Multicomponent Synthesis of Heterocycles. Eur. J. Org. Chem. 2003, 2003, 4101–4111. 10.1002/ejoc.200300378. [DOI] [Google Scholar]; b Shen C.; Wu X.-F. Palladium-Catalyzed Carbonylative Multicomponent Reactions. Chem. - Eur. J. 2017, 23, 2973–2987. 10.1002/chem.201603623. [DOI] [PubMed] [Google Scholar]; c Saranya S.; Rohit K. R.; Radhika S.; Anilkumar G. Palladium-catalyzed multicomponent reactions: an overview. Org. Biomol. Chem. 2019, 17, 8048–8061. 10.1039/C9OB01538H. [DOI] [PubMed] [Google Scholar]
- Zhao L.; Lu X. Palladium(II)-Catalyzed Three-Component Coupling Reaction Initiated by Acetoxypalladation of Alkynes: An Efficient Route to γ,δ-Unsaturated Carbonyls. Org. Lett. 2002, 4, 3903–3906. 10.1021/ol026741h. [DOI] [PubMed] [Google Scholar]
- a Shibata K.; Satoh T.; Miura M. Palladium-Catalyzed Intermolecular Three-Component Coupling of Aryl Iodides, Alkynes, and Alkenes to Produce 1,3-Butadiene Derivatives. Org. Lett. 2005, 7, 1781–1783. 10.1021/ol0503743. [DOI] [PubMed] [Google Scholar]; b Horiguchi H.; Hirano K.; Satoh T.; Miura M. Palladium-Catalyzed Three-Component 1:2:1 Coupling of Aryl Iodides, Alkynes, and Alkenes to Produce 1,3,5-Hexatriene Derivatives. Adv. Synth. Catal. 2009, 351, 1431–1436. 10.1002/adsc.200900096. [DOI] [Google Scholar]
- Shibata K.; Satoh T.; Miura M. Palladium-Catalyzed Intermolecular Three-Component Coupling of Organic Halides with Alkynes and Alkenes: Efficient Synthesis of Oligoene Compounds. Adv. Synth. Catal. 2007, 349, 2317–2325. 10.1002/adsc.200700171. [DOI] [Google Scholar]
- a Drent E.; Budzelaar P. H. M. Palladium-Catalyzed Alternating Copolymerization of Alkenes and Carbon Monoxide. Chem. Rev. 1996, 96, 663–682. 10.1021/cr940282j. [DOI] [PubMed] [Google Scholar]; b Barlow G. K.; Boyle J. D.; Cooley N. A.; Ghaffar T.; Wass D. F. Mechanistic Studies of Alkene/CO Polymerization with Palladium Complexes Promoted by B(C6F5)3. Organometallics 2000, 19, 1470–1476. 10.1021/om990911l. [DOI] [Google Scholar]; c Bianchini C.; Meli A. Alternating copolymerization of carbon monoxide and olefins by single-site metal catalysis. Coord. Chem. Rev. 2002, 225, 35–66. 10.1016/S0010-8545(01)00405-2. [DOI] [Google Scholar]; d Bianchini C.; Meli A.; Oberhauser W. Catalyst design and mechanistic aspects of the alternating copolymerisation of ethene and carbon monoxide by diphosphine-modified palladium catalysis. Dalton Trans. 2003, 2627–2635. 10.1039/b303502f. [DOI] [Google Scholar]
- a Ittel S. D.; Johnson L. K.; Brookhart M. Late-Metal Catalysts for Ethylene Homo- and Copolymerization. Chem. Rev. 2000, 100, 1169–1204. 10.1021/cr9804644. [DOI] [PubMed] [Google Scholar]; b Nakamura A.; Ito S.; Nozaki K. Coordination-Insertion Copolymerization of Fundamental Polar Monomers. Chem. Rev. 2009, 109, 5215–5244. 10.1021/cr900079r. [DOI] [PubMed] [Google Scholar]; c Ito S.; Nozaki K. Coordination-Insertion Copolymerization of Polar Vinyl Monomers by Palladium Catalysts. Chem. Rec. 2010, 10, 315–325. 10.1002/tcr.201000032. [DOI] [PubMed] [Google Scholar]; d Guo L.; Dai S.; Sui X.; Chen C. Palladium and Nickel Catalyzed Chain Walking Olefin Polymerization and Copolymerization. ACS Catal. 2016, 6, 428–441. 10.1021/acscatal.5b02426. [DOI] [Google Scholar]; e Tan C.; Chen C. Emerging Palladium and Nickel Catalysts for Copolymerization of Olefins with Polar Monomers. Angew. Chem., Int. Ed. 2019, 58, 7192–7200. 10.1002/anie.201814634. [DOI] [PubMed] [Google Scholar]
