

SUMMARY
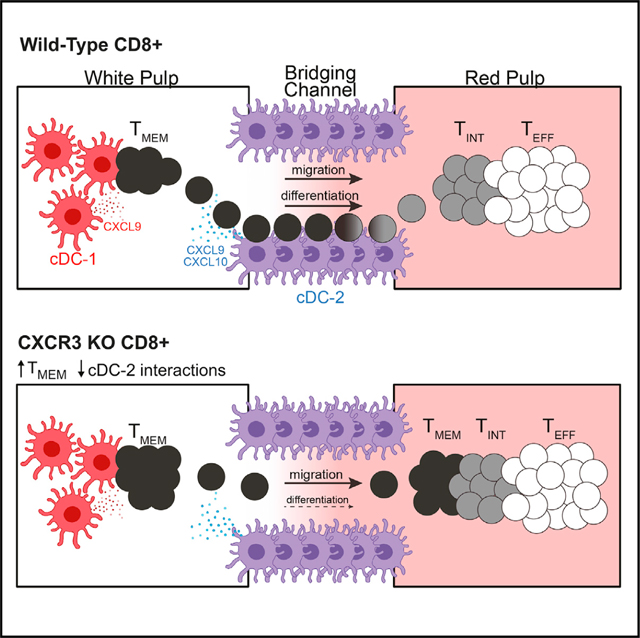
Production of effector CD8+ T cells during persistent infection requires a stable pool of stem-like cells that can give rise to effector cells via a proliferative intermediate population. In infection models marked by T cell exhaustion, this process can be transiently induced by checkpoint blockade but occurs spontaneously in mice chronically infected with the protozoan intracellular parasite Toxoplasma gondii. We observe distinct locations for parasite-specific T cell subsets, implying a link between differentiation and anatomical niches in the spleen. Loss of the chemokine receptor CXCR3 on T cells does not prevent white pulp-to-red pulp migration but reduces interactions with CXCR3 ligand-producing dendritic cells (DCs) and impairs memory-to-intermediate transition, leading to a buildup of memory T cells in the red pulp. Thus, CXCR3 increases T cell exposure to differentiation-inducing signals during red pulp migration, providing a dynamic mechanism for modulating effector differentiation in response to environmental signals.
In brief
Bangs et al. report that distinct subsets of CD8+ T cells found during chronic infection occupy distinct regions of the spleen. CXCR3 regulates differentiation of T cells but not their migration. Instead, CXCR3 promotes the interaction of T cells with ligand-producing DCs in bridging channels, resulting in effector differentiation.
Graphical Abstract
INTRODUCTION
Upon infection, naive pathogen-specific CD8+ T cells rapidly expand and differentiate to give rise to functionally heterogeneous populations of effector and memory T cells (Williams and Bevan, 2007). Effector cells directly control infection by efficiently killing invaded target cells, whereas memory cells demonstrate less effector capacity but confer long-term immunological protection. The fate determination of CD8+ T cells towards effector or memory differentiation is regulated by cell-intrinsic processes, including asymmetric cell division (Chang et al., 2007; Lin et al., 2015), but can also be influenced by environmental signals, including the strength of T cell receptor signaling, costimulatory signals, and cytokines (Chang et al., 2014). The signals T cells receive in vivo are largely dependent on their local environment and their interaction with specific populations of antigen-presenting cells (APCs). Although some of the signals directing effector and memory differentiation have been identified, our understanding of the anatomical niches that control T cell fate remains limited. An important component of these niches are dendritic cells (DCs), specialized APCs that are critical for T cell priming and activation. DCs are heterogenous in phenotype and function, and recent evidence suggests that T cell fate is influenced by interactions with distinct lineages of DCs (Li et al., 2016; Brown et al., 2019; Eisenbarth, 2019; Kotov et al., 2019; Shin et al., 2019; Opejin et al., 2020).
The chemokine receptor CXCR3 is expressed by populations of antigen-experienced CD8+ T cells and plays a critical role in determining how T cells migrate and interact with APCs within tissues. The CXCR3 ligands CXCL9 and CXCL10 (CXCL11, the third ligand, is not expressed in C57BL/6J mice) are expressed by a variety of cells upon exposure to inflammatory cytokines and can help guide T cells into inflamed tissues. In the spleen, CD4+ and CD8+ T cells utilize CXCR3 to migrate to and interact with APCs during T cell priming and recall responses (Sung et al., 2012; Goldberg et al., 2018; Maurice et al., 2019; Duckworth et al., 2021). In particular, the marginal zone and bridging channels as well as the red pulp of the spleen are important sites of antigen presentation during infection because of their high exposure to circulation, and CXCR3 mediates migration of activated CD8+ T cells into these compartments during infection, promoting generation of short-lived effector cells at the expense of the long-lived memory precursor pool (Hu et al., 2011; Kurachi et al., 2011).
Although the role of CXCR3 in directing T cell differentiation during acute infection is well established, much less is known about its role during chronic infection. After the establishment of chronic infection, armed effector T cells can be generated from a stem-like memory population via a proliferative intermediate state. This process was first characterized in a well-controlled model of mouse infection with the intracellular protozoan parasite Toxoplasma gondii. In this setting, a long-lived progenitor population with stem-like functions (TMEM cells; CXCR3+ KLRG1−) continuously feeds into a proliferative intermediate state (TINT cells; CXCR3+ KLRG1+) that produces large numbers of terminally differentiated effector T cells (TEFF cells; CXCR3− KLRG1+) (Chu et al., 2016). Similar stem-like and proliferative intermediate populations have subsequently been defined in other chronic infection models, including cytomegalovirus (CMV) in mice and humans (Gordon et al., 2018; Welten et al., 2020) and (lymphocytic choriomeningitis virus (LCMV) in mice (Im et al., 2016; Utzschneider et al., 2016; Hudson et al., 2019). However, unlike the LCMV chronic infection model, in which T cells express the inhibitor receptor PD-1 and exhibit functional exhaustion, T cells in the T. gondii infection model retain potent effector function and lack expression of inhibitory receptors. As a result, proliferative intermediate cells are only robustly produced following PD-1 blockade in the LCMV infection model, whereas TMEM, TINT, and TEFF cell populations are produced spontaneously and continuously in T. gondii-infected mice (Chu et al., 2016; Tsitsiklis et al., 2020). These features make T. gondii infection a valuable model to investigate differentiation of stem, intermediate, and TEFF cells in the setting of chronic infection. Advancing our understanding of this model should shed light on similar populations in other infections and may lead to improved strategies to manipulate CD8+ T cell responses during chronic infection and cancer.
Here we showed that TMEM, TINT, and TEFF cells expressed unique gene expression programs correlating with their location within distinct regions of the spleen. We also identified a critical role of the chemokine receptor CXCR3 in promoting conversion of TMEM cells into TINT cells. Interestingly, although CXCR3 was critical for T cell differentiation, it was not required for the migration from white pulp to red pulp that normally accompanies effector differentiation. Instead, CXCR3 mediates interactions between T cells and populations of CXCR3 ligand-producing type 2 conventional DCs (cDC-2s) located within the bridging channel of the spleen. Our data support a model in which CXCR3 promotes transient interactions between TMEM cells and cDC-2s during their migration from the white pulp to the red pulp, resulting in a balanced differentiation signal that allows TINT cell development and further clonal expansion prior to terminal effector differentiation.
RESULTS
TMEM and TINT cells are transcriptionally distinct precursors of effector cells
We have previously described a highly protective, immunodominant CD8+ T cell response mounted against a peptide derived from the parasite protein GRA6 presented by the major histocompatibility complex (MHC) class I molecule H-2Ld (Blanchard et al., 2008; Feliu et al., 2013; Chu et al., 2016; Salvioni et al., 2019; Tsitsiklis et al., 2020). During chronic infection in H-2Ld-expressing mice, GRA6-specific CD8+ T cells experience continuous antigen stimulation in the spleen and exhibit ongoing proliferation and differentiation. To understand the factors regulating CD8+ T cell differentiation, we infected B6xB6.c H-2b/d mice with T. gondii, and 6 weeks after infection, we isolated GRA6-tetramer+ TMEM, TINT, and TEFF cells from the spleen based on CXCR3 and KLRG1 expression and performed RNA sequencing (RNA-seq) (Figure 1A). Principal-component analysis (PCA) of the transcriptional data showed that the populations segregated into separate clusters, confirming that they represent transcriptionally distinct subsets (Figure 1B). Examination of differentially expressed transcripts revealed a large set of genes that increased with effector differentiation (Figure 1C, groups 1 and 2), including canonical genes related to cytotoxic T lymphocyte (CTL) activity (e.g., GzmB, Prf1, and Fasl) (Figure S1). Other groups of genes were selectively upregulated in the TMEM (gene group 5) or TINT (gene group 7) populations, indicative of two distinct differentiation states amongst antigen-experienced progenitors to TEFF cells. To gain a broad understanding of the pathways defining each population, we performed gene set enrichment analysis (GSEA) (Figures 1D and S1). TMEM cells were enriched for pathways related to cytokine signaling and inflammation, including receptors for cytokines with known roles in regulating T cell fate, such as Il21r, Il2rb, and Il7r (Figures 1D and S1). In contrast, TINT cells were enriched for cell cycle-associated genes (Figure 1D). TINT cells also showed enrichment for several metabolic pathways, with oxidative phosphorylation showing the strongest enrichment relative to the TMEM and TEFF populations (Figure 1D). Interestingly, TINT cells had reduced expression of genes associated with transforming growth factor β (TGF-β) and wnt/β-catenin signaling and were enriched in complement pathway genes relative to TMEM and TEFF populations (Figure 1D). Finally, TMEM, TINT, and TEFF cells showed differential expression of several transcription factors known to be critical for driving T cell fate (Figure 1E). TINT cells showed elevated expression of Eomes mRNA, consistent with previous measurements at the protein level (Chu et al., 2016). TMEM cells expressed elevated Tcf7 (encoding TCF1) and Id3 but lacked Id2 and Prdm1 (encoding Blimp-1), similar to stem-like CD8+ T cells, which arise during chronic viral infection (Figure S1).
Figure 1. TMEM, TINT, and TEFF cells express distinct transcriptional profiles.
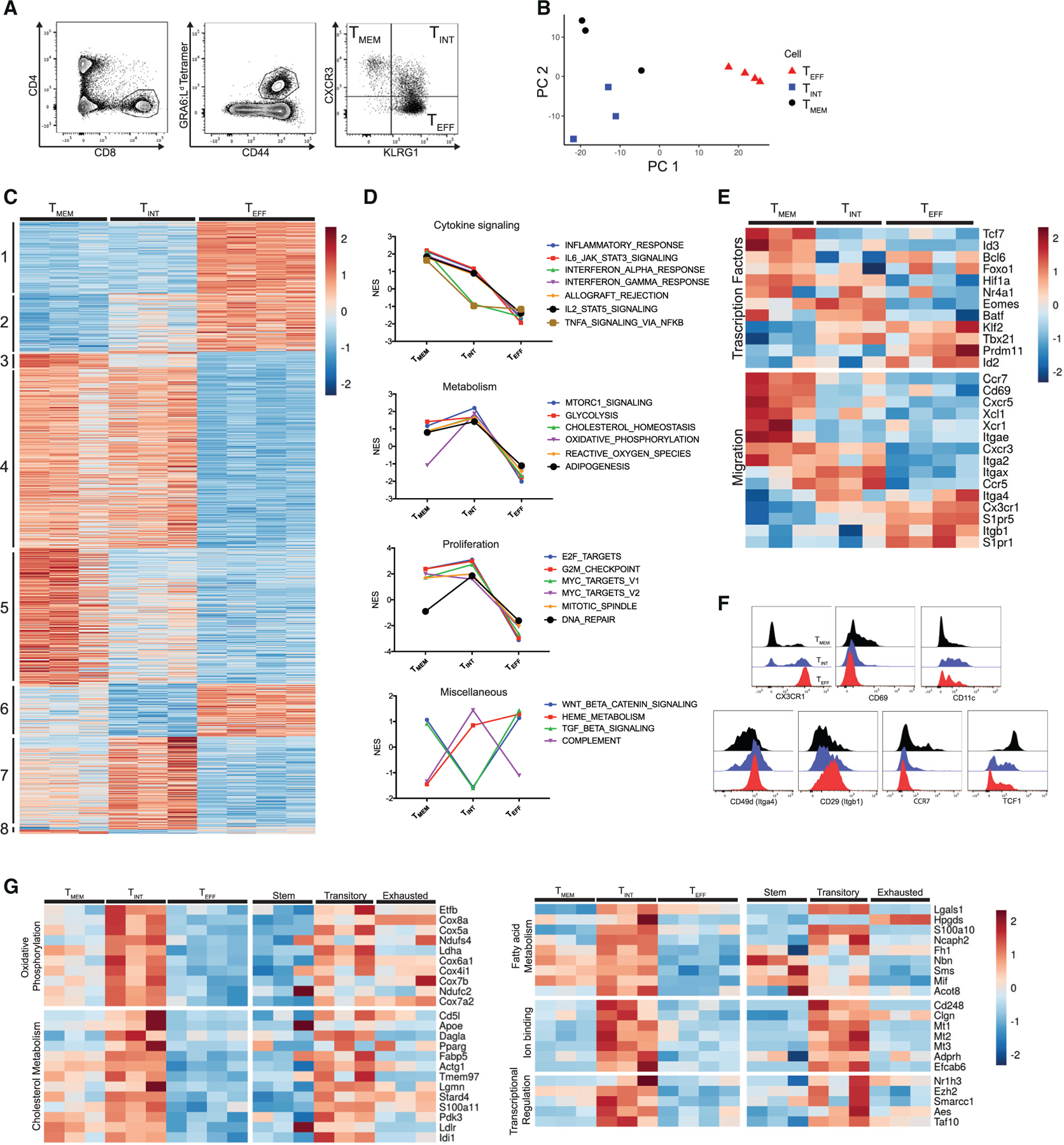
(A) Gating strategy for cell sorting prior to RNA-seq.
(B) The first two principal components of the sorted GRA6-specific populations.
(C) Heatmap displaying the normalized expression for all genes differentially expressed in pairwise comparison (TMEM versus TINT, TINT versus TEFF, and TMEM versus TEFF cells). Genes were ordered and grouped based on the results of differential expression tests. Each column represents independent biological replicates (n = 3 mice for TMEM and TINT cells, n = 4 mice for TEFF cells).