- Lindhardt A. T.; Mantel M. L. H.; Skrydstrup T. Palladium-Catalyzed Intermolecular Ene-Yne Coupling: Development of an Atom-Efficient Mizoroki-Heck-Type Reaction. Angew. Chem., Int. Ed. 2008, 47, 2668–2672. 10.1002/anie.200705558. [DOI] [PubMed] [Google Scholar]
- a Zhou P.; Jiang H.; Huang L.; Li X. Acetoxypalladation of unactivated alkynes and capture with alkenes to give 1-acetoxy-1,3-dienes taking dioxygen as terminal oxidant. Chem. Commun. 2011, 47, 1003–1005. 10.1039/C0CC03723K. [DOI] [PubMed] [Google Scholar]; b Kawashima T.; Ohashi M.; Ogoshi S. Nickel-Catalyzed Formation of 1,3-Dienes via a Highly Selective Cross-Tetramerization of Tetrafluoroethylene, Styrenes, Alkynes, and Ethylene. J. Am. Chem. Soc. 2017, 139, 17795–17798. 10.1021/jacs.7b12007. [DOI] [PubMed] [Google Scholar]; c Kanbayashi N.; Okamura T.-A.; Onitsuka K. Living Cyclocopolymerization through Alternating Insertion of Isocyanide and Allene via Controlling the Reactivity of the Propagation Species: Detailed Mechanistic Investigation. J. Am. Chem. Soc. 2019, 141, 15307–15317. 10.1021/jacs.9b07431. [DOI] [PubMed] [Google Scholar]
- Wen Y.; Huang L.; Jiang H. Access to C(sp3)-C(sp2) and C(sp2)-C(sp2) Bond Formation via Sequential Intermolecular Carbopalladation of Multiple Carbon-Carbon Bonds. J. Org. Chem. 2012, 77, 5418–5422. 10.1021/jo300662x. [DOI] [PubMed] [Google Scholar]
- a Milde B.; Leibeling M.; Hecht A.; Jones P. G.; Visscher A.; Stalke D.; Grunenberg J.; Werz D. B. Oligoene-Based π-Helicenes or Dispiranes? Winding up Oligoyne Chains by a Multiple Carbopalladation/Stille/(Electrocyclization) Cascade. Chem. - Eur. J. 2015, 21, 16136–16146. 10.1002/chem.201501797. [DOI] [PubMed] [Google Scholar]; b Zhu C.; Yang B.; Bäckvall J.-E. Highly Selective Cascade C-C Bond Formation via Palladium-Catalyzed Oxidative Carbonylation-Carbocyclization-Carbonylation-Alkynylation of Enallenes. J. Am. Chem. Soc. 2015, 137, 11868–11871. 10.1021/jacs.5b06828. [DOI] [PubMed] [Google Scholar]; c Blouin S.; Gandon V.; Blond G.; Suffert J. Synthesis of Cyclooctatetraenes through a Palladium-Catalyzed Cascade Reaction. Angew. Chem., Int. Ed. 2016, 55, 7208–7211. 10.1002/anie.201602586. [DOI] [PubMed] [Google Scholar]; d Qiu Y.; Yang B.; Zhu C.; Bäckvall J.-E. Highly Efficient Cascade Reaction for Selective Formation of Spirocyclobutenes from Dienallenes via Palladium-Catalyzed Oxidative Double Carbocyclization-Carbonylation-Alkynylation. J. Am. Chem. Soc. 2016, 138, 13846–13849. 10.1021/jacs.6b09240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- García-López J.-A.; Oliva-Madrid M.-J.; Saura-Llamas I.; Bautista D.; Vicente J. Reactivity toward CO of Eight-Membered Palladacycles Derived from the Insertion of Alkenes into the Pd-C Bond of Cyclopalladated Primary Arylalkylamines of Pharmaceutical Interest. Synthesis of Tetrahydrobenzazocinones, Ortho-Functionalized Phenethylamines, Ureas, and an Isocyanate. Organometallics 2012, 31, 6351–6364. 10.1021/om300593x. [DOI] [Google Scholar]
- García-López J.-A.; Oliva-Madrid M.-J.; Saura-Llamas I.; Bautista D.; Vicente J. Room-Temperature Isolation of Palladium(II) Organocarbonyl Intermediates in the Synthesis of Eight-Membered Lactams after Alkyne/CO Sequential Insertions. Organometallics 2013, 32, 1094–1105. 10.1021/om301241n. [DOI] [Google Scholar]