(D) Selected results of GSEA with Hallmark gene sets using one-versus-all comparisons (TMEM versus TINT and TEFF cells, TINT versus TMEM and TEFF cells, TMEM versus TEFF and TINT cells). The full set of enriched Hallmark gene sets is displayed in Figure S1. Plots show normalized enrichment score (NES).
(E) Heatmap displaying the normalized expression of selected differentially expressed genes related to the indicated function.
(F) Representative flow cytometry histograms showing protein staining on TMEM, TINT, and TEFF cells.
(G) Heatmap displaying the expression of selected genes from group 7, arranged by the indicated functional categories.
Values are from sorted GRA6-specific subsets (current study) or published transcriptional data on stem, transitory, and exhausted cells during LCMV infection (Hudson et al., 2019).
To further compare the TMEM and TINT populations in the Toxoplasma infection model with the progenitors of exhausted effector T cells described in other settings, we performed GSEA analyses using gene signatures derived from published transcriptional data on stem-like and transitory proliferative CD8+ T cells during LCMV infection (Utzschneider et al., 2016; Hudson et al., 2019). We observed that TMEM cells showed an enrichment for stem-like signatures (Figure S1), and both populations exhibited elevated expression of key co-stimulatory molecules (including Cd28 and Icos) and Tlr genes (Figure S1). In addition, most of the genes that were specifically upregulated in TINT cells (Figure 1C, group 7) were also selectively expressed in the transitory T population from LMCV relative to stem-like and exhausted effector cells (Figure S1). This included genes involved in oxidative phosphorylation, fatty acid, and cholesterol metabolism (including Ndufs4, Tmem97, Cd5l, and Fabp5) as well as a set of transcriptional regulators and genes involved in binding divalent metal cations (Figure 1G). Thus, TMEM cells resemble the stem-like cells described in other models of infection and cancer, and the TINT population represents a distinct proliferative and metabolically active transitional population that parallels the transitory intermediate cells that arise during LCMV infection.
TMEM-to-TEFF cell differentiation in the spleen coincides with white pulp-to-red pulp migration
In line with their divergent gene expression patterns, the three GRA6-specific T cell populations also expressed distinct levels of many genes important for localization within the spleen, including Ccr7, Cd69, Xcl1, S1pr1, and S1pr5 (Figures 1E and 1F). We hypothesized that this may result in differences in the location of these cells in the spleen, which, in turn, may control to which signals these cells are exposed in vivo. To investigate this, we generated partial hematopoietic chimeras in which a small percentage of T cells expressed rearranged T cell receptor (TCR) α and β transgenes specific for the GRA6 peptide/H2-Ld complex, referred to as TG6 (Chu et al., 2016), along with a GFP transgene (Figure S2). We infected adult TG6-GFP chimeric mice with T. gondii, and 6 weeks after infection, the spleens of these mice were analyzed by confocal microscopy (Figure 2). The majority (80%) of TG6-GFP cells were found in the red pulp of the spleen, whereas a smaller fraction (20%) resided in the white pulp (Figures 2A–2D). To identify TMEM cells, we stained sections with antibodies specific for TCF1, a transcription factor associated with stem-like T cells in other chronic infections (Im et al., 2016; Utzschneider et al., 2016; Welten et al., 2020) whose expression at the RNA and protein levels correlated well with TMEM cells, as defined by CXCR3 and KLRG1 expression (Figures 1E and 1F). We observed that the majority of TCF1+ (TMEM) cells (72%) were localized to the white pulp of the spleen, specifically in the T cell zone (Figures 2A and 2D). Proliferating Ki67+ cells (enriched for TINT cells) were located primarily in the red pulp (81%) (Figures 2B and 2D), and KLRG1+ cells (marking TINT and TEFF cells) were localized almost entirely (99%) in the red pulp (Figures 2C and 2D). To confirm these results and extend our conclusions to a polyclonal T cell population, we utilized intravenous (i.v.) anti-CD8 antibody labeling to measure the blood exposure of each GRA6 tetramer-positive population in the spleens of wild-type infected mice (Anderson et al., 2014). Cells in the marginal zone/bridging channels and red pulp are more blood exposed than cells in the white pulp, allowing us to infer the localization of these cells based on the relative amount of antibody labeling. TMEM cells showed the lowest proportion of i.v. antibody labeling, with TINT and TEFF cells showing significantly higher labeling (Figure 2E). This suggests that, similar to TCR transgenic cells, polyclonal TMEM cells reside in the white pulp, whereas TINT and TEFF cells reside in the red pulp. The transcriptional profiles of polyclonal TINT and TEFF cells showed a specific enrichment for heme metabolism (Figure 1D), further supporting the notion that these cells reside within the blood-exposed compartments of the spleen.
Figure 2. TMEM, TINT, and TEFF cells reside in distinct areas of the spleen.
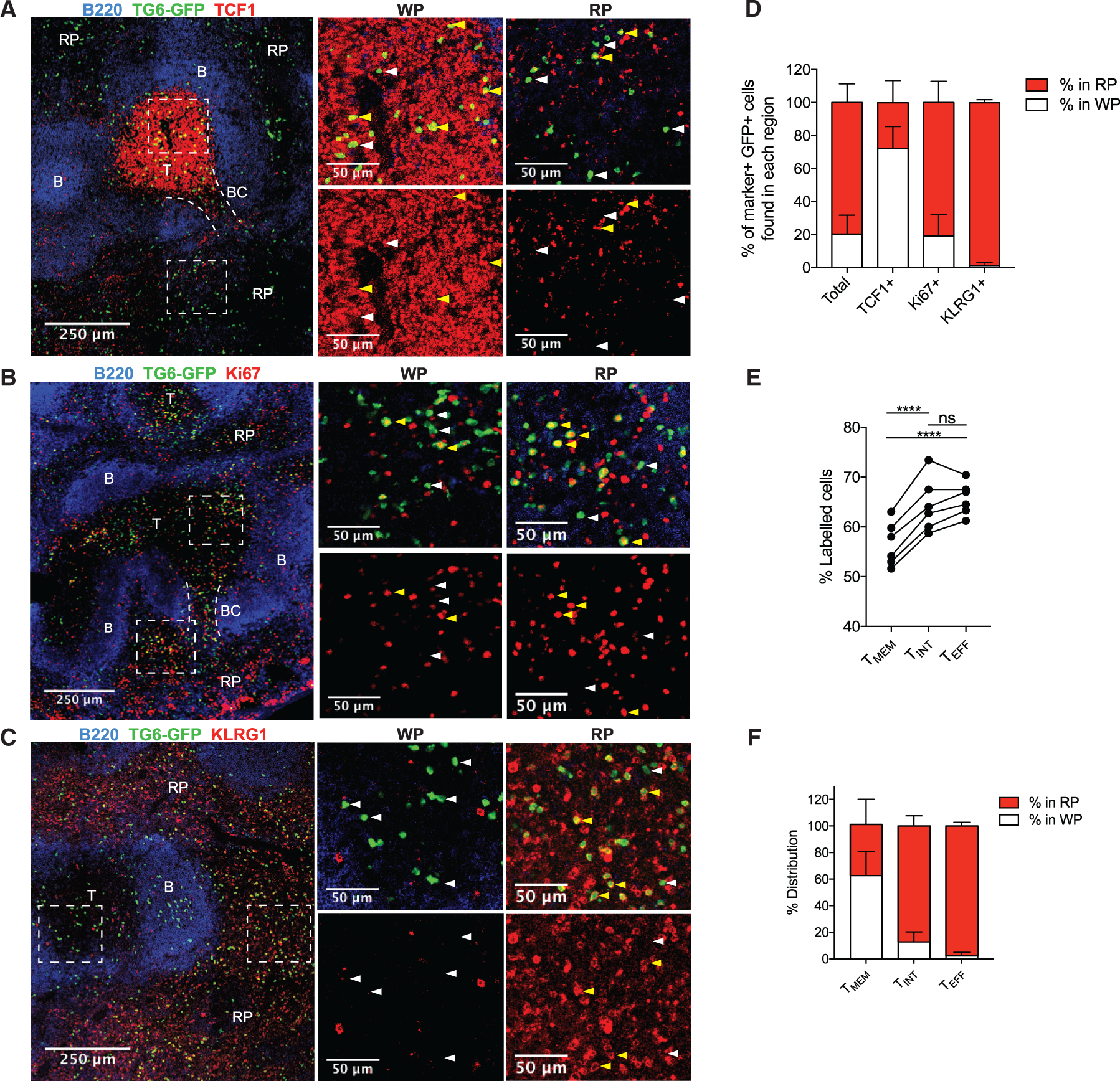
(A–C) TG6-GFP chimeric mice infected with T. gondii and their spleens were analyzed by confocal microscopy after 6 weeks. Sections were stained with antibodies against TCF1 (A), Ki67 (B), and KLRG1 (C) to identify the location of TMEM, T, and TINT/TEFF TG6-GFP cells, respectively. B220 staining was used to identify B cell zones (B) and T cell zones (T) to map the white pulp (WP) and red pulp (RP) regions. In some images, bridging channels (BCs) are marked with dotted lines. Dashed rectangles indicate the area in the magnified regions on the right. Examples of cells staining positively with the indicated marker are highlighted with yellow arrows and negative examples with white arrows.
(D) Data summarizing the location of TG6-GFP cells that stained positively for each protein.
(E) Data showing the proportion of each GRA6-specific population labeled after i.v. anti-CD8 antibody injection.
(F) Summary data showing the location of TMEM, TINT, and TEFF cells in the spleen after adoptive transfer.
Images in (A)–(C) and data in (D) are representative of images taken from three or more separate mice. Data in (E) are taken from two separate infections and are representative of data from three separate experiments. Data in (F) are summarized from multiple recipient mice across three infections (TMEM cells, n = 3; TINT cells, n = 3; TEFF cells, n = 4).
As an alternative approach to determine the localization of GRA6-specific subsets, we utilized an adoptive transfer method (Figure 2F). TMEM, TINT, and TEFF cells from chronically infected TG6-GFP neonatal chimeric mice were sorted based on CXCR3 and KLRG1 expression, adoptively transferred into separate infection-matched wild-type (WT) recipients, and analyzed 48 h later. Flow cytometry analysis confirmed that the transferred cells retained their CXCR3 and KLRG1 phenotypes at this time point, consistent with previous findings (Chu et al., 2016). We observed that a large fraction of TMEM cells were found within the T cell zone, whereas TINT cells and TEFF cells were found in the red pulp (Figures 2F and S3). These data indicate that TMEM cells have specific access and residency in a niche within the splenic white pulp and that migration into the red pulp coincides with differentiation into TINT and TEFF subsets.
CXCR3-deficient CD8+ T cells exhibit an inflated TMEM pool and impaired TMEM-to-TINT transition
CXCR3 is a chemokine receptor expressed by populations of antigen-experienced CD8+ T cells, including TMEM and TINT cells, and has been implicated in driving T cells into the splenic red pulp and toward an effector fate in settings of early acute infection and vaccination (Hu et al., 2011; Kohlmeier et al., 2011; Kurachi et al., 2011; Shah et al., 2015). To interrogate the role of CXCR3 during chronic T. gondii infection, we co-transferred equal numbers of WT and CXCR3 knockout (KO) naive TG6 CD8+ T cells and tracked the two populations at different time points after infection (Figure 3A). By the peak of the acute phase of infection (2 weeks), both populations had robustly expanded, consistent with previous reports that loss of CXCR3 does not impair T cell priming (Kurachi et al., 2011; Figure 3B). However, CXCR3 KO T cells outnumbered WT cells in the spleen 3 weeks after infection, and by 6 weeks after infection, the CXCR3 KO cells were completely dominant, with many mice showing no detectable WT populations (Figures 3B–3D). To understand the competitive advantage of the KO population, we investigated the phenotype of the WT and KO cells. Because of the absence of CXCR3 as a surface marker on these cells, we utilized Ki67 and KLRG1 staining to identify TMEM (KLRG1−), TINT (Ki67+, KLRG1+), and TEFF cells (Ki67−, KLRG1+) (Chu et al., 2016). Compared with the WT cells, CXCR3 KO cells showed a significant increase in the proportion and number of TMEM cells 3 weeks and 6 weeks after infection (Figures 3E–3G). In addition, the ratio of TMEM to TINT cells was increased in the CXCR3 KO population, raising the possibility that CXCR3 KO TMEM cells were less efficient at differentiating into the TINT stage. Despite this, CXCR3 KO cells generated similar or greater absolute numbers of total TINT and TEFF cells compared with the WT, likely because of the inflated precursor pool of TMEM cells (Figure 3G).
Figure 3. CXCR3-deficient CD8+ T cells develop an inflated TMEM cell pool.
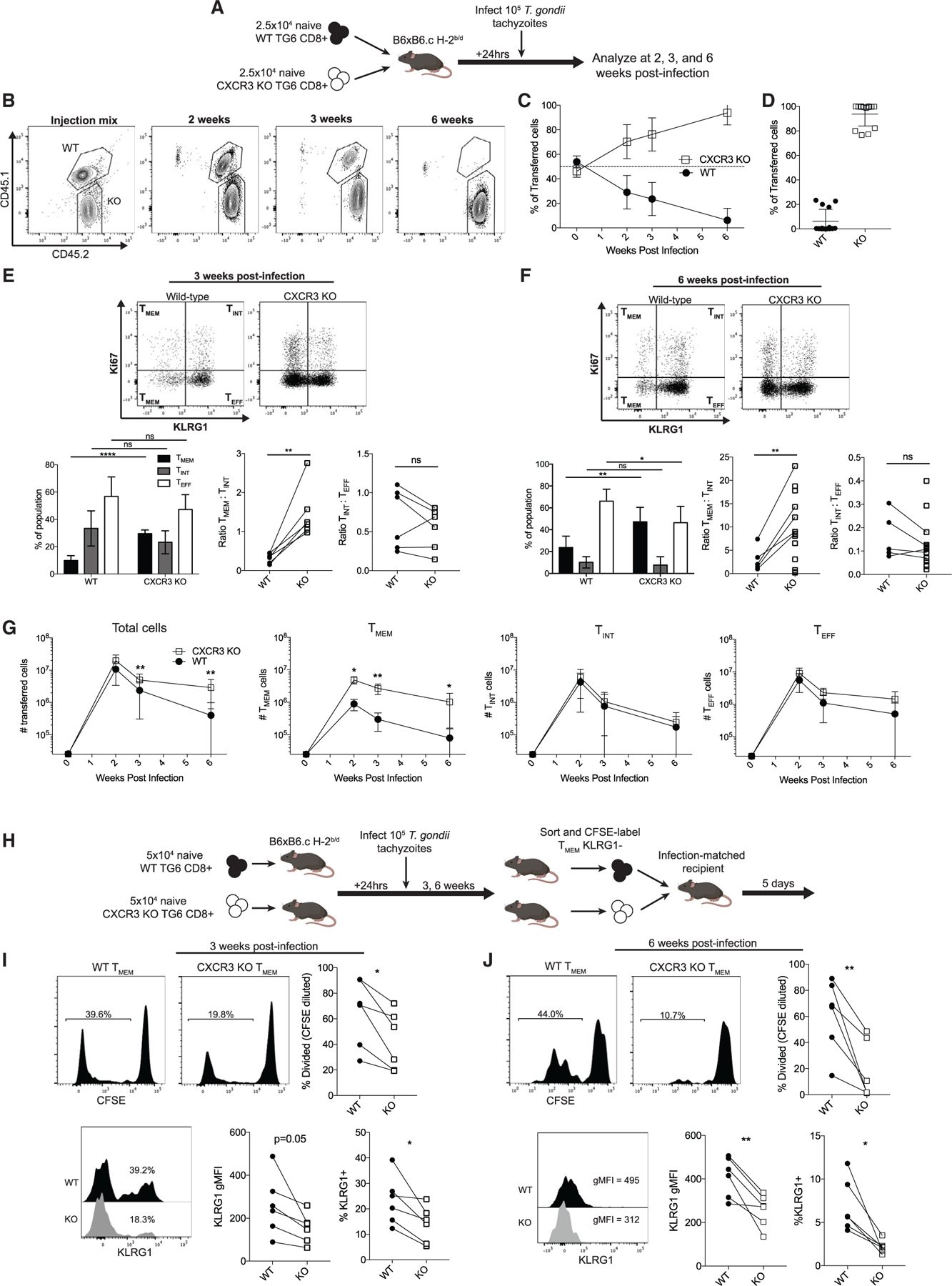
(A) Experimental design.