- Oliva-Madrid M.-J.; García-López J.-A.; Saura-Llamas I.; Bautista D.; Vicente J. Reactivity of Eight-Membered Palladacycles Arising from Monoinsertion of Alkynes into the Pd-C bond of Ortho-Palladated Phenethylamines toward Unsaturated Molecules. Synthesis of Dihydro-3-Benzazocinones, N7-amino Acids, N7-amino Esters, and 3-Benzazepines. Organometallics 2014, 33, 19–32. 10.1021/om4010059. [DOI] [Google Scholar]
- Oliva-Madrid M. J.; García-López J. A.; Saura-Llamas I.; Bautista D.; Vicente J. Insertion Reactions on Carbopalladated Benzyne: From Eight- to Nine- and Ten-Membered Palladacycles. Applications to the Synthesis of N-Heterocycles. Organometallics 2014, 33, 6420–6430. 10.1021/om500772p. [DOI] [Google Scholar]
- a Vicente J.; Abad J.-A.; Förtsch W.; López-Sáez M.-J.; Jones P. G. Reactivity of Ortho-Palladated Phenol Derivatives with Unsaturated Molecules. 1. Insertion of CO, Isocyanides, Alkenes, and Alkynes. CO/Alkene, Alkyne/Isocyanide, and Isocyanide/Alkene Sequential Insertion Reactions. Organometallics 2004, 23, 4414–4429. 10.1021/om0496131. [DOI] [Google Scholar]; b Vicente J.; Chicote M. T.; Martínez-Martínez A. J.; Abellán-López A.; Bautista D. Reactivity of ortho-β-Enaminone-phenyl Palladium Complexes. Insertion of CO into the Pd-C Bond to Give the First Acyl C,N,O-Pincer Complexes. Sequential Insertion of Dimethylacetylenedicarboxylate into the Enaminone C-H Bond and of Isocyanide into the Pd-C Bond. A New Photooxigenation/Cyclization Process. Organometallics 2010, 29, 5693–5707. 10.1021/om1005403. [DOI] [Google Scholar]; c Frutos-Pedreño R.; González-Herrero P.; Vicente J.; Jones P. G. Sequential Insertion of Alkynes and CO or Isocyanides into the Pd-C Bond of Cyclopalladated Phenylacetamides. Synthesis of Eight-Membered Palladacycles, Benzo[d]azocine-2,4(1H,3H)-diones, and Highly Functionalized Acrylonitrile and Acrylamide Derivatives. Organometallics 2012, 31, 3361–3372. 10.1021/om300151n. [DOI] [Google Scholar]; d García-López J.-A.; Frutos-Pedreño R.; Bautista D.; Saura-Llamas I.; Vicente J. Norbornadiene as a Building Block for the Synthesis of Linked Benzazocinones and Benzazocinium Salts through Tetranuclear Carbopalladated Intermediates. Organometallics 2017, 36, 372–383. 10.1021/acs.organomet.6b00795. [DOI] [Google Scholar]
- Vicente J.; Saura-Llamas I.; Bautista D. Regiospecific Functionalization of Pharmaceuticals and Other Biologically Active Molecules Through Cyclopalladated Compounds. 2-Iodination of Phentermine and L-Tryptophan Methyl Ester. Organometallics 2005, 24, 6001–6004. 10.1021/om0506522. [DOI] [Google Scholar]
- a Vicente J.; Saura-Llamas I.; García-López J. A.; Calmuschi-Cula B.; Bautista D. Ortho Palladation and Functionalization of L-Phenylalanine Methyl Ester. Organometallics 2007, 26, 2768–2776. 10.1021/om070127y. [DOI] [Google Scholar]; b Vicente J.; Saura-Llamas I.; García-López J.-A.; Bautista D. Insertion of Isocyanides, Isothiocyanates, and Carbon Monoxide into the Pd-C Bond of Cyclopalladated Complexes Containing Primary Arylalkylamines of Biological and Pharmaceutical Significance. Synthesis of Lactams and Cyclic Amidinium Salts Related to the Isoquinoline, Benzo[g]isoquinoline, and β-Carboline Nuclei. Organometallics 2009, 28, 448–464. 10.1021/om800951k. [DOI] [Google Scholar]
- Oliva-Madrid M.-J.; García-López J.-A.; Saura-Llamas I.; Bautista D.; Vicente J. Ortho Palladation of the Phenethylamines of Biological Relevance L-Tyrosine Methyl Ester and Homoveratrylamine. Reactivity of the Palladacycles toward CO and Isocyanides. Synthesis of the Natural Alkaloid Corydaldine. Organometallics 2012, 31, 3647–3660. 10.1021/om300141b. [DOI] [Google Scholar]
- Frutos-Pedreño R.; García-Sánchez E.; Oliva-Madrid M. J.; Bautista D.; Martínez-Viviente E.; Saura-Llamas I.; Vicente J. C-H Activation in Primary 3-Phenylpropylamines: Synthesis of Seven-Membered Palladacycles through Orthometalation. Stoichiometric Preparation of Benzazepinones and Catalytic Synthesis of Ureas. Inorg. Chem. 2016, 55, 5520–5533. 10.1021/acs.inorgchem.6b00542. [DOI] [PubMed] [Google Scholar]
- a Heck R. F. Palladium-catalyzed reactions of organic halides with olefins. Acc. Chem. Res. 1979, 12, 146–151. 10.1021/ar50136a006. [DOI] [Google Scholar]; b Brisdon B. J.; Nair P.; Dyke S. F. Palladium Assisted Organic Reactions-II: Ortho-Aminomethylstilbene Derivatives. Tetrahedron 1981, 37, 173–177. 10.1016/S0040-4020(01)97731-2. [DOI] [Google Scholar]; c Ryabov A. D.; Sakodinskaya I. K.; Yatsimirsky A. K. Kinetics and Mechanism of Vinylation of ortho-Palladated N,N-Dialkylbenzylamines by para-Substituted Styrenes. J. Chem. Soc., Perkin Trans. 2 1983, 1511–1518. 10.1039/p29830001511. [DOI] [Google Scholar]; d Cabri W.; Candiani I. Recent Developments and New Perspectives in the Heck Reaction. Acc. Chem. Res. 1995, 28, 2–7. 10.1021/ar00049a001. [DOI] [Google Scholar]; e Crisp G. T. Variations on a theme-recent developments on the mechanism of the Heck reaction and their implications for synthesis. Chem. Soc. Rev. 1998, 27, 427–436. 10.1039/a827427z. [DOI] [Google Scholar]; f Ludwig M.; Strömberg S.; Svensson M.; Åkermark B. An Exploratory Study of Regiocontrol in the Heck Type Reaction. Influence of Solvent Polarity and Bisphosphine Ligands. Organometallics 1999, 18, 970–975. 10.1021/om9803265. [DOI] [Google Scholar]; g Albéniz A. C.; Espinet P.; López-Fernández R. Polymerization of Acrylates by Neutral Palladium Complexes. Isolation of Complexes at the Initial Steps. Organometallics 2003, 22, 4206–4212. 10.1021/om030507t. [DOI] [Google Scholar]; h Cai G.; Fu Y.; Li Y.; Wan X.; Shi Z. Indirect ortho Functionalization of Substituted Toluenes through ortho Olefination of N,N-Dimethylbenzylamines Tuned by the Acidity of Reaction Conditions. J. Am. Chem. Soc. 2007, 129, 7666–7673. 10.1021/ja070588a. [DOI] [PubMed] [Google Scholar]; i McConville M.; Saidi O.; Blacker J.; Xiao J. Regioselective Heck Vinylation of Electron-Rich Olefins with Vinyl Halides: Is the Neutral Pathway in Operation?. J. Org. Chem. 2009, 74, 2692–2698. 10.1021/jo802781m. [DOI] [PubMed] [Google Scholar]
- a Zhang Y.; Negishi E. Metal-promoted cyclization. 25. Palladium-catalyzed cascade carbometalation of alkynes and alkenes as an efficient route to cyclic and polycyclic structures. J. Am. Chem. Soc. 1989, 111, 3454–3456. 10.1021/ja00191a066. [DOI] [Google Scholar]; b Yanada R.; Obika S.; Inokuma T.; Yanada K.; Yamashita M.; Ohta S.; Takemoto Y. Stereoselective Synthesis of 3-Alkylideneoxindoles via Palladium-Catalyzed Domino Reactions. J. Org. Chem. 2005, 70, 6972–6975. 10.1021/jo0508604. [DOI] [PubMed] [Google Scholar]