(B) Representative flow cytometry plots of gated GRA6 tetramer+ CD8+ splenocytes at the indicated time points.
(C) Summary data showing the proportion of the transferred cells belonging to the WT or CXCR3 KO population.
(D) Individual data points for the time point 6 weeks after infection in (C).
(E and F) The differentiation profile of WT and CXCR3 KO TG6 cells 3 weeks (E) or 6 weeks (F) after infection. TMEM, TINT, and TEFF cell subsets were identified using Ki67 and KLRG1 staining. Bar graphs display the proportion of the transferred population belonging to each subset. Adjacent plots display the ratios of TMEM:TINT and TINT:TEFF cells in the WT and KO populations. Lines connect populations from the same mouse.
(G) Absolute numbers of cells belonging to each population.
(H) Experimental design.
(I and J) Representative flow plots and summary data for CFSE dilution and KLRG1 expression within the population of transferred WT and KO TMEM cells 3 weeks (I) and 6 weeks (J) after infection.
Data in (A)–(G) are combined from three independent experiments. Data in (H)–(J) are combined from two independent experiments.
To test whether loss of CXCR3 leads to a block of TMEM-to-TINT cell differentiation, we performed adoptive transfer experiments. We sorted WT and CXCR3 KO TMEM cells from infected mice and co-transferred both populations into infection-matched recipients 3 or 6 weeks after infection. We analyzed the donor population by flow cytometry 5 days after transfer (Figure 3H), a time point when transferred TMEM cells give rise to TINT cells but do not yet produce a large pool of TEFF cells (Chu et al., 2016). We measured the conversion of TMEM cells into TINT cells using KLRG1 expression and proliferation (measured by dye dilution), two hallmarks of TINT cell fate. After adoptive transfer, the WT TMEM cell population produced a larger KLRG1+ population and divided significantly more than the KO population, demonstrating that they were more efficient at producing TINT cells (Figures 3I and 3J). These results indicate that loss of CXCR3 impairs differentiation of TMEM cells into TINT cells, leading to an expanded TMEM cell population.
CXCR3 KO cells migrate to the red pulp while retaining a TCF1+ TMEM cell phenotype
Given the role of CXCR3 in promoting TMEM cell differentiation, and given our observation that the differentiation of TMEM into TINT cells correlates with their migration from the white pulp into the red pulp, we predicted that cells lacking CXCR3 would be “trapped” in the white pulp niche, similar to the observed localization of CXCR3-deficient CD8+ T cells during the early stages of acute infection (Kurachi et al., 2011). To test this, we performed confocal imaging on spleen sections from chronically infected TG6-GFP CXCR3 KO chimeric mice. Surprisingly, we observed no global defect in red pulp migration in CXCR3 KO cells (Figures 4A–4D). Compared with the WT chimeras, the KO cells had a similar distribution in the spleen, with approximately 80% of the total GFP population found in the red pulp and 20% in the white pulp.
Figure 4. CXCR3-deficient CD8+ T cells retain a memory phenotype in the RP.
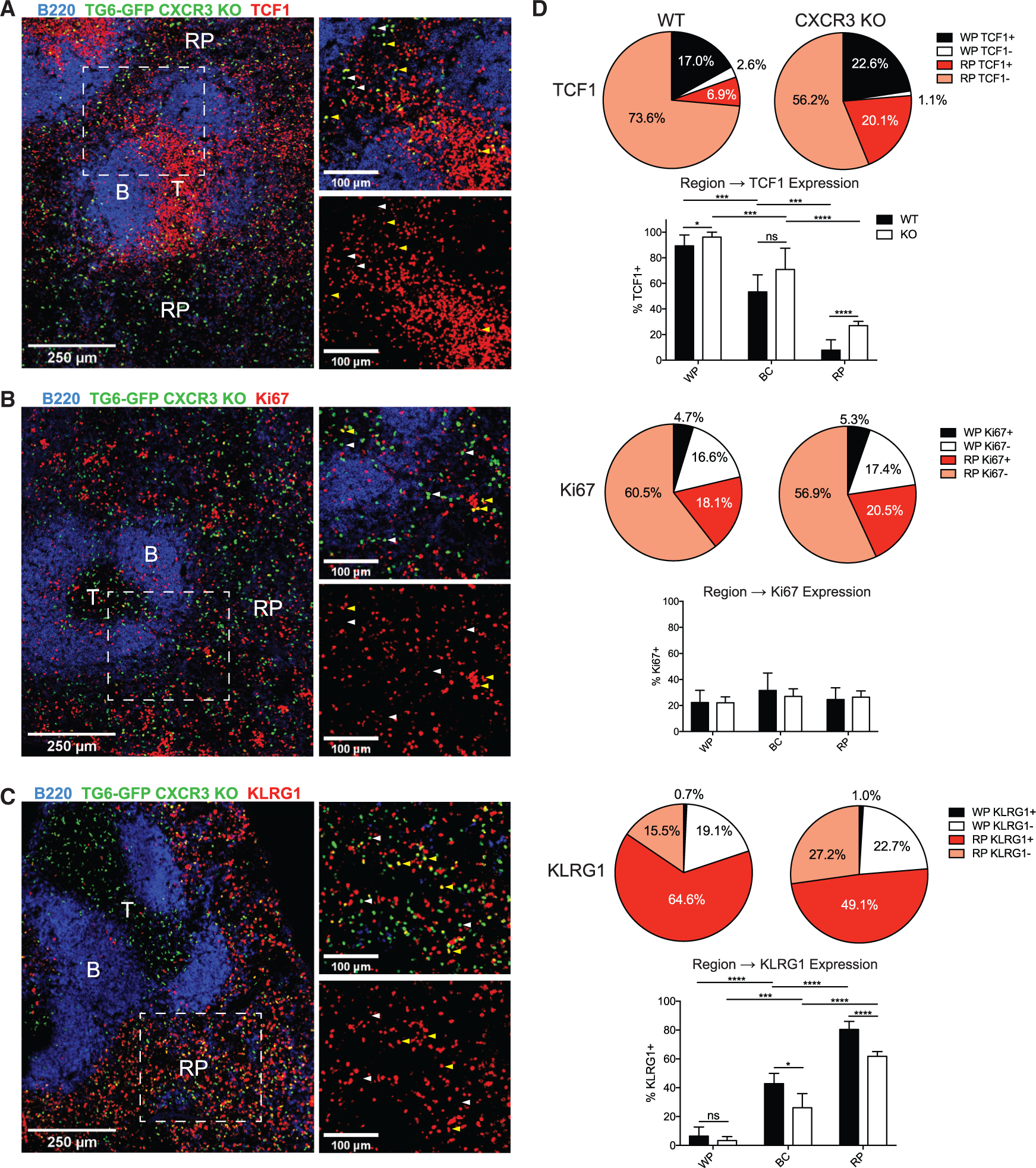
(A–C) CXCR3 KO TG6-GFP neonatal chimeras were infected with T. gondii and analyzed by confocal microscopy after 6 weeks. Sections were stained with antibodies against TCF1 (A), Ki67 (B), and KLRG1 (C). B220 staining was used to identify B cell zones (B) to map the T cell zone (T) and RP regions. Yellow arrows highlight examples of cells staining positively for the specified marker; white arrows highlight examples of negative staining.
(D) Summary data showing the proportion of GFP cells found in the WP or RP that are identified as positive or negative for expression of the indicated marker. Bar graphs display the proportion of WT or CXCR3 KO GFP cells expressing the protein of interest (TCF1, Ki67, or KLRG1) within the WP, BC, or RP.
Data for WT cells are combined from at least three mice across two different experiments, and data for KO cells are combined from four mice taken across two experiments.
Next we investigated the effect of CXCR3 deletion on the location of CD8+ subsets defined by expression of TCF1, Ki67, and KLRG1, as shown in Figure 2. Similar to WT mice, we observed that GFP+ cells in the white pulp retained a uniformly TCF1+ (TMEM) phenotype (Figure 4A). However, contrary to our initial expectations, we observed a large increase in the proportion of TCF1+ cells in the red pulp. This indicated that TMEM cells lacking CXCR3 were able to efficiently migrate into the red pulp but no longer uniformly converted into TINT cells during that transition. The presence of a large population of red pulp TMEM cells was also indicated by KLRG1 staining; a large fraction of red-pulp-resident TG6-GFP CXCR3 KO T cells were KLRG1 negative (Figure 4C). Ki67+ cells remained primarily localized to the red pulp and did not show a significant change between the WT and KO populations (Figures 4B and 4D). These data indicate that, in the absence of CXCR3, TMEM cells still migrate normally into the red pulp. However, although migration of CXCR3-sufficient T cells to the red pulp is accompanied by their transition into a TCF1− KLRG1+ population, CXCR3 KO T cells retain a TCF1+ KLRG1− phenotype after migrating to the red pulp.
Bridging channels are splenic structures that connect the T cell zone to the red pulp and are an important migratory pathway for T cells in the spleen (Khanna et al., 2007; Chauveau et al., 2020). We analyzed the phenotype of GFP+ cells occupying bridging channels and observed that the proportion of cells expressing TCF1 and KLRG1 was intermediate compared with the white pulp and red pulp, suggesting that migration along bridging channels to the red pulp coincided with conversion of TMEM cells into a TINT phenotype (Figures 4D and S4). CXCR3 KO cells in the bridging channels also had a mixed phenotype but with a slightly higher percentage of TCF1+ cells and a lower percentage of KLRG1+ cells compared with WT cells (Figure 4D), although these differences were not as pronounced as in the red pulp. These data suggest that bridging channels may be critical sites for the CXCR3-dependent transition of TMEM cells into TINT cells.
cDCs are an important source of CXCR3 ligands
The observation that CXCR3 plays a critical role in differentiation of CD8+ T cells but has no obvious role in their migration within the spleen suggests that CXCR3 may regulate localized interactions between T cells and other cell types. To characterize expression of the CXCR3 ligands CXCL9 and CXCL10, we utilized the REX3 strain of dual reporter mice, which contains a transgenic construct with coding regions for red fluorescent protein (RFP) and blue fluorescent protein (BFP) linked to the CXCL9and CXCL10 genes, respectively (Groom et al., 2012). Uninfected REX3 mice displayed a small amount of basal CXCL9 and CXCL10 production in the spleen, but the proportion and numbers of chemokine-producing splenocytes substantially increased during acute infection and remained elevated during chronic infection (Figures 5A–5C). To identify the cell types producing CXCL9 and CXCL10 during infection, we stained splenocytes isolated from chronically infected REX3 mice with surface markers to delineate myeloid and lymphoid immune cell types (Figures 5D and 5E). Although CD11c+ cells comprised only a small fraction of the total splenocyte population, they made up a majority of the cells within the CXCL9+ and CXCL10+ subsets. CXCL9+ CXCL10− cells and CXCL9+ CXCL10+ cells were almost entirely CD11c+. The CXCL9− CXCL10+ fraction consisted predominantly of lymphoid cells, including CD4+ T cells, and also contained a substantial portion of CD11c+ cells. CD11c is a marker expressed primarily by DCs, which can be divided into populations of cDCs (cDC-1s and cDC-2s), monocyte-derived DCs (mDCs), and plasmacytoid DCs (pDCs). We stained for markers to identify these different DC subsets in splenocytes from chronically infected REX3 mice (Figure S5). We found that cDC-1s produced high levels of CXCL9 and moderate levels of CXCL10, with very little change in the proportion or mean fluorescence of CXCL9 or CXCL10 after infection (Figure 5F). mDCs expressed some CXCL9 and CXCL10 after infection, whereas pDCs expressed very little of either chemokine. Interestingly, cDC-2s made no CXCL9 or CXCL10 in uninfected mice but became prominent producers of both chemokines during infection. In addition, cDC-2s upregulated FcεR1 (MAR-1) upon infection, indicative of an inflammatory phenotype (Bosteels et al., 2020), and expressed high levels of CD80 (B7–1) and PDL1 before and after infection (Figure S5). These results indicate that cDCs are an important source of CXCR3 ligands in the spleen and that cDC-2s with an inflammatory phenotype show the greatest infection-induced increase in CXCR3 ligand expression.
Figure 5. DCs are a significant source of CXCR3 ligands during chronic infection.