- a Engelin C.; Fristrup P. Palladium Catalyzed Allylic C-H Alkylation: A Mechanistic Perspective. Molecules 2011, 16, 951–969. 10.3390/molecules16010951. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Le Bras J.; Muzart J. Production of Csp3-Csp3 Bonds through Palladium-Catalyzed Tsuji-Trost-Type Reactions of (Hetero)Benzylic Substrates. Eur. J. Org. Chem. 2016, 2016, 2565–2593. 10.1002/ejoc.201600094. [DOI] [Google Scholar]; c Orcel U.; Waser J. In situ tether formation from amines and alcohols enabling highly selective Tsuji-Trost allylation and olefin functionalization. Chem. Sci. 2017, 8, 32–39. 10.1039/C6SC04366F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Maassarani F.; Pfeffer M.; Le Borgne G. Controlled Synthesis of Heterocyclic Compounds through Ring Enlargements by Alkyne Insertion into the Pd-C Bonds of Cyclopalladated Amines Followed by Subsequent Ring Closure. Organometallics 1987, 6, 2029–2043. 10.1021/om00153a002. [DOI] [Google Scholar]; b Dupont J.; Pfeffer M.; Daran J.-C.; Gouteron J. Reactivity of Cyclopalladated Compounds. Part 18. Compared Reactivity of the Pd-C Bonds of Two Closely Related Six-membered Palladocyclic Rings with Substituted Alkynes. X-Ray and Molecular Structures of [Pd{C(Ph)=C(R)C(Ph)=C(R)(o-C6H4N = CMeNHPh)}Cl] (R = CO2Et) and [Pd{C(R)[C(CO2Me)C(R)=C(R)C(R)=C(R)][o-C6H4N = CMe(OH)]}Cl] (R = CO2Me). J. Chem. Soc., Dalton Trans. 1988, 2421–2429. 10.1039/DT9880002421. [DOI] [Google Scholar]; c Tao W.; Silverberg L. J.; Rheingold A. L.; Heck R. F. Alkyne Reactions with Arylpalladium Compounds. Organometallics 1989, 8, 2550–2559. 10.1021/om00113a006. [DOI] [Google Scholar]; d Vicente J.; Saura-Llamas I.; Turpín J.; Bautista D.; de Arellano C. R.; Jones P. G. Insertion of One, Two, and Three Molecules of Alkyne into the Pd-C Bond of Ortho-palladated Primary and Secondary Arylalkylamines. Organometallics 2009, 28, 4175–4195. 10.1021/om9002895. [DOI] [Google Scholar]
- a Hartog T. D.; Toro J. M. S.; Chen P. A Palladium-Catalyzed Methylenation of Olefins Using Halomethylboronate Reagents. Org. Lett. 2014, 16, 1100–1103. 10.1021/ol403695b. [DOI] [PubMed] [Google Scholar]; b Mao J.; Zhang S.-Q.; Shi B.-F.; Bao W. Palladium(0)-catalyzed cyclopropanation of benzyl bromides via C(sp3)-H bond activation. Chem. Commun. 2014, 50, 3692–3694. 10.1039/C3CC49231A. [DOI] [PubMed] [Google Scholar]; c Tanioka Y.; Tsukada N. Palladium-Catalyzed α-Ketocyclopropanation of Norbornenes with Propargyl Acetates. J. Org. Chem. 2014, 79, 5301–5304. 10.1021/jo500357f. [DOI] [PubMed] [Google Scholar]
- a Liu C.-H.; Cheng C.-H.; Cheng M.-C.; Peng S.-M. Palladium-Catalyzed Addition of Alkyne to Norbornene Derivatives. Unusual Ring Formation and Expansion Reactions. Organometallics 1994, 13, 1832–1839. 10.1021/om00017a046. [DOI] [Google Scholar]; b Bigeault J.; Giordano L.; Buono G. [2 + 1] Cycloadditions of Terminal Alkynes to Norbornene Derivatives Catalyzed by Palladium Complexes with Phosphinous Acid Ligands. Angew. Chem., Int. Ed. 2005, 44, 4753–4757. 10.1002/anie.200500879. [DOI] [PubMed] [Google Scholar]; c Mo D.-L.; Yuan T.; Ding C.-H.; Dai L.-X.; Hou X.-L. Palladacycle-Catalyzed Methylenecyclopropanation of Bicyclic Alkenes with Propiolates. J. Org. Chem. 2013, 78, 11470–11476. 10.1021/jo402024v. [DOI] [PubMed] [Google Scholar]