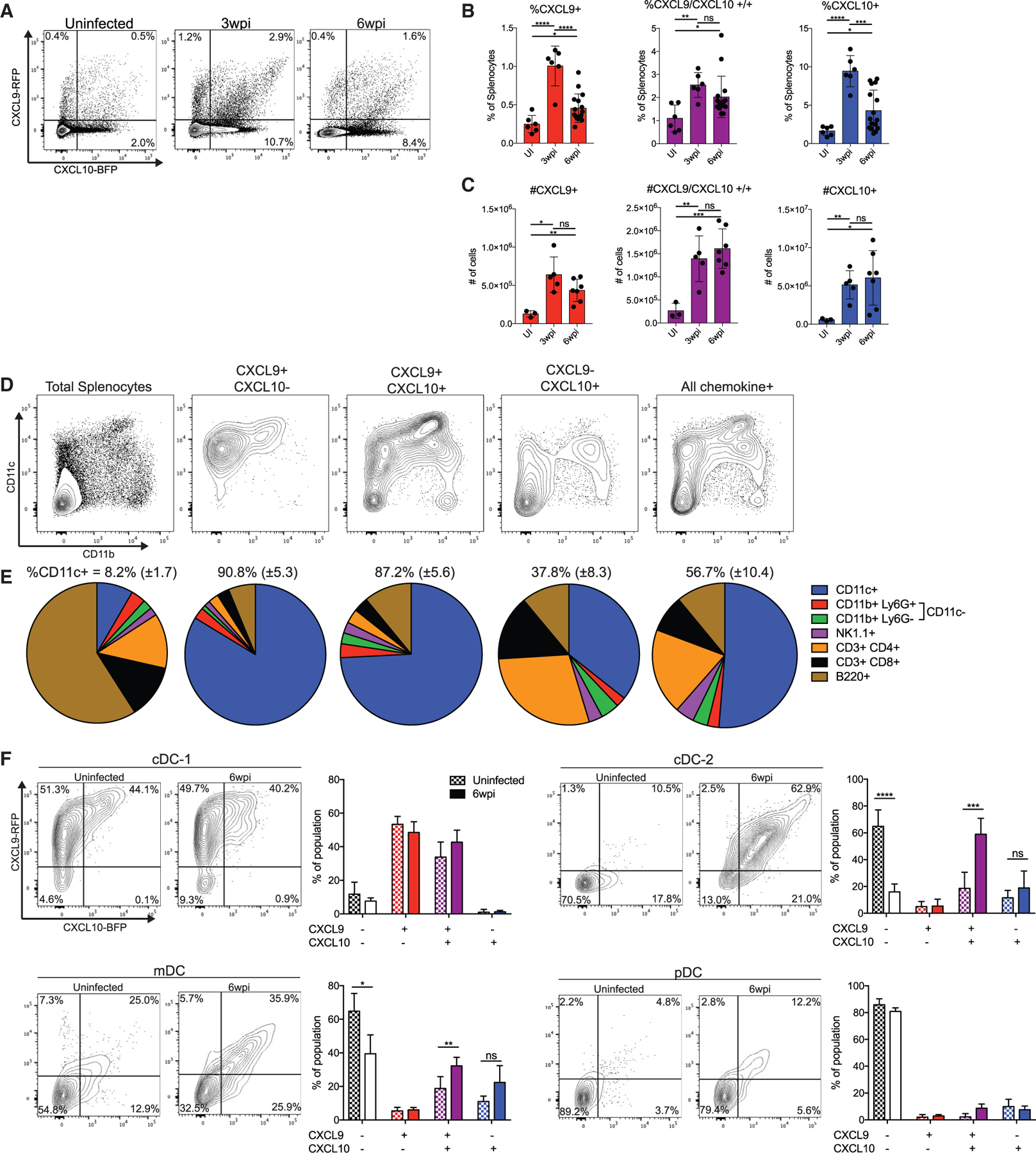
(A) Representative flow cytometry plots of chemokine expression in REX3 spleens before infection, 3 weeks after infection, and 6 weeks after infection.
(B and C) Summary data of the percentages (B) and absolute numbers (C) of chemokine-producing splenocytes during infection.
(D and E) Splenocytes from REX3 mice were stained with the indicated surface markers. Cells were then gated on chemokine expression, and the phenotype of different chemokine-expressing subsets was measured. Shown are (D) representative plots of CD11c × CD11b staining and (E) summary data. The mean and standard deviation of the percent of CD11c+ cells in each population is displayed above each chart.
(F) Subsets of DCs were identified (Figure S6), and their chemokine expression was analyzed in uninfected mice and 6 weeks after infection.
Data in (A)–(E) are pooled from three separate experiments. Data in (F) are combined from two separate experiments (n = 3 mice for uninfected, n = 8 mice for 6 weeks post infection).
GRA6-specific CD8+ T cells form CXCR3-dependent interactions with cDC-2s in the bridging channels
T cell:DC interactions are critical for regulating the activation and differentiation of CD8+ T cells, and CXCR3 can promote more frequent and long-lasting interactions between these two cell types (Groom et al., 2012). Because cDCs are significant producers of CXCR3 ligands in vivo during chronic infection, we sought to characterize interactions between TMEM cells and chemokine-producing DCs in the spleen. We imaged spleen sections of chronically infected REX3 mice to determine the location of chemokine-producing cells during chronic infection (Figure 6A). CXCL9 expression was observed throughout the spleen, with a slightly higher signal localized to the marginal zone and red pulp (Figures 6A and 6B). Much of the CXCL9 expression was co-localized with CXCL10, although a population of CXCL9 single-positive cells was evident primarily in the T cell zone. CXCL10 was not evident in the white pulp but was highly expressed in the red pulp, with a maximum signal in the marginal zone (Figure 6B). Marginal-zone CXCL10 appeared to primarily come from clusters of CXCL9+ CXCL10+ cells.
Figure 6. CXCL9/10+ DCs occupy distinct regions in the spleen.
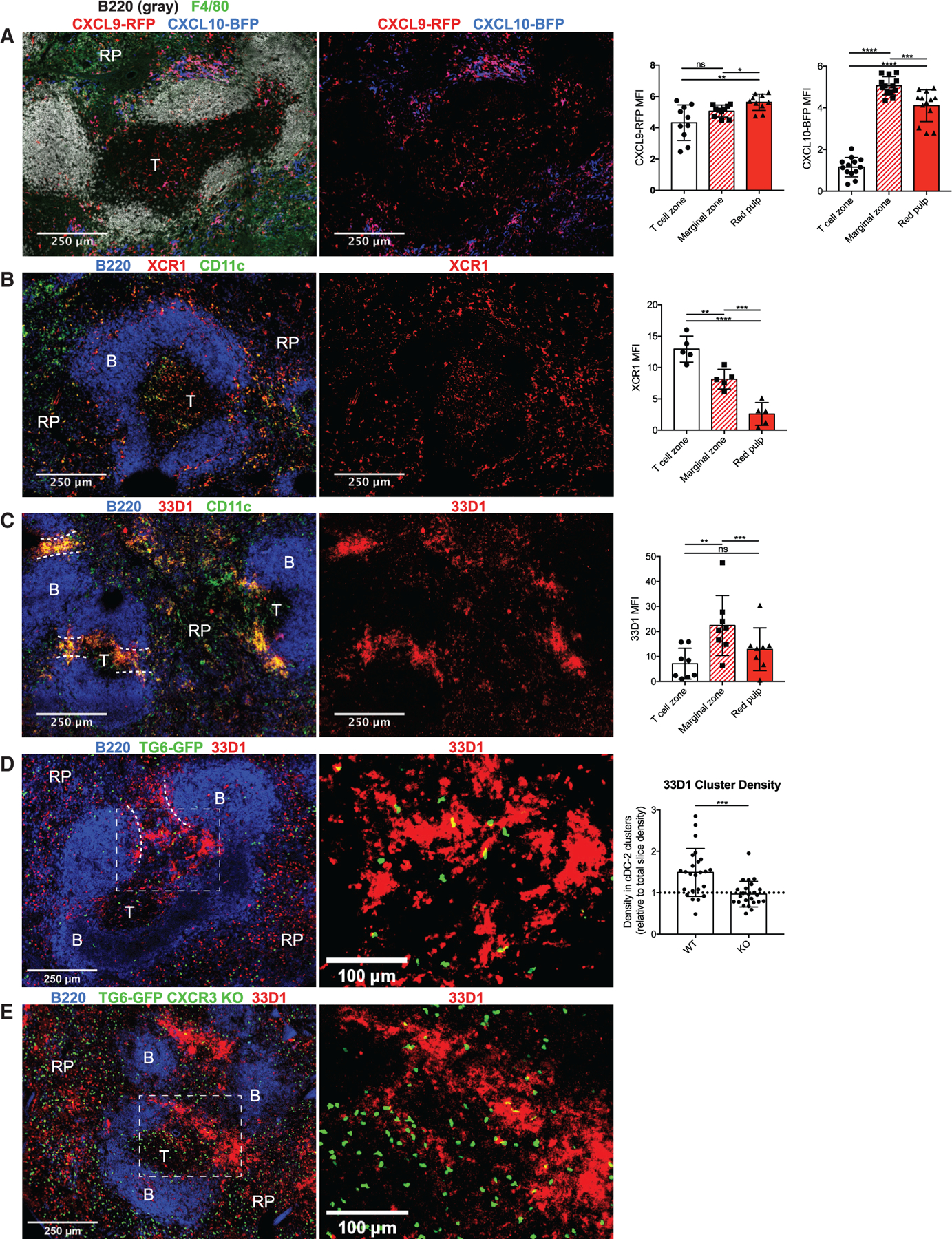
(A) Spleens from REX3 mice were imaged 6 weeks after infection with T. gondii. Sections were stained with F4/80 to highlight the RP and B220 to highlight B cell follicles. The mean fluorescence intensity was measured for CXCL9-RFP and CXCL10-BFP in each splenic region. Data were normalized to the B cell zone to adjust for background fluorescence.
(B and C) Spleens from WT mice infected for 6 weeks were stained with XCR1 (B) or 33D1 (C) together with CD11c to identify cDC-1s and cDC-2s, respectively. Summary data show mean fluorescence for the indicated marker in each splenic region. Data were normalized to the B cell follicle as in (A).
(D and E) WT (D) and CXCR3 KO (E) TG6-GFP neonatal chimeras were stained with 33D1 to identify cDC-2 clusters. The density of GFP cells within cDC-2 clusters was then measured and normalized to the total density of GFP cells in the slice.
Data in (A) are representative of three mice, and data in (B)–(E) are representative of two mice.
The distinct pattern of chemokine expression observed by flow cytometry (Figure 5F) combined with the localization pattern of chemokines suggested that cDC-1s and cDC-2s occupy distinct splenic niches during chronic T. gondii infection. To more specifically localize each population of DCs, we stained chronically infected mice with antibodies against XCR1 (Figure 6B) and DCIR2 (clone 33D1; Figure 6C) together with CD11c to identify the location of cDC-1s and cDC-2s, respectively. XCR1+ CD11c+ cells (cDC-1s) were found scattered throughout the spleen, with maximal XCR1 signal coming from the white pulp but considerable fluorescence in the marginal zone and red pulp as well (Figure 6B). This is consistent with previous reports suggesting that splenic cDC-1s patrol the marginal zone and red pulp before migrating into the T cell zone to present blood-derived antigens (Backer et al., 2019; Eisenbarth, 2019). In contrast, 33D1+ CD11c+ cells (cDC-2s) were enriched in dense clusters in the bridging channels and marginal zones, two connected splenic compartments that serve as transition regions between the white and red pulp (Figure 6C). Flow cytometry analysis of REX3 reporter mice shows that most XCR1+CD11c+ cDC-1s in chronically infected mice express CXCL9, whereas most 33D1+CD11c+ cDC-2s express CXCL10, and a substantial fraction express CXCL9 (Figure S5). These results suggest that TMEM cells primarily encounter CXCL9-expressing cDC-1s in the white pulp but encounter CXCL9- and CXCL10-expressing cDC-2 networks during their migration through bridging channels.
The localization of CXCL10-expressing cDC-2 clusters at the bridging channels made them a particularly interesting population because they co-localized with the CXCR3-dependent conversion of TMEM cells into TINT cells. To investigate whether CD8+ T cells formed CXCR3-dependent interactions with cDC-2s, we quantified the occupancy of TG6-GFP cells in 33D1+ clusters in the spleens of chronically infected mice. We measured the density of WT and CXCR3 KO TG6-GFP cells in 33D1+ clusters and normalized to the density of cells in the total slice to account for the overall increase in the numbers of CXCR3 KO TG6-GFP cells. We observed that WT TG6-GFP cells were enriched in 33D1+ clusters, whereas CXCR3 KO cells showed no enrichment (Figures 6D and 6E), demonstrating that CD8+ T cells experience CXCR3-dependent interactions with cDC-2s in the bridging channels.
cDC-2s confer a CD25hi phenotype on TMEM cells in vitro
Evidence showing that CXCR3 mediates the interaction of CD8+ T cells with cDC-2s and also affects their differentiation raises the possibility that CXCR3-dependent interactions with cDC-2s may directly modulate effector differentiation. To explore this possibility, we devised an ex vivo co-culture system to investigate how different DC populations from infected mice affect TMEM differentiation and the contribution of CXCR3 to this effect. WT or CXCR3 KO TMEM cells were sorted from chronically infected mice. In parallel, DC subsets (cDC-1s, cDC-2s, or mDCs) were isolated from infection-matched mice and loaded with the GRA6 peptide (Figure S6). TMEM cells and peptide-loaded DCs were co-cultured in vitro for 3 days, and the activation of the TMEM cells was measured by dilution of a proliferation dye (carboxyfluorescein succinimidyl ester: CFSE) and expression of surface markers. To maximize the cells’ ability to migrate and adhere, cells were plated in flat-bottom wells at a low density. As expected, all three subsets of DCs were capable of inducing robust proliferation of WT TMEM cells, but cDC-2s induced slightly less division compared with cDC-1s and mDCs (Figures 7B and 7C). Although we did not observe KLRG1 expression in any of the conditions tested (data not shown), we observed significant differences in the ability of the DCs to induce CD25 expression in TMEM cells. cDC-1s and mDCs produced modest CD25 expression among the CFSElo population (Figure 7B). In contrast, cells stimulated by cDC-2s were almost uniformly CD25+ and had higher CD25 geometic mean fluorescence intensity (gMFI). Increased CD25 expression and interleukin-2 (IL-2) signaling can bias CD8+ T cells towards effector differentiation (Kalia et al., 2010; Pipkin et al., 2010). These data demonstrate that cDC-2s can provide a distinct stimulation to TMEM cells in vitro compared with other DC subsets and suggests that cDC-2s may be more effective at promoting effector differentiation compared with cDC-1s.
Figure 7. DC populations have distinct capacities to stimulate TMEM cells in vitro.