- a Mao J.; Bao W. Palladium(0)-Catalyzed Methylenecyclopropanation of Norbornenes with Vinyl Bromides. Org. Lett. 2014, 16, 2646–2649. 10.1021/ol500829t. [DOI] [PubMed] [Google Scholar]; b Mao J.; Xie H.; Bao W. Palladium(0)-Catalyzed Methylcyclopropanation of Norbornenes with Vinyl Bromides and Mechanism Study. Org. Lett. 2015, 17, 3678–3681. 10.1021/acs.orglett.5b01603. [DOI] [PubMed] [Google Scholar]
- Oh C. H.; Park D. I.; Ryu J. H.; Cho J. H.; Han J.-S. Syntheses and Characterization of Cyclopropane-fused Hydrocarbons as New High Energetic Materials. Bull. Korean Chem. Soc. 2007, 28, 322–324. 10.5012/bkcs.2007.28.2.322. [DOI] [Google Scholar]
- Arai Y.; Matsui M.; Koizumi T. An enantiodivergent synthesis of fused bicyclo[2.2.1]heptane lactones via an asymmetric Diels-Alder reaction. J. Chem. Soc., Perkin Trans. 1 1990, 1233–1234. 10.1039/P19900001233. [DOI] [Google Scholar]
- Teixeira M. G.; Alvarenga E. S.; Pimentel M. F.; Picanço M. C. Synthesis and Insecticidal Activity of Lactones Derived from Furan-2(5H)-one. J. Braz. Chem. Soc. 2015, 26, 2279–2289. 10.5935/0103-5053.20150217. [DOI] [Google Scholar]
- a Tomioka H.; Oshima K.; Nozaki H. Cerium catalyzed selective oxidation of secondary alcohols in the presence of primary ones. Tetrahedron Lett. 1982, 23, 539–542. 10.1016/S0040-4039(00)86883-5. [DOI] [Google Scholar]; b Jhulki S.; Seth S.; Mondal M.; Moorthy J. N. Facile organocatalytic domino oxidation of diols to lactones by in situ-generated TetMe-IBX. Tetrahedron 2014, 70, 2286–2293. 10.1016/j.tet.2014.01.034. [DOI] [Google Scholar]
- a Lok K. P.; Jakovac I. J.; Jones J. B. Enzymes in Organic Synthesis. 34. Preparations of Enantiomerically Pure Exo- and Endo-Bridged Bicyclic [2.2.1] and [2.2.2] Chiral Lactones via Stereospecific Horse Liver Alcohol Dehydrogenase Catalyzed Oxidations of Meso Diols. J. Am. Chem. Soc. 1985, 107, 2521–2526. 10.1021/ja00294a052. [DOI] [Google Scholar]; b Mihovilovic M. D.; Kapitán P. Regiodivergent Baeyer-Villiger oxidation of fused ketone substrates by recombinant whole-cells expressing two monooxygenases from Brevibacterium. Tetrahedron Lett. 2004, 45, 2751–2754. 10.1016/j.tetlet.2004.02.036. [DOI] [Google Scholar]
- a Aït-Mohand S.; Hénin F.; Muzart J. Palladium-Catalyzed Oxidations: Inhibition of a Pd-H Elimination by Coordination of a Remote Carbon-Carbon Double Bond. Organometallics 2001, 20, 1683–1686. 10.1021/om001028m. [DOI] [Google Scholar]; b Suzuki T.; Morita K.; Matsuo Y.; Hiroi K. Catalytic asymmetric oxidative lactonizations of meso-diols using a chiral iridium complex. Tetrahedron Lett. 2003, 44, 2003–2006. 10.1016/S0040-4039(03)00228-4. [DOI] [Google Scholar]
- Pérez-Gómez M.; Hernández-Ponte S.; García-López J.-A.; Frutos-Pedreño R.; Bautista D.; Saura-Llamas I.; Vicente J. Palladium-Mediated Functionalization of Benzofuran and Benzothiophene Cores. Organometallics 2018, 37, 4648–4663. 10.1021/acs.organomet.8b00682. [DOI] [Google Scholar]
- a Sheldrick G. M. Crystal structure refinement with SHELXL. Acta Crystallogr., Sect. C: Struct. Chem. 2015, 71, 3–8. 10.1107/S2053229614024218. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Sheldrick G. M.SHELXL-2018/3, Program for the Refinement of Crystal Structure; University of Göttingen: Göttingen, Germany, 2018.
- Flack H. D. On enantiomorph-polarity estimation. Acta Crystallogr., Sect. A: Found. Crystallogr. 1983, 39, 876–881. 10.1107/S0108767383001762. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.