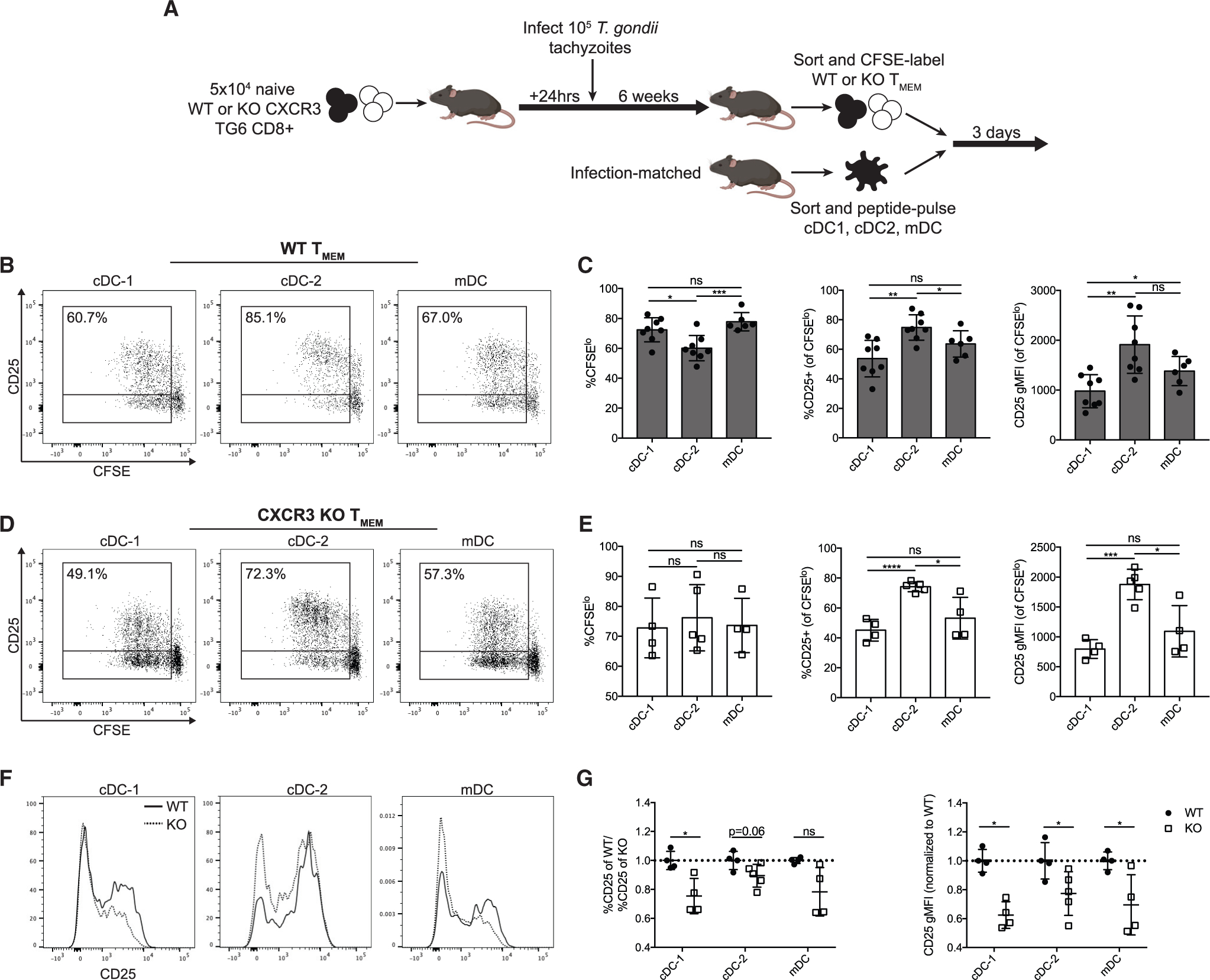
(A) Co-culture experiment layout.
(B and C) Representative flow plots and compiled data showing CD25 expression and CFSE dilution of WT TMEM cells after 3 days of culture with the indicated DCs. Percentages represent the portion of CFSElo cells expressing CD25.
(D and E) Representative flow cytometry plots and compiled data from CXCR3 KO TMEM cells after 3 days of culture with the indicated DCs.
(F and G) Representative histograms and compiled data of CD25 expression on WT and CXCR3 KO TMEM cells cultured with the indicated DCs. Data are normalized to the WT TMEM cells within each experiment.
Data in (C) are from three independent experiments and in (E) and (G) from two independent experiments.
CXCR3 KO TMEM cells showed similar trends after activation with all three DC subsets, with cDC-2s inducing the highest levels of CD25 within the population of divided cells (Figures 7D and 7E). Interestingly, we observed that CXCR3 KO TMEM cells had reduced expression of CD25 compared with WT TMEM cells after stimulation with all three DC subsets (Figures 7F and 7G). This suggests that CXCR3 KO cells experience altered DC interaction and are thus less capable of adopting a CD25hi effector phenotype. Additionally, we observed that WT TMEM cells treated with anti-CXCL9 or anti-CXCR3 (clone CXCR3–173, which specifically blocks the activity of CXCL10 binding) showed reduced activation by cDC-2s regarding their ability to divide and express CD25 (Figure S6). These results indicate that loss of CXCR3 results in altered stimulation of TMEM cells by DCs in vitro, resulting in a bias towards a CD25lo phenotype.
DISCUSSION
Generation of a protective CD8 T cell response during chronic infection requires a long-term pool of stem-like memory cells to drive continuous production of short-lived TEFF cells, but the signals that balance effector differentiation with memory preservation remain unclear. Here we provide evidence that CXCR3-dependent cDC-2 interactions in bridging channels promote TMEM cell differentiation into a transcriptionally distinct, proliferative TINT cell population that expands before eventually giving rise to armed TEFF cells. Our data reveal how transient interactions with cDC-2 populations during migration between TMEM and TEFF cell niches in the spleen can deliver balanced differentiation signals that allow efficient TEFF production and robust memory preservation.
Although early studies of CXCR3 focused on its role in driving entry of activated T cells into inflamed tissues, more recent studies have highlighted its role in local T cell positioning and effector differentiation within lymph nodes and the spleen (Kohlmeier et al., 2011; Kurachi et al., 2011; Sung et al., 2012; Shah et al., 2015; Maurice et al., 2019). CXCR3 ligands are induced by inflammatory signals such as IL-12 and type I interferons (IFNs) (Groom and Luster, 2011), leading to accumulation and effector differentiation of antigen-experienced CD8+ T cells near sources of CXCR3 ligand induction in the splenic red pulp and marginal zone (Kurachi et al., 2011; Shah et al., 2015). However, in mice chronically infected with T. gondii, CXCR3 does not mediate CD8+ T cell migration to the red pulp, nor does it lead directly to terminal effector differentiation. Instead, the CXCR3:CXCL9/10 axis appears to exert a transient and localized differentiation signal by promoting interactions with DCs, similar to its proposed function in lymph nodes (Groom et al., 2012; Duckworth et al., 2021) and tumors (Chow et al., 2019). Given that CXCR3 ligands are regulated by inflammatory signals generated in response to infection, this system may allow dynamic modulation of effector cell production during chronic infections so that transient increases in pathogen burden are met with transient increases in effector T cell production.
T cell migration to the red pulp still occurs in the absence of CXCR3, implying that other signals must regulate this migration step. CCR7 and S1P are known regulators of lymphocyte migration in and out of the white pulp, respectively (Förster et al., 1999; Arnon and Cyster, 2014), and our transcriptional analyses implicate these pathways in migration of GRA6-specific CD8+ T cells. TMEM cells had little expression of the genes encoding the S1P receptors S1pr1 and S1pr5 but elevated expression of Ccr7 as well as Cd69, which can oppose the action of S1P (Mackay et al., 2015). This expression pattern allows entry and occupancy of the white pulp while resisting exit signals. Downregulation of Ccr7 and Cd69 together with the upregulation of S1pr1 and S1pr5 observed in TINT and TEFF cells may initiate white pulp-to-red pulp migration. CXCR3 ligands (especially CXCL10, which has a higher binding affinity for CXCR3 than CXCL9; Weng et al., 1998) may enhance migration as well as promote transient interactions with cDC-2s as T cells pass through the bridging channels. Interestingly, loss of CXCR3 on T cells uncouples differentiation and red pulp migration, further supporting the notion that differentiation-inducing signals come primarily from bridging channels/marginal zones rather than the red pulp. Although our data indicate that defective differentiation during white pulp-to-red pulp migration contributes to accumulation of CXCR3 KO TMEM cells in the red pulp, it is interesting to consider that changes in the composition of cells in the blood may also further enhance this phenotype through recirculation.
The localization of WT TMEM cells in the white pulp raises the following question: what aspects of the white pulp may support a stem-like fate? One defining feature of the white pulp niche is elevated TGF-β expression, which promotes memory fate during acute and chronic viral infection (Seo et al., 2016; Gabriel et al., 2021). Our transcriptional data revealed a drop in TGF-β signaling in parallel with an increase in mechanistic target of rapamycin (mTOR) activity at the TINT stage. This is in line with recent data from LCMV chronic infection showing that TGF-β signaling suppresses mTOR activity to preserve the quiescent stem-like memory population and that ablation of TGF-β signaling results in an increase in transitory intermediate cells (Gabriel et al., 2021). These observations imply that T cell migration out of the white pulp relieves the metabolic constrains imposed by TGF-β, allowing T cells to transition into a proliferative intermediate stage. TMEM cells in the white pulp are also exposed to cDC-1s, and we showed that stimulation by cDC-1s in vitro resulted in the lowest CD25 expression on TMEM cells. Although CD25 expression and enhanced IL-2 signaling are associated with effector differentiation in many settings (Kalia et al., 2010; Pipkin et al., 2010), the mechanistic significance of CD25 expression and IL-2 signaling during chronic T. gondii infection requires further study. Nevertheless, our data suggests that cDC-1s may have an inherent capacity to promote memory fate, which may contribute to the TMEM niche. In support of this proposed function, recent evidence demonstrated that cDC-1s are critical for maintaining a reservoir of stem-like cells in tumor-draining lymph nodes (Schenkel et al. 2021). It is interesting to note that cDC-1s can bias CD4+ T cells towards the TREG lineage, which can be potent sources of TGF-β, whereas cDC-2s tend to induce an effector T helper 1 (TH1) fate (Brown et al., 2019; Opejin et al., 2020). Thus, cDC-1s and CD4+ T cells in the white pulp may collaborate to provide a TGF-β-rich environment supporting stem-like CD8+ T cells, in contrast to the tendency of DCs in the marginal zone and bridging channel (BC) to promote differentiation.
Although, initially, cDC-1s and cDC-2s were identified as having separate roles in priming CD8+ and CD4+ T cells, respectively (Den Haan and Bevan, 2002; Eisenbarth, 2019), recent studies have demonstrated that cDC-2s can acquire the ability to cross-present antigens on MHC class I and can influence CD8+ T cell activation (Calabro et al., 2016; Bosteels et al., 2020). In line with these data, we observed that cDC-2s during chronic T. gondii infection increased MAR-1 staining, a phenotype that coincides with the ability to cross-present antigen to CD8+ T cells (Bosteels et al., 2020). This raises the possibility that cDC-2s may influence CD8+ fate through direct antigenic stimulation. We observe that cDC-2s have a distinct costimulatory profile with elevated CD80 (B7–1) expression. CD80 binds to CD28 with higher avidity compared with CD86 (B7–2) (Sansom et al., 2003; Thomas et al., 2007), suggesting that cDC-2s may provide more intense co-stimulatory signals to T cells, promoting effector differentiation. Some studies have also implicated cell-intrinsic CXCR3 signaling in activating components of the TCR signaling cascade (Dar and Knechtle, 2007; Newton et al., 2009), suggesting that cDC-2s may directly contribute to T cell activation through this pathway as well. These features of cDC-2s agree with the results of our in vitro stimulations, where they were able to induce the highest expression of CD25 on stimulated TMEM cells. MAR-1+ cDC-2s have been reported to be potent IL-12 producers (Bosteels et al., 2020), and populations of cDC-2s express high levels of transcripts for other inflammatory cytokines, such as Il1B and Il27 (Brown et al., 2019), suggesting that cDC-2s may directly contribute to a cytokine environment that favors differentiation. Thus, the CXCR3 axis represents a tunable system whereby the immune response can tailor generation of effector cells by favoring interactions with cDC-1s or cDC-2s, depending on the inflammatory environment, analogous to what has been proposed to occur during acute viral infection (Duckworth et al., 2021).
Although cDC-2s are equipped to promote T effector differentiation, they also have high levels of PD-L1, suggesting that they may have differential effects on the fate of stem and proliferative populations, depending on the specific setting. We have shown previously that GRA6-specific CD8+ T cells are resistant to exhaustion and do not express PD-1, partially because of unusually low reactivity to self-peptide-MHC complexes (Tsitsiklis et al., 2020; Lutes et al., 2021). Thus, GRA6-specific cells are poised to receive activating signals from cDC-2s while remaining ignorant of the inhibitory signals provided by PD-1. In contrast, chronic infections that induce widespread T cell exhaustion, such as LCMV infection, result in a pool of stem-like cells that express high levels of PD-1 (Im et al., 2016; Utzschneider et al., 2016). These PD-1+ stem-like cells would likely simultaneously receive inhibitory and differentiation-inducing signals upon encountering DCs, leading to development of exhausted effector cells and reduced proliferation. Interestingly, PD-1 blockade during chronic LCMV infection results in a burst of intermediate proliferating cells emerging from the exhausted TCF1+ precursor pool (Im et al., 2016; Hudson et al., 2019), suggesting that this treatment might block the inhibitory signal while leaving the differentiation-inducing signal intact. Our data here indicate that CXCR3 is likely critical in driving this proliferative burst, analogous to the requirement for CXCR3 to achieve effective checkpoint blockade treatment in the tumor setting (Chow et al., 2019). Mouse infection with murine cytomegalovirus (MCMV), another model of chronic viral infection, results in a well-controlled infection characterized by ongoing antigen stimulation. This continuous antigenic stimulation of CD8+ T cells causes long-term expansion of virus-specific CD8+ T cells in the spleen, a process called “inflation memory.” Inflation memory CD8+ T cells in this model share many similarities with GRA6-specific CD8+ T cells, including a lack of functional exhaustion, the presence of a long-lived TCF1+ subset that localizes to the white pulp, and continuous generation of red-pulp-localized KLRG1+ proliferative intermediate cells (Gordon et al., 2018; Smith et al., 2014; Welten et al., 2020). Given these similarities, it seems likely that many of the dynamics observed here affect CD8+ fate during MCMV infection as well. Advancing our understanding of this system will be critical for improving therapeutic strategies for a wide range of chronic infections because stem-like cells and proliferative intermediate cells are critical for sustaining CD8+ responses to many chronic pathogens in mice and humans (Wieland et al., 2017; Gordon et al., 2018; Hudson et al., 2019; Kefalakes et al., 2019; Welten et al., 2020; Rutishauser et al., 2021).
Limitations of the study
The role of stromal cells
The analysis of CXCL9 and CXCL10 expression performed in this study focused on the hematopoietic compartment. Although our results indicate a critical role of DCs as CXCL9/10 sources, we cannot rule out that some effects of CXCR3 deletion are dependent on the stromal compartment.
Limitations of static imaging
Many of the conclusions in this study infer migratory patterns in the spleen based on static imaging data together with known T cell migration patterns determined from time-lapse imaging from earlier studies. Definite proof of these migratory patterns would require time-lapse live imaging of the spleens of infected mice.
In addition, we conclude that CXCR3 KO T cells experience reduced interactions with cDC-2s because of the absence of CXCR3-mediating signaling. However, the experimental setup used results in production of dramatically more CXCR3 KO T cells compared with the WT. This raises the alternative possibility that the reduced interactions observed between CXCR3 KO T cells and cDC-2s may also be due to increased competition for this niche with other T cells.
The role of CD25 in effector differentiation
We used ex vivo induction of CD25 by DCs as an indication of the propensity of DCs to induce a T effector cell fate. However, in vivo expression of CD25 is undetectable in GRA6-specific CD8 T cells from chronically infected mice. This is in line with previous work indicating differential requirements for CD25 expression in vivo and in vitro (Wong and Pamer, 2004), and the mechanistic significance of CD25 expression and IL-2 signaling during chronic T. gondii infection requires further study.
STAR★METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Ellen Robey (erobey@berkeley.edu).
Materials availability
This study did not generate new unique reagents.
Data and code availability
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
All original code has been deposited on Zenodo and is publicly available as of the date of publication (see key resources table).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-mouse CD3ε (145-2C11) | BioLegend | Cat #100203, RRID:AB_312660 |
| Anti-mouse CD4 (GK1.5) | BioLegend | Cat #100405, RRID:AB_312690 |
| Anti-mouse CD8b (YTS156.7.7) | BioLegend | Cat #126619, RRID:AB_2563950 |
| Anti-mouse CD11b (M1/70) | BioLegend | Cat #101229, RRID:AB_2129375 |
| Anti-mouse CD11c (N418) | BioLegend | Cat #117313, RRID:AB_492849 |
| Anti-mouse CD25 (PC61) | BioLegend | Cat #102029, RRID:AB_893291 |
| Anti-mouse CD44 (1M7) | ThermoFisher | Cat #25-0441-82, RRID:AB_469623 |
| Anti-mouse CD45.1 (A20) | BioLegend | Cat #110713, RRID:AB_313502 |
| Anti-mouse CD45.2 (104) | BioLegend | Cat #109829, RRID:AB_1186103 |
| Anti-mouse/human B220 (RA3-6B2) | BioLegend | Cat #103229, RRID:AB_492875 |
| Anti-mouse CXCL9 (MIG-2F5.5) | BioXCell | Cat #BE0309, RRID:AB_2736989 |
| Anti-mouse CXCR3 (CXCR3-173) | BioLegend | Cat #126521, RRID:AB_10900974 |
| Anti-mouse CXCR3 (CXCR3-173) | BioXCell | Cat #BE0249, RRID:AB_2687730 |
| Anti-mouse DCIR2 (33D1) | BioLegend | Cat #124914, RRID:AB_1227625 |
| Anti-mouse F4/80 (BM8) | BioLegend | Cat #123120, RRID:AB_893479 |
| Anti-mouse KLRG1 (2F1/KLRG1) | BioLegend | Cat #138415, RRID:AB_2561735 |
| Anti-mouse/human Ki67 (SolA15) | ThermoFisher | Cat #17-5698-82, RRID:AB_2688057 |
| Anti-mouse Ly6G (1A8) | BioLegend | Cat #127624, RRID:AB_10640819 |
| Anti-mouse MHC-II (M5/114.15.2) | BioLegend | Cat #107620, RRID:AB_493527 |
| Anti-mouse NK1.1 (PK136) | BioLegend | Cat #108710, RRID:AB_313397 |
| Anti-mouse/human TCF1 (C63D9) | Cell Signaling | Cat #2203S, RRID:AB_2199302 |
| Anti-mouse/rat XCR1 (ZET) | BioLegend | Cat #148204, RRID:AB_2563843 |
| Polyclonal anti-GFP | ThermoFisher | Cat #A-21311, RRID:AB_221477 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| CAS-Block Histochemical Reagent | ThermoFisher | Cat #008120 |
| Ghost Dye Violet 510 | Tonbo Biosciences | Cat #13-0870-T500 |
| CFSE proliferation dye | ThermoFisher | Cat #C34554 |
| Collagenase D | Sigma-Aldrich | Cat #11088866001 |
| Tissue-Tek O.C.T. compound | Sakura | Product code 4583 |
| Opti-Prep | StemCell Technologies | Cat #07820 |
| GRA6 Peptide (HPGSVNEFDF) | Peptide 2.0 Inc. | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| FoxP3 staining kit | ThermoFisher | Cat #00-5523-00 |
| CD8a+ T cell isolation kit, mouse | Miltenyi Biotec. | Cat #130-104-075 |
| Quick-RNA Microprep Kit | Zymo Research | Cat #R1050 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed data | N/A | GEO: GSE187545 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6J | The Jackson Laboratory | Stock No: 000644 |
| B6.c (B6.C-H2d/bByJ) | The Jackson Laboratory | Stock No: 000359 |
| UBI-GFP (C57BL/6-Tg(UBC-GFP)30Scha/J) | The Jackson Laboratory | Stock No: 004353 |
| CXCR3 KO B6 (B6.129P2-Cxcr3tm1Dgen/J) | The Jackson Laboratory | Stock No: 005796 |
| REX3 B6 | Groom et al., 2012 | N/A |
| TG6 TCR Transgenic | Chu et al., 2016 | N/A |
| T. gondii type II Prugniuad-tomato-OVA | Chtanova et al., 2008 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo | FlowJo LLC | Version 10 |
| DISC (Dynamic in situ cytometry) | Moreau et al., 2012 | N/A |
| ImageJ | (Schneider et al., 2012) | N/A |
| Imaris Software | Bitplane Scientific | N/A |
| Trimmomatic | Bolger et al. (2014) | N/A |
| Bowtie2 | Langmead and Salzberg, 2012 | N/A |
| RSEM | Li and Dewey, 2011 | N/A |
| DESeq2 | Love et al. (2014) | N/A |
| FGSEA | Korotkevich et al., 2016 | N/A |
| Prism (Version 7) | GraphPad Software | N/A |
| RNA-seq analysis code | N/A | Zenodo: https://doi.org/10.5281/zenodo.5794236 |
EXPERIMENTAL MODELS AND SUBJECT DETAILS
Animals
C57BL/6, B6c (B6.C-H2d/bByJ), Ubi-GFP, and CXCR3 KO B6 mice were originally purchased from The Jackson Laboratory. TG6 transgenic mice were generated in our lab as previously described (Chu et al., 2016). REX3 transgenic mice were received from the lab of Andrew Luster. All mice were bred in the UC Berkeley animal facility and were used with the approval of the Animal Care and Use Committee (ACUC) of the University of California. All experiments were performed on B6xB6c H-2b/d F1 mice between the ages of six weeks to six months. For experiments involving the comparison of WT to CXCR3 KO mice, male mice were used to allow for rapid crossing due to the X-linked nature of CXCR3. All other experiments used a roughly equal proportion of male and female mice in infection cohorts.
Microbe strains
For all infections, mice received 105 tachyzoites of the type II Prugniuad-tomato-OVA strain i.p. as previously described (Chu et al., 2016).
METHOD DETAILS
Neonatal chimeras
Neonatal chimeric mice were generated as previously described (Ladi et al., 2008). Briefly, donor bone marrow was isolated and RBC lysed using ACK lysis buffer (0.15M NH4CL, 1mM KHC3, 0.1mM Na2EDTA) for 5 minutes at room temperature. Bone marrow cells were resuspended in PBS, and 106 cells were injected into 5–7 day old recipient mice intraperitoneally. Adult chimeric mice were then infected with T. gondii, and TG6 GFP+ populations were expanded.
Spleen tissue cryosectioning
Spleens from infected mice were removed and cut in half laterally. Spleens were fixed in 4% paraformaldehyde at room temperature in the dark for one hour, washed twice with PBS, then dehydrated in 10% sucrose, 20% sucrose, and 30% sucrose for at least 6 hours each at 4°C. Spleens were imbedded in OCT (Fisher) cut side-down and frozen in a slurry of dry ice and ethanol, then stored at −80°C until sectioning. Spleens were cut into 12-micron sections on a cryostat microtome (Leica Biosystems) and stored at −80°C until used for immunofluorescence. REX3 transgenic reporter spleens were fixed for 6 hours in 4% paraformaldehyde at room temperature instead of 1 hour.
Immunofluorescence
Spleen sections were thawed at room temperature in the dark until dry. Sections were rehydrated with PBS for 10 minutes, fixed with 4% paraformaldehyde for 3 minutes, and permeabilized in 0.5% Triton X- for 15 minutes. Samples were blocked with CAS-Block (ThermoFisher) at 4°C for 1 hour, then stained with antibodies overnight. Samples were stained with antibodies against B220 (RA3–6B2), F4/80 (BM8), GFP (polyclonal), TCF1 (C63D9), Ki67 (SolA15), KLRG1 (2F1), CD11c (N418), XCR1 (ZET), and DCIR2 (33D1). Samples were imaged using a LSM 880 NLO AxioExaminer (Zeiss). Analysis of T cell location was performed using ImageJ and Imaris software (Bitplane Scientific Software). For analysis of intracellular expression by lymphocytes (i.e. TCF1, Ki67) Imaris software was used. In Imaris, GFP + cells in images were identified and the mean fluorescence of TCF1 and Ki67 was reported for cells classified as localized in each splenic region. Dynamic in situ cytometry (DISC) was used to convert data to an FCS format (Moreau et al., 2012), and FlowJo software was used to classify cells as positive or negative for protein expression. For surface staining (KLRG1, XCR1, 33D1) and REX3 CXCL9-RFP/CXCL10-BFP analysis, ImageJ was used. KLRG1 expression on GFP cells was scored by hand, and results were confirmed by an independent and blinded third party. To measure the density of GFP + cells in 33D1+ clusters, Imaris software was used. 33D1-rich regions were manually identified, and the number of GFP+ spots in each region was measured and divided by the area of the 33D1-rich region. The process was repeated to identify the area and number of GFP+ spots for the entire slice, and each 33D1-rich region was then normalized to the density of cells in the corresponding slice.
Flow cytometry
Spleens were dissociated in FACS buffer (0.5% BSA in PBS) to generate single-cell suspensions. Splenocytes were passed through a 70um filter and then RBC lysed using ACK lysis buffer (0.15M NH4CL, 1mM KHC3, 0.1mM Na2EDTA) for 5 minutes at room temperature. Samples were stained with Ghost Dye Violet 510 (Tonbo) for 20 minutes at 4°C, then stained with the following antibodies: CD4 (GK1.5), B220 (RA3–6B2), CD8b (YTS156.7.7), CXCR3 (CXCR3–173), KLRG1 (2F1/KLRG1), Ki67 (SolA15), CD44 (1M7), CD3 (145–2C11), CD11c (N418), MHC-II (M5/114.15.2), CD11b (M1/70), XCR1 (ZET), DCIR2 (33D1), CD25 (PC61), CD45.1 (A20), CD45.2 (104), NK1.1 (PK136), Ly6G (1A8). In some experiments, cells were labeled with CFSE proliferation dye (ThermoFisher) following the manufacturer’s protocol. For intracellular staining, samples were fixed and permeabilized using the eBioscience FoxP3 staining kit (ThermoFisher). Samples were processed using an LSR Fortessa Analyzer (BD) and analyzed using FlowJo software.
Adoptive transfers
For adoptive transfer of naïve CD8+ T cells, spleens were isolated from donor mice and single-cell suspensions were generated. CD8+ T cells were isolated by negative enrichment using a magnetic column (Miltenyi Biotec.) according to manufacturer’s instruction. 5 × 104 naïve CD8+ T cells were transferred into recipient mice intravenously (i.v.) and recipient mice were infected 24 hours later. In experiments where two naïve CD8+ T cell populations were transferred, 2.5 × 104 of each population was injected. For adoptive transfer of TG6-GFP antigen-experienced populations, CD8+ T cells were isolated from infected TG6-GFP chimeras and enriched through magnetic bead selection as described above. Cells were then sorted for GFP-expression and CXCR3xKLRG1 phenotype using a BD influx cell sorter (BD Biosciences) and adoptively transferred into separate mice through i.v. injection. For adoptive transfer of WT and CXCR3 KO TMEM cells, CD8+ T cells were isolated from infected mice and enriched through magnetic bead isolation as described above. TMEM cells were then sorted (CD4- B220- CD45.1+/− KLRG1-) using a BD influx cell sorter and 105 cells were injected i.v. into infected recipient mice.
T cell:dendritic cell co-culture
Dendritic cells were enriched prior to cell sorting using density gradient selection (Bosteels et al., 2018). Briefly, spleens from infected mice were injected and flushed out with collagenase D and DNAse I, cut into small pieces, and incubated at 37°C for 1 hour. Tissues were then dissociated and filtered. Filtered splenocytes were placed over a density column (Opti-Prep) and centrifuged at 400g for 20 minutes with no brake, and the interphase was removed. Cells were stained with Live/Dead and antibodies for CD3, B220, MHC-II, CD11c, XCR1, CD11b, and 33D1. Cells were sorted using an Influx Cell Sorter (BD) through a 100-micron nozzle. Sorted cells were then pulsed with 1uM GRA6 peptide (Peptide2.0 Inc) at 37°C for 1 hour and plated in flat bottom 96-well plates at a density of 5,000 cells per well. In parallel, TG6 donor mice spleens were enriched for CD8+ T cells using a magnetic column (Miltenyi) and stained with antibodies for CD4, B220, CD45.1, CD45.2, KLRG1. TMEM cells were sorted (CD4/B220-, CD45.1+/−, KLRG1-) through an 85-micron nozzle and co-cultured with dendritic cells at a density of 25,000 cells per well. Media was refreshed on day 2 of culture, and cells were harvested and stained on day 3. In some experiments, anti-CXCL9 (MIG-2F5.5, BioXCell) and anti-CXCR3 (CXCR3–173, BioXCell) were added at a concentration of 40ug/mL.
RNA sequencing
Splenocytes were isolated from mice infected with T. gondii for 6 weeks. GRA6-specific TMEM, TINT, and TEFF cells were sorted using a BD FACSAria Fusion by CD44+ GRA6 Tetramer+ and CXCR3xKLRG1 staining. RNA was harvested using a Quick-RNA Microprep Kit from Zymo Research (Cat. No. R1050) according to manufacturer’s instructions. RNA integrity was confirmed via Bioanalyzer and Qbit. RNA was sent to BGI Genomics for library generation and RNA sequencing on an Illumina HiSeq2500/4000 to a depth of 20 million reads.
RNA-seq analysis
Sequencing reads were processed with Trimmomatic (Bolger et al., 2014) to remove adapter sequences and trim low-quality bases. Reads were aligned to the mm10 genome using Bowtie2 (Langmead and Salzberg, 2012) and transcripts were quantified using RSEM (Li and Dewey, 2011). PCA was performed on RNA-seq data following normalization by variance stabilizing transform (VST) with DESeq2 (Love et al., 2014). Differential expression testing was performed using DESeq2, which produced adjusted p-values corrected by the Benjamini-Hochberg procedure. In each test, genes with an adjusted p-value < 0.05 and |log2 fold change| > 1 were considered differentially expressed. Gene set enrichment analysis (GSEA) was performed for each DESeq2 comparison using FGSEA (Korotkevich et al., 2016) with gene sets downloaded from MsigDB (Liberzon et al., 2011). GSEA was also performed on gene sets generated from Utzschneider et al. (2016) and Hudson et al. (2019). To generate these gene sets, published data was filtered to include significantly differentially expressed genes (adjusted p-value < 0.05 and |log2 fold change| > 1) in each relevant comparison. Significantly upregulated and significantly downregulated genes both contributed towards the GSEA enrichment score by accounting for the signed expression (i.e., upregulated genes in the gene set contribute towards the enrichment score when upregulated in the population of interest, while downregulated genes contribute towards the enrichment score when downregulated in the population of interest).
QUANTIFICATION AND STATISTICAL ANALYSIS
Data were analyzed using Prism 7 (GraphPad Software). Statistical significance was determined using either an unpaired or paired t-test. Plots show mean ± SEM. (*p <0.05, **p <0.01, ***p <0.001, ****p <0.0001).
Supplementary Material
Highlights.
TMEM, TINT, and TEFF cells reside in distinct splenic regions during chronic infection
CXCR3 regulates differentiation of TMEM into TINT cells but not their bulk migration
cDC-2s are potent producers of CXCR3 ligands CXCL9/10 found near bridging channels
CXCR3 KO T cells have reduced interaction with cDC-2s and retain a memory phenotype
ACKNOWLEDGMENTS
We thank Andrew Luster for providing the REX3 mouse line. We thank H. Nolla and A. Valero for help with cell sorting, H. Aaron and F. Ives from the UC Berkeley MIC for help with imaging studies, and H. Chu and members of the Robey Lab for helpful comments. BioRender was used to construct the graphical abstract and some portions of the figures. Funding was provided by the National Institutes of Health (RO1AI065537 and AI093132).
Footnotes
DECLARATION OF INTERESTS
The authors declare no competing interests.
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.celrep.2021.110266.
REFERENCES
- Anderson KG, Mayer-Barber K, Sung H, Beura L, James BR, Taylor JJ, Qunaj L, Griffith TS, Vezys V, Barber DL, and Masopust D (2014). Intravascular staining for discrimination of vascular and tissue leukocytes. Nat. Protoc. 9, 209–222. 10.1038/nprot.2014.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arnon TI, and Cyster JG (2014). Blood, sphingosine-1-phosphate and lymphocyte migration dynamics in the spleen. Curr. Top. Microbiol. Immunol. 378, 107–128. 10.1007/978-3-319-05879-5_5. [DOI] [PubMed] [Google Scholar]
- Backer RA, Diener N, and Clausen BE (2019). Langerin+CD8+ dendritic cells in the splenic marginal zone: not so marginal after all. Front. Immunol. 10, 741. 10.3389/fimmu.2019.00741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blanchard N, Gonzalez F, Schaeffer M, Joncker NT, Cheng T, Shastri AJ, Robey EA, and Shastri N (2008). Immunodominant, protective response to the parasite Toxoplasma gondii requires antigen processing in the endoplasmic reticulum. Nat. Immunol. 9, 937–944. 10.1038/ni.1629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bolger AM, Lohse M, and Usadel B (2014). Trimmomatic: a flexible trimmer for Illumina sequence data. Bioinformatics 30, 2114–2120. 10.1093/bioinformatics/btu170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosteels C, Neyt K, Vanheerswynghels M, van Helden MJ, Sichien D, Debeuf N, De Prijck S, Bosteels V, Vandamme N, Martens L, et al. (2020). Inflammatory type 2 cDCs acquire features of cDC1s and macrophages to orchestrate immunity to respiratory virus infection. Immunity 52, 1039–1056.e9. 10.1016/j.immuni.2020.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bosteels C, Lambrecht BN, and Hammad H (2018). Isolation of conventional murine lung dendritic cell subsets. Curr. Protoc. Immunol. 2018, 3.7B.1–3.7B.16. 10.1002/cpim.39. [DOI] [PubMed] [Google Scholar]
- Brown CC, Gudjonson H, Pritykin Y, Deep D, Lavallée VP, Mendoza A, Fromme R, Mazutis L, Ariyan C, Leslie C, et al. (2019). Transcriptional basis of mouse and human dendritic cell heterogeneity. Cell 179, 846–863.e24. 10.1016/j.cell.2019.09.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calabro S, Liu D, Gallman A, Nascimento MS, Yu Z, Zhang TT, Chen P, Zhang B, Xu L, Gowthaman U, et al. (2016). Differential intrasplenic migration of dendritic cell subsets tailors adaptive immunity. Cell Rep. 16, 2472–2485. 10.1016/j.celrep.2016.07.076. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chang JT, Palanivel VR, Kinjyo I, Schambach F, Intlekofer AM, Banerjee A, Longworth SA, Vinup KE, Mrass P, Oliaro J, et al. (2007). Asymmetric T lymphocyte division in the initiation of adaptive immune responses. Science 315, 1687–1691. 10.1126/science.1139393. [DOI] [PubMed] [Google Scholar]
- Chang JT, Wherry EJ, and Goldrath AW (2014). Molecular regulation of effector and memory T cell differentiation. Nat. Rev. Immunol. 15, 1104–1115. 10.1038/ni.3031.1104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chauveau A, Pirgova G, Cheng HW, De Martin A, Zhou FY, Wideman S, Rittscher J, Ludewig B, and Arnon TI (2020). Visualization of T Cell migration in the spleen reveals a network of perivascular pathways that guide entry into T zones. Immunity 52, 794–807.e7. 10.1016/j.immuni.2020.03.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow MT, Ozga AJ, Servis RL, Frederick DT, Lo JA, Fisher DE, Freeman GJ, Boland GM, and Luster AD (2019). Intratumoral activity of the CXCR3 chemokine system is required for the efficacy of anti-PD-1 therapy. Immunity 50, 1498–1512.e5. 10.1016/j.immuni.2019.04.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chtanova T, Schaeffer M, Han S-J, van Dooren GG, Nollmann M, Herzmark P, Chan SW, Satija H, Camfield K, Aaron H, et al. (2008). Dynamics of neutrophil migration in lymph nodes during infection. Immunity 29, 487–496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chu HH, Chan SW, Gosling JP, Blanchard N, Tsitsiklis A, Lythe G, Shastri N, Molina-París C, and Robey EA (2016). Continuous effector CD8+ T cell production in a controlled persistent infection is sustained by a proliferative intermediate population. Immunity 45, 159–171. 10.1016/j.immuni.2016.06.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dar W, and Knechtle S (2007). CXCR3-mediated T-cell chemotaxis involves ZAP-70 and is regulated by signalling through the T-cell receptor. Immunology 120, 467–485. 10.1111/j.1365-2567.2006.02534.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Duckworth BC, Lafouresse F, Wimmer VC, Broomfield BJ, Dalit L, Alexandre YO, Sheikh AA, Qin RZ, Alvarado C, Mielke LA, et al. (2021). Effector and stem-like memory cell fates are imprinted in distinct lymph node niches directed by CXCR3 ligands. Nat. Immunol. 22, 434–448. 10.1038/s41590-021-00878-5. [DOI] [PubMed] [Google Scholar]
- Eisenbarth SC (2019). Dendritic cell subsets in T cell programming: location dictates function. Nat. Rev. Immunol. 19, 89–103. 10.1038/s41577-018-0088-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Feliu V, Vasseur V, Grover HS, Chu HH, Brown MJ, Wang J, Boyle JP, Robey EA, Shastri N, and Blanchard N (2013). Location of the CD8 T cell epitope within the antigenic precursor determines immunogenicity and protection against the Toxoplasma gondii parasite. PLoS Pathog. 9, e1003449. 10.1371/journal.ppat.1003449. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Förster R, Schubel A, Breitfeld D, Kremmer E, Renner-Müller I, Wolf E, and Lipp M (1999). CCR7 coordinates the primary immune response by establishing functional microenvironments in secondary lymphoid organs. Cell 99, 23–33. 10.1016/S0092-8674(00)80059-8. [DOI] [PubMed] [Google Scholar]
- Gabriel SS, Tsui C, Chisanga D, Weber F, Llano-León M, Gubser PM, Bartholin L, Souza-Fonseca-Guimaraes F, Huntington ND, Shi W, et al. (2021). Transforming growth factor- β -regulated mTOR activity preserves cellular metabolism to maintain long-term T cell responses in chronic infection. Immunity 54, 1698–1714.e5. 10.1016/j.immuni.2021.06.007. [DOI] [PubMed] [Google Scholar]
- Goldberg MF, Roeske EK, Ward LN, Pengo T, Dileepan T, Kotov DI, and Jenkins MK (2018). Salmonella persist in activated macrophages in T cell-sparse granulomas but are contained by surrounding CXCR3 ligand-positioned Th1 cells. Immunity 49, 1090–1102.e7. 10.1016/j.immuni.2018.10.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gordon CL, Lee LN, Swadling L, Hutchings C, Zinser M, Highton AJ, Capone S, Folgori A, Barnes E, and Klenerman P (2018). Induction and maintenance of CX3CR1-intermediate peripheral memory CD8+ T cells by persistent viruses and vaccines. Cell Rep. 23, 768–782. 10.1016/j.celrep.2018.03.074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom JR, Richmond J, Murooka TT, Sorensen EW, Sung JH, Bankert K, von Andrian UH, Moon JJ, Mempel TR, and Luster AD (2012). CXCR3 chemokine receptor-ligand interactions in the lymph node optimize CD4+ T helper 1 cell differentiation. Immunity 37, 1091–1103. 10.1016/j.immuni.2012.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Groom JR, and Luster AD (2011). CXCR3 ligands: redundant, collaborative and antagonistic functions. Immunol. Cell Biol. 89, 207–215. 10.1038/icb.2010.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Den Haan JMM, and Bevan MJ (2002). Constitutive versus activation-dependent cross-presentation of immune complexes by CD8+ and CD8− dendritic cells in vivo. J. Exp. Med. 196, 817–827. 10.1084/jem.20020295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hu JK, Kagari T, Clingan JM, and Matloubian M (2011). Expression of chemokine receptor CXCR3 on T cells affects the balance between effector and memory CD8 T-cell generation. Proc. Natl. Acad. Sci. U S A 108, E118–E127. 10.1073/pnas.1101881108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudson WH, Gensheimer J, Hashimoto M, Wieland A, Valanparambil RM, Li P, Lin JX, Konieczny BT, Im SJ, Freeman GJ, et al. (2019). Proliferating transitory T cells with an effector-like transcriptional signature emerge from PD-1 + stem-like CD8 + T Cells during Chronic infection. Immunity, 1–16. 10.1016/j.immuni.2019.11.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Im SJ, Hashimoto M, Gerner MY, Lee J, Kissick HT, Burger MC, Shan Q, Hale JS, Lee J, Nasti TH, et al. (2016). Defining CD8+ T cells that provide the proliferative burst after PD-1 therapy. Nature 537, 417–421. 10.1038/nature19330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalia V, Sarkar S, Subramaniam S, Haining WN, Smith KA, and Ahmed R (2010). Prolonged interleukin-2Rα expression on virus-specific CD8+ T cells favors terminal-effector differentiation in vivo. Immunity 32, 91–103. 10.1016/j.immuni.2009.11.010. [DOI] [PubMed] [Google Scholar]
- Kefalakes H, Koh C, Sidney J, Amanakis G, Sette A, Heller T, and Rehermann B (2019). Hepatitis D virus-specific CD8+ T cells have a memory-like phenotype Associated with viral immune escape in patients with chronic hepatitis D virus infection. Gastroenterology 156, 1805–1819.e9. 10.1053/j.gastro.2019.01.035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khanna KM, McNamara JT, and Lefrançois L (2007). In situ imaging of the endogenous CD8 T cell response to infection. Science 318, 116–120. 10.1126/science.1146291. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kohlmeier JE, Reiley WW, Perona-Wright G, Freeman ML, Yager EJ, Connor LM, Brincks EL, Cookenham T, Roberts AD, Burkum CE, et al. (2011). Inflammatory chemokine receptors regulate CD8 + T cell contraction and memory generation following infection. J. Exp. Med. 208, 1621–1634. 10.1084/jem.20102110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Korotkevich G, Sukhov V, Budin N, Shpak B, Artyomov MN, and Sergushichev A (2016). Fast gene set enrichment analysis. bioRxiv, 1–29. 10.1101/060012. [DOI] [Google Scholar]
- Kotov DI, Mitchell JS, Pengo T, Ruedl C, Way SS, Langlois RA, Fife BT, and Jenkins MK (2019). TCR affinity biases Th cell differentiation by regulating CD25, Eef1e1, and Gbp2. J. Immunol. 202, 2535–2545. 10.4049/jimmunol.1801609. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurachi M, Kurachi J, Suenaga F, Tsukui T, Abe J, Ueha S, Tomura M, Sugihara K, Takamura S, Kakimi K, and Matsushima K (2011). Chemokine receptor CXCR3 facilitates CD8(+) T cell differentiation into short-lived effector cells leading to memory degeneration. J. Exp. Med. 208, 1605–1620. 10.1084/jem.20102101. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ladi E, Herzmark P, and Robey E (2008). In situ imaging of the mouse thymus using 2-photon microscopy. J. Vis. Exp. 166, 2008. 10.3791/652. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. 10.1038/nmeth.1923. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-Seq data with or without a reference genome. Bioinformatics, 21–40. 10.1186/1471-2105-12-323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li J, Lu E, Yi T, and Cyster JG (2016). EBI2 augments Tfh cell fate by promoting interaction with IL-2-quenching dendritic cells. Nature 533, 110–114. 10.1038/nature17947. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liberzon A, Subramanian A, Pinchback R, Thorvaldsdóttir H, Tamayo P, and Mesirov JP (2011). Molecular signatures database (MSigDB) 3.0. Bioinformatics 27, 1739–1740. 10.1093/bioinformatics/btr260. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin W-HW, Nish SA, Yen B, Chen YH, Adams WC, Kratchmarov R, Rothman NJ, Bhandoola A, Xue HH, and Reiner SL (2015). CD8+ T Lymphocyte Self-renewal during Effector Cell Determination. Cell Rep. 344, 1173–1178. 10.1126/science.1249098.Sleep. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 15, 1–21. 10.1186/s13059-014-0550-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lutes LK, Steier Z, McIntyre LL, Pandey S, Kaminski J, Hoover AR, Ariotti S, Streets A, Yosef N, and Robey EA (2021). T cell self-reactivity during thymic development dictates the timing of positive selection. bioRxiv. 10.1101/2021.01.18.427079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mackay LK, Braun A, Macleod BL, Collins N, Tebartz C, Bedoui S, Carbone FR, and Gebhardt T (2015). Cutting edge: CD69 interference with sphingosine-1-phosphate receptor function regulates peripheral T cell retention. J. Immunol. 194, 2059–2063. 10.4049/jimmunol.1402256. [DOI] [PubMed] [Google Scholar]
- Maurice NJ, McElrath MJ, Andersen-Nissen E, Frahm N, and Prlic M (2019). CXCR3 enables recruitment and site-specific bystander activation of memory CD8+ T cells. Nat. Commun. 10, 1–15. 10.1038/s41467-019-12980-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moreau HD, Lemaître F, Terriac E, Azar G, Piel M, Lennon-Dumenil AM, and Bousso P (2012). Dynamic in situ cytometry uncovers t cell receptor signaling during immunological synapses and kinapses in vivo. Immunity 37, 351–363. 10.1016/j.immuni.2012.05.014. [DOI] [PubMed] [Google Scholar]
- Newton P, O’Boyle G, Jenkins Y, Ali S, and Kirby JA (2009). T cell extravasation: demonstration of synergy between activation of CXCR3 and the T cell receptor. Mol. Immunol. 47, 485–492. 10.1016/j.molimm.2009.08.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Opejin A, Surnov A, Misulovin Z, Pherson M, Gross C, Iberg CA, Fallahee I, Bourque J, Dorsett D, and Hawiger D (2020). A two-step process of effector programming governs CD4+ T cell fate determination induced by antigenic activation in the steady state. Cell Rep. 33, 108424. 10.1016/j.celrep.2020.108424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pipkin ME, Sacks JA, Cruz-Guilloty F, Lichtenheld MG, Bevan MJ, and Rao A (2010). Interleukin-2 and inflammation induce distinct transcriptional programs that promote the differentiation of effector cytolytic T cells. Immunity 32, 79–90. 10.1016/j.immuni.2009.11.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rutishauser RL, Deguit CDT, Hiatt J, Blaeschke F, Roth TL, Wang L, Raymond KA, Starke CE, Mudd JC, Chen W, et al. (2021). TCF-1 regulates HIV-specific CD8+ T cell expansion capacity. JCI Insight 6, 1–16. 10.1172/jci.insight.136648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salvioni A, Belloy M, Lebourg A, Bassot E, Cantaloube-Ferrieu V, Vasseur V, Blanié S, Liblau RS, Suberbielle E, Robey EA, and Blanchard N (2019). Robust control of a brain-persisting parasite through MHC I presentation by infected neurons. Cell Rep. 27, 3254–3268.e8. 10.1016/j.celrep.2019.05.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sansom DM, Manzotti CN, and Zheng Y (2003). What’s the difference between CD80 and CD86? Trends Immunol. 24, 313–318. 10.1016/S1471-4906(03)00111-X. [DOI] [PubMed] [Google Scholar]
- Schneider CA, Rasband WS, and Eliceiri KW (2012). NIH Image to ImageJ: 25 years of image analysis. Nature Methods 9, 671–675. 10.1038/nmeth.2089. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seo YJ, Jothikumar P, Suthar MS, Zhu C, and Grakoui A (2016). Local cellular and cytokine cues in the spleen regulate in situ T cell receptor affinity, function, and fate of CD8+ T cells. Immunity 45, 988–998. 10.1016/j.immuni.2016.10.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schenkel JM, Herbst RH, Canner D, Li A, Hillman M, Shanahan SL, Gibbons G, Smith OC, Kim JY, Westcott P, et al. (2021). Conventional type I dendritic cells maintain a reservoir of proliferative tumor-antigen specific TCF-1+ CD8+ T cells in tumor-draining lymph nodes. Immunity 54, 2338–2353.e6. 10.1016/j.immuni.2021.08.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shah S, Grotenbreg GM, Rivera A, and Yap GS (2015). An extrafollicular pathway for the generation of effector CD8+T cells driven by the proinflammatory cytokine, IL-12. Elife 4, 1–21. 10.7554/eLife.09017. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shin KS, Jeon I, Kim BS, Kim IK, Park YJ, Koh CH, Song B, Lee JM, Lim J, Bae EA, et al. (2019). Monocyte-derived dendritic cells dictate the memory differentiation of CD8+ T cells during acute infection. Front. Immunol. 10, 5–7. 10.3389/fimmu.2019.01887. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith CJ, Turula H, and Snyder CM (2014). Systemic hematogenous maintenance memory inflation by MCMV infection. PLos Pathog. 10. 10.1371/journal.ppat.1004233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sung JH, Zhang H, Moseman EA, Alvarez D, Iannacone M, Henrickson SE, de la Torre JC, Groom JR, Luster AD, and von Andrian UH (2012). Chemokine guidance of central memory T cells is critical for antiviral recall responses in lymph nodes. Cell 150, 1249–1263. 10.1016/j.cell.2012.08.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thomas IJ, Petrich de Marquesini LG, Ravanan R, Smith RM, Guerder S, Flavell RA, Wraith DC, Wen L, and Wong FS (2007). CD86 has sustained costimulatory effects on CD8 T cells. J. Immunol. 179, 5936–5946. 10.4049/jimmunol.179.9.5936. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tsitsiklis A, Bangs DJ, Lutes LK, Chan SW, Geiger KM, Modzelewski AJ, Labarta-Bajo L, Wang Y, Zuniga EI, Dai S, and Robey EA (2020). An unusual MHC molecule generates protective CD8+ T cell responses to chronic infection. Front. Immunol. 11, 1–13. 10.3389/fimmu.2020.01464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Utzschneider DT, Charmoy M, Chennupati V, Pousse L, Ferreira DP, Calderon-Copete S, Danilo M, Alfei F, Hofmann M, Wieland D, et al. (2016). T cell factor 1-expressing memory-like CD8+ T cells sustain the immune response to chronic viral infections. Immunity 45, 415–427. https://doi. org/10.1016/j.immuni.2016.07.021. [DOI] [PubMed] [Google Scholar]
- Welten SPM, Yermanos A, Baumann NS, Wagen F, Oetiker N, Sandu I, Pedrioli A, Oduro JD, Reddy ST, Cicin-Sain L, et al. (2020). Tcf1+ cells are required to maintain the inflationary T cell pool upon MCMV infection. Nat. Commun. 11, 1–14. 10.1038/s41467-020-16219-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Weng Y, Siciliano SJ, Waldburger KE, Sirotina-Meisher A, Staruch MJ, Daugherty BL, Gould SL, Springer MS, and DeMartino JA (1998). Binding and functional properties of recombinant and endogenous CXCR3 chemokine receptors. J. Biol. Chem. 273, 18288–18291. 10.1074/jbc.273.29.18288. [DOI] [PubMed] [Google Scholar]
- Wieland D, Kemming J, Schuch A, Emmerich F, Knolle P, Neumann-Haefelin C, Held W, Zehn D, Hofmann M, and Thimme R (2017). TCF1+ hepatitis C virus-specific CD8+ T cells are maintained after cessation of chronic antigen stimulation. Nat. Commun. 8, 1–13. 10.1038/ncomms15050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Williams MA, and Bevan MJ (2007). Effector and memory CTL differentiation. Annu. Rev. Immunol. 25, 171–192. 10.1146/annurev.immunol.25.022106.141548. [DOI] [PubMed] [Google Scholar]
- Wong P, and Pamer EG (2004). Disparate in vitro and in vivo requirements for IL-2 during antigen-independent CD8 T cell expansion. J. Immunol. 172, 2171–2176. 10.4049/jimmunol.172.4.2171. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq data have been deposited at GEO and are publicly available as of the date of publication. Accession numbers are listed in the key resources table.
All original code has been deposited on Zenodo and is publicly available as of the date of publication (see key resources table).
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
KEY RESOURCES TABLE.
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
|
| ||
| Antibodies | ||
|
| ||
| Anti-mouse CD3ε (145-2C11) | BioLegend | Cat #100203, RRID:AB_312660 |
| Anti-mouse CD4 (GK1.5) | BioLegend | Cat #100405, RRID:AB_312690 |
| Anti-mouse CD8b (YTS156.7.7) | BioLegend | Cat #126619, RRID:AB_2563950 |
| Anti-mouse CD11b (M1/70) | BioLegend | Cat #101229, RRID:AB_2129375 |
| Anti-mouse CD11c (N418) | BioLegend | Cat #117313, RRID:AB_492849 |
| Anti-mouse CD25 (PC61) | BioLegend | Cat #102029, RRID:AB_893291 |
| Anti-mouse CD44 (1M7) | ThermoFisher | Cat #25-0441-82, RRID:AB_469623 |
| Anti-mouse CD45.1 (A20) | BioLegend | Cat #110713, RRID:AB_313502 |
| Anti-mouse CD45.2 (104) | BioLegend | Cat #109829, RRID:AB_1186103 |
| Anti-mouse/human B220 (RA3-6B2) | BioLegend | Cat #103229, RRID:AB_492875 |
| Anti-mouse CXCL9 (MIG-2F5.5) | BioXCell | Cat #BE0309, RRID:AB_2736989 |
| Anti-mouse CXCR3 (CXCR3-173) | BioLegend | Cat #126521, RRID:AB_10900974 |
| Anti-mouse CXCR3 (CXCR3-173) | BioXCell | Cat #BE0249, RRID:AB_2687730 |
| Anti-mouse DCIR2 (33D1) | BioLegend | Cat #124914, RRID:AB_1227625 |
| Anti-mouse F4/80 (BM8) | BioLegend | Cat #123120, RRID:AB_893479 |
| Anti-mouse KLRG1 (2F1/KLRG1) | BioLegend | Cat #138415, RRID:AB_2561735 |
| Anti-mouse/human Ki67 (SolA15) | ThermoFisher | Cat #17-5698-82, RRID:AB_2688057 |
| Anti-mouse Ly6G (1A8) | BioLegend | Cat #127624, RRID:AB_10640819 |
| Anti-mouse MHC-II (M5/114.15.2) | BioLegend | Cat #107620, RRID:AB_493527 |
| Anti-mouse NK1.1 (PK136) | BioLegend | Cat #108710, RRID:AB_313397 |
| Anti-mouse/human TCF1 (C63D9) | Cell Signaling | Cat #2203S, RRID:AB_2199302 |
| Anti-mouse/rat XCR1 (ZET) | BioLegend | Cat #148204, RRID:AB_2563843 |
| Polyclonal anti-GFP | ThermoFisher | Cat #A-21311, RRID:AB_221477 |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| CAS-Block Histochemical Reagent | ThermoFisher | Cat #008120 |
| Ghost Dye Violet 510 | Tonbo Biosciences | Cat #13-0870-T500 |
| CFSE proliferation dye | ThermoFisher | Cat #C34554 |
| Collagenase D | Sigma-Aldrich | Cat #11088866001 |
| Tissue-Tek O.C.T. compound | Sakura | Product code 4583 |
| Opti-Prep | StemCell Technologies | Cat #07820 |
| GRA6 Peptide (HPGSVNEFDF) | Peptide 2.0 Inc. | N/A |
|
| ||
| Critical commercial assays | ||
|
| ||
| FoxP3 staining kit | ThermoFisher | Cat #00-5523-00 |
| CD8a+ T cell isolation kit, mouse | Miltenyi Biotec. | Cat #130-104-075 |
| Quick-RNA Microprep Kit | Zymo Research | Cat #R1050 |
|
| ||
| Deposited data | ||
|
| ||
| Raw and analyzed data | N/A | GEO: GSE187545 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| C57BL/6J | The Jackson Laboratory | Stock No: 000644 |
| B6.c (B6.C-H2d/bByJ) | The Jackson Laboratory | Stock No: 000359 |
| UBI-GFP (C57BL/6-Tg(UBC-GFP)30Scha/J) | The Jackson Laboratory | Stock No: 004353 |
| CXCR3 KO B6 (B6.129P2-Cxcr3tm1Dgen/J) | The Jackson Laboratory | Stock No: 005796 |
| REX3 B6 | Groom et al., 2012 | N/A |
| TG6 TCR Transgenic | Chu et al., 2016 | N/A |
| T. gondii type II Prugniuad-tomato-OVA | Chtanova et al., 2008 | N/A |
|
| ||
| Software and algorithms | ||
|
| ||
| FlowJo | FlowJo LLC | Version 10 |
| DISC (Dynamic in situ cytometry) | Moreau et al., 2012 | N/A |
| ImageJ | (Schneider et al., 2012) | N/A |
| Imaris Software | Bitplane Scientific | N/A |
| Trimmomatic | Bolger et al. (2014) | N/A |
| Bowtie2 | Langmead and Salzberg, 2012 | N/A |
| RSEM | Li and Dewey, 2011 | N/A |
| DESeq2 | Love et al. (2014) | N/A |
| FGSEA | Korotkevich et al., 2016 | N/A |
| Prism (Version 7) | GraphPad Software | N/A |
| RNA-seq analysis code | N/A | Zenodo: https://doi.org/10.5281/zenodo.5794236 